
Abstract
Introduzione
Lo stress è noto da tempo per essere un fattore precipitante per l’abuso di droghe che creano dipendenza. Modelli animali hanno dimostrato che i fattori di stress acuti e ripetuti possono aumentare l’assunzione di sostanze che creano dipendenza (Piazza et al., 1990; Ramsey e Van Ree, 1993; Goeders e Guerin, 1994; Shaham e Stewart, 1994; Haney et al., 1995), e che lo stress acuto può ripristinare la ricerca di droghe in animali che hanno subito un addestramento all’estinzione (Shaham et al., 1994, 1995; Conrad et al., 2010; Mantsch et al., 2016). Negli ultimi anni, i neuroni dopaminergici del VTA sono emersi come un luogo significativo per gli effetti sovrapposti delle droghe da abuso e da stress (Polter e Kauer, 2014). Gli input sinaptici, modellando l’attività di questi neuroni, sono pronti a svolgere un ruolo importante nella ricerca di droghe. Sia lo stress acuto che l’esposizione alle droghe di abuso inducono un potenziamento concomitante delle sinapsi eccitatorie e la perdita del potenziamento a lungo termine delle sinapsi inibitorie (Ungless et al., 2001; Saal et al., 2003; Kauer e Malenka, 2007; Nugent et al., 2007; Chen et al., 2008; Niehaus et al., 2010; Polter e Kauer, 2014). La comprensione di come queste sinapsi siano alterate dallo stress fornirà informazioni chiave sulla ricerca di farmaci indotti dallo stress e fornirà obiettivi per il trattamento dei disturbi da uso di sostanze.
Un importante mediatore dei cambiamenti indotti dallo stress nelle sinapsi VTA inibitorie è il sistema del recettore degli oppioidi dinorfina/kappa (κOR). κOR e il loro ligando endogeno, la dinorfina, si trovano in tutto il cervello e sono stati altamente associati con esperienze stressanti, avverse e disforiche (Bruchas et al., 2010; Wee e Koob, 2010; Van’t Veer e Carlezon, 2013; Crowley e Kash, 2015). All’interno del VTA, gli κOR hanno una serie di effetti fisiologici. Gli κOR riducono la velocità di combustione dei neuroni della dopamina attraverso l’attivazione dei canali GIRK (Margolis et al., 2003, 2006), inibiscono la trasmissione sinaptica eccitatoria sui neuroni dopaminergici e non dopaminergici VTA (Margolis et al., 2005), riducono la trasmissione sinaptica inibitoria in un sottoinsieme di neuroni dopaminergici (Ford et al., 2006) e inibiscono gli IPSC dopaminergici somatodendritici (Ford et al., 2007). I VTA κORs possono anche controllare le interazioni tra stress e ricompensa. Il nostro lavoro precedente ha identificato una forma di plasticità sinaptica sensibile allo stress a sinapsi inibitorie sui neuroni dopaminergici VTA (LTPGABA; Nugent et al., 2007, 2009; Niehaus et al., 2010). LTPGABA è indotta attraverso l’attivazione dell’ossido nitrico sintasi nel neurone della dopamina, che porta al rilascio di ossido nitrico (NO) e al miglioramento del rilascio di GABA attraverso la segnalazione cGMP (Nugent et al., 2007, 2009).
Più recentemente, abbiamo dimostrato che lo stress acuto blocca l’LTPGABA attraverso l’attivazione di κOR, e che impedendo questa attivazione attraverso la somministrazione intra-VTA dell’antagonista κOR, nor-binaltorfimina (norBNI), impedisce il ripristino della ricerca di cocaina indotto dallo stress (Graziane et al., 2013). È sorprendente che una singola esposizione allo stress porti a una perdita di LTPGABA che dura almeno cinque giorni ed è mediata dall’attivazione persistente di VTA κORs (Polter et al., 2014). Abbiamo anche dimostrato che il trattamento con l’antagonista κOR dopo lo stress può salvare la reintegrazione indotta dallo stress. Questi studi evidenziano l’importanza della regolazione κOR-mediata dell’LTP presso GABAergic sinapsi nella ricerca di farmaci indotti dallo stress e sottolineano la necessità di comprendere meglio il meccanismo di questa regolazione unica e persistente.
Nel presente studio, abbiamo ora identificato il meccanismo attraverso il quale l’attivazione dell’κOR e la soppressione dell’LTPGABA nel VTA viene mantenuta per più giorni dopo un grave e acuto fattore di stress. Presentiamo la prova che lo stress blocca l’LTPGABA inducendo l’attivazione costitutiva di κORs alle sinapsi inibitorie del VTA piuttosto che attraverso persistenti aumenti del rilascio di dinorfina. Questa attività costitutiva è probabile che venga innescata inizialmente attraverso la segnalazione attraverso la dinorfina ligando endogena, ma poi viene mantenuta in modo persistente indipendentemente dal rilascio di dinorfina. In parallelo, troviamo che la persistente ricerca di droga indotta da una singola esposizione a stress acuto dipende anche dall’attività costitutiva dell’κORs. I nostri risultati rivelano un nuovo meccanismo di regolazione della funzione κOR che dipende dall’esperienza e sottolineano il ruolo essenziale dell’κOR nel mediare i cambiamenti indotti dallo stress nella plasticità sinaptica e nel comportamento di ricerca di farmaci.
Risultati
JNK-dipendente salvataggio di LTPGABA da norBNI acuta
Come precedentemente mostrato, l’applicazione a bagno del donatore di ossido nitrico SNAP potenzia le sinapsi GABAergiche sui neuroni della dopamina nel VTA, in modo simile alla stimolazione ad alta frequenza delle afferenze VTA; questo potenziamento è bloccato da una singola esposizione a più farmaci di abuso o stress acuto da nuoto in acqua fredda (LTPGABA; Nugent et al., 2007; Niehaus et al., 2010; Graziane et al., 2013; Polter et al., 2014; Figura 1A-B). I nostri studi recenti indicano che il blocco di κORs con norNBI previene e inverte gli effetti dello stress acuto su LTPGABA, anche se somministrato diversi giorni dopo lo stress (Graziane et al., 2013; Polter et al., 2014). Abbiamo quindi indagato se l’attivazione persistente e indotta da stress di κORs possa essere rilevata ex vivo nella sezione del mesencefalo. Abbiamo sottoposto i ratti a stress acuto di nuoto forzato in acqua fredda e preparato fette di mesencefalo 24 ore dopo. Se dopo lo stress, κORs nel VTA sono persistentemente segnalazione in vitro, abbiamo ragionato che il norBNI applicato al bagno potrebbe essere utilizzato per salvare SNAP-indotta LTPGABA. L’applicazione del bagno di norBNI (100 nM) ci ha infatti permesso di ottenere LTPGABA NO-dipendente in fette da animali stressati (Figura 1E), indicando che l’attività indotta da stress di κORs nel VTA persiste attraverso la preparazione di fette di cervello e il recupero. Sembrava improbabile che sufficiente dinorfina endogena potrebbe essere rilasciato tonicamente dalle fette di cervello denervato per mantenere un blocco di LTPGABA in vitro. Abbiamo quindi cercato di stabilire il meccanismo con cui norBNI ha salvato questa plasticità. Oltre a competere con gli agonisti presso il sito di legame dell’κOR, norBNI agisce come un agonista inverso o collaterale, e le sue interazioni con l’κOR possono inibire in modo non competitivo l’ulteriore attività dell’κOR attraverso l’attivazione della cascata di segnalazione JNK (Bruchas et al., 2007; Melief et al., 2010, 2011). Abbiamo ipotizzato che il salvataggio di LTPGABA da parte di norBNI possa avvenire anche in modo non competitivo attraverso la segnalazione JNK (Figura 1C). Le fette sono state trattate con l’inibitore JNK SP600125 (20 µM) per 10 minuti prima dell’applicazione del bagno di norBNI (Figura 1D). In contrasto con il robusto potenziamento indotta da SNAP osservato in fette trattate con norBNI da solo, abbiamo trovato che LTPGABA è rimasto bloccato in fette pretrattate con SP600125 (Figura 1F-H). È importante notare che l’applicazione del bagno di SP600125 non ha interferito con l’espressione di LTPGABA nelle fette provenienti da animali ingenui o la perdita di LTPGABA nelle fette provenienti da animali stressati (Figura 1-figure supplement 1A-B). Pertanto, l’attività di JNK non ha alcun ruolo nell’induzione di LTPGABA o nel blocco di questa plasticità da parte di κORs, ma è necessaria per norBNI per salvare LTPGABA dopo lo stress.10.7554/eLife.23785.003Figure 1.norBNI salva LTPGABA attraverso l’attivazione di JNK.(A) Dati riassuntivi che mostrano il blocco di LTPGABA dopo lo stress. (B) Confronto della grandezza di LTPGABA10-15 min dopo l’applicazione SNAP. (Ampiezze IPSC, controllo: 140 ± 5% dei valori di base, n = 13; stress: 94 ± 11% dei valori di base, n = 6; t-test non accoppiato, *p=0,0005. (C) Schema dell’inibizione competitiva e non competitiva della segnalazione κOR da parte della norBNI. (D) Progettazione sperimentale. (E) Rappresentante singolo esperimento che mostra che l’applicazione bagno di norBNI (100 nM) salva LTPGABA in una fetta preparata 24 ore dopo lo stress. (F) Esperimento rappresentativo singolo esperimento da una fetta preparata 24 ore dopo lo stress che mostra che norBNI non salva LTPGABA in presenza dell’inibitore JNK SP600125 (20 µM). (G) Dati di sintesi da entrambi i gruppi. (H) Confronto della grandezza di LTPGABA10-15 min dopo l’applicazione SNAP. (Ampiezze IPSC, norBNI solo: 139 ± 7% dei valori di base, n = 6; norBNI+SP600125: 106 ± 9% dei valori di base, n = 11; t-test non accoppiato, *p=0,029). Inserti per questo e per tutte le cifre: IPSC prima (traccia nera, controllo) e 15 minuti dopo l’applicazione del farmaco (traccia rossa, SNAP, 400 µM). Barre di scala: 20 ms, 100 pA. Gli inserti sono medie di 12 IPSC. DOI:http://dx.doi.org/10.7554/eLife.23785.00310.7554/eLife.23785.004Figure supplemento a 1 cifra 1.L’inibizione di JNK non influisce su LTPGABA o il suo blocco da stress in assenza di norBNI.(A) Dati riassuntivi che mostrano che LTPGABA è espresso in fette da animali ingenui in presenza dell’inibitore JNK SP600125 (20 µM). Magnitudo media di LTPGABA10-15 min dopo SNAP = 144 ± 13% dei valori di base, n = 5; un campione t-test, p=0,0257. (B) Dati di sintesi che mostrano che LTPGABA rimane bloccato in fette di animali stressati in presenza dell’inibitore JNK SP600125 (20 µM). Magnitudo media di LTPGABA10-15 min dopo SNAP = 111 ± 11% dei valori di base, n = 6; un campione t-test, p=0.38.DOI:http://dx.doi.org/10.7554/eLife.23785.004
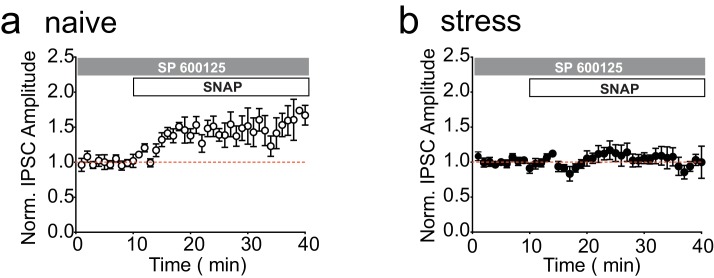
