
Abstract
Introduzione
È stato dimostrato che il microbiota intestinale influenza criticamente una moltitudine di funzioni fisiologiche dell’ospite, spesso attraverso la modulazione del sistema immunitario(Gevers et al., 2014; Paun et al., 2016). L’evidenza include studi in topi privi di germi, che mostrano che sia le cellule T-helper 17 (Th17) pro-infiammatorie che le cellule T regolatorie anti-infiammatorie (Treg) sono ridotte in numero nella lamina propria intestinale rispetto ai topi convenzionali o specifici privi di patogeni(Atarashi et al., 2008), e che il ripopolamento con batteri specifici può ricostituirli(Atarashi et al., 2015; Ivanov et al., 2009). Una via per capire come il microbiota influenza l’attivazione del sistema immunitario è la produzione di piccole molecole(Donia e Fischbach, 2015). È stato dimostrato che gli acidi grassi a catena corta (SCFAs) prodotti dai microbi facilitano la differenziazione extrattimica del sistema immunitario modulatorioTreg(Arpaia et al., 2013; Atarashi et al., 2013; Smith et al., 2013) e sono implicati nelle proprietà antinfiammatoriedipendenti daTreg di una miscela di 17 ceppi di Clostridia di origine umana (Atarashi et al.,2013; Tanoue et al., 2016). È interessante notare che i singoli ceppi di Clostridia hanno solo un modesto effetto sull’induzione diTreg – l’induzione ottimale si basa sull’interazione sinergica di diversi ceppi (Atarashiet al., 2013; Belkaid e Hand, 2014). A causa di queste proprietà, c’è attualmente un grande interesse nella manipolazione di questa comunità per il trattamento di malattie infiammatorie e allergiche(Honda e Littman, 2012; Olle, 2013; Vieira et al., 2016). Gli sforzi che coinvolgono il trapianto di batteri da esseri umani sani a esseri umani con infezioni C. difficile o con malattie metaboliche hanno fornito la prova che il ripopolamento dei microbioti potrebbe essere utilizzato come possibile strategia per la prevenzione e il trattamento delle malattie(van Nood et al., 2013; Vrieze et al., 2012). Per questo motivo, l’integrazione intestinale di composizioni definite di batteri intestinali per il trattamento di una serie di malattie è attualmente perseguita da diverse aziende biofarmaceutiche(Garber, 2015). Determinare quali consorzi microbici possono colonizzare l’ospite e coesistere stabilmente con una comunità microbica già residente, inducendo al contempo la risposta immunitaria desiderata, è ancora una grande sfida che ostacola la traduzione di questi sforzi nella clinica(Maldonado-Gómez et al., 2016; Weil e Hohmann, 2015).
Nel lavoro precedente, alcuni di noi hanno identificato un insieme di 17 ceppi di Clostridi che inducono i Treg-inducenti potenti per selezionare i sottoinsiemi dei candidati (Atarashi et al., 2013). La determinazione di combinazioni al massimo immunofenotipo inducenti da questi 17 ceppi è, tuttavia, sperimentalmente impossibile in quanto richiederebbe il test di 217-1 =131071 possibili sottoinsiemi nei topi (Faith et al., 2014). Pertanto, un approccio computazionale per dare priorità ai sottoinsiemi da convalidare sperimentalmente avrebbe una grande utilità. A nostra conoscenza, a tutt’oggi non esiste un modello che permetta di prevedere simultaneamente la dinamica sia del microbiota che della risposta immunitaria dell’ospite. Basandoci sull’approccio di selezione delle combinazioni batteriche ottimali(Faith et al., 2014) e su altri sforzi volti a identificare potenziali microbi immunomodulanti(Geva-Zatorsky et al., 2017; Schirmer et al., 2016), superiamo questo problema proponendo una struttura basata sulla modellazione matematica che, risolvendo le interazioni tra sistema microbico e sistema immunitario con parametri vincolati alle osservazioni sperimentali degli effettori microbici e immunitari, permette l’ottimizzazione computazionale delle combinazioni batteriche immunostimolanti. Per raggiungere questo obiettivo, utilizziamo una serie di analisi logicamente connesse che catturano l’accumulo di CD4+FOXP3+Treg nella lamina propria del colon(Omenetti e Pizarro, 2015; Round e Mazmanian, 2010) in risposta alla dinamica dei ceppi di Clostridia di derivazione umana nei topi privi di germi, che in precedenza avevano dimostrato di indurre potentemente l’espansione di Treg (Atarashiet al., 2013) (Figura 1). Per limitare questo quadro modello proposto ai dati sperimentali, combiniamo i dati precedentemente pubblicati(Atarashi et al., 2013) e i dati di nuova generazione della scansione cellulare attivata dalla fluorescenza (FACS) con i dati di colonizzazione simulati e di nuova generazione delle serie temporali dei topi privi di germi(Figura 2A). Successivamente, applicando un modello matematico microbiome-Treg a questo insieme di dati combinato, si deduce il contributo individuale di ogni ceppo al pool di Treg CD4+FOXP3+( Figura 2B) e si ottiene una stima del potenziale di induzione Treg di ogni possibile consorzio. Per giustificare l’uso di concentrazioni di mono-colonizzazione previste da un modello ecologico di microbioma sviluppato in precedenza(Bucci et al., 2016), ne validiamo la capacità di prevedere la dinamica temporale in risposta a diversi sottoinsiemi di ceppi diTreg(Figura 3A) e quantifichiamo la deviazione dei dati e delle previsioni(Figura 3B). L’introduzione del TrIS, che assegna un punteggio ad ogni composizione microbica allo stato stazionario previsto, ci permette di identificare combinazioni che massimizzano fortemente l’induzione diTreg(Figura 4A). Analizziamo la relazione tra TrIS e biomassa(Figura 4B) così come le caratteristiche metaboliche dei consorzi(Figura 4C e D). La cosa più importante, dimostriamo l’utilità del nostro approccio nel prevedere la potenza di combinazioni microbiche selezionate convalidando l’induzione di Treg e la capacità di colonizzazione di consorzi modello-predetto forti, intermedi e deboli che inducono Treg in vivo ( Figura 4E ).
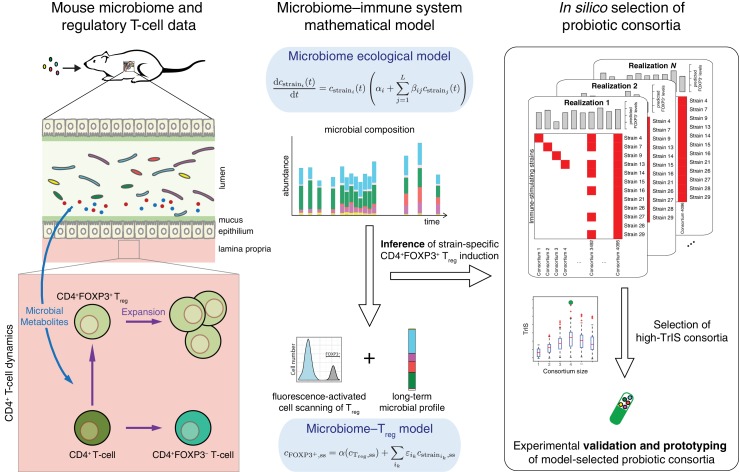
Figura 1.Figura 1. Figura concettuale.Un modello matematico del sistema microbiomo-immune che descrive l’attivazione delle cellule T regolatorie (Treg)in risposta ai profili di colonizzazione dei ceppi di Clostridia stimolanti Treg è l’elemento centrale di questo lavoro. Esso consiste in un modello ecologico microbiomico precedentemente derivato che descrive la dinamica temporale a breve e lungo termine dei ceppi di Clostridia nei topi privi di germi (Bucci et al.,2016) ed è integrato da un modello microbiome-Treg di attivazione CD4+FOXP3+ Treg in risposta a composizioni a lungo termine nel microbioma. Il contributo individuale di ogni ceppo all’attivazione diTreg misurata viene dedotto usando i dati di colonizzazione a lungo termine provenienti da esperimenti sui topi con sottoinsiemi di questi ceppi e le corrispondenti misurazioni dell’induzione di Treg. Il punteggio diinduzione di Treg, TrIS, che rappresenta la stabilità ecologica e l’attivazione immunitaria, assegna un punteggio ad ogni possibile combinazione di ceppi e quindi identifica i consorzi probiotici candidati per la validazione sperimentale.
