
Abstract
Introduzione
È opinione diffusa che la vita basata sull’RNA abbia preceduto la vita basata sul DNA e sulle proteine durante la storia della vita sulla Terra (Gilbert, 1986; Joyce, 2002). Forse la prova più forte a sostegno di questa ipotesi è il ribosoma, presente in tutte le forme di vita esistenti, che è un enzima RNA che catalizza la sintesi di polipeptidi con RNA (Nissen et al., 2000). Questo presunto residuo del “mondo dell’RNA” non doveva necessariamente persistere nella biologia moderna, ma sarebbe stato necessario per l’invenzione della macchina di traduzione. L’altra molecola chiave di transizione tra l’RNA e la vita basata su DNA/proteine è la trascrittasi inversa, che catalizza la polimerizzazione del DNA dipendente dall’RNA ed è responsabile del mantenimento dell’informazione genetica nella forma più stabile del DNA.
La prima trascrittasi inversa può essere stata o un RNA o un enzima proteico. Il primo sembra plausibile perché un tale enzima potrebbe essere derivato da un RNA-dipendente RNA polimerasi, che sarebbe stato un componente essenziale della vita basata su RNA. È stato sostenuto che la macchina di traduzione richiede più informazioni ereditarie di quante possano essere mantenute dai genomi dell’RNA (Maynard Smith e Szathmáry, 1995), ponendo così l’invenzione del DNA prima dell’invenzione delle proteine. Al contrario, è stato sostenuto che la riduzione biochimica dei ribonucleotidi in deossinucleotidi va oltre le capacità catalitiche dell’RNA (Freeland, 1999), ponendo le proteine prima del DNA, anche se è stata proposta anche una via fotoreducente ai deossinucleotidi (Ritson e Sutherland, 2014). Il presente studio dimostra che un enzima RNA con attività altamente evoluta della RNA polimerasi RNA-dipendente può anche funzionare come trascrittasi inversa, fornendo così un ponte tra il materiale genetico ancestrale e quello contemporaneo senza la necessità di proteine.
Ci sono diversi esempi di enzimi RNA con attività RNA polimerasi RNA-dipendente RNA, che sono stati tutti ottenuti dall’evoluzione in vitro (Ekland e Bartel, 1996; Johnston et al., 2001; McGinness e Joyce, 2002; Sczepanski e Joyce, 2014). La più sofisticata di queste è la polimerasi di classe I, che deriva da una RNA ligasi (Bartel e Szostak, 1993) e catalizza la polimerizzazione dei nucleosidici 5´-trifosfati (NTP). Negli ultimi due decenni, l’attività di questo enzima è stata notevolmente migliorata (Zaher e Unrau, 2007; Wochner et al., 2011), acquisendo più recentemente la capacità di sintetizzare una varietà di RNA funzionali e di catalizzare l’amplificazione esponenziale di RNA brevi (Horning and Joyce, 2016).
La chimica della polimerizzazione del DNA è più impegnativa di quella della polimerizzazione dell’RNA perché la mancanza di un 2´-idrossile nel DNA riduce la nucleofilia del 3´-idrossile adiacente. Sulla base dei valori relativi pKa del 3´-idrossile in RNA o DNA, questa differenza è di ~ 100 volte (Åström et al., 2004), mentre l’aggiunta non enzimatica di monomeri attivati ad un primer RNA o DNA indica una differenza di ~ 10 volte (Wu e Orgel, 1992). Per tutti, tranne che per la forma più recentemente evoluta della RNA polimerasi, un singolo deossinucleoside 5´-trifosfato (dNTP) può essere aggiunto ad un modello di primer RNA legato al modello, ma la successiva aggiunta di dNTP al deossinucleotide 3´-terminale non si verifica (Attwater et al., 2013). La forma più recente della polimerasi ha ~ 100 volte più veloce tasso catalitico e la generalizzazione della sequenza molto maggiore rispetto al suo predecessore (Horning e Joyce, 2016). Come riportato qui, questo enzima è in grado di compensare la minore reattività chimica di un deossinucleotide a 3´terminali e di aggiungere più dNTPs successive in modo temperato dall’RNA.
Una seconda importante differenza tra RNA e DNA è la forte tendenza del primo ad adottare un C3´-endo pucker di zucchero, mentre il secondo favorisce un C2´-endo pucker. Tuttavia, quando parte di un primer legato ad un modello di RNA, sia i ribo- e deossiribonucleotidi tendono ad adottare una conformazione C3´-endo, quindi questo problema è meno probabile che sia un ostacolo nella transizione da una RNA polimerasi ad una DNA polimerasi. Una terza differenza tra RNA e DNA è la presenza di uracile nel primo rispetto alla timina nel secondo, che fornisce un mezzo per distinguere la timina dalla deossiuridina che risulta dalla deaminazione spontanea della deossicitidina. La maggior parte delle polimerasi dell’RNA, compresa la polimerasi di classe I (Attwater et al., 2013), sono indifferenti alla presenza di una sostituzione C5-metile sull’uracile, quindi anche questo non è probabilmente un ostacolo allo sviluppo di una trascrittasi inversa. Una volta che anche un modesto livello di attività della DNA polimerasi dipendente dall’RNA, ci si aspetta che si verifichi un’ottimizzazione evolutiva di tale attività.
Risultati
Attraverso molte generazioni successive di evoluzione in vitro, il ribozima della polimerasi di classe I è stato progressivamente perfezionato in modo da poter aggiungere molti NTP successivi, operare con un rapido tasso catalitico e accettare una vasta gamma di sequenze di template. Tra le innovazioni chiave vi sono state: (1) installazione e ottimizzazione evolutiva di un dominio accessorio per aumentare l’efficienza catalitica (Johnston et al., 2001; Zaher e Unrau, 2007); (2) aggiunta di un dominio di accoppiamento Watson-Crick tra l’estremità 5´ dell’enzima e l’estremità 5´ del template per migliorare il legame del complesso template-primer (Wochner et al., 2011); e (3) scoperta di una costellazione di mutazioni per migliorare la velocità di reazione e la generalità delle sequenze (Horning and Joyce, 2016). Questa forma più recente dell’enzima, la ’24-3 polimerasi’, ha un tasso iniziale di aggiunta di NTP >2 min-1 e può copiare la maggior parte delle sequenze di template.
