
Abstract
Dichiarazione di rilevanza
L’allele ε4 dell’apolipoproteina E (APOE4)èil più forte fattore di rischio genetico per la malattia di Alzheimer (AD), ma non tutti i portatori diAPOE4 svilupperanno la malattia suggerendo che il genotipo APOE interagisce con altri fattori per modulare il rischio di Alzheimer. Qui, mostriamo che l’obesità indotta dalla dieta (DIO) interagisce con il genotipo APOE4per aumentare la patologia simile all’Alzheimer in un modello murino transgenico di Alzheimer che contiene isoforme umane APOE3 controAPOE4. È interessante notare che i topi con APOE3non mostrano aumenti della patologia indotti dalla dieta, suggerendo che gli effetti negativi dell’obesità sul rischio di Alzheimer possono essere limitati ai portatori di APOE4. Questi risultati identificano un’importante interazione gene-ambiente che può avere un impatto significativo per la comprensione del rischio e dell’eziologia dell’Alzheimer e per promuovere lo sviluppo di approcci terapeutici mirati che incorporano sia l’obesità che il genotipo dell’APOE.
Introduzione
Il morbo di Alzheimer (AD) è un disturbo neurodegenerativo progressivo, le cui cause sottostanti sono attualmente incomplete. Sia i fattori genetici e ambientali sono importanti nel determinare il rischio individuale per la malattia di Alzheimer. Il più forte fattore di rischio genetico per l’AD ad insorgenza tardiva è l’allele ε4 dell’apolipoproteina E (APOE4;Strittmatter et al., 1993; Liu et al., 2013). Negli Stati Uniti, circa il 12% della popolazione ha l’allele ε4, ma la sua frequenza aumenta fino al 7% nei pazienti affetti da AD (Rebeck etal., 1993). L’APOE4non solo aumenta il rischio, ma accelera anche l’età di insorgenza della DA(Corder et al., 1993; van der Flier et al., 2011). Tuttavia, poiché i portatori omozigoti di APOE4hanno un rischio di APOE4 per tutta la vita, un numero significativo di portatori di APOE4 non sviluppa mai la malattia (Genin et al., 2011). Pertanto, l ‘APOE4probabilmente interagisce con altri fattori genetici e/o ambientali per guidare il rischio di AD.
Un significativo fattore di rischio modificabile per la demenza è l’obesità. L’obesità ha numerosi effetti neurali negativi(Lee e Mattson, 2013) e aumenta il rischio di demenza fino a tre volte(Whitmer et al., 2008). L’indice di massa corporea, una misura comunemente usata per misurare l’obesità, ha dimostrato di essere associato al rischio di AD(Profenno et al., 2010) così come alla riduzione del volume cerebrale nei pazienti con AD(Ho et al., 2010). Diversi studi indicano che l’obesità può essere particolarmente problematica a metà della vita(Fitzpatrick et al., 2009; Profenno et al., 2010; Meng et al., 2014; Emmerzaal et al., 2015), suggerendo che l’obesità contribuisce allo sviluppo dell’AD. Relazioni simili sono state osservate in modelli animali. In particolare, l’obesità indotta dalla dieta (DIO) accelera la patologia correlata all’AD nei modelli murini di AD(Ho et al., 2004; Julien et al., 2010; Kohjima et al., 2010; Barron et al., 2013; Orr et al., 2014). Inoltre, i modelli genetici dell’obesità e del diabete di tipo 2 presentano caratteristiche della neuropatologia di tipo AD(Kim et al., 2009; Jung et al., 2013; Ramos-Rodriguez et al., 2013).
Non è chiaro in che misura l’APOE4e l’obesità interagiscono per regolare il rischio di AD. È interessante notare che i portatori di APOE4possono essere più sensibili alle conseguenze metaboliche associate all’obesità(de-Andrade et al., 2000; Kypreos et al., 2009; Niu et al., 2009; Atabek et al., 2012; Zarkesh et al., 2012; Guan et al., 2013). Anche se alcuni studi non riportano un pregiudizio APOE4nel rischio di AD associato all’obesità(Profenno e Faraone, 2008; Luchsinger et al., 2012), altri hanno scoperto che il rischio di AD è aumentato dall’obesità(Peila et al., 2002; Ghebranious et al., 2011) e da diete ricche di calorie e acidi grassi(Luchsinger et al., 2002) solo nei portatori di APOE4. Anche se la letteratura umana suggerisce un’interazione gene-ambiente tra APOE e obesità nel regolare lo sviluppo di AD, questa questione non è stata affrontata in modelli sperimentali. Per studiare queste relazioni, abbiamo utilizzato i topi transgenici EFAD, che combinano i transgeni dell’AD con la sostituzione mirata dell’APOE dei topi con l’APOE umano(Youmans et al., 2012). Abbiamo confrontato gli effetti metabolici e AD-correlati della dieta occidentale nei topi maschi APOE3(E3FAD) e APOE4(E4FAD). Qui, riportiamo che la DIO aumenta la patologia amiloide e la gliosi quasi esclusivamente nei topi E4FAD. I nostri dati rivelano un’interazione gene-ambiente tra genotipo APOE e obesità, suggerendo che i portatori di APOE4possono essere più suscettibili agli aumenti associati all’obesità nel rischio di AD.
Materiali e metodi
Procedure per gli animali
Una colonia di topi EFAD, eterozigoti per i transgeni 5xFAD e omozigoti per gli umani APOE3o APOE4(Youmans et al., 2012), sono stati mantenuti presso le strutture di vivarium della University of Southern California da topi allevatori generosamente forniti dalla Dr. Mary Jo LaDu (University of Illinois at Chicago). Tutti gli animali sono stati alloggiati in un ciclo luce/buio di 12 ore con luci accese alle 6 del mattino e accesso ad libitum al cibo e all’acqua. A tre mesi di età, i topi maschi E3FAD e E4FAD sono stati randomizzati a gruppi di trattamento dietetico(N = 7-11/gruppo): dieta di controllo (10% di grassi, 7% di saccarosio; #D12450J Research Diets) o dieta occidentale (45% di grassi, 17% di saccarosio; #D12451, Research Diets). I topi EFAD sono stati mantenuti su diete sperimentali per settimane 12, un periodo di esposizione precedentemente stabilito per produrre obesità-indotta da alterazioni metaboliche nei topi APOE(Arbones-Mainar et al., 2010; Segev et al., 2016). Il peso corporeo e il consumo di cibo sono stati registrati settimanalmente.
Alla fine del periodo di trattamento, i topi sono stati anestetizzati con isoflurano per via inalatoria e perfusi transcardialmente con ghiaccio 0,1 M PBS. Il cervello è stato rapidamente rimosso e fissato per 48 ore in paraformaldeide 4% paraformaldeide / 0,1 M PBS, poi conservato a 4 ° C in 0,1 M PBS / 0,3% NaN3 fino al trattamento per immunoistochimica. Gonadal e cuscinetti di grasso retroperitoneale sono stati sezionati e pesati come misura di adiposità, e scattare congelati per l’estrazione di RNA. Tutte le procedure animali sono state condotte in base a protocolli approvati dalla University of Southern California Institutional Animal Care and Use Committee e in conformità con gli standard del National Institute of Health.
Misurazioni di glucosio, colesterolo e trigliceridi
I valori della glicemia sono stati misurati dopo il digiuno notturno (16 ore) ogni quattro settimane a partire dalla settimana 0 del periodo di trattamento di 12 settimane. Il sangue è stato raccolto dalla vena della coda laterale e immediatamente valutato per i livelli di glucosio utilizzando il sistema di controllo del glucosio e dei chetoni di precisione Xtra Blood Glucose and Ketone Monitoring System (Abbott Diabetes Care).
Il test di tolleranza al glucosio (GTT) è stato eseguito alla settimana 11. A digiuno, sono state effettuate letture del glucosio di base, dopo di che ai topi è stato somministrato un bolo di glucosio (2 g/kg di peso corporeo) per via orale. I livelli di glucosio nel sangue sono stati registrati 15, 30, 60 e 120 minuti dopo la somministrazione del bolo di glucosio. È stata calcolata l’area sotto la curva (AUC).
I livelli di colesterolo plasmatico e di trigliceridi sono stati determinati enzimaticamente alla conclusione dell’esperimento utilizzando i kit disponibili in commercio (LabAssay Triglycerides #290-63701, Wako Chemicals; Total Cholesterol Colorimetric Assay kit, #K603, BioVision). Tutti i campioni sono stati eseguiti in duplicato secondo le istruzioni del produttore.
Tioflavina-S (Tio-S) colorazione e quantificazione
Gli emi-cervelli fissi sono stati completamente sezionati sul piano orizzontale a 40 μm utilizzando un vibratore (Leica Biosystems). Ogni ottava sezione è stata colorata per Thio-S (#230456, Sigma-Aldrich) utilizzando la metodologia standard. Le sezioni sono state montate e lasciate asciugare durante la notte, dopo di che sono state lavate tre volte in 50% di etanolo per 5 minuti ciascuna, poi lavate inH2Oin doppia distillazione prima di essere incubate per 10 minuti in 1% di Thio-S disciolto inH2O. Vetrini macchiati sono stati poi risciacquati in etanolo al 70% prima di essere disidratati e coprivetrato in mezzo di montaggio acquoso anti-dissolvenza (Vector Laboratories). Le immagini digitali sono state catturate con ingrandimento 20× utilizzando un microscopio Olympus BX50 dotato di una fotocamera DP74 e del software CellSens (Olympus). Il numero di depositi sferici tioflavina positivi sono stati contati utilizzando NIH ImageJ 1.50i (United States National Institutes of Health) con il plugin del contatore di cellule per contrassegnare le strutture simili a placche colorate. Tioflavina depositi positivi sono stati contati nella corteccia entorinale (tre campi / sezione), subiculum (due campi / sezione), e ippocampo sottocampi CA1 (tre campi / sezione) e CA2/3 (tre campi / sezione), attraverso quattro sezioni per animale, per un totale di ∼44 campi per cervello.
Immunoistochimica
L’immunoistochimica è stata eseguita utilizzando un approccio standard di avidina / biotina perossidasi con i kit ABC Vector Elite (Vector Laboratories). Immunoistochimica Aβ è stata eseguita su ogni ottava sezione utilizzando sezioni immediatamente adiacenti a quelle trattate per Thio-S. In breve, le sezioni sono state pretrattate con acido formico al 95% per 5 min, poi risciacquate in TBS prima di essere trattati con una soluzione di blocco endogeno perossidasi per 10 min. Dopo tre lavaggi di 10 minuti in Triton-X/TBS allo 0,1%, le sezioni sono state incubate per 30 minuti in una soluzione bloccante composta dal 2% di siero albumina bovina in TBS. Sezioni bloccate sono stati incubati durante la notte a 4 ° C in anticorpo primario diretto contro Aβ (# 71-5800, diluizione 1:300, Invitrogen) che è stato diluito in soluzione di blocco. Successivamente, le sezioni sono state risciacquate e incubate in anticorpo secondario biotinilato diluito in soluzione bloccante. Immunoreattività è stata visualizzata utilizzando 3,3′-diamminobenzidina (laboratori vettoriali). Ulteriori sezioni sono stati analogamente immunocolorazione senza pretrattamento con acido formico utilizzando IBA-1 (#019-19741, diluizione 1:2000, Wako) e GFAP (#ab7260, diluizione 1:1000, Abcam).
Per quantificare l’area percentuale occupata da immunoreattività Aβ (carico Aβ), immagini di campi non sovrapposti sono state prese a 20× ingrandimento nella corteccia entorinale (tre campi / sezione), subiculum (tre campi / sezione), e ippocampo sottocampi CA1 (cinque campi / sezione) e CA2/3 (tre campi / sezione) attraverso 4 sezioni di tessuto, per un totale di ∼56 immagini per cervello. Le immagini sono state catturate digitalmente utilizzando un microscopio Olympus BX50 e una fotocamera DP74 abbinati a un computer con il software CellSens (Olympus). Le immagini sono state convertite in immagini in scala di grigi e soglia utilizzando NIH ImageJ 1.50i per produrre immagini binarie che separano l’immunocolorazione positiva e negativa. Il carico Aβ è stato calcolato come percentuale dell’area totale che è stata immuno-colorata positivamente.
L’attivazione di microglia e astrocittà è stata quantificata utilizzando immagini dal vivo (Olympus BX50, software CASTGrid, Olympus) con ingrandimento 40×. Ogni cellula è stata classificata come a riposo o reattiva in base alla sua morfologia, come riportato in studi precedenti(Ayoub e Salm ,2003; Wilhelmsson et al., 2006). In particolare, le microglia sono state classificate come a riposo (tipo 1) se avevano corpi cellulari sferici, con numerosi processi sottili e altamente ramificati. Le cellule sono state segnate come cellule di tipo 2 se presentavano corpi cellulari ingrossati a forma di bacchetta con meno processi più corti e più spessi, e sono state segnate come cellule di tipo 3 se avevano pochi o nessun processo o diversi processi filopodiali. Sia le morfologie di tipo 2 che quelle di tipo 3 erano considerate un fenotipo a microglia attivata. Gli astrociti sono stati visualizzati con immunocolorazione GFAP e classificati come non reattivi (corpi cellulari di dimensioni normali con poche proiezioni piuttosto corte) o reattivi (sia i corpi cellulari che le proiezioni sono ingrandite) fenotipi morfologici. Corteccia entorinale (quattro campi / sezione), subiculum (quattro campi / sezione), e ippocampo sottocampi CA-1 (cinque campi / sezione) e CA-2/3 (tre campi / sezione) sono stati quantificati sia per microglia e astrociti. Il numero di cellule attraverso le regioni cerebrali segnato per ogni animale in media ∼700 microglia e ∼600 astrociti.
