
Introduzione
I dati storici degli studi preclinici e delle sperimentazioni cliniche supportano gli effetti anti-neoplastici degli analoghi 1,2:5,6-dianhydrogalactitol (DAG) in una varietà di tipi di cancro, tra cui la leucemia, i tumori cerebrali, cervicali, ovarici e polmonari.1–6. In Cina, il DAG è approvato per il trattamento del cancro ai polmoni7. In tutto il mondo, il cancro ai polmoni è la principale causa di morte per cancro. Il tasso di sopravvivenza relativa a 5 anni per il cancro ai polmoni è del 15% per gli uomini e del 21% per le donne. Ci sono due tipi principali di cancro ai polmoni, il cancro ai polmoni non a piccole cellule (NSCLC) e il cancro ai polmoni a piccole cellule (SCLC). L’NSCLC rappresenta l’80-85% di tutti i tumori polmonari e circa il 57% dei pazienti con NSCLC di nuova diagnosi presenti con malattia metastatica allo stadio IV. La sopravvivenza globale mediana per i pazienti con NSCLC in stadio IV è di 4 mesi e il tasso di sopravvivenza a 5 anni è solo del 4%.8–10. Le metastasi cerebrali si verificano frequentemente nei pazienti affetti da NSCLC, contribuendo alla cattiva prognosi di questa malattia11. Attualmente, i trattamenti di base delle NSCLC primarie e metastatiche includono chirurgia, radioterapia, chemioterapia e terapie mirate con anticorpi monoclonali o inibitori della tirosina chinasi (TKI) in pazienti che presentano mutazioni dei recettori del fattore di crescita epidermico.12–15. Tuttavia, l’esito dei pazienti NSCLC rimane scarso soprattutto a causa della chemioterapia a base di platino acquisita e della resistenza al trattamento TKI16.
DAG è una piccola molecola solubile in acqua che attraversa facilmente la barriera emato-encefalica (BBB) e si accumula nei tumori cerebrali primari e secondari4,17. Forse per questo motivo, DAG mostra una forte attività nei modelli animali di NSCLC metastatico, incluso l’NSCLC resistente alla TKI18. Informato da studi preclinici, il DAG può avere un vantaggio terapeutico rispetto ad altri agenti reticolanti del DNA3,5.
Grazie alla sua capacità di attraversare il BBB, il DAG è attualmente testato in pazienti con glioblastoma multiforme refrattario temozolomide (TMZ).19,20. Uno studio clinico di fase I/II recentemente completato in pazienti adulti con GBM refrattari ha stabilito un regime di dosaggio ben tollerato del DAG e ha confermato la mielosoppressione come tossicità limitante il dosaggio con completa reversione alla fine del trattamento.21. Tuttavia, nonostante gli incoraggianti dati preclinici e clinici in NSCLC e GBM, il tempestivo avanzamento degli analoghi del DAG verso l’arena clinica è ostacolato da una comprensione inadeguata dei meccanismi molecolari responsabili della citotossicità mediata dal DAG nelle cellule tumorali. Abbiamo quindi utilizzato l’NSCLC come sistema modello per studiare i meccanismi di citotossicità imposti dall’analogo DAG di grado clinico VAL-08322.
Risultati
Perdita di vitalità delle cellule tumorali polmonari dopo il trattamento con DAG
Per studiare gli effetti del DAG sulle cellule tumorali del polmone, abbiamo valutato le attività citotossiche del VAL-083 in un pannello di linee cellulari NSCLC. Il trattamento delle cellule A549, H2122, e H1792 con 10 μM VAL-083 per 72 h ha portato a cambiamenti morfologici drammatici come il gonfiore e il distacco delle cellule (Fig. 1a). Per caratterizzare ulteriormente l’effetto del DAG sulle cellule tumorali, abbiamo trattato linee cellulari H1792, H2122, H23, e A549 NSCLC con diverse concentrazioni di VAL-083 per 72 e successivamente determinato la vitalità di ogni linea cellulare. L’analisi ha mostrato una perdita di vitalità in funzione della concentrazione in tutte le linee cellulari trattate con VAL-083 con valori di concentrazione inibitoria semimassimale (IC50) nel campo di concentrazione basso µM (Fig. 1b). In sintesi, questi dati dimostrano gli effetti citotossici del DAG sulle cellule NSCLC.Fig. 1Citotossicità del DAG nelle linee cellulari NSCLC. sono state mostrate immagini in campo luminoso di cellule A549, H2122, e H1792 coltivate in 10 % FBS DMEM o RPMI 1640 medium per 72 ore con o senza 10 μM VAL-083. La barra di scala rappresenta 100μm. b Quattro linee di cellule NSCLC A549, H23, H1792, e H2122 cellule sono stati seminati in 96 pozzetti di coltura piastre e trattati con diverse concentrazioni di VAL-083 (0, 100 nM, 500 nM, 1μM, 2.5 μM, 5 μM, 10 μM, 25 μM, 50 μM e 100 μM) per 72 ore. Dopo il trattamento, è stato eseguito il test del cristallo violetto per rilevare l’assorbanza a 560 nm di lunghezza d’onda. Il valore IC50 del VAL-083 è stato determinato applicando una curva dose-risposta sigmoidale ai dati utilizzando GraphPad Prism 6. I dati sulla curva sono presentati come errore medio-±standard. Ogni linea cellulare è stata testata in tre o quattro singoli esperimenti
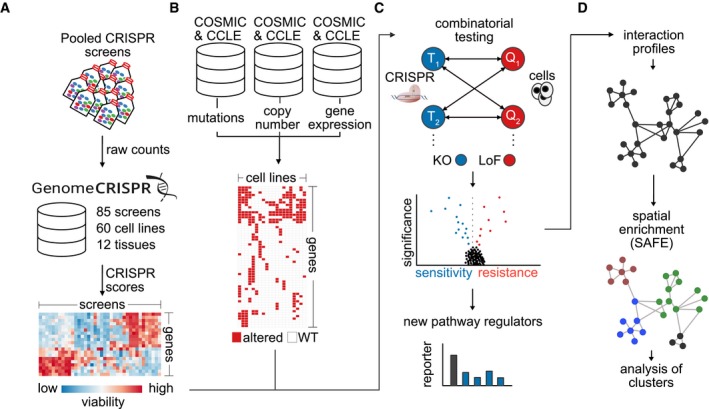
