
Introduzione
La risposta cellulare allo stress intrinseco, compresi i danni al DNA, lo squilibrio metabolico e la fame, si traduce prima di tutto nel tentativo di riparare o correggere il problema e quando la correzione non ha successo porta all’induzione della morte cellulare programmata o all’apoptosi. Molteplici eventi e meccanismi di segnalazione dedicati sono stati caratterizzati per governare queste decisioni sul destino delle cellule e, in caso di insuccesso, promuovere l’attivazione del fattore di trascrizione e l’espressione delle proteine chiave pro-apoptotiche. Queste proteine pro-apoptotiche in ultima analisi si interfacciano con una singola famiglia di proteine, la famiglia del linfoma a cellule B 2 (BCL2) di proteine pro- e anti-apoptotiche, che fungono da gatekeeper dell’apoptosi1. La famiglia BCL2 è suddivisa in tre gruppi funzionali: (1) Effettori pro-apoptotici (BAK, BAX, BOK); (2) proteine anti-apoptotiche (BCL2, BCLxL, BCLW, MCL1, BFL1/A1); e (3) proteine pro-apoptotiche solo BH3 (ad esempio, BIM, BAD, BID, BID, PUMA, NOXA). La famiglia BCL2 regola l’apoptosi attraverso una serie di interazioni tra queste sottofamiglie utilizzando un motivo alfa-elica anfpatica a 3 (BH3) a omologia condivisa BCL2 (BH3) comune a tutti i membri della famiglia BCL2 (recensione in refs. 2,3). Questo motivo BH3 si trova al centro della regolazione della famiglia BCL2 con i membri della famiglia anti-apoptotica posizionati per legarsi ai motivi BH3 delle sottofamiglie pro-apoptotiche e sequestrarle in complessi dimerici inattivi. Quando non c’è un’espressione sufficiente dei membri della famiglia BCL2 anti-apoptotici per sequestrare gli effettori pro-apoptotici, allora BAK e BAX omooligomerizzano nelle membrane esterne mitocondriali. Questo avvia la permeabilizzazione della membrana esterna mitocondriale (MOMP), il rilascio di citocromo C, e l’attivazione della caspasi. Due modelli sono emersi che dettaglio come queste interazioni in ultima analisi inducono oligomerizzazione degli effettori pro-apoptotici attraverso i loro motivi BH3, anche se in ogni modello, oligomerizzazione può essere soppressa attraverso anti-apoptotico BCL2 legame BCL2 per l’elica BH3 di BAK o BAX rendendoli incapaci di oligomerizzare. Separatamente, i soli membri della famiglia BH3 agiscono o per promuovere l’oligomerizzazione di BAK/BAX o per sequestrare le proteine BCL2 anti-apoptotiche. In tutti i modelli, la funzione della famiglia BCL2 è regolata dalle interazioni tra le proteine anti-apoptotiche e le eliche BH3 di proteine pro-apoptotiche in quanto queste interazioni servono come fulcro tra la vitalità cellulare e l’inizio dell’apoptosi4,5.
Il posizionamento centrale delle proteine antiapoptotiche della famiglia BCL2 nella regolazione della morte cellulare, in particolare la risposta cellulare allo stress intrinseco, le ha collocate come regolatori chiave della risposta terapeutica tumorigenesi e/o antitumorale. L’interpretazione dei membri della famiglia delle BCL2 anti-apoptotiche nei tumori è un evento comune a vari tipi di cancro che impiegano una o un sottoinsieme di queste proteine per tollerare lo stress genomico ed energetico che spesso accompagna la tumorigenesi.6. La sovraespressione di una delle proteine antiapoptotiche della famiglia BCL2, MCL1, è stata identificata come un meccanismo utilizzato dai tumori per eludere una serie di chemioterapie standard, compresi i taxani, gli alcaloidi della vinca, i composti contenenti platino e le radiazioni.5,7–11. Più recentemente, i mimetici BH3 sono stati sviluppati per sopprimere le proteine antiapoptotiche della famiglia BCL2 come metodo per trattare questo meccanismo chiave associato alla sopravvivenza al cancro.12–14. Così, le interazioni con la famiglia degli anti-apoptotici BCL2 sono ben riconosciute per la loro importanza nella regolazione della risposta acuta allo stress cellulare. A causa del posizionamento di MCL1 nel cancro, abbiamo inizialmente cercato di identificare peptidi in grado di mirare in modo specifico MCL1. Questi studi hanno portato alla nostra identificazione di un motivo di sequenza romanzo, un’inversione del motivo tradizionale BH3, che abbiamo definito un motivo inverso BH3 (rBH3), in quanto mantiene il consenso chiave residui acidi e idrofobi.15. Questa sequenza ha evidenziato l’importanza di puntare alla tasca P2 per ottenere la specificità MCL1 e ha inoltre fornito la possibilità che altre proteine possano avere un impatto diretto sulla regolazione della famiglia BCL2.15. Una proteina che è stata identificata per contenere presumibilmente un motivo rBH3 è il regolatore del ciclo cellulare G1/S, P18 (P18INK4C, CDKN2C). Qui, dimostriamo che il motivo rBH3 è più di una sequenza peptidica unica, ma che è un motivo proteico naturale che è in grado di mediare le interazioni dirette proteina-proteina tra MCL1 e una proteina contenente rBH3.