Isolamento dell’RNA e PCR in tempo reale
Per le estrazioni di RNA, i cuscinetti di grasso gonadico e gli ippocampi sono stati omogeneizzati utilizzando il reagente TRIzol (Invitrogen), seguendo il protocollo del produttore. Il pellet di RNA è stato trattato con RNase-free DNasi I (Epicentro) per 30 minuti a 37 ° C, e un fenolo / cloroformio estrazione è stata eseguita per isolare l’RNA. Il sistema di sintesi iScript cDNA (Bio-Rad) è stato utilizzato per invertire la trascrizione di cDNA da 1 μg di RNA purificato. PCR quantitativa in tempo reale è stata eseguita sul cDNA risultante utilizzando SsoAdvanced Universal SYBR Green Supermix (Bio-Rad) e un Bio-Rad CFX Connect Thermocycler. Tutte le misurazioni sono state eseguite in duplicato. La quantificazione dei prodotti PCR è stata condotta normalizzando con una combinazione di corrispondenti iposantina-guanina fosforibosiltransferasi (HPRT) e succinato deidrogenasi [ubichinone] subunità flavoproteica, mitocondriale (SDHA) livelli di espressione dai campioni di grasso gonadico, e con livelli di espressione β-actina dall’ippocampo, utilizzando il metodo ΔΔ-CT per ottenere livelli relativi di mRNA. Grasso gonadico è stato sondato per i livelli di cluster di fattore di differenziazione 68 (CD68) e EGF-come modulo contenente mucina-come recettore ormonale 1 (F4/80), mentre l’ippocampo è stato sondato per β-secretasi 1 (BACE1), neprilisina, insulina degradante enzima (IDE), CD68, proteina acida fibrillare gliale (GFAP), e cluster di fattore di differenziazione 74 (CD74). Le sequenze di coppie di primer sono mostrate nella Tabella 1.
| Gene target | Sequenza |
|---|---|
| CD68 | Avanti: 5′-TTCTGCTGCTGTGGAAATGCAATGCAAG-3’Reverse: 5′-AGAGGGGGGGCTGGTAGGTTGAT-3′. |
| F4/80 | Avanti: 5′-TGCATCATCTAGCAATGGACAGC-3’Reverse: 5′-GCCTTTTTTTTGGATCCATTTGAA-3′. |
| HPRT | Avanti: 5′-AAGCTTGTGGGGTGAAAAAGGA-3’Reverse: 5′-TTGGGCCCCTCATCTTAGGTTTTTTT-3′. |
| SDHA | Avanti: 5′-ACACAGACCTGGGTGGAGACC-3’Reverse: 5′-GGATGGGCTTTTGGAGTAATCA-3′. |
| Neprilysin | Avanti: 5′-GAGAAAAAGCCCACTTGGCTTG-3’Reverse: 5′-GAAAGACAAAATGGGGCAGA-3′. |
| BACE1 | Avanti: 5′-TCGCTGCTGTCTCACAGTCATCC-3’Reverse: 5′-AACAAACGGGGTTTTTTCCCCATTOO3′. |
| IDE | Avanti: 5′-TGTTTCCACACACACAGGCAAT-3’Reverse: 5′-ACCTGTGTGAAAAGCCGAGAGA-3′. |
| CD74 | Avanti: 5′-CAAGTACGGGCAACATGACACCC-3’Reverse: 5′-GCACTTTTTTTTTAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAAA… |
| GFAP | Avanti: 5′-AACGACTATCGCCCCGGCCAACTG-3’Reverse: 5′-CTCTTTTTTTTTTTTCGGGGCATTTG-3′. |
| β-Actin | Avanti: 5′-AGCCATGTACGTAGGATCCATCC-3’Reverse: 5′-CTCTCCAGCTGTGTGGTGGTGAA-3′. |
Analisi statistiche
Per l’analisi dei dati sul peso corporeo e sulla tolleranza al glucosio, sono state eseguite misure ripetute a due vie ANOVA utilizzando il pacchetto statistico per le scienze sociali (SPSS; versione 23, IBM). Tutti gli altri dati sono stati analizzati da ANOVA bidirezionali utilizzando Prism (versione 5, GraphPad Software). Nel caso di effetti principali significativi, i confronti pianificati tra i gruppi di interesse sono stati effettuati utilizzando la correzione Bonferroni. Tutti i dati sono presentati come media ± SEM. La significatività è stata fissata ad una soglia di p < 0,05. I risultati statistici sono presentati nelle tabelle 2, 3.
| Figura | Test di Kolmogorov-Smirnov per la normalità( valorep ) | Significato statistico |
|---|---|---|
|
Figura 1A peso corporeo |
Tutti i gruppi in tutti i punti di tempo sono normalmente distribuiti(p > 0,05). | Genotipo: F(1,29) = 0, 10, p = 0,759diet: F(1,29) = 10,51, p = 0,003interazione: F(1,29) = 2,68, p = 0,112 |
|
Figura 1B colesterolo plasmatico |
E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo: F(1,29) = 2, 86, p = 0,103diet: F(1,29) = 1,58, p = 0,221 interazione: F(1,29) = 2,60, p = 0,119 |
|
Figura 1C trigliceridi plasmatici |
E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo: F(1,29) = 0,56, p = 0,46diet: F(1,29) = 2,87, p = 0,102 interazione: F(1,29) = 1,91, p = 0,179 |
|
Figura 1D peso grasso gonadico |
E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo: F(1,29) = 0,18, p = 0,673diet: F(1,29) = 37,04, p < 0,001 interazione: F(1,29) = 5,01, p = 0,033 |
|
Figura 1E CD68 |
E3FAD CTL N/AE3FAD WD > 0.10E4FAD CTL = 0.004E4FAD WD > 0.10 | Genotipo: F(1,21) = 0.90, p = 0.353diet: F(1,21) = 11,54, p = 0,003interazione: F(1,21) = 0,85, p = 0,366 |
|
Figura 1F F4/80 |
E3FAD CTL N/AE3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo: F(1,21) = .09, p = .768diet: F(1,21) = 7,02, p = 0,015 interazione: F(1,21) = 1,19, p = 0,288 |
|
Figura 1G glucosio (GTT) |
Tutti i gruppi in tutti i punti di tempo sono normalmente distribuiti(p > 0,05), tranne: E4FAD CTL 0 min = 0,002 E4FADWD 15 min = 0,025 E3FAD WD 30 min = 0,011 E4FAD WD 30 min = 0,008 | Genotipo: F(1,29) = 0,02, p = 0,886 diet: F(1,29) = 5,03, p = 0,033interazione: F(1,29) = 0,10, p = 0,750 |
|
Figura 1H GTT AUC |
E3FAD CTL = 0.07E3FAD WD = 0.097E4FAD CTL > 0.10E4FAD WD = 0.033 | Genotipo: F(1,29) = .06, p = 0,817diet: F(1,29) = 5,73, p = 0,023 interazione: F(1,29) = 0,12, p = 0,737 |
|
Figura 1I variazione percentuale di glucosio |
E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo: F(1,29) = .83, p = 0.371diet: F(1,29) = 3,84, p = 0,059 interazione: F(1,29) = 0,90, p = 0,352 |
| Figura 2BThio-S: corteccia entorinale | E3FAD CTL > 0.10E3FAD WD = 0.049E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo: F(1,29) = 50, 30, p < 0,001diet: F(1,29) = 6,62, p = 0,016 interazione: F(1,29) = 4,09, p = 0,053 |
| Figura 2CThio-S: subiculum | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo: F(1,29) = 59, 40, p < 0,001diet: F(1,29) = 2,98, p = 0,095 interazione: F(1,29) = 9,75, p = 0,004 |
| Figura 2DThio-S: CA1 | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo: F(1,29) = 80,58, p < 0,001diet: F(1,29) = 4,95, p = 0,034 interazione: F(1,29) = 8,41, p = 0,007 |
| Figura 2EThio-S: CA2/3 | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo F(1,29) = 46, 39, p < 0,001diet: F(1,29) = 7,41, p = 0,011 interazione: F(1,29) = 7,32, p = 0,011 |
| Figura 3BAβcarico: corteccia entorinale | E3FAD CTL > 0.10E3FAD WD = 0.002E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo F(1,29) = 21, 38, p < 0,001diet: F(1,29) = 7,83, p = 0,009 interazione: F(1,29) = 4,91, p = 0,035 |
| Figura 3CAβcarico:subiculum | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo F(1,29) = 25, 40, p < 0,001diet: F(1,29) = 11,19, p = 0,002interazione: F(1,29) = 0,11, p = 0,742 |
| Figura 3DAβcarico:CA1 | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD = 0.036 | Genotipo F(1,29) = 37,66, p < 0,001diet: F(1,29) = 2,91, p = 0,099 interazione: F(1,29) = 2,71, p = 0,110 |
| Figura 3E Carico Aβ:CA2/3 | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo F(1,29) = 47, 27, p < 0,001diet: F(1,29) = 10,36, p = 0,003 interazione: F(1,29) = 4,48, p = 0,043 |
| Figura 4Bmicroglianumero: corteccia entorinale | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo F(1,27) = 9, 78, p = 0,004diet: F(1,27) = 2,31, p = 0,141 interazione: F(1,27) = 1,05, p = 0,316 |
| Figura 4Cmicroglianumero:subiculum | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo F(1,27) = 42, 77, p < 0,001diet: F(1,27) = 4,20, p = 0,050 interazione: F(1,27) = 4,75, p = 0,038 |
| Figura 4Dmicroglianumero:CA1 | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo F(1,27) = 51, 42, p < 0,001diet: F(1,27) = 10,78, p = 0,003 interazione: F(1,27) = 7,97, p = 0,009 |
| Figura 4Numero di microglia:CA2/3 | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo F(1,27) = 21, 64, p < 0,001diet: F(1,27) = 1,97, p = 0,172 interazione: F(1,27) = 1,90, p = 0,180 |
| Figura 4Fmicrogliareattività: corteccia entorinale | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo F(1,27) = 109, 10, p < 0,001diet: F(1,27) = 1,64, p = 0,212 interazione: F(1,27) = 5,52, p = 0,027 |
| Figura 4Greattivitàmicrogliale: subiculum | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL = 0.07E4FAD WD < 0.001 | Genotipo F(1,27) = 19, 70, p < 0,001diet: F(1,27) = 0,00, p = 0,995 interazione: F(1,27) = 0,51, p = 0,480 |
| Figura 4Hreattivitàmicrogliale:CA1 | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD = 0.04 | Genotipo F(1,27) = 78, 70, p < 0,001diet: F(1,27) = 5,00, p = 0,034 interazione: F(1,27) = 11,58, p = 0,002 |
| Figura 4Reatività microgliale: CA2/3 | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo F(1,27) = 165, 70, p < 0,001diet: F(1,27) = 21,04, p < 0,001 interazione: F(1,27) = 32,66, p < 0,001 |
| Figura 5Numero di gastrociti: corteccia entorinale | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo F(1,29) = 3, 82, p = 0,060diet: F(1,29) = 0,29, p = 0,593 interazione: F(1,29) = 0,41, p = 0,528 |
| Figura 5Numero di catastrofe: subiculum | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo F(1,29) = 9, 95, p = 0,004diet: F(1,29) = 4,79, p = 0,037 interazione: F(1,29) = 1,04, p = 0,316 |
| Figura 5Numero di catastrofe:CA1 | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo F(1,29) = 5, 88, p = 0,022diet: F(1,29) = 3,55, p = 0,069 interazione: F(1,29) = 0,49, p = 0,489 |
| Figura 5Numero di catastrofe:CA2/3 | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo F(1,29) = 1,82, p = 0,188diet: F(1,29) = 4,26, p = 0,048interazione: F(1,29) = 0,02, p = 0,894 |
| Figura 5Reatività delle catastrofi: corteccia entorinale | E3FAD CTL > 0.10E3FAD WD = 0.004E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo F(1,29) = 46, 97, p < 0,001diet: F(1,29) = 5,75, p = 0,023 interazione: F(1,29) = 4,82, p = 0,036 |
| Figura 5Reatività dei gastrociti: subiculum | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL = 0.045E4FAD WD > 0.10 | Genotipo F(1,29) = 27, 72, p < 0,001diet: F(1,29) = 3,13, p = 0,088 interazione: F(1,29) = 0,00, p = 0,989 |
| Figura 5Reatività degliastrociti:CA1 | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo F(1,29) = 87, 49, p < 0,001diet: F(1,29) = 23,82, p < 0,001 interazione: F(1,29) = 2,08, p = 0,160 |