Fig. 1.Citotossicità del DAG nelle linee cellulari NSCLC.sono state mostrate immagini a campo luminoso di cellule A549, H2122, e H1792 coltivate in 10 % FBS DMEM o RPMI 1640 medium per 72 ore con o senza 10μM VAL-083. La barra di scala rappresenta 100μm. b Quattro linee di cellule NSCLC A549, H23, H1792, e H2122 cellule sono stati seminati in 96 pozzetti di coltura piastre e trattati con diverse concentrazioni di VAL-083 (0, 100 nM, 500 nM, 1μM, 2.5 μM, 5 μM, 10 μM, 25 μM, 50 μM e 100 μM) per 72 ore. Dopo il trattamento, è stato eseguito il test del cristallo violetto per rilevare l’assorbanza a 560 nm di lunghezza d’onda. Il valore IC50 del VAL-083 è stato determinato applicando una curva dose-risposta sigmoidale ai dati utilizzando GraphPad Prism 6. I dati sulla curva sono presentati come errore medio-±standard. Ogni linea cellulare è stata testata in tre o quattro singoli esperimenti
DAG induce danni persistenti al DNA nelle cellule tumorali del polmone
Molti farmaci chemioterapici funzionano inducendo diversi tipi di danni al DNA nelle cellule tumorali a rapida divisione. DAG è stato segnalato per avere attività bifunzionale DNA-targeting bifunzionale che porta alla formazione di N7-monoalchilguanina e inter-filo di DNA crosslinks22. Per indagare gli effetti del DAG sull’integrità del DNA, abbiamo esaminato le cellule NSCLC trattate con VAL-083 per la variante dell’istone fosforilato H2AX (ɣH2AX), un marcatore surrogato ampiamente utilizzato di rotture a doppio filamento del DNA (DSBs)23,24. La valutazione biochimica delle cellule A549, H1792 e H2122 trattate con VAL-083 per 24 ore seguita dalla rimozione del farmaco ha mostrato una forte espressione ɣH2AX che persisteva per 72 ore dopo la rimozione del farmaco (Fig. 2a). Ciò è stato supportato da un’analisi di imaging di immunofluorescenza (IF) che ha mostrato una formazione sostenuta di focolai ɣH2AX nel 90-100 % delle cellule (Fig. 2b) in un arco di tempo simile (Fig. 2c). Questi dati suggeriscono che il DAG induce il DNA DSBs nelle cellule NSCLC che non possono essere riparate entro un periodo di recupero di 72 ore. Inoltre, VAL-083 ha indotto una forte espressione ɣH2AX con almeno 10 ore di incubazione (Fig. Supplementare S1a) e ha mostrato un pattern dose-dipendente (Fig. Supplementare S1b).Fig. 2DAG induce danni al DNA nelle cellule NSCLC. un A549, H1792, e H2122 cellule sono state trattate con 20 μM VAL-083 per 24 ore, seguite da lavaggio e sostituzione con mezzo completo in coltura per vari periodi di tempo (0, 4, 8, 24, 48, o 72 ore). Poi, le cellule sono state raccolte per l’estrazione delle proteine, e 50 mg è stato analizzato per l’espressione fosforilato e totale H2AX da western blot utilizzando specifici anticorpi policlonali di coniglio con anticorpi policlonali come descritto nella sezione “Materiali e metodi”. Immagini rappresentative sono mostrati per l’effetto del corso del tempo di VAL-083 su ɣH2AX espressione. Totale H2AX e GAPDH servito come controlli di carico. b Le cellule coltivate A549 sono stati trattati con 20μM VAL-083 per 24 ore. Dopo di che, le cellule sono state lavate e sostituite con terreno completo per un ulteriore tempo di incubazione di 0, 4, 8, 24, 48, o 72 ore. Poi, le cellule sono state fissate, permeabilizzate e immunocolorazione con anticorpo anti-ɣH2AX. Quantificazione di ɣH2AX focolai (cellule con >10 focolai sono stati considerati come “foci-positivi”) da 40 a 50 cellule per campione è stato mostrato. c Immagini rappresentative confocale da ogni condizione sperimentale in b sono stati mostrati con ɣH2AX in rosso. La barra della scala rappresenta 5 μm
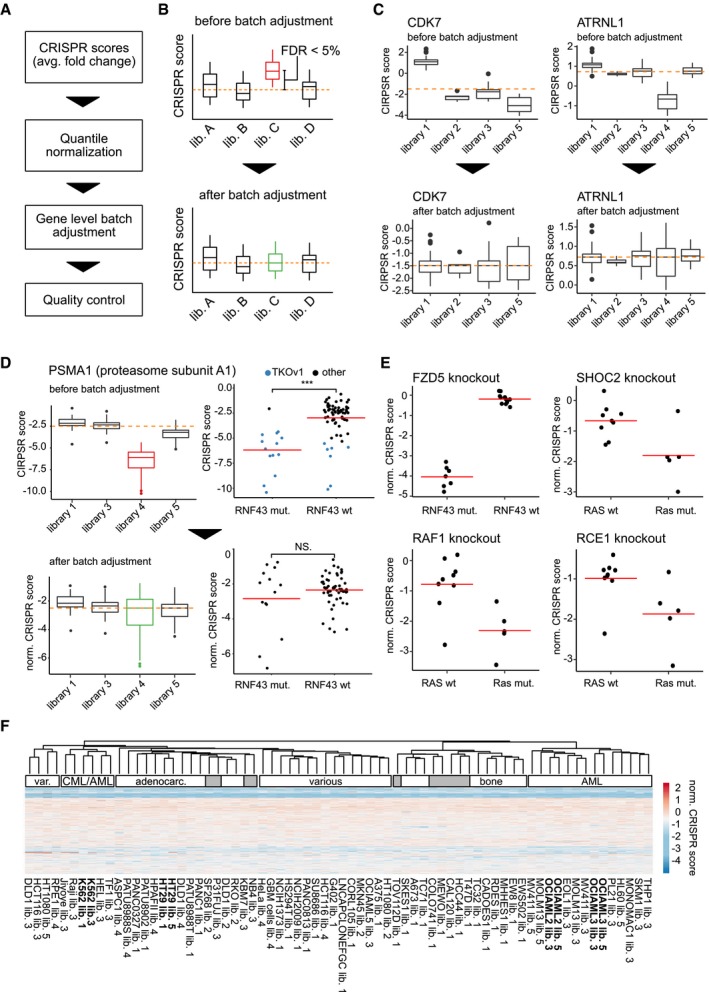
Fig. 2.DAG induce danni al DNA nelle cellule NSCLC.una cella A549, H1792 e H2122 è stata trattata con 20 μM VAL-083 per 24 ore, seguita da lavaggio e sostituzione con mezzo completo in coltura per vari periodi di tempo (0, 4, 8, 24, 48 o 72 ore). Poi, le cellule sono state raccolte per l’estrazione delle proteine, e 50 mg è stato analizzato per l’espressione fosforilato e totale H2AX da western blot utilizzando specifici anticorpi policlonali di coniglio con anticorpi policlonali come descritto nella sezione “Materiali e metodi”. Immagini rappresentative sono mostrati per l’effetto del corso del tempo di VAL-083 su ɣH2AX espressione. Totale H2AX e GAPDH servito come controlli di carico. b Le cellule coltivate A549 sono stati trattati con 20μM VAL-083 per 24 ore. Dopo di che, le cellule sono state lavate e sostituite con terreno completo per un ulteriore tempo di incubazione di 0, 4, 8, 24, 48, o 72 ore. Poi, le cellule sono state fissate, permeabilizzate e immunocolorazione con anticorpo anti-ɣH2AX. Quantificazione di ɣH2AX focolai (cellule con >10 focolai sono stati considerati come “foci-positivi”) da 40 a 50 cellule per campione è stato mostrato. c Immagini rappresentative confocale da ogni condizione sperimentale in b sono stati mostrati con ɣH2AX in rosso. La barra della scala rappresenta 5 μm
Danni al DNA dipendenti dalla replicazione delle cellule NSCLC durante il trattamento con DAG
