
Abstract
Introduzione
Un paradigma emergente in biologia è che gli eventi nella tradizionale cultura cellulare bidimensionale spesso differiscono da quelli della cultura tridimensionale (3-D)(Benam et al., 2015; Pampaloni et al., 2007). Inoltre, la matrice extracellulare regola la biologia cellulare(Yamada e Cukierman ,2007; Schwartz e Chen, 2013; Parker et al., 2014) e la biologia delle infezioni differisce tra i sistemi 2-D e 3-D (Cheng et al., 2011; Barrila et al., 2010). La malattia umana si verifica in 3-D e nel contesto della matrice extracellulare. Di conseguenza, le conclusioni tratte dai sistemi di coltura cellulare bidimensionale possono non riflettere pienamente gli eventi in vivo(Yamada e Cukierman, 2007). Ciò rappresenta una sfida per il progresso dai sistemi di coltura standard, dove le cellule sono cresciute in 2-D su plastica, a sistemi più avanzati che replicano più fedelmente gli eventi nell’uomo(Yamada e Cukierman, 2007). Queste difficoltà tecniche sono particolarmente marcate nello studio delle malattie infettive, dove gli esperimenti devono avere ulteriori livelli di contenimento per prevenire il rilascio di agenti patogeni(Barrila et al., 2010).
Ilmicobatterio tubercolosi (Mtb) è un agente patogeno di rilevanza mondiale che continua a uccidere 1,5 milioni di persone all’anno(O’Garra et al., 2013; Horsburgh et al., 2015). Purtroppo, nonostante i notevoli investimenti nella ricerca, i recenti studi clinici e gli studi sui vaccini per ridurre il carico globale della tubercolosi (TB) non hanno avuto successo(Tameris et al., 2013; Ndiaye et al., 2015; Warner e Mizrahi, 2014), indicando che i sistemi modello che hanno informato questi studi richiedono un ulteriore perfezionamento. Nella TBC, l’interazione ospite-patogeno è molto complessa, con la risposta immunitaria necessaria per il contenimento dell’infezione, ma paradossalmente anche la guida immunopatologia che porta alla distruzione e alla trasmissione polmonare(Russell, 2011; Elkington e Friedland, 2015). Il topo è il sistema modello principale per studiare la TBC, ma le condizioni infiammatorie nel topo sono diverse da quelle dell’uomo(Seok et al., 2013), e la patologia polmonare è diversa nell’infezione murina da Mtb(Young, 2009). La Mtb è un patogeno umano obbligato e ha un’interazione molto prolungata con le cellule ospiti, sopravvivendo all’interno di fagociti professionali(Russell, 2011). Pertanto, sono necessari esperimenti di coltura umana a lungo termine per indagare la patogenesi. Un vantaggio specifico della coltura cellulare 3-D che incorpora matrice extracellulare è che la sopravvivenza cellulare è molto prolungata(Buchheit et al., 2012; Mueller-Klieser, 1997). Inoltre, la segnalazione infiammatoria nei granulomi della TBC è organizzata spazialmente(Marakalala et al., 2016), con microambienti specifici(Mattila et al., 2013), e la matrice extracellulare regola la sopravvivenza cellulare nella TBC(Al Shammari et al., 2015), indicando che un sistema ottimale per studiare la malattia umana dovrà essere 3-D con matrice extracellulare.
Abbiamo ipotizzato che per comprendere appieno l’interazione ospite-patogeno nella TBC, è necessario un sistema di coltura cellulare 3-D che incorpori cellule umane primarie, matrice extracellulare, Mtb completamente virulenta, e letture longitudinali multiparametriche. Mentre i modelli cellulari umani di formazione del granuloma umano sono stati sviluppati, nessuno ha tutte queste caratteristiche(Puissegur et al., 2004; Lay et al., 2007; Kapoor et al., 2013; Parasa et al., 2014). Abbiamo affrontato le sfide tecniche dell’esecuzione di questi esperimenti al terzo livello di contenimento della biosicurezza adottando un approccio di bioingegneria(Workman et al., 2014). Abbiamo sviluppato un sistema modello che permette di interrogare l’interazione ospite-patogeno in 3-D nel contesto della matrice extracellulare. Dimostriamo che le caratteristiche cardinali della malattia umana si sviluppano e che la risposta immunitaria dell’ospite è significativamente diversa quando le cellule sono aderenti al collagene, favorendo l’ospite rispetto all’agente patogeno. Studiamo gli approcci terapeutici emergenti nel sistema e dimostriamo che ogni intervento ha effetti sia benefici che probabilmente dannosi. Il modello permette l’analisi simultanea di molteplici esiti e quindi può essere utilizzato per sviluppare approcci ottimali per affrontare la pandemia di TBC, e può essere applicato a diverse malattie infettive, infiammatorie e neoplastiche.
Risultati
Le caratteristiche principali della tubercolosi umana si sviluppano nel modello bio-elettrospray
Per affrontare le sfide dello studio dell’infezione delle cellule umane primarie con un agente patogeno virulento all’interno di una matrice extracellulare 3-D, abbiamo ottimizzato i parametri bio-elettrospray per la generazione di microsfere stabili. I PBMC sono stati isolati da donatori sani, contati e poi infettati con Mtb che era stato coltivato in brodo di Middlebrook 7H9 a una molteplicità di infezione di 0,1. Dopo l’infezione durante la notte, le cellule sono state staccate, risospese e pellettate per centrifugazione, e poi risospese in alginato o alginato-collagene matrice prima di bioelettrospray in microsfere utilizzando un incapsulatore di cellule Nisco(Video 1, Figura 1-figure supplemento 1 e 2). La caratterizzazione di diversi alginati indicava che l’alginato ultrapuro a media viscosità guluronato (MVG)-dominante ha proprietà biofisiche ottimali per l’elettrospray e la minima immunogenicità(Figura 1-figure supplement 3).Video 1.Generation of microspheres.Durante il processo di bio-elettrospray, una telecamera Phantom v7 ad alta velocità, in grado di catturare 150000 fps in combinazione con una lente per microscopio a lunga distanza, è stata attivata simultaneamente con un sistema di illuminazione a fibre ottiche. Velocità video relativa 0,15 s.DOI:http://dx.doi.org/10.7554/eLife.21283.00210.7554/eLife.21283.002
Subito dopo la generazione, le cellule sono distribuite uniformemente all’interno di microsfere e di giorno iniziano a formarsi sette aggregati cellulari(Figura 1A e B). La quantità ha dimostrato un numero significativamente maggiore di aggregati nelle microsfere infette rispetto alle microsfere unificate(Figura 1C). Un giorno dopo l’infezione, un quarto dei monociti aveva fagocitato Mtb, analizzato mediante l’analisi citometrica a flusso delle cellule infettate con GFP-espressione di Mtb(Figura 1D). Dopo 14 giorni di incubazione, si osservano grandi aggregati cellulari(Figura 1E). La sopravvivenza delle cellule infette da Mtb in microsfere di collagene in fusione 3-D è stata molto maggiore che in coltura 2-D, come analizzato dal rilascio di LDH(Figura 1F) e dai saggi di citotossicità cellulare(Figura 1G). Un vantaggio del modello è che le cellule possono essere rilasciate dalla microsfera per decapsulazione mediante chelazione cationica divalente con EDTA e citrato di sodio in HBSS per 10 minuti, che provoca la dissoluzione delle sfere e rilascia le cellule per i saggi a valle. Decapsulazione non ha influenzato in modo significativo la vitalità cellulare, con oltre il 90% di vitalità cellulare dopo la decapsulazione(Figura 1-figure supplemento 4). I monociti maturano in macrofagi nelle microsfere infette, con una maggiore espressione di CD68(Figura 1H). All’interno degli aggregati, si sviluppano cellule giganti multinucleate tipiche della TBC umana, colorate per CD68 dalla immunoistochimica(Figura 1I). Queste cellule giganti multinucleate sono simili a quelle che si verificano nei pazienti umani con TB polmonare(Figura 1J). La differenziazione delle cellule T avviene all’interno di microsfere, con un progressivo aumento della proporzione di cellule T CD4+, mentre la percentuale di cellule T CD8+ diminuisce(Figura 1-figure supplement 5). La proliferazione delle cellule T non differisce tra le microsfere infette da Mtb e non infette.10.7554/eLife.21283.003Figure 1.Primary cellule umane hanno una maggiore sopravvivenza in 3-D e aggregato, differenziare e fondere in cellule giganti multinucleate.(A) Microscopia a contrasto di fase con sovrapposizione di Hoeschst 33256 (blu) al settimo giorno dimostra PBMCs formando aggregati all’interno di microsfere. Scala 300 µm.(B) Aggregazione cellulare in Mtb-infetto PBMC-collagene-collagene-algama microsfere al giorno 7. Scala 50 µm.(C) L’aggregazione cellulare è maggiore nelle microsfere infettate con Mtb rispetto alle microsfere non infettate. Aggregati di cellule sono stati definiti come otto o più cellule viste sotto 20x ingrandimento. I dati sono rappresentativi di un minimo di 10 campi di vista per gruppo.(D) Le cellule sono state infettate con GFP+ Mtb e poi rilasciate per decapsulazione. A 24 ore dopo l’infezione, il 24,2% dei monociti aveva fagocitato GFP-espressione Mtb fagocitante mediante analisi citometrica a flusso.(E) Ematossilina e colorazione eosina di microsfere fissate con paraffina dimostra aggregati cellulari in microsfere infette da Mtb al giorno 14. Scala 20 µm.(F) La sopravvivenza delle cellule ospiti è significativamente maggiore nelle microsfere 3-D rispetto alla coltura cellulare 2-D, come dimostrato dal saggio LDH. Clear box 2-D coltura cellulare 2-D, riempito box 3-D cultura; un numero uguale di cellule uccise con digitonina (30 µg/ml) nella rispettiva cultura 3D e 2D è stato utilizzato come denominatore. Media ± valori SE (n = 4).(G) La citotossicità misurata dal saggio CytoTox Glo è significativamente inferiore nella cultura 3D rispetto alla cultura 2D.(H) L’espressione CD68 è aumentata nei macrofagi nelle microsfere infette da Mtb analizzate con la citometria a flusso. Controllo dell’isotipo nero, cellule blu non infette, cellule rosse infette da Mtb.(I) Le cellule giganti multinucleate si formano all’interno delle microsfere al 14° giorno, immunocolorizzate con CD68 (marrone) e contro-colorate con ematossilina (blu). Scala 20 µm.(J) Nei pazienti con TB polmonare, cellule giganti simili sono osservate nei granulomi polmonari. Un’immagine a bassa potenza del granuloma polmonare umano (G), con numerose cellule giganti multinucleate che circondano il centro casoso (scatola, area ingrandita). Barra di scala: 1000 μm.DOI:http://dx.doi.org/10.7554/eLife.21283.00310.7554/eLife.21283.004Figuresupplemento a 1 cifra 1.Equipment set-up all’interno del laboratorio di tubercolosi livello di contenimento tre. (A) Bioelectrosprayer alloggiato all’interno di un doppio formato di classe I/III Microbiological Cabinet di sicurezza.