| Figura 5Reatività degliastrociti:CA2/3 | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo F(1,29) = 11, 68, p = 0,002diet: F(1,29) = 7,83, p = 0,009interazione: F(1,29) = 2,405, p = 0,132 |
| Gene | Media ± SEM | Test di Kolmogorov-Smirnov per la normalità( valorep ) | Significato statistico |
|---|---|---|---|
| BACE1 | E3FAD CTL = 1 ± N/AE3FAD WD = 1,53 ± 0,31E4FAD CTL = 1,32 ± 0,19E4FAD WD = 1,76 ± 0,41 | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo: F(1,28) = 1, 10, p = 0,304diet: F(1,28) = 3,44, p = 0,074 interazione: F(1,28) = 0,03, p = 0,874 |
| Neprilysin | E3FAD CTL = 1 ± N/AE3FAD WD = 1,61 ± 0,79E4FAD CTL = 0,94 ± 0,30E4FAD WD = 1,79 ± 0,63 | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo: F(1,28) = 0, 02, p = 0,902diet: F(1,28) = 2,49, p = 0,126 interazione: F(1,28) = 0,06, p = 0,802 |
| IDE | E3FAD CTL = 1 ± N/AE3FAD WD = 1,27 ± 0,39E4FAD CTL = 1,30 ± 0,39E4FAD WD = 1,12 ± 0,35 | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL = 0.01E4FAD WD > 0.10 | Genotipo: F(1,28) = 0, 08, p = 0,785diet: F(1,28) = 0,00, p = 0,955interazione: F(1,28) = 0,49, p = 0,489 |
| CD68 | E3FAD CTL = 1 ± N/AE3FAD WD = 1,21 ± 0,29E4FAD CTL = 1,74 ± 0,30E4FAD WD = 2,30 ± 0,29 | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo: F(1,28) = 10, 75, p = 0,003diet: F(1,28) = 1,91, p = 0,178 interazione: F(1,28) = 0,40, p = 0,532 |
| GFAP | E3FAD CTL = 1 ± N/AE3FAD WD = 1,02 ± 0,11E4FAD CTL = 1,56 ± 0,21E4FAD WD = 2,70 ± 0,04 | E3FAD CTL > 0.10E3FAD WD > 0.10E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo: F(1,28) = 14, 26, p < 0,001diet: F(1,28) = 0,23, p = 0,634 interazione: F(1,28) = 0,14, p = 0,712 |
| CD74 | E3FAD CTL = 1 ± N/AE3FAD WD = 1,28 ± 0,28E4FAD CTL = 3,32 ± 0,62E4FAD WD = 5,04 ± 1,30 | E3FAD CTL > 0.10E3FAD WD = 0.01E4FAD CTL > 0.10E4FAD WD > 0.10 | Genotipo: F(1,28) = 16, 98, p < 0,001diet: F(1,28) = 1,86, p = 0,184 interazione: F(1,28) = 0,96, p = 0,335 |
Risultati
Risultati dell’obesità nella dieta occidentale
Per iniziare a indagare se ci sono interazioni dell’ambiente del gene X tra APOE e dieta occidentale, abbiamo prima confrontato le misure di DIO in E3FAD contro i topi E4FAD dopo l’esposizione di 12 settimane alle diete di controllo e occidentale. La dieta di controllo è stata associata ad un aumento <1% del peso corporeo in entrambi i topi E3FAD e E4FAD, mentre la dieta occidentale ha prodotto un aumento del 39 ± 7,7% del peso corporeo in E3FAD e un aumento del 24 ± 7,21% nei topi E4FAD (Fig. 1A), in modo tale che gli effetti della dieta non variano significativamente tra i genotipi(p = 0,112; Fig. 1A Tabella 2). A 2 × 2 misure ripetute ANOVA ha rivelato un effetto principale significativo della dieta sul peso corporeo (F =10,51, p = 0 , 003; Fig. 1A) in cui la dieta occidentale era associata ad un aumento di peso. Il genotipo APOE non ha influito in modo significativo sul peso corporeo(p = 0,759; Fig. 1A). Tra i confronti di gruppo ha rivelato che i topi E3FAD alimentati con una dieta occidentale pesavano significativamente di più rispetto ai topi E3FAD alimentati con una dieta di controllo a 4, 8 e 12 settimane(p < 0,05). Non ci sono state differenze statisticamente significative nel peso corporeo in qualsiasi momento tra i gruppi di controllo e dieta occidentale nei topi E4FAD.
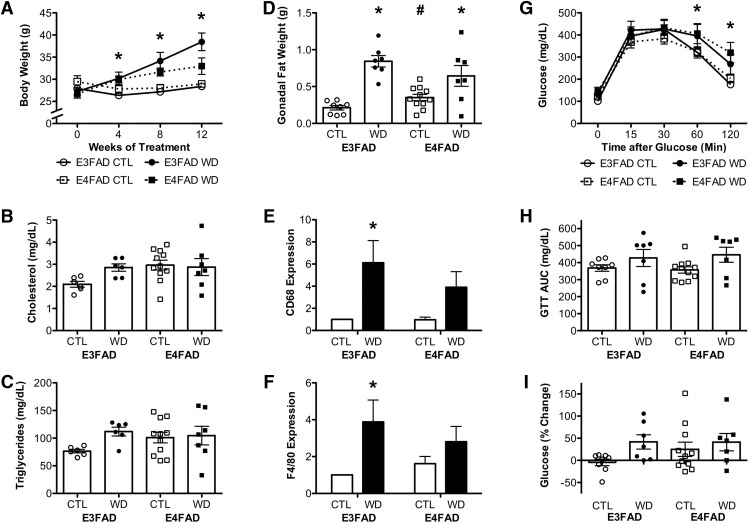
Figura 1.
Risultati metabolici associati alla DIO nei topi E3FAD e E4FAD. A, Pesi corporei nei topi maschi E3FAD e E4FAD mantenuto su controllo (CTL) e Western (WD) diete prese al basale (settimana 0) e intervalli di quattro settimane per tutto il periodo di 12 settimane sperimentale. Livelli plasmatici di colesterolo (B) e i livelli di trigliceridi (C) in topi E3FAD e E4FAD su controllo e diete occidentali alla fine del periodo sperimentale. D, Peso dei cuscinetti di grasso gonadico attraverso i gruppi. Espressione relativa mRNA di marcatori macrofagi (E) CD68 e (F) F4/80 in grasso gonadico, come determinato dalla PCR in tempo reale. I dati mostrano le differenze di piega rispetto al gruppo di dieta E3FAD + controllo. GGTT che mostra i livelli di glucosio nel sangue nel tempo dopo un bolo di glucosio. H, AUC per il GTT. I, Variazione percentuale dei livelli di glicemia a digiuno rispetto alla linea di base dopo 12 settimane di controllo o dieta occidentale. I dati sono presentati come valori medi (±SEM); n = 7-11/gruppo. I topi E3FAD sono mostrati come cerchi, i topi E4FAD sono mostrati come quadrati; i gruppi di dieta di controllo sono indicati come simboli o barre aperte, mentre i gruppi di dieta occidentale sono simboli o barre riempite. *, p < 0,05 rispetto ai topi con corrispondenza del genotipo in condizioni di dieta di controllo. #, p < 0,05 rispetto ai topi E3FAD nella stessa condizione di dieta.
Abbiamo poi esaminato i livelli plasmatici di colesterolo e trigliceridi come misure degli effetti negativi della dieta occidentale. Abbiamo trovato che i livelli plasmatici di colesterolo sono stati significativamente influenzati da né genotipo(p = 0,103) né dieta(p = 0,221), e non abbiamo trovato un effetto di interazione(p = 0,119; Fig. 1B Tabella 2). Allo stesso modo, non ci sono stati effetti né del genotipo(p = 0,46) o della dieta( p = 0,102), né un effetto di interazione(p = 0,179) sui livelli di trigliceridi plasmatici(Fig. 1C).
Poiché i danni metabolici associati all’obesità sono stati collegati all’adiposità, abbiamo valutato la deposizione di grasso in gruppi. Abbiamo osservato un effetto di interazione significativo(F = 5,01, p = 0,033; Tabella 2), tale che sulle diete di controllo, i topi E4FAD avevano più grasso gonadico di E3FADs(p = 0,027), ma non c’era alcuna differenza tra i topi E3FAD e E4FAD sulla dieta occidentale(p = 0,230; Fig. 1D). Inoltre, c’è stato un significativo effetto principale della dieta(F = 37,04, p < 0,001) sul peso dei cuscinetti di grasso gonadico, in modo che sia i topi E3FAD e E4FAD avevano aumentato i cuscinetti di grasso con la dieta occidentale (Fig.1D). Risultati paralleli sono stati osservati nei cuscinetti di grasso retroperitoneali (dati non mostrati). Poiché l’infiammazione è una caratteristica consolidata dell’obesità, abbiamo esaminato l’espressione genica dei marcatori macrofagi CD68 e F4/80 mediante PCR nel tessuto adiposo. Abbiamo trovato un significativo effetto principale della dieta sull’espressione CD68(F = 11,54, p = 0,003), anche se questo effetto ha raggiunto una significatività statistica solo in E3FAD ma non nei topi E4FAD(Fig. 1E). Non c’è stato un effetto statisticamente significativo del genotipo(p = 0,353), né vi è stata un’interazione tra dieta e genotipo(p = 0,366) sull’espressione CD68. La dieta ha avuto un effetto principale sull’espressione adiposa F4/80(F = 7,02, p = 0,015), e ancora una volta, questo effetto ha raggiunto la significatività statistica solo nei topi E3FAD(Fig. 1F). Non c’è stato alcun effetto statisticamente significativo del genotipo(p = .768), e nessun effetto di interazione(p = 0,288) sull’espressione F4/80(Tabella 2).
Oltre ad aumentare il peso corporeo e l’adiposità, la dieta occidentale può indurre disturbi metabolici, tra cui la disregolazione dell’omeostasi del glucosio. Esaminando la clearance del glucosio nel GTT, abbiamo trovato un effetto principale significativo della dieta(F = 5,03, p = 0,033), tale che sia i topi E3FAD e E4FAD alimentati con una dieta occidentale sono stati compromessi a cancellare il glucosio(Fig. 1G Tabella 2). Non c’è stato alcun effetto principale del genotipo(p = 0,886), o effetto di interazione tra dieta e genotipo(p = 0,750) sulla clearance del glucosio. Abbiamo anche calcolato l’area sotto la curva (AUC) per GTT, e abbiamo trovato che c’era un effetto principale significativo della dieta(F = 5,73, p = 0,023), ma non del genotipo(p = 0,817) su GTT AUC(Fig. 1H). Tuttavia, l’effetto della dieta non ha raggiunto la significatività statistica se esaminato separatamente nei topi E3FAD e E4FAD. Non vi è stata alcuna interazione tra genotipo e dieta su GTT AUC(p = 0,737). I cambiamenti nei livelli di glucosio a digiuno durante il periodo di trattamento con la dieta hanno mostrato una tendenza verso un effetto principale della dieta(F = 3,84, p = 0,059; Fig. 1I). Non vi è stato alcun effetto del genotipo(p = 0,371) né vi è stata un’interazione tra dieta e genotipo(p = 0,352) sulle variazioni dei livelli di glucosio (Tabella 2).
Figura 1.
Risultati metabolici associati a DIO nei topi E3FAD e E4FAD. A, Pesi corporei in topi maschi E3FAD e E4FAD mantenuto su controllo (CTL) e occidentale (WD) diete prese al basale (settimana 0) e quattro settimane intervalli di quattro settimane per tutto il periodo di 12 settimane sperimentale. Livelli plasmatici di colesterolo (B) e i livelli di trigliceridi (C) in topi E3FAD e E4FAD su controllo e diete occidentali alla fine del periodo sperimentale. D, Peso dei cuscinetti di grasso gonadico attraverso i gruppi. Espressione relativa mRNA di marcatori macrofagi (E) CD68 e (F) F4/80 in grasso gonadico, come determinato dalla PCR in tempo reale. I dati mostrano le differenze di piega rispetto al gruppo di dieta E3FAD + controllo. GGTT che mostra i livelli di glucosio nel sangue nel tempo dopo un bolo di glucosio. H, AUC per il GTT. I, Variazione percentuale dei livelli di glicemia a digiuno rispetto alla linea di base dopo 12 settimane di controllo o dieta occidentale. I dati sono presentati come valori medi (±SEM); n = 7-11/gruppo. I topi E3FAD sono mostrati come cerchi, i topi E4FAD sono mostrati come quadrati; i gruppi di dieta di controllo sono indicati come simboli o barre aperte, mentre i gruppi di dieta occidentale sono simboli o barre riempite. *, p < 0,05 rispetto ai topi con corrispondenza del genotipo in condizioni di dieta di controllo. #, p < 0,05 rispetto ai topi E3FAD nella stessa condizione di dieta.