La 24-3 polimerasi è stato testato per la sua capacità di catalizzare l’RNA-templato aggiunta di dNTPs alla fine 3 ‘di un RNA o primer DNA (Figura 1A). L’enzima è stato trovato per essere in grado di più aggiunte successive dNTP, che non è il caso per i suoi predecessori evolutivi (Attwater et al., 2013). Utilizzando un primer 15mer che si lega ad un modello di RNA complementare, il primer può essere esteso per generare prodotti a tutta lunghezza, insieme ad una scala di prodotti ad estensione parziale (Figura 1B). Per i modelli brevi ricchi di C, come 3´-GCCCCCACCAC-5´ (modello 1) o 3´-GCCCCCACGCCCCCCCUC-3´ (modello 2), una parte sostanziale dei prodotti è a lunghezza intera, mentre per i modelli meno ricchi di C e/o contenenti regioni di struttura secondaria stabile (modelli 3 e 4), il prodotto a lunghezza intera è scarso o nullo. I modelli lunghi e non strutturati ricchi di C, come i modelli 3´-GCCCCCCCACGCCCCCCCCCUCGCCCCCCCCCCCACGCCCCCUC-3´ (modello 5), possono dare origine a prodotti a lunghezza intera, in questo caso richiedendo l’aggiunta di due o più residui di ciascuno dei quattro dNTP.
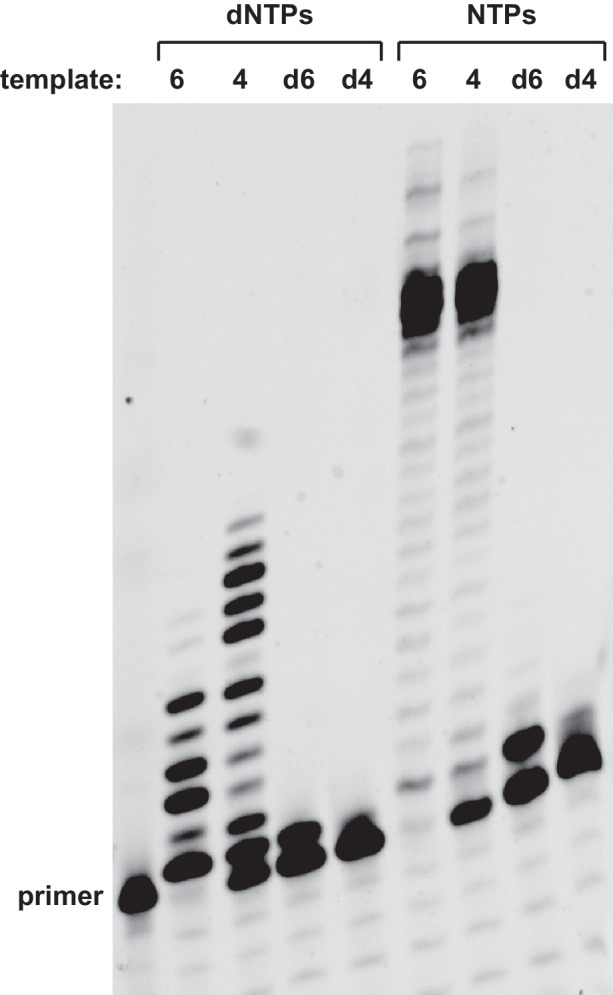
Figura 1-figure supplement 2.Figura 1—figura 2. Attività trascrittasi inversa della trascrittasi del 24-3 ribozyme.attività trascrittasi inversa del 24-3 ribozyme.Lack di attività polimerasi DNA-dipendente del 24-3 ribozyme.(A) Struttura secondaria del complesso formato da ribozima, template e primer (le sequenze nucleotidiche sono elencate nel file supplementare 1). Il template è costituito da quattro regioni: sito di legame del primer, sequenza da copiare, distanziatore A3 o A5 e dominio di accoppiamento del ribozyme (elencati 3´ → 5´). Il ribozyme è stato testato per la sua capacità di copiare cinque diverse sequenze di template (1-5). Per le sequenze di altre regioni del template, vedere il file supplementare 1. (B) Estensione di un primer RNA terminato con deossinucleotide su un template RNA. 2. Condizioni di reazione: 100 nM ribozima, 125 nM template, 125 nM primer, 2 mM ciascuno dNTP, 200 mM MgCl2, pH 8,3 o 9,0, 20°C, 3 o 22 ore. I punti neri indicano la posizione prevista dei prodotti a lunghezza piena.Estensione di (A) primer tutto l’RNA, (B) primer deossinucleotide-terminato RNA, o (C) primer tutto l’RNA su un modello di RNA. Condizioni di reazione: 100 nM ribozima, 125 nM template 1, 125 nM primer, 2 mM ciascuno dNTP, 200 mM MgCl2, pH 8,3 o 9,0, 20°C, 3 o 22 ore. I punti neri indicano la posizione prevista dei prodotti a lunghezza piena.Estensione di un primer all-RNA su un modello di RNA o DNA, utilizzando dNTP o NTP. Le sequenze dei modelli di RNA (6 e 4) e i corrispondenti modelli di DNA (d6 e d4) sono elencati nel file supplementare 1. 2. Condizioni di reazione: 100 nM ribozima, 125 nM template, 125 nM primer, 2 mM ciascuno dNTP o NTP, 200 mM MgCl2, pH 8,3, 20°C, 21 ore.
Per tutti i modelli testati, la reazione procede in modo simile utilizzando un primer tutto RNA o un primer RNA che ha un singolo deossinucleotide a 3´terminali (Figura 1-figure supplement 1). Quando si usa un primer all-DNA, la reazione procede in modo simile per i modelli più favorevoli, ma è meno efficiente per i modelli più impegnativi. Questo comportamento riflette presumibilmente la maggiore difficoltà dell’ibridazione tra DNA e RNA quando il legame del primer deve competere con la struttura secondaria nella regione di legame del primer del template. L’enzima 24-3 ha un’attività trascurabile nella polimerizzazione DNA-temperata di dNTP o NTP (Figura 1-figure supplement 2). Questo è vero anche quando si usa un primer all-RNA, che permette l’aggiunta di un singolo nucleotide, ma quasi nessuna aggiunta successiva di nucleotidi.
