
Abstract
Contesto
In questa revisione, esaminiamo i dati in continua espansione e sempre più convincenti che collegano le radiazioni e le varie sostanze chimiche presenti nel nostro ambiente all’attuale elevata incidenza del cancro al seno. Riconosciamo l’importanza di molti fattori di rischio ampiamente conosciuti per il cancro al seno, tra cui: mutazioni genetiche primarie, storia riproduttiva e fattori legati allo stile di vita come l’aumento di peso, il consumo di alcol e la mancanza di esercizio fisico [1, 2]. Tuttavia, partiamo dal presupposto che, in totale, questi fattori non affrontano una parte considerevole del rischio della malattia [2, 5]. Un corpus sostanziale di prove scientifiche indica che l’esposizione alle comuni sostanze chimiche e alle radiazioni, singolarmente e in combinazione, contribuisce anche all’incidenza sempre più elevata del cancro al seno osservata negli ultimi decenni. Sebbene i tassi si siano complessivamente stabilizzati negli ultimi anni per alcuni sottoinsiemi di donne, c’è stato un significativo e progressivo aumento dell’incidenza del cancro al seno nei decenni successivi alla seconda guerra mondiale [6, 7], gli stessi decenni che hanno visto un aumento esponenziale dell’uso di sostanze chimiche per la produzione di pesticidi, erbicidi, plastiche, cosmetici e altri materiali e prodotti di uso comune [8- 10].
Questo rapporto si concentra su questi temi ambientali. Negli 8 anni trascorsi dall’ultima pubblicazione di una rassegna completa della letteratura in materia [11], sono stati pubblicati centinaia di nuovi articoli a sostegno di questo link, e le prove su questo argomento sono più ampie e di migliore qualità rispetto a quelle precedentemente disponibili. Dopo aver descritto la nostra metodologia per la selezione dei rapporti scientifici e il resoconto dei risultati statistici, presentiamo sezioni introduttive sulle statistiche e sui sottotipi del cancro al seno, nonché concetti critici per inquadrare i complessi dati che stiamo esplorando. Esaminiamo poi la letteratura sulle esposizioni a sostanze tossiche ambientali e sul rischio di sviluppare il cancro al seno, dividendo la discussione sulle prove in sette sezioni principali: (1) Ormoni: Agenti farmaceutici e prodotti per la cura personale; (2) Composti che alterano il sistema endocrino (EDC); (3) Ormoni negli alimenti: Naturale e additivi; (4) Prodotti chimici industriali non EDC; (5) Fumo di tabacco: Attivo e passivo; (6) lavoro a turni, luce notturna e melatonina; e (7) radiazioni. Concludiamo con una breve sinossi e una riflessione sullo stato delle prove, comprese le limitazioni metodologiche e le promesse, nonché le direttive per le future esigenze di ricerca.
Metodologia
Processo di selezione dell’articolo
L’obiettivo di questa revisione è di offrire un’ampia panoramica della letteratura scientifica che esamina le potenziali connessioni tra l’esposizione a sostanze tossiche ambientali e i cambiamenti nel rischio di sviluppare il cancro al seno, aggiornando la nostra ultima revisione di questo argomento pubblicata nel 2009. Per incorporare pienamente i materiali pertinenti, abbiamo inserito i seguenti termini di ricerca sia in PubMed che in Scopus: “cancro al seno” e “tumori mammari” in combinazione con “ambiente”, “perturbatori endocrini/composti perturbatori endocrini” e tutti i singoli agenti tossici trattati in questo rapporto.
Nella selezione degli studi epidemiologici, abbiamo sottolineato il lavoro degli ultimi 10 anni. Quando gli studi erano rapporti di follow-up di studi longitudinali di grandi dimensioni, abbiamo anche riportato i dati precedenti in quanto il tempo che intercorreva tra le esposizioni e le valutazioni dei risultati poteva portare a conclusioni diverse, o al riconoscimento di risultati diversi man mano che i partecipanti allo studio raggiungevano età più avanzate e, soprattutto, progredivano dallo stato pre-menopausa a quello post-menopausa.
Negli 8 anni trascorsi dal nostro ultimo rapporto, c’è stato un sostanziale aumento della quantità di informazioni focalizzate sui meccanismi alla base delle complesse relazioni tra esposizione e rischio di sviluppare il cancro al seno. Ciò è particolarmente vero nel crescente campo dell’esame delle esposizioni a composti che alterano il sistema endocrino e del rischio di malattia. Ci siamo quindi concentrati su articoli degli ultimi 8 anni. Anche se non abbiamo riportato tutti i geni la cui espressione potrebbe essere influenzata da una particolare esposizione, abbiamo cercato di dare una panoramica completa della comprensione attuale dei processi fisiologici, evolutivi, genetici, epigenetici ed endocrini che sono influenzati da esposizioni rilevanti per un cambiamento del rischio di sviluppare il cancro al seno. Anche se l’enfasi è stata posta sui dati più recenti, abbiamo incluso i risultati precedenti quando erano necessari come sfondo o per fornire un quadro più completo delle prove.
Eccezioni alla nostra primaria dipendenza dalla letteratura molto recente si trovano nelle sezioni sui prodotti chimici industriali non perturbatori endocrini e su alcuni pesticidi ed erbicidi. Gran parte dei dati rilevanti per questi agenti tossici provengono da studi condotti da 25 a 30 anni fa, quando il Programma Nazionale di Tossicologia (NTP) e l’Agenzia Internazionale per la Ricerca sul Cancro (IARC) determinavano la possibile cancerogenicità di queste sostanze chimiche.
Infine, nella selezione degli studi da riportare, abbiamo avuto cura di includere gli studi che hanno avuto risultati negativi, cioè quelli che non hanno riportato alcuna relazione significativa tra l’esposizione e il rischio di sviluppare il cancro al seno. Dove possibile, abbiamo poi esplorato le possibili differenze nella progettazione dello studio o nei metodi che potrebbero spiegare le differenze nei risultati tra gli studi.
Segnalazione di statistiche per studi epidemiologici
Riportiamo le statistiche (ad esempio, RR, OR, HR, ecc.), insieme ai livelli di fiducia del 95%, come offerto dagli autori dei singoli rapporti. Laddove sono stati effettuati aggiustamenti espliciti, notiamo il tipo di statistica utilizzata e la variabile di aggiustamento. Più spesso, però, fattori quali l’età, lo stato della menopausa, il sottotipo di cancro al seno (per stato dei recettori, duttale vs. lobulare, in situ vs. invasivo, ecc.), l’identità razziale/etnica, sono riportati come fattori principali da analizzare, insieme agli effetti di particolari esposizioni. I principali effetti e le interazioni significative tra le esposizioni e queste altre variabili sono riportati in questa rassegna.
Introduzione
In questa sezione introduttiva, forniamo le statistiche di base e una breve esplorazione dei diversi sottotipi di cancro al seno – riconoscendo che il termine “cancro al seno” è spesso usato come proxy di diversi profili genetici, istopatologici e ormonali distinti per la malattia. Introduciamo quindi una serie di concetti chiave di inquadramento necessari per apprezzare la complessa evidenza a sostegno (o meno) di una crescente comprensione dei dati che implicano specifici agenti tossici ambientali in un aumento del rischio di sviluppare il cancro al seno. Questi concetti di inquadramento includono: (a) risposte a basse dosi e non monotoniche; (b) interazioni tra tossicanti ambientali; (c) interazioni gene-ambiente e cambiamenti epigenetici; (d) interazioni cellula-cellula e la teoria di campo dell’organizzazione dei tessuti; e (e) tempi di esposizione. Concludiamo con un modello schematico della complessità dei fattori che influenzano il rischio di sviluppare il cancro al seno, con un’enfasi sui fattori ambientali.
Statistiche sul cancro al seno
Il programma di sorveglianza, epidemiologia e risultati finali (SEER) del National Cancer Institute (NCI) prevedeva che nel 2015 negli Stati Uniti, 40.290 donne e 440 uomini sarebbero morti di cancro al seno e 231.840 donne e 2350 uomini avrebbero ricevuto una diagnosi di cancro al seno invasivo; altre 60.290 donne avrebbero ricevuto una diagnosi di cancro al seno in situ. All’inizio del 2016, il NSC ha stimato che circa 3.560.570 donne statunitensi vivono con una diagnosi di cancro al seno [12].
L’anno più recente per il quale esistono dati accurati relativi all’incidenza e alla mortalità del cancro al seno è il 2012. Oltre ai rapporti di incidenza e mortalità totali nazionali, i dati SEER sono suddivisi per categorie di razza/etnicità autodescritte nei principali censimenti. I tassi di incidenza medi (numero di donne diagnosticate per 100.000 donne, adattate per età e normalizzate alla popolazione statunitense standardizzata del 2000) nei 5 anni dal 2008 al 2012 differiscono tra le diverse categorie del censimento, così come le tendenze nel tempo. I tassi di incidenza media quinquennale per i bianchi sono stati i più alti (126,1), mentre i tassi per le donne di colore sono stati solo leggermente inferiori (124,1). Tuttavia, nel 2012, per la prima volta da quando il SEER ha iniziato a raccogliere dati nel 1975, l’incidenza per questi due gruppi è convergente; storicamente le donne nere avevano un tasso di incidenza significativamente più basso. I tassi di incidenza media a 5 anni erano più bassi per le donne americane indiane e nordamericane (91,9), ispaniche (91,9) e delle isole dell’Asia e del Pacifico (88,3) [12].
In tutti i gruppi razziali ed etnici degli Stati Uniti, i tassi di mortalità (decessi per 100.000 donne, adeguati all’età e normalizzati alla popolazione statunitense standardizzata del 2000) per cancro al seno sono diminuiti dal loro picco della metà della fine degli anni ’90. Nonostante questa apparente buona notizia, significative disparità razziali/etniche sono rimaste costanti negli ultimi decenni. Negli Stati Uniti, le donne di colore hanno il più alto tasso di mortalità per cancro al seno (31,0) di qualsiasi altro gruppo razziale/etnico. Le donne dell’Asia e delle isole del Pacifico hanno il più basso tasso di mortalità (11,4), mentre le donne bianche (21,9), ispaniche (14,5) e americane indiane e nordamericane (15,0) hanno tassi intermedi. Nonostante il calo universale dei tassi di mortalità negli ultimi due decenni e la somiglianza dei tassi di incidenza, nello stesso periodo le disparità tra i tassi di mortalità delle donne bianche e nere sono cresciute in modo significativo; il tasso di mortalità delle donne nere con diagnosi di cancro al seno è superiore del 42% rispetto al tasso comparabile delle donne bianche [12, 13].
Sottotipi di cancro al seno
Il cancro al seno non è una malattia singolare, e sarà importante in tutto questo rapporto esaminare, ove possibile, il sottotipo o i sottotipi della malattia più colpiti dall’esposizione a sostanze tossiche ambientali. Sono stati sviluppati diversi sistemi di classificazione per distinguere diversi sottotipi della malattia, tra cui l’età della paziente (di solito divisa per la pre o post-menopausa, con l’età di 50 anni spesso come proxy per lo spostamento tra le fasi riproduttive); presentazione in situ, localizzata, regionale o metastatica; caratteristiche morfologiche; grado istologico e tasso di proliferazione cellulare; o profilo di espressione genica [14-17].
Di particolare rilevanza per la discussione delle esposizioni ambientali, in particolare ai composti che alterano il sistema endocrino (EDC), è la classificazione basata sull’espressione del recettore degli estrogeni (ER), del progesterone (PR) o dell’oncogene HER2. Due sottotipi luminali (A e B) esprimono ER ma non HER2, con Luminal A co-espressivo PR e che ha un basso tasso di proliferazione e Luminal B che ha un alto tasso di proliferazione o bassa espressione PR. Luminal B-like (HER2 positivo) esprime ER e alti livelli di HER2, con qualsiasi profilo di proliferazione e PR. Il sottotipo HER2 positivo ha una sovraespressione di HER2 ma senza ER o PR. Il tumore al seno triplo negativo non ha alcuna espressione di ER, PR o HER2 [18].
I sottotipi di cancro al seno non sono distribuiti in modo casuale tra la popolazione e si riscontrano differenze quando le diagnosi sono stratificate per età, razza/etnia, storia riproduttiva, indice di massa corporea, stato socioeconomico o posizione geografica [17, 19-22]. Per esempio, le donne più giovani in generale, e le donne di colore più giovani in particolare, hanno maggiori probabilità di presentare il sottotipo triplo negativo (ER-, PR-, e HER2-) della malattia, una diagnosi che è sia più aggressiva che meno reattiva al trattamento rispetto ai tumori ER+/PR+ o HER2+ [12, 23, 24]. Come le giovani donne di colore, anche i latini sono colpiti in modo sproporzionato da tumori triplo-negativi aggressivi [17, 24, 25].
Razza ed etnia
L’esistenza di differenze tra categorie razziali ed etniche auto-identificate non implica necessariamente differenze genetiche. Infatti, esse riflettono la complessità della posizione geografica; lo status sociale e socioeconomico; lo stress e la sicurezza personale e della comunità; i fattori dello stile di vita, tra cui la dieta, l’esercizio fisico, l’uso di alcol e farmaci; le risposte fisiologiche ai fattori della vita; le interazioni gene-ambiente; i cambiamenti epigenetici – tutti fattori che possono cambiare nel corso della vita dell’individuo e possono variare considerevolmente tra le persone che si identificano in una particolare categoria razziale/etnica [26, 27]. A causa della confusione di fattori economici e sociali, persone di diverse identità razziali/etniche possono anche sperimentare diverse esposizioni ambientali e professionali a sostanze tossiche che colpiscono la malattia [28- 30].
Le minoranze razziali ed etniche sono spesso esposte a livelli e varietà di inquinanti ambientali sproporzionatamente elevati negli Stati Uniti [31], così come le persone che vivono in povertà [32]. Ci sono differenze razziali/etniche nel carico corporeo delle diverse sostanze chimiche ambientali che sono state associate ad un aumento del rischio di cancro al seno. I neri hanno livelli di carico corporeo più elevati rispetto ai bianchi o agli americani messicani di molte sostanze chimiche, tra cui molti bifenili policlorurati (PCB), mercurio, idrocarburi poliaromatici (IPA) e ftalati. I messicani americani hanno livelli più elevati di diclorodifeniltricloroetano (DDT) [33]. Il carico corporeo variabile di alcune sostanze chimiche, tra cui il bisfenolo A (BPA), i polifluorurati (PFC) e il triclosan, tutti comunemente presenti nei prodotti per la casa, sono associati sia alla razza/etnicità che allo stato socioeconomico [27, 34, 35]. Tuttavia, come sottolinea Nelson, lo status socioeconomico e la razza/etnicità servono molto probabilmente in modo indipendente come marcatori per altre attività o circostanze che influenzano il livello di esposizione a sostanze chimiche potenzialmente tossiche [27].
Concetti di inquadramento
Costruendo ed estendendo il quadro dei ‘Hallmarks of Cancer’ proposto da Hanahan e Weinberg [36], un team internazionale di 170 scienziati che partecipano al progetto Halifax ha recentemente valutato i contributi alla cancerogenesi delle esposizioni a basse dosi a singoli composti e miscele di sostanze chimiche ambientali su ciascuno dei fenotipi di Halifax proposti [37]. Altre recenti revisioni si sono concentrate sull’importanza di valutare: le relazioni dose-risposta non monotoniche, specialmente tra CDE e risultati sanitari [38]; la tempistica delle esposizioni a sostanze tossiche ambientali, con particolare attenzione alle esposizioni fetali ad adolescenti a CDE e al successivo sviluppo di malattie [39- 41]; la carcinogenesi ambientale dal punto di vista delle alterazioni della cellula-cellula (ad es, interazioni stromale-epiteliali) [42, 43]; interazioni gene-ambiente [44, 45]; l’importanza di usare i principi dell’endocrinologia di base per stabilire modelli meccanicistici per l’esame degli impatti sulla salute delle esposizioni agli EDC [46, 47] e la rilevanza per questi meccanismi nella comprensione del crescente apprezzamento dei legami tra i tossici ambientali e l’aumento del rischio per molte malattie, incluso il cancro al seno [48- 52].
Nel presente documento non offriremo una panoramica completa di questi concetti di inquadramento, ma rimanderemo il lettore alle recensioni sopra citate. Introdurremo invece brevemente i concetti principali con un paio di esempi rilevanti per esplorare le seguenti evidenze che collegano l’esposizione a sostanze chimiche ambientali tossiche con l’aumento del rischio di sviluppo del cancro al seno. Mentre alcune delle sostanze chimiche che destano preoccupazione sono tradizionalmente definite cancerogene, molte altre rientrano nella classe dei composti che alterano il sistema endocrino (EDC), un gruppo di composti esogeni che esercitano almeno in parte il loro impatto sui risultati sanitari alterando l’attività del sistema endocrino.
Risposte a basse dosi e non monotoniche
Gli EDC disturbano il sistema endocrino. In quanto tali, i loro meccanismi d’azione e le loro proprietà sono diversi dalla maggior parte delle sostanze cancerogene non EDC per le quali il modello tossicologico è che dosi più alte sono più dannose di dosi più basse; la relazione tra dose e danno è funzionalmente lineare; e ci possono essere livelli di sicurezza al di sotto dei quali non si osserva alcun impatto negativo (il livello senza effetti collaterali o NOAEL) [53, 54]. Invece, sotto molti aspetti, gli EDC agiscono molto come gli ormoni naturali: a dosi molto basse, specialmente durante i periodi critici di sviluppo, e spesso seguendo curve di risposta non monotonica (NMR) [38, 47]. Così le risposte subcellulari e fisiologiche alle esposizioni a basse dosi possono essere maggiori o almeno diverse dalle esposizioni a dosi più elevate.
Per esempio, molti studi su animali hanno dimostrato che le esposizioni prenatali o neonatali al bisfenolo A (BPA) portano a cambiamenti nello sviluppo dei tessuti mammari che aumentano la probabilità di un successivo sviluppo di tumori mammari. Eppure alcuni di questi effetti dipendono dalla dose, ma non in modo lineare. In uno studio, le esposizioni prenatali a basse dosi (e rilevanti per l’ambiente) di BPA hanno avuto effetti significativi sul profilo di espressione del gene della ghiandola mammaria appena prima dell’inizio della pubertà, mentre livelli di esposizione più elevati hanno alterato l’espressione di diversi geni e in un’età molto più avanzata [55]. In un altro rapporto, le dighe di ratto sono state esposte via gavage a nessun BPA, o dosi che vanno da 0,025 a 50 mg BPA/kg bw/d dal settimo giorno di gestazione fino allo svezzamento dei loro cuccioli. Come adulti, la prole femmina che era stata esposta alla dose di 0,25 mg aveva aumentato l’incidenza di iperplasia intraduttale, anche se non sono stati trovati effetti simili sia per le esposizioni più elevate o inferiori [56].
Blei et al. hanno esaminato l’effetto a vita delle esposizioni alimentari a due diverse quantità di isoflavoni derivati dalla soia, scegliendo dosi che hanno prodotto concentrazioni simili ai livelli plasmatici più alti e più bassi di isoflavoni nelle donne asiatiche. Anche se sia bassi che alti livelli di esposizione hanno portato ad un inizio precoce della pubertà, solo bassi livelli di esposizione hanno portato ad una maggiore espressione del marcatore di proliferazione Ki67 nelle ghiandole mammarie degli adulti di 97 giorni. D’altra parte, solo livelli di esposizione più elevati hanno portato a significative diminuzioni nell’espressione del marcatore di proliferazione PCNA nel tessuto mammario da ratti ovariectomizzati che erano stati trattati con estradiolo. In questi animali, la somministrazione di estradiolo ha portato alla stimolazione additiva dell’induzione di PR negli animali che sono stati esposti alle esposizioni a basse dosi, mentre i livelli di esposizione ad alte dosi hanno inibito l’espressione di PR indotta da estradiolo nella ghiandola mammaria [57].
Interazioni tra i tossici ambientali
Numerosi studi sugli animali indicano che i tipi di miscele a cui un animale è esposto sono determinanti per la determinazione del rischio finale [58]. Sono state testate solo relativamente poche combinazioni e dosi di sostanze chimiche. Questo forse non è sorprendente: Una stima prevede che sarebbero necessari 166 milioni di esperimenti per testare tutte le combinazioni di tre delle 1000 più comuni sostanze chimiche di sintesi attualmente in uso [59]. Mentre solo una piccola parte di questi studi è stata effettivamente condotta, ci sono diversi rapporti che dimostrano che le miscele di sostanze chimiche ambientali o di sostanze chimiche e radiazioni, possono alterare i processi biologici e possibilmente portare ad un aumento del rischio di cancro al seno.
Per esempio, il saggio E-screen test utilizza le cellule tumorali del cancro al seno umano ER+ (cellule MCF-7) che dipendono dagli estrogeni per la crescita e la proliferazione cellulare [60], e singoli studi possono esaminare gli effetti di decine di sostanze chimiche a dosi multiple, da sole e in combinazione sulla proliferazione delle cellule del cancro al seno [61, 62]. Un esame degli effetti combinati di 11 diversi contaminanti ambientali – tutti aggiunti a concentrazioni di NOAEL – ha mostrato che le sostanze chimiche avevano effetti additivi tra loro e anche con l’estradiolo presente in natura [63]. A livelli riscontrati nel nostro ambiente, l’onnipresente plastificante bisfenolo A ha anche aumentato significativamente gli effetti dell’estradiolo [64].
Payne et al. hanno usato lo screening degli estrogeni del lievito (SÌ), un test in vitro di attivazione del recettore degli estrogeni, per esaminare gli effetti combinati di un residuo di pesticida (o,p’-DDT), di un estrogeno vegetale (genestio, che si trova nella soia) e di due tensioattivi alchilfenolo (agenti schiumogeni e disperdenti chimici; 4-n-ottilfenolo e 4-nonilfenolo). Sono stati trovati chiari effetti additivi dei quattro prodotti chimici [65].
Rivero et al. hanno esaminato gli effetti di due miscele di pesticidi organoclorurati, la prima composta per imitare il profilo chimico trovato in donne sane e la seconda per imitare il profilo del pesticida trovato in pazienti con cancro al seno. Entrambe le miscele di geni down-regolati la cui espressione è coinvolta nel legame dell’ATP nelle normali cellule epiteliali mammarie umane, ma ci sono stati effetti molto diversi dei due profili di miscela sull’espressione degli oncogeni e dei geni soppressori dei tumori [66, 67]. Allo stesso modo, combinazioni di diversi pesticidi organoclorurati, miscelati per imitare le combinazioni trovate in campioni umani, hanno aumentato gli effetti citotossici in una linea cellulare derivata dalle normali cellule epiteliali del seno umano [68].
In uno studio sullo sviluppo dei tessuti mammari, miscele di sostanze chimiche comunemente presenti nell’ambiente hanno reso i tessuti mammari di ratto più suscettibili all’esposizione agli estrogeni alimentari dopo la nascita, portando ad anomalie tissutali che sono state associate a tumori mammari [69]. E il pretrattamento di ratti giovani con una bassa dose di radiazioni ha portato ad un’insorgenza precoce e ad un aumento della frequenza di tumori mammari mutati dopo la successiva esposizione ad una nota sostanza chimica cancerogena [70].
Interazioni gene-ambiente e cambiamenti epigenetici
Diversi studi hanno riportato un aumento del rischio di sviluppare il cancro al seno nelle donne con mutazioni BRCA1 o BRCA2 in seguito all’esposizione a radiazioni mediche, attraverso la mammografia o la radioterapia [71- 74]. Un altro rapporto ha rilevato che una combinazione di molteplici varianti nei geni associati ai meccanismi di riparazione del DNA ha portato ad un aumento del rischio di sviluppare il cancro al seno associato alla mammografia [75].
Altri studi hanno riportato un’interazione tra varie varianti geniche associate al rischio di cancro al seno e all’esposizione all’ambiente [76]. Ma nel complesso, la letteratura in materia è mista, con diversi polimorfismi mononucleotidici (SNP) e diverse sostanze tossiche ambientali sottoposte a test. Una panoramica completa del settore ha concluso che questi studi sono troppo pochi e insufficienti per una chiara dimostrazione delle interazioni tra particolari SNP o cluster di SNP e fattori ambientali nell’influenzare il rischio di cancro al seno, dato che la maggior parte dei grandi studi epidemiologici produce, nella migliore delle ipotesi, effetti molto piccoli che spesso non sono replicabili [45]. Tuttavia, gli autori hanno concluso che, “Attualmente, dovremmo considerare le varianti ereditarie e i fattori ambientali come fattori moltiplicativi/additivi nella previsione del rischio di cancro al seno” [45].
Oltre ai polimorfismi genetici che influenzano gli effetti dei tossici ambientali sulle risposte inter e intracellulari, le sostanze chimiche ambientali, in particolare gli EDC, possono alterare la regolazione dei geni coinvolti nella proliferazione cellulare, le vie di segnalazione dell’apoptosi, ecc. attraverso processi epigenetici [77, 78]. Attraverso meccanismi quali la metilazione del DNA alterato, le modifiche degli istoni e l’espressione di piccoli RNA regolatori (microRNA), l’esposizione chimica e alle radiazioni può avere effetti profondi sulla struttura e la funzione della ghiandola mammaria in via di sviluppo [79- 82].
Per esempio, Kutanzi e Kovalchuk hanno riferito che il trattamento concomitante di ratti adulti ACI con fonti esogene di estradiolo e radiazioni ha portato ad un aumento della metilazione delle ghiandole mammarie e dell’acetilazione degli istoni H3 e H4, e ad un significativo aumento dell’induzione delle vie MAPK e p38, biomarcatori noti per l’instabilità cromosomica [83]. E nel normale MCF-7 umana ER + linea cellulare umana, l’aggiunta del promotore della crescita, zeranolo, ha portato a effetti stimolatori sulla crescita cellulare. Questi risultati sono stati guidati, almeno in parte, dal down-regolazione del gene soppressore tumorale p53, un processo che è stato accompagnato da up-regolazione del DNA-metiltransferasi 1 [84].
Hussain et al. hanno esplorato gli effetti del BPA sull’espressione di HOXC6, un gene omeocontenuto che è associato alla crescita e allo sviluppo delle cellule mammarie e che è sovraespresso in molti tumori al seno. Sia nelle linee cellulari MCF-7 che nel tessuto mammario di ratti adulti Sprague-Dawley ovariectomizzati, l’esposizione al BPA ha aumentato la metilazione dell’istone e l’acetilazione e ha reclutato l’RNA polimerasi II presso il promotore dell’HOXC6, con conseguente sovraespressione dell’HOXC6 [85]. Allo stesso modo, Doherty et al. hanno dimostrato in entrambe le cellule MCF-7 e nelle ghiandole mammarie da topi esposti neonatalmente che sia il trattamento BPA o dietilstilbestrolo (DES) ha portato ad un aumento di 2-3 volte l’espressione del cancro al seno associato istone metiltransferasi, Enhancer di Zeste Homolog 2 (EZH2) espressione mRNA e la successiva sintesi EZH2. Questi cambiamenti sono stati accompagnati da un aumento della trimetilazione dell’istone H3, sia in vivo che in vitro [86].
Le interazioni cellule-cellule e la teoria del campo dell’organizzazione dei tessuti
Piuttosto che modellare lo sviluppo del cancro come risultato di mutazioni del DNA accumulate, con conseguenti cambiamenti nella fisiologia cellulare che si basano sull’instabilità genetica iniziale [36, 37], la Teoria di Campo dell’Organizzazione dei Tessuti (TOFT) della carcinogenesi [87, 88] si basa su una visione più ecologica del funzionamento cellulare e dell’organizzazione dei tessuti. La TOFT inizia riconoscendo che la proliferazione cellulare è lo stato di default per le cellule, con processi e segnali chimici che regolano criticamente il tasso di proliferazione, e anche che le cellule lavorano in costante interazione con le cellule vicine nei vari tessuti all’interno di un organo [87]. Le perturbazioni dei segnali reciproci e l’interruzione delle interazioni da cellula a cellula, in particolare tra il mesenchima/stroma e i compartimenti parenchima/epiteliali della ghiandola mammaria in via di sviluppo, possono essere alla base dello sviluppo del cancro al seno [39].
Gran parte del lavoro di esplorazione di questo modello è stato fatto esaminando gli effetti dell’esposizione prenatale o neonatale al BPA e i cambiamenti morfologici nei compartimenti stromali ed epiteliali della ghiandola mammaria dei roditori [40, 89- 92]. Ad esempio, Wadia et al. hanno esplorato gli effetti delle esposizioni prenatali a basse dosi di BPA su cambiamenti morfologici nelle ghiandole mammarie dei topi fetali utilizzando livelli di esposizione che hanno dimostrato in precedenza di indurre tumori preneoplastici e tumori cancerogeni in età adulta. Esposizioni neonatali BPA neonatale ha portato a cambiamenti nell’espressione genica in entrambi i compartimenti epiteliali e stromali di sviluppo delle ghiandole mammarie in sviluppo dal giorno gestazionale 19 topi. Espressione alterata nella frazione stromale è stato trovato per i geni coinvolti in percorsi che mediano l’adesione focale e l’adipogenesi, mentre nella frazione epiteliale ci sono stati cambiamenti nell’espressione dei geni coinvolti in apoptosi. Ne derivano cambiamenti morfologici dovuti all’esposizione al BPA, tra cui lo sviluppo avanzato del cuscinetto di grasso e la formazione ritardata del lume epiteliale, effetti che vengono eliminati in assenza di ERα. Insieme questi dati hanno portato gli autori a proporre che il BPA (e gli estrogeni, più in generale) agiscono direttamente sullo stroma dove sono espressi i recettori degli estrogeni prenatali (ERα, ERβ e GPR30). A sua volta, i segnali dello stroma alterano l’espressione del gene epiteliale e, in definitiva, la prima programmazione morfologica per la ghiandola mammaria in via di sviluppo [89].
Tempistica delle esposizioni
Un ampio corpus di ricerche dimostra che la tempistica delle esposizioni lungo tutto l’arco della vita può avere un’enorme influenza sul se, quanto e come un’esposizione ambientale possa influenzare il rischio di un successivo sviluppo del cancro al seno. Le cellule mammarie sono più suscettibili agli effetti cancerogeni degli ormoni, delle sostanze chimiche e delle radiazioni durante le prime fasi dello sviluppo, dal periodo prenatale alla pubertà e all’adolescenza, fino alla prima gravidanza a termine. Particolari preoccupazioni sono state dimostrate per l’esposizione durante il periodo prenatale e la prima infanzia. Molti di questi dati provengono dall’uso di modelli animali (esaminati in sezioni appropriate all’interno di questo rapporto), ma ci sono anche diverse fonti di dati che supportano questa affermazione dalla letteratura clinica umana.
Ad esempio, le figlie di madri che hanno sofferto di preeclampsia durante la gravidanza, associata a livelli più bassi di estrogeni materni, hanno diminuito il rischio di sviluppare il cancro al seno in età adulta [93, 94]. Alla nascita, i livelli del cordone ombelicale di estriolo (E3) e di estetrol (E4) – ma non di estradiolo (E2) o di estrone (E1) – hanno dimostrato di essere diminuiti nei neonati partoriti da gravidanze associate alla preeclampsia [95]. D’altra parte, le ragazze che nascono con un peso alla nascita inferiore, associato a una maggiore esposizione agli estrogeni fetali, hanno aumentato il rischio di una successiva diagnosi di cancro al seno [96, 97].
E anche se è raro avere l’esposizione a sostanze chimiche esogene solo durante lo sviluppo fetale, tra il 1938 e il 1971 milioni di feti sono stati esposti agli estrogeni sintetici, il dietilstilbestrolo (DES), quando alle loro madri in gravidanza è stato prescritto il farmaco al fine di prevenire aborti spontanei e altre complicazioni della gravidanza. DES è stato vietato quando le figlie di donne che hanno preso il farmaco durante la gravidanza sono stati trovati per avere aumentato i tassi di un adenocarcinoma vaginale a cellule chiare estremamente raro. L’esposizione al DES è stata anche associata ad un aumento del rischio di cancro al seno nelle madri [98-100].
In uno studio di follow-up su figlie che sono state esposte prenatalmente al DES, è stato osservato un aumento quasi doppio del rischio di cancro al seno nelle donne di età superiore ai 40 anni. Un effetto ancora maggiore è stato riscontrato per le donne oltre i 50 anni, anche se relativamente poche delle figlie avevano ancora raggiunto quell’età al momento dello studio [101, 102]. Anche le donne esposte in utero che presentavano le anomalie più gravi delle loro cellule epiteliali vaginali (un indicatore dell’esposizione a dosi più elevate di DES) presentavano un rischio maggiore di sviluppare il cancro al seno [99]. Ora sembra che anche le nipoti delle donne a cui è stato prescritto il DES durante la gravidanza stiano sperimentando un’elevata incidenza di cancro al seno [100].
In uno studio prospettico caso-controllo su 9300 donne in una coorte di gravidanza, sono stati analizzati campioni di sangue materno post-partum memorizzati per i livelli di diclorodifenil-tricloroetano (DDT). Le figlie sono state seguite per 52 anni e la diagnosi di cancro al seno in questa coorte è stata determinata. Livelli più elevati di DDT materno sono stati associati a un aumento di quasi 4 volte della presenza di cancro al seno nelle figlie all’età di 52 anni [103].
Uno studio prospettico e nidificante caso-controllo su 258 donne ha esplorato i loro livelli storici stimati di DDT sulla base di dati aggregati dell’anno di nascita e dei livelli di DDT nel sangue al momento in cui le donne hanno dato alla luce il loro primo figlio. L’esposizione al DDT durante l’infanzia e la prima adolescenza (al di sotto dei 14 anni) è stata associata a un aumento di 5 volte del rischio di sviluppare il cancro al seno prima dei 50 anni. Più giovani erano le donne quando l’uso pesante del DDT è iniziato nel 1945, maggiore era il rischio [104].
Altri studi hanno dimostrato che l’infanzia e l’adolescenza sono particolarmente suscettibili all’esposizione alle radiazioni mediche e al successivo sviluppo del cancro al seno. Decenni di ricerche hanno confermato il legame tra le radiazioni e il cancro al seno nelle donne che sono state irradiate per molte diverse condizioni mediche, tra cui tubercolosi [105], malattia mammaria benigna [106, 107], mastite acuta post-partum [108], timo ingrossato [109, 110], emangiomi cutanei [111], scoliosi [112], Hodgkin’.malattia di Hodgkin [113-116], linfoma non Hodgkin [117], acne [118] e cure dentistiche profilattiche [119]. L’evidenza di quasi tutte le condizioni suggerisce che l’esposizione alle radiazioni ionizzanti durante l’infanzia e l’adolescenza è particolarmente pericolosa rispetto all’aumento del rischio di cancro al seno più tardi nella vita [73, 120, 121].
Riassunto della sezione: questi concetti di inquadramento rivelano la complessità della ricerca che esamina le relazioni tra i tossici ambientali e i rischi di sviluppare il cancro al seno. Il cancro al seno non presenta un singolo profilo di biomarcatore; i tassi di incidenza differiscono tra i gruppi etnici/razziali e a livello di risorse; le concentrazioni delle esposizioni possono fare la differenza, così come le possibili miscele e interazioni. E la tempistica e la durata specifica delle esposizioni, soprattutto quando si verificano all’inizio dello sviluppo, possono causare effetti più dannosi rispetto alle esposizioni successive.
Mentre ci muoviamo nell’esaminare la letteratura scientifica che affronta la relazione tra i vari agenti tossici e il rischio di cancro al seno, offriamo un modello interattivo per aiutare a collocare questi dati (Fig. 1). Pur non volendo essere completamente esaustivo, questo modello sfida il lettore a considerare gli effetti delle esposizioni ambientali sul rischio di malattia all’interno di una complessa struttura di tipo web di fattori spesso interconnessi, ognuno dei quali può esercitare effetti diretti, indiretti e interattivi sui processi cellulari nei tessuti mammari [11].Fig. 1La complessità dei fattori che influenzano il rischio di sviluppare il cancro al seno. Questa sintesi di gran parte delle prove descritte in questo rapporto dimostra la complessità delle potenziali connessioni tra l’esposizione a sostanze tossiche ambientali e lo sviluppo del cancro al seno, il tutto inserito in un quadro di fattori interconnessi. Le frecce solide indicano le connessioni che sono state dimostrate direttamente tra le esposizioni e il rischio di cancro al seno, o, se del caso, mediate attraverso i fattori descritti nella sezione di inquadramento di questa revisione. Queste relazioni riflettono i risultati degli studi epidemiologici umani e/o animali discussi. Le frecce tratteggiate indicano connessioni più ambigue tra le esposizioni e il rischio di cancro al seno, con prove provenienti da studi non umani o su animali, ma senza i dati in vivo a supporto più diretto del collegamento. Le frecce non sono ponderate per indicare la forza relativa dei collegamenti. Lo scopo di questo modello è piuttosto quello di dimostrare la complessità delle relazioni tra fattori ambientali e cancro al seno. (Aggiornato e modificato da Gray et al. 2009 [11])
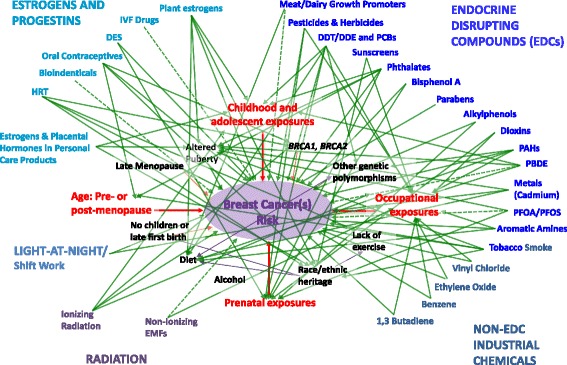
Fig. 1.Complessità dei fattori che influenzano il rischio di sviluppare il cancro al seno. Questa sinossi di gran parte delle prove descritte in questo rapporto dimostra la complessità delle potenziali connessioni tra l’esposizione a sostanze tossiche ambientali e lo sviluppo del cancro al seno, il tutto inserito in una struttura simile a una rete di fattori interconnessi. Le frecce solide indicano le connessioni che sono state dimostrate direttamente tra le esposizioni e il rischio di cancro al seno, o, se del caso, mediate attraverso i fattori descritti nella sezione di inquadramento di questa revisione. Queste relazioni riflettono i risultati degli studi epidemiologici umani e/o animali discussi. Le frecce tratteggiate indicano connessioni più ambigue tra le esposizioni e il rischio di cancro al seno, con prove provenienti da studi non umani o su animali, ma senza i dati in vivo a supporto più diretto del collegamento. Le frecce non sono ponderate per indicare la forza relativa dei collegamenti. Lo scopo di questo modello è piuttosto quello di dimostrare la complessità delle relazioni tra fattori ambientali e cancro al seno. (Aggiornato e modificato da Gray et al. 2009 [11])
Prove che collegano i fattori ambientali e il cancro al seno
Passiamo ora alle prove che riguardano le possibili connessioni tra l’esposizione a sostanze tossiche ambientali e il rischio di sviluppare il cancro al seno. Nell’esplorare la letteratura scientifica, attingiamo da studi su esseri umani, animali, colture cellulari e studi ad alto rendimento. Dove possibile, affrontiamo esplicitamente i temi complicati sollevati nella sezione di inquadramento di cui sopra. E, ove opportuno, presentiamo dati contrastanti, soprattutto dalle letterature epidemiologiche, che rendono chiare le sfumature della metodologia e dei risultati che complicano queste relazioni.
Ormoni: prodotti farmaceutici e per la cura della persona
Per decenni, gli scienziati hanno apprezzato il rapporto positivo tra l’esposizione agli estrogeni nel corso della vita e il rischio di sviluppare il cancro al seno [122]. Più recentemente è diventato chiaro che anche le esposizioni a lungo termine al progesterone possono influenzare il possibile sviluppo del cancro al seno [123]. Queste esposizioni sono spesso raggruppate nella categoria dei “fattori di rischio riproduttivi” (ad esempio, l’età alla menarca, le mestruazioni, la prima gravidanza a termine e il fatto che i bambini siano stati allattati o meno al seno) nello sviluppo di modelli e di semplici test di valutazione per determinare il rischio di cancro al seno [124, 125].
Oltre alle variazioni delle esposizioni a livelli endogeni sia di estrogeni che di progesterone, esistono diverse altre fonti di steroidi naturali e sintetici, tra cui quelli presenti in diversi prodotti farmaceutici e prodotti per la cura della persona. La maggior parte di questi agenti ormonali sono stati designati come cancerogeni dall’IARC e dall’NTP (vedi Tabella 1). Questa sezione esamina le relazioni tra l’uso di questi composti e le possibili variazioni del rischio di sviluppare il cancro al seno.Tabella 1Classificazioni della cancerogenicità e fonti di esposizione agli ormoni nei prodotti farmaceutici e nei prodotti per l’igiene personaleProdottoIARCNTFonte di esposizione Dietilstilbestrolo1KSempre prescritto alle donne in gravidanza per sostenere le gravidanze vitali Terapia ormonale sostitutiva1Trattamento dei sintomi sperimentati in menopausaConiugati di estrogeni equini2AMedrossiprogesterone acetato Ormoni biologicamente identici1ContraccezioneDroghetti per il trattamento dell’infertilitàTrattamento dell’infertilitàTrattamento dell’infertilitàClomifene citrato1GonadotropineOrmoni nei prodotti per la cura personale1Utilizzo di estratti placentali nei prodotti per la cura personale, in particolare nei prodotti commercializzati alle donne delle classificazioni dell’Agenzia Internazionale per la Ricerca sul Cancro (IARC): 1 = cancerogeno per l’uomo, 2A = probabilmente cancerogeno per l’uomo, 2B = probabilmente cancerogeno per l’uomo, 3 = non classificabile per quanto riguarda la sua cancerogenicità per l’uomo; classificazioni del Programma Nazionale di Tossicologia degli Stati Uniti (NTP): K = Noto come cancerogeno per l’uomo, RA = Ragionevolmente prevedibile come cancerogeno per l’uomo. L’elenco delle fonti di esposizione contiene le fonti di esposizione più comuni
Dietilstilbestrolo
La prova più evidente che un estrogeno sintetico può aumentare il rischio di cancro al seno decenni dopo deriva dalla tragica esperienza con il dietilstilbestrolo (DES). Dagli anni ’40 fino al 1971, i medici prescrissero il DES a milioni di donne incinte per prevenire aborti spontanei e altre complicazioni della gravidanza. Il farmaco fu proibito quando si scoprì che le figlie delle donne che lo assumevano avevano un tasso di adenosarcoma vaginale a cellule chiare estremamente raro rispetto a quelle che non erano esposte al DES nell’utero. L’esposizione al DES era anche associata a un aumento del rischio di cancro al seno nelle madri [98, 126, 127].
In uno studio di follow-up su figlie che sono state esposte prenatalmente al DES, è stato osservato un aumento quasi doppio del rischio di cancro al seno in donne di età superiore ai 40 anni (HR = 1,82; 95% CI = 1,04-3,18) [99]. Un effetto ancora maggiore (triplice) è stato riscontrato per le donne oltre i 50 anni, anche se relativamente poche delle figlie avevano ancora raggiunto quell’età al momento dello studio [101, 102]. Le donne esposte in utero che presentavano le anomalie più gravi delle loro cellule epiteliali vaginali (un indicatore dell’esposizione a dosi più elevate di DES) avevano anche un rischio maggiore di sviluppare il cancro al seno [99].
Gli studi sono appena iniziati sulle nipoti delle donne a cui è stato prescritto il DES durante la gravidanza, ma poiché queste donne stanno solo ora raggiungendo l’età in cui aumenta l’incidenza del cancro al seno, i set di dati sono troppo piccoli per raggiungere la significatività statistica [128]. I modelli di roditori rilevanti, tuttavia, indicano che la generazione F2 (nipoti) delle madri esposte a basse dosi di DES durante la gravidanza ha sviluppato anche diversi tumori, compresi i tumori mammari, a tassi significativamente più alti del previsto [129].
Studi che esaminano i meccanismi con cui il DES potrebbe esercitare i suoi effetti cancerogeni indicano che il composto attiva le stesse vie subcellulari che l’estradiolo, sia alterando il metabolismo cellulare e l’interazione con il DNA [130] sia aumentando il tasso di proliferazione delle cellule epiteliali del seno [131, 132]. Nei ratti femmine adulte, l’esposizione al DES ha aumentato l’induzione della trascrizione di HOTAIR che produce una proteina di silenziamento genico che risponde agli estrogeni implicata nello sviluppo del cancro al seno [133]. Il DES disregola ulteriormente l’espressione dell’espressione genica regolata dall’estradiolo nelle femmine adulte, contribuendo, anche in questo caso, ad aumentare il rischio di cancro al seno [134].
Le esposizioni prenatali al DES portano a cambiamenti nell’epigenoma della ghiandola mammaria adulta attraverso alterazioni dell’istone metilazione, un processo che porta ad un’alterazione dell’espressione genica nella pubertà e nell’età adulta [86, 133, 135]. Questi cambiamenti epigenetici potrebbero fornire un meccanismo per gli effetti transgenerazionali del DES sullo sviluppo del cancro al seno [128, 136].
Terapia ormonale sostitutiva (HRT)
L’iniziativa per la salute delle donne (WHI) è un grande (n = 16.608 donne) studio randomizzato di controllo del caso progettato per esplorare i benefici e i rischi di estrogeni combinati (estrogeni equini coniugati) più progestinici (acetato di medrossiprogesterone) HRT nelle donne in post-menopausa. Nel 2002, è stato interrotto dopo un follow-up mediano di 5,5 anni e mezzo, tre anni e mezzo prima della fine prevista del periodo di studio, perché i ricercatori hanno osservato un aumento significativo del rischio relativo di cancro al seno (HR = 1,26; 95% CI = 1,00-1,59) oltre a significativi aumenti del rischio di malattie cardiache, ictus e coaguli di sangue [137].
Le analisi di un secondo braccio dello studio WHI hanno chiarito che l’aumento del rischio di cancro al seno nello studio WHI si è verificato nelle donne che assumevano la formula combinata estrogeno-progestinica, ma non per quelle donne che assumevano integratori HRT solo estrogeni [138, 139] dove è stato riscontrato un rischio ridotto di sviluppare il cancro al seno (HR = 0,77; 95% CI = 0,62-0,95). È fondamentale notare che l’opzione dei soli estrogeni può essere offerta solo alle donne che sono state precedentemente sottoposte a isterectomie chirurgiche perché il trattamento con soli estrogeni comporta un aumento altamente significativo del rischio di cancro all’utero [140]. Una differenza tra i contraccettivi solo estrogeni e le forme combinate è nel tipo di estrogeni presenti nella formulazione. Il più delle volte l’estrogeno nella pillola mista è il composto semisintetico, l’etinilestradiolo, mentre quello nella pillola solo estrogeno è un estrogeno equino coniugato. La forma coniugata è associata a tassi più bassi di proliferazione epiteliale nei seni in post-menopausa, fornendo un unico meccanismo attraverso il quale i due tipi di intervento possono avere effetti diversi [141].
Il follow-up a lungo termine (mediana di 13 anni) di entrambi i bracci dello studio WHI indica che per le donne nel braccio ormonale combinato, c’è stato un aumento dipendente dal tempo e significativo del rischio di sviluppare il cancro al seno (HR = 0.71; 95% IC = 0,47-1,08 al primo anno di intervento; 1,36 95% IC = 0,94-1,94 durante il terzo anno di intervento; 1,65; 95% IC = 1,17-2,32 durante il quinto anno di intervento). Sebbene ci sia stata una netta diminuzione del rischio dopo il primo anno di interruzione dell’uso della formulazione mista HRT, per l’intero periodo di follow-up di 8 anni dopo l’interruzione del trattamento ormonale, i valori di HR sono stati superiori a 1 (HR = 1,32; 95% CI = 1,08-1,61) [142]. Il risultato iniziale, a breve termine, è coerente con il rapido calo dell’incidenza del cancro al seno in post-menopausa nella popolazione statunitense dal 2002, una diminuzione che è stata attribuita al precipitoso calo delle prescrizioni di HRT in popolazioni selezionate di donne (bianche, di classe medio/alta, in post-menopausa, tumori ER+) in seguito al rilascio dei dati di questi grandi studi [143, 144].
Per il braccio solo estrogeno, il rischio ridotto di cancro al seno è rimasto per la fase iniziale dopo l’intervento (HR = 0,55; 95% IC = 73-1,87 per i primi 3 anni dopo l’intervento), anche se il beneficio scompare nei successivi 5 anni (HR = 1,17; 95% CI = .73-1,87) [142].
Da quando i risultati del WHI originale sono stati inizialmente pubblicati, altri grandi studi ne hanno sostenuto le conclusioni principali. Nel 2003, i ricercatori svedesi hanno interrotto uno studio sulla TOS nelle donne con una precedente storia di cancro al seno. Originariamente pianificato come studio di 5 anni, lo studio svedese è stato interrotto dopo 2 anni perché le donne che assumevano la TOS combinata estrogeno-progestinica avevano un tasso significativamente aumentato di recidive o nuovi tumori rispetto alle donne che ricevevano altri trattamenti per i sintomi della menopausa ((HR = 3,5; 95% CI = 1,5-8,1) [145].
Sempre nel 2003, i ricercatori dello studio Million Women Study (MWS) nel Regno Unito hanno riferito che l’uso attuale di tutti i tipi di HRT post-menopausa ha aumentato significativamente il rischio di cancro al seno (RR = 1,66; 95% CI = 1,58-1,75). Anche in questo caso, il rischio era maggiore tra gli utenti della terapia combinata estrogeno-progestinica (RR = 2,00; 95% CI = 1,88-2,12) [146].
Altre ricerche hanno confermato il risultato di base che l’uso della HRT combinata aumenta il rischio di cancro al seno nelle donne in post-menopausa e che l’interruzione dell’uso della pillola combinata porta ad una diminuzione del rischio di sviluppare il cancro al seno. Uno studio in California ha trovato che la diminuzione dell’incidenza del cancro al seno in tutta la contea è stata più alta (22,6%) nelle contee con il maggior calo nell’uso della TOS, intermedia (13,9%) nelle contee con una moderata diminuzione dell’uso della TOS, e più piccola (8,8%) nelle contee con il minor calo nell’uso della TOS [147].
Uno studio ha esaminato l’incidenza del cancro al seno nei portatori di mutazioni BRCA1 che erano stati sottoposti a ooforectomia per prevenire l’insorgenza del cancro ovarico. L’uso a breve termine (mediana = 4,27 anni) della TOS non è stato associato ad alcun cambiamento nel rischio di sviluppare il cancro al seno (OR = 0,80; 95% CI = 0,55-1,16), indipendentemente dalla formulazione della TOS (estrogeni da soli o estrogeni + progestinici) [148].
Un altro studio che esamina le possibili interazioni tra l’uso della TOS e la razza, il peso e la densità del seno ha trovato che l’uso della TOS aumenta il rischio di cancro al seno in bianco (OR = 1,21; 95% CI = 1,14-1).28), asiatici (OR = 1,58; 95% CI = 1,18-2,11) e ispanici (OR = 1,35; 95% CI = 1,09-1,67), ma non donne nere (OR = 0,91; 95% CI = 0,72-1,14). Non c’è stata interazione tra l’uso della HRT e l’IMC o la densità del seno [149].
Una meta-analisi che includeva 116.304 casi di tumore al seno ha dimostrato che le donne che svolgono attività fisica ad alto livello hanno un rischio significativamente ridotto di sviluppare il tumore al seno (SRR = 0,88; 95% CI = 0,85-0,90), con una diminuzione che si riscontra sia nei tumori ER+/PR+ che in quelli ER-/PR-. Tuttavia, le donne che hanno usato l’HRT non hanno avuto una diminuzione del rischio di cancro al seno quando si sono impegnate in un esercizio fisico vigoroso [150].
L’esame dell’istologia del cancro nelle donne che assumono la TOS combinata al momento della diagnosi rivela una maggiore presentazione del cancro al seno di origine lobulare [151- 153], ma anche di tumori con bassi tassi di proliferazione (indici mitotici) e un esito prognostico favorevole [153, 154].
Ormoni bioidentici
A seguito dei risultati dei principali studi che implicano la TOS come causalmente correlata al cancro al seno in post-menopausa, molte donne si sono rivolte a fonti alternative di terapia ormonale per trattare i loro sintomi della menopausa con la speranza di trovare opzioni più sicure. Per molte donne, ciò significava utilizzare “ormoni bioidentici” di qualche tipo, sperando di imitare gli effetti degli ormoni naturali senza soccombere agli esiti negativi per la salute associati alla TOS tradizionale [155]. Purtroppo ci sono stati pochissimi studi che hanno esaminato la relazione tra l’assunzione di ormoni bioidentici e il successivo sviluppo del cancro al seno. Forse ancora più importante, e confondendo la conversazione su questo argomento, il termine “ormoni bioidentici” è usato in molti modi diversi con implicazioni potenzialmente diverse per le associazioni con gli esiti sanitari [156]. La definizione più conservatrice, adottata dalla Endocrine Society, è per i composti che “hanno esattamente la stessa struttura chimica e molecolare degli ormoni prodotti nel corpo umano” [156]. Gli ormoni bioidentici possono essere sintetizzati o derivati da fonti vegetali.
Alcuni tipi di composti ormonali bioidentici o singoli componenti sono stati testati e approvati dalla Food and Drug Administration (FDA). Ma l’uso sempre più comune di regimi ormonali bioidentici composti individualmente non è stato testato per la sicurezza o per i risultati sanitari associati e la consistenza della prescrizione e della fornitura di formule composte individualizzate varia enormemente [156, 157].
La prova più forte della mancanza di associazione tra l’uso di ormoni bioidentici e il possibile sviluppo del cancro al seno deriva dai dati che esaminano l’uso del progesterone ormonale naturale, invece dell’MPA o di altri progestinici sintetici, come parte del regime HRT [158]. La ricerca indica che l’aumento dell’esposizione al progesterone naturale non ha aumentato il rischio di cancro al seno e, in alcune circostanze, potrebbe anche essere protettivo [159, 160]. In un unico studio di coorte su larga scala che esamina i rischi per il cancro al seno nelle donne che assumono regimi di sostituzione ormonale con progesterone naturale o progestinici sintetici composti con estrogeni, l’uso di una sostituzione a base di progesterone non è stato associato ad alcun rischio aggiunto per il cancro al seno rispetto ai controlli (RR = 1.).00; 95% CI = 0,83-1,22), mentre le donne che hanno assunto HRT combinate che includevano progestinici sintetici avevano aumentato significativamente il rischio di sviluppare la malattia (RR = 1,69; 95% CI = 1,50-1,91) [161]. Questa differenza era particolarmente prevalente nell’incidenza dei tumori ER+, in particolare delle masse ER+/PR- (RR = 2,6; 95% CI = 1,9-3,5) [162].
Notizie meno positive provengono da uno studio che confronta gli effetti degli estrogeni equini coniugati, la componente estrogenica principale nella HRT tradizionale combinata estrogeno-progestinica, con estradiolo naturale in un modello di primate di cancro al seno in postmenopausa. In questo studio, l’estradiolo naturale ha indotto una maggiore proliferazione delle cellule epiteliali del seno rispetto alla forma coniugata [141].
Contraccettivi orali
Numerosi studi hanno dimostrato un aumento del rischio di cancro al seno nelle donne che usano contraccettivi orali. Il rischio di cancro al seno è maggiore tra le attuali e recenti utilizzatrici di contraccettivi orali, in particolare quelle che li hanno usati per più di 5 anni e ne hanno iniziato l’uso in giovane età [163- 168]. Ad esempio, in un ampio studio di coorte, è stata riscontrata una maggiore incidenza di cancro al seno in donne che avevano meno di 50 anni al momento della diagnosi e che avevano iniziato a usare i contraccettivi orali prima dei 20 anni – rispetto a quelle che avevano iniziato più tardi (HR = 3,26; 95% CI = 1,06-10,01). Le donne che avevano iniziato l’uso prima dei 20 anni di età ed erano più vecchie di 50 anni al momento della diagnosi non hanno mostrato un aumento del rischio rispetto ai casi simili di età che hanno iniziato l’uso più tardi (HR = 0,70; 95% CI = 0,33-1,46). Le donne in questo studio hanno assunto contraccettivi per una media di 6 anni, anche se la durata dell’uso variava da 2 ½ a 12 anni [169].
