
Abstract
Introduzione
La sclerosi laterale amiotrofica (SLA) è una malattia neurodegenerativa che progredisce senza sosta da un sottile declino della funzione motoria a una paralisi respiratoria letale entro pochi anni dalla diagnosi (Pasinelli e Brown, 2006; Taylor et al., 2016). La malattia può essere familiare e causata da mutazioni dominanti in uno dei vari geni, tra cui SOD1, C9orf72, TDP43 e FUS (Taylor et al., 2016). Più comunemente, tuttavia, la malattia è idiopatica.
Sebbene la morte delle cellule motoneuronali sia una caratteristica distintiva della SLA, la perdita delle sinapsi neuromuscolari avviene prima della perdita dei motoneuroni ed è la causa primaria della paralisi motoria sia nelle forme familiari che sporadiche della SLA (Fischer et al., 2004; Schaefer et al., 2005; Pun et al., 2006). Il distacco dei terminali dei nervi motori e il ritiro degli assoni motori ha ricevuto meno attenzione rispetto alla successiva perdita dei motoneuroni, ma gli approcci terapeutici progettati per preservare le sinapsi neuromuscolari hanno il potenziale di mantenere la funzione motoria, soprattutto durante le prime fasi della malattia, e di fornire un beneficio alla qualità della vita del paziente e della famiglia.
I topi transgenici portatori di mutazioni dominanti nel gene SOD1 umano, compresi i topi SOD1-G93A, ricapitolano le caratteristiche distintive della SLA e forniscono il modello animale più studiato per la SLA (Vinsant et al., 2013a; Vinsant et al., 2013b). Inoltre, poiché il distacco dei terminali dei nervi motori è la causa principale della paralisi nei topi SOD1-G93A, i topi SOD1-G93A rappresentano un modello clinicamente rilevante per la SLA.
I percorsi di segnalazione che controllano l’attacco dei terminali degli assoni motori al muscolo stanno solo iniziando a essere compresi, ma due geni, Lrp4 e Muschio, espressi dal muscolo, giocano un ruolo importante. Lrp4, un membro della famiglia dei recettori LDL, è il recettore muscolare per il ligando neuronale critico, Agrin (Kim et al., 2008; Zhang et al., 2008). Dopo aver legato Agrin, Lrp4 si associa con MuSK, un recettore tirosina chinasi, stimolando MuSK e portando all’ancoraggio e alla migliore espressione delle proteine critiche postsinaptiche, tra cui Lrp4 (Burden et al., 2013). Clustered Lrp4 poi segnala di nuovo agli assoni motori per stimolare il loro attaccamento e la differenziazione (Yumoto et al., 2012).
Mutazioni recessive in Agrin, Lrp4 o Musk causano miastenia congenita, un gruppo di disturbi neuromuscolari, distinti dalla SLA, che compromettono la struttura e la funzione delle sinapsi neuromuscolari e portano a debolezza e fatica muscolare (Engel et al., 2015). Inoltre, gli autoanticorpi di Agrin, Lrp4, o MuSK causano miastenia gravis (MG), che è anche distinta dalla SLA (Gilhus e Verschuuren, 2015). Nel MuSK MG, gli anticorpi patogeni sono di solito diretti verso il primo dominio Ig-like in MuSK e riducono la fosforilazione MuSK compromettendo il legame tra Lrp4 e MuSK (Huijbers et al., 2013; Koneczny et al., 2013).
Anche se i difetti nel percorso di segnalazione MuSK non sono associati alla SLA, l’aumento dell’espressione del gene MuSK stabilizza le sinapsi neuromuscolari nei topi SOD1-G93A, riducendo l’estensione della denervazione muscolare e migliorando la funzione motoria (Pérez-García e Burden, 2012). Tuttavia, questi esperimenti hanno utilizzato topi transgenici per aumentare modestamente l’espressione del MuSK dal muscolo, iniziando durante lo sviluppo precoce, diversi mesi prima dell’insorgenza della malattia. Pertanto, il potenziale terapeutico dell’aumento della segnalazione di MuSK come strategia per ridurre la denervazione e migliorare la funzione motoria nei pazienti con diagnosi di SLA è rimasto poco chiaro. In questo caso, abbiamo cercato di determinare se un approccio farmacologico per aumentare l’attività della MuSK in vivo avrebbe preservato le sinapsi neuromuscolari nei topi SOD1-G93A quando il dosaggio è stato avviato dopo l’insorgenza della malattia. Questo tipo di approccio avrebbe migliorato sostanzialmente il potenziale di traduzione nei pazienti affetti da SLA senza i complessi requisiti della terapia genica (Miyoshi et al., 2017).
Risultati
Anticorpi agonisti per MuSK
Uno studio precedente ha identificato ventuno anticorpi a catena singola (scFvs) che riconoscono il MuSK dei topi e ha sollevato l’idea che un sottoinsieme di questi anticorpi possa funzionare come agonisti MuSK in vivo (Xie et al., 1997). Abbiamo studiato l’attività di due anticorpi, #13 e #22, riportati per stimolare il MuSK nei miotoubi in coltura, così come l’anticorpo #21, riportato per legare ma non per stimolare il MuSK. Abbiamo confermato che gli anticorpi #13 e #22, re-ingegnerizzati come molecole umane IgG1, hanno stimolato la fosforilazione della tirosina MuSK nella linea di cellule muscolari di topo C2 (Figura 1A), mentre l’anticorpo #21, così come un anticorpo IgG1 di controllo per il polline di ambrosia, non è riuscito a stimolare la fosforilazione MuSK (Figura 1A).
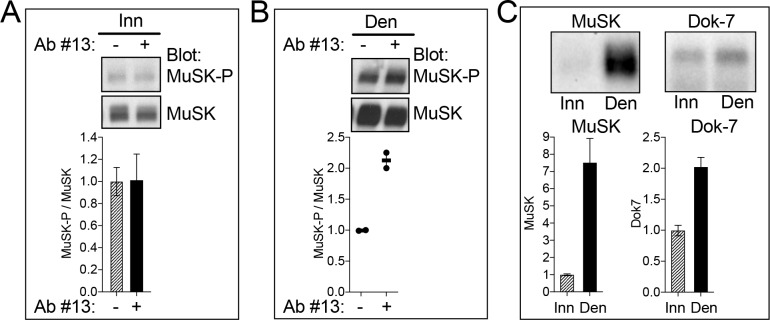
Figura 1-figura supplemento 1.Gli anticorpi agonisti MuSK attivano MuSK, indipendentemente da Lrp4, legando il dominio Fz-like in MuSK.L’anticorpo agonista MuSK #13 stimola la fosforilazione della tirosina MuSK in vivo.(A) I miotoubi C2 sono stati trattati con Agrin neurale o anticorpi per i tempi indicati. Il MuSK è stato immunoprecipitato e le macchie occidentali sono state sondate per il MuSK o la fosfotirosina. Gli anticorpi neurali Agrin e gli anticorpi MuSK #13 e #22 stimolano la fosforilazione della tirosina MuSK nei miotoubi C2, mentre l’anticorpo MuSK #21 e un anticorpo di controllo del polline di ambrosia (Rw) non sono riusciti a stimolare la fosforilazione MuSK. (B) Abbiamo usato un test di legame in fase solida per misurare il legame delle sue proteine MuSK marcate a pozzetti di microtitolazione rivestiti con l’anticorpo agonista MuSK #13. Il diagramma di dispersione mostra che ecto-MuSK a tutta lunghezza (), così come il dominio MuSK Fz-come solo () legano l’anticorpo MuSK # 13 in modo dose-dipendente e saturabile; al contrario, i primi tre domini Ig-like in MuSK () non riescono a legare l’anticorpo agonista MuSK (n = 3). (C) I miotoubi mutanti di tipo selvaggio e Lrp4 sono stati trattati con l’anticorpo agonista neurale Agrin o l’anticorpo agonista MuSK #13. Agrin stimola la fosforilazione MuSK in wild type ma non i miotobi mutanti Lrp4, mentre l’anticorpo agonista MuSK #13 stimola la fosforilazione MuSK sia in wild type che nei miotobi mutanti Lrp4.(A) L’anticorpo agonista MuSK (Ab #13) non è riuscito a stimolare la fosforilazione MuSK nel muscolo innervato (Inn), suggerendo che il MuSK può essere fosforilato al massimo alle sinapsi da Agrin e Lrp4. (B) Dopo la denervazione (Den), le regioni non sinaptiche del muscolo esprimono MuSK, ma non Agrin neurale. Ab #13 ha stimolato la fosforilazione MuSK nel muscolo denervato di 2,2 volte, dimostrando che Ab #13 attiva MuSK in vivo. (C) A seguito della denervazione, l’espressione del MuSK aumenta di 7,5 volte, ma l’espressione del Dok-7, un attivatore essenziale, dentro e fuori del MuSK, aumenta solo di 2,0 volte. Così, l’attività di Ab # 13 nel muscolo denervato può essere limitata da bassa non sinaptica Dok-7 espressione non sinaptica. (A, B) Il rapporto di MuSK-P/MuSK in assenza di Ab #13 è stato assegnato un valore di 1,0. (C) Il livello di espressione di MuSK e Dok-7 nel muscolo innervato è stato assegnato un valore di 1.0. I SEM (n = 3) sono mostrati in (A, C); i valori e le medie di due esperimenti sono mostrati in (B).
Agrin stimola la fosforilazione MuSK legando Lrp4, che promuove l’associazione tra Lrp4 e MuSK, richiedendo il primo dei tre domini Ig-like in MuSK (Zhang et al., 2011). In contrasto con il meccanismo Agrin-dipendente per l’attivazione di MuSK, l’anticorpo agonista lega il dominio Fz-like in MuSK, forza-dimerizzando e stimolando la fosforilazione MuSK, indipendentemente dalla Lrp4 (Figura 1B,C e Figura 1-figure supplement 1). È importante notare che il dominio Fz-like è dispensabile per la formazione di sinapsi nei topi (Remédio et al., 2016).
Figura 1-figure supplement 1.Gli anticorpi agonisti MuSK attivano il MuSK, indipendentemente da Lrp4, legando il dominio Fz-like in MuSK.MuSK l’anticorpo agonista #13 stimola la fosforilazione tirosina MuSK in vivo.(A) I miotoubi C2 sono stati trattati con Agrin neurale o anticorpi per i tempi indicati. Il MuSK è stato immunoprecipitato, e le macchie occidentali sono state sondate per il MuSK o la fosfotirosina. Gli anticorpi neurali Agrin e gli anticorpi MuSK #13 e #22 stimolano la fosforilazione della tirosina MuSK nei miotoubi C2, mentre l’anticorpo MuSK #21 e un anticorpo di controllo del polline di ambrosia (Rw) non sono riusciti a stimolare la fosforilazione MuSK. (B) Abbiamo usato un test di legame in fase solida per misurare il legame delle sue proteine MuSK marcate a pozzetti di microtitolazione rivestiti con l’anticorpo agonista MuSK #13. Il diagramma di dispersione mostra che ecto-MuSK a tutta lunghezza (), così come il dominio MuSK Fz-come solo () legano l’anticorpo MuSK # 13 in modo dose-dipendente e saturabile; al contrario, i primi tre domini Ig-like in MuSK () non riescono a legare l’anticorpo agonista MuSK (n = 3). (C) I miotoubi mutanti di tipo selvaggio e Lrp4 sono stati trattati con l’anticorpo agonista neurale Agrin o l’anticorpo agonista MuSK #13. Agrin stimola la fosforilazione MuSK in wild type ma non i miotobi mutanti Lrp4, mentre l’anticorpo agonista MuSK #13 stimola la fosforilazione MuSK sia in wild type che nei miotobi mutanti Lrp4.(A) L’anticorpo agonista MuSK (Ab # 13) non è riuscito a stimolare la fosforilazione MuSK nel muscolo innervato (Inn), suggerendo che il MuSK può essere fosforilato al massimo alle sinapsi da Agrin e Lrp4. (B) Dopo la denervazione (Den), le regioni non sinaptiche del muscolo esprimono MuSK, ma non Agrin neurale. Ab #13 ha stimolato la fosforilazione MuSK nel muscolo denervato di 2,2 volte, dimostrando che Ab #13 attiva MuSK in vivo. (C) A seguito della denervazione, l’espressione del MuSK aumenta di 7,5 volte, ma l’espressione del Dok-7, un attivatore essenziale, dentro e fuori del MuSK, aumenta solo di 2,0 volte. Così, l’attività di Ab # 13 nel muscolo denervato può essere limitata da bassa non sinaptica Dok-7 espressione non sinaptica. (A, B) Il rapporto di MuSK-P/MuSK in assenza di Ab #13 è stato assegnato un valore di 1,0. (C) Il livello di espressione di MuSK e Dok-7 nel muscolo innervato è stato assegnato un valore di 1.0. I SEM (n = 3) sono mostrati in (A, C); i valori e le medie di due esperimenti sono mostrati in (B).