Sono stati adottati due approcci per confermare l’identità dei prodotti di trascrizione inversa ottenuti utilizzando il modello 1. In primo luogo, i presunti materiali a lunghezza totale, iniziati da un primer tutto RNA o tutto DNA, sono stati purificati mediante elettroforesi in gel di poliacrilamide denaturante e sottoposti a digestione parziale con DNasi I. Questo enzima degrada i legami 3´,5´-phosphodiester nel DNA ma non l’RNA. Per i prodotti con RNA, la porzione estesa è stata degradata dalla DNasi e la porzione di primer è rimasta intatta, mentre per i prodotti con DNA è stata degradata l’intera molecola (Figura 2A). Gli standard autentici sono stati trattati in modo parallelo e hanno dato origine allo stesso schema di prodotti di degradazione.

Figura 2-figure supplement 1-source data 1.Analisi dei prodotti di trascrizione inversa.analisi LC/MS di prodotti a lunghezza intera.analisi LC/MS di prodotti a lunghezza intera.analisi MS/MS ad alta risoluzione MS/MS di prodotti di trascrizione inversa.analisi MS/MS ad alta risoluzione di prodotti di trascrizione inversa (in relazione alla Figura 2-figure supplement 1).analisi MS/MS ad alta risoluzione di prodotti di trascrizione inversa (in relazione alla Figura 2-figure supplement 1).(A) parziale DNasi I digestione parziale di prodotti a lunghezza intera ottenuti utilizzando il modello 1, in confronto ai materiali autentici, con un primer RNA (a sinistra 7 corsie) o primer DNA (a destra 7 corsie). Per la reazione preparata con RNA, viene scissa solo la porzione estesa; per la reazione preparata con DNA, sia il primer che la porzione estesa vengono scisse. (B,C) Analisi LC/MS di prodotti purificati a lunghezza intera ottenuti utilizzando il modello 1 e o un RNA o un primer del DNA, rispettivamente.10.7554/eLife.31153.008Cifra dati a 2 fonti 1.LC/MS analisi di prodotti a lunghezza intera.L’estensione di un primer tutto RNA sul modello quattro ha prodotto un prodotto con una massa calcolata di 8874.432 e una massa osservata di 8874.441. Lo ione genitore è stato frammentato a legami internucleotidici all’interno della porzione di DNA della molecola per generare ioni secondari che sono stati analizzati da MS tandem. I frammenti successivi a-h corrispondono a 3´terminali successive all’interno della porzione di DNA della molecola.10.7554/eLife.31153.009Figure 2-figure supplement 1-source data 1.High-resolution MS/MS analysis of reverse transcription product (related to Figure 2-figure supplement 1).
Il secondo approccio di conferma ha riguardato l’analisi dei materiali purificati in gel, a tutta lunghezza, mediante cromatografia liquida / spettrometria di massa. L’RNA o il primer del DNA conteneva un’etichetta di 5´-fluoresceina per consentire la visualizzazione nel gel e la reazione comportava l’aggiunta di residui deossinucleotidi aventi la sequenza 5´-CGGGGGGTG-3´. Per la reazione RNA-primed la massa calcolata era 8252.4 e la massa osservata era 8252.4 (Figura 2B); per la reazione DNA-primed la massa calcolata era 8068.5 e la massa osservata era 8068.1 (Figura 2C). La trappola ionica tandem MS ad alta risoluzione è stata utilizzata per confermare la sequenza della trascrizione inversa 10mer ottenuta utilizzando il modello 4. Questo prodotto a lunghezza parziale contiene tutti e quattro i deossinucleotidi e ha la sequenza 5´-GCGGAGGAGTG-3´. Per la reazione RNA-primed, la massa calcolata era 8874.432 e la massa osservata era 8874.441. Dallo ione genitore, sono stati generati frammenti di 3´terminali che contenevano 2-9 deossinucleotidi e avevano osservato masse corrispondenti alle masse calcolate per questi materiali (Figura 2-figure supplement 1).
Per indagare ulteriormente la fedeltà della trascrizione inversa, la reazione è stata effettuata in presenza di tutti e quattro i dNTP o in una miscela priva di dGTP, dATP, TTP o dCTP. Sono stati testati i modelli 3 e 4, entrambi contenenti tutti e quattro i nucleotidi e diretti alla sintesi di prodotti a lunghezza parziale contenenti rispettivamente fino a 6 o 12 deossinucleotidi. Per entrambi i modelli, la distribuzione dimensionale dei prodotti è stata la stessa quando sono stati utilizzati tutti e quattro i dNTP o una miscela che mancava di dATP (Figura 3). Per le reazioni senza dATP, tuttavia, la mobilità del gel è stata alterata nel sito di incorporazione dATP, coerente con l’errata incorporazione di dGTP come una coppia G-U traballante. La 24-3 polimerasi è nota per tollerare l’accoppiamento traballante del G-U durante la polimerizzazione con RNA temperato (Horning and Joyce, 2016), quindi non sorprende che ciò avvenga anche durante la trascrizione inversa. Al contrario, l’omissione di dGTP, TTP, o dCTP ha portato alla cessazione della polimerizzazione del DNA nel sito del dNTP mancante.