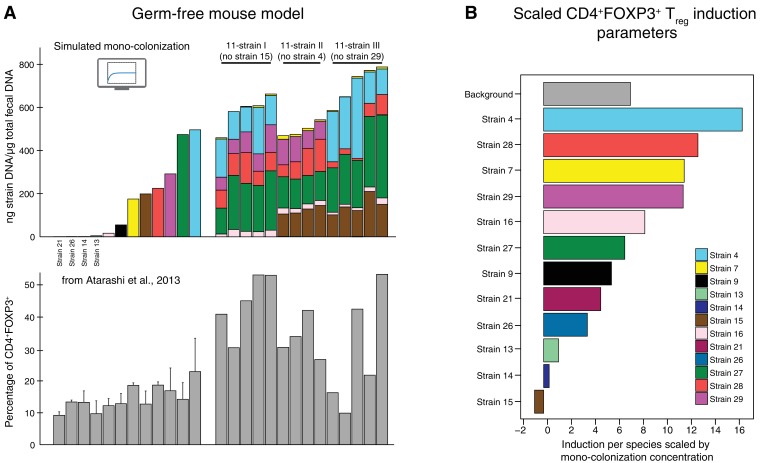
Figura 2 – Dati fonte 1.Dati utilizzati per l’inferenza dei parametri di induzione CD4+FOXP3+Treg.dati contenenti le composizioni microbicromatiche, le corrispondenti proporzioni CD4+FOXP3+ misurate e i parametri di induzione derivati.dati contenenti le composizioni microbicromatiche, le corrispondenti proporzioni CD4+FOXP3+ misurate e i parametri di induzione derivati.(A) Per dedurre i parametri di induzione CD4+FOXP3+Treg risolti dal ceppo CD4+FOXP3+ parametri di induzione CD4+FOXP3+Treg si usano le misurazioni dell’abbondanza CD4+FOXP3+ e i corrispondenti dati di colonizzazione del microbioma. Idati CD4+FOXP3+ Treg di singoli ceppi provengono da misurazioni precedentemente pubblicate da Atarashi et al. (2013). Poiché in Atarashi et al. (2013) i livelli di mono-colonizzazione microbica non sono stati misurati, per simulare questi esperimenti è stato utilizzato un modello predittivo precedentemente pubblicato(Bucci et al., 2016). Inoltre, i dati generati di recente CD4+FOXP3+Treg e le feci di topo di colonizzazione da tre combinazioni di 11 ceppi sono stati inclusi nell’analisi (vedi anche Figura 3). Le combinazioni di 11 ceppi sono state scelte in base ai risultati dell’analisi della ‘keystoneness’ precedentemente descritta in Bucci et al. (2016). Le composizioni microbicromatiche di queste tre combinazioni di 11 ceppi sono state stimate da qPCR specifico per ceppo.(B) I risultanti parametri di induzione CD4+FOXP3+Treg quantificano il contributo di ogni singolo ceppo all’induzioneTreg. (I coefficienti sono scalati dalle concentrazioni microbiche di mono-colonizzazione per ragioni di visualizzazione).10.7554/eLife.30916.005Figure 2-source data 1.Data contenente le composizioni microbiche, le corrispondenti proporzioni CD4+FOXP3+ misurate e i parametri di induzione derivati.
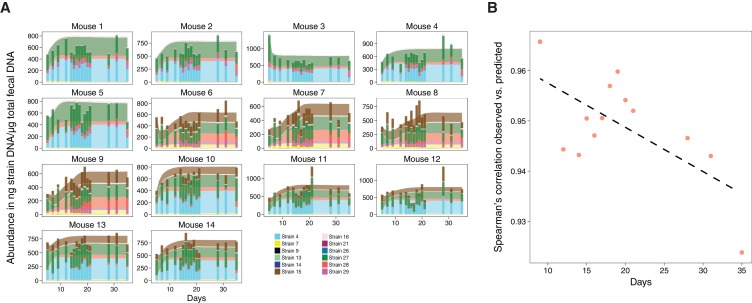
Figura 3.Figura 3. Convalida del modello microbico che descrive la dinamica dei ceppi di Clostridia nei topi privi di germi.(A) Le dinamiche previste dal modello di tre combinazioni di 11 ceppi dei 13 ceppi di Clostridia descritti in Bucci et al. (2016) (aree) sono confrontate con i dati misurati da topi privi di germi (barre impilate) e ogni pannello corrisponde ad un singolo topo(Figura 2A). Le previsioni sono state ottenute integrando numericamente le corrispondenti equazioni generalizzate di Lotka-Volterra con i parametri di Bucci et al. (2016) utilizzando solo la composizione microbica iniziale di ogni topo. Nelle linee temporali 1-5, il ceppo 15 è assente con riferimento al set di 11 ceppi I (cinque repliche biologiche); nelle linee temporali 6-9, il ceppo 4 è assente (set di 11 ceppi II; quattro repliche biologiche), e nelle linee temporali 10-14, il ceppo 29 è assente (set di 11 ceppi III; cinque repliche biologiche). I dati sono stati ottenuti mediante qPCR di geni specifici per ogni ceppo. Le densità sono calcolate come medie di tre repliche tecniche. Il numero di topi utilizzati in ogni condizione è stato scelto in modo coerente con i precedenti lavori sperimentali(Atarashi et al., 2013; Bucci et al., 2016) e combinato con test approfonditi in silico dell’errore di inferenza in funzione della frequenza di campionamento e dei livelli di rumore(Bucci et al., 2016).(B) Il coefficiente di correlazione del rango di Spearman tra i dati osservati e quelli previsti è stato calcolato in diversi punti temporali. Tutti i coefficienti visualizzati hanno un valore p inferiore a 10-16.
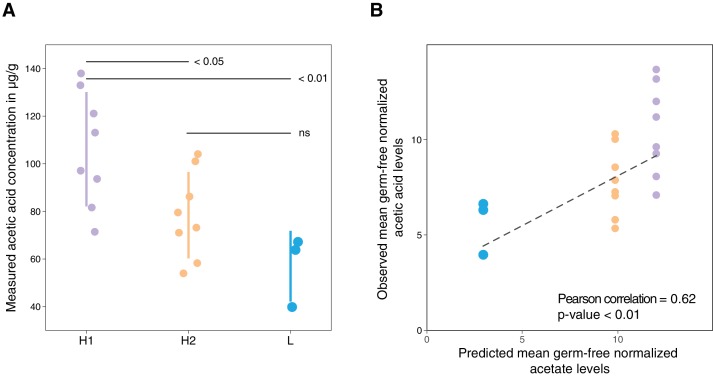
Figura 4-figure supplemento 2.Identificazione e validazione sperimentale dei consorzistimolatori di Treg previsti dal modello.TrIS, rango TrIS e concentrazioni mediane previste per lo stato stazionario dei consorzi microbici selezionati dalla Figura 4E.TrIS, rango TrIS e concentrazioni mediane previste per lo stato stazionario dei consorzi microbici selezionati dalla Figura 4E.Profilo metabolico di tre consorzi microbici selezionati e confronto dei profili metabolici previsti e misurati di tre consorzi microbici selezionati.(A) Punteggio di induzione diTreg previsto (TrIS) in topi privi di germi in funzione delle dimensioni del consorzio probiotico. Ogni punto dati rappresenta uno dei 2N-1 possibili consorzi non banali di dimensione N. I cinque consorzi TrIS più alti e più bassi di dimensione sette sono evidenziati rispettivamente da punti pieni di viola e ciano, così come, i consorzi a 4 ceppi utilizzati per la validazione sperimentale nel sotto-pannello E. (B) Distribuzione dei TrIS tracciati rispetto alla densità di popolazione totale di ciascun consorzio previsto dal modello. Questa analisi mostra una forte correlazione (Spearman’s Rank Correlation con il coefficiente 0,88 e p<0,05) tra TrIS e la densità microbica totale. Il colore di ogni punto rappresenta l’abbondanza del ceppo 27, che si prevede fortemente colonizzato in sottoinsiemi ad alto TrIS.(C) Heatmap di appartenenza al consorzio dei cinque più alti e più bassi consorzi TrIS di dimensione sette e la loro induzioneTreg prevista e la produzione stimata di butirrato utilizzando i dati di Atarashi et al. (2013).(D) Stima della produzione potenziale di SCFA basata sul profilo metabolico in vivo di un singolo ceppo in vivo di Atarashi et al. (2013). Si prevede che i cinque maggiori consorzi TrIS di dimensione sette (da 4C) avranno una produzione di SCFA significativamente più elevata rispetto ai cinque più bassi.(E) Cinque consorzi di 4-strain sono stati utilizzati per la validazione sperimentale: i due consorzi di più alto rango (H1 e H2, rispettivamente), il consorzio di più basso rango (L) e due intermedi (M1, M2). M1 e M2 sono stati inclusi a causa dell’interesse per altre aree di malattia. I ceppi introdotti sperimentalmente sono elencati accanto a ciascun bar/consorzio. Ceppi con numeri in nero sono stati rilevati dal sequenziamento 16S rRNA in campioni di feci di topo, ceppi con numeri in grigio sono stati introdotti, ma non è riuscito a colonizzare. Una correlazione di Pearson di 0,97 tra il TrIS previsto e la media della percentuale di Treg di ciascun consorzio misurata CD4+FOXP3+ Treg dimostra la capacità della selezione basata su TrIS di recuperare correttamente l’induzione di Treg osservata sperimentalmente. (La correlazione di Pearson di tutti i punti ha un valore di 0,54.) È importante notare che il consorzio H2 mostra un aumento medio dell’attività immunitaria del 107% rispetto alla media del controllo del mouse privo di germi. Otto repliche biologiche sono state utilizzate per GF, H1 e H2, cinque repliche biologiche sono state utilizzate per M1, M2 e tre repliche biologiche sono state utilizzate per L. La replica e la progettazione dell’esperimento di convalida per la valutazione dell’induzione diTreg è coerente con il lavoro svolto da noi e da altri(Atarashi et al., 2013; Narushima et al., 2014).10.7554/eLife.30916.010Figure 4-source data 1.TrIS, TrIS classifica e prevede le concentrazioni mediane allo stato stazionario dei consorzi microbici selezionati dalla Figura 4E.In contrasto con la Figura 4B, che evidenzia solo l’abbondanza del ceppo 27 in ogni consorzio, la densità di popolazione totale prevista dei consorzi microbici viene confrontata con il TrIS, mentre il colore in ogni sotto-pannello rappresenta l’abbondanza del ceppo corrispondente.(A) Risultati del profilo metabolico del contenuto fecale dei consorzi selezionati rispetto all’acido acetico.(B) Confronto tra le concentrazioni medie previste di acetato normalizzato privo di germi in base alle misurazioni del singolo ceppo nel contenuto cecale da(Atarashi et al., 2013) con i livelli medi osservati di acido acetico normalizzato privo di germi nelle feci di topo da (A).
Immaginiamo che il nostro quadro, mentre in questo studio su misura per trovare combinazioni che migliorano le condizioni auto-infiammatorie, può anche avere una rilevanza diretta per altre applicazioni che migliorano il sistema immunitario, come la consegna ottimale di immunoterapie antitumorali basate su probiotici(Garrett, 2015).