Figura 1-figure supplement 1.L’anticorpo agonista MuSK #13 stimola la fosforilazione della tirosina MuSK in vivo.(A) L’anticorpo agonista MuSK (Ab #13) non è riuscito a stimolare la fosforilazione MuSK nel muscolo innervato (Inn), suggerendo che il MuSK può essere fosforilato al massimo alle sinapsi da Agrin e Lrp4. (B) Dopo la denervazione (Den), le regioni non sinaptiche del muscolo esprimono MuSK, ma non Agrin neurale. Ab #13 ha stimolato la fosforilazione MuSK nel muscolo denervato di 2,2 volte, dimostrando che Ab #13 attiva MuSK in vivo. (C) A seguito della denervazione, l’espressione del MuSK aumenta di 7,5 volte, ma l’espressione del Dok-7, un attivatore essenziale, dentro e fuori del MuSK, aumenta solo di 2,0 volte. Così, l’attività di Ab # 13 nel muscolo denervato può essere limitata da bassa non sinaptica Dok-7 espressione non sinaptica. (A, B) Il rapporto di MuSK-P/MuSK in assenza di Ab #13 è stato assegnato un valore di 1,0. (C) Il livello di espressione di MuSK e Dok-7 nel muscolo innervato è stato assegnato un valore di 1.0. I SEM (n = 3) sono mostrati in (A, C); i valori e le medie di due esperimenti sono mostrati in (B).
Gli anticorpi agonisti MuSK impegnano il MuSK in vivo
Per determinare se l’anticorpo agonista n. 13 potesse coinvolgere il MuSK in vivo, abbiamo iniettato per via intraperitoneale (IP) quantità variabili dell’anticorpo agonista MuSK su una spina dorsale IgG1 umana, o un anticorpo IgG1 umano di controllo per il polline di ambrosia, in topi di tipo selvatico. Alcuni giorni dopo, abbiamo colorato interi montaggi del muscolo del diaframma per determinare se l’anticorpo agonista ha assunto l’anticorpo MuSK alla sinapsi. Figura 2A mostra che le sinapsi neuromuscolari sono stati etichettati specificamente dall’anticorpo agonista MuSK. MuSK colorazione era evidente già 3 giorni (Figura 2Aiv-vi), e la colorazione persisteva per almeno 7 giorni dopo la singola iniezione (Figura 2A, vii-ix). L’organizzazione di AChR e terminali nervosi apparve normale (Figura 2A,ii,v,viii), indicando che l’anticorpo agonista MuSK non ha disturbato le caratteristiche principali della differenziazione sinaptica. Inoltre, l’osservazione visiva dell’anticorpo iniettato nei topi non ha rivelato anomalie comportamentali evidenti, indicando che l’anticorpo agonista MuSK era ben tollerato dai topi. Due mg / kg di anticorpo agonista era sufficiente per saturare l’etichettatura MuSK alla sinapsi (Figura 2B) e aumentare MuSK tirosina fosforilazione in vivo (Figura 1-figure supplemento 1).
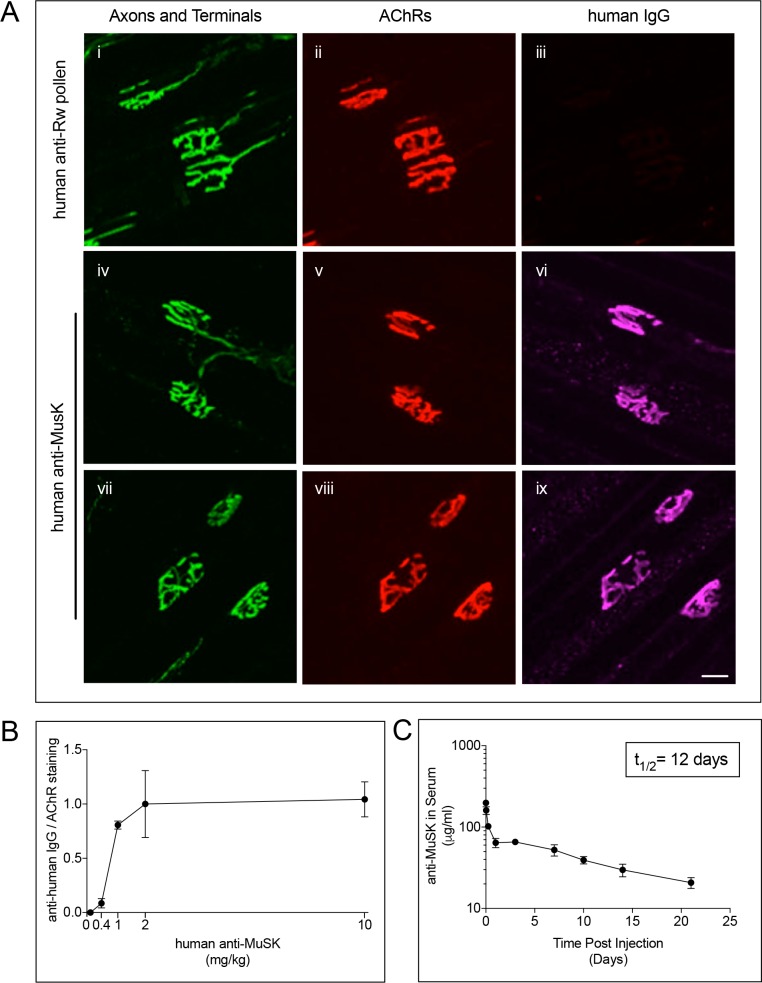
Figura 2.L’anticorpo agonista MuSK #13 innesta il MuSK alla sinapsi poco dopo l’iniezione dell’IP.(A) La colorazione per l’anticorpo MuSK iniettato (10 mg/kg) era evidente già tre giorni (iv-vi) e persisteva per almeno sette giorni dopo una singola iniezione di anticorpo (vii-ix). Un anticorpo umano per il polline di ambrosia (Rw) (10 mg/kg) non è riuscito a macchiare le sinapsi (i-iii) (barra di scala = 20 μm). (B) 2 mg/kg dell’anticorpo agonista MuSK iniettato MuSK saturo di anticorpi alla sinapsi. Il rapporto di colorazione MuSK/AChR a 2 mg / kg di anticorpo è stato assegnato un valore di 1,0 (±SEM, n = 3), ei valori ad altre dosi sono stati espressi in relazione a questo valore. (C) A seguito di una singola iniezione di anticorpo MuSK umano #13 (10mg/kg) il livello di anticorpo nel siero diminuisce con un tempo di dimezzamento di 12,1 giorni. n = 3.
Abbiamo misurato le proprietà farmacocinetiche dell’anticorpo iniettato e abbiamo trovato che l’emivita dell’anticorpo iniettato nel sangue era di ~12 giorni (Figura 2C). L’anticorpo ha mostrato una clearance lineare per 21 giorni dopo l’iniezione dell’anticorpo, indicando che l’esposizione poteva essere mantenuta per diverse settimane. Inoltre, questi risultati hanno dimostrato che il sistema immunitario del topo non riconosceva e cancellava l’anticorpo, che conteneva una regione Fc umana, dalla circolazione durante questo periodo di tre settimane (Figura 2C).
Figura 2.L’anticorpo agonista MuSK # 13 si impegna MuSK alla sinapsi poco dopo l’iniezione di IP.(A) La colorazione per l’anticorpo MuSK iniettato (10 mg/kg) era evidente già tre giorni (iv-vi) e persisteva per almeno sette giorni dopo una singola iniezione di anticorpo (vii-ix). Un anticorpo umano per il polline di ambrosia (Rw) (10 mg/kg) non è riuscito a macchiare le sinapsi (i-iii) (barra di scala = 20 μm). (B) 2 mg/kg dell’anticorpo agonista MuSK iniettato MuSK saturo di anticorpi alla sinapsi. Il rapporto di colorazione MuSK/AChR a 2 mg / kg di anticorpo è stato assegnato un valore di 1,0 (±SEM, n = 3), ei valori ad altre dosi sono stati espressi in relazione a questo valore. (C) A seguito di una singola iniezione di anticorpo MuSK umano #13 (10mg/kg) il livello di anticorpo nel siero diminuisce con un tempo di dimezzamento di 12,1 giorni. n = 3.
Una singola dose di anticorpo agonista MuSK diminuisce la denervazione nei topi SOD1-G93A
Abbiamo studiato i topi SOD1-G93A femmina e maschio, su uno sfondo C57BL/6, con 21-26 copie del gene SOD1-G93A umano (Figura 3-figure supplemento 1). Nei topi SOD1-G93A, denervazione dei muscoli degli arti inizia a P50, mentre la denervazione del muscolo del diaframma inizia un mese dopo (Pun et al., 2006; Rocha et al., 2013). Poiché la denervazione del muscolo del diaframma è responsabile della paralisi respiratoria letale, abbiamo concentrato la nostra analisi sull’innervazione di questo muscolo. Abbiamo prima quantificato l’entità dell’innervazione nel muscolo del diaframma a P90 colorando per i terminali nervosi e AChR postsinaptici, che rimangono anche nei siti sinaptici denervati (Figura 3A). Denervazione era evidente nei topi SOD1-G93A già nel P90 (Figura 3B,C). Da P90 a P110, l’estensione della innervazione completa, definita come perfetta applicazione dei terminali nervosi e la membrana postsinaptica ricca di AChR, è diminuita dal 77,3% al 18,1% nella femmina e dal 53,1% al 16,1% nei topi maschi SOD1-G93A (Figura 3B). Allo stesso modo, l’entità della denervazione completa è aumentata dal 2,3% al 41% nelle femmine e dal 16,7% al 24,4% nei topi maschi SOD1-G93A in questo periodo di venti giorni (Figura 3C).
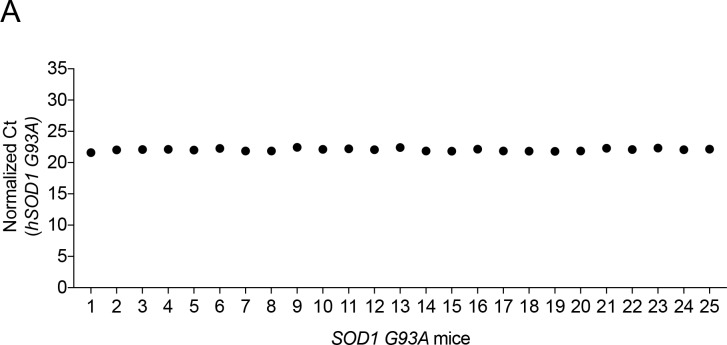
Figura 3-figure supplement 1.Una singola iniezione dell’anticorpo agonista MuSK a P90 ha ridotto la perdita sinaptica per venti giorni nei topi SOD1-G93A. hSOD1-G93A numero di copia è rimasto invariato nel corso delle generazioni e durante gli esperimenti.(A) Le AChR sono concentrate nella membrana postsinaptica a sinapsi innervate e completamente denervate (barra di scala = 20 μm). (B, C) La denervazione è evidente nella femmina ed estesa nei topi maschi SOD1-G93A a P90 (). Nei successivi venti giorni, l’estensione della denervazione completa aumenta e il numero di sinapsi completamente innervate diminuisce (). Una singola iniezione di anticorpo agonista # 13 () riduce l’estensione della denervazione e la perdita di innervazione. Il diagramma di dispersione mostra i valori per i singoli topi (n = 4 o 5), così come i valori medi e SEM; **p<0,01, ****p<0,0001.(A) Il numero di copie del gene SOD1-G93A umano è stato quantificato mediante PCR in tempo reale e normalizzato a GAPDH. Tutti i topi inclusi in questo studio avevano 21-26 copie di hSOD1-G93A. I valori Ct hSOD1-G93A normalizzati sono mostrati per 25 campioni (cerchi riempiti).