Figura 1-figura supplemento 1.norBNI salva LTPGABA attraverso l’attivazione di JNK.l’inibizione di JNK non influenza LTPGABA o il suo blocco da stress in assenza di norBNI.(A) Dati di sintesi che mostrano il blocco di LTPGABA dopo lo stress. (B) Confronto della grandezza di LTPGABA10-15 min dopo l’applicazione SNAP. (Ampiezze IPSC, controllo: 140 ± 5% dei valori di base, n = 13; stress: 94 ± 11% dei valori di base, n = 6; t-test non accoppiato, *p=0,0005. (C) Schema dell’inibizione competitiva e non competitiva della segnalazione κOR da parte della norBNI. (D) Progettazione sperimentale. (E) Rappresentante singolo esperimento che mostra che l’applicazione bagno di norBNI (100 nM) salva LTPGABA in una fetta preparata 24 ore dopo lo stress. (F) Esperimento rappresentativo singolo esperimento da una fetta preparata 24 ore dopo lo stress che mostra che norBNI non salva LTPGABA in presenza dell’inibitore JNK SP600125 (20 µM). (G) Dati di sintesi da entrambi i gruppi. (H) Confronto della grandezza di LTPGABA10-15 min dopo l’applicazione SNAP. (Ampiezze IPSC, norBNI solo: 139 ± 7% dei valori di base, n = 6; norBNI+SP600125: 106 ± 9% dei valori di base, n = 11; t-test non accoppiato, *p=0,029). Inserti per questo e per tutte le cifre: IPSC prima (traccia nera, controllo) e 15 minuti dopo l’applicazione del farmaco (traccia rossa, SNAP, 400 µM). Barre di scala: 20 ms, 100 pA. Gli inserti sono medie di 12 IPSC.DOI:
http://dx.doi.org/10.7554/eLife.23785.003(A) Dati riassuntivi che mostrano che l’LTPGABA è espresso in fette di animali ingenui in presenza dell’inibitore JNK SP600125 (20 µM). Magnitudo media di LTPGABA10-15 min dopo SNAP = 144 ± 13% dei valori di base, n = 5; un campione t-test, p=0,0257. (B) Dati di sintesi che mostrano che LTPGABA rimane bloccato in fette di animali stressati in presenza dell’inibitore JNK SP600125 (20 µM). Magnitudo media di LTPGABA10-15 min dopo SNAP = 111 ± 11% dei valori di base, n = 6; un campione t-test, p = 0,38.DOI:
http://dx.doi.org/10.7554/eLife.23785.004
Figura 1-figure supplement 1.L’inibizione di JNK non influisce su LTPGABA o sul suo blocco da stress in assenza di norBNI.(A) Dati riassuntivi che mostrano che l’LTPGABA è espresso in fette di animali ingenui in presenza dell’inibitore JNK SP600125 (20 µM). Magnitudo media di LTPGABA10-15 min dopo SNAP = 144 ± 13% dei valori di base, n = 5; un campione t-test, p=0,0257. (B) Dati di sintesi che mostrano che LTPGABA rimane bloccato in fette di animali stressati in presenza dell’inibitore JNK SP600125 (20 µM). Magnitudo media di LTPGABA10-15 min dopo SNAP = 111 ± 11% dei valori di base, n = 6; un campione t-test, p = 0,38.DOI:
http://dx.doi.org/10.7554/eLife.23785.004
LTPGABA non viene salvato da un antagonista neutrale
I nostri dati indicano che in seguito a stress, l’attivazione dell’κOR persiste anche nella fetta di cervello, e viene salvata in modo dipendente da JNK. Questo suggerisce che le azioni non competitive di norBNI, piuttosto che il suo blocco di legame con la dinorfina, sono rilevanti per la perdita di LTPGABA. Per testare ulteriormente questa ipotesi, abbiamo nuovamente utilizzato strumenti farmacologici in fette di animali stressati. Abbiamo trattato tali fette con norBNI o 6β-naltrexolo, un antagonista neutro che inibisce solo l’attività κOR stimolata dagli agonisti (Figura 2A-B; Wang et al., 2007). Se norBNI salva LTPGABA solo perché può attivare la segnalazione JNK, prevediamo che un antagonista neutro che inibisce solo il legame agonista κOR sarebbe inefficace (Figura 2A, Wang et al., 2007). Mentre il trattamento norBNI ha salvato l’LTPGABA, l’applicazione a bagno dell’antagonista neutro non ha invertito il blocco indotto dallo stress dell’LTPGABA (Figura 2C-F). L’applicazione a bagno del 6β-naltrexolo è stata sufficiente a prevenire la depressione dell’EPSC sui neuroni della dopamina VTA indotta dall’agonista κOR U50488 (Figura 2-figure supplement 1B, Margolis et al., 2005), indicando che questa concentrazione del farmaco è sufficiente a bloccare l’κOR nella fetta di VTA. 6β-naltrexolo non ha avuto alcun effetto sulla trasmissione basale inibitoria in fette da ratti stressati o ingenui (Figura 2-figure supplement 1A). Questi risultati mostrano che un antagonista competitivo dell’κOR non è in grado di salvare efficacemente l’LTPGABA in seguito a stress, e suggeriscono che il blocco persistente dell’LTPGABA è mantenuto dall’attivazione costitutiva dell’κOR nel VTA piuttosto che da un aumento prolungato del rilascio di dinorfina.10.7554/eLife.23785.005Figure 2.L’antagonista neutro 6β-naltrexolo non riesce a salvare LTPGABA in fette da animali stressati.(A) Schema di norBNI e 6β-naltrexolo inibizione della segnalazione κOR. (B) Progettazione sperimentale. (C) Esperimento rappresentativo che mostra che l’applicazione bagno di norBNI (100 nM) salva LTPGABA in una fetta preparata 24 ore dopo lo stress. (D) Esperimento rappresentativo da una cella 24 ore dopo lo stress che mostra che il 6β-naltrexolo (10 µM) non riesce a salvare LTPGABA. (E) I dati di sintesi da entrambi i gruppi. (F) Confronto della grandezza di LTPGABA10-15 min dopo l’applicazione SNAP. (IPSC ampiezze, norBNI: 141 ± 20% dei valori di base, n = 10; 6β-naltrexolo: 100 ± 8% dei valori di base, n = 10; t-test non accoppiato, *p=0,048). DOI:http://dx.doi.org/10.7554/eLife.23785.00510.7554/eLife.23785.006Figure L’integratore a 2 cifre 1.6β-naltrexolo non influisce sulla trasmissione sinaptica basale inibitoria ma blocca κORs.(A) Dati di sintesi che mostrano che il 6β-naltrexolo (10 µM) non influisce sulla trasmissione basale inibitoria nelle cellule da ratti di controllo o stressati. Normalizzato IPSC ampiezza 5-10 min dopo 6β-naltrexolo: controllo = 97 ± 6% dei valori di base, n = 6 (4 animali); stress = 92 ± 17% dei valori di base, n = 4 (2 animali); t-test non accoppiato p = 0,79. (B) Dati di sintesi che mostrano che U50488 deprime le ampiezze dell’EPSC dai neuroni della dopamina VTA e che il 6β-naltrexolo (10 µM) previene questa depressione. L’ampiezza dell’EPSC normalizzata 5-10 minuti dopo U50488: U50488 da solo = 75 ± 2% dei valori di base, n = 3; U50488 + 6β-naltrexolo = 95 ± 1% dei valori di base, n = 4; t-test non accoppiato *p=0.0003.DOI:http://dx.doi.org/10.7554/eLife.23785.006
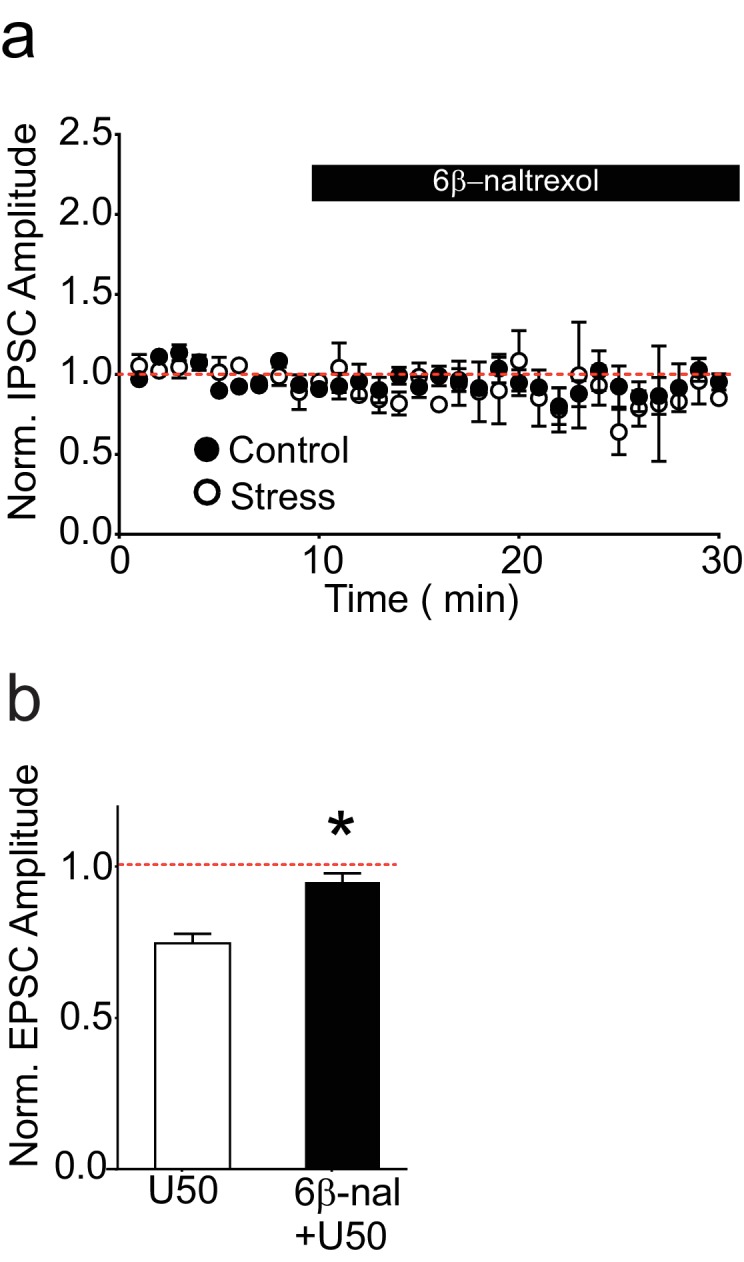
Figura 2-figure supplement 1.L’antagonista neutro 6β-naltrexolo non riesce a salvare LTPGABA in fette da animali stressati.6β-naltrexolo non influisce sulla trasmissione sinaptica basale inibitoria ma blocca κORs.(A) Schema di norBNI e 6β-naltrexolo inibizione della segnalazione κOR. (B) Progettazione sperimentale. (C) Esperimento rappresentativo che mostra che l’applicazione bagno di norBNI (100 nM) salva LTPGABA in una fetta preparata 24 ore dopo lo stress. (D) Esperimento rappresentativo da una cella 24 ore dopo lo stress che mostra che il 6β-naltrexolo (10 µM) non riesce a salvare LTPGABA. (E) I dati di sintesi da entrambi i gruppi. (F) Confronto della grandezza di LTPGABA10-15 min dopo l’applicazione SNAP. (IPSC ampiezze, norBNI: 141 ± 20% dei valori di base, n = 10; 6β-naltrexolo: 100 ± 8% dei valori di base, n = 10; t-test non accoppiato, *p=0,048).DOI:
http://dx.doi.org/10.7554/eLife.23785.005(A) Dati di sintesi che mostrano che il 6β-naltrexolo (10 µM) non influisce sulla trasmissione basale inibitoria nelle cellule da ratti di controllo o stressati. Normalizzato IPSC ampiezza 5-10 min dopo 6β-naltrexolo: controllo = 97 ± 6% dei valori di base, n = 6 (4 animali); stress = 92 ± 17% dei valori di base, n = 4 (2 animali); t-test non accoppiato p = 0,79. (B) Dati di sintesi che mostrano che U50488 deprime le ampiezze dell’EPSC dai neuroni della dopamina VTA e che il 6β-naltrexolo (10 µM) previene questa depressione. L’ampiezza dell’EPSC normalizzata 5-10 minuti dopo U50488: U50488 da solo = 75 ± 2% dei valori di base, n = 3; U50488 + 6β-naltrexolo = 95 ± 1% dei valori di base, n = 4; t-test non accoppiato *p=0,0003.DOI:
http://dx.doi.org/10.7554/eLife.23785.006
Figura 2-figure supplement 1.Il 6β-naltrexolo non influisce sulla trasmissione sinaptica basale inibitoria ma blocca gli κOR.(A) Dati di sintesi che mostrano che il 6β-naltrexolo (10 µM) non influisce sulla trasmissione basale inibitoria nelle cellule da ratti di controllo o stressati. Normalizzato IPSC ampiezza 5-10 min dopo 6β-naltrexolo: controllo = 97 ± 6% dei valori di base, n = 6 (4 animali); stress = 92 ± 17% dei valori di base, n = 4 (2 animali); t-test non accoppiato p = 0,79. (B) Dati di sintesi che mostrano che U50488 deprime le ampiezze dell’EPSC dai neuroni della dopamina VTA e che il 6β-naltrexolo (10 µM) previene questa depressione. L’ampiezza dell’EPSC normalizzata 5-10 minuti dopo U50488: U50488 da solo = 75 ± 2% dei valori di base, n = 3; U50488 + 6β-naltrexolo = 95 ± 1% dei valori di base, n = 4; t-test non accoppiato *p=0,0003.DOI:
http://dx.doi.org/10.7554/eLife.23785.006
L’attivazione transitoria dell’κOR porta ad un’attività κOR persistente
In che modo lo stress acuto può causare l’attivazione costitutiva dell’κOR? Mentre i risultati dei nostri esperimenti su slice escludono la necessità di un’elevata dinorfina per mantenere l’attività persistente di VTA κORs dopo lo stress, il rilascio di dinorfina durante o immediatamente dopo lo stress può essere necessario per innescare un cambiamento nel recettore che porta ad un’attivazione costitutiva prolungata. Se questo modello è corretto, impedendo il legame della dinorfina all’κOR durante lo stress si eviterebbe la perdita di LTPGABA. Tuttavia, dopo lo stress, quando il blocco di LTPGABA non è più dipendente dalla dinorfina, prevenire il legame della dinorfina non salverebbe LTPGABA. Per testare questa idea, abbiamo trattato gli animali con l’antagonista competitivo 6β-naltrexolo 30 minuti prima o un giorno dopo la FSS (Figura 3A). Coerente con la nostra ipotesi, le cellule di animali trattati con 6β-naltrexol prima dello stress hanno mostrato LTPGABA, mentre quelli trattati un giorno dopo lo stress non lo ha fatto, in modo simile agli animali trattati con il veicolo (Figura 3B-F). Al contrario, i nostri studi precedenti hanno dimostrato che il trattamento dei ratti con norBNI nello stesso momento dopo lo stress (un giorno) salva LTPGABA (Polter et al., 2014). Questi dati supportano fortemente l’idea che il blocco persistente di LTPGABA a seguito di stress da nuoto acuto è mediato da un’attivazione del κOR dipendente dalla dinorfina seguita da una transizione all’attività costitutiva indipendente dalla dinorfina del recettore.10.7554/eLife.23785.007Figure 3.6β-naltrexol salva LTPGABA quando somministrato prima dello stress, ma non dopo lo stress.(A) Progettazione sperimentale. (B) Esperimento rappresentativo che mostra che una cellula da un veicolo trattato con un animale stressato non mostra LTPGABA. (C) Esperimento rappresentativo che mostra che una cellula da un animale trattato con 6β-naltrexolo (10 mg / kg) 30 min pre-stress mostra LTPGABA. (D) Esperimento rappresentativo che mostra che una cella da un animale trattato con 6β-naltrexol 24 ore dopo lo stress non mostra LTPGABA. (E) I dati di sintesi che mostra i dati compilati da tutti i gruppi. (F) Confronto della grandezza di LTPGABA 10-15 min dopo l’applicazione SNAP. (1-way ANOVA seguito dal test di confronto multiplo di Dunnett. F2, 30=4,231,p=0,024. Ampiezze IPSC, 6β-naltrexolo pre-stress: 136 ± 12% dei valori di base, n = 12, p<0,05 dal veicolo; 6β-naltrexolo post-stress: 100 ± 9% dei valori di base, n = 11, n.s. dal veicolo; veicolo+stress: 102 ± 8% dei valori di base, n = 10). DOI:http://dx.doi.org/10.7554/eLife.23785.007