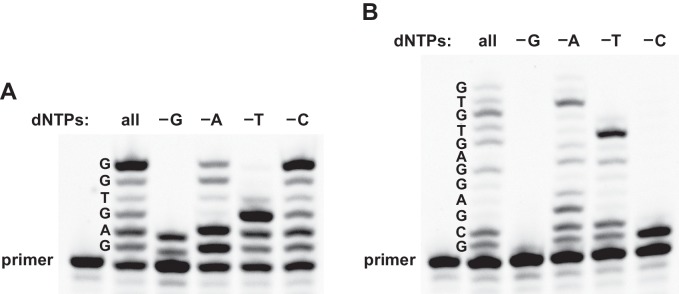
Figura 3.Trascrizione inversa in miscele di reazione prive di uno dei quattro dNTPs.(A) Per la sequenza 5´-GAGTGG-3´ (modello 3), il materiale di lunghezza prevista è stato ottenuto in presenza di tutti e quattro i dNTP o di una miscela priva di dCTP, il materiale di lunghezza prevista di mobilità alterata è stato ottenuto in una miscela priva di dATP, e solo il materiale di lunghezza parziale è stato ottenuto in una miscela priva di dGTP o TTP. (B) Per la sequenza 5´-GCGGAGGAGTGGTGTG-3´ (modello 4), il materiale di lunghezza prevista è stato ottenuto in presenza di tutti e quattro i dNTP, il materiale di lunghezza prevista di mobilità alterata è stato ottenuto in una miscela priva di dATP, e solo materiale di lunghezza parziale è stato ottenuto in una miscela priva di dGTP, TTP, o dCTP. Condizioni di reazione: 100 nM ribozyme, 125 nM template, 125 nM primer, 2 mM dNTPs, 200 mM MgCl2, pH 8,3, 20°C, 22 hr.
Sono stati effettuati esperimenti di durata per determinare il tasso di trascrizione inversa, estendendo un primer RNA con un singolo deossinucleotide a 3′ terminali, rispetto al tasso di polimerizzazione dell’RNA dipendente dall’RNA, estendendo un primer tutto RNA. Questi esperimenti hanno impiegato 100 nM primer, 125 nM modello 1, 125 nM enzima nM, 2 mM ciascuno dei quattro dNTP o NTP, e 200 mM MgCl2, e sono stati effettuati a pH 8,3 ° C e 20 ° C. La reazione ha una rapida fase iniziale di scoppio, seguita da una seconda fase più lenta che continua fino a >90% delle molecole del primer sono estese (Figura 4). Per la polimerizzazione del DNA, la velocità della fase iniziale di scoppio è di 1,1 min-1, procedendo in una misura di ~ 35%, seguita da una seconda fase con un tasso di 0,029 min-1. Per la polimerizzazione dell’RNA, il tasso di scoppio iniziale è >2,0 min-1, procedendo in misura di ~20%, seguito da una seconda fase con un tasso di 0,073 min-1. Il motivo della cinetica bifasica non è chiaro. La fase veloce riflette presumibilmente la frazione di complessi enzimatico-templato-primerici presenti all’inizio della reazione, mentre la seconda fase più lenta può riflettere la formazione di ulteriori complessi reattivi.
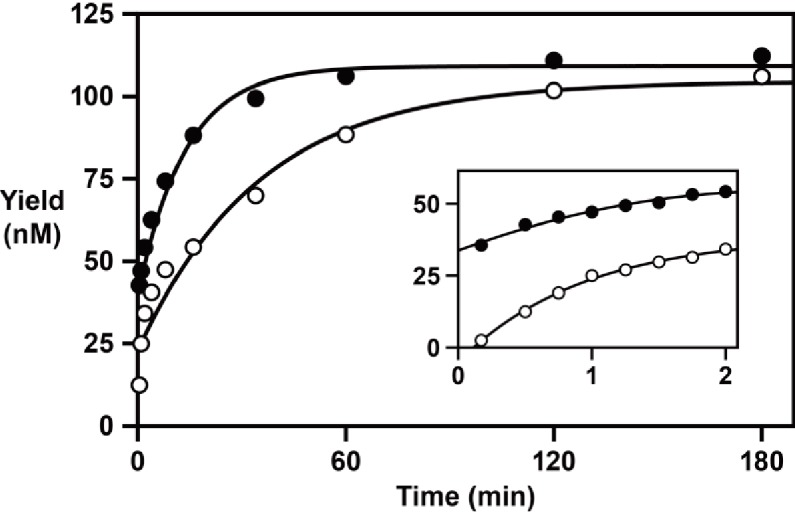
Figura 4 dati fonte 1.RNA-dipendente RNA e attività della DNA polimerasi del 24-3 ribozyme.RNA-dipendente RNA e attività della DNA polimerasi.RNA-dipendente RNA e attività della DNA polimerasi.Il corso temporale della reazione utilizzando NTP (cerchi riempiti) o dNTP (cerchi aperti), misurando la velocità di aggiunta di un singolo nucleotide a un primer deossinucleotide-terminato sul modello 1. I dati erano adatti ad un doppio aumento esponenziale al massimo (r = 0,996 per la polimerizzazione dell’RNA; r = 0,983 per la polimerizzazione del DNA). L’inserto illustra i dati relativi ai primi 2 minuti della reazione. Condizioni di reazione: 100 nM ribozyme, 125 nM template, 125 nM primer, 2 mM NTPs o dNTPs, 200 mM MgCl2, pH 8,3, 20°C.10,7554/eLife.31153.012Cifra dati a 4 fonti 1.RNA-dipendente dall’RNA e dall’attività della DNA polimerasi.
Trascrizione inversa è accelerata a pH 9,0 rispetto a pH 8,3 (Figura 1B). Per le reazioni avviate da un primer RNA, il pH più alto si traduce in una certa degradazione della porzione di primer dei prodotti estesi. L’enzima RNA ha un elevato fabbisogno di Mg2+, tipicamente 200 mM, che promuove anche la degradazione dell’RNA. Tuttavia, una volta che le informazioni sulla sequenza sono state copiate dall’RNA al DNA, il prodotto DNA può essere mantenuto in condizioni di alta H, alto Mg2+.