Figura 1.Figura 1. Figura concettuale.Un modello matematico del sistema microbiomo-immune che descrive l’attivazione delle cellule T regolatorie (Treg)in risposta ai profili di colonizzazione dei ceppi di Clostridia stimolanti Treg è l’elemento centrale di questo lavoro. Esso consiste in un modello ecologico microbiomico precedentemente derivato che descrive le dinamiche temporali a breve e lungo termine dei ceppi di Clostridia nei topi privi di germi (Bucci et al.,2016) ed è integrato da un modello microbiome-Treg di attivazione CD4+FOXP3+ Treg in risposta a composizioni a lungo termine nel microbioma. Il contributo individuale di ogni ceppo all’attivazione diTreg misurata viene dedotto usando i dati di colonizzazione a lungo termine provenienti da esperimenti sui topi con sottoinsiemi di questi ceppi e le corrispondenti misurazioni dell’induzione di Treg. Il punteggio diinduzione di Treg, TrIS, che rappresenta la stabilità ecologica e l’attivazione immunitaria, assegna un punteggio ad ogni possibile combinazione di ceppi e quindi identifica i consorzi probiotici candidati per la validazione sperimentale.
Figura 2 – Dati fonte 1.Dati utilizzati per l’inferenza dei parametri di induzione CD4+FOXP3+Treg.dati contenenti le composizioni microbicromatiche, le corrispondenti proporzioni CD4+FOXP3+ misurate e i parametri di induzione derivati.dati contenenti le composizioni microbicromatiche, le corrispondenti proporzioni CD4+FOXP3+ misurate e i parametri di induzione derivati.(A) Per dedurre i parametri di induzione CD4+FOXP3+Treg risolti dal ceppo CD4+FOXP3+ parametri di induzione CD4+FOXP3+Treg si usano le misurazioni dell’abbondanza CD4+FOXP3+ e i corrispondenti dati di colonizzazione del microbioma. Idati CD4+FOXP3+ Treg di singoli ceppi provengono da misurazioni precedentemente pubblicate da Atarashi et al. (2013). Poiché in Atarashi et al. (2013) i livelli di mono-colonizzazione microbica non sono stati misurati, per simulare questi esperimenti è stato utilizzato un modello predittivo precedentemente pubblicato(Bucci et al., 2016). Inoltre, i dati generati di recente CD4+FOXP3+Treg e le feci di topo di colonizzazione da tre combinazioni di 11 ceppi sono stati inclusi nell’analisi (vedi anche Figura 3). Le combinazioni di 11 ceppi sono state scelte in base ai risultati dell’analisi della ‘keystoneness’ precedentemente descritta in Bucci et al. (2016). Le composizioni microbicromatiche di queste tre combinazioni di 11 ceppi sono state stimate da qPCR specifico per ceppo.(B) I risultanti parametri di induzione CD4+FOXP3+Treg quantificano il contributo di ogni singolo ceppo all’induzioneTreg. (I coefficienti sono scalati dalle concentrazioni microbiche di mono-colonizzazione per ragioni di visualizzazione).10.7554/eLife.30916.005Figure 2-source data 1.Data contenente le composizioni microbiche, le corrispondenti proporzioni CD4+FOXP3+ misurate e i parametri di induzione derivati.
Figura 3.Figura 3. Convalida del modello microbico che descrive la dinamica dei ceppi di Clostridia nei topi privi di germi.(A) Le dinamiche previste dal modello di tre combinazioni di 11 ceppi dei 13 ceppi di Clostridia descritti in Bucci et al. (2016) (aree) sono confrontate con i dati misurati da topi privi di germi (barre impilate) e ogni pannello corrisponde ad un singolo topo(Figura 2A). Le previsioni sono state ottenute integrando numericamente le corrispondenti equazioni generalizzate di Lotka-Volterra con i parametri di Bucci et al. (2016) utilizzando solo la composizione microbica iniziale di ogni topo. Nelle linee temporali 1-5, il ceppo 15 è assente con riferimento al set di 11 ceppi I (cinque repliche biologiche); nelle linee temporali 6-9, il ceppo 4 è assente (set di 11 ceppi II; quattro repliche biologiche), e nelle linee temporali 10-14, il ceppo 29 è assente (set di 11 ceppi III; cinque repliche biologiche). I dati sono stati ottenuti mediante qPCR di geni specifici per ogni ceppo. Le densità sono calcolate come medie di tre repliche tecniche. Il numero di topi utilizzati in ogni condizione è stato scelto in modo coerente con i precedenti lavori sperimentali(Atarashi et al., 2013; Bucci et al., 2016) e combinato con test approfonditi in silico dell’errore di inferenza in funzione della frequenza di campionamento e dei livelli di rumore(Bucci et al., 2016).(B) Il coefficiente di correlazione del rango di Spearman tra i dati osservati e quelli previsti è stato calcolato in diversi punti temporali. Tutti i coefficienti visualizzati hanno un valore p inferiore a 10-16.
Figura 4-figure supplemento 2.Identificazione e validazione sperimentale dei consorzistimolatori di Treg previsti dal modello.TrIS, rango TrIS e concentrazioni mediane previste per lo stato stazionario dei consorzi microbici selezionati dalla Figura 4E.TrIS, rango TrIS e concentrazioni mediane previste per lo stato stazionario dei consorzi microbici selezionati dalla Figura 4E.Profilo metabolico di tre consorzi microbici selezionati e confronto dei profili metabolici previsti e misurati di tre consorzi microbici selezionati.(A) Punteggio di induzione diTreg previsto (TrIS) nei topi privi di germi in funzione delle dimensioni del consorzio probiotico. Ogni punto di dati rappresenta uno dei 2N-1 possibili consorzi non banali di dimensione N. I cinque consorzi TrIS più alti e più bassi di dimensione sette sono evidenziati rispettivamente da punti pieni di viola e ciano, così come, i consorzi a 4 ceppi utilizzati per la convalida sperimentale nel sotto-pannello E. (B) Distribuzione dei TrIS tracciati rispetto alla densità di popolazione totale di ciascun consorzio previsto dal modello. Questa analisi mostra una forte correlazione (Spearman’s Rank Correlation con il coefficiente 0,88 e p<0,05) tra TrIS e la densità microbica totale. Il colore di ogni punto rappresenta l’abbondanza del ceppo 27, che si prevede fortemente colonizzato in sottoinsiemi ad alto TrIS.(C) Heatmap di appartenenza al consorzio dei cinque più alti e più bassi consorzi TrIS di dimensione sette e la loro induzioneTreg prevista e la produzione stimata di butirrato utilizzando i dati di Atarashi et al. (2013).(D) Stima della produzione potenziale di SCFA basata sul profilo metabolico in vivo di un singolo ceppo in vivo di Atarashi et al. (2013). Si prevede che i cinque maggiori consorzi TrIS di dimensione sette (da 4C) avranno una produzione di SCFA significativamente più elevata rispetto ai cinque più bassi.(E) Cinque consorzi di 4-strain sono stati utilizzati per la validazione sperimentale: i due consorzi di più alto rango (H1 e H2, rispettivamente), il consorzio di più basso rango (L) e due intermedi (M1, M2). M1 e M2 sono stati inclusi a causa dell’interesse per altre aree di malattia. I ceppi introdotti sperimentalmente sono elencati accanto a ciascun bar/consorzio. Ceppi con numeri in nero sono stati rilevati dal sequenziamento 16S rRNA in campioni di feci di topo, ceppi con numeri in grigio sono stati introdotti, ma non è riuscito a colonizzare. Una correlazione di Pearson di 0,97 tra il TrIS previsto e la media della percentuale di Treg di ciascun consorzio misurata CD4+FOXP3+ Treg dimostra la capacità della selezione basata su TrIS di recuperare correttamente l’induzione di Treg osservata sperimentalmente. (La correlazione di Pearson di tutti i punti ha un valore di 0,54.) È importante notare che il consorzio H2 mostra un aumento medio dell’attività immunitaria del 107% rispetto alla media del controllo del topo privo di germi. Otto repliche biologiche sono state utilizzate per GF, H1 e H2, cinque repliche biologiche sono state utilizzate per M1, M2 e tre repliche biologiche sono state utilizzate per L. La replica e la progettazione dell’esperimento di convalida per la valutazione dell’induzione diTreg è coerente con il lavoro svolto da noi e da altri(Atarashi et al., 2013; Narushima et al., 2014).10.7554/eLife.30916.010Figure 4-source data 1.TrIS, TrIS classifica e prevede le concentrazioni mediane allo stato stazionario dei consorzi microbici selezionati dalla Figura 4E.In contrasto con la Figura 4B, che evidenzia solo l’abbondanza del ceppo 27 in ogni consorzio, la densità di popolazione totale prevista dei consorzi microbici viene confrontata con il TrIS, mentre il colore in ogni sotto-pannello rappresenta l’abbondanza del ceppo corrispondente.(A) Risultati del profilo metabolico del contenuto fecale dei consorzi selezionati rispetto all’acido acetico.(B) Confronto tra le concentrazioni medie previste di acetato normalizzato privo di germi in base alle misurazioni del singolo ceppo nel contenuto cecale da(Atarashi et al., 2013) con i livelli medi osservati di acido acetico normalizzato privo di germi nelle feci di topo da (A).
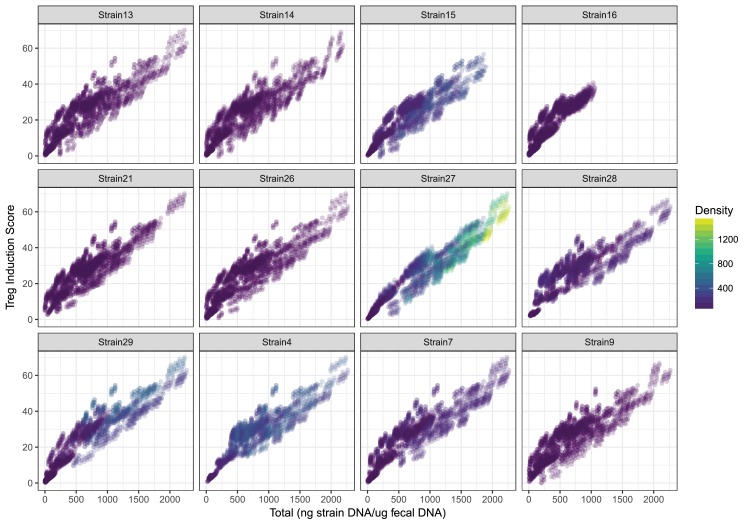
Figura 4-figure supplemento 1.TrIS rispetto alla densità media stimata della popolazione.In contrasto con la Figura 4B, che evidenzia solo l’abbondanza del ceppo 27 in ogni consorzio, la densità di popolazione totale prevista dei consorzi microbici viene confrontata con il TrIS, mentre il colore in ogni sottopannello rappresenta l’abbondanza del ceppo corrispondente.