(B) Close-up del bioelectrosprayer pulito dopo un esperimento, con un driver della siringa sulla parte superiore. Le porte esterne devono essere chiuse per l’inizio della generazione di microsfere, fornendo il primo livello di contenimento, con il flusso d’aria della classe I MSC che fornisce il secondo livello di contenimento. L’armadio viene formalizzato dopo ogni esperimento.DOI:http://dx.doi.org/10.7554/eLife.21283.00410.7554/eLife.21283.005Figure1-figure supplemento supplemento 2.Microsfere posto in una piastra di coltura di tessuto 12 pozzo immediatamente dopo la generazione dimostrando l’aspetto non magnetizzato.DOI:http://dx.doi.org/10.7554/eLife.21283.00510.7554/eLife.21283.006Figure1-figure supplement 3.Medium viscosity guluronate (MVG) alginato è l’alginato ottimale per gli studi immunologici. (A)IL-8 secrezione da microsfere composto da diversi alginati misurati da ELISA. MVM: mannuronato a media viscosità, LVG: guluronato a bassa viscosità. Inoltre, LVG aveva proprietà biofisiche molto scarse per il biolelettrospray.(B) La vitalità cellulare analizzata dal rilascio di LDH non era diversa tra i diversi tipi di alginato.(C) Nell’alginato sterile, la secrezione di IL-8 era più alta nelle microsfere contenenti SLG rispetto alla SLM. SLG: guluronato sterile, SLM: mannuronato sterile. Quadrati, linee riempite: Microsfere infette da Mtb, cerchi e linee spezzate: microsfere non infette.DOI:http://dx.doi.org/10.7554/eLife.21283.00610.7554/eLife.21283.007FigureSupplemento a 1 cifra 4.La vitalità delle PBMC rimane oltre il 90% dopo la decapsulazione.Le PBMC sono state incapsulate in microsfere di alginato e collagene. Dopo 1 ora di incubazione a 37 ° C in HBSS, sono stati decapsulati in soluzione di 55 mM di citrato di sodio e 5 mM EDTA preparato in HBSS (pH = 7,4) per 10 min. Le cellule sono state poi lavate con HBSS due volte prima della sospensione in 5 ml di RPMI e la colorazione con ioduro di propidio (1 mg / ml) a 1:100 diluizione e analisi per citometria a flusso. Per confronto, è stata utilizzata la vitalità delle PBMC tenute in RPMI a 4°C durante questo periodo. I dati sono un rappresentante di due esperimenti indipendenti.DOI:http://dx.doi.org/10.7554/eLife.21283.00710.7554/eLife.21283.008Figure1-figure supplemento 5.T composizione cellulare 5.T di microsfere. La proporzione di cellule T CD4 + aumenta tra il giorno 0 e il giorno sette in entrambe le microsfere non infette e infette (A), mentre la proporzione di cellule T CD8 + scende (B). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 rispetto al giorno 0.DOI:http://dx.doi.org/10.7554/eLife.21283.008
Successivamente, abbiamo misurato la crescita di Mtb all’interno di microsfere longitudinalmente infettando le cellule con Mtb luminescenti che esprimono l’operone Lux, che è geneticamente modificato per luminescenziare costantemente(Andreu et al., 2010). Mtb in microsfere senza cellule umane cresce relativamente lentamente, mentre in presenza di PBMCs Mtb prolifera per 24 giorni, raggiungendo la stessa luminescenza della crescita in brodo di Middlebrook 7H9 (Figura 2A). Le proteasi implicate nella patogenesi della TB sono upregolate, con l’espressione del gene MMP-1 aumentata all’interno delle sfere 4 giorni dopo l’infezione(Figura 2B) e l’accumulo di MMP-9 nei media che circondano le sfere che raggiungono il picco al giorno 7(Figura 2C). Questa attività della proteasi ha un effetto funzionale, causando una maggiore degradazione del collagene marcato fluorescente all’interno di microsfere(Figura 2D). Infezione Mtb anche upregulated espressione genica di IFN-γ (Figura 2E) e guida la secrezione di citochine pro-infiammatorie multiple, tra cui IL-1β, IL-12, GM-CSF, IP-10 e MCP-1 analizzati da Luminex multiplex array (Figura 2Fe Figura 2-figure supplement 1), dimostrando che citochine simili upregulated nella malattia umana sono espressi all’interno delle microsfere.10.7554/eLife.21283.009Figure 2.Mtb cresce all’interno di microsfere contenenti PBMC e upregola l’espressione di MMP e citochine.(A) Mtb prolifera lentamente nelle microsfere senza cellule (linea verde), ma progressivamente nelle microsfere contenenti PBMC (linea rossa), raggiungendo una luminescenza simile alla coltura di brodo Middlebrook 7H9 a 24 giorni (linea nera). Linea blu, microsfere non infette.(B) L’infezione da Mtb upregola l’espressione del gene MMP-1 e(C) la secrezione di MMP-9 nelle microsfere.(D) MMP upregulation ha un effetto funzionale, causando la degradazione del collagene. DQ La degradazione del collagene è più alta nelle microsfere infette da Mtb (linea rossa) che non infette (linea blu). Triangoli, microsfere senza PBMC.(E) L’infezione da Mtb aumenta l’accumulo cellulare di mRNA IFN-γ mRNA rispetto alle cellule non infette al quarto giorno nelle microsfere (n = 4).(F) La secrezione di citochine da microsfere infette da Mtb (quadrati) è significativamente più alta che nelle microsfere contenenti PBMC non infette (cerchi). ****p<0.0001 mediante test t (B ed E) e ANOVA (A,C, D, F).DOI:http://dx.doi.org/10.7554/eLife.21283.00910.7554/eLife.21283.010Figuresupplemento a 2 cifre 1.Mtb infezione 1.Mtb upregulates secrezione di fattori di crescita multipli, citochine e chemochine da microsfere misurata da Luminex array.*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 rispetto alle microsfere non infette in ogni punto di tempo.DOI:http://dx.doi.org/10.7554/eLife.21283.010

Video 1.1. Generazione di microsfere.Durante il processo di bio-elettrospray, una telecamera Phantom v7 ad alta velocità, in grado di catturare 150000 fps in combinazione con un obiettivo per microscopio a lunga distanza, è stata attivata simultaneamente con un sistema di illuminazione a fibre ottiche. Velocità video relativa 0,15 s.DOI:
http://dx.doi.org/10.7554/eLife.21283.002
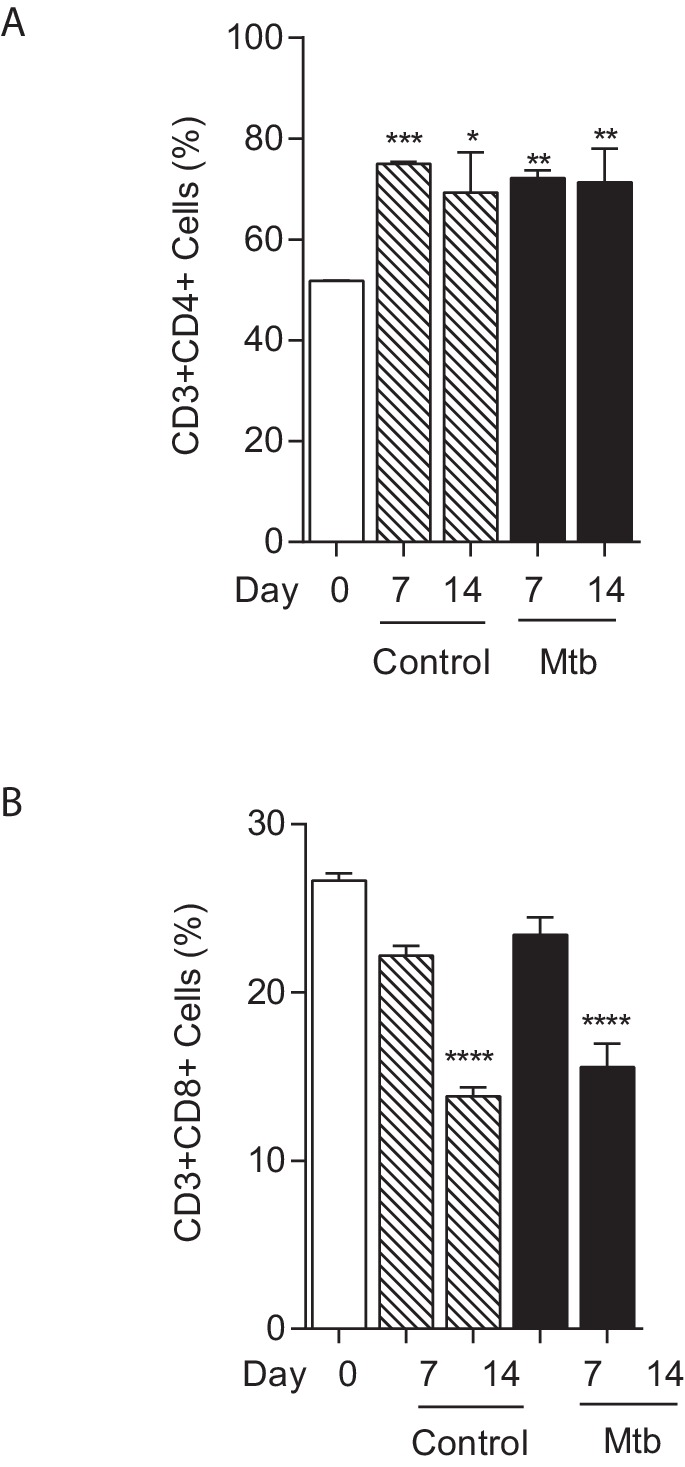
Figura 1-figura supplemento 5.Le cellule umane primarie hanno una maggiore sopravvivenza in 3-D e aggregati, differenziano e si fondono in cellule giganti multinucleate.apparecchiature set-up all’interno del laboratorio di contenimento livello tre tubercolosi.microsfere posto in una piastra di coltura dei tessuti 12 pozzo immediatamente dopo la generazione dimostrando l’aspetto non magnetizzato.alginato guluronato media viscosità (MVG) è l’alginato ottimale per gli studi immunologici.vitalità di PBMC rimane oltre il 90% dopo la decapsulazione.composizione cellulare T di microsfere.(A) La microscopia a contrasto di fase con sovrapposizione di Hoeschst 33256 (blu) al settimo giorno dimostra che le PBMC formano aggregati all’interno di microsfere. Scala 300 µm.(B) Aggregazione cellulare in Mtb-infetto PBMC-collagene-collagene-algama microsfere al giorno 7. Scala 50 µm.(C) L’aggregazione cellulare è maggiore nelle microsfere infettate con Mtb rispetto alle microsfere non infettate. Aggregati di cellule sono stati definiti come otto o più cellule viste sotto 20x ingrandimento. I dati sono rappresentativi di un minimo di 10 campi di vista per gruppo.(D) Le cellule sono state infettate con GFP+ Mtb e poi rilasciate per decapsulazione. A 24 ore dopo l’infezione, il 24,2% dei monociti aveva fagocitato GFP-espressione Mtb fagocitante mediante analisi citometrica a flusso.(E) Ematossilina e colorazione eosina di microsfere fissate con paraffina dimostra aggregati cellulari in microsfere infette da Mtb al giorno 14. Scala 20 µm.(F) La sopravvivenza delle cellule ospiti è significativamente maggiore nelle microsfere 3-D rispetto alla coltura cellulare 2-D, come dimostrato dal saggio LDH. Clear box 2-D coltura cellulare 2-D, riempito box 3-D cultura; un numero uguale di cellule uccise con digitonina (30 µg/ml) nella rispettiva cultura 3D e 2D è stato utilizzato come denominatore. Media ± valori SE (n = 4).(G) La citotossicità misurata dal saggio CytoTox Glo è significativamente inferiore nella cultura 3D rispetto alla cultura 2D.(H) L’espressione CD68 è aumentata nei macrofagi nelle microsfere infette da Mtb analizzate con la citometria a flusso. Controllo dell’isotipo nero, cellule blu non infette, cellule rosse infette da Mtb.(I) Le cellule giganti multinucleate si formano all’interno delle microsfere al 14° giorno, immunocolorizzate con CD68 (marrone) e contro-colorate con ematossilina (blu). Scala 20 µm.(J) Nei pazienti con TB polmonare, cellule giganti simili sono osservate nei granulomi polmonari. Un’immagine a bassa potenza del granuloma polmonare umano (G), con numerose cellule giganti multinucleate che circondano il centro casoso (scatola, area ingrandita). Barra di scala: 1000 μm.DOI:
http://dx.doi.org/10.7554/eLife.21283.003(A) Bioelectrosprayer alloggiato all’interno di un doppio armadio di sicurezza microbiologica di classe I/III.(B) Close-up del bioelectrosprayer pulito dopo un esperimento, con un driver della siringa sulla parte superiore. Le porte esterne devono essere chiuse per l’inizio della generazione di microsfere, fornendo il primo livello di contenimento, con il flusso d’aria della classe I MSC che fornisce il secondo livello di contenimento. L’armadio viene formalizzato dopo ogni esperimento.DOI:
http://dx.doi.org/10.7554/eLife.21283.004DOI:
http://dx.doi.org/10.7554/eLife.21283.005(A) Secrezione IL-8 da microsfere composte da diversi alginati misurati con ELISA. MVM: mannuronato a media viscosità, LVG: guluronato a bassa viscosità. Inoltre, LVG aveva proprietà biofisiche molto scarse per il biolelettrospray.(B) La vitalità cellulare analizzata dal rilascio di LDH non era diversa tra i diversi tipi di alginato.(C) Nell’alginato sterile, la secrezione di IL-8 era più alta nelle microsfere contenenti SLG rispetto alla SLM. SLG: guluronato sterile, SLM: mannuronato sterile. Quadrati, linee riempite: Microsfere infette da Mtb, cerchi e linee spezzate: microsfere non infette.DOI:
http://dx.doi.org/10.7554/eLife.21283.006Le PBMC sono state incapsulate in microsfere di alginato e collagene. Dopo 1 ora di incubazione a 37°C in HBSS, sono state decapulate in soluzione di 55 mM di citrato di sodio e 5 mM di EDTA preparato in HBSS (pH = 7,4) per 10 min. Le cellule sono state poi lavate con HBSS due volte prima della sospensione in 5 ml di RPMI e la colorazione con ioduro di propidio (1 mg / ml) a 1:100 diluizione e analisi per citometria a flusso. Per confronto, è stata utilizzata la vitalità delle PBMC tenute in RPMI a 4°C durante questo periodo. I dati sono un rappresentante di due esperimenti indipendenti.DOI:
http://dx.doi.org/10.7554/eLife.21283.007La proporzione di cellule T CD4+ aumenta tra il giorno 0 e il settimo giorno sia nelle microsfere non infette che in quelle infette(A), mentre la proporzione di cellule T CD8+ diminuisce(B). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 rispetto al giorno 0.DOI:
http://dx.doi.org/10.7554/eLife.21283.008

Figura 1-figure supplement 1.1. Attrezzatura allestita all’interno del laboratorio per la tubercolosi di livello di contenimento tre.(A) Bioelectrosprayer alloggiato all’interno di una doppia classe di dimensioni I/III Microbiological Cabinet di sicurezza.(B) Close-up del bioelectrosprayer pulito dopo un esperimento, con un driver siringa sulla parte superiore. Le porte esterne devono essere chiuse per l’inizio della generazione di microsfere, fornendo il primo livello di contenimento, con il flusso d’aria della classe I MSC che fornisce il secondo livello di contenimento. L’armadio viene formalizzato dopo ogni esperimento.DOI:
http://dx.doi.org/10.7554/eLife.21283.004
Figura 1-figure supplement 2.Microsfere posizionate in una piastra di coltura tissutale a 12 pozzetti immediatamente dopo la generazione, dimostrando un aspetto non illuminato.DOI:
http://dx.doi.org/10.7554/eLife.21283.005
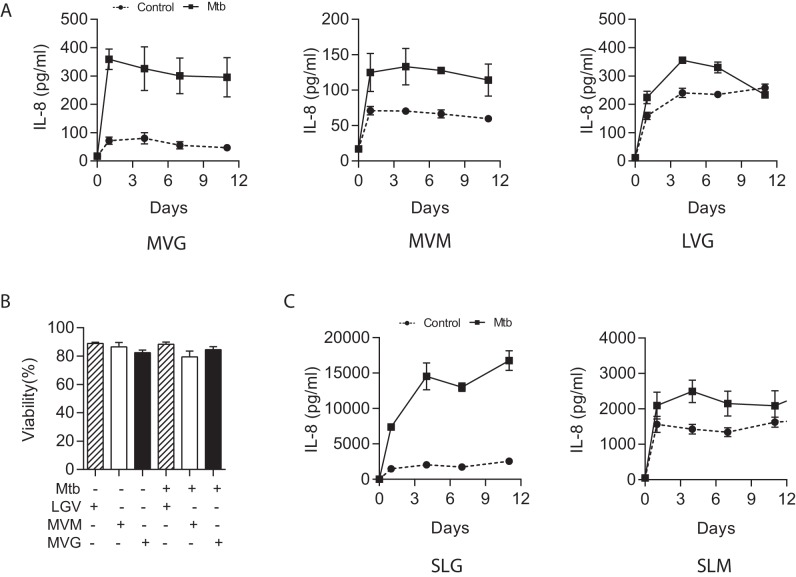
Figura 1-figure supplement 3.L’alginato guluronato a media viscosità (MVG) è l’alginato ottimale per gli studi immunologici.(A) IL-8 secrezione da microsfere composte da diversi alginati misurati con ELISA. MVM: mannuronato a media viscosità, LVG: guluronato a bassa viscosità. Inoltre, LVG aveva proprietà biofisiche molto scarse per il biolelettrospray.(B) La vitalità cellulare analizzata dal rilascio di LDH non era diversa tra i diversi tipi di alginato.(C) Nell’alginato sterile, la secrezione di IL-8 era più alta nelle microsfere contenenti SLG rispetto alla SLM. SLG: guluronato sterile, SLM: mannuronato sterile. Quadrati, linee riempite: Microsfere infette da Mtb, cerchi e linee spezzate: microsfere non infette.DOI:
http://dx.doi.org/10.7554/eLife.21283.006
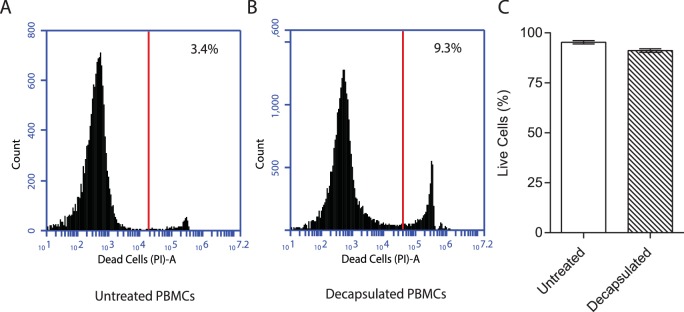
Figura 1-figure supplement 4.La vitalità dei PBMC rimane superiore al 90% dopo la decapsulazione.Le PBMC sono state incapsulate in microsfere di alginato e collagene. Dopo 1 ora di incubazione a 37°C in HBSS, sono stati decapsulati in soluzione di 55 mM di citrato di sodio e 5 mM di EDTA preparato in HBSS (pH = 7,4) per 10 min. Le cellule sono state poi lavate con HBSS due volte prima della sospensione in 5 ml di RPMI e la colorazione con ioduro di propidio (1 mg / ml) a 1:100 diluizione e analisi per citometria a flusso. Per confronto, è stata utilizzata la vitalità delle PBMC tenute in RPMI a 4°C durante questo periodo. I dati sono un rappresentante di due esperimenti indipendenti.DOI:
http://dx.doi.org/10.7554/eLife.21283.007
Figura 1-figura supplemento 5.Composizione delle cellule T delle microsfere.La proporzione di cellule T CD4+ aumenta tra il giorno 0 e il settimo giorno sia nelle microsfere non infette che in quelle infette(A), mentre la proporzione di cellule T CD8+ diminuisce(B). *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 rispetto al giorno 0.DOI:
http://dx.doi.org/10.7554/eLife.21283.008
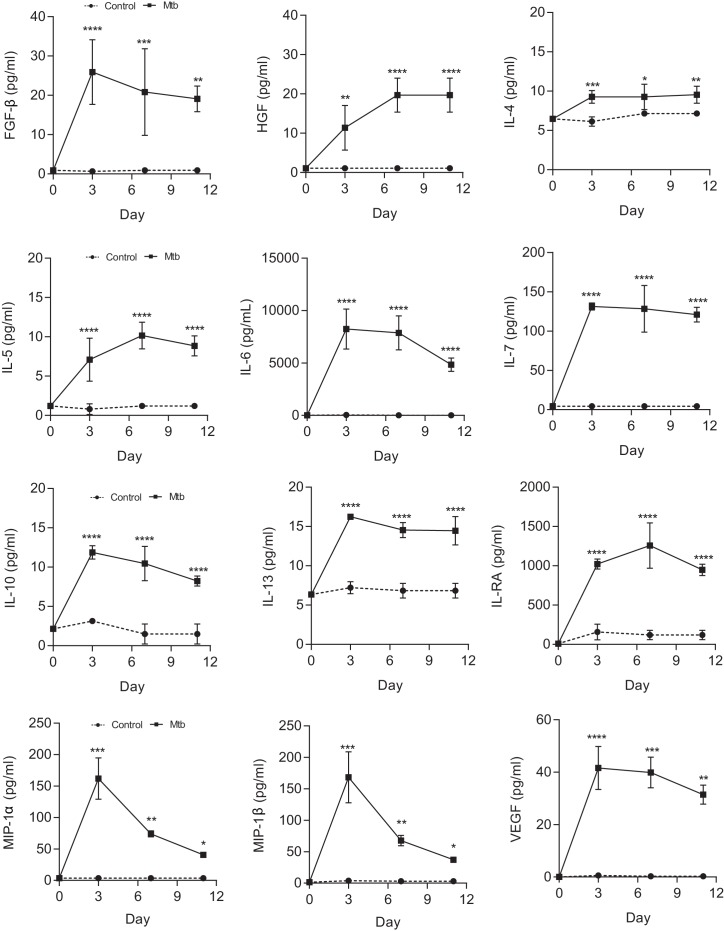
Figura 2-figure supplement 1.Mtb cresce all’interno di microsfere contenenti PBMC e upregola l’espressione di MMP e citochine.l’infezione da Mtb upregola la secrezione di fattori di crescita multipli, citochine e chemochine da microsfere misurate da Luminex array.(A) Mtb prolifera lentamente nelle microsfere senza cellule (linea verde), ma progressivamente nelle microsfere contenenti PBMC (linea rossa), raggiungendo una luminescenza simile alla coltura di brodo Middlebrook 7H9 a 24 giorni (linea nera). Linea blu, microsfere non infette.(B) L’infezione da Mtb upregola l’espressione del gene MMP-1 e(C) la secrezione di MMP-9 nelle microsfere.(D) MMP upregulation ha un effetto funzionale, causando la degradazione del collagene. DQ La degradazione del collagene è più alta nelle microsfere infette da Mtb (linea rossa) che non infette (linea blu). Triangoli, microsfere senza PBMC.(E) L’infezione da Mtb aumenta l’accumulo cellulare di mRNA IFN-γ mRNA rispetto alle cellule non infette al quarto giorno nelle microsfere (n = 4).(F) La secrezione di citochine da microsfere infette da Mtb (quadrati) è significativamente più alta che nelle microsfere contenenti PBMC non infette (cerchi). ****p<0.0001 mediante test t (B ed E) e ANOVA (A,C, D, F).DOI:
http://dx.doi.org/10.7554/eLife.21283.009*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 rispetto alle microsfere non infette in ogni punto temporale.DOI:
http://dx.doi.org/10.7554/eLife.21283.010
Figura 2-figure supplement 1.L’infezione da Mtb upregola la secrezione di molteplici fattori di crescita, citochine e chemochine da microsfere misurate da Luminex array.*p<0.05, **p<0.01, ***p<0.001, ****p<0.0001 rispetto alle microsfere non infette in ogni punto di tempo.DOI:
http://dx.doi.org/10.7554/eLife.21283.010
L’integrità della matrice extracellulare regola l’interazione ospite-agente patogeno
Nei pazienti affetti da TBC, l’interazione ospite-patogeno si verifica nel contesto della matrice extracellulare polmonare ricca di collagene(Elkington et al., 2011), ma la maggior parte degli studi di laboratorio si verifica in assenza di matrice. La matrice regola molteplici aspetti della biologia cellulare(Pampaloni et al., 2007), e quindi per determinare se l’incorporazione della matrice nel sistema bio-elettrospray era un componente critico del modello, abbiamo generato microsfere senza collagene o con collagene uniformemente distribuito(Figura 3A). L’incorporazione di collagene di tipo I ha ridotto significativamente la morte cellulare dopo l’infezione da Mtb, mentre l’adesione all’elastina ha aumentato la morte cellulare(Figura 3B). Abbiamo quindi studiato ulteriormente il fenotipo nelle microsfere contenenti collagene di tipo I. Le PBMC nelle microsfere contenenti collagene avevano una capacità significativamente maggiore di controllare la proliferazione di Mtb, con una proliferazione di Mtb inferiore dal settimo giorno in presenza di collagene (Figura 3C), dimostrando ulteriormente che l’integrità della matrice regola l’interazione ospite-patogeno.10.7554/eLife.21283.011Figure 3.Incorporazione di collagene nelle microsfere limita la crescita di Mtb e aumenta la sopravvivenza delle cellule ospiti.(A) Le microsfere sono state create senza collagene (i),o incorporando collagene con etichetta FITC (ii)per dimostrare la distribuzione. Subito dopo la bioelettrospirazione, il collagene è omogeneo in tutte le microsfere.(B) L’incorporazione di collagene di tipo I nelle microsfere migliora la sopravvivenza cellulare a 72 ore dopo l’infezione da Mtb, mentre l’elastina non lo ha fatto, analizzato dal saggio CytoTox-Glo.(C) PBMCs controllare la crescita Mtb in microsfere contenenti collagene (quadrati) meglio di cellule senza collagene (cerchi). Quadrati aperti, PBMCs non infetti.(D) Il livello di apoptosi e il rapporto NADP+/NADPH(E) sono più alti nelle microsfere contenenti collagene al settimo giorno. L’incorporazione del collagene ha causato una maggiore secrezione di IL-1β (F),TNF-α(G), IFN-γ (H), IL-6 (I),MCP-1(J) e IL-8 (K) al giorno 7. Ogni esperimento è stato eseguito un minimo di 2 volte e i grafici rappresentano i valori medi + SEM di un esperimento rappresentativo eseguito in triplice copia. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.DOI:http://dx.doi.org/10.7554/eLife.21283.011
Per studiare i meccanismi alla base della ridotta crescita in presenza di collagene, abbiamo sviluppato una lettura multiparametrica. L’apoptosi, considerata un meccanismo di protezione dell’ospite, è stata aumentata nelle sfere contenenti collagene rispetto alle sfere prive di collagene(Figura 3D). Inoltre, il rapporto NADP-NADPH era più alto nelle sfere contenenti collagene(Figura 3E), dimostrando omeostasi divergente energia cellulare. Secrezione di citochine proinfiammatorie multiple in microsfere è stato aumentato in presenza di collagene, tra cui IL-1β, TNF-α, IFN-γ, IL-6, IL-8 e MCP-1 (Figura 3F-K). Pertanto, l’interazione ospite-patogeno è nettamente diversa in presenza di collagene, con un migliore controllo della crescita di Mtb, una maggiore sopravvivenza cellulare e un alterato equilibrio energetico e la secrezione di citochine.
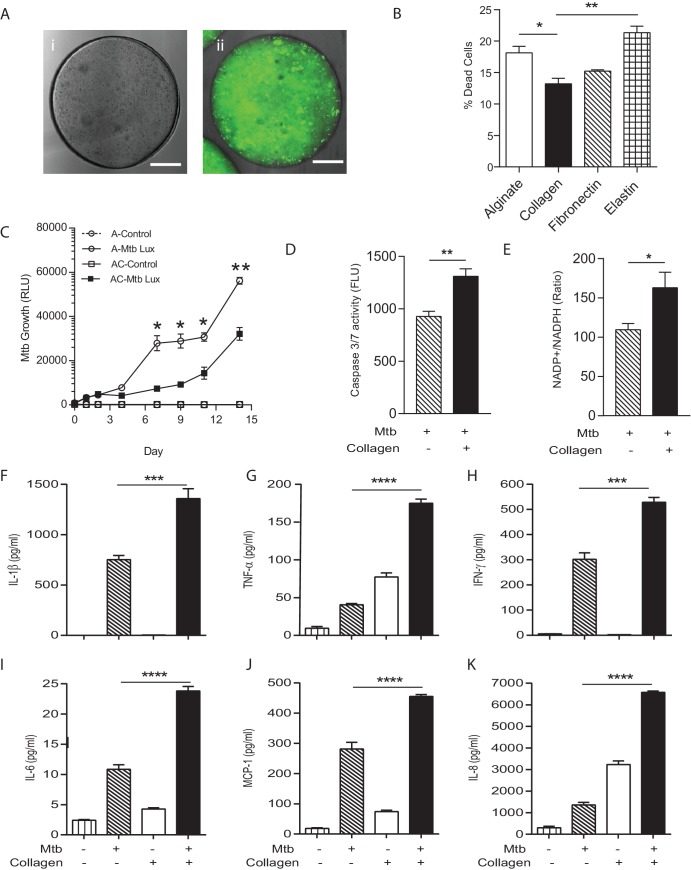
Figura 3.L’incorporazione del collagene nelle microsfere limita la crescita delle Mtb e aumenta la sopravvivenza delle cellule ospiti.(A) Le microsfere sono state create senza collagene (i),o incorporando collagene marcato FITC (ii)per dimostrare la distribuzione. Subito dopo la bioelettrospirazione, il collagene è omogeneo in tutte le microsfere.(B) L’incorporazione di collagene di tipo I nelle microsfere migliora la sopravvivenza cellulare a 72 ore dopo l’infezione da Mtb, mentre l’elastina non lo ha fatto, analizzato dal saggio CytoTox-Glo.(C) PBMCs controllare la crescita Mtb in microsfere contenenti collagene (quadrati) meglio di cellule senza collagene (cerchi). Quadrati aperti, PBMCs non infetti.(D) Il livello di apoptosi e il rapporto NADP+/NADPH(E) sono più alti nelle microsfere contenenti collagene al settimo giorno. L’incorporazione del collagene ha causato una maggiore secrezione di IL-1β (F),TNF-α(G), IFN-γ (H), IL-6 (I),MCP-1(J) e IL-8 (K) al giorno 7. Ogni esperimento è stato eseguito un minimo di 2 volte e i grafici rappresentano i valori medi + SEM di un esperimento rappresentativo eseguito in triplice copia. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.DOI:
http://dx.doi.org/10.7554/eLife.21283.011
Definizione del ruolo delle singole citochine
Un paradigma emergente nel campo della TBC è che è necessaria una risposta immunitaria ottimale e che un deficit o un eccesso di mediatori specifici può essere deleterio dal punto di vista dell’ospite (O’Garra et al., 2013). Pertanto, abbiamo studiato l’effetto di integrare le citochine sia sull’ospite che sull’agente patogeno nelle microsfere contenenti collagene, studiando IFN-γ e IFN-β. La completa assenza di IFN-γ porta all’infezione da Mtb diffusa nell’uomo e nel topo (O’Garraet al., 2013), mentre IFN-β è di importanza emergente da analisi imparziali, ma ha un meccanismo d’azione non definito (Cliff etal., 2015). L’aggiunta di IFN-β esogeno ha portato ad una minore ma significativa soppressione della crescita di Mtb (Figura 4A), mentre al contrario l’aggiunta di IFN-γ ha aumentato costantemente la crescita (Figura 4B). Abbiamo studiato i meccanismi di questa divergenza. Sia IFN-β che IFN-γ hanno ridotto la tossicità cellulare nelle microsfere infette da Mtb (Figura 4Ce D). Tuttavia, l’attività del collagenasi è stata regolata in modo divergente, come IFN-β soppresso MMP-1 espressione mRNA mentre IFN-γ aumentato MMP-1 espressione (Figura 4E), suggerendo che IFN-β ha un ruolo di matrice-protettivo. Mtb infezione Mtb upregulated IL-1β, TNF-α e MCP-1 secrezione, ma l’aumento di IFN non ha modulato in modo significativo questo (Figura 4F, G e H). Infezione Mtb aumentato IFN-γ secrezione, e questo è stato ulteriormente aumentato da IFN-β (Figura 4I), dimostrando complesso cross-talk tra queste due citochine.10.7554/eLife.21283.012Figure 4.IFN-β e IFN-γ hanno effetti divergenti sulla crescita batterica all’interno microsfere. (A) Esogeno IFN-β sopprime la crescita Mtb dopo 24 giorni di cultura. La linea nera rappresenta i PBMC infetti da Mtb. L’integrazione di IFN-β a 0,02 nM (blu), 0,2 nM (verde) e 2 nM (rosso) sopprime la luminescenza della Mtb.(B) IFN-γ aumenta la crescita di Mtb rispetto alle PBMC infette senza citochina addizionale. L’IFN-γ esogeno a 0,02 nM (blu), 0,2 nM (verde) e 2 nM (rosso) aumenta la luminescenza della Mtb rispetto alle PBMC infette senza supplementazione di citochine (linea nera).(C, D) Sia IFN-β che IFN-γ riducono la tossicità nelle PBMC infette da Mtb, analizzate dal saggio CytoTox-Glo.(E) IFNs divergenti regolano MMP-1, con IFN-β soppressione espressione genica IFN-β in microsfere infette, mentre IFN-γ aumenta l’espressione MMP-1. (F- H). Mtb upregola la secrezione di citochine, ma questo non è modulato da IFN. IFN-β guida TNF-α e MCP-1 come un unico stimolo (E e G), ma non ha alcun effetto sinergico significativo con Mtb.(I) Mtb upregola la secrezione di IFN-γ, e questo è ulteriormente aumentato con l’aggiunta di IFN-β. Media + SEM di un esperimento rappresentativo eseguito in triplice copia è mostrato, e sono rappresentativi di un minimo di 2 esperimenti fatti in triplice copia. *p<0.05, **p<0.01, ***p<0.001 e ****p<0.0001.DOI:http://dx.doi.org/10.7554/eLife.21283.012