SOD1-G93A topi sono stati iniettati con l’anticorpo agonista MuSK a P90. Poiché l’anticorpo aveva un tempo di dimezzamento di 12 giorni e 2 mg / kg di anticorpi saturi MuSK alla sinapsi (Figura 2B,C), abbiamo iniettato topi SOD1-G93A con 10 mg / kg di anticorpo agonista, garantendo che la concentrazione di anticorpi nel sangue rimarrebbe a livelli di saturazione per il legame MuSK nel periodo di 20 giorni. Abbiamo scoperto che una singola dose dell’anticorpo agonista MuSK ha aumentato il numero di sinapsi completamente innervate di 2,7- e 2,5 volte nei topi SOD1-G93A femmina e maschio, rispettivamente, e diminuito il numero di sinapsi completamente denervate di 3,7- e 2,3 volte nei topi SOD1-G93A femmina e maschio, rispettivamente (Figura 3B,C). Questi risultati hanno dimostrato che l’anticorpo agonista MuSK, introdotto dopo l’insorgenza della malattia, ha diminuito il ritiro dell’assone motorio dal muscolo del diaframma.
Figura 3-figure supplemento 1.Una singola iniezione dell’anticorpo agonista MuSK a P90 ha ridotto la perdita sinaptica per venti giorni nei topi SOD1-G93A. hSOD1-G93A numero di copia è rimasto invariato nel corso delle generazioni e nel corso degli esperimenti.(A) Le AChR sono concentrate nella membrana postsinaptica a sinapsi innervate e completamente denervate (barra di scala = 20 μm). (B, C) La denervazione è evidente nella femmina ed estesa nei topi maschi SOD1-G93A a P90 (). Nei successivi venti giorni, l’estensione della denervazione completa aumenta e il numero di sinapsi completamente innervate diminuisce (). Una singola iniezione di anticorpo agonista # 13 () riduce l’estensione della denervazione e la perdita di innervazione. Il diagramma di dispersione mostra i valori per i singoli topi (n = 4 o 5), così come i valori medi e SEM; **p<0,01, ****p<0,0001.(A) Il numero di copie del gene SOD1-G93A umano è stato quantificato mediante PCR in tempo reale e normalizzato a GAPDH. Tutti i topi inclusi in questo studio avevano 21-26 copie di hSOD1-G93A. I valori Ct hSOD1-G93A normalizzati sono mostrati per 25 campioni (cerchi riempiti).
Figura 3-figure supplement 1.hSOD1-G93A numero di copie è rimasto invariato per generazioni e durante gli esperimenti.(A) Il numero di copie del gene SOD1-G93A umano è stato quantificato mediante PCR in tempo reale e normalizzato a GAPDH. Tutti i topi inclusi in questo studio avevano 21-26 copie di hSOD1-G93A. I valori Ct hSOD1-G93A normalizzati sono mostrati per 25 campioni (cerchi riempiti).
Il dosaggio cronico con l’anticorpo agonista MuSK arresta l’ulteriore denervazione nei topi SOD1-G93A per oltre due mesi
Per determinare se l’anticorpo agonista MuSK potesse preservare le sinapsi neuromuscolari per un periodo di tempo più lungo, abbiamo dosato cronicamente i topi SOD1-G93A. Per evitare il riconoscimento dell’ospite e la clearance dell’anticorpo durante l’esposizione cronica, abbiamo usato un anticorpo MuSK #13 su una spina dorsale IgG2a murina che mancava anche di funzioni effettori (Lo et al., 2017). La capacità di questa ‘chimera inversa’ di legare e stimolare il MuSK era simile all’anticorpo con una spina dorsale IgG umana (Figura 4-figure supplement 1). Inoltre, la ‘chimera inversa’ aveva un tempo di dimezzamento simile all’anticorpo umano agonista in vivo (Figura 4-figure supplement 2).
SOD1-G93A topi sono stati iniettati con 10 mg / kg di anticorpo agonista chimera inversa a P90 e ogni 24 giorni dopo, e abbiamo sacrificato topi cronicamente iniettato ogni 24 giorni per quantificare l’innervazione del muscolo del diaframma (Figura 4A). Poiché 2 mg / kg di anticorpo saturo MuSK alla sinapsi e perché l’anticorpo aveva un emivita di 11 giorni nel sangue, questo programma di dosaggio ha assicurato che i livelli di saturazione dell’anticorpo agonista MuSK sono stati mantenuti in ogni momento (Figura 4-figure supplemento 2).
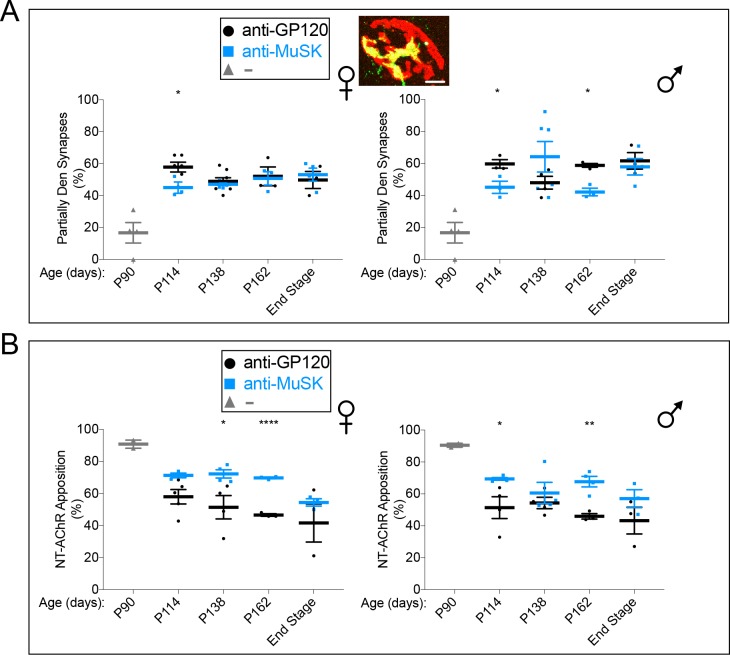
Figura 4-figure supplemento 3.Figura 4— figura 3. Il dosaggio cronico con l’anticorpo agonista MuSK previene l’ulteriore denervazione nei topi SOD1-G93A.le versioni umane e chimera inversa dell’anticorpo agonista MuSK # 13 inducono il raggruppamento del recettore dell’acetilcolina (AChR) nei miotoubi C2C12, mentre un Fab dell’anticorpo MuSK # 13 non riesce a stimolare il raggruppamento AChR.L’anticorpo agonista MuSK #13 ha un tempo di dimezzamento di 11 giorni e il dosaggio cronico con questo anticorpo mantiene l’anticorpo agonista a livelli sufficienti a saturare il MuSK alla sinapsi.il dosaggio cronico con l’anticorpo agonista MuSK aumenta l’estensione della copertura terminale del nervo alle sinapsi parzialmente innervate nei topi SOD1-G93A.(A) L’anticorpo agonista chimera inversa MuSK #13 è stato iniettato a P90 () (B) L’estensione dell’innervazione completa diminuisce progressivamente da P90 a P162 nella femmina e nel maschio SOD1-G93A iniettato con un anticorpo di controllo a GP120 (). L’anticorpo agonista chimera inversa MuSK #13 () arresta questa perdita progressiva, in quanto il numero di sinapsi completamente innervate è invariato tra P114 e P162. (C) La denervazione completa aumenta progressivamente da P90 a P162 nella SOD1-G93A femminile e maschile iniettata con un anticorpo di controllo a GP120 (). L’anticorpo agonista MuSK n. 13 della chimera inversa MuSK impedisce questo progressivo aumento della denervazione, in quanto il numero di sinapsi completamente denervate è invariato tra P114 e P162 (). Allo stadio finale della malattia, il numero di sinapsi completamente innervate e denervate era identico nei topi SOD1-G93A iniettati con l’anticorpo agonista o di controllo MuSK. Il grafico di dispersione mostra i valori per i singoli topi (n = da 3 a 8), così come i valori medi e SEM; *p<0.05, ***p<0.001, ****p<0.0001.(A) Gli anticorpi umani (h) e i chimera inversa (rc) MuSK #13 sono altrettanto efficaci nello stimolare il clustering AChR nei miotoubi C2C12 (n = 3). (B,C) C2C12 miotubi sono stati trattati con rc MuSK # 13 o un Fab da MuSK # 13 per 16 ore alle concentrazioni indicate e colorato con α-BGT. Abbiamo trovato che il frammento Fab dell’anticorpo #13, a differenza delle IgG intatte o dello scFv, non era in grado di stimolare il clustering di AChR, indicando che gli anticorpi devono essere dimerici e forzare-dimerizzare il MuSK e promuovere un orientamento favorevole alla trans-fosforilazione. (C) L’anticorpo rc #13 stimola il clustering di AChR in modo dose-dipendente e saturabile, mentre Fab #13 non riesce a stimolare il clustering di AChR (n = 3).L’anticorpo Rc MuSK #13 è stato prodotto per ridurre al minimo la comparsa di una risposta immunitaria ad un anticorpo umano, così come per eliminare il pericolo di suscitare una risposta immunitaria alla sinapsi neuromuscolare. (A) A seguito di una singola iniezione di 10 mg / kg di anticorpo agonista MuSK inversa chimera MuSK anticorpo #13 nei topi di tipo selvatico, la quantità di anticorpo nel sangue è diminuita nel tempo come una singola esponenziale con un tempo di dimezzamento di 11 giorni. (B) Ripetute iniezioni di 10 mg / kg di anticorpo agonista chimera inversa MuSK anticorpo agonista # 13, ogni 24 giorni, nei topi SOD1-G93A ripristinato i livelli di anticorpi e mantenuto l’anticorpo a livelli che erano sufficienti a saturare MuSK alla sinapsi. Sono mostrati i valori medi per i singoli topi (n = 5) e il SEM.(A) Il numero (~ 50%) di sinapsi parzialmente innervate nei topi SOD1-G93A non è alterato da iniezione cronica di chimera inversa MuSK anticorpo agonista # 13 (). L’inserto mostra una sinapsi parzialmente innervata rappresentativa (barra della scala = 20 μm). (B) L’estensione della membrana postsinaptica ricca di AChR che è apposta dai terminali nervosi (NT) è maggiore nei topi SOD1-G93A iniettati con l’anticorpo agonista MuSK () che con un anticorpo di controllo a GP120 (). Il grafico di dispersione mostra i valori per i singoli topi, così come i valori medi e SEM; *p<0.05, **p<0.01, ****p<0.0001.
Nei topi SOD1-G93A iniettati con un anticorpo di controllo contro GP120, la perdita sinaptica ha continuato a diminuire da P114 a P162, cosicché solo l’11% delle sinapsi è stato completamente innervato a P162 (Figura 4B). Questa perdita progressiva è stata arrestata dall’iniezione dell’anticorpo agonista MuSK, in quanto il numero di sinapsi completamente innervate è rimasto sostanzialmente invariato (40-50%) da P114 a P162 nei topi SOD1-G93A iniettati con l’anticorpo agonista MuSK (Figura 4B). Allo stesso modo, il numero di sinapsi completamente denervate ha continuato ad aumentare da P114 a P162 nei topi SOD1-G93A iniettati con l’anticorpo di controllo, mentre questo aumento progressivo è stato impedito dall’anticorpo agonista MuSK (Figura 4C). Questi risultati indicano che l’anticorpo agonista MuSK ha impedito un’ulteriore perdita sinaptica e ha preservato le sinapsi per almeno 50 giorni dopo che i segni di denervazione e la malattia erano evidenti nei topi SOD1-G93A.