Per indagare se una breve attivazione di κORs è sufficiente per produrre κORs persistentemente attivati, abbiamo trattato i ratti con una singola dose dell’agonista κOR, U50488, e misurato LTPGABA in vari punti temporali dopo l’iniezione (Figura 4A). Dopo l’iniezione, U50488 entra rapidamente nel SNC ed è metabolizzato e non rilevabile nel cervello da 24 ore dopo la somministrazione (Russell et al., 2014), e ci aspettiamo quindi che entro il nostro periodo di tempo sperimentale, U50488 non occupava più l’κOR. Nei neuroni da animali trattati con la soluzione salina, l’applicazione a bagno di SNAP IPSC fortemente potenziato (Figura 4B). Al contrario, SNAP non è stato in grado di suscitare LTPGABA nei neuroni da ratti uno o cinque giorni dopo la somministrazione U50488 (Figura 4C-F). In particolare, questo corso di tempo riflette da vicino quello del blocco in vivo di LTPGABA a seguito di stress acuto (Polter et al., 2014).10.7554/eLife.23785.008Figure 4.Single trattamento con un agonista κOR porta al blocco prolungato di LTPGABA.(A) Progettazione sperimentale. (B) Esperimento rappresentativo che mostra che una cellula da un animale trattato con sale mostra LTPGABA. (C) Esperimento rappresentativo singolo che mostra una cella preparato 24 ore dopo un singolo trattamento con U50488 (5 mg / kg) non mostra LTPGABA. (D) Esperimento rappresentativo che mostra che una cella preparato cinque giorni dopo un singolo trattamento con U50488 non mostra LTPGABA. (E) Dati di sintesi da tutti i gruppi. (F) Confronto della grandezza di LTPGABA10-15 min dopo l’applicazione SNAP. (1-way ANOVA seguito dal test di confronto multiplo di Dunnett. F2, 27=12,21, p=0,0002. Ampiezze IPSC, Salina: 137 ± 6% dei valori di base, n = 11; U50488 1 giorno: 108 ± 6% dei valori di base, n = 9, p<0,05 vs. soluzione fisiologica; U50488 5 giorni: 100 ± 5% dei valori di base, n = 10, p<0,05 vs. salina). DOI:http://dx.doi.org/10.7554/eLife.23785.008
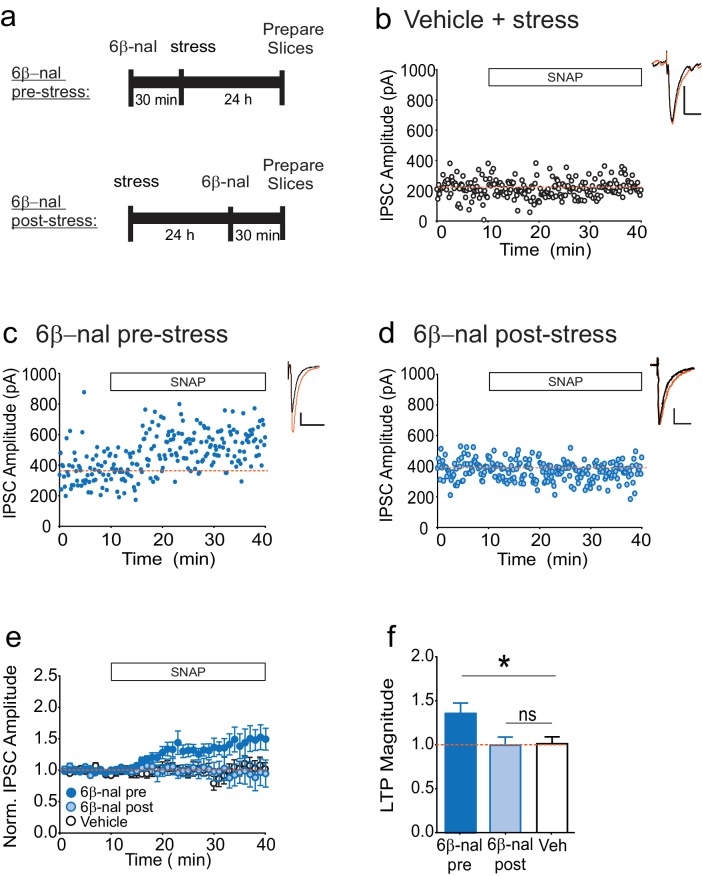
Figura 3.Il 6β-naltrexol salva il LTPGABA quando viene somministrato prima dello stress, ma non dopo lo stress.(A) Progettazione sperimentale. (B) Esperimento rappresentativo che mostra che una cellula da un veicolo trattato con un animale stressato non mostra LTPGABA. (C) Esperimento rappresentativo che mostra che una cellula da un animale trattato con 6β-naltrexolo (10 mg / kg) 30 min pre-stress mostra LTPGABA. (D) Esperimento rappresentativo che mostra che una cella da un animale trattato con 6β-naltrexol 24 ore dopo lo stress non mostra LTPGABA. (E) I dati di sintesi che mostra i dati compilati da tutti i gruppi. (F) Confronto della grandezza di LTPGABA 10-15 min dopo l’applicazione SNAP. (1-way ANOVA seguito dal test di confronto multiplo di Dunnett. F2, 30=4,231,p=0,024. Ampiezze IPSC, 6β-naltrexolo pre-stress: 136 ± 12% dei valori di base, n = 12, p<0,05 dal veicolo; 6β-naltrexolo post-stress: 100 ± 9% dei valori di base, n = 11, n.s. dal veicolo; veicolo+stress: 102 ± 8% dei valori di base, n = 10).DOI:
http://dx.doi.org/10.7554/eLife.23785.007
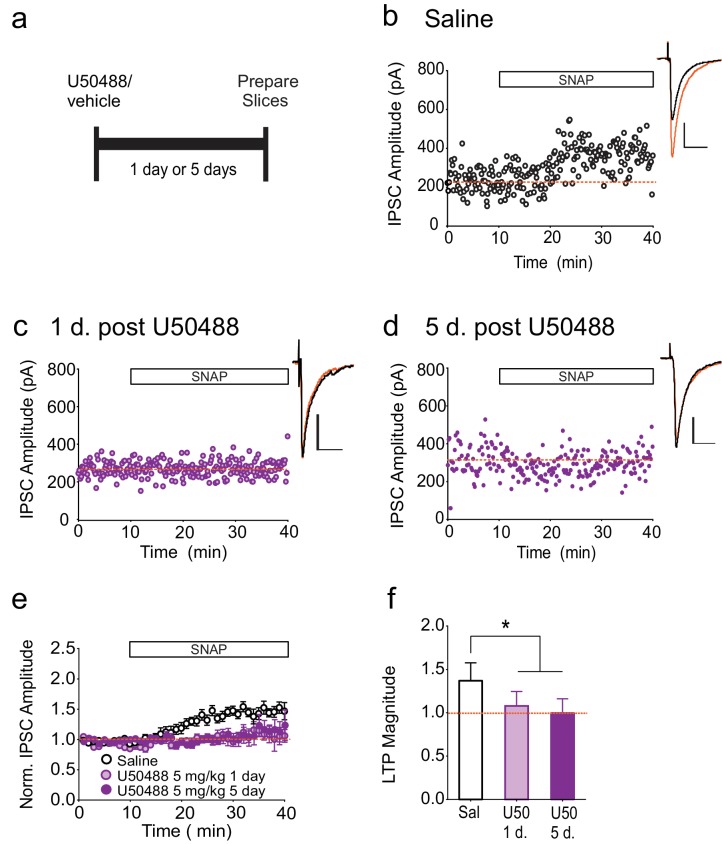
Figura 4.Il trattamento singolo con un agonista κOR porta al blocco prolungato di LTPGABA.(A) Progettazione sperimentale. (B) Esperimento rappresentativo che mostra che una cellula da un animale trattato con sale mostra LTPGABA. (C) Esperimento rappresentativo singolo che mostra una cella preparato 24 ore dopo un singolo trattamento con U50488 (5 mg / kg) non mostra LTPGABA. (D) Esperimento rappresentativo che mostra che una cella preparato cinque giorni dopo un singolo trattamento con U50488 non mostra LTPGABA. (E) Dati di sintesi da tutti i gruppi. (F) Confronto della grandezza di LTPGABA10-15 min dopo l’applicazione SNAP. (1-way ANOVA seguito dal test di confronto multiplo di Dunnett. F2, 27=12,21, p=0,0002. Ampiezze IPSC, Salina: 137 ± 6% dei valori di base, n = 11; U50488 1 giorno: 108 ± 6% dei valori di base, n = 9, p<0,05 vs. soluzione fisiologica; U50488 5 giorni: 100 ± 5% dei valori di base, n = 10, p<0,05 vs. salina).DOI:
http://dx.doi.org/10.7554/eLife.23785.008
Specificità per inibire le sinapsi VTA
Abbiamo poi affrontato la questione se le κOR in altre sinapsi cerebrali sono attivate in modo persistente anche dopo uno stress acuto. L’applicazione del bagno dell’agonista κOR U69593 è stata riportata per deprimere l’ampiezza delle EPSC glutammatergiche sia in VTA Ih positivo (neuroni presuntivi della dopamina) che Ih negativo (neuroni presuntivi non dopaminergici), e norBNI inverte questa depressione (Margolis et al., 2005). Pertanto, se le κOR a sinapsi eccitatorie si attivano costitutivamente dopo lo stress da nuoto, la riduzione della loro attività con norBNI dovrebbe essere rilevabile come potenziamento delle sinapsi eccitatorie VTA. Per testare questo, abbiamo preparato fette di VTA 24 ore dopo FSS. Abbiamo registrato EPSC da Ih positivo e Ih negativo da neuroni sia da animali stressati e non stressati che da norBNI applicati al bagno. NorBNI non ha avuto alcun effetto sull’ampiezza dell’EPSC nei neuroni Ih-positivi in fette provenienti da animali ingenui o stressati (Figura 5A-C), e norBNI non ha aumentato l’ampiezza dell’EPSC nei neuroni VTA Ih-negativi in fette provenienti da animali ingenui o stressati (Figura 5D-F). Pertanto, la persistente attivazione costitutiva κOR che osserviamo nelle sinapsi GABAergiche dopo lo stress acuto non si verifica affatto κOR, anche all’interno del VTA.10.7554/eLife.23785.009Figure 5.κORs a sinapsi eccitatorie VTA non sono attivate costitutivamente dallo stress.(A) Esperimento rappresentativo che mostra che norBNI (100 nM) non potenzia le sinapsi eccitatorie sui neuroni Ih+ VTA in una fetta preparata da un animale di controllo. (B) Esperimento rappresentativo che mostra che norBNI non potenziare sinapsi eccitatorie sui neuroni Ih + VTA in una fetta preparata da un animale stressato. (C) Dati di sintesi da Ih + neuroni. Nessuna differenza significativa in ampiezza IPSC 10-15 min dopo l’applicazione norBNI (t-test p = 0,81 p = 0,81 ampiezze IPSC, controllo: 94 ± 2% dei valori di base, n = 5; stressato: 92 ± 6% dei valori di base, n = 6). (D) Esperimento rappresentativo che mostra che norBNI non potenziare sinapsi eccitatorie su Ih- VTA neuroni in una fetta preparata da un animale di controllo. (E) Esperimento rappresentativo singolo che mostra che norBNI non potenziare sinapsi eccitatorie su Ih- VTA neuroni in una fetta preparata da un animale stressato. (F) Dati di sintesi da Ih- neuroni. Nessuna differenza significativa in ampiezza IPSC 10-15 min dopo l’applicazione norBNI (t-test p=0,49 ampiezze IPSC, controllo: 112 ± 2% dei valori di base, n = 5; stressato: 110 ± 4% dei valori di base, n = 5). DOI:http://dx.doi.org/10.7554/eLife.23785.009
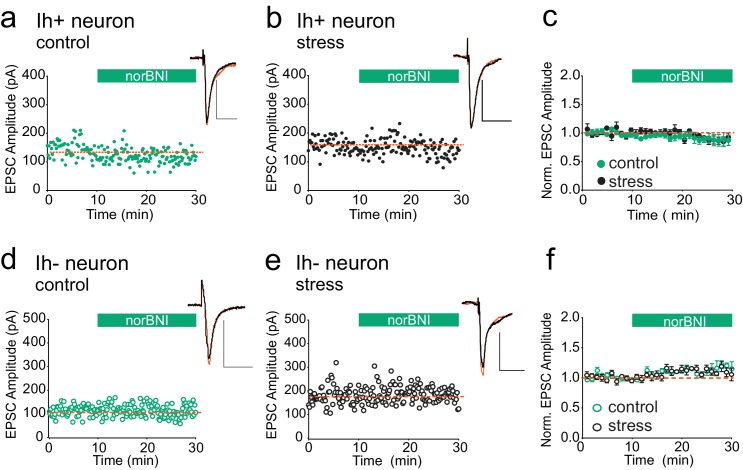
Figura 5.κOR alle sinapsi eccitatorie VTA non sono attivate costitutivamente dallo stress.(A) Esperimento rappresentativo che mostra che norBNI (100 nM) non potenziare sinapsi eccitatorie su Ih + neuroni VTA in una fetta preparata da un animale di controllo. (B) Esperimento rappresentativo che mostra che norBNI non potenziare sinapsi eccitatorie sui neuroni Ih + VTA in una fetta preparata da un animale stressato. (C) Dati di sintesi da Ih + neuroni. Nessuna differenza significativa in ampiezza IPSC 10-15 min dopo l’applicazione norBNI (t-test p = 0,81 p = 0,81 ampiezze IPSC, controllo: 94 ± 2% dei valori di base, n = 5; stressato: 92 ± 6% dei valori di base, n = 6). (D) Esperimento rappresentativo che mostra che norBNI non potenziare sinapsi eccitatorie su Ih- VTA neuroni in una fetta preparata da un animale di controllo. (E) Esperimento rappresentativo singolo che mostra che norBNI non potenziare sinapsi eccitatorie su Ih- VTA neuroni in una fetta preparata da un animale stressato. (F) Dati di sintesi da Ih- neuroni. Nessuna differenza significativa in ampiezza IPSC 10-15 min dopo l’applicazione norBNI (t-test p=0,49 ampiezze IPSC, controllo: 112 ± 2% dei valori di base, n = 5; stressato: 110 ± 4% dei valori di base, n = 5).DOI:
http://dx.doi.org/10.7554/eLife.23785.009
L’attività costitutiva dell’κORs è necessaria per la ricerca prolungata di cocaina indotta dallo stress
Numerosi studi del nostro laboratorio e di altri hanno dimostrato una stretta associazione tra l’attivazione dell’κOR e il comportamento di ricerca di droga indotto dallo stress (McLaughlin et al., 2003; Redila e Chavkin, 2008; Land et al., 2009; Wee e Koob, 2010; Graziane et al., 2013; Zhou et al., 2013; Polter et al., 2014). Abbiamo recentemente riportato che il blocco di κORs con norBNI inverte il modesto ma prolungato ripristino della ricerca di cocaina indotto dallo stress da nuoto (Conrad et al., 2010; Graziane et al., 2013). Questo salvataggio si vede anche quando il norBNI viene somministrato due ore dopo lo stress (Polter et al., 2014). Questi risultati sono coerenti con l’ipotesi che il ripristino della ricerca di cocaina dopo lo stress da nuoto richieda l’attivazione del VTA κORs e la soppressione del LTPGABA. Avendo ora dimostrato che il blocco dell’LTPGABA da stress da nuoto dipende dall’attività costitutiva dell’κORs, abbiamo poi verificato se il ripristino della ricerca di cocaina dipende anche dall’attività costitutiva dell’κORs.