La dieta occidentale aumenta la deposizione di β-amiloide in E4FAD ma non nei topi E3FAD
Il primario cambiamento neuropatologico correlato AD primario nei topi EFAD a questa età è l’accumulo di proteina β-amiloide, in gran parte sotto forma di depositi extracellulari, molti dei quali mostrano una colorazione positiva Thio-S che è indicativo di amiloide. Così, per iniziare a valutare la neuropatologia AD-correlata, Thio-S placche positive sono stati contati nella corteccia entorinale e nelle sottoregioni dell’ippocampo. L’ispezione visiva delle sezioni colorate qualitativamente ha mostrato non solo l’aumento previsto dei depositi amiloidi nei topi E4FAD, ma anche la sorprendente scoperta che la dieta occidentale ha aumentato Thio-S placche positive solo nei topi E4FAD(Fig. 2A). In particolare, ci sono stati significativi effetti di interazione tra genotipo e dieta su placche tio-s positive nel subiculum(F = 9,75, p = 0,004; Fig. 2C), CA1(F = 8,41, p = 0,007; Fig. 2D), e CA2/3(F = 7,32, p = 0,011; Fig. 2E), e una tendenza non significativa verso una interazione nella corteccia entorinale(F = 4,09, p = 0,053; Fig. 2B Tabella 2). Ulteriori analisi hanno rivelato che la dieta ha aumentato significativamente i conteggi di placca Thio-S positivo in E4FAD ma non E3FAD maschi in tutte le regioni cerebrali campionati(p <0,01). Inoltre, vi è stato un significativo effetto principale di genotipo anche in assenza di dieta, in modo tale che i topi E4FAD aveva un numero maggiore di Thio-S placche positive nella corteccia entorinale(F = 50,30, p <0,001; Fig. 2B), subiculum(F = 59,40, p < 0,001; Fig. 2C), CA1(F = 80,58, p < 0,001; Fig. 2D), e CA2/3(F = 46,39, p < 0,001; Fig. 2E), rispetto ai topi E3FAD.
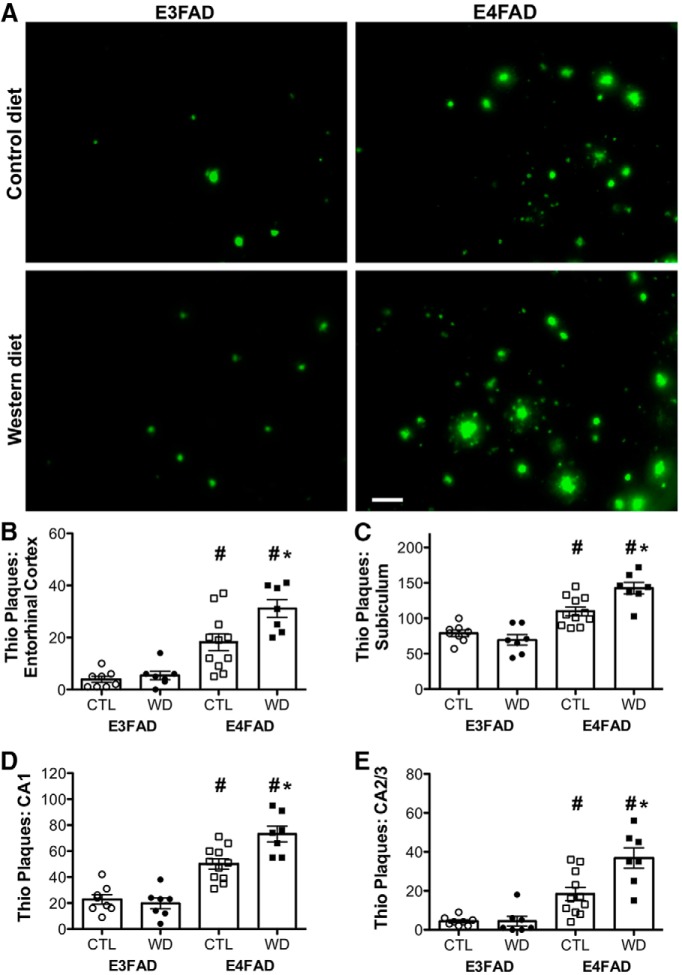
Figura 2.Accumulo di depositi amiloidogenici valutati con la colorazione Thio-S nei topi E3FAD e E4FAD attraverso i trattamenti dietetici. A, Immagini rappresentative di Thio-S colorazione Thio-S nel subiculum di E3FAD e E4FAD maschi alimentati controllo e diete occidentali. Barra di scala, 50 µm. I numeri di Thio-S numeri placca positiva in E3FAD e topi E4FAD mantenuto su controllo e le diete occidentali sono stati quantificati in (B) corteccia entorinale, e subregioni ippocampali (C) subiculum, (D) CA1, e (E) CA2/3. I dati sono presentati come valori medi (±SEM); n = 7-11/gruppo. I topi E3FAD sono mostrati come cerchi, i topi E4FAD sono mostrati come quadrati; i gruppi di dieta di controllo sono indicati come simboli aperti, e i gruppi di dieta occidentali come simboli pieni. *, p < 0,05 rispetto ai topi con corrispondenza del genotipo in condizioni di dieta di controllo. #, p < 0,05 rispetto ai topi E3FAD nella stessa condizione di dieta.
Come seconda misura della patologia di tipo AD, abbiamo valutato il carico totale di β-amiloide da immunoistochimica. Questo fornisce una misura della β-amiloide completa, in quanto l’anticorpo riconosce gli accumuli intra ed extracellulari di Aβ, anche quelli che non sono progrediti verso depositi amiloidi tio-positivi. I risultati hanno ripetuto lo stesso schema generale osservato con la colorazione Tio-S. Cioè, (1) i topi E4FAD mostrano un maggiore carico di β-amiloide, e (2) i topi E4FAD ma non E3FAD mostrano un aumento dell’accumulo di β-amiloide con la dieta occidentale (Fig. 3A). Abbiamo trovato effetti significativi di interazione tra genotipo e dieta nella corteccia entorinale(F = 4,91, p = 0,035; Fig. 3B) e in CA2/3(F = 4,48, p = 0,043; Fig. 3E), ma non nel subiculum(F = 0,11, p = 0,742; Fig. 3C) o in CA1(F = 2,71, p = 0,110; Fig. 3D Tabella 2). Bonferroni test post hoc hanno mostrato che la dieta occidentale ha aumentato significativamente il carico Aβ in E4FAD ma non nei topi E3FAD in tutte le regioni cerebrali esaminate (p <0,05). C’è stato un effetto significativo principale del genotipo con topi E4FAD con un carico Aβ maggiore rispetto ai topi E3FAD nella corteccia entorinale (F = 21,38, p <0,001; Fig. 3B), subiculum(F = 25,40, p < 0,001; Fig. 3C), CA1(F = 37,66, p < 0,001; Fig. 3D), e CA2/3(F = 47,27, p < 0,001; Fig. 3E).
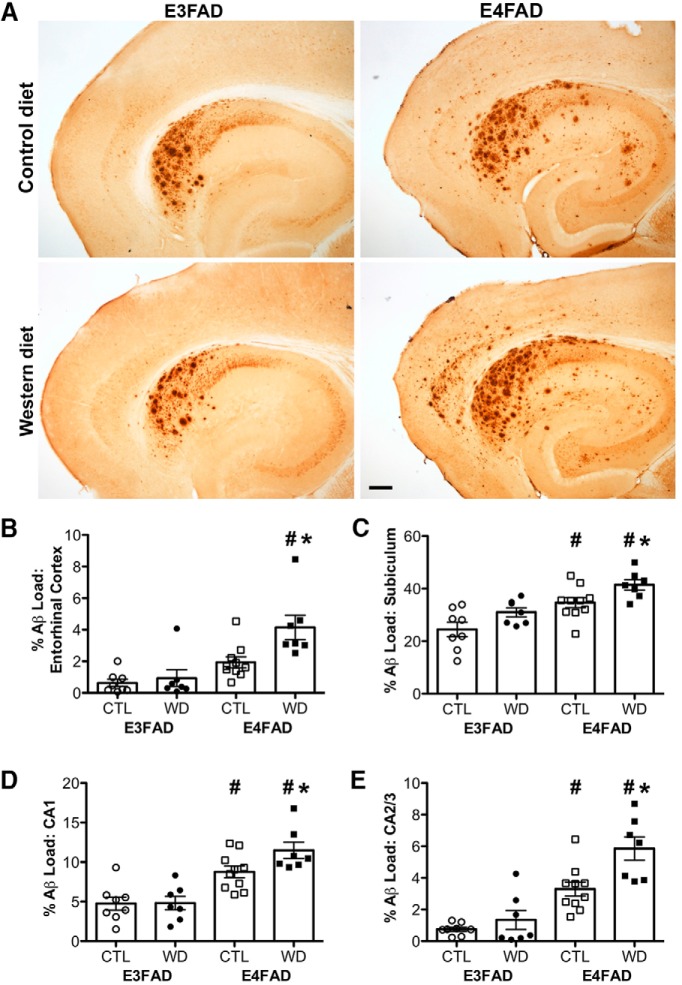
Figura 3.Accumulo di depositi di β-amiloide valutati dalla immunoistochimica nei topi E3FAD e E4FAD attraverso i trattamenti dietetici. A, Immagini rappresentative di β-amiloide immunoreattività β-amiloide nella corteccia entorinale e ippocampo in E3FAD e maschi E4FAD mantenuto sul controllo e le diete occidentali. Barra di scala, 100 µm. β-amiloide carico è stato quantificato come carico di immunoreattività nei topi E3FAD e E4FAD nei gruppi di controllo e diete occidentali in (B) corteccia entorinale, e subregioni ippocampali (C) subiculum, (D) CA1, e (E) CA2/3. I dati sono presentati come valori medi (±SEM); n = 7-11/gruppo. I topi E3FAD sono mostrati come cerchi, i topi E4FAD sono mostrati come quadrati; i gruppi di dieta di controllo sono indicati come simboli aperti, e i gruppi di dieta occidentali come simboli pieni. *, p < 0,05 rispetto ai topi con corrispondenza del genotipo in condizioni di dieta di controllo. #, p < 0,05 rispetto ai topi E3FAD nella stessa condizione di dieta.
Figura 2.Accumulo di depositi amiloidogenici valutati dalla colorazione Thio-S nei topi E3FAD e E4FAD attraverso i trattamenti dietetici. A, Immagini rappresentative di Thio-S colorazione Thio-S nel subiculum di E3FAD e E4FAD maschi alimentati controllo e diete occidentali. Barra di scala, 50 µm. I numeri di Thio-S numeri placca positiva in E3FAD e topi E4FAD mantenuto su controllo e le diete occidentali sono stati quantificati in (B) corteccia entorinale, e subregioni ippocampali (C) subiculum, (D) CA1, e (E) CA2/3. I dati sono presentati come valori medi (±SEM); n = 7-11/gruppo. I topi E3FAD sono mostrati come cerchi, i topi E4FAD sono mostrati come quadrati; i gruppi di dieta di controllo sono indicati come simboli aperti, e i gruppi di dieta occidentali come simboli pieni. *, p < 0,05 rispetto ai topi con corrispondenza del genotipo in condizioni di dieta di controllo. #, p < 0,05 rispetto ai topi E3FAD nella stessa condizione di dieta.
Figura 3.Accumulo di depositi di β-amiloide valutati dalla immunoistochimica nei topi E3FAD e E4FAD attraverso i trattamenti dietetici. A, Immagini rappresentative di β-amiloide immunoreattività β-amiloide nella corteccia entorinale e ippocampo in E3FAD e maschi E4FAD mantenuto sul controllo e le diete occidentali. Barra di scala, 100 µm. β-amiloide carico è stato quantificato come carico di immunoreattività nei topi E3FAD e E4FAD nei gruppi di controllo e diete occidentali in (B) corteccia entorinale, e subregioni ippocampali (C) subiculum, (D) CA1, e (E) CA2/3. I dati sono presentati come valori medi (±SEM); n = 7-11/gruppo. I topi E3FAD sono mostrati come cerchi, i topi E4FAD sono mostrati come quadrati; i gruppi di dieta di controllo sono indicati come simboli aperti, e i gruppi di dieta occidentali come simboli pieni. *, p < 0,05 rispetto ai topi con corrispondenza del genotipo in condizioni di dieta di controllo. #, p < 0,05 rispetto ai topi E3FAD nella stessa condizione di dieta.
La dieta occidentale aumenta la gliosi più fortemente in E4FAD che nei topi E3FAD
La gliosi è un’importante caratteristica neuropatologica dell’AD che è anche associata sia all’obesità che all’APOE4. Per valutare la gliosi, abbiamo confrontato sia il numero relativo di cellule che lo stato di attivazione morfologica della microglia e degli astrociti nei vari gruppi. Abbiamo trovato che, rispetto ai topi E3FAD, topi E4FAD aveva costantemente un numero totale più elevato di cellule gliali così come una maggiore percentuale di cellule gliali con fenotipi reattivi rispetto a riposo. Inoltre, gli effetti della dieta sul numero di gliali e sulla reattività erano più forti nei topi E4FAD che nei topi E3FAD.