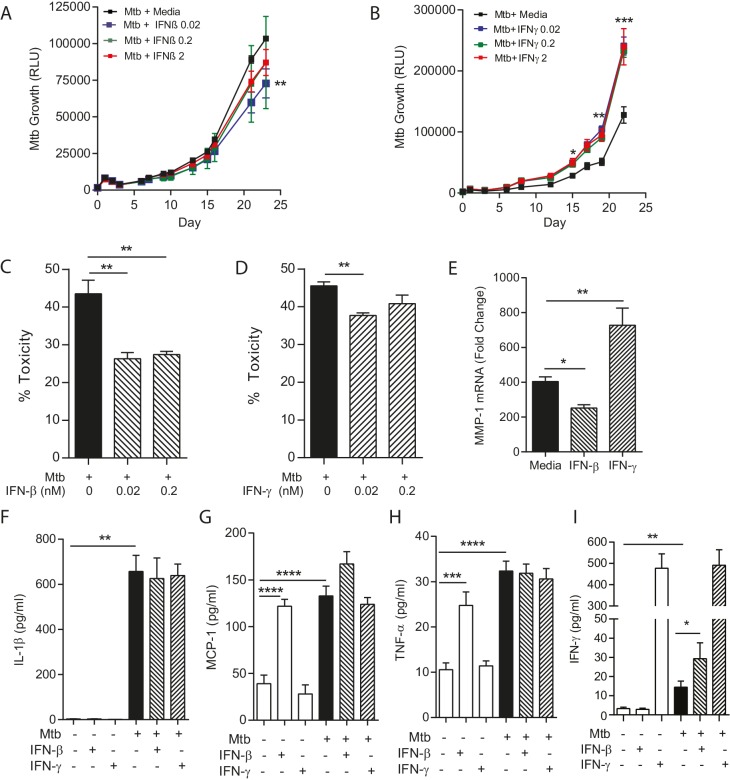
Figura 4.IFN-β e IFN-γ hanno effetti divergenti sulla crescita batterica all’interno delle microsfere.(A) IFN-β esogeno sopprime la crescita di Mtb dopo 24 giorni di coltura. La linea nera rappresenta i PBMC infetti da Mtb. L’integrazione di IFN-β a 0,02 nM (blu), 0,2 nM (verde) e 2 nM (rosso) sopprime la luminescenza della Mtb.(B) IFN-γ aumenta la crescita di Mtb rispetto alle PBMC infette senza citochina addizionale. L’IFN-γ esogeno a 0,02 nM (blu), 0,2 nM (verde) e 2 nM (rosso) aumenta la luminescenza della Mtb rispetto alle PBMC infette senza supplementazione di citochine (linea nera).(C, D) Sia IFN-β che IFN-γ riducono la tossicità nelle PBMC infette da Mtb, analizzate dal saggio CytoTox-Glo.(E) IFNs divergenti regolano MMP-1, con IFN-β soppressione espressione genica IFN-β in microsfere infette, mentre IFN-γ aumenta l’espressione MMP-1. (F- H). Mtb upregola la secrezione di citochine, ma questo non è modulato da IFN. IFN-β guida TNF-α e MCP-1 come un unico stimolo (E e G), ma non ha alcun effetto sinergico significativo con Mtb.(I) Mtb upregola la secrezione di IFN-γ, e questo è ulteriormente aumentato con l’aggiunta di IFN-β. Media + SEM di un esperimento rappresentativo eseguito in triplice copia è mostrato, e sono rappresentativi di un minimo di 2 esperimenti fatti in triplice copia. *p<0.05, **p<0.01, ***p<0.001 e ****p<0.0001.DOI:
http://dx.doi.org/10.7554/eLife.21283.012
Indagare la terapia diretta dall’ospite
La terapia diretta dall’ospite è un paradigma emergente per migliorare l’esito dell’infezione da TBC(Hawn et al., 2015). Tuttavia, la risposta immunitaria dell’ospite alla Mtb ha effetti sia benefici che dannosi, e quindi tale terapia può inavvertitamente guidare l’immunopatologia limitando al contempo la proliferazione dei micobatteri(Elkington e Friedland, 2015). La modulazione della via della cicloossigenasi è stata proposta come obiettivo chiave per limitare la crescita della Mtb(Mayer-Barber et al., 2014). L’aumento con PGE2 esogeno ha soppresso la crescita di Mtb in modo dose-dipendente(Figura 5A), coerente con i risultati del modello murino di Mtb(Mayer-Barber et al., 2014). La ridotta luminescenza di Mtb correlata con i conteggi delle colonie quando le microsfere sono state lisate e placcate su Middlebrook 7H11 agar al giorno 22(Figura 5B). Tuttavia, questo migliore controllo della crescita batterica non era privo di potenziali effetti nocivi. La secrezione di IL-6 e IL-8 proinfiammatorio, un potente chemioattrattore neutrofilo, è stato aumentato di PGE2(Figura 5C e D). Al contrario, la secrezione di IFN-γ è stata soppressa da alte dosi di PGE2 (Figura 5E). Inoltre, la PGE2 ha aumentato la tossicità cellulare(Figura 5F) e ha soppresso la vitalità cellulare totale(Figura 5G). La PGE2 ha ridotto l’attività della caspasi 3/7, indicando la soppressione dell’apoptosi(Figura 5H). Pertanto, la lettura multiparametrica ha il potenziale di prevedere gli effetti protettivi e dannosi della terapia diretta dall’ospite.10.7554/eLife.21283.013L’aumento della PGE2 limita la crescita batterica ma aumenta la secrezione pro-infiammatoria delle citochine e la tossicità cellulare.(A) L’aggiunta di PGE2 esogenasopprime la crescita di Mtb nelle microsfere in modo dose-dipendente. PBMC infetti da Mtb (linea nera), 0,2 µg/ml PGE2 (linea verde), 2 µg/ml PGE2 (linea rossa), 20 µg/ml PGE2 (linea blu).(B) I conteggi delle colonie di microsfere decapitate al giorno 24 e poi placcate su Middlebrook 7H11 agar sono correlati con la luminescenza.(C, D ed E) PGE2 aumenta la secrezione di IL-6 e IL-8, ma diminuisce significativamente la secrezione di IFN-γ, da microsfere infette da Mtb.(F) La tossicità cellulare è aumentata in PGE2 microsfere trattate al 3 ° giorno, analizzato dal rilascio di LDH, e(G) la vitalità cellulare totale è stata ridotta al 7 ° giorno, analizzato dal saggio CytoTox-Glo.(H) PGE2 riduce l’attività della caspasi 3/7 al giorno 7. *p<0.05, **p<0.01, ***p<0.001.DOI:http://dx.doi.org/10.7554/eLife.21283.013
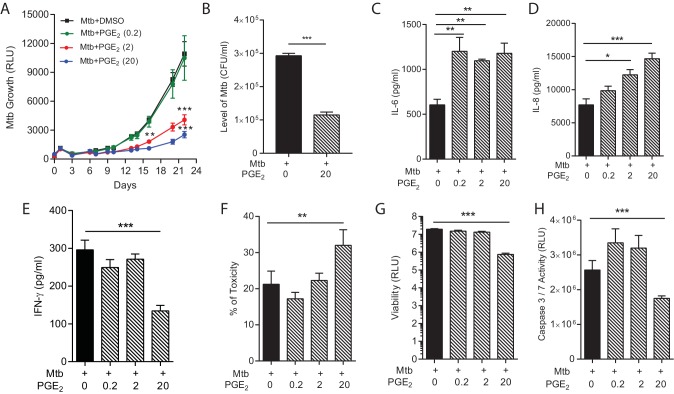
Figura 5.L’aumento del PGE2 limita la crescita batterica ma aumenta la secrezione di citochine pro-infiammatorie e la tossicità cellulare.(A) L’aggiunta di PGE2 esogenasopprime la crescita di Mtb in microsfere in modo dose-dipendente. PBMC infetti da Mtb (linea nera), 0,2 µg/ml PGE2 (linea verde), 2 µg/ml PGE2 (linea rossa), 20 µg/ml PGE2 (linea blu).(B) I conteggi delle colonie di microsfere decapitate al giorno 24 e poi placcate su Middlebrook 7H11 agar sono correlati con la luminescenza.(C, D ed E) PGE2 aumenta la secrezione di IL-6 e IL-8, ma diminuisce significativamente la secrezione di IFN-γ, da microsfere infette da Mtb.(F) La tossicità cellulare è aumentata in PGE2 microsfere trattate al 3 ° giorno, analizzato dal rilascio di LDH, e(G) la vitalità cellulare totale è stata ridotta al 7 ° giorno, analizzato dal saggio CytoTox-Glo.(H) PGE2 riduce l’attività della caspasi 3/7 al giorno 7. *p<0.05, **p<0.01, ***p<0.001.DOI:
http://dx.doi.org/10.7554/eLife.21283.013
Immunoaugmentazione con linee cellulari T specifiche per Mtb
La risposta delle cellule T è fondamentale per il controllo della Mtb ma guida anche la patologia(Kaufmann e Dorhoi, 2013), e quindi una questione critica è quali aspetti della risposta immunitaria adattiva sono protettivi rispetto a quelli immunopatogeni(Jasenosky et al., 2015). Abbiamo usato la trattabilità del modello bio-elettrospray per studiare l’aumento delle cellule T, integrando PBMC con linee cellulari T specifiche di antigene autologo che erano state proliferate ex vivo(Figura 6-figure supplement 1). Quattro giorni dopo il bio-elettrospray, aggregati multicellulari hanno iniziato a formare aggregati contenenti PBMC, Mtb e cellule T aumentate(Figura 6A). ESAT-6 o CFP-10 specifiche linee di cellule T, che rispondono agli antigeni secreti attraverso il locus RD1 patogenicità, proliferavano nelle microsfere infette da Mtb ma non sfere non infette(Figura 6B), che era statisticamente significativo dal settimo giorno sulla quantificazione(Figura 6C). Sorprendentemente, l’immunoaugmentazione con linee cellulari ESAT-6 o CFP-10 T ha portato ad un aumento della crescita di Mtb all’interno delle microsfere rispetto alle sole PBMC infette(Figura 6D). L’aumento con una linea cellulare innata autologa iNKT non ha alcun effetto sulla crescita di Mtb(Figura 6E), dimostrando che questa non era una risposta generica all’integrazione di cellule T all’interno delle microsfere. L’immunoaugmentazione con le linee ESAT-6 e CFP-10 ha aumentato significativamente la secrezione di citochine multiple, tra cui TNF-α e IL-1β nei media intorno alle microsfere (Figura 6Fe G e Figura 6-figure supplement 2). Al contrario, l’aumento non ha influenzato in modo significativo la tossicità cellulare all’interno di microsfere infette(Figura 6H).10.7554/eLife.21283.014Figure 6.Immunoaugmentation con le cellule T Mtb-specifiche Mtb aumenta la crescita Mtb.(A) Microsfere riprese dopo 4 giorni mostrano la formazione precoce di granuloma.(i) PBMCs etichettati con CellTrace CFSE (verde),(ii) Mtb che esprime mCherry (rosso),(iii) autologo ESAT-6 specifiche cellule T etichettati con CellTracker Blu,(iv) immagine congiunta mostra granulomi contenenti Mtb, PBMCs e cellule T aumentate (giallo).(B) La proliferazione cellulare è aumentata nelle microsfere infette con cellule T autologhe immunoaugate, analizzate mediante colorazione CFSE. Giorno 1, linea nera; Giorno 7, linea rossa;(i) Non infetto,(ii) Mtb-infetto.(C) Analisi quantitativa della capacità proliferativa dei PBMC augumentati ESAT-6 al primo giorno e al settimo giorno. Le barre mostrano la percentuale di cellule CD4 proliferanti dopo il gating su linfociti CD3+CD4+. Le differenze tra il primo e il settimo giorno sono state valutate per tre esperimenti con il t-test.(D) L’aggiunta di cellule T reattive ESAT-6 (rosso) o di cellule T reattive CFP-10 (blu) aumenta la crescita di Mtb rispetto alle PBMC infette senza cellule T integrate (nero). Simboli aperti, microsfere non infette.(E) L’integrazione con una linea di cellule T autologhe iNKT (triangolo blu) non ha influito in modo significativo sulla crescita di Mtb rispetto alle sole PBMC infette (quadrato nero).(F, G) Secrezione di TNF-α e IL-1β è aumentata in microsfere immunoaugate al giorno 7.(H) L’immunoaugmentazione non ha modulato in modo significativo la tossicità cellulare nelle microsfere infette al terzo giorno analizzate dal rilascio di LDH.DOI:http://dx.doi.org/10.7554/eLife.21283.01410.7554/eLife.21283.015Figuresupplemento a 6 cifre 1.Conferma della specificità delle cellule T espanse in vitro. (A) Il grafico a punti mostra la percentuale di cellule T che sono o CD4+/CD8+, CD4+/CD8-, CD4-/CD8+, CD4-/CD8+, CD4-/CD8- da ESAT-6 (i) e CFP-10 (ii) linee cellulari T specifiche.(B) PBMC sono stati coltivati con ESAT-6 (i) e CFP-10(ii) peptidi e dopo due settimane di coltura, la specificità delle cellule T è stata confermata dal rilascio di IFN-γ dopo l’esposizione al loro antigene rispettivo. L’antigene iNKT KRN7000 è stato utilizzato come controllo negativo e PMA / Ionomicina come mitogeno policlonale delle cellule T. I dati sono rappresentativi di tre donatori con ogni co-cultura eseguita in triplice copia.(C) Percentuale di ESAT-6(i) e CFP-10(ii) cellule T specifiche misurate mediante colorazione intracellulare delle citochine. Le cellule dendritiche derivate da monociti (moDC) sono state pulsate con il peptide ESAT-6 o CFP-10 per 12 ore prima dell’aggiunta di cellule T. Dopo 8 ore di stimolazione con il loro antigene corrispondente, ogni linea è stata colorata per citochine intracellulari e le cellule sono state acquisite su un FACSaria.(D) Dot trame che rappresentano l’espansione iNKT in vitro antigene indotta da antigene in un donatore. Live CD3 + CD1d-K7 tet + Vα24 + cellule iNKT sono stati colorati (i) ex vivo e (ii)due settimane dopo la stimolazione con KRN7000. I grafici mostrano la % di cellule iNKT ex vivo (0,68%) e dopo la cultura in vitro (71,1%), confermando la loro espansione.DOI:http://dx.doi.org/10.7554/eLife.21283.01510.7554/eLife.21283.016Figuresupplemento a 6 cifre 2.Augmentation 6-figure supplemento 2.Augmentation di PBMCs con ESAT-6/CFP-10 specifiche linee di cellule T in microsfere provoca secrezione differenziale di citochine dopo l’infezione Mtb rispetto ai PBMCs solo. (A- D) secrezione di citochine in CFP-10 e ESAT-6 specifiche cellule T microsfere integrate è stato normalizzato per la secrezione di PBMCs infetti all’interno PBMC solo microsfere ei risultati sono espressi come unità relative.(E) Rappresentazione grafica del profilo delle citochine (n = 3). I dati mostrano la media ± SEM di tre esperimenti indipendenti fatti in triplice copia. *p<0.05, **p<0.01, ***p<0.001, p<0.0001.DOI:http://dx.doi.org/10.7554/eLife.21283.016
