
Introduzione
La scoperta (Schulze, 1883) e la riscoperta a metà del XX secolo (Grell e Benwitz, 1971) dell’enigmatico Trichoplax adhaerens placozoico, simile ad ameba, ha fatto molto per accendere l’immaginazione degli zoologi interessati alla prima evoluzione animale (Bütschli, 1884). Come animali microscopici adattati al pascolo extracellulare sui biofilm sui quali si insinuano (Wenderoth, 1986), i placozoi hanno un’anatomia semplice adatta a sfruttare la diffusione passiva per molte esigenze fisiologiche, con solo sei tipi di cellule morfologiche riconoscibili anche al microscopio intensivo (Grell e Ruthmann, 1991; Smith et al., 2014), anche se una maggiore diversità di tipi di cellule è evidente attraverso RNA-seq monocellulare (Sebé-Pedrós, 2018a). Non hanno un sistema muscolare, digestivo o nervoso convenzionale, ma mostrano un comportamento strettamente coordinato regolato da una segnalazione peptidergica (Smith et al., 2015; Senatore et al., 2017; Varoqueaux, 2018; Armon et al., 2018). In condizioni di laboratorio, essi proliferano attraverso la fissione e la crescita somatica. Le prove della riproduzione sessuale rimangono elusive, nonostante le prove genetiche della ricombinazione (Srivastava et al., 2008) e le descrizioni dell’embriogenesi abortiva precoce (Eitel et al., 2011; Grell, 1972), con la possibilità che le fasi sessuali del ciclo vitale si verifichino solo in condizioni di campo poco chiare (Pearse e Voigt, 2007; McFall-Ngai et al., 2013).
Data la loro morfologia semplice e sconcertante e la scarsità di indizi embriologici, i dati molecolari sono cruciali nel collocare filogeneticamente i placogeni. La posizione di Placozoa nell’albero animale si è dimostrata recalcitrante alle prime analisi standard (Kim et al., 1999; Silva et al., 2007; Wallberg et al., 2004), anche se questo paradigma ha rivelato un ampio grado di diversità molecolare negli isolati di placozoa di tutto il mondo, indicando chiaramente l’esistenza di molte specie criptiche (Pearse e Voigt, 2007; Eitel et al., 2013; Signorovitch et al., 2007) con una distanza genetica fino al 27% in allineamenti di rRNA 16S (Eitel e Schierwater, 2010). Un’apparente risposta alla questione delle affinità placozoiche è stata fornita dall’analisi di un assemblaggio di genoma nucleare (Srivastava et al., 2008), che ha fortemente sostenuto una posizione come gruppo gemello di un clade di Cnidaria + Bilateria (talvolta chiamato Planulozoa). Tuttavia, questo sforzo ha anche rivelato un kit di strumenti genetici di sviluppo sorprendentemente bilateriano (Dunn et al., 2015) nei placozoi, un paradosso per un animale così semplice.
Mentre la filogenetica metazoica si è spinta verso l’era genomica, forse la più grande polemica è stata il dibattito sull’identità del gruppo gemello ai rimanenti metazoi, tradizionalmente considerati Porifera, ma considerati Ctenophora da Dunn et al, 2008). e successivamente da altri studi (Hejnol et al., 2009; Moroz et al., 2014; NISC Comparative Sequencing Program et al., 2013; Whelan et al., 2015; Whelan et al., 2017). Altri hanno suggerito che questo risultato deriva da artefatti con effetti potenzialmente additivi, come un campionamento inadeguato del taxon, un allevamento della matrice difettoso (paralogia o contaminazione non rilevata) e l’uso di modelli di sostituzione poco adatti (Philippe et al., 2009; Pick et al., 2010; Pisani et al., 2015; Simion et al., 2017; Feuda et al., 2017). Un terzo punto di vista ha sottolineato che l’utilizzo di diversi insiemi di geni può portare a conclusioni diverse, con solo un piccolo numero a volte sufficiente a guidare un risultato o l’altro (Nosenko et al., 2013; Shen et al., 2017). Questa controversia, indipendentemente dalla sua eventuale risoluzione, ha stimolato una seria contemplazione di possibili origini indipendenti di diversi tratti distintivi come i muscoli striati, l’apparato digerente e il sistema nervoso (Moroz et al., 2014; Dayraud et al., 2012; Hejnol e Martín-Durán, 2015; Liebeskind et al., 2017; Moroz e Kohn, 2016; Presnell et al., 2016; Steinmetz et al., 2012).
Spinti da questa controversia, si sono accumulati nuovi dati genomici e trascrittomici provenienti da spugne, ctenofori e gruppi di metazoi, mentre nuove sequenze e analisi incentrate sulla posizione di Placozoa sono state lente ad emergere. Qui, forniamo un nuovo test della posizione filogenetica dei placogeni, aggiungendo genomi di progetto da tre specie putativo che abbracciano la radice della diversità nota di questo clade (Eitel et al., 2013) (Tabella 1), e valutando criticamente il ruolo dell’errore sistematico nella collocazione di questi organismi enigmatici (Laumer, 2018).
| H11 | H4 | H6 | H1 | |
|---|---|---|---|---|
| campata di montaggio (Mbp) | 56.63 | 83.39 | 76.7 | 98.06 |
| numero dell’impalcatura | 5813 | 5337 | 8310 | 1415 |
| impalcatura N50 (kbp) | 12.738 | 25.97 | 12.84 | 5790 |
| GC% | 30.76 | 30.84 | 29.9 | 29.37 |
| BUSCO2 Eukaryota completo (di 303) | 220 | 276 | 239 | 294 |
| BUSCO2 Eukaryota completo + parziale (di 303) | 246 | 282 | 265 | 298 |
| Numero medio di contatti per BUSCO | 1.00 | 1.04 | 1.00 | 1.00 |
| % di BUSCO con più di una corrispondenza | 0.45 | 3.99 | 0.42 | 0.34 |
Risultati e discussione
L’assegnazione dell’ortografia su insiemi di proteomi previsti derivati da 59 gruppi di genomi e trascrittomi ha dato origine a 4294 alberi genetici con almeno 20 sequenze ciascuno, campionando tutti e cinque i cinque principali cladi e gruppi di metazoi, dai quali abbiamo ottenuto 1388 ortologi ben allineati. All’interno di questo set, sono stati costruiti alberi genetici di massima probabilità individuale (ML) e sono stati selezionati 430 ortologi più informativi sulla base dei punteggi di probabilità degli alberi (Misof et al., 2013). Questo ha prodotto una matrice amminoacidica di 73.547 residui con il 37,55% di lacune o dati mancanti, con una media di 371,92 e 332,75 ortologi rappresentati per Cnidaria e Placozoa, rispettivamente (con un massimo di 383 ortologi presenti per il rappresentante del placozoan H4 clade appena sequenziato; Figura 1).
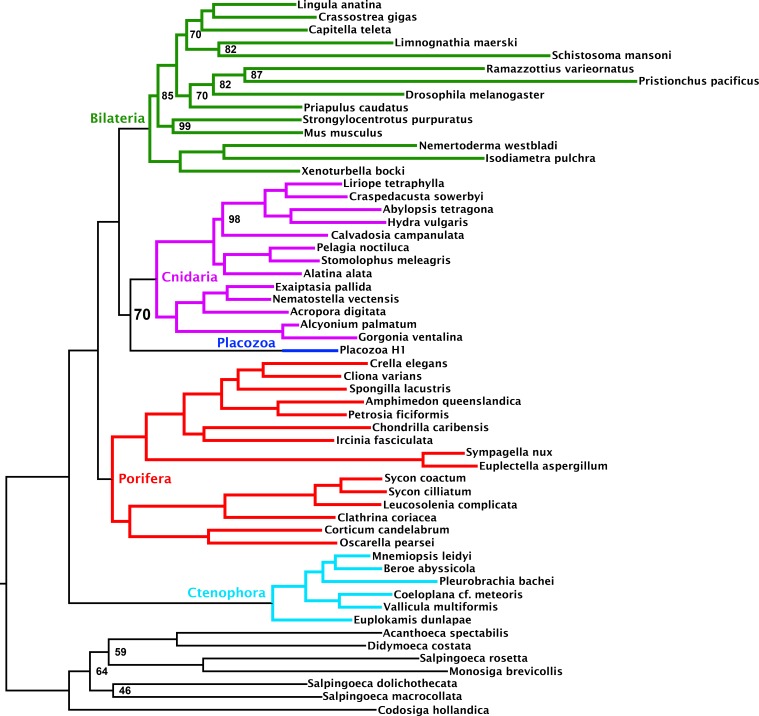
Figura 1-figure supplemento 2.Filogramma di consenso che mostra le interrelazioni metazoiche profonde sotto l’inferenza filogenetica bayesiana della matrice amminoacidica 430-ortologo, utilizzando il modello di miscela CAT + GTR + Г4.Albero di probabilità massima sotto il modello di miscela di profilo C60 +LG + FO + R4, dedotto dalla matrice 430-ortologo con campionamento completo del taxon.albero di probabilità massima sotto un modello di miscela di profilo dedotto dalla matrice 430-ortologo, con solo Placozoa H1 usato per rappresentare questo clade.Tutti i nodi hanno ricevuto la probabilità posteriore completa. Le annotazioni numeriche di determinati nodi rappresentano i punteggi di Extended Quadripartition Internode Certainty (EQP-IC), descrivendo l’accordo among-gene-tree sia per la monofilia dei cinque principali clade metazoici che per le relazioni date tra di essi in questo albero di riferimento. Un diagramma a barre a destra rappresenta la proporzione dell’ortologo totale impostato da ciascun taxon terminale nella matrice concatenata: “Placozoa H1” in questa e in tutte le altre figure si riferisce all’isolato GRELL sequenziato in Srivastava et al., 2008, che lì e altrove è stato indicato come Trichoplax adhaerens, nonostante l’assenza di materiale tipografico che colleghi questo nome ad un qualsiasi isolato moderno. I disegni delle linee dei rappresentanti del clade sono tratti dal database BIODIDAC (http://biodidac.bio.uottawa.ca/).Nodi annotati con supporti bootstrap ultraveloci con correzione NNI; i nodi non annotati hanno ricevuto pieno supporto.Nodi annotati con supporti bootstrap ultraveloci con correzione NNI; i nodi non annotati hanno ricevuto pieno supporto.