Abbiamo esaminato per la prima volta il numero di microglia e la morfologia con la colorazione IBA-1. Figura 4A mostra una cellula microgliale a riposo con processi sottili e ramificati (tipo 1), e cellule attivate con corpi cellulari a forma di bacchetta e meno, processi più spessi (tipo 2), e cellule ameboidi (tipo 3). Abbiamo trovato interazioni significative tra genotipo e dieta quando si esamina il numero totale di microglia per mm2 nel subiculum(F = 4,75, p = 0,038; Fig. 4C) e in CA1(F = 7,97, p = 0,009; Fig. 4D), con Bonferroni test post hoc che dimostrano che la dieta occidentale ha aumentato il numero di microglia in E4FAD ma non in topi E3FAD in queste regioni cerebrali(p < 0,05; Tabella 2). Non ci sono stati effetti di interazione sul numero di microglia nella corteccia entorinale(p = 0,316; Fig. 4B), o in CA2/3(p = 0,180; Fig. 4E). C’è stato un effetto significativo del genotipo sul numero totale di microglia per mm2 nella corteccia entorinale(F = 9,78, p = 0,004; Fig. 4B), subiculum(F = 42,77, p < 0,001; Fig. 4C), CA1(F = 51,42, p < 0,001; Fig. 4D), e CA2/3(F = 21,64, p < 0,001; Fig. 4E), in modo tale che i topi E4FAD avevano un numero totale maggiore di microglia in queste regioni cerebrali rispetto ai topi E3FAD. Tuttavia, nella corteccia entorinale, l’effetto del genotipo è stato significativo solo negli animali con una dieta occidentale.
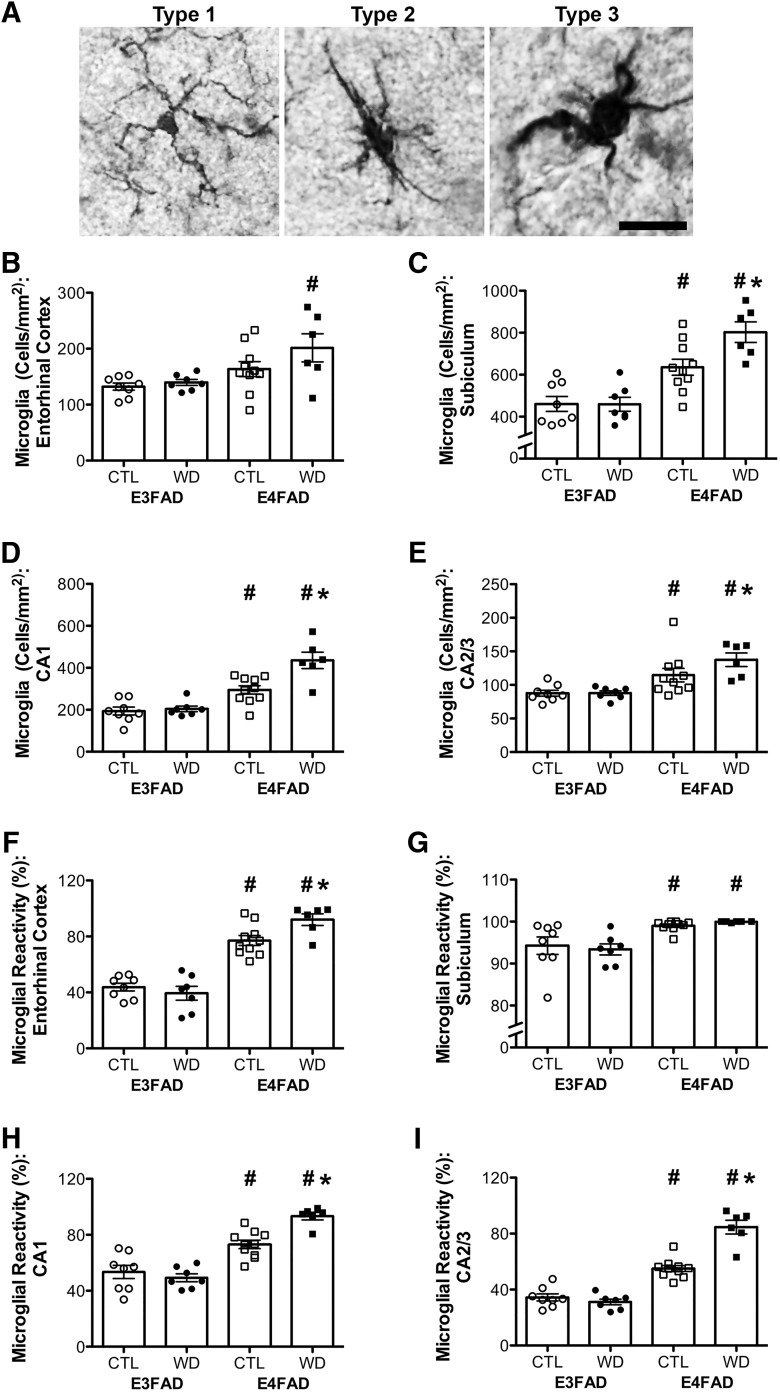
Figura 4.Numero di microglia e stato morfologico valutato dalla immunoistochimica IBA-1 nei topi E3FAD e E4FAD attraverso i trattamenti dietetici. A, Immagini rappresentative della morfologia microgliale associata a riposo (tipo 1) e reattivi (tipi 2 e 3) fenotipi. Barra di scala, 40 µm. B-E, Densità (cellule / mm2) di IBA-1-immunoreattive cellule in E3FAD e E4FAD topi sul controllo e le diete occidentali sono stati quantificati in (B) corteccia entorinale, e subregioni ippocampali (C) subiculum, (D) CA1, e E) CA2/3. F-I) Le percentuali di tutte le cellule IBA-1-immunoreattive valutate come aventi fenotipo reattivo (tipi 2 e 3) sono state quantificate in (F) corteccia entorinale, e subregioni ippocampali (G) subiculum, (H) CA1, e (I) CA2/3. I dati sono presentati come valori medi (±SEM); n = 7-11/gruppo. I topi E3FAD sono mostrati come cerchi, i topi E4FAD sono mostrati come quadrati; i gruppi di dieta di controllo sono indicati come simboli aperti, e i gruppi di dieta occidentali come simboli pieni. *, p < 0,05 rispetto ai topi con corrispondenza del genotipo in condizioni di dieta di controllo. #, p < 0,05 rispetto ai topi E3FAD nella stessa condizione di dieta.
Misure di reattività microgliale ha mostrato risultati simili come numero microgliale. Significativi effetti di interazione tra genotipo e dieta sono stati osservati nella corteccia entorinale(F = 5,52, p = 0,027; Fig. 4F), CA1(F = 11,58, p = 0,002; Fig. 4H), e CA2/3(F = 32,66, p < 0,001; Fig. 4I), ma non nel subiculum(p = 0,480; Fig. 4G; Tabella 2). Bonferroni test post hoc ha rivelato che la dieta occidentale ha aumentato la percentuale di microglia reattiva nella corteccia entorinale, CA1, e CA2/3 di E4FAD, ma non E3FAD, topi maschi. C’è stato un significativo effetto principale di genotipo anche in assenza di dieta, in modo tale che i topi E4FAD aveva una maggiore percentuale di microglia reattiva rispetto ai topi E3FAD nella corteccia entorinale(F = 109,10, p <0,001; Fig . 4F), subiculum(F = 19,70, p < 0,001; Fig. 4G), CA1(F = 78,70, p < 0,001; Fig. 4H), e CA2/3(F = 165,70, p < 0,001; Fig. 4I).
Abbiamo poi esaminato il numero di astrociti e l’attivazione tramite colorazione GFAP. Figura 5A mostra esempi di un astrocittà non reattivo con un soma di dimensioni normali contro un fenotipo reattivo con soma ingrandito e proiezioni. Per la misura del numero di astrociti, gli effetti della dieta non differiscono a seconda del genotipo per nessuna delle regioni cerebrali campionate(Tabella 2). Abbiamo trovato significativi effetti principali del genotipo sul numero totale di astrociti nel subiculum(F = 9,95, p = 0,004; Fig. 5C), anche se questo effetto è stato solo statisticamente significativo negli animali con una dieta occidentale. C’è stato un effetto principale del genotipo sul numero di astrociti nel CA1(F = 5,88, p = 0,022; Fig. 5D), ma questo non ha raggiunto la significatività statistica se esaminato separatamente negli animali di controllo e in quelli alimentati con la dieta occidentale. C’è stata una tendenza verso un effetto significativo del genotipo nella corteccia entorinale(F = 3,82, p = 0,060; Fig. 5B), ma nessun effetto in CA2/3(p = 0,188; Fig. 5E). La dieta ha avuto significativi effetti principali sul numero di astrociti nel subiculum(F = 4,79, p = 0,037; Fig. 5C), e CA2/3(F = 4,26, p = 0,048; Fig. 5E), con una tendenza verso un effetto principale in CA1(F = 3,55, p = 0,069; Fig. 5D), anche se questo effetto non ha raggiunto la significatività statistica se esaminato separatamente nei topi E3FAD e E4FAD in qualsiasi regione cerebrale. Non vi è stato alcun effetto della dieta sul numero di astrociti nella corteccia entorinale(p = 0,593; Fig. 5B).
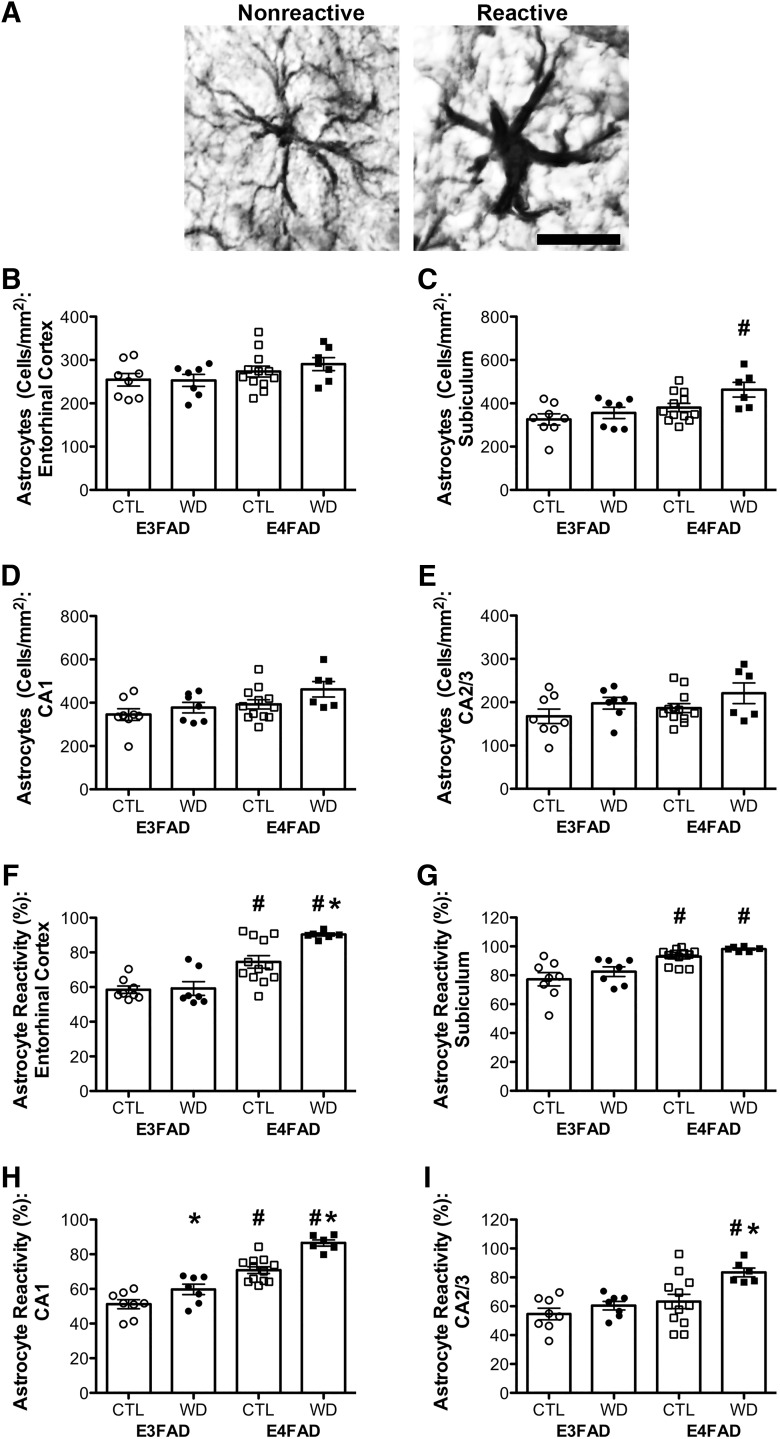
Figura 5.Numero di astrociti e stato morfologico valutato dalla immunoistochimica GFAP nei topi E3FAD e E4FAD attraverso i trattamenti dietetici. AImmagini rappresentative della morfologia degli astrociti associati con fenotipi a riposo e reattivi. Barra di scala, 50 µm. B-E, Densità (cellule / mm2) di GFAP-immunoreattive cellule in E3FAD e topi E4FAD sul controllo e le diete occidentali sono stati quantificati in (B) corteccia entorinale, e subregioni ippocampali (C) subiculum, (D) CA1, e (E) CA2/3. F-I, Percentuali di tutte le cellule GFAP-immunoreattive valutate come aventi fenotipo reattivo (tipo 2) sono state quantificate in (F) corteccia entorinale, e subregioni ippocampali (G) subiculum, (H) CA1, e (I) CA2/3. I dati sono presentati come valori medi (±SEM); n = 7-11/gruppo. I topi E3FAD sono mostrati come cerchi, i topi E4FAD sono mostrati come quadrati; i gruppi di dieta di controllo sono indicati come simboli aperti, e i gruppi di dieta occidentali come simboli pieni. *, p < 0,05 rispetto ai topi con corrispondenza del genotipo in condizioni di dieta di controllo. #, p < 0,05 rispetto ai topi E3FAD nella stessa condizione di dieta.