Sweeney et al. hanno esaminato i possibili effetti dell’uso di contraccettivi orali sul rischio successivo di cancro al seno nelle donne bianche ispaniche e non ispaniche. Statisticamente, le donne ispaniche hanno tassi di tumore al seno leggermente inferiori rispetto alle donne bianche e hanno maggiori probabilità di avere tumori al pronto soccorso. Tuttavia, l’uso di contraccettivi orali durante i 5 anni precedenti ha portato ad un aumento significativo dell’incidenza del cancro al seno in entrambi i gruppi. L’effetto è stato amplificato per le donne di entrambi i gruppi quando l’uso di contraccettivi orali è continuato per più di 20 anni (OR = 2,23; 95% CI = 1,17-4,25 per i tumori ER). A riprova di altre prove di studio, e ancora una volta per le donne bianche sia ispaniche che non ispaniche, sono stati osservati aumenti significativi dei tumori ER+ [170].
I ricercatori del Black Women’s Health Study, un ampio studio prospettico (oltre 53.000 donne) sulle donne negli Stati Uniti, riferiscono che l’uso di contraccettivi orali da parte di donne afroamericane è stato associato a un rischio più elevato di tumori recettoriali negativi (ER-, PR-) rispetto alle donne che non hanno usato la pillola (IRR = 1,65; 95% CI = 1,19-2,30). Il rischio di una diagnosi successiva di cancro al seno ER/PR è aumentato in quanto la durata dell’uso di contraccettivi è stata prolungata tra le donne che hanno preso la pillola e che la stavano ancora usando negli ultimi 5 anni (tendenza p = 0,001). L’unico effetto significativo dell’uso di contraccettivi orali sullo sviluppo dei tumori ER+/PR+ in questa coorte è stato per le donne che hanno preso la pillola per più di 10 anni (IRR = 1,45; 95% CI = 1,02-2,07) [171].
Le donne con mutazioni BRCA1 o BRCA2, così come le donne con storie familiari di cancro al seno o alle ovaie, hanno una maggiore suscettibilità agli effetti di induzione di rischio dell’uso di contraccettivi orali [166, 172, 173]. Il contributo paterno (rispetto al contributo materno) della mutazione BRCA conferisce un rischio maggiore alle donne con questa variazione genetica che utilizzano anche contraccettivi orali (HR = 1,84; 95% CI = 1,46-2,34) [174]. Un meccanismo attraverso il quale l’interazione tra lo stato del gene BRCA e l’uso di contraccettivi orali può influenzare il rischio di cancro al seno, è l’alterazione della sensibilità e dell’attività del progesterone nelle cellule del cancro al seno, sia aumentando la sintesi di PR nelle cellule sia aumentando la reattività dei geni regolati dal progesterone [175].
L’uso di contraccettivi orali è associato ad un aumento dei tumori della mammella in stadio avanzato (tipo II o superiore) [176], dei tumori che hanno origine nel tessuto lobulare [171], nonché al profilo ER della malattia [171, 177]. Associazioni significative tra l’uso di contraccettivi orali e lo sviluppo della forma aggressiva tripla negativa (ER-/PR-/Her-2R-) della malattia sono state trovate in una coorte principalmente bianca (OR = 2.5; 95% CI = 1,4-4,3) [178] e in una coorte di donne afroamericane (OR = 1,78; 95% CI = 1,25-2,53) [179]. L’uso di contraccettivi orali per 10 o più anni è stato anche associato a una diagnosi di DCIS comedo (OR = 1,31; 95% CI = 0,70-2,47) [180], la forma più aggressiva di DCIS che a volte viene confusa con le forme precoci di cancro al seno invasivo [181].
Le donne in post-menopausa che hanno usato contraccettivi orali per otto o più anni, ma che hanno interrotto l’uso per almeno un decennio, non mostrano un aumento significativo dei tassi di cancro al seno [182, 183].
Due studi hanno esaminato la relazione tra l’uso di contraccettivi progestinici iniettabili e l’incidenza del cancro al seno. Entrambi gli studi hanno trovato aumenti significativi del rischio di cancro al seno, ma i tassi sono scesi alla normalità nel giro di pochi anni dopo la cessazione dell’uso dei farmaci [184, 185].
Farmaci per il trattamento dell’infertilità
Nonostante le prove sostanziali che collegano l’HRT e l’uso di contraccettivi orali con l’aumento dell’incidenza del cancro al seno, né la condizione di subfertilità né l’uso di farmaci per il trattamento dell’infertilità (ovulazione-stimolazione) sembrano avere un chiaro legame con la malattia [186- 189]. Questo è vero anche quando lo studio coinvolge donne infertili che sono anche portatrici di BRCA [190]. Laddove è stato trovato un collegamento, è stato per le donne che hanno dato alla luce più di un neonato a seguito del loro trattamento di FIVET (HR = 1,44; 95% CI = 1,06-1,97) [191] e per quelle che sono state trattate con alte dosi di citrato di clomifene.
Due studi hanno trovato un aumento del rischio di cancro al seno per le donne che sono state trattate per l’infertilità ovarica con farmaci, tra cui gonadotropine o citrato di clomifene. Tuttavia, i risultati sono stati significativi solo quando l’incidenza del cancro al seno è stata confrontata con la popolazione generale delle donne, ma non con il controllo più appropriato delle donne con infertilità ovarica che non sono state trattate con farmaci per la fertilità [192, 193]. Altri due studi, tuttavia, hanno rilevato aumenti statisticamente significativi dei tassi di cancro al seno nelle donne che assumono citrato di clomifene rispetto ai tassi delle donne sterili che non assumono alcun trattamento per l’infertilità (HR = 1,42; 95% CI = 0,99-2,55) [194]; (OR = 2,7; 95% CI = 1,3-5,7]. [195]. Un sottogruppo più piccolo di donne la cui infertilità non era di origine ovarica e che si sottoponeva a trattamenti multipli con alte dosi di citrato di clomifene, aveva aumentato il rischio di sviluppare successivamente il cancro al seno rispetto alle donne della popolazione generale (OR = 3,0; 95% CI = 1,35-6,67) [188].
Un altro studio complica la storia, tuttavia. All’interno della coorte delle donne con problemi di fertilità, non c’era alcuna differenza nel tasso di tumore al seno quando si facevano confronti generali tra le donne che avevano assunto farmaci per la fertilità e quelle che non li avevano assunti. Ma quando è stata presa in considerazione l’età del trattamento, è stato trovato un significativo aumento del rischio di cancro al seno nelle donne che avevano iniziato i trattamenti con farmaci per l’infertilità prima dei 24 anni, rispetto alle donne sterili della stessa età che non si erano sottoposte alla FIVET e ai trattamenti farmacologici associati (HR = 1,59; 95% CI = .1,05-2,42). L’aumento del rischio di cancro al seno non era associato al trattamento dell’infertilità nelle donne anziane (dopo i 40 anni) sottoposte a protocolli di FIVET [196]. Questi dati sono coerenti con un modello in cui le cellule mammarie adulte più giovani sono più sensibili alle perturbazioni e/o alle protezioni derivanti da esposizioni alterate a fonti ormonali sia endogene che esogene.
Ormoni nei prodotti per la cura della persona
Gli estratti placentari, probabilmente con alte concentrazioni di progesterone [197] e di sostanze chimiche estrogeniche [198], sono talvolta utilizzati nei cosmetici e nei prodotti per la cura dei capelli, in particolare nei prodotti commercializzati alle donne di colore. L’aggiunta di ormoni ed estratti è pubblicizzata per promuovere la crescita e lo spessore dei capelli. Tuttavia, la ricerca indica che l’uso di questi prodotti nei neonati e nei bambini può anche essere legato alla pubertà precoce o alla maturazione sessuale precoce [191, 199, 200], un fattore di rischio per il cancro al seno in età avanzata [201]. Gli scienziati hanno proposto che l’uso di questi prodotti con alterazioni ormonali potrebbe contribuire all’aumento dell’incidenza del cancro al seno, soprattutto tra le giovani donne afroamericane che usano questi prodotti più delle loro controparti bianche [202, 203].
Sette degli otto estratti di prodotti per la pelle e i capelli comunemente usati dalle donne afroamericane hanno avuto effetti sulla proliferazione delle cellule MCF-7 in coltura; quattro dei sette erano estrogenici mentre tre mostravano attività antiestrogenica [204].
Gli ormoni, specialmente gli estrogeni, sono anche regolarmente aggiunti alle creme anti-invecchiamento [205], a causa della loro efficacia nell’aumentare il conteggio del collagene, così come l’idratazione della pelle. Insieme, questi due fattori si pensa che diminuiscano le rughe della pelle [206], ma possono anche aumentare l’esposizione totale delle donne agli estrogeni per tutta la vita.
Sommario della sezione: Ci sono chiare prove che l’esposizione al DES durante la gestazione aumenta il rischio di sviluppare il cancro al seno nelle donne che sono state esposte in utero, e anche per le loro madri ed eventualmente le loro figlie. L’uso post-menopausa di HRT composta da estrogeni e progestinici sintetici aumenta anche la probabilità di sviluppare il cancro al seno, anche se l’uso di HRT solo estrogeni ha effetti protettivi per quelle donne che hanno subito un’isterectomia. L’uso di farmaci HRT composti con l’ormone naturale, il progesterone, non sembra avere effetti dannosi sul rischio di cancro al seno, anche se l’uso di estrogeni naturali, l’estradiolo, può aumentare la proliferazione delle cellule mammarie e il conseguente rischio di sviluppare il cancro al seno. Ci sono poche prove coerenti che l’uso di farmaci ormonali nelle procedure di fecondazione in vitro alteri il rischio di cancro al seno, anche se ci sono numerosi problemi metodologici in questi studi. Infine, diversi prodotti per la cura della persona, soprattutto quelli commercializzati principalmente alle comunità di colore, hanno additivi estrogenici e progestinici, aumentando l’esposizione a questi ormoni per tutta la vita.
Composti perturbatori endocrini (EDC)
Sebbene l’uso intenzionale di ormoni naturali e sintetici sia stato una pratica per decenni, se non secoli, è solo negli ultimi due decenni che gli scienziati sono giunti a riconoscere che molti prodotti comuni contengono anche sostanze chimiche che perturbano il sistema endocrino squisitamente sensibile [207]. Queste sostanze chimiche, che si trovano in prodotti così diversi come le materie plastiche, i pesticidi, i ritardanti di fiamma e le creme solari, sono state aggiunte ai prodotti fabbricati per ragioni non intenzionalmente legate alle loro proprietà endocrine. Tuttavia, è stato dimostrato che molti composti si adattano alla definizione della Endocrine Society di un composto perturbatore endocrino (EDC), “una sostanza chimica esogena, o miscela di sostanze chimiche, che interferisce con qualsiasi aspetto dell’azione ormonale” [47].
Interferendo con le azioni degli ormoni naturali, è stato dimostrato che le esposizioni agli EDC contribuiscono allo sviluppo di un’ampia varietà di stati patologici [49, 51]. Spesso questi effetti sono più profondi quando le esposizioni sono a basse dosi [38] e durante lo sviluppo precoce [48]. Questa sezione affronta la crescente letteratura sulle connessioni tra diversi importanti CDE e il rischio di sviluppare il cancro al seno, principalmente – ma non esclusivamente – da modelli non umani. Anche se per lo più trattiamo le sostanze chimiche in modo indipendente, come è vero per la letteratura di ricerca, riconosciamo l’importanza dell’esposizione a miscele di EDC, poiché queste sostanze infondono i prodotti che usiamo, ma anche l’aria che respiriamo, l’acqua che beviamo e le superfici su cui lavoriamo e giochiamo. Mentre la maggior parte di questi EDC non è stata formalmente valutata per la cancerogenicità, la Tabella 2 dimostra la presenza quasi onnipresente di queste sostanze chimiche nel nostro ambiente.Tabella 2Classificazioni della cancerogenicità e fonti di esposizione per i composti che alterano il sistema endocrino (EDC)EDCIARCNTPS Fonti di esposizioneBisfenolo APolicarbonato di plastica, resine epossidiche legate a lattine di cibo, sigillanti dentali, ricevute termicheFtalatiFragranza ingredienti nei prodotti per la cura personale e la pulizia, plastica. Anche prodotti farmaceutici, materiali da costruzione, insetticidi e imballaggi/lavorazione di alimenti.2BRA di (2-etilesile) ftalato (DEHP) 2BRA di-n-butilftalato (DNP/DBP), ftalato di monoetile (MEHP), ftalato di etile (DEP), benzilftalato di butile, (BBP) 3 di-n-ottile ftalato, (DOP), di-i-butilftalato (DiBP).monometil ftalato di metileParabeniConservanti antimicrobici in alimenti, prodotti per la cura della persona, saponi e detergenti e prodotti farmaceutici a base di metil-parabene, propil-parabeni di butile-parabeniAlchilpenoliDetergenti e prodotti per la pulizia, antiossidanti in prodotti di plastica e gomma4-nonilfenolo (4-NP)■4-ottilfenolo (4-OP)■Triclosan & TriclocarbanAntimicrobici nel sapone liquido per le mani, altri prodotti per la cura personale e articoli per la casaEDC che si trovano nei filtri solariUV 3-(4-metilbenzilidene)■camfora (4-MBC)ottil-metossicinnamato di ottile-dimetil-PABA (OD-PABA)■benzofenone-3 (Bp-3)■omosalato (HMS)■acido perfluoroottanoico (PFOA) ■solfato perfluoroottanoico (PFOS)2 ■rivestimenti resistenti alle macchie, rivestimenti antiaderenti, prodotti commerciali, comprese le schiume antincendio.Idrocarburi aromatici policiclici (IPA)RABiprodotti della combustione derivanti dalla produzione di combustibili fossili, gas di scarico del gasolio, carni alla griglia, sigarette.■Benz[a]antracene2BRA[a]antracene2BRA[a]pirene1RATriazina erbicida Controllo delle erbacce per le colture di mais e sorgo.ÁrazinaSimazina CianazinaAltri pesticidi ed erbicidi Eptaloclor2BInsetticida, ora proibito Dieldrin e Aldrin2AInsetticida per mais e cotone, ora proibito Clordano2BHtermiti domestiche, pesticida generale per colture Malathion2AResidenziale, ricreativo, pesticida per coltureErbicida per malerbe a foglia larga 2,4-D2BErbicida per malerbe a foglia larga 2,4,5-acido triclorofenossipropionico (2,4,5-TP)Erbicida per piante legnose e malerbe a foglia larga, ora vietatoOrganoclorurati persistentiDicloro-difenil-tricloroetano (DDT)/DDE2ARAInsecticida, ora vietatoPCBs1RAEisolamento elettrico, liquidi refrigeranti, plastificante in vernici, coloranti e inchiostri Diossine: 2,3,7,8-tetra clorodibenzo-para-diossina (TCDD)1Prodotto della combustione di prodotti chimici a base di cloro – Eteri di difenile polibromurati (PBDE)-Ritardanti di fiamma, precedentemente usati nei mobili e nell’elettronica; la maggior parte sono stati vietati o eliminati volontariamente in modo graduale -Aromatic amineso-toluidine1KHair dyes4-aminodifenile (ABP)1KAzo coloranti in prodotti tessili p-fenilendiamminaTinture per capelli 2-amino-fenilimidazo[4,5-b]piridina (PhIP)Carni cotteAmmine aromatiche eterociclicheAmmine per capelliTinture per capelliMetalliElementi naturali; contaminanti in coloranti di derivazione naturale, argille e altri metalli, che si trovano in cosmetici, giocattoli e altri prodotti.Classificazioni dell’Agenzia Internazionale per la Ricerca sul Cancro (IARC) dell’Agenzia Internazionale per la Ricerca sul Cancro (IARC) di Rame-CobaltPORA-NickelPOLead2BRA-Mercury-Methylmercury2BTin-Cadmium1KZinc-Iron1: 1 = cancerogeno per l’uomo, 2A = probabilmente cancerogeno per l’uomo, 2B = probabilmente cancerogeno per l’uomo, 3 = non classificabile per quanto riguarda la sua cancerogenicità per l’uomo; classificazioni del Programma Nazionale di Tossicologia degli Stati Uniti (NTP): K = Noto come cancerogeno per l’uomo, RA = Ragionevolmente prevedibile come cancerogeno per l’uomo. L’elenco delle fonti di esposizione contiene le fonti di esposizione più comuni
Bisfenolo A (BPA)
L’onnipresente sostanza chimica sintetica bisfenolo A (BPA) è il componente principale utilizzato nella produzione di plastica in policarbonato e si trova in molti comuni prodotti per la casa. Si trova anche nei sigillanti dentali, nelle ricevute termiche, negli imballaggi alimentari e nelle resine epossidiche che rivestono i barattoli di cibo. Livelli significativi di BPA sono stati misurati nell’aria ambiente [208], nella polvere di casa [209], nel fiume e nell’acqua potabile [210].
Il BPA è un composto instabile e lipofilo che può lisciviare nei prodotti alimentari, specialmente se riscaldato [211], e si ritiene che una delle principali fonti di esposizione al BPA sia dovuta a prodotti alimentari contaminati con la sostanza chimica [212, 213]. Due studi hanno esplorato gli effetti dell’aumento dell’ingestione di cibi e bevande confezionati in materiali contenenti BPA. Entrambi hanno trovato rapidi aumenti dei livelli di BPA nelle urine e/o nei campioni di sangue prelevati da soggetti che hanno intenzionalmente aumentato l’assunzione di cibi e bevande comuni confezionati in prodotti contenenti BPA [214, 215]. Un altro studio ha adottato l’approccio opposto e ha dimostrato che solo un periodo di 3 giorni di limitazione dell’assunzione di alimenti confezionati ha diminuito le concentrazioni di BPA trovate nelle urine di una media del 65% [216].
I campioni prelevati da persone a digiuno indicano che anche fonti diverse dagli alimenti possono essere responsabili dell’esposizione pervasiva al BPA, in quanto i livelli della sostanza chimica non sono diminuiti così rapidamente come si sarebbe potuto prevedere se gli alimenti fossero stati l’unica fonte di contaminazione [217]. Di crescente preoccupazione sono gli alti livelli di BPA che vengono trasferiti alla nostra pelle e poi rapidamente assorbiti trattenendo le ricevute termiche contenenti BPA [218].
L’eliminazione del BPA dal corpo è abbastanza rapida, con la sua emivita urinaria dell’ordine delle ore e dei giorni [217]. Nonostante il suo rapido tasso di eliminazione, il BPA è stato trovato nel 93% dei circa 2500 campioni di urina di un ampio campione nazionale di adulti attraverso lo studio NHANES [219]. Il BPA è stato trovato nel sangue e nelle urine di donne incinte [220-222], e nel latte materno subito dopo il parto [223, 224]. Il BPA è stato trovato anche in campioni di sangue provenienti da feti in via di sviluppo e nel liquido amniotico circostante [225]; nel tessuto placentare e nel sangue del cordone ombelicale alla nascita [226, 227]; e nelle urine di neonati prematuri alloggiati in terapia intensiva neonatale [228].
Molti studi che utilizzano sia modelli di ratto che di topo hanno dimostrato che anche brevi esposizioni a dosi di BPA rilevanti per l’ambiente durante la gestazione o intorno al momento della nascita portano a cambiamenti nella struttura del tessuto mammario che predicono lo sviluppo successivo dei tumori [90, 229, 230]. L’esposizione precoce al BPA ha portato ad anomalie nello sviluppo dei tessuti mammari che erano osservabili durante la gestazione e sono state mantenute in età adulta [92, 231, 232]. Molti di questi cambiamenti sono simili a quelli osservati dopo l’esposizione prenatale al DES [132]. L’esposizione prenatale dei ratti al BPA ha portato ad un aumento del numero di lesioni precancerose e di carcinomi in situ [233, 234], così come un aumento del numero di tumori mammari a seguito di esposizioni adulte a dosi sotto soglia di agenti cancerogeni noti [235, 236] o senza l’aggiunta dell’agente cancerogeno aggiuntivo [234].
L’esposizione prenatale al BPA modifica la trascrizione genica sia nel compartimento epiteliale che in quello stromale della ghiandola fetale del topo, attraverso meccanismi che sono mediati sia attraverso le vie ER-dipendenti che ER-indipendenti [237, 238]. Sia le esposizioni BPA e DES alterano l’espressione di diversi geni coinvolti nella formazione della matrice extracellulare, così come l’adipogenesi e la formazione del lume [237]. BPA agisce sulle vie estrogeno-indipendenti per alterare la transizione epitelio-mesenchimale (EMT) attraverso la regolazione del down-regolazione di FOXA1, un regolatore chiave delle risposte ormonali nelle cellule tumorali della mammella [238]. Questi dati suggeriscono che durante la gestazione, il BPA agisce sulle cellule stromali per alterare il contenuto di fibre di collagene e l’espressione di diverse proteine, compresi i recettori che mediano le vie di segnalazione, che poi alterano l’espressione genica epiteliale e la proliferazione cellulare [237, 239].
L’esposizione neonatale dei topi al BPA aumenta la sensibilità allo sviluppo mediato dall’estradiolo delle strutture della ghiandola mammaria durante la pubertà [240] e aumenta la sintesi del recettore del progesterone e l’attivazione della proliferazione delle cellule mammarie regolate dal progesterone [132].
Cambiamenti nello sviluppo mammario paragonabili a quelli osservati nei modelli di roditori sono stati osservati anche quando le scimmie rhesus femmine sono state esposte a dosi di BPA rilevanti per l’ambiente durante la gestazione [241].
Alcuni degli effetti a lungo termine delle esposizioni neonatali al BPA possono essere dose-dipendenti, con esposizioni a basse e alte dosi con conseguenti tempi e profili diversi di cambiamenti nell’espressione genica nelle cellule della ghiandola mammaria. In uno studio, le esposizioni a basse dosi hanno avuto l’effetto più profondo sulle ghiandole mammarie di ratto durante il periodo appena prima che gli animali raggiungessero la maturità riproduttiva, mentre dosi più alte hanno avuto effetti più ritardati, alterando l’espressione genica nei tessuti mammari degli adulti maturi [55]. L’esposizione prenatale a basse dosi di BPA alterato lo sviluppo delle ghiandole mammarie nei ratti adulti, mentre dosi più elevate non [56]. In uno studio sull’esposizione cronica di topi adulti a diverse concentrazioni di BPA, solo basse dosi hanno diminuito la latenza dell’aspetto tumorale e aumentato il numero di tumori mammari e il loro tasso di metastasi. Tutte le dosi hanno aumentato il tasso di proliferazione delle cellule mammarie, ma solo dosi relativamente più alte hanno contrastato questa proliferazione aumentata con aumenti paralleli della morte cellulare programmata [242]. E in una valutazione delle esposizioni prenatali al BPA nei ratti maschi, sono stati trovati effetti dose-risposta non lineari del BPA per lo sviluppo delle strutture della ghiandola mammaria [243].
Oltre alle anomalie fisiche nel tessuto mammario in via di sviluppo dei roditori trattati perinatalmente con bassi livelli di BPA, ci sono anche deficit funzionali. Le femmine di ratti esposti al BPA durante la gestazione e l’allattamento hanno avuto anomalie fisiche nel loro tessuto mammario adulto, così come la diminuzione della produzione e il contenuto proteico alterato del loro latte quando, come neo-mamme, stavano nutrendo i loro cuccioli. Le differenze osservate in seguito all’esposizione al BPA erano simili a quelle riscontrate nei ratti che erano stati esposti in modo simile al DES, un noto agente cancerogeno per il seno [244].
Studi che utilizzano colture di cellule umane di cancro al seno dimostrano che il BPA agisce, in parte, attraverso le stesse vie di risposta cellulare dell’estradiolo estrogeno naturale [245, 246]. Il BPA si lega debolmente alla ER intracellulare e influenza anche le funzioni cellulari attraverso le interazioni con la membrana ER (mER) [247, 248]. Ma il BPA esercita anche effetti dirompenti sui processi cellulari, compresi i cambiamenti nell’attivazione dei percorsi di trasduzione del segnale nelle linee cellulari ER [238, 249]. Oltre a legarsi a ER, il BPA si lega al recettore nucleare orfano del recettore gamma degli estrogeni (ERRγ), una proteina alla quale l’estradiolo non si lega [250- 253]. La famiglia dei recettori nucleari è coinvolta in un ampio spettro di processi biologici, dallo sviluppo embrionale e la differenziazione attraverso il normale mantenimento dei sistemi omeostatici alla disregolazione di questi processi coinvolti nello sviluppo dei tumori [254]. Il BPA si lega anche al recettore degli androgeni (AR) e al recettore degli ormoni tiroidei (TR) [255- 257], alterando le attività di questi sistemi ormonali.
L’esposizione prenatale dei topi al BPA ha anche portato alla disregolazione delle citochine infiammatorie nei tessuti mammari adulti, un processo che può portare ad un’alterazione della crescita cellulare attraverso l’inibizione delle risposte immunitarie che comunemente prendono di mira le cellule tumorali in via di sviluppo [258].
L’esposizione di cellule mammarie umane normali e cancerose a bassi livelli di BPA ha portato all’alterazione dell’espressione di centinaia di geni, tra cui molti coinvolti in processi ormono-recettore-mediato, proliferazione cellulare e apoptosi e carcinogenesi [259- 261]. In presenza di BPA, le cellule derivate dal seno non canceroso di donne con diagnosi di cancro al seno avevano un profilo di risposta genica associato allo sviluppo di tumori altamente aggressivi [262].
Gli effetti del BPA sullo sviluppo dei tessuti mammari possono anche manifestarsi attraverso meccanismi epigenetici, portando a cambiamenti nella regolazione dei geni nel corso della vita. L’esposizione prenatale dei ratti a bassi livelli di BPA ha alterato l’epigenoma nel tessuto mammario con diversi profili osservati allo svezzamento e alla post-pubertà [135]. Esposizioni a BPA o DES portano a cambiamenti simili nell’epigenoma della ghiandola mammaria adulta attraverso alterazioni della metilazione dell’istone e del silenziamento genico, processi che portano all’espressione genica alterata nella pubertà e nell’età adulta [86, 133- 135].
Il BPA riduce l’efficacia dei comuni agenti chemioterapici (cisplatino, doxirubicina e vinblastina) nel bloccare la proliferazione delle cellule tumorali umane del seno quando sono testati in vitro [263, 264].
Ftalati
Gli ftalati sono un gruppo di sostanze chimiche che alterano il sistema endocrino comunemente usate per rendere la plastica morbida e flessibile. Si trovano in un’ampia varietà di prodotti comuni, tra cui plastica (ad esempio, giocattoli per bambini), cosmetici, prodotti farmaceutici, prodotti per la cura dei bambini, materiali da costruzione, argilla per modellare, automobili, materiali per la pulizia e insetticidi [265]. Gli ftalati sono prontamente assorbiti attraverso la pelle [266] e possono anche entrare nel corpo attraverso procedure di inalazione o di iniezione medica [267]. Altre fonti principali di almeno uno ftalato, il di(2-etilesil)ftalato (DEHP), sono gli imballaggi alimentari [268, 269] e i fast food [270]. Uno studio di intervento dietetico ha dimostrato che solo un periodo di 3 giorni di limitazione dell’assunzione di alimenti confezionati è diminuito della metà delle concentrazioni di DEHP presenti nelle urine [216]. Un altro intervento dietetico in cui i partecipanti allo studio hanno seguito uno stile di vita monastico di 5 giorni, compresa una dieta vegetariana, ha portato a una significativa diminuzione dei livelli di ftalati urinari [271]. Livelli significativi di DEHP e di un altro ftalato usato nel confezionamento degli alimenti, il di-n-butilftalato (DNP), sono stati trovati in alimenti cotti, sia prima che dopo il confezionamento, che sono stati serviti ai bambini attraverso programmi di pasti scolastici [272]. Molti vini e liquori, così come le spezie, sono contaminati da ftalati derivanti dalla fuoriuscita delle sostanze chimiche dai contenitori di stoccaggio [273, 274].
Gli ftalati sono stati trovati nell’aria interna e nella polvere [275] e in campioni di urina umana e di sangue di bambini, adolescenti e adulti [216, 276- 278], così come nel liquido amniotico delle donne in gravidanza [279]. Sono stati rilevati ftalati anche nel latte materno e nell’urina umana [280, 281]. Gli ftalati attraversano la placenta umana, esponendo i feti ai rischi associati all’esposizione a un’importante classe di EDC durante questo periodo critico di sviluppo [282]. Anche i giovani neonati sono esposti a livelli elevati di ftalati, con livelli misurabili di sette diversi ftalati nei neonati nati tra il 2000 e il 2005 [283].
Uno studio del 2012 ha esaminato se esiste o meno una relazione tra i livelli urinari di nove diversi ftalati e l’incidenza del cancro al seno. In questo studio, i metaboliti degli ftalati urinari sono stati rilevati nell’82% delle donne, indipendentemente dal fatto che fosse stato loro diagnosticato o meno un cancro al seno. Elevati livelli di ftalato monoetilico (MEP), un metabolita urinario del composto madre dietil ftalato (DEP; spesso usato in profumeria), è stato associato ad un aumento del rischio di cancro al seno (OR = 2,20; 95% CI = 1,33-3,63). Questa associazione era più alta nelle donne in premenopausa (OR = 4,13; 95% CI = 1,60-10,70). I metaboliti di altri due ftalati comuni (butilbenzilftalato, BBP, e di-n-ottilftalato, DOP) sono stati associati negativamente al rischio di cancro al seno in questo studio (BBP: OR = 0,46; 95% CI = 0,27-0,79 e DOP: O = 0,44; 95% CI = 0,24-0,80) [284]. Livelli più alti di mono(2-etilesil)ftalato urinario (MEHP), un marcatore del carico corporeo DEP, sono stati associati ad una maggiore perdita di gravidanza in uno studio sulle donne danesi [285].
Gli ftalati sono considerati perturbatori endocrini a causa dei loro complessi effetti su diversi sistemi ormonali, compresi i sistemi ormonali degli estrogeni e degli androgeni. Alcuni ftalati, tra cui BBP e DBP, agiscono come estrogeni deboli nei sistemi di coltura cellulare. Essi possono legarsi ai recettori degli estrogeni (ER), indurre risposte cellulari appropriate agli estrogeni e agire in modo additivo con l’estradiolo nell’alterazione di questi sistemi [286, 287]. Anche il DBP, il di-i-butil ftalato di butile (DiBP) e il BBP si legano debolmente al recettore degli androgeni (AR), perturbando le azioni cellulari normalmente avviate dagli androgeni [288, 289]. Nelle linee cellulari del cancro al seno, il BBP promuove la crescita delle cellule staminali tumorali attraverso l’attivazione del recettore degli idrocarburi arilici (AhR) [290]. Gli ftalati possono anche indurre proliferazione, invasione maligna e formazione di tumori nelle linee cellulari del cancro al seno che sono recettori negativi, indicando che almeno alcuni effetti di questi composti sono indipendenti dai loro effetti diretti estrogenici o androgeni [291, 292].
Le proprietà di interferenza endocrina di questa classe di sostanze chimiche sono state ben stabilite nella prole di ratti madre che erano stati trattati con ftalati durante la gravidanza. Gli ftalati disturbano lo sviluppo e il funzionamento del sistema riproduttivo maschile e femminile interferendo con la produzione di testosterone ed estradiolo, rispettivamente [293, 294]. Anomalie nella prole maschile esposta prenatale incluso la ritenzione del capezzolo, ridotta distanza ano-genitale e aumento del criptorchidismo [295, 296]. L’esposizione delle madri umane agli ftalati, misurata attraverso l’analisi dei loro campioni di urina, è stata anche associata a distanze ano-genitali ridotte nei loro figli neonati – una misura della femminilizzazione degli organi genitali esterni [297, 298].
Uno studio caso-controllo ha esaminato i livelli di ftalati in ragazze apparentemente sane che hanno attraversato la laringe (sviluppo del seno) prima degli 8 anni di età, rispetto a ragazze che hanno subito una pubertà precoce a causa di anomalie nel loro sistema neuroendocrino e a ragazze che stavano progredendo nella pubertà in età normale. L’aumento dei livelli di monometil ftalato (MMP) è stato associato al gruppo dei primi anni di età, ma non a nessuno dei due gruppi di confronto [299]. Lo sviluppo precoce del seno in ragazze altrimenti sane è associato ad un aumento del rischio di cancro al seno [300].
L’esposizione di ratti molto giovani al BBP ha portato ad un aumento della proliferazione cellulare nelle gemme terminali del tessuto mammario. I cambiamenti indotti dal BBP nel profilo di espressione dei geni delle cellule mammarie erano coerenti con le anomalie nella differenziazione cellulare e nella comunicazione cellula-cellula [301]. Simili irregolarità strutturali sono state osservate nello sviluppo post-natale dei tessuti mammari quando i ratti sono stati esposti al BBP solo in utero quando le loro madri sono state alimentate con bassi livelli del composto durante la seconda metà delle loro gravidanze [302].
È stato dimostrato che il DEHP altera i meccanismi cellulari a diversi livelli, tra cui l’induzione di danni al DNA che portano a tassi alterati di mitosi e apoptosi, l’aumento della proliferazione cellulare, della mobilità e dell’invasività delle cellule tumorali e la diminuzione della comunicazione intercellulare nei punti di intersezione. DEHP migliora anche la transizione delle cellule epiteliali verso le cellule mesenchimali, guadagnando così sia potenziali migratori che invasivi [303]. L’esposizione delle normali cellule epiteliali del seno umano al DBP ha portato a cambiamenti nell’espressione genica nei percorsi legati ad un certo numero di sistemi, comprese le risposte immunitarie, la regolazione del ciclo cellulare e lo stato antiossidante della cellula [304].
BBP, DBP e DEHP hanno tutti aumentato significativamente la proliferazione cellulare nelle cellule del cancro al seno MCF-7. Inoltre, questi tre ftalati hanno inibito l’azione antitumorale del tamoxifene nelle cellule del cancro al seno MCF-7 [305]. Il BBP ha anche diminuito l’efficacia degli agenti chemioterapici, la doxorubicina e la ciclofosfamide [306].
Parabeni
I parabeni sono un gruppo di composti ampiamente utilizzati come conservanti antimicrobici in prodotti alimentari, farmaceutici e cosmetici. I parabeni vengono assorbiti attraverso la pelle intatta e dal tratto gastrointestinale e dal sangue. I parabeni sono stati trovati in quasi tutti i campioni di urina esaminati da un campione demograficamente diversificato di adulti statunitensi attraverso lo studio National Health and Nutrition Examination Survey (NHANES). Gli adolescenti e le femmine adulte avevano livelli più alti di parabeni metilici urinari e propilparabeni rispetto ai maschi di età simile [307]. I parabeni si trovano anche in campioni amniotici durante il secondo trimestre di gravidanza [308].
Concentrazioni misurabili di sei diversi parabeni sono state identificate in campioni bioptici di tumori al seno [309]. I parabeni particolari sono stati riscontrati in concentrazioni relative che si avvicinano strettamente al loro utilizzo nella sintesi di prodotti cosmetici [310]. Livelli più alti di n-propylparaben sono stati trovati nel quadrante ascellare del seno [311], la regione in cui si trova la più alta percentuale di tumori al seno, anche se le concentrazioni non sono state correlate alla reale localizzazione dei tumori nel seno delle singole donne. Diversi ricercatori hanno notato l’importanza di studiare gli effetti delle miscele di parabeni, nei profili di concentrazione che sono rilevanti per l’esposizione naturale ai composti, per comprendere i complessi effetti di questa classe di sostanze chimiche sull’aumento del rischio di sviluppare il cancro al seno [312- 314].
I parabeni sono deboli mimatori di estrogeni, e la potenza della risposta agonistica è correlata alla struttura del gruppo alchilico [315, 316]. Possono legarsi sia all’ERα che all’ERβ, con una maggiore affinità con il sito ERβ [316], e aumentano l’espressione di diversi geni che reagiscono agli estrogeni coinvolti nella crescita e proliferazione delle cellule, così come l’inibizione dell’apoptosi [317- 320]. L’aggiunta di miscele di parabeni a concentrazioni e combinazioni misurate nel tessuto della biopsia del seno ha portato ad un aumento della crescita e della proliferazione di MCF-7 [321].
I parabeni di metile, propil- e butil-parabeni hanno tutti stimolato la proliferazione nelle cellule del cancro al seno umano ER+ (MCF-7), così come nelle cellule epiteliali non maligne del seno umano (MCF-10A). I parabeni hanno aumentato la secrezione di estrogeni nelle cellule MCF-7, ma l’hanno diminuita nelle cellule MCF-10A [321]. Il lavoro di follow-up ha dimostrato che l’effetto proliferativo dei parabeni sulle cellule MCF-7 era indipendente dagli effetti diretti sul ciclo cellulare o sull’espressione del gene dell’apoptosi. D’altra parte, nelle cellule MCF-10A, i parabeni imitavano l’estradiolo nell’alterare l’espressione dei geni coinvolti sia nella progressione del ciclo cellulare che nell’apoptosi [322].
Se sommati a colture di ERα- e HER2-positivo umano BT-474 cellule tumorali del seno, butilparaben ed eregulina, un ligando naturale di HER, hanno portato ad un aumento sinergico dell’espressione dell’oncogene Myc mRNA e dell’attività cellulare. Questi dati indicano che i ligandi per i due recettori possono impegnarsi in diafonia nelle cellule del cancro al seno, aumentando gli effetti dell’esposizione ai parabeni ambientali [323]. A concentrazioni inferiori a quelle riscontrate nei campioni di cancro al seno, i parabeni esercitano anche effetti antagonisti inversi, mimando così gli effetti della stimolazione estrogenica, alla membrana ERRY [324].
Diciassette giorni di trattamento di cellule umane non maligne del seno (MCF-10A) con metil-, propil-, o butil-parabeni hanno portato all’induzione di un fenotipo trasformato legato al processo di carcinogenesi delle cellule del seno [325]. Il trattamento a lungo termine (>20 settimane) delle cellule MCF-7 con gli stessi parabeni alla concentrazione che porta al massimo aumento della proliferazione cellulare ha migliorato le risposte migratorie e invasive [326].
Nelle cellule epiteliali del seno derivate da donne ad alto rischio di sviluppare il cancro al seno, il metilparaben ha contrastato l’effetto apoptotico del tamoxifene, un importante trattamento adiuvante del cancro al seno [318].
Alchilfenoli
Gli alchilfenoli sono prodotti chimici industriali utilizzati nella produzione di detergenti e altri prodotti per la pulizia e come antiossidanti nei prodotti in plastica e gomma. Si trovano anche nei prodotti per la cura della persona, in particolare nei prodotti per capelli, e come componente attivo in molti spermicidi. Nello studio del Silent Spring Institute sui contaminanti domestici, gli alchilfenoli, in particolare il 4-nonilfenolo (4-NP) e i suoi prodotti di degradazione, sono stati trovati in tutti i campioni di aria domestica e nell’80% dei campioni di polvere domestica [202]. Concentrazioni sostanziali di queste sostanze chimiche sono state trovate anche nelle acque reflue associate alle acque grigie domestiche e alle fognature, nelle acque reflue urbane e nelle discariche municipali [327- 330].
I dati NHANES che esaminano i livelli chimici nelle urine di adulti americani hanno trovato 4-NP nel 51% dei campioni valutati [331] e 4-ottilfenolo (4-OP) nel 57,4% dei campioni [219]. Risultati simili sono stati trovati in campioni di siero di donne svedesi che allattano 3 settimane dopo il parto [332], e livelli significativi di 4-NP e 4-OP sono stati trovati in campioni di latte materno di donne taiwanesi [333].
Alchilfenoli, tra cui 4-NP, hanno dimostrato di imitare le azioni dell’estradiolo, mediando i loro effetti attraverso il recettore cellulare degli estrogeni. Essi si legano anche alla membrana cellulare ER e mimare le risposte di segnalazione cellulare di solito controllato da estradiolo [334]. In uno studio che esamina gli effetti del 4-NP nelle cellule tumorali del seno umano (MCF-7) in vitro, sono stati osservati cambiamenti nell’espressione genica in diversi geni coinvolti nella proliferazione cellulare, nella trascrizione del DNA e nella segnalazione cellulare – tutti sistemi che sono disturbati nella formazione del tumore [335- 337].
L’esposizione prenatale dei ratti a 4-NP ha causato lo sviluppo alterato della ghiandola mammaria così come i cambiamenti nelle popolazioni di recettori steroidei in diversi tessuti riproduttivi [338]. Il trattamento dei topi con 4-NP ha portato ad una maggiore sintesi di estriolo, un estrogeno naturale debole, da parte dei fegati degli animali trattati. Rispetto ai topi trattati con quantità equivalenti di estradiolo, i topi esposti al 4-NP avevano un aumentato rischio di cancro mammario [339].
Triclsoan e triclocarbano
Triclosan e triclocarban sono agenti antimicrobici che negli ultimi 40 anni sono stati ampiamente utilizzati in una vasta gamma di prodotti per la cura della persona, per la casa e industriali [340]. La struttura chimica del triclosan ha somiglianze sia con l’ormone tiroideo (T4) sia con diversi noti perturbatori endocrini tra cui PCB, DES e bisfenolo A, mentre il triclocarbano ha proprietà chimiche simili a quelle di diversi pesticidi e prodotti farmaceutici [341]. Entrambi si trovano in campioni di acqua dolce, specialmente nei laghi e a valle degli impianti di trattamento delle acque reflue, in concentrazioni note per essere dannose per la fauna selvatica [342- 344].
In uno studio di campioni di urine americane adulte nell’ambito del protocollo di studio del CDC NHANES, il 75% dei campioni è risultato avere livelli significativi di triclosan e dei suoi metaboliti. Livelli più alti sono stati trovati in adulti più giovani e più ricchi [34]. Un’analisi della tendenza a 10 anni dei livelli di triclosan urinario di NHANES ha rilevato una piccola diminuzione dei livelli nei 6 anni successivi al picco del 2006. Uno studio parallelo di NHANES che esamina i livelli chimici nelle donne in gravidanza ha trovato livelli misurabili di triclosan nell’87% dei campioni di urina esaminati [345]. In uno studio più piccolo di campioni di adulti americani, il triclocarbano e i suoi metaboliti sono stati rilevati in un terzo dei campioni di urina e in metà dei campioni di siero esaminati [346]. L’analisi dell’autopsia umana rivela che il triclosan bioaccumula nel fegato e nel tessuto adiposo, ma non nel cervello, i tre tessuti esaminati [347].
Sebbene ci sia stato un lavoro molto limitato nell’esaminare gli effetti diretti del triclosan o del triclocarban sullo sviluppo del sistema mammario o sul rischio di sviluppare il cancro al seno [348], una considerevole ricerca dimostra che questi due composti esercitano effetti sui sistemi ormonali in modi simili ai meccanismi stabiliti per perturbare il normale sviluppo del seno e la salute. A seconda della concentrazione della sostanza chimica testata e del sistema modello utilizzato, il triclosan e il triclocarban esercitano una complessa combinazione di effetti estrogenici e antiestrogenici, androgeni e antiandrogeni, tutti mediati almeno in parte attraverso interazioni con il recettore degli estrogeni (ER) e il recettore degli androgeni (AR) delle cellule target [340- 342, 350, 351].
Anche a dosi molto basse, il triclosan era estrogeno-simile nell’aumentare i tassi di proliferazione delle cellule tumorali umane coltivate (linea MCF-7 BOS), ma in combinazione con basse dosi di estradiolo, il triclosan era antiestrogenico nel sopprimere l’aumento completo dell’estradiolo indotto dalla crescita e dalla proliferazione delle cellule. A concentrazioni più elevate, ma ancora rilevanti dal punto di vista ambientale, il triclosan ha diminuito la vitalità delle cellule [351]. Anche nelle cellule MCF-7, il triclosan ha significativamente migliorato sia l’attività della ciclina D1 e D2 che l’aumento della proliferazione cellulare. Questi effetti sono stati bloccati dal trattamento concomitante con ICI-182.780, un antagonista specifico ER [352].
Oltre ai suoi effetti esercitati attraverso i sistemi di recettori steroidei, il triclosan ha dimostrato di alterare i livelli di ormone tiroideo nei ratti puberali [353, 354]. Il trattamento dei ratti madre con triclosan durante il periodo di allattamento ha portato ad una diminuzione sostenuta di 3 settimane di ormone tiroideo nelle dighe. Tuttavia, i cuccioli, solo aveva soppresso i livelli di T4 per i primi giorni di allattamento, con livelli normali di essere registrato più tardi nel periodo, nonostante la continua esposizione al triclosan materno ingerito [355].
Sostanze chimiche ormonalmente attive presenti negli schermi solari (filtri UV)
La preoccupazione per l’esposizione ai raggi ultravioletti (UV) del sole e il rischio di cancro della pelle ha portato ad un uso diffuso delle protezioni solari. Molti schermi solari contengono sostanze chimiche che non sono solo estrogeniche ma anche lipofile. Gli studi dimostrano che queste sostanze chimiche si stanno accumulando nella fauna selvatica [356], e si trovano nelle urine umane e nei campioni di latte materno [357, 358]. I dati NHANES indicano che oltre il 96% degli adulti americani ha livelli rilevabili di benzofenone-3 (Bp-3) nelle urine [219], e che i livelli urinari sono aumentati negli anni tra il 2006 e il 2012. I livelli erano più alti nelle donne e nei bianchi non ispanici rispetto ad altri gruppi [351]. Un recente studio occupazionale dei vigili del fuoco ha rilevato che i loro livelli di Bp-3 erano cinque volte superiori a quelli riportati negli studi NHANES [359].
Di sei comuni prodotti chimici per la protezione solare, cinque di essi esercitavano una significativa attività estrogenica, misurata dall’aumento dei tassi di proliferazione delle cellule umane del cancro al seno (cellule MCF-7) cresciute in vitro. Queste sostanze chimiche erano 3-(4-metilbenzilidene)-camfora (4-MBC), ottil-metossicinnamato (OMC), ottil-dimetil-PABA (OD-PABA), benzofenone-3 (Bp-3) e omosalato (HMS) [360, 361]. I risultati per 4-MBC sono stati replicati in un altro laboratorio [362].
In un saggio comune di lievito che misura la forza della risposta estrogenica di un composto, miscele di basse concentrazioni (al di sotto delle “concentrazioni senza effetto osservato” o NOEC) di sostanze chimiche simili a quelle che si trovano nelle creme solari hanno dimostrato effetti sinergici additivi. Altri studi indicano che oltre ad agire come gli estrogeni in molte vie cellulari, i composti che si trovano nelle creme solari possono anche antagonizzare gli effetti dell’estradiolo naturale in altre vie [360].
L’applicazione di OMC sulla pelle dei ratti ha migliorato la penetrazione dell’erbicida perturbatore endocrino 2,4-D [363].
Acido perfluoroottanoico (PFOA) e solfato perfluoroottanoico (PFOS)
L’acido perfluorooattanoico (PFOA) e il solfato perfluoroottanoico (PFOS) sono ampiamente utilizzati in applicazioni commerciali per le loro proprietà chimiche di essere altamente stabili e di avere una bassa tensione superficiale. Il PFOA si trova in composti come il Teflon® e il Gore-tex® così come in altri prodotti, tra cui i protettori per tappeti e mobili. Il PFOS è l’ingrediente principale di Scotchguard® e di altri prodotti destinati come trattamenti per fornire resistenza allo sporco o alle macchie, specialmente nei tessuti.
Il PFOA e i PFOS sono onnipresenti, con livelli misurabili che si trovano nella fauna selvatica di tutto il pianeta [364]. Queste sostanze chimiche si trovano in campioni di siero provenienti da oltre il 95% degli adulti statunitensi testati in uno studio NHANES, anche se i livelli di PFOS sono diminuiti nell’ultimo decennio, poiché la sostanza chimica è stata gradualmente messa fuori uso negli Stati Uniti [365]. Un altro studio ha trovato il PFOA e i PFOS in campioni di siero del sangue prelevati da adulti provenienti da nove paesi che rappresentano quattro continenti [366]. In uno studio su campioni di sangue del cordone ombelicale di neonati di Baltimora, il 100% dei campioni aveva livelli misurabili di PFOA e PFOS [367]. Livelli più alti di sostanze chimiche nel sangue del cordone ombelicale erano associati sia a un peso inferiore alla nascita che a dimensioni più piccole, indicando un effetto del PFOA sullo sviluppo prenatale [368]. Ci sono forti correlazioni tra il siero materno e i livelli di liquido amniotico del PFOA. Anche il PFOS è stato rilevato nel liquido amniotico, ma solo quando i livelli materni erano relativamente alti [369].
Le esposizioni prenatali al PFOA e ai PFOS sono state associate a pesi più bassi alla nascita, ma a pesi più alti all’età di 20 mesi nelle ragazze che partecipano allo Studio Avon Longitudinale dei Genitori e dei Bambini [370]. Il follow-up di queste stesse ragazze all’età di 15 anni ha indicato che queste esposizioni prenatali erano associate ad aumenti dei livelli sierici di testosterone negli adolescenti [371]. Il testosterone e altri androgeni inibiscono il normale sviluppo mammario durante l’adolescenza [372].
Nelle adolescenti dell’Ohio sud-orientale esposte alle sostanze chimiche perfluorinate, livelli più alti nel siero del sangue erano associati a un ritardo nell’inizio delle mestruazioni nelle ragazze [373]. Altri studi sulle ragazze dell’Ohio hanno dimostrato che l’esposizione a livelli più elevati di PFOA era associata sia al menarca successivo [374] che al successivo sviluppo del seno [375]. Mentre lo sviluppo precoce del seno è un noto fattore di rischio per il cancro al seno, questi dati supportano un potenziale effetto di interferenza endocrina del PFOA, che può portare ad altri effetti sulla salute più tardi nella vita. Per esempio, livelli più elevati di PFOA (o PFOS) nel siero sono associati a ritardi più lunghi nella gravidanza nelle donne che cercano di concepire [376].
Livelli sierici più elevati di PFOA e PFOS nel sangue, così come altri composti perfluorurati, sono stati associati a un aumento dell’incidenza del cancro al seno in uno studio sulle donne Inuit in Groenlandia. Anche i livelli di policlorobifenili (PCB) sono stati elevati nelle donne a cui era stato diagnosticato il cancro al seno [377].
In una serie di studi che esaminano gli effetti dell’esposizione gestazionale o neonatale dei topi al PFOA, sono state riscontrate anomalie nella formazione di tessuto mammario nelle dighe durante l’allattamento e nei cuccioli quando sono maturati. Le esposizioni a basse dosi di PFOA durante la gravidanza hanno portato ad una differenziazione alterata durante l’allattamento, un processo necessario per la normale produzione e il rilascio del latte. Nei cuccioli femminili, le ghiandole mammarie hanno mostrato uno sviluppo stentato dei rami epiteliali prima ancora che gli animali fossero stati svezzati [378]. In uno studio di follow-up che ha utilizzato un cross-fostering design, i cuccioli esposti sia in utero o nei primi anni di vita postnatale avevano anomalie durature nello sviluppo dei loro tessuti mammari, e queste anomalie sono rimasti almeno attraverso il tempo della pubertà, l’ultimo tempo valutato [379]. In un terzo studio, le esposizioni gestazionali al PFOA hanno mostrato di alterare lo sviluppo mammario in tre generazioni. In un altro gruppo di topi, le esposizioni croniche al PFOA nell’acqua potabile a livelli simili a quelli che si trovano in alcune riserve di acqua umana contaminata, hanno portato a simili esiti negativi dello sviluppo nei tessuti mammari dei cuccioli in via di sviluppo [378].
La complessità degli effetti del PFOA è sottolineata da uno studio che esamina l’esposizione a basse dosi di diversi ceppi di topi alla sostanza chimica nel periodo tra lo svezzamento e la pubertà. In un ceppo (Balb/c), l’esposizione alla sostanza chimica ha portato a deficit nel normale sviluppo mammario attraverso la pubertà, mentre nell’altro ceppo (C57/BL6), basse dosi di esposizione al PFOA hanno aumentato lo sviluppo mammario, ma dosi più alte sono state inibitorie [380]. Non è ancora noto quali siano i fattori alla base del ceppo e delle differenze di dose.
D’altra parte, quando i topi CD-1 o C57/BL6 sono stati esposti a basse dosi di PFOA prima della nascita, sono stati osservati ritardi nello sviluppo mammario in entrambi i ceppi, anche se non sono state osservate differenze nei livelli ormonali ovarici o nel tempo di inizio della pubertà. È importante sottolineare che questi risultati indicano che la ghiandola mammaria è più sensibile alle perturbazioni prenatali del PFOA rispetto ad altre misure dello stato puberale [381].
Nelle cellule tumorali della mammella umana T47D ormono-dipendenti, né il PFOA né il PFOS, di per sé, colpiscono la proliferazione cellulare. Tuttavia, in presenza di estradiolo, entrambe le sostanze chimiche hanno migliorato gli effetti dell’estradiolo sulla crescita cellulare, nonché l’espressione di diversi geni che reagiscono agli estrogeni e l’attivazione dell’ERK1/2 [382].
Idrocarburi policiclici aromatici (IPA)
Gli idrocarburi policiclici aromatici (IPA) sono onnipresenti sottoprodotti della combustione, che entrano nel corpo da fonti diverse come i bruciatori di carbone e coke, i motori a gasolio, le carni alla griglia e le sigarette. I residui di IPA sono spesso associati al particolato in sospensione nell’aria, quindi l’inalazione è uno dei principali mezzi di esposizione agli IPA [383]. In uno studio del Silent Spring Institute sui contaminanti ambientali presenti nella polvere delle case, tre PAH (pirene, benz[a]antracene e benz[a]pirene) sono stati trovati in più di tre quarti delle case testate [209]. Sebbene si trovino ancora ampiamente nel particolato in sospensione, le norme imposte a livello federale sulle emissioni dei veicoli hanno portato a una significativa diminuzione del rilascio di IPA da parte dei veicoli rispetto ai loro livelli più alti degli anni ’70 [384].
Come molte altre sostanze chimiche ambientali associate al rischio di cancro al seno, gli IPA sono lipofili e sono immagazzinati nel tessuto adiposo del seno [385]. È stato dimostrato che gli IPA aumentano il rischio di cancro al seno attraverso una varietà di meccanismi. I PAH più comuni sono debolmente estrogenici [386]. Tuttavia, la via principale diretta dai recettori è attraverso l’interazione con una proteina chiamata il recettore degli idrocarburi arilici (AhR), dando inizio ad una serie di cambiamenti cellulari che portano ad un’alterazione della segnalazione cellulare e, in ultima analisi, ad un aumento delle mutazioni del DNA [387, 388]. Anche se attualmente non è chiaro quale sia il ligando naturale per l’AhR, l’evidenza suggerisce che il sistema AhR è importante nel regolare le risposte allo stress cellulare che può portare all’interruzione del normale funzionamento delle cellule [389]. Almeno alcuni degli effetti degli IPA sono mediati attraverso complesse interazioni tra le vie regolate dall’AhR e quelle regolate dal recettore degli estrogeni [390]. Gli IPA possono anche essere direttamente genotossici, interagendo direttamente con il genoma e causando danni al DNA [391].
Diversi studi epidemiologici hanno implicato l’esposizione alla PAH in un aumento del rischio di cancro al seno. Ad esempio, utilizzando le stime di esposizione al traffico di uno dei più potenti IPA (benzo[a]pirene) come proxy per tutti gli IPA correlati al traffico, i ricercatori hanno confrontato l’incidenza del cancro al seno nelle donne esposte al 5% dell’esposizione al traffico con quelle con livelli di esposizione inferiori alla mediana. Le maggiori esposizioni agli IPA generati dal traffico sono state associate a un aumento dell’incidenza del cancro al seno nelle donne che hanno mangiato bassi livelli di frutta e verdura (OR = 1,46; 95% CI = 0,89-2,40). Non è stata trovata un’associazione significativa per le donne che hanno mangiato livelli elevati di questi alimenti. L’associazione con una maggiore incidenza di cancro al seno è stata trovata solo per i tumori ER/PR [392].
In uno studio caso-controllo, anche la combustione di tronchi sintetici, ma non di soli tronchi di legno, in stufe a legna o caminetti è stata associata ad un aumento del rischio di cancro al seno (OR = 1,42; 95% CI = 1,11-1,84). L’associazione era più forte nelle donne la cui esposizione era dopo i 20 anni di età e per almeno 7 anni di durata d’uso. Le donne che hanno bruciato tronchi sintetici e sviluppato il cancro al seno avevano più probabilità di avere almeno due varianti genetiche nei geni che regolano la glutatione S-transferasi, enzimi che sono importanti come disintossicanti cellulari (OR = 1,71; 95% CI = 1,09-2,69) [393].