Durante la progressione della malattia, la transizione sinapsi attraverso una fase parzialmente innervata, quando solo una parte della membrana postsinaptica ricca di AChR è apposto da terminali del nervo motore (Figura 4-figure supplemento 3). Anche se il numero di sinapsi parzialmente innervate era simile in topi SOD1-G93A iniettato con il controllo o anticorpo agonista MuSK (Figura 4-figure supplemento 3), l’estensione della copertura dei terminali nervosi era del 34% in più a sinapsi parzialmente innervate in topi iniettati con l’anticorpo agonista MuSK (Figura 4-figure supplemento 3). Così, l’anticorpo agonista MuSK agonista ha aumentato sia l’innervazione completa così come la copertura terminale del nervo a sinapsi parzialmente innervate in topi SOD1-G93A.
Figura 4-figure supplement 3.Il dosaggio cronico con l’anticorpo agonista MuSK impedisce l’ulteriore denervazione nei topi SOD1-G93A.le versioni umane e chimera inversa dell’anticorpo agonista MuSK # 13 inducono il clustering del recettore dell’acetilcolina (AChR) nei miotoubi C2C12 mentre un Fab dall’anticorpo MuSK # 13 non riesce a stimolare il clustering AChR.L’anticorpo agonista MuSK #13 ha un tempo di dimezzamento di 11 giorni e il dosaggio cronico con questo anticorpo mantiene l’anticorpo agonista a livelli sufficienti a saturare il MuSK alla sinapsi.il dosaggio cronico con l’anticorpo agonista MuSK aumenta l’estensione della copertura terminale del nervo alle sinapsi parzialmente innervate nei topi SOD1-G93A.(A) L’anticorpo agonista chimera inversa MuSK #13 è stato iniettato a P90 () (B) L’estensione dell’innervazione completa diminuisce progressivamente da P90 a P162 nella femmina e nel maschio SOD1-G93A iniettato con un anticorpo di controllo a GP120 (). L’anticorpo agonista chimera inversa MuSK #13 () arresta questa perdita progressiva, in quanto il numero di sinapsi completamente innervate è invariato tra P114 e P162. (C) La denervazione completa aumenta progressivamente da P90 a P162 nella SOD1-G93A femminile e maschile iniettata con un anticorpo di controllo a GP120 (). L’anticorpo agonista MuSK n. 13 della chimera inversa MuSK impedisce questo progressivo aumento della denervazione, in quanto il numero di sinapsi completamente denervate è invariato tra P114 e P162 (). Allo stadio finale della malattia, il numero di sinapsi completamente innervate e denervate era identico nei topi SOD1-G93A iniettati con l’anticorpo agonista o di controllo MuSK. Il grafico di dispersione mostra i valori per i singoli topi (n = da 3 a 8), così come i valori medi e SEM; *p<0.05, ***p<0.001, ****p<0.0001.(A) Gli anticorpi umani (h) e i chimera inversa (rc) MuSK #13 sono altrettanto efficaci nello stimolare il clustering AChR nei miotoubi C2C12 (n = 3). (B,C) C2C12 miotubi sono stati trattati con rc MuSK # 13 o un Fab da MuSK # 13 per 16 ore alle concentrazioni indicate e colorato con α-BGT. Abbiamo trovato che il frammento Fab dell’anticorpo #13, a differenza delle IgG intatte o dello scFv, non era in grado di stimolare il clustering di AChR, indicando che gli anticorpi devono essere dimerici e forzare-dimerizzare il MuSK e promuovere un orientamento favorevole alla trans-fosforilazione. (C) L’anticorpo rc #13 stimola il clustering di AChR in modo dose-dipendente e saturabile, mentre Fab #13 non riesce a stimolare il clustering di AChR (n = 3).L’anticorpo Rc MuSK #13 è stato prodotto per minimizzare il verificarsi di una risposta immunitaria ad un anticorpo umano, così come per eliminare il pericolo di suscitare una risposta immunitaria alla sinapsi neuromuscolare. (A) A seguito di una singola iniezione di 10 mg / kg di anticorpo agonista MuSK inversa chimera MuSK anticorpo #13 nei topi di tipo selvatico, la quantità di anticorpo nel sangue è diminuita nel tempo come una singola esponenziale con un tempo di dimezzamento di 11 giorni. (B) Ripetute iniezioni di 10 mg / kg di anticorpo agonista chimera inversa MuSK anticorpo agonista # 13, ogni 24 giorni, nei topi SOD1-G93A ripristinato i livelli di anticorpi e mantenuto l’anticorpo a livelli che erano sufficienti a saturare MuSK alla sinapsi. Sono mostrati i valori medi per i singoli topi (n = 5) e il SEM.(A) Il numero (~ 50%) di sinapsi parzialmente innervate nei topi SOD1-G93A non è alterato dall’iniezione cronica dell’anticorpo agonista MuSK #13 (). L’inserto mostra una sinapsi parzialmente innervata rappresentativa (barra della scala = 20 μm). (B) L’estensione della membrana postsinaptica ricca di AChR che è apposta dai terminali nervosi (NT) è maggiore nei topi SOD1-G93A iniettati con l’anticorpo agonista MuSK () che con un anticorpo di controllo a GP120 (). Il grafico di dispersione mostra i valori per i singoli topi, così come i valori medi e SEM; *p<0.05, **p<0.01, ****p<0.0001.
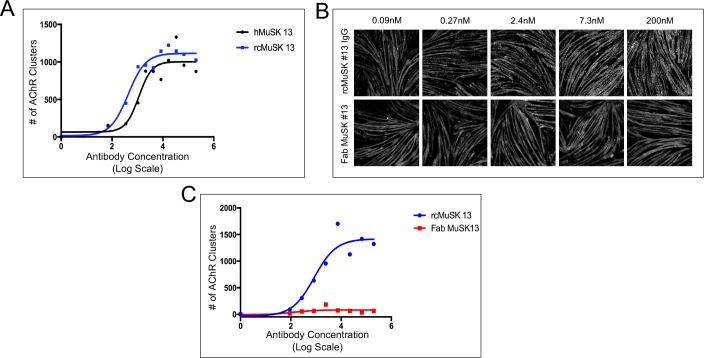
Figura 4-figure supplemento 1.Le versioni umane e chimera inversa dell’anticorpo agonista MuSK # 13 dell’anticorpo agonista MuSK inducono il clustering del recettore dell’acetilcolina (AChR) nei miotoubi C2C12, mentre un Fab dell’anticorpo MuSK # 13 non riesce a stimolare il clustering AChR.(A) Umano (h) e chimera inversa (rc) anticorpi MuSK # 13 sono altrettanto efficaci nello stimolare il clustering AChR nei miotoubi C2C12 (n = 3). (B,C) C2C12 miotubi sono stati trattati con rc MuSK # 13 o un Fab da MuSK # 13 per 16 ore alle concentrazioni indicate e colorato con α-BGT. Abbiamo trovato che il frammento Fab dell’anticorpo #13, a differenza delle IgG intatte o dello scFv, non era in grado di stimolare il clustering di AChR, indicando che gli anticorpi devono essere dimerici e forzare-dimerizzare il MuSK e promuovere un orientamento favorevole alla trans-fosforilazione. (C) L’anticorpo rc #13 stimola il clustering di AChR in modo dose-dipendente e saturabile, mentre Fab #13 non riesce a stimolare il clustering di AChR (n = 3).
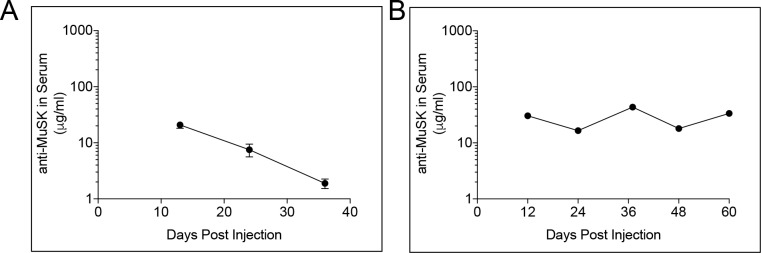
Figura 4-figure supplemento 2.L’anticorpo agonista MuSK n. 13 della chimera inversa ha un tempo di dimezzamento di 11 giorni e il dosaggio cronico con questo anticorpo mantiene l’anticorpo agonista a livelli sufficienti a saturare il MuSK alla sinapsi.L’anticorpo Rc MuSK #13 è stato prodotto per ridurre al minimo la comparsa di una risposta immunitaria ad un anticorpo umano, così come per eliminare il pericolo di suscitare una risposta immunitaria alla sinapsi neuromuscolare. (A) A seguito di una singola iniezione di 10 mg / kg di anticorpo agonista MuSK inversa chimera MuSK anticorpo #13 nei topi di tipo selvatico, la quantità di anticorpo nel sangue è diminuita nel tempo come una singola esponenziale con un tempo di dimezzamento di 11 giorni. (B) Ripetute iniezioni di 10 mg / kg di anticorpo agonista chimera inversa MuSK anticorpo agonista # 13, ogni 24 giorni, nei topi SOD1-G93A ripristinato i livelli di anticorpi e mantenuto l’anticorpo a livelli che erano sufficienti a saturare MuSK alla sinapsi. Sono mostrati i valori medi per i singoli topi (n = 5) e il SEM.
Figura 4-figure supplement 3.Il dosaggio cronico con l’anticorpo agonista MuSK aumenta l’estensione della copertura dei terminali nervosi alle sinapsi parzialmente innervate nei topi SOD1-G93A.(A) Il numero (~ 50%) di sinapsi parzialmente innervate nei topi SOD1-G93A non è alterato dall’iniezione cronica dell’anticorpo agonista MuSK n. 13 (). L’inserto mostra una sinapsi parzialmente innervata rappresentativa (barra della scala = 20 μm). (B) L’estensione della membrana postsinaptica ricca di AChR che è apposta dai terminali nervosi (NT) è maggiore nei topi SOD1-G93A iniettati con l’anticorpo agonista MuSK () che con un anticorpo di controllo a GP120 (). Il grafico di dispersione mostra i valori per i singoli topi, così come i valori medi e SEM; *p<0.05, **p<0.01, ****p<0.0001.
Migliorata l’uscita del sistema motorio del muscolo del diaframma
Per determinare se il mantenimento delle sinapsi neuromuscolari ha portato ad un miglioramento dell’uscita del sistema motorio, abbiamo usato un ex-vivo preparazione del nervo frenico e del muscolo diaframma per misurare i potenziali d’azione del muscolo composto (CMAP), in seguito alla stimolazione del nervo frenico. Abbiamo studiato i topi SOD1-G93A tre o quattro settimane prima della fase finale (Figura 5). Abbiamo stimolato il nervo frenico al muscolo del diaframma e registrato CMAPs, che provocano la contrazione muscolare (Figura 5-figure supplemento 1). Abbiamo trovato nessuna differenza significativa nell’ampiezza del primo CMAP tra i topi SOD1-G93A iniettati con l’anticorpo agonista MuSK o l’anticorpo di controllo per GP120 (anti-GP120 maschi trattati: 5.95 ± 1,14 mV; maschi trattati anti-MuSK: 5,93 ± 0,52 mV; femmine trattate anti-GP120: 4,95 ± 0,54 mV; femmine trattate anti-MuSK: 5,81 ± 0,63 mV). Abbiamo poi misurato l’affidabilità della trasmissione sinaptica alla giunzione neuromuscolare stimolando ripetutamente il nervo frenico ad una frequenza fisiologica (20 Hz). Abbiamo trovato un rapido e grave declino nell’ampiezza del CMAP, indicativo di disfunzione sinaptica e denervazione, in topi SOD1-G93A cronicamente iniettato con l’anticorpo di controllo GP120. Al contrario, il declino dell’ampiezza del CMAP è stato molto meno grave nei topi SOD1-G93A trattati con l’anticorpo agonista MuSK, dimostrando che l’anticorpo agonista MuSK ha migliorato la funzione neuromuscolare (Figura 5). Inoltre, la stimolazione ripetitiva del nervo frenico ad una frequenza più impegnativa (50 Hz) ha portato a frequenti fallimenti di suscitare un CMAP nei topi SOD1-G93A iniettati con l’anticorpo di controllo GP120. Tali fallimenti sono stati meno frequenti nei topi SOD1-G93A iniettati con l’anticorpo agonista MuSK, simile ai topi selvatici (Figura 5). Questi guasti CMAP sono probabilmente dovuti a meccanismi presinaptici, come il blocco di conduzione o il rilascio di neurotrasmettitori compromessi, piuttosto che l’incapacità delle piastre terminali del motore per generare un potenziale d’azione. In entrambi i casi, il mantenimento delle sinapsi neuromuscolari, stimolato dall’anticorpo agonista MuSK, ha portato ad una migliore affidabilità della trasmissione sinaptica e l’uscita del muscolo diaframmatico di importanza critica nei topi SOD1-G93A.