Abbiamo poi studiato l’effetto del DAG sulla progressione del ciclo cellulare utilizzando l’analisi citometrica a flusso di ioduro di propidio (PI)-celle NSCLC macchiate. Il trattamento VAL-083 ha indotto un forte arresto del ciclo cellulare dose-dipendente S/G2-fase in A549 (Fig. 3a) e H1792 (Fig. 3b) cellule NSCLC. Questo risultato suggerisce che la citotossicità mediata dal DAG dipende probabilmente dalla replicazione. Per determinare ulteriormente il ruolo della replicazione per la citotossicità indotta da DAG, abbiamo sincronizzato la maggior parte delle cellule A549 in fase G0/G1 con la fame nel siero per 24 ore (Fig. 3c). Le cellule sono state poi rilasciate dall’arresto del ciclo cellulare con l’aggiunta di siero con o senza VAL-083 5μM e seguita da citometria a flusso. Mentre le cellule non trattate hanno rapidamente assunto un normale profilo del ciclo cellulare dopo l’aggiunta di siero, le cellule sottoposte a VAL-083 hanno mostrato un arresto di fase S/G2 dipendente dal tempo visivo a e dopo 19 ore in mezzo completo (Fig. 3c). In particolare, questa concentrazione di trattamento VAL-083 non ha aumentato la popolazione di cellule sub-G1 (il contenuto di DNA <2 N riflette i detriti cellulari e le cellule apoptotiche), indicando che la diminuzione dipendente dal tempo di G0/G1 non era dovuta ad un aumento della morte cellulare (Fig. 3c). In parallelo l’analisi western blot dell’espressione della caspasi 3 scissione ha confermato la mancanza di apoptosi nelle cellule trattate con VAL-083 (Supplementare Fig. S2).Fig. 3DAG danno indotta da DNA si verifica nella fase S del ciclo cellulare. un A549 cellule A549 sono state trattate con 5 o 25μM VAL-083 per 24 ore. Dopo di che, le cellule sono state raccolte per l’analisi del ciclo cellulare con colorazione PI mediante citometria a flusso. b Dopo il trattamento con diverse concentrazioni di VAL-083 (1, 2,5, 5 o 10 μM) in cellule H1792 per 48 h, l’analisi del ciclo cellulare con colorazione PI è stata eseguita utilizzando citometria a flusso. c A549 cellule A549 sono state sincronizzate dalla fame nel siero (ST) per 24 ore prima del trattamento con o senza 5μM VAL-083 per i periodi di tempo indicati (1, 4, 19, 24, 44, o 49 ore). Le cellule sono state poi raccolte, fissate e successivamente colorate con PI per l’analisi della distribuzione del ciclo cellulare mediante citometria a flusso. Per ulteriori dettagli sperimentali, vedere “Materiali e metodi”. I grafici rappresentativi citometrici a flusso da due esperimenti individuali sono mostrati. d Diagramma di flusso che delinea le condizioni sperimentali utilizzati in e. e A549 cellule (sincronizzato da 24 h di fame siero) sono stati trattati con 50μM VAL-083 per 1 h e sostituito con terreno completo per un altro 24 ore di incubazione. Poi, le cellule sono state fissate, permeabilizzate e immunocolorazione con anticorpi anti-ciclina A2 e anti-ɣH2AX. Immagini rappresentative IF sono mostrati con la ciclina A2 in verde e ɣH2AX in rosso. La barra della scala rappresenta 10 μm. f In tutto, 100-120 cellule sono state esaminate da ogni condizione di trattamento come in e. Le percentuali di due categorie (ciclina A2-/ɣH2AX- e ciclina A2+/ɣH2AX+) di cellule A549 sono mostrati con l’analisi statistica corrispondente (*p ≤ 0.05; **p≤≤ 0,01; Test dello studente). g Le cellule A549 sono state seminate con terreno sieroso o completo in piastre di coltura a 96 pozzetti. Dopo 24 ore di incubazione, le cellule sono state trattate con 0, 100 mm, 500 mm, 1 mm, 2,5 mm, 5 mm, 10 mm, 25 mm, 50 mm o 100 mm VAL-083 per 72 ore. Dopo il trattamento, la percentuale di cellule di sopravvivenza rispetto alla condizione non trattata è stata determinata con il saggio crystal violet. I dati sono presentati come errore medio-±standard. H1792 e H2122 cellule sono state anche testate nella stessa condizione sperimentale
Cicli e chinasi ciclo-dipendenti (CDK) sono i regolatori chiave della progressione del ciclo cellulare eucariotico. L’attivazione dei complessi chinasi B-Cdc2 della ciclina innesca l’ingresso in fase M, mentre i complessi A-Cdk2 della ciclina controllano la progressione attraverso la fase S25–27. Di conseguenza, l’espressione della ciclina è strettamente regolata durante tutto il ciclo della cella dove la ciclina A è espressa esclusivamente in fase S in fase G228. Utilizzando la ciclina A come marcatore dell’ingresso della fase S, abbiamo eseguito la colorazione IF per la ciclina A2 e ɣH2AX su celle A549 con o senza trattamento a impulsi VAL-083 (50 μM VAL-083 per 1 h) (Fig. 3d). L’analisi ha mostrato che le cellule A549 sincronizzate hanno mostrato un drammatico accumulo di ciclina A2 (verde) e ɣH2AX (rosso) espressione dopo il trattamento con VAL-083 seguito da 24 ore in mezzo completo (Fig. 3e). È importante notare che la popolazione di cellule doppio-positive per la ciclina A2+/ɣH2AX+ è stata significativamente aumentata dopo il trattamento con VAL-083 per 1 h seguito da un lavaggio per 24 h (VAL 1 h++WO 24 h) mentre le cellule doppio-negative della ciclina A2-/ɣH2AX- sono diminuite (Fig. 3f). Ciò indica che VAL-083 indotta da DNA N7-guanina N7-guanina crosslinks si traducono in DSBs in fase S / G2 del ciclo cellulare. Per convalidare ulteriormente la citotossicità osservata replicazione dipendente da VAL-083 nelle linee cellulari NSCLC, abbiamo confrontato i tassi di sopravvivenza delle cellule coltivate in mezzo completo con le cellule coltivate in mezzo siero-deprivato, sfidati con concentrazioni crescenti di VAL-083. Infatti, le cellule A549, H1792, e H2122 non potevano entrare nella fase S a causa della privazione del siero, tutte hanno mostrato resistenza alla citotossicità indotta dalla VAL-083 (Fig. 3g). Combinati, questi dati suggeriscono che la citotossicità indotta da DAG si materializza man mano che le cellule NSCLC progrediscono nella fase S del ciclo cellulare con lesioni non riparate di N7-guanina DNA crosslink non riparate.