Uno degli studi del Long Island Breast Cancer Study Project ha rilevato che le donne con il più alto livello di addotti PAH-DNA avevano un rischio aumentato del 50% di cancro al seno [394]. Più specificamente, quando gli IPA interagiscono con il DNA e formano un addotto, il risultato è la perdita di una delle basi delle purine che, se non corretta, porta a mutazioni geniche. Questi addotti instabili della IPA sono stati collegati allo sviluppo del cancro [395].
In un precedente rapporto, i ricercatori hanno esplorato la presenza di addotti PAH-DNA in campioni di seno prelevati da donne a cui era stato diagnosticato il cancro rispetto a quelle a cui era stata diagnosticata una malattia mammaria benigna. I campioni cancerogeni avevano il doppio di probabilità di avere addotti di PAH-DNA rispetto ai campioni benigni [396]. Il lavoro di follow-up indica che quelle donne che avevano livelli più alti di addotti di PAH-DNA potrebbero non avere necessariamente avuto esposizioni più elevate agli IPA, ma erano invece più sensibili alle esposizioni agli IPA perché avevano particolari profili genetici che incoraggiavano i deficit nella riparazione del DNA [397]. Altri studi sostengono l’esistenza di diversi profili genetici nelle donne che hanno aumentato il numero di addotti PAH-DNA, compresi i polimorfismi nei geni coinvolti nel metabolismo cellulare, nei meccanismi di soppressione dei tumori e nella riparazione del DNA [397, 398]. Non sono state riscontrate differenze nei profili dei geni i cui prodotti sono coinvolti nell’attivazione e nella disattivazione degli stessi IPA [399]. Uno studio caso-controllo basato sulla popolazione ha trovato che le esposizioni agli IPA erano associate a mutazioni specifiche del gene soppressore del tumore p53 in campioni di tumore al seno [400].
Gli studi sull’esposizione professionale hanno esaminato le lavoratrici esposte regolarmente ai fumi della benzina e ai gas di scarico dei veicoli, fonti principali di IPA (oltre che di benzene). Queste esposizioni professionali sono associate ad un aumento del rischio di cancro al seno per le donne in pre-menopausa (esposizione a basso livello, OR = 1,56; 95% IC = 0,78-3,12; esposizione ad alte dosi, OR = 2,40; 95% CI = 0,96-8,01) [401] e anche per gli uomini. Nel caso del cancro al seno maschile, gli IPA possono aumentare il rischio di cancro al seno in particolare negli uomini portatori di una mutazione BRCA1 o BRCA2. In un piccolo studio condotto su 23 uomini affetti da cancro al seno, quattro dei quali erano portatori di una mutazione BRCA1/2, lo stato di portatore di BRCA1/2 ha interagito con la storia di aver mai guidato camion in modo professionale per aumentare il rischio di sviluppare il cancro al seno (COR = 25,5; 95% CI = 1,1-1415) [402].
Uno studio caso-controllo nella parte occidentale di New York ha indicato che l’esposizione molto precoce della vita (intorno al momento della nascita) ad alti livelli di particolato totale sospeso, una misura proxy dei livelli di IPA, è associata ad un aumento del rischio di cancro al seno nelle donne in post-menopausa [375]. Un’estensione di questo studio, esaminando l’esposizione alla IPA nei momenti critici della storia riproduttiva delle donne, ha dimostrato una relazione tra l’esposizione al particolato intorno al primo periodo mestruale e l’incidenza del cancro al seno in pre-menopausa (OR = 2).05; 95% CI = 0,92-4,54), e una relazione tra il livello di esposizione al momento del primo parto di una donna e il suo rischio di cancro al seno in post-menopausa (OR = 2,57; 95% CI = 1,16-5,69) [403].
Gli studi hanno riguardato soprattutto l’incidenza del cancro al seno. Due relazioni hanno esaminato il rapporto tra le esposizioni alla IPA e la mortalità. Utilizzando un modello ecologico che esplora l’associazione tra le polveri sottili sospese in diversi comuni di Taiwan, i ricercatori hanno scoperto che le donne che vivono in aree con alti livelli di particolato nell’aria avevano una maggiore probabilità di morire di cancro al seno, rispetto a quelle che vivono in aree più pulite (RR = 1,19; 95% CI = 1,03-1,38) [404]. Un altro rapporto ha esaminato i livelli di addotto di PAH-DNA e la mortalità tra le donne a cui era stato diagnosticato il cancro al seno. In un’estensione dello studio di Long Island descritto sopra, i ricercatori non hanno trovato alcuna relazione complessiva tra sopravvivenza e livelli di addotti di PAH-DNA. Guardando più da vicino i gruppi di donne che avevano subito diversi tipi di trattamenti, tuttavia, ha rivelato un doppio aumento dei tassi di mortalità per cancro al seno in base all’età tra le donne con alti livelli di addotto di PAH-DNA che avevano ricevuto il trattamento con radiazioni (HR = 2,47; 95% CI = 0,74-8,21). Tuttavia, c’è stato un aumento del tasso di sopravvivenza per le donne con addotti che hanno ricevuto una terapia ormonale come parte del trattamento per i loro tumori al seno (HR = 0,52; 95% CI = 0,24-1,13) [405].
Pesticidi ed erbicidi
Un rapporto del 2007 del progetto di studio sul cancro al seno di Long Island ha dimostrato che l’uso di pesticidi residenziali per tutta la vita, auto-riferito, è stato associato ad un aumento del rischio di cancro al seno. L’aumento è stato riscontrato per le donne che avevano segnalato l’uso di pesticidi nell’aggregato (avendo mai usato pesticidi residenziali; OR = 1,39; 95% CI = 1,15-1,68), così come specificamente per l’uso del prato (OR = 1.48; 95% CI = 1,20-1,82) e pesticidi da giardino (OR = 1,58; 95% CI = 1,12-2,22), sebbene non ci fossero relazioni tra le dosi percepite di esposizione e il rischio di cancro [406]. Questi risultati sono importanti perché riguardano l’esposizione a sostanze chimiche nel corso della vita ordinaria, con tutte le complessità delle miscele e i molteplici tipi di utilizzo. Molti altri studi si concentrano su singole sostanze chimiche o classi di sostanze chimiche, e i risultati sono spesso contraddittori a seconda della durata e della tempistica delle esposizioni, dei tipi di sostanze chimiche oggetto di studio e così via. Nonostante ciò, molti pesticidi ed erbicidi sono stati etichettati come cancerogeni per l’uomo o per gli animali. Molti si trovano nelle riserve d’acqua [406] così come campioni di aria e polvere provenienti dalle case [209, 407].
Erbicidi triazinici: atrazina
Gli erbicidi triazinici (tra cui atrazina, simazina e cianazina) sono i prodotti chimici per l’agricoltura più usati negli Stati Uniti. L’atrazina e la simazina sono state vietate nell’Unione Europea, e la cianazina è etichettata come un mutageno altamente tossico, a causa della loro elevata presenza nell’acqua potabile, hanno dimostrato effetti nocivi sulla fauna selvatica, e potenziali effetti sulla salute degli esseri umani. La cianazina è stata gradualmente eliminata dall’uso negli Stati Uniti a partire dal 1996, anche se la simazina e l’atrazina sono ancora approvate per l’uso negli Stati Uniti. Più di 75 milioni di libbre di atrazina, la più usata tra le sostanze chimiche, sono state applicate ogni anno negli Stati Uniti nel 2008 (l’anno più recente per il quale l’Environmental Protection Agency (EPA) dispone di dati), principalmente per controllare le malerbe a foglia larga nelle colture di mais e sorgo nel Midwest [408].
Livelli elevati di atrazina si trovano ogni primavera ed estate sia nell’acqua potabile che nelle falde acquifere delle aree agricole [409- 411]. L’atrazina è un noto perturbatore endocrino, che causa danni drammatici alle strutture riproduttive di rane, pesci e altri animali selvatici [412, 413]. Sebbene sia stato dimostrato che tutte e tre le triazine causano il cancro mammario nei ratti da laboratorio [414] e l’aumento della proliferazione delle linee cellulari del seno umano in vitro [415], ci sono relativamente pochi dati scientifici che esplorano la relazione tra simazina o cianazina e il cancro al seno umano. La letteratura sull’atrazina è un po’ più consistente.
Alti livelli di triazine, principalmente atrazina, in acque contaminate erano associati ad un aumento del rischio (OR = 1,20; 95% CI = 1,13-1,28) di cancro al seno [416], anche se un’ulteriore espansione di questo studio ha concluso che non c’era alcuna relazione tra l’esposizione all’atrazina e il rischio di sviluppare il cancro al seno [417]. Risultati altrettanto contraddittori sono stati trovati in un altro studio ecologico in cui livelli più elevati di pesticidi misti, tra cui l’atrazina, sono stati associati ad un aumento del cancro al seno in una contea rurale del Regno Unito, ma non nella contea vicina [418]. Poiché questi studi ecologici tendono a confrontare i livelli medi di contaminazione da atrazina e i tassi di incidenza in tutta la contea, piuttosto che le storie di esposizione individuale e i risultati sanitari, è difficile capire chiaramente la differenza dei risultati [419].
Una revisione del peso dell’evidenza di sette studi epidemiologici, finanziati da Syngenta – il produttore di atrazina, ha concluso che attraverso i disegni di studio, non c’era alcuna relazione causale tra l’esposizione all’atrazina e lo sviluppo del cancro al seno. Tuttavia, la disponibilità di pochi studi, la mancanza di attenzione per i sottotipi di cancro al seno e altre complicazioni metodologiche rendono impossibile escludere un’associazione [420].
La ricerca sui roditori ha dimostrato che l’esposizione all’atrazina perturba la funzione pituitaria-ovarica, con conseguente diminuzione della prolattina in circolazione e dei livelli di ormone luteinizzante, cambiamenti che contribuiscono agli effetti di questo erbicida sull’aumento dei tumori mammari [414, 421]. L’atrazina esercita anche effetti di interferenza endocrina aumentando l’attività dell’enzima aromatasi [422, 423], un enzima che catalizza la conversione del testosterone e di altri androgeni in estrogeni, tra cui l’estradiolo.
L’esposizione all’atrazina o a miscele di metaboliti di atrazina durante la gestazione ritarda lo sviluppo della ghiandola mammaria di ratto nella pubertà, allargando la finestra di sensibilità alle sostanze cancerogene del seno [424- 426]. Allo stesso modo, l’esposizione dei ratti in ritardo di gravidanza a una miscela di metaboliti di atrazina comunemente formati porta anche a cambiamenti persistenti nello sviluppo della ghiandola mammaria nei cuccioli esposti durante la gestazione. Queste anomalie persistono in età adulta. L’esposizione di ratti con tumori mammari esistenti a atrazina aumenta il tasso di proliferazione cellulare in quei tumori [427]. I cambiamenti precoci nello sviluppo della ghiandola mammaria possono riflettere un effetto indiretto dell’atrazina sulla salute materna, in particolare la restrizione calorica indotta dall’atrazina, durante il periodo di esposizione ai pesticidi [428].
Sebbene l’atrazina sia un perturbatore endocrino nelle vie dirette agli estrogeni, diversi studi hanno indicato che l’atrazina non esercita i suoi effetti attraverso il legame al pronto soccorso [429, 430]. Altri meccanismi proposti con cui l’atrazina può alterare le vie degli estrogeni includono attraverso il legame e l’aumento dell’espressione del recettore GPR30 legato alla membrana [407, 423], l’attivazione del gene steroidogenico fattore-1 (SF-1), l’attivazione della fosforilazione ERK, e l’amplificazione diretta o indiretta del cAMP [430- 432].
Eptacloro
L’eptacloro è un insetticida che è stato ampiamente utilizzato negli Stati Uniti negli anni ’80, soprattutto per il controllo delle termiti. Nel 1988, l’U.S. EPA ha limitato l’uso dell’eptacloro ad alcune applicazioni per il controllo delle formiche rosse, ma l’uso agricolo è continuato fino al 1993 perché ai coltivatori è stato permesso di utilizzare le scorte esistenti [433]. L’uso dell’eptacloro era particolarmente elevato nelle Hawaii, dove veniva impiegato in modo estensivo sulle colture di ananas e di conseguenza contaminava sia le colture agricole locali che le scorte di latte. I tassi di cancro al seno nelle Hawaii sono aumentati drammaticamente per le donne di tutti i gruppi etnici negli ultimi quattro decenni [434]. In uno studio di controllo dei casi relativamente piccolo (96 casi) che esplorava le possibili relazioni tra i livelli sierici di pesticidi organoclorurati, incluso l’eptacloro, e lo sviluppo del cancro al seno, è stata trovata una tendenza (P = .078) tra le concentrazioni di eptacloro e il rischio di cancro al seno.
L’eptacloro contamina ancora sia il suolo che l’uomo. Il suo prodotto di degradazione, l’eptacloro epossidico (HE), è noto per accumularsi nel grasso, compreso il tessuto mammario. I livelli sono più alti nelle donne dai 20 anni in su, ma l’HE si trova anche nei corpi di adolescenti dai 12 ai 19 anni [435] e in otto dei dieci campioni di sangue ombelicale di neonati [436]. È stato dimostrato che alti livelli di eptacloro nel latte materno [437] e nel tessuto adiposo delle biopsie mammarie [438] sono associati a un aumento dell’incidenza del cancro al seno.
Sebbene l’HE non agisca come gli estrogeni, influisce sul modo in cui il fegato elabora gli ormoni, permettendo così ai livelli di estrogeni in circolazione di aumentare e aumentando il rischio di cancro al seno. È stato anche dimostrato che l’HE disturba la comunicazione da cellula a cellula nelle cellule del seno umano in coltura [439] e aumenta la produzione di ossido nitrico, una sostanza chimica che si trova naturalmente nelle cellule e che è nota per causare danni al DNA [438].
Dieldrin e aldrin
Dagli anni ’50 fino al 1970, i pesticidi dieldrin e aldrin (che si scompone in dieldrin, il principio attivo) sono stati ampiamente utilizzati per le colture, tra cui il mais e il cotone. A causa delle preoccupazioni per i danni all’ambiente e, potenzialmente, alla salute umana, nel 1975 l’EPA ha vietato tutti gli usi dell’aldrin e del dieldrin ad eccezione del controllo delle termiti; l’EPA ha vietato del tutto questi pesticidi nel 1987 [440]. Così, la maggior parte del carico corporeo umano di questa sostanza chimica deriva o da esposizioni passate o da una persistente contaminazione ambientale.
Hoyer et al. hanno mostrato una chiara relazione tra l’incidenza del cancro al seno e il dieldrin nell’esame di una rara banca di campioni di sangue prelevati da donne prima dello sviluppo del cancro al seno [441]. Alla fine degli anni ’70 e all’inizio degli anni ’80, sono stati prelevati campioni di sangue da circa 7500 donne danesi di età compresa tra i 30 e i 75 anni. I ricercatori hanno rilevato composti organoclorurati nella maggior parte delle 240 donne a cui era stato diagnosticato un tumore al seno prima della pubblicazione dello studio nel 2000. Hanno trovato dieldrin nel 78% delle donne a cui è stato successivamente diagnosticato il cancro al seno, con le donne che avevano i più alti livelli di dieldrin prima della diagnosi avendo più del doppio delle probabilità di sviluppare la malattia rispetto alle donne con i livelli più bassi. L’esposizione al dieldrin è correlata all’aggressività del cancro al seno: i livelli più alti di dieldrin sono stati associati a una maggiore mortalità per cancro al seno (RR = 2,61; 95% CI + 0,97-7,01; Ptrend = <,01) [442].
Il trattamento di topi prenatale e neonatale a dosi di dieldrin rilevanti per l’ambiente ha aumentato il numero e la dimensione dei tumori mammari. Questi effetti possono essere stati mediati attraverso cambiamenti nell’espressione cellulare del fattore di crescita BDNF e del recettore del segnale cellulare Trks. Entrambi questi sono stati elevati in tumori dagli animali trattati con dieldrin [443].
Come molti altri pesticidi trovati nell’ambiente, il dieldrin ha dimostrato di essere un perturbatore endocrino, sia stimolando i sistemi regolati dagli estrogeni che interferendo con le vie regolate dagli androgeni. L’aggiunta di dieldrin alle cellule del cancro al seno umano (MCF-7) in vitro può stimolare la loro crescita e proliferazione [444].
Altri pesticidi
Uno studio caso-controllo di 128 lavoratori agricoli di Latina a cui è stato recentemente diagnosticato un cancro al seno in California e 640 controlli senza cancro, ha identificato tre pesticidi -clordano, malathion e 2,4-D – associati ad un aumento del rischio di malattia. Gli scienziati hanno scoperto che i rischi associati all’uso di queste sostanze chimiche erano più elevati nelle donne giovani e in quelle con cancro al seno ad insorgenza precoce rispetto alle donne non esposte [445].
Engel et al. hanno studiato l’associazione tra l’uso di pesticidi e il rischio di cancro al seno nelle mogli degli agricoltori nello Studio sulla salute in agricoltura del NCI. Questo grande studio prospettico di coorte ha arruolato più di 30.000 donne in Iowa e North Carolina. I ricercatori hanno trovato prove di un aumento dell’incidenza del cancro al seno nelle donne che utilizzano l’acido 2,4,5-triclorofenossipropionico (2,4,5-TP) (RR = 2,0; 95% CI = 1,2-3,2); un’associazione non significativa è stata trovata per dieldrin e captan. L’incidenza è stata anche modestamente elevata nelle donne le cui case erano più vicine alle aree di applicazione dei pesticidi (RR = 1,7; 95% CI = 1,0-1,9) [446].
I bambini piccoli dei contadini che usano il 2,4,5-TP nelle loro fattorie avevano alti livelli di pesticida nei loro campioni di urina subito dopo l’applicazione del prodotto chimico nei campi [447]. Ciò è preoccupante, data l’evidenza di una maggiore suscettibilità dei bambini e dei giovani adolescenti agli effetti cancerogeni delle sostanze chimiche che alterano il sistema endocrino.
Il trattamento delle femmine di ratto con malathion ha portato ad un aumento anormale della proliferazione dei dotti mammari e dell’induzione di tumori mammari quando gli animali sono stati testati a 8 mesi di età [448].
Organoclorurati persistenti
DDT/DDE
Il dicloro-difenil-tricloroetano (DDT) è stato il primo pesticida sintetico ampiamente utilizzato. È accreditato da un lato con l’eradicazione della malaria negli Stati Uniti e in Europa e dall’altro con effetti devastanti a lungo termine sul successo riproduttivo nella fauna selvatica ed effetti negativi per la salute umana [449]. Vietato nella maggior parte dei Paesi per uso agricolo, il DDT è ancora usato per il controllo della malaria in molti Paesi, specialmente nell’Africa subsahariana [450]. A causa del suo uso continuato e della sua persistenza nell’ambiente, il DDT e il suo principale metabolita, il DDE, si trovano in tutto il mondo. La maggior parte degli animali, compresi gli esseri umani, ingerisce il DDT e gli alimenti contaminati dal DDE e ne trattiene le sostanze chimiche. Concentrazioni significative di DDT e DDE si trovano nel grasso corporeo dell’uomo e degli animali, così come nel latte materno e nella placenta, anche in regioni dove non è stato usato per lungo tempo [452- 454].
I dati epidemiologici sono contrastanti per quanto riguarda gli effetti del DDT/DDE sul rischio di cancro al seno [454]. Uno studio caso-controllo dalla Tunisia ha trovato associazioni positive tra i livelli di DDE nel siero e il rischio di cancro al seno (OR = 9,65; 95% CI = 1,81-63,33; trend dose-risposta p = .02). D’altra parte, uno studio del Long Island Breast Cancer Study Project non ha trovato un’associazione tra DDT/DDE (o policlorobifenili, PCB) e cancro al seno [456]. Entrambi questi studi hanno misurato i livelli di contaminanti intorno al momento della diagnosi del cancro al seno, senza considerare le possibili esposizioni durante i periodi critici di sviluppo precoce del seno [457].
Due studi critici che hanno esaminato le esposizioni al DDT nei primi anni di vita (prenatale e infantile) hanno dimostrato una chiara associazione tra le esposizioni al DDT e l’aumento del rischio di sviluppare il cancro al seno [103, 104]. Uno studio prospettico e nidificante su 129 donne a cui era stato diagnosticato un tumore al seno prima dei 50 anni e 129 controlli in base all’età, tutte partecipanti al Child Health and Development Studies (CHDS), ha esplorato i livelli storici stimati di DDT delle donne sulla base di dati aggregati dell’anno di nascita e dei livelli di DDT nel sangue al momento in cui le donne hanno dato alla luce il loro primo figlio. I ricercatori hanno poi monitorato le donne per i successivi 2 decenni, notando quando alle donne è stato diagnosticato un tumore al seno (invasivo o non invasivo) prima dei 50 anni o sono morte di cancro al seno prima dei 50 anni. L’esposizione al DDT durante l’infanzia e la prima adolescenza (meno di 14 anni) è stata associata a un aumento di 5 volte del rischio di sviluppare il cancro al seno prima dei 50 anni [104].
In uno studio prospettico caso-controllo su 9300 donne della coorte CHDS in gravidanza (figlie delle madri della coorte CHDS più grande), sono stati analizzati campioni di sangue materno post-partum memorizzati per i livelli di DDT. Le figlie sono state seguite per 52 anni e la diagnosi di cancro al seno in questa coorte è stata determinata. I livelli di DDT nel sangue perinatale delle madri di casi di tumore al seno sono stati confrontati con i livelli nei campioni di sangue perinatale delle madri di controlli di età corrispondente. Livelli più alti di DDT materno sono stati associati ad un aumento significativo della presenza di cancro al seno nelle loro figlie all’età di 52 anni (OR = 3,7; 95% CI = 1,5-9,0) [103].
Un confronto tra l’associazione tra il rischio di malattia e l’uso del DDT nei paesi sviluppati (dove il DDT è stato vietato per diversi decenni) e nei paesi in via di sviluppo (dove l’uso del DDT è ancora prevalente) sostiene la premessa che le esposizioni al DDT sono associate ad un aumento del rischio di cancro al seno. L’associazione tra i livelli di DDT e il cancro al seno è stata molto più forte nei Paesi in via di sviluppo, dove anche le donne dell’età in cui viene loro diagnosticato il cancro al seno sarebbero state esposte al DDT durante i periodi critici di sviluppo [458].
Uno studio del Long Island Breast Cancer Study Project ha esaminato la mortalità post-diagnosi e i livelli sierici di DDT al momento della diagnosi. Livelli più alti di DDT 5 anni dopo la diagnosi sono stati associati a un aumento della mortalità (HR = 2,72; 95% CI = 1,04-2,12) anche se l’effetto non era significativo a 15 anni dopo la diagnosi [459].
Studi di laboratorio hanno scoperto che oltre ad essere direttamente genotossica o cancerogena [460], la forma estrogeno-simile del DDT migliora la crescita dei tumori mammari ER+ [461- 463]. La percentuale di tumori al seno negli Stati Uniti che sono ER+ è passata dal 73% nel 1973 al 78% nel 1992 [464]. Sebbene non sia possibile dedurre una relazione diretta, questo cambiamento corrisponde al periodo in cui ci si aspettava che le donne esposte al DDT da giovani ragazze mostrassero un’incidenza ambientale alterata nel cancro al seno in relazione all’esposizione al DDT. Un altro studio, che ha esaminato i livelli chimici nel tessuto adiposo del seno, non ha trovato un’associazione del DDT/DDE con i tumori ER+. Tuttavia, i dati di questo studio hanno indicato una significativa associazione di concentrazioni più elevate di questi composti nel tessuto mammario con tumori più aggressivi e con prognosi più sfavorevole (OR = 2,40; 95% CI = 1,0-5,4) [465].
PCB
Sebbene l’EPA abbia vietato l’uso dei PCB nei nuovi prodotti nel 1976, quantità sostanziali di fluidi isolanti, plastica, adesivi, carta, inchiostri, vernici, coloranti e altri prodotti contenenti PCB fabbricati prima del divieto sono rimasti in uso per decenni [466]. Circa un terzo fu scartato, il che significa che questi composti tossici finirono nelle discariche e nelle discariche [467]. I PCB si trovano nell’aria e nelle falde acquifere e nei fiumi, dove si accumulano nei sedimenti, ma poi vengono nuovamente disciolti nell’acqua dove si contaminano e si bioaccumulano attraverso la catena alimentare [468, 469].
I livelli di PCB erano alti prima di essere proibiti negli Stati Uniti, ma in generale la loro presenza nell’ambiente e nei tessuti umani è diminuita lentamente negli ultimi decenni [470, 471]. Choi et al. hanno scoperto che i livelli di PCB nel siero del cordone neonatale erano correlati alla distanza delle abitazioni delle madri da un sito Superfund; i livelli erano più bassi dopo il risanamento del sito [472]. Tuttavia, i livelli di esposizione erano elevati tra l’infanzia e la giovane età adulta per molte donne che ora si trovano ad affrontare la diagnosi di cancro al seno.
Gli oltre 200 singoli PCB sono classificati in tre tipi in base ai loro effetti cellulari. I PCB del gruppo I sono estrogenici; i composti del gruppo II sono anti-estrogenici; e i PCB del gruppo III non sembrano essere ormonalmente attivi, ma possono stimolare i sistemi enzimatici degli animali, compresi gli esseri umani, in modo simile a certi farmaci (come il fenobarbital) e altre sostanze chimiche tossiche [473, 474]. Inoltre, i metaboliti idrossilati dei PCB alterano l’espressione dei geni coinvolti nella sintesi ormonale, indicando che questi composti possono agire come perturbatori endocrini attraverso meccanismi che non coinvolgono direttamente il recettore degli estrogeni [475].
Ci sono diversi studi epidemiologici che hanno implicato l’esposizione ai PCB come fattore di rischio per il successivo sviluppo del cancro al seno. Le donne che mangiavano regolarmente lucci o persici contaminati da PCB avevano un rischio maggiore di cancro al seno rispetto alle donne che non mangiavano mai questi pesci (persici): OR = 7,90; 95% CI = 1,01-61,9; luccio: O =9 .07; 95% CI = 1.10-74.4) [476]. Un altro studio ha coinvolto i PCB nella recidiva del cancro al seno tra le donne con tumore al seno non metastatico. Lo studio ha trovato che le donne con i più alti livelli di PCB totali (RR = 2,91; 95% CI = 1,0-8,2), così come di PCB 118 (RR = 4,0; 95% CI = 1,3-4,9), nei loro tessuti adiposi avevano quasi tre volte più probabilità di avere una recidiva di cancro al seno rispetto alle donne con livelli più bassi [477].
La maggior parte degli studi ha esaminato i livelli totali di PCB senza identificare i singoli tipi. Alcuni studi, tuttavia, hanno esaminato le relazioni tra lo stato del cancro e particolari PCB o gruppi di PCB. Per esempio, una recente meta-analisi ha dimostrato che non c’era alcuna relazione tra le esposizioni ai PCB del gruppo I, ma c’era un rischio significativamente aumentato di cancro al seno con esposizioni ai PCB del gruppo II (OR = 1,23; 95% IC = 1,08-1,40) o del gruppo III (OR = 1,25; 95% CI = 1,09-1,43) [474]. Un altro studio ha esaminato i livelli di PCB nel tessuto mammario di donne prive di malattia e di donne con tumore al seno metastatico. In tutti i campioni esaminati sono stati trovati livelli più alti di PCB del Gruppo III, seguiti dai composti del Gruppo II e poi del Gruppo I. Questi risultati erano indipendenti dallo stato di malattia e non erano associati ad alcuna caratteristica patologica nelle donne a cui era stato diagnosticato il cancro al seno [478]. Una recente meta-analisi specifica per i congeneri ha esaminato l’associazione tra congeneri rappresentativi di tre sottogruppi di PCB e ha rilevato che i congeneri del gruppo III PCB 99 e PCB 183 conferivano un rischio maggiore rispetto ai PCB 187 del gruppo I (rispettivamente OR): 1,36; 95% CI = 1,02-1,80; 1,56; 95% CI = 1,25-1,95; 1,18; 95% CI = 1,01-1,39) [479].
In uno studio caso-controllo, Aronson et al. hanno misurato diversi tipi di PCB, insieme al DDE, in campioni di tessuto di donne sottoposte a biopsia del seno. L’aumento del rischio di cancro al seno è stato associato a concentrazioni più elevate di PCB del Gruppo II 105 e 118, con gli OR per questi due PCB che aumentano linearmente con concentrazioni più elevate (p per trend <0,01) [480].
D’altra parte, alcuni studi non hanno trovato alcun legame tra PCB e cancro al seno [481]. Una revisione della letteratura del 2009 ha concluso che il quadro complessivo era che i PCB, come classe (non considerando i tipi di PCB), non erano associati ad un aumento del rischio di cancro al seno [482]. In uno studio che esaminava l’esposizione professionale ai PCB nelle lavoratrici produttrici di condensatori elettrici e la successiva incidenza del cancro al seno, non è stata osservata alcuna relazione complessiva tra i livelli di esposizione o la durata e l’incidenza della malattia per le lavoratrici in generale. Ma per le donne non bianche, è stata trovata una relazione significativa tra l’incidenza del cancro al seno e la precedente durata dell’esposizione ai PCB, nonché le quantità di esposizione cumulative (confrontando le categorie di esposizione più alte rispetto a quelle più basse, HR = 22,3; 95% CI = 2,38-209) [483]. Più recentemente, Artacho-Cordón non ha trovato alcuna correlazione tra i livelli sierici o adiposi di PCB e il rischio di una diagnosi di cancro al seno [484].
Alcuni di questi composti possono avere il loro maggiore impatto sulle donne con particolari suscettibilità e guardando a grandi campioni non si può dire l’intera storia del rischio di cancro come influenzato dall’esposizione ai PCB. Ad esempio, i ricercatori che hanno valutato i dati dello Studio sulla salute degli infermieri hanno rivisto la questione dei PCB e del rischio di cancro al seno e hanno rivisto le loro conclusioni sul legame tra PCB, DDE e cancro al seno. Negli studi sui PCB e sulla DDE nel sangue, essi avevano precedentemente concluso che l’esposizione a queste sostanze chimiche difficilmente può spiegare gli alti tassi di cancro al seno [485]. Prove più recenti hanno trovato che la complessa interazione tra elevati livelli sierici di PCB e una particolare variante (esone 7) del gene CYP1A1 era associata ad un aumento del rischio di cancro al seno (HR = 2,78; 95% CI = 0,99-7,82, rispetto alle donne con livelli di PCB più bassi e il genotipo wild-type) [486].
Come è stato vero per le critiche degli studi sul DDT sopra citati, i metodi utilizzati per testare queste relazioni non tengono conto dell’esposizione ai PCB durante i primi tempi dello sviluppo, quando il tessuto mammario è particolarmente sensibile agli effetti tossici di molte sostanze chimiche ambientali [487]. I risultati del lavoro di Cohn sul DDT e sul cancro al seno chiariscono che si tratta di una questione metodologica critica [457].
Diossine
Le diossine si formano dall’incenerimento di prodotti contenenti PVC, PCB e altri composti clorurati, nonché da processi industriali che utilizzano cloro e dalla combustione di gasolio e benzina [488]. Una delle diossine (2,3,7,8-tetra clorodibenzo-para-diossina; TCDD) è stata classificata dallo IARC [489] e dall’U.S. EPA [490] come cancerogena.
Le diossine si accumulano nel grasso corporeo della fauna selvatica e dell’uomo, e si scompongono molto lentamente, con un’emivita di 7-11 anni nei tessuti corporei [491]. Le persone sono esposte alle diossine principalmente attraverso il consumo di prodotti animali e di latte materno umano [488, 489]. La diossina entra nella catena alimentare quando i gas di scarico dei veicoli o la fuliggine dei composti clorurati inceneriti cadono sulle coltivazioni dei campi che poi vengono mangiate dagli animali da allevamento. Viene poi trasmessa all’uomo attraverso i prodotti caseari e i prodotti a base di carne. Si pensa che il grasso corporeo di ogni essere umano, incluso ogni neonato, contenga diossina [492].
C’è una sostanziale diminuzione della quantità di diossina che rimane nel tessuto adiposo del seno di una donna dopo l’allattamento, perché le sostanze chimiche sono state trasmesse al neonato attraverso il latte materno [493]. Sebbene la presenza di sostanze chimiche tossiche nel latte materno sia potenzialmente pericolosa, si ritiene che le sostanze nutritive benefiche e gli stimolatori del sistema immunitario che vengono trasferiti dalla madre al neonato siano di gran lunga superiori ai potenziali trasferimenti tossici [494]. Ma oltre al potenziale trasferimento di diossine ai neonati che allattano al seno, il rilascio delle sostanze chimiche dalla conservazione nelle cellule di grasso mammario, iniziato dal processo di sintesi del latte, può effettivamente innescare effetti genotossici nel tessuto mammario della madre. L’aggiunta di estratti di latte materno alle cellule MCF-7 ha portato ad una riprogrammazione dell’espressione genica ad un modello che si trova tipicamente in seguito alla stimolazione degli estrogeni, specialmente nei geni CYP1A1 e CPY1B1 [495].
Uno studio su donne esposte alla diossina durante l’esplosione di un impianto chimico nel 1976 a Seveso, in Italia, ha dimostrato un’associazione dipendente dal tempo tra l’esposizione alla diossina e il cancro al seno [496, 497]. Un aumento di dieci volte dei livelli di TCDD nei campioni di sangue prelevati al momento dell’esplosione è stato associato a più del doppio del rischio di cancro al seno nelle donne che, nel 1996, avevano in media 40 anni (HR = 2,1; 95% CI = 1,0-4,6) e il cui cancro al seno era stato diagnosticato prima della menopausa. Il follow-up della coorte nel 2008 ha rivelato un aumento non significativo del rischio di sviluppare il cancro al seno tra il momento in cui le donne avevano 40 anni e 12 anni dopo, quando avevano 52 anni, in media (HR = 1,44; 95% CI = 0,89-2,33). Per tutti i casi di cancro al seno per i quali esistevano dati di profilo tumorale, più dell’80% erano tumori ER+/PR+ [497]. Il follow-up continuo di questa coorte esaminerà il rischio di sviluppare il cancro al seno man mano che le donne entrano e continuano negli anni post-menopausa.
Uno studio retrospettivo sulla mortalità in Germania ha esaminato i decessi per cancro tra le persone che avevano lavorato in una fabbrica chimica in cui erano state esposte ad alti livelli di TCDD. Rispetto ai tassi di mortalità nazionali della Germania occidentale, 5 anni dopo la chiusura dello stabilimento, non si è registrato un aumento della mortalità complessiva per cancro per le lavoratrici, anche se c’è stato un aumento significativo dei decessi per cancro al seno tra le persone che hanno lavorato in regioni ad alta esposizione della fabbrica (SMR = 2,15) [498]. Un follow-up successivo, 23 anni dopo la chiusura dello stabilimento, ha riscontrato un tasso di mortalità inferiore per tutte le cause per le lavoratrici dello stabilimento (SMR = 0,91; 95% CI = 0,78-1,05), ma un aumento significativo della mortalità per cancro al seno (SMR = 1,86; 95% CI = 1,12-2,91) in questa coorte [499].
Diversi studi di laboratorio hanno dimostrato che la tempistica delle esposizioni alle diossine è importante. Sebbene l’esposizione degli animali alle diossine in età adulta possa non influenzare i tassi di cancro, le esposizioni precedenti possono avere effetti profondi. La somministrazione di di diossine (specialmente TCDD) a ratti gravidi porta ad anomalie strutturali nello sviluppo dei tessuti mammari dei loro cuccioli e ad una maggiore incidenza di tumori quando i cuccioli raggiungono l’età adulta [500- 504]. La TCDD può esercitare i suoi effetti cancerogeni sia diminuendo l’efficacia dei meccanismi di soppressione dei tumori, sia potenziando la segnalazione estrogenica all’interno delle cellule mammarie [505].
Il TCDD ha dimostrato di inibire la crescita e la proliferazione delle cellule indotte dall’estradiolo e altre vie regolate dagli estrogeni, compresa la metilazione del CYP1A1, in una varietà di linee di coltura di cellule tumorali della mammella umana [506, 507]. Come gli IPA descritti sopra, le diossine come il TCDD esercitano i loro effetti attivando sia il recettore degli idrocarburi arilici (AhR), un importante mediatore della crescita e della proliferazione cellulare [508], sia le vie anti-apoptosi [509], così come altre vie indipendenti dall’AhR, comprese le vie mediate dalle PR [506, 510, 511]. Prove crescenti indicano che molti degli effetti della TCDD e di altre diossine sono il risultato di un dialogo incrociato tra i sistemi AhR ed ER, PR e persino AR [512, 513].
Il trattamento prenatale dei ratti con basse dosi di TCDD ha portato ad un aumento del numero di gemme terminali (TEB) contate nelle preparazioni di montatura intera del giorno postnatale di 71 femmine. Il trattamento ha anche portato ad una soppressione dell’espressione BRCA-1, risultato dell’aumento dell’ipermetilazione del promotore BRCA-1 [514].
Ritardanti di fiamma polibromurati di etere di difenile (PBDE)
I PBDE sono un gruppo complesso di sostanze chimiche strutturalmente simili ai policlorobifenili (PCB). I prodotti contenenti PBDE comprendono la schiuma poliuretanica nei mobili (penta-BDE) e i prodotti elettronici e plastici (octa- e deca-BDE) [515].
Sebbene sia il penta-BDE che l’octa-BDE siano stati vietati nell’Unione Europea e non siano più prodotti negli Stati Uniti dal 2004, i prodotti che li contengono rimangono in tutto il mondo. I PBDE si trovano onnipresenti nell’ambiente e vengono rilevati nell’aria, nella polvere, nel suolo, nel cibo e in molte specie selvatiche. Sebbene le esposizioni domestiche (misurate in base ai livelli di polvere) siano diminuite nell’ultimo decennio, i livelli rimangono abbastanza alti da rimanere una seria preoccupazione per la salute [516].
C’è una notevole variabilità geografica nelle esposizioni alle sostanze chimiche; le persone in California, con i suoi standard di infiammabilità dei mobili storicamente rigorosi, hanno livelli di esposizione al PBDE molto più alti rispetto alle persone in Massachusetts. All’interno del gruppo della California, uno status socioeconomico inferiore è stato associato a livelli più alti di PBDE [488, 517]. I messicani americani che vivono in California hanno livelli significativamente più alti di PBDE nel siero del sangue rispetto ai messicani che vivono nella loro patria [518].
I dati delle giovani ragazze (dai 6 ai 9 anni) della California e dell’Ohio confermano questi risultati. Sebbene i PBDE siano stati trovati in quasi tutti i campioni esaminati, le ragazze californiane avevano livelli di PBDE nel sangue significativamente più alti rispetto alle ragazze dell’Ohio, e le giovani ragazze afroamericane di colore avevano livelli più alti rispetto alle ragazze bianche o ispaniche [519]. In queste coorti, le esposizioni al PBDE sono associate a ritardi nella pubertà nelle ragazze adolescenti (RR = 1,05; 95% CI = 1,02-1,08), un effetto non moderato dall’aggiustamento dell’IMC [520].
I PBDE sono stati trovati nel tessuto adiposo umano, così come nel siero del sangue, nel tessuto mammario e nel latte [521- 523]. I PBDE attraversano la placenta, con conseguente esposizione ai feti in via di sviluppo [524]. I PBDE sono composti che alterano il sistema endocrino, esercitando effetti su una serie di sistemi ormonali, compresi gli androgeni, i progestinici e gli estrogeni, anche se il sistema principale interessato dai PBDE è il sistema ormonale tiroideo [486]. La maggior parte degli studi sui risultati sanitari dopo l’esposizione a PBDE si sono concentrati sullo sviluppo neurale, dato il ruolo preminente degli ormoni tiroidei (specialmente il T4) nella regolazione dello sviluppo cerebrale [525, 526].
Pochissimi dati riguardano direttamente i possibili effetti dei PBDE sul rischio di cancro al seno. Uno studio caso-controllo non ha trovato alcuna relazione tra i livelli di PBDE nel grasso del seno e il rischio di cancro al seno, ma il campione era piccolo e l’analisi chimica è stata fatta intorno al momento della diagnosi del cancro al seno nelle donne che hanno sviluppato la malattia [527]. Tuttavia, almeno alcuni PBDE si sono dimostrati efficaci quanto molti degli altri composti che alterano il sistema endocrino nel promuovere la proliferazione di tipo estrogenico delle cellule umane del cancro al seno in vitro [528]. Il penta-BDE migliora la proliferazione delle cellule tumorali nelle cellule MCF-7 attraverso effetti simili agli estrogeni sulle vie cellulari che interrompono l’apoptosi [529]. Gli effetti proteiferativi e antiapoptotici dei PBDE sono additivi con quelli dell’estradiolo naturale [530] e contrastano gli effetti antitumorali del tamoxifene nelle cellule del tumore al seno in coltura [531]. Data l’ampia sovrapposizione e l’interazione di risposte estrogeno-mediate e tiroide-mediate nella regolazione del cancro al seno [532], i PBDE saranno una classe di sostanze chimiche di continua preoccupazione per gli scienziati interessati a comprendere i legami ambientali con il cancro al seno [533].
Anche se i PBDE sono utilizzati meno spesso come ritardanti di fiamma nei comuni prodotti di consumo, è ora dimostrato che le sostanze chimiche utilizzate come sostituti – tra cui Firemaster 550®, un comune sostituto con ingredienti proprietari – stanno contaminando sempre più il nostro ambiente [489, 534]. Anche se gli effetti fisiologici delle esposizioni a Firemaster 550® non sono ancora stati studiati a fondo, uno studio ha dimostrato che l’alimentazione delle dighe per ratti a basse dosi durante la gravidanza e l’allattamento ha portato a cambiamenti associati all’esposizione ad altri composti che alterano il sistema endocrino. Questi effetti includevano cambiamenti nei livelli di ormoni tiroidei nelle madri, e cambiamenti nel comportamento, nell’aumento di peso e nella pubertà precoce nei cuccioli di femmina [507].
Ammine aromatiche
Le ammine aromatiche sono una classe di prodotti chimici che si trovano nelle industrie plastiche e chimiche come sottoprodotti della produzione di composti come schiume poliuretaniche, coloranti, pesticidi, prodotti farmaceutici e semiconduttori [535]. Si trovano anche nell’inquinamento ambientale come i gas di scarico del diesel, la combustione di trucioli di legno e gomma, il fumo di tabacco e la carne e il pesce alla griglia [536, 537]. Esistono tre tipi di ammine aromatiche: monociliche, policicliche ed eterocicliche.
Tre ammine monocicliche, tra cui l’o-toluidina, sono state identificate nel latte materno di donne in allattamento sane [536], così come nelle urine della maggior parte delle persone [535]. σ-Toluidina è nota per causare tumori mammari nei roditori [538, 539]. Le ammine aromatiche cancerogene, la 2-amino-fenilimidazo [4,5-b]piridina (PhIP) e 4-aminodifenile (ABP) si trovano anche nel latte materno umano, così come i DNA-addotti di questi composti [540].
L’esposizione professionale delle lavoratrici della fabbrica di gomma ad un’altra serie di ammine aromatiche monocicliche derivate dalla p-fenilendiammina è associata ad un aumento del rischio di cancro al seno nei prossimi anni. L’ammontare dell’aumento del rischio è stato correlato con i livelli di esposizione cumulativi totali alle ammine aromatiche, con livelli più bassi che portano ad un aumento di 3,7 volte il rischio di cancro e i livelli più alti di esposizione che aumentano il rischio di più di dieci volte [541].
Uno studio di controllo dei casi di donne che hanno usato tinture per capelli, rispetto a quelle che non l’hanno fatto, ha rivelato un aumento del rischio di cancro al seno nelle utilizzatrici di tinture (OR = 1,15; 95% CI = 1,06-1,24). Oltre all’inclusione intenzionale di p-fenilendiammina [542], i principali contaminanti in molte tinture per capelli includono PhIP e ABP [543].
Le ammine aromatiche eterocicliche (HAA) si formano (insieme agli IPA) quando le carni o il pesce vengono grigliati o altrimenti cotti ad alte temperature. In uno studio di controllo del caso, Steck et al. hanno trovato un’associazione tra un maggiore consumo di carne e pesce alla griglia per tutta la vita e una maggiore incidenza di cancro al seno in post-menopausa (OR = 1,42; 95% CI = 1,12-1,92) [544]. Studi sia sul latte che sulle cellule dei dotti mammari delle donne hanno rivelato la presenza di addotti del DNA in associazione con l’HAA [545, 546].
Le ammine aromatiche sono metabolizzate da N-acetiltrasferasi. Questo processo metabolico porta alla fine ad altri composti che si pensa siano i veri e propri prodotti chimici cancerogeni. Esiste un’ampia letteratura che esamina se i profili genetici che alterano l’efficacia o la velocità dell’attivazione della N-acetiltrasferasi, in particolare attraverso il percorso regolato dalla N-acetiltrasferasi 2 (NAT2), possano alterare la suscettibilità al cancro al seno. Gli studi hanno raggiunto conclusioni diverse sul ruolo dei possibili polimorfismi del gene NAT2 sulla suscettibilità al cancro al seno. Uno studio del 2010 ha cercato di districarsi tra molti dei possibili fattori di confusione e ha scoperto che mangiare carne alla griglia (e bere caffè) ha comportato un rischio maggiore per la diagnosi di tumori al seno in donne con la forma “a lenta acetilazione” del gene NAT2 [547].
Studi di laboratorio di HAA in sistemi che utilizzano cellule tumorali del seno in coltura hanno dimostrato che queste sostanze chimiche possono imitare gli estrogeni, e possono anche avere effetti diretti sui processi di divisione cellulare in modi che potrebbero migliorare lo sviluppo dei tumori [548].
Metalli
Nelle biopsie del seno cancerose sono stati riscontrati accumuli più elevati di ferro, nichel, cromo, zinco, cadmio, mercurio e piombo, rispetto alle biopsie effettuate su donne senza cancro al seno. Questi metalli sono stati trovati anche in concentrazioni più elevate nel siero e nelle urine di donne con diagnosi di cancro rispetto a quelle di donne sane [549- 552].
Studi di laboratorio hanno dimostrato che un certo numero di metalli tra cui rame, cobalto, nichel, piombo, mercurio, metilmercurio, stagno, cadmio e cromo hanno effetti estrogenici sulle cellule tumorali della mammella (MCF-7) coltivate in vitro [553- 555], con il cadmio che esprime il più alto livello di attività estrogenica [555]. Il lavoro più esteso sulla relazione tra il cancro al seno e i metalli è stato fatto esaminando questo metallo-estrogeno, il cadmio.
Diversi studi epidemiologici hanno dimostrato un’associazione tra i livelli più alti di cadmio nelle urine (OR = 2,29; 95% CI = 1,3-4,2) [556, 557] o nel sangue [558] e l’aumento del rischio di cancro al seno, anche se in un ampio studio prospettico che utilizza la coorte WHI non è stata trovata alcuna associazione tra i livelli di cadmio nelle urine e il rischio di sviluppare il cancro al seno [559]. Ciononostante, una recente meta-analisi ha riportato un significativo aumento del rischio di sviluppare il cancro al seno con l’aumento dei livelli di cadmio urinario (OR = 2,24; 95% CI = 1,49-3,35) [560].
Le differenze nell’efficacia di stabilire relazioni tra il cancro al seno e le esposizioni al cadmio, determinate dalle misure dietetiche rispetto a quelle urinarie, possono riflettere l’osservazione che i livelli di cadmio urinario sono un marker più forte delle esposizioni al metallo nel corso della vita, dato il tempo di dimezzamento del cadmio di 12-30 anni, mentre i livelli di esposizione dietetica riflettono un marker a più breve termine e potenzialmente più variabile [560].
Studi prospettici sull’assunzione di cadmio da parte delle donne con la dieta e la successiva diagnosi di tumori hanno dimostrato una relazione significativa tra i livelli più elevati di esposizione al cadmio con la dieta e l’incidenza di entrambi i tumori endometriali (RR = 1.39; 95% CI = 1,04-1,86) [561] e tumori del seno in post-menopausa (RR = 1,21; 95% CI = 1,07-1,36) [562]. Per quanto riguarda il cancro al seno, l’effetto è stato significativo per tutti i sottotipi combinati, ma più pronunciato per i tumori ER+ (OR = 1,94; 95% CI = 1,04-3,63 [563]; RR = 1,23; 95% CI = 1,02-1,49) [562]. D’altra parte, gli studi che esaminano l’assunzione di cadmio nella dieta delle donne giapponesi [563, 564] e danesi [565] e il loro rischio di sviluppare il cancro al seno non hanno trovato alcuna relazione. Uno studio del 2012 negli Stati Uniti che ha esaminato i livelli di cadmio nella dieta e il rischio di cancro al seno non ha trovato una relazione significativa [566], né due recenti meta-analisi che hanno studiato questa relazione [567, 568].
Nei ratti giovani, il trattamento con basse dosi di cadmio ha portato ad un aumento della ramificazione e della formazione di gemme nel tessuto mammario, e l’induzione di diverse proteine associate agli estrogeni. L’esposizione prenatale dei ratti al cadmio ha portato all’inizio precoce della pubertà e ad un maggior numero di gemme terminali mammarie, entrambi fattori di rischio noti per il cancro al seno [569].
Gli effetti estrogenici del cadmio sono stati studiati in dettaglio, ed è stato dimostrato che il metallo interferisce con una serie di normali vie sensibili agli estrogeni [570]: il cadmio può legarsi e attivare i recettori degli estrogeni delle cellule mammarie; inoltre interagisce e regola la trascrizione dei geni estrogeno-dipendenti che influenzano la sintesi delle proteine e/o l’attività delle vie di segnalazione delle cellule in modo simile all’estrogeno naturale, l’estradiolo [570, 571]. L’esposizione al cadmio può anche portare alla trasformazione maligna delle normali cellule epiteliali del seno umano (MCF-10A) attraverso un meccanismo che non richiede la presenza di ERα [572]. Altri studi sostengono la possibilità che il cadmio possa anche esercitare effetti cellulari attraverso meccanismi che vanno oltre le vie convenzionali dirette al nucleare-ER [573, 574], anche eventualmente attraverso il legame con il recettore di membrana, GPR30 [575].
Oltre agli effetti ormonali mediati, il cadmio può anche promuovere lo sviluppo del cancro attraverso processi epigenetici che includono cambiamenti nei modelli di metilazione del DNA e possibili modifiche degli istoni associati ai geni [576].
Come molti altri interferenti endocrini che imitano gli estrogeni, il cadmio interferisce con l’efficacia di un comune agente chemioterapico spesso prescritto alle donne a cui è stato diagnosticato un tumore al seno. In uno studio che esamina gli effetti del cadmio, del 5-fluorouracile (5-FU) e dei due combinati sulla struttura subcellulare e sull’attività metabolica nelle cellule coltivate di tumore al seno MCF-7, la co-somministrazione di cadmio ha annullato gli effetti anti-cancro del 5-FU [577].
Sintesi della sezione: la crescente letteratura sulle esposizioni agli EDC, soprattutto nelle prime fasi di sviluppo, indica un aumento del rischio di sviluppare il cancro al seno in seguito all’esposizione a molti di questi composti, da soli o in combinazione. I dati epidemiologici umani più significativi a sostegno di questa relazione provengono da studi prospettici su donne esposte al DDT durante la gestazione o la prima infanzia e da un aumento dello sviluppo del cancro al seno in premenopausa [103, 104]. La letteratura non umana più vasta collega le esposizioni precoci e a basse dosi di BPA ad un aumento del rischio di sviluppare tumori mammari. Sia gli studi in situ che quelli in vitro hanno contribuito in modo sostanziale alla nostra comprensione dei complessi meccanismi alla base di queste relazioni. Anche se non così solidi come per le sostanze chimiche di cui sopra, sono stati documentati anche i collegamenti tra le esposizioni, soprattutto nelle prime fasi di sviluppo, e molti altri EDC.
Ormoni negli alimenti: naturali e additivi
Le prove prevalenti contro gli estrogeni sintetici devono essere comprese insieme alle prove sugli effetti degli estrogeni vegetali (fitoestrogeni) sul rischio di sviluppare il cancro al seno. Mentre la maggior parte della ricerca in questo settore si è concentrata sulle possibili associazioni protettive con gli isoflavoni a base di soia in un regime alimentare normale, una crescente letteratura sta anche esaminando i potenziali effetti protettivi dei lignani, dell’enterolattone e dell’acido α-linolenico.
I micoestrogeni (estrogeni che si trovano nelle specie fungine) possono contaminare i prodotti agricoli e a base di carne, e questa contaminazione può aumentare la suscettibilità allo sviluppo del cancro al seno. Inoltre, l’esposizione ai composti che favoriscono la crescita dati agli animali da carne e da latte è stata collegata ad un aumento del rischio di sviluppare il cancro al seno. Questa sezione esplora i complicati profili di esposizione agli estrogeni alimentari e il rischio di sviluppare il cancro al seno. Non vi è stata alcuna derminazione sulla potenziale cancerogenicità di queste sostanze da parte di IARC o NTP.
Fitoestrogeni (estrogeni vegetali)
Gli alimenti come i cereali integrali, i fagioli secchi, i piselli, la frutta, i broccoli, il cavolfiore e soprattutto i prodotti a base di soia sono ricchi di fitoestrogeni. Mentre la maggior parte della ricerca in questo settore si è concentrata sulle possibili associazioni con gli isoflavoni a base di soia. Inoltre, c’è anche una crescente letteratura che sta esaminando il possibile ruolo dei lignani, compresi l’enterolattone e l’acido α-linolenico, nell’influenzare il rischio di cancro al seno.
Lignani
I lignani sono composti polifenolici che si trovano ampiamente nei semi e nei grani comuni ad una dieta occidentale. Essi sono rapidamente metabolizzati nell’intestino ai composti estrogenici, enterolattone ed enterodiolo [578].
Uno studio caso-controllo di casi combinati di donne in premenopausa nella coorte dello Studio sulla salute degli infermieri II non ha trovato alcuna relazione complessiva tra i livelli plasmatici di enterolattone e il rischio di cancro al seno. Tuttavia, in quelle donne i cui livelli di estradiolo a circolazione follicolare erano al di sotto della mediana, un enterolattone più alto è stato associato ad una significativa riduzione del rischio di cancro al seno (OR = 0,49; 95% CI = 0,27-0,91) [579]. Una meta-analisi che esamina la possibile relazione tra i livelli sierici di enterolattone e la mortalità totale o la mortalità associata al cancro al seno ha trovato correlazioni negative tra i livelli di lignan in circolazione ed entrambe le misure di esito (HR = 0.57; 95% CI = 0,42-0,67 per la mortalità per tutte le cause e HR = 0,54; 95% CI = 0,39-0,75 per la mortalità per cancro al seno [580].
Un recente esame dei possibili meccanismi alla base dell’effetto protettivo dell’enterolattone ha utilizzato la linea cellulare aggressiva e altamente invasiva MDA-MB-321 per il cancro al seno umano. L’aggiunta del lignano alla coltura cellulare ha portato ad una diminuzione dell’attività nei livelli di mRNA di diversi geni associati alla proliferazione cellulare, così come ad una maggiore ritenzione delle cellule mitotiche nella fase S, e ad una diminuzione della migrazione e dell’invasione attraverso l’interferenza con il citoscheletro cellulare [580].
Una revisione sistematica della letteratura sulle associazioni tra l’acido α-linolenico e il rischio di cancro al seno ha trovato significative relazioni negative tra una maggiore assunzione di lino, una delle principali fonti di acido α-linolenico, e l’incidenza del cancro al seno (OR = 0.82; 95% CI = 0,69-0,97) e la mortalità (HR = 0,69; 95% CI = 0,50-0,95) [581]. Nelle donne con diagnosi recenti di tumore al seno, una maggiore assunzione di lino è stata associata a profili tumorali meno aggressivi che avevano indici apoptotici più alti, e una minore espressione di HER2 e tassi proliferativi più bassi [581].