Le nostre analisi bayesiane di questa matrice collocano Cnidaria e Placozoa come gruppi fratelli con piena probabilità posteriore sotto il modello generale eterogeneo CAT + GTR + Г4 (Figura 1). Sotto l’inferenza ML con il modello di miscela di profili C60 +LG + FO + R4 (Wang et al., 2018) (Figura 1-figure supplement 1), recuperiamo nuovamente Cnidaria + Placozoa, anche se con un supporto di ricampionamento più marginale. Sia l’analisi Bayesiana che quella ML mostrano una scarsa diversità di ramo interno all’interno di Placozoa. Di conseguenza, l’eliminazione di tutti i genomi placozoici aggiunti di recente dalla nostra analisi non ha alcun effetto sulla topologia e solo un effetto marginale sul supporto nell’analisi ML (Figura 1-figure supplement 2). Le analisi di concordanza basate sui quartetti (Zhou, 2017) non mostrano alcuna evidenza di forti conflitti filogenetici tra gli alberi del gene ML in questo insieme di 430 geni (Figura 1), sebbene le metriche di certezza internodica siano vicine a 0 per molti cladi chiave tra cui Cnidaria + Placozoa, indicando che il supporto per alcune relazioni antiche può essere mascherato da errori di stima dei geni, emersi solo nell’analisi combinata (Gatesy e Baker, 2005).
L’eterogeneità compositiva delle frequenze amminoacide lungo l’albero è una fonte di errore filogenetico non modellato da modelli sostitutivi anche complessi ed eterogenei del sito come CAT+GTR (Blanquart e Lartillot, 2008; Foster, 2004; Lartillot e Philippe, 2004; Lartillot et al., 2013). Inoltre, analisi precedenti (Nosenko et al., 2013) hanno dimostrato che i placozoi e i choanoflagellati in particolare, entrambi i taxa nostri campioni di matrice si discostano fortemente dalla composizione amminoacidica media di Metazoa, forse a causa di discrepanze genomiche del contenuto di GC. Come misura per migliorare almeno in parte tale sostituzione non stazionaria, abbiamo ricodificato la matrice amminoacidica nelle 6 categorie ‘Dayhoff’, una strategia comune precedentemente dimostrata per ridurre l’effetto della variazione compositiva tra i taxa, sebbene i gruppi Dayhoff-6 rappresentino solo una delle tante strategie di ricodifica plausibili, tutte sacrificando informazioni (Feuda et al., 2017; Nesnidal et al., 2010; Rota-Stabelli et al., 2013; Susko e Roger, 2007). L’analisi di questa matrice ricodificata sotto il modello CAT + GTR ha nuovamente recuperato il pieno supporto (pp = 1) per Cnidaria + Placozoa (Figura 2). Infatti, sotto la ricodifica Dayhoff-6, l’unico cambiamento importante è nelle posizioni relative di Ctenophora e Porifera, con quest’ultimo che qui costituisce il gruppo gemello a tutti gli altri animali con pieno supporto. Simili effetti di ricodifica sulle posizioni relative di Porifera e Ctenophora sono stati osservati anche in altri lavori recenti (Feuda et al., 2017), e sono stati interpretati per indicare un ruolo per il pregiudizio compositivo nel collocare erroneamente Ctenophora come gruppo gemello a tutti gli altri animali.
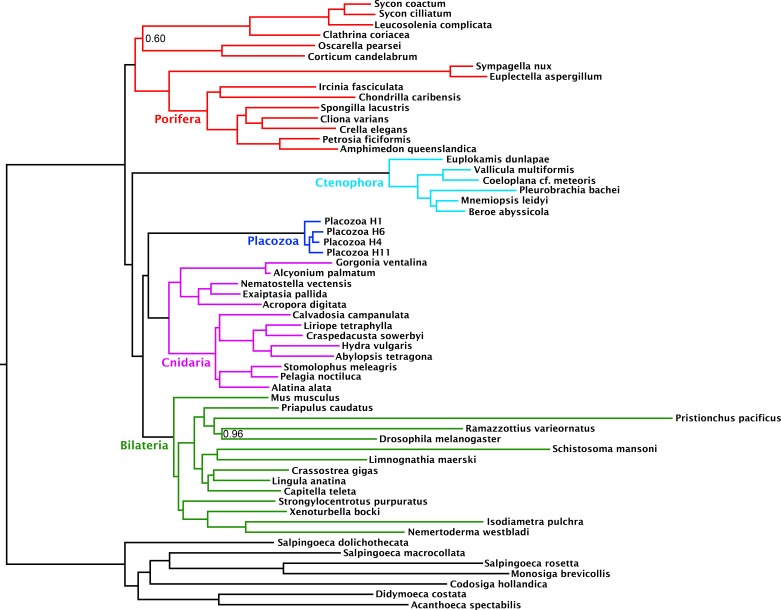
Figura 2.Filogramma di consenso sotto l’inferenza filogenetica bayesiana sotto il modello di miscela CAT + GTR + Г4, sul 430-orthologue concatenato matrice amminoacidica, ricodificato in 6 gruppi Dayhoff.Nodi annotati con probabilità posteriore; i nodi non annotati hanno ricevuto pieno supporto.
Molti gruppi di ricerca, utilizzando un buon campionamento dei taxon e set di dati a scala genomica, e anche recentemente includendo dati di una nuova specie di placoceno divergente (Whelan et al., 2017; Feuda et al., 2017; Eitel, 2017), hanno costantemente riportato un forte sostegno per i planulozoi sotto il modello CAT + GTR. Infatti, quando costruiamo una supermatrice dai nostri cataloghi peptidici previsti utilizzando una strategia diversa, basandoci su sequenze complete di 303 pan-eukaryote ‘Benchmarking Universal Single-Copy Orthologs’ (BUSCOs) (Simão et al., 2015), vediamo anche il pieno supporto in un’analisi CAT + GTR + Г per Planulozoa, sia in alfabeto amminoacido (Figura 3a) che Dayhoff-6 ricodificato (Figura 3b). Quale filogenesi è corretta e quale processo guida il supporto per la topologia non corretta? I test predittivi posteriori, che confrontano l’uso osservato dell’among-taxon delle frequenze di amminoacido con le distribuzioni attese simulate utilizzando la distribuzione posteriore campionata e un singolo vettore di composizione, possono fornire un’intuizione (Feuda et al., 2017; Lartillot e Philippe, 2004). Sia la matrice iniziale del 430-gene che la matrice BUSCO del 303-gene falliscono questi test, ma la matrice BUSCO fallisce più profondamente, con punteggi z-score (che misurano l’eterogeneità media quadrettata attraverso il tassonomio) nell’intervallo 330-340, in contrasto con l’intervallo 176-187 visto nella matrice del 430-gene (Tabella 2). Inoltre, l’ispezione di z-scores per i singoli taxa in catene rappresentative di entrambe le matrici mostra che una grande quantità di questa differenza globale di z-scores può essere attribuita ai placozoi, con contributi aggiuntivi di choanoflagellati e selezionare rappresentanti isolati di altri clades (Figura 3C).
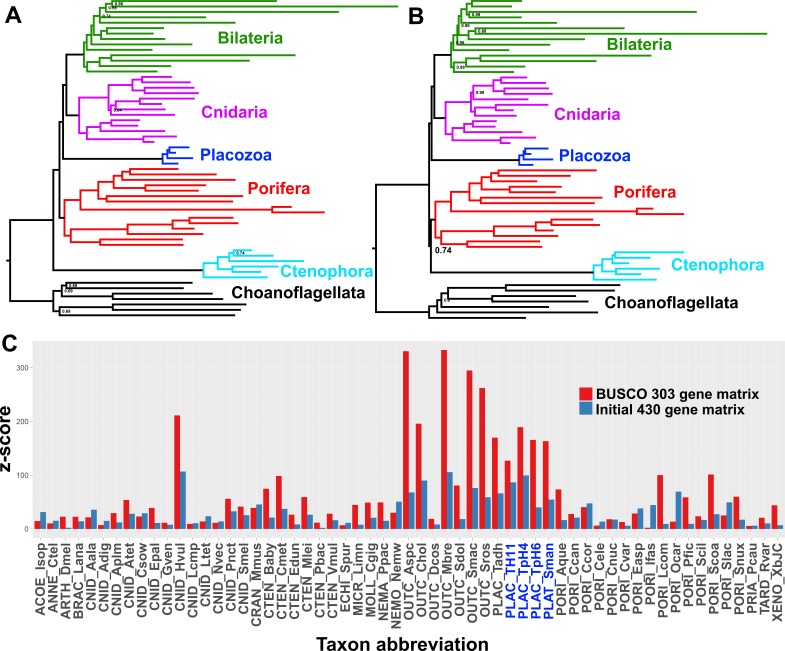
Figura 3.Alberi di consenso posteriori da CAT + GTR + Г4 analisi del modello di miscela di una supermatrice di 94.444 amminoacidi derivati da 303 ortologi eucarioti BUSCO eucarioti conservati in copia singola, analizzati in A.spazio aminoacido o (B) lo spazio alfabetico ridotto Dayhoff-6. I valori di supporto nodale comprendono le probabilità posteriori; nodi con supporto completo non annotato. Colorazione del taxon come nelle figure precedenti. (C) Trama di z-score (somma della distanza assoluta tra le frequenze empiriche specifiche del taxon e quelle globali) da prove predittive posteriori rappresentative della polarizzazione compositiva degli amminoacidi, sia dalla matrice ortolografica BUSCO 303 (rosso) che dalla matrice ortolografica iniziale 430 (blu). Le abbreviazioni del taxon di Placozoan sono mostrate in carattere blu.
| Diversità | Composizione (media) | Composizione (massimo) | |
|---|---|---|---|
| Matrice 430 | 1.94 (0.09) | 181.35 (7.50) | 105.04 (3.13) |
| Matrice del gene BUSCO 303 | 11.27 (0.73) | 334.98 (4.56) | 107.56 (6.17) |
| matrice compilata | 2.51 (0.19) | 270.16 (12.03) | 173.87 (9.15) |
| matrice a bussola | 0.81 (0.18) | 107.67 (10.10) | 63.19 (6.95) |
Come misura finale per descrivere l’influenza dell’eterogeneità compositiva in questo set di dati, abbiamo applicato un test di simulazione nullo per la polarizzazione compositiva ad ogni allineamento nel nostro set di 1388 ortologi. Questo test, che confronta i dati reali con una distribuzione nulla di frequenze amminoacidiche simulate lungo gli alberi genetici ipotizzati con un modello di sostituzione che utilizza un singolo vettore di composizione, è meno soggetto ad errori di Tipo II rispetto al più convenzionale test X (Grell e Benwitz, 1971) (Foster, 2004). Notevolmente, ad una soglia di significatività conservativa di α = 0,10, la maggioranza (764 geni o ~55%) di questo insieme di geni è identificata come influenzata da questo test, evidenziando l’importanza di utilizzare test statistici appropriati per controllare questa fonte di errore sistematico, piuttosto che applicare tagli euristici arbitrari (Kück e Struck, 2014). Costruendo matrici informative da insiemi di geni su entrambi i lati di questa soglia di significatività, e ancora una volta applicando sia modelli di miscele CAT + GTR che miscele di profili ML, vediamo un forte supporto per Cnidaria + Placozoa nella supermatrice che passa il test, e, al contrario, un forte supporto per Cnidaria + Bilateria nella supermatrice che non passa il test (Figura 4, Figura 4-figure supplement 1, Figura 4-figure supplement 2). È interessante notare che negli alberi costruiti attraverso l’analisi CAT + GTR + Г4 della supermatrice non conforme al test (Figura 4A,C), sia in alfabeto amminoacido che Dayhoff-6, si osserva anche il pieno supporto di Porifera come sorella di tutti gli altri animali. Al contrario, l’analisi di questa matrice amminoacidica sotto un modello di miscela di profilo recupera il supporto per Ctenophora in questa posizione (Figura 4-figure supplement 1), indicando che, almeno per questo allineamento, l’eterogeneità compositiva non deve essere invocata per spiegare perché i risultati differiscono tra le analisi, come alcuni hanno sostenuto (Feuda et al., 2017): sia il CAT + GTR che il modello di miscela di profili C60 +LG + FO + R4 assumono un unico vettore di composizione nel tempo, ma il modello CAT + GTR è meglio in grado di accogliere modelli di sostituzione eterogenei (Lartillot et al., 2013; Quang et al., 2008). Nel contesto di questo esperimento, la ricodifica Dayhoff-6 appare d’impatto solo per la supermatrice che passa il test (Figura 4B,D), dove evita il supporto per Ctenophora-sister (Figura 4B, Figura 4-figura 2) a favore di (anche se con supporto marginale) Porifera-sister (Figura 4D), e diminuisce anche il supporto per Placozoa + Cnidaria (in contrasto con la matrice 430-gene; Figura 2), forse riflettendo la perdita di informazione intrinseca dell’uso di un alfabeto amminoacido ridotto per questa matrice relativamente più corta.