Figura 1-figure supplement 2.Figura 1—figura 2. Attività trascriptasi inversa del ribozyme 24-3. Attività trascriptasi inversa del ribozyme 24-3. Mancanza di attività polimerasi DNA-dipendente del ribozyme 24-3.(A) Struttura secondaria del complesso formato da ribozyme, template e primer (le sequenze nucleotidiche sono elencate nel file supplementare 1). Il template è costituito da quattro regioni: sito di legame del primer, sequenza da copiare, distanziatore A3 o A5 e dominio di accoppiamento del ribozyme (elencati 3´ → 5´). Il ribozyme è stato testato per la sua capacità di copiare cinque diverse sequenze di template (1-5). Per le sequenze di altre regioni del template, vedere il file supplementare 1. (B) Estensione di un primer RNA terminato con deossinucleotide su un template RNA. 2. Condizioni di reazione: 100 nM ribozima, 125 nM template, 125 nM primer, 2 mM ciascuno dNTP, 200 mM MgCl2, pH 8,3 o 9,0, 20°C, 3 o 22 ore. I punti neri indicano la posizione prevista dei prodotti a lunghezza piena.Estensione di (A) primer all-RNA, (B) primer deossinucleotide-terminato RNA, o (C) primer all-DNA su un modello di RNA. Condizioni di reazione: 100 nM ribozima, 125 nM template 1, 125 nM primer, 2 mM ciascuno dNTP, 200 mM MgCl2, pH 8,3 o 9,0, 20°C, 3 o 22 ore. I punti neri indicano la posizione prevista dei prodotti a lunghezza piena.Estensione di un primer all-RNA su un modello di RNA o DNA, utilizzando sia dNTPs o NTPs. Le sequenze dei modelli di RNA (6 e 4) e i corrispondenti modelli di DNA (d6 e d4) sono elencati nel file supplementare 1. 2. Condizioni di reazione: 100 nM ribozima, 125 nM template, 125 nM primer, 2 mM ciascuno dNTP o NTP, 200 mM MgCl2, pH 8,3, 20°C, 21 ore.
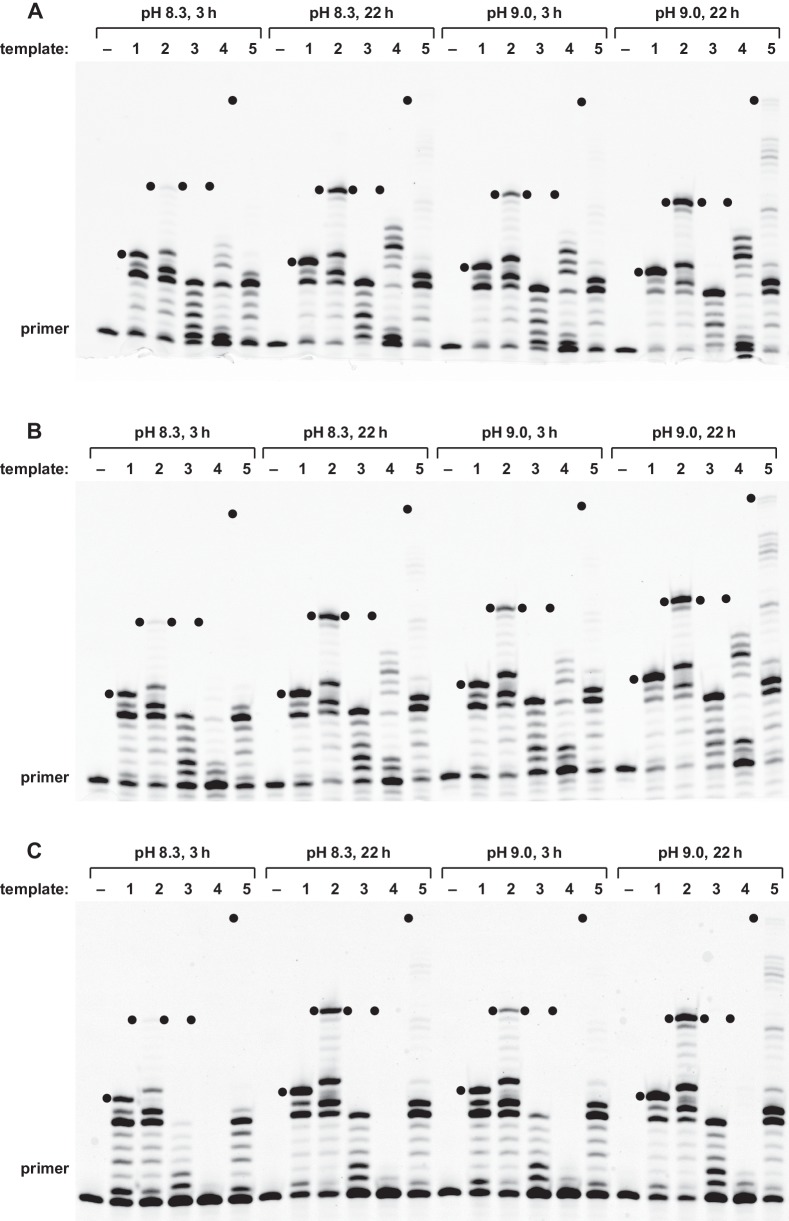
Figura 1-figura supplemento 1.Attività trascrittasi inversa del ribozima 24-3.Estensione di (A) primer all-RNA, (B) primer deossinucleotide-terminato RNA, o (C) primer all-DNA su un modello di RNA. Condizioni di reazione: 100 nM ribozima, 125 nM template 1, 125 nM primer, 2 mM ciascuno dNTP, 200 mM MgCl2, pH 8,3 o 9,0, 20°C, 3 o 22 ore. I punti neri indicano la posizione prevista dei prodotti a lunghezza piena.
Figura 1-figure supplement 2.Mancanza di attività della polimerasi DNA-dipendente dal DNA del ribozima 24-3.3. Estensione di un primer tutto-RNA su un modello di RNA o DNA, utilizzando sia dNTPs o NTPs. Le sequenze dei modelli di RNA (6 e 4) e i corrispondenti modelli di DNA (d6 e d4) sono elencati nel file supplementare 1. 2. Condizioni di reazione: 100 nM ribozima, 125 nM template, 125 nM primer, 2 mM ciascuno dNTP o NTP, 200 mM MgCl2, pH 8,3, 20°C, 21 ore.