Soia e derivati della soia
Sebbene le prove scientifiche suggeriscono che gli alimenti derivati dalla soia offrono benefici nutrizionali e sono associati a diete sane [582, 583], i dati sono più contrastanti sul fatto che le diete a base di soia siano benefiche, dannose o neutre quando si tratta di influenzare il rischio di cancro al seno [584, 585].
Alcune delle disparità possono essere correlate al tipo di prodotti a base di soia consumati dagli individui. Nelle diete asiatiche e occidentali, la soia può essere trattata in decine di modi diversi, con conseguenti differenze fino a 100 volte superiori nei livelli di particolari fitoestrogeni tra i prodotti [583] e livelli di esposizione molto diversi per i consumatori. È stato dimostrato che le diete ricche di prodotti che contengono livelli più elevati sia di isoflavone di soia, genisteina, sia del suo metabolita genistina, influenzano la crescita del tumore al seno in diversi modelli. Al contrario, la farina di soia altamente lavorata che non contiene isoflavoni non ha alcun effetto. Gli isolati di proteine di soia purificate sono spesso elaborati per contenere diverse concentrazioni di isoflavoni, e la loro influenza sui tumori mammari è legata alla quantità di isoflavone fitoestrogeno, non alla quantità totale di proteine di soia consumate [586].
Diversi studi epidemiologici hanno dimostrato che il consumo regolare di prodotti a base di soia, o di altre verdure ad alto contenuto di fitoestrogeni, nell’ambito di una normale dieta equilibrata, può esercitare un’influenza protettiva per quanto riguarda il successivo sviluppo del cancro al seno. Questo effetto è stato ampiamente studiato in Cina, dove l’assunzione di soia è una parte regolare della dieta culturale. Lì, prove sostanziali indicano che una maggiore assunzione di soia in età adulta o nell’adolescenza è associata a una diminuzione del rischio di cancro al seno in pre-menopausa (OR = 0,41; 95% CI = 0.21-0.70 per l’assunzione di soia; OR = 0.44; 95% CI = 0.26-0.73 per l’assunzione di isoflavone [587]; OR = 0.68; 95% CI = 0.50-.93 per l’assunzione di soia) [588]. Altri studi hanno trovato effetti protettivi dell’assunzione di soia per il cancro sia prima che dopo la menopausa, indipendentemente dal profilo del recettore (ER e PR positivo o negativo) dei tumori (OR che vanno da 0,30 a 0,43) con una relazione inversa dose-dipendente trovata attraverso sottotipi di cancro (tendenza p < .0001) [589].
Per le donne cinesi a cui era stato precedentemente diagnosticato il cancro al seno, un maggiore consumo di soia nelle sue numerose forme che si trovano regolarmente nella dieta di una donna, è stato correlato con una diminuzione delle recidive di cancro (OR = 0,68; 95% CI = 0,54-0,92) e una maggiore sopravvivenza (Or = 0,71; 95% CI = 0,54-0,87) [590]. A complicare ulteriormente il quadro è uno studio sulle donne coreane a cui era stato precedentemente diagnosticato e trattato il cancro al seno. L’assunzione di soia dietetica è stata associata a una diminuzione del tasso di ricorrenza nelle donne i cui tumori erano HER-2 negativi (OR = 0,27; 95% CI = 0,13-0,57), e a un aumento del tasso di ricorrenza del cancro nelle donne i cui tumori originali erano HER-2 positivi (tendenza p < .02) [591].
D’altra parte, in uno studio prospettico su donne di età compresa tra 43 e 55 anni che non avevano mai avuto una diagnosi di cancro al seno ma che erano considerate ad alto rischio, l’assunzione di 6 mesi di isoflavone alimentare (PTIG-2535, contenente 150 mg di genisteina, 74 mg di daidzeina e 11 mg di gliciteina) è stata associata ad un aumento della proliferazione delle cellule del seno. L’effetto è stato più pronunciato nelle donne in pre-menopausa [592].
Una serie di rapporti dal Giappone che esaminano la relazione tra l’assunzione di soia e il rischio di cancro al seno hanno trovato una relazione inversa [593- 595], con forti relazioni dose-risposta che sono state trovate per le donne in postmenopausa (tendenza, p = .023 per l’assunzione di soia e tendenza p = .046 per l’assunzione di isoflavone) [596].
Uno studio su donne asiatiche-americane che vivono in California e nelle Hawaii ha trovato significative diminuzioni del rischio di cancro al seno associate ad una maggiore assunzione di soia durante l’infanzia (RR = 0.40 95% CI = 0,18-,83), adolescenza (RR = 0,80 95% CI = 0,59-1,09), o età adulta (RR = 0,76 95% CI = 0,56-1,02) [597]. L’effetto protettivo dell’assunzione regolare di soia durante l’infanzia è stato il più forte, e non è stato attenuato quando altre variabili come il luogo di nascita (paesi asiatici o Stati Uniti), il grado di continuità dello stile di vita e delle pratiche culturali asiatiche, i fattori riproduttivi o la storia familiare del cancro al seno sono stati presi in considerazione nell’analisi. In generale, gli effetti protettivi dell’assunzione di soia con l’alimentazione sono risultati essere più forti in associazione con l’assunzione nell’infanzia e nella prima adolescenza [598], soprattutto in relazione allo sviluppo del cancro al seno in post-menopausa ER+/PR+ (OR = 0,79; 95% CI = 0,65-0,96) [599]. Una possibile spiegazione di questa associazione è che le esposizioni peri-pubertali alla genisteina e ad altri fitoestrogeni possono imitare i cambiamenti protettivi nello sviluppo del seno che si osservano di solito durante la prima gravidanza [600, 601].
Diversi studi hanno confrontato direttamente gli effetti del consumo di diete a base di soia culturalmente appropriate nelle donne asiatiche e occidentali. Una meta-analisi del 2012 che ha combinato i dati di sei studi ha trovato che l’assunzione regolare di soia durante l’adolescenza ha diminuito l’incidenza di tutti i tumori al seno successivi (OR = 0,82 95% CI = 0,67-0,99), ed è stata particolarmente efficace nel diminuire l’incidenza del cancro nelle donne in pre-menopausa (OR = 0,66 95% CI = 0,55-0,80). Non è stata riportata alcuna differenza negli effetti dell’assunzione di soia durante l’adolescenza tra donne asiatiche e americane/europee [602]. D’altra parte, una meta-analisi del 2014 che esamina l’assunzione di isoflavone nelle donne in pre e post-menopausa provenienti da paesi asiatici e occidentali ha trovato effetti protettivi della soia in entrambe le fasi della premenopausa (OR = 0,59; 95% CI = 0.48-0,69) e in post-menopausa (OR = 0,59; 95% CI = 0,47-0,74) donne asiatiche, con solo effetti molto piccoli e non significativi in entrambe le donne occidentali in premenopausa e post-menopausa [603]. Uno studio multietnico condotto alle Hawaii nel 2009 ha dimostrato che la quantità di soia nella dieta potrebbe interagire con le associazioni tra altri fitoestrogeni e la protezione contro il cancro al seno. Per i giapponesi americani che avevano un alto contenuto di soia nella loro dieta regolare, c’era un forte, significativo ma non monotono rapporto per i livelli di genisteina urinaria entro i due quartili medi (OR = 0,88; 95% CI = 0,78-0,99) con un rischio ridotto di cancro al seno. Una simile forte relazione non è stata trovata per le donne bianche nello studio che tendevano a mangiare diete a basso contenuto di soia [604]. Le differenze nelle risposte tra donne asiatiche e occidentali possono riflettere sia differenze nel contenuto della dieta sia differenze culturali nella capacità di metabolizzare gli isoflavoni, un effetto che può derivare sia da differenze genetiche sia da interazioni con altri fattori dietetici [584].
I dati degli studi sugli animali da laboratorio e sui modelli di coltura cellulare hanno indicato una storia più complicata. In diversi studi, le esposizioni ai fitoestrogeni hanno portato ad un aumento della proliferazione e della crescita dei tumori mammari. Nei ratti ACI, l’esposizione alimentare al contenuto totale di isoflavone della soia dal concepimento fino all’età adulta ha diminuito l’incidenza dei tumori mammari negli animali adulti del 20% e la molteplicità del 56%, diminuendo anche la latenza all’insorgenza del tumore del 20% e quasi triplicando i volumi tumorali [605]. L’esposizione dei ratti Wistar alla genisteina dal concepimento attraverso lo svezzamento ha portato a una diminuzione del numero di tumori indotti dal DMBA, della molteplicità e dell’incidenza al giorno postnatale 50 [606].
I fitoestrogeni di soia, la genisteina e la daidzeina, così come i loro metaboliti, causano danni ossidativi al DNA, un processo che si pensa abbia un ruolo nell’insorgenza del tumore. Altri dati suggeriscono che questi due fitoestrogeni a base di soia possono avere effetti opposti sull’efficacia del farmaco per il cancro al seno, il tamoxifene [607, 608].
Gli effetti dei fitoestrogeni possono essere correlati ai particolari componenti e alle dosi della dieta [609], e gli effetti cellulari possono variare a seconda della concentrazione e della tempistica. In uno studio che esamina gli effetti di diversi tipi e concentrazioni di fitoestrogeni sull’espressione dell’attività genica estrogeno-dipendente nelle cellule tumorali della mammella umana cresciute in vitro (cellule MCF-7), basse dosi di genisteina hanno portato ad un modello di espressione che indicava un aumento della proliferazione cellulare, mentre concentrazioni un po’ più alte hanno portato ad un aumento dell’apoptosi [610].
Gli isoflavoni contenuti nella farina di soia naturale hanno effetti diversi rispetto all’aggiunta di isoflavoni purificati dalla farina di soia sui modelli di espressione genica nei tumori indotti cresciuti da cellule MCF-7 nei topi nudi atimici. Quando i topi sono stati alimentati con isoflavoni isolati, il modello di espressione genica era simile a quello trovato nei topi che erano stati trattati con estradiolo, mentre il modello in animali trattati con isoflavoni inclusi all’interno di una farina di soia completa erano più come il controllo negativo, suggerendo un effetto inibitorio delle proteine della farina di soia su alcuni degli effetti proliferativi degli isoflavoni isolati [611]. Oltre ad alterare i modelli genici associati alla proliferazione cellulare e alla carcinogenesi, l’esposizione alla genisteina è anche un forte inibitore dell’angiogenesi, un importante processo associato alla crescita tumorale e alle metastasi. D’altra parte, 30 giorni di trattamento con gavage di ratti peripubertali e adulti ovariectomizzati con integratori di isoflavone non hanno avuto alcun effetto sulla proliferazione cellulare o l’angiogenesi [612].
Nelle cellule coltivate MCF-7, il fitoestrogeno di soia daidzeina leggermente migliorata la proliferazione cellulare in assenza di estrogeni naturali (un possibile modello per il cancro al seno in post-menopausa), mentre il resveratrolo (che si trova in uva e vino rosso) ha diminuito significativamente la proliferazione delle cellule tumorali [602]. Questi ultimi dati sono coerenti con altri studi che trovano effetti anti-cancerogeni del resveratrolo in diversi modelli [613, 614].
È stata sollevata la preoccupazione per l’esposizione dei neonati ai prodotti a base di soia, principalmente attraverso le formule per neonati. Sebbene uno studio abbia dimostrato che l’alimentazione con sole formule a base di soia per i primi 4 mesi di vita è stata associata ad una diminuzione dello sviluppo successivo del cancro al seno [615], studi su animali hanno indicato effetti deleteri dell’esposizione neonatale alla soia sullo sviluppo del sistema riproduttivo femminile e sulla successiva fertilità [616].
Micoestrogeni (estrogeni fungini)
Le micotossine sono composti prodotti da diverse specie fungine che contaminano i prodotti agricoli e i mangimi, tra cui l’insilato di mais e il fieno, sia prima del raccolto che durante il successivo stoccaggio [617- 619]. Le persone sono esposte a questi composti direttamente, mangiando cereali contaminati dai funghi, e indirettamente, mangiando carne proveniente da animali che hanno consumato mangimi contaminati [620].
La contaminazione degli alimenti da parte dello zearalenone (ZEA) e dei suoi metaboliti naturali è stata associata allo sviluppo della pubertà precoce, un noto fattore di rischio per il cancro al seno, nelle giovani ragazze [621, 622]. D’altra parte, le ragazze dello studio Jersey Girl Study che avevano livelli di ZEA urinaria più elevati, derivanti dalla recente assunzione di carne di manzo o popcorn, tendevano ad essere meno propense a raggiungere l’inizio dello sviluppo del seno e ad essere di statura più bassa. Quasi l’80% delle ragazze dello studio aveva livelli rilevabili di micoestrogeni nelle urine [623].
Uno studio caso-controllo sui cani ha rivelato che una maggiore esposizione alimentare alle micotossine (aflatossina G1 o G2) ha portato ad un significativo aumento del numero di tumori mammari (OR 2,74; 95% IC = 1,13-6,60 e OR 4,6; 95% CI = 2,2-7,8, rispettivamente) [624].
Nelle dighe per ratti alimentati con diete contenenti ZEA, sia il composto che i suoi metaboliti hanno attraversato la barriera placentare e sono apparsi anche nel latte materno [625]. L’esposizione di cuccioli di femmina a dosi di ZEA rilevanti per l’ambiente durante le ultime 2 (di 3) settimane di sviluppo fetale e i primi giorni postnatali ha portato ad alterazioni a lungo termine nello sviluppo della ghiandola mammaria del tipo associato ad un aumento del rischio di sviluppo di tumori mammari [626].
I ratti trattati nei giorni post-parto 15-19 con ZEA e poi con la sostanza cancerogena, N-metil-N-nitrosurea (MNU), alla pubertà hanno sviluppato meno tumori mammari, con una minore molteplicità, rispetto ai controlli abbinati trattati solo con l’agente cancerogeno NMU, anche se non c’è stata alcuna differenza nella latenza alla comparsa di tumori in entrambi i gruppi [627].
Nei modelli di coltura cellulare del cancro al seno umano, è stato dimostrato che le micotossine, tra cui Fusarin C e ZEA e i loro metaboliti, sono estrogeniche [628]. Ad esempio, Fusarin C stimola la crescita e la proliferazione delle cellule tumorali del seno MCF-7 attraverso processi mediati dal sistema ER [629]. Allo stesso modo, ZEA migliora anche la proliferazione delle cellule MCF-7 in vitro attraverso vie mediate dagli estrogeni e l’attivazione dell’espressione genica indotta dall’estradiolo [630, 631].
Ormoni naturali, sintetici e geneticamente modificati utilizzati nella produzione alimentare
Zeranolo (Ralgro®)
Il composto sintetico, lo zeranolo (Ralgro®), è un potente promotore di crescita non steroideo che imita molti degli effetti dell’estradiolo ormonale naturale. Lo zeranolo (ZER) è ampiamente utilizzato negli Stati Uniti e in Canada per promuovere tassi di crescita rapidi e più efficienti negli animali utilizzati come fonte di carne [632].
Come per il composto naturale ZEA, lo ZER è una sostanza chimica fortemente estrogenica, come dimostrato dalla sua capacità di stimolare la crescita e la proliferazione delle cellule tumorali della mammella umana in vitro a potenze simili agli ormoni naturali, l’estradiolo, e il noto agente cancerogeno, il dietilstilbestrolo (DES) [633]. Uno studio del 2007 ha dimostrato che l’aggiunta di ZER alle cellule epiteliali del seno in coltura ha portato ad un aumento della proliferazione cellulare, accompagnato da un upregulation o dalla stimolazione dell’attività della proteina disolfuro isomerasi, un enzima la cui attività è spesso aumentata nei tessuti tumorali [634].
Il trattamento di topi femmine adulte giovani con ZER ha portato ad un aumento della crescita e della ramificazione delle ghiandole mammarie, simile a quello che si trova nei topi trattati con l’estradiolo ormonale naturale [635]. L’aumento della proliferazione duttile, in assenza di una completa maturazione dei dotti durante la gravidanza e l’allattamento, è associato ad un aumento del rischio di tumori mammari (del seno).
Breve (4 giorni) esposizione prepuberale di topi o ratti a ZEA o ZER ha accelerato l’inizio della pubertà, ma non ha influito sullo sviluppo delle strutture della ghiandola mammaria attraverso l’età adulta precoce [636, 637].
Una serie di studi ha esaminato l’attività estrogenica nelle normali cellule epiteliali del seno e nelle cellule tumorali del seno trattate con ZER. La crescita cellulare anormale è stata significativa anche a livelli di ZER quasi 30 volte inferiori al limite stabilito dalla FDA nella carne bovina [638]. Il lavoro di follow-up ha dimostrato che lo ZER è paragonabile agli estrogeni naturali (estradiolo) e all’estrogeno sintetico dietilstilbestrolo (DES) nella sua capacità di trasformare le cellule epiteliali del seno umano MCF-10A in un profilo pre-cancro in vitro [639]. I dati preliminari indicano che il siero di bovini da carne bovina trattati con ZER può stimolare la proliferazione delle normali cellule epiteliali del seno e la trasformazione delle cellule tumorali della mammella in vitro [640, 641].
Ormone della crescita bovina (rBGH)/ Somatotropina bovina ricombinante (rBST)
Nonostante l’opposizione di medici, scienziati e gruppi di difesa dei consumatori, la Food and Drug Administration nel 1993 ha approvato il prodotto ormonale geneticamente modificato della Monsanto, l’ormone ricombinante della crescita bovina (successivamente rinominato somatotropina bovina ricombinante, rBST), da iniettare nelle vacche da latte per aumentare la produzione di latte [642]. Questo ormone ha trovato rapidamente la sua strada (senza etichettatura) nella fornitura di latte negli Stati Uniti, e da lì in gelato, latticello, formaggio, yogurt e altri prodotti lattiero-caseari. Dalla sua introduzione, l’rBST si è dimostrato controverso a causa dei suoi potenziali effetti cancerogeni.
Bere qualsiasi tipo di latte vaccino aumenta notevolmente i livelli corporei del fattore di crescita dell’insulina 1 (IGF-1), un ormone naturale sia nelle vacche che negli esseri umani. L’iniezione di rBST nelle vacche porta ad un aumento dei livelli di IGF-1 nel latte [643], anche se è possibile che l’aumento della produzione di latte da parte degli animali trattati possa diluire l’eccesso di produzione di ormone [644]. Il contenuto di IGF-1 nel latte non viene alterato dalla pastorizzazione [645].
Sebbene i dati siano complessi, con alcuni studi che giungono a conclusioni diverse, diversi studi epidemiologici hanno indicato una relazione tra il consumo di latte e il rischio di cancro al seno nelle donne in pre-menopausa [646]. Livelli elevati di IGF-1, in particolare, sono stati associati ad un aumento del rischio di cancro al seno [647- 650]. Uno studio caso-controllo nidificante all’interno di un più ampio studio prospettico sulle donne americane ha trovato che le donne in pre-menopausa con i più alti livelli di IGF-1 nel sangue (prelevati prima dello sviluppo del cancro) avevano sette volte più probabilità di sviluppare il cancro al seno rispetto alle donne con i livelli più bassi quando i risultati sono stati corretti per le concentrazioni plasmatiche della proteina legante IGF [647]. Non è stato rilevato alcun aumento del rischio nelle donne in post-menopausa. Tre studi riportati nel 2005 da scienziati in Svezia, Regno Unito [651] e Stati Uniti [652] hanno anche mostrato un’associazione tra i livelli circolanti di IGF-1 e il rischio di cancro al seno nelle donne in pre-menopausa.
Un meccanismo con cui l’IGF-1 può aumentare il rischio nelle donne più giovani è l’aumento della densità del seno nelle donne in pre-menopausa, un noto fattore di rischio per il cancro [653]. Inoltre, studi di laboratorio hanno dimostrato che l’IGF-1 può regolare la crescita e aumentare la proliferazione delle cellule tumorali del seno (MCF-7) cresciute in vitro [654] e diminuire la morte delle cellule tumorali mammarie negli animali da laboratorio [655].
I sostenitori della rBST sostengono che l’IGF-1 è innocuo perché si verifica naturalmente nell’uomo, è contenuto nella saliva umana e si scompone durante la digestione. Tuttavia, l’evidenza animale indica che la digestione non scompone l’IGF-1 nel latte perché la caseina, la principale proteina del latte vaccino, protegge l’IGF-1 dall’azione degli enzimi digestivi [656].
Sommario della sezione: Quando incorporato in diete nutrienti regolari, i lignani e gli alimenti a base di soia hanno dimostrato di essere protettivi contro il cancro al seno in numerosi studi epidemiologici. Questa protezione è particolarmente chiara quando l’assunzione con la dieta inizia nell’infanzia. D’altra parte, sia i micoestrogeni che lo zeranolo, uno stimolatore della crescita degli animali, sono estrogeni nelle loro interazioni con le cellule del seno umano, comprese le cellule derivate dal cancro, in ambienti di coltura cellulare. I dati sono più ambigui sui possibili effetti di elevati livelli di IGF-1, riscontrati dopo aver bevuto latte vaccino.
Sostanze chimiche industriali non EDC
Non tutte le sostanze chimiche associate all’aumento del rischio di cancro al seno esercitano i loro effetti attraverso meccanismi di interferenza endocrina. Questa sezione esamina le letterature che collegano un aumento del rischio di sviluppare il cancro al seno ad alcune sostanze chimiche industriali, tutte determinate come cancerogene dallo IARC (vedi Tabella 3). Questi composti e/o gli addotti del DNA che si formano a seguito di esposizioni ai composti, sono direttamente mutageni.Tabella 3Classificazioni della cancerogenicità e fonti di esposizione di sostanze chimiche che si trovano in sostanze chimiche industriali non ECDCSTRUZIONI CHIMICHE FONDAMENTALI Fonte di esposizione Sostanze chimiche industriali non ECDSBenzene1KSolvente petrolchimicoCloruro di vinile1KMonomero utilizzato nella plastica di cloruro di polivinile (PVC) 1,3-Butadiene1KBiprodotto della combustione Ossido di etilene1KSterilizzatore, contaminante sottoprodotto in alcuni cosmetici Classificazioni dell’Agenzia internazionale per la ricerca sul cancro (IARC): 1 = cancerogeno per l’uomo, 2A = probabilmente cancerogeno per l’uomo, 2B = probabilmente cancerogeno per l’uomo, 3 = non classificabile per quanto riguarda la sua cancerogenicità per l’uomo; classificazioni del Programma Nazionale di Tossicologia degli Stati Uniti (NTP): K = Noto come cancerogeno per l’uomo, RA = Ragionevolmente prevedibile come cancerogeno per l’uomo. L’elenco delle fonti di esposizione contiene le fonti di esposizione più comuniTabella 4Classificazioni di cancerogenicità delle esposizioni chimiche riscontrate nel fumo di sigarettaChemicalIARCNTPT fumo di tabacco: Idrocarburi aromatici attivi e passiviKPoliciclici (IPA)RAPolonio-210Benzene1KCloruro di vinile1K1,3-butadiene1KNitrosamina chetone (NNK)Classificazioni dell’Agenzia Internazionale per la Ricerca sul Cancro (IARC): 1 = cancerogeno per l’uomo, 2A = probabilmente cancerogeno per l’uomo, 2B = probabilmente cancerogeno per l’uomo, 3 = non classificabile per quanto riguarda la sua cancerogenicità per l’uomo; classificazioni del Programma Nazionale di Tossicologia degli Stati Uniti (NTP): K = Noto come cancerogeno per l’uomo, RA = Ragionevolmente prevedibile come cancerogeno per l’uomo. L’elenco delle fonti di esposizione contiene le fonti di esposizione più comuni
Benzene
Il benzene è uno dei più grandi volumi di solventi petrolchimici attualmente in produzione e si prevede che i tassi di produzione globale continueranno a crescere nei prossimi anni. Le industrie chimiche stimano che più di 46 milioni di tonnellate metriche (più di 115 miliardi di libbre) di benzene saranno consumate a livello globale entro il 2020 [657]. L’esposizione al benzene deriva dall’inalazione di fumi di benzina, dai gas di scarico delle automobili o dal fumo di sigaretta (primario e secondario) e dalla combustione industriale. Il benzene rappresenta un grave rischio professionale per le persone esposte attraverso il loro lavoro nelle industrie chimiche, della gomma, della produzione di calzature, della raffinazione del petrolio e della benzina. Sia l’NTP che lo IARC hanno designato il benzene come cancerogeno per l’uomo [658, 659].
Gli studi epidemiologici sugli effetti del benzene sul rischio di cancro al seno sono difficili da condurre, soprattutto perché l’esposizione al benzene avviene in concomitanza con l’esposizione ad altre sostanze chimiche che vengono rilasciate anche nei processi di combustione e di produzione. Inoltre, pochi degli studi occupazionali che si concentrano sulle industrie chimiche e automobilistiche hanno incluso le donne in numero sostanziale per trarre conclusioni significative. Uno studio che ha esaminato le occupazioni rilevanti tra le lavoratrici cinesi, ha esaminato l’incidenza per occupazione come standardizzato dai tassi di incidenza generale del cancro al seno a Shanghai e il numero di donne in ogni occupazione secondo il censimento del 1982. Tra le professioni in cui sono stati riscontrati rischi elevati di cancro al seno vi sono i ricercatori scientifici (SIR + 3,3); i medici che praticano la medicina occidentale (SIR = 14,7, 95% CI = 5).9-30,3) o medicina cinese occidentale (SIR = 7,2; 95% IC = 4,4-11,4); così come i lavoratori con esposizioni inferiori previste, come insegnanti, bibliotecari e contabili (SIR 2,3-2,7). Nello stesso studio, guardando tra le varie professioni, l’esposizione al benzene è stata associata a un elevato rischio di cancro al seno [660]. Uno studio su un campione piuttosto piccolo di donne per le quali i ricercatori hanno dati di esposizione al benzene provenienti dal loro lavoro in una fabbrica di scarpe a Firenze, Italia, sostiene anche una relazione tra l’esposizione al benzene e il successivo sviluppo del cancro al seno [661].
Il più grande studio che coinvolge il benzene e le sostanze chimiche associate proviene da uno studio occupazionale su uomini a cui è stato diagnosticato un cancro al seno. Gli uomini che avevano lavorato in professioni che prevedevano l’esposizione ai fumi della benzina e alla combustione avevano aumentato significativamente i tassi di cancro al seno. L’effetto è stato più pronunciato tra gli uomini che hanno iniziato a lavorare prima dei 40 anni [662].
La somministrazione di benzene ai topi da laboratorio induce tumori mammari. I topi esposti al benzene hanno frequenti mutazioni dei geni che sono responsabili della soppressione dello sviluppo dei tumori [663, 664].
Cloruro di vinile
I produttori utilizzano ampiamente il cloruro di polivinile (PVC) per produrre imballaggi alimentari, prodotti medici, elettrodomestici, automobili, giocattoli, carte di credito e indumenti per la pioggia. Quando il PVC viene prodotto, il cloruro di vinile può essere rilasciato nell’aria o nelle acque reflue. Il cloruro di vinile è stato trovato anche nell’aria vicino a siti di rifiuti pericolosi e discariche e nel fumo di tabacco.
Il cloruro di vinile è stata una delle prime sostanze chimiche designate come cancerogene per l’uomo dall’NTP [665, 666]. Il cloruro di vinile è stato collegato all’aumento della mortalità per cancro al seno e al fegato tra i lavoratori coinvolti nella sua produzione [667, 668]. Nel grande studio prospettico della coorte dei docenti della California Teachers Study, l’esposizione al cloruro di vinile è stata associata ad un aumento del rischio di cancro al seno. Le analisi dei sottoinsiemi all’interno della coorte rivelano associazioni significative tra l’esposizione al cloruro di vinile (per la più alta esposizione al cloruro di vinile quintile) e i tumori ER+/PR+ (HR = 1,08; 95% CI = 0,98-1,19), e nelle donne che non avevano mai, o non stavano attualmente, utilizzando HRT (HR = 1,27; 95% CI =1,04-1,54) [669]. In uno studio caso-controllo su pazienti di sesso maschile affette da tumore al seno, che avevano tutte vissuto a Camp LeJeune durante i decenni in cui l’acqua potabile era contaminata da diversi solventi tossici, l’esposizione al cloruro di vinile era associata ad un rischio più elevato di sviluppare il cancro al seno (OR = 1,20 (95% CI = .16-5,89) e ad un inizio precoce della malattia (OR = 2,14; 95% CI = 0,31-14,81) [670].
Gli animali esposti a lungo termine a bassi livelli di cloruro di vinile trasportato dall’aria mostrano un aumento del rischio di tumori mammari [671].
1,3-butadiene
L’1,3-butadiene è un inquinante dell’aria creato dai motori a combustione interna e dalle raffinerie di petrolio. È anche una sostanza chimica utilizzata nella fabbricazione e nella lavorazione di prodotti in gomma sintetica e di alcuni fungicidi. Inoltre, l’1,3-butadiene si trova nel fumo di tabacco.
L’EPA ha determinato che l’1,3-butadiene è cancerogeno per l’uomo, con la principale via di esposizione attraverso l’inalazione. Le donne che lavorano nell’industria della gomma sintetica che hanno avuto un’elevata esposizione all’1,3-butadiene hanno aumentato il rischio di morire di cancro al seno (RR = 2,6 m 95% CI = .9-7,3) [672]. L’NTP classifica l’1,3-butadiene come noto agente cancerogeno umano [673].
I dati della ricerca sugli animali indicano che le femmine possono essere più vulnerabili agli effetti cancerogeni dell’1,3-butadiene [674], che è noto per causare tumori mammari e ovarici nelle femmine di topi e ratti. Questo inquinante produce effetti tossici ancora maggiori nelle popolazioni di roditori più giovani [675].
Ossido di etilene
L’ossido di etilene è un fumigante utilizzato per sterilizzare gli strumenti chirurgici ed è anche usato in alcuni prodotti cosmetici [676]. L’ossido di etilene è classificato come cancerogeno per l’uomo [677, 678] e uno dei 221 prodotti chimici identificati dai ricercatori del Silent Spring Institute come associati a tumori mammari negli animali [197].
Gli scienziati del National Institute for Occupational Safety and Health (NIOSH) hanno studiato l’incidenza del cancro al seno in 7576 donne esposte all’ossido di etilene mentre lavoravano in impianti di sterilizzazione commerciali. Hanno trovato un aumento dell’incidenza del cancro al seno tra queste donne in proporzione diretta alla loro esposizione cumulativa all’ossido di etilene [679]. Sebbene ci siano dati contraddittori nella letteratura recente [678], altri studi occupazionali sostengono la conclusione che l’esposizione all’ossido di etilene è associata ad un aumento del rischio di cancro al seno nelle donne [680, 681].
Studi in cui le cellule mammarie umane cresciute in vitro sono state esposte a basse dosi di ossido di etilene hanno dimostrato che l’esposizione chimica ha portato ad un significativo aumento del danno al DNA delle cellule [682]. Questi risultati sono supportati dai risultati di uno studio che esamina le mutazioni geniche nei tumori mammari indotte nei topi dall’esposizione all’ossido di etilene. Le mutazioni comuni includevano quelle del gene soppressore del tumore, p53, e del gene regolatore della proliferazione cellulare, H-ras [664].
Riassunto della sezione: gli studi epidemiologici di uomini e donne esposti per motivi professionali al benzene o al cloruro di vinile hanno un rischio maggiore di sviluppare un tumore al seno. Le limitate esposizioni umane indicano inoltre che anche le esposizioni all’1,3-butadiene hanno un rischio maggiore per il cancro al seno, mentre le prove a sostegno di questa relazione sono più solide per l’ossido di etilene.
Fumo di tabacco: attivo e passivo
Prove sempre più evidenti indicano4 che l’esposizione alle numerose sostanze chimiche incluse nel fumo di tabacco, sia attraverso mezzi attivi (di prima mano) che passivi (di seconda mano), può aumentare il rischio di sviluppare il cancro al seno. Discutiamo questa letteratura in questa sezione. Mentre le esposizioni al fumo sono spesso associate al consumo di alcol e ad altri fattori legati allo stile di vita, in questa categoria ci concentriamo solo sulle esposizioni al fumo di tabacco, poiché queste sono dovute a sostanze chimiche che inquinano l’ambiente, e le esposizioni sono spesso involontarie.
Il fumo di tabacco contiene idrocarburi policiclici aromatici (IPA), così come centinaia di altre sostanze chimiche [683], tra cui tre noti agenti cancerogeni per l’uomo (polonio-210, un elemento radioattivo; benzene; e cloruro di vinile) così come 1,3-butadiene e nitrosammina chetone derivato dalla nicotina (NNK), che sono tutti noti per causare tumori mammari negli animali. NNK è una sostanza cancerogena specifica del tabacco che ha dimostrato di aumentare la proliferazione delle cellule tumorali e la trasformazione di cellule epiteliali del seno sane in cellule tumorali [684- 686], almeno in parte attraverso il recettore nicotinico dell’acetilcolina [687] (vedi Tabella 4).
Un ampio studio di insegnanti californiani ha rivelato un aumento del rischio di cancro al seno tra i fumatori, in particolare tra coloro che hanno iniziato a fumare durante l’adolescenza (HR = 1,17; 95% CI = 1,05-1).30), almeno 5 anni prima della prima gravidanza a termine (HR = 1,13; 95% CI = 1,00-1,28), o che erano fumatrici di lunga durata o fumatrici incallite (HR = 1,32; 95% CI = 1,10-1,57) [688]. Diversi studi precedenti suggeriscono inoltre che le donne che iniziano a fumare sigarette in età adolescenziale sono esposte a maggiori rischi di cancro al seno [689- 693].
I risultati del Canadian National Breast Screening Study hanno indicato che l’aumento dell’incidenza del cancro al seno è associato a una maggiore durata del fumo (RR = 1,50; 95% CI = 1,19-1,89), numero di sigarette fumate al giorno (per 40 sigarette al giorno): RR = 1,20; 95% CI = 1,00-1,44), e l’esposizione cumulativa al fumo di sigaretta (per 40 pacchetti/anno: RR = 1,17; 95% CI = 1,02-1,34) [694]. Risultati simili sono stati registrati nei rapporti di due grandi studi prospettici: lo studio sulla salute degli infermieri [695] e lo studio WHI [696], che ha coinvolto rispettivamente circa 110.000 e 80.000 partecipanti.
Sebbene diversi studi più recenti abbiano riportato che l’inizio del fumo prima di una prima gravidanza a termine (indipendentemente dall’età di inizio del fumo) può rendere una donna sempre più suscettibile a diagnosi successive di cancro al seno [691, 695, 697], una meta-analisi del 2011 di 23 ricerche pertinenti non ha trovato una relazione statisticamente significativa [698]. A complicare questo quadro è un rapporto della coorte EPIC che riporta che l’impatto più importante del fumo di sigaretta attivo è stato il numero di anni-pacco (1 anno-pacco = 20 sigarette/giorno per un anno intero) fumati dal menarca alla prima gravidanza a termine (HR = 1,73, 95% CI = 1,29-2,32 per ogni aumento di 20 anni-pacco). D’altra parte, il numero di anni-pacco fumati dopo la menopausa è stato significativamente associato a una diminuzione del rischio di sviluppare il cancro al seno (HR = 0,53; 95% CI = 0,34-0,82) [699]. Risultati molto diversi sono stati riportati in un ampio studio dell’African American Breast Cancer Epidemiology and Risk (AMBER): rispetto alle donne che non hanno mai fumato, l’aumento dei pacchetti di anni di fumo attivo nelle donne in pre-menopausa è stato associato a un rischio ridotto di cancro al seno (OR = 0.80; 95% CI = 0,68-0,96), mentre gli anni di confezione più alti di fumo attivo nelle fumatrici in post-menopausa erano associati ad un aumentato rischio di cancro al seno (OR = 1,16; 95% CI = 1,01-1,33) [700]. L’effetto post menopausa è stato più forte nelle donne che hanno sviluppato tumori ER+. Nelle donne in post-menopausa è stata riscontrata una relazione inversa tra il fumo attivo e la densità mammografica del seno, con un effetto amplificato per le donne che hanno iniziato a fumare prima dei 16 anni (OR = 0,79; 95% CI = 0,64-0,96) [701]. Una minore densità del seno è associata ad un minore rischio di sviluppare il cancro al seno [702].
Uno studio di casi di controllo basato sulla popolazione ha esaminato gli effetti del fumo attivo e il rischio di sviluppare il cancro al seno in base al fatto che i tumori fossero di tipo luminale (ER+ e/o PR+) o basale (ER-, PR-, HER2-). Il fumo ha portato ad un aumento del rischio di sviluppare un tumore di tipo luminale (OR = 1,12; 95% CI = 0,92-1,33), ma non di tipo basale, un effetto che è stato più pronunciato nelle donne nere. Un altro studio sull’incidenza del cancro al seno infiammatorio ha riportato effetti del fumo attivo sul luminal (OR = 2,37; 95% CI = 1,24-4,52), ma non altri tipi di cancro al seno [703]. Tuttavia, l’aumento della durata del fumo è stato associato ad un aumento del rischio di sviluppare tumori di tipo basale (OR = 1,51; 95% CI = 1,19-1,93), ma non di tipo luminale [704].
Differenze etniche sono state riportate in uno studio su donne bianche messicane e statunitensi non ispaniche. Per le donne messicane, è stato riscontrato un aumento significativo del rischio di cancro al seno per le ex fumatrici (OR = 1,43; 95% CI = 1,04-1,96 vs. non fumatrici), e questo effetto è stato aumentato per le ex fumatrici con una storia di consumo di alcolici (OR = 2,30; 95% CI = 1,01-5,21). Per le donne bianche non ispaniche statunitensi, il fumo corrente di oltre 20 sigarette al giorno è stato associato a un aumento del rischio (OR = 1,61; 95% CI = 1,07-2,41). Non sono stati riscontrati effetti significativi per le donne bianche ispaniche statunitensi [705].
Uno studio prospettico di coorte di 186.150 membri femminili dell’AARP, 7486 dei quali hanno sviluppato il cancro al seno, ha trovato un aumento del rischio di sviluppare il cancro al seno nelle attuali fumatrici attive (OR = 1,19; 95% CI = 1,10-1,28) e nelle ex fumatrici attive (OR = 1,07; 95% CI = 1,01-1,13). Per le fumatrici attuali, l’effetto è stato significativo nelle donne senza una storia familiare di cancro al seno (OR = 1,24; 95% CI = 1,15-1,35), ma non nelle donne con una storia familiare. L’età successiva del menarca era anche associata a un rischio più elevato di sviluppare il cancro al seno nelle fumatrici attive (età del menarca x interazione dello stato di fumo, p < .03) [706].
Diversi studi recenti hanno esaminato gli effetti del fumo al momento della diagnosi del cancro al seno e i successivi esiti. Bérubé ha riferito che il fumo al momento della diagnosi ha portato ad un aumento della mortalità per tutte le cause, così come un aumento significativo della mortalità per cancro al seno (HR = 1,33; 95% CI = 1,12-1,58) [707]. Sono stati riportati effetti simili sulla mortalità per cancro al seno (HR = 1,10; 95% CI = 0,73-1,68) in donne con diagnosi di cancro al seno localizzato [708]. Il fumo attivo continuato dopo la diagnosi è stato associato ad un aumento dei decessi dovuti al cancro al seno (HR = 1,72; 95% CI = 1,13-2,60) [709].
In 309 pazienti donne affette da tumore al seno ER+ in trattamento con inibitori dell’aromatasi, il fumo prima del trattamento chirurgico per la loro malattia era associato ad un aumento del numero di eventi di tumore al seno (ad es, recidiva, nuova diagnosi di cancro al seno, metastasi; HR = 2,97; 95% CI = 1,44-6,13), metastasi a distanza (HR = 4.19; 95% IC = 1,81-9,72), e mortalità (HR = 3,52; 95% CI = 1,59-7,61). Non vi era alcuna relazione tra il fumo e gli esiti correlati al cancro al seno nelle donne sottoposte ad altre forme di terapia adiuvante [710].
Due studi recenti hanno esaminato lo stato del fumo e gli esiti del cancro al seno negli uomini. In un consorzio di studio congiunto con 2378 casi di tumore al seno maschile in Florida, Cook et al. non hanno trovato alcuna prova di associazione tra la condizione di fumatore, gli anni di consumo, la durata o l’età al momento dell’inizio del fumo e il rischio di sviluppare il tumore al seno [711]. Un altro studio sui casi di cancro al seno maschile in Florida ha esaminato i tassi di sopravvivenza a seguito della diagnosi e ha stratificato la loro analisi in base all’etnia razziale e allo stato socioeconomico. Nel complesso, rispetto ai non fumatori mai fumatori, i fumatori attuali hanno avuto tassi di mortalità più elevati (HR = 1,63; 95% CI = 1,23-2,16), anche se non vi è stato alcun effetto per i fumatori del passato che avevano abbandonato l’abitudine. C’era un rapporto dose-risposta tra quantità fumata e rischio di mortalità (tendenza, p < .001). Effetti simili sono stati riscontrati sia per i bianchi (ma non per i neri) che per i non ispanici (ma non per gli ispanici) [712].
Oltre agli effetti del fumo attivo sull’incidenza del cancro al seno e sulla mortalità, una crescente letteratura implica l’esposizione al fumo di seconda mano (fumo passivo) ad un rischio maggiore per la malattia. Fino a poco tempo fa, più prove collegavano il fumo di seconda mano che il fumo attivo al rischio di cancro al seno. Le prove attuali suggeriscono che entrambe le esposizioni aumentano il rischio di cancro al seno di circa la stessa quantità, anche se le donne che sono esposte al fumo passivo ricevono una dose molto più bassa di agenti cancerogeni rispetto ai fumatori attivi [699, 713, 714]. I ricercatori del National Cancer Center del Giappone hanno riportato i risultati di uno studio che ha coinvolto 21.000 donne di età compresa tra i 40 e i 59 anni. Hanno trovato che il rischio di cancro al seno era elevato nelle donne in pre-menopausa che erano fumatrici attive (RR = 3,9; 95% CI = 1,5-9,9) o esposte al fumo ambientale di seconda mano (RR = 2,6; 95% CI = 1,3-5,2) [715]. Altri studi importanti, tra cui il WHI, sostengono la scoperta di un legame tra l’esposizione prolungata al fumo passivo di oltre 10 anni e l’aumento del rischio di cancro al seno (HR = 1,32; 95% CI = 1,04-1,67) [696]. L’esposizione al fumo passivo a casa, ma non al lavoro, aumenta il rischio di sviluppare il cancro al seno (OR = 1,30; 95% CI = 1,05-1,61) e la quantità di esposizione a casa è collegata in modo dose-risposta (p = .009) [716].
Una meta-analisi di otto studi su donne cinesi esposte al fumo passivo, che non sono mai state fumatrici attive, ha mostrato un aumento significativo del rischio di sviluppare il cancro al seno (OR = 1,67; 95% CI = 1,27-2,21) [717]. Un’analisi più dettagliata delle donne cinesi che non hanno mai fumato ha mostrato un effetto significativo di oltre 4 anni di esposizione al fumo passivo (OR = 1,71; 95% CI = 1,17-2,50). L’effetto è stato riscontrato in donne con tumori ER+/PR+, ma non per altri sottotipi tumorali [718].
Nel cercare di capire i meccanismi attraverso i quali il fumo attivo e/o passivo può influenzare il rischio di sviluppare un tumore al seno, sono stati condotti diversi studi sull’espressione genica. Studi che esplorano i legami tra il fumo e l’incidenza, la ricorrenza e la mortalità del cancro al seno hanno identificato diversi polimorfismi associati ad un aumento del rischio. I dati più consistenti implicano un fenotipo n-acetiltransferasi 2 (NAT2) “acetilatore lento” [719, 720], sebbene anche specifici polimorfismi dei geni BRCA1 [719, 721] e CYP1A1 e COMT [719] siano stati riportati come associati a un aumento dell’incidenza e/o della mortalità nei fumatori attivi.
Altri disturbi fisiologici derivanti da esposizioni al fumo includono il danneggiamento della struttura e della funzione delle ovaie, abbassando così i livelli di estrogeni nelle donne in pre-menopausa. Mentre livelli più bassi di estrogeni ridurrebbero il rischio di cancro al seno, allo stesso tempo gli agenti cancerogeni nel fumo di sigaretta aumenterebbero il rischio di sviluppare il cancro al seno [722].
Uno studio trasversale, che fa parte di un ampio progetto prospettico in corso (la coorte EPIC), ha esaminato l’associazione tra il fumo di tabacco e i livelli di ormoni sessuali nelle donne in post-menopausa, le cui ovaie non sono più la fonte principale dei loro ormoni circolanti. Il fumo è stato correlato a livelli più elevati di testosterone, estradiolo e altri ormoni steroidei [723]. L’aumento dei livelli di estradiolo circolante era statisticamente significativo solo per le donne che erano notevolmente in sovrappeso. Di per sé, l’obesità è un noto fattore di rischio per il cancro al seno in post-menopausa. Il tessuto adiposo è il principale sito di aromatizzazione del testosterone all’estradiolo negli uomini e nelle donne in postmenopausa, e l’aumento del tessuto adiposo può quindi contribuire ad aumentare gli estrogeni circolanti. Una maggiore attivazione delle vie metaboliche delle cellule adipose del seno da parte di sostanze chimiche contenenti tabacco può migliorare lo sviluppo del cancro al seno [724].
Sommario della sezione: Esiste ora una letteratura sostanziale che indica che il fumo di sigaretta attivo passato e presente è associato ad un rischio più elevato di sviluppare il cancro al seno. Per le donne che fumano al momento della diagnosi, vi è anche un aumento del rischio di mortalità per cancro al seno. Questi effetti sono complicati dalle interazioni con la razza/etnicità, dalla storia del consumo di alcol e dal sottotipo di cancro al seno in corso di valutazione.
Lavoro a turni, luce a notte fonda e melatonina
Nel 2007, IARC ha concluso che il lavoro a turni è “probabilmente cancerogeno per l’uomo”, basandosi in gran parte sulla crescente associazione tra il lavoro a turni e l’aumento dell’incidenza del cancro al seno [725] (vedi Tabella 5). Diversi studi occupazionali hanno dimostrato che le donne che lavorano costantemente a turni di notte hanno aumentato il rischio di cancro al seno, anche se non tutti i rapporti hanno trovato prove di questa relazione. Le differenze metodologiche tra gli studi, comprese le varie definizioni di “lavoro a turni” e “notturno”, così come la mancanza di un’attenzione coerente ai fattori di confusione possono spiegare alcune delle differenze nei risultati tra i singoli studi [726, 727].Tabella 5Classificazioni della cancerogenicità e fonti di esposizione di luce di notte e di radiazioneEsposizioneIARCNTPUseShift Work, Light-at-NightPRShift work o l’inquinamento della luce ambienteIonizing RadiationKKDiagnostic test medici; procedure di medicina nucleare; centrali nucleari, protocolli di ricercaNon-ionizzanti radiazioni (campi elettromagnetici)Illuminazione, computer, telefoni cellulari e altre fonti elettronicheInternational Agency for Research on Cancer (IARC) classificazioni: 1 = cancerogeno per l’uomo, 2A = probabilmente cancerogeno per l’uomo, 2B = probabilmente cancerogeno per l’uomo, 3 = non classificabile per quanto riguarda la sua cancerogenicità per l’uomo; classificazioni del Programma Nazionale di Tossicologia degli Stati Uniti (NTP): K = Noto come cancerogeno per l’uomo, RA = Ragionevolmente prevedibile come cancerogeno per l’uomo. L’elenco delle fonti di esposizione contiene le fonti di esposizione più comuni
Le associazioni tra il lavoro a lungo termine (> 20-30 anni) con turni di notte e il cancro al seno sono state riportate in una revisione completa di 13 studi del 2008 [728]. Quattro altre quattro revisioni che includevano meta-analisi hanno raggiunto conclusioni simili, anche se la forza delle associazioni variava considerevolmente. Sulla base dell’analisi di 13 studi, Megdal e colleghi hanno riportato una stima aggregata del rischio di cancro al seno (RR) sia per le assistenti di volo sia per le altre che lavorano nel turno di notte come 1,48 (95% CI = 1,36-1,61) [729]. Kamdar e colleghi hanno incluso 15 studi caso-controllo e di coorte che hanno esaminato la possibile relazione tra il lavoro notturno e il rischio di cancro al seno, e hanno riportato un RR comune di 1,21 (95% CI = 1,00-1,47) per gli individui con qualsiasi esperienza di lavoro notturno rispetto a quelli senza tale esperienza [730]. Egli et al. hanno incluso un gruppo più eterogeneo di 28 studi che hanno esaminato le interruzioni del ritmo circadiano, ma definite in vari modi (lavoro a turni, breve durata del sonno, occupazione come assistente di volo, esposizione alla luce notturna). Hanno riportato un RR aggregato di 1,14 (95% CI = 1,08-1,21), con RRs simili quando le analisi erano limitate a soli studi che esaminavano il lavoro a turni (RR = 1,19; 95% CI = 1,08-1.32) o esposizione alla luce di notte (RR = 1,12; 95% CI = 1,12-1,12), ma un rischio notevolmente più elevato quando gli studi di soli assistenti di volo sono stati analizzati (RR = 1,56 (95% CI = 1,10-2,56). La minore durata del sonno non ha conferito un cambiamento nel rischio di sviluppare il cancro al seno in questa analisi [731]. Lin, et al. hanno analizzato 16 studi di coorte prospettici e hanno riportato un RR aggregato di 1,09 (95% CI = 1,02-1,17) per le lavoratrici del turno di notte rispetto alle lavoratrici diurne. Una tendenza lineare (p = 0,010) è stata trovata per l’aumento della durata dell’esposizione (< 5, 5-10, 10-20 e >20 anni) e il rischio di sviluppare il cancro al seno [732]. D’altra parte, una nuova meta-analisi di 10 studi prospettici, per un totale di 1,4 milioni di donne, non ha trovato alcun effetto sul rischio di cancro al seno per l’impegno in qualsiasi lavoro a turni (RR = 0,99; 95% CI = 0.95-1.03), per 20 o più anni di lavoro a turni di notte (RR = 1.01; 95% CI = 0.93-1.10), o per 30 o più anni di lavoro a turni di notte (RR = 1.00; 95% CI = 0.87-1.14) [733]. Una più recente valutazione di questa meta-analisi ha messo in discussione diversi criteri metodologici e interpretazioni di dati all’interno del rapporto [734].
Uno studio record di collegamento tra occupazione e cancro in Gran Bretagna ha calcolato che per il 2012, il fattore attribuibile alla popolazione (PAF) del lavoro notturno può rappresentare il 4,5% (95% CI = 3,2-5,9) delle diagnosi di cancro al seno (1957 casi; 95% CI = 1395-2547) e dei decessi (552 casi; 95% CI = 393-724) [735]. Un’analisi simile nel 2015 ha calcolato un PAF per il lavoro a turni del 5,7% (95% CI = 0,0-11,9) delle donne statunitensi a cui è stato diagnosticato il cancro al seno (casi di cancro al seno attribuibili = 11.777; 95% CI = 0-24.625) [736].
In uno studio condotto su infermiere danesi, gli effetti del lavoro notturno sul rischio di cancro al seno sono stati maggiori per le donne che hanno lavorato a rotazione con orari che includono il turno di notte, rispetto al turno serale (OR = 1.8; 95% CI = 1,2-2,8) e per coloro che hanno lavorato a turni di 12 ore che alternavano il lavoro diurno e quello notturno, rispetto a periodi di lavoro più brevi (OR = 2,9; 95% CI = 1,1-8,0) [737].
Il rischio di sviluppare il cancro al seno è aumentato per le donne che hanno lavorato in turni di notte per più di 4,5-5 anni (OR = 1,40; 95% CI = 1,01-1).92), e per coloro che hanno svolto regolarmente lavoro notturno per almeno 4 anni prima della loro prima gravidanza (OR = 1,95; 95% CI = 1,13-3,35), quindi prima del momento in cui le loro cellule mammarie si erano completamente differenziate [738].
Il meccanismo più studiato per spiegare questi effetti del lavoro notturno è l’ipotesi della luce di notte (LAN) [739]. Aumentando l’esposizione alla luce, soprattutto alla luce interna brillante, a volte al di fuori delle normali ore diurne, diminuisce la secrezione di melatonina da parte della ghiandola pineale. I normali livelli elevati di melatonina durante le ore notturne sono importanti per la regolazione degli ormoni sia pituitari che ovarici, per sopprimere la produzione locale di estrogeni derivante dall’aromatizzazione degli androgeni nelle cellule tumorali della mammella e per mantenere i normali profili metabolici e il peso corporeo [740- 743].
Studi clinici hanno dimostrato che c’è una diminuzione del picco di melatonina secreta nelle donne con tumore metastatico, rispetto alle donne sane, e che i tumori più grandi sono associati a livelli più bassi di melatonina [740]. Le donne cieche che non sono completamente incapaci di percepire la presenza di luce ambientale, e che contemporaneamente non hanno diminuzioni giornaliere dei livelli di melatonina, hanno un rischio significativamente più basso di diagnosi di cancro al seno rispetto alle donne cieche che percepiscono la luce e hanno cambiamenti regolari nella secrezione di melatonina durante il normale ciclo di 24 ore (OR = 0,52; 95% CI = 0,27-1,01) [744].
Una via proposta per ridurre la melatonina che potrebbe influenzare il rischio di cancro al seno è il miglioramento della produzione o della secrezione di estradiolo e di altri ormoni ovarici. Nagata et al. hanno riferito che le donne in postmenopausa che lavoravano in turni di notte che andavano oltre la mezzanotte avevano aumentato significativamente le concentrazioni sieriche di estradiolo sia durante le fasi del turno di notte che quando ruotavano verso i normali periodi di veglia diurna, rispetto ai controlli che non facevano il turno di notte [745]. Davis et al. hanno testato i livelli urinari di 6-sulfatossimelatonina (il metabolita principale della melatonina), LH, FSH e coniugato estrone attraverso il sonno e i cicli di lavoro per le infermiere in premenopausa che lavorano sia di giorno che di notte. Rispetto agli infermieri che lavorano in turni di giorno, nei turni di notte i livelli di LH e FSH erano entrambi significativamente più alti (35 e 38% più alti, rispettivamente), mentre i livelli di 6-sulfatossimelatonina erano significativamente diminuiti (69% più bassi); non sono state riscontrate differenze significative nel coniugato estrone [746]. Tuttavia, uno studio che ha esaminato il possibile legame tra le variazioni dei livelli di melatonina e le variazioni dei livelli di ormoni riproduttivi non ha trovato una relazione una volta che altri fattori come l’età, lo stato mestruale e l’indice di massa corporea sono stati presi in considerazione nell’analisi [747].
Nei modelli di roditori, livelli più elevati di melatonina sono associati a una diminuzione dell’incidenza e delle dimensioni dei tumori mammari, e quando si verificano, il periodo di latenza dello sviluppo del tumore si allunga [739]. Nei tumori mammari umani che erano stati innestati nei topi, la perfusione con il sangue prelevato dalle donne di notte (quando la melatonina è alta) diminuisce la proliferazione e la crescita dei tumori mammari, rispetto all’uso di campioni raccolti durante il giorno quando i livelli di melatonina sono naturalmente più bassi [748].
Meccanicamente, gli impulsi notturni di melatonina aumentano l’attività delle vie endocrine, metaboliche e immunitarie che possono prevenire lo sviluppo del cancro [749]. Questi effetti protettivi della melatonina sono mediati da cambiamenti epigenetici in molti dei geni coinvolti nella regolazione della crescita e della proliferazione cellulare, così come nella sintesi e nell’attivazione del recettore degli estrogeni [750]. I geni che sono associati alla regolazione del ciclo giornaliero della melatonina regolano anche altre vie che possono essere coinvolte nello sviluppo del cancro al seno. La variazione strutturale di uno di questi geni, il Per3, è associata a tassi più elevati di cancro al seno nelle giovani donne [751]. Anche il Per2, un altro gene associato al controllo dei ritmi giornalieri, è scarsamente regolato in molte donne affette da cancro al seno, con la normale struttura ed espressione di questo gene associata a una minore efficacia dell’estradiolo nell’alterazione dell’attività cellulare. Nelle cellule sane, il Per2 può anche agire direttamente come un gene soppressore di tumori, diminuendo l’attività delle vie associate alla formazione del tumore [752]. Un raro polimorfismo del gene CLOCK è stato associato ad un aumento del rischio di sviluppare il cancro al seno (OR = 3,53; 95% CI +1,09-11,42), e c’è stata un’interazione positiva tra la presenza di questo genotipo e il lavoro notturno sul rischio di sviluppare il cancro al seno (p = .02) [753]. Tuttavia, uno studio caso-controllo che ha valutato 100 SNPs di 14 geni legati all’orologio nell’interazione con la storia del lavoro a turni non ha trovato alcuna associazione per nessuno degli SNPs [754].