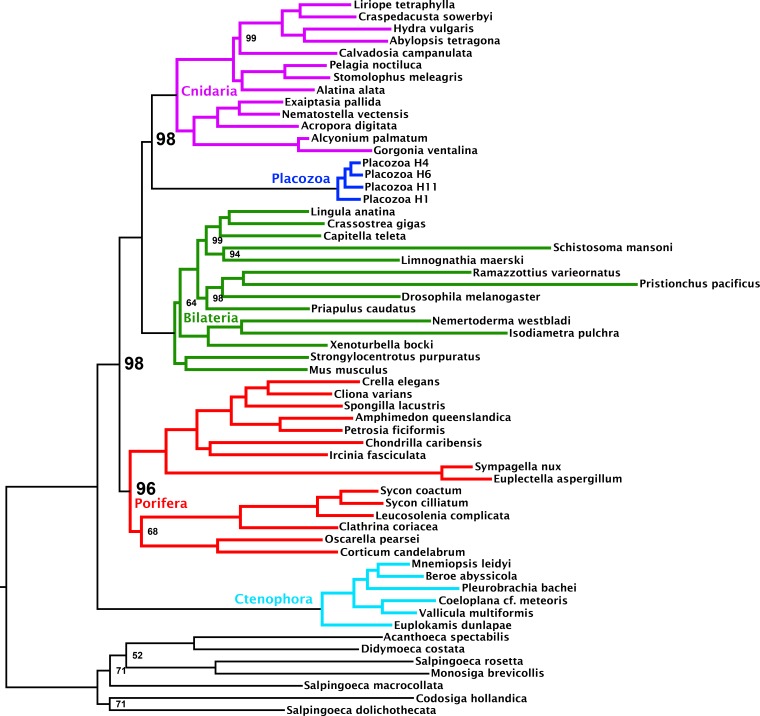
Figura 4-figure supplement 2.Rappresentazione schematica delle interrelazioni metazoiche profonde negli alberi di consenso posteriori da CAT + GTR + Г4 analisi del modello di miscela di matrici fatte da sottoinsiemi di geni che passano o non superano un test di simulazione sensibile di eterogeneità compositiva.Albero di probabilità massima sotto un modello di miscela di profilo dedotto dalla matrice 349-ortologo composta dal sottoinsieme di geni binned come fallito il test di bias composizionale di null-simulation.albero di probabilità massima sotto un modello di miscela di profilo dedotto dalla matrice 348-ortologo composta dal sottoinsieme di geni binned come fallito il test di bias composizionale di null-simulation.albero di probabilità massima sotto un modello di miscela di profilo dedotto dalla matrice 348-ortologo composta dal sottoinsieme di geni binned come fallito il test di bias composizionale di null-simulation.I pannelli corrispondono a (A) la matrice amminoacidica realizzata all’interno dell’insieme fallito; (B) la matrice amminoacidica derivata dall’insieme passante; (C) la matrice ricodificata Dayhoff-6 dall’insieme fallito; (D) la matrice ricodificata Dayhoff-6 dall’insieme passante. Solo i nodi con probabilità posteriore inferiore a 1,00 sono annotati numericamente.Nodi annotati con supporti bootstrap ultraveloci con correzione NNI; i nodi non annotati hanno ricevuto pieno supporto.Nodi annotati con supporti bootstrap ultraveloci con correzione NNI; i nodi non annotati hanno ricevuto pieno supporto.
Una possibile variabile nascosta relativa alla discordanza filogenetica che descriviamo, il cui significato preciso non è chiaro, è la lunghezza media dell’allineamento tagliata: sia la matrice del 430gene originale che quella del 430 sono composte da allineamenti notevolmente più brevi rispetto a quelli del 303gene BUSCO (vedi Materiali e metodi). In effetti, la lunghezza di allineamento è stata precedentemente dimostrata essere predittiva di una serie di altre metriche di rilevanza filogenetica (Shen et al., 2016); la generalità e la direzionalità di tali relazioni in set di dati empirici a diverse scale di divergenza è chiaramente degna di ulteriore approfondimento.
Il legame filogenetico, precedentemente criptico, tra cnidari e placocani visto in insiemi genetici meno influenzati da bias composizionali richiederà ulteriori test con altre analisi e modalità di dati, come le rare modifiche genomiche, che dovrebbero essere sempre più visibili in quanto gli assemblaggi altamente contigui continuano ad essere riportati da animali non-bilateriani (Eitel et al., 2018; Kamm et al., 2018; Jiang, 2018; Leclère, 2018). Tuttavia, se convalidata, questa relazione deve continuare a sollevare interrogativi sull’omologia di alcune caratteristiche dei non-bilateriani. Molti lavoratori, citando lo sviluppo incompleto (Eitel et al., 2011; Pearse e Voigt, 2007) e il contenuto genico relativamente bilateriano dei placocani (Srivastava et al., 2008; Eitel, 2017), presumono che questi organismi debbano avere uno sviluppo e un ciclo di vita ancora non osservato e più tipico (DuBuc et al., 2018), oppure sono semplicemente bizzarrie che hanno sperimentato una semplificazione secondaria all’ingrosso, avendo scarso significato per qualsiasi percorso evolutivo al di fuori del proprio. In effetti, si è tentati di interpretare questa nuova posizione filogenetica come un ulteriore rafforzamento di tali ipotesi, in quanto molto lavoro sui modelli cnidariani nel paradigma evo-devo si basa sulla nozione che cnidari e bilateri condividono, più o meno, molte caratteristiche morfologiche omologhe, vale a dire l’organizzazione assiale (Genikhovich e Technau, 2017; DuBuc et al., 2018), il sistema nervoso (Liebeskind et al., 2017; Moroz e Kohn, 2016; Kelava et al., 2015; Kristan, 2016; Arendt et al., 2016), epitelio a membrana basamentale (Fidler et al., 2017; Leys e Riesgo, 2012), muscolatura (Steinmetz et al., 2012), organizzazione dello strato germinale embrionale (Steinmetz et al., 2017) e digestione interna (Presnell et al., 2016; Putnam et al., 2007; Hejnol e Martindale, 2008; Martindale e Hejnol, 2009). Anche se non sosteniamo, come alcuni hanno fatto (Schierwater, 2005; Syed e Schierwater, 2002), che i placocani assomiglino ad ipotetici antenati metazoici, esitiamo a respingerli a priori come irrilevanti per la comprensione della prima evoluzione bilateriana in particolare: sebbene apparentemente più semplici e meno diversificati, i placocani hanno comunque lo stesso status degli cnidari come gruppo immediatamente esistente. Piuttosto, vediamo il valore nella sperimentazione di ipotesi assunte di omologia, carattere per carattere, estendendo i confronti a coppie tra bilateriani e cnidari per includere i placozoi, un’agenda che richiede di ridurre la grande disparità di conoscenze genetiche embriologiche, fisiologiche e molecolari tra questi taxa, verso cui sono stati fatti recenti progressi utilizzando entrambi i metodi stabiliti, come l’ibridazione in situ (DuBuc et al.., 2018) e l’analisi delle immagini (Varoqueaux, 2018), così come le nuove tecnologie come l’RNA-seq monocellulare (Sebé-Pedrós, 2018a; Sebé-Pedrós et al., 2018b). Al contrario, sottolineiamo un’altra implicazione di questa filogenesi: i caratteri che possono essere validati come omologhi a qualsiasi livello tra Bilateria e Cnidaria devono avere avuto origine prima nell’evoluzione animale rispetto a quanto precedentemente apprezzato, e dovrebbero o verificarsi cripticamente nei moderni placogeni o essere andati perduti a un certo punto della loro ascendenza. In questa luce, scenari paleobiologici dell’evoluzione animale precoce fondati su interpretazioni intrinsecamente filogenetiche delle forme fossili ediacarine (Cavalier-Smith, 2018; Cavalier-Smith, 2017; Dufour e McIlroy, 2018; Sperling e Vinther, 2010; Evans et al, 2017) e le stime dell’orologio molecolare (Cunningham et al., 2017; dos Reis et al., 2015; Dohrmann e Wörheide, 2017; Erwin et al., 2011) possono richiedere un riesame.
Figura 1-figure supplement 2.Figura 1—figura 2. Filgramma di consenso che mostra profonde interrelazioni metazoiche sotto l’inferenza filogenetica bayesiana della matrice amminoacidica 430-ortologo, utilizzando il modello di miscela CAT + GTR + Г4.Albero di probabilità massima sotto il modello di miscela di profilo C60 +LG + FO + R4, dedotto dalla matrice 430-ortologo con campionamento completo del taxon.albero di probabilità massima sotto un modello di miscela di profilo dedotto dalla matrice 430-ortologo, con solo Placozoa H1 usato per rappresentare questo clade.Tutti i nodi hanno ricevuto la probabilità posteriore completa. Le annotazioni numeriche di determinati nodi rappresentano i punteggi di Extended Quadripartition Internode Certainty (EQP-IC), descrivendo l’accordo among-gene-tree sia per la monofilia dei cinque principali clade metazoici che per le relazioni date tra di essi in questo albero di riferimento. Un diagramma a barre a destra rappresenta la proporzione dell’ortologo totale impostato da ciascun taxon terminale nella matrice concatenata: “Placozoa H1” in questa e in tutte le altre figure si riferisce all’isolato GRELL sequenziato in Srivastava et al., 2008, che lì e altrove è stato indicato come Trichoplax adhaerens, nonostante l’assenza di materiale tipografico che colleghi questo nome ad un qualsiasi isolato moderno. I disegni delle linee dei rappresentanti del clade sono tratti dal database BIODIDAC (http://biodidac.bio.uottawa.ca/).Nodi annotati con supporti bootstrap ultraveloci con correzione NNI; i nodi non annotati hanno ricevuto pieno supporto.Nodi annotati con supporti bootstrap ultraveloci con correzione NNI; i nodi non annotati hanno ricevuto pieno supporto.