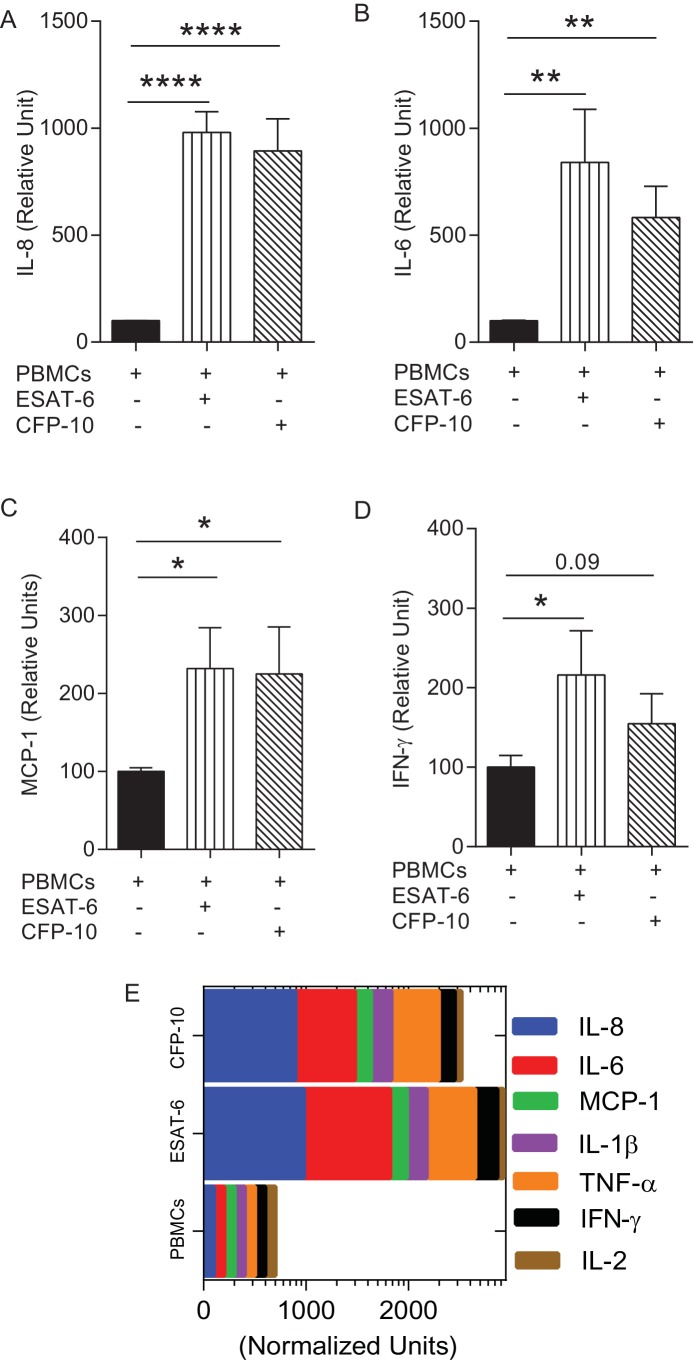
Figura 6-figure supplement 2.L’immunoaugmentazione con cellule T specifiche per le Mtb aumenta la crescita delle Mtb. conferma della specificità delle cellule T espanse in vitro. l’aumento delle PBMC con linee di cellule T specifiche per le PBMC con ESAT-6/CFP-10 nelle microsfere provoca una secrezione differenziale di citochine dopo l’infezione da Mtb rispetto alle PBMC da sole.(A) Le microsfere riprese dopo 4 giorni mostrano la formazione precoce di granuloma.(i) PBMCs etichettate con CellTrace CFSE (verde),(ii) Mtb che esprime mCherry (rosso), (iii) cellule T autologhe ESAT-6 specifiche etichettate con CellTracker Blue, (iv) L’immagine unita mostra granulomi contenenti Mtb, PBMCs e cellule T aumentate (giallo).(B) La proliferazione cellulare è aumentata nelle microsfere infette con cellule T autologhe immunoaugate, analizzate mediante colorazione CFSE. Giorno 1, linea nera; Giorno 7, linea rossa;(i) Non infetto,(ii) Mtb-infetto.(C) Analisi quantitativa della capacità proliferativa dei PBMC augumentati ESAT-6 al primo giorno e al settimo giorno. Le barre mostrano la percentuale di cellule CD4 proliferanti dopo il gating su linfociti CD3+CD4+. Le differenze tra il primo e il settimo giorno sono state valutate per tre esperimenti con il t-test.(D) L’aggiunta di cellule T reattive ESAT-6 (rosso) o di cellule T reattive CFP-10 (blu) aumenta la crescita di Mtb rispetto alle PBMC infette senza cellule T integrate (nero). Simboli aperti, microsfere non infette.(E) L’integrazione con una linea di cellule T autologhe iNKT (triangolo blu) non ha influenzato significativamente la crescita di Mtb rispetto alle PBMC infette da sole (quadrato nero).(F, G) Secrezione di TNF-α e IL-1β è aumentata in microsfere immunoaugate al giorno 7.(H) L’immunoaugmentazione non ha modulato in modo significativo la tossicità cellulare nelle microsfere infette al terzo giorno analizzate dal rilascio di LDH.DOI:
http://dx.doi.org/10.7554/eLife.21283.014(A) Il diagramma a punti mostra la percentuale di celle T che sono o CD4+/CD8+, CD4+/CD8-, CD4-/CD8+, CD4-/CD8+, CD4-/CD8- da ESAT-6 (i)e CFP-10 (ii) linee di celle T specifiche.(B) PBMC sono stati coltivati con ESAT-6 (i) e CFP-10(ii) peptidi e dopo due settimane di coltura, la specificità delle cellule T è stata confermata dal rilascio di IFN-γ dopo l’esposizione al loro antigene rispettivo. L’antigene iNKT KRN7000 è stato utilizzato come controllo negativo e PMA / Ionomicina come mitogeno policlonale delle cellule T. I dati sono rappresentativi di tre donatori con ogni co-cultura eseguita in triplice copia.(C) Percentuale di ESAT-6(i) e CFP-10(ii) cellule T specifiche misurate mediante colorazione intracellulare delle citochine. Le cellule dendritiche derivate da monociti (moDC) sono state pulsate con il peptide ESAT-6 o CFP-10 per 12 ore prima dell’aggiunta di cellule T. Dopo 8 ore di stimolazione con il loro antigene corrispondente, ogni linea è stata colorata per citochine intracellulari e le cellule sono state acquisite su un FACSaria.(D) Dot trame che rappresentano l’espansione iNKT in vitro antigene indotta da antigene in un donatore. Live CD3 + CD1d-K7 tet + Vα24 + cellule iNKT sono stati colorati (i) ex vivo e (ii)due settimane dopo la stimolazione con KRN7000. I grafici mostrano la % di cellule iNKT ex vivo (0,68%) e dopo la cultura in vitro (71,1%), confermando la loro espansione.DOI:
http://dx.doi.org/10.7554/eLife.21283.015(A- D) La secrezione dicitochine inCFP-10 e ESAT-6 specifiche cellule T microsfere integrate con cellule T è stata normalizzata alla secrezione da PBMC infette all’interno di microsfere solo PBMC e i risultati sono espressi come unità relative.(E) Rappresentazione grafica del profilo delle citochine (n = 3). I dati mostrano la media ± SEM di tre esperimenti indipendenti fatti in triplice copia. *p<0.05, **p<0.01, ***p<0.001, p<0.0001.DOI:
http://dx.doi.org/10.7554/eLife.21283.016
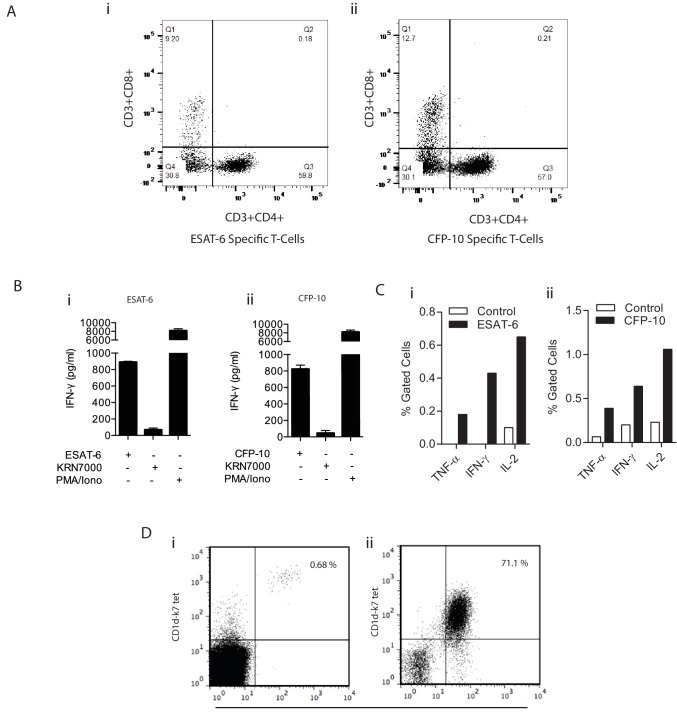
Figura 6-figure supplement 1.2. Conferma della specificità delle cellule T espanse in vitro.(A) Il grafico a punti mostra la percentuale di cellule T che sono o CD4+/CD8+, CD4+/CD8-, CD4-/CD8+, CD4-/CD8+, CD4-/CD8- da ESAT-6 (i)e CFP-10 (ii) linee di cellule T specifiche.(B) PBMC sono stati coltivati con ESAT-6 (i) e CFP-10(ii) peptidi e dopo due settimane di coltura, la specificità delle cellule T è stata confermata dal rilascio di IFN-γ dopo l’esposizione al loro antigene rispettivo. L’antigene iNKT KRN7000 è stato utilizzato come controllo negativo e PMA / Ionomicina come mitogeno policlonale delle cellule T. I dati sono rappresentativi di tre donatori con ogni co-cultura eseguita in triplice copia.(C) Percentuale di ESAT-6(i) e CFP-10(ii) cellule T specifiche misurate mediante colorazione intracellulare delle citochine. Le cellule dendritiche derivate da monociti (moDC) sono state pulsate con il peptide ESAT-6 o CFP-10 per 12 ore prima dell’aggiunta di cellule T. Dopo 8 ore di stimolazione con il loro antigene corrispondente, ogni linea è stata colorata per citochine intracellulari e le cellule sono state acquisite su un FACSaria.(D) Dot trame che rappresentano l’espansione iNKT in vitro antigene indotta da antigene in un donatore. Live CD3 + CD1d-K7 tet + Vα24 + cellule iNKT sono stati colorati (i) ex vivo e (ii)due settimane dopo la stimolazione con KRN7000. I grafici mostrano la % di cellule iNKT ex vivo (0,68%) e dopo la cultura in vitro (71,1%), confermando la loro espansione.DOI:
http://dx.doi.org/10.7554/eLife.21283.015
Figura 6-figure supplement 2.L’aumento di PBMCs con linee di cellule T specifiche ESAT-6/CFP-10 in microsfere provoca una secrezione differenziale di citochine dopo l’infezione da Mtb rispetto alle sole PBMCs.(A- D) La secrezione dicitochine in CFP-10 e ESAT-6 specifiche cellule T microsfere completate è stata normalizzata alla secrezione da PBMC infette all’interno di PBMC solo microsfere e i risultati sono espressi come unità relative.(E) Rappresentazione grafica del profilo delle citochine (n = 3). I dati mostrano la media ± SEM di tre esperimenti indipendenti fatti in triplice copia. *p<0.05, **p<0.01, ***p<0.001, p<0.0001.DOI:
http://dx.doi.org/10.7554/eLife.21283.016
Discussione
Sono urgentemente necessari nuovi approcci alla pandemia globale di tubercolosi. La Mtb è un patogeno umano obbligato caratterizzato da una prolungata interazione con l’ospite(Russell, 2011; Cambier et al., 2014), una risposta immunitaria organizzata spazialmente(Marakalala et al., 2016; Mattila et al., 2013; Egen et al., 2008) e un ampio turnover della matrice extracellulare(Elkington et al., 2011). Pertanto, studi estesi di cellule umane aderenti alla matrice extracellulare in 3-D sono probabilmente essenziali per comprendere appieno l’interazione ospite-patogeno. Abbiamo sviluppato un modello di TBC umana utilizzando la tecnologia bio-elettrospray che replica le caratteristiche chiave della malattia clinica e ottimizzato una lettura multiplex per indagare sia le risposte dell’ospite che quelle del patogeno. Abbiamo dimostrato che la matrice extracellulare regola la risposta immunitaria dell’ospite, in linea con i rapporti della ECM che regola l’infiammazione(Sorokin, 2010), e abbiamo scoperto che il collagene favorisce il controllo dell’ospite della Mtb. Abbiamo poi utilizzato il modello per indagare nuovi approcci terapeutici. Il sistema ha permesso la coltura prolungata di cellule umane primarie, e abbiamo scoperto che differenze significative tra le condizioni sperimentali spesso emergono solo dopo più di 7 giorni, cosa che non sarebbe stata osservata nei sistemi di coltura 2D dove 3-4 giorni è la durata sperimentale standard. I nostri risultati sono coerenti con altre infezioni, dove gli adattamenti cellulari al loro contesto determinano l’esito(Snijder et al., 2009). Questa piattaforma di coltura cellulare è altamente flessibile sia per la matrice che per la composizione cellulare all’interno delle sfere e quindi ha un’ampia applicabilità potenziale all’interno del campo biomedico.
Il nostro approccio di bioingegneria si differenzia in modo significativo dai sistemi di modelli tradizionali per indagare la TBC, che si basano prevalentemente sulla coltura di cellule umane in sistemi di coltura 2D senza matrice extracellulare, sull’infezione delle larve di pesce zebra con M. marinum o sull’infezione dei topi con Mtb(Young, 2009; Guirado e Schlesinger, 2013; Vogt e Nathan, 2011). Il modello murino della TBC ha molti vantaggi e i risultati chiave nel topo sono stati replicati nell’uomo, come i ruoli critici per le cellule T CD4+, TNF-α e IFN-γ (Flynn eChan, 2001), ma la patologia nel topo si differenzia dalla TBC umana (Young,2009) e i topi umanizzati sono necessari per generare lesioni caseanti (Calderonet al., 2013; Heuts et al., 2013). Sono stati sviluppati altri modelli cellulari umani avanzati di TBC. Ad esempio, il gruppo di Altare ha studiato un modello prolungato di coltura PBMC con Mtb e ha dimostrato la formazione di aggregati cellulari (Puisseguret al., 2004; Lay et al., 2007), ma questo modello manca di matrice extracellulare. Un modello contenente matrice di collagene è stato sviluppato da Kapoor, mostrando l’aggregazione e la dormienza della TB(Kapoor et al., 2013), ma manca l’alto potenziale di throughput del sistema bioelectrospray e il rapido recupero cellulare per la lettura multiparametrica. La generazione di una struttura completa del granuloma umano richiederà cellule stromali come i fibroblasti, che sono presenti nella periferia dei granulomi della TBC(O’Kane et al., 2010). Un passo successivo fondamentale sarà quello di confrontare i pazienti con TBC latente con TBC attiva, e quelli con e senza infezione da HIV, per determinare se il modello può differenziare l’immunità protettiva alla TBC direttamente ex vivo.