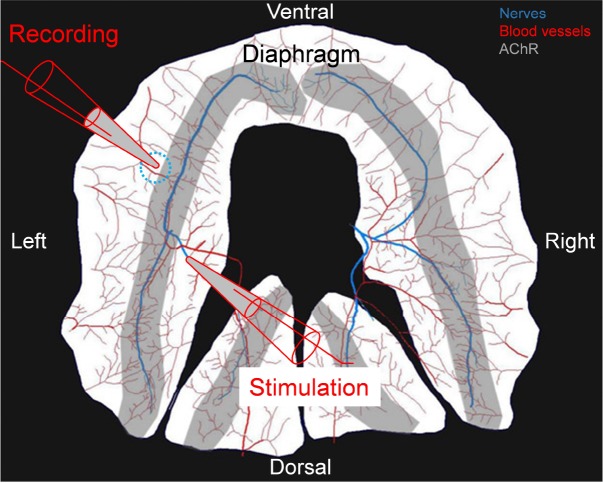
Figura 5-figure supplemento 1.L’anticorpo agonista MuSK migliora la produzione del sistema motorio nel muscolo del diaframma. disegno del protocollo sperimentale per stimolare il nervo frenico e registrare CMAPs nel muscolo del diaframma.(A,B) 20 Hz stimolazione (freccia) del nervo frenico da topi SOD1-G93A iniettato con l’anticorpo di controllo a GP120 ha portato ad un rapido e grave declino dell’ampiezza CMAP. Al contrario, l’ampiezza CMAP è diminuita gradualmente e modestamente nei topi SOD1-G93A iniettati con l’anticorpo agonista MuSK. Dopo 5 s, l’agonista MuSK agonista ha migliorato l’ampiezza CMAP del 13,6% nelle femmine e del 31,7% nei maschi (n = 6-7; p<0,0001). Le deboli linee grigie e blu indicano i SEM. (C,D) 50 Hz stimolazione (S, freccia) del nervo frenico nei topi SOD1-G93A iniettato con l’anticorpo di controllo a GP120 ha portato a frequenti fallimenti (F, freccia) per suscitare un CMAP, mentre CMAP sono stati attendibilmente suscitato in topi SOD1-G93A iniettato con l’anticorpo agonista MuSK, simile al numero di fallimenti visto nei topi di tipo selvaggio. L’anticorpo agonista MuSK ha ridotto il numero di fallimenti dell’88% nelle femmine e del 70% nei maschi durante 1 minuto di stimolazione. Il grafico di dispersione mostra i valori per i singoli topi, così come i valori medi e SEM; *p<0.05, **p<0.01, ***p<0.001. I dati di base dell’ampiezza CMAP sono i seguenti: maschi trattati anti-GP120, 5,95 ± 1,14 mV; maschi trattati con anticorpi agonisti MuSK, 5.93 ± 0,52 mV; femmine trattate con anticorpi anti-GP120, 4,95 ± 0,54 mV; femmine trattate con anticorpi agonisti MuSK, 5,81 ± 0,63 mV.Il nervo frenico è stato posto in un elettrodo di aspirazione bipolare per la stimolazione. CMAP sono stati registrati da un elettrodo di registrazione di aspirazione posto nello stesso luogo (cerchio tratteggiato) attraverso i diversi animali utilizzati in questo studio. Il nervo frenico è mostrato in blu, i vasi sanguigni in rosso e la zona con i recettori dell’acetilcolina in grigio.
Figura 5-figure supplement 1.L’anticorpo agonista MuSK agonista migliora l’uscita del sistema motorio nel muscolo del diaframma. disegno del protocollo sperimentale per stimolare il nervo frenico e registrare CMAPs nel muscolo del diaframma.(A,B) 20 Hz stimolazione (freccia) del nervo frenico da topi SOD1-G93A iniettato con l’anticorpo di controllo a GP120 ha portato ad un rapido e grave declino dell’ampiezza CMAP. Al contrario, l’ampiezza CMAP è diminuita gradualmente e modestamente nei topi SOD1-G93A iniettati con l’anticorpo agonista MuSK. Dopo 5 s, l’agonista MuSK agonista ha migliorato l’ampiezza CMAP del 13,6% nelle femmine e del 31,7% nei maschi (n = 6-7; p<0,0001). Le deboli linee grigie e blu indicano i SEM. (C,D) 50 Hz stimolazione (S, freccia) del nervo frenico nei topi SOD1-G93A iniettato con l’anticorpo di controllo a GP120 ha portato a frequenti fallimenti (F, freccia) per suscitare un CMAP, mentre CMAP sono stati attendibilmente suscitato in topi SOD1-G93A iniettato con l’anticorpo agonista MuSK, simile al numero di fallimenti visto nei topi di tipo selvaggio. L’anticorpo agonista MuSK ha ridotto il numero di fallimenti dell’88% nelle femmine e del 70% nei maschi durante 1 minuto di stimolazione. Il grafico di dispersione mostra i valori per i singoli topi, così come i valori medi e SEM; *p<0.05, **p<0.01, ***p<0.001. I dati di base dell’ampiezza CMAP sono i seguenti: maschi trattati anti-GP120, 5,95 ± 1,14 mV; maschi trattati con anticorpi agonisti MuSK, 5.93 ± 0,52 mV; femmine trattate con anticorpi anti-GP120, 4,95 ± 0,54 mV; femmine trattate con anticorpi agonisti MuSK, 5,81 ± 0,63 mV.Il nervo frenico è stato posto in un elettrodo di aspirazione bipolare per la stimolazione. CMAP sono stati registrati da un elettrodo di registrazione di aspirazione posto nello stesso luogo (cerchio tratteggiato) attraverso i diversi animali utilizzati in questo studio. Il nervo frenico è mostrato in blu, i vasi sanguigni in rosso e la zona con i recettori dell’acetilcolina in grigio.
Figura 5-figure supplement 1.Disegno del protocollo sperimentale per stimolare il nervo frenico e registrare CMAPs nel muscolo del diaframma.Il nervo frenico è stato posto in un elettrodo bipolare di aspirazione per la stimolazione. CMAP sono stati registrati da un elettrodo di registrazione di aspirazione posto nello stesso luogo (cerchio tratteggiato) attraverso i diversi animali utilizzati in questo studio. Il nervo frenico è mostrato in blu, i vasi sanguigni in rosso e la zona con i recettori dell’acetilcolina in grigio.
Anticorpo agonista MuSK diminuisce la perdita di motoneuroni nei topi SOD1-G93A
Abbiamo poi valutato se la conservazione delle sinapsi neuromuscolari nei topi SOD1-G93A ha ridotto la morte dei motoneuroni. Durante lo sviluppo embrionale la morte del motoneurone è regolata dall’innervazione e ridotta quando i motoneuroni fanno sinapsi aggiuntive con il muscolo (Hollyday e Hamburger, 1976; Tanaka e Landmesser, 1986; Landmesser, 1992), mentre la sopravvivenza dei motoneuroni adulti è meno dipendente dall’innervazione muscolare (Lowrie e Vrbová, 1992). Abbiamo quantificato il numero di motoneuroni, colorati per l’acetiltrasferasi colina (ChAT), nel midollo spinale lombare dei topi SOD1-G93A iniettati cronicamente con l’anticorpo di controllo per GP120 o l’anticorpo agonista MuSK (Figura 6A). L’anticorpo agonista MuSK ha aumentato il numero di motoneuroni del 31% al 57% a P138 (Figura 6B), durante il periodo di picco della morte delle cellule motoneuronali nei topi SOD1-G93A quando circa la metà dei motoneuroni spinali sono stati persi (Vinsant et al., 2013a). Questi risultati dimostrano che l’aumento della segnalazione retrograda dopo l’insorgenza della malattia non solo preserva le sinapsi neuromuscolari, ma promuove anche la sopravvivenza dei motoneuroni spinali nei topi SOD1-G93A.
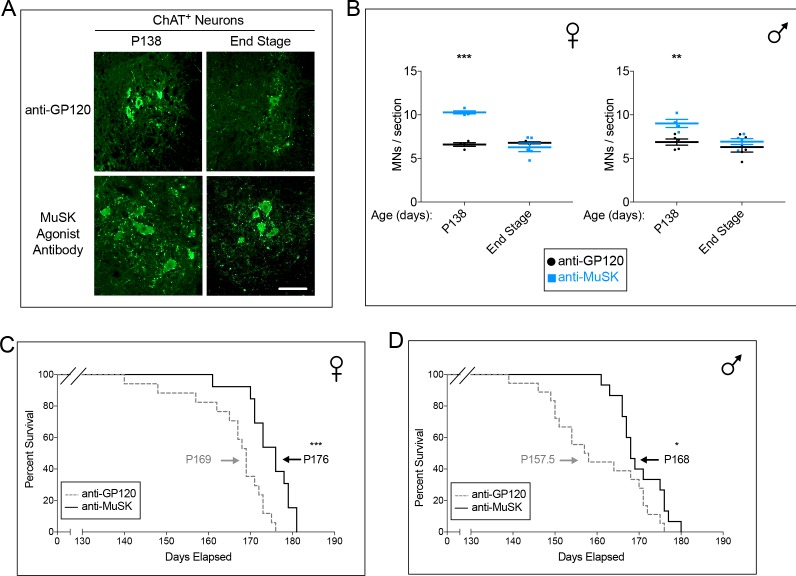
Figura 6.Il dosaggio cronico con l’anticorpo agonista MuSK aumenta la sopravvivenza dei motoneuroni e prolunga la durata della vita dei topi SOD1-G93A.(A) Immagini rappresentative di midollo spinale lombare macchiato con anticorpi per ChAT (barra della scala = 100 μm). (B) A P138, durante il periodo di picco di morte delle cellule motoneuronali, il numero di motoneuroni spinali nell’ingrandimento lombare è maggiore nei topi SOD1-G93A trattati con l’anticorpo agonista per MuSK () che nei topi trattati con l’anticorpo di controllo per GP120 (). Il grafico di dispersione mostra i valori per i singoli topi (n = da 3 a 5), così come i valori medi e SEM; *p<0,05, **p<0,01, ***p<0,001. (C, D) I topi SOD1-G93A femminili e maschi iniettati cronicamente con l’anticorpo di controllo per GP120 hanno una durata di vita rispettivamente di 169 e 157,5 giorni (linea tratteggiata). L’iniezione cronica dell’anticorpo agonista MuSK chimera inversa prolunga la longevità di 7 e 10 giorni nei topi SOD1-G93A femmina e maschio, rispettivamente (linea tratteggiata). n ≥ 13; *p<0,05, **p<0,01, ***p<0,001.