Figura 2-figure supplement 1-source data 1.Analisi dei prodotti di trascrizione inversa.analisi LC/MS di prodotti a lunghezza intera.analisi LC/MS di prodotti a lunghezza intera.analisi MS/MS ad alta risoluzione del prodotto di trascrizione inversa.analisi MS/MS ad alta risoluzione del prodotto di trascrizione inversa (relativa alla Figura 2-figure supplement 1).analisi MS/MS ad alta risoluzione del prodotto di trascrizione inversa (relativa alla Figura 2-figure supplement 1).(A) Digestione parziale DNasi I di prodotti a tutta lunghezza ottenuti utilizzando il modello 1, rispetto ai materiali autentici, con un primer RNA (7 corsie a sinistra) o un primer DNA (7 corsie a destra). Per la reazione preparata con RNA, viene scissa solo la porzione estesa; per la reazione preparata con DNA, sia il primer che la porzione estesa vengono scisse. (B,C) Analisi LC/MS di prodotti purificati a lunghezza intera ottenuti utilizzando il modello 1 e o un RNA o un primer del DNA, rispettivamente.10.7554/eLife.31153.008Figure 2-source data 1.LC/MS analisi di prodotti full-length.L’estensione di un primer tutto RNA sul modello quattro ha prodotto un prodotto con una massa calcolata di 8874,432 e una massa osservata di 8874,441. Lo ione genitore è stato frammentato a legami internucleotidici all’interno della porzione di DNA della molecola per generare ioni secondari che sono stati analizzati da MS tandem. I frammenti successivi a-h corrispondono a 3´terminali successive all’interno della porzione di DNA della molecola.10.7554/eLife.31153.009Figure 2-figure supplement 1-source data 1.High-resolution MS/MS analysis of reverse transcription product (correlato alla figura 2-figure supplement 1).
Figura 2-figure supplemento 1-figure dati fonte 1.Figura 2—supplemento figura 1—dati fonte 1. Analisi MS/MS ad alta risoluzione del prodotto di trascrizione inversa.analisi MS/MS ad alta risoluzione del prodotto di trascrizione inversa (relativa alla figura 2-figure supplement 1).analisi MS/MS ad alta risoluzione del prodotto di trascrizione inversa (relativa alla figura 2-figure supplement 1).L’estensione di un primer tutto RNA sul modello quattro ha prodotto un prodotto con una massa calcolata di 8874.432 e una massa osservata di 8874.441. Lo ione genitore è stato frammentato a legami internucleotidici all’interno della porzione di DNA della molecola per generare ioni secondari che sono stati analizzati da MS tandem. I frammenti successivi a-h corrispondono a 3´terminali successive all’interno della porzione di DNA della molecola.10.7554/eLife.31153.009Figure 2-figure supplement 1-figure data 1-source data 1.High-resolution MS/MS analysis of reverse transcription product (correlato alla figura 2-figure supplement 1).
Figura 3.Figura 3. Trascrizione inversa nelle miscele di reazione prive di uno dei quattro dNTP.(A) Per la sequenza 5´-GAGTGG-3´ (modello 3), il materiale di lunghezza prevista è stato ottenuto in presenza di tutti e quattro i dNTP o di una miscela priva di dCTP, il materiale di lunghezza prevista di mobilità alterata è stato ottenuto in una miscela priva di dATP, e solo parzialmente il materiale di lunghezza è stato ottenuto in una miscela priva di dGTP o TTP. (B) Per la sequenza 5´-GCGGAGGAGTGGTGTG-3´ (modello 4), il materiale di lunghezza prevista è stato ottenuto in presenza di tutti e quattro i dNTP, il materiale di lunghezza prevista di mobilità alterata è stato ottenuto in una miscela priva di dATP, e solo materiale di lunghezza parziale è stato ottenuto in una miscela priva di dGTP, TTP, o dCTP. Condizioni di reazione: 100 nM ribozyme, 125 nM template, 125 nM primer, 2 mM dNTPs, 200 mM MgCl2, pH 8,3, 20°C, 22 hr.
Figura 4 dati fonte 1.RNA-dipendente RNA e attività della DNA polimerasi del 24-3 ribozyme.RNA-dipendente RNA e attività della DNA polimerasi.RNA-dipendente RNA e attività della DNA polimerasi.Il corso temporale della reazione utilizzando NTP (cerchi riempiti) o dNTP (cerchi aperti), misurando la velocità di aggiunta di un singolo nucleotide a un primer deossinucleotide-terminato sul modello 1. I dati erano adatti ad un doppio aumento esponenziale al massimo (r = 0,996 per la polimerizzazione dell’RNA; r = 0,983 per la polimerizzazione del DNA). L’inserto illustra i dati relativi ai primi 2 minuti della reazione. Condizioni di reazione: 100 nM ribozyme, 125 nM template, 125 nM primer, 2 mM NTPs o dNTPs, 200 mM MgCl2, pH 8,3, 20°C.10.7554/eLife.31153.012Cifra dati a 4 fonti 1.RNA-dipendente dall’RNA e dall’attività della DNA polimerasi.
Discussione
La sintesi di RNA temperata dall’RNA, catalizzata da un ribozima, è nota da 20 anni (Ekland e Bartel, 1996). Trasportare tale attività alla sintesi di DNA temperato con RNA è sempre sembrato plausibile (Joyce, 2002), ma richiedeva un ribozima con sufficiente attività di polimerizzazione per compensare la reattività chimica intrinsecamente inferiore del desossiriboso rispetto al ribosio 3´-idrossile. Un percorso storico simile può essere immaginato per il passaggio dall’RNA ai genomi del DNA durante la storia della vita sulla Terra. La stabilità del DNA rispetto all’RNA supera di gran lunga la differenza nella reattività chimica dei rispettivi gruppi 3´-idrossile. Il DNA è più incline alla depurazione rispetto all’RNA, ma questa debolezza è di gran lunga superata dalla maggiore stabilità della spina dorsale del DNA. La principale vulnerabilità chimica del DNA è la sua propensione a subire la deaminazione spontanea della citosina all’uracile. Questa carenza è stata presumibilmente affrontata da un successivo adattamento evolutivo che coinvolge 5-metilazione di uracile (timina) e l’escissione-riparazione di residui di uracile non metilato che derivano dalla citosina.