Molti dei nostri risultati sono coerenti con le conclusioni tratte da questi sistemi. Per esempio, un ruolo significativo per l’IFN-β nella risposta immunitaria dell’ospite alla TBC sta emergendo dagli studi genomici, anche se rimane controverso se questo sia protettivo o dannoso (Cliffet al., 2015). I nostri dati suggeriscono un effetto prevalentemente protettivo, e i dati emergenti supportano questa conclusione(Moreira-Teixeira et al., 2016). Analogamente, abbiamo confermato che l’aumento della PGE2 migliora il controllo dell’ospite sulla proliferazione dei micobatteri, in linea con i risultati ottenuti nel topo(Mayer-Barber et al., 2014). Infine, le cellule T che rispondono a specifici antigeni Mtb proliferavano in microsfere infette e citochine secrete note per essere importanti nella risposta immunitaria dell’ospite alla Mtb(O’Garra et al., 2013).
Tuttavia, mentre alcuni risultati sono stati come ci si aspettava, diversi risultati nel sistema 3-D non possono essere previsti dai paradigmi attuali della malattia. Ad esempio, IFN-γ in alte concentrazioni aumenta la crescita della TBC, mentre gli esperimenti murini prevedono un migliore controllo. Coerentemente con i nostri risultati, diversi studi precedenti hanno dimostrato che IFN-γ aumenta la crescita di Mtb nelle cellule umane primarie (Vogte Nathan, 2011; Douvas et al., 1985; Rook et al., 1986; Crowle e Elkins, 1990). L’evidenza di un ruolo benefico dell’IFN-γ negli esseri umani è principalmente supportata da individui dove c’è una completa assenza di segnalazione attraverso il percorso IL-12/IFN (Karpet al., 2015), e questo effetto protettivo è stato chiaramente dimostrato nel topo sia attraverso studi di knock-out che di vaccinazione (Flynne Chan, 2001; Aagaard et al., 2011). Tuttavia, studi di coorte hanno dimostrato che un’elevata risposta PPD, o un’elevata risposta IFN-γ ad ESAT-6 o CFP-10, si associa al successivo sviluppo della TBC (Comstocket al., 1974; Higuchi et al., 2008; del Corral et al., 2009), suggerendo che un’eccessiva risposta IFN-γ può essere deleteria. Abbiamo cercato di determinare se ci fosse un punto di ribasso della concentrazione di IFN-γ aggiungendo anticorpi IFN-γ neutralizzanti alla matrice microsfera, e anche se siamo stati in grado di dimostrare un aumento della crescita con la neutralizzazione di IFN-γ, abbiamo trovato un effetto simile con gli anticorpi di controllo degli isotipi, quindi è stato impossibile determinare se si trattasse di un effetto specifico. Le nostre osservazioni longitudinali della crescita di Mtb in modo non distruttivo con l’uso di micobatteri luminescenti sostengono il concetto emergente che una risposta immunitaria equilibrata è essenziale, e che un deficit o un eccesso di un mediatore specifico può favorire il patogeno(O’Garra et al., 2013; Gideon et al., 2015). La combinazione del modello con l’editing del gene CRISPR/Cas9 permetterà di interrogare ulteriormente ogni componente citochina della risposta immunitaria.
L’aumento della PGE2 è stato proposto come una nuova terapia diretta dall’ospite per migliorare l’esito della TBC, e dimostriamo una ridotta crescita della Mtb. Tuttavia, l’analisi multiparamterica nel nostro sistema umano ha dimostrato che la PGE2 aumenta la secrezione di IL-8, che probabilmente guiderà la migrazione dei neutrofili, e la PGE2 ha anche ridotto la vitalità delle cellule ospiti. Il reclutamento dei neutrofili è stato descritto per avere un effetto deleterio sul controllo dell’infezione da parte dell’ospite(Kimmey et al., 2015; Nouailles et al., 2014) e quindi c’è il potenziale che questo possa favorire la Mtb, causando un aumento della patologia, della distruzione polmonare e della trasmissione. Il picco di concentrazione PGE2 che abbiamo studiato è simile a quello riportato nei tessuti umani(Reikerås et al., 2009). Abbiamo anche scoperto che l’immunoaugmentazione con ESAT-6 o CFP-10 linee cellulari T reattive ha portato ad un aumento della crescita di Mtb. I micobatteri patogeni esprimono il locus RD1, ma il meccanismo preciso che lega RD1 alla patologia non è pienamente compreso(Majlessi et al., 2015). I nostri risultati sono coerenti con le recenti osservazioni che gli epitopi delle cellule T sono iperconservati nei micobatteri patogeni(Comas et al., 2010; Coscolla et al., 2015; Lindestam Arlehamn et al., 2015), indicando un vantaggio evolutivo per il patogeno di stimolare specificamente i componenti della risposta immunitaria dell’ospite per facilitare la trasmissione(Orme et al., 2015). Tuttavia, una spiegazione alternativa è che le condizioni di espansione in vitro hanno generato un fenotipo di cellule T che era permissivo alla crescita di Mtb, distorcendo un fenotipo inizialmente protettivo ad uno deleterio, e quindi è necessaria un’ulteriore conferma attraverso diverse linee cellulari T. Le cellule iNKT aumentate non hanno aumentato la crescita di Mtb dopo l’espansione ex vivo. La conclusione chiave di questi esperimenti è che il modello di immunoaugmentazione ha il potenziale di sezionare le risposte immunitarie protettive rispetto a quelle patologiche dell’ospite.
I recenti risultati negativi di entrambi gli studi sui vaccini(Tameris et al., 2013; Ndiaye et al., 2015) e i regimi di accorciamento del trattamento(Warner e Mizrahi, 2014) dimostrano che le osservazioni nei sistemi di modelli attuali potrebbero non tradursi in modo affidabile nella malattia umana ed evidenziano la necessità di approcci più sfumati che riflettano l’infezione da TBC umana. I nostri dati suggeriscono che il semplice fatto di guidare una maggiore risposta immunitaria alla Mtb non migliorerà il controllo della crescita dei micobatteri. Aumentare il rilascio di PGE2 modulando il percorso dei leucotrieni può ridurre la proliferazione dei micobatteri, ma può avere un costo in termini di aumento della patologia. La vaccinazione con ESAT-6 come antigene, che è entrata negli studi umani(Luabeya et al., 2015), può effettivamente favorire la crescita di Mtb in determinate circostanze, dimostrando il sottile equilibrio tra la risposta immunitaria dell’ospite e il controllo della crescita del patogeno. Criticamente, i nostri dati del modello bioingegnerizzato sono coerenti con i fenomeni clinici osservati nella TBC umana. Ad esempio, una risposta immunitaria eccessiva nei pazienti è associata a una maggiore patologia polmonare(Kaufmann e Dorhoi, 2013; Comstock et al., 1974; Philips e Ernst, 2012; Nunes-Alves et al., 2014). Allo stesso modo, i nostri studi di immunoaugmentazione concordano con l’espressione di ESAT-6 e CFP-10 da micobatteri patogeni, che implica un ruolo critico nel causare la malattia(Brites e Gagneux, 2015). Il modello può essere utilizzato per indagare gli approcci attualmente in fase di sviluppo, come i vaccini basati sul targeting delle cellule T con restrizione CD1(Van Rhijn et al., 2015) e le emergenti terapie dirette dagli ospiti(Hawn et al., 2013; Zumla et al., 2015), per determinare se conferiscono una maggiore protezione senza probabili effetti dannosi.
Il modello di coltura cellulare bio-elettrospray ha un ampio potenziale, affrontando la complessità tecnica dell’esecuzione di colture cellulari primarie 3-D all’interno di diverse matrici extracellulari. Il sistema può essere facilmente applicato per lo studio di diverse malattie infettive e infiammatorie, o per l’immunoterapia del cancro, e può essere sviluppato per applicazioni ad alta produttività combinando il sistema a microsfere con la microfluidica. L’integrazione con l’editing del gene CRISPR/Cas9 permetterà la manipolazione genetica sia dell’ospite che del patogeno(Chakraborty et al., 2014). Le letture multiparametriche definiscono il potenziale traslazionale di nuovi interventi nel tempo con l’acquisizione longitudinale dei dati, identificando sia gli effetti benefici che quelli deleteri. Pertanto, questo sistema sviluppato per sezionare l’interazione ospite-patogeno nella TBC umana può essere applicato per identificare nuovi approcci terapeutici a molteplici malattie umane.
Materiali e metodi
Approvazione etica
Per l’analisi del sangue di donatori sani e di individui sani esposti alla tubercolosi, questo lavoro è stato approvato dal National Research Ethics Service Committee South Central – Southampton A, titolo dello studio “An investigation into the immun response to tuberculosis infection and development of novel diagnostic markers”, riferimento 13/SC/0043. Tutti i donatori hanno dato il loro consenso informato per iscritto. Per l’analisi istologica, i campioni utilizzati in questo studio sono stati prelevati dal Southampton Research Biorepository, University Hospital Southampton NHS Foundation Trust e University of Southampton, Mailpoint 218, Tremona Road, Southampton, SO16 6YD. Il tessuto della biopsia polmonare è stato prelevato come parte delle cure cliniche di routine e in questo studio sono stati analizzati i blocchi di tessuto in eccesso rispetto ai test diagnostici. Il progetto è stato approvato dall’Institutional Review Board (riferimento 12/NW/0794 SRB04_14). Il comitato etico ha approvato l’analisi di questo tessuto senza il consenso informato dell’individuo, poiché si trattava di tessuto archiviato in eccesso prelevato come parte della cura di routine.
Isolamento delle cellule PBMC dal sangue umano
I PBMC sono stati isolati da coni leucociti di donatori singoli (National Health Service Blood and Transfusion, Southampton, UK) o sangue fresco di volontari mediante centrifugazione a gradiente di densità su Ficoll-Paque (GE Healthcare Life Sciences). Questi donatori sani sono stati tutti reclutati da una regione con un’incidenza di tubercolosi molto bassa. Per gli esperimenti di immunoaugmentazione che richiedono Mycobacterium tuberculosis-responsivecellule T, sono state studiate cellule provenienti da donatori con una documentata esposizione alla tubercolosi. Tutti gli esperimenti sono stati eseguiti con cellule umane primarie; nello studio non sono state utilizzate linee cellulari immortalate.
M. coltura della tubercolosi
M. tubercolosi H37Rv (Mtb) è stato coltivato in Middlebrook 7 H9 medio (integrato con 10% ADC, 0,2% glicerolo e 0,02% Tween 80) (BD Biosciences, Oxford) e bioluminescente M. tuberc olosi H37Rv(Andreu et al., 2010), GFP o mCherry che esprimono la M. tubercolosi H37Rv(Carroll et al., 2010) sono stati coltivati rispettivamente con kanamicina 25 μg/ml e igromicina 50 μg/ml. Bioluminescente Mtb H37Rv Mtb H37Rv è stato utilizzato per tutti gli esperimenti, a parte l’imaging confocale. Culture a 1 × 108 CFU / ml Mtb (OD = 0,6) è stato utilizzato per tutti gli esperimenti a molteplicità di infezione (M.O.I) di 0,1. Per il conteggio delle colonie, Mtb è stato rilasciato da microsfere da EDTA / dissoluzione del citrato di sodio, le cellule e batteri extracellulari sono stati pellettato per centrifugazione a 3000 g, lisato con l’1% di saponina e poi placcato su Middlebrook 7H11 agar. Le colonie sono state contate a tre settimane.
Incapsulamento delle cellule
Le microsfere sono state generate con un generatore elettrostatico (Nisco, Zurigo, Svizzera) come descritto in precedenza(Workman et al., 2014). Per visualizzare la formazione delle microsfere, una telecamera Phantom v7 ad alta velocità, in grado di catturare 150000 fps in combinazione con un obiettivo per microscopio a lunga distanza, è stata attivata simultaneamente con un sistema di illuminazione a fibre ottiche per catturare il processo di jetting. Per l’incapsulamento cellulare, i PBMC sono stati infettati con Mtb durante la notte in una fiaschetta T75. Le cellule sono state poi staccate, lavate e pellettate per centrifugazione a 320 g e miscelate con alginato sterile 1,5% (alginato Pronova UP MVG, Nova Matrix, Norvegia) o alginato con collagene umano, fibronectina (entrambi da Advanced BioMatrix) o elastina umana (Sigma Aldrich, Gillingham, Regno Unito) ad una concentrazione finale di 5 × 106 cellule / ml. La matrice standard utilizzata negli esperimenti era alginato-collagene umano, se non diversamente specificato. Tutti i reagenti sono stati confermati esenti da endotossine.
Il protocollo per l’incapsulamento di PBMCs in matrice a base di alginato è descritto in dettaglio a Bio-protocollo (Tezeraet al., 2017). La cella-alginato sospensione è stato redatto in una siringa sterile e iniettato nel generatore di perline a 10 mL / ora attraverso un driver siringa Harvard attraverso un ugello di 0,7 mm di diametro esterno. Microsfere di 600 μm di diametro formato in un bagno di gelificazione ionotropo di 100 mM CaCl2 in HBSS posto sotto l’anello elettrostatico che ha accelerato le microsfere dalla testa dell’ago. Dopo il lavaggio due volte con HBSS con Ca / mg, le microsfere sono state trasferite in mezzo RPMI 1640 contenente il 10% di siero AB umano e incubato a 37 ° C, 21% O2 e 5% di CO2. Non sono stati effettuati cambiamenti di terreno, e il supernatante è stato raccolto in punti temporali definiti per l’analisi. La crescita di Mtb all’interno di microsfere è stata monitorata longitudinalmente dalla luminescenza (GloMax 20/20 Luminometro, Promega).
Immunofluorescenza e imaging confocale
In esperimenti specifici di imaging, i PBMC sono stati preetichettati con CellTracker Blue, CellTrace CFSE o Hoechst 33342 (ThermoFisher Scientific, Regno Unito) secondo le raccomandazioni del produttore. Le microsfere sono state fissate in paraformaldeide al 4% e lavate in HBSS con Ca/Mg. Le immagini Confocal sono state acquisite su un microscopio Leica TCS SP5 Confocal ed elaborate utilizzando l’immagine J 1.5 0d (NIH, USA).