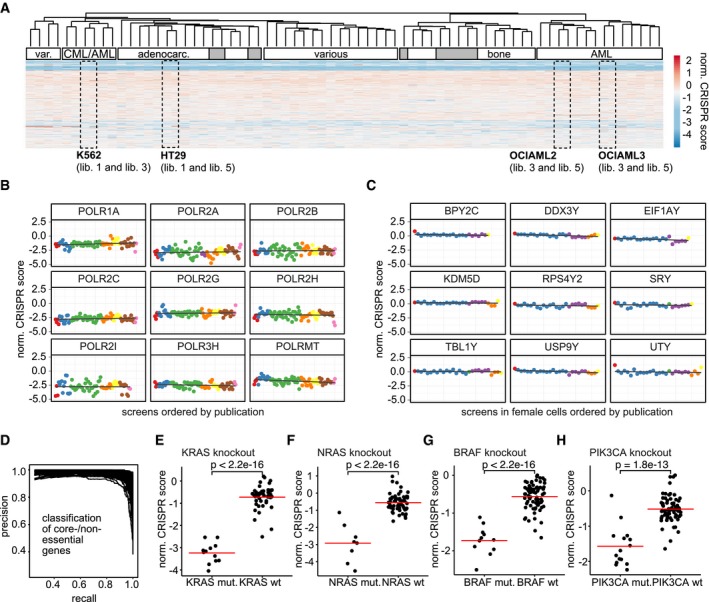
Fig. 3.Danni al DNA indotti da DAG si verificano nella fase S del ciclo cellulare.a A549 cellule sono state trattate con 5 o 25μM VAL-083 per 24 ore. Dopo di che, le cellule sono state raccolte per l’analisi del ciclo cellulare utilizzando la colorazione PI con citometria a flusso. b Dopo il trattamento con diverse concentrazioni di VAL-083 (1, 2,5, 5, o 10 μM) in cellule H1792 per 48 h, l’analisi del ciclo cellulare con colorazione PI è stata eseguita con citometria a flusso. c A549 cellule A549 sono state sincronizzate dalla fame nel siero (ST) per 24 ore prima del trattamento con o senza 5μM VAL-083 per i periodi di tempo indicati (1, 4, 19, 24, 44, o 49 ore). Le cellule sono state poi raccolte, fissate e successivamente colorate con PI per l’analisi della distribuzione del ciclo cellulare mediante citometria a flusso. Per ulteriori dettagli sperimentali, vedere “Materiali e metodi”. I grafici rappresentativi citometrici a flusso da due esperimenti individuali sono mostrati. d Diagramma di flusso che delinea le condizioni sperimentali utilizzati in e. e A549 cellule (sincronizzato da 24 h di fame siero) sono stati trattati con 50μM VAL-083 per 1 h e sostituito con terreno completo per un altro 24 ore di incubazione. Poi, le cellule sono state fissate, permeabilizzate e immunocolorazione con anticorpi anti-ciclina A2 e anti-ɣH2AX. Immagini rappresentative IF sono mostrati con la ciclina A2 in verde e ɣH2AX in rosso. La barra della scala rappresenta 10 μm. f In tutto, 100-120 cellule sono state esaminate da ogni condizione di trattamento come in e. Le percentuali di due categorie (ciclina A2-/ɣH2AX- e ciclina A2+/ɣH2AX+) di cellule A549 sono mostrati con l’analisi statistica corrispondente (*p ≤ 0.05; **p≤≤ 0,01; Test dello studente). g Le cellule A549 sono state seminate con terreno sieroso o completo in piastre di coltura a 96 pozzetti. Dopo 24 ore di incubazione, le cellule sono state trattate con 0, 100 mm, 500 mm, 1 mm, 2,5 mm, 5 mm, 10 mm, 25 mm, 50 mm o 100 mm VAL-083 per 72 ore. Dopo il trattamento, la percentuale di cellule di sopravvivenza rispetto alla condizione non trattata è stata determinata con il saggio crystal violet. I dati sono presentati come errore medio-±standard. H1792 e H2122 cellule sono state anche testate nella stessa condizione sperimentale
I danni al DNA indotti da DAG sono preferibilmente riparati mediante ricombinazione omologa
Poiché il DAG induce il DNA DSBs nella fase S, la citotossicità nelle cellule tumorali probabilmente dipende almeno parzialmente dalla loro capacità di riparare il DNA nella fase S/G2 del ciclo cellulare. I DSBs del DNA possono essere riparati sia con l’unione finale non omologa (NHEJ) che con la ricombinazione omologa (HR). Mentre NHEJ può verificarsi durante tutto il ciclo cellulare, HR è limitato alle fasi S e G2 dove i cromatidi gemelli sono disponibili come modelli per la sequenza di riparazione guidata dall’omologia.29. Poiché il DNA DSBs indotta da DAG si è materializzato specificamente in S/G2, abbiamo ipotizzato che le cellule NSCLC avrebbero riparato queste lesioni con l’HR. Per testare questa ipotesi, abbiamo monitorato i sensori e gli effettori del DNA DSB coinvolti nel percorso HR nelle cellule NSCLC dopo il trattamento VAL-083. Il trattamento a impulsi VAL-083 seguito da 20-48 incubazione in mezzo completo senza VAL-083 ha innescato l’attivazione della chinasi ataxia telangiectasia mutata (ATM) nelle cellule A549, H1792 e H2122 (Fig. 4a e Supplementare Fig. S3). Questo è stato associato alla fosforilazione della trifosforilazione 68 della fase S della chinasi Chk2 e alla presenza di ɣH2AX. All’attivazione della riparazione HR, il DNA che circonda il DSBs viene elaborato in una fase enzimatica che dipende dal CtIP e dai co-fattori della famiglia dei fattori di crescita derivati dall’epatoma per creare un modello di DNA a filamento singolo (ssDNA) per l’accoppiamento di omologia30–33. L’ssDNA resecato viene successivamente occupato dalla proteina A (RPA32) di replicazione ssDNA-binding replication, che viene fosforilata sulla serina 33 dall’atassia telangiectasia e dalla proteina ATR (Rad3-correlata alla chinasi) in risposta all’esposizione a ssDNA.34. Questo evento fa scattare la fase finale del percorso HR in cui la riparazione dell’omologia viene completata in modo dipendente da Rad5135. Infatti, il trattamento VAL-083 ha indotto la fosforilazione indotta da RPA32 così come la chinasi Chk1, un noto obiettivo fosforetico dell’ATR (Fig. 4a). In aggregato, questi dati suggeriscono che le cellule NSCLC riparano i DSB indotti da DAG da HR. Per corroborare ulteriormente questo risultato, abbiamo analizzato il reclutamento di proteine chiave di riparazione HR a ɣH2AX focolai in cellule NSCLC trattati con VAL-083 da microscopia confocale. L’analisi ha mostrato che le proteine di riparazione HR BRCA1, RPA32, e Rad5135 co-localizzata con ɣH2AX indotta da VAL-083 (Fig. 4b) e queste co-localizzazioni sono state statisticamente significative (Fig. 4c). Combinati, questi dati dimostrano che le cellule NSCLC riparano i DSBs indotti da DSBs indotti da HR repair.Fig. 4DAG danni al DNA indotti da DSB sono riparati da ricombinazione omologa. un A549 cellule A549 sono state sincronizzate con la fame di 24 ore di siero. Dopo di che, le cellule sono state incubate in mezzo completo con il trattamento di 50μM VAL-083 per 1 h. Poi, le cellule sono state lavate e sostituite con terreno completo per un ulteriore tempo di incubazione di 20, 24 o 48 ore. I lisati cellulari sono stati poi estratti per l’analisi western blot dei sensori e degli effettori coinvolti nel percorso di risposta al danno del DNA HR utilizzando i seguenti anticorpi: phospho-ATM (Ser1981), phospho-Chk2 (Thr68), phospho-Chk1 (Ser345 e Ser317), phospho-RPA32 (Ser33), e ɣH2AX. Immagini rappresentative sono mostrate da tre a quattro esperimenti indipendenti. b A549 cellule A549 sono state sincronizzate con 24 ore di fame di siero. Dopo di che, le cellule sono state incubate in mezzo completo con il trattamento di 50μM VAL-083 per 1 h. Poi, le cellule