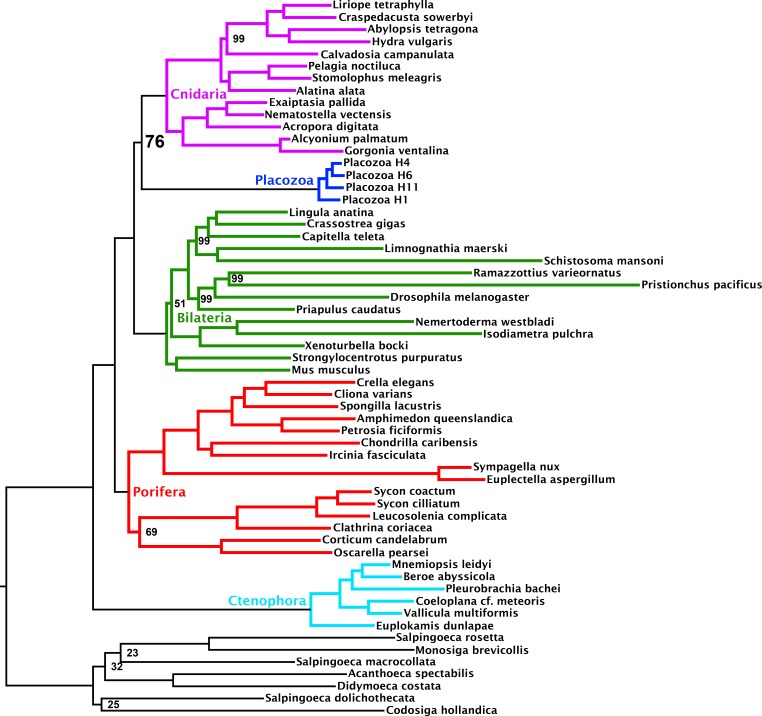
Figura 1-figure supplement 1.Albero di probabilità massima sotto il modello di miscela di profilo C60 +LG + FO + R4, dedotto dalla matrice 430-ortologo con campionamento completo del taxon.Nodi annotati con supporti bootstrap ultraveloci con correzione NNI; i nodi non annotati hanno ricevuto pieno supporto.
Figura 1-figura supplemento 2.Albero di probabilità massima sotto un modello di miscela di profilo dedotto dalla matrice 430-ortologo, con solo Placozoa H1 utilizzato per rappresentare questo clade.Nodi annotati con supporti bootstrap ultraveloci con correzione NNI; i nodi non annotati hanno ricevuto pieno supporto.
Figura 2.Figura 2. Filogramma di consenso sotto l’inferenza filogenetica bayesiana sotto il modello di miscela CAT + GTR + Г4, sulla matrice amminoacidica concatenata 430-ortologo, ricodificata in 6 gruppi Dayhoff.Nodi annotati con probabilità posteriore; i nodi non annotati hanno ricevuto pieno supporto.
Figura 3.Figura 3. Alberi di consenso posteriori da CAT + GTR + Г4 analisi del modello di miscela di una supermatrice di 94.444 amminoacidi derivati da 303 ortologi eucariotici BUSCO conservati in copia singola, analizzati in A.spazio amminoacido o (B) lo spazio alfabetico ridotto Dayhoff-6. I valori di supporto nodale comprendono le probabilità posteriori; i nodi con supporto completo non annotato. Colorazione del taxon come nelle figure precedenti. (C) Trama di z-score (somma della distanza assoluta tra le frequenze empiriche specifiche del taxon e quelle globali) da prove predittive posteriori rappresentative della polarizzazione compositiva degli amminoacidi, sia dalla matrice ortolografica BUSCO 303 (rosso) che dalla matrice ortolografica iniziale 430 (blu). Le abbreviazioni del taxon di Placozoan sono mostrate in carattere blu.
Figura 4-figure supplement 2.Rappresentazione schematica delle interrelazioni metazoiche profonde negli alberi di consenso posteriori da CAT + GTR + Г4 analisi del modello di miscela di matrici fatte da sottoinsiemi di geni che passano o non superano un test di simulazione sensibile di eterogeneità compositiva.Albero di probabilità massima sotto un modello di miscela di profilo dedotto dalla matrice 349-ortologo composta dal sottoinsieme di geni binned come fallito il test di bias composizionale di null-simulation.albero di probabilità massima sotto un modello di miscela di profilo dedotto dalla matrice 348-ortologo composta dal sottoinsieme di geni binned come fallito il test di bias composizionale di null-simulation.albero di probabilità massima sotto un modello di miscela di profilo dedotto dalla matrice 348-ortologo composta dal sottoinsieme di geni binned come fallito il test di bias composizionale di null-simulation.I pannelli corrispondono a (A) la matrice amminoacidica realizzata all’interno dell’insieme fallito; (B) la matrice amminoacidica derivata dall’insieme fallito; (C) la matrice ricodificata Dayhoff-6 dall’insieme fallito; (D) la matrice ricodificata Dayhoff-6 dall’insieme fallito. Solo i nodi con probabilità posteriore inferiore a 1,00 sono annotati numericamente.I nodi annotati con supporti bootstrap ultraveloci con correzione NNI; i nodi non annotati hanno ricevuto pieno supporto.Nodi annotati con supporti bootstrap ultraveloci con correzione NNI; i nodi non annotati hanno ricevuto pieno supporto.
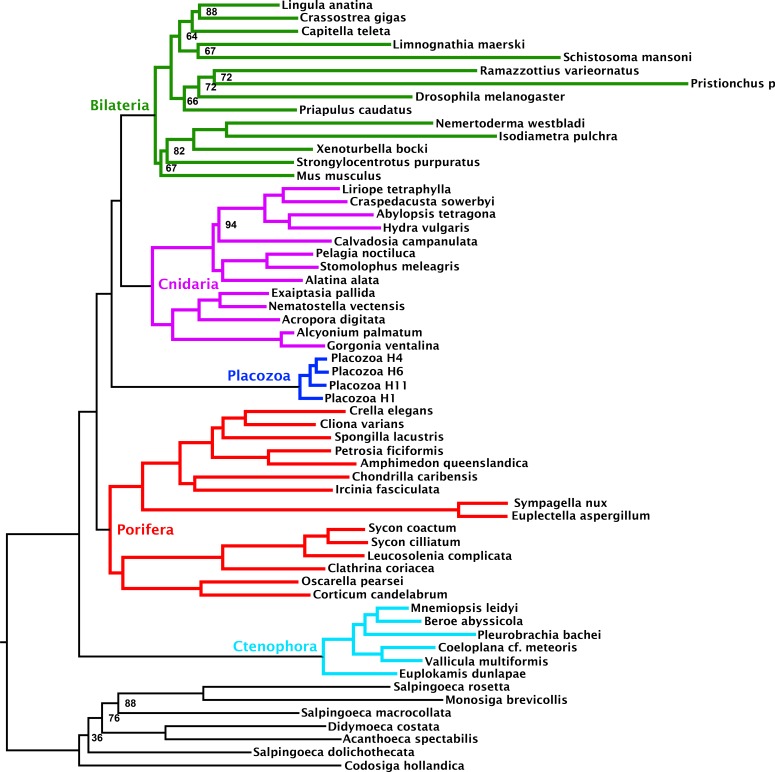
Figura 4-figure supplement 1.Albero di probabilità massima sotto un modello di miscela di profili dedotto dalla matrice 349-ortologo composto dal sottoinsieme di geni binned come fallendo il test di bias composizionale null-simulazione di bias.Nodi annotati con supporti bootstrap ultraveloci con correzione NNI; i nodi non annotati hanno ricevuto pieno supporto.
Figura 4-figure supplement 2.Figura 4—supplemento di figura 2. Albero di probabilità massima sotto un modello di miscela di profili dedotto dalla matrice 348-ortologo composta dal sottoinsieme dei geni binned come superamento del test di bias composizionale null-simulazione.Nodi annotati con supporti bootstrap ultraveloci con correzione NNI; i nodi non annotati hanno ricevuto pieno supporto.
Materiali e metodi
Campionamento, sequenziamento e assemblaggio di genomi di riferimento da placogeni precedentemente non campionati
Gli aplotipi Haplotype H4 e H6 placozoani sono stati raccolti dalle falde acquifere presso il Kewalo Marine Laboratory, University of Hawaii-Manoa, Honolulu, Hawaii, nell’ottobre 2016. I placogeni di aplotipo H11 sono stati raccolti dalla vasca di esposizione mediterranea ‘Anthias‘ nell’acquario di Palma di Maiorca, Mallorca, Spagna, nel giugno 2016. Tutti i placogeni sono stati campionati inserendo nelle vasche per 10 giorni scivoli di vetro sospesi liberamente o montati in supporti per scivoli di plastica tagliati (Pearse e Voigt, 2007). I placozoiani sono stati identificati al microscopio di dissezione e singoli individui sono stati trasferiti a 500 µl di RNAlater, conservati secondo le raccomandazioni del produttore.
Il DNA è stato estratto da 3 individui di aplotipo H11 e 5 individui di aplotipo H6 utilizzando il kit DNeasy Blood and Tissue Kit (Qiagen, Hilden, Germania). Il DNA e l’RNA da tre individui di aplotipo H4 sono stati estratti utilizzando il kit AllPrep DNA/RNA Micro Kit (Qiagen), con entrambi i kit utilizzati secondo i protocolli del produttore.
La preparazione della biblioteca Illumina e il sequenziamento è stata eseguita dal Centro Max Planck Genoma di Colonia, Germania, come parte di un progetto in corso di metagenomica in simbiosi marina. In breve, la qualità del DNA/RNA è stata valutata con il Bioanalizzatore Agilent 2100 (Agilent, Santa Clara, USA) e il DNA genomico è stato frammentato fino a una dimensione media del frammento di 500 bp. Per i campioni di DNA, la concentrazione è stata aumentata (MinElute PCR kit di purificazione MinElute PCR; Qiagen, Hilden, Germania) e una biblioteca compatibile con Illumina è stata preparata utilizzando il kit Ovation Ultralow Library Systems (NuGEN, Leek, Paesi Bassi) secondo il protocollo del produttore. Per i campioni di aplotipo H4 RNA, il sistema Ovation RNA-seq System V2 (NuGen, 376 San Carlos, CA, USA) è stato utilizzato per sintetizzare il cDNA e le librerie di sequenziamento sono state poi generate con il kit di preparazione della libreria del DNA per Illumina (BioLABS, Francoforte sul Meno, Germania). Tutte le biblioteche sono state selezionate per elettroforesi su gel di agarosio, e la qualità dei frammenti recuperati è stata valutata e quantificata con la fluorometria. Per ogni biblioteca del DNA sono stati sequenziati 14 – 75 milioni di 100 bp o 150 bp di letture paired-end su Illumina HiSeq 2500 o 4000 macchine (Illumina, San Diego, U.S.A); per le biblioteche di aplotipo H4 RNA 32 – 37 milioni di singole letture a 150 bp sono state ottenute.