Altri studi recenti hanno notevolmente complicato la storia della luce a notte e del cancro al seno. Le relazioni tra il lavoro a turni notturni e i livelli di melatonina possono essere mediate dalla razza o dal background etnico. In un ampio studio sulle donne cinesi basato sulla popolazione, non è stata riportata alcuna associazione tra il lavoro a turni e l’incidenza del cancro al seno [755]. Le donne asiatiche e asiatiche-americane che fanno il turno di notte hanno una minore soppressione della melatonina rispetto alle loro controparti bianche [756]. Gli effetti del lavoro a turni di notte possono anche essere limitati a specifici tipi di tumore al seno. In uno studio caso-controllo basato sulla popolazione delle lavoratrici del turno di notte, l’aggiustamento per il cronotipo (punto a metà del sonno nei giorni in cui le partecipanti potevano scegliere quando dormire), ha portato ad un aumento del rischio di invasione (rispetto a quello che si è verificato nei turni di notte). in situ) tumori (OR = 1,23; 95% IC = 1,05-1,99) e un aumentato rischio di tumori premenopausali ER+/PR+ (OR = 1,44; 95% IC = 1,05-1,99) [757].
Diversi autori hanno proposto che i fattori associati al lavoro notturno, oltre alla diminuzione dei livelli di melatonina, devono essere considerati per comprendere meglio i legami con l’aumento del rischio di cancro al seno. Altre possibili conseguenze del lavoro a turni, tra cui il disturbo del sonno a turni, i fattori legati allo stile di vita, i cambiamenti nel metabolismo, la desincronizzazione tra sistemi neurali centrali e periferici, o la diminuzione della produzione di vitamina D, possono anche essere collegati ad un aumento dei tassi di cancro. Questi fattori devono essere studiati come fattori singoli e possibilmente interagenti nel rischio alterato di sviluppare il cancro al seno [758- 762].
Infine, come per diverse sostanze chimiche che alterano il sistema endocrino, la luce notturna riduce significativamente l’efficacia dei principali agenti chemioterapici utilizzati nel trattamento del cancro al seno. Nei modelli di ratto con xenotrapianti di cellule tumorali mammarie MCF-7, l’aggiunta di esposizioni alla luce fioca durante la fase oscura del ciclo ha portato a una diminuzione della secrezione di melatonina durante la fase oscura, a una diminuzione della latenza alla progressione tumorale, a un aumento della crescita tumorale e a una completa resistenza sia al tamoxifene che alla doxorubicina [763, 764].
Riassunto della sezione: è stato dimostrato che una vasta esperienza con il lavoro notturno, e quindi una maggiore esposizione alla luce notturna (LAN), aumenta il rischio di cancro al seno, anche se ci possono essere differenze etniche in questa risposta. Il meccanismo di base più studiato per l’effetto dell’esposizione alla luce notturna è il cambiamento dei modelli di secrezione di melatonina. Sono stati proposti anche altri stili di vita e fattori fisiologici associati al lavoro a turni per modificare il rischio di sviluppare il cancro al seno.
Radiazioni
L’esposizione alle radiazioni ionizzanti, provenienti sia da fonti militari che mediche, è la causa ambientale più nota e più longeva del cancro al seno sia nelle donne che negli uomini. Le esposizioni all’inizio della vita, durante l’infanzia e l’adolescenza, sono particolarmente importanti. I dati relativi ai potenziali legami tra i campi elettromagnetici, o radiazioni non ionizzanti, e il cancro al seno sono misti e non conclusivi.
Radiazioni ionizzanti
La radiazione ionizzante è qualsiasi forma di radiazione con energia sufficiente a separare gli elettroni dagli atomi (per ionizzare gli atomi). Questa radiazione può rompere i legami chimici nelle molecole, comprese le molecole di DNA, disturbando così il loro normale funzionamento. I raggi X e i raggi gamma sono le uniche forme principali di radiazione con energia sufficiente per penetrare e danneggiare il tessuto corporeo sotto la superficie della pelle.
Tra le molte fonti di radiazioni ionizzanti vi sono i tradizionali raggi X, la tomografia computerizzata (TAC), la fluoroscopia e altre procedure radiologiche mediche. Tra le fonti di raggi gamma vi sono le emissioni delle centrali nucleari, la ricerca scientifica che coinvolge i radionuclidi, i test sulle armi militari e le procedure di medicina nucleare come le scansioni ossee, tiroidee e polmonari [765].
Nel 2005, il Programma Nazionale di Tossicologia ha classificato le radiazioni X e le radiazioni gamma come noti agenti cancerogeni per l’uomo [766] (vedi Tabella 5). Sebbene alcuni scienziati contestino questa premessa [767], la maggior parte concorda sul fatto che non è stata identificata alcuna dose sicura di radiazioni [768, 769]. I danni da radiazioni ai geni sono cumulativi nell’arco della vita [770]. Le esposizioni ripetute a basse dosi nel tempo possono avere gli stessi effetti dannosi di una singola esposizione ad alte dosi.
L’esposizione alle radiazioni ionizzanti è la migliore e più longeva causa ambientale del cancro al seno umano, sia nelle donne che negli uomini. Il legame tra l’esposizione alle radiazioni e il cancro al seno è stato dimostrato nei sopravvissuti alla bomba atomica [771- 774]. I tassi di cancro al seno erano più alti tra le donne che avevano meno di 20 anni quando gli Stati Uniti sganciarono le bombe atomiche su Hiroshima e Nagasaki [773]. Inoltre, Ron et al. hanno riportato una significativa associazione tra l’esposizione alle radiazioni ionizzanti e l’incidenza del cancro al seno maschile nelle sopravvissute alle bombe atomiche giapponesi [775].
Le radiazioni ionizzanti possono aumentare il rischio di cancro al seno attraverso una serie di meccanismi diversi, tra cui la mutagenesi diretta, l’instabilità genomica [776, 777] e i cambiamenti nei microambienti delle cellule del seno che possono portare a una danneggiata regolazione delle interazioni cellula-cellula all’interno del seno [778- 780]. Le radiazioni ionizzanti non solo colpiscono le cellule direttamente esposte, ma possono anche alterare il DNA, la crescita cellulare e le interazioni cellula-cellula delle cellule vicine, definite “effetto spettatore” [767, 781]. Un saggio G2 micronucleo di campioni di sangue da donne asintomatiche che portano la mutazione BRCA1 hanno deficit in molti di questi processi cellulari e una maggiore sensibilità agli effetti delle esposizioni alle radiazioni, rispetto ai campioni di donne sane senza la mutazione [782].
Interazioni tra radiazioni e altri fattori. Ci sono una serie di fattori che possono interagire con le radiazioni per aumentare la potenza dei loro effetti cancerogeni. Alcuni di questi fattori includono l’età della donna al momento dell’esposizione, il profilo genetico ed eventualmente i livelli di estrogeni. Studi su donne esposte a fonti di radiazioni militari, accidentali o mediche hanno dimostrato chiaramente che i bambini e gli adolescenti esposti sono più gravemente colpiti nel loro rischio successivo di cancro al seno rispetto alle donne più anziane [769]. Inoltre, dati genetici recenti indicano che le donne con alcune mutazioni genetiche (ad esempio, ATM, TP53 e BRCA1/2) hanno maggiori probabilità di sviluppare il cancro al seno e possono essere particolarmente sensibili agli effetti cancerogeni delle esposizioni alle radiazioni ionizzanti [71, 783- 785].
Gli studi che utilizzano modelli di coltura di cellule tumorali della mammella umana animali e in vitro hanno dimostrato che gli effetti delle radiazioni sulla carcinogenesi mammaria possono essere additivi con gli effetti degli estrogeni [786-788]. Ciò è particolarmente preoccupante data la diffusa esposizione a sostanze chimiche che imitano gli estrogeni nel nostro ambiente e le molteplici fonti di radiazioni ionizzanti.
Esposizioni professionali
Le radiologhe che avevano sostenuto esposizioni giornaliere a radiazioni ionizzanti hanno dimostrato un aumento del rischio di cancro al seno per le donne che hanno iniziato a lavorare durante l’adolescenza o, indipendentemente dall’età, a lavorare sul campo prima degli anni ’40, quando i livelli di esposizione erano sostanzialmente più alti di quelli degli ultimi decenni [789, 790]. Il follow-up di questa coorte per un altro decennio ha rivelato un aumento del tasso di mortalità per i tecnologi che hanno iniziato a lavorare prima del 1950, con una tendenza significativa (P = .01) per la correlazione con l’inizio dell’anno precedente. I tecnologi che hanno iniziato a lavorare prima del 1950 e che hanno lavorato per almeno 5 anni hanno avuto un aumento del tasso di mortalità per cancro al seno (HR = 2,25; 95% CI = .95-6,68) [791]. In un sottoinsieme di questa coorte che aveva lavorato con procedure interventistiche guidate fluoroscopicamente, c’era una maggiore incidenza di cancro al seno rispetto ai tecnologi che non si erano impegnati in questo lavoro (HR = 1,166; 95% CI = 1,02-1,32), ma nessun effetto sulla mortalità per cancro al seno [792]. La suscettibilità dei radiologi alla diagnosi successiva di cancro al seno può essere influenzata da varianti comuni nei geni che sono coinvolti nel metabolismo degli estrogeni circolanti [793].
Una revisione e un’analisi di tutti gli studi correlati esistenti ha rilevato che le donne che lavorano come assistenti di volo delle compagnie aeree hanno aumentato i livelli di cancro al seno [794]. Una meta-analisi di sette studi che esaminano l’incidenza del cancro nelle assistenti di volo ha riportato un’elevata incidenza di cancro al seno (SIR = 1,40; 95% CI = 1,19-1,65) [795]. I fattori che potrebbero spiegare questo aumento possono includere lo stile di vita e le storie riproduttive, le esposizioni alla luce di notte, così come l’aumento delle esposizioni alle radiazioni ionizzanti cosmiche (atmosferiche).
Radiazioni mediche: rischi e benefici
Radiografie mediche: L’uso dei raggi X per esaminare la colonna vertebrale, il cuore, i polmoni, le costole, le spalle e l’esofago espone anche parti del seno alle radiazioni. I raggi X e la fluoroscopia dei neonati irradiano tutto il corpo [796]. Decenni di ricerche hanno confermato il legame tra le radiazioni e il cancro al seno nelle donne che sono state irradiate per molte diverse condizioni mediche, tra cui tubercolosi [105], malattia benigna del seno [106, 107], mastite acuta post-partum [108], timo ingrossato [109, 110], emangiomi cutanei [111], scoliosi [112], Hodgkin’.malattia di Hodgkin [113-116], linfoma non Hodgkin [117], acne [118] e cure dentistiche profilattiche [119].
Ogni volta che l’uso di radiografie diagnostiche del torace prima dei 50 anni di età nelle donne portatrici di mutazioni del gene BRCA1/2 è associato ad un aumento del rischio di cancro al seno (BRCA1 OR = 1,16; 95% CI = .64-2,11; BRCA2 OR = 1,22; 95% CI = .62-2,42) [797].
L’evidenza di quasi tutte le condizioni suggerisce che l’esposizione alle radiazioni ionizzanti durante l’infanzia e l’adolescenza è particolarmente pericolosa rispetto all’aumento del rischio di cancro al seno più tardi nella vita [75, 120, 121, 126] e che esiste una relazione dose-risposta significativa tra il dosaggio delle radiazioni infantili e l’aumento dell’incidenza del cancro al seno (tendenza p < .001) [798]. È importante notare che l’uso di radiazioni in medicina pediatrica porta a una dose efficace più elevata per i bambini che per gli adulti, data l’esposizione equivalente alle radiazioni, un riflesso delle loro dimensioni corporee più piccole [799, 800].
Tomografia computerizzata (TC) Scansioni: Ci sono prove credibili che le radiografie mediche (comprese la mammografia, la fluoroscopia e la TAC) sono una causa importante e controllabile del cancro al seno [119, 801]. Anche se negli ultimi decenni c’è stata una sostanziale diminuzione delle esposizioni a radiazioni ionizzanti da singole radiografie negli ultimi decenni, c’è stato un aumento di sei volte l’esposizione a fonti di radiazioni mediche dalla metà degli anni ’80 fino al 2007, con un aumento annuale del 16%, principalmente a causa dell’aumento dell’uso delle TAC e della medicina nucleare [802, 803]. Nel 2007, circa 72 milioni di TAC sono state effettuate negli Stati Uniti [804]. Quando una TAC è diretta al torace, l’individuo riceve l’equivalente di 30-442 raggi X al torace [805]. Le stime dei modelli hanno indicato che l’uso di TAC toracica e angiografia TC nel solo 2007 porterà ad altri 5300 casi di cancro ai polmoni e al seno nei prossimi due o tre decenni [804]. Altre modellazioni suggeriscono che 1 donna su 150 che ha 20 anni quando si sottopone ad angiogrammi TC del torace, e 1 donna su 270 di tutte le età che si sottopone all’intervento, svilupperà successivamente tumori del torace, incluso il cancro al seno [806].
L’angiografia TC, una fonte di radiazioni relativamente elevate al torace, è stata associata ad un significativo aumento del rischio di sviluppare il cancro al seno, specialmente nelle donne in pre-menopausa [807, 808].
Mammografia: Molti esperti ritengono che le esposizioni a basse dosi di radiazioni ricevute a seguito di procedure mammografiche non siano sufficienti ad aumentare il rischio di cancro al seno. Tuttavia, i danni derivanti da fonti di raggi X a bassa energia, compresi quelli utilizzati nella mammografia, non possono essere previsti stimando il rischio da modelli basati su dosi più elevate [75, 809]. Le prove indicano che le radiografie a bassa energia fornite dalla mammografia hanno causato danni al DNA sostanzialmente maggiori di quelli previsti da tali modelli. Le prove suggeriscono anche che il rischio di cancro al seno causato dall’esposizione alle radiazioni mammografiche può essere notevolmente sottovalutato [808].
Come per altri fattori di rischio per il cancro al seno, sia l’età al momento dell’esposizione che il profilo genetico dell’individuo influenzano il grado di aumento del rischio di malattia nelle donne esposte a mammografie multiple. Per esempio, le donne che hanno fatto la mammografia multipla più di 5 anni prima della diagnosi hanno avuto un aumento del rischio di cancro al seno, ma l’effetto è stato statisticamente significativo solo per le donne la cui prima mammografia è iniziata prima dei 35 anni [119].
Questo effetto dell’età è particolarmente preoccupante, poiché spesso si raccomanda alle donne ad alto rischio, comprese le portatrici di una delle due mutazioni BRCA, di iniziare lo screening mammografico annuale all’età compresa tra i 25 e i 30 anni. Ma le giovani donne con queste mutazioni sono in realtà più vulnerabili agli effetti cancerogeni delle mammografie precoci e ripetute. Questa maggiore vulnerabilità è stata segnalata nelle donne con mutazioni BRCA1/2 [71, 72] e nelle donne con altre variazioni relativamente rare di geni noti per essere coinvolti nel processo di riparazione del DNA [75]. Per le donne con mutazioni di BRCA2, la meta-analisi di sette articoli trovati esposti a basse dosi da mammografia o radiografia toracica ha portato ad un aumento del rischio di cancro al seno (OR = 1,3; 95% CI = .9-1,8). L’esposizione prima dei 20 anni di età ha aumentato il rischio (OR = 2,0; 95% IC = 1,3-3,1), così come l’esposizione a più di 5 anni (OR = 1,8; 95% CI = 1,1-3,0) [67]. È stato dimostrato che le radiazioni diagnostiche aumentano il rischio di sviluppare il cancro al seno in modo dose-dipendente [73].
I rischi dannosi della mammografia potrebbero anche aumentare nelle donne più anziane, le cui cellule epiteliali del seno hanno attraversato diversi decenni di divisione cellulare. Le cellule derivate dal tessuto mammario delle donne più anziane erano più sensibili agli effetti dannosi per il DNA delle radiazioni a bassa energia, aumentando la probabilità di una successiva conversione in cellule cancerose [810].
Nel 2009, l’US Preventive Services Task Force (USPSTF) ha raccomandato di non utilizzare lo screening mammografico di routine prima dei 50 anni (Nelson, [811]; USPSTF, 2009), ma ha sostenuto l’uso dello screening biennale tra i 50 e i 75 anni [811]. Queste raccomandazioni si basavano su modelli che utilizzavano una serie di fattori, tra cui i risultati positivi e negativi dei test e le conseguenze psicologiche di tali risultati sulle donne, il numero di procedure di imaging di follow-up e di biopsie, le diagnosi effettive e, in ultima analisi, i tassi di mortalità per cancro al seno. Non è stato considerato nell’analisi il contributo delle radiazioni provenienti da mammografie singole o ripetute o da altri test di follow-up [812]. Diverse analisi suggeriscono che per le donne di età superiore ai 40 anni che non sono ad alto rischio, i compromessi tra l’efficacia diagnostica della mammografia e l’esposizione alle radiazioni sono più favorevoli a uno screening mammografico regolare [813- 815]. Nel 2016, l’USPSTF ha aggiornato le raccomandazioni per le donne tra i 40 e i 49 anni, lasciando la decisione se iniziare o meno lo screening mammografico alla singola donna [816]. Poiché le donne si trovano ora ad affrontare la necessità di decidere da sole se sottoporsi o meno allo screening mammografico di routine, è fondamentale che sia i medici che le donne siano meglio istruiti sui potenziali danni della mammografia, insieme ai suoi potenziali benefici [72, 817].
La radioterapia: Alcuni studi suggeriscono che medici e pazienti dovrebbero valutare attentamente i rischi e i benefici della radioterapia per le sopravvissute al cancro al seno in fase iniziale, in particolare le donne più anziane. Le donne di età superiore ai 55 anni traggono meno benefici dalla radioterapia in termini di riduzione del tasso di recidiva locale [818] e possono affrontare maggiori rischi di complicazioni cardiovascolari indotte dalle radiazioni [819], nonché di tumori secondari come le leucemie e i tumori del polmone, dell’esofago, dello stomaco e del seno [820, 821]. Utilizzando i dati di sorveglianza, epidemiologia e risultati finali (SEER) del NCI, i ricercatori hanno mostrato un aumento di 16 volte del rischio relativo di angiosarcoma della mammella e della parete toracica in seguito all’irradiazione di un tumore primario al seno [822]. Gli angiosarcomi del seno sono associati ad una prognosi relativamente scarsa [823].
Dati più recenti indicano che le donne di età inferiore ai 45 anni che hanno ricevuto la maggiore esposizione alle radiazioni associata alla radioterapia post-lumpectomia (rispetto alla radioterapia post-mastectomia) hanno avuto un aumento di 1,5-2,5 volte superiore nelle successive diagnosi di tumore mammario controlaterale. Questo effetto è stato particolarmente evidente nelle donne più giovani con una storia familiare di cancro al seno [824- 826]. Bernstein et al. hanno studiato una coorte di donne, annidate all’interno del grande studio WECARE, che avevano sviluppato un tumore al seno controlaterale (rispetto alle pazienti con tumore al seno che non avevano sviluppato un tumore al seno controlaterale). Hanno trovato gli effetti principali sia per lo stato genico che per il trattamento, con elevazioni significative per i portatori di BRCA1/2 (RR = 4,5; 95% CI = 3,0-6,6), e mai il trattamento con la radioterapia (RR = 1,2; 95% CI = 1,0-6,6), ma nessuna interazione significativa tra i due fattori [74].
Radiazioni non ionizzanti (campi elettromagnetici)
Le onde elettromagnetiche sono un tipo di radiazione a bassa frequenza, non ionizzante, senza energia sufficiente per separare gli elettroni dalle loro orbite intorno agli atomi e ionizzare gli atomi. Le microonde, le onde radio, i radar e le radiazioni prodotte dalla trasmissione elettrica sono esempi di sorgenti di radiazioni che generano campi elettromagnetici (CEM). L’illuminazione fluorescente, i computer e molti altri tipi di apparecchiature elettroniche cablate e senza fili (ad esempio, i telefoni cellulari) creano campi elettromagnetici di varia intensità.
Sia IARC e il National Institute of Environmental Health Sciences (NIEHS) EMF Working Group hanno classificato le esposizioni ai campi elettromagnetici come possibile cancerogeno per l’uomo sulla base della letteratura scientifica relativa ai campi elettromagnetici e alle leucemie infantili [827]. Più recentemente, i dati hanno suggerito un collegamento tra l’esposizione ai campi elettromagnetici, in particolare dall’uso del telefono cellulare, e lo sviluppo del cancro al cervello e dei neuromi acustici, anche se la forza di queste connessioni rimane controversa [828]. Il consenso è stato ancora più difficile da raggiungere sulla relazione tra i campi elettromagnetici e il cancro al seno.
Anche se molti studi epidemiologici o occupazionali non hanno trovato relazioni significative tra l’esposizione ai campi elettromagnetici e il rischio di cancro al seno, altri hanno riportato dati a sostegno di questi effetti [829, 830]. Questioni metodologiche possono spiegare alcune delle discrepanze, dati gli effetti relativamente piccoli che si trovano e la natura onnipresente dei campi elettromagnetici “di fondo” nella nostra vita quotidiana [831].
Kliukiene et al. hanno riportato un aumento del rischio di cancro al seno tra le operatrici radiotelegrafiche norvegesi esposte a radiofrequenza (un tipo di CEM) e a campi elettromagnetici a bassissima frequenza. Le donne in pre-menopausa hanno mostrato un aumento del rischio di tumori positivi al recettore degli estrogeni (OR = 1,78; 95% CI = 0,59-5,41) e le donne in post-menopausa hanno avuto un aumento del rischio di tumori negativi al recettore degli estrogeni (OR = 2,37; 95% CI = 0,88-6,36) [832].
In uno studio occupazionale che ha esaminato le donne in ambiente di lavoro con il potenziale di alte, medie o basse esposizioni elettromagnetiche, le esposizioni elevate sono state associate ad un aumento del rischio di sviluppare un tumore al seno (OR = 1,43; 95% CI = 0,99-2,09). Le donne in pre-menopausa sembrano essere più a rischio (OR = 1,98; 95% CI = 1,04-3,78) rispetto alle donne in post-menopausa (OR = 1,33; 95% CI = 0,82-2,17) [833].
Studi sull’esposizione residenziale e professionale ai campi elettromagnetici hanno rilevato un aumento significativo (OR = 1,58; 95% CI = 1,30-1,92) del rischio di cancro al seno tra le donne di tutte le età che vivono in prossimità di linee elettriche ad alta tensione. Effetti simili sono stati riscontrati per le donne con tumori ER+ e ER-. Anche l’esposizione professionale ha aumentato il rischio (OR = 1,13; 95% CI = 0,91-1,40) [834]. Le donne di età inferiore ai 50 anni che sono state esposte ai campi elettromagnetici sia a casa che al lavoro hanno avuto un modesto aumento del rischio di cancro al seno [835].
Tuttavia, le meta-analisi di 15 studi hanno concluso che non esiste una chiara relazione tra l’esposizione ai campi elettromagnetici e il cancro al seno nelle donne (OR = 0.99; 95% CI = 0.90-1.09) [836]. Un’altra meta-analisi che ha esaminato un sottoinsieme di studi pubblicati che hanno specificato la modalità di esposizione ha riportato un piccolo aumento dei tassi di cancro al seno nelle donne in premenopausa associato ad una maggiore esposizione residenziale ai campi elettromagnetici (OR = 1,18; 95% CI = 1,02-1,37) [837].
Sebbene il cancro al seno sia raro negli uomini, numerosi studi sull’esposizione professionale indicano una connessione tra l’esposizione ai campi elettromagnetici e il cancro al seno maschile [838- 842].
In laboratorio, i campi elettromagnetici possono causare un aumento dei tumori mammari negli animali e la proliferazione nei sistemi in cui i tumori delle cellule mammarie umane sono cresciuti in coltura. È importante notare che gli effetti nei roditori si trovano in alcuni ceppi di animali ma non in altri, indicando che sottili differenze nel background genetico potrebbero rendere alcuni animali più suscettibili agli effetti cancerogeni dei campi elettromagnetici [843]. In un sistema cellulare in vitro, l’esposizione ai campi elettromagnetici delle cellule del tumore al seno umano (MCF-7) ha portato ad un’attivazione di geni che sono stati associati con l’induzione di metastasi nelle cellule del cancro al seno [844].
Sommario della sezione: L’esposizione alle radiazioni ionizzanti è una causa nota di aumento del rischio di cancro al seno. Le vittime dell’uso militare delle bombe nucleari hanno aumentato il rischio, così come le donne che hanno subito trattamenti a raggi X a scopo medico, specialmente quando erano giovani. Le donne portatrici delle mutazioni BRCA1/2 sono particolarmente sensibili agli effetti dei raggi X, compresi quelli emessi dalla mammografia di routine.
Risultati più eterogenei provengono da studi su donne esposte a radiazioni non ionizzanti, sia a causa di esposizioni professionali che residenziali.
Discussione e conclusioni
Negli otto anni trascorsi dall’ultima pubblicazione di un’ampia rassegna della letteratura in materia, sono apparsi centinaia di nuovi articoli che affrontano il legame tra l’esposizione a sostanze tossiche ambientali e l’aumento del rischio di sviluppare il cancro al seno; la maggior parte degli studi sostiene l’esistenza di questo legame per gli agenti discussi in questa rassegna. Non solo il corpus della letteratura si è ampliato negli ultimi anni, ma è stato anche potenziato da una maggiore profondità, ampiezza e complessità. La crescente letteratura sulle esposizioni allo sviluppo di EDC e sul successivo sviluppo del cancro al seno è particolarmente forte.
I dati epidemiologici supportano fortemente il legame tra l’aumento del rischio di sviluppare il cancro al seno e le esposizioni precoci allo sviluppo al DES, al DDT e alle radiazioni, così come le esposizioni degli adulti ai contraccettivi orali e alla HRT. Una letteratura in crescita implica anche l’impegno nel lavoro notturno come fattore importante che porta ad un aumento del rischio di cancro al seno. D’altra parte, una consistente letteratura che esamina gli effetti del consumo di prodotti a base di soia e lignani nell’ambito di una dieta regolare, soprattutto all’inizio della vita, indica che essi possono avere un effetto protettivo contro lo sviluppo successivo del cancro al seno.
I modelli animali e altri modelli in vitro sostengono l’ipotesi che molte altre sostanze chimiche presenti nei prodotti di consumo di uso comune, così come nell’aria, nell’acqua e nella polvere, sono tutte associate ad un aumento del rischio di predisposizione dei tessuti mammari a sviluppare tumori. Questi dati supportano i forti legami sopra descritti per gli EDC. I dati degli studi epidemiologici suggeriscono connessioni tra le esposizioni e il successivo sviluppo del cancro, anche se i limiti metodologici spesso limitano le conclusioni che si possono trarre. Di particolare preoccupazione per la maggior parte degli studi epidemiologici in questo campo è la mancanza di una misurazione diretta dei livelli di esposizione ai tossici negli individui, specialmente negli anni (o decenni) precedenti la diagnosi di cancro al seno [845]. Come la letteratura ha documentato chiaramente, spesso c’è una lunga latenza tra le esposizioni e la diagnosi, e le prime esposizioni allo sviluppo possono essere particolarmente potenti nell’influenzare lo sviluppo del cancro al seno, anche decenni dopo [457]. Per ampliare il corpus di lavori rilevanti, sarà importante che i grandi studi di coorte raccolgano regolarmente informazioni sull’esposizione per gran parte della durata della vita e sviluppino le tecnologie necessarie per quantificare i livelli di esposizione, i biomarcatori e gli esiti sanitari su larga scala [845, 846].
È importante sottolineare che, attraverso la coltura animale, la coltura cellulare, l’alta produttività e altri modelli non epidemiologici, si stanno delineando i meccanismi attraverso i quali l’esposizione a vari agenti tossici può portare a un aumento del rischio di sviluppare il cancro. Questa letteratura è stata lenta a svilupparsi, perché gli studi tossicologici di regolamentazione che esaminano le conseguenze riproduttive e di sviluppo delle esposizioni a vari farmaci o potenziali tossici non hanno richiesto l’esame degli endpoint dei tessuti mammari [847]. Non esistono protocolli standardizzati per determinare i tempi appropriati di esposizione, i range di dosi o gli endpoint delle ghiandole mammarie da studiare e, in seguito, i potenziali effetti cancerogeni, genotossici o di interferenza endocrina di queste esposizioni. Al fine di rivendicare in modo più definitivo le connessioni tra le molte sostanze chimiche che sono state implicate nell’aumento del rischio di sviluppo del cancro al seno e i legami causali con la malattia, sarà importante sviluppare una serie di endpoint da studiare di routine. Gli endpoint critici da valutare includono lo sviluppo alterato della ghiandola mammaria, l’attività di vari biomarcatori tra cui PR, LEI, altri fattori endocrini, e diversi sottotipi di vari recettori ormonali, ognuno dei quali può avere effetti diversi sull’attività cellulare quando attivato [848].
Nonostante questi limiti e preoccupazioni metodologiche critiche, l’ampiezza e la forza delle prove citate in questa revisione, se considerate nel loro insieme, rafforzano la conclusione che l’esposizione a un’ampia varietà di sostanze tossiche – molte delle quali si trovano in comuni prodotti e sottoprodotti di uso quotidiano – può portare a un aumento del rischio di sviluppo del cancro al seno. Come concluso dai rapporti del Presidential Cancer Panel [4] e dell’Interagency Breast Cancer and Environment Research Coordinating Committee [2], è fondamentale riconoscere la crescente letteratura che dimostra le connessioni tra l’esposizione a sostanze tossiche ambientali e il successivo sviluppo di malattie, incluso il cancro al seno, e dare priorità alla prevenzione sia a livello di ricerca che di salute pubblica.
References
- Colditz GA, Bohlke K. Priorities for the primary prevention of breast cancer. CA Cancer J Clin. 2014; 64:186-194. DOI | PubMed
- . Accessed 25 Oct 2016.Publisher Full Text
- Kruk J, Aboul-Enein HY. Environmental exposure, and other behavioral risk factors in breast cancer. Curr Cancer Ther Rev. 2006; 2:3-21. DOI
- . Accessed 25 Oct 2016.Publisher Full Text
- Fenton SE. A special issue dedicated to a complex tissue. Reprod Toxicol. 2015; 54:1-5. DOI | PubMed
- American Cancer Society. Breast Cancer Facts & Figures 2015–2016, vol. 2015. Atlanta: American Cancer Society, Inc.
- Sondik EJ. Breast cancer trends: Incidence, mortality, and survival. Cancer. 1994; 74(Suppl 1):995-999. DOI | PubMed
- Timmons FL. A history of weed control in the United States and Canada. Weed Sci. 2005; 53:748-761. DOI
- . Accessed 16 May 2017.Publisher Full Text
- . Accessed 16 May 2017.Publisher Full Text
- Gray J, Evans N, Taylor B, Rizzo J, Walker M. State of the evidence: The connection between breast cancer and the environment. Int J Occup Environ Health. 2009; 15:43-78. DOI | PubMed
- DeSantis CE, Fedewa SA, Goding Sauer A, Kramer JL, Smith RA, Jemal A. Breast cancer statistics, 2015: Convergence of incidence rates between black and white women: Breast cancer statistics, 2015. CA Cancer J Clin. 2016; 66:31-42. DOI | PubMed
- DeSantis CE, Siegel RL, Sauer AG, Miller KD, Fedewa SA, Alcaraz KI. Cancer statistics for African Americans, 2016: Progress and opportunities in reducing racial disparities: Cancer statistics for African Americans, 2016. CA Cancer J Clin. 2016; 66:290-308. DOI | PubMed
- Alizart M, Saunus J, Cummings M, Lakhani SR. Molecular classification of breast carcinoma. Diagn Histopathol. 2012; 18:97-103. DOI
- Curtis C, Shah SP, Chin S-F, Turashvili G, Rueda OM, Dunning MJ. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature. 2012; 486:346-352. PubMed
- Tao Z, Shi A, Lu C, Song T, Zhang Z, Zhao J. Breast cancer: Epidemiology and etiology. Cell Biochem Biophys. 2015; 72:333-338. DOI | PubMed
- Kohler BA, Sherman RL, Howlader N, Jemal A, Ryerson AB, Henry KA, et al. Annual report to the nation on the status of cancer, 1975–2011, featuring incidence of breast cancer subtypes by race/ethnicity, poverty, and state. J Natl Cancer Inste. 2015;107:djv048.
- Inic Z, Zegarac M, Inic M, Markovic I, Kozomara Z, Djurisic I. Difference between luminal a and luminal B subtypes according to Ki-67, tumor size, and progesterone receptor negativity providing prognostic information. Clin Med Insights Oncol. 2014; 8:107-111. DOI | PubMed
- Yang XR, Sherman ME, Rimm DL, Lissowska J, Brinton LA, Peplonska B. Differences in risk factors for breast cancer molecular subtypes in a population-based study. Cancer Epidemiol Biomarkers Prev. 2007; 16:439-443. DOI | PubMed
- Akinyemiju TF, Pisu M, Waterbor JW, Altekruse SF. Socioeconomic status and incidence of breast cancer by hormone receptor subtype. SpringerPlus. 2015;4:508.
- Turkoz FP, Solak M, Petekkaya I, Keskin O, Kertmen N, Sarici F. Association between common risk factors and molecular subtypes in breast cancer patients. Breast. 2013; 22:344-350. DOI | PubMed
- Anderson KN, Schwab RB, Martinez ME. Reproductive risk factors and breast cancer subtypes: A review of the literature. Breast Cancer Res Treat. 2014; 144:1-10. DOI | PubMed
- Bowen R, Stebbing J, Jones L. A review of the ethnic differences in breast cancer. Pharmacogenomics. 2006; 7:935-942. DOI | PubMed
- Chen J-Q, Russo J. ERα-negative and triple negative breast cancer: Molecular features and potential therapeutic approaches. Biochim Biophys Acta. 2009; 1796:162-175. PubMed
- Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V. Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: A population-based study from the California cancer registry. Cancer. 2007; 109:1721-1728. DOI | PubMed
- Lorusso L, Bacchini F. A reconsideration of the role of self-identified races in epidemiology and biomedical research. Stud Hist Philos Biol Biomed Sci. 2015; 52:56-64. DOI | PubMed
- Nelson JW, Scammell MK, Hatch EE, Webster TF. Social disparities in exposures to bisphenol a and polyfluoroalkyl chemicals: A cross-sectional study within NHANES 2003–2006. Environ Health. 2012; 11:1. DOI | PubMed
- Evans GW, Kantrowitz E. Socioeconomic status and health: The potential role of environmental risk exposure. Annu Rev Public Health. 2002; 23:303-331. DOI | PubMed
- Forastiere F, Stafoggia M, Tasco C, Picciotto S, Agabiti N, Cesaroni G. Socioeconomic status, particulate air pollution, and daily mortality: Differential exposure or differential susceptibility. Am J Ind Med. 2007; 50:208-216. DOI | PubMed
- Quinn MM, Sembajwe G, Stoddard AM, Kriebel D, Krieger N, Sorensen G. Social disparities in the burden of occupational exposures: Results of a cross-sectional study. Am J Ind Med. 2007; 50:861-875. DOI | PubMed
- Brulle RJ, Pellow DN. Environmental justice: Human health and environmental inequalities. Annu Rev Public Health. 2006; 27:103-124. DOI | PubMed
- Rauh VA, Landrigan PJ, Claudio L. Housing and health: Intersection of poverty and environmental exposures. Ann N Y Acad Sci. 2008; 1136:276-288. DOI | PubMed
- . Accessed 25 Oct 2016.Publisher Full Text
- Calafat AM, Ye X, Wong L-Y, Reidy JA, Needham LL. Urinary concentrations of triclosan in the U.S. population: 2003-2004. Environ Health Perspect. 2008; 116:303-307. DOI | PubMed
- Mortensen ME, Calafat AM, Ye X. Urinary concentrations of environmental phenols in pregnant women in a pilot study of the National Children’s study. Environ Res. 2014; 129:32-38. DOI | PubMed
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000; 100:57-70. DOI | PubMed
- Goodson WH, Lowe L, Carpenter DO, Gilbertson M, Ali AM, de CS. Assessing the carcinogenic potential of low-dose exposures to chemical mixtures in the environment: The challenge ahead. Carcinogenesis. 2015; 36:S254-S296. DOI | PubMed
- Vandenberg LN, Colborn T, Hayes TB, Heindel JJ, Jacobs DR, Lee D-H. Hormones and endocrine-disrupting chemicals: Low-dose effects and nonmonotonic dose responses. Endocr Rev. 2012; 33:378-455. DOI | PubMed
- Soto AM, Brisken C, Schaeberle C, Sonnenschein C. Does cancer start in the womb? Altered mammary gland development and predisposition to breast cancer due to in utero exposure to endocrine disruptors. J Mammary Gland Biol Neoplasia. 2013; 18:199-208. DOI | PubMed
- Paulose T, Speroni L, Sonnenschein C, Soto AM. Estrogens in the wrong place at the wrong time: Fetal BPA exposure and mammary cancer. Reprod Toxicol. 2015; 54:58-65. DOI | PubMed
- Fenton SE, Birnbaum LS. Timing of environmental exposures as a critical element in breast cancer risk. J Clin Endocrinol Metab. 2015; 100:3245-3250. DOI | PubMed
- Sonnenschein C, Soto AM. The aging of the 2000 and 2011 hallmarks of cancer reviews: A critique. J Biosci. 2013; 38:651-663. DOI | PubMed
- Soto AM, Sonnenschein C. The tissue organization field theory of cancer: A testable replacement for the somatic mutation theory. BioEssays. 2011; 33:332-340. DOI | PubMed
- Mechanic LE, Hutter CM. Environmental Epigenetics. Springer London: London; 2015.
- Rudolph A, Chang-Claude J, Schmidt MK. Gene–environment interaction and risk of breast cancer. Br J Cancer. 2016; 114:125-133. DOI | PubMed
- Vandenberg LN, Colborn T, Hayes TB, Heindel JJ, Jacobs DR, Lee D-H. Regulatory decisions on endocrine disrupting chemicals should be based on the principles of endocrinology. Reprod Toxicol. 2013; 38:1-15. DOI | PubMed
- Zoeller RT, Brown TR, Doan LL, Gore AC, Skakkebaek NE, Soto AM. Endocrine-disrupting chemicals and public health protection: A statement of principles from the Endocrine Society. Endocrinology. 2012; 153:4097-4110. DOI | PubMed
- Diamanti-Kandarakis E, Bourguignon J-P, Giudice LC, Hauser R, Prins GS, Soto AM. Endocrine-disrupting chemicals: An Endocrine Society scientific statement. Endocr Rev. 2009; 30:293-342. DOI | PubMed
- De Coster S, van Larebeke N. Endocrine-disrupting chemicals: Associated disorders and mechanisms of action. J Environ Public Health. 2012; 2012:1-52. DOI
- Di Renzo GC, Conry JA, Blake J, DeFrancesco MS, DeNicola N, Martin JN. International Federation of Gynecology and Obstetrics Opinion on reproductive health impacts of exposure to toxic environmental chemicals. Int J Gynecol Obstet. 2015; 13:218-225.
- Gore AC, Chappell VA, Fenton SE, Flaws JA, Nadal A, Prins GS. EDC-2: The Endocrine Society’s second scientific statement on endocrine-disrupting chemicals. Endocrinol Rev. 2015; 36:1-150. DOI
- Bergman Å, Heindel JJ, Kasten T, Kidd KA, Jobling S, Neira M. The impact of endocrine disruption: A consensus statement on the state of the science. Environ Health Perspect. 2013; 121:A101-A106. DOI
- . Accessed 25 Oct 2016.Publisher Full Text
- DeSesso J, Watson R. The case for integrating low dose, beneficial responses into US EPA risk assessments. Hum Exp Toxicol. 2006; 25:7-10. DOI | PubMed
- Moral R, Wang R, Russo IH, Lamartiniere CA, Pereira J, Russo J. Effect of prenatal exposure to the endocrine disruptor bisphenol a on mammary gland morphology and gene expression signature. J Endocrinol. 2008; 196:101-112. DOI | PubMed
- Mandrup K, Boberg J, Isling LK, Christiansen S, Hass U. Low-dose effects of bisphenol a on mammary gland development in rats. Andrology. 2016; 4:673-683. DOI | PubMed
- Blei T, Soukup ST, Schmalbach K, Pudenz M, Möller FJ, Egert B. Dose-dependent effects of isoflavone exposure during early lifetime on the rat mammary gland: Studies on estrogen sensitivity, isoflavone metabolism, and DNA methylation. Mol Nutr Food Res. 2015; 59:270-283. DOI | PubMed
- Kortenkamp A. Breast cancer, oestrogens and environmental pollutants: A re-evaluation from a mixture perspective. Int J Androl. 2006; 29:193-198. DOI | PubMed
- Koppe J, Bartonova A, Bolte G, Bistrup ML, Busby C, Butter M. Exposure to multiple environmental agents and their effect. Acta Paediatr Suppl. 2006; 95:106-113. DOI | PubMed
- Soto AM, Chung KL, Olea N. The E-SCREEN assay as a tool to identify estrogens: An update on estrogenic environmental pollutants. Environ Health Perspect. 1995; 103:113-122. DOI
- Silva E, Scholze M, Kortenkamp A. Activity of xenoestrogens at nanomolar concentrations in the E-screen assay. Environ Health Perspect. 2007; 115:91-97. DOI | PubMed
- van Meeuwen JA, ter Burg W, Piersma AH, van den Berg M, Sanderson JT. Mixture effects of estrogenic compounds on proliferation and pS2 expression of MCF-7 human breast cancer cells. Food Chem Toxicol. 2007; 45:2319-2330. DOI | PubMed
- Rajapakse N, Silva E, Kortenkamp A. Combining xenoestrogens at levels below individual no-observed-effect concentrations dramatically enhances steroid hormone action. Environ Health Perspect. 2002; 110:917-921. DOI | PubMed
- Rajapakse N, Ong D, Kortenkamp A. Defining the impact of weakly estrogenic chemicals on the action of steroidal estrogens. Toxicol Sci. 2001; 60:296-304. DOI | PubMed
- Payne J, Rajapakse N, Wilkins M, Kortenkamp A. Prediction and assessment of the effects of mixtures of four xenoestrogens. Environ Health Perspect. 2000; 108:983. DOI | PubMed
- Boada LD, Zumbado M, Henríquez-Hernández LA, Almeida-González M, Álvarez-León EE, Serra-Majem L. Complex organochlorine pesticide mixtures as determinant factor for breast cancer risk: A population-based case–control study in the Canary Islands (Spain). Environ Health. 2012; 11:1. DOI | PubMed
- Rivero J, Luzardo OP, Henríquez-Hernández LA, Machín RP, Pestano J, Zumbado M. In vitro evaluation of oestrogenic/androgenic activity of the serum organochlorine pesticide mixtures previously described in a breast cancer case–control study. Sci Total Environ. 2015; 537:197-202. DOI | PubMed
- Valerón PF, Pestano JJ, Luzardo OP, Zumbado ML, Almeida M, Boada LD. Differential effects exerted on human mammary epithelial cells by environmentally relevant organochlorine pesticides either individually or in combination. Chem Biol Interact. 2009; 180:485-491. DOI | PubMed
- Foster WG. Mammary gland morphology in Sprague-Dawley rats following treatment with an organochlorine mixture in utero and neonatal genistein. Toxicol Sci. 2003; 77:91-100. DOI | PubMed
- Imaoka T, Nishimura M, Teramoto A, Nishimura Y, Ootawara M, Osada H. Cooperative induction of rat mammary cancer by radiation and 1-methyl-1-nitrosourea via the oncogenic pathways involving c-Myc activation and H-ras mutation. Int J Cancer. 2005; 115:187-193. DOI | PubMed
- Berrington de Gonzalez A, Berg CD, Visvanathan K, Robson M. Estimated risk of rradiation-induced breast cancer from mammographic screening for young BRCA mutation carriers. J Natl Cancer Inst. 2009; 101:205-209. DOI | PubMed
- Jansen-van der Weide MC, Greuter MJW, Jansen L, Oosterwijk JC, Pijnappel RM, de Block GH. Exposure to low-dose radiation and the risk of breast cancer among women with a familial or genetic predisposition: A meta-analysis. Eur Radiol. 2010; 20:2547-2556. DOI | PubMed
- Pijpe A, Andrieu N, Easton DF, et al. Exposure to diagnostic radiation and risk of breast cancer among carriers of BRCA1/2 mutations: Retrospective cohort study (GENE-RAD-RISK). Brit Med J. 2012;345:e5660.
- Bernstein JL, Thomas DC, Shore RE, Robson M, Boice JD, Stovall M. Contralateral breast cancer after radiotherapy among BRCA1 and BRCA2 mutation carriers: A WECARE study report. Eur J Cancer. 2013; 49:2979-2985. DOI | PubMed
- Millikan RC. Polymorphisms in DNA repair genes, medical exposure to ionizing radiation, and breast cancer risk. Cancer Epidemiol Biomarkers Prev. 2005; 14:2326-2334. DOI | PubMed
- Fletcher O, Dudbridge F. Candidate gene-environment interactions in breast cancer. BMC Med. 2014; 12:195. DOI | PubMed
- Bernal AJ, Jirtle RL. Epigenomic disruption: The effects of early developmental exposures: Epigenomic reactions to early exposures. Birth Defects Res A Clin Mol Teratol. 2010; 88:938-944. DOI | PubMed
- Wild CP, Scalbert A, Herceg Z. Measuring the exposome: A powerful basis for evaluating environmental exposures and cancer risk. Environ Mol Mutagen. 2013; 54:480-499. DOI | PubMed
- Treviño LS, Wang Q, Walker CL. Hypothesis: Activation of rapid signaling by environmental estrogens and epigenetic reprogramming in breast cancer. Reprod Toxicol. 2015; 54:136-140. DOI | PubMed
- Luzhna L, Kutanzi K, Kovalchuk O. Gene expression and epigenetic profiles of mammary gland tissue: Insight into the differential predisposition of four rat strains to mammary gland cancer. Mutat Res. 2015; 779:39-56. DOI
- Samantarrai D, Dash S, Chhetri B, Mallick B. Genomic and epigenomic cross-talks in the regulatory landscape of miRNAs in breast cancer. Mol Cancer Res. 2013; 11:315-328. DOI | PubMed
- Khan SI, Aumsuwan P, Khan IA, Walker LA, Dasmahapatra AK. Epigenetic events associated with breast cancer and their prevention by dietary components targeting the epigenome. Chem Res Toxicol. 2012; 25:61-73. DOI | PubMed
- Kutanzi K, Kovalchuk O. Exposure to estrogen and ionizing radiation causes epigenetic dysregulation, activation of mitogen-activated protein kinase pathways, and genome instability in the mammary gland of ACI rats. Cancer Biol Ther. 2013; 14:564-573. DOI | PubMed
- Ye W, Xu P, Jen R, Feng E, Zhong S, Li H. Zeranol down-regulates p53 expression in primary cultured human breast cancer epithelial cells through epigenetic modification. Int J Mol Sci. 2011; 12:1519-1532. DOI | PubMed
- Hussain I, Bhan A, Ansari KI, Deb P, Bobzean SAM, Perrotti LI. Bisphenol-a induces expression of HOXC6, an estrogen-regulated homeobox-containing gene associated with breast cancer. Biochim Biophys Acta. 2015; 1849:697-708. DOI | PubMed
- Doherty LF, Bromer JG, Zhou Y, Aldad TS, Taylor HS. In utero exposure to diethylstilbestrol (DES) or bisphenol-a (BPA) increases EZH2 expression in the mammary gland: An epigenetic mechanism linking endocrine disruptors to breast cancer. Horm Cancer. 2010; 1:146-155. DOI | PubMed
- Soto AM, Sonnenschein C. The somatic mutation theory of cancer: Growing problems with the paradigm?. BioEssays. 2004; 26:1097-1107. DOI | PubMed
- Soto AM, Maffini MV, Sonnenschein C. Neoplasia as development gone awry: The role of endocrine disruptors. Int J Androl. 2008; 31:288-293. DOI | PubMed
- Wadia PR, Cabaton NJ, Borrero MD, et al. Low-dose BPA exposure alters the mesenchymal and epithelial transcriptomes of the mouse fetal mammary gland. PLoS One. 2013;8:e63902.
- Markey CM, Luque EH, Munoz de Toro M, Sonneschein C, Soto AM. In utero exposure to bisphenol a alters the development and tissue organization of the mouse mammary gland. Biol Reprod. 2001; 65:1215-1223. DOI | PubMed
- Vandenberg LN, Wadia PR, Schaeberle CM, Rubin BS, Sonnenschein C, Soto AM. The mammary gland response to estradiol: Monotonic at the cellular level, non-monotonic at the tissue-level of organization?. J Steroid Biochem Mol Biol. 2006; 101:263-274. DOI | PubMed
- Vandenberg LN, Maffini MV, Wadia PR, Sonnenschein C, Rubin BS, Soto AM. Exposure to environmentally relevant doses of the xenoestrogen bisphenol-a alters development of the fetal mouse mammary gland. Endocrinology. 2007; 148:116-127. DOI | PubMed
- Tamimi R, Lagiou P, Vatten LJ, Mucci L, Trichopoulos D, Hellerstein S. Pregnancy hormones, pre-eclampsia, and implications for breast cancer risk in the offspring. Cancer Epidemiol Biomarkers Prev. 2003; 12:647-650. PubMed
- Vatten LJ, Forman MR, Nilsen TIL, Barrett JC, Romundstad PR. The negative association between pre-eclampsia and breast cancer risk may depend on the offspring’s gender. Br J Cancer. 2007; 180:1436-1438.
- Hickey M, Hart R, Keelan JA. The relationship between umbilical cord estrogens and perinatal characteristics. Cancer Epidemiol Biomarkers Prev. 2014; 23:946-952. DOI | PubMed
- Ekbom A, Adami H-O, Lan S-J, Trichopoulos D, Hsieh C-C. Evidence of prenatal influences on breast cancer risk. Lancet. 1992; 340:1015-1018. DOI | PubMed
- Trichopoulos D. Is breast cancer initiated in utero?. Epidemiology. 1990; 1:95-96. DOI | PubMed
- Titus-Ernstoff L, Hatch E, Hoover R, Palmer J, Greenberg E, Ricker W. Long-term cancer risk in women given diethylstilbestrol (DES) during pregnancy. Br J Cancer. 2001; 84:126-133. DOI | PubMed
- Hoover RN, Hyer M, Pfeiffer RM, Adam E, Bond B, Cheville AL. Adverse health outcomes in women exposed in utero to diethylstilbestrol. N Engl J Med. 2011; 365:1304-1314. DOI | PubMed
- Hilakivi-Clarke L. Maternal exposure to diethylstilbestrol during pregnancy and increased breast cancer risk in daughters. Breast Cancer Res. 2014; 16:208. DOI | PubMed
- Palmer JR. Prenatal diethylstilbestrol exposure and risk of breast cancer. Cancer Epidemiol Biomarkers Prev. 2006; 15:1509-1514. DOI | PubMed
- Troisi R, Hatch EE, Titus-Ernstoff L, Hyer M, Palmer JR, Robboy SJ. Cancer risk in women prenatally exposed to diethylstilbestrol. Int J Cancer. 2007; 121:356-360. DOI | PubMed
- Cohn BA, La M, Krigbaum NY, Yeh G, Park J-S, Zimmermann L. DDT exposure in utero and breast cancer. J Clin Endocrinol Metab. 2015; 100:2865-2872. DOI | PubMed
- Cohn BA, Wolff MS, Cirillo PM, Sholtz RI. DDT and breast cancer in young women: New data on the significance of age at exposure. Environ Health Perspect. 2007; 115:1406-1414. PubMed
- Mackenzie I. Breast cancer following multiple fluoroscopies. Br J Cancer. 1965; 19:1-8. DOI | PubMed
- Golubicic I, Borojevic N, Pavlovic T. Risk factors for breast cancer: Is ionizing radiation among them?. J BUON. 2008; 13:487-494. PubMed
- Mattson A, Ruden BI, Palmgren J, Rutgvist LE. Dose- and time-response for breast cancer risk after radiation therapy for benign breast disease. Br J Cancer. 1995; 72:1054-1061. DOI | PubMed
- Shore RE, Hildreth N, Woodard E, Dvoretsky P, Hempelmann L, Pasternack B. Breast cancer among women given X-ray therapy for acute postpartum mastitis. J Natl Cancer Inst. 1986; 77:689-696. DOI | PubMed
- Adams MJ, Dozier A, Shore RE, Lipshultz SE, Schwartz RG, Constine LS. Breast cancer risk 55+ years after irradiation for an enlarged thymus and its implications for early childhood medical irradiation today. Cancer Epidemiol Biomarkers Prev. 2010; 19:48-58. DOI | PubMed
- Hildreth NG, Shore RE, Dvoretsky PM. The risk of breast cancer after irradiation of the thymus in infancy. N Engl J Med. 1989; 321:1281-1284. DOI | PubMed
- Lundell M, Mattsson A, Karlsson P, Holmberg E, Gustafsson A, Holm L-E. Breast ancer risk after radiotherapy in infancy: A pooled analysis of two Swedish cohorts of 17,202 infants. Radiat Res. 1999; 151:626. DOI | PubMed
- Morin Doody M, Lonstein JE, Stovall M, Hacker DG, Luckyanov N, Land CE. Breast cancer mortality after diagnostic radiography: Findings from the U.S. scoliosis cohort study. Spine. 2000; 25:2052-2063. DOI | PubMed
- Bhatia S. High risk of subsequent neoplasms continues with extended follow-up of childhood Hodgkin’s disease: Report from the late effects study group. J Clin Oncol. 2003; 21:4386-4394. DOI | PubMed
- Guibout C, Adjadj E, Rubino C, Shamsaldin A, Grimaud E, Hawkins M. Malignant breast tumors after radiotherapy for a first cancer during childhood. J Clin Oncol. 2005; 23:197-204. DOI | PubMed
- Horwich A, Swerdlow AJ. Second primary breast cancer after Hodgkin’s disease. Br J Cancer. 2004; 90:294-298. DOI | PubMed
- Wahner-Roedler DL, Petersen IA. Risk of breast cancer and breast cancer characteristics in women after treatment for Hodgkin lymphoma. Drugs Today (Barc). 2004; 40:865-879. DOI | PubMed
- Tward JD, Wendland MMM, Shrieve DC, Szabo A, Gaffney DK. The risk of secondary malignancies over 30 years after the treatment of non-Hodgkin lymphoma. Cancer. 2006; 107:108-115. DOI | PubMed
- El-Gamal H, Bennett RG. Increased breast cancer risk after radiotherapy for acne among women with skin cancer. J Am Acad Dermatol. 2006; 55:981-989. DOI | PubMed
- Ma H, Hill CK, Bernstein L, Ursin G. Low-dose medical radiation exposure and breast cancer risk in women under age 50 years overall and by estrogen and progesterone receptor status: Results from a case-control and a case-case comparison. Breast Cancer Res Treat. 2008; 109:77-90. DOI | PubMed
- John EM, Phipps AI, Knight JA, Milne RL, Dite GS, Hopper JL. Medical radiation exposure and breast cancer risk: Findings from the breast cancer family registry. Int J Cancer. 2007; 121:386-394. DOI | PubMed
- Koo E, Henderson MA, Dwyer M, Skandarajah AR. Management and prevention of breast cancer after radiation to the chest for childhood, adolescent, and young adulthood malignancy. Ann Surg Oncol. 2015; 22:545-551. DOI
- Russo J, Russo IH. Molecular basis of breast cancer. Springe-Verlag: Berlin; 2004.