I ratti sono stati addestrati ad autosomministrare cocaina per un minimo di 10 giorni. Gli animali sono stati poi sottoposti ad un addestramento per l’estinzione, e dopo l’ultima sessione di estinzione, sono stati sottoposti a stress da nuoto forzato, per poi tornare alle loro gabbie di casa. Ventiquattro ore dopo lo stress, un gruppo di animali è stato trattato con norBNI e un secondo gruppo con soluzione fisiologica. Un terzo gruppo è stato trattato con 6β-naltrexolo 2 giorni dopo lo stress e 60 minuti prima del test di reintegrazione (Figura 6A). A causa dei diversi profili farmacocinetici del norBNI e del 6β-naltrexolo, il tempo di somministrazione è stato variato per ottimizzare il blocco dell’κOR durante il test di reintegrazione e per garantire che tutti gli animali fossero testati per la reintegrazione nello stesso momento (Endoh et al., 1992; Raehal et al., 2005); così, tutti gli animali sono stati testati per la reintegrazione 48 ore dopo lo stress.10.7554/eLife.23785.010Figure 6.Post-stress salvataggio di reintegrazione da norBNI ma non 6β-naltrexol.(A) Progettazione sperimentale. (B) Premendo la leva durante la sessione di estinzione finale (barra bianca) e la sessione di reintegrazione (barra colorata). Salina (nero): ultima sessione di estinzione: 6,4 ± 1,7 pressioni della leva; sessione di reintegro: 13,8 ± 2,4 pressioni della leva; n = 8, *p=0,011, t-test abbinato. norBNI (verde): ultima sessione di estinzione: 4,8 ± 1,4 pressioni della leva; sessione di ripristino: 5,2 ± 1,1 pressioni della leva; n = 6, p=0,76, t-test abbinato. 6β-naltrexolo (blu): ultima sessione di estinzione: 5,7 ± 1,8 pressioni della leva; sessione di reintegro: 11,2 ± 3,4 pressioni della leva; n = 11, *p=0,033, t-test abbinato. DOI:http://dx.doi.org/10.7554/eLife.23785.010
Come precedentemente mostrato, dopo lo stress acuto, gli animali trattati con veicoli hanno mostrato un significativo aumento della pressione della leva rispetto alla sessione di estinzione finale (Figura 6B). Anche se il ripristino è stato modesto, questo è stato misurato due giorni interi dopo lo stress, dimostrando l’aumento prolungato della ricerca di cocaina (Conrad et al., 2010). Al contrario, gli animali sottoposti a norBNI 24 ore dopo lo stress non hanno aumentato la pressione della leva due giorni dopo lo stress (Figura 6B). Inoltre, l’antagonista neutro 6β-naltrexolo non ha impedito la reintegrazione, in quanto gli animali trattati con 6β-naltrexolo hanno aumentato significativamente la pressione della leva rispetto alla sessione finale di estinzione (Figura 6B). Questi dati suggeriscono che, mentre l’attivazione persistente di κORs è alla base della reintegrazione prolungata indotta dallo stress da nuoto, questa è mediata da recettori costitutivamente attivi piuttosto che da aumenti a lungo termine del livello di dinorfina ligando endogena.
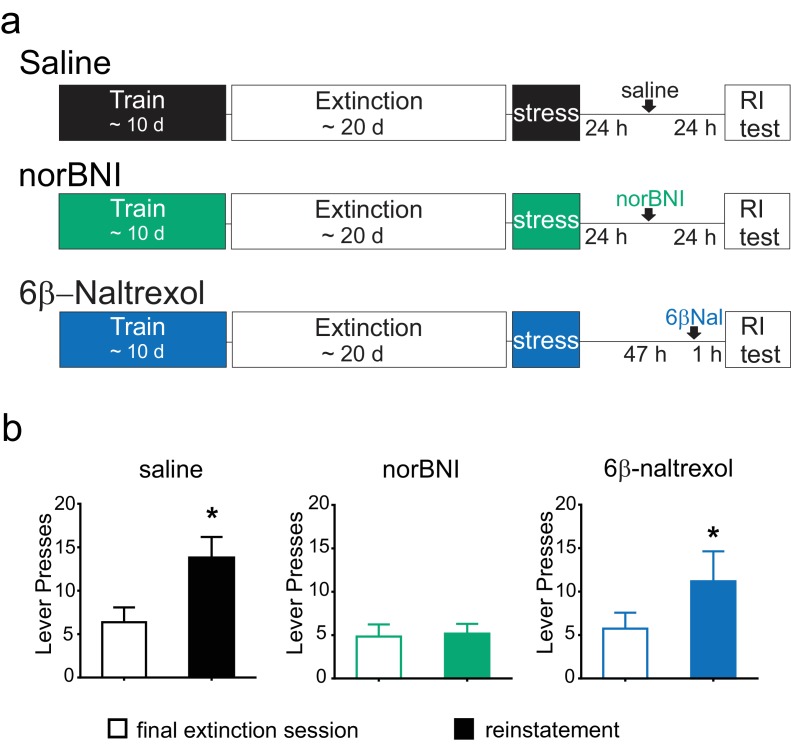
Figura 6.Salvataggio post-stress del reintegro da norBNI ma non da 6β-naltrexolo.(A) Progettazione sperimentale. (B) Premendo la leva durante la sessione finale di estinzione (barra bianca) e la sessione di reintegrazione (barra colorata). Salina (nero): ultima sessione di estinzione: 6,4 ± 1,7 pressioni della leva; sessione di reintegro: 13,8 ± 2,4 pressioni della leva; n = 8, *p=0,011, t-test abbinato. norBNI (verde): ultima sessione di estinzione: 4,8 ± 1,4 pressioni della leva; sessione di ripristino: 5,2 ± 1,1 pressioni della leva; n = 6, p=0,76, t-test accoppiato. 6β-naltrexolo (blu): ultima sessione di estinzione: 5,7 ± 1,8 pressioni della leva; sessione di reintegro: 11,2 ± 3,4 pressioni della leva; n = 11, *p=0,033, t-test abbinato.DOI:
http://dx.doi.org/10.7554/eLife.23785.010
Discussione
Lo stress acuto causa una perdita di plasticità alle sinapsi di VTA GABAAA che dura per giorni ed è causato dall’attivazione persistente di κORs (Graziane et al., 2013; Polter et al., 2014). Questa attivazione potrebbe essere causata o da un aumento prolungato della dinorfina o da un aumento dell’attività costitutiva di κORs. In questo studio, i nostri dati supportano quest’ultimo meccanismo: una singola esposizione ad un fattore di stress acuto provoca un’attivazione costitutiva duratura del VTA κORs che sopprime la plasticità alle sinapsi inibitorie correlate con lo stress indotto dalla ricerca di farmaci (Figura 7). Mentre studi precedenti hanno dimostrato l’attività costitutiva di κORs nelle cellule coltivate e nel cervello di ratto (Wang et al., 2007; Sirohi e Walker, 2015), la nostra è la prima dimostrazione di cambiamenti indotti dall’esperienza nell’attività costitutiva di questi recettori. Questo rappresenta un nuovo meccanismo di regolazione da stress acuto del sistema dynorphin-κOR, e getta nuova luce sui percorsi di segnalazione coinvolti nel ripristino della ricerca di farmaci.10.7554/eLife.23785.011Cifra 7.Attivazione costitutiva dell’κOR da stress.(A) Durante lo stress, il legame della dinorfina all’κOR innesca uno spostamento verso uno stato costitutivo attivo. Bloccando il legame della dinorfina, sia norBNI che 6β-naltrexolo impediscono la perdita di LTPGABA durante questo tempo. (B) Dopo lo stress, il blocco di LTPGABA viene mantenuto dall’attività costitutiva di κORs e non dipende più dal legame con la dinorfina. (C) norBNI inverte il blocco di LTPGABA indotto dallo stress attivando il percorso di segnalazione JNK che riduce in modo non competitivo l’attività κOR. DOI:http://dx.doi.org/10.7554/eLife.23785.011
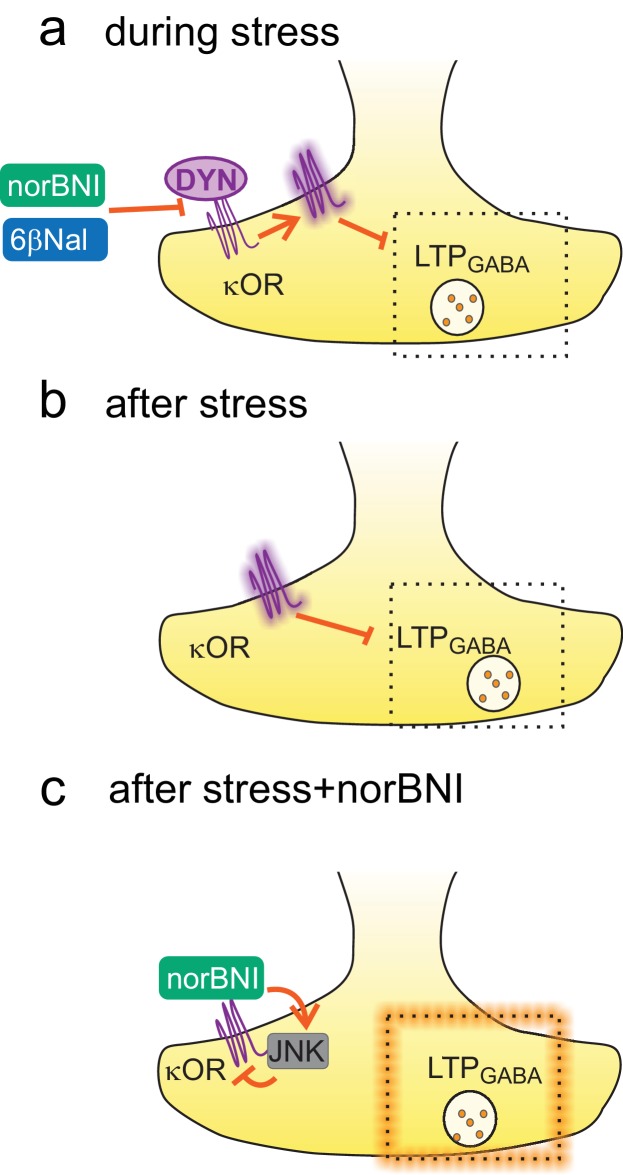
Figura 7.Attivazione costitutiva di κORs da stress.(A) Durante lo stress, il legame della dinorfina all’κOR innesca uno spostamento verso uno stato costitutivamente attivo. Bloccando il legame dinorfina, sia norBNI e 6β-naltrexolo prevenire la perdita di LTPGABA durante questo tempo. (B) Dopo lo stress, il blocco di LTPGABA viene mantenuto dall’attività costitutiva di κORs e non dipende più dal legame con la dinorfina. (C) norBNI inverte il blocco di LTPGABA indotto dallo stress attivando il percorso di segnalazione JNK che riduce in modo non competitivo l’attività κOR.DOI:
http://dx.doi.org/10.7554/eLife.23785.011
Attivazione costitutiva di VTA κORs
È ampiamente accettato che i GPCR possono adottare conformazioni indipendenti dall’agonista che sono costituzionalmente attive (Seifert e Wenzel-Seifert, 2002; Sadée et al., 2005; Young et al., 2013Meye et al., 2014). Oltre agli κOR, gli altri membri della sottofamiglia dei recettori degli oppioidi, µOR e δOR, hanno entrambi dimostrato di mostrare attività costitutiva (Wang et al., 1994, 2004; Chiu et al., 1996; Wang et al., 1999; Liu e Prather, 2001; Wang et al, Gli stessi κOR hanno mostrato attività costitutiva, sia nei sistemi di espressione eterologhi (Becker et al., 1999; Wang et al., 2007) sia nel ratto PFC (Sirohi e Walker, 2015). È stata anche riportata una diminuzione dei comportamenti di paura e ansia nei ratti dopo uno shock acuto, che si riduce con il norBNI post-shock, suggerendo la possibilità di un’attivazione costitutiva dell’κOR, sebbene sia stata richiesta una dose elevata e l’effetto del norBNI non sia stato paragonato a quello di un antagonista neutrale (Rogala et al., 2012). Molto poco si sa, tuttavia, sui processi che regolano le transizioni tra stati costitutivamente attivi e inattivi, che presumibilmente rappresentano conformazioni recettoriali distinte (Seifert e Wenzel-Seifert, 2002; Sadée et al., 2005).