sono state lavate e sostituite con terreno completo per un ulteriore tempo di incubazione di 24 ore. Immagini rappresentative confocale di proteine indicate (BRCA1, RPA32, Rad51, e ɣH2AX) sono stati mostrati. La barra di scala rappresenta 5 mg. c Quantificazione di cellule A549 foci-positive presentate in b da 60-80 cellule per condizione sono stati mostrati. Le analisi statistiche sono state ottenute da tre esperimenti indipendenti (**p ≤ 0,01; ***p ≤ 0,001; Student’s t test). d A549 cellule A549 sono state trasfettate con controllo negativo (C) o tre BRCA1-targeting siRNA (B1, siBRCA1-2; B2, siBRCA1-15; o B3, siBRCA1-17) per 24 ore. Le cellule sono state poi seminate in piastre di coltura a 96 pozzetti e trattate con diverse concentrazioni di VAL-083 (0, 100 nM, 500 nM, 1μM, 1.5 μM, 2,5 μM, 5 μM, 10 μM, 25 μM, 50 μM, 100 μM e 200 μM) per 5 giorni. Dopo il trattamento, è stato eseguito il test del cristallo violetto per rilevare l’assorbanza a 560 nm di lunghezza d’onda. Il valore IC50 del VAL-083 è stato determinato applicando una curva dose-risposta sigmoidale ai dati utilizzando GraphPad Prism 6. I valori IC50 della VAL-083 nelle celle di controllo o BRCA1-knockdown sono presentati come errore medio-±standard (*p≤ 0,05; **p≤ 0,01; ***p≤ 0,001; test t dello studente). I lisati delle cellule del controllo o cellule BRCA1-knockdown sono stati analizzati da western blot con anticorpo contro BRCA1, e GAPDH è stato utilizzato come controllo di carico. Le immagini rappresentative sono mostrate da tre esperimenti indipendenti
I nostri dati contengono una previsione che le cellule tumorali carenti nella riparazione delle risorse umane avrebbero aumentato la sensibilità al DAG. Per confermare questa previsione, abbiamo abbattuto la proteina essenziale per la riparazione delle risorse umane BRCA136,37 in cellule NSCLC utilizzando piccoli RNA interferenti (siRNA) prima del trattamento con VAL-083. Infatti, l’abbattimento del BRCA1 con tre siRNA non sovrapposti ha sensibilizzato in modo significativo le cellule A549 alla VAL-083 (Fig. 4d). Questi dati confermano la previsione che le cellule tumorali carenti nella riparazione di HR non sono in grado di risolvere i DSB indotti dal DAG.
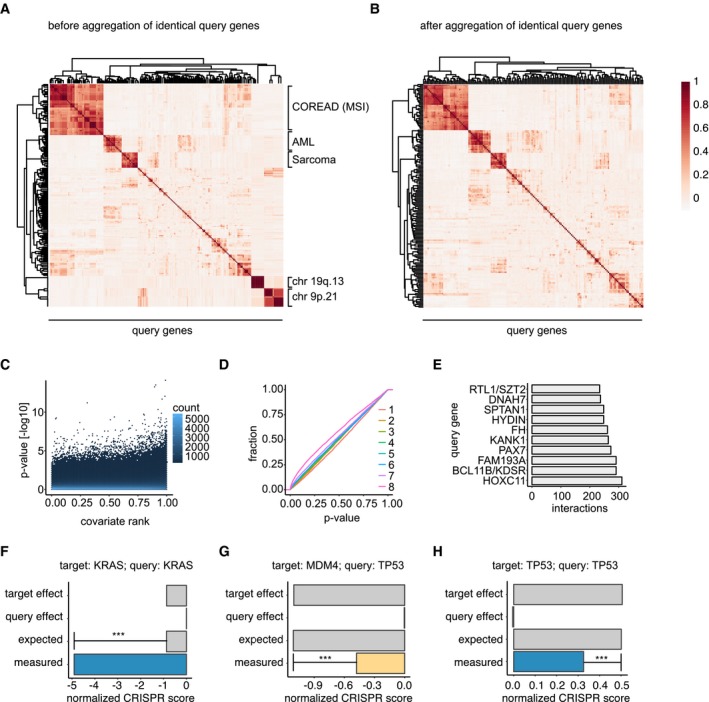
Fig. 4.I danni al DNA indotti dal DAG vengono riparati mediante ricombinazione omologa.un A549 cellule A549 sono state sincronizzate con 24 sieri affamati. Dopo di che, le cellule sono state incubate in mezzo completo con il trattamento di 50μM VAL-083 per 1 h. Poi, le cellule sono state lavate e sostituite con terreno completo per un ulteriore tempo di incubazione di 20, 24 o 48 ore. I lisati cellulari sono stati poi estratti per l’analisi western blot dei sensori e degli effettori coinvolti nel percorso di risposta al danno del DNA HR utilizzando i seguenti anticorpi: phospho-ATM (Ser1981), phospho-Chk2 (Thr68), phospho-Chk1 (Ser345 e Ser317), phospho-RPA32 (Ser33), e ɣH2AX. Immagini rappresentative sono mostrate da tre a quattro esperimenti indipendenti. b A549 cellule A549 sono state sincronizzate con 24 ore di fame di siero. Dopo di che, le cellule sono state incubate in mezzo completo con il trattamento di 50μM VAL-083 per 1 h. Poi, le cellule sono state lavate e sostituite con terreno completo per un ulteriore tempo di incubazione di 24 ore. Immagini rappresentative confocale di proteine indicate (BRCA1, RPA32, Rad51, e ɣH2AX) sono stati mostrati. La barra di scala rappresenta 5 mg. c Quantificazione di cellule A549 foci-positive presentate in b da 60-80 cellule per condizione sono stati mostrati. Le analisi statistiche sono state ottenute da tre esperimenti indipendenti (**p ≤ 0,01; ***p ≤ 0,001; Student’s t test). d A549 cellule A549 sono state trasfettate con controllo negativo (C) o tre BRCA1-targeting siRNA (B1, siBRCA1-2; B2, siBRCA1-15; o B3, siBRCA1-17) per 24 ore. Le cellule sono state poi seminate in piastre di coltura a 96 pozzetti e trattate con diverse concentrazioni di VAL-083 (0, 100 nM, 500 nM, 1μM, 1.5 μM, 2,5 μM, 5 μM, 10 μM, 25 μM, 50 μM, 100 μM e 200 μM) per 5 giorni. Dopo il trattamento, è stato eseguito il test del cristallo violetto per rilevare l’assorbanza a 560 nm di lunghezza d’onda. Il valore IC50 del VAL-083 è stato determinato applicando una curva dose-risposta sigmoidale ai dati utilizzando GraphPad Prism 6. I valori IC50 della VAL-083 nelle celle di controllo o BRCA1-knockdown sono presentati come errore medio-±standard (*p≤ 0,05; **p≤ 0,01; ***p≤ 0,001; test t dello studente). I lisati delle cellule del controllo o cellule BRCA1-knockdown sono stati analizzati da western blot con anticorpo contro BRCA1, e GAPDH è stato utilizzato come controllo di carico. Le immagini rappresentative sono mostrate da tre esperimenti indipendenti
Discussione
Gli agenti di reticolazione del DNA sono ampiamente utilizzati come chemioterapia in una varietà di tumori38. Mentre tutti mirano al DNA, ci sono piccole differenze che distinguono questi agenti l’uno dall’altro. Ad esempio, la maggior parte degli agenti hanno più di una funzione ed è spesso difficile disaccoppiare queste funzioni in termini di meccanismo d’azione. Il DAG è un agente bersaglio del DNA bifunzionale che causa N7-monoalchilguanina e legami incrociati tra i fili del DNA22ma è stato anche segnalato per inibire l’angiogenesi7. È stato dimostrato che il DAG interagisce con il DNA producendo 7-(1-deossigalattit-1-il)guanina, 7-(1-deossianidrogalattit-1-il)guanina, e 1,6-di(guanina-7-il)-1,6-dideoxigalattilo, di cui l’ultimo prodotto indica la formazione di legami incrociati inter o intra-filo39. Fino a questa data, negli Stati Uniti sono stati condotti più di 40 studi clinici di fase I e II sponsorizzati da NCI che coinvolgono DAG. I dati degli studi clinici e preclinici suggeriscono un’attività antitumorale del DAG in diversi tumori maligni, tra cui il cancro ai polmoni, i tumori cerebrali, la leucemia, il cancro al collo dell’utero e il cancro alle ovaie.1–6. Inoltre, uno studio clinico post-marketing aperto in Cina indaga l’attività del VAL-083 in pazienti affetti da NSCLC recidivanti o refrattari.40.