Figura 6.Il dosaggio cronico con l’anticorpo agonista MuSK aumenta la sopravvivenza dei motoneuroni e prolunga la durata della vita dei topi SOD1-G93A.(A) Immagini rappresentative di midollo spinale lombare colorato con anticorpi anti-CHAT (barra di scala = 100 μm). (B) A P138, durante il periodo di picco della morte delle cellule motoneuronali, il numero di motoneuroni spinali nell’ingrandimento lombare è maggiore nei topi SOD1-G93A trattati con l’anticorpo agonista per MuSK () che nei topi trattati con l’anticorpo di controllo per GP120 (). Il grafico di dispersione mostra i valori per i singoli topi (n = da 3 a 5), così come i valori medi e SEM; *p<0,05, **p<0,01, ***p<0,001. (C, D) I topi SOD1-G93A femminili e maschi iniettati cronicamente con l’anticorpo di controllo per GP120 hanno una durata di vita rispettivamente di 169 e 157,5 giorni (linea tratteggiata). L’iniezione cronica dell’anticorpo agonista MuSK chimera inversa prolunga la longevità di 7 e 10 giorni nei topi SOD1-G93A femmina e maschio, rispettivamente (linea tratteggiata). n ≥ 13; *p<0,05, **p<0,01, ***p<0,001.
L’anticorpo agonista MuSK estende la durata della vita dei topi SOD1-G93A
La denervazione del muscolo del diaframma è responsabile della paralisi respiratoria letale nei topi SOD1-G93A e nella SLA. Abbiamo quindi chiesto se il mantenimento delle sinapsi neuromuscolari e il miglioramento della produzione del muscolo del diaframma hanno prolungato la durata della vita dei topi SOD1-G93A. Femmina SOD1-G93A topi SOD1-G93A iniettati con l’anticorpo di controllo per GP120 aveva una durata media di 169 giorni (vedi Materiali e metodi), mentre i topi maschi SOD1-G93A iniettati con l’anticorpo di controllo aveva una durata media di 157,5 giorni (Figura 6C,D). L’iniezione cronica con l’anticorpo agonista MuSK ha prolungato la sopravvivenza dei topi SOD1-G93A femmina e maschio di 7 (p<0,05) e 10 giorni (p<0,001), rispettivamente (Figura 6C,D). Così, l’anticorpo agonista MuSK, introdotto dopo l’insorgenza della malattia, ha rallentato lo smontaggio delle sinapsi neuromuscolari, ha migliorato la potenza motoria del muscolo del diaframma e ha prolungato la durata della vita dei topi SOD1-G93A.
Discussione
La SLA è una malattia devastante che progredisce in modo inesorabile dal distacco dei terminali dei nervi motori alla paralisi respiratoria letale entro diversi anni dalla diagnosi. Attualmente, c’è un bisogno insoddisfatto di terapie che alterano significativamente il corso della malattia. Qui, descriviamo un approccio terapeutico progettato per rallentare la perdita dell’innervazione motoria al muscolo, mirando ad una molecola ben definita e ad un meccanismo per la formazione e il mantenimento delle sinapsi neuromuscolari. Mostriamo che un anticorpo agonista contro la MuSK, introdotto dopo l’insorgenza della malattia, diminuisce la denervazione muscolare, migliora la produzione del sistema motorio, riduce la perdita di motoneuroni e prolunga la sopravvivenza in un modello murino aggressivo di SLA. Se questa strategia, qui descritta per un modello murino aggressivo di SLA, avesse un successo simile nel preservare l’innervazione nella SLA sporadica e familiare, questo approccio terapeutico avrebbe il potenziale di migliorare la qualità della vita dei pazienti SLA, e come tale merita un ulteriore studio.
L’RNA anti-senso diretto verso la SOD1 è attualmente in fase di test come promettente terapeutico per la SLA causata da mutazioni nella SOD1 (Miller et al., 2013). Un approccio simile potrebbe in definitiva essere efficace per altre forme dominanti e familiari di SLA (Reddy e Miller, 2015; van Zundert e Brown, 2017). Tuttavia, a più dell’80% dei pazienti affetti da SLA viene diagnosticata una SLA sporadica, per cui le strategie per inattivare un singolo gene colpevole non sono sostenibili per la maggior parte dei casi di SLA. Invece, saranno probabilmente necessari interventi terapeutici multipli e simultanei che affrontino efficacemente la patologia e i sintomi della SLA per alterare il decorso della malattia (Brown e Al-Chalabi, 2017).
Poiché la perdita sinaptica e la denervazione muscolare sono comuni a forme sporadiche e familiari di SLA, l’approccio qui descritto ha il potenziale per essere efficace per entrambe le forme di SLA. Inoltre, l’aumento dell’attività MuSK e la segnalazione retrograda può anche rallentare il deterioramento delle sinapsi neuromuscolari in altre malattie neuromuscolari e durante l’invecchiamento (Engel et al., 2015; Gilhus e Verschuuren, 2015; Valdez et al., 2012; Poort et al., 2016). Coerentemente con questa idea, l’espressione adenovirale di Dok-7, un attivatore interno-esterno del MuSK, non solo estende la longevità dei topi SOD1-G93A, ma fornisce anche benefici in altri modelli murini di malattie neuromuscolari, tra cui la miastenia congenita e la distrofia muscolare Emery-Dreifuss (Miyoshi et al., 2017; Arimura et al., 2014). Inoltre, è sempre più evidente che la perdita sinaptica si verifica precocemente durante la progressione della malattia in altre malattie neurodegenerative, come il morbo di Alzheimer, il morbo di Huntington, il morbo di Parkinson e la demenza frontotemporale e l’atrofia muscolare spinale (Henstridge et al., 2016), per cui strategie simili, progettate per preservare le sinapsi, possono rallentare la progressione anche in queste malattie.
I nostri esperimenti di proof of concept sono stati progettati per determinare se l’aumento della segnalazione retrograda in vivo con l’anticorpo agonista MuSK potrebbe rallentare il ritiro dell’assone motorio e la denervazione muscolare nei topi SOD1-G93A. Come tale, abbiamo introdotto l’anticorpo agonista MuSK dopo che la denervazione era già evidente, durante la fase iniziale della denervazione nei topi SOD1-G93A femmina e nella fase intermedia nei topi maschi SOD1-G93A, ma prima che i topi SOD1-G93A mostrassero evidenti e gravi deficit nella funzione motoria degli arti. Questa tempistica per la consegna dell’anticorpo agonista MuSK può essere pertinente e significativa per la SLA, in quanto la denervazione è la causa delle fibrillazioni muscolari, un segno clinico precoce nella SLA. Poiché la segnalazione retrograda dipendente dalla MuSK è probabile che agisca in modo focale sui terminali nervosi e sugli assoni che si trovano vicino alla membrana postsinaptica e che sia meno efficace nel promuovere la rigenerazione degli assoni che si sono completamente ritirati, la consegna precoce di un agonista MuSK è probabilmente più efficace rispetto alla consegna successiva nella SLA.
Tuttavia, la SLA è una diagnosi di esclusione, che porta a ritardi nella diagnosi. Tuttavia, anche negli stadi avanzati della malattia, la maggior parte delle sinapsi nei topi SOD1-G93A sono parzialmente innervate, e l’anticorpo agonista MuSK ha migliorato la copertura terminale del nervo in queste sinapsi parzialmente innervate. Questi risultati suggeriscono che l’anticorpo agonista MuSK può anche essere efficace se introdotto successivamente durante la malattia. Tuttavia, poiché i deficit motori evidenti diventano evidenti nei topi SOD1-G93A solo un mese prima della morte, questo modello di topo aggressivo di SLA può non essere il modello ottimale e più informativo per dedurre se l’introduzione successiva dell’anticorpo agonista MuSK può stabilizzare le sinapsi e le disfunzioni motorie lente nella SLA.
La perdita di motoneuroni durante lo sviluppo embrionale è regolata, almeno in parte, dalla formazione di sinapsi (Hollyday and Hamburger, 1976; Tanaka e Landmesser, 1986; Landmesser, 1992). L’aumento della sopravvivenza dei motoneuroni nei topi agonisti MuSK con anticorpi SOD1-G93A ad iniezione di SOD1-G93A indica che i motoneuroni adulti possono anche ricevere supporto trofico dal muscolo. Così, preservando le sinapsi neuromuscolari non solo mantiene l’attaccamento essenziale del nervo al muscolo, ma fornisce anche il beneficio aggiunto di promuovere la sopravvivenza dei motoneuroni.
Anche se abbiamo usato un anticorpo agonista al MuSK per stimolare la segnalazione retrograda dal muscolo, si possono prevedere altri approcci per stimolare il MuSK o migliorare la segnalazione retrograda al fine di mantenere l’attaccamento degli assoni motori al muscolo. L’anticorpo agonista MuSK è efficace nel mantenere le sinapsi neuromuscolari nei topi SOD1-G93A fino a P162, ma entro la prossima settimana le sinapsi si perdono e i topi muoiono. Poiché l’anticorpo agonista MuSK è progettato per mantenere le sinapsi neuromuscolari e non mira direttamente o affrontare la causa sottostante della malattia e di altre patologie nei topi SOD1-G93A e nella SLA, il beneficio di aumentare la segnalazione retrograda dal muscolo al nervo e di promuovere l’attaccamento terminale del nervo è limitato. Tuttavia, anche se l’anticorpo non può annullare le numerose vie patologiche che si verificano nel motoneurone e nelle cellule non neuronali, questo approccio terapeutico ha un potente effetto sul decorso della malattia, riducendo la perdita sinaptica, migliorando la potenza motoria e prolungando la durata della vita dei topi SOD1-G93A più a lungo del riluzolo, l’antico trattamento approvato dalla FDA per la SLA (Jablonski et al., 2014). La morte delle cellule motoneuronali è una caratteristica critica nella SLA, ma l’eliminazione del Bax, che previene la morte delle cellule apoptotiche, non riesce a preservare le sinapsi neuromuscolari e aumenta la sopravvivenza dei topi SOD1-G93A di soli 20 giorni (Gould et al., 2006). Insieme ai nostri studi, questi risultati danno credito all’idea che gli interventi terapeutici combinatoriali, compresi quelli che preservano le sinapsi neuromuscolari, saranno necessari per affrontare pienamente la complessa patologia e i sintomi della SLA e contribuire ad una migliore qualità di vita per il paziente e la famiglia.
Materiali e metodi
| Tipo di reagente (specie) o risorsa | Designazione | Fonte o riferimento | Identificatori | Ulteriori informazioni |
|---|---|---|---|---|
| sforzo, sforzo di fondo (topo) | Il topo: B6.Cg-Tg(SOD1*G93A)1Gur/J | Il Laboratorio Jackson | RRID:IMSR_JAX:004435 | |
| deformazione, sfondo di deformazione (mouse) | Il topo: C57BL/6J | Il Laboratorio Jackson | RRID:IMSR_JAX000664 | |
| linea cellulare (mouse) | Mouse C2C12 cellule muscolari scheletriche del muscolo scheletrico | Laboratorio degli oneri | ATCC Cat# CRL-1772, RRID:CVCL_0188 | |
| linea cellulare (mouse) | Il topo: Cellule muscolari di tipo selvaggio immortalate | Laboratorio di carico | PMID: 18848351 | |
| linea cellulare (topo) | Il topo: Cellule muscolari mutanti Lrp4 immortalate | Laboratorio oneri | PMID: 18848351 | |
| anticorpo | Coniglio anti-Neurofilame-L | Sistemi sinaptici | Cat# 171 002, RRID:AB_887743 | |
| anticorpo | Coniglio anti-Synapsin 1/2 | Sistemi sinaptici | Cat# 106 002, RRID:AB_887804 | |
| anticorpo | Alexa 647-anti-human IgG | Tecnologie per la vita | Cat# A-21445, RRID: AB_2535862 | |
| anticorpo | anti-colina acetiltrasferasi | Millipore | Cat# AB144P-200UL, RRID:AB_90661 | |
| anticorpo | anti-NeuNEuN | Millipore | Cat# MAB377, RRID: AB_2298772 | |
| anticorpo | Coniglio anti-MuSK | Laboratorio oneri | PMID: 10781064 | |
| anticorpo | Capra anti-MuSK | Sistemi R e D | Cat# AF3904, RRID:AB_2147242 | |
| anticorpo | Coniglio anti-Dok-7 | Laboratorio oneri | PMID: 18848351 | |
| anticorpo | Antifosfotrosina per topi 4G10 | Millipore | ||
| peptide, proteina ricombinante | Alexa 594-alfa-bungarotossina | Tecnologie per la vita | Cat#B13423 | |
| peptide, proteina ricombinante | Alexa 488-alfa-bungarotossina | Tecnologie per la vita | Cat#B13422 | |
| peptide, proteina ricombinante | Hoechst 33342 | Thermo Fisher | Cat# 62249 | |
| peptide, proteina ricombinante | Proteina Agrin ricombinante di ratto | Sistemi R e D | Cat#550-AG-100 | |
| saggio commerciale o kit | GAPDH TaqMan Assay Mm00186822_cn | Thermo Fisher | Cat# 4400291 | |
| software, algoritmo | Prisma 7.0 | http://www.graphpad.com/scientific-software/prism/ | RRID:SCR_002798 | |
| software, algoritmo | Software di analisi delle immagini 3D Volocity | http://www.perkinelmer.com/pages/020/cellularimaging/products/volocity.xhtml | RRID:SCR_002668 | |
| software, algoritmo | Suite di software pCLAMP | https://www.moleculardevices.com/systems/axon-conventional-patch-clamp/pclamp-11-software-suite | ||
| altro | DietGel 76A | ClearH20 | Cat#72-07-5022 |
Studio di progettazione
Gli investigatori non erano in grado di sapere se i topi erano stati trattati con l’agonista MuSK o con l’anticorpo di controllo durante l’acquisizione e l’analisi iniziale dei dati. I dati sono presentati come medi ±SEM. I confronti statistici tra i gruppi sono stati analizzati utilizzando un t-test dello studente non accoppiato e a due code, un test log-rank (sopravvivenza), una regressione lineare (CMAP), o un ANOVA a due vie (fallimenti). Le analisi statistiche sono state condotte utilizzando il software Prism 7.0 (software GraphPad). Il numero (n) di topi utilizzati per calcolare la media, i valori SEM e i limiti di confidenza (valori p) sono indicati nelle legende delle figure.