Quando si esamina la reattività degli astrociti, abbiamo trovato tendenze simili a quelle della reattività microgliale. Cioè, c’è stato un effetto significativo di interazione tra genotipo e dieta sulla reattività degli astrociti nella corteccia entorinale(F = 4,82, p = 0,036; Fig. 5F), con la dieta occidentale che aumenta la reattività solo nei topi E4FAD(Tabella 2). Non ci sono stati effetti significativi di interazione tra genotipo e dieta nel subiculum(p = 0,989 p = 0,989; Fig. 5G), CA1(p = 0,160; Fig. 5H), o CA2/3( p = 0,132; Fig. 5I). Inoltre, in assenza di dieta, il genotipo ha avuto un effetto significativo sulla reattività degli astrociti, con i topi E4FAD che hanno una percentuale maggiore di astrociti reattivi nella corteccia entorinale(F = 46,97, p < 0,001; Fig. 5F), subiculum(F = 27,72, p < 0,001; Fig. 5G), CA1(F = 87,49, p < 0,001; Fig. 5H), e CA2/3(F = 11,68, p = 0,002; Fig. 5I). In CA2/3 l’effetto del genotipo è stato significativo solo negli animali alimentati con la dieta occidentale. Inoltre, la dieta occidentale ha aumentato significativamente la reattività degli astrociti in CA1(F = 23,82, p < 0,001; Fig . 5H), e CA2/3(F = 7,83, p = 0,009; Fig. 5I), sebbene questo effetto sia stato significativo solo nei topi E4FAD in CA2/3. C’è stata una tendenza non significativa verso un effetto della dieta nel subiculum(F = 3,13, p = 0,088; Fig. 5G).
Figura 4.Numero di microglia e stato morfologico valutato dalla immunoistochimica IBA-1 nei topi E3FAD e E4FAD attraverso i trattamenti dietetici. A, Immagini rappresentative della morfologia microgliale associata a riposo (tipo 1) e reattivi (tipi 2 e 3) fenotipi. Barra di scala, 40 µm. B-E, Densità (cellule / mm2) di IBA-1-immunoreattive cellule in E3FAD e E4FAD topi sul controllo e le diete occidentali sono stati quantificati in (B) corteccia entorinale, e subregioni ippocampali (C) subiculum, (D) CA1, e E) CA2/3. F-I) Le percentuali di tutte le cellule IBA-1-immunoreattive valutate come aventi fenotipo reattivo (tipi 2 e 3) sono state quantificate in (F) corteccia entorinale, e subregioni ippocampali (G) subiculum, (H) CA1, e (I) CA2/3. I dati sono presentati come valori medi (±SEM); n = 7-11/gruppo. I topi E3FAD sono mostrati come cerchi, i topi E4FAD sono mostrati come quadrati; i gruppi di dieta di controllo sono indicati come simboli aperti, e i gruppi di dieta occidentali come simboli pieni. *, p < 0,05 rispetto ai topi con corrispondenza del genotipo in condizioni di dieta di controllo. #, p < 0,05 rispetto ai topi E3FAD nella stessa condizione di dieta.
Figura 5.Numero di astrociti e stato morfologico valutato dalla immunoistochimica GFAP nei topi E3FAD e E4FAD attraverso i trattamenti dietetici. AImmagini rappresentative della morfologia degli astrociti associati con fenotipi a riposo e reattivi. Barra di scala, 50 µm. B-E, Densità (cellule / mm2) di GFAP-immunoreattive cellule in E3FAD e topi E4FAD sul controllo e le diete occidentali sono stati quantificati in (B) corteccia entorinale, e subregioni ippocampali (C) subiculum, (D) CA1, e (E) CA2/3. F-I, Percentuali di tutte le cellule GFAP-immunoreattive valutate come aventi fenotipo reattivo (tipo 2) sono state quantificate in (F) corteccia entorinale, e subregioni ippocampali (G) subiculum, (H) CA1, e (I) CA2/3. I dati sono presentati come valori medi (±SEM); n = 7-11/gruppo. I topi E3FAD sono mostrati come cerchi, i topi E4FAD sono mostrati come quadrati; i gruppi di dieta di controllo sono indicati come simboli aperti, e i gruppi di dieta occidentali come simboli pieni. *, p < 0,05 rispetto ai topi con corrispondenza del genotipo in condizioni di dieta di controllo. #, p < 0,05 rispetto ai topi E3FAD nella stessa condizione di dieta.
I topi E4FAD hanno aumentato l’espressione genica dei marcatori infiammatori
Per iniziare ad affrontare i possibili meccanismi alla base degli effetti interattivi dell’APOE4e della dieta occidentale, abbiamo esaminato l’espressione del gene dell’ippocampo di diversi marcatori relativi alla produzione e alla clearance di Aβ, così come l’infiammazione. Nel complesso, i nostri risultati indicano che l’espressione genica dei fattori coinvolti nella clearance e nella produzione di Aβ non è significativamente alterata dal genotipo o dalla dieta, e che l’espressione genica infiammatoria è aumentata nei topi E4FAD, senza essere alterata dalla dieta occidentale (Tabella 3).
Per il BACE1, i livelli relativi di mRNA non hanno mostrato evidenza di una interazione tra la dieta e i genotipi APOE(p = 0,874). Non c’era un genotipo significativo effetto principale(p = 0,304), ma c’era una tendenza non significativa di aumento dei livelli di BACE1 con la dieta occidentale(p = 0,074). L’espressione del fattore di clearance Aβ neprilisina non è stata influenzata in modo significativo dal genotipo (p =0,902) o dalla dieta (p =0,126), e non vi è stata alcuna interazione tra genotipo e dieta (p =0,802). Allo stesso modo, l’espressione genica dell’IDE non è stata alterata dal genotipo( p = 0,785), dalla dieta(p = 0,955), o dall’interazione tra genotipo e dieta( p = 0,489).
Nel valutare l’espressione genica dei marcatori infiammatori abbiamo scoperto che i topi E4FAD avevano livelli significativamente maggiori dei marcatori microgliali CD68(F = 10,75, p = 0,003), del marker astrocitario GFAP(F = 14,26, p < 0,001), e del marker immunitario innato CD74 (F = 16,98, p < 0,001), rispetto ai topi E3FAD. Tuttavia, non ci sono stati effetti significativi della dieta sui livelli di CD68(p = 0,178), GFAP(p = 0,634), o CD74( p = 0,184). Inoltre, non ci sono state interazioni significative tra genotipo e dieta sui livelli di CD68(p = 0,532), GFAP(p = 0,712), o CD74( p = 0,335).
Discussione
L’obiettivo di questo studio è quello di esaminare se il genotipo APOE e l’obesità interagiscono per promuovere la patogenesi dell’AD. Confrontando i topi E3FAD e E4FAD mantenuti su diete standard rispetto a quelle occidentali, dimostriamo una significativa interazione gene-ambiente in cui la DIO guida la patologia AD-correlata principalmente nei topi APOE4. I nostri risultati sono coerenti con i risultati precedenti negli esseri umani(Fitzpatrick et al., 2009; Profenno et al., 2010), e confermano studi su modelli di roditori(Ho et al., 2004; Julien et al., 2010; Kohjima et al., 2010; Barron et al., 2013) che l’obesità aumenta il rischio di sviluppo di AD. Analogamente, i nostri risultati replicano i dati precedenti sui roditori(Fryer et al., 2005; Castellano et al., 2011; Youmans et al., 2012; Rodriguez et al., 2014; Cacciottolo et al., 2016) che modellano l’osservazione umana secondo cui l’APOE4aumenta il rischio e o accelera l’insorgenza della patologia dell’AD(Corder et al., 1993; Saunders et al., 1993; Strittmatter et al., 1993; Morris et al., 2010; Jack et al., 2015). È importante notare che i nostri dati indicano che gli effetti di DIO e APOE4non sono strettamente additivi. Anche se lo stato di APOE4è associato ad una maggiore patologia AD-come patologia sia sul controllo e le diete occidentali, l’obesità aumentata AD-come patologia in E4FAD ma non E3FAD topi. La nostra scoperta che i topi E3FAD non ha mostrato un aumento della patologia AD-correlata alla dieta è simile ai risultati nulli in alcuni modelli di roditori dell’obesità(Zhang et al., 2013; Knight et al., 2014; Niedowicz et al., 2014), suggerendo che gli effetti deleteri dell’obesità possono essere regolati da fattori genetici oltre che da APOE4. Così, questi dati suggeriscono un’importante interazione ambientale del gene X in cui i portatori di APOE4sono più suscettibili agli effetti di promozione dell’AD dell’obesità.
Come i risultati neurali nelle popolazioni umane sono influenzati dalla relazione tra il genotipo APOE e i fattori di rischio metabolico rimane incompletamente definito. Molti studi si limitano a controllare il genotipo dell’APOE piuttosto che considerare il suo potenziale ruolo moderatore nel rapporto tra obesità e rischio di AD(Vanhanen et al., 2006; Luchsinger et al., 2012). Quando lo stato di APOE è stato considerato come un modulatore del rischio di AD associato a fattori metabolici, i risultati sono stati misti. In alcuni studi, i portatori di APOE4hanno mostrato un deterioramento cognitivo significativamente maggiore in associazione a condizioni metaboliche avverse, tra cui l’aterosclerosi, le malattie vascolari periferiche, il diabete di tipo 2(Haan et al., 1999) e l’alta pressione sistolica a metà vita(Peila et al., 2001). Inoltre, i livelli di placche senili e di grovigli neurofibrillari erano più alti negli uomini obesi che erano anche portatori di APOE4(Peila et al., 2002). Tuttavia, diversi altri studi hanno riportato che il rischio di AD associato all’obesità e alla sindrome metabolica è più forte nei portatori di APOE3(Dixit et al., 2005; Leiva et al., 2005; Singh et al., 2006; Profenno e Faraone, 2008).
Una considerazione importante nell’interpretare questi risultati apparentemente discordanti è il ruolo potenziale delle differenze di sesso. Anche se l’impatto delle differenze di sesso nelle interazioni tra obesità, APOE, e il rischio di AD non è stato completamente affrontato, AD è caratterizzato da numerose differenze di sesso(Li e Singh, 2014; Pike, 2017). Inoltre, il rischio associato all’AD dell’APOE4sembra colpire in modo sproporzionato le donne(Payami et al., 1994; Farrer et al., 1997; Altmann et al., 2014). Inoltre, ci sono differenze di sesso in vari aspetti dell’obesità(Lovejoy et al., 2009; Mauvais-Jarvis, 2015; Moser e Pike, 2016), comprese le osservazioni che le donne mostrano una relativa protezione contro l’obesità fino alla menopausa(Meyer et al., 2011; Sugiyama e Agellon, 2012; Bloor e Symonds, 2014). Dato che sono state riscontrate differenze di sesso in ciascuno di questi fattori, gli studi futuri dovrebbero affrontare il sesso come possibile mediatore nella relazione tra APOE4e obesità. I progetti in corso nel nostro laboratorio hanno iniziato ad affrontare questo problema utilizzando topi femmine E3FAD e E4FAD.