Per il montaggio, sono stati rimossi con bbduk (https://sourceforge.net/projects/bbmap/) adattatori e letture di bassa qualità con un valore di qualità minimo di due e una lunghezza minima di 36 e sono state escluse dall’analisi le singole letture. Ogni libreria è stata corretta con BayesHammer (Nikolenko et al., 2013). Un assemblaggio combinato di tutte le librerie per ogni aplotipo è stato eseguito utilizzando SPAdes 3.62 (Bankevich et al., 2012). I dati di aplotipo quattro e H11 sono stati assemblati dal set di lettura completo con parametri standard e kmer 21, 33, 55, 77, 99. I dati di aplotipo H6 sono stati preprocessati per rimuovere tutte le letture con una copertura media di kmer <5 utilizzando bbnorm e poi assemblati con kmer 21, 33, 55 e 77.
Le letture di ogni libreria sono state mappate sulle impalcature assemblate utilizzando bbmap (https://sourceforge.net/projects/bbmap/) con l’opzione fast = t. I ponteggi sono stati mappati sulla base dei dati di lettura mappati utilizzando MetaBAT (Kang et al., 2015) con le impostazioni di default e l’opzione di binning dell’insieme attivata (switch -B 20). I bins dell’host Trichoplax sono stati valutati utilizzando metawatt (Strous et al., 2012) sulla base della densità di codifica e della somiglianza di sequenza con l’assieme di riferimento Trichoplax H1 (NZ_ABGP0000000000.1). Le metriche della qualità del bin sono state calcolate con BUSCO2 (Simão et al., 2015) (Tabella 1) e QUAST (Gurevich et al., 2013). Sia la rigorosa procedura di binning metagenomico (una procedura opportuna anche in altri organismi olobionti (Celis et al., 2018)) sia la bassissima percentuale di riscontri di ortologia multipli nella valutazione di BUSCO2 (Tabella 1) attestano la mancanza di prove di contaminazione residua non placozoica all’interno delle impalcature utilizzate per la previsione genica.
2. Previsione di proteomi da assemblaggi di trascrittomi e genomi
Nel giugno 2017 sono stati scaricati dal portale NCBI Genoma o dall’Ensemble Metazoa del giugno 2017 i proteomi previsti delle specie con le bozze di genoma pubblicate. Per i placogeni Clade A sono stati utilizzati direttamente per l’annotazione dei geni. Per i rappresentanti dell’H6 e dell’H11, l’annotazione è stata interamente ab initio, eseguita con GeneMark-ES (Ter-Hovhannisyan et al., 2008); per il rappresentante dell’H4, le librerie RNA-seq totali ottenute da tre isolati separati (BioProject PRJNA505163) sono state mappate in contiguità genomiche con STAR v2.5.3a (Dobin et al., 2013) con impostazioni predefinite; i file bam uniti sono stati poi utilizzati per annotare i contig genomici e ricavare i peptidi previsti con BRAKER v1.9 (Hoff et al., 2016) con impostazioni predefinite. Le previsioni del proteoma di Choanoflagellate (Simion et al., 2017) sono state fornite come dati non pubblicati da Dan Richter. I peptidi di una Calvadosia (in precedenza Leucosolenia) complicata transcriptome assembly sono stati scaricati da compagen.org. Le previsioni dei peptidi da Nemertoderma westbladi e Xenoturbella bocki come usati in Cannon et al 2016 (Cannon et al., 2016) sono stati forniti direttamente dagli autori. L’assemblaggio del trascrittoma (grezzo legge inedito) da Euplectella aspergillum è stato fornito dal gruppo Satoh, scaricato da (http://marinegenomics.oist.jp/kairou/viewer/info?project_id=62). I peptidi previsti sono stati derivati dagli assemblaggi Trinity RNA-seq (versioni multiple rilasciate nel 2012-2016) come descritto da Laumer et al (Laumer et al., 2015). per le seguenti fonti/adesioni all’ARA:: Porifera: Petrosia ficiformis: SRR504688, varianti Cliona: SRR1391011, Crella elegans: SRR648558, Corticium candelabro: SRR504694-SRR499820-SRR499817, Spongilla lacustris: SRR1168575, Clathrina coriacea: SRR3417192, Sycon coactum: SRR504689-SRR504690, Sycon ciliatum: ERR466762, Ircinia fasciculata, Chondrilla caribensis (originariamente erroneamente identificata come Chondrilla nucula) e Pseudospongosorites suberitoides da (https://dataverse.harvard.edu/dataverse/spotranscriptomes); Cnidaria: Abylopsis tetragona: SRR871525, Stomolophus meleagris: SRR1168418, Craspedacusta sowerbyi: SRR923472, Gorgonia ventalina: SRR935083; Ctenophora: Vallicula multiformis: SRR786489, Pleurobrachia bachei: SRR777663, Beroe abyssicola: SRR777787; Bilateria: Limnognathia maerski: SRR2131287. Tutte le altre previsioni peptidiche sono state derivate attraverso l’assemblaggio del trascrittoma come librerie paired-end, unstranded con Trinity v2.4.0 (Haas et al., 2013), con il flag -trimmomatic abilitato (e tutti gli altri parametri come default), con l’estrazione peptidica dalle trascrizioni assemblate usando TransDecoder v4.0.1 con impostazioni predefinite. Per queste specie non è stata effettuata alcuna selezione di isoforme ad hoc: eventuali isoforme ridondanti sono state rimosse durante la potatura degli alberi nella pipeline di determinazione dell’ortologo (vedi sotto).
Identificazione e allineamento degli ortologi
I proteomi previsti sono stati raggruppati in ortogruppi di primo livello con OrthoFinder v1.0.6 (Emms e Kelly, 2015), eseguiti come un lavoro a 200 fili, diretto a fermarsi dopo l’assegnazione dell’ortogruppo, e stampare sequenze raggruppate e non allineate come file FASTA con il flag ‘-os’. Uno script pitone personalizzato (‘renamer.py’) è stato usato per rinominare tutte le intestazioni in ogni file FASTA di ortogruppo nella convenzione [abbreviazione taxon] + ‘@’ + [numero di sequenza come assegnato da OrthoFinder SequenceIDs.txt file], e per selezionare solo quegli ortogruppi con appartenenza che comprendono almeno uno di tutti e cinque i cinque principali clades metazoan più outgroups, di cui esattamente 4300 di un iniziale 46.895 sono stati mantenuti. Gli script della pipeline di costruzione del Dataset Filogenomico (Yang e Smith, 2014) sono stati utilizzati per le successive fasi di data grooming come segue: Gli alberi genetici per gli ortogruppi di livello superiore sono stati derivati chiamando lo script fasta_to_tree.py come job array, senza repliche bootstrap; sei ortogruppi molto grandi non hanno completato questo processo. Nella stessa directory, gli script trim_tips.py, mask_tips_by_taxonID_transcripts.py, e cut_long_internal_branches.py sono stati chiamati in successione, con ‘./. tre 10 10’, ‘././y’, e ‘./. mm 1 20. /’ passati come argomenti, rispettivamente. I 4267 sottoalberi generati attraverso questo processo sono stati concatenati in un unico file Newick e 1419 ortologi sono stati estratti con UPhO (Ballesteros e Hormiga, 2016). L’allineamento degli ortologi è stato eseguito utilizzando l’algoritmo MAFFT v7.271 ‘E-INS-i’, e sono stati assegnati punteggi di mascheramento probabilistico con ZORRO (Wu et al., 2012), rimuovendo tutti i siti in ogni allineamento con punteggi inferiori a cinque come descritto in precedenza (Laumer et al., 2015). 31 ortologi con lunghezze trattenute inferiori a 50 amminoacidi sono stati scartati, lasciando 1388 ortologi ben allineati.
Assemblaggio della matrice
Una concatenazione completa di tutti i 1388 ortogruppi conservati è stata eseguita con lo script ‘geneStitcher.py’ distribuito con l’UPhO disponibile su https://github.com/ballesterus/PhyloUtensils. Tuttavia, una tale matrice sarebbe troppo grande per poter dedurre in modo tracciabile una filogenesi sotto modelli di miscele ben aderenti come CAT + GTR; pertanto abbiamo usato MARE v0.1.2 (Misof et al., 2013) per estrarre un sottoinsieme informativo di geni utilizzando i punteggi di somiglianza degli alberi, eseguendo con ‘-t 100’ per mantenere tutti i taxa e utilizzando ‘-d 1’ come parametro di sintonia sulla lunghezza dell’allineamento. Questo ha prodotto il nostro 430-ortologo, 73.547 matrice del sito, con una lunghezza media della partizione di 202,24 (s.d. 116,96) residui.
Come verifica della procedura di cui sopra, che è agnostica per l’identità dei geni assegnati in gruppi di ortologi, abbiamo anche cercato di costruire una matrice utilizzando sequenze complete a copia singola identificate dall’algoritmo BUSCO v3.0.1 (Simão et al., 2015), utilizzando il set di ortologi 303-gene eukaryote_odb9. BUSCO è stato eseguito in modo indipendente su ogni file peptidico FASTA utilizzato come input per OrthoFinder, ed è stato utilizzato uno script pitone personalizzato (‘extract.py’) per analizzare l’intera tabella di output di ogni specie, selezionando solo quelle voci identificate come rappresentanti a lunghezza completa, a copia singola di ogni ortologo BUSCO, e raggruppandole in directory unix, facilitando l’allineamento a valle, il mascheramento probabilistico e la concatenazione, come descritto per la matrice OrthoFinder. Questa matrice BUSCO 303-gene aveva una lunghezza totale di 94.444 amminoacidi, con il 39,6% dei siti che rappresentano lacune o dati mancanti, con una lunghezza media di partizione 311,70 (deviazione standard 202,78).
All’interno dei bins del gene nominati dal test di eterogeneità composizionale (vedi sotto), le matrici sono state costruite nuovamente concatenando e riducendo le matrici con MARE, utilizzando ‘-t 100’ per mantenere tutti i taxa e impostando ‘-d 0,5’ per produrre una matrice di dimensioni ottimali per dedurre una filogenesi sotto il modello CAT + GTR. Questa procedura ha dato una matrice di 349-geni di 80.153 amminoacidi (lunghezze medie di partizione 228,67 ± s.d. 136,19, gap del 41,64%) all’interno del set di geni che non hanno superato il test, e una matrice di 348-geni di 55.426 amminoacidi (lunghezze medie di partizione 158,27 ± s.d. 79,06, gap del 38,92%), all’interno del set per il passaggio del test (Figura 4).