Figura 4-figure supplemento 2.Profilo metabolico di tre consorzi microbici selezionati e confronto dei profili metabolici previsti e misurati di tre consorzi microbici selezionati.A) Risultati del profilo metabolico del contenuto fecale di consorzi selezionati rispetto all’acido acetico.(B) Confronto delle concentrazioni medie previste di acetato normalizzato privo di germi in base alle misurazioni del singolo ceppo nel contenuto cecale da(Atarashi et al., 2013) con i livelli medi osservati di acido acetico normalizzato privo di germi nelle feci di topo da (A).
Risultati
Generazione di un set di dati multimodale Microbiome-Treg
L’obiettivo di questo studio è quello di sviluppare una struttura basata sulla modellazione matematica per selezionare rapidamente e sistematicamente i consorzi microbici che massimizzano il risultato immunitario desiderato quando vengono introdotti in uno specifico background microbico dell’ospite. Per raggiungere questo obiettivo, combiniamo (i) un modello ecologico microbico che descrive accuratamente la dinamica di questi batteri nell’ospite con (ii) un modello matematico microbiome-Treg che caratterizza il contributo di ogni ceppo al fenotipo immunitario di interesse dati corrispondenti di colonizzazione microbica (Figura 1). Per (i), abbiamo raccolto i dati di colonizzazione della reazione a catena della polimerasi quantitativa della polimerasi (qPCR) di nuova produzione da topi gnotobiotici e li abbiamo combinati con un modello microbiomo-ecologico precedentemente pubblicato della dinamica di 12 ceppi di ClostridiaTreg-inducenti che fanno parte di un consorzio originale di 17 ceppiTreg-inducenti scoperti da alcuni di noi(Atarashi et al., 2013) (vedi anche la Tabella 1 per una ripartizione dei ceppi utilizzati in ogni studio). A differenza dello studio originale di(Bucci et al., 2016), abbiamo incluso solo 12 dei 13 ceppi utilizzati perché, sulla base della modellazione e dell’analisi di Bucci et al. (2016), si prevedeva che il ceppo 6 del set di 13 ceppi non avrebbe colonizzato in modo stabile in presenza degli altri 12 ceppi. I dati pubblicati sulla colonizzazione sono stati riportati nel nostro lavoro precedente(Bucci et al., 2016) e comprendono misurazioni in serie temporali delle abbondanze microbiche da parte del qPCR sotto perturbazioni alimentari. Questi sono utilizzati per ricavare un modello di ecologia microbica predittiva in condizioni gnotobiotiche basato su un’estensione delle equazioni generalizzate di Lotka-Volterra (gLV) (Hofbauere Sigmund, 1998) come introdotto in Stein et al. (2013). Per il dataset di nuova generazione, abbiamo gavaged 14 topi con uno dei tre possibili sottoinsiemi di 11 ceppi del sottoinsieme di 12 ceppi originali di 13 ceppi(Figura 2) e abbiamo utilizzato le misure delle feci derivate per convalidare la capacità del nostro modello matematico di prevedere condizioni non viste. I tre sottoinsiemi di 11 deformazioni sono stati scelti in base alla nostra definizione di ‘keystoneness’, una misura che descrive l’effetto quantitativo marginale previsto della rimozione di ogni deformazione dall’intera comunità (Bucci etal., 2016). In particolare, abbiamo incluso due combinazioni di 11 ceppi, ognuna delle quali mancava uno dei due ceppi con il punteggio più alto di keystoneness (VE202 ceppo 15 e VE202 ceppo 4) e una combinazione di 11 ceppi che mancava del ceppo con il punteggio più basso di keystoneness (VE202 ceppo 29). In analogia a Bucci et al. (2016), la densità di ogni deformazione è stata profilata nel tempo da qPCR con primer specifici per la deformazione (vedere Materiali e metodi). Per risolvere il contributo di ciascuno di questi all’induzione diTreg per il punto (ii) abbiamo accoppiato i dati di colonizzazione dal contenuto fecale con le misurazioni FACS appena raccolte e pubblicate della popolazione di CD4+FOXP3+Treg nella lamina propria di questi topi(Figura 2A). Poiché è fondamentale catturare il contributo di ogni ceppo da solo e in combinazione con altri, abbiamo anche incluso le misurazioni CD4+FOXP3+ Treg dei nostri esperimenti di monocolonizzazione precedentemente pubblicati (Atarashiet al., 2013) (Figura 2A). A causa del fatto che le concentrazioni di mono-colonizzazione a singolo ceppo non sono state misurate in questi esperimenti(Atarashi et al., 2013), le abbiamo simulate utilizzando il modello ecologico del microbioma e i parametri di(Bucci et al., 2016). Questa scelta è stata supportata dalla capacità del modello di prevedere dati di convalida non visti(Figura 3A). Il coefficiente di correlazione di ordine di rango di Spearman varia da 0,92 a 0,98 (p-valore<10-16) tra osservazioni e previsioni a seconda del punto temporale (Figura 3B).
| Atarashi et al., 2013 | Bucci et al., 2016 | 11-str. I | 11-str. II | 11-str. III | H1 | H2 | M1 | M2 | L | |
|---|---|---|---|---|---|---|---|---|---|---|
| Ceppo 1 | x | |||||||||
| Ceppo 3 | x | |||||||||
| Ceppo 4 | x | x | x | x | x | x | ||||
| Ceppo 6 | x | x | ||||||||
| Ceppo 7 | x | x | x | x | x | x | x | x | ||
| Ceppo 8 | x | |||||||||
| Ceppo 9 | x | x | x | x | x | x | ||||
| Ceppo 13 | x | x | x | x | x | |||||
| Ceppo 14 | x | x | x | x | x | x | x | |||
| Ceppo 15 | x | x | x | x | x | |||||
| Ceppo 16 | x | x | x | x | x | x | x | |||
| Ceppo 18 | x | |||||||||
| Ceppo 21 | x | x | x | x | x | |||||
| Ceppo 26 | x | x | x | x | x | |||||
| Ceppo 27 | x | x | x | x | x | x | x | x | ||
| Ceppo 28 | x | x | x | x | x | x | x | |||
| Ceppo 29 | x | x | x | x | x | x | x | x |
Derivazione del modello matematico microbiome-Treg
Abbiamo usato i dati di colonizzazione del microbioma descritti e le corrispondenti misurazioni CD4+FOXP3+Treg per determinare il contributo di ciascun ceppo alpool Treg. Iniziamo suddividendo la popolazione di CD4+ cellule T in due sottopopolazioni principali, a seconda della loro espressione intracellulare FOXP3: CD4+FOXP3+Treg e il resto tra le CD4+ T-cellule, le convenzionali CD4+FOXP3- T-cellule (Bilatee Lafaille, 2012; Rudensky , 2011). La concentrazione di CD4+ T-cellule T al tempo t nella lamina propria del colon, cT(t), è quindi la combinazione di queste due popolazioni di cellule T, cT(t)=cFOXP3+(t)+cFOXP3-(t). Per includere una varietà di effetti nel nostro modello, assumiamo la dinamica delle cellule T per seguire le equazioni gLV(Gerber, 2014; Hofbauer e Sigmund, 1998), che tengono conto anche dell’effetto dei ceppi microbici nel lume sulla sottopopolazione delle cellule T CD4+FOXP3-. Inoltre, utilizziamo un’estensione delle equazioni standard gLV per includere l’impatto dei ceppi di Clostridia in termini di perturbazioni esterne (Stein etal., 2013). Il modello matematico microbiome-Treg risultante si trova come,(1)dcFOXP3+(t)dt=cFOXP3+(t)(αFOXP3++βFOXP3+FOXP3+FOXP3+cFOXP3+cFOXP3+(t)∑k=1K+βFOXP3+FOXP3+FOXP3-cFOXP3-(t)+∑k=1Kεikcstrainik(t))dove αFOXP3+ indica il tasso di crescita basale e βFOXP3+FOXP3+ il termine di autointerazione della popolazioneCD4+FOXP3+ Treg. I termini di interazione βFOXP3+FOXP3+FOXP3- e βFOXP3-FOXP3+ rappresentano l’effetto del CD4+FOXP3- cellula T sullapopolazione CD4+FOXP3+ Treg e del CD4+FOXP3+ Tregsulla popolazione CD4+FOXP3- cellula T, rispettivamente (d’Onofrio, 2005). Di conseguenza, i parametri di interazione positivi corrispondono all’attivazione, quelli negativi all’inibizione. Inoltre, εik denota l’effetto dello strain ik sulla popolazione CD4+FOXP3+ Treg. Per osservazioni a lungo termine, t→∞, la dinamica del cFOXP3+(t) è data dalla sua soluzione (non banale) allo stato stazionario, che semplifica il microbioma allora statico-Treg modello matematico della relativa proporzione CD4+FOXP3+ in stato stazionario, rFOXP3+,ss, ad un problema di regressione lineare,(2)rFOXP3+,ss=α~+∑k=1Kε~ikcstrainik,ss.
Qui, usiamo che l’abbondanza assoluta e relativa della popolazione CD4+FOXP3+Treg sono accoppiate da cFOXP3+/-,ss=cT,ss⋅rFOXP3+/-,ss (vedi Materiali e metodi). Si assume che la concentrazione costante delle cellule T CD4+ CD4+ sia costante, cT,ss=const., in tutte le composizioni microbiche. Giustifichiamo questo perché abbiamo a che fare con topi geneticamente simili e un insieme di Clostridi strettamente correlati. Questo presupposto non è tuttavia giustificabile quando si confrontano topi non colonizzati e topi privi di germi non colonizzati(Faith et al., 2014).