Colorazione istologica e immunoistochimica
Il settimo e il quattordicesimo giorno di incubazione, le microsfere sono state fissate con il 4% di paraformaldeide durante la notte e inserite nella paraffina utilizzando il sistema Shandon Cytoblock (ThermoFisher Scientific, UK). I blocchi sono stati sezionati e macchiati di ematossilina ed eosina. Per l’immunoistochimica CD68 (Dako, Clone PG-M1), sono state colorate sezioni di 0,5 μm. Analisi del tessuto polmonare umano preso come parte della cura clinica di routine è stata approvata dal Consiglio di revisione istituzionale (riferimento 12/NW/0794 SRB04_14). Le sezioni sono state decerate, bloccate (Envision FLEX), colorate con Anti-Human CD68 (Dako, Clone PG-M1), rilevate con HRP e colorate con ematossilina.
Analisi citometrica a flusso
Le cellule sono state estratte sciogliendo le microsfere in 55 mM di citrato di sodio e 10 mM di EDTA in PBS per 10 minuti a 37°C. Le cellule sono state poi sospese in PBS. La colorazione superficiale e intracellulare è stata fatta in un’analisi a tre colori con combinazioni di fluoresceina isotiocianato (FITC), ficoeritrina (PE) e alloficocianina (APC). Gli anticorpi utilizzati comprendevano anti-CD3, anti-CD4, anti-CD8, anti-CD14 e anti-CD68 (ImmunoTools, Germania). Per la proliferazione delle cellule T, i PBMC sono stati colorati con CellTrace CFSE Cell Proliferation Kit per citometria a flusso (ThermoFisher Scientific, UK) prima dell’infezione da Mtb. La fluorescenza è stata poi analizzata con citometria a flusso (citometro a flusso BD Accuri C6).
Espressione del gene MMP-1 e IFN-γ
Le microsfere sono state decapulate utilizzando 55 mM di soluzione di citrato di sodio e le cellule sono state lisate immediatamente utilizzando TRIzol Reagent (Life Technologies, Paisley, UK). 1 μg di RNA è stato trascritto al contrario utilizzando il kit di trascrizione inversa del cDNA ad alta capacità (Life Technologies Ltd, Paisley, UK). Taqman Universal master mix e primer specifici per il gene GAPDH, β-Actin, MMP1 e IFN-γ sono stati utilizzati per la qPCR seguendo le istruzioni del produttore (Applied Biosystems, USA) e il metodo della soglia comparativa (CT) è stato impiegato per analizzare tutti i dati qPCR.
Analisi Luminex
I campioni sono stati sterilizzati per filtrazione attraverso una membrana Durapore da 0,22 μM (Millipore) (Elkingtonet al., 2006). Le concentrazioni di citochine (Life Technologies, UK) e MMP (R and D Systems, UK) sono state determinate utilizzando una piattaforma Bioplex 200 (Bio-Rad, UK) secondo il protocollo del produttore.
DQ test di degradazione del collagene
Per l’analisi della degradazione della matrice extracellulare, sono state generate microsfere da una soluzione di alginato al 3% (alginato Pronova UP MVG, Nova Matrix, Norvegia), 1 mg/ml di collagene umano di tipo I (Advanced BioMatrix, San Diego, California) e 100 µg/ml di collagene DQ (Invitrogen, Paisley, UK). Le microsfere sono state poi collocate in terreno libero di siero macrofago (Life Technologies) e incubate a 37°C. La fluorescenza è stata letta su un GloMax Discover (Promega) a un massimo di assorbimento di 495 nm e un massimo di emissione di fluorescenza di 515 nm.
Saggio di vitalità cellulare
Le microsfere sono state incubate in piastre a 96 pozzetti a 37°C. La vitalità cellulare è stata analizzata utilizzando il CellTiter-Glo 3D Cell Viability Assay (Promega) secondo le istruzioni del produttore, analizzando le cellule vitali in base alla quantificazione ATP. La luminescenza è stata analizzata dal lettore di placche GloMax Discover (Promega).
Saggi di tossicità cellulare
Il test di citotossicità CytoTox-Glo Cytotoxicity Assay (Promega) ha misurato la necrosi cellulare nelle microsfere. Il kit misura l’attività della proteasi a cellule morte rilasciata dalle cellule senza integrità della membrana utilizzando un substrato peptidico luminogenico, il substrato AAF-Glo. La luminescenza è stata analizzata da GloMax Discover (Promega). La morte totale delle cellule è stata causata dalla digitonina su numeri di cellule uguali per fornire il denominatore. Come seconda misura di tossicità, il rilascio di lattato deidrogenasi (LDH) è stato analizzato da un test di attività colorimetrica (Roche, Burgess Hill, Regno Unito). Il confronto tra la coltura cellulare 2D e la vitalità della coltura cellulare 3D è stato effettuato placcando un numero uguale di cellule nei pozzetti di una piastra a 96 pozzetti, e poi misurando la tossicità con LDH e CytoTox-Glo. La tossicità totale è stata normalizzata ai pozzetti trattati con digitonina placcati in parallelo.
Apoptosi cellulare
Le microsfere sono state incubate in piastre a 96 pozzetti. Le attività di proteasi Caspase-3/7 sono state misurate come indicatori di apoptosi utilizzando Apo-ONE Homogeneous Caspase-3/7 Assay (Promega) o Caspase-Glo 3/7 Assay Systems (Promega) secondo le istruzioni del produttore.
Rapporto NADP/NADPH
Il kit bioluminescente NADP/NADPH (Promega) è stato utilizzato secondo le istruzioni del produttore.
Integrazione di citochina e PG
Le microsfere sono state incubate in RPMI 1640 con il 10% di siero AB con IFN-β, IFN-γ (eBioscience) o PGE2 (sistemi R eD ) a 37°C.
Immunoaugmentazione con cellule T autologhe
Per generare linee di cellule T specifiche espanse, le cellule specifiche ESAT-6 o CFP-10 sono state derivate da PBMC di individui esposti a Mtb(Wölfl e Greenberg, 2014). I monociti sono stati isolati dalle PBMC utilizzando la separazione delle cellule magnetiche (Miltenyi Biotec, UK) e parzialmente differenziati in cellule dendritiche derivate da monociti (moDC) per 3 giorni utilizzando mezzi completi integrati con GM-CSF (20 ng/ml) e IL-4 (25 ng/ml). moDCs sono stati poi caricati con pool di antigene peptidico derivati da ESAT-6 o CFP-10 proteine in presenza di IFN-γ e LPS. moDCs sono stati poi esposti a CD14- T frazioni cellulari in un rapporto 1:2 per 7 giorni, dopo di che IL-2 (400IU / ml, Proleukin, Chiron), IL-15 (2 ng / ml) e IL-7 (2 ng / ml, Immunotools) sono stati aggiunti. La specificità delle cellule T è stata confermata dalla secrezione di IFN-γ in seguito all’esposizione delle cellule T a moDC autologhi caricati con CFP-10 o ESAT-6. Umani iNK cellule T sono stati derivati da PBMCs secondo il metodo precedentemente descritto(Mansour et al., 2015). In breve, le linee di cellule T iNK iNK sono state generate incubando le PBMC con 200 ng/ml KRN7000 (AXXORA) per 7 giorni prima dell’aggiunta di IL-2, IL-7 e IL-15. Dopo due settimane di coltura, l’espansione delle cellule iNK T è stata confermata tramite la colorazione tetrametrica CD3, Va24 e CD1d-K7. Le cellule sono state acquisite utilizzando FACSAria (Becton Dickinson, UK). ESAT-6, CFP-10 specifiche linee cellulari T o cellule T iNK iNK sono stati contati e poi PBMCs sono stati integrati con il 20% di cellule immunoaugmentate aggiuntive immediatamente prima dell’infezione Mtb. Dopo l’incubazione notturna, le cellule combinate sono state bioelettrospruzzate con il nostro protocollo standard.
Analisi statistica
Tutti gli esperimenti sono stati eseguiti in almeno 2 occasioni da donatori separati come repliche biologiche e in ogni occasione con un minimo di 3 repliche tecniche. I dati presentati provengono da un donatore rappresentativo e includono la media e il SEM. L’analisi è stata eseguita in Graphpad Prism v6.0. Il t-test degli studenti è stato utilizzato per confrontare le coppie e l’ANOVA per gruppi di 3 o più persone.
References
- Aagaard C, Hoang T, Dietrich J, Cardona PJ, Izzo A, Dolganov G, Schoolnik GK, Cassidy JP, Billeskov R, Andersen P. A multistage tuberculosis vaccine that confers efficient protection before and after exposure. Nature Medicine. 2011; 17:189-194. DOI | PubMed
- Al Shammari B, Shiomi T, Tezera L, Bielecka MK, Workman V, Sathyamoorthy T, Mauri F, Jayasinghe SN, Robertson BD, D’Armiento J, Friedland JS, Elkington PT. The extracellular matrix regulates granuloma necrosis in tuberculosis. Journal of Infectious Diseases. 2015; 212:463-473. DOI | PubMed
- Andreu N, Zelmer A, Fletcher T, Elkington PT, Ward TH, Ripoll J, Parish T, Bancroft GJ, Schaible U, Robertson BD, Wiles S. Optimisation of bioluminescent reporters for use with mycobacteria. PLoS One. 2010; 5DOI | PubMed
- Barrila J, Radtke AL, Crabbé A, Sarker SF, Herbst-Kralovetz MM, Ott CM, Nickerson CA. Organotypic 3D cell culture models: using the rotating wall vessel to study host-pathogen interactions. Nature Reviews Microbiology. 2010; 8:791-801. DOI | PubMed
- Benam KH, Dauth S, Hassell B, Herland A, Jain A, Jang KJ, Karalis K, Kim HJ, MacQueen L, Mahmoodian R, Musah S, Torisawa YS, van der Meer AD, Villenave R, Yadid M, Parker KK, Ingber DE. Engineered in vitro disease models. Annual Review of Pathology: Mechanisms of Disease. 2015; 10:195-262. DOI | PubMed
- Brites D, Gagneux S. Co-evolution of Mycobacterium tuberculosis and Homo sapiens. Immunological Reviews. 2015; 264:6-24. DOI | PubMed
- Buchheit CL, Rayavarapu RR, Schafer ZT. The regulation of cancer cell death and metabolism by extracellular matrix attachment. Seminars in Cell & Developmental Biology. 2012; 23:402-411. DOI | PubMed
- Calderon VE, Valbuena G, Goez Y, Judy BM, Huante MB, Sutjita P, Johnston RK, Estes DM, Hunter RL, Actor JK, Cirillo JD, Endsley JJ. A humanized mouse model of tuberculosis. PLoS One. 2013; 8DOI | PubMed
- Cambier CJ, Falkow S, Ramakrishnan L. Host evasion and exploitation schemes of Mycobacterium tuberculosis. Cell. 2014; 159:1497-1509. DOI | PubMed
- Carroll P, Schreuder LJ, Muwanguzi-Karugaba J, Wiles S, Robertson BD, Ripoll J, Ward TH, Bancroft GJ, Schaible UE, Parish T. Sensitive detection of gene expression in mycobacteria under replicating and non-replicating conditions using optimized far-red reporters. PLoS One. 2010; 5DOI | PubMed
- Chakraborty S, Ji H, Kabadi AM, Gersbach CA, Christoforou N, Leong KW. A CRISPR/Cas9-based system for reprogramming cell lineage specification. Stem Cell Reports. 2014; 3:940-947. DOI | PubMed
- Cheng F, Pekkonen P, Laurinavicius S, Sugiyama N, Henderson S, Günther T, Rantanen V, Kaivanto E, Aavikko M, Sarek G, Hautaniemi S, Biberfeld P, Aaltonen L, Grundhoff A, Boshoff C, Alitalo K, Lehti K, Ojala PM. KSHV-initiated notch activation leads to membrane-type-1 matrix metalloproteinase-dependent lymphatic endothelial-to-mesenchymal transition. Cell Host & Microbe. 2011; 10:577-590. DOI | PubMed
- Cliff JM, Kaufmann SH, McShane H, van Helden P, O’Garra A. The human immune response to tuberculosis and its treatment: a view from the blood. Immunological Reviews. 2015; 264:88-102. DOI | PubMed
- Comas I, Chakravartti J, Small PM, Galagan J, Niemann S, Kremer K, Ernst JD, Gagneux S. Human T cell epitopes of Mycobacterium tuberculosis are evolutionarily hyperconserved. Nature Genetics. 2010; 42:498-503. DOI | PubMed
- Comstock GW, Livesay VT, Woolpert SF. The prognosis of a positive tuberculin reaction in childhood and adolescence. American Journal of Epidemiology. 1974; 99:131-138. PubMed
- Coscolla M, Copin R, Sutherland J, Gehre F, de Jong B, Owolabi O, Mbayo G, Giardina F, Ernst JD, Gagneux S. M. tuberculosis T cell epitope analysis reveals paucity of antigenic variation and identifies rare variable TB antigens. Cell Host & Microbe. 2015; 18:538-548. DOI | PubMed
- Crowle AJ, Elkins N. Relative permissiveness of macrophages from black and white people for virulent tubercle bacilli. Infection and Immunity. 1990; 58:632-638. PubMed
- del Corral H, París SC, Marín ND, Marín DM, López L, Henao HM, Martínez T, Villa L, Barrera LF, Ortiz BL, Ramírez ME, Montes CJ, Oquendo MC, Arango LM, Riaño F, Aguirre C, Bustamante A, Belisle JT, Dobos K, Mejía GI, Giraldo MR, Brennan PJ, Robledo J, Arbeláez MP, Rojas CA, García LF. IFNgamma response to Mycobacterium tuberculosis, risk of infection and disease in household contacts of tuberculosis patients in Colombia. PLoS One. 2009; 4DOI | PubMed
- Douvas GS, Looker DL, Vatter AE, Crowle AJ. Gamma interferon activates human macrophages to become tumoricidal and leishmanicidal but enhances replication of macrophage-associated mycobacteria. Infection and Immunity. 1985; 50:1-8. PubMed
- Egen JG, Rothfuchs AG, Feng CG, Winter N, Sher A, Germain RN, Macrophage GRN. Macrophage and T cell dynamics during the development and disintegration of mycobacterial granulomas. Immunity. 2008; 28:271-284. DOI | PubMed
- Elkington PT, D’Armiento JM, Friedland JS. Tuberculosis immunopathology: the neglected role of extracellular matrix destruction. Science Translational Medicine. 2011; 3:71ps6. DOI | PubMed
- Elkington PT, Friedland JS. Permutations of time and place in tuberculosis. The Lancet Infectious Diseases. 2015; 15:1357-1360. DOI | PubMed