Presentiamo due dati critici che indicano che lo stress induce l’attività costitutiva degli κOR. In primo luogo, breve applicazione (~ 15 min) di un agonista κOR inversa a fette VTA da ratti stressati salva LTPGABA in un modo JNK-dipendente. In secondo luogo, l’antagonista neutro non salva LTPGABA. Si ritiene che la segnalazione attraverso il percorso JNK sia responsabile della lunga inibizione non competitiva dell’κOR (Bruchas et al., 2007; Melief et al., 2010, 2011). Nei nostri esperimenti, il requisito di JNK per il norBNI di salvare l’LTPGABA è la prova che il norBNI agisce attraverso mezzi non competitivi, e suggerisce che l’attivazione persistente dell’κOR a seguito di stress non richiede il legame continuo dei recettori da parte del ligando. È importante notare che l’inibizione della segnalazione JNK da sola non ha impedito l’induzione di LTPGABA in fette da animali ingenui, né ha ripristinato LTPGABA in fette da animali stressati, indicando che il ruolo di JNK è limitato all’inibizione del recettore da parte del norBNI. Il fallimento dell’inibitore JNK nel salvare l’LTPGABA indica che l’inibizione dell’LTPGABA da parte dell’κORs non è mediata dal percorso JNK, ma molto probabilmente attraverso uno degli altri percorsi a valle dell’κORs, come i percorsi p38 o ERK MAPK, o attraverso l’attivazione di Gα i (Bruchas e Chavkin, 2010; Iñiguez et al., 2010; Ehrich et al., 2015).
L’incapacità dell’antagonista neutro, il 6β-naltrexolo, di salvare l’LTPGABA è coerente con lo stress che promuove l’attività costitutiva dell’κOR alle sinapsi VTA inibitorie. NorBNI, attraverso l’attivazione di JNK, riduce la capacità di segnalazione dell’κOR indipendentemente dal fatto che ciò avvenga attraverso l’attività costitutiva o l’aumento del legame con la dinorfina. Al contrario, un antagonista neutro come il 6β-naltrexolo potrebbe solo invertire la perdita di LTPGABA impedendo il legame agonista al recettore. In contrasto con il rapido ripristino del LTPGABA mediante l’applicazione di norBNI in bagno, l’applicazione di 6β-naltrexol in bagno non ha salvato il LTPGABA. Questa discrepanza non può essere spiegata da una concentrazione insufficiente o dal tempo di applicazione del 6β-naltrexol, in quanto una simile perfusione in bagno del 6β-naltrexol è stata sufficiente a bloccare la depressione di EPSC sui neuroni della dopamina VTA indotta dall’applicazione in bagno dell’agonista κOR, U50488. La spiegazione più semplice dei nostri dati è che lo stress acuto induce l’attività costitutiva dell’κOR. In alternativa, norBNI può promuovere la segnalazione di JNK attraverso un meccanismo sconosciuto indipendente dai recettori dell’κOR.
Attività costitutiva indotta dall’esperienza
Lo stress acuto sembra innescare uno spostamento verso κOR costitutivamente attivi attraverso un rilascio transitorio del ligando endogeno κOR, la dinorfina (Figura 7). La nostra prova più forte per questo modello è la capacità dell’antagonista neutro 6β-naltrexolo di prevenire la perdita di LTPGABA quando somministrato prima, ma non dopo lo stress. Sebbene il 6β-naltrexol abbia un’affinità equivalente per gli OR µ e κ, (Wang et al., 2007) il nostro lavoro precedente ha dimostrato che il blocco di LTPGABA da stress non è influenzato dalla somministrazione pre-stress dell’antagonista µOR cyprodime (Graziane et al., 2013). Pertanto, la capacità del 6β-naltrexolo di prevenire la perdita di LTPGABA indotta dallo stress è improbabile che coinvolga la segnalazione µOR e si verifica invece bloccando il legame della dinorfina all’κOR. Una singola somministrazione sistemica in vivo dell’agonista κOR U50488 blocca anche l’LTPGABA per almeno cinque giorni, sostenendo l’idea che una breve esposizione agonistica da sola è sufficiente per innescare un’attività costitutiva duratura dell’κOR.
Come può l’attivazione di κORs da parte del suo ligando endogeno spostare il recettore verso l’attività costitutiva? In un sistema di coltura cellulare eterologo, la precedente esposizione ad un agonista κOR da solo ha aumentato significativamente l’attività costitutiva del recettore (Wang et al., 2007). Si sa di più per quanto riguarda la regolazione dell’attività costitutiva dei µOR. In cellule coltivate che esprimono eterologicamente µORs (Wang et al., 1994, 2000; Liu e Prather, 2001) o in animali intatti (Wang et al., 2004; Shoblock e Maidment, 2006; Meye et al., 2012), l‘esposizione alla morfina agonista µOR innesca un aumento dell’attività costitutiva dei µORs. L’attività costitutiva indotta da morfina di µORs è regolata da calmodulina e protein chinasi. In condizioni basali, il legame della calmodulina al µORs impedisce l’associazione costitutiva con le proteine G. A seguito di esposizione alla morfina, la calmodulina si dissocia dal µOR, permettendo l’attivazione costitutiva (Wang et al., 1999, 2000). Anche se non si sa se la calmodulina regola l’attività dell’κOR, un intricato complesso di impalcature regola la segnalazione dell’κOR (Bruchas e Chavkin, 2010), e studi futuri che indagano il ruolo di questi complessi di segnalazione nell’attività dell’κOR in risposta allo stress saranno importanti e intriganti. La maggior parte del lavoro di indagine sull’attivazione costitutiva dei GPCR si è concentrato sul miglioramento dell’attività costitutiva attraverso la somministrazione di ligandi esogeni (Meye et al., 2014), mentre molto meno si sa sull’induzione di stati costitutivi attivi dei GPCR attraverso la segnalazione endogena. Tuttavia, è stato recentemente riportato che il dolore infiammatorio aumenta i µORs costitutivamente attivi nel midollo spinale del topo, portando a iperalgesia e dipendenza cellulare (Corder et al., 2013).
I nostri dati indicano che il trattamento con il solo agonista κOR U50488 è sufficiente per produrre κOR costitutivamente attivi sulle sinapsi VTA inibitorie. Un altro rompicapo è il motivo per cui l’attivazione dell’κOR da parte del ligando endogeno può produrre recettori costitutivamente attivi in alcune sinapsi ma non nei loro vicini, e in risposta a certi segnali ambientali (stress acuto) ma non ad altri durante i quali può essere rilasciata anche la dinorfina. Una possibilità è che i recettori in diversi tipi di cellule possono accoppiarsi a diverse cascate di segnalazione o molecole di ponteggio. Un’altra possibilità è che sia necessaria una segnalazione coordinata tra κOR e un altro sistema di neurotrasmettitori. Il nostro lavoro precedente indica che l’attivazione dei recettori dei glucocorticoidi è necessaria per il blocco di LTPGABA da stress (Niehaus et al., 2010; Polter et al., 2014). Anche se l’attivazione persistente di questi recettori non si vede dopo lo stress, è possibile che l’attivazione coincidente dei recettori dei glucocorticoidi e degli oppioidi kappa porti all’attivazione costitutiva di questi ultimi. Inoltre, è stato riportato che il recettore dell’orexin-1 attenua l’inibizione dell’κOR nella produzione di cAMP, ma migliora il reclutamento di β-arrestina e l’attivazione del MAPK p38, ed entrambi gli effetti sono prevenuti dall’inibitore JNK SP600125 utilizzato nel nostro studio (Robinson e McDonald, 2015). Sia l’orexina che la dinorfina sono co-sviluppate dalle proiezioni ipotalamiche al VTA (Chou et al., 2001; Muschamp et al., 2014; Baimel et al., 2015). Questa disposizione solleva la possibilità che il rilascio di entrambi i peptidi insieme, o forse il rilascio simultaneo di dinorfina e di un neurotrasmettitore sconosciuto che agisce in modo simile all’orexina, possa avviare eventi di segnalazione non innescati dalla sola dinorfina. La presunta segnalazione del doppio recettore potrebbe essere un modo per indurre una sinapsi o un’attività costitutiva selettiva dell’κOR.
Regolamentazione del LTPGABA da parte dell’κORs
Una domanda senza risposta è come l’κOR sopprime l’espressione di LTPGABA. I nostri studi precedenti hanno dimostrato che l’LTPGABA è innescato dall’attivazione mediata dall’ossido nitrico della cGMP-proteina chinasi G (PKG) (Nugent et al., 2007, 2009; Niehaus et al., 2010). Poiché una fonte esogena di ossido nitrico (SNAP) non salva il potenziamento a seguito di stress, è probabile che il blocco si verifichi nel terminale presinaptico tra l’attivazione della ciclasi di guanilato e il miglioramento del rilascio GABAergico. Mentre è possibile che l’attivazione di κOR in genere deprima il rilascio di GABA, il nostro lavoro precedente (Graziane et al., 2013) non ha trovato alcun cambiamento nella frequenza mIPSC a seguito di stress da nuoto in acqua fredda. Questi dati suggeriscono che gli κOR non alterano il rilascio basale del GABA. Inoltre, LTPGABA è anche perso 24 ore dopo una singola esposizione alla morfina, e in questa situazione un cGMP analogico o una forte attivazione di sGC potenziano il rilascio di GABA (Nugent et al., 2007; Niehaus et al., 2010). Siamo quindi a favore di un meccanismo per cui, dopo uno stress acuto, gli κOR costitutively-active κOR agiscono in modo simile su un substrato che limita l’induzione della plasticità senza influenzare i meccanismi di rilascio basale, forse attraverso il downregulation della ciclasi guanylyl solubile, o i cambiamenti dell’impalcatura che sequestrano la PKG dai suoi substrati.
Sebbene non si sappia in quali condizioni il LTPGABA sia attivato in un animale intatto, i nostri studi precedenti hanno fatto luce sui suoi potenziali ruoli. LTPGABA è una forma eterosinaptica di plasticità che può essere attivata da un tetano ad alta frequenza che attiva l’attivazione NMDAR-dipendente dell’ossido nitrico sintasi sensibile al calcio (Nugent et al., 2007). Ci aspettiamo quindi che l’LTPGABA sia indotto quando c’è un’attivazione robusta degli input eccitatori sui neuroni della dopamina. LTPGABA può svolgere un ruolo omeostatico per migliorare l’inibizione dei neuroni della dopamina dopo forte eccitazione NMDAR-attivante. La perdita di LTPGABA, quindi, si tradurrebbe in uno squilibrio tra input inibitori ed eccitatori sul neurone della dopamina. Come sinapsi GABAergiche sui neuroni della dopamina controllano fortemente la loro cottura spontanea (van Zessen et al., 2012), la perdita di LTPGABA è suscettibile di prolungare o migliorare la cottura in risposta a stimoli salienti.
Attività costitutiva di κORs nel VTA e comportamento di ricerca di droga
Il ruolo critico del VTA nella reintegrazione della ricerca di droga è stato ripetutamente sottolineato (McFarland et al., 2004; Briand et al., 2010; Graziane et al., 2013; Mantsch et al., 2016), e all’interno del VTA, le sinapsi GABAergiche sui neuroni della dopamina VTA regolano fortemente la cottura delle cellule DA (Tan et al., 2012; van Zessen et al., 2012; Polter e Kauer, 2014).