TMZ è un agente alchilante del DNA che agisce sulle posizioni N7 e O6 della guanina ed è attualmente utilizzato come trattamento di prima linea di GBM41. È interessante notare che, mentre TMZ e DAG hanno proprietà almeno parzialmente sovrapposte in termini di interazioni del DNA, i meccanismi di citotossicità sembrano essere diversi. Ad esempio, le cellule GBM resistenti a TMZ mostrano ancora sensibilità al DAG42, che indica funzioni non sovrapposte tra i due agenti. La conoscenza precisa dei meccanismi molecolari alla base della citotossicità delle cellule tumorali è essenziale per il posizionamento ottimale dei farmaci chemioterapici in un contesto clinico. Per questo motivo, abbiamo deciso di sezionare e descrivere i meccanismi citotossici del DAG utilizzando l’NSCLC come sistema modello. Abbiamo scoperto che il VAL-083, un DAG di grado clinico prodotto secondo una buona pratica di fabbricazione, è ben tollerato dalle cellule fintanto che si trovano nella fase G1 del ciclo cellulare. Le cellule hanno diversi sistemi in atto che possono risolvere i legami incrociati del DNA e le metilazioni come la riparazione dell’escissione nucleotidica (intra-filo) e il sistema di anemia Fanconi (inter-filo)38,43. La riparazione dei crosslinks del DNA in fase G1 è supportata dal macchinario di controllo della fase G1/S che mantiene le cellule in G1 fino al completamento della riparazione.44. Tuttavia, il punto di controllo della fase G1/S è spesso compromesso nelle cellule tumorali e in questo scenario, le cellule possono entrare nella fase S con lesioni del DNA non riparate.45. Quando le forchette di replicazione si scontrano con le lesioni del DNA, la replicazione viene bloccata esponendo tratti di ssDNA che saranno successivamente legati da RPA. La violenta collisione tra la lesione di DNA e la macchina di replicazione spesso si traduce in DSBs DNA, che sono catastrofici per le cellule46. Infatti, abbiamo trovato l’effetto citotossico del DAG sulle cellule NSCLC a svilupparsi in fase S, indicando che queste cellule progrediscono dalla fase G1 alla fase S con lesioni di DNA non riparate. Nella fase S, le cellule trattate con DAG mostrano una risposta profonda ɣH2AX indicativa del DNA DSBs. Abbiamo inoltre trovato che questi DSBs DAG-indotti indotta da DAG attivato il percorso HR e l’inibizione di HR drammaticamente sensibilizzati cellule NSCLC a DAG. Come tale, i nostri dati suggeriscono che le lesioni del DNA indotte da DAG si traducono in DNA DSBs replica-dipendenti in fase S che vengono successivamente riparati da HR (Fig. 5). È importante notare che il nostro lavoro fornisce una logica clinica per il posizionamento della chemioterapia a base di DAG nei tumori carenti nella riparazione dell’HR.Fig. 5Modello del meccanismo d’azione del DAG nelle cellule tumorali polmonari.il trattamento con DAG induce legami incrociati di DNA tra i filamenti attraverso l’alchilazione della N7-guanina, portando a rotture a doppio filamento di DNA replicazione-dipendente (DSB). Le cellule tumorali polmonari rispondono al DNA DSB indotto dal DAG attraverso l’attivazione della riparazione del DNA HR
Fig. 5.Modello del meccanismo d’azione del DAG nelle cellule tumorali polmonari.Il trattamento DAG induce legami incrociati tra i fili del DNA attraverso l’alchilazione della N7-guanina, portando a rotture a doppio filo del DNA replica-dipendente (DSB). Le cellule tumorali polmonari rispondono al DNA DSB indotto dal DAG attraverso l’attivazione della riparazione del DNA HR
Materiali e metodi
Reagenti e coltura cellulare
Il VAL-083 è stato ottenuto da DelMar Pharmaceuticals, Inc. (Vancouver, Canada e Menlo Park, CA, USA). La soluzione PI (1 mg/ml) e la soluzione di glutaraldeide (grado I, 50% in H2O) sono state acquistate da Sigma-Aldrich (Oakville, Canada). La soluzione di Sorenson è stata preparata con 9 mg di citrato trisodico, 195 ml 0,1 N HCl, 500 ml di etanolo al 90% e 305 ml di acqua distillata. Tutte le linee cellulari sono state mantenute a 37°C in atmosfera al 5% di CO2. A549 cellule A549 sono state coltivate nel mezzo di Dulbecco modificato Eagle’s Aquila integrato con il 10% di siero bovino fetale. H2122, H1792, e cellule H23 sono state coltivate in RPMI 1640 con il 10% di siero fetale bovino.
Saggio di proliferazione delle cellule di cristallo violetto
Dopo 72 h di diverse concentrazioni di trattamento VAL-083, le cellule sono state fissate in soluzione di glutaraldeide all’1% per 5 minuti. Dopo il risciacquo con acqua distillata, le cellule sono state incubate con 0,1% di soluzione di cristallo violetto colorante per 10 minuti. Le cellule sono state poi delicatamente lavate con acqua distillata e asciugate all’aria. I cristalli sulla piastra sono stati sciolti nella soluzione di Sorenson prima di leggere l’assorbanza a 560 nm di lunghezza d’onda con un lettore di micropiastra. Le cellule di sopravvivenza sono stati espressi come la percentuale rispetto alle cellule non trattate.
Analisi del ciclo cellulare utilizzando la colorazione PI
La distribuzione del ciclo cellulare è stata valutata in base al contenuto di DNA utilizzando la colorazione PI. Le cellule sieriche affamate (24 ore) sono state trattate con 5 μM VAL-083 per 1, 4, 19, 24, 44 e 49 ore. Le cellule sono state poi tripsinizzate, lavate in soluzione salina tamponata con fosfato (PBS) e centrifugate a 1000 giri al minuto per 5 minuti. I pellet delle cellule sono stati fissati in etanolo al 70% almeno per una notte a 4°C. Dopo il lavaggio con PBS, le cellule sono state incubate con 500 ml di soluzione PI in PBS contenente 50 mg / ml di PI, 100 mg / ml di RNase A, e 0,05% di Triton X-100 per 40 minuti a 37 ° C al buio. Successivamente, le cellule sono state lavate e risospese in PBS. Il contenuto di DNA sono stati analizzati con citometria a flusso (FACS Canto II), e gli istogrammi e le analisi quantitative delle proporzioni delle cellule in G0/G1, S e G2/M fasi sono state fatte utilizzando il software FlowJo. Le cellule non trattate sono state incluse come controllo.