- Briskin C. Progesterone signaling in breast cancer: A neglected hormone coming into the limelight. Nat Rev Cancer. 2013; 13:385-396. DOI | PubMed
- Gail MH, Brinton LA, Byar DP, Corle DK, Green SB, Schairer C. Projecting individualized probabilities of developing breasat cancer for white females who are being examined annually. J Natl Cancer Inst. 1989; 81:1879-1886. DOI | PubMed
- Gail MH. Twenty-five years of breast cancer risk models and their applications. J Natl Cancer Inst. 2015; 107:djv042. DOI | PubMed
- Hoover RN, Troisi RJ. Understanding mechanisms of breast cancer prevention. J Natl Cancer Inst. 2001; 93:1119-1120. DOI | PubMed
- Hilakivi-Clarke L, de Assis S, Warri A. Exposures to synthetic estrogens at different times during the life, and their effect on breast cancer risk. J Mammary Gland Biol Neoplasia. 2013; 18:25-42. DOI | PubMed
- Reed CE, Fenton SE. Exposure to diethylstilbestrol during sensitive life stages: A legacy of heritable health effects. Birth Defects Res C Embryo Today. 2013; 99:134-146. DOI | PubMed
- Walker BE, Haven MI. Intensity of multigenerational carcinogenesis from diethylstilbestrol in mice. Carcinogenesis. 1997; 18:791-793. DOI | PubMed
- Saeed M, Rogan E, Cavalieri E. Mechanism of metabolic activation and DNA adduct formation by the human carcinogen diethylstilbestrol: The defining link to natural estrogens. Int J Cancer. 2009; 124:1276-1284. DOI | PubMed
- Larson PS, Ungarelli RA, de Las MA, Cupples LA, Rowlings K, Palmer JR. In utero exposure to diethylstilbestrol (DES) does not increase genomic instability in normal or neoplastic breast epithelium. Cancer. 2006; 107:2122-2126. DOI | PubMed
- Ayyanan A, Laribi O, Schuepbach-Mallepell S, Schrick C, Gutierrez M, Tanos T. Perinatal exposure to bisphenol a increases adult mammary gland progesterone response and cell number. Mol Endocrinol. 2011; 25:1915-1923. DOI | PubMed
- Bhan A, Hussain I, Ansari KI, Bobzean SAM, Perrotti LI, Mandal SS. Bisphenol-a and diethylstilbestrol exposure induces the expression of breast cancer associated long noncoding RNA HOTAIR in vitro and in vivo. J Steroid Biochem Mol Biol. 2014; 141:160-170. DOI | PubMed
- Bhan A, Hussain I, Ansari KI, Bobzean SAM, Perrotti LI, Mandal SS. Histone methyltransferase EZH2 is transcriptionally induced by estradiol as well as estrogenic endocrine disruptors bisphenol-a and diethylstilbestrol. J Mol Biol. 2014; 426:3426-3441. DOI | PubMed
- Dhimolea E, Wadia PR, Murray TJ. Prenatal exposure to BPA alters the epigenome of the rat mammary gland and increases the propensity to neoplastic development. PLoS One. 2014; 9:e99800. DOI | PubMed
- Knower KC, To SQ, Leung Y-K, Ho S-M, Clyne CD. Endocrine disruption of the epigenome: A breast cancer link. Endocr Relat Cancer. 2014; 21:T33-T55. DOI | PubMed
- Rossouw JE, Anderson GL, Prentice RL. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: Principal results from the women’s health initiative randomized controlled trial. JAMA. 2002; 288:321-333. DOI | PubMed
- Anderson GL, Chlebowski RT, Aragaki AK, Kuller LH, Manson JE, Gass M. Conjugated equine oestrogen and breast cancer incidence and mortality in postmenopausal women with hysterectomy: Extended follow-up of the Women’s health initiative randomised placebo-controlled trial. Lancet Oncol. 2012; 13:476-486. DOI | PubMed
- Anderson GL, Limacher M, Assaf AR, Bassford T, Beresford SAA, Black H. Effects of conjugated equine estrogen in postmenopausal women with hysterectomy: The Women’s health initiative randomized controlled trial. J Am Med Assoc. 2004; 291:1701-1712. DOI
- Montgomery BE, Daum GS, Dunton CJ. Endometrial hyperplasia: A review. Obstet Gynecol Surv. 2004; 59:368-378. DOI | PubMed
- Wood CE, Clarkson TB, Chen H, Veenstra TD, Xu X, Scott L. Comparative effects of oral conjugated equine estrogens and micronized 17beta-estradiol on breast proliferation: A retrospective analysis. Menopause. 2008; 15:890-898. DOI | PubMed
- Chlebowski RT, Rohan TE, Manson JE, Aragaki AK, Kaunitz A, Stefanick ML. Breast cancer after use of estrogen plus progestin and estrogen alone: Analyses of data from 2 Women’s health initiative randomized clinical trials. JAMA Oncol. 2015; 1:296-305. DOI | PubMed
- Krieger N, Chen JT, Waterman PD. Decline in US breast cancer rates after the Women’s health initiative: Socioeconomic and racial/ethnic differentials. Am J Public Health. 2010; 100(Suppl 1):S132-S139. DOI | PubMed
- Verkooijen HM, Bouchardy C, Vinh-Hung V, Rapiti E, Hartman M. The incidence of breast cancer and changes in the use of hormone replacement therapy: A review of the evidence. Maturitas. 2009; 64:80-85. DOI | PubMed
- Holmberg L, Anderson H, HABITS steering and data monitoring committees. HABITS (hormonal replacement therapy after breast cancer—is it safe?), a randomized comparison: Trial stopped. Lancet. 2004; 363:453-455. DOI | PubMed
- Million Women Study Collaborators. Breast cancer and hormone replacement therapy in the million women study. Lancet. 2003; 362:419-427. DOI | PubMed
- Robbins AS, Clarke CA. Re: Declines in invasive breast cancer and use of postmenopausal hormone therapy in a screening mammography population. J Natl Cancer Inst. 2007; 99:1815. DOI
- Kotsopoulos J, Huzarski T, Gronwald J, Moller P, Lynch HT, Neuhausen SL. Hormone replacement therapy after menopause and risk of breast cancer in BRCA1 mutation carriers: A case–control study. Breast Cancer Res Treat. 2016; 155:365-373. DOI | PubMed
- Hou N, Hong S, Wang W, Olopade OI, Dignam JJ, Huo D. Hormone replacement therapy and breast cancer: Heterogeneous risks by race, weight, and breast density. J Natl Cancer Inst. 2013; 105:1365-1372. DOI | PubMed
- Pizot C, Boniol M, Mullie P, Koechlin A, Boniol M, Boyle P. Physical activity, hormone replacement therapy and breast cancer risk: A meta-analysis of prospective studies. Eur J Cancer. 2016; 52:138-154. DOI | PubMed
- Biglia N, Sgro L, Defabiani E, De Rosa G, Ponzone R, Marenco D. The influence of hormone replacement therapy on the pathology of breast cancer. Eur J Surg Oncol. 2005; 31:467-472. DOI | PubMed
- Borgquist S, Anagnostaki L, Jirström K, Landberg G, Manjer J. Breast tumours following combined hormone replacement therapy express favourable prognostic factors. Int J Cancer. 2007; 120:2202-2207. DOI | PubMed
- Reeves GK, Beral V, Green J, Gathani T, Bull D, Million Women Study Collaborators. Hormonal therapy for menopause and breast-cancer risk by histological type: A cohort study and meta-analysis. Lancet Oncol.. 2006; 7:910-918. DOI | PubMed
- Schuetz F, Diel IJ, Pueschel M, von Holst T, Solomayer EF, Lange S. Reduced incidence of distant metastases and lower mortality in 1072 patients with breast cancer with a history of hormone replacement therapy. Am J Obstet Gynecol. 2007; 196:342.e1-342.e9. DOI | PubMed
- Davis R, Batur P, Thacker HL. Risks and effectiveness of compounded bioidentical hormone therapy: A case series. J Women’s Health (Larchmt). 2014; 23:642-648. DOI | PubMed
- Files JA, Ko MG, Pruthi S. Bioidentical hormone therapy. Mayo Clin Proc. 2011; 86:673-680. DOI | PubMed
- Rosenthal MS. The Wiley protocol: An analysis of ethical issues. Menopause. 2008; 15:1014-1022. DOI | PubMed
- L’hermite M, Simoncini T, Fuller S, Genazzani AR. Could transdermal estradiol + progesterone be a safer postmenopausal HRT? A review. Maturitas. 2008; 60:185-201. DOI | PubMed
- Campagnoli C, Clavel-Chapelon F, Kaaks R, Peris C, Berrino F. Progestins and progesterone in hormone replacement therapy and the risk of breast cancer. J Steroid Biochem Mol Biol. 2005; 96:95-108. DOI | PubMed
- Holtorf K. The bioidentical hormone debate: Are bioidentical hormones (estradiol, estriol, and progesterone) safer or more efficacious than commonly used synthetic versions in hormone replacement therapy?. Postgrad Med. 2009; 121:73-85. DOI
- Fournier A, Berrino F, Clavel-Chapelon F. Unequal risks for breast cancer associated with different hormone replacement therapies: Results from the E3N cohort study. Breast Cancer Res Treat. 2008; 107:103-111. DOI | PubMed
- Fournier A, Fabre A, Mesrine S, Boutron-Ruault M-C, Berrino F, Clavel-Chapelon F. Use of different postmenopausal hormone therapies and risk of histology- and hormone receptor-defined invasive breast cancer. J Clin Oncol. 2008; 26:1260-1268. DOI | PubMed
- Althuis MD, Brogan DR, Coates RJ, Daling JR, Gammon MD, Malone KE. Hormonal content and potency of oral contraceptives and breast cancer risk among young women. Br J Cancer. 2003; 88:50-57. DOI | PubMed
- Delort L, Kwiatkowski F, Chalabi N, Satih S, Bignon Y-J, Bernard-Gallon DJ. Risk factors for early age at breast cancer onset—the “COSA program” population-based study. Anticancer Res. 2007; 27:1087-1094. PubMed
- Dai Q, Liu B, Du Y. Meta-analysis of the risk factors of breast cancer concerning reproductive factors and oral contraceptive use. Front Med China. 2009; 3:452-458. DOI
- Pasanisi P, Hédelin G, Berrino J, Chang-Claude J, Hermann S, Steel M. Oral contraceptive use and BRCA penetrance: A case-only study. Cancer Epidemiol Biomark Prev. 2009; 18:2107-2113. DOI
- Kumle M, Weiderpass E, Braaten T, Persson I, Adami H-O, Lund E. Use of oral contraceptives and breast cancer risk: The Norwegian-Swedish Women’s lifestyle and health cohort study. Cancer Epidemiol Biomark Prev. 2002; 11:1375-1381.
- Rosenberg L, Boggs DA, Wise LA, Adams-Campbell LL, Palmer JR. Oral contraceptive use and estrogen/progesterone receptor-negative breast cancer among African American women. Cancer Epidemiol Biomark Prev. 2010; 19:2073-2079. DOI
- Huzell L, Persson M, Simonsson M, Markkula A, Ingvar C, Rose C. History of oral contraceptive use in breast cancer patients: Impact on prognosis and endocrine treatment response. Breast Cancer Res Treat. 2015; 149:505-515. DOI | PubMed
- Sweeney C, Giuliano AR, Baumgartner KB, Byers T, Herrick JS, Edwards SL. Oral, injected and implanted contraceptives and breast cancer risk among U.S. Hispanic and non-Hispanic white women. Int J Cancer. 2007; 121:2517-2523. DOI | PubMed
- Rosenberg L, Zhang Y, Coogan PF, Strom BL, Palmer JR. A case-control study of oral contraceptive use and incident breast cancer. Am J Epidemiol. 2009; 169:473-479. DOI | PubMed
- Haile RW, Thomas DC, McGuire V, Felberg A, John EM, Milne RL. BRCA1 and BRCA2 mutation carriers, oral contraceptive use, and breast cancer before age 50. Cancer Epidemiol Biomark Prev. 2006; 15:1863-1870. DOI
- Narod SA, Dubé M-P, Klijn J. Oral contraceptives and the risk of breast cancer in BRCA1 and BRCA2 mutation carriers. J Natl Cancer Inst. 2002; 94:1773-1779. DOI | PubMed
- Bernholtz S, Laitman Y, Kaufman B, Paluch Shimon S, Friedman E. Cancer risk in Jewish BRCA1 and BRCA2 mutation carriers: Effects of oral contraceptive use and parental origin of mutation. Breast Cancer Res Treat. 2011; 129:557-563. DOI | PubMed
- Calvo V, Beato M. BRCA1 counteracts progesterone action by ubiquitination leading to progesterone receptor degradation and epigenetic silencing of target promoters. Cancer Res. 2011; 71:3422-3431. DOI | PubMed
- Jia X, Liu G, Mo M, Cheng J, Shen Z, Shao Z. Reproductive factors and hormone receptor status among very young (<35 years) breast cancer patients. Oncotarget. 2015; 6:24571-24580. DOI | PubMed
- Newcomer LM, Newcomb PA, Trentham-Dietz A, Longnecker MP, Greenberg ER. Oral contraceptive use and risk of breast cancer by histologic type. Int J Cancer. 2003;106:961–4.
- Dolle JM, Daling JR, White E, Brinton LA, Doody DR, Porter PL. Risk factors for triple-negative breast cancer in women under the age of 45 years. Cancer Epidemiol Biomark Prev. 2009; 18:1157-1166. DOI
- Bethea TN, Rosenberg L, Hong C-C, Troester MA, Lunetta KL, Bandera EV. A case-control analysis of oral contraceptive use and breast cancer subtypes in the African American breast cancer epidemiology and risk consortium. Breast Cancer Res. 2015; 17:22. DOI | PubMed
- Phillips LS, Millikan RC, Schroeder JC, Barnholtz-Sloan JS, Levine BJ. Reproductive and hormonal risk factors for ductal carcinoma in situ of the breast. Cancer Epidemiol Biomark Prev. 2009; 18:1507-1515. DOI
- Pervez S, Khan H. Infiltrating ductal carcinoma breast with central necrosis closely mimicking ductal carcinoma in situ (comedo type): A case series. J Med Case Rep. 2007; 1:83. DOI | PubMed
- Calle EE, Heath J, Miracle-McMahill HL, Coates RJ, Liff JM, Franceschi S. Breast cancer and hormonal contraceptives: Collaborative reanalysis of individual data on 53 297 women with breast cancer and 100 239 women without breast cancer from 54 epidemiological studies. Lancet. 1996; 347:1713-1727. DOI | PubMed
- Vessey M, Painter R. Oral contraceptive use and cancer. Findings in a large cohort study, 1968-2004. Br J Cancer. 2006; 95:385-389. DOI | PubMed
- Beaber EF, Buist DSM, Barlow WE, Malone KE, Reed SD, Li CI. Recent oral contraceptive use by formulation and breast cancer risk among women 20 to 49 years of age. Cancer Res. 2014; 74:4078-4089. DOI | PubMed
- Urban M, Banks E, Egger S, Canfell K, O’Connell D, Beral V. Injectable and oral contraceptive use and cancers of the breast, cervix, ovary, and endometrium in black south African women: Case-control study. PLoS Med. 2012; 9:e1001182. DOI | PubMed
- Gauthier E, Paoletti X, Clavel-Chapelon F, E3N group. Breast cancer risk associated with being treated for infertility: Results from the French E3N cohort study. Hum Reprod. 2004; 19:2216-2221. DOI | PubMed
- Klip H, Burger CW, Kenemans P, van Leeuwen FE. Cancer risk associated with subfertility and ovulation induction: A review. Cancer Causes Control. 2000; 11:319-344. DOI | PubMed
- Orgéas CC, Sanner K, Hall P, Conner P, Holte J, Nilsson SJ. Breast cancer incidence after hormonal infertility treatment in Sweden: A cohort study. Am J Obstet Gynecol. 2009; 200:72.e1-72.e7. DOI | PubMed
- Kessous R, Davidson E, Meirovitz M, Sergienko R, Sheiner E. The risk of female malignancies after fertility treatments: A cohort study with 25-year follow-up. J Cancer Res Clin Oncol. 2016; 142:287-293. DOI | PubMed
- Kotsopoulos J, Librach CL, Lubinski J, Gronwald J, Kim-Sing C, Ghadirian P. Infertility, treatment of infertility, and the risk of breast cancer among women with BRCA1 and BRCA2 mutations: A case-control study. Cancer Causes Control. 2008; 19:1111-1119. DOI | PubMed
- Krul IM, Groeneveld E, Spaan M, van den Belt-Dusebout AW, Mooij TM, Hauptmann M. Increased breast cancer risk in in vitro fertilisation treated women with a multiple pregnancy: A new hypothesis based on historical in vitro fertilisation treatment data. Eur J Cancer. 2015; 51:112-120. DOI | PubMed
- Brinton LA, Scoccia B, Moghissi KS, Westhoff CL, Althuis MD, Mabie JE. Breast cancer risk associated with ovulation-stimulating drugs. Hum Reprod. 2004; 19:2005-2013. DOI | PubMed
- Silva I dos S, Wark PA, McCormack VA, Mayer D, Overton C, Little V. Ovulation-stimulation drugs and cancer risks: A long-term follow-up of a British cohort. Br J Cancer. 2009; 100:1824-1831. DOI | PubMed
- Calderon-Margalit R, Friedlander Y, Yanetz R, Kleinhaus K, Perrin MC, Manor O. Cancer risk after exposure to treatments for ovulation induction. Am J Epidemiol. 2009; 169:365-375. DOI | PubMed
- Lerner-Geva L, Keinan-Boker L, Blumstein T, Boyko V, Olmar L, Mashiach S. Infertility, ovulation induction treatments and the incidence of breast cancer—a historical prospective cohort of Israeli women. Breast Cancer Res Treat. 2006; 100:201-212. DOI | PubMed
- Stewart LM, Holman CDJ, Hart R, Bulsara MK, Preen DB, Finn JC. In vitro fertilization and breast cancer: Is there cause for concern?. Fertil Steril. 2012; 98:334-340. DOI | PubMed
- Rudel RA, Attfield KR, Schifano JN, Brody JG. Chemicals causing mammary gland tumors in animals signal new directions for epidemiology, chemicals testing, and risk assessment for breast cancer prevention. Cancer. 2007; 109:2635-2666. DOI | PubMed
- Tiwary CM. Premature sexual development in children following the use of estrogen- or placenta-containing hair products. Clin Pediatr (Phila). 1998; 37:733-739. DOI | PubMed
- Li S-TT, Lozano P, Grossman DC, Graham E. Hormone-containing hair product use in prepubertal children. Arch Pediatr Adolesc Med. 2002; 156:85-86. DOI | PubMed
- Tiwary CM, Ward JA. Use of hair products containing hormone or placenta by US military personnel. J Pediatr Endocrinol Metab. 2003; 16:1025-1032. DOI | PubMed
- Hsieh CC, Trichopoulos D, Katsouyanni K, Yuasa S. Age at menarche, age at menopause, height and obesity as risk factors for breast cancer: Associations and interactions in an international case-control study. Int J Cancer. 1990; 46:796-800. DOI | PubMed
- Donovan M, Tiwary CM, Axelrod D, Sasco AJ, Jones L, Hajek R. Personal care products that contain estrogens or xenoestrogens may increase breast cancer risk. Med Hypotheses. 2007; 68:756-766. DOI | PubMed
- James-Todd T, Senie R, Terry MB. Racial/ethnic differences in hormonally-active hair product use: A plausible risk factor for health disparities. J Immigr Minor Health. 2012; 14:506-511. DOI | PubMed
- Myers SL, Yang CZ, Bittner GD, Witt KL, Tice RR, Baird DD. Estrogenic and anti-estrogenic activity of off-the-shelf hair and skin care products. J Expo Sci Environ Epidemiol. 2015; 25:271-277. DOI | PubMed
- Olson AC, Link JS, Waisman JR, Kupiec TC. Breast cancer patients unknowingly dosing themselves with estrogen by using topical moisturizers. J Clin Oncol. 2009; 27:e103-e104. DOI | PubMed
- Draelos ZD. Topical and oral estrogens revisited for antiaging purposes. Fertil Steril. 2005; 84:291-292. DOI | PubMed
- Marty MS, Carney EW, Rowlands JC. Endocrine disruption: Historical perspectives and its impact on the future of toxicology testing. Toxicol Sci. 2010; 120:S93-108. DOI | PubMed
- Matsumoto H, Adachi S, Suzuki Y. Bisphenol a in ambient air particulates responsible for the proliferation of MCF-7 human breast cancer cells and its concentration changes over 6 months. Arch Environ Contam Toxicol. 2005; 48:459-466. DOI | PubMed
- Rudel RA, Camann DE, Spengler JD, Korn LR, Brody JG. Phthalates, alkylphenols, pesticides, polybrominated diphenyl ethers, and other endocrine-disrupting compounds in indoor air and dust. Environ Sci Technol. 2003; 37:4543-4553. DOI | PubMed
- Rodriguez-Mozaz S, de Alda ML, Barceló D. Analysis of bisphenol a in natural waters by means of an optical immunosensor. Water Res. 2005; 39:5071-5079. DOI | PubMed
- Brotons JA, Olea-Serrano MF, Villalobos M, Pedraza V, Olea N. Xenoestrogens released from lacquer coatings in food cans. Environ Health Perspect. 1995; 103:608-612. DOI | PubMed
- Lakind JS, Naiman DQ. Daily intake of bisphenol a and potential sources of exposure: 2005-2006 National Health and nutrition examination survey. J Expo Sci Environ Epidemiol. 2011; 21:272-279. DOI | PubMed
- Vandenberg LN, Hauser R, Marcus M, Olea N, Welshons WV. Human exposure to bisphenol a (BPA). Reprod Toxicol. 2007; 24:139-177. DOI | PubMed
- Carwile JL, Luu HT, Bassett LS, Driscoll D, Yuan C, Chang J. Polycarbonate bottle use and urinary bisphenol a concentrations. Environ Health Perspect. 2009; 117:1368-1372. DOI | PubMed
- Smith R, Lourie Slow death by rubber duck: The secret danger of everyday things. Counterpoint: Berkeley; 2009.
- Rudel RA, Gray JM, Engel CL, Rawsthorne TW, Dodson RE, Ackerman JM. Food packaging and bisphenol a and bis(2-ethyhexyl) phthalate exposure: Findings from a dietary intervention. Environ Health Perspect. 2011; 119:914-920. DOI | PubMed
- Stahlhut RW, Welshons WV, Swan SH. Bisphenol a data in NHANES suggest longer than expected half-life, substantial nonfood exposure, or both. Environ Health Perspect. 2009; 117:784-789. DOI | PubMed
- Vom Saal FS, Welshons WV. Evidence that bisphenol a (BPA) can be accurately measured without contamination in human serum and urine and that BPA causes numerous hazards from multiple routes of exposure. Mol Cell Endocrinol. 2014; 398:101-113. DOI | PubMed
- Calafat AM, Ye X, Wong L-Y, Reidy JA, Needham LL. Exposure of the U.S. population to bisphenol a and 4-tertiary-octylphenol: 2003–2004. Environ Health Perspect. 2008; 116:39-44. DOI | PubMed
- Ye X, Pierik FH, Angerer J, Meltzer HM, Jaddoe VWV, Tiemeier H. Levels of metabolites of organophosphate pesticides, phthalates, and bisphenol a in pooled urine specimens from pregnant women participating in the Norwegian mother and child cohort study (MoBa). Int J Hyg Environ Health. 2009; 212:481-491. DOI | PubMed
- Padmanabhan V, Siefert K, Ransom S, Johnson T, Pinkerton J, Anderson L. Maternal bisphenol-a levels at delivery: A looming problem?. J Perinatol. 2008; 28:258-263. DOI | PubMed
- Teeguarden JG, Twaddle NC, Churchwell MI, Doerge DR. Urine and serum biomonitoring of exposure to environmental estrogens I: Bisphenol a in pregnant women. Food Chem Toxicol. 2016; 92:129-142. DOI | PubMed
- Kuruto-Niwa R, Ito T, Goto H, Nakamura H, Nozawa R, Terao Y. Estrogenic activity of the chlorinated derivatives of estrogens and flavonoids using a GFP expression system. Environ Toxicol Pharmacol. 2007; 23:121-128. DOI | PubMed
- Sun Y, Irie M, Kishikawa N, Wada M, Kuroda N, Nakashima K. Determination of bisphenol a in human breast milk by HPLC with column-switching and fluorescence detection. Biomed Chromatogr. 2004; 18:501-507. DOI | PubMed
- Ikezuki Y, Tsutsumi O, Takai Y, Kamei Y, Taketani Y. Determination of bisphenol a concentrations in human biological fluids reveals significant early prenatal exposure. Hum Reprod. 2002; 17:2839-2841. DOI | PubMed
- Environmental Working Group. Guide to BPA. 2009.
- Schönfelder G, Wittfoht W, Hopp H, Talsness CE, Paul M, Chahoud I. Parent bisphenol a accumulation in the human maternal-fetal-placental unit. Environ Health Perspect. 2002; 110:A703-A707. DOI | PubMed
- Calafat AM, Weuve J, Ye X, Jia LT, Hu H, Ringer S. Exposure to bisphenol a and other phenols in neonatal intensive care unit premature infants. Environ Health Perspect. 2009; 117:639-644. DOI | PubMed
- Maffini MV, Rubin BS, Sonnenschein C, Soto AM. Endocrine disruptors and reproductive health: The case of bisphenol-a. Mol Cell Endocrinol. 2006; 254–255:179-186. DOI
- Muñoz-de-Toro M, Markey CM, Wadia PR, Luque EH, Rubin BS, Sonnenschein C. Perinatal exposure to bisphenol-a alters peripubertal mammary gland development in mice. Endocrinology. 2005; 146:4138-4147. DOI | PubMed
- Vandenberg LN, Maffini MV, Schaeberle CM, Ucci AA, Sonnenschein C, Rubin BS. Perinatal exposure to the xenoestrogen bisphenol-a induces mammary intraductal hyperplasias in adult CD-1 mice. Reprod Toxicol. 2008; 26:210-219. DOI | PubMed
- Betancourt AM, Mobley JA, Russo J, Lamartiniere CA. Proteomic analysis in mammary glands of rat offspring exposed in utero to bisphenol a. J Proteome. 2010; 73:1241-1253. DOI
- Murray TJ, Maffini MV, Ucci AA, Sonnenschein C, Soto AM. Induction of mammary gland ductal hyperplasias and carcinoma in situ following fetal bisphenol a exposure. Reprod Toxicol. 2007; 23:383-390. DOI | PubMed
- Acevedo N, Davis B, Schaeberle CM, Sonnenschein C, Soto AM. Perinatally administered bisphenol a as a potential mammary gland carcinogen in rats. Environ Health Perspect. 2013; 121:1040-1046. PubMed
- Durando M, Kass L, Piva J, Sonnenschein C, Soto AM, Luque EH. Prenatal bisphenol a exposure induces preneoplastic lesions in the mammary gland in Wistar rats. Environ Health Perspect. 2007; 115:80-86. DOI | PubMed
- Jenkins S, Raghuraman N, Eltoum I, Carpenter M, Russo J, Lamartiniere CA. Oral exposure to bisphenol a increases dimethylbenzanthracene-induced mammary cancer in rats. Environ Health Perspect. 2009; 117:910-915. DOI | PubMed
- Wadia PR, Cabaton NJ, Borrero MD, Rubin BS, Sonnenschein C, Shioda T. Low-dose BPA exposure alters the mesenchymal and epithelial transcriptomes of the mouse fetal mammary gland. PLoS One. 2013; 8:e63902. DOI | PubMed
- Zhang X-L, Wang H-S, Liu N, Ge L-C. Bisphenol a stimulates the epithelial mesenchymal transition of estrogen negative breast cancer cells via FOXA1 signals. Arch Biochem Biophys. 2015; 585:10-16. DOI | PubMed
- Ibrahim MAA, Elbakry RH, Bayomy NA. Effect of bisphenol a on morphology, apoptosis and proliferation in the resting mammary gland of the adult albino rat. Int J Exp Pathol. 2016; 97:27-36. DOI | PubMed
- Wadia PR, Vandenberg LN, Schaeberle CM, Rubin BS, Sonnenschein C, Soto AM. Perinatal bisphenol a exposure increases estrogen sensitivity of the mammary gland in diverse mouse strains. Environ Health Perspect. 2007; 115:592-598. DOI | PubMed
- Tharp AP, Maffini MV, Hunt PA, VandeVoort CA, Sonnenschein C, Soto AM. Bisphenol a alters the development of the rhesus monkey mammary gland. Proc Natl Acad Sci U S A. 2012; 109:8190-8195. DOI | PubMed
- Jenkins S, Wang J, Eltoum I, Desmond R, Lamartiniere CA. Chronic oral exposure to bisphenol a results in a nonmonotonic dose response in mammary carcinogenesis and metastasis in MMTV-erbB2 mice. Environ Health Perspect. 2011; 119:1604-1609. DOI | PubMed
- Vandenberg LN, Schaeberle CM, Rubin BS, Sonnenschein C, Soto AM. The male mammary gland: A target for the xenoestrogen bisphenol a. Reprod Toxicol. 2013; 37:15-23. DOI | PubMed
- Kass L, Altamirano GA, Bosquiazzo VL, Luque EH, Muñoz-de-Toro M. Perinatal exposure to xenoestrogens impairs mammary gland differentiation and modifies milk composition in Wistar rats. Reprod Toxicol. 2012; 33:390-400. DOI | PubMed
- Rivas A, Lacroix M, Olea-Serrano F, Laíos I, Leclercq G, Olea N. Estrogenic effect of a series of bisphenol analogues on gene and protein expression in MCF-7 breast cancer cells. J Steroid Biochem Mol Biol. 2002; 82:45-53. DOI | PubMed
- Welshons WV, Nagel SC, vom Saal FS. Large effects from small exposures. III. Endocrine mechanisms mediating effects of bisphenol a at levels of human exposure. Endocrinology. 2006; 147:S56-S69. DOI | PubMed
- Watson CS, Bulayeva NN, Wozniak AL, Finnerty CC. Signaling from the membrane via membrane estrogen receptor-alpha: Estrogens, xenoestrogens, and phytoestrogens. Steroids. 2005; 70:364-371. DOI | PubMed
- Wozniak AL, Bulayeva NN, Watson CS. Xenoestrogens at picomolar to nanomolar concentrations trigger membrane estrogen receptor-alpha-mediated Ca2+ fluxes and prolactin release in GH3/B6 pituitary tumor cells. Environ Health Perspect. 2005; 113:431-439. DOI | PubMed
- Castillo Sanchez R, Gomez R, Perez Salaezar E. Bisphenol a induces migration through a GPER-, FAK-, Src-, and ERK2-dependent pathway in MDA-MB-231 breast cancer cells. Chem Res Toxicol. 2016; 29:285-295. DOI | PubMed
- Okada H, Tokunaga T, Liu X, Takayanagi S, Matsushima A, Shimohigashi Y. Direct evidence revealing structural elements essential for the high binding ability of bisphenol a to human estrogen-related receptor-gamma. Environ Health Perspect. 2008; 116:32-38. DOI | PubMed
- Matsushima A, Teramoto T, Okada H, Liu X, Tokunaga T, Kakuta Y. ERRgamma tethers strongly bisphenol a and 4-alpha-cumylphenol in an induced-fit manner. Biochem Biophys Res Commun. 2008; 373:408-413. DOI | PubMed
- Takayanagi S, Tokunaga T, Liu X, Okada H, Matsushima A, Shimohigashi Y. Endocrine disruptor bisphenol a strongly binds to human estrogen-related receptor gamma (ERRgamma) with high constitutive activity. Toxicol Lett. 2006; 167:95-105. DOI | PubMed
- Song H, Zhang T, Yang P, Li M, Yang Y, Wang Y. Low doses of bisphenol a stimulate the proliferation of breast cancer cells via ERK1/2/ERRγ signals. Toxicol in Vitro. 2015; 30:521-528. DOI | PubMed
- Riggins RB, Mazzotta MM, Maniya OZ, Clarke R. Orphan nuclear receptors in breast cancer pathogenesis and therapeutic response. Endocr Relat Cancer. 2010; 17:R213-R231. DOI | PubMed
- Hofmann PJ, Schomburg L, Köhrle J. Interference of endocrine disrupters with thyroid hormone receptor-dependent transactivation. Toxicol Sci. 2009; 110:125-137. DOI | PubMed
- Roy P, Salminen H, Koskimies P, Simola J, Smeds A, Saukko P. Screening of some anti-androgenic endocrine disruptors using a recombinant cell-based in vitro bioassay. J Steroid Biochem Mol Biol. 2004; 88:157-166. DOI | PubMed
- Sun H, Xu L-C, Chen J-F, Song L, Wang X-R. Effect of bisphenol a, tetrachlorobisphenol a and pentachlorophenol on the transcriptional activities of androgen receptor-mediated reporter gene. Food Chem Toxicol. 2006; 44:1916-1921. DOI | PubMed
- Fischer C, Mamillapalli R, Goetz LG, Jorgenson E, Ilagan Y, Taylor HS. Bisphenol a (BPA) exposure in utero leads to Immunoregulatory cytokine dysregulation in the mouse mammary gland: A potential mechanism programming breast cancer risk. Horm Cancer. 2016; 7:241-251. DOI | PubMed
- Goodson WH, Luciani MG, Sayeed SA, Jaffee IM, Moore DH, Dairkee SH. Activation of the mTOR pathway by low levels of xenoestrogens in breast epithelial cells from high-risk women. Carcinogenesis. 2011; 32:1724-1733. DOI | PubMed
- Tilghman SL, Bratton MR, Segar HC, Martin EC, Rhodes LV, Li M. Endocrine disruptor regulation of microRNA expression in breast carcinoma cells. PLoS One. 2012; 7:e32754. DOI | PubMed
- Weng Y-I, Hsu P-Y, Liyanarachchi S, Liu J, Deatherage DE, Huang Y-W. Epigenetic influences of low-dose bisphenol a in primary human breast epithelial cells. Toxicol Appl Pharmacol. 2010; 248:111-121. DOI | PubMed
- Dairkee SH, Seok J, Champion S, Sayeed A, Mindrinos M, Xiao W. Bisphenol a induces a profile of tumor aggressiveness in high-risk cells from breast cancer patients. Cancer Res. 2008; 68:2076-2080. DOI | PubMed
- Lapensee EW, Tuttle TR, Fox SR, Ben-Jonathan N. Bisphenol a at low nanomolar doses confers chemoresistance in estrogen receptor-alpha-positive and -negative breast cancer cells. Environ Health Perspect. 2009; 117:175-180. DOI | PubMed
- LaPensee EW, LaPensee CR, Fox S, Schwemberger S, Afton S, Ben-Jonathan N. Bisphenol a and estradiol are equipotent in antagonizing cisplatin-induced cytotoxicity in breast cancer cells. Cancer Lett. 2010; 290:167-173. DOI | PubMed
- . Accessed 25 Oct 2016.Publisher Full Text
- Janjua NR, Frederiksen H, Skakkebaek NE, Wulf HC, Andersson A-M. Urinary excretion of phthalates and paraben after repeated whole-body topical application in humans. Int J Androl. 2008; 31:118-130. DOI | PubMed
- Schettler T. Human exposure to phthalates via consumer products. Int J Androl. 2005; 29:134-139. DOI
- Fromme H, Gruber L, Schlummer M, Wolz G, Böhmer S, Angerer J. Intake of phthalates and di(2-ethylhexyl)adipate: Results of the integrated exposure assessment survey based on duplicate diet samples and biomonitoring data. Environ Int. 2007; 33:1012-1020. DOI | PubMed
- Birnbaum LS, Schug TT. Phthalates in our food. Endocr Disrupt. 2013; 1(1):e25078. DOI
- Zota AR, Phillips CA, Mitro SD. Recent fast food consumption and bisphenol a and phthalates exposures among the U.S. population in NHANES, 2003-2010. Environ Health Perspect. 2016; doi:10.1289/ehp.1510803.
- Ji K, Lim Kho Y, Park Y, Choi K. Influence of a five-day vegetarian diet on urinary levels of antibiotics and phthalate metabolites: A pilot study with “temple stay” participants. Environ Res. 2010; 110:375-382. DOI | PubMed
- Cirillo T, Fasano E, Castaldi E, Montuori P, Amodio Cocchieri R. Children’s exposure to di(2-ethylhexyl)phthalate and dibutylphthalate plasticizers from school meals. J Agric Food Chem. 2011; 59:10532-10538. DOI | PubMed
- Chatonnet P, Boutou S, Plana A. Contamination of wines and spirits by phthalates: Types of contaminants present, contamination sources and means of prevention. Food Addit Contam Part A Chem Anal Control Expo Risk Assess. 2014; 31:1605-1615. DOI | PubMed
- Sathyanarayana S, Alcedo G, Saelens BE, Zhou C, Dills RL, Yu J. Unexpected results in a randomized dietary trial to reduce phthalate and bisphenol a exposures. J Expo Sci Environ Epidemiol. 2013; 23:378-384. DOI | PubMed
- Rudel RA, Brody JG, Spengler JD, Vallarino J, Geno PW, Sun G. Identification of selected hormonally active agents and animal mammary carcinogens in commercial and residential air and dust samples. J Air Waste Manag Assoc. 2001; 51:499-513. DOI | PubMed
- Calafat AM, Wong L-Y, Silva MJ, Samandar E, Preau JL, Jia LT. Selecting adequate exposure biomarkers of diisononyl and diisodecyl phthalates: Data from the 2005-2006 National Health and nutrition examination survey. Environ Health Perspect. 2011; 119:50-55. DOI | PubMed
- Frederiksen H, Aksglaede L, Sorensen K, Skakkebaek NE, Juul A, Andersson A-M. Urinary excretion of phthalate metabolites in 129 healthy Danish children and adolescents: Estimation of daily phthalate intake. Environ Res. 2011; 111:656-663. DOI | PubMed
- Zota AR, Calafat AM, Woodruff TJ. Temporal trends in phthalate exposures: Findings from the National Health and nutrition examination survey, 2001–2010. Environ Health Perspect. 2014; 122:235-241. DOI | PubMed
- Jensen MS, Nørgaard-Pedersen B, Toft G, Hougaard DM, Bonde JP, Cohen A. Phthalates and perfluorooctanesulfonic acid in human amniotic fluid: Temporal trends and timing of amniocentesis in pregnancy. Environ Health Perspect. 2012; 120:897-903. DOI | PubMed
- Hines EP, Calafat AM, Silva MJ, Mendola P, Fenton SE. Concentrations of phthalate metabolites in milk, urine, saliva, and serum of lactating North Carolina women. Environ Health Perspect. 2009; 117:86-92. DOI | PubMed
- Meeker JD, Sathyanarayana S, Swan SH. Phthalates and other additives in plastics: Human exposure and associated health outcomes. Philos Trans R Soc Lond Ser B Biol Sci. 2009; 364:2097-2113. DOI | PubMed
- Wittassek M, Angerer J, Kolossa-Gehring M, Schäfer SD, Klockenbusch W, Dobler L. Fetal exposure to phthalates—a pilot study. Int J Hyg Environ Health. 2009; 212:492-498. DOI | PubMed
- Sathyanarayana S, Karr CJ, Lozano P, Brown E, Calafat AM, Liu F. Baby care products: Possible sources of infant phthalate exposure. Pediatrics. 2008; 121:e260-e268. DOI | PubMed
- López-Carrillo L, Hernández-Ramírez RU, Calafat AM, Torres-Sánchez L, Galván-Portillo M, Needham LL. Exposure to phthalates and breast cancer risk in northern Mexico. Environ Health Perspect. 2010; 118:539-544. DOI | PubMed
- Toft G, Jönsson BAG, Lindh CH, Jensen TK, Hjollund NH, Vested A. Association between pregnancy loss and urinary phthalate levels around the time of conception. Environ Health Perspect. 2012; 120:458-463. DOI | PubMed
- Jobling S, Reynolds T, White R, Parker MG, Sumpter JP. A variety of environmentally persistent chemicals, including some phthalate plasticizers, are weakly estrogenic. Environ Health Perspect. 1995; 103:582-587. DOI | PubMed
- Kang SC, Lee BM. DNA methylation of estrogen receptor alpha gene by phthalates. J Toxicol Environ Health A. 2005; 68:1995-2003. DOI | PubMed
- Borch J, Metzdorff SB, Vinggaard AM, Brokken L, Dalgaard M. Mechanisms underlying the anti-androgenic effects of diethylhexyl phthalate in fetal rat testis. Toxicology. 2006; 223:144-155. DOI | PubMed
- Fang H, Tong W, Branham WS, Moland CL, Dial SL, Hong H. Study of 202 natural, synthetic, and environmental chemicals for binding to the androgen receptor. Chem Res Toxicol. 2003; 16:1338-1358. DOI | PubMed
- Wang Y-C, Tsai C-F, Chuang H-L, Chang Y-C, Chen H-S, Lee J-N. Benzyl butyl phthalate promotes breast cancer stem cell expansion via SPHK1/S1P/S1PR3 signaling. Oncotarget. 2016; 7:29563-29576. DOI | PubMed
- Das MT, Singh MK, Thakur IS. Differential toxicological endpoints of di(2-ethylhexyl) phthalate (DEHP) exposure in MCF-7 and MDA-MB-231 cell lines: Possible estrogen receptor α (ERα) independent modulations. Indian J Exp Biol. 2014; 52:1052-1061. PubMed
- Hsieh T-H, Tsai C-F, Hsu C-Y, Kuo P-L, Lee J-N, Chai C-Y. Phthalates induce proliferation and invasiveness of estrogen receptor-negative breast cancer through the AhR/HDAC6/c-Myc signaling pathway. FASEB J. 2012; 26:778-787. DOI | PubMed
- Jiang J, Ma L, Yuan L, Wang X, Zhang W. Study on developmental abnormalities in hypospadiac male rats induced by maternal exposure to di-n-butyl phthalate (DBP). Toxicology. 2007; 232:286-293. DOI | PubMed
- Lovekamp-Swan T, Davis BJ. Mechanisms of phthalate ester toxicity in the female reproductive system. Environ Health Perspect. 2003; 111:139-145. DOI | PubMed
- Foster PMD. Disruption of reproductive development in male rat offspring following in utero exposure to phthalate esters. Int J Androl. 2006; 29:140-147. DOI | PubMed
- Latini G, Del Vecchio A, Massaro M, Verrotti A, De Felice C. Phthalate exposure and male infertility. Toxicology. 2006; 226:90-98. DOI | PubMed
- Swan SH, Main KM, Liu F, Stewart SL, Kruse RL, Calafat AM. Decrease in anogenital distance among male infants with prenatal phthalate exposure. Environ Health Perspect. 2005; 113:1056-1061. DOI | PubMed
- Bornehag C-G, Carlstedt F, Jönsson BAG, Lindh CH, Jensen TK, Bodin A. Prenatal phthalate exposures and anogenital distance in Swedish boys. Environ Health Perspect. 2015; 123:101-107. PubMed
- Chou Y-Y, Huang P-C, Lee C-C, Wu M-H, Lin S-J. Phthalate exposure in girls during early puberty. J Pediatr Endocrinol Metab. 2009; 22:69-77. DOI | PubMed
- Steingraber S. The falling age of puberty in U.S. girls: What we know, what we need to know. Breast Cancer Fund: San Francisco; 2007.
- Moral R, Wang R, Russo IH, Mailo DA, Lamartiniere CA, Russo J. The plasticizer butyl benzyl phthalate induces genomic changes in rat mammary gland after neonatal/prepubertal exposure. BMC Genomics. 2007; 8:453. DOI | PubMed
- Moral R, Santucci-Pereira J, Wang R, Russo IH, Lamartiniere CA, Russo J. In utero exposure to butyl benzyl phthalate induces modifications in the morphology and the gene expression profile of the mammary gland: An experimental study in rats. Environ Health. 2011; 10:5. DOI | PubMed
- Oral D, Erkekoglu P, Kocer-Gumusel B, Chao M-W. Epithelial-mesenchymal transition: A special focus on phthalates and bisphenol a. J Environ Pathol Toxicol Oncol. 2016; 35:43-58. DOI | PubMed
- Gwinn MR, Whipkey DL, Tennant LB, Weston A. Gene expression profiling of di-n-butyl phthalate in normal human mammary epithelial cells. J Environ Pathol Toxicol Oncol. 2007; 26:51-61. DOI | PubMed
- Kim IY, Han SY, Moon A. Phthalates inhibit tamoxifen-induced apoptosis in MCF-7 human breast cancer cells. J Toxicol Environ Health A. 2004; 6:2025-2035. DOI
- Hsu Y-L, Hung J-Y, Tsai E-M, Wu C-Y, Ho Y-W, Jian S-F. Benzyl butyl phthalate increases the chemoresistance to doxorubicin/cyclophosphamide by increasing breast cancer-associated dendritic cell-derived CXCL1/GROα and S100A8/A9. Oncol Rep. 2015; 34:2889-2900. DOI | PubMed
- Calafat AM, Ye X, Wong L-Y, Bishop AM, Needham LL. Urinary concentrations of four parabens in the U.S. population: NHANES 2005–2006. Environ Health Perspect. 2010; 118:679-685. DOI | PubMed
- Philippat C, Wolff MS, Calafat AM, Ye X, Bausell R, Meadows M. Prenatal exposure to environmental phenols: Concentrations in amniotic fluid and variability in urinary concentrations during pregnancy. Environ Health Perspect. 2013; 121:1225-1231. PubMed
- Darbre P, Aljarrah A, Miller W. Concentrations of parabens in human breast tumours. J Appl Toxicol. 2004; 24:5-13. DOI | PubMed
- Rastogi S, Schouten A, De Kruijf N, Weijland J. Contents of methylparaben, ethylparaben, propylparaben, butylparaben and benzylparaben in cosmetic products. Contact Dermatitis. 1995; 32:28-30. DOI | PubMed
- Barr L, Metaxas G, Harbach CAJ, Savoy LA, Darbre PD. Measurement of paraben concentrations in human breast tissue at serial locations across the breast from axilla to sternum. J Appl Toxicol. 2012; 32:219-232. DOI | PubMed
- Darbre PD, Harvey PW. Parabens can enable hallmarks and characteristics of cancer in human breast epithelial cells: A review of the literature with reference to new exposure data and regulatory status. J Appl Toxicol. 2014; 34:925-938. DOI | PubMed
- Karpuzoglu E, Holladay SD, Gogal RM. Parabens: Potential impact of low-affinity estrogen receptor binding chemicals on human health. J Toxicol Environ Health B Crit Rev. 2013; 16:321-335. DOI | PubMed
- Klopčič I, Kolšek K, Dolenc MS. Glucocorticoid-like activity of propylparaben, butylparaben, diethylhexyl phthalate and tetramethrin mixtures studied in the MDA-kb2 cell line. Toxicol Lett. 2015; 232:376-383. DOI | PubMed
- Darbre PD, Harvey PW. Paraben esters: Review of recent studies of endocrine toxicity, absorption, esterase, and discussion of possible health risks. J Appl Toxicol. 2008; 28:561-578. DOI | PubMed
- Watanabe Y, Kojima H, Takeuchi S, Uramaru N, Ohta S, Kitamura S. Comparative study on transcriptional activity of 17 parabens mediated by estrogen receptor α and β and androgen receptor. Food Chem Toxicol. 2013; 57:227-234. DOI | PubMed
- Byford J, Shaw L, Drew M, Pope G, Sauer M, Darbre P. Oestrogenic activity of parabens in MCF7 human breast cancer cells. J Steroid Biochem Mol Biol. 2002; 80:49-60. DOI | PubMed
- Gopalakrishan K, Teitelbaum SL, Lambertini L, Wetmur J, Manservisi F, Luciani MG, Falcioni L, et al. Changes in mammary histology and transcriptome profils by low-dose exposure to environmental phenols at critical periods of development. Environ Res. 2017;152:232–43.
- Pugazhendhi D, Sadler A, Darbre P. Comparison of the global gene expression profiles produced by methylparaben, n-butylparaben and 17B-oestradiol in mcf7 human breast cancer cells. J Appl Toxicol. 2007; 27:67-77. DOI | PubMed
- Wróbel A, Gregoraszczuk EL. Effects of single and repeated in vitro exposure of three forms of parabens, methyl-, butyl-and propylparabens on the proliferation and estradiol secretion in MCF-7 and MCF-10A cells. Pharmacol Rep. 2013; 65:484-493. DOI | PubMed
- Charles AK, Darbre PD. Combinations of parabens at concentrations measured in human breast tissue can increase proliferation of MCF-7 human breast cancer cells. J Appl Toxicol. 2013; 33:390-398. DOI | PubMed
- Wróbel AM, Gregoraszczuk EL. Differential effect of methyl-, butyl- and propylparaben and 17β-estradiol on selected cell cycle and apoptosis gene and protein expression in MCF-7 breast cancer cells and MCF-10A non-malignant cells. J App Toxicol. 2014; 34:1041-1050. DOI
- Pan S, Yuan C, Tagmount A, Rudel RA, Ackerman JM, Yaswen P. Parabens and human epidermal growth factor receptor ligand cross-talk in breast cancer cells. Environ Health Perspect. 2016; 124:563-569. PubMed
- Zhang Z, Sun L, Hu Y, Jiao J, Hu J. Inverse antagonist activities of parabens on human oestrogen-related receptor γ (ERRγ): In vitro and in silico studies. Toxicol Appl Pharmacol. 2013; 270:16-22. DOI | PubMed
- Khanna S, Darbre PD. Parabens enable suspension growth of MCF-10A immortalized, non-transformed human breast epithelial cells. J Appl Toxicol. 2013; 33:378-382. DOI | PubMed
- Khanna S, Dash PR, Darbre PD. Exposure to parabens at the concentration of maximal proliferative response increases migratory and invasive activity of human breast cancer cells in vitro. J Appl Toxicol. 2014; 34:1051-1059. DOI | PubMed
- Deshayes S, Eudes V, Droguet C, Bigourie M, Gasperi J, Moilleron R. Alkylphenols and phthalates in greywater from showers and washing machines. Water Air Soil Pollut. 2015;226 doi:10.1007/s11270-015-2652-7.
- Slack RJ, Gronow JR, Voulvoulis N. Household hazardous waste in municipal landfills: Contaminants in leachate. Sci Total Environ. 2005; 337:119-137. DOI | PubMed
- Pasquini L, Munoz J-F, Pons M-N, Yvon J, Dauchy X, France X. Occurrence of eight household micropollutants in urban wastewater and their fate in a wastewater treatment plant. Statistical evaluation. Sci Total Environ. 2014; 481:459-468. DOI | PubMed
- Swartz CH, Reddy S, Benotti MJ, Yin H, Barber LB, Brownawell BJ. Steroid estrogens, nonylphenol ethoxylate metabolites, and other wastewater contaminants in groundwater affected by a residential septic system on cape cod, MA. Environ Sci Technol. 2006; 40:4894-4902. DOI | PubMed
- Calafat AM, Kuklenyik Z, Reidy JA, Caudill SP, Ekong J, Needham LL. Urinary concentrations of bisphenol a and 4-nonylphenol in a human reference population. Environ Health Perspect. 2005; 113:391-395. DOI | PubMed
- Gyllenhammar I, Glynn A, Darnerud PO, Lignell S, van Delft R, Aune M. 4-Nonylphenol and bisphenol a in Swedish food and exposure in Swedish nursing women. Environ Int. 2012; 43:21-28. DOI | PubMed
- Chen G-W, Ding W-H, Ku H-Y, Chao H-R, Chen H-Y, Huang M-C. Alkylphenols in human milk and their relations to dietary habits in central Taiwan. Food Chem Toxicol. 2010; 48:1939-1944. DOI | PubMed
- Thomas P, Dong J. Binding and activation of the seven-transmembrane estrogen receptor GPR30 by environmental estrogens: A potential novel mechanism of endocrine disruption. J Steroid Biochem Mol Biol. 2006; 102:175-179. DOI | PubMed
- Oh M. Comparative analysis of gene expression pattern after exposure to nonylphenol in human cell lines. Biochip J. 2009:2:261–8.
- Amaro AA, Esposito AI, Mirisola V, Mehilli A, Rosano C, Noonan DM. Endocrine disruptor agent nonyl phenol exerts an estrogen-like transcriptional activity on estrogen receptor positive breast cancer cells. Curr Med Chem. 2014; 2:630-640. DOI
- Lee H-R, Hwang K-A, Nam K-H, Kim H-C, Choi K-C. Progression of breast cancer cells was enhanced by endocrine-disrupting chemicals, triclosan and octylphenol, via an estrogen receptor-dependent signaling pathway in cellular and mouse xenograft models. Chem Res Toxicol. 2014; 27:834-842. DOI | PubMed
- Moon HJ, Han SY, Shin J-H, Kang IH, Kim TS, Hong JH. Gestational exposure to nonylphenol causes precocious mammary gland development in female rat offspring. J Reprod Dev. 2007; 53:333-344. DOI | PubMed
- Acevedo R, Parnell PG, Villanueva H, Chapman LM, Gimenez T, Gray SL. The contribution of hepatic steroid metabolism to serum estradiol and estriol concentrations in nonylphenol treated MMTVneu mice and its potential effects on breast cancer incidence and latency. J Appl Toxicol. 2005; 25:339-353. DOI | PubMed
- Dann AB, Hontela A. Triclosan: Environmental exposure, toxicity and mechanisms of action. J Appl Toxicol. 2011; 31:285-311. DOI | PubMed
- Ahn KC, Zhao B, Chen J, Cherednichenko G, Sanmarti E, Denison MS. In vitro biologic activities of the antimicrobials triclocarban, its analogs, and triclosan in bioassay screens: Receptor-based bioassay screens. Environ Health Perspect. 2008; 116:1203-1210. DOI | PubMed
- Brausch JM, Rand GM. A review of personal care products in the aquatic environment: Environmental concentrations and toxicity. Chemosphere. 2011; 82:1518-1532. DOI | PubMed
- Venkatesan AK, Pycke BFG, Barber LB, Lee KE, Halden RU. Occurrence of triclosan, triclocarban, and its lesser chlorinated congeners in Minnesota freshwater sediments collected near wastewater treatment plants. J Hazard Mater. 2012; 229–230:29-35. DOI
- Yueh M-F, Tukey RH. Triclosan: A widespread environmental toxicant with many biological effects. Annu Rev Pharmacol Toxicol. 2016; 56:251-272. DOI | PubMed
- Woodruff TJ, Zota AR, Schwartz JM. Environmental chemicals in pregnant women in the united states: NHANES 2003–2004. Environ Health Perspect. 2011; 119:878-885. DOI | PubMed
- Ye X, Zhou X, Furr J, Ahn KC, Hammock BD, Gray EL. Biomarkers of exposure to triclocarban in urine and serum. Toxicology. 2011; 286:69-74. DOI | PubMed
- Geens T, Neels H, Covaci A. Distribution of bisphenol-a, triclosan and n-nonylphenol in human adipose tissue, liver and brain. Chemosphere. 2012; 87:796-802. DOI | PubMed
- Dinwiddie MT, Terry PD, Chen J. Recent evidence regarding triclosan and cancer risk. Int J Environ Res Public Health. 2014; 11:2209-2217. DOI | PubMed
- Christen V, Crettaz P, Oberli-Schrämmli A, Fent K. Some flame retardants and the antimicrobials triclosan and triclocarban enhance the androgenic activity in vitro. Chemosphere. 2010; 81:1245-1252. DOI | PubMed
- Gee RH, Charles A, Taylor N, Darbre PD. Oestrogenic and androgenic activity of triclosan in breast cancer cells. J Appl Toxicol. 2008; 28:78-91. DOI | PubMed
- Henry ND, Fair PA. Comparison of in vitro cytotoxicity, estrogenicity and anti-estrogenicity of triclosan, perfluorooctane sulfonate and perfluorooctanoic acid. J Appl Toxicol. 2013; 33:265-272. DOI | PubMed
- Lee H-R, Hwang K-A, Nam K-H, Kim H-C, Choi K-C. Progression of breast cancer cells was enhanced by endocrine-disrupting chemicals, triclosan and octylphenol, via an estrogen receptor-dependent signaling pathway in cellular and mouse xenograft models. Chem Res Toxicol. 2014; 27:834-842. DOI | PubMed
- Kim J-Y, Yi B-R, Go R-E, Hwang K-A, Nam K-H, Choi K-H. Methoxychlor and triclosan stimulates ovarian cancer growth by regulating cell cycle- and apoptosis-related genes via an estrogen-dependent pathway. Environ Toxicol Pharmacol. 2014;37:1264–74.