Assegnando a rFOXP3+,ss le proporzioni FACS derivate dal CD4+FOXP3+ Tregdopo 35 giorni e a cstrainik,ss i corrispondenti profili microbici, si deduce il contributo di ogni ceppo al pool CD4+FOXP3+ Treg( Figura 2B) risolvendol‘equazione (2) con un ℓ 2-penalizzata regressione al minimo quadrato con un parametro di restringimento, che viene determinato in una convalida incrociata di un solo campione (Stein etal., 2013). La risultante deviazione quadratica normalizzata di radice, cioè la deviazione quadrata di radice su campioni lasciati fuori, è risultata essere del 12%.
Derivazione del punteggio di induzione di Treg (TrIS) e selezione dei consorzi di induzione di Treg
Dopo aver elaborato un modello per prevedere la dinamica dei ceppi candidati in condizioni di assenza di germi e dopo aver risolto il contributo di ciascun ceppo all’espansione di Treg, ci proponiamo di utilizzare queste informazioni per selezionare in modo computazionale i consorzi che massimizzano l’induzione di Treg e che sono ecologicamente robusti (Bucciet al., 2016; Stein et al., 2013). Per specificare una misura della robustezza ecologica e del potenziale di induzione immunitaria per i consorzi microbici nei topi privi di germi, definiamo ilTreg Induction Score (TrIS) come la media prevista dell’attivazione regolatoria delle cellule T di un determinato consorzio di ceppi K1,⋯, strainiK ignorando i contributi dell’ospite,(3)TrIS({straini1,⋯,strainiK})=1N∑n=1N∑k=1Kε~ikcstrainik,ss(n).
Se lo stato stazionario previsto del consorzio microbico (cstraini1(n),…,cstrainiK(n))ss che è calcolato dalla stima del parametro n-esimo Markov Chain Monte Carlo (MCMC) (Bucciet al., 2016) è biologicamente significativo, cioè positivo, e stabile, quindi cstrainik,ss(n) denota la concentrazione allo stato stazionario della deformazione ik; altrimenti cstrainik,ss(n) è impostato a 0. Quindi, il valore del TrIS è indicativo dell’induzione CD4+FOXP3+Treg prevista (dopo aver rimosso il contributo dell’ospite) ed è della stessa unità delle misure FACS. Abbiamo valutato TrIS per ogni possibile combinazione di deformazione che colonizzerebbe stabilmente l’intestino in background privo di germi. Nel nostro calcolo, delle 212-1 = 4095 possibili configurazioni di deformazione allo stato stazionario valutate in N = 22.500 stime dei parametri MCMC, l’84% è risultato biologicamente significativo e stabile. È interessante notare che, mentre il TrIS medio aumenta con la dimensione del consorzio, la nostra analisi mostra che una dimensione del sottoinsieme di sette contiene già combinazioni batteriche che massimizzano l’induzione(Figura 4A). Inoltre, oltre alla forte correlazione tra TrIS e l’abbondanza batterica totale prevista nel consorzio, abbiamo osservato che i consorzi ad alta induzione mostrano un arricchimento particolarmente grande nell’abbondanza del ceppo 27(Figura 4B, Figura 4-figure supplement 1). Poiché gli acidi grassi a catena corta sono stati precedentemente associati con l’induzione delTreg del colon(Arpaia et al., 2013) e l’aumento della densità al momento dell’integrazione con questi ceppi(Atarashi et al., 2013), abbiamo deciso di testare se i consorzi ad alta induzione diTreg previsti dalla modellazione sono stati arricchiti anche in SCFA. Abbiamo quindi confrontato i primi 5 consorzi microbici di dimensioni 7 con la loro controparte della stessa dimensione in basso 5(Figura 4C). Abbiamo previsto la concentrazione di SCFA per ciascuna delle composizioni previste sommando le uscite metaboliche scalari di ciascun ceppo misurate in esperimenti di mono-colonizzazione(Atarashi et al., 2013; Narushima et al., 2014) e normalizzate dalla densità di mono-colonizzazione prevista dal modello del ceppo. Abbiamo eseguito un test di Welch a due campioni t-test per ciascuna delle concentrazioni di SCFAs previste e abbiamo trovato un arricchimento significativo per tutti gli SCFAs stimati (p<0,05, una coda) nei consorzi ad alto TrIS rispetto a quelli bassi (Figura 4D).
Validazione sperimentale delle previsioni del modello matematico
Abbiamo deciso di testare sperimentalmente la capacità del nostro approccio di predire correttamente la classifica del consorzio rispetto alla Treg-induzione. A causa dei vincoli normativi sui ceppi probiotici utilizzati – limitandoci a un massimo di quattro ceppi alla volta negli esperimenti successivi – abbiamo selezionato cinque combinazioni di 4 ceppi con una varietà di effetti immunitari previsti. Abbiamo misurato l’induzione CD4+FOXP3+Treg per cinque consorzi microbici di taglia 4 e il controllo privo di germi. Abbiamo scelto i due consorzi TrIS più alti (H1, H2 con rango 1 e 2, rispettivamente), quello più basso (L con rango 495) e due consorzi di interesse TrIS-intermediato (M1, M2 con rango 129 e 452, rispettivamente). I ceppi contenuti in ciascuno dei cinque consorzi sono dettagliati nella Tabella 1. Abbiamo correlato il punteggio TrIS con la percentuale media osservata di CD4+FOXP3+Treg e abbiamo trovato una significativa correlazione Pearson con il coefficiente di 0,97 e il valore p<0,01 (Figura 4E). È importante notare che, quando si utilizza il sequenziamento 16S rRNA per indagare i profili di colonizzazione risultanti per queste combinazioni, abbiamo osservato che i consorzi ad alto punteggio TrIS (H1, H2, M1) tutti stabilmente colonizzati, mentre i due consorzi a basso punteggio hanno mostrato solo un sottoinsieme dei ceppi introdotti. Questo risultato riflette notevolmente la natura del nostro sistema di punteggio che, oltre al potenziale di attivazione immunitaria, incorpora anche il successo della colonizzazione(Equazione 3).
Poiché l’analisi di arricchimento SCFA da misurazioni in vivo dei singoli ceppi(Figura 4C) ha previsto un aumento significativo dell’acetato nei cinque consorzi di dimensioni sette, abbiamo deciso di confrontare le concentrazioni di acido acetico misurate(Figura 4-figure supplement 2A) con le previsioni dell’acetato nei due consorzi a 4 ceppi ad alto (H1 e H2) e a basso (L) che inducono ilTreg-inducing(Figura 4-figure supplement 2B). ANOVA e successivo post-hoc Tukey test post-hoc ha mostrato significato nell’arricchimento misurato di H1 rispetto a L (regolato p-valore<0,05) e di H1 rispetto a H2 (regolato p-valore<0,05) (Figura4-figure supplemento 2A). Tuttavia, non è stata osservata alcuna differenza statistica tra le misurazioni in H2 e L (valore p corretto<0,05). Notevolmente, il modello previsto dall’arricchimento dell’acetato normalizzato senza germi e la concentrazione di acido acetico osservata (normalizzata dalla media in condizioni di assenza di germi) sono ben correlati (Figura 4-figuresupplement 2B).
Discussione
La manipolazione dei microbioti intestinali con consorzi batterici definiti per il trattamento delle malattie è una via promettente per la futura terapeutica(Hansen e Sartor, 2015). Tuttavia, la scelta di combinazioni batteriche dal vasto spazio combinatorio dei microbi che colonizzano efficacemente un ospite (possibilmente con un microbiota disbiotico) e allo stesso tempo massimizzano un fenotipo di ospite desiderato richiede un numero enorme di prove sperimentali costose e che richiedono tempo(Faith et al., 2014). Mentre il data mining e gli approcci statistici potrebbero aiutare in questo processo, la maggior parte dei metodi di analisi dei microbiomi disponibili è ancora basata su correlazioni(Gerber, 2014; Morgan et al., 2015) e incapace di prevedere fenotipi non visti. L’identificazione delle associazioni causali tra i microbi e il fenotipo ospite è stata recentemente ottenuta utilizzando una triangolazione dei fenotipi microbici basata su esperimenti (Surana e Kasper, 2017). Tuttavia, questo approccio rimane impraticabile quando l’obiettivo è quello di esplorare e classificare tutte le possibili combinazioni del consorzio microbico rispetto a un fenotipo ospite di interesse. In questo studio, abbiamo fatto leva sui nostri precedenti metodi di calcolo per prevedere le dinamiche temporali del microbioma e fare previsioni sulla stabilità nel contesto delle malattie infettive e infiammatorie, tra cui la colonizzazione di C. difficile e la malattia infiammatoria intestinale(Bucci et al., 2016; Buffie et al., 2015; Stein et al., 2013).