Il nostro lavoro mostra che lo stress produce un’attività costitutiva κOR di lunga durata che si limita ad inibire le sinapsi delle cellule dopaminergiche, influenzando così le informazioni immagazzinate o elaborate qui per molto più tempo rispetto alle sinapsi eccitatorie. Potremmo quindi prevedere due fasi di attivazione dell’κOR indotta dallo stress. Si ipotizza che la dinorfina viene rilasciata durante e / o immediatamente dopo lo stress, deprimendo EPSCs sui neuroni dopaminergici e iperpolarizzanti neuroni dopaminergici, sul saldo diminuendo l’eccitabilità dei neuroni dopaminergici (Margolis et al., 2003, 2005; Ford et al., 2006). Tuttavia, poiché la dinorfina è degradata, ci aspetteremmo che la forza delle sinapsi eccitatorie ritornasse a livelli normali, mentre l’LTPGABA si bloccherebbe a causa dell’attività costitutiva dell’κOR, uno stato che dura almeno cinque giorni dopo lo stress da nuoto. Questo aumenterebbe invece il tasso di cottura dei neuroni dopaminergici, soprattutto in risposta a stimoli eccitatori. Questa maggiore eccitabilità potrebbe contribuire all’aumento della spinta verso un comportamento di ricerca di droga in seguito all’esposizione a stimoli spaziali associati all’esperienza della droga del passato (ad esempio, il ritorno alla camera operatoria), e potrebbe creare uno stato di vulnerabilità ad ulteriori fattori di stress. Infatti, nei ratti sottoposti allo stesso stress da acqua fredda utilizzato in questo studio, la velocità di combustione dei neuroni dopaminergici rimane elevata per diversi giorni dopo (Marinelli, 2007). È interessante notare che una singola dose dell’agonista κOR, la salvinorina A, ha effetti bifasici sulla funzione di ricompensa: subito dopo la somministrazione, i ratti mostrano un aumento anedonico delle soglie di ricompensa all’autostimolazione intracranica. Tuttavia, 24 ore dopo la somministrazione di salvinorina A, i ratti mostrano una diminuzione delle soglie di ricompensa, indicando un aumento della sensibilità alla ricompensa (Potter et al., 2011). Questo effetto bifasico è coerente con una divisione tra gli effetti a breve e a lungo termine dell’attivazione di κOR, forse a causa di meccanismi differenziali di regolazione e di attivazione costitutiva di sottoinsiemi di recettori.
Il circuito del VTA è altamente complesso, e neuroni della dopamina all’interno del VTA mostrano eterogeneità fisiologica e funzionale che si correla con l’obiettivo di proiezione. Mentre rimane il disaccordo sui marcatori farmacologici, fisiologici e anatomici più appropriati di diverse sottoclassi di neuroni dopaminergici (Ford et al., 2006; Margolis et al., 2006; Lammel et al., 2008, 2011; Ungless e Grace, 2012; Baimel et al., 2017), i marcatori elettrofisiologici qui utilizzati e la posizione laterale delle nostre registrazioni all’interno del VTA ci suggeriscono che la nostra popolazione di cellule può comprendere in gran parte neuroni dopaminergici che proiettano al nucleo accumbens. Questo può essere significativo per la ricompensa della droga, in quanto l’attivazione di questi neuroni ha dimostrato di essere gratificante nei topi (Lammel et al., 2012). Pertanto, il nostro studio indica che un fattore di stress acuto induce una perdita duratura di plasticità inibitoria nei circuiti che può guidare un comportamento gratificante.
GABAergico afferisce sui neuroni della dopamina VTA può rilasciare GABAA su entrambi i recettori GABAA o GABABAB. Studi precedenti, tra cui approcci optogenetici più recenti, hanno suggerito che i neuroni bersaglio dei recettori GABAB nascono dal nucleo accumbens e regolano i comportamenti indotti dalla droga (Sugita et al., 1992; Cameron e Williams, 1993; McCall et al., 2017; Edwards et al., 2017). Tuttavia, il nostro lavoro precedente non ha trovato alcuna LTPGABA nelle sinapsi di GABABAB sui neuroni della dopamina (Nugent et al., 2009), suggerendo che gli effetti di κORs persistentemente attivati sono improbabili per coinvolgere il nucleo accumbens-VTA GABAergic afferents.
I nostri dati forniscono la prima dimostrazione che gli κOR costituzionalmente attivi nel VTA sono necessari per il ripristino della ricerca di cocaina indotta dallo stress. La somministrazione post-stress (almeno 24 ore) di norBNI impedisce la reintegrazione, mentre la somministrazione post-stress dell’antagonista neutro 6β-naltrexolo non lo fa. La capacità del norBNI di modificare il comportamento di ricerca di farmaci anche se somministrato significativamente dopo lo stressor è notevole, e indica il potenziale terapeutico del targeting κORs per invertire i neuroadattamenti indotti dallo stress. L’incapacità del 6β-naltrexolo di prevenire il reintegro nei momenti in cui il norBNI è efficace suggerisce fortemente che il persistente aumento della ricerca di farmaci indotti dallo stress da nuoto è mediato dall’attività costitutiva di κORs piuttosto che da un aumento prolungato del rilascio di dinorfina. Inoltre, questo risultato è coerente con un ruolo importante per la plasticità delle sinapsi GABAergiche nel comportamento di ricerca di farmaci indotti dallo stress. Mentre una notevole attenzione è stata data al ruolo dell’LTP nelle sinapsi eccitatorie nel VTA, il blocco κOR di norBNI non impedisce allo stress di potenziare le sinapsi eccitatorie sui neuroni della dopamina (Graziane et al., 2013). Il nostro lavoro attuale conferma che la perdita di LTP alle sinapsi GABAergiche nel VTA è altamente correlata con la ricerca di farmaci indotti dallo stress.
κORs come bersaglio per l’abuso di sostanze
Gli κOR hanno dimostrato di essere promettenti come potenziale bersaglio di droga per l’uso di sostanze e disturbi dell’umore (Bruchas et al., 2010; Van‘t Veer e Carlezon, 2013; Crowley e Kash, 2015) e il nostro lavoro suggerisce un nuovo modo in cui la segnalazione dell’κOR può andare storta. Nei modelli preclinici, gli antagonisti dell’κOR hanno mostrato una potenziale efficacia per la depressione e per l’uso compulsivo e indotto da stress (Bruchas et al., 2010; Wee e Koob, 2010). Sono in corso numerosi studi clinici che utilizzano i ligandi κOR per trattare i disturbi da uso di sostanze e la depressione (Ehrich et al., 2015; Karp et al., 2014; Chavkin e Koob, 2016; Ling et al., 2016; Nasser et al., 2016). Tuttavia, molti di questi studi utilizzano la buprenorfina, un agonista parziale dell’κORs, o nuovi composti che possono mancare di attività agonista inversa, nessuno dei quali ridurrebbe l’attività degli κORs costitutivi (Karp et al., 2014; Rorick-Kehn et al., 2014).
Il nostro studio suggerisce che il futuro sviluppo di farmaci dovrebbe considerare l’attività κOR in eccesso attraverso la segnalazione dei recettori e a livello di legatura dei leganti. Allo stesso modo, risultati deludenti o effetti minimi negli studi clinici possono non rappresentare un fallimento dell’κOR come bersaglio farmaceutico, ma la necessità di considerare farmaci che mirano a specifiche conformazioni dell’κOR che promuovono la segnalazione costitutiva. Una strategia alternativa sarebbe quella di mirare alla segnalazione di JNK nel VTA, dato che norBNI sembra salvare la funzione κOR attivando JNK. Un’intrigante implicazione dei nostri studi deriva dai nostri dati che l’attività costitutiva degli κOR alle sinapsi inibitorie nel VTA dura solo da cinque a dieci giorni dopo lo stress acuto (Polter et al., 2014), un periodo di tempo considerevolmente più breve rispetto ai 14-21 giorni tipici per il turnover degli κOR (McLaughlin et al., 2004). Questo suggerisce che l’attività costitutiva dell’κORs può essere interrotta da un meccanismo attivo non identificato. Studi futuri che indagano su tale meccanismo potrebbero identificare obiettivi che potrebbero essere reclutati per promuovere la resilienza allo stress. Il nostro lavoro dimostra un nuovo meccanismo di regolazione dell’κORs che dipende dall’esperienza ed evidenzia la capacità di modulazione dell’κORs di invertire i neuroadattamenti indotti dallo stress e le carenze comportamentali ben dopo che il fattore di stress si è verificato. Un ulteriore studio dei meccanismi di attivazione costitutiva di κORs può produrre numerosi obiettivi potenziali per il trattamento dei disturbi da uso di sostanze e altre malattie legate allo stress.
Materiali e metodi
Animali
Tutte le procedure sono state effettuate in conformità con le linee guida dei National Institutes of Health per la cura e l’uso degli animali, e sono state approvate dal Comitato Istituzionale per la cura e l’uso degli animali della Brown University o dal Comitato Istituzionale per la cura e l’uso degli animali della University of Wyoming. Per gli studi di elettrofisiologia a fette, i ratti Sprague-Dawley maschi e femmine (P16-25) sono stati mantenuti su un ciclo di 12 ore luce/scuro e hanno fornito cibo e acqua ad libitum. Per gli studi di auto-somministrazione, i ratti maschi Sprague-Dawley (350-450g) sono stati allevati in casa e individualmente ospitati in una stanza a temperatura controllata con un 12 ore di luce inversa / ciclo di luce / buio. Tutti gli animali hanno avuto accesso ad libitum all’acqua durante tutta la sperimentazione, tranne nei periodi in cui si trovavano nelle camere operatorie (descritte di seguito). I ratti erano 60-70 giorni all’inizio degli esperimenti comportamentali.
Acuto stress da nuoto forzato
Lo stress è stato somministrato da un compito di nuoto forzato Porsolt modificato (Niehaus et al., 2010). I ratti sono stati messi per 5 minuti in acqua fredda (4-6°C), poi essiccati e lasciati recuperare in una gabbia riscaldata per due ore prima di tornare alla gabbia di casa. U50488 (5 mg / kg) e 6β-naltrexolo (10 mg / kg) sono stati sciolti in soluzione fisiologica o 10% DMSO in soluzione fisiologica, rispettivamente. Agli animali iniettati dal veicolo è stata somministrata un’iniezione del volume equivalente. Per alcuni esperimenti, gli animali dato iniezioni veicolo iniettato in vari punti di tempo sono stati collassati in un unico gruppo. Fette di cervello sono stati preparati in diversi punti di tempo dopo l’esposizione allo stress, come descritto di seguito.
Preparazione di fette di cervello
Le fette orizzontali del mesencefalo (250 µm) sono state preparate come descritto in precedenza da ratti Sprague-Dawley profondamente anestetizzati (Nugent et al., 2007; Niehaus et al., 2010; Polter et al., 2014). Le fette sono state conservate per almeno 1 ora a 34°C in una soluzione contenente HEPES ossigenata (in mM): 86 NaCl, 2,5 KCl, 1,2 NaH2PO4, 35 NaHCO3, 20 HEPES, 25 glucosio, 5 ascorbato di sodio, 2 tiourea, 3 piruvato di sodio, 1 MgSO4.7H2O, 2 CaCl2.2H2O (Ting et al., 2014). Le fette sono state poi trasferite in una camera di registrazione dove sono state immerse in ACSF contenente (in mM): 126 NaCl, 21,4 NaHCO3, 2,5 KCl, 1,2 NaH2PO4, 2,4 CaCl2, 1,0 MgSO4, 11,1 glucosio.
Elettrofisiologia
I metodi generali sono quelli precedentemente riportati (Niehaus et al., 2010; Polter et al., 2014). Le fette di mesencefalo sono state continuamente perfuse a 1,5-2 mL ⁄ min. Le pipette patch sono state riempite con (in mM): 125 KCl, 2,8 NaCl, 2 MgCl2, 2 ATP-Na+, 0,3 GTP-Na+, 0,6 EGTA e 10 HEPES. Per registrare gli IPSC, la soluzione extracellulare era ACSF (28-32°C) contenente: 6,7-dinitrochinossalina-2,3-dione (DNQX; 10 µM) e stricnina (1 µM), per bloccare i recettori AMPA e glicina rispettivamente. Per registrare gli EPSC, all’ACSF sono stati aggiunti 100 µM di picrotossina. I neuroni dopaminergici, che comprendono circa il 70% di tutti i neuroni VTA, sono stati identificati dalla presenza di una grande Ih (>50 pA) durante un passaggio di tensione da -50 mV a -100 mV. Gli IPSC mediati dal recettore GABAA sono stati stimolati utilizzando un elettrodo bipolare in acciaio inossidabile stimolante posto 100-300 µm rostrale al sito di registrazione nel VTA. Le cellule sono stati fissati in tensione a -70 mV e resistenza di ingresso e resistenza in serie sono stati monitorati per tutta la durata dell’esperimento, le cellule sono state scartate se questi valori sono cambiati di oltre il 15% durante l’esperimento.
NO-triggered LTP
La 3-isobutil-1-metilxantina (IBMX; 100 µM) è stata utilizzata per inibire la degradazione mediata dalla fosfodiesterasi del cGMP e applicata tramite ACSF perfuso per almeno 10 minuti prima dell’induzione di LTPGABA mediante l’applicazione del donatore NO, SNAP (S-nitroso-N-acetilpenicillamina, 400 µM). Animali di controllo (veicolo iniettato animali stressati o non stressati) sono stati interleaved con animali da esperimento (farmaco-iniettato animali stressati). Dove indicato, NorBNI (100 nM), 6β-naltrexolo (10 µM), e SP600135 (20 µM) sono stati applicati a bagno su fette almeno 10 minuti prima dell’induzione di LTPGABA.