Topi
Il numero di copie del gene SOD1-G93A umano è stato quantificato di routine dalla PCR in tempo reale TaqMan e normalizzato a GAPDH (Life Technologies Assay# Mm00186822_cn). Tutti i topi inclusi in questo studio avevano 21-26 copie di hSOD1-G93A (Figura 3-figure supplement 1). DietGel 76A (ClearH20) è stato posizionato sul pavimento della gabbia in modo che i topi avessero accesso al nutrimento. Altri hanno misurato la durata della vita dei topi SOD1-G93A mettendo i topi sul loro fianco e sacrificando i topi se non erano in grado di raddrizzarsi in 15 s. Poiché eravamo preoccupati che questo saggio riferito sulla funzione muscolare degli arti e non può essere temporalmente allineato con l’ora della morte, abbiamo usato un saggio variante, che ha fornito una misura accurata della longevità. Quando i topi non sono stati in grado di raddrizzarsi per mangiare o bere nel corso di diverse ore, hanno invariabilmente ceduto nel giro di un giorno; abbiamo definito questo tempo come punto finale della malattia e abbiamo sacrificato i topi in questo momento. I topi sono stati alloggiati e mantenuti secondo le linee guida dell’Institutional Animal Use and Care Committee (IACUC).
Istologia
I muscoli del diaframma sono stati macchiati con α-bungarotossina coniugata con Alexa 594 (α-BGT) (Life Technologies, Carlsbad, CA) per marcare gli AChR e gli anticorpi di coniglio contro Neurofilament-L (SYnaptic Systems, Goettingen, Germania) e Synapsin 1/2 (SYynaptic Systems, Goettingen, Germania) per etichettare assoni e terminali nervosi, come descritto in precedenza (Jaworski e Burden, 2006; Friese et al., 2007). Alle sinapsi completamente innervate, la colorazione dei terminali nervosi si è completamente sovrapposta alle AChR postsinaptiche, mentre i terminali nervosi erano assenti dai siti sinaptici originali, contrassegnati dalle AChR, alle sinapsi completamente denervate. Alle sinapsi parzialmente innervate, i terminali nervosi occupavano solo una parte della membrana postsinaptica. Abbiamo esaminato un minimo di 50 sinapsi nel muscolo del diaframma di ogni topo e abbiamo designato ogni sinapsi come completamente innervate, parzialmente innervate o completamente denervate. Ad ogni sinapsi parzialmente innervata, la percentuale di membrana postsinaptica macchiata di AChR che è stata applicata da terminali nervosi macchiati di sinapsi è stata quantificata utilizzando il software di imaging Volocity (PerkinElmer, Waltham, MA). Per visualizzare e quantificare la colorazione dell’anticorpo agonista, contenente Fc umano, alla giunzione neuromuscolare, abbiamo usato un secondario antiumano coniugato con Alexa 647 (Life Technologies, Carlsbad, CA). Interi montaggi di muscoli sono stati ripresi con un microscopio confocale Zeiss LSM800, e il segnale fluorescente è stato quantificato come descritto in precedenza (Jaworski and Burden, 2006; Friese et al., 2007).
I cordoni spinali sono stati sezionati da topi perfetti con il 4% di formaldeide. Sezioni congelate (20 μm) della regione lombare sono stati macchiati con anticorpi contro l’acetiltrasferasi colina (ChAT) (AB144P-200UL da Millipore, Billerica, MA). Abbiamo definito i motoneuroni come cellule nel corno ventrale del midollo spinale lombare che erano positivi per la ChAT, escludendo i neuroni pregangliari ChAT-positivi e Pitx2-positivi. Abbiamo contato solo le cellule macchiate di ChAT con un nucleo chiaramente definito, al fine di evitare il doppio conteggio dei motoneuroni in sezioni multiple. Abbiamo analizzato ~10 sezioni, uniformemente distanziate nell’ingrandimento lombare, che insieme contenevano >50 motoneuroni in ogni topo.
Legame anticorpale e fosforilazione MuSK
Gli anticorpi chimerici sono stati prodotti trasferendo i cDNA che codificano le regioni variabili dell’anticorpo agonista MuSK #13 ai vettori di espressione che contengono la regione costante kappa e IgG2a del mouse. Gli anticorpi agonisti MuSK sono stati prodotti in cellule CHO e purificati con la proteina A e la cromatografia ad esclusione della dimensione. L’attività dell’anticorpo chimera inversa per la stimolazione del clustering di AChRs in miotoubi C2 era simile a quella dell’anticorpo agonista umano per MuSK (Figura 4-figure supplement 1). Frammenti Fab sono stati preparati da digestione proteasi di IgG1 umana IgG1 seguito dalla rimozione di frammenti di IgG e Fc non salvati su una colonna di Proteina A Sepharose e cromatografia esclusione dimensioni.
Abbiamo usato un test di legame in fase solida per misurare il legame tra l’anticorpo agonista MuSK e la regione extracellulare (ecto) (da E22 a T494), i primi tre domini Ig-like (da E22 a I103) o il dominio Frizzled-like (da D312 a K456) del mouse MuSK (Zhang et al., 2011). Maxisorp piastre sono state rivestite con anticorpo agonista MuSK # 13 (5μg/ml), e successivamente incubato con 8-His-tagliato MuSK proteine, seguito da un anticorpo coniugato perossidasi di rafano (HRP) coniugato con 8-His. L’HRP legato è stato quantificato misurando l’attività dell’HRP (Thermo scientific#34028).
C2C12 cellule muscolari sono stati acquistati presso l’ATCC e non sono stati testati per micoplasma prima dell’uso. Queste cellule muscolari C2C12, così come immortalato tipo selvaggio o Lrp4 cellule muscolari mutanti Lrp4, sono stati differenziati e trattati con Agrin neurale (1 nM) o anticorpi (10 nM). MuSK è stato immunoprecipitato da lisati, e l’espressione MuSK e la fosforilazione tirosina MuSK sono stati misurati sondando macchie occidentali, come descritto in precedenza (Herbst e Burden, 2000). Le cellule C2C12 sono state coltivate in piastre di coltura a 24 pozzetti in DMEM con il 10% di siero bovino fetale (FBS) fino a quando i mioblasti erano il 70% confluenti. I mioblasti sono stati poi permesso di differenziare in miotubi sostituendo il FBS con il 2% di siero di cavallo. Dopo 7 giorni, le colture sono state trattate per 16 ore con concentrazioni variabili di anticorpo rcMuSK # 13 o un Fab dall’anticorpo MuSK # 13. Le cellule sono state fissate in paraformaldeide al 4% e colorate con Alexa 488 coniugato – α-BGT. Da ogni pozzetto sono state raccolte da due a quattro immagini e il numero di cluster AChR è stato analizzato con il software imageJ. Neural Agrin (10 nM) (R e D Systems, Minneapolis, MN) è stato utilizzato come controllo positivo per il clustering AChR (dati non mostrati).
I muscoli degli arti posteriori sono stati denervati tagliando il nervo sciatico, come descritto in precedenza (Simon et al., 1992). Quattro giorni dopo la denervazione, i topi sono stati iniettati con l’anticorpo agonista MuSK # 13, e abbiamo misurato l’espressione MuSK e la fosforilazione tirosina MuSK 3 giorni dopo. MuSK e Dok-7 sono stati immunoprecipitati dai lisati, e i loro livelli di espressione sono stati determinati da Western blotting (Herbst and Burden, 2000; Hallock et al., 2010). La fosforilazione della tirosina MuSK è stata misurata sondando Western blotting con l’anticorpo 4G10, come descritto in precedenza (Herbst and Burden, 2000; Hallock et al., 2010).
Registrazione e valutazione dei potenziali d’azione dei muscoli composti (CMAP) dal muscolo del diaframma
Per valutare la funzione della giunzione neuromuscolare nel muscolo del diaframma del topo (Figura 5-figure supplement 1), abbiamo sviluppato una preparazione ex vivo del nervo frenico-diaframma. Abbiamo studiato il muscolo del diaframma da ~ P140 maschi e ~ P150 topi femmina, che è di tre o quattro settimane prima della fase finale, rispettivamente. Non abbiamo utilizzato la preparazione in vivo, descritto da altri (Lepore et al., 2011), perché temevamo che la stimolazione in vivo del nervo frenico, a frequenze da moderate ad alte, avrebbe portato a registrazioni CMAP variabili e inaffidabili, probabilmente a causa di cambiamenti nella posizione degli elettrodi causati dalla contrazione muscolare. Inoltre, un metodo correlato, riportato per registrare dal muscolo del diaframma del topo, utilizza un elettrodo di registrazione di superficie, e probabilmente controlla l’attività di più muscoli toracici (Martin et al., 2015). Così, a seguito di anestesia con il 5% di isoflurano, i topi sono stati decapitati, e il muscolo del diaframma, insieme al nervo frenico, è stato rapidamente isolato e trasferito in una camera di registrazione personalizzata. La camera è stato perfuso continuamente con ossigenato (95% O2 e 5% di CO2) soluzione artificiale liquido cerebrospinale (128,25 mM NaCl, 4 mM KCl, 0,58 mM NaH2PO4, 21 mM NaHCO3, 30 mM D-glucosio, 1,5 mM CaCl2, e 1 mM MgSO4) ad una velocità di ~ 10 ml / min a temperatura ambiente (~ 20-24 ° C). Il nervo frenico che innerva il muscolo emi-diaframma sinistro è stato stimolato disegnando la parte distale del nervo frenico sinistro in un elettrodo di aspirazione (Figura 5-figure supplemento 1). Abbiamo convalidato il corretto posizionamento dell’elettrodo stimolante ispezionando visivamente le contrazioni muscolari dopo la stimolazione del nervo frenico. L’attività EMG è stata registrata utilizzando un elettrodo di aspirazione posto nel quadrante superiore sinistro del muscolo, 1 mm verso il lato costale del nervo intramuscolare principale e la zona della piastra terminale al centro del muscolo. Una leggera aspirazione è stata applicata all’elettrodo di registrazione per assicurare una tenuta ermetica tra la punta dell’elettrodo e le fibre muscolari. In questo modo, il danno al muscolo del diaframma è stato evitato, il che è stato confermato osservando le contrazioni muscolari durante la stimolazione. Il nervo frenico è stato stimolato con impulsi quadrati (0,2 ms di durata) a diverse frequenze (da 1 Hz a 50 Hz) per 60 s. L’intensità della stimolazione è stata progressivamente aumentata dalla soglia, definita come la risposta minima in tre prove su cinque, fino a quando il CMAP ha raggiunto una risposta massima.