In particolare, abbiamo presentato un nuovo metodo basato sulla modellazione che combina le previsioni dinamiche da modelli di dati-driven del microbioma e le interazioni tra i microbi e la risposta immunitaria (come CD4 +FOXP3 +Treg) e quindi permette la progettazione razionale di consorzi batterici immunomodulanti. Limitando il nostro campo d’azione alle dinamiche stazionarie a lungo termine, siamo stati in grado di ricavare un modello matematico microbiome-Treg che è vincolato da osservazioni sperimentali a partire da dati multimodali. Nel lavoro presentato, le modalità di dati utilizzati sono i dati di sequenziamento qPCR e 16S rRNA per stimare le abbondanze microbiche e FACS per valutare l’induzione CD4+FOXP3+Treg, rispettivamente. Tuttavia, il quadro proposto è naturalmente estensibile ad altri dati dell’ospite, ad esempio i dati di profilazione dei metaboliti, le letture immunitarie (ad esempio l’attivazione delle cellule T CD8, l’esaurimento delle celluleTh1/Th17) o la profilazione del trascrittoma dell’ospite(Atarashi et al., 2017; Morgan et al., 2015). Per valutare i consorzi candidati che potrebbero essere rilevanti per il trattamento delle malattie auto-infiammatorie, abbiamo introdotto ilTreg Induction Score, TrIS, come una nuova metrica che rappresenta sia la stabilità ecologica che l’efficacia nella modulazione immunitaria. Questa metrica ci ha permesso di identificare razionalmente le combinazioni che avrebbero colonizzato in modo stabile e allo stesso tempo prodotto una sostanziale induzione diTreg nei topi privi di germi. È interessante notare che, negli esperimenti di validazione di cinque distinti consorzi guidati da modelli, con un potenziale di induzione previsto molto diverso, abbiamo dimostrato la capacità del nostro approccio di selezionare con successo le combinazioni microbiche con un’attività terapeutica desiderata. A nostra conoscenza, questo è ad oggi il primo studio in cui l’osservazione limitata nella modellazione in silico del microbo e del fenotipo ospite ha guidato con successo la progettazione razionale dei farmaci di determinati consorzi batterici.
Il nostro lavoro si basa sull’ipotesi del modello gLV sia per il microbicromo che per la dinamica regolatoria delle cellule T CD4+ che, pur essendo molto versatile, ha alcuni limiti tra cui la mancanza di interazioni di terzo ordine o di ordine superiore o di effetti di saturazione (Wangersky, 1978). Inoltre, con i dati a nostra disposizione, abbiamo dovuto supporre che la densità complessiva delle cellule T allo stato stazionario sia costante tra i topi e le diverse composizioni microbiche. Mentre questa ipotesi era ragionevole per i dati che abbiamo analizzato (vedi Risultati), l’aggiunta di misurazioni future sull’abbondanza totale delle cellule T migliorerà probabilmente l’accuratezza di previsione del nostro modello, oltre a fornire importanti informazioni sulla variabilità delle dimensioni della popolazione delle cellule T in risposta all’induzione immunitaria microbica.
Abbiamo eseguito le nostre analisi di dati e modellizzazione nei topi privi di germi. Mentre il lavoro precedente di alcuni di noi ha dimostrato che la quantità di induzione di Treg è indipendente dal background privo di germi (IQI, Balb/c o C57BL/6) (Atarashiet al., 2015), è degno di nota che il nostro modello, che è stato addestrato sui dati IQI dei topi, ha predetto in modo robusto sia l’induzione di Treg che l’arricchimento in acetato dei topi colonizzati C57BL/6 come usato negli esperimenti di validazione.
Le nostre previsioni suggeriscono un certo grado di coerenza in termini di risultato funzionale (ad esempio l’arricchimento in acetato) dei consorzi ad alto punteggio rispetto a quelli a basso punteggio nei topi privi di germi. Tuttavia, prima di tradurre questi risultati in sviluppo terapeutico, gli studi futuri dovranno essere eseguiti in ambienti non privi di germi (ad esempio, topi SPF, topi umanizzati), al fine di rendere conto degli effetti di una flora già consolidata. Le caratteristiche microbiologiche specifiche di ogni singolo individuo possono limitare il potenziale di colonizzazione dei ceppi selezionati a causa di specifici effetti di rete ecologici(Smits et al., 2016), il che suggerisce la necessità di un’attenta caratterizzazione delle interazioni ecologiche tra il prodotto probiotico proposto e una specifica comunità ricevente (ad es. un singolo paziente affetto da colite ulcerosa(Atarashi et al., 2013)).
Il trattamento delle malattie autoimmuni nel loro complesso non si ottiene necessariamente attraverso l’ottimizzazione esclusiva di una funzione obiettivo (ad esempio, per la colite ulcerosa, la massimizzazione dell’attivazione diTreg ), ma può richiedere la manipolazione simultanea di uno spettro immunitario ospite multiprocesso, che potrebbe includere la concomitante riduzione dei fenotipi pro-infiammatori(Atarashi et al., 2015). Data la nuova disponibilità di dati e l’attenta progettazione sperimentale, riteniamo che il quadro di modellazione proposto in questo studio potrebbe superare questi problemi introducendo dei vincoli nella metrica di punteggio che tengono conto delle caratteristiche del modello di topo pre-colonizzato. Al limite, in presenza di un ampio set di dati formativi che includano informazioni sufficienti sulle variazioni dei singoli pazienti, il nostro flusso di lavoro ci darà il potenziale senza precedenti di testare la manipolazione dei microbiota a livello personalizzato in silico.
I metodi basati sulla modellazione matematica hanno il potenziale di accelerare notevolmente lo sviluppo del trattamento delle malattie umane (Michore Beal, 2015). In questo lavoro, sviluppiamo e dimostriamo in una prima convalida sperimentale l’utilità di un modello matematico del sistema immunitario microbico, che può essere considerato un trampolino di lancio per la prototipazione accelerata e la progettazione razionale delle terapie microbicotiche.
Materiali e metodi
Derivazione del modello matematico a induzione di microbiome-Treg
Si assume la seguente dinamica per la popolazione di cellule T regolamentare CD4+FOXP3+:dcFOXP3+(t)dt=cFOXP3+(t)(αFOXP3+++βFOXP3+FOXP3+FOXP3+cFOXP3+cFOXP3+(t)+ βFOXP3+FOXP3-cFOXP3-(t)+∑k=1Kεikcstrainik(t)).
Qui, αFOXP3+ indica il tasso di crescita basale e βFOXP3+FOXP3+ il termine di autointerazione della popolazione CD4+FOXP3+ Treg, mentre i parametri di interazione βFOXP3+FOXP3- e βFOXP3- e βFOXP3-FOXP3+ caratterizzano l’effetto delle CD4+FOXP3- cellule Tsulla popolazione CD4+FOXP3+ Treg e delle CD4+FOXP3+ Treg sullapopolazione CD4+FOXP3-cellule T, rispettivamente (d’Onofrio, 2005).Inoltre, εik denota l’effetto dello strain ik sulla popolazione CD4+FOXP3+ Treg. La soluzione non banale dello stato stazionario (cioè la soluzione algebrica del lato destro dell’equazione (1) impostata a 0 con cFOXP3+≠0) si trova come,cFOXP3+,ss=-1βFOXP3+FOXP3+FOXP3+(αFOXP3++βFOXP3+FOXP3+FOXP3-cFOXP3-, ss+∑k=1Kεikcstrainik,ss).
Utilizzando cT,ss=cFOXP3+,ss+cFOXP3-,ss, le concentrazioni allo stato stazionario delle popolazioni CD4+FOXP3+ Trege CD4+FOXP3-, cFOXP3+/-, ss, sono derivate dalle abbondanze relative basate sul FACS rFOXP3+/-,ss da, cFOXP3+/-,ss= cT,ss⋅rFOXP3+/-, ss=cT,ss(1-rFOXP3-/+,ss). Infine, la relazione lineare tra le abbondanze relative, rFOXP3+,ss, e le densità di deformazione, cstrainik,ss, si trova come,rFOXP3+,ss=1βFOXP3+FOXP3+FOXP3–βFOXP3+FOXP3+FOXP3+(αFOXP3+cT,ss+βFOXP3+FOXP3+FOXP3-+1cT,ss∑k=1Kεikcstrainik,ss)≡α~+∑k=1Kε~ikcstrainik,ssassumendo una concentrazione costante di CD4+ cellule T, cT,ss=const.,in tutte le possibili composizioni microbiche. I parametri sconosciuti α~ e ε~ik sono stimati in un ℓ2-penalizzato regressione al minimo quadrato (la cosiddetta regressione di cresta) con un parametro di restringimento positivo λ, che viene determinato in una validazione incrociata lasci-one-campione-out come λ*=2.
Raccolta di dati sperimentali per l’addestramento del modello di sistema microbicoimmune
Profilazione dell’abbondanza di ceppi
Nel nostro lavoro precedente(Bucci et al., 2016), abbiamo dedotto un modello matematico che descrive la dinamica di un sottoinsieme di 13 ceppi del consorzio Clostridia originale di Atarashi et al. (2013) in topi privi di germi. Utilizzando i dati generati di recente dalla stessa configurazione sperimentale, siamo ora in grado di valutare la sua qualità predittiva per tre distinti sottoinsiemi a 11 ceppi. Le tre composizioni a 11 ceppi sono state selezionate in base alla loro capacità di mantenere la stabilità nelle simulazioni quando uno dei due ceppi alti (ceppo 15 e ceppo 4, qui indicati come casi I e II, rispettivamente) o uno basso (ceppo 29; III) è stato rimosso(Bucci et al., 2016). Per la convalida sperimentale, i topi IQI privi di germi sono stati acquistati da Sankyo Laboratories (Giappone), randomizzati e mantenuti in isolatori vinilici privi di germi nella struttura animale di Riken. Dodici ceppi di ClostridiaTreg-inducing Clostridia sono stati selezionati dal consorzio VE202 precedentemente segnalato, composto da 17 ceppiTreg-inducing(4, 7, 9, 13-16, 21, 26-29) e sono stati coltivati individualmente in brodo modificato Eggerth Gagnon in condizioni strettamente anaerobiche (80% N2, 10%H2, 10% CO2) a 37°C in una camera anaerobica (Coy Laboratory Products, Grass Lake, MI) alla confluenza. I ceppi batterici coltivati sono stati poi mescolati e le tre miscele di 11 ceppi (descritti sopra) sono stati inoculati per via orale in cinque topi adulti IQI privi di germi ciascuno. Un topo per la condizione II è morto ed è stato quindi scartato dallo studio. Dopo un iniziale intervallo di 9 giorni di acclimatazione, abbiamo raccolto pellet fecale a 2-4 giorni di intervallo fino al giorno 35, momento in cui i topi sono stati eutanasia e analizzati per induzione CD4 +FOXP3 come in Atarashi et al. (2013). I livelli di colonizzazione per ogni ceppo sono stati valutati amplificando le regioni specifiche del ceppo con qPCR come descritto in Bucci et al. (2016). Il DNA genomico batterico è stato estratto da 1 a 2 pellet fecali utilizzando QIAamp DNA Stool Mini Kit (Qiagen, Hilden, Germania). La quantità di DNA è stata quantificata utilizzando un kit di analisi Qubit dsDNA HS e un fluorimetro Qubit (Invitrogen, Carlsbad, CA). Il DNA è stato poi sottoposto a qPCR utilizzando Thunderbird SYBR qPCR Mix (TOYOBO, Osaka, Giappone) e un LightCycler 480 (Roche) con primer specifici per i geni 16S dell’RNA ribosomiale (rRNA) dei 12 ceppi di Clostridia come in Bucci et al. (2016). La quantificazione di ogni ceppo in ogni campione è stata effettuata utilizzando curve standard di concentrazioni note di DNA purificato da ogni ceppo coltivato individualmente in vitro. Le densità dei ceppi in ogni campione sono state calcolate dividendo i numeri di quantificazione assoluta di cui sopra per il peso del DNA fecale estratto. Gli esperimenti di 11 ceppi sono stati eticamente approvati dalle università di Riken, Keio e Azabu con il protocollo H24-9(14) di Riken.