- Zorrilla LM, Gibson EK, Jeffay SC, Crofton KM, Setzer WR, Cooper RL. The effects of triclosan on puberty and thyroid hormonezorillas in male wistar rats. Toxicol Sci. 2009; 107:56-64. DOI | PubMed
- Paul KB, Hedge JM, DeVito MJ, Crofton KM. Developmental triclosan exposure decreases maternal and neonatal thyroxine in rats. Environ Toxicol Chem. 2010; 29:2840-2844. DOI | PubMed
- Hayden C, Roberts M, Benson H. Systemic absorption of sunscreen after topical application. Lancet. 1997; 350:853-864. DOI
- Felix T, Hall BJ, Brodbelt JS. Determination of benzophenone-3 and metabolites in water and human urine by solid-phase microextraction and quadrupole ion trap GC-MS. Anal Chim Acta. 1998; 371:195-203. DOI
- Kunz PY, Fent K. Estrogenic activity of UV filter mixtures. Toxicol Appl Pharmacol. 2006; 217:86-99. DOI | PubMed
- Waldman JM, Gavin Q, Anderson M, Hoover S, Alvaran J, Ip HSS. Exposures to environmental phenols in Southern California firefighters and findings of elevated urinary benzophenone-3 levels. Environ Int. 2016; 88:281-287. DOI | PubMed
- Krause M, Klit A, Blomberg Jensen M, Søeborg T, Frederiksen H, Schlumpf M. Sunscreens: Are they beneficial for health? An overview of endocrine disrupting properties of UV-filters. Int J Androl. 2012; 35:424-436. DOI | PubMed
- Schlumpf M, Cotton B, Conscience M, Haller V, Steinmann B, Lichtensteiger W. In vitro and in vivo estrogenicity of UV screens. Environ Health Perspect. 2001; 109:239-244. DOI
- Klann A, Levy G, Lutz I, Muller C, Kloas W, Hildebrandt J. Estrogen-like effects of ultraviolet screen 3-(4methylbenzylidene)-camphor (Eusolex 6300) on cell proliferation and gene induction in mammalian and amphibian cells. Environ Res. 2005; 97:274-281. DOI | PubMed
- Brand R, McMahon L, Jendrzejewski J, Charron A. Transdermal absorption of the herbicide 2,4-dichlorophenoxyacetic acid is enhanced by both ethanol consumption and sunscreen application. Food Chem Toxicol. 2007; 45:93-97. DOI | PubMed
- Jensen AA, Leffers H. Emerging endocrine disrupters: Perfluoroalkylated substances. Int J Androl. 2008; 31:161-169. DOI | PubMed
- Kato K, Wong L-Y, Jia LT, Kuklenyik Z, Calafat AM. Trends in exposure to polyfluoroalkyl chemicals in the U.S. p[o]pulation:1999–2008. Environ Sci Technol. 2001; 45:8037-8045. DOI
- Kannan K, Corsolini S, Falandysz J, Fillmann G, Kumar KS, Loganathan BG. Perfluorooctanesulfonate and related fluorochemicals in human blood from several countries. Environ Sci Technol. 2004; 38:4489-4495. DOI | PubMed
- Apelberg BJ, Goldman LR, Calafat AM, Herbstman JB, Kuklenyik Z, Heidler J. Determinants of fetal exposure to polyfluoroalkyl compounds in Baltimore, Maryland. Environ Sci Technol. 2007; 41:3891-3897. DOI | PubMed
- Apelberg BJ, Witter FR, Herbstman JB, Calafat AM, Halden RU, Needham LL. Cord serum concentrations of perfluorooctane sulfonate (PFOS) and perfluorooctanoate (PFOA) in relation to weight and size at birth. Environ Health Perspect. 2007; 115:1670-1676. DOI | PubMed
- Stein CR, Wolff MS, Calafat AM, Kato K, Engel SM. Comparison of polyfluoroalkyl compound concentrations in maternal serum and amniotic fluid: A pilot study. Reprod Toxicol. 2012; 34:312-316. DOI | PubMed
- Maisonet M, Terrell ML, McGeehin MA, Christensen KY, Holmes A, Calafat AM. Maternal concentrations of polyfluoroalkyl compounds during pregnancy and fetal and postnatal growth in British girls. Environ Health Perspect. 2012; 120:1432-1437. DOI | PubMed
- Maisonet M, Calafat AM, Marcus M, Jaakkola JJK, Lashen H. Prenatal exposure to Perfluoroalkyl acids and serum testosterone concentrations at 15 years of age in female ALSPAC study participants. Environ Health Perspect. 2015; 123:1325-1330. DOI | PubMed
- Dimitrakakis C, Bondy C. Androgens and the breast. Breast Cancer Res. 2009; 11:212. DOI | PubMed
- Lopez-Espinosa M-J, Fletcher T, Armstrong B, Genser B, Dhatariya K, Mondal D. Association of Perfluorooctanoic Acid (PFOA) and Perfluorooctane sulfonate (PFOS) with age of puberty among children living near a chemical plant. Environ Sci Technol. 2011; 45:8160-8166. DOI | PubMed
- Kristensen SL, Ramlau-Hansen CH, Ernst E, Olsen SF, Bonde JP, Vested A. Long-term effects of prenatal exposure to perfluoroalkyl substances on female reproduction. Hum Reprod. 2013; 28:3337-3348. DOI | PubMed
- Pinney SM, Windham GC, Biro FM, Kushi LH, Yaghjyan L, Calafat A. Perfluorooctanoic acid (PFOA) and pubertal maturation in young girls. Epidemiology. 2009; 20:S80. DOI
- Fei C, McLaughlin JK, Lipworth L, Olsen J. Maternal levels of perfluorinated chemicals and subfecundity. Hum Reprod. 2009; 24:1200-1205. DOI | PubMed
- Bonefeld-Jorgensen EC, Long M, Bossi R, Ayotte P, Asmund G, Krüger T. Perfluorinated compounds are related to breast cancer risk in Greenlandic Inuit: A case control study. Environ Health. 2011; 10:88. DOI | PubMed
- White SS, Calafat AM, Kuklenyik Z, Villanueva L, Zehr RD, Helfant L. Gestational PFOA exposure of mice is associated with altered mammary gland development in dams and female offspring. Toxicol Sci. 2007; 96:133-144. DOI | PubMed
- White SS, Kato K, Jia LT, Basden BJ, Calafat AM, Hines EP. Effects of perfluorooctanoic acid on mouse mammary gland development and differentiation resulting from cross-foster and restricted gestational exposures. Reprod Toxicol. 2009; 27:289-298. DOI | PubMed
- Yang C, Tan YS, Harkema JR, Haslam SZ. Differential effects of peripubertal exposure to perfluorooctanoic acid on mammary gland development in C57Bl/6 and Balb/c mouse strains. Reprod Toxicol. 2009; 27:299-306. DOI | PubMed
- Tucker DK, Macon MB, Strynar MJ, Dagnino S, Andersen E, Fenton SE. The mammary gland is a sensitive pubertal target in CD-1 and C57Bl/6 mice following perinatal perfluorooctanoic acid (PFOA) exposure. Reprod Toxicol. 2015; 54:26-36. DOI | PubMed
- Sonthithai P, Suriyo T, Thiantanawat A, Watcharasit P, Ruchirawat M, Satayavivad J. Perfluorinated chemicals, PFOS and PFOA, enhance the estrogenic effects of 17β-estradiol in T47D human breast cancer cells. J Appl Toxicol. 2016; 36:790-801. DOI | PubMed
- Bonner M, Han D, Nie J, Rogerson P, Vena J, Muti P. Breast cancer risk and exposure in early life to polycyclic aromatic hydrocarbons using total suspended particulates as a proxy measure. Cancer Epidemiol Biomark Prev. 2005; 14:53-60.
- Beyea J, Stellman S, Hatch M, Gammon M. Airborne emissions from 1961 to 2004 of benzo(a)pyrene from U.S. vehicles per km of travel based on tunnel studies. Environ Sci Technol. 2008; 47:7315-7320. DOI
- Morris JJ, Seifter E. The role of aromatic hydrocarbons in the genesis of breast cancer. Med Hypoth. 1992; 38:177-184. DOI
- Pliskova M, Vondracek J, Vojtesek B, Kozubik A, Machala M. Deregulation of cell proliferation by polycyclic aromatic hydrocarbons in human breast carcinoma MCF-7 cells reflects both genotoxic and nongenotoxic events. Toxicol Sci. 2005; 83:246. DOI | PubMed
- Kemp M, Liu W, Thorne P, Kane M, Selmin O, Romagnolo D. Induction of the transferring receptor gene by benzo[a]pyrene in breast cancer MCF-7 cells: Potential as a biomarker of PAH exposure. Environ Mol Mutagen. 2006; 47:518-526. DOI | PubMed
- Santodonato J. Review of the estrogenic and antiestrogenic activity of polycyclic aromatic hydrocarbons: Relationship to carcinogenicity. Chemosphere. 1997; 34:835-848. DOI | PubMed
- Kung T, Murphy KA, White LA. The aryl hydrocarbon receptor (AhR) pathway as a regulatory pathway for cell adhesion and matrix metabolism. Biochem Pharmacol. 2009; 77:536-546. DOI | PubMed
- Ohura T, Morita M, Kuruto-Niwa R, Amagai T, Sakakibara H, Shimoi K. Differential action of chlorinated polycyclic aromatic hydrocarbons on aryl hydrocarbon receptor-mediated signaling in breast cancer cells. Environ Toxicol. 2010; 25:180-187. PubMed
- Ralston S, Coffing S, Seidel A, Luch A, Platt K, Baird W. Stereoselective activation of dibenzo[a,l]pyrene and its trans-11,12-dihydrodiol to fjord region 11,12-diol 13,14-epoxides in a human mammary carcinoma MCF-7 cell-mediated V79 cell mutation assay. Chem Res Toxciol. 1997; 10:687-693. DOI
- Mordukhovich I, Beyea J, Herring AH, Hatch M, Stellman SD, Teitelbaum SL. Vehicular traffic–related polycyclic aromatic hydrocarbon exposure and breast cancer incidence: The long island breast cancer study project (LIBCSP). Environ Health Perspect. 2016; 124:30-38. PubMed
- White AJ, Teitelbaum SL, Stellman SD, Beyea J, Steck SE, Mordukhovich I. Indoor air pollution exposure from use of indoor stoves and fireplaces in association with breast cancer: A case-control study. Environ Health. 2014; 13:108. DOI | PubMed
- Gammon MD, Santella RM, Neugut AI. Environmental toxins and breast cancer on Long Island. I. Polycyclic aromatic hydrocarbon DNA adducts. Cancer Epidemiol Biomark Prev. 2002; 11:677-685.
- Cavalieri E, Rogan E. The molecular etiology and prevention of estrogen-initiated cancers: Ockham’s razor: Pluralitas non est ponenda sine necessitate. Plurality should not be posited without necessity. Mol Asp Med. 2014; 36:1-55. DOI
- Rundle A, Tang D, Hibshoosh H, Estabrook A, Schnabel F, Cao C. The relationship between genetic damage from polycyclic hydrocarbons in breast tissue and breast cancer. Carcinogenesis. 2000; 21:1281-1289. DOI | PubMed
- Gammon MD, Santella RM. PAH, genetic susceptibility and breast cancer risk: An update from the Long Island breast cancer study project. Eur J Cancer. 2008; 44:636-640. DOI | PubMed
- Mahadevan B, Keshava C, Musafia-Jeknic T, Pecaj A, Weston A, Baird W. Altered gene expression patterns in MCF-7 cells induced by the urban dust particulate complex mixture standard reference material 1649a. Cancer Res. 2005; 65:1251-1258. DOI | PubMed
- McCarty K, Santella R, Steck S, Cleveland R, Ahn J, Ambrosone C. PAH-DNA adducts, cigarette smoking, GST polymorphisms and breast cancer risk. Environ Health Perspect. 2009; 117:552-555. DOI | PubMed
- Mordukhovich I, Pavel R, Terry MB, Santella R, Zhang Y-J, Hibshoosh H. Associations between polycyclic aromatic hydrocarbon-related exposures and p53 mutations in breast tumors. Environ Health Perspect. 2010; 118:511-518. DOI | PubMed
- Petralia SA, Vena JE, Freudenheim JL, Dosemeci M, Michalek A, Goldberg MS. Risk of premenopausal breast cancer in association with occupational exposure to polycyclic aromatic hydrocarbons and benzene. Scand J Work Environ Health. 1999; 25:215-221. DOI | PubMed
- Palli D, Masala G, Mariani-Costantini R, Zanna I, Saieva C, Sera F. A gene-environment interaction between occupation and BRCA1/BRCA2 mutations in male breast cancer. Eur J Cancer. 2004; 40:2474-2479. DOI | PubMed
- Nie J, Beyea J, Bonner M, Han D, Vena J, Rogerson P. Exposure to traffic emissions throughout life and risk of breast cancer: The western New York exposures and breast cancer (web) study. Cancer Causes Control. 2007; 18:947-955. DOI | PubMed
- Hung L-J, Tsai S-S, Chen P-S, Yang Y-H, Liou S-H, Wu T-N. Traffic air pollution and risk of death from breast cancer in Taiwan: Fine particulate matter (PM 2.5) as a proxy marker. Aerosol Air Qual Res. 2012; 12:275-282.
- Sagiv SK, Gaudet MM, Eng SM, Abrahamson PE, Shantakumar S, Teitelbaum SL. Polycyclic aromatic hydrocarbon-DNA adducts and survival among women with breast cancer. Environ Res. 2009; 109:287-291. DOI | PubMed
- Teitelbaum SL, Gammon MD, Britton JA, Neugut AI, Levin B, Stellman SD. Reported residential pesticide use and breast cancer risk on Long Island, New York. Am J Epidemiol. 2007; 165:643-651. DOI | PubMed
- Dodson RE, Camann DE, Morello-Frosch R, Brody JG, Rudel RA. Semivolatile organic compounds in homes: Strategies for efficient and systematic exposure measurement based on empirical and theoretical factors. Environ Sci Technol. 2015; 49:113-122. DOI | PubMed
- . Accessed 24 Sept 2016.Publisher Full Text
- Hua W, Bennett E, Maio X, Metcalfe C, Letcher R. Seasonality effects on pharmaceuticals and S-triazine herbicides in wastewater effluent and surface water from the Canadian side of the upper Detroit River. Environ Toxicol Chem. 2006; 25:2356-2365. DOI | PubMed
- Miller S, Sweet C, Depinto J, Hornbuckle K. Atrazine and nutrients in precipitation from the Lake Michigan mass balance study. Environ Sci Technol. 2000; 34:55-61. DOI
- Villanueva C, Durand G, Coutté M, Chevrier C, Cordier S. Atrazine in municipal drinking water and risk of low birth weight, preterm delivery, and small-for-gestational-age status. Occup Environ Med. 2005; 62:400-405. DOI | PubMed
- Hayes TB, Anderson LL, Beasley VR. Demasculinization and feminization of male gonads by atrazine: Consistent effects across vertebrate classes. J Steroid Biochem Mol Biol. 2011; 127:64-73. DOI | PubMed
- Rohr JR, McCoy KA. A qualitative meta-analysis reveals consistent effects of atrazine on freshwater fish and amphibians. Environ Health Perspect. 2010; 118:20-32. DOI | PubMed
- O’Connor J, Plowchalk D, Van-Pelt C, Davis L, Cook J. Role of prolactin in chloro-S-triazine rat mammary tumorigenesis. Drug Chem Toxicol. 2000; 23:575-601. DOI | PubMed
- Florian CP, Mansfield SR, Schroeder JR. Differences in GPR30 regulation by Chlorotriazine herbicides in human breast cells. Biochem Res Int. 2016;2016:2984081.
- Kettles MA, Browning SR, Prince TS, Horstman SW. Triazine herbicide exposure and breast cancer incidence: An ecologic study of Kentucky counties. Environ Health Perspect. 1997; 105:1222-1227. DOI | PubMed
- Hopenhayn-Rich C, Stump ML, Browning SR. Regional assessment of atrazine exposure and incidence of breast and ovarian cancers in Kentucky. Arch Environ Contam Toxicol. 2002; 42:127-136. DOI | PubMed
- Muir K, Rattanamongkolgul S, Smallman-Raynor M, Thomas M, Downer S, Jenkinson C. Breast cancer incidence and its possible spatial association with pesticide application in two counties of England. Public Health. 2004; 118:513-520. DOI | PubMed
- Hunter L, Gadbury G, Huang Y. Atrazine exposure and breast cancer incidence: An ecologic study of Missouri counties. Toxicol Environ Chem. 2008; 90:367-376. DOI
- Boffetta P, Adami H-O, Berry SC, Mandel JS. Atrazine and cancer: A review of the epidemiologic evidence. Eur J Cancer Prev. 2013; 22:169-180. DOI | PubMed
- Cooper R, Stoker T, Tyrey L, Goldman J, McElroy W. Atrazine disrupts the hypothalamic control of pituitary-ovarian function. Toxicol Sci. 2000; 53:297-307. DOI | PubMed
- Fan WQ, Yanase T, Morinaga H, Gondo S, Okabe T, Nomura M. Atrazine-induced aromatase expression is SF-1 dependent: Implications for endocrine disruption in wildlife and reproductive cancers in humans. Environ Health Perspect. 2007; 115:720-727. DOI | PubMed
- Sanderson J, Letcher R, Heneweer M, Giesy J, van den Berg M. Effects of chloro-S-triazine herbicides and metabolites on aromatase activity in various human cell lines and on vitellogenin production in male carp hepatocytes. Environ Health Perspect. 2001; 109:1027-1031. DOI | PubMed
- Enoch R, Stanko J, Greiner S. Mammary gland development as a sensitive end point after acute prenatal exposure to an atrazine metabolite mixture in female long-Evans rats. Environ Health Perspect. 2007;115:541–7.
- Raynor J, Enoch R, Fenton S. Adverse effects of prenatal exposure to atrazine during a critical period of mammary gland growth. Toxicol Sci. 2005;87:255–66.
- Fenton SE, Reed C, Newbold RR. Perinatal environmental exposures affect mammary development, function, and cancer risk in adulthood. Ann Rev Pharmacol Toxicol. 2012;52:455–79.
- Ueda M, Imai T, Takizawa T, Onodera H, Mitsumori K, Matsui T. Possible enhancing effects of atrazine on growth of 7, 12-dimethylbenz(a) anthracene-induced mammary tumors in ovariectomized Sprague-Dawley rats. Cancer Sci. 2005; 96:19-25. DOI | PubMed
- Hovey RC, Coder PS, Wolf JC, Sielken RL, Tisde MO, Breckenridge CB. Quantitative assessment of mammary gland development in female long Evans rats following in utero exposure to atrazine. Toxicol Sci. 2011; 119:380-390. DOI | PubMed
- Eldridge JC, Stevens JT, Breckenridge CB. Atrazine interaction with estrogen expression systems. Journal or something? 2008;196:147–156.
- Albanito L, Lappano R, Madeo A. Effects of atrazine on estrogen receptor α– And G protein–coupled receptor 30–mediated signaling and proliferation in cancer cells and cancer-associated fibroblasts. Environ Health Perspect. 2015; 123:493-499. PubMed
- Kucka M, Pogrmic-Majkic K, Fa S, Stojilkovic SS, Kovacevic R. Atrazine acts as an endocrine disrupter by inhibiting cAMP-specific phosphodiesterase-4. Toxicol Appl Pharmacol. 2012; 265:19-26. DOI | PubMed
- Suzawa M, Ingraham HA. The herbicide atrazine activates endocrine gene networks via non-steroidal NR5A nuclear receptors in fish and mammalian cells. PLoS One. 2008;3:e2117.
- Siegel BZ. Pesticide Hazard Assessment 1981–1984. Pacific Biomedical Research Center, University of Hawaii: Honolulu; 1995.
- Maskarinec G, Zhang Y, Takata Y, Pagano I, Shumay D, Goodman M. Trends of breast cancer incidence and risk factor prevalence over 25 years. Breast Cancer Res Treat. 2006; 98:45-54. DOI | PubMed
- Arrebola JP, Belhassen H, Artacho-Cordón F, Ghali R, Ghorbel H, Boussen H, et al. Risk of female breast cancer and serum concentrations of organochlorine pesticides and polychlorinated biphenyls: A case-control study in Tunisia. Sci Total Environ. 2015;520:106–13.
- Center for Disease Control and Prevention (CDC): Third National Report on Human Exposure to Environmental Chemicals. 2005. http://www.jhsph.edu/research/centers-and-institutes/center-for-excellence-in-environmental-health-tracking/Third_Report.pdf. Accessed 25 Oct 2016.
- Samra NM, Selim AA. Organochlorine pesticides concentrations in maternal serum and their effects on umbilical cord serum pesticides concentrations, neonatal birth weight and gestational age. Aust J Basic Appl Sci. 2009;3:1972–83.
- Khanjani N, English DR, Sim MR. An ecological study of organochlorine pesticides and breast cancer in rural Victoria, Astralie. Arch Environ Contam Toxicol. 2006;50:452–61.
- Cassidy RA, Natarajan S, Vaughan GM. The link between the insecticide heptachlor epoxide, estradiol, and breast cancer. Breast Cancer Res Treat. 2005; 90:55-64. DOI | PubMed
- Dich J, Zahm S, Hanberg A, Adami H. Pesticides (heptachlor) and cancer. Cancer Causes Control. 1997; 8:420-443. DOI | PubMed
- . Access date 10 Oct 2016.Publisher Full Text
- Hoyer A, Grandjean P, Jorgensen T, Brock J, Hartvig H. Organochlorine exposure and risk of breast cancer. Lancet. 1998; 352:1816-1820. DOI | PubMed
- Hoyer A, Jorgensen T, Brock J, Grandjean P. Organochlorine exposure and breast cancer survival. J Clin Epidemiol. 2000; 53:323-330. DOI | PubMed
- Cameron H, Foster W. Developmental and lactational exposure to dieldrin alters mammary tumorigenesis in her2/neu transgenic mice. PLoS One. 2009; 4:4303. DOI
- Andersen H, Vinggaard A, Rasmussen T, Gjermandsen I, Bonefeld-Jorgensen E. Effects of currently used pesticides in assays for estrogenicity, androgenicity, and aromatase activity in vitro. Toxicol Appl Pharmacol. 2002; 179:1-12. DOI | PubMed
- Mills P, Yang R. Breast cancer risk in Hispanic agricultural workers in California. Int J Occup Environ Health. 2005; 11:123-131. DOI | PubMed
- Engel LS, Hill DA, Hoppin JA, Lubin JH, Lynch CF, Pierce J. Pesticide use and breast cancer risk among farmers’ wives in the agricultural health study. Am J Epidemiol. 2005; 161:121-135. DOI | PubMed
- Alexander B, Mandel J, Baker B, Burns C, Bartels M, Acquavella J. Biomonitoring of 2,4-dichlorophenoxyacetic acid exposure and dose in farm families. Environ Health Perspect. 2007; 115:370-376. DOI | PubMed
- Calaf GM, Garrido F. Catechol estrogens as biomarkers for mammary gland cancer. Int J Oncol. 2011; 39:177-183. PubMed
- Beard J. DDT and human health. Sci Total Environ. 2006; 355:78-89. DOI | PubMed
- . Accessed 25 Oct 2016.Publisher Full Text
- Rogan WJ, Ragan NB. Some evidence of effects of environmental chemicals on the endocrine system in children. Int J Hyg Environ Health.. 2007; 21:659-667. DOI
- Shen H, Main K, Virtanen H, Damggard I, Haavisto A, Kaleva M. From mother to child: Investigation of prenatal and postnatal exposure to persistent bioaccumulating toxicants using breast milk and placenta biomonitoring. Chemosphere. 2007; 67:236-262. DOI
- Zheng T, Holford T, Mayne S, Ward B, Carter D, Owens P. DDE and DDT in breast adipose tissue and risk of female breast cancer. Am J Epidemiol. 1999; 150:453-458. DOI | PubMed
- Brody JG, Moysich KB, Humblet O, Attfield KR, Beehler GP, Rudel RA. Environmental pollutants and breast cancer: Epidemiologic studies. Cancer. 2007; 109:2667-2711. DOI | PubMed
- Gammon MD, Santella RM, Neugut AI. Environmental toxins and breast cancer on Long Island. II. Organochlorine compound levels in Blood. Cancer Epidemiol Biomark Prev. 2002; 11:686-697.
- Cohn BA. Developmental and environmental origins of breast cancer: DDT as a case study. Reprod Toxicol. 2011; 31:302-311. DOI | PubMed
- Shakeel MK, George PS, Jose J, Jose J, Mathew A. Pesticides and breast cancer risk: A comparison between developed and developing countries. Asian Pac J Cancer Prev. 2010; 11:173-180. PubMed
- Parada H, Wolff MS, Engel LS, White AJ, Eng SM, Cleveland RJ. Organochlorine insecticides DDT and chlordane in relation to survival following breast cancer. Int J Cancer. 2016; 138:565-575. DOI | PubMed
- Canales-Aguirre A, Padilla-Camberos E, Gómez-Pinedo U, Salado-Ponce H, Feria-Velasco A, De Celis R. Genotoxic effect of chronic exposure to DDT on lymphocytes, oral mucosa and breast cells of female parrats. Int J Environ Res Public Health. 2011; 8:540-553. DOI | PubMed
- Robison AK, Sirbasku DA, Stancel GM. DDT supports the growth of an estrogen-responsive tumor. Toxicol Lett. 1985; 27:109-113. DOI | PubMed
- Scribner J, Mottet N. DDT acceleration of mammary gland tumors induced in the male Sprague-Dawley rat by 2-acetamidophenanthrene. Carcinogenesis. 1981; 2:1235-1239. DOI | PubMed
- Uppala PT, Roy SK, Tousson A, Barnes S, Uppala GR, Eastmond DA. Induction of cell proliferation, micronuclei and hyperdiploidy/polyploidy in the mammary cells of DDT- and DMBA-treated pubertal rats. Environ Mol Mutagen. 2005; 46:43-52. DOI | PubMed
- Pujol P, Hilsenbeck S, Chamness G, Elledge R. Rising levels of estrogen receptor in breast cancer over 2 decades. Cancer. 1994; 74:1601-1606. DOI | PubMed
- Woolcott C, Aronson K, Hanna W, SenGupta S, McCready D, Sterns E. Organochlorines and breast cancer risk by receptor status, tumor size and grade. Canada. Cancer Causes Control. 2001; 12:395-404. DOI | PubMed
- Davies H, Delistraty D. Evaluation of PCB sources and releases for identifying priorities to reduce PCBs in Washington state (USA). Environ Sci Pollut Res Int. 2016; 23:2033-2041. DOI | PubMed
- Weber R, Watson A, Forter M, Oliaei F. Review article: Persistent organic pollutants and landfills – a review of past experiences and future challenges. Waste Manag Res. 2011; 29:107-121. DOI | PubMed
- . Accessed 24 Oct 2016.Publisher Full Text
- Martinez A, Schnoebelen DJ, Hornbuckle KC. Polychlorinated biphenyl congeners in sediment cores from the upper Mississippi River. Chemosphere. 2016; 144:1943-1949. DOI | PubMed
- Shunthirasingham C, Gawor A, Hung H, Brice KA, Su K, Alexandrou N. Atmospheric concentrations and loadings of organochlorine pesticides and polychlorinated biphenyls in the Canadian Great Lakes Basin (GLB): Spatial and temporal analysis (1992–2012). Environ Pollut. 2016; 217:124-133. DOI | PubMed
- Hagmar L, Wallin E, Vessby B, Jönsson BA, Bergman A, Rylander L. Intra-individual variations and time trends 1991–2001 in human serum levels of PCB, DDE and hexachlorobenzene. Chemosphere. 2006; 64:1507-1513. DOI | PubMed
- Choi AL, Levy JI, Dockery DW, Ryan LM, Tolbert PE, Altshul LM. Does living near a superfund site contribute to higher polychlorinated biphenyl (PCB) exposure?. Environ Health Perspect. 2006; 114:1092-1098. PubMed
- Wolff MS, Camann D, Gammon M, Stellman SD. Proposed PCB congener groupings for epidemiological studies. Environ Health Perspect. 1997; 105:13-14. DOI | PubMed
- Zhang J, Huang Y, Wang X, Lin K, Wu K. Environmental polychlorinated biphenyl exposure and breast cancer risk: A meta-analysis of observational studies. PLoS One. 2015; 10:e0142513. DOI | PubMed
- Braathen M, Mortensen AS, Sandvik M, Skåre JU, Arukwe A. Estrogenic effects of selected hydroxy polychlorinated biphenyl congeners in primary culture of Atlantic Salmon (Salmo Salar) hepatocytes. Arch Environ Contam Toxicol. 2009; 56:111-122. DOI | PubMed
- Helmfrid I, Berglund M, Löfman O, Wingren G. Health effects and exposure to polychlorinated biphenyls (PCBs) and metals in a contaminated community. Environ Int. 2012; 44:53-58. DOI | PubMed
- Muscat JE, Britton JA, Djordjevic MV, Citron ML, Kemeny M, Busch-Devereaux E. Adipose concentrations of organochlorine compounds and breast cancer recurrence in Long Island, New York. Cancer Epidemiol Biomark Prev. 2003; 12:1474-1478.
- Ellsworth RE, Mamula KA, Costantino NS, Deyarmin B, Kostyniak PJ, Chi L-H. Abundance and distribution of polychlorinated biphenyls (PCBs) in breast tissue. Environ Res. 2015; 138:291-297. DOI | PubMed
- Leng L, Li J, Luo X-M, Kim J-Y, Li Y-M, Guo X-M. Polychlorinated biphenyls and breast cancer: A congener-specific meta-analysis. Environ Int. 2016; 88:133-141. DOI | PubMed
- Aronson KJ, Miller AB, Woolcott CG, Sterns EE, McCready DR, Lickley LA. Breast adipose tissue concentrations of polychlorinated biphenyls and other organochlorines and breast cancer risk. Cancer Epidemiol Biomark Prev. 2000; 9:55-63.
- Salehi F, Turner MC, Phillips KP, Wigle DT, Krewski D, Aronson KJ. Review of the etiology of breast cancer with special attention to organochlorines as potential endocrine disruptors. J Toxicol Environ Health B Crit Rev. 2008; 11:276-300. DOI | PubMed
- Golden R, Kimbrough R. Weight of evidence evaluation of potential human cancer risks from exposure to polychlorinated biphenyls: An update based on studies published since 2003. Crit Rev Toxicol. 2009; 39:299-331. DOI | PubMed
- Silver SR, Whelan EA, Deddens JA, Steenland NK, Hopf NB, Waters MA. Occupational exposure to polychlorinated biphenyls and risk of breast cancer. Environ Health Perspect. 2009; 117:276-282. DOI | PubMed
- Artacho-Cordón F, Fernández-Rodríguez M, Garde C, Salamanca E, Iribarne-Durán LM, Torné P. Serum and adipose tissue as matrices for assessment of exposure to persistent organic pollutants in breast cancer patients. Environ Res. 2015; 142:633-643. DOI | PubMed
- Laden F, Collman G, Iwamoto K, Alberg AJ, Berkowitz GS, Freudenheim JL. 1,1-Dichloro-2,2-bis(p-chlorophenyl)ethylene and polychlorinated biphenyls and breast cancer: Combined analysis of five U.S. studies. J Natl Cancer Inst. 2001; 93:768-776. DOI | PubMed
- Laden F, Ishibe N, Hankinson SE, Wolff MS, Gertig DM, Hunter DJ. Polychlorinated biphenyls, cytochrome P450 1A1, and breast cancer risk in the nurses’ health study. Cancer Epidemiol Biomark Prev. 2002; 11:1560-1565.
- Verner M-A, Bachelet D, McDougall R, Charbonneau M, Guénel P, Haddad S. A case study addressing the reliability of polychlorinated biphenyl levels measured at the time of breast cancer diagnosis in representing early-life exposure. Cancer Epidemiol Biomark Prev. 2011; 20:281-286. DOI
- Kulkarni PS, Crespo JG, Afonso CAM. Dioxins sources and current remediation technologies—a review. Environ Int. 2008; 34:139-153. DOI | PubMed
- . Accessed 25 Oct 2016.Publisher Full Text
- . Accessed 25 Oct 2016.Publisher Full Text
- . Accessed 24 Oct 2016.Publisher Full Text
- Schecter A, Birnbaum L, Ryan JJ, Constable JD. Dioxins: An overview. Environ Res. 2006; 101:419-428. DOI | PubMed
- Massart F, Harrell JC, Federico G, Saggese G. Human breast milk and xenoestrogen exposure: A possible impact on human health. J Perinatol. 2005; 25:282-288. DOI | PubMed
- Nickerson K. Environmental contaminants in breast milk. J Midwifery Womens Health. 2006; 51:26-34. DOI | PubMed
- Dip R, Buterin T, Wenger D, Schmid P, Naegeli H. Genetic reprogramming of human mammary cells by environmental carcinogens released into breast milk. CHIMIA Int J Chem. 2008; 62:410-416. DOI
- Warner M, Eskenazi B, Mocarelli P, Gerthoux PM, Samuels S, Needham L. Serum dioxin concentrations and breast cancer risk in the Seveso Women’s health study. Environ Health Perspect. 2002; 110:625-628. DOI
- Warner M, Mocarelli P, Samuels S, Needham L, Brambilla P, Eskenazi B. Dioxin exposure and cancer risk in the Seveso Women’s health study. Environ Health Perspect. 2011; 119:1700-1705. DOI | PubMed
- Manz A, Berger J, Dwyer JH, Flesch-Janys D, Nagel S, Waltsgott H. Cancer mortality among workers in chemical plant contaminated with dioxin. Lancet. 1991; 338:959-964. DOI | PubMed
- Manuwald U, Velasco Garrido M, Berger J, Manz A, Baur X. Mortality studies of chemical workers exposed to dioxins: Follow-up 23 years after chemical plant closure. Br Med J. 2012; 69:636-642.
- Brown NM, Manzolillo PA, Zhang JX, Wang J, Lamartiniere CA. Prenatal TCDD and predisposition to mammary cancer in the rat. Carcinogenesis. 1998; 19:1623-1629. DOI | PubMed
- Fenton SE, Hamm JT, Birnbaum LS, Youngblood GL. Persistent abnormalities in the rat mammary gland following gestational and lactational exposure to 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD). Toxicol Sci. 2002; 67:63-74. DOI | PubMed
- Lewis BC, Hudgins S, Lewis A, Schorr K, Sommer R, Peterson RE. In utero and lactational treatment with 2,3,7,8-tetrachlorodibenzo-p-dioxin impairs mammary gland differentiation but does not block the response to exogenous estrogen in the postpubertal female rat. Toxicol Sci. 2001; 62:46-53. DOI | PubMed
- Jenkins S, Rowell C, Wang J, Lamartiniere C. Prenatal TCDD exposure predisposes for mammary cancer in rats. Reprod Toxicol. 2007; 23:391-396. DOI | PubMed
- La Merrill M, Kuruvilla BS, Pomp D, Birnbaum LS, Threadgill DW. Dietary fat alters body composition, mammary development, and cytochrome p450 induction after maternal TCDD exposure in DBA/2J mice with low-responsive aryl hydrocarbon receptors. Environ Health Perspect. 2009; 117:1414-1419. DOI | PubMed
- Seifert A, Taubert H, Hombach-Klonisch S, Fischer B, Navarrete Santos A. TCDD mediates inhibition of p53 and activation of ERalpha signaling in MCF-7 cells at moderate hypoxic conditions. Int J Oncol. 2009; 35:417-424. PubMed
- Yoshioka H, Hiromori Y, Aoki A, Kimura T, Fujii-Kuriyama Y, Nagase H. Possible aryl hydrocarbon receptor-independent pathway of 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced antiproliferative response in human breast cancer cells. Toxicol Lett. 2012; 211:257-265. DOI | PubMed
- Marques M, Laflamme L, Gaudreau L. Estrogen receptor α can selectively repress dioxin receptor-mediated gene expression by targeting DNA methylation. Nucleic Acids Res. 2013; 41:8094-8106. DOI | PubMed
- Marlowe JL, Puga A. Aryl hydrocarbon receptor, cell cycle regulation, toxicity, and tumorigenesis. J Cell Biochem. 2005; 96:1174-1184. DOI | PubMed
- Bekki K, Vogel H, Li W, Ito T, Sweeney C, Haarmann-Stemmann T. The aryl hydrocarbon receptor (AhR) mediates resistance to apoptosis induced in breast cancer cells. Pestic Biochem Physiol. 2015; 120:5-13. DOI | PubMed
- Chen Y-J, Hung C-M, Kay N, Chen C-C, Kao Y-H, Yuan S-S. Progesterone receptor is involved in 2,3,7,8-tetrachlorodibenzo-p-dioxin-stimulated breast cancer cells proliferation. Cancer Lett. 2012; 319:223-231. DOI | PubMed
- Labrecque MP, Takhar MK, Hollingshead BD, Prefontaine GG, Perdew GH, Beischlag TV. Distinct roles for aryl hydrocarbon receptor nuclear translocator and ah receptor in estrogen-mediated signaling in human cancer cell lines. PLoS One. 2012; 7:e29545. DOI | PubMed
- Lo R, Matthews J. The aryl hydrocarbon receptor and estrogen receptor alpha differentially modulate nuclear factor erythroid-2-related factor 2 transactivation in MCF-7 breast cancer cells. Toxicol Appl Pharmacol. 2013; 270:139-148. DOI | PubMed
- Ohtake F, Fujii-Kuriyama Y, Kawajiri K, Kato S. Cross-talk of dioxin and estrogen receptor signals through the ubiquitin system. J Steroid Biochem Mol Biol. 2011; 127:1027. DOI
- Papoutis AJ, Selmin OI, Borg JL, Romagnolo DF. Gestational exposure to AhR agonist 2,3,7,8-tetrachlorodibenzo-p-dioxin induces BRCA-1 promoter hypermethylation and reduces BRCA-1 expression in mammary tissue of rat offspring: Preventive effects of resveratrol. Mol Carcinogen. 2015; 54:261-269. DOI
- Zota A, Rudel R, Morello-Frosch R, Brody J. Elevated house dust and serum concentrations of PBDEs in California: Unintended consequences of furniture flammability standards?. Environ Sci Technol. 2008; 42:8158-8164. DOI | PubMed
- Dodson RE, Perovich LJ, Covaci A, Van Den Eede N, Ionas AC, Dirtu AC. After the PBDE phase-out: A broad suite of flame retardants in repeat house dust samples from California. Environ Sci Technol. 2012; 46:13056-13066. DOI | PubMed
- Petreas M, Nelson D, Brown FR, Goldberg D, Hurley S, Reynolds P. High concentrations of polybrominated diphenylethers (PBDEs) in breast adipose tissue of California women. Environ Int. 2011; 37:190-197. DOI | PubMed
- Eskenazi B, Fenster L, Castorina R, Marks AR, Sjödin A, Rosas LG. A comparison of PBDE serum concentrations in Mexican and Mexican-American children living in California. Environ Health Perspect. 2011; 119:1442-1448. DOI | PubMed
- Windham G, Pinney S, Sjodin A, Lum R, Jones R, Needham L. Body burdens of brominated flame retardants and other persistent organo-halogenated compounds and their descriptors in US girls. Environ Res. 2010; 110:251-257. DOI | PubMed
- Windham GC, Pinney SM, Voss RW, Sjödin A, Biro FM, Greenspan LC. Brominated flame retardants and other persistent organohalogenated compounds in relation to timing of puberty in a longitudinal study of girls. Environ Health Perspect. 2015; 123:1046-1052. DOI | PubMed
- Costa L, Giordano G, Tagliaferri S, Caglieri A, Mutti A. Polybrominated diphenyl ether (PBDE) flame retardants: Environmental contamination, human body burden and potential adverse health effects. Acta Biomed. 2008; 79:172-183. PubMed
- Darnerud PO, Eriksen GS, Jóhannesson T, Larsen PB, Viluksela M. Polybrominated diphenyl ethers: Occurrence, dietary exposure, and toxicology. Environ Health Perspect. 2001; 109(Suppl 1):49-68. DOI | PubMed
- De Wit C. An overview of brominated flame retardants in the environment. Chemosphere. 2002; 46:583-624. DOI | PubMed
- Frederiksen M, Thomsen C, Froshaug M, Vorkamp K, Thomsen M, Becher G. Polybrominated diphenyl ethers in paired samples of maternal and umbilical cord blood plasma and assocaitions with house dust in a Danish cohort. Int J Hyg Environ Health. 2010; 13:233-242. DOI
- Costa LG, Giordano G. Developmental neurotoxicity of polybrominated diphenyl ether (PBDE) flame retardants. Neurotoxicology. 2007; 28:1047-1067. DOI | PubMed
- Talsness CE. Overview of toxicological aspects of polybrominated diphenyl ethers: A flame-retardant additive in several consumer products. Environ Res. 2008; 108:158-167. DOI | PubMed
- Hurley S, Reynolds P, Goldberg D, Nelson DO, Jeffrey SS, Petreas M. Adipose levels of polybrominated diphenyl ethers and risk of breast cancer. Breast Cancer Res Treat. 2011; 129:505-511. DOI | PubMed
- Meerts IA, Letcher RJ, Hoving S, Marsh G, Bergman A, Lemmen JG. In vitro estrogenicity of polybrominated diphenyl ethers, hydroxylated PDBEs, and polybrominated bisphenol a compounds. Environ Health Perspect. 2001; 109:399-407. DOI | PubMed
- Yu L, Zhan P. Molecular mechanisms underlying proliferation and apoptosis in breast cancer MCF-7 cells induced by pentabrominated diphenyl ethers. Toxicol Environ Chem. 2009; 91:665-670. DOI
- Kwiecińska P, Wróbel A, Gregoraszczuk EŁ. Combinatory effects of PBDEs and 17β-estradiol on MCF-7 cell proliferation and apoptosis. Pharmacol Rep. 2011; 63:189-194. DOI | PubMed
- Li Z-H, Liu X-Y, Wang N, Chen J-S, Chen Y-H, Huang J-T. Effects of decabrominated diphenyl ether (PBDE-209) in regulation of growth and apoptosis of breast, ovarian, and cervical cancer cells. Environ Health Perspect. 2012; 120:541-546. DOI | PubMed
- Davis F, Lin H-Y, Luidens M, Zhou M, Mousa S. Similar and shared nongenomic mechanisms of action of estrogen and thyroid hormone. Immunol Endocr Metabol Agents Med Chem. 2009; 9:84-89. DOI
- Birnbaum L. Halogenated flame retardants: Does the benefit justify the risk?. Environ Health Perspect. 2009; 117:A478. DOI | PubMed
- Patisaul HB, Roberts SC, Mabrey N, Mccaffrey KA, Gear RB, Braun J. Accumulation and endocrine disrupting effects of the flame retardant mixture Firemaster® 550 in rats: An exploratory assessment. J Biochem Mol Toxicol. 2013; 27:124-136. DOI | PubMed
- Baan R, Straif K, Grosse Y, Secretan B, El G, Bouvard V. Carcinogenicity of some aromatic amines, organic dyes, and related exposures. Lancet Oncol. 2008; 9:322-323. DOI | PubMed
- DeBruin LS, Pawliszyn JB, Josephy PD. Detection of monocyclic aromatic amines, possible mammary carcinogens, in human milk. Chem Res Toxicol. 1999; 12:78-82. DOI | PubMed
- DeBruin LS, Josephy PD. Perspectives on the chemical etiology of breast cancer. Environ Health Perspect. 2002; 110(Suppl 1):119-128. DOI
- National Toxicology Program (NTP). Report on carcinogens. U.S. Department of Health and Human Services, Public Health Service: Research Triangle Park; 2014.
- Layton D, Bogen K, Knize M, Hatch F, Johnson V, Felton J. Cancer risk of heterocyclic amines in cooked foods: An analysis and implications for research. Carcinogenesis. 1995; 16:39-52. DOI | PubMed
- Ambrosone CB, Abrams SM, Gorlewska-Roberts K, Kadlubar FF. Hair dye use, meat intake, and tobacco exposure and presence of carcinogen-DNA adducts in exfoliated breast ductal epithelial cells. Arch Biochem Biophys. 2007; 464:169-175. DOI | PubMed
- de Vocht F, Sobala W, Wilczynska U, Kromhout H, Szeszenia-Dabrowska N, Peplonska B. Cancer mortality and occupational exposure to aromatic amines and inhalable aerosols in rubber tire manufacturing in Poland. Cancer Epidemiol. 2009; 33:94-102. DOI | PubMed
- Meyer A, Fischer K. Oxidative transformation processes and products of para-phenylenediamine (PPD) and para-tolyenediamine (PTD) – A review. Environ Sci Eur. 2015; 27:11. DOI
- Heikkinen S, Pitkäniemi J, Sarkeala T, Malila N, Koskenvuo M. Does hair dye use increase the risk of breast cancer? A population-based case-control study of Finnish women. PLoS One. 2015; 10:e0135190. DOI | PubMed
- Steck SE, Gaudet MM, Eng SM, Britton JA, Teitelbaum SL, Neugut AI. Cooked meat and risk of breast cancer—lifetime versus recent dietary intake. Epidemiology. 2007; 18:373-382. DOI | PubMed
- Thompson PA, DeMarini DM, Kadlubar FF, McClure GY, Brooks LR, Green BL. Evidence for the presence of mutagenic arylamines in human breast milk and DNA adducts in exfoliated breast ductal epithelial cells. Environ Mol Mutagen. 2002; 39:134-142. DOI | PubMed
- Turesky RJ. Formation and biochemistry of carcinogenic heterocyclic aromatic amines in cooked meats. Toxicol Lett. 2007; 168:219-227. DOI | PubMed
- Rabstein S, Brüning T, Harth V, Fischer H-P, Haas S, Weiss T. N-acetyltransferase 2, exposure to aromatic and heterocyclic amines, and receptor-defined breast cancer. Eur J Cancer Prev. 2010; 19:100-109. DOI | PubMed
- Gooderham NJ, Creton S, Lauber SN, Zhu H. Mechanisms of action of the carcinogenic heterocyclic amine PhIP. Toxicol Lett. 2007; 168:269-277. DOI | PubMed
- Ionescu JG, Novotny J, Stejskal V, Lätsch A, Blaurock-Busch E, Eisenmann-Klein M. Increased levels of transition metals in breast cancer tissue. Neuroendocrinol Lett. 2006; 27:36-39. PubMed
- Peng L, Huang Y, Zhang J, Peng Y, Lin X, Wu K. Cadmium exposure and the risk of breast cancer in Chaoshan population of southeast China. Environ Sci Pollut Res. 2015; 22:19870-19878. DOI
- Strumylaite L, Bogusevicius A, Abdrachmanovas O, Baranauskiene D, Kregzdyte R, Pranys D. Cadmium concentration in biological media of breast cancer patients. Breast Cancer Res Treat. 2011; 125:511-7. DOI | PubMed
- Wu H-DI, Chou S-Y, Chen D-R, Kuo H-W. Differentiation of serum levels of trace elements in normal and malignant breast patients. Biol Trace Elem Res. 2006;113:9–18.
- Brama M, Gnessi L, Basciani S, Cerulli N, Politi L, Spera G, et al. Cadmium induces mitogenic signaling in breast cancer cell by an ERα-dependent mechanism. Mol Cell Endocrinol. 2007;264:102–8.
- Martin MB, Reiter R, Pham T, Avellanet YR, Camara J, Lahm M. Estrogen-like activity of metals in MCF-7 breast cancer cells. Endocrinology. 2003; 144:2425-2436. DOI | PubMed
- Sukocheva OA, Yang Y, Gierthy JF, Seegal RF. Methyl mercury influences growth-related signaling in MCF-7 breast cancer cells. Environ Toxicol. 2005; 20:32-44. DOI | PubMed
- Choe S-Y, Kim S-J, Kim H-G, Lee JH, Choi Y, Lee H. Evaluation of estrogenicity of major heavy metals. Sci Total Environ. 2003; 312:15-21. DOI | PubMed
- McElroy JA, Shafer MM, Trentham-Dietz A, Hampton JM, Newcomb PA. Cadmium exposure and breast cancer risk. J Natl Cancer Inst. 2006; 98:869-873. DOI | PubMed
- Gallagher C, Chen JJ, Kovach JS. Environmental cadmium and breast cancer. Aging. 2010; 1:804-814. DOI
- Saleh F, Behbehani A, Asfar S, Khan I, Ibrahim G. Abnormal blood levels of trace elements and metals, DNA damage, and breast cancer in the state of Kuwait. Biol Trace Elem Res. 2011; 141:96-109. DOI | PubMed
- Adams SV, Shafer MM, Bonner MR, LaCroix AZ, Manson JE, Meliker JR. Urinary cadmium and risk of invasive breast cancer in the Women’s health initiative. Am J Epidemiol. 2016; 183:815-823. DOI | PubMed
- Lin J, Zhang F, Lei Y. Dietary intake and urinary level of cadmium and breast cancer risk: A meta-analysis. Cancer Epidemiol. 2016; 42:101-107. DOI | PubMed
- Åkesson A, Julin B, Wolk A. Long-term dietary cadmium intake and postmenopausal endometrial cancer incidence: A population-based prospective cohort study. Cancer Res. 2008; 68:6435-6441. DOI | PubMed
- Julin B, Wolk A, Bergkvist L, Bottai M, Åkesson A. Dietary cadmium exposure and risk of postmenopausal breast cancer: A population-based prospective cohort study. Cancer Res. 2012; 72:1459-1466. DOI | PubMed
- Itoh H, Iwasaki M, Sawada N, Takachi R, Kasuga Y, Yokoyama S. Dietary cadmium intake and breast cancer risk in Japanese women: A case-control study. Int J Hyg Environ Health. 2014; 217:70-77. DOI | PubMed
- Sawada N, Iwasaki M, Inoue M, Takachi R, Sasazuki S, Yamaji T. Long-term dietary cadmium intake and cancer incidence. Epidemiology. 2012; 23:368-376. DOI | PubMed
- Eriksen KT, Halkjær J, Sørensen M, Meliker JR, McElroy JA, Tjønneland A, et al. Dietary cadmium intake and risk of breast, endometrial and ovarian cancer in danish postmenopausal women: A prospective cohort study. PLoS One. 2014;9:e100815.
- Adams SV, Newcomb PA, White E. Dietary cadmium and risk of invasive postmenopausal breast cancer in the VITAL cohort. Cancer Causes Control. 2012; 23:845-854. DOI | PubMed
- Van Maele-Fabry G, Lombaert N, Lison D. Dietary exposure to cadmium and risk of breast cancer in postmenopausal women: A systematic review and meta-analysis. Environ Int. 2016; 86:1-13. DOI | PubMed
- Johnson MD, Kenney N, Stoica A. Cadmium mimics the in vivo effects of estrogen in the uterus and mammary gland. Nat Med. 2003; 9:1081-1084. DOI | PubMed
- Byrne C, Divekar SD, Storchan GB, Parodi DA, Martin MB. Metals and breast cancer. J Mammary Gland Biol Neoplasia. 2013; 18:63-73. DOI | PubMed
- Silva N, Peiris-John R, Wickremasinghe R, Senanayake H, Sathiakumar N. Cadmium a metalloestrogen: Are we convinced?. J Appl Toxicol. 2012; 32:318-332. DOI | PubMed
- Benbrahim-Tallaa L, Tokar EJ, Diwan BA, Dill AL, Coppin J-F, Waalkes MP. Cadmium malignantly transforms normal human breast epithelial cells into a basal-like phenotype. Environ Health Perspect. 2009; 117:1847-1852. DOI | PubMed
- Ali I, Penttinen-Damdimopoulou PE, Mäkelä SI, Berglund M, Stenius U, Åkesson A. Estrogen-like effects of cadmium in vivo do not appear to be mediated via the classical estrogen receptor transcriptional pathway. Environ Health Perspect. 2010; 118:1389-1394. DOI | PubMed
- Ali I, Damdimopoulou P, Stenius U, Adamsson A, Mäkelä SI, Åkesson A. Cadmium-induced effects on cellular signaling pathways in the liver of transgenic estrogen reporter mice. Toxicol Sci. 2012; 127(1):66-75. DOI | PubMed
- Yu X, Filardo EJ, Shaikh ZA. The membrane estrogen receptor GPR30 mediates cadmium-induced proliferation of breast cancer cells. Toxicol Appl Pharmacol. 2010; 245:83-90. DOI | PubMed
- Martinez-Zamudio R, Ha HC. Environmental epigenetics in metal exposure. Epigenetics. 2011; 6:820-827. DOI | PubMed
- Asara Y, Marchal JA, Bandiera P, Mazzarello V, Delogu LG, Sotgiu MA. Cadmium influences the 5-fluorouracil cytotoxic effects on breast cancer cells. Eur J Histocyhem. 2012; 56:1-6. DOI
- Seibold P, Vrieling A, Heinz J, Obi N, Sinn H-P, Flesch-Janys D. Pre-diagnostic smoking behaviour and poorer prognosis in a german breast cancer patient cohort – differential effects by tumour subtype, NAT2 status, BMI and alcohol intake. Cancer Epidemiol. 2014; 38:419-426. DOI | PubMed
- Xie J, Tworoger SS, Franke AA, Terry KL, Rice MS, Rosner BA. Plasma enterolactone and breast cancer risk in the nurses’ health study II. Breast Cancer Res Treat. 2013; 139:801-809. DOI | PubMed
- Xiong X-Y, Hu X-J, Li Y, Liu C-M. Inhibitory effects of Enterolactone on growth and metastasis in human breast cancer. Nutr Cancer. 2015; 67:1324-1332. DOI | PubMed
- Flower G, Fritz H, Balneaves LG, Verma S, Skidmore B, Fernandes R. Flax and breast cancer: A systematic review. Integr Cancer Ther. 2014; 13:181-192. DOI | PubMed
- Cederroth C, Nef S. Soy, phytoestrogens and metabolism: A review. Mol Cell Endocrinol. 2009; 304:30-42. DOI | PubMed
- He F-J, Chen J-Q. Consumption of soybean, soy foods, soy isoflavones and breast cancer incidence: Differences between Chinese women and women in western countries and possible mechanisms. Food Sci Human Wellness. 2013; 2:146-161. DOI
- Andres S, Abraham K, Appel KE, Lampen A. Risks and benefits of dietary isoflavones for cancer. Crit Rev Toxicol. 2011; 41:463-506. DOI | PubMed
- Rice S, Whitehead SA. Phytoestrogens and breast cancer – promoters or protectors?. Endocr Relat Cancer. 2006; 13:995-1015. DOI | PubMed
- Helferich WG, Andrade JE, Hoagland MS. Phytoestrogens and breast cancer: A complex story. Inflammopharmacology. 2008; 16:219-226. DOI | PubMed
- Lee S-A, Shu X-O, Li H, Yang G, Cai H, Wen W. Adolescent and adult soy food intake and breast cancer risk: Results from the shanghai Women’s health study. Am J Clin Nutr. 2009; 89:1920-1926. DOI | PubMed
- Wu Y-C, Zheng D, Sun J-J, Zou Z-K, Ma Z-L. Meta-analysis of studies on breast cancer risk and diet in Chinese women. Int J Clin Exp Med. 2015; 8:73-85. PubMed
- Zhang M, Yang H, Holman CDJ. Dietary intake of isoflavones and breast cancer risk by estrogen and progesterone receptor status. Breast Cancer Res Treat. 2009; 118:553-563. DOI | PubMed
- Shu XO, Zheng Y, Cai H, Gu K, Chen Z, Zheng W. Soy food intake and breast cancer survival. JAMA. 2009; 302:2437-2443. DOI | PubMed
- Woo HD, Park KS, Ro J, Kim J. Differential influence of dietary soy intake on the risk of breast cancer recurrence related to HER2 status. Nutr Cancer. 2012; 64:198-205. DOI | PubMed
- Khan SA, Chatterton RT, Michel N, Bryk M, Lee O, Ivancic D. Soy isoflavone supplementation for breast cancer risk reduction: A randomized phase ii trial. Cancer Prev Res. 2012; 5:309-319. DOI
- Nagata C, Mizoue T, Tanaka K, Tsuji I, Tamakoshi A, Matsuo K. Soy intake and breast cancer risk: An evaluation based on a systematic review of epidemiologic evidence among the Japanese population. Jap J Clin Oncol. 2014; 44:282-295. DOI | PubMed
- Takagi A, Kano M, Kaga C. Possibility of breast cancer prevention: Use of soy isoflavones and fermented soy beverage produced using probiotics. Int J Mol Sci. 2015; 16:10907-10920. DOI | PubMed
- Toi M, Hirota S, Tomotaki A, Sato N, Hozumi Y, Anan K. Probiotic beverage with soy isoflavone consumption for breast cancer prevention: A case-control study. Curr Nutr Food Sci. 2013; 9:194-200. DOI | PubMed
- Wada K, Nakamura K, Tamai Y, Tsuji M, Kawachi T, Hori A. Soy isoflavone intake and breast cancer risk in Japan: From the Takayama study. Int J Cancer. 2013; 133:952-960. DOI | PubMed
- Korde LA, Wu AH, Fears T, Nomura AMY, West DW, Kolonel LN. Childhood soy intake and breast cancer risk in Asian American women. Cancer Epidemiol Biomarkers Prev. 2009; 18:1050-1059. DOI | PubMed
- Adlercreutz H. Phytoestrogens and breast cancer. J Ster Biochem Mol Biol. 2002; 83:113-118. DOI
- Anderson LN, Cotterchio M, Boucher BA, Kreiger N. Phytoestrogen intake from foods, during adolescence and adulthood, and risk of breast cancer by estrogen and progesterone receptor tumor subgroup among Ontario women. Int J Cancer. 2013; 132:1683-1692. DOI | PubMed
- Messina M, Wu A. Perspectives on the soy-breast cancer relation. Am Soc Nutr. 2009; 89:1673-1679.