Macchia occidentale
Le cellule sono state lisate in tampone EBC (50 mM Tris-HCl, pH 8,0, 120 mM NaCl, 1% NP-40, e 1 mM EDTA) integrate con inibitore della fosfatasi e inibitore della proteasi (Roche, Mississauga, Canada). Le proteine cellulari sono state separate mediante elettroforesi in gel di SDS-poliacrilammide e trasferite sulla membrana di fluoruro di polivinilidene. Dopo l’incubazione con tampone di bloccaggio per 1 ora, la membrana è stata incubata con anticorpi primari designati per una notte a 4°C. Poi, la membrana è stata lavata tre volte per 10 minuti con TBST e incubata con anticorpi anti-topo o anti-coniugati con perossidasi di rafano (Santa Cruz Biotechnology, Dallas, TX, USA) per 1-2 ore. La membrana è stata lavata con TBST tre volte e sviluppata con il sistema di substrato ECL Pierce (ThermoFisher Scientific, Burlington, Canada) secondo le istruzioni del produttore. I seguenti anticorpi primari sono stati utilizzati per l’immunoblotting: ɣH2AX (Cell Signaling Technology, Danvers, MA, USA, 2577); H2AX (Abcam, Toronto, Canada, ab11175); phospho-ATM (Ser1981) (Rockland Antibodies and Assays, Limerick, PA, USA, 200-301-400); ATM (Cell Signaling Technology, 2873); GAPDH (Cell Signaling Technology, 5174); phospho-RPA32 (Ser33) (Bethyl Laboratories, Montgomery, TX, USA, A300-246A); phospho-Chk1 (Ser345) (Tecnologia di segnalazione cellulare, 2348); phospho-Chk1 (Ser317) (Tecnologia di segnalazione cellulare, 12302); phospho-Chk2 (Thr68) (Tecnologia di segnalazione cellulare, 2661); RPA32 (Abcam, ab2175); BRCA1 (Novus Biologicals, Oakville, Canada, NB 100-404); e caspase 3 scissa (Tecnologia di segnalazione cellulare, 9661). Immagini rappresentative di blotting sono state mostrate da tre a quattro esperimenti indipendenti.
IF e microscopio
Le cellule sono state coltivate su coprioggetti in vetro per almeno 16 ore prima di morire di fame per 24 ore. Le cellule sincronizzate sono state trattate con 50 μM VAL-083 per 1 h seguita da lavaggio e incubazione con mezzo completo per altri 24 h. Successivamente, le cellule sono state lavate una volta con PBS e fissate per 30 minuti con il 4% di paraformaldeide in PBS a temperatura ambiente. Per la rilevazione dei focolai di danno al DNA, le cellule sono state preestratte con tampone citoscheletrico (25mM HEPES, pH 7.4, 50 mM NaCl, 1 mM EDTA, 3 mM MgCl2, 300 mM saccarosio, e 0,5% Tritone X-100) per 5 minuti a 4°C prima della fissazione con soluzione di paraformaldeide al 4%. Le cellule fisse sono stati lavati tre volte con PBS e permeabilizzato per 20 minuti con 0,5% Triton X-100 in PBS. Dopo il lavaggio con PBS per tre volte e il blocco con il 3% di siero albumina bovina in PBS per 1 ora a temperatura ambiente, le cellule sono state incubate durante la notte a 4°C con anticorpi primari corrispondenti diluiti in soluzione bloccante. Il giorno successivo, le cellule sono state lavate tre volte con PBS e incubate con anticorpi secondari appropriati marcati con fluoroforo per 1 h a temperatura ambiente. Dopo il lavaggio con PBS per tre volte, i coprioggetto sono stati montati con Vectashield mezzo di montaggio (con 4′,6-diamidino-2-fenilindolo). Le immagini sono state acquisite utilizzando il microscopio Zeiss AxioObserver e il microscopio confocale LSM-780. Per l’analisi delle immagini è stato utilizzato il software LSM-ZEN. I seguenti anticorpi primari e secondari sono stati utilizzati nella colorazione IF: ɣH2AX (Cell Signaling Technology, 2577); cyclin A2 (Abcam, ab16726); ɣH2AX (EMD Millipore, Etobicoke, Canada, 05-636); BRCA1 (Abcam, ab16780); Rad51 (Santa Cruz Biotechnology, H8349); RPA32 (Abcam, ab2175); asino anti-coniglio Alexa-Fluor 594 (ThermoFisher Scientific, A21207); asino anti-coniglio Alexa-Fluor 488 (ThermoFisher Scientific, A21206); asino anti-topo Alexa-Fluor 594 (ThermoFisher Scientific, A21203); e asino anti-topo Alexa-Fluor 488 (ThermoFisher Scientific, A21202). Sono state mostrate immagini rappresentative di tre esperimenti indipendenti.
transfezione di siRNA
Le cellule A549 sono state trasfettate con un siRNA di controllo o con siRNA di target BRCA1 utilizzando il reagente di trasfezione RNAiMAX (ThermoFisher Scientific) secondo le istruzioni del produttore. Dopo 24 ore di trasfezione, le cellule A549 sono state seminate in piastre di coltura a 96 pozzetti e trattate con diverse concentrazioni di VAL-083 per 5 giorni, seguite da un saggio violetto di cristallo. In parallelo lisati cellulari sono stati raccolti per la verifica occidentale blot verifica di BRCA1 knockdown. I siRNA utilizzati in questo studio sono stati i seguenti: C, siCon (mezzo di controllo negativo GC duplex, Invitrogen, 462001); B1, siBRCA1-2 (Qiagen, Toronto, Canada, SI00096313); B2, siBRCA1-15 (Qiagen, SI02664368); e B3, siBRCA1-17 (Qiagen, SI03103975).
Analisi statistica
Dove indicato, i valori p sono stati calcolati utilizzando il test t dello studente. I dati sono stati presentati come media±SD di tre esperimenti indipendenti.