Stima di CD4+FOXP3+ Treg – Isolamento dei linfociti della lamina propria intestinale e citometria a flusso
I colon sono stati raccolti e aperti longitudinalmente, lavati con PBS per rimuovere tutto il contenuto luminale e agitati nella soluzione salina bilanciata di Hanks (HBSS) contenente 5 mM EDTA per 20 minuti a 37°C. Dopo aver rimosso le cellule epiteliali, gli strati muscolari e il tessuto adiposo con pinze, gli strati di lamina propria sono stati tagliati in piccoli pezzi e incubati con RPMI1640 contenente il 4% di siero bovino fetale, 0,5 mg/ml collagenasi D, 0,5 mg/ml dispase e 40 mg/ml DNasi I (tutti Roche Diagnostics, Risch-Rotkreuz, Svizzera) per 1 ora a 37 ° C in un bagno d’acqua di agitazione. I tessuti digeriti sono stati lavati con HBSS contenente 5 mM EDTA, risospesi in 5 ml di Percoll al 40% (GE Healthcare, Boston, MA) e sovrapposti su 2,5 ml di Percoll all’80% in una provetta Falcon da 15 ml. Percoll separazione gradiente è stata eseguita per centrifugazione a 850 g per 25 min a 25 ° C. I linfociti lamina propria linfociti sono stati raccolti dall’interfaccia del gradiente Percoll e sospesi in PBS ghiacciato. Per l’analisi diTreg, i linfociti isolati sono stati etichettati con il kit LIVE/DEAD fixable dead cell stain kit (Life Technologies, Carlsbad, CA) per escludere le cellule morte dall’analisi. Successivamente, la colorazione superficiale e intracellulare di CD3, CD4 e FOXP3 è stata eseguita utilizzando il kit di colorazione anti-CD3 marcato BV605 (17A2, Biolegend, San Diego, CA), l’anti-CD4 marcato BV421 (RM4-5, Biolegend), l’anticorpo anti-FOXP3 marcato Alexa700 (FJK-16 s, eBioscience, San Diego, CA) e il buffer di colorazione FOXP3 (eBioscience). Le cellule macchiate di anticorpi sono state analizzate con LSR Fortessa e i dati sono stati analizzati utilizzando il software FlowJo (Tree Star, Ashland, OR).
Misurazione degli acidi organici
Le concentrazioni di acido organico nel contenuto cecale sono state determinate mediante gascromatografia-spettrometria di massa (GC-MS). Il contenuto cecale (10 mg) è stato perturbato utilizzando perle di zirconia/silice da 3 mm (BioSpec Products) e omogeneizzato in soluzione di estrazione contenente 100 ml di standard interno (100 mM di acido crotonico), 50 ml di HCl e 200 ml di etere. Dopo una vigorosa agitazione con uno Shakemaster neo (Bio Medical Science) a 1500 rpm per 10 minuti, gli omogeneizzati sono stati centrifugati a 1000 g per 10 minuti e poi lo strato superiore di etere è stato raccolto e trasferito in nuove fiale di vetro. Aliquote (80 ml) degli estratti di etere sono stati miscelati con 16 ml di N-tert-butildimetilsililil-N-metiltrifluoroacetammide (MTBSTFA). Le fiale sono state sigillate ermeticamente mediante avvitamento e riscaldate a 80°C per 20 minuti in un bagno d’acqua, e lasciate a temperatura ambiente per 48 ore per la derivatizzazione. I campioni sono stati poi fatti passare attraverso un sistema GC di rete 6890N (Agilent Technologies) dotato di colonna HP-5MS (0,25 mm 330 m 30,25 mm) e 5973 Network Mass Selective Detector (Agilent Technologies, Santa Clara, CA). L’elio puro (99,9999%) è stato utilizzato come gas di trasporto ed erogato ad una portata di 1,2 ml/min. La pressione della testa è stata impostata a 10 psi con divisione 10:1. Le temperature della linea di ingresso e di trasferimento erano rispettivamente di 250 µC e 260 µC. Il seguente programma di temperatura è stato utilizzato: 60 µC (3 min), 60-120°C (5°C/min), 120-300°C (20°C/min). Una quantità di microlitro di ogni campione è stata iniettata con un tempo di esecuzione di 30 min. Le concentrazioni di acido organico sono state quantificate confrontando le loro aree di picco con gli standard.
Simulazioni numeriche dei tre sottoinsiemi 11-strain utilizzati per la formazione modello microbiome-Treg
Abbiamo utilizzato 22.500 set di set di parametri Markov Chain Monte Carlo generalizzati Lotka-Volterra determinati applicando l’algoritmo Bayesian Variable Selection all’interno di MDSINE ai dati di Bucci et al. (2016), e il primo punto temporale dei profili microbici misurati per ciascuno dei 14 topi di validazione come condizione iniziale, per simulare il sistema gLV di equazioni differenziali corrispondenti a ciascun microbioma del topo (Figura 3A). L’accuratezza della previsione è stata valutata calcolando il coefficiente di correlazione Spearman tra i dati osservati e quelli previsti(Figura 3B).
Simulazione delle abbondanze di mono-colonizzazione
Poiché i dati sperimentali di Atarashi et al. (2013) hanno fornito solo i livelli delle cellule T CD4+FOXP3+ per gli esperimenti di colonizzazione mono-deformazione a 35 giorni dall’inoculazione, ma nessuna misurazione delle concentrazioni microbiche a lungo termine nell’intestino, abbiamo usato invece le corrispondenti densità di colonizzazione mono-deformazione stimate ottenute dal modello gLV (sezione sopra e figura 2A) . In generale, il comportamento a lungo termine del sistema gLV è determinato dai suoi stati stazionari che sono definiti in modo univoco dai parametri del modello dedotti(Stein et al., 2013). Abbiamo calcolato la mediana del parametro in ogni singola variabile del modello a partire dai 22.500 set di parametri MCMC. Questi parametri mediani sono stati poi utilizzati per dedurre le densità di stato stazionario, che erano insieme alle proporzioni CD4+FOXP3 misurate incluse nell’addestramento del modello microbiome-Treg.
Raccolta di dati CD4+FOXP3+ Treg da esperimenti a 4 ceppi per la validazione delle previsioni basate sulla modellazione
I ceppi batterici 4, 7, 9, 14, 15, 16, 27, 28, 29 sono stati coltivati in modo anaerobico in brodo PYG (brodo di peptone, lievito e glucosio della Anaerobe Systems, Cat no: AS-822) fino a raggiungere la fase stazionaria (48 ore per i ceppi 27 e 29, 24 ore per i restanti ceppi). Ogni dose di 200 µl-mouse di un LBP a 4 ceppi conteneva 50 µl di coltura in fase stazionaria concentrata 20 volte. Germ-free C57BL/6 topi di età compresa tra 6-8 settimane sono stati randomizzati e gavaged con una dose totale di 5-107-2-108 batteri in un 200µl, e mantenuto in condizioni gnotobiotiche per quattro settimane. L’uso di un background C57BL/6 per questi esperimenti è stato motivato dalla disponibilità di animali presso la struttura in cui abbiamo effettuato la convalida e giustificato dal fatto che il lavoro precedente da noi svolto ha dimostrato che l’induzione diTreg da parte dei nostri ceppi Clostridia non differisce tra Balb/c, IQI, e topi C57BL/6(Atarashi et al., 2015). I topi sono stati poi sacrificati, i leucociti sono stati raccolti e i leucociti lamina propria isolati e colorati per CD3+CD4+FOXP3+Treg come descritto sopra. Otto topi ciascuno sono stati utilizzati per i consorzi High 1 (ceppi H1: 7, 27, 28, 29) e High 2 (ceppi H2: 4, 7, 27, 29). Cinque topi ciascuno sono stati utilizzati per l’alto intermedio (ceppi M1: 4, 7, 14, 28) e per i consorzi intermedi bassi (ceppi M2: 9, 16, 27, 29). Tre topi sono stati utilizzati per il basso 1 (ceppi L1: 14, 15, 16, 29). Il profilo di colonizzazione è stato determinato attraverso il sequenziamento dell’rRNA 16S (come sopra) e verificato facendo saltare le sequenze rappresentative in un database fasta 16S VE202. Gli esperimenti di convalida a 4 ceppi sono stati eseguiti nel Massachusetts Host Microbiome Center sotto il protocollo IACUC 2016N000141.