Autosomministrazione
I ratti sono stati anestetizzati con ketamina HCl (87 mg / kg, i.m.) e xilazina (13 mg / kg, i.m.) e impiantati con cateteri intravenosi giugulare. Al fine di proteggere contro le infezioni e mantenere la pervietà del catetere, i cateteri sono stati lavati quotidianamente con 0,2 ml di una soluzione mista di cefazolina (0,1 gm/ml) ed eparina (100 UI). I ratti sono stati autorizzati a recuperare per una settimana prima del test comportamentale. Tutte le procedure di autosomministrazione sono state condotte in camere operatorie standard (Med Associates, St. Albans, VT; 30,5 cm x 24,1 cm x 21,0 cm). Ogni scatola conteneva una luce di casa (illuminata durante il test comportamentale), due leve retrattili, un segnale luminoso e un generatore di toni. Prima di iniziare l’addestramento all’autosomministrazione della cocaina, gli animali sono stati privati del cibo per 24 ore e successivamente collocati nelle camere operatorie durante la notte per 14 ore. Durante questa sessione, una risposta alla leva attiva (la leva sinistra) ha portato alla consegna di un singolo pellet di cibo da 45 mg (#F0165, Bio-Serv, Flemington, NJ) e alla presentazione di una stecca composta (illuminazione della luce sopra la leva attiva +5 s tono, 2900 Hz), seguita da un periodo di timeout di 25 s. Le risposte alla leva inattiva (la leva destra) non hanno avuto conseguenze programmate ma sono state registrate. Sono state registrate anche le ricompense totali ricevute. Il giorno successivo è iniziato l’addestramento all’autosomministrazione di cocaina. Durante questo periodo, una risposta alla leva attiva ha prodotto un’infusione di 0,05 ml di 0,20 mg di cocaina (sciolta in soluzione salina allo 0,9%) e la presentazione della stecca composta (luce + tono). L’autosomministrazione è continuata (2 ore al giorno) fino a quando gli animali non hanno premuto in modo affidabile la leva attiva (3d con almeno 10 infusioni di cocaina ricevute). In seguito all’acquisizione dell’autosomministrazione di cocaina, tutti gli animali sono stati sottoposti ad un addestramento all’estinzione, durante il quale le risposte alla leva precedentemente attiva hanno prodotto la stecca del composto ma non hanno più prodotto l’infusione di droga. Durante l’addestramento all’autosomministrazione, gli animali sono stati sottoposti a restrizioni alimentari fino all’80% del loro peso corporeo. Durante l’estinzione, gli animali hanno avuto accesso ad libitum al cibo nella gabbia domestica. Le procedure di estinzione sono continuate fino a quando gli animali non hanno raggiunto i criteri di estinzione (3d con meno di 10 presse a leva attive).
Stress da nuoto forzato e reintegro
Il giorno successivo all’ultima sessione di estinzione, i ratti sono stati sottoposti a 5 minuti di nuoto forzato in acqua fredda (4-6˚C, Saal et al., 2003; Niehaus et al., 2010). I ratti sono stati poi divisi in tre gruppi, ricevendo (i.p.) iniezioni di salina (1 ml / kg), norBNI (10 mg / kg), o 6β-Naltrexolo (10 mg / kg). 24 ore dopo lo stress da nuoto, i ratti nei gruppi salini e norBNI sono stati iniettati e lasciati indisturbati nella loro gabbia di casa per un giorno. I ratti del gruppo 6β-naltrexol sono stati iniettati un’ora prima del test di reintegrazione. A 48 ore dopo lo stress da nuoto, tutti gli animali sono stati sottoposti a test di reintegrazione, che in modo simile all’estinzione ha prodotto la stecca composta, ma non l’infusione di farmaci.
Analisi
L’ampiezza dell’LTP è stata determinata come ampiezza media dell’IPSC per 5 minuti appena prima dell’applicazione dello SNAP rispetto all’ampiezza media dell’IPSC da 10-15 minuti dopo l’applicazione dello SNAP, salvo diversa indicazione. I dati sono presentati come media ± SEM dell’ampiezza percentuale di IPSC normalizzata in IPSC nei 10 minuti precedenti l’applicazione dello SNAP. Per determinare la dimensione del campione non sono stati utilizzati metodi statistici. La dimensione del campione si basava sulle nostre precedenti esperienze e su studi pubblicati in precedenza (Graziane et al., 2013; Polter et al., 2014). Tutte le n riportate sono il numero di animali (repliche biologiche), se non diversamente specificato. La significatività è stata determinata utilizzando un t-test per studenti a due code o un ANOVA a senso unico con un livello di significatività di p<0,05. Tutti i confronti post-hoc sono stati fatti utilizzando il test di Dunnett, se non diversamente specificato. I dati di autosomministrazione sono stati analizzati utilizzando t-test abbinati.
Materiali
IBMX è stato ottenuto da Enzo Life Sciences. NorBNI, U50488 e SNAP sono stati ottenuti da Tocris Biosciences. DNQX, picrotossina, stricnina e 6β-naltrexolo sono stati ottenuti da Sigma-Aldrich. SP600125 è stato ottenuto da Calbiochem.
References
- Baimel C, Bartlett SE, Chiou LC, Lawrence AJ, Muschamp JW, Patkar O, Tung LW, Borgland SL. Orexin/hypocretin role in reward: implications for opioid and other addictions. British Journal of Pharmacology. 2015; 172:334-348. DOI | PubMed
- Baimel C, Lau BK, Qiao M, Borgland SL. Projection-Target-Defined effects of orexin and dynorphin on VTA dopamine neurons. Cell Reports. 2017; 18:1346-1355. DOI | PubMed
- Becker JA, Wallace A, Garzon A, Ingallinella P, Bianchi E, Cortese R, Simonin F, Kieffer BL, Pessi A. Ligands for kappa-opioid and ORL1 receptors identified from a conformationally constrained peptide combinatorial library. Journal of Biological Chemistry. 1999; 274:27513-27522. DOI | PubMed
- Briand LA, Vassoler FM, Pierce RC, Valentino RJ, Blendy JA. Ventral tegmental afferents in stress-induced reinstatement: the role of cAMP response element-binding protein. Journal of Neuroscience. 2010; 30:16149-16159. DOI | PubMed
- Bruchas MR, Chavkin C. Kinase cascades and ligand-directed signaling at the kappa opioid receptor. Psychopharmacology. 2010; 210:137-147. DOI | PubMed
- Bruchas MR, Land BB, Chavkin C. The dynorphin/kappa opioid system as a modulator of stress-induced and pro-addictive behaviors. Brain Research. 2010; 1314:44-55. DOI | PubMed
- Bruchas MR, Yang T, Schreiber S, Defino M, Kwan SC, Li S, Chavkin C. Long-acting kappa opioid antagonists disrupt receptor signaling and produce noncompetitive effects by activating c-Jun N-terminal kinase. Journal of Biological Chemistry. 2007; 282:29803-29811. DOI | PubMed
- Cameron DL, Williams JT. Dopamine D1 receptors facilitate transmitter release. Nature. 1993; 366:344-347. DOI | PubMed
- Chavkin C, Koob GF. Dynorphin, dysphoria, and dependence: the stress of addiction. Neuropsychopharmacology. 2016; 41:373-374. DOI | PubMed
- Chen BT, Bowers MS, Martin M, Hopf FW, Guillory AM, Carelli RM, Chou JK, Bonci A. Cocaine but not natural reward self-administration nor passive cocaine infusion produces persistent LTP in the VTA. Neuron. 2008; 59:288-297. DOI | PubMed
- Chiu TT, Yung LY, Wong YH. Inverse agonistic effect of ICI-174,864 on the cloned delta-opioid receptor: role of G protein and adenylyl cyclase activation. Molecular Pharmacology. 1996; 50:1651-1657. PubMed
- Chou TC, Lee CE, Lu J, Elmquist JK, Hara J, Willie JT, Beuckmann CT, Chemelli RM, Sakurai T, Yanagisawa M, Saper CB, Scammell TE. Orexin (hypocretin) neurons contain dynorphin. Journal of Neuroscience. 2001; 21PubMed
- Conrad KL, McCutcheon JE, Cotterly LM, Ford KA, Beales M, Marinelli M. Persistent increases in cocaine-seeking behavior after acute exposure to cold swim stress. Biological Psychiatry. 2010; 68:303-305. DOI | PubMed
- Corder G, Doolen S, Donahue RR, Winter MK, Jutras BL, He Y, Hu X, Wieskopf JS, Mogil JS, Storm DR, Wang ZJ, McCarson KE, Taylor BK. Constitutive μ-opioid receptor activity leads to long-term endogenous analgesia and dependence. Science. 2013; 341:1394-1399. DOI | PubMed
- Crowley NA, Kash TL. Kappa opioid receptor signaling in the brain: circuitry and implications for treatment. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2015; 62:51-60. DOI | PubMed
- Edwards NJ, Tejeda HA, Pignatelli M, Zhang S, McDevitt RA, Wu J, Bass CE, Bettler B, Morales M, Bonci A. Circuit specificity in the inhibitory architecture of the VTA regulates cocaine-induced behavior. Nature Neuroscience. 2017; 20:438-448. DOI | PubMed
- Ehrich E, Turncliff R, Du Y, Leigh-Pemberton R, Fernandez E, Jones R, Fava M. Evaluation of opioid modulation in major depressive disorder. Neuropsychopharmacology. 2015; 40:1448-1455. DOI | PubMed
- Ehrich JM, Messinger DI, Knakal CR, Kuhar JR, Schattauer SS, Bruchas MR, Zweifel LS, Kieffer BL, Phillips PE, Chavkin C. Kappa opioid Receptor-Induced aversion requires p38 MAPK activation in VTA dopamine neurons. Journal of Neuroscience. 2015; 35:12917-12931. DOI | PubMed
- Endoh T, Matsuura H, Tanaka C, Nagase H. Nor-binaltorphimine: a potent and selective kappa-opioid receptor antagonist with long-lasting activity in vivo. Archives Internationales De Pharmacodynamie Et De Therapie. 1992; 316:30-42. PubMed
- Ford CP, Beckstead MJ, Williams JT. Kappa opioid inhibition of somatodendritic dopamine inhibitory postsynaptic currents. Journal of Neurophysiology. 2007; 97:883-891. DOI | PubMed
- Ford CP, Mark GP, Williams JT. Properties and opioid inhibition of mesolimbic dopamine neurons vary according to target location. Journal of Neuroscience. 2006; 26:2788-2797. DOI | PubMed
- Goeders NE, Guerin GF. Non-contingent electric footshock facilitates the acquisition of intravenous cocaine self-administration in rats. Psychopharmacology. 1994; 114:63-70. DOI | PubMed
- Graziane NM, Polter AM, Briand LA, Pierce RC, Kauer JA. Kappa opioid receptors regulate stress-induced cocaine seeking and synaptic plasticity. Neuron. 2013; 77:942-954. DOI | PubMed
- Haney M, Maccari S, Le Moal M, Simon H, Piazza PV. Social stress increases the acquisition of cocaine self-administration in male and female rats. Brain Research. 1995; 698:46-52. DOI | PubMed
- Iñiguez SD, Vialou V, Warren BL, Cao JL, Alcantara LF, Davis LC, Manojlovic Z, Neve RL, Russo SJ, Han MH, Nestler EJ, Bolaños-Guzmán CA. Extracellular signal-regulated kinase-2 within the ventral tegmental area regulates responses to stress. Journal of Neuroscience. 2010; 30:7652-7663. DOI | PubMed
- Karp JF, Butters MA, Begley AE, Miller MD, Lenze EJ, Blumberger DM, Mulsant BH, Reynolds CF. Safety, tolerability, and clinical effect of low-dose buprenorphine for treatment-resistant depression in midlife and older adults. The Journal of Clinical Psychiatry. 2014; 75:e785-e793. DOI | PubMed
- Kauer JA, Malenka RC. Synaptic plasticity and addiction. Nature Reviews. Neuroscience. 2007; 8:844-858. DOI | PubMed
- Lammel S, Hetzel A, Häckel O, Jones I, Liss B, Roeper J. Unique properties of mesoprefrontal neurons within a dual mesocorticolimbic dopamine system. Neuron. 2008; 57:760-773. DOI | PubMed
- Lammel S, Ion DI, Roeper J, Malenka RC. Projection-specific modulation of dopamine neuron synapses by aversive and rewarding stimuli. Neuron. 2011; 70:855-862. DOI | PubMed
- Lammel S, Lim BK, Ran C, Huang KW, Betley MJ, Tye KM, Deisseroth K, Malenka RC. Input-specific control of reward and aversion in the ventral tegmental area. Nature. 2012; 491:212-217. DOI | PubMed
- Land BB, Bruchas MR, Schattauer S, Giardino WJ, Aita M, Messinger D, Hnasko TS, Palmiter RD, Chavkin C. Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking. PNAS. 2009; 106:19168-19173. DOI | PubMed
- Ling W, Hillhouse MP, Saxon AJ, Mooney LJ, Thomas CM, Ang A, Matthews AG, Hasson A, Annon J, Sparenborg S, Liu DS, McCormack J, Church S, Swafford W, Drexler K, Schuman C, Ross S, Wiest K, Korthuis PT, Lawson W, Brigham GS, Knox PC, Dawes M, Rotrosen J. buprenorphine + naloxone plus naltrexone for the treatment of cocaine dependence: the Cocaine Use Reduction with Buprenorphine (CURB) study. Addiction. 2016; 111:1416-1427. DOI | PubMed