Materiale elettronico supplementare
Figura supplementare LeggendeFigura supplementare S1Figura supplementare S1Figura supplementare S2Figura supplementare S3
References
- Nemeth L. Pharmacologic and antitumor effects of 1,2:5,6-dianhydrogalactitol (NSC-132313). Cancer Chemother. Rep.. 1972; 56:593-602. PubMed
- Haas CD, Stephens RL, Hollister M, Hoogstraten B. Phase I evaluation of dianhydrogalactitol (NSC-132313). Cancer Treat. Rep.. 1976; 60:611-614. PubMed
- Eagan RT. Platinum-based polychemotherapy versus dianhydrogalactitol in advanced non-small cell lung cancer. Cancer Treat. Rep.. 1977; 61:1339-1345. PubMed
- Eagan RT. Dianhydrogalactitol and radiation therapy. Treatment of supratentorial glioma. JAMA. 1979; 241:2046-2050. DOI | PubMed
- Eagan RT. Phase II study of the combination of dianhydrogalactitol, doxorubicin, and cisplatin (DAP) in patients with advanced squamous cell lung cancer. Cancer Treat. Rep.. 1981; 65:517-519. PubMed
- Haas CD, Baker L, Thigpen T. Phase II evaluation of dianhydrogalactitol in lung cancer: a Southwest Oncology Group Study. Cancer Treat. Rep.. 1981; 65:115-117. PubMed
- Jiang X. Dianhydrogalactitol, a potential multitarget agent, inhibits glioblastoma migration, invasion, and angiogenesis. Biomed. Pharmacother.. 2017; 91:1065-1074. DOI | PubMed
- (2017).Publisher Full Text
- Cetin K, Ettinger DS, Hei YJ, O’Malley CD. Survival by histologic subtype in stage IV nonsmall cell lung cancer based on data from the Surveillance, Epidemiology and End Results Program. Clin. Epidemiol.. 2011; 3:139-148. DOI | PubMed
- (2016).Publisher Full Text
- Barnholtz-Sloan JS. Incidence proportions of brain metastases in patients diagnosed (1973 to 2001) in the Metropolitan Detroit Cancer Surveillance System. J. Clin. Oncol.. 2004; 22:2865-2872. DOI | PubMed
- Pfister DG. American Society of Clinical Oncology treatment of unresectable non-small-cell lung cancer guideline: update 2003. J. Clin. Oncol.. 2004; 22:330-353. DOI | PubMed
- Pisters KM. Cancer Care Ontario and American Society of Clinical Oncology adjuvant chemotherapy and adjuvant radiation therapy for stages I-IIIA resectable non small-cell lung cancer guideline. J. Clin. Oncol.. 2007; 25:5506-5518. DOI | PubMed
- Azzoli CG. American Society of Clinical Oncology Clinical Practice Guideline update on chemotherapy for stage IV non-small-cell lung cancer. J. Clin. Oncol.. 2009; 27:6251-6266. DOI | PubMed
- Minari R, Bordi P, Tiseo M. Third-generation epidermal growth factor receptor-tyrosine kinase inhibitors in T790M-positive non-small cell lung cancer: review on emerged mechanisms of resistance. Transl. Lung Cancer Res.. 2016; 5:695-708. DOI | PubMed
- Chang A. Chemotherapy, chemoresistance and the changing treatment landscape for NSCLC. Lung Cancer. 2011; 71:3-10. DOI | PubMed
- Eckhardt S. Uptake of labeled dianhydrogalactitol into human gliomas and nervous tissue. Cancer Treat. Rep.. 1977; 61:841-847. PubMed
- Steino A. In vivo efficacy of VAL-083 in the treatment of non-small cell lung cancer. Cancer Res. 2014; 74:824. DOI
- Fouse SD. Dianhydrogalactitol inhibits the growth of glioma stem and non-stem cultures, including temozolomide-resistant cell lines, in vitro and in vivo. Cancer Res.. 2015; 75:2562. DOI
- Shih KC. Phase I/II study of VAL-083 in patients with recurrent glioblastoma. J. Clin. Oncol.. 2016; 34:2063. DOI
- . (2011–2016).Publisher Full Text
- Institoris E, Szikla K, Otvos L, Gal F. Absence of cross-resistance between two alkylating agents: BCNU vs bifunctional galactitol. Cancer Chemother. Pharmacol.. 1989; 24:311-313. DOI | PubMed
- Kuo LJ, Yang LX. Gamma-H2AX—a novel biomarker for DNA double-strand breaks. Vivo. 2008; 22:305-309.
- , Article ID 920161 (2010) (PMID: 20811597).
- Heichman KA, Roberts JM. Rules to replicate by. Cell. 1994; 79:557-562. DOI | PubMed
- King RW, Jackson PK, Kirschner MW. Mitosis in transition. Cell. 1994; 79:563-571. DOI | PubMed
- Sherr CJ. G1 phase progression: cycling on cue. Cell. 1994; 79:551-555. DOI | PubMed
- Yam CH, Fung TK, Poon RY. Cyclin A in cell cycle control and cancer. Cell. Mol. Life Sci.. 2002; 59:1317-1326. DOI | PubMed
- Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol. Cell. 2012; 47:497-510. DOI | PubMed
- Sartori AA. Human CtIP promotes DNA end resection. Nature. 2007; 450:509-514. DOI | PubMed
- You Z. CtIP links DNA double-strand break sensing to resection. Mol. Cell. 2009; 36:954-969. DOI | PubMed
- Daugaard M. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nat. Struct. Mol. Biol.. 2012; 19:803-810. DOI | PubMed
- Baude A. Hepatoma-derived growth factor-related protein 2 promotes DNA repair by homologous recombination. Nucleic Acids Res.. 2016; 44:2214-2226. DOI | PubMed
- Liu S. Distinct roles for DNA-PK, ATM and ATR in RPA phosphorylation and checkpoint activation in response to replication stress. Nucleic Acids Res.. 2012; 40:10780-10794. DOI | PubMed
- Feng L, Fong KW, Wang J, Wang W, Chen J. RIF1 counteracts BRCA1-mediated end resection during DNA repair. J. Biol. Chem.. 2013; 288:11135-11143. DOI | PubMed
- Jasin M. Homologous repair of DNA damage and tumorigenesis: the BRCA connection. Oncogene. 2002; 21:8981-8993. DOI | PubMed
- Konstantinopoulos PA, Ceccaldi R, Shapiro GI, D’Andrea AD. Homologous recombination deficiency: exploiting the fundamental vulnerability of ovarian cancer. Cancer Discov.. 2015; 5:1137-1154. DOI | PubMed
- Helleday T, Petermann E, Lundin C, Hodgson B, Sharma RA. DNA repair pathways as targets for cancer therapy. Nat. Rev. Cancer. 2008; 8:193-204. DOI | PubMed
- Institoris E. In vivo study on alkylation site in DNA by the bifunctional dianhydrogalactitol. Chem. Biol. Interact.. 1981; 35:207-216. DOI | PubMed
- (2015).Publisher Full Text
- Fan CH. O6-methylguanine DNA methyltransferase as a promising target for the treatment of temozolomide-resistant gliomas. Cell Death Dis.. 2013; 4:e876. DOI | PubMed
- Steino A. The unique mechanism of action of VAL-083 may provide a new treatment option for some chemo-resistant cancers. Mol. Cancer Ther.. 2014; 12:B252. DOI
- Ceccaldi R, Sarangi P, D’Andrea AD. The Fanconi anaemia pathway: new players and new functions. Nat. Rev. Mol. Cell Biol.. 2016; 17:337-349. DOI | PubMed
- Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev.. 2001; 15:2177-2196. DOI | PubMed
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature. 2009; 461:1071-1078. DOI | PubMed
- Jackson SP. Sensing and repairing DNA double-strand breaks. Carcinogenesis. 2002; 23:687-696. DOI | PubMed
Fonte
Zhai B, Steinø A, Bacha J, Brown D, Daugaard M, et al. (2018) Dianhydrogalactitol induces replication-dependent DNA damage in tumor cells preferentially resolved by homologous recombination. Cell Death & Disease 9(10): 1016. https://doi.org/10.1038/s41419-018-1069-9