Materiali e metodi
Espressione proteica
Umano MCL1 [Uniprot: Q07820] (residui 163-326), umano P18 [Uniprot: P42773] (residui 1-168), A1-4 (residui P18 1-140), A4-5 (residui P18 106-168), P16 [Uniprot: P42771] (residui 1-156), e AC (residui P16 1-113 e residui P18 106-168) sono stati ottimizzati in sequenza per l’espressione batterica e clonati nei siti di restrizione di NdeI e HindIII in un vettore pET28a (EMD Millipore) per incorporare un tag N-terminale esa-istidina (His6) e trasformati in BL21(DE3) E. Coli (New England Bio Labs). Le colture sono state coltivate sotto selezione di kanamicina nel brodo di Luria ad una densità ottica di 600 nm di 0,6 e l’espressione è stata indotta dall’aggiunta di 1mM isopropil β-d-1-tiogalattopiranoside (Thermo Fisher). Le colture sono state raccolte 3-4 ore dopo l’induzione per centrifugazione a 4700 × g. I pellet batterici risultanti sono stati conservati a -80 °C. I pellet batterici His6-MCL1 sono stati risospesi (10 ml/g di pellet bagnato) nel tampone A (1× PBS, pH 6,8) con l’aggiunta di 1× lisozima (0,25 mg/ml) (Thermo Fisher) e di 1 compressa di inibitore della proteasi (Pierce #88266), sonicati, ed eliminati a 14.000 g. Il lisato risultante è stato purificato su NGC FPLC (Bio-Rad Laboratories) utilizzando una colonna di nichel HiTrap HP da 1 ml di HiTrap HP (GE Healthcare) bilanciata con il tampone A. La proteina è stata eluita con un gradiente di imidazolo di 2 mM-1000 mM. His-MCL1 contenente frazioni, come identificato da denaturazione elettroforesi gel di poliacrilamide denaturato, sono stati ulteriormente purificati da filtrazione gel nel buffer B (1 × PBS, pH 6,8) su una colonna HiPrep 16/60 sefacril S-100 (GE Healthcare). L’identità della proteina risultante è stata confermata dal desorbimento laser matrix-assistito / ionizzazione laser (MALDI) spettrometria di massa.
Sintesi peptidica
I peptidi sono stati sintetizzati utilizzando una strategia di sintesi di peptidi standard, a doppia aggiunta, fluorenylmethyloxycarbonyl (FMOC), in fase solida, sul sistema Prelude (Gyros Protein Technologies). La resina amminica 4-(2′,4′- dimetossifenil-fmocaminmetil)-fenossiacetammido-metilbenzidrilica (resina ammidica di pista MBHA, Anaspec) è stata gonfiata in N,N-dimetilformammide (DMF, Fisher Scientific) seguita da cloruro di metilene (DCM, Fisher Scientific) per aumentare la disponibilità di superficie per l’incollaggio. Utilizzando una strategia di doppia aggiunta di FMOC, l’N-terminale FMOC sulla catena peptidica crescente è stato deprotetto con 0,8 M piperidina (Fisher Scientific) in DMF per 2 minuti e 30 s, poi il seguente aminoacido (200 mM) da aggiungere al N-terminale è stato attivato con 0.4-M O-(1H-6-Clorobenzotriazolo-1-il)-1,1,3,3,3-tetrametiluronio esafluorofosfato (HCTU, Anaspec) in DMF per l’attacco nucleofilo della peptidina N-terminale. Successivamente, 800 mM 4-Methylmorpholine (NMM, Fisher Scientific) in DMF è stato aggiunto, e il peptidyl-resina, HCTU, NMMM slurry è stato mescolato per 30 min seguita da 4 x 30 lavaggi DMF s. La resina peptidilica è stata scissa utilizzando 88% di acido trifluoroacetico, 5% di acqua, 5% di fenolo e 2% di triisopropilsilano per 180 minuti. Il peptide scisso è stato filtrato a mano utilizzando i vasi di reazione Prelude lontano dalla resina. Il peptide filtrato e scisso è stato precipitato con etere a freddo e centrifugato a 14.000×× g per pellet il peptide grezzo scisso dalla resina. I peptidi grezzi sono stati liofilizzati e risospesi in acqua all’80%/20% acetonitrile e purificati su una colonna Zorbax Eclipse XDB-C18 (Agilent) su un 1260 Infinity HPLC (Agilent) con un gradiente di acetonitrile 5-60%. La massa peptidica è stata confermata da MALDI-MS. La concentrazione di peptide è stata determinata da una risonanza magnetica nucleare unidimensionale (1D) (NMR) confrontando le concentrazioni peptidiche approssimate con gli standard noti. L’N-termino di BAK peptidilresina