Inferenza filogenetica
I singoli alberi genetici ML sono stati costruiti su tutti i 1388 ortologi in IQ-tree v1.6beta, con ‘-m MFP -b 100’ passati come parametri per eseguire la selezione automatica del modello e 100 bootstraps standard non parametrici su ogni albero genetico.
Per dedurre la matrice iniziale di 430 geni, si è proceduto come segue: L’inferenza ML sulla matrice concatenata (Figura 1-figure supplement 1) è stata eseguita con IQ-tree v1.6beta, passando ‘-m C60 +LG + FO + R4 bb 1000’ come parametri per specificare un modello di miscela di profilo e mantenere 1000 alberi per il bootstrapping ultraveloce; il flag ‘-bnni’ è stato utilizzato per incorporare la correzione NNI durante il bootstrapping UF, un approccio mostrato per controllare il supporto gonfiato fuorviante derivante da una errata specificazione del modello (Hoang et al., 2018). L’inferenza ML utilizzando solo l’aplotipo H1 come rappresentante di Placozoa (Figura 1-figure supplement 2) è stata intrapresa in modo simile, anche se utilizzando un modello di miscela di profilo leggermente meno complesso (C20 +LG + FO + R4). L’inferenza bayesiana sotto il modello CAT + GTR + Г4 è stata eseguita in PhyloBayes MPI v1.6j (Lartillot et al., 2013) con 20 core ciascuno dedicato a quattro catene separate, eseguito per 2885-3222 generazioni con il flag ‘-dc’ applicato per rimuovere i siti costanti dall’analisi, e utilizzando un albero di partenza derivato dal programma FastTree2 (Price et al., 2010). Le due catene usate per generare l’albero di consenso posteriore riassunto nella Figura 1 convergono esattamente sullo stesso albero in tutti i campioni MCMC dopo aver rimosso le prime 2000 generazioni come burn-in. L’analisi delle matrici ricodificate Dayhoff-6-state in CAT + GTR + Г4 è stata eseguita con il programma seriale PhyloBayes v4.1c, con ‘-dc -recode dayhoff6’ passato come flag. Sei catene sulla matrice del gene 430 sono state eseguite da 1441 a 1995 generazioni; due catene hanno mostrato una discrepanza bipartizione massima (maxdiff) di 0,042 dopo aver rimosso le prime 1000 generazioni come burn-in (Figura 2). QuartetScores (Zhou, 2017) è stato utilizzato per misurare le metriche di certezza degli internodi, compreso l’EQP-IC riportato, utilizzando gli alberi del gene 430 da quegli ortologi utilizzati per derivare la matrice come alberi di valutazione, e utilizzando l’albero dell’amminoacido CAT + GTR + Г4 come riferimento da annotare (Figura 1).
Per l’inferenza sul set di geni BUSCO 303, abbiamo eseguito 4 catene del modello di miscela CAT + GTR + Г4 con PhyloBayes MPI v1.7a, applicando di nuovo il flag -dc per rimuovere i siti costanti, ma qui non specificando un albero di partenza; le catene sono state eseguite dal 1873 a 2361 generazioni. Sfortunatamente, nessuna coppia di catene ha raggiunto una stretta convergenza sulla versione amminoacidica di questa matrice (con tutte le coppie che mostrano una maxdiff = 1 ad ogni proporzione di burn-in esaminata), forse indicando problemi di miscelazione tra le quattro catene che abbiamo eseguito. Tuttavia, tutte le catene hanno mostrato un pieno supporto posteriore per relazioni identiche tra i cinque gruppi animali principali, con differenze tra le catene assegnabili a differenze minori nelle relazioni interne all’interno di Choanoflagellata e Bilateria. Di conseguenza, l’albero di consenso posteriore della Figura 3A è riassunto da tutte e quattro le catene, con un burn-in di 1000 generazioni, campionamento ogni 10 generazioni. Per la versione codificata Dayhoff di questa matrice, abbiamo eseguito di nuovo sei catene separate con CAT + GTR + Г4 con la bandiera -dc, per 5433 – 6010 generazioni; due catene sono state giudicate convergenti, dando una maxdiffusione di 0,141157 durante il riepilogo dei consensi posteriori con un burn-in di 2500, campionamento ogni 10 generazioni (Figura 3B).
Per l’inferenza sulle matrici geniche 348 e 349 prodotte all’interno dei bins genici definiti dal test di null-simulazione di bias composizionale (vedi sotto), abbiamo eseguito sei catene ciascuna per l’amminoacido e le versioni ricodificate di ciascuna matrice, sotto CAT + GTR + Г4 con siti costanti rimossi. Nella matrice amminoacidica, le catene andavano da 2709 a 3457 e da 1423 a 1475 generazioni per le matrici di test falliti e di test passanti, rispettivamente. Nella matrice ricodificata, le catene andavano da 3893 a 4480 e da 4350 a 4812 generazioni per le matrici test-failing e test-passing, rispettivamente. Nella selezione delle catene da inserire per la presentazione dell’albero di consenso posteriore (Figura 4A-D), abbiamo scelto coppie di catene e burn-ins che hanno prodotto i valori di maxdiff più bassi possibili (tutti <0,1 con le prime 500 generazioni scartate come burn-in, ad eccezione della matrice con codifica amminoacidica test-failing, la cui coppia di catene più simile ha dato un maxdiff di 0,202 con 1000 generazioni scartate come burn-in). Sottolineiamo che le topologie e i supporti visualizzati nella Figura 4A-D sono simili quando tutte le catene (e i valori conservativi di burn-in) sono usati per generare alberi di consenso. Per gli alberi ML che utilizzano modelli di miscela di profili per le matrici geniche non conformi al test (Figura 4-figure supplement 1) e per il test-passing (Figura 4-figure supplement 2), abbiamo usato IQ-tree 1.6rc, chiamando nello stesso modo (con C60 +LG + FO + R4) come usato sulla nostra matrice 430-gene (vedi sopra).
Test di eterogeneità compositiva
Per i test predittivi posteriori di eterogeneità compositiva e diversità dei residui utilizzando campioni MCMC sotto CAT + GTR (Tabella 2), abbiamo usato PhyloBayes MPI v1.8 per testare due catene della matrice iniziale di 430 geni, tre catene della matrice BUSCO di 303 geni, e sei catene ciascuna delle matrici dei geni 348 (test-failing) e 349 (test-passing), eliminando 2000 generazioni dalla prima matrice e 1000 dalle altre come burn-in. I risultati dei test su catene rappresentative sono stati selezionati per la tracciatura nella Figura 3C e il riassunto nella Tabella 2; tuttavia, i risultati di tutte le catene testate sono depositati nell’adesione Data Dryad.
Per i test di simulazione null per gene di bias composizionale (Foster, 2004), abbiamo usato il pacchetto p4 (https://github.com/pgfoster/p4-phylogenetics), inserendo gli alberi ML dedotti da IQ-tree per ciascuno dei 1388 allineamenti, e assumendo un modello di sostituzione LG+Γ4 con un singolo vettore di frequenza empirico per ogni gene; questo test è stato implementato con un semplice script wrapper (‘p4_compo_test_multiproc.py’) sfruttando il modulo python multiprocessing. Abbiamo scelto di non eseguire il model-test di ogni singolo gene in p4, sia perché la gamma di modelli implementati in p4 è più limitata rispetto a quelli testati in IQ-tree, sia perché, in pratica, LG (di solito con variante del modello FreeRates di eterogeneità di tasso) è stato scelto come il modello più adatto nei test su modelli IQ-tree per una grande maggioranza di geni, suggerendo che LG+Γ4 sarebbe una ragionevole approssimazione ai fini di questo test. Abbiamo selezionato una soglia α di 0,10 per la suddivisione dei geni in bins per il superamento dei test e per il mancato superamento dei bins come misura conservativa; tuttavia, sottolineiamo che anche con un α meno conservativo = 0,05, il 47% dei geni verrebbe comunque rilevato come al di fuori dell’aspettativa nulla.
Disponibilità dei dati di partenza
I codici di adesione SRA, ove utilizzati, e tutte le fonti alternative per i dati di sequenza (ad es. siti web ospitati individualmente, comunicazioni personali), sono elencati sopra nella sezione Materiali e metodi. Un’adesione DataDryad è disponibile all’indirizzo https://doi.org/10.5061/dryad.6cm1166, che rende disponibili tutti gli script di aiuto, gli ortogruppi, gli allineamenti di sequenze multiple, l’output del programma filogenetico e i proteomi host grezzi inseriti in OrthoFinder. In questa adesione vengono forniti anche i contenitori metagenomici contenenti contigui di host placozoani e annotazioni geniche da isolati H4, H6 e H11. PhyloBayes. file di catena, a causa delle loro grandi dimensioni, sono separatamente adesione a Zenodo a https://doi.org/10.5281/zenodo.1197272.
References
- Arendt D, Tosches MA, Marlow H. From nerve net to nerve ring, nerve cord and brain–evolution of the nervous system. Nature Reviews Neuroscience. 2016; 17:61-72. DOI | PubMed
- Armon S, Bull MS, Aranda-Diaz AJ, Prakash M. Ultra-fast cellular contractions in the epithelium of T. adhaerens and the ‘active cohesion’ hypothesis. bioRxiv. 2018. DOI
- Ballesteros JA, Hormiga G. A new orthology assessment method for phylogenomic data: unrooted phylogenetic orthology. Molecular Biology and Evolution. 2016; 33:2117-2134. DOI | PubMed
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology. 2012; 19:455-477. DOI | PubMed
- Blanquart S, Lartillot N. A site- and time-heterogeneous model of amino acid replacement. Molecular Biology and Evolution. 2008; 25:842-858. DOI | PubMed
- Bütschli O. Bemerkungen zur Gastraea-Theorie. Morphologisches Jahrbuch. 1884; 9:415-427.