- Liu JG, Prather PL. Chronic exposure to mu-opioid agonists produces constitutive activation of mu-opioid receptors in direct proportion to the efficacy of the agonist used for pretreatment. Molecular Pharmacology. 2001; 60:53-62. PubMed
- Mantsch JR, Baker DA, Funk D, Lê AD, Shaham Y. Stress-Induced reinstatement of drug seeking: 20 years of progress. Neuropsychopharmacology : Official Publication of the American College of Neuropsychopharmacology. 2016; 41:1-22. DOI | PubMed
- Margolis EB, Hjelmstad GO, Bonci A, Fields HL. Kappa-opioid agonists directly inhibit midbrain dopaminergic neurons. Journal of Neuroscience. 2003; 23:9981-9986. PubMed
- Margolis EB, Hjelmstad GO, Bonci A, Fields HL. Both kappa and mu opioid agonists inhibit glutamatergic input to ventral tegmental area neurons. Journal of Neurophysiology. 2005; 93:3086-3093. DOI | PubMed
- Margolis EB, Lock H, Chefer VI, Shippenberg TS, Hjelmstad GO, Fields HL. Kappa opioids selectively control dopaminergic neurons projecting to the prefrontal cortex. PNAS. 2006; 103:2938-2942. DOI | PubMed
- Marinelli M. Stress and Addiction. 2007. DOI
- McCall NM, Kotecki L, Dominguez-Lopez S, Marron Fernandez de Velasco E, Carlblom N, Sharpe AL, Beckstead MJ, Wickman K. Selective ablation of GIRK channels in dopamine neurons alters behavioral effects of cocaine in mice. Neuropsychopharmacology. 2017; 42:707-715. DOI | PubMed
- McFarland K, Davidge SB, Lapish CC, Kalivas PW. Limbic and motor circuitry underlying footshock-induced reinstatement of cocaine-seeking behavior. Journal of Neuroscience. 2004; 24:1551-1560. DOI | PubMed
- McLaughlin JP, Marton-Popovici M, Chavkin C. Kappa opioid receptor antagonism and prodynorphin gene disruption block stress-induced behavioral responses. Journal of Neuroscience. 2003; 23PubMed
- McLaughlin JP, Myers LC, Zarek PE, Caron MG, Lefkowitz RJ, Czyzyk TA, Pintar JE, Chavkin C. Prolonged kappa opioid receptor phosphorylation mediated by G-protein receptor kinase underlies sustained analgesic tolerance. Journal of Biological Chemistry. 2004; 279:1810-1818. DOI | PubMed
- Melief EJ, Miyatake M, Bruchas MR, Chavkin C. Ligand-directed c-Jun N-terminal kinase activation disrupts opioid receptor signaling. PNAS. 2010; 107:11608-11613. DOI | PubMed
- Melief EJ, Miyatake M, Carroll FI, Béguin C, Carlezon WA, Cohen BM, Grimwood S, Mitch CH, Rorick-Kehn L, Chavkin C. Duration of action of a broad range of selective κ-opioid receptor antagonists is positively correlated with c-Jun N-terminal kinase-1 activation. Molecular Pharmacology. 2011; 80:920-929. DOI | PubMed
- Meye FJ, Ramakers GM, Adan RA. The vital role of constitutive GPCR activity in the mesolimbic dopamine system. Translational Psychiatry. 2014; 4DOI | PubMed
- Meye FJ, van Zessen R, Smidt MP, Adan RA, Ramakers GM. Morphine withdrawal enhances constitutive μ-opioid receptor activity in the ventral tegmental area. Journal of Neuroscience. 2012; 32:16120-16128. DOI | PubMed
- Muschamp JW, Hollander JA, Thompson JL, Voren G, Hassinger LC, Onvani S, Kamenecka TM, Borgland SL, Kenny PJ, Carlezon WA. Hypocretin (orexin) facilitates reward by attenuating the antireward effects of its cotransmitter dynorphin in ventral tegmental area. PNAS. 2014; 111:E1648-E1655. DOI | PubMed
- Nasser AF, Greenwald MK, Vince B, Fudala PJ, Twumasi-Ankrah P, Liu Y, Jones JP, Heidbreder C. Sustained-Release buprenorphine (RBP-6000) Blocks the effects of opioid challenge with hydromorphone in subjects with opioid use disorder. Journal of Clinical Psychopharmacology. 2016; 36:18-26. DOI | PubMed
- Niehaus JL, Murali M, Kauer JA. Drugs of abuse and stress impair LTP at inhibitory synapses in the ventral tegmental area. European Journal of Neuroscience. 2010; 32:108-117. DOI | PubMed
- Nugent FS, Niehaus JL, Kauer JA. PKG and PKA signaling in LTP at GABAergic synapses. Neuropsychopharmacology. 2009; 34:1829-1842. DOI | PubMed
- Nugent FS, Penick EC, Kauer JA. Opioids block long-term potentiation of inhibitory synapses. Nature. 2007; 446:1086-1090. DOI | PubMed
- Piazza PV, Deminiere JM, le Moal M, Simon H. Stress- and pharmacologically-induced behavioral sensitization increases vulnerability to acquisition of amphetamine self-administration. Brain Research. 1990; 514:22-26. DOI | PubMed
- Polter AM, Bishop RA, Briand LA, Graziane NM, Pierce RC, Kauer JA. Poststress block of kappa opioid receptors rescues long-term potentiation of inhibitory synapses and prevents reinstatement of cocaine seeking. Biological Psychiatry. 2014; 76:785-793. DOI | PubMed
- Polter AM, Kauer JA. Stress and VTA synapses: implications for addiction and depression. European Journal of Neuroscience. 2014; 39:1179-1188. DOI | PubMed
- Potter DN, Damez-Werno D, Carlezon WA, Cohen BM, Chartoff EH. Repeated exposure to the κ-opioid receptor agonist salvinorin A modulates extracellular signal-regulated kinase and reward sensitivity. Biological Psychiatry. 2011; 70:744-753. DOI | PubMed
- Raehal KM, Lowery JJ, Bhamidipati CM, Paolino RM, Blair JR, Wang D, Sadée W, Bilsky EJ. In vivo characterization of 6beta-naltrexol, an opioid ligand with less inverse agonist activity compared with naltrexone and naloxone in opioid-dependent mice. Journal of Pharmacology and Experimental Therapeutics. 2005; 313:1150-1162. DOI | PubMed
- Ramsey NF, Van Ree JM. Emotional but not physical stress enhances intravenous cocaine self-administration in drug-naive rats. Brain Research. 1993; 608:216-222. DOI | PubMed
- Redila VA, Chavkin C. Stress-induced reinstatement of cocaine seeking is mediated by the kappa opioid system. Psychopharmacology. 2008; 200:59-70. DOI | PubMed
- Robinson JD, McDonald PH. The orexin 1 receptor modulates kappa opioid receptor function via a JNK-dependent mechanism. Cellular Signalling. 2015; 27:1449-1456. DOI | PubMed
- Rogala B, Li Y, Li S, Chen X, Kirouac GJ. Effects of a post-shock injection of the kappa opioid receptor antagonist norbinaltorphimine (norBNI) on fear and anxiety in rats. PLoS One. 2012; 7DOI | PubMed
- Rorick-Kehn LM, Witkin JM, Statnick MA, Eberle EL, McKinzie JH, Kahl SD, Forster BM, Wong CJ, Li X, Crile RS, Shaw DB, Sahr AE, Adams BL, Quimby SJ, Diaz N, Jimenez A, Pedregal C, Mitch CH, Knopp KL, Anderson WH, Cramer JW, McKinzie DL. LY2456302 is a novel, potent, orally-bioavailable small molecule kappa-selective antagonist with activity in animal models predictive of efficacy in mood and addictive disorders. Neuropharmacology. 2014; 77:131-144. DOI | PubMed
- Russell SE, Rachlin AB, Smith KL, Muschamp J, Berry L, Zhao Z, Chartoff EH. Sex differences in sensitivity to the depressive-like effects of the kappa opioid receptor agonist U-50488 in rats. Biological Psychiatry. 2014; 76:213-222. DOI | PubMed
- Saal D, Dong Y, Bonci A, Malenka RC. Drugs of abuse and stress trigger a common synaptic adaptation in dopamine neurons. Neuron. 2003; 37:577-582. DOI | PubMed
- Sadée W, Wang D, Bilsky EJ. Basal opioid receptor activity, neutral antagonists, and therapeutic opportunities. Life Sciences. 2005; 76:1427-1437. DOI | PubMed
- Seifert R, Wenzel-Seifert K. Constitutive activity of G-protein-coupled receptors: cause of disease and common property of wild-type receptors. Naunyn-Schmiedeberg’s Archives of Pharmacology. 2002; 366:381-416. DOI | PubMed
- Shaham Y, Rodaros D, Stewart J. Reinstatement of heroin-reinforced behavior following long-term extinction: implications for the treatment of relapse to drug taking. Behavioural Pharmacology. 1994; 5:360-364. DOI | PubMed
- Shaham Y, Stewart J. Exposure to mild stress enhances the reinforcing efficacy of intravenous heroin self-administration in rats. Psychopharmacology. 1994; 114:523-527. DOI | PubMed
- Shaham Y, Stewart J. Stress reinstates heroin-seeking in drug-free animals: an effect mimicking heroin, not withdrawal. Psychopharmacology. 1995; 119:334-341. DOI | PubMed
- Shoblock JR, Maidment NT. Constitutively active micro opioid receptors mediate the enhanced conditioned aversive effect of naloxone in morphine-dependent mice. Neuropsychopharmacology : Official Publication of the American College of Neuropsychopharmacology. 2006; 31:171-177. DOI | PubMed
- Sirohi S, Walker BM. Maturational alterations in constitutive activity of medial prefrontal cortex kappa-opioid receptors in Wistar rats. Journal of Neurochemistry. 2015; 135:659-665. DOI | PubMed
- Sugita S, Johnson SW, North RA. Synaptic inputs to GABAA and GABAB receptors originate from discrete afferent neurons. Neuroscience Letters. 1992; 134:207-211. DOI | PubMed
- Tan KR, Yvon C, Turiault M, Mirzabekov JJ, Doehner J, Labouèbe G, Deisseroth K, Tye KM, Lüscher C. GABA neurons of the VTA drive conditioned place aversion. Neuron. 2012; 73:1173-1183. DOI | PubMed
- Ting JT, Daigle TL, Chen Q, Feng G. Acute brain slice methods for adult and aging animals: application of targeted patch clamp analysis and optogenetics. Methods in Molecular Biology. 2014; 1183:1-21. DOI | PubMed
- Ungless MA, Grace AA. Are you or aren’t you? challenges associated with physiologically identifying dopamine neurons. Trends in Neurosciences. 2012; 35:422-430. DOI | PubMed
- Ungless MA, Whistler JL, Malenka RC, Bonci A. Single cocaine exposure in vivo induces long-term potentiation in dopamine neurons. Nature. 2001; 411:583-587. DOI | PubMed
- van Zessen R, Phillips JL, Budygin EA, Stuber GD. Activation of VTA GABA neurons disrupts reward consumption. Neuron. 2012; 73:1184-1194. DOI | PubMed
- Van’t Veer A, Carlezon WA. Role of kappa-opioid receptors in stress and anxiety-related behavior. Psychopharmacology. 2013; 229:435-452. DOI | PubMed
- Wang D, Raehal KM, Lin ET, Lowery JJ, Kieffer BL, Bilsky EJ, Sadée W. Basal signaling activity of mu opioid receptor in mouse brain: role in narcotic dependence. Journal of Pharmacology and Experimental Therapeutics. 2004; 308:512-520. DOI | PubMed
- Wang D, Sadée W, Quillan JM. Calmodulin binding to G protein-coupling domain of opioid receptors. Journal of Biological Chemistry. 1999; 274:22081-22088. DOI | PubMed
- Wang D, Sun X, Sadee W. Different effects of opioid antagonists on mu-, delta-, and kappa-opioid receptors with and without agonist pretreatment. Journal of Pharmacology and Experimental Therapeutics. 2007; 321:544-552. DOI | PubMed
- Wang D, Surratt CK, Sadée W. Calmodulin regulation of basal and agonist-stimulated G protein coupling by the mu-opioid receptor (OP(3)) in morphine-pretreated cell. Journal of Neurochemistry. 2000; 75:763-771. DOI | PubMed
- Wang Z, Bilsky EJ, Porreca F, Sadée W. Constitutive mu opioid receptor activation as a regulatory mechanism underlying narcotic tolerance and dependence. Life Sciences. 1994; 54:PL339-PL350. DOI | PubMed
- Wee S, Koob GF. The role of the dynorphin-kappa opioid system in the reinforcing effects of drugs of abuse. Psychopharmacology. 2010; 210:121-135. DOI | PubMed
- Young SR, Chuang SC, Zhao W, Wong RK, Bianchi R. Persistent receptor activity underlies group I mGluR-mediated cellular plasticity in CA3 neuron. Journal of Neuroscience. 2013; 33:2526-2540. DOI | PubMed
- Zhou Y, Leri F, Grella SL, Aldrich JV, Kreek MJ. Involvement of dynorphin and kappa opioid receptor in yohimbine-induced reinstatement of heroin seeking in rats. Synapse. 2013; 67:358-361. DOI | PubMed
Fonte
Polter AM, Barcomb K, Chen RW, Dingess PM, Graziane NM, et al. () Constitutive activation of kappa opioid receptors at ventral tegmental area inhibitory synapses following acute stress. eLife 6e23785. https://doi.org/10.7554/eLife.23785