L’intensità di stimolazione è stata impostata al doppio dell’intensità richiesta per la risposta massima per garantire un’intensità di stimolazione sovramassimale (da 25µA a 200µA). Le registrazioni sono state accettate per l’analisi solo quando l’ampiezza CMAP (picco-picco) è rimasto invariato dopo 1 Hz stimolazione. Le ampiezze dei CMAP evocati a frequenze più elevate sono stati espressi come percentuale del primo CMAP evocato per tutta la durata della stimolazione. Le registrazioni sono state alimentate ad un’interfaccia A / D (Digidata 1440A, Dispositivi Molecolari) e acquisite con Clampex (v10.2, Dispositivi Molecolari) ad una frequenza di campionamento di 50 kHz. I dati sono stati analizzati off-line utilizzando Clampfit (v10.2, Dispositivi Molecolari). Abbiamo definito i guasti CMAP come l’assenza di una risposta evocata distinguibile dal rumore di fondo registrato prima della stimolazione.
References
- Arimura S, Okada T, Tezuka T, Chiyo T, Kasahara Y, Yoshimura T, Motomura M, Yoshida N, Beeson D, Takeda S, Yamanashi Y. Neuromuscular disease. DOK7 gene therapy benefits mouse models of diseases characterized by defects in the neuromuscular junction. Science. 2014; 345:1505-1508. DOI | PubMed
- Brown RH, Al-Chalabi A. Amyotrophic Lateral Sclerosis. New England Journal of Medicine. 2017; 377:162-172. DOI | PubMed
- Burden SJ, Yumoto N, Zhang W. The role of MuSK in synapse formation and neuromuscular disease. Cold Spring Harbor Perspectives in Biology. 2013; 5DOI | PubMed
- Engel AG, Shen XM, Selcen D, Sine SM. Congenital myasthenic syndromes: pathogenesis, diagnosis, and treatment. The Lancet Neurology. 2015; 14:420-434. DOI | PubMed
- Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Experimental Neurology. 2004; 185:232-240. DOI | PubMed
- Friese MB, Blagden CS, Burden SJ. Synaptic differentiation is defective in mice lacking acetylcholine receptor beta-subunit tyrosine phosphorylation. Development. 2007; 134:4167-4176. DOI | PubMed
- Gilhus NE, Verschuuren JJ. Myasthenia gravis: subgroup classification and therapeutic strategies. The Lancet Neurology. 2015; 14:1023-1036. DOI | PubMed
- Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM, Milligan CE, Oppenheim RW. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. Journal of Neuroscience. 2006; 26:8774-8786. DOI | PubMed
- Hallock PT, Xu CF, Park TJ, Neubert TA, Curran T, Burden SJ. Dok-7 regulates neuromuscular synapse formation by recruiting Crk and Crk-L. Genes & Development. 2010; 24:2451-2461. DOI | PubMed
- Henstridge CM, Pickett E, Spires-Jones TL. Synaptic pathology: A shared mechanism in neurological disease. Ageing Research Reviews. 2016; 28:72-84. DOI | PubMed
- Herbst R, Burden SJ. The juxtamembrane region of MuSK has a critical role in agrin-mediated signaling. The EMBO Journal. 2000; 19:67-77. DOI | PubMed
- Hollyday M, Hamburger V. Reduction of the naturally occurring motor neuron loss by enlargement of the periphery. The Journal of Comparative Neurology. 1976; 170:311-320. DOI | PubMed
- Huijbers MG, Zhang W, Klooster R, Niks EH, Friese MB, Straasheijm KR, Thijssen PE, Vrolijk H, Plomp JJ, Vogels P, Losen M, Van der Maarel SM, Burden SJ, Verschuuren JJ. MuSK IgG4 autoantibodies cause myasthenia gravis by inhibiting binding between MuSK and Lrp4. PNAS. 2013; 110:20783-20788. DOI | PubMed
- Jablonski MR, Markandaiah SS, Jacob D, Meng NJ, Li K, Gennaro V, Lepore AC, Trotti D, Pasinelli P. Inhibiting drug efflux transporters improves efficacy of ALS therapeutics. Annals of Clinical and Translational Neurology. 2014; 1:996-1005. DOI | PubMed
- Jaworski A, Burden SJ. Neuromuscular synapse formation in mice lacking motor neuron- and skeletal muscle-derived Neuregulin-1. Journal of Neuroscience. 2006; 26:655-661. DOI | PubMed
- Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, Huang JH, Hubbard SR, Dustin ML, Burden SJ. Lrp4 is a receptor for Agrin and forms a complex with MuSK. Cell. 2008; 135:334-342. DOI | PubMed
- Koneczny I, Cossins J, Waters P, Beeson D, Vincent A. MuSK myasthenia gravis IgG4 disrupts the interaction of LRP4 with MuSK but both IgG4 and IgG1-3 can disperse preformed agrin-independent AChR clusters. PLoS One. 2013; 8DOI | PubMed
- Landmesser L. The relationship of intramuscular nerve branching and synaptogenesis to motoneuron survival. Journal of Neurobiology. 1992; 23:1131-1139. DOI | PubMed
- Lepore AC, O’Donnell J, Kim AS, Williams T, Tuteja A, Rao MS, Kelley LL, Campanelli JT, Maragakis NJ. Human glial-restricted progenitor transplantation into cervical spinal cord of the SOD1 mouse model of ALS. PLoS One. 2011; 6DOI | PubMed
- Lo M, Kim HS, Tong RK, Bainbridge TW, Vernes JM, Zhang Y, Lin YL, Chung S, Dennis MS, Zuchero YJ, Watts RJ, Couch JA, Meng YG, Atwal JK, Brezski RJ, Spiess C, Ernst JA. Effector-attenuating substitutions that maintain antibody stability and reduce toxicity in mice. Journal of Biological Chemistry. 2017; 292:3900-3908. DOI | PubMed
- Lowrie MB, Vrbová G. Dependence of postnatal motoneurones on their targets: review and hypothesis. Trends in Neurosciences. 1992; 15:80-84. DOI | PubMed
- Martin M, Li K, Wright MC, Lepore AC. Functional and morphological assessment of diaphragm innervation by phrenic motor neurons. Journal of Visualized Experiments. 2015. DOI | PubMed
- Miller TM, Pestronk A, David W, Rothstein J, Simpson E, Appel SH, Andres PL, Mahoney K, Allred P, Alexander K, Ostrow LW, Schoenfeld D, Macklin EA, Norris DA, Manousakis G, Crisp M, Smith R, Bennett CF, Bishop KM, Cudkowicz ME. An antisense oligonucleotide against SOD1 delivered intrathecally for patients with SOD1 familial amyotrophic lateral sclerosis: a phase 1, randomised, first-in-man study. The Lancet Neurology. 2013; 12:435-442. DOI | PubMed
- Miyoshi S, Tezuka T, Arimura S, Tomono T, Okada T, Yamanashi Y. DOK7 gene therapy enhances motor activity and life span in ALS model mice. EMBO Molecular Medicine. 2017; 9:880-889. DOI | PubMed
- Pasinelli P, Brown RH. Molecular biology of amyotrophic lateral sclerosis: insights from genetics. Nature Reviews Neuroscience. 2006; 7:710-723. DOI | PubMed
- Pérez-García MJ, Burden SJ. Increasing MuSK activity delays denervation and improves motor function in ALS mice. Cell Reports. 2012; 2:497-502. DOI | PubMed
- Poort JE, Rheuben MB, Breedlove SM, Jordan CL. Neuromuscular junctions are pathological but not denervated in two mouse models of spinal bulbar muscular atrophy. Human Molecular Genetics. 2016; 25:3768-3783. DOI | PubMed
- Pun S, Santos AF, Saxena S, Xu L, Caroni P. Selective vulnerability and pruning of phasic motoneuron axons in motoneuron disease alleviated by CNTF. Nature Neuroscience. 2006; 9:408-419. DOI | PubMed
- Reddy LV, Miller TM. RNA-targeted Therapeutics for ALS. Neurotherapeutics. 2015; 12:424-427. DOI | PubMed
- Remédio L, Gribble KD, Lee JK, Kim N, Hallock PT, Delestrée N, Mentis GZ, Froemke RC, Granato M, Burden SJ. Diverging roles for Lrp4 and Wnt signaling in neuromuscular synapse development during evolution. Genes & Development. 2016; 30:1058-1069. DOI | PubMed
- Rocha MC, Pousinha PA, Correia AM, Sebastião AM, Ribeiro JA. Early changes of neuromuscular transmission in the SOD1(G93A) mice model of ALS start long before motor symptoms onset. PLoS One. 2013; 8DOI | PubMed
- Schaefer AM, Sanes JR, Lichtman JW. A compensatory subpopulation of motor neurons in a mouse model of amyotrophic lateral sclerosis. The Journal of Comparative Neurology. 2005; 490:209-219. DOI | PubMed
- Simon AM, Hoppe P, Burden SJ. Spatial restriction of AChR gene expression to subsynaptic nuclei. Development. 1992; 114:545-553. PubMed
- Tanaka H, Landmesser LT. Cell death of lumbosacral motoneurons in chick, quail, and chick-quail chimera embryos: a test of the quantitative matching hypothesis of neuronal cell death. Journal of Neuroscience. 1986; 6:2889-2899. PubMed
- Taylor JP, Brown RH, Cleveland DW. Decoding ALS: from genes to mechanism. Nature. 2016; 539:197-206. DOI | PubMed
- Valdez G, Tapia JC, Lichtman JW, Fox MA, Sanes JR. Shared resistance to aging and ALS in neuromuscular junctions of specific muscles. PLoS One. 2012; 7DOI | PubMed
- van Zundert B, Brown RH. Silencing strategies for therapy of SOD1-mediated ALS. Neuroscience Letters. 2017; 636:32-39. DOI | PubMed
- Vinsant S, Mansfield C, Jimenez-Moreno R, Del Gaizo Moore V, Yoshikawa M, Hampton TG, Prevette D, Caress J, Oppenheim RW, Milligan C. Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: part II, results and discussion. Brain and Behavior. 2013a; 3:431-457. DOI | PubMed
- Vinsant S, Mansfield C, Jimenez-Moreno R, Del Gaizo Moore V, Yoshikawa M, Hampton TG, Prevette D, Caress J, Oppenheim RW, Milligan C. Characterization of early pathogenesis in the SOD1(G93A) mouse model of ALS: part I, background and methods. Brain and Behavior. 2013b; 3:335-350. DOI | PubMed
- Xie M-H, Yuan J, Adams C, Gurney A. Direct demonstration of MuSK involvement in acetylcholine receptor clustering through identification of agonist ScFv. Nature Biotechnology. 1997; 15:768-771. DOI
- Yumoto N, Kim N, Burden SJ. Lrp4 is a retrograde signal for presynaptic differentiation at neuromuscular synapses. Nature. 2012; 489:438-442. DOI | PubMed
- Zhang B, Luo S, Wang Q, Suzuki T, Xiong WC, Mei L. LRP4 serves as a coreceptor of agrin. Neuron. 2008; 60:285-297. DOI | PubMed
- Zhang W, Coldefy AS, Hubbard SR, Burden SJ. Agrin binds to the N-terminal region of Lrp4 protein and stimulates association between Lrp4 and the first immunoglobulin-like domain in muscle-specific kinase (MuSK). Journal of Biological Chemistry. 2011; 286:40624-40630. DOI | PubMed
Fonte
Cantor S, Zhang W, Delestrée N, Remédio L, Mentis GZ, et al. () Preserving neuromuscular synapses in ALS by stimulating MuSK with a therapeutic agonist antibody. eLife 7e34375. https://doi.org/10.7554/eLife.34375