- Elkington PT, Green JA, Friedland JS. Filter sterilization of highly infectious samples to prevent false negative analysis of matrix metalloproteinase activity. Journal of Immunological Methods. 2006; 309:115-119. DOI | PubMed
- Flynn JL, Chan J. Immunology of tuberculosis. Annual Review of Immunology. 2001; 19:93-129. DOI | PubMed
- Gideon HP, Phuah J, Myers AJ, Bryson BD, Rodgers MA, Coleman MT, Maiello P, Rutledge T, Marino S, Fortune SM, Kirschner DE, Lin PL, Flynn JL. Variability in tuberculosis granuloma T cell responses exists, but a balance of pro- and anti-inflammatory cytokines is associated with sterilization. PLoS Pathogens. 2015; 11DOI | PubMed
- Guirado E, Schlesinger LS. Modeling the mycobacterium tuberculosis granuloma – the critical battlefield in host immunity and disease. Frontiers in Immunology. 2013; 4:98. DOI | PubMed
- Hawn TR, Matheson AI, Maley SN, Vandal O. Host-directed therapeutics for tuberculosis: can we harness the host?. Microbiology and Molecular Biology Reviews. 2013; 77:608-627. DOI | PubMed
- Hawn TR, Shah JA, Kalman D. New tricks for old dogs: countering antibiotic resistance in tuberculosis with host-directed therapeutics. Immunological Reviews. 2015; 264:344-362. DOI | PubMed
- Heuts F, Gavier-Widén D, Carow B, Juarez J, Wigzell H, Rottenberg ME. CD4+ cell-dependent granuloma formation in humanized mice infected with mycobacteria. PNAS. 2013; 110:6482-6487. DOI | PubMed
- Higuchi K, Harada N, Fukazawa K, Mori T. Relationship between whole-blood interferon-gamma responses and the risk of active tuberculosis. Tuberculosis. 2008; 88:244-248. DOI | PubMed
- Horsburgh CR, Barry CE, Lange C. Treatment of Tuberculosis. New England Journal of Medicine. 2015; 373:2149-2160. DOI | PubMed
- Jasenosky LD, Scriba TJ, Hanekom WA, Goldfeld AE. T cells and adaptive immunity to Mycobacterium tuberculosis in humans. Immunological Reviews. 2015; 264:74-87. DOI | PubMed
- Kapoor N, Pawar S, Sirakova TD, Deb C, Warren WL, Kolattukudy PE. Human granuloma in vitro model, for TB dormancy and resuscitation. PLoS One. 2013; 8DOI | PubMed
- Karp CL, Wilson CB, Stuart LM. Tuberculosis vaccines: barriers and prospects on the quest for a transformative tool. Immunological Reviews. 2015; 264:363-381. DOI | PubMed
- Kaufmann SH, Dorhoi A. Inflammation in tuberculosis: interactions, imbalances and interventions. Current Opinion in Immunology. 2013; 25:441-449. DOI | PubMed
- Kimmey JM, Huynh JP, Weiss LA, Park S, Kambal A, Debnath J, Virgin HW, Stallings CL. Unique role for ATG5 in neutrophil-mediated immunopathology during M. tuberculosis infection. Nature. 2015; 528:565-569. DOI | PubMed
- Lay G, Poquet Y, Salek-Peyron P, Puissegur MP, Botanch C, Bon H, Levillain F, Duteyrat JL, Emile JF, Altare F. Langhans giant cells from M. tuberculosis-induced human granulomas cannot mediate mycobacterial uptake. The Journal of Pathology. 2007; 211:76-85. DOI | PubMed
- Lindestam Arlehamn CS, Paul S, Mele F, Huang C, Greenbaum JA, Vita R, Sidney J, Peters B, Sallusto F, Sette A. Immunological consequences of intragenus conservation of Mycobacterium tuberculosis T-cell epitopes. PNAS. 2015; 112:E147-E155. DOI | PubMed
- Luabeya AK, Kagina BM, Tameris MD, Geldenhuys H, Hoff ST, Shi Z, Kromann I, Hatherill M, Mahomed H, Hanekom WA, Andersen P, Scriba TJ, Schoeman E, Krohn C, Day CL, Africa H, Makhethe L, Smit E, Brown Y, Suliman S, Hughes EJ, Bang P, Snowden MA, McClain B, Hussey GD, H56-032 Trial Study Group. First-in-human trial of the post-exposure tuberculosis vaccine H56:IC31 in Mycobacterium tuberculosis infected and non-infected healthy adults. Vaccine. 2015; 33:4130-4140. DOI | PubMed
- Majlessi L, Prados-Rosales R, Casadevall A, Brosch R. Release of mycobacterial antigens. Immunological Reviews. 2015; 264:25-45. DOI | PubMed
- Mansour S, Tocheva AS, Sanderson JP, Goulston LM, Platten H, Serhal L, Parsons C, Edwards MH, Woelk CH, Elkington PT, Elliott T, Cooper C, Edwards CJ, Gadola SD. Structural and functional changes of the invariant NKT clonal repertoire in early rheumatoid arthritis. The Journal of Immunology. 2015; 195:5582-5591. DOI | PubMed
- Marakalala MJ, Raju RM, Sharma K, Zhang YJ, Eugenin EA, Prideaux B, Daudelin IB, Chen PY, Booty MG, Kim JH, Eum SY, Via LE, Behar SM, Barry CE, Mann M, Dartois V, Rubin EJ. Inflammatory signaling in human tuberculosis granulomas is spatially organized. Nature Medicine. 2016; 22:531-538. DOI | PubMed
- Mattila JT, Ojo OO, Kepka-Lenhart D, Marino S, Kim JH, Eum SY, Via LE, Barry CE, Klein E, Kirschner DE, Morris SM, Lin PL, Flynn JL. Microenvironments in tuberculous granulomas are delineated by distinct populations of macrophage subsets and expression of nitric oxide synthase and arginase isoforms. The Journal of Immunology. 2013; 191:773-784. DOI | PubMed
- Mayer-Barber KD, Andrade BB, Oland SD, Amaral EP, Barber DL, Gonzales J, Derrick SC, Shi R, Kumar NP, Wei W, Yuan X, Zhang G, Cai Y, Babu S, Catalfamo M, Salazar AM, Via LE, Barry CE, Sher A. Host-directed therapy of tuberculosis based on interleukin-1 and type I interferon crosstalk. Nature. 2014; 511:99-103. DOI | PubMed
- Moreira-Teixeira L, Sousa J, McNab FW, Torrado E, Cardoso F, Machado H, Castro F, Cardoso V, Gaifem J, Wu X, Appelberg R, Castro AG, O’Garra A, Saraiva M. Type I IFN inhibits alternative macrophage activation during Mycobacterium tuberculosis infection and leads to enhanced protection in the absence of IFN-γ signaling. The Journal of Immunology. 2016; 197:4714-4726. DOI | PubMed
- Mueller-Klieser W. Three-dimensional cell cultures: from molecular mechanisms to clinical applications. The American Journal of Physiology. 1997; 273:C1109-C1123. PubMed
- Ndiaye BP, Thienemann F, Ota M, Landry BS, Camara M, Dièye S, Dieye TN, Esmail H, Goliath R, Huygen K, January V, Ndiaye I, Oni T, Raine M, Romano M, Satti I, Sutton S, Thiam A, Wilkinson KA, Mboup S, Wilkinson RJ, McShane H, MVA85A 030 trial investigators. Safety, immunogenicity, and efficacy of the candidate tuberculosis vaccine MVA85A in healthy adults infected with HIV-1: a randomised, placebo-controlled, phase 2 trial. The Lancet Respiratory Medicine. 2015; 3:190-200. DOI | PubMed
- Nouailles G, Dorhoi A, Koch M, Zerrahn J, Weiner J, Faé KC, Arrey F, Kuhlmann S, Bandermann S, Loewe D, Mollenkopf HJ, Vogelzang A, Meyer-Schwesinger C, Mittrücker HW, McEwen G, Kaufmann SH. CXCL5-secreting pulmonary epithelial cells drive destructive neutrophilic inflammation in tuberculosis. Journal of Clinical Investigation. 2014; 124:1268-1282. DOI | PubMed
- Nunes-Alves C, Booty MG, Carpenter SM, Jayaraman P, Rothchild AC, Behar SM. In search of a new paradigm for protective immunity to TB. Nature Reviews Microbiology. 2014; 12:289-299. DOI | PubMed
- O’Garra A, Redford PS, McNab FW, Bloom CI, Wilkinson RJ, Berry MP. The immune response in tuberculosis. Annual Review of Immunology. 2013; 31:475-527. DOI | PubMed
- O’Kane CM, Elkington PT, Jones MD, Caviedes L, Tovar M, Gilman RH, Stamp G, Friedland JS. STAT3, p38 MAPK, and NF-kappaB drive unopposed monocyte-dependent fibroblast MMP-1 secretion in tuberculosis. American Journal of Respiratory Cell and Molecular Biology. 2010; 43:465-474. DOI | PubMed
- Orme IM, Robinson RT, Cooper AM. The balance between protective and pathogenic immune responses in the TB-infected lung. Nature Immunology. 2015; 16:57-63. DOI | PubMed
- Pampaloni F, Reynaud EG, Stelzer EH. The third dimension bridges the gap between cell culture and live tissue. Nature Reviews Molecular Cell Biology. 2007; 8:839-845. DOI | PubMed
- Parasa VR, Rahman MJ, Ngyuen Hoang AT, Svensson M, Brighenti S, Lerm M. Modeling Mycobacterium tuberculosis early granuloma formation in experimental human lung tissue. Disease Models & Mechanisms. 2014; 7:281-288. DOI | PubMed
- Parker MW, Rossi D, Peterson M, Smith K, Sikström K, White ES, Connett JE, Henke CA, Larsson O, Bitterman PB. Fibrotic extracellular matrix activates a profibrotic positive feedback loop. Journal of Clinical Investigation. 2014; 124:1622-1635. DOI | PubMed
- Philips JA, Ernst JD. Tuberculosis pathogenesis and immunity. Annual Review of Pathology: Mechanisms of Disease. 2012; 7:353-384. DOI | PubMed
- Puissegur MP, Botanch C, Duteyrat JL, Delsol G, Caratero C, Altare F. An in vitro dual model of mycobacterial granulomas to investigate the molecular interactions between mycobacteria and human host cells. Cellular Microbiology. 2004; 6:423-433. DOI | PubMed
- Reikerås O, Helle A, Krohn CD, Brox JI. Effects of high-dose corticosteroids on post-traumatic inflammatory mediators. Inflammation Research. 2009; 58:891-897. DOI | PubMed
- Rook GA, Steele J, Ainsworth M, Champion BR. Activation of macrophages to inhibit proliferation of Mycobacterium tuberculosis: comparison of the effects of recombinant gamma-interferon on human monocytes and murine peritoneal macrophages. Immunology. 1986; 59:333-338. PubMed
- Russell DG. Mycobacterium tuberculosis and the intimate discourse of a chronic infection. Immunological Reviews. 2011; 240:252-268. DOI | PubMed
- Schwartz MA, Chen CS. Cell biology. Deconstructing dimensionality. Science. 2013; 339:402-404. DOI | PubMed
- Seok J, Warren HS, Cuenca AG, Mindrinos MN, Baker HV, Xu W, Richards DR, McDonald-Smith GP, Gao H, Hennessy L, Finnerty CC, López CM, Honari S, Moore EE, Minei JP, Cuschieri J, Bankey PE, Johnson JL, Sperry J, Nathens AB, Billiar TR, West MA, Jeschke MG, Klein MB, Gamelli RL, Gibran NS, Brownstein BH, Miller-Graziano C, Calvano SE, Mason PH, Cobb JP, Rahme LG, Lowry SF, Maier RV, Moldawer LL, Herndon DN, Davis RW, Xiao W, Tompkins RG, Inflammation and Host Response to Injury, Large Scale Collaborative Research Program. Genomic responses in mouse models poorly mimic human inflammatory diseases. PNAS. 2013; 110:3507-3512. DOI | PubMed
- Snijder B, Sacher R, Rämö P, Damm EM, Liberali P, Pelkmans L. Population context determines cell-to-cell variability in Endocytosis and virus infection. Nature. 2009; 461:520-523. DOI | PubMed
- Sorokin L. The impact of the extracellular matrix on inflammation. Nature Reviews Immunology. 2010; 10:712-723. DOI | PubMed
- Tameris MD, Hatherill M, Landry BS, Scriba TJ, Snowden MA, Lockhart S, Shea JE, McClain JB, Hussey GD, Hanekom WA, Mahomed H, McShane H, MVA85A 020 Trial Study Team. Safety and efficacy of MVA85A, a new tuberculosis vaccine, in infants previously vaccinated with BCG: a randomised, placebo-controlled phase 2b trial. The Lancet. 2013; 381:1021-1028. DOI | PubMed
- Tezera L, Bielecka M, Elkington P. Bioelectrospray Methodology for Dissection of the Host-pathogen Interaction in Human Tuberculosis. BIO-PROTOCOL. 2017; 7DOI
- Van Rhijn I, Moody DB, Rhijn V, Moody DB. CD1 and mycobacterial lipids activate human T cells. Immunological Reviews. 2015; 264:138-153. DOI | PubMed
- Vogt G, Nathan C. In vitro differentiation of human macrophages with enhanced antimycobacterial activity. Journal of Clinical Investigation. 2011; 121:3889-3901. DOI | PubMed
- Warner DF, Mizrahi V. Shortening treatment for tuberculosis–to basics. New England Journal of Medicine. 2014; 371:1642-1643. DOI | PubMed
- Workman VL, Tezera LB, Elkington PT, Jayasinghe SN. Controlled generation of microspheres incorporating extracellular matrix fibrils for Three-Dimensional cell culture. Advanced Functional Materials. 2014; 24:2648-2657. DOI | PubMed
- Wölfl M, Greenberg PD. Antigen-specific activation and cytokine-facilitated expansion of naive, human CD8+ T cells. Nature Protocols. 2014; 9:950-966. DOI | PubMed
- Yamada KM, Cukierman E. Modeling tissue morphogenesis and cancer in 3D. Cell. 2007; 130:601-610. DOI | PubMed
- Young D. Animal models of tuberculosis. European Journal of Immunology. 2009; 39:2011-2014. DOI | PubMed
- Zumla A, Maeurer M, Chakaya J, Hoelscher M, Ntoumi F, Rustomjee R, Vilaplana C, Yeboah-Manu D, Rasolof V, Munderi P, Singh N, Aklillu E, Padayatchi N, Macete E, Kapata N, Mulenga M, Kibiki G, Mfinanga S, Nyirenda T, Maboko L, Garcia-Basteiro A, Rakotosamimanana N, Bates M, Mwaba P, Reither K, Gagneux S, Edwards S, Mfinanga E, Abdulla S, Cardona PJ, Russell JB, Gant V, Noursadeghi M, Elkington P, Bonnet M, Menendez C, Dieye TN, Diarra B, Maiga A, Aseffa A, Parida S, Wejse C, Petersen E, Kaleebu P, Oliver M, Craig G, Corrah T, Tientcheu L, Antonio M, Rao M, McHugh TD, Sheikh A, Ippolito G, Ramjee G, Kaufmann SH, Churchyard G, Steyn A, Grobusch M, Sanne I, Martinson N, Madansein R, Wilkinson RJ, Mayosi B, Schito M, Wallis RS, Host-Directed Therapies Network. Towards host-directed therapies for tuberculosis. Nature Reviews Drug Discovery. 2015; 14:511-512. DOI | PubMed
Fonte
Tezera LB, Bielecka MK, Chancellor A, Reichmann MT, Shammari BA, et al. () Dissection of the host-pathogen interaction in human tuberculosis using a bioengineered 3-dimensional model. eLife 6e21283. https://doi.org/10.7554/eLife.21283