- Cannon JT, Vellutini BC, Smith J, Ronquist F, Jondelius U, Hejnol A. Xenacoelomorpha is the sister group to nephrozoa. Nature. 2016; 530:89-93. DOI | PubMed
- Cavalier-Smith T. Origin of animal multicellularity: precursors, causes, consequences—the choanoflagellate/sponge transition, neurogenesis and the Cambrian explosion. Philosophical Transactions of the Royal Society B: Biological Sciences. 2017; 372
- Cavalier-Smith T. Vendozoa and selective forces on animal origin and early diversification: reply to Dufour and McIlroy (2017). Philosophical Transactions of the Royal Society B: Biological Sciences. 2018; 373DOI
- Celis JS, Wibberg D, Ramírez-Portilla C, Rupp O, Sczyrba A, Winkler A, Kalinowski J, Wilke T. Binning enables efficient host genome reconstruction in cnidarian holobionts. GigaScience. 2018; 7DOI | PubMed
- Cunningham JA, Liu AG, Bengtson S, Donoghue PCJ. The origin of animals: can molecular clocks and the fossil record be reconciled?. BioEssays. 2017; 39DOI
- Dayraud C, Alié A, Jager M, Chang P, Le Guyader H, Manuel M, Quéinnec E. Independent specialisation of myosin II paralogues in muscle vs. non-muscle functions during early animal evolution: a ctenophore perspective. BMC Evolutionary Biology. 2012; 12DOI | PubMed
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15-21. DOI | PubMed
- Dohrmann M, Wörheide G. Dating early animal evolution using phylogenomic data. Scientific Reports. 2017; 7DOI | PubMed
- dos Reis M, Thawornwattana Y, Angelis K, Telford MJ, Donoghue PC, Yang Z. Uncertainty in the timing of origin of animals and the limits of precision in molecular timescales. Current Biology. 2015; 25:2939-2950. DOI | PubMed
- DuBuc T, Bobkov Y, Ryan JF, Martindale M. The radial expression of dorsal-ventral patterning genes in Placozoans, Trichoplax adhaerens. argues for an oral-aboral Axis.. bioRxiv. 2018. DOI
- Dufour SC, McIlroy D. An ediacaran pre-placozoan alternative to the pre-sponge route towards the cambrian explosion of animal life: a comment on Cavalier-Smith 2017. Philosophical Transactions of the Royal Society B: Biological Sciences. 2018; 373DOI
- Dunn CW, Hejnol A, Matus DQ, Pang K, Browne WE, Smith SA, Seaver E, Rouse GW, Obst M, Edgecombe GD, Sørensen MV, Haddock SH, Schmidt-Rhaesa A, Okusu A, Kristensen RM, Wheeler WC, Martindale MQ, Giribet G. Broad phylogenomic sampling improves resolution of the animal tree of life. Nature. 2008; 452:745-749. DOI | PubMed
- Dunn CW, Leys SP, Haddock SH. The hidden biology of sponges and ctenophores. Trends in Ecology & Evolution. 2015; 30:282-291. DOI | PubMed
- Eitel M, Schierwater B. The phylogeography of the placozoa suggests a taxon-rich phylum in tropical and subtropical waters. Molecular Ecology. 2010; 19:2315-2327. DOI | PubMed
- Eitel M, Guidi L, Hadrys H, Balsamo M, Schierwater B. New insights into placozoan sexual reproduction and development. PLoS ONE. 2011; 6DOI | PubMed
- Eitel M, Osigus HJ, DeSalle R, Schierwater B. Global diversity of the placozoa. PLoS ONE. 2013; 8DOI | PubMed
- Eitel M. A taxogenomics approach uncovers a new genus in the phylum placozoa. bioRxiv. 2017. DOI
- Eitel M, Francis WR, Varoqueaux F, Daraspe J, Osigus HJ, Krebs S, Vargas S, Blum H, Williams GA, Schierwater B, Wörheide G. Comparative genomics and the nature of placozoan species. PLOS Biology. 2018; 16DOI | PubMed
- Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biology. 2015; 16DOI | PubMed
- Erwin DH, Laflamme M, Tweedt SM, Sperling EA, Pisani D, Peterson KJ. The cambrian conundrum: early divergence and later ecological success in the early history of animals. Science. 2011; 334:1091-1097. DOI | PubMed
- Evans SD, Droser ML, Gehling JG. Highly regulated growth and development of the Ediacara macrofossil Dickinsonia costata. PLOS ONE. 2017; 12DOI | PubMed
- Feuda R, Dohrmann M, Pett W, Philippe H, Rota-Stabelli O, Lartillot N, Wörheide G, Pisani D. Improved modeling of compositional heterogeneity supports sponges as sister to all other animals. Current Biology. 2017; 27:3864-3870. DOI | PubMed
- Fidler AL, Darris CE, Chetyrkin SV, Pedchenko VK, Boudko SP, Brown KL, Gray Jerome W, Hudson JK, Rokas A, Hudson BG. Collagen IV and basement membrane at the evolutionary dawn of metazoan tissues. eLife. 2017; 6DOI | PubMed
- Foster PG. Modeling compositional heterogeneity. Systematic Biology. 2004; 53:485-495. DOI | PubMed
- Gatesy J, Baker RH. Hidden likelihood support in genomic data: can forty-five wrongs make a right?. Systematic Biology. 2005; 54:483-492. DOI | PubMed
- Genikhovich G, Technau U. On the evolution of bilaterality. Development. 2017; 144:3392-3404. DOI | PubMed
- Grell KG, Benwitz B. Die Ultrastruktur Von Trichoplax adhaerens F. E. Schultze. Cytobiologie. 1971; 4:216-240.
- Grell KG. Eibildung und Furchung Von Trichoplax adhaerens F. E. Schulze (Placozoa). Zeitschrift F R Morphologie Der Tiere. 1972; 73:297-314. DOI
- Grell KG, Ruthmann A. Placozoa. in Microscopic Anatomy of Invertebrates. Book Depository; 1991.
- Gurevich A, Saveliev V, Vyahhi N, Tesler G. QUAST: quality assessment tool for genome assemblies. Bioinformatics. 2013; 29:1072-1075. DOI | PubMed
- Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, MacManes MD, Ott M, Orvis J, Pochet N, Strozzi F, Weeks N, Westerman R, William T, Dewey CN, Henschel R, LeDuc RD, Friedman N, Regev A. De novo transcript sequence reconstruction from RNA-seq using the trinity platform for reference generation and analysis. Nature Protocols. 2013; 8:1494-1512. DOI | PubMed
- Hejnol A, Martín-Durán JM. Getting to the bottom of anal evolution. Zoologischer Anzeiger – a Journal of Comparative Zoology. 2015; 256:61-74. DOI
- Hejnol A, Martindale MQ. Acoel development indicates the independent evolution of the bilaterian mouth and anus. Nature. 2008; 456:382-386. DOI | PubMed
- Hejnol A, Obst M, Stamatakis A, Ott M, Rouse GW, Edgecombe GD, Martinez P, Baguna J, Bailly X, Jondelius U, Wiens M, Muller WEG, Seaver E, Wheeler WC, Martindale MQ, Giribet G, Dunn CW. Assessing the root of bilaterian animals with scalable phylogenomic methods. Proceedings of the Royal Society B: Biological Sciences. 2009; 276:4261-4270. DOI
- Hoang DT, Chernomor O, von Haeseler A, Minh BQ, Vinh LS. UFBoot2: improving the ultrafast bootstrap approximation. Molecular Biology and Evolution. 2018; 35:518-522. DOI | PubMed
- Hoff KJ, Lange S, Lomsadze A, Borodovsky M, Stanke M. BRAKER1: unsupervised RNA-Seq-Based genome annotation with GeneMark-ET and AUGUSTUS. Bioinformatics. 2016; 32:767-769. DOI | PubMed
- Jiang J. A hybrid de novo assembly of the sea pansy (Renilla muelleri) Genome. bioRxiv. 2018. DOI
- Kamm K, Osigus HJ, Stadler PF, DeSalle R, Schierwater B. Trichoplax genomes reveal profound admixture and suggest stable wild populations without bisexual reproduction. Scientific Reports. 2018; 8DOI | PubMed
- Kang DD, Froula J, Egan R, Wang Z. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. PeerJ. 2015; 3DOI | PubMed
- Kelava I, Rentzsch F, Technau U. Evolution of eumetazoan nervous systems: insights from cnidarians. Philosophical Transactions of the Royal Society B: Biological Sciences. 2015; 370DOI
- Kim J, Kim W, Cunningham CW. A new perspective on lower metazoan relationships from 18S rDNA sequences. Molecular Biology and Evolution. 1999; 16:423-427. DOI | PubMed
- Kristan WB. Early evolution of neurons. Current Biology. 2016; 26:R949-R954. DOI | PubMed
- Kück P, Struck TH. BaCoCa–a heuristic software tool for the parallel assessment of sequence biases in hundreds of gene and taxon partitions. Molecular Phylogenetics and Evolution. 2014; 70:94-98. DOI | PubMed
- Lartillot N, Philippe H. A bayesian mixture model for across-site heterogeneities in the amino-acid replacement process. Molecular Biology and Evolution. 2004; 21:1095-1109. DOI | PubMed
- Lartillot N, Rodrigue N, Stubbs D, Richer J. PhyloBayes MPI: phylogenetic reconstruction with infinite mixtures of profiles in a parallel environment. Systematic Biology. 2013; 62:611-615. DOI | PubMed
- Laumer CE, Hejnol A, Giribet G. Nuclear genomic signals of the ‘microturbellarian’ roots of platyhelminth evolutionary innovation. eLife. 2015; 4DOI
- Laumer CE. Inferring ancient relationships with genomic data: a commentary on current practices. Integrative and Comparative Biology. 2018; 58:623-639. DOI | PubMed
- Leclère L. The genome of the jellyfish Clytia hemisphaerica and the evolution of the cnidarian life-cycle. bioRxiv. 2018. DOI
- Leys SP, Riesgo A. Epithelia, an evolutionary novelty of metazoans. Journal of Experimental Zoology Part B: Molecular and Developmental Evolution. 2012; 318:438-447. DOI
- Liebeskind BJ, Hofmann HA, Hillis DM, Zakon HH. Evolution of animal neural systems. Annual Review of Ecology, Evolution, and Systematics. 2017; 48:377-398. DOI
- Martindale MQ, Hejnol A. A developmental perspective: changes in the position of the blastopore during bilaterian evolution. Developmental Cell. 2009; 17:162-174. DOI | PubMed
- McFall-Ngai M, Hadfield MG, Bosch TCG, Carey HV, Domazet-Lošo T, Douglas AE, Dubilier N, Eberl G, Fukami T, Gilbert SF, Hentschel U, King N, Kjelleberg S, Knoll AH, Kremer N, Mazmanian SK, Metcalf JL, Nealson K, Pierce NE, Rawls JF, Reid A, Ruby EG, Rumpho M, Sanders JG, Tautz D, Wernegreen JJ. Animals in a bacterial world, a new imperative for the life sciences. Proceedings of the National Academy of Sciences. 2013; 110:3229-3236. DOI
- Misof B, Meyer B, von Reumont BM, Kück P, Misof K, Meusemann K. Selecting informative subsets of sparse supermatrices increases the chance to find correct trees. BMC Bioinformatics. 2013; 14DOI | PubMed