Resta da determinare come l’obesità e l’APOE interagiscono per regolare la patogenesi dell’AD. Un meccanismo candidato collegato ad entrambi i fattori è il deterioramento metabolico. L’obesità è fortemente associata allo sviluppo di glucosio alterato e il metabolismo dell’insulina(Kahn et al., 2006; Singla et al., 2010), che sono anche caratteristici dei pazienti AD e sono stati proposti come possibili meccanismi di guida della patogenesi AD(Craft, 2005; Martins et al., 2006; Craft, 2009). In particolare, il genotipo APOE influenza le risposte metaboliche alla dieta(Snook et al., 1999; Barberger-Gateau et al., 2011), e diversi studi dimostrano che i portatori di APOE4sono a rischio aumentato per una serie di disturbi metabolici(de-Andrade et al., 2000; Oh e Barrett-Connor, 2001; Elosua et al., 2003; Marques-Vidal et al., 2003; Sima et al., 2007; Kypreos et al., 2009; Niu et al., 2009; Atabek et al., 2012; Zarkesh et al., 2012; Guan et al., 2013), sebbene alcuni studi non trovino alcun effetto del genotipo APOE sugli esiti metabolici(Meigs et al., 2000; Ragogna et al., 2011). I nostri risultati suggeriscono che i topi E3FAD possono essere più suscettibili ad alcuni effetti metabolici della dieta occidentale, anche se i topi E4FAD tendono ad avere disturbi metabolici anche in assenza di una dieta occidentale. In particolare, rispetto ai topi E4FAD, i topi E3FAD hanno mostrato un maggiore aumento di peso corporeo indotto dalla dieta, un’espressione infiammatoria delle citochine gonadiche e livelli di glucosio più elevati nella dieta occidentale. Al contrario, i topi E4FAD avevano un maggiore peso del cuscinetto di grasso gonadico e una tendenza verso livelli di glucosio a digiuno più elevati rispetto ai topi E3FAD sotto la condizione di dieta di controllo. Questi risultati sono coerenti con diversi rapporti precedenti che mostrano che i topi con APOE3umano guadagnano più peso in risposta a una dieta ad alto contenuto di grassi rispetto ai topi con APOE4umano(Arbones-Mainar et al., 2008; Segev et al., 2016) o topi APOE(Karagiannides et al., 2008). È importante notare che la dieta occidentale utilizzata in questo studio ha elevati livelli di grassi saturi, colesterolo e saccarosio, che sono stati tutti indipendentemente associati con un aumento della patologia AD-correlata(Refolo et al., 2000; Oksman et al., 2006; Cao et al., 2007; Takechi et al., 2010). La comprensione di come il genotipo APOE interagisce con vari componenti dietetici dovrebbe essere uno degli obiettivi dei futuri studi. Anche se i fattori metabolici possono avere un ruolo nella patogenesi di AD, i nostri risultati che i risultati metabolici di DIO erano maggiori in E3FAD rispetto ai topi E4FAD sostengono contro la possibilità che il deterioramento metabolico contribuisce in modo significativo alla distorsione APOE4osservata nella dieta indotta da aumenti della patologia AD-like.
Ci sono diversi altri meccanismi oltre al deterioramento metabolico che possono contribuire alle interazioni osservate tra obesità, APOE, e AD-come patologia. Una conseguenza consolidata delle diete obesogene è l’alterazione pro-amiloidogenica nell’espressione e/o nell’attività dei fattori che regolano la generazione e la clearance di Aβ, tra cui BACE1, neprilisina e IDE (Standeven et al., 2010; Maesako et al., 2012; Brandimarti et al., 2013; Wei et al., 2014; Maesako et al., 2015). Anche se non possiamo escludere un ruolo significativo di tali percorsi nelle nostre osservazioni, non abbiamo osservato che i livelli di mRNA di BACE1, neprilisina e IDE sono stati significativamente alterati dagli effetti semplici o interattivi della dieta occidentale e dell’APOE. Un altro meccanismo candidato convincente è la neuroinfiammazione, che è ampiamente implicato come un regolatore significativo del rischio di AD e lo sviluppo di patologia AD(Glass et al., 2010; Wyss-Coray e Rogers, 2012; Heneka et al., 2015). In particolare, sia l’obesità che l’APOE4sono associati ad un aumento dell’infiammazione cerebrale e sistemica. Ad esempio, l’obesità è legata ad una maggiore infiltrazione di cellule immunitarie nel cervello(Buckman et al., 2014), così come una maggiore attivazione della glia(Koga et al., 2014; Dorfman e Thaler, 2015; Douglass et al., 2017). Inoltre, l’obesità aumenta l’infiammazione degli organi periferici, compreso il tessuto adiposo(Weisberg et al., 2003; Zeyda e Stulnig, 2009) e del fegato(Park et al., 2010). L’APOE4è anche associato a maggiori livelli di infiammazione nel cervello(Ophir et al., 2005; Vitek et al., 2009) e in tutto il corpo(Colton et al., 2004; Gale et al., 2014). Inoltre, la stimolazione dell’infiammazione innata in presenza di apoE4 aumenta la morte cellulare e i danni nei macrofagi(Cash et al., 2012), e nei microglia e nei neuroni(Maezawa et al., 2006a; 2006b). Nel contesto della patologia dell’AD, l ‘APOE4è associata a una maggiore attivazione gliale nei topi EFAD(Rodriguez et al., 2014). Allo stesso modo, abbiamo trovato che sia il numero totale che il livello relativo di attivazione morfologica della microglia e degli astrociti erano più alti in E4FAD rispetto ai topi E3FAD. Inoltre, abbiamo osservato che i topi E4FAD esprimono livelli di mRNA significativamente più elevati di marker gliali rispetto ai topi E3FAD sotto controllo e alle diete occidentali. Questi marcatori gliali sono stati significativamente aumentati in diverse regioni del cervello in risposta alla DIO in E4FAD ma non nei topi E3FAD. Forse in contrasto con i nostri risultati, topi di mezza età femminile APOE4topi di mezza età hanno mostrato livelli più elevati di neuroinfiammazione nell’ippocampo sotto controllo dieta, ma è diminuita la neuroinfiammazione con dieta ad alto contenuto di grassi, rispetto all’età e sesso accoppiato topi wild-type(Janssen et al., 2016). Anche se la presenza di transgeni AD familiari e la patologia Aβ nel modello EFAD può spiegare questi risultati divergenti, ci possono essere anche differenze di età e sesso nelle risposte infiammatorie sia alla dieta e APOE4 .Inoltre, poiché gli astrociti reattivi e la microglia sono associati alle placche Aβ, i cambiamenti nella gliosi che osserviamo con APOE4 e DIO possono essere una conseguenza, piuttosto che un contributo alla patologia Aβ. Pertanto, sono necessarie ulteriori ricerche per valutare direttamente il potenziale ruolo meccanicistico della gliosi nell’interazione tra APOE4e obesità in AD.
A nostra conoscenza, questa è la prima indagine sperimentale che esamina l’interazione tra APOE4e obesità nel contesto di AD. Le interazioni tra i fattori di rischio genetico come l ‘APOE4e i fattori di rischio ambientale e di stile di vita modificabile in AD non sono stati finora ben studiati, anche se esistono alcuni studi epidemiologici coerenti con questa possibilità(Dufouil et al., 2000; Hanson et al., 2013; Rajan et al., 2014; Wirth et al., 2014; Ishioka et al., 2016; Zheng e Li, 2016). I nostri risultati suggeriscono che il genotipo APOE influenza il rapporto tra obesità e AD, in modo tale che i portatori di APOE4possono essere più sensibili ai rischi associati all’obesità rispetto ai portatori di APOE3. Ciò illustra un’importante interazione gene-ambiente e indica la necessità di ulteriori ricerche che esplorino tali relazioni nel contesto dell’AD, oltre a identificare i meccanismi sottostanti. Inoltre, questi risultati identificano un’ampia popolazione che può essere a maggior rischio di AD, ma la cui possibilità di sviluppare la malattia può essere ridotta da cambiamenti preventivi dello stile di vita.
References
- Alzheimer’s Disease Neuroimaging Initiative Investigators. Sex modifies the APOE-related risk of developing Alzheimer disease. Ann Neurol. 2014; 75:563-573. DOI | PubMed
- Differential modulation of diet-induced obesity and adipocyte functionality by human apolipoprotein E3 and E4 in mice. Int J Obes Relat Metab Disord. 2008; 32:1595-1605. DOI
- Impaired adipogenic response to thiazolidinediones in mice expressing human apolipoproteinE4. FASEB J. 2010; 24:3809-3818. DOI | PubMed
- Association between apolipoprotein E polymorphism and subclinic atherosclerosis in patients with type 1 diabetes mellitus. J Clin Res Pediatr Endocrinol. 2012; 4:8-13. PubMed
- Increased morphological diversity of microglia in the activated hypothalamic supraoptic nucleus. J Neurosci. 2003; 23:7759-7766. PubMed
- Dietary omega 3 polyunsaturated fatty acids and Alzheimer’s disease: interaction with apolipoprotein E genotype. Curr Alzheimer Res. 2011; 8:479-491. PubMed
- Sex-specific effects of high fat diet on indices of metabolic syndrome in 3xTg-AD mice: implications for Alzheimer’s Disease. PLoS One. 2013; 8:e78554. DOI | PubMed
- Sexual dimorphism in white and brown adipose tissue with obesity and inflammation. Horm Behav. 2014; 66:95-103. DOI | PubMed
- Cafeteria diet inhibits insulin clearance by reduced insulin-degrading enzyme expression and mRNA splicing. J Endocrinol. 2013; 219:173-182. DOI | PubMed
- Obesity induced by a high-fat diet is associated with increased immune cell entry into the central nervous system. Brain Behav Immun. 2014; 35:33-42. DOI | PubMed
- Alzheimer’s Disease Neuroimaging Initiative. The APOE4 allele shows opposite sex bias in microbleeds and Alzheimer’s disease of humans and mice. Neurobiol Aging. 2016; 37:47-57. DOI | PubMed
- Intake of sucrose-sweetened water induces insulin resistance and exacerbates memory deficits and amyloidosis in a transgenic mouse model of Alzheimer disease. J Biol Chem. 2007; 282:36275-36282. DOI | PubMed
- Apolipoprotein E4 impairs macrophage efferocytosis and potentiates apoptosis by accelerating endoplasmic reticulum stress. J Biol Chem. 2012; 287:27876-27884. DOI | PubMed
- Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci Transl Med. 2011; 3:89ra57. DOI | PubMed
- APOE genotype-specific differences in human and mouse macrophage nitric oxide production. J Neuroimmunol. 2004; 147:62-67. PubMed
- Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science. 1993; 261:921-923. PubMed
- Insulin resistance syndrome and Alzheimer’s disease: age- and obesity-related effects on memory, amyloid, and inflammation. Neurobiol Aging. 2005; 26:65-69. DOI | PubMed
- The role of metabolic disorders in Alzheimer disease and vascular dementia: two roads converged. Arch Neurol. 2009; 66:300-305. DOI | PubMed
- Association of apolipoprotein E polymorphism with plasma lipids and Alzheimer’s disease in a Southern Brazilian population. Braz J Med Biol Res. 2000; 33:529-537. DOI | PubMed
- Association of CETP TaqI and APOE polymorphisms with type II diabetes mellitus in North Indians: a case control study. BMC Endocr Disord. 2005; 5:7. DOI | PubMed
- Hypothalamic inflammation and gliosis in obesity. Curr Opin Endocrinol Diabetes Obes. 2015; 22:325-330. DOI | PubMed
- Glia: silent partners in energy homeostasis and obesity pathogenesis. Diabetologia. 2017; 60:226-236. DOI | PubMed
- Influence of apolipoprotein E genotype on the risk of cognitive deterioration in moderate drinkers and smokers. Epidemiology. 2000; 11:280-284. PubMed
- Obesity modulates the association among APOE genotype, insulin, and glucose in men. Obes Res. 2003; 11:1502-1508. DOI | PubMed
- 2003-2013: a decade of body mass index, Alzheimer’s disease, and dementia. J Alzheimers Dis. 2015; 43:739-755. DOI | PubMed
- Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta-analysis. APOE and Alzheimer Disease Meta Analysis Consortium. JAMA. 1997; 278:1349-1356. PubMed
- Midlife and late-life obesity and the risk of dementia: cardiovascular health study. Arch Neurol. 2009; 66:336-342. DOI | PubMed
- Human apolipoprotein E4 alters the amyloid-beta 40:42 ratio and promotes the formation of cerebral amyloid angiopathy in an amyloid precursor protein transgenic model. J Neurosci. 2005; 25:2803-2810. DOI | PubMed
- APOε4 is associated with enhanced in vivo innate immune responses in human subjects. J Allergy Clin Immunol. 2014; 134:127-134. DOI | PubMed
- APOE and Alzheimer disease: a major gene with semi-dominant inheritance. Mol Psychiatry. 2011; 16:903-907. DOI | PubMed
- A pilot study of gene/gene and gene/environment interactions in Alzheimer disease. Clin Med Res. 2011; 9:17-25. DOI | PubMed