- Warri A, Saarinen N, Makela S, Hilakivi-Clarke L. The role of early life genistein exposures in modifying breast cancer risk. Br J Cancer. 2008; 98:1485-1493. DOI | PubMed
- Zhao Y, Pan J-H, Zhang L-L. Soy foods intake in adolescence and the risk of breast cancer: A systematic review. Chin J Evid-Based Med. 2012; 12:550-556.
- Chen M, Rao Y, Zheng Y, Wei S, Li Y, Guo T. Association between soy isoflavone intake and breast cancer risk for pre- and post-menopausal women: A meta-analysis of epidemiological studies. PLoS One. 2014; 9:e89288. DOI | PubMed
- Goodman M, Shvetsov Y, Wilkens L, Franke A, Le Marchand L, Kakazu K. Urinary phytoestrogen excretion and postmenopausal breast cancer risk: The multiethnic cohort study. Cancer Prev Res. 2009; 2:887-894. DOI
- Möller FJ, Pemp D, Soukup ST, Wende K, Zhang X, Zierau O. Soy isoflavone exposure through all life stages accelerates 17β-estradiol-induced mammary tumor onset and growth, yet reduces tumor burden, in ACI rats. Arch Toxicol. 2016; 90:1907-1916. DOI | PubMed
- Phrakonkham P, Brouland JP, Saad HES, Bergès R, Pimpie C, Pocard M. Dietary exposure in utero and during lactation to a mixture of genistein and an anti-androgen fungicide in a rat mammary carcinogenesis model. Reprod Toxicol. 2015; 54:101-109. DOI | PubMed
- Liu B, Edgerton S, Yang X, Kim A, Ordonez-Ercan D, Mason T. Low-dose dietary phytoestrogen abrogates tamoxifen-associated mammary tumor prevention. Cancer Res. 2005; 65:879-886. PubMed
- Constantinou A, White B, Tonetti D, Yang Y, Liang W, Li W. The soy isoflavone daidzein improves the capacity of tamoxifen to prevent mammary tumors. Eur J Cancer. 2005; 41:647-654. DOI | PubMed
- Dip R, Lenz S, Gmuender H, Naegeli H. Pleiotropic combinatorial transcriptomes of human breast cancer cells exposed to mixtures of dietary phytoestrogens. Food Chem Toxicol. 2009; 47:787-795. DOI | PubMed
- Sakamoto T, Horiguchi H, Oguma E, Kayama F. Effects of diverse dietary phytoestrogens on cell growth, cell cycle and apoptosis in estrogen-receptor-positive breast cancer cells. J Nutr Biochem. 2010; 21:856-864. DOI | PubMed
- Liu Y, Hilakivi-Clarke L, Zhang Y, Wang X, Pan Y-X, Xuan J. Isoflavones in soy flour diet have different effects on whole-genome expression patterns than purified isoflavone mix in human MCF-7 breast tumors in ovariectomized athymic nude mice. Mol Nutr Food Res. 2015; 59:1419-1430. DOI | PubMed
- Aparecida Santos M, Florencio-Silva R, Teixeira CP, Rodrigues Da Silva Sasso G, Souza Marinho D, Simões RS. Effects of early and late treatment with soy isoflavones in the mammary gland of ovariectomized rats. Climacteric. 2016; 19:77-84. DOI | PubMed
- Garvin S, Ollinger K, Dabrosin C. Resveratrol induces apoptosis and inhibits angiogenesis in human breast cancer xenografts in vivo. Cancer Lett. 2006; 231:113-122. DOI | PubMed
- Athar M, Back J, Kopelovich L, Bickers D, Kim A. Multiple molecular targets of resveratrol: Anti-carcinogenic mechanisms. Arch Biochem Biophys. 2009; 486:95-102. DOI | PubMed
- Boucher B, Cotterchio M, Krieger N, Thompson L. Soy formula and breast cancer risk. Epidemiology. 2008; 19:165-168. DOI | PubMed
- Jefferson W, Doerge D, Padilla-Banks E, Woodling K, Kissling G, Newbold R. Oral exposure to genistin, the glycosylated form of genistein, during neonatal life adversely affects the female reproductive system. Environ Health Perspect. 2009; 117:1883-1889. DOI | PubMed
- Benzoni E, Minervini F, Giannoccaro A, Fornelli F, Vigo D, Visconti A. Influence of in vitro exposure to mycotoxin zearalenone and its derivatives on swine sperm quality. Reprod Toxicol. 2008; 25:461-467. DOI | PubMed
- Mirocha C, Schauerhamer B, Christensen C, Niku-Paavola M, Nummi M. Incidence of zearalenol (fusarium mycotoxin) in animal feed. Appl Environ Microbiol. 1979; 38:749-750. PubMed
- Aiko V, Mehta A. Occurrence, detection and detoxification of mycotoxins. J Biosci. 2015; 40:943-954. DOI | PubMed
- Mukherjee D, Royce SG, Alexander JA, Buckley B, Isukapalli SS, Bandera EV. Physiologically-based toxicokinetic modeling of zearalenone and its metabolites: Application to the Jersey girl study. PLoS One. 2014; 9:e113632. DOI | PubMed
- Massart F, Meucci V, Saggese G, Soldani G. High growth rate of girls with precocious puberty exposed to estrogenic mycotoxins. J Pediatr. 2008; 152:690-5.e1. DOI | PubMed
- Massart F, Saggese G. Oestrogenic mycotoxin exposures and precocious pubertal development. Int J Androl. 2010; 33:369-376. DOI | PubMed
- Bandera EV, Chandran U, Buckley B, Lin Y, Isukapalli S, Marshall I. Urinary mycoestrogens, body size and breast development in New Jersey girls. Sci Total Environ. 2011; 409:5221-5227. DOI | PubMed
- Frehse MS, Martins MIM, Ono EYS, Bracarense APFRL, Bissoqui LY, Teixeira EMK. Aflatoxins ingestion and canine mammary tumors: There is an association?. Food Chem Toxicol. 2015; 84:74-78. DOI | PubMed
- Videmann B, Koraichi F, Mazallon M, Lecoeur S. Effect of gender, pregnancy and exposure conditions on metabolism and distribution of zearalenone in rats. World Mycotox J. 2012; 5:57-69. DOI
- Belli P, Bellaton C, Durand J, Balleydier S, Milhau N, Mure M. Fetal and neonatal exposure to the mycotoxin zearalenone induces phenotypic alterations in adult rat mammary gland. Food Chem Toxicol. 2010; 48:2818-2826. DOI | PubMed
- Nikaido Y, Yoshizawa K, Pei R-J, Yuri T, Danbara N, Hatano T. Prepubertal zearalenone exposure suppresses N-methyl-N-nitrosourea-induced mammary tumorigenesis but causes severe endocrine disruption in female Sprague-Dawley rats. Nutr Cancer. 2003; 47:164-170. DOI | PubMed
- Belhassen H, Jiménez-Díaz I, Arrebola JP, Ghali R, Ghorbel H, Olea N. Zearalenone and its metabolites in urine and breast cancer risk: A case-control study in Tunisia. Chemosphere. 2015; 128:1-6. DOI | PubMed
- Sondergaard TE, Hansen FT, Purup S, Nielsen AK, Bonefeld-Jørgensen EC, Giese H. Fusarin C acts like an estrogenic agonist and stimulates breast cancer cells in vitro. Toxicol Lett. 2011; 205:116-121. DOI | PubMed
- Parveen M, Zhu Y, Kiyama R. Expression profiling of the genes responding to zearalenone and its analogues using estrogen-responsive genes. FEBS Lett. 2009; 583:2377-2384. DOI | PubMed
- Khosrokhavar R, Rahimifard N, Shoeibi S, Hamedani MP, Hosseini M-J. Effects of zearalenone and α-Zearalenol in comparison with Raloxifene on T47D cells. Toxicol Mech Methods. 2009; 19:246-250. DOI | PubMed
- Al-Dobaib S, Mousa H. Benefits and risks of growth promoters in animal production. J Food Agric Environ. 2009; 7:202-208.
- Leffers H, Næsby M, Vendelbo B, Skakkebæk NE, Jørgensen M, Grandjean P. Oestrogenic potencies of Zeranol, oestradiol, diethylstilboestrol, bisphenol-a and genistein: Implications for exposure assessment of potential endocrine disrupters. APMIS Suppl. 2001; 109:S463-S472. DOI
- Updike MS, Sawdy JC, Wang L-S, Liu S, Huang Y-W, Ye W. Primary cultured human breast epithelial cells up-regulate protein disulfide isomerase in response to zeranol. Anticancer Res. 2007; 27:407-410. PubMed
- Sheffield LG, Welsch CW. Zeranol (β-resorcylic acid lactone), a common residous component of natural foodstuffs, stimulates developmental growth of the mouse mammary Gland. Cancer Lett. 1985; 28:77-83. DOI | PubMed
- Nikaido Y, Danbara N, Tsujita-Kyutoku M, Yuri T, Uehara N, Tsubura A. Effects of prepubertal exposure to xenoestrogen on development of estrogen target organs in female CD-1 mice. In Vivo. 2005; 19:487-494. PubMed
- Yuri T, Nikaido Y, Shimano N, Uehara N, Shikata N, Tsubura A. Effects of prepubertal zeranol exposure on estrogen target organs and N-methyl-N-nitrosourea-induced mammary tumorigenesis in female Sprague-Dawley rats. In Vivo. 2004; 18:755-762. PubMed
- Liu S, Kulp SK, Sugimoto Y, Jiang J, Chang H-L, Lin YC. Involvement of breast epithelial-stromal interactions in the regulation of protein tyrosine phosphatase-γ (PTPγ) mRNA expression by estrogenically active agents. Breast Cancer Res Treat. 2002; 71:21-35. DOI | PubMed
- Liu S, Lin Y. Transformation of MCF-10A human breast epithelial cells by zeranol and estradiol-17B. Breast J. 2004; 10:514-521. DOI | PubMed
- Xu P, Ye W, Jen R, Lin S, Kuo C, Lin Y. Mitogenic activity of zeranol in human breast cancer cells is enhanced by leptin and suppressed by gossypol. Anticancer Res. 2009; 29:4621-4628. PubMed
- Ye W, Xu P, Zhong S, Threlfall WR, Frasure C, Feng E. Serum harvested from heifers one month post-zeranol implantation stimulates MCF-7 breast cancer cell growth. Exp Ther Med. 2010; 1:963-968. DOI | PubMed
- Eaton ML. Ethics and business of bioscience. Stanford University Press: Palo Alto; 2004.
- Daxenberger A, Breier BH, Sauerwein H. Increased milk levels of insulin-like growth factor 1 (IGF-1) for the identification of bovine somatotropin (bST) treated cows. Analyst. 1998; 123:2429-2435. DOI | PubMed
- Collier R, Miller M, McLaughlin C, Johnson H, Baile C. Effects of recombinant bovine somatotropin (rbST) and season on plasma and milk insulin-like growth factors I (IGF-I) and II (IGF-II) in lactating dairy cows. Domest Anim Endocrinol. 2008; 35:16-23. DOI | PubMed
- Collier R, Miller M, Hildebrandt J, Torkelson A, White T, Madsen K. Factors affecting insulin-like growth factor-I concentration in bovine milk. J Dairy Sci. 1991; 74:2095-2911. DOI
- Outwater JL, Nicholson A, Barnard N. Dairy products and breast cancer: The IGF-I, estrogen, and bGH hypothesis. Med Hypotheses. 1997; 48:453-461. DOI | PubMed
- Hankinson SE, Willett WC, Colditz GA, Hunter DJ, Michaud DS, Deroo B. Circulating concentrations of insulin-like growth factor-I and risk of breast cancer. Lancet. 1998; 351:1393-1396. DOI | PubMed
- Ciftci K, Su J, Trovitch PB. Growth factors and chemotherapeutic modulation of breast cancer cells. J Pharm Pharmacol. 2003; 55:1135-1141. DOI | PubMed
- Fürstenberger G, Morant R, Senn HJ. Insulin-like growth factors and breast cancer. Onkologie. 2003; 26:290-294. PubMed
- Holly J. Insulin-like growth factor-I and new opportunities for cancer prevention. Lancet. 1998; 351:1373-1375. DOI | PubMed
- Allen N, Roddam A, Allen D, Fentiman I, Peto J, Holly J. A prospective study of serum insulin-like growth factor-I (IGF-1), IGF-II, IGF-binding protein-3 and breast cancer risk. Br J Cancer. 2005; 92:1283-1287. DOI | PubMed
- Schernhammer ES, Holly JM, Pollak MN, Hankinson SE. Circulating levels of insulin-like growth factors, their binding proteins, and breast cancer risk. Cancer Epidemiol Biomark Prev. 2005; 14:699-704. DOI
- Frydenberg H, Flote VG, Iversen A, Finstad SE, Furberg A-S, Torjesen PA. Insulin-like growth factor-1, growth hormone, and daily cycling estrogen are associated with mammographic density in premenopausal women. Cancer Causes Control. 2014; 25:891-903. DOI | PubMed
- Macaulay VM. Insulin-lie growth factors and cancer. Br J Cancer. 1992; 65:311-320. DOI | PubMed
- Resnicoff M, Abraham D, Yutanawiboonchai W, Rotman HL, Kajstura J, Rubin R. The insulin-like growth factor I receptor protects tumor cells from apoptosis in vivo. Cancer Res. 1995; 55:2463-2469. PubMed
- Xian C. Degradation of IGF-1 in the adult rat gastrointestinal tract is limited by a specific antiserum or the dietary protein casein. J Endocrinol. 1995; 146:215-225. DOI | PubMed
- . Accessed 25 Oct 2016.Publisher Full Text
- . Accessed 25 Oct 2016.Publisher Full Text
- IARC. Benzene. IARC Monog Eval Carcinog Risk Chem Hum. 1987; Supl 7:120-122.
- Petralia SA, Chow WH, McLaughlin J. Occupational risk factors for breast cancer among women in shanghai. Am J Ind Med. 1998; 34:477-483. DOI | PubMed
- Costantini A, Gorini G, Consonni D, Miligi L, Giovannetti L, Quinn M. Exposure to benzene and risk of breast cancer among shoe factory workers in Italy. Tumori. 2009; 95:8-12. PubMed
- Hansen J. Elevated risk for male breast cancer after occupational exposure to gasoline and vehicular combustion products. Am J Ind Med. 2000; 37:349-352. DOI | PubMed
- Huff JE, Haseman JK, DeMarini DM, Eustis S, Maronpot RR, Peters AC. Multiple-site carcinogenicity of benzene in fisher 344 rats and B6C3F1 mice. Environ Health Perspect. 1989; 82:125-163. PubMed
- Houle CD, Ton T-VT, Clayton N, Huff J, Hong H-HL, Sills RC. Frequent p53 and H-ras mutations in benzene- and ethylene oxide-induced mammary gland carcinomas from B6C3F1 mice. Toxicol Pathol. 2006; 34:752-762. DOI | PubMed
- .Publisher Full Text
- IARC. Vinyl chloride (group 1). IARC Monog Eval Carcinog Risk Chem Hum. 1987; Suppl 7:383.
- Chiazze L, Ference LD. Mortality among PVC-fabricating employees. Environ Health Perspect. 1981; 41:137-143. DOI | PubMed
- Infante PF, Pesák J. A historical perspective of some occupationally related diseases of women. J Occup Med. 1994; 36:826-831. PubMed
- Garcia E, Hurley S, Nelson DO, Hertz A, Reynolds P. Hazardous air pollutants and breast cancer risk in California teachers: A cohort study. Environ Health. 2015; 14:14. DOI | PubMed
- Ruckart PZ, Bove FJ, Shanley E, Maslia M. Evaluation of contaminated drinking water and male breast cancer at marine Corps Base camp Lejeune, North Carolina: A case control study. Environ Health. 2015; 14:74. DOI | PubMed
- Drew RT, Boorman GA, Haseman JK, McConnell EE, Busey WM, Moore JA. The effect of age and exposure duration on cancer induction by a known carcinogen in rats, mice, and hamsters. Toxicol Appl Pharmacol. 1983; 68:120-130. DOI | PubMed
- Sathiakumar N, Delzell E. A follow-up of women in the synthetic rubber industry. Chem Biol Interact. 2007; 166:25-28. DOI | PubMed
- . Accessed 25 Oct 2016.Publisher Full Text
- . Accessed 24 Sept 2016.Publisher Full Text
- Melnick RL, Sills RC, Portier CJ, Roycroft JH, Chou BJ, Grumbein SL. Multiple organ carcinogenicity of inhaled chloroprene (2-chloro-1,3-butadiene) in F344/N rats and B6C3F1 mice and comparison of dose-response with 1,3-butadiene in mice. Carcinogenesis. 1999; 20:867-878. DOI | PubMed
- . Accessed 25 Oct 2016.Publisher Full Text
- . Accessed 25 Oct 2016.Publisher Full Text
- . Accessed 25 Oct 2016.Publisher Full Text
- Steenland K, Whelan E, Deddens J, Stayner L, Ward E. Ethylene oxide and breast cancer incidence in a cohort study of 7576 women (United States). Cancer Causes Control. 2003; 14:531-539. DOI | PubMed
- Norman SA, Berlin JA, Soper KA, Middendorf BF, Stolley PD. Cancer incidence in a group of workers potentially exposed to ethylene oxide. Int J Epidemiol. 1995; 24:276-284. DOI | PubMed
- Mikoczy Z, Tinnerberg H, Björk J, Albin M. Cancer incidence and mortality in Swedish Sterilant workers exposed to ethylene oxide: Updated cohort study findings 1972–2006. Inl J Environ Res Public Health. 2011; 8:2009-2019. DOI
- Adám B, Bárdos H, Adány R. Increased genotoxic susceptibility of breast epithelial cells to ethylene oxide. Mutat Res. 2005; 585:120-126. DOI | PubMed
- . Accessed 26 Oct 2016.Publisher Full Text
- Chen Z, An Y, Wang Z, Zhang B, Liu L. Tobacco-specific carcinogen 4-(Methylnitrosoamino)-1-(3-pyridyl)-1-butanone(NNK) activating ERK1/2 MAP kinases and stimulating proliferation of human mammary epithelial cells. Chem Res Chin Univ. 2007; 23:76-80. DOI
- Mei J, Hu H, McEntee M, Plummer H, Song P, Wang H-CR. Transformation of non-cancerous human breast epithelial cell line MCF10A by the tobacco-specific carcinogen NNK. Breast Cancer Res Treat. 2003; 79:95-105. DOI | PubMed
- Siriwardhana N, Choudhary S, Wang H-CR. Precancerous model of human breast epithelial cells induced by NNK for prevention. Breast Cancer Res Treat. 2008; 109:427-441. DOI | PubMed
- Kalantari-Dehaghi M, Parnell EA, Armand T, Bernard H-U, Grando SA. The nicotinic acetylcholine receptor-mediated reciprocal effects of the tobacco nitrosamine NNK and SLURP-1 on human mammary epithelial cells. Int Immunopharmacol. 2015; 29:99-104. DOI | PubMed
- Reynolds P, Hurley S, Goldberg DE, Anton-Culver H, Bernstein L, Deapen D. Active smoking, household passive smoking, and breast cancer: Evidence from the California teachers study. J Natl Cancer Inst. 2004; 96:29-37. DOI | PubMed
- Band PR, Le ND, Fang R, Deschamps M. Carcinogenic and endocrine disrupting effects of cigarette smoke and risk of breast cancer. Lancet. 2002; 360:1044-1049. DOI | PubMed
- Calle EE, Miracle-McMahill HL, Thun MJ, Heath CW. Cigarette smoking and risk of fatal breast cancer. Am J Epidemiol. 1994; 139:1001-1007. DOI | PubMed
- Gram IT, Park S-Y, Kolonel LN, Maskarinec G, Wilkens LR, Henderson BE. Smoking and risk of breast cancer in a racially/ethnically diverse population of mainly women who do not drink alcohol: The MEC study. Am J Epidemiol. 2015; 182:917-925. DOI | PubMed
- Johnson KC, Hu J, Mao Y, Canadian Cancer Registries Epidemiology Research Group. Passive and active smoking and breast cancer risk in Canada, 1994-97. Cancer Causes Control. 2000; 11:211-221. DOI | PubMed
- Marcus PM, Newman B, Millikan RC, Moorman PG, Baird DD, Qaqish B. The associations of adolescent cigarette smoking, alcoholic beverage consumption, environmental tobacco smoke, and ionizing radiation with subsequent breast cancer risk (United States). Cancer Causes Control. 2000; 11:271-278. DOI | PubMed
- Cui Y, Miller AB, Rohan TE. Cigarette smoking and breast cancer risk: Update of a prospective cohort study. Breast Cancer Res Treat. 2006; 100:293-299. DOI | PubMed
- Xue F, Willett WC, Rosner BA, Hankinson SE, Michels KB. Cigarette smoking and the incidence of breast cancer. Arch Intern Med. 2011; 171:125-133. PubMed
- Luo J, Margolis KL, Wactawski-Wende J, Horn K, Messina C, Stefanick ML. Association of active and passive smoking with risk of breast cancer among postmenopausal women: A prospective cohort study. Br Med J. 2011; 342:536. DOI
- Bjerkaas E, Parajuli R, Engeland A, Maskarinec G, Weiderpass E, Gram IT. Social inequalities and smoking-associated breast cancer — Results from a prospective cohort study. Prev Med. 2015; 73:125-129. DOI | PubMed
- DeRoo LA, Cummings P, Mueller BA. Smoking before the first pregnancy and the risk of breast cancer: A meta-analysis. Am J Epidemiol. 2011; 174:390-402. DOI | PubMed
- Dossus L, Boutron-Ruault M-C, Kaaks R, Gram IT, Vilier A, Fervers B. Active and passive cigarette smoking and breast cancer risk: Results from the EPIC cohort. Int J Cancer. 2014; 134:1871-1888. DOI | PubMed
- Park S-Y, Palmer JR, Rosenberg L, Haiman CA, Bandera EV, Bethea TN. A case-control analysis of smoking and breast cancer in African American women: Findings from the AMBER consortium. Carcinogenesis. 2016; 37:607-615. DOI | PubMed
- Jacobsen KK, Lynge E, Vejborg I, Tjønneland A, von E-C, Andersen ZJ. Cigarette smoking and mammographic density in the Danish diet, cancer and health cohort. Cancer Causes Control. 2016; 27:271-280. DOI | PubMed
- McCormack VA, dos Santos Silva I. Breast density and parenchymal patterns as markers of breast cancer risk: A meta-analysis. Cancer Epidemiol Biomark Prev. 2006; 15:1159-1169. DOI
- Atkinson RL, El-Zein R, Valero V, Lucci A, Bevers TB, Fouad T. Epidemiological risk factors associated with inflammatory breast cancer subtypes. Cancer Causes Control. 2016; 27:359-366. DOI | PubMed
- Butler EN, Tse C-K, Bell ME, Conway K, Olshan AF, Troester MA. Active smoking and risk of luminal and basal-like breast cancer subtypes in the Carolina breast cancer study. Cancer Causes Control. 2016; 27:775-786. DOI | PubMed
- Connor AE, Baumgartner KB, Baumgartner RN, Pinkston CM, Boone SD, John EM. Cigarette smoking and breast cancer risk in Hispanic and non-Hispanic white women: The breast cancer health disparities study. J Women’s Health. 2016; 25:299-310. DOI
- Nyante SJ, Gierach GL, Dallal CM, Freedman ND, Park Y, Danforth KN. Cigarette smoking and postmenopausal breast cancer risk in a prospective cohort. Br J Cancer. 2014; 110:2339-2347. DOI | PubMed
- Bérubé S, Lemieux J, Moore L, Maunsell E, Brisson J. Smoking at time of diagnosis and breast cancer-specific survival: New findings and systematic review with meta-analysis. Breast Cancer Res. 2014; 16:R42. DOI | PubMed
- Izano M, Satariano WA, Hiatt RA, Braithwaite D. Smoking and mortality after breast cancer diagnosis: The health and functioning in women study. Cancer Med. 2015; 4:315-324. DOI | PubMed
- Passarelli MN, Newcomb PA, Hampton JM, Trentham-Dietz A, Titus LJ, Egan KM. Cigarette smoking before and after breast cancer diagnosis: Mortality from breast cancer and smoking-related diseases. J Clin Oncol. 2016; 34:1315-1322. DOI | PubMed
- Persson M, Simonsson M, Markkula A, Rose C, Ingvar C, Jernström H. Impacts of smoking on endocrine treatment response in a prospective breast cancer cohort. Br J Cancer. 2016; 115:382-390. DOI | PubMed
- Cook MB, Guénel P, Gapstur SM, van den Brandt PA, Michels KB, Casagrande JT. Tobacco and alcohol in relation to male breast cancer: An analysis of the male breast cancer pooling project consortium. Cancer Epidemiol Biomark Prev. 2015; 24:520-531. DOI
- Padron-Monedero A, Koru-Sengul T, Tannenbaum SL, Miao F, Hansra D, Lee DJ. Smoking and survival in male breast cancer patients. Breast Cancer Res Treat. 2015; 153:679-687. DOI | PubMed
- Ambrosone CB, Freudenheim JL, Graham S, Marshall JR, Vena JE, Brasure JR. Cigarette smoking, N-acetyltransferase 2 genetic polymorphisms, and breast cancer risk. JAMA. 1996; 276:1494-1501. DOI | PubMed
- Macacu A, Autier P, Boniol M, Boyle P. Active and passive smoking and risk of breast cancer: A meta-analysis. Breast Cancer Res Treat. 2015; 154:213-224. DOI | PubMed
- Hanaoka T, Yamamoto S, Sobue T, Sasaki S, Tsugane S, Japan Public Health Center-Based Prospective Study on Cancer and Cardiovascular Disease Study Group. Active and passive smoking and breast cancer risk in middle-aged Japanese women. Int J Cancer. 2005; 114:317-322. DOI | PubMed
- Li B, Wang L, Lu M-S, Mo X-F, Lin F-Y, Ho SC. Passive smoking and breast cancer risk among non-smoking women: A case-control study in China. PLoS One. 2015; 10:e0125894. DOI | PubMed
- Chen Z, Shao J, Gao X, Li X. Effect of passive smoking on female breast cancer in China: A meta-analysis. Asia Pac J Public Health. 2015; 27:NP58-NP64. DOI | PubMed
- Tong J, Li Z, Shi J, Li H, Wang Y, Fu L. Passive smoking exposure from partners as a risk factor for ER+/PR+ double positive breast cancer in never-smoking Chinese urban women: A hospital-based matched case control study. PLoS One. 2014; 9:e97498. DOI | PubMed
- Andres SA, Bickett KE, Alatoum MA, Kalbfleisch TS, Brock GN, Wittliff JL. Interaction between smoking history and gene expression levels impacts survival of breast cancer patients. Breast Cancer Res Treat. 2015; 152:545-556. DOI | PubMed
- Kasajova P, Holubekova V, Mendelova A, Lasabova Z, Zubor P, Kudela E. Active cigarette smoking and the risk of breast cancer at the level of N-acetyltransferase 2 (NAT2) gene polymorphisms. Tumour Biol. 2016; 37:7929-7937. DOI | PubMed
- Ricks-Santi LJ, Nie J, Marian C, Ochs-Balcom HM, Trevisan M, Edge SB. BRCA1 polymorphisms and breast cancer epidemiology in the western New York exposures and breast cancer (WEB) study. Genet Epidemiol. 2013; 37:504-511. DOI | PubMed
- Dechanet C, Anahory T, Mathieu Daude JC, Quantin X, Reyftmann L, Hamamah S. Effects of cigarette smoking on reproduction. Hum Reprod Update. 2011; 17:76-95. DOI | PubMed
- Brand JS, Chan M-F, Dowsett M, Folkerd E, Wareham NJ, Luben RN. Cigarette smoking and endogenous sex hormones in postmenopausal women. J Clin Endocrinol Metab. 2011; 96:3184-3192. DOI | PubMed
- Wang Y-Y, Lehuédé C, Laurent V, Dirat B, Dauvillier S, Bochet L. Adipose tissue and breast epithelial cells: A dangerous dynamic duo in breast cancer. Cancer Lett. 2012; 324:142-151. DOI | PubMed
- . 2007. Accessed 25 July 2017.Publisher Full Text
- Bonde JP, Hansen J, Kolstad HA, Mikkelsen S, Olsen JH, Blask DE. Work at night and breast cancer—report on evidence-based options for preventive actions. Scand J Work Environ Health. 2012; 38:380-390. DOI | PubMed
- Costa G, Haus E, Stevens R. Shift work and cancer–considerations on rationale, mechanisms, and epidemiology. Scand J Work Environ Health. 2010; 36:163-179. DOI | PubMed
- Kolstad HA. Nightshift work and risk of breast cancer and other cancers—a critical review of the epidemiologic evidence. Scand J Work Environ Health. 2008; 34:5-22. DOI | PubMed
- Megdal SP, Kroenke CH, Laden F, Pukkala E, Schernhammer ES. Night work and breast cancer risk: A systematic review and meta-analysis. Eur J Cancer. 2005; 41:2023-2032. DOI | PubMed
- Kamdar BB, Tergas AI, Mateen FJ, Bhayani NH, Oh J. Night-shift work and risk of breast cancer: A systematic review and meta-analysis. Breast Cancer Res Treat. 2013; 138:291-301. DOI | PubMed
- He C, Anand ST, Ebell MH, Vena JE, Robb SW. Circadian disrupting exposures and breast cancer risk: A meta-analysis. Int Arch Occup Environ Health. 2015; 88:533-547. DOI | PubMed
- Lin X, Chen W, Wei F, Ying M, Wei W, Xie X. Night-shift work increases morbidity of breast cancer and all-cause mortality: A meta-analysis of 16 prospective cohort studies. Sleep Med. 2015; 16:1381-1387. DOI | PubMed
- Travis RC, Balkwill A, Fensom GK, Appleby PN, Reeves GK, Wang X-S. Night shift work and breast cancer incidence: Three prespective studies and meta-analysis of the published studies. J Natl Cancer Inst. 2016; 108:djw241. DOI
- . Hazards Magazine. Accessed 9 Feb 2017.Publisher Full Text
- Slack R, Young C, Rushton L, British Occupational Cancer Burden Study Group. Occupational cancer in Britain. Female cancers: Breast, cervix and ovary. Br J Cancer. 2012; 107(Suppl 1):S27-S32. DOI | PubMed
- Purdue MP, Hutchings SJ, Rushton L, Silverman DT. The proportion of cancer attributable to occupational exposures. Ann Epidemiol. 2015; 25:188-192. DOI | PubMed
- Hansen J, Stevens RG. Case-control study of shift-work and breast cancer risk in Danish nurses: Impact of shift systems. Eur J Cancer. 2012; 48:1722-1729. DOI | PubMed
- Menegaux F, Truong T, Anger A, Cordina-Duverger E, Lamkarkach F, Arveux P. Night work and breast cancer: A population-based case-control study in France (the CECILE study). Int J Cancer. 2013; 132:924-931. DOI | PubMed
- Stevens RG. Working against our endogenous circadian clock: Breast cancer and electric lighting in the modern world. Mutat Res. 2009; 680:106-108. DOI | PubMed
- Cos S, González A, Güezmes A, Mediavilla MD, Martínez-Campa C, Alonso-González C. Melatonin inhibits the growth of DMBA-induced mammary tumors by decreasing the local biosynthesis of estrogens through the modulation of aromatase activity. Int J Cancer. 2006; 118:274-278. DOI | PubMed
- Cos S, Sánchez-Barceló EJ. Melatonin and mammary pathological growth. Front Neuroendocrinol. 2000; 21:133-170. DOI | PubMed
- Knower KC, To SQ, Takagi K, Miki Y, Sasano H, Simpson ER. Melatonin suppresses aromatase expression and activity in breast cancer associated fibroblasts. Breast Cancer Res Treat. 2012; 132:765-771. DOI | PubMed
- Fonken LK, Nelson RJ. The effects of light at night on circadian clocks and metabolism. Endocr Rev. 2014; 35:648-670. DOI | PubMed
- Flynn-Evans EE, Stevens RG, Tabandeh H, Schernhammer ES, Lockley SW. Total visual blindness is protective against breast cancer. Cancer Causes Control. 2009; 20:1753-1756. DOI | PubMed
- Nagata C, Nagao Y, Yamamoto S, Shibuya C, Kashiki Y, Shimizu H. Light exposure at night, urinary 6-sulfatoxymelatonin, and serum estrogens and androgens in postmenopausal Japanese women. Cancer Epidemiol Biomark Prev. 2008; 17:1418-1423. DOI
- Davis S, Mirick DK, Chen C, Stanczyk FZ. Night shift work and hormone levels in women. Cancer Epidemiol Biomark Prev. 2012; 21:609-618. DOI
- Langley AR, Graham CH, Grundy AL, Tranmer JE, Richardson H, Aronson KJ. A cross-sectional study of breast cancer biomarkers among shift working nurses. BMJ Open. 2012; 2:e000532. DOI
- Blask DE, Hill SM, Dauchy RT, Xiang S, Yuan L, Duplessis T. Circadian regulation of molecular, dietary, and metabolic signaling mechanisms of human breast cancer growth by the nocturnal melatonin signal and the consequences of its disruption by light at night. J Pineal Res. 2011; 51:259-269. DOI | PubMed
- Blask DE, Brainard GC, Dauchy RT, Hanifin JP, Davidson LK, Krause JA. Melatonin-depleted blood from premenopausal women exposed to light at night stimulates growth of human breast cancer xenografts in nude rats. Cancer Res. 2005; 65:11174-11184. DOI | PubMed
- Zhu Y, Stevens RG, Hoffman AE, Tjonneland A, Vogel UB, Zheng T. Epigenetic impact of long-term shiftwork: Pilot evidence from circadian genes and whole-genome methylation analysis. Chronobiol Int. 2011; 28:852-861. DOI | PubMed
- Zhu Y, Stevens RG, Leaderer D, Hoffman A, Holford T, Zhang Y. Non-synonymous polymorphisms in the circadian gene NPAS2 and breast cancer risk. Breast Cancer Res Treat. 2008; 107:421-425. DOI | PubMed
- Gery S, Virk RK, Chumakov K, Yu A, Koeffler HP. The clock gene Per2 links the circadian system to the estrogen receptor. Oncogene. 2007; 26:7916-7920. DOI | PubMed
- Rabstein S, Harth V, Justenhoven C, Pesch B, Plöttner S, Heinze E. Polymorphisms in circadian genes, night work and breast cancer: Results from the GENICA study. Chronobiol Int. 2014; 31:1115-1122. DOI | PubMed
- Grundy A, Schuetz JM, Lai AS, Janoo-Gilani R, Leach S, Burstyn I. Shift work, circadian gene variants and risk of breast cancer. Cancer Epidemiol. 2013; 37:606-612. DOI | PubMed
- Pronk A, Ji B-T, Shu X-O, Xue S, Yang G, Li H-L. Night-shift work and breast cancer risk in a cohort of Chinese women. Am J Epidemiol. 2010; 171:953-959. DOI | PubMed
- Bhatti P, Mirick DK, Davis S. Racial differences in the association between night shift work and melatonin levels among women. Am J Epidemiol. 2013; 177:388-393. DOI | PubMed
- Papantoniou K, Castaño-Vinyals G, Espinosa A, Aragonés N, Pérez-Gómez B, Ardanaz E. Breast cancer risk and night shift work in a case-control study in a Spanish population. Eur J Epidemiol. 2015; 31:867-878. DOI | PubMed
- Anjum B. Associations of circadian disruption of sleep and nutritional factors with risk of cancer. Open Nutraceuticals J. 2012; 5:124-135. DOI
- Fritschi L, Glass DC, Heyworth JS, Aronson K, Girschik J, Boyle T. Hypotheses for mechanisms linking shiftwork and cancer. Med Hypotheses. 2011; 77:430-436. DOI | PubMed
- Sharat A, Tamrin ShBM, Daneshjoo A, Sadeghi H. The adverse health effects of shift work in relation to health/disease: A review. Acta Medica Bulg. 2015;42:63–72.
- Aronson K, Grundy A, Korsiak J, Spinelli JJ. Causes of breast cancer: Could work at night really be a cause?. Breast Cancer Manag. 2015; 4:125-127. DOI
- Van Dycke KCG, Rodenburg W, van Oostrom CTM, van Kerkhof LWM, Pennings JLA, Roenneberg T. Chronically alternating light cycles increase breast cancer risk in mice. Curr Biol. 2015; 25:1932-1937. DOI | PubMed
- Dauchy RT, Xiang S, Mao L, Brimer S, Wren MA, Yuan L. Circadian and melatonin disruption by exposure to light at night drives intrinsic resistance to tamoxifen therapy in breast cancer. Cancer Res. 2014; 74:4099-4110. DOI | PubMed
- Xiang S, Dauchy RT, Hauch A, Mao L, Yuan L, Wren MA. Doxorubicin resistance in breast cancer is driven by light at night-induced disruption of the circadian melatonin signal. J Pineal Res. 2015; 59:60-69. DOI | PubMed
- . Accessed 25 Oct 2016.Publisher Full Text
- . Accessed 25 Oct 2016.Publisher Full Text
- Harbron RW. Cancer risks from low dose exposure to ionising radiation – Is the linear no-threshold model still relevant?. Radiography. 2012; 18:28-33. DOI
- Brenner DJ, Doll R, Goodhead DT, Hall EJ, Land CE, Little JB. Cancer risks attributable to low doses of ionizing radiation: Assessing what we really know. Proc Natl Acad Sci U S A. 2003; 100:13761-13766. DOI | PubMed
- National Radiological Protection Board (NRPB) (Britain). Risk of radiation-induced cancer at low doses and low dose rates for radiation protection purposes. 1995.
- Boice JD. Radiation and breast carcinogenesis. Med Pediatr Oncol. 2001; 36:508-513. DOI | PubMed
- Goto H, Watanabe T, Miyao M, Fukuda H, Sato Y, Oshida Y. Cancer mortality among atomic bomb survivors exposed as children. Environ Health Prev Med. 2012; 17:228-234. DOI | PubMed
- Pierce DA, Shimizu Y, Preston DL, Vaeth M, Mabuchi K. Studies of the mortality of atomic bomb survivors. Report 12, part I. Cancer: 1950–1990. Radiat Res. 1996; 146:1-27. DOI | PubMed
- Tokunaga M, Land CE, Tokuoka S, Nishimori I, Soda M, Akiba S. Incidence of female breast cancer among atomic bomb survivors, 1950–1985. Radiat Res. 1994; 138:209-223. DOI | PubMed
- Land CE. Studies of cancer and radiation dose among atomic bomb survivors. The example of breast cancer. JAMA. 1995; 274:402-407. DOI | PubMed
- Ron E, Ikeda T, Preston DL, Tokuoka S. Male breast cancer incidence among atomic bomb survivors. J Natl Cancer Inst. 2005; 97:603-605. DOI | PubMed
- Goldberg Z, Lehnert BE. Radiation-induced effects in unirradiated cells: A review and implications in cancer. Int J Oncol. 2002; 21:337-349. PubMed
- Morgan WF. Non-targeted and delayed effects of exposure to ionizing radiation: II. Radiation-induced genomic instability and bystander effects in vivo, clastogenic factors and transgenerational effects. Radiat Res. 2003; 159:581-596. DOI | PubMed
- Tsai KKC, Chuang EY-Y, Little JB, Yuan Z-M. Cellular mechanisms for low-dose ionizing radiation-induced perturbation of the breast tissue microenvironment. Cancer Res. 2005; 65:6734-6744. DOI | PubMed
- Borrego-Soto G, Ortiz-López R, Rojas-Martínez A. Ionizing radiation-induced DNA injury and damage detection in patients with breast cancer. Genet Mol Biol. 2015; 38:420-432. DOI | PubMed
- Maier P, Hartmann L, Wenz F, Herskind C. Cellular pathways in response to ionizing radiation and their targetability for tumor radiosensitization. Int J Mol Sci. 2016; 17:102. DOI
- Little JB. Genomic instability and radiation. J Radiol Prot. 2003; 23:173-181. DOI | PubMed
- Baert A, Depuydt J, Van Maerken T, Poppe B, Malfait F, Storm K. Increased chromosomal radiosensitivity in asymptomatic carriers of a heterozygous BRCA1 mutation. Breast Cancer Res. 2016; 18:52. DOI | PubMed
- Andrieu N, Easton DF, Chang-Claude J, Rookus MA, Brohet R, Cardis E. Effect of chest X-rays on the risk of breast cancer among BRCA1/2 mutation carriers in the international BRCA1/2 carrier cohort study: A report from the EMBRACE, GENEPSO, GEO-HEBON, and IBCCS collaborators’ group. J Clin Oncol. 2006; 24:3361-3366. DOI | PubMed
- Pepe S, Pensabene M, Condello C. Modifiers of risk in BRCA1/2 mutation carriers. Curr Women’s Health Rev. 2012; 8:23-29. DOI
- Turnbull C, Mirugaesu N, Eeles R. Radiotherapy and genetic predisposition to breast cancer. Clin Oncol (R Coll Radiol). 2006; 1:257. DOI
- Calaf GM, Hei TK. Establishment of a radiation- and estrogen-induced breast cancer model. Carcinogenesis. 2000; 21:769-776. DOI | PubMed
- Imaoka T, Nishimura M, Iizuka D, Daino K, Takabatake T, Okamoto M. Radiation-induced mammary carcinogenesis in rodent models: What’s different from chemical carcinogenesis?. J Radiat Res. 2009; 50:281-293. DOI | PubMed
- Segaloff A, Maxfield WS. The synergism between radiation and estrogen in the production of mammary cancer in the rat. Cancer Res. 1971; 31:166-168. PubMed
- Doody MM, Freedman DM, Alexander BH, Hauptmann M, Miller JS, Rao RS. Breast cancer incidence in U.S. radiologic technologists. Cancer. 2006; 106:2707-2715. DOI | PubMed
- Simon SL, Weinstock RM, Doody MM, Neton J, Wenzl T, Stewart P. Estimating historical radiation doses to a cohort of U.S. radiologic technologists. Radiat Res. 2006; 166:174-192. DOI | PubMed
- Liu JJ, Freedman DM, Little MP, Doody MM, Alexander BH, Kitahara CM. Work history and mortality risks in 90,268 US radiological technologists. Occup Environ Med. 2014; 71:819-835. DOI | PubMed
- Rajaraman P, Doody MM, Yu CL, Preston DL, Miller JS, Sigurdson AJ. JOURNAL CLUB: Cancer risks in U.S. radiologic technologists working with fluoroscopically guided interventional procedures, 1994. Am J Roentgenol. 2016; 206:1101-1109. DOI | PubMed
- Sigurdson AJ, Bhatti P, Chang S, Rajaraman P, Doody MM, Bowen L. Polymorphisms in estrogen biosynthesis and metabolism-related genes, ionizing radiation exposure, and risk of breast cancer among US radiologic technologists. Breast Cancer Res Treat. 2009; 118:177-184. DOI | PubMed
- Ballard T, Lagorio S, De Angelis G, Verdecchia A. Cancer incidence and mortality among flight personnel: A meta-analysis. Aviat Space Environ Med. 2000; 71:216-224. PubMed
- Buja A, Mastrangelo G, Perissinotto E, Grigoletto F, Frigo AC, Rausa G. Cancer incidence among female flight attendants: A meta-analysis of published data. J Women’s Health (Larchmt). 2006; 15:98-105. DOI | PubMed
- Gofman J. Preventing breast cancer: The story of a major proven, preventable cause of this disease. CNR Division, Committee for Nuclear Responsibility: San Francisco; 1996.
- John EM, McGuire V, Thomas D, Haile R, Ozcelik H, Milne RL. Diagnostic chest X-rays and breast cancer risk before age 50 years for BRCA1 and BRCA2 mutation carriers. Cancer Epidemiol Biomarkers Prev. 2013; 22:1547-1556. DOI | PubMed
- Inskip PD, Sigurdson AJ, Veiga L, Bhatti P, Ronckers C, Rajaraman P. Radiation-related new primary solid cancers in the childhood cancer survivor study: Comparative radiation dose response and modification of treatment effects. Int J Radiat Oncol Biol Phys. 2016; 94:800-807. DOI | PubMed
- Miglioretti DL, Johnson E, Williams A, Greenlee RT, Weinmann S, Solberg LI. The use of computed tomography and the associated radiation exposure and estimated cancer risk. JAMA Pediatr. 2013; 167:700-707. DOI | PubMed
- Islam SMS, Abru AF, Al Obaidani S, Al-Shabibi S, Al FS. Trends in CT request and related outcomes in a pediatric emergency department. Oman Med J. 2016; 31:365-369. DOI | PubMed
- Gofman JW. Radiation from medical procedures in the pathogenesis of cancer and ischemic heart disease: Dose-response studies with physicians per 100,000 population. CNR Division, Committee for Nuclear Responsibility: San Francisco; 1999.
- Larson DB, Johnson LW, Schnell BM, Salisbury SR, Forman HP. National trends in CT use in the emergency department: 1995-2007. Radiology. 2011; 258:164-173. DOI | PubMed
- Linet MS, Slovis TL, Miller DL, Kleinerman R, Lee C, Rajaraman P. Cancer risks associated with external radiation from diagnostic imaging procedures. CA Cancer J Clin. 2012; 62:75-100. DOI | PubMed
- Berrington de González A, Mahesh M, Kim K-P, Bhargavan M, Lewis R, Mettler F. Projected cancer risks from computed tomographic scans performed in the United States in 2007. Arch Intern Med. 2009; 169:2071-2077. DOI | PubMed
- Redberg RF. Cancer risks and radiation exposure from computed tomographic scans: How can we be sure that the benefits outweigh the risks?. Arch Intern Med. 2009; 169:2049. DOI | PubMed
- Smith-Bindman R. Radiation dose associated with common computed tomography examinations and the associated lifetime attributable risk of cancer. Arch Intern Med. 2009; 169:2078. DOI | PubMed
- Huda W, Schoepf UJ, Abro JA, Mah E, Costello P. Radiation-related cancer risks in a clinical patient population undergoing cardiac CT. Am J Roetenol. 2011; 196:W159-W165. DOI
- Einstein AJ, Henzlova MJ, Rajagopalan S. Estimating risk of cancer associated with radiation exposure from 64-slice computed tomography coronary angiography. J Am Med Assoc. 2007; 298:317-323. DOI
- Heyes GJ, Mill AJ, Charles MW. Mammography—Oncogenecity at low doses. J Radiol Protect. 2009; 29:A123-A132. DOI
- Soler D, Pampalona J, Tusell L, Genescà A. Radiation sensitivity increases with proliferation-associated telomere dysfunction in nontransformed human epithelial cells. Aging Cell. 2009; 8:414-425. DOI | PubMed
- Nelson HD, Tyne K, Naik A, Bougatsos C, Chan BK, Humphrey L. Screening for breast cancer: An update for the U.S. preventive services task force. Ann Intern Med. 2009; 151:727-737. DOI | PubMed
- Nelson HD, Pappas M, Cantor A, Griffin J, Daeges M, Humphrey L. Harms of breast cancer screening: Systematic review to update the 2009 U.S. preventive services task force recommendation. Ann Intern Med. 2016; 164:256. DOI | PubMed
- de Gelder R, Draisma G, Heijnsdijk EA, de Koning HJ. Population-based mammography screening below age 50: Balancing radiation-induced vs prevented breast cancer deaths. Br J Cancer. 2011; 104:1214-1220. DOI | PubMed
- Yaffe MJ, Mainprize JG. Risk of radiation-induced breast cancer from mammographic screening. Radiology. 2011; 258:98-105. DOI | PubMed
- Miglioretti DL, Lange J, van den Broek JJ, Lee CI, van Ravesteyn NT, Ritley D. Radiation-induced breast cancer incidence and mortality from digital mammography screening: A modeling study. Ann Intern Med. 2016; 164:205. DOI | PubMed
- Siu AL. Screening for breast cancer: U.S preventive task forces recommendation statement. Ann Intern Med. 2016; 164:279-296. DOI | PubMed
- Gotzsche PC, Hartling OJ, Nielsen M, Brodersen J, Jorgensen KJ. Breast screening: The facts—or maybe not. Br Med J. 2009; 338:b86. DOI | PubMed
- Veronesi U, Luini A, Del Vecchio M. Radiotherapy after breast-preserving surgery in women with localized cancer of the breast. N Engl J Med. 1993; 328:1587-1591. DOI | PubMed
- Early Breast Cancer Trialists’ Collaborative Group (EBCTCG). Favourable and unfavourable effects on long-term survival of radiotherapy for early breast cancer: An overview of the randomised trials. Early breast cancer Trialists’ collaborative group. Lancet. 2000; 355:1757-1770. DOI | PubMed
- Mellemkjær L, Friis S, Olsen JH, Scélo G, Hemminki K, Tracey E. Risk of second cancer among women with breast cancer. Int J Cancer. 2006; 118:2285-2292. DOI | PubMed
- Roychoudhuri R, Evans H, Robinson D, Møller H. Radiation-induced malignancies following radiotherapy for breast cancer. Brit J Cancer. 2004; 91:868-872. PubMed
- Huang J, Mackillop WJ. Increased risk of soft tissue sarcoma after radiotherapy in women with breast carcinoma. Cancer. 2001; 92:172-180. DOI | PubMed
- Shah S, Rosa M. Radiation-associated angiosarcoma of the breast: Clinical and pathologic features. Arch Pathol Lab Med. 2016; 140:477-481. DOI | PubMed
- Hooning MJ, Aleman BMP, Hauptmann M, Baaijens MHA, Klijn JGM, Noyon R. Roles of radiotherapy and chemotherapy in the development of contralateral breast cancer. J Clin Oncol. 2008; 26:5561-5568. DOI | PubMed
- Ng AK, Travis LB. Radiation therapy and breast cancer risk. J Natl Compr Cancer Netw. 2009; 7:1121-1128. DOI
- Stovall M, Smith SA, Langholz BM, Boice JD, Shore RE, Andersson M. Dose to the contralateral breast from radiotherapy and risk of second primary breast cancer in the WECARE study. Int J Radiat Oncol Biol Phys. 2008; 72:1021-1030. DOI | PubMed
- National Institute of Environmental Heal Sciences (NIEHS). Working group report: Assessment of health effects from exposure to power-line frequency electric and magnetic fields. National Institute of Environmental Health: Brooklyn Park; 1998.
- Carpenter DO. Electromagnetic fields and cancer: The cost of doing nothing. Rev Environ Health. 2010; 25:75-80. DOI | PubMed
- McElroy JA, Egan KM, Titus-Ernstoff L, Anderson HA, Trentham-Dietz A, Hampton JM. Occupational exposure to electromagnetic field and breast cancer risk in a large, population-based, case-control study in the United States. J Occup Environ Med. 2007; 49:266-264. DOI | PubMed
- Peplonska B, Stewart P, Szeszenia-Dabrowska N, Rusiecki J, Garcia-Closas M, Lissowska J. Occupation and breast cancer risk in polish women: A population-based case-control study. Am J Ind Med. 2007; 50:97-111. DOI | PubMed
- Ahlbom IC, Cardis E, Green A, Linet M, Savitz D, Swerdlow A. Review of the epidemiologic literature on EMF and health. Environ Health Perspect. 2001; 109:911-933. DOI | PubMed
- Kliukiene J, Tynes T, Andersen A. Follow-up of radio and telegraph operators with exposure to electromagnetic fields and risk of breast cancer. Eur J Cancer Prev. 2003; 12:301-307. DOI | PubMed
- Coogan PF, Clapp RW, Newcomb PA, Wenzl TB, Bogdan G, Mittendorf R. Occupational exposure to 60-hertz magnetic fields and risk of breast cancer in women. Epidemiology. 1996; 7:459-464. DOI | PubMed
- Kliukiene J, Tynes T, Andersen A. Residential and occupational exposures to 50-Hz magnetic fields and breast cancer in women: A population-based study. Am J Epidemiol. 2004; 159:852-861. DOI | PubMed
- Feychting M, Forssén U, Rutqvist LE, Ahlbom A. Magnetic fields and breast cancer in Swedish adults residing near high-voltage power lines. Epidemiology. 1998; 9:392-397. DOI | PubMed
- Chen C, Ma X, Zhong M, Yu Z. Extremely low-frequency electromagnetic fields exposure and female breast cancer risk: A meta-analysis based on 24,338 cases and 60,628 controls. Breast Cancer Res Treat. 2010; 123:569-576. DOI | PubMed
- Zhang Y, Lai J, Ruan G, Chen C, Wang DW. Meta-analysis of extremely low frequency electromagnetic fields and cancer risk: A pooled analysis of epidemiologic studies. Environ Int. 2016; 88:36-43. DOI | PubMed
- Loomis D. Cancer of breast among men in electrical occupations. Lancet. 1992; 339:1482-1483. DOI
- Matanoski GM, Breysse PN, Elliott EA. Electromagnetic field exposure and male breast cancer. Lancet. 1991; 337:737. DOI | PubMed
- Milham S. Most cancer in firefighters is due to radio-frequency radiation exposure not inhaled carcinogens. Med Hypotheses. 2009; 73:788-789. DOI | PubMed
- Milham S. A cluster of male breast cancer in office workers. Am J Ind Med. 2004; 46:86-87. DOI | PubMed
- Tynes T, Andersen A, Langmark F. Incidence of cancer in Norwegian workers potentially exposed to electromagnetic fields. Am J Epidemiol. 1992; 136:81-88. DOI | PubMed
- Fedrowitz M, Kamino K, Löscher W. Significant differences in the effects of magnetic field exposure on 7,12-dimethylbenz(a)anthracene-induced mammary carcinogenesis in two substrains of Sprague-Dawley rats. Cancer Res. 2004; 64:243-251. DOI | PubMed
- Girgert R, Emons G, Hanf V, Gründker C. Exposure of mcf-7 breast cancer cells to electromagnetic fields up-regulates the plasminogen activator system. Int J Gynecol Cancer. 2009; 19:334-338. DOI | PubMed
- Rudel RA, Ackerman JM, Attfield KR, Brody JG. New exposure biomarkers as tools for breast cancer epidemiology, biomonitoring, and prevention: A systematic approach based on animal evidence. Environ Health Perspect. 2014; 122:881-895. PubMed
- Khoury MJ, Lam TK, Ionnidis JPA, Hartge P, Spitz MR, Buring JE. Transforming epidemiology for 21st century medicine and public health. Cancer Epidemiol Biomark Prev. 2013; 22:508-516. DOI
- Osborne G, Rudel R, Schwarzmann Evaluating chemical effects on mammary gland development: A critical need in disease prevention. Reprod Toxicol. 2014; 54:148-155. DOI | PubMed
- Schwarzman MR, Ackerman JM, Dairkee SH, Fenton SE, Johnson D. Screening for chemical contributions to breast cancer risk: A case study for chemical safety evaluation. Environ Health Perspect. 2015; 123:1255-1264. DOI | PubMed
Fonte
Gray JM, Rasanayagam S, Engel C, Rizzo J (2017) State of the evidence 2017: an update on the connection between breast cancer and the environment. Environmental Health 1694. https://doi.org/10.1186/s12940-017-0287-4