- Moroz LL, Kocot KM, Citarella MR, Dosung S, Norekian TP, Povolotskaya IS, Grigorenko AP, Dailey C, Berezikov E, Buckley KM, Ptitsyn A, Reshetov D, Mukherjee K, Moroz TP, Bobkova Y, Yu F, Kapitonov VV, Jurka J, Bobkov YV, Swore JJ, Girardo DO, Fodor A, Gusev F, Sanford R, Bruders R, Kittler E, Mills CE, Rast JP, Derelle R, Solovyev VV, Kondrashov FA, Swalla BJ, Sweedler JV, Rogaev EI, Halanych KM, Kohn AB. The ctenophore genome and the evolutionary origins of neural systems. Nature. 2014; 510:109-114. DOI | PubMed
- Moroz LL, Kohn AB. Independent origins of neurons and synapses: insights from ctenophores. Philosophical Transactions of the Royal Society B: Biological Sciences. 2016; 371DOI | PubMed
- Nesnidal MP, Helmkampf M, Bruchhaus I, Hausdorf B. Compositional heterogeneity and phylogenomic inference of metazoan relationships. Molecular Biology and Evolution. 2010; 27:2095-2104. DOI | PubMed
- Nikolenko SI, Korobeynikov AI, Alekseyev MA. BayesHammer: bayesian clustering for error correction in single-cell sequencing. BMC Genomics. 2013; 14DOI | PubMed
- NISC Comparative Sequencing Program, Ryan JF, Pang K, Schnitzler CE, Nguyen AD, Moreland RT, Simmons DK, Koch BJ, Francis WR, Havlak P, Smith SA, Putnam NH, Haddock SH, Dunn CW, Wolfsberg TG, Mullikin JC, Martindale MQ, Baxevanis AD. The genome of the ctenophore Mnemiopsis leidyi and its implications for cell type evolution. Science. 2013; 342DOI | PubMed
- Nosenko T, Schreiber F, Adamska M, Adamski M, Eitel M, Hammel J, Maldonado M, Müller WE, Nickel M, Schierwater B, Vacelet J, Wiens M, Wörheide G. Deep metazoan phylogeny: when different genes tell different stories. Molecular Phylogenetics and Evolution. 2013; 67:223-233. DOI | PubMed
- Pearse VB, Voigt O. Field biology of placozoans (Trichoplax): distribution, diversity, biotic interactions. Integrative and Comparative Biology. 2007; 47:677-692. DOI | PubMed
- Philippe H, Derelle R, Lopez P, Pick K, Borchiellini C, Boury-Esnault N, Vacelet J, Renard E, Houliston E, Quéinnec E, Da Silva C, Wincker P, Le Guyader H, Leys S, Jackson DJ, Schreiber F, Erpenbeck D, Morgenstern B, Wörheide G, Manuel M. Phylogenomics revives traditional views on deep animal relationships. Current Biology. 2009; 19:706-712. DOI | PubMed
- Pick KS, Philippe H, Schreiber F, Erpenbeck D, Jackson DJ, Wrede P, Wiens M, Alié A, Morgenstern B, Manuel M, Wörheide G. Improved phylogenomic taxon sampling noticeably affects nonbilaterian relationships. Molecular Biology and Evolution. 2010; 27:1983-1987. DOI | PubMed
- Pisani D, Pett W, Dohrmann M, Feuda R, Rota-Stabelli O, Philippe H, Lartillot N, Wörheide G. Genomic data do not support comb jellies as the sister group to all other animals. Proceedings of the National Academy of Sciences. 2015; 112:15402-15407. DOI
- Presnell JS, Vandepas LE, Warren KJ, Swalla BJ, Amemiya CT, Browne WE. The presence of a functionally tripartite Through-Gut in ctenophora has implications for metazoan character trait evolution. Current Biology. 2016; 26:2814-2820. DOI | PubMed
- Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS ONE. 2010; 5DOI | PubMed
- Putnam NH, Srivastava M, Hellsten U, Dirks B, Chapman J, Salamov A, Terry A, Shapiro H, Lindquist E, Kapitonov VV, Jurka J, Genikhovich G, Grigoriev IV, Lucas SM, Steele RE, Finnerty JR, Technau U, Martindale MQ, Rokhsar DS. Sea Anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science. 2007; 317:86-94. DOI | PubMed
- Quang leS, Gascuel O, Lartillot N. Empirical profile mixture models for phylogenetic reconstruction. Bioinformatics. 2008; 24:2317-2323. DOI | PubMed
- Rota-Stabelli O, Lartillot N, Philippe H, Pisani D. Serine codon-usage Bias in deep phylogenomics: pancrustacean relationships as a case study. Systematic Biology. 2013; 62:121-133. DOI | PubMed
- Schierwater B. My favorite animal, Trichoplax adhaerens. BioEssays. 2005; 27:1294-1302. DOI | PubMed
- Schulze FE. Trichoplax adhaerens, nov. gen., nov. spec. Zoologischer Anzeiger. 1883; 6:92-97.
- Sebé-Pedrós A. Early metazoan cell type diversity and the evolution of multicellular gene regulation. nat. Ecology and Evolution. 2018a; 2:1176-1188. PubMed
- Sebé-Pedrós A, Saudemont B, Chomsky E, Plessier F, Mailhé MP, Renno J, Loe-Mie Y, Lifshitz A, Mukamel Z, Schmutz S, Novault S, Steinmetz PRH, Spitz F, Tanay A, Marlow H. Cnidarian cell type diversity and regulation revealed by Whole-Organism Single-Cell RNA-Seq. Cell. 2018b; 173:1520-1534. DOI | PubMed
- Senatore A, Reese TS, Smith CL. Neuropeptidergic integration of behavior in Trichoplax adhaerens, an animal without synapses. The Journal of Experimental Biology. 2017; 220:3381-3390. DOI | PubMed
- Shen XX, Salichos L, Rokas A. A Genome-Scale investigation of how sequence, function, and Tree-Based gene properties influence phylogenetic inference. Genome Biology and Evolution. 2016; 8:2565-2580. DOI | PubMed
- Shen X-X, Hittinger CT, Rokas A. Contentious relationships in phylogenomic studies can be driven by a handful of genes. Nature Ecology & Evolution. 2017; 1DOI
- Signorovitch AY, Buss LW, Dellaporta SL. Comparative genomics of large mitochondria in placozoans. PLoS Genetics. 2007; 3DOI | PubMed
- Silva FBda, Muschner VC, Bonatto SL. Phylogenetic position of placozoa based on large subunit (LSU) and small subunit (SSU) rRNA genes. Genetics and Molecular Biology. 2007; 30:127-132. DOI
- Simão FA, Waterhouse RM, Ioannidis P, Kriventseva EV, Zdobnov EM. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics. 2015; 31:3210-3212. DOI | PubMed
- Simion P, Philippe H, Baurain D, Jager M, Richter DJ, Di Franco A, Roure B, Satoh N, Quéinnec É, Ereskovsky A, Lapébie P, Corre E, Delsuc F, King N, Wörheide G, Manuel M. A large and consistent phylogenomic dataset supports sponges as the sister group to all other animals. Current Biology. 2017; 27:958-967. DOI | PubMed
- Smith CL, Varoqueaux F, Kittelmann M, Azzam RN, Cooper B, Winters CA, Eitel M, Fasshauer D, Reese TS. Novel cell types, neurosecretory cells, and body plan of the early-diverging metazoan Trichoplax adhaerens. Current Biology. 2014; 24:1565-1572. DOI | PubMed
- Smith CL, Pivovarova N, Reese TS. Coordinated feeding behavior in Trichoplax, an animal without synapses. Plos One. 2015; 10DOI | PubMed
- Sperling EA, Vinther J. A placozoan affinity for dickinsonia and the evolution of late Proterozoic metazoan feeding modes. Evolution & Development. 2010; 12:201-209. DOI | PubMed
- Srivastava M, Begovic E, Chapman J, Putnam NH, Hellsten U, Kawashima T, Kuo A, Mitros T, Salamov A, Carpenter ML, Signorovitch AY, Moreno MA, Kamm K, Grimwood J, Schmutz J, Shapiro H, Grigoriev IV, Buss LW, Schierwater B, Dellaporta SL, Rokhsar DS. The Trichoplax genome and the nature of placozoans. Nature. 2008; 454:955-960. DOI | PubMed
- Steinmetz PR, Kraus JE, Larroux C, Hammel JU, Amon-Hassenzahl A, Houliston E, Wörheide G, Nickel M, Degnan BM, Technau U. Independent evolution of striated muscles in cnidarians and bilaterians. Nature. 2012; 487:231-234. DOI | PubMed
- Steinmetz PRH, Aman A, Kraus JEM, Technau U. Gut-like ectodermal tissue in a sea Anemone challenges germ layer homology. Nature Ecology & Evolution. 2017; 1:1535-1542. DOI | PubMed
- Strous M, Kraft B, Bisdorf R, Tegetmeyer HE. The binning of metagenomic contigs for microbial physiology of mixed cultures. Frontiers in Microbiology. 2012; 3DOI | PubMed
- Susko E, Roger AJ. On reduced amino acid alphabets for phylogenetic inference. Molecular Biology and Evolution. 2007; 24:2139-2150. DOI | PubMed
- Syed T, Schierwater B. Trichoplax adhaerens: discovered as a missing link, forgotten as a Hydrozoan, re-discovered as a key to metazoan evolution. Vie Et Milieu. 2002; 52:177-187.
- Ter-Hovhannisyan V, Lomsadze A, Chernoff YO, Borodovsky M. Gene prediction in novel fungal genomes using an ab initio algorithm with unsupervised training. Genome Research. 2008; 18:1979-1990. DOI | PubMed
- Varoqueaux F. High cell diversity and complex peptidergic signalling underlie placozoan behaviour. bioRxiv. 2018. DOI
- Wallberg A, Thollesson M, Farris JS, Jondelius U. The phylogenetic position of the comb jellies (Ctenophora) and the importance of taxonomic sampling. Cladistics. 2004; 20:558-578. DOI
- Wang HC, Minh BQ, Susko E, Roger AJ. Modeling site heterogeneity with posterior mean site frequency profiles accelerates accurate phylogenomic estimation. Systematic Biology. 2018; 67:216-235. DOI | PubMed
- Wenderoth H. Transepithelial cytophagy by Trichoplax adhaerens FE. Schulze (Placozoa) Feeding on Yeast.. Z. Für Naturforschung. 1986; 41:343-347. DOI
- Whelan NV, Kocot KM, Moroz LL, Halanych KM. Error, signal, and the placement of ctenophora sister to all other animals. PNAS. 2015; 112:5773-5778. DOI | PubMed
- Whelan NV, Kocot KM, Moroz TP, Mukherjee K, Williams P, Paulay G, Moroz LL, Halanych KM. Ctenophore relationships and their placement as the sister group to all other animals. Nature Ecology & Evolution. 2017; 1:1737-1746. DOI | PubMed
- Wu M, Chatterji S, Eisen JA. Accounting for alignment uncertainty in phylogenomics. PLoS One. 2012; 7DOI | PubMed
- Yang Y, Smith SA. Orthology inference in nonmodel organisms using transcriptomes and low-coverage genomes: improving accuracy and matrix occupancy for phylogenomics. Molecular Biology and Evolution. 2014; 31:3081-3092. DOI | PubMed
- Zhou X. Quartet-based computations of internode certainty provide accurate and robust measures of phylogenetic incongruence. bioRxiv. 2017. DOI
Fonte
Laumer CE, Gruber-Vodicka H, Hadfield MG, Pearse VB, Riesgo A, et al. () Support for a clade of Placozoa and Cnidaria in genes with minimal compositional bias. eLife 7e36278. https://doi.org/10.7554/eLife.36278