Il ribozima trascriptasi inversa ha un ragionevole tasso catalitico, ma è significativamente limitato per quanto riguarda le sequenze di template che può accettare. Fa fatica ad aggiungere più residui A o T ed è ostacolato dalla struttura secondaria all’interno del template RNA. Il ribozima inoltre incorpora erroneamente il dGTP come una coppia traballante G-U quando viene privato del dATP, sebbene questo comportamento non permetta alla polimerasi di attraversare le posizioni U su template difficili (Figura 1B). Analoghe limitazioni esistevano per le versioni precedenti del ribozyme RNA polimerasi, che sono state superate da molti cicli di evoluzione in vitro. È probabile che anche l’attività della trascrittasi inversa possa essere migliorata attraverso l’evoluzione. Una DNA polimerasi altamente ottimizzata dipendente dall’RNA potrebbe avere diminuito l’attività della RNA polimerasi dipendente dall’RNA, a meno che entrambe le funzioni non siano state esplicitamente mantenute attraverso la selezione. Potrebbe anche essere possibile utilizzare l’enzima 24-3 come punto di partenza per far evolvere una polimerasi DNA-dipendente che sintetizza l’RNA o il DNA, consentendo la trascrizione in avanti o la replicazione del DNA, rispettivamente. L’emergere storico di queste attività avrebbe segnato la fine dell’era mondiale dell’RNA.
Il livello iniziale di attività della trascrittasi inversa nel mondo dell’RNA potrebbe essere stato modesto, permettendo la copia di soli brevi segmenti di RNA, come è stato il caso nel presente studio. Affinché questo tratto venga mantenuto, sarebbe necessario conferire un vantaggio selettivo, e per essere ottimizzato ci sarebbe bisogno di un ulteriore vantaggio derivante dal potenziamento di questa attività. Un potenziale vantaggio di generare anche brevi segmenti di DNA potrebbe essere quello di proteggere i termini o altre regioni critiche dell’RNA, estendendosi in ultima analisi alla protezione dell’intero genoma. All’inizio, è probabile che l’RNA sia servito come primer per la trascrizione inversa, forse attraverso l’autoadescamento da una forcina a 3´terminali, anche se sarebbe stato possibile anche l’adescamento da un 2´-idrossile interno o una modifica chimica adeguata. L’ibridazione di un primer separato dovrebbe competere con la struttura secondaria che comprende il 3´terminale del modello di RNA, che potrebbe essere evitata con l’autoadescamento.
Tutte le discussioni relative al passaggio dall’RNA ai genomi del DNA sono speculative, anche se probabilmente questo evento è uno dei più significativi nella storia della vita. Senza il passaggio a un materiale genetico più stabile, la lunghezza dei genomi ereditabili e quindi la complessità della vita sarebbe stata fortemente limitata. La modesta differenza chimica tra ribosio e desossiriboso ha un profondo effetto sia sulla reattività chimica dei mononucleotidi che sulla stabilità della spina dorsale dei polinucleotidi. Genomi con il contenuto informativo dei moderni organismi cellulari probabilmente non sarebbero stati possibili senza l’invenzione della trascrittasi inversa.
Materiali e metodi
Materiali
Tutti gli oligonucleotidi utilizzati in questo studio sono elencati nel file supplementare 1. Gli oligonucleotidi sintetici sono stati acquistati da Integrated DNA Technologies (Coralville, IA) o preparati per sintesi in fase solida utilizzando un sintetizzatore di DNA/RNA Expedite 8909, con reagenti e fosforamiditi acquistati da Glen Research (Sterling, VA). I modelli di RNA sono stati preparati mediante trascrizione in vitro da modelli di DNA sintetico. I ribozimi della polimerasi sono stati preparati mediante trascrizione in vitro di modelli di DNA a doppio filamento generati dalla PCR da DNA plasmidico corrispondente. Tutti i modelli di RNA e ribozimi sono stati purificati mediante elettroforesi in gel di poliacrilamide denaturante (PAGE) e precipitazione di etanolo prima dell’uso. NTP sono stati acquistati da Sigma-Aldrich (St. Louis, MO) e dNTPs erano da Denville Scientific (Holliston, MA). TURBO DNase I, Superscript II trascriptasi inversa e streptavidina C1 Dynabeads erano di ThermoFisher (Grand Island, NY).
Trascrizione in vitro
Modelli di RNA sono stati trascritti da 0,5 μM DNA a filamento singolo che era stato ricotto con 0,5 µM di un oligodeossinucleotide sintetico che codifica il secondo filamento del promotore della polimerasi T7 RNA polimerasi. La trascrizione è stata effettuata in una miscela contenente 15 U /μL T7 RNA polimerasi, 0,002 U /μL pirofosfatasi inorganica, 5 mM ogni NTP, 25 mM MgCl2, 2 mM spermidina, 10 mM DTT, e 40 mM Tris (pH 8,0), che è stato incubato a 37 ° C per 2 ore. Il DNA è stato poi digerito aggiungendo 0,1 U/μL TURBO DNasi I e continuando l’incubazione per 1 ora. I ribozimi sono stati trascritti da modelli di DNA completamente a doppio filamento (20 µg/mL) che sono stati ottenuti mediante amplificazione PCR del DNA plasmidico che codifica il ribozima 24-3 (per gentile concessione di David Horning).