References
- Arpaia N, Campbell C, Fan X, Dikiy S, van der Veeken J, deRoos P, Liu H, Cross JR, Pfeffer K, Coffer PJ, Rudensky AY. Metabolites produced by commensal bacteria promote peripheral regulatory T-cell generation. Nature. 2013; 504:451-455. DOI | PubMed
- Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, Yagita H, Ishii N, Evans R, Honda K, Takeda K. ATP drives lamina propria T(H)17 cell differentiation. Nature. 2008; 455:808-812. DOI | PubMed
- Atarashi K, Suda W, Luo C, Kawaguchi T, Motoo I, Narushima S, Kiguchi Y, Yasuma K, Watanabe E, Tanoue T, Thaiss CA, Sato M, Toyooka K, Said HS, Yamagami H, Rice SA, Gevers D, Johnson RC, Segre JA, Chen K, Kolls JK, Elinav E, Morita H, Xavier RJ, Hattori M, Honda K. Ectopic colonization of oral bacteria in the intestine drives TH1 cell induction and inflammation. Science. 2017; 358:359-365. DOI | PubMed
- Atarashi K, Tanoue T, Ando M, Kamada N, Nagano Y, Narushima S, Suda W, Imaoka A, Setoyama H, Nagamori T, Ishikawa E, Shima T, Hara T, Kado S, Jinnohara T, Ohno H, Kondo T, Toyooka K, Watanabe E, Yokoyama S, Tokoro S, Mori H, Noguchi Y, Morita H, Ivanov II, Sugiyama T, Nuñez G, Camp JG, Hattori M, Umesaki Y, Honda K. Th17 cell induction by adhesion of microbes to intestinal epithelial Cells. Cell. 2015; 163:367-380. DOI | PubMed
- Atarashi K, Tanoue T, Oshima K, Suda W, Nagano Y, Nishikawa H, Fukuda S, Saito T, Narushima S, Hase K, Kim S, Fritz JV, Wilmes P, Ueha S, Matsushima K, Ohno H, Olle B, Sakaguchi S, Taniguchi T, Morita H, Hattori M, Honda K. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013; 500:232-236. DOI | PubMed
- Belkaid Y, Hand TW. Role of the microbiota in immunity and inflammation. Cell. 2014; 157:121-141. DOI | PubMed
- Bilate AM, Lafaille JJ. Induced CD4+Foxp3+ regulatory T cells in immune tolerance. Annual Review of Immunology. 2012; 30:733-758. DOI | PubMed
- Bucci V, Tzen B, Li N, Simmons M, Tanoue T, Bogart E, Deng L, Yeliseyev V, Delaney ML, Liu Q, Olle B, Stein RR, Honda K, Bry L, Gerber GK. MDSINE: microbial dynamical systems inference engine for microbiome time-series analyses. Genome Biology. 2016; 17:1-17. DOI | PubMed
- Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, Littmann E, van den Brink MR, Jenq RR, Taur Y, Sander C, Cross JR, Toussaint NC, Xavier JB, Pamer EG. Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature. 2015; 517:205-208. DOI | PubMed
- d’Onofrio A. A general framework for modeling tumor-immune system competition and immunotherapy: Mathematical analysis and biomedical inferences. Physica D: Nonlinear Phenomena. 2005; 208:220-235. DOI
- Donia MS, Fischbach MA. Small molecules from the human microbiota. Science. 2015; 349DOI | PubMed
- Faith JJ, Ahern PP, Ridaura VK, Cheng J, Gordon JI. Identifying gut microbe-host phenotype relationships using combinatorial communities in gnotobiotic mice. Science Translational Medicine. 2014; 6DOI | PubMed
- Garber K. Drugging the gut microbiome. Nature Biotechnology. 2015; 33:228-231. DOI | PubMed
- Garrett WS. Cancer and the microbiota. Science. 2015; 348:80-86. DOI | PubMed
- Gerber GK. The dynamic microbiome. FEBS Letters. 2014; 588:4131-4139. DOI | PubMed
- Geva-Zatorsky N, Sefik E, Kua L, Pasman L, Tan TG, Ortiz-Lopez A, Yanortsang TB, Yang L, Jupp R, Mathis D, Benoist C, Kasper DL. Mining the human gut microbiota for immunomodulatory organisms. Cell. 2017; 168:928-943. DOI | PubMed
- Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M, Morgan XC, Kostic AD, Luo C, González A, McDonald D, Haberman Y, Walters T, Baker S, Rosh J, Stephens M, Heyman M, Markowitz J, Baldassano R, Griffiths A, Sylvester F, Mack D, Kim S, Crandall W, Hyams J, Huttenhower C, Knight R, Xavier RJ. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host & Microbe. 2014; 15:382-392. DOI | PubMed
- Hansen JJ, Sartor RB. Therapeutic manipulation of the microbiome in IBD: Current results and future approaches. Current Treatment Options in Gastroenterology. 2015; 13:105-120. DOI | PubMed
- Hofbauer J, Sigmund K. Evolutionary Games and Population Dynamics. Cambridge University Press; 1998.
- Honda K, Littman DR. The microbiome in infectious disease and inflammation. Annual Review of Immunology. 2012; 30:759-795. DOI | PubMed
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umesaki Y, Honda K, Littman DR. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009; 139:485-498. DOI | PubMed
- Maldonado-Gómez MX, Martínez I, Bottacini F, O’Callaghan A, Ventura M, van Sinderen D, Hillmann B, Vangay P, Knights D, Hutkins RW, Walter J. Stable engraftment of bifidobacterium longum ah1206 in the human gut depends on individualized features of the resident microbiome. Cell Host & Microbe. 2016; 20:515-526. DOI | PubMed
- Michor F, Beal K. Improving cancer treatment via mathematical modeling: surmounting the challenges is worth the effort. Cell. 2015; 163:1059-1063. DOI | PubMed
- Morgan XC, Kabakchiev B, Waldron L, Tyler AD, Tickle TL, Milgrom R, Stempak JM, Gevers D, Xavier RJ, Silverberg MS, Huttenhower C. Associations between host gene expression, the mucosal microbiome, and clinical outcome in the pelvic pouch of patients with inflammatory bowel disease. Genome Biology. 2015; 16DOI | PubMed
- Narushima S, Sugiura Y, Oshima K, Atarashi K, Hattori M, Suematsu M, Honda K. Characterization of the 17 strains of regulatory T cell-inducing human-derived Clostridia. Gut Microbes. 2014; 5:333-339. DOI | PubMed
- Olle B. Medicines from microbiota. Nature Biotechnology. 2013; 31:309-315. DOI | PubMed
- Omenetti S, Pizarro TT. The Treg/Th17 Axis: a dynamic balance regulated by the gut microbiome. Frontiers in Immunology. 2015; 6DOI | PubMed
- Paun A, Yau C, Danska JS. Immune recognition and response to the intestinal microbiome in type 1 diabetes. Journal of Autoimmunity. 2016; 71:10-18. DOI | PubMed
- Round JL, Mazmanian SK. Inducible Foxp3+ regulatory T-cell development by a commensal bacterium of the intestinal microbiota. PNAS. 2010; 107:12204-12209. DOI | PubMed
- Rudensky AY. Regulatory T cells and Foxp3. Immunological Reviews. 2011; 241:260-268. DOI | PubMed
- Schirmer M, Smeekens SP, Vlamakis H, Jaeger M, Oosting M, Franzosa EA, Ter Horst R, Jansen T, Jacobs L, Bonder MJ, Kurilshikov A, Fu J, Joosten LAB, Zhernakova A, Huttenhower C, Wijmenga C, Netea MG, Xavier RJ. Linking the human gut microbiome to inflammatory cytokine production capacity. Cell. 2016; 167:1125-1136. DOI | PubMed
- Smith PM, Howitt MR, Panikov N, Michaud M, Gallini CA, Bohlooly-Y M, Glickman JN, Garrett WS. The microbial metabolites, short-chain fatty acids, regulate colonic Treg cell homeostasis. Science. 2013; 341:569-573. DOI | PubMed
- Smits SA, Marcobal A, Higginbottom S, Sonnenburg JL, Kashyap PC. Individualized Responses of Gut Microbiota to Dietary Intervention Modeled in Humanized Mice. mSystems. 2016; 1DOI | PubMed
- Stein RR, Bucci V, Toussaint NC, Buffie CG, Rätsch G, Pamer EG, Sander C, Xavier JB. Ecological modeling from time-series inference: insight into dynamics and stability of intestinal microbiota. PLoS Computational Biology. 2013; 9DOI | PubMed
- Surana NK, Kasper DL. Moving beyond microbiome-wide associations to causal microbe identification. Nature. 2017; 552:244-247. DOI | PubMed
- Tanoue T, Atarashi K, Honda K. Development and maintenance of intestinal regulatory T cells. Nature Reviews Immunology. 2016; 16:295-309. DOI | PubMed
- van Nood E, Vrieze A, Nieuwdorp M, Fuentes S, Zoetendal EG, de Vos WM, Visser CE, Kuijper EJ, Bartelsman JF, Tijssen JG, Speelman P, Dijkgraaf MG, Keller JJ. Duodenal infusion of donor feces for recurrent Clostridium difficile. New England Journal of Medicine. 2013; 368:407-415. DOI | PubMed
- Vieira AT, Fukumori C, Ferreira CM. New insights into therapeutic strategies for gut microbiota modulation in inflammatory diseases. Clinical & Translational Immunology. 2016; 5DOI | PubMed
- Vrieze A, Van Nood E, Holleman F, Salojärvi J, Kootte RS, Bartelsman JF, Dallinga-Thie GM, Ackermans MT, Serlie MJ, Oozeer R, Derrien M, Druesne A, Van Hylckama Vlieg JE, Bloks VW, Groen AK, Heilig HG, Zoetendal EG, Stroes ES, de Vos WM, Hoekstra JB, Nieuwdorp M. Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012; 143:913-916. DOI | PubMed
- Wangersky PJ. Lotka-volterra population models. Annual Review of Ecology and Systematics. 1978; 9:189-218. DOI
- Weil AA, Hohmann EL. Fecal microbiota transplant: benefits and risks. Open Forum Infectious Diseases. 2015; 2:ofv005. DOI | PubMed
Fonte
Stein RR, Tanoue T, Szabady RL, Bhattarai SK, Olle B, et al. () Computer-guided design of optimal microbial consortia for immune system modulation. eLife 7e30916. https://doi.org/10.7554/eLife.30916