- Mechanisms underlying inflammation in neurodegeneration. Cell. 2010; 140:918-934. DOI | PubMed
- Histopathological correlations of islet amyloidosis with apolipoprotein E polymorphisms in type 2 diabetic Chinese patients. Pancreas. 2013; 42:1129-1137. DOI | PubMed
- The role of APOE epsilon4 in modulating effects of other risk factors for cognitive decline in elderly persons. JAMA. 1999; 282:40-46. PubMed
- Effect of apolipoprotein E genotype and diet on apolipoprotein E lipidation and amyloid peptides. JAMA Neurol. 2013; 70:972-980. DOI | PubMed
- Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015; 14:388-405. DOI | PubMed
- Obesity is linked with lower brain volume in 700 AD and MCI patients. Neurobiol Aging. 2010; 31:1326-1339. DOI | PubMed
- Diet-induced insulin resistance promotes amyloidosis in a transgenic mouse model of Alzheimer’s disease. FASEB J. 2004; 18:902-904. PubMed
- Effects of the APOE ε4 allele and education on cognitive function in Japanese centenarians. Age (Dordr). 2016; 38:495-503. DOI | PubMed
- Age, sex, and APOE ε4 effects on memory, brain structure, and β-Amyloid across the adult life span. JAMA Neurol. 2015; 72:511-519. DOI | PubMed
- The effect of a high-fat diet on brain plasticity, inflammation and cognition in female ApoE4-knockin and ApoE-knockout mice. PLoS One. 2016; 11:e0155307. DOI | PubMed
- High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiol Aging. 2010; 31:1516-1531. DOI | PubMed
- Age-dependent increases in tau phosphorylation in the brains of type 2 diabetic rats correlate with a reduced expression of p62. Exp Neurol. 2013; 248:441-450. DOI | PubMed
- Mechanisms linking obesity to insulin resistance and type 2 diabetes. Nature. 2006; 444:840-846. DOI | PubMed
- Apolipoprotein E predisposes to obesity and related metabolic dysfunctions in mice. FEBS J. 2008; 275:4796-4809. DOI | PubMed
- Increased tau phosphorylation and cleavage in mouse models of type 1 and type 2 diabetes. Endocrinology. 2009; 150:5294-5301. DOI | PubMed
- High-fat diet-induced memory impairment in triple-transgenic Alzheimer’s disease (3xTgAD) mice is independent of changes in amyloid and tau pathology. Neurobiol Aging. 2014; 35:1821-1832. DOI | PubMed
- Immunohistochemical analysis of tau phosphorylation and astroglial activation with enhanced leptin receptor expression in diet-induced obesity mouse hippocampus. Neurosci Lett. 2014; 571:11-16. DOI | PubMed
- Increased food intake leads to obesity and insulin resistance in the Tg2576 Alzheimer’s disease mouse model. Endocrinology. 2010; 151:1532-1540. DOI | PubMed
- Mechanisms of obesity and related pathologies: role of apolipoprotein E in the development of obesity. FEBS J. 2009; 276:5720-5728. DOI | PubMed
- The neuropathology of obesity: insights from human disease. Acta Neuropathol. 2013; 127:3-28. DOI | PubMed
- Apolipoprotein E polymorphism in type 2 diabetic patients of Talca, Chile. Diabetes Res Clin Pract. 2005; 68:244-249. DOI | PubMed
- Sex differences in cognitive impairment and Alzheimer’s disease. Front Neuroendocrinol. 2014; 35:385-403. DOI | PubMed
- Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat Rev Neurol. 2013; 9:106-118. DOI | PubMed
- Stock Conference 2008 Working Group. Sex differences in obesity and the regulation of energy homeostasis. Obes Rev. 2009; 10:154-167. DOI | PubMed
- Central obesity in the elderly is related to late-onset Alzheimer disease. Alzheimer Dis Assoc Disord. 2012; 26:101-105. DOI | PubMed
- Caloric intake and the risk of Alzheimer disease. Arch Neurol. 2002; 59:1258-1263. PubMed
- Exercise is more effective than diet control in preventing high fat diet-induced β-amyloid deposition and memory deficit in amyloid precursor protein transgenic mice. J Biol Chem. 2012; 287:23024-23033. DOI | PubMed
- High fat diet enhances β-Site cleavage of amyloid precursor protein (APP) via promoting β-Site APP cleaving enzyme 1/adaptor protein 2/clathrin complex formation. PLoS One. 2015; 10:e0131199. DOI | PubMed
- Apolipoprotein E-specific innate immune response in astrocytes from targeted replacement mice. J Neuroinflammation. 2006a; 3:10. PubMed
- Apolipoprotein E isoform-dependent dendritic recovery of hippocampal neurons following activation of innate immunity. J Neuroinflammation. 2006b; 3:21. PubMed
- Obesity and alcohol modulate the effect of apolipoprotein E polymorphism on lipids and insulin. Obes Res. 2003; 11:1200-1206. DOI | PubMed
- Apolipoprotein E, cholesterol metabolism, diabetes, and the convergence of risk factors for Alzheimer’s disease and cardiovascular disease. Mol Psychiatry. 2006; 11:721-736. DOI | PubMed
- Sex differences in metabolic homeostasis, diabetes, and obesity. Biol Sex Differ. 2015; 6:14. DOI | PubMed
- Apolipoprotein E isoform polymorphisms are not associated with insulin resistance: the Framingham Offspring Study. Diabetes Care. 2000; 23:669-674. DOI | PubMed
- Midlife vascular risk factors and the risk of Alzheimer’s disease: a systematic review and meta-analysis. J Alzheimers Dis. 2014; 42:1295-1310. DOI | PubMed
- Obesity, insulin resistance and diabetes: sex differences and role of oestrogen receptors. Acta Physiol (Oxf). 2011; 203:259-269. DOI | PubMed
- APOE predicts amyloid-beta but not tau Alzheimer pathology in cognitively normal aging. Ann Neurol. 2010; 67:122-131. DOI | PubMed
- Obesity and sex interact in the regulation of Alzheimer’s disease. Neurosci Biobehav Rev. 2016; 67:102-118. DOI | PubMed
- Obesity and diabetes cause cognitive dysfunction in the absence of accelerated β-amyloid deposition in a novel murine model of mixed or vascular dementia. Acta Neuropathol Commun. 2014; 2:64. DOI | PubMed
- The relationship between apolipoprotein E e2/e3/e4 polymorphisms and hypertension: a meta-analysis of six studies comprising 1812 cases and 1762 controls. Hypertens Res. 2009; 32:1060-1066. DOI | PubMed
- Apolipoprotein E polymorphism and lipid levels differ by gender and family history of diabetes: the Rancho Bernardo Study. Clin Genet. 2001; 60:132-137. DOI | PubMed
- Impact of different saturated fatty acid, polyunsaturated fatty acid and cholesterol containing diets on beta-amyloid accumulation in APP/PS1 transgenic mice. Neurobiol Dis. 2006; 23:563-572. DOI | PubMed
- Apolipoprotein E4 enhances brain inflammation by modulation of the NF-κB signaling cascade. Neurobiol Dis. 2005; 20:709-718. DOI | PubMed
- Mammalian target of rapamycin hyperactivity mediates the detrimental effects of a high sucrose diet on Alzheimer’s disease pathology. Neurobiol Aging. 2014; 35:1233-1242. DOI | PubMed
- Dietary and genetic obesity promote liver inflammation and tumorigenesis by enhancing IL-6 and TNF expression. Cell. 2010; 140:197-208. DOI | PubMed
- Alzheimer’s disease, apolipoprotein E4, and gender. JAMA. 1994; 271:1316-1317. PubMed
- Type 2 diabetes, APOE gene, and the risk for dementia and related pathologies: the Honolulu-Asia Aging Study. Diabetes. 2002; 51:1256-1262. PubMed
- Joint effect of the APOE gene and midlife systolic blood pressure on late-life cognitive impairment: the Honolulu-Asia aging study. Stroke. 2001; 32:2882-2889. PubMed
- Sex and the development of Alzheimer’s disease. J Neurosci Res. 2017; 95:671-680. DOI | PubMed
- Diabetes and overweight associate with non-APOE4 genotype in an alzheimer’s disease population. Am J Med Genet. 2008; 147B:822-829. DOI | PubMed
- Meta-analysis of Alzheimer’s disease risk with obesity, diabetes, and related disorders. Biol Psychiatry. 2010; 67:505-512. DOI | PubMed
- Lack of association of apoE ε4 allele with insulin resistance. Acta Diabetol. 2011; 49:25-32. DOI | PubMed
- Gene-environment interaction of body mass index and apolipoprotein E ε4 allele on cognitive decline. Alzheimer Dis Assoc Disord. 2014; 28:134-140. DOI | PubMed
- Differential central pathology and cognitive impairment in pre-diabetic and diabetic mice. Psychoneuroendocrinology. 2013; 38:2462-2475. DOI | PubMed
- Apolipoprotein E in sporadic Alzheimer’s disease: allelic variation and receptor interactions. Neuron. 1993; 11:575-580. PubMed
- Hypercholesterolemia accelerates the Alzheimer’s amyloid pathology in a transgenic mouse model. Neurobiol Dis. 2000; 7:321-331. DOI | PubMed
- Human APOE4 increases microglia reactivity at Aβ plaques in a mouse model of Aβ deposition. J Neuroinflammation. 2014; 11:111. DOI | PubMed
- Association of apolipoprotein E allele epsilon 4 with late-onset familial and sporadic Alzheimer’s disease. Neurology. 1993; 43:1467-1472. PubMed
- Concurrence of high fat diet and APOE gene induces allele specific metabolic and mental stress changes in a mouse model of Alzheimer’s disease. Front Behav Neurosci. 2016; 10:170. DOI | PubMed
- Apolipoprotein E polymorphism–a risk factor for metabolic syndrome. Clin Chem Lab Med. 2007; 45:1149-1153. DOI | PubMed
- Association of APOE (Hha1) and ACE (I/D) gene polymorphisms with type 2 diabetes mellitus in North West India. Diabetes Res Clin Pract. 2006; 74:95-102. DOI | PubMed
- Metabolic effects of obesity: a review. World J Diabetes. 2010; 1:76-88. DOI | PubMed
- Effect of synthetic triglycerides of myristic, palmitic, and stearic acid on serum lipoprotein metabolism. Eur J Clin Nutr. 1999; 53:597-605. PubMed
- Neprilysin, obesity and the metabolic syndrome. Int J Obes Relat Metab Disord. 2010; 35:1031-1040. DOI
- Apolipoprotein E: high-avidity binding to beta-amyloid and increased frequency of type 4 allele in late-onset familial Alzheimer disease. Proc Natl Acad Sci USA. 1993; 90:1977-1981. PubMed
- Sex differences in lipid metabolism and metabolic disease risk. Biochem Cell Biol. 2012; 90:124-141. DOI | PubMed
- Differential effects of dietary fatty acids on the cerebral distribution of plasma-derived apo B lipoproteins with amyloid-beta. Br J Nutr. 2010; 103:652-662. DOI | PubMed
- Early-onset versus late-onset Alzheimer’s disease: the case of the missing APOE ε4 allele. Lancet Neurol. 2011; 10:280-288. DOI | PubMed
- Association of metabolic syndrome with Alzheimer disease: a population-based study. Neurology. 2006; 67:843-847. DOI | PubMed
- APOE genotype-specific differences in the innate immune response. Neurobiol Aging. 2009; 30:1350-1360. DOI | PubMed
- Regulation of insulin degrading enzyme activity by obesity-associated factors and pioglitazone in liver of diet-induced obese mice. PLoS One. 2014; 9:e95399. DOI | PubMed
- Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest. 2003; 112:1796-1808. DOI | PubMed
- Central obesity and increased risk of dementia more than three decades later. Neurology. 2008; 71:1057-1064. DOI | PubMed
- Redefining the concept of reactive astrocytes as cells that remain within their unique domains upon reaction to injury. Proc Natl Acad Sci USA. 2006; 103:17513-17518. DOI | PubMed
- Gene-environment interactions: lifetime cognitive activity, APOE genotype, and beta-amyloid burden. J Neurosci. 2014; 34:8612-8617. DOI | PubMed
- Inflammation in Alzheimer disease-a brief review of the basic science and clinical literature. Cold Spring Harb Perspect Med. 2012; 2:a006346. DOI | PubMed
- APOE4-specific changes in amyloid-beta accumulation in a new transgenic mouse model of Alzheimer disease. J Biol Chem. 2012; 287:41774-41786. DOI | PubMed
- Is there any association of apolipoprotein E gene polymorphism with obesity status and lipid profiles? Tehran Lipid and Glucose Study (TLGS). Gene. 2012; 509:282-285. DOI | PubMed
- Obesity, inflammation, and insulin resistance–a mini-review. Gerontology. 2009; 55:379-386. DOI | PubMed
- Prolonged diet induced obesity has minimal effects towards brain pathology in mouse model of cerebral amyloid angiopathy: implications for studying obesity-brain interactions in mice. Biochim Biophys Acta. 2013; 1832:1456-1462. DOI | PubMed
- Impact of apolipoprotein E gene polymorphism and additional gene-obesity interaction on type 2 diabetes risk in a Chinese Han old population. Obes Res Clin Pract. 2016.
Fonte
Moser VA, Pike CJ (2017) Obesity Accelerates Alzheimer-Related Pathology in . eNeuro 4(3): ENEURO.0077-17.2017. https://doi.org/10.1523/ENEURO.0077-17.2017