Polimerizzazione catalizzata dall’RNA
La polimerizzazione temperata con RNA di RNA e DNA è stata effettuata utilizzando 100 nM ribozyme, 125 nM template e 125 nM primer. Il primer, che consisteva di RNA, DNA o RNA con un singolo deossinucleotide a 3´terminali, conteneva sia un’etichetta di fluoresceina che una parte di biotina alla sua estremità a 5´. Il ribozyme, template e primer sono stati prima riscaldati a 80 ° C per 2 minuti, poi raffreddato a 17 ° C per 5 minuti e aggiunto alla miscela di reazione, che conteneva anche 2 mM ogni NTP o dNTP, 200 mM MgCl2, 0,05% TWEEN20, e 50 mM Tris (pH 8,3 o 9,0). La polimerizzazione è stata effettuata a 20°C e si è estinta con l’aggiunta di 250 mM EDTA. I primer biotinilati e prodotti estesi sono stati catturati su streptavidina C1 Dynabeads, lavato due volte con alcali (25 mM NaOH, 1 mM EDTA, e 0,05% TWEEN20) e una volta con TE-urea (1 mM EDTA, 0,05% TWEEN20, 10 mM Tris (pH 8,0), e 8 M urea), poi eluito con 98% formamide e 10 mM EDTA (pH 8,0) a 95 ° C per 15 min. I prodotti di reazione sono stati analizzati denaturando la PAGINA.
I prodotti di estensione a lunghezza definita per l’analisi mediante digestione DNasi o LC/MS sono stati preparati utilizzando 1 µM di ribozima, 1 µM di template e 0,8 μM di RNA o DNA primer. La reazione è stata effettuata come descritto sopra a pH 8,3 per 21 ore. Presunti materiali a tutta lunghezza sono stati purificati per elettroforesi in un gel di poliacrilamide denaturato al 15%, estratto dal gel, eluito con 200 mM NaCl, 1 mM EDTA, e 10 mM Tris (pH 7,5), ed etanolo precipitato.
Digestione DNasi
I prodotti di estensione purificati sono stati sottoposti a parziale digestione DNasi in una miscela contenente 1 μM oligonucleotide, 0,1 U/μL TURBO DNasi I, 10 mM MgCl2, 0,5 mM CaCl2, e 20 mM Tris (pH 7,5), che è stato incubato a 37 ° C per 30 min, poi estinto con 20 mM EDTA, seguita da inattivazione dell’enzima a 75 ° C per 10 min. I prodotti risultanti sono stati analizzati per elettroforesi in un gel di poliacrilamide denaturante al 15%.
Analisi LC/MS
L’analisi di cromatografia liquida / spettrometria di massa è stata eseguita da Novatia LLC (Newtown, PA) utilizzando 50 pmol di prodotti di estensione purificati. Le analisi standard sono state eseguite mediante ionizzazione elettrospray LC/MS sulla piattaforma Oligo HTCS, che raggiunge una precisione di massa dello 0,01-0,02%. La conferma della sequenza oligonucleotidica è stata eseguita da una trappola ionica ad alta risoluzione tandem MS su uno spettrometro di massa ionico LTQ-Orbitrap, che raggiunge una risoluzione di massa dello 0,003% (FWHM). Lo ione genitore è stato utilizzato per generare uno spettro di frammenti risultanti da scissione a legami fosfodiesterici all’interno della porzione di DNA della molecola. Il software di deconvoluzione ReSpect (Positive Probability Ltd.) è stato utilizzato per deisotopare lo spettro MS/MS e per ottenere uno spettro frammentato semplificato con masse esatte.
References
- Attwater J, Tagami S, Kimoto M, Butler K, Kool ET, Wengel J, Herdewijn P, Hirao I, Holliger P. Chemical fidelity of an RNA polymerase ribozyme. Chemical Science. 2013; 4:2804-2814. DOI
- Åström H, Limén E, Strömberg R. Acidity of secondary hydroxyls in ATP and adenosine analogues and the question of a 2′,3′-hydrogen bond in ribonucleosides. Journal of the American Chemical Society. 2004; 126:14710-14711. DOI | PubMed
- Bartel DP, Szostak JW. Isolation of new ribozymes from a large pool of random sequences [see comment]. Science. 1993; 261:1411-1418. DOI | PubMed
- Ekland EH, Bartel DP. RNA-catalysed RNA polymerization using nucleoside triphosphates. Nature. 1996; 382:373-376. DOI | PubMed
- Freeland SJ. Molecular evolution:do proteins predate DNA?. Science. 1999; 286:690-692. DOI | PubMed
- Gilbert W. Origin of life: The RNA world. Nature. 1986; 319DOI
- Horning DP, Joyce GF. Amplification of RNA by an RNA polymerase ribozyme. PNAS. 2016; 113:9786-9791. DOI | PubMed
- Johnston WK, Unrau PJ, Lawrence MS, Glasner ME, Bartel DP. RNA-catalyzed RNA polymerization: accurate and general RNA-templated primer extension. Science. 2001; 292:1319-1325. DOI | PubMed
- Joyce GF. The antiquity of RNA-based evolution. Nature. 2002; 418:214-221. DOI | PubMed
- Maynard Smith J, Szathmáry E. The Major Transitions in Evolution. Freeman Press: Oxford; 1995.
- McGinness KE, Joyce GF. RNA-catalyzed RNA ligation on an external RNA template. Chemistry & Biology. 2002; 9:297-307. DOI | PubMed
- Nissen P, Hansen J, Ban N, Moore PB, Steitz TA. The structural basis of ribosome activity in peptide bond synthesis. Science. 2000; 289:920-930. DOI | PubMed
- Ritson DJ, Sutherland JD. Conversion of biosynthetic precursors of RNA to those of DNA by photoredox chemistry. Journal of Molecular Evolution. 2014; 78:245-250. DOI | PubMed
- Sczepanski JT, Joyce GF. A cross-chiral RNA polymerase ribozyme. Nature. 2014; 515:440-442. DOI | PubMed
- Wochner A, Attwater J, Coulson A, Holliger P. Ribozyme-catalyzed transcription of an active ribozyme. Science. 2011; 332:209-212. DOI | PubMed
- Wu T, Orgel LE. Nonenzymatic template-directed synthesis on oligodeoxycytidylate sequences in hairpin oligonucleotides. Journal of the American Chemical Society. 1992; 114:317-322. DOI | PubMed
- Zaher HS, Unrau PJ. Selection of an improved RNA polymerase ribozyme with superior extension and fidelity. RNA. 2007; 13:1017-1026. DOI | PubMed
Fonte
Samanta B, Joyce GF, Ellington AD () A reverse transcriptase ribozyme. eLife 6e31153. https://doi.org/10.7554/eLife.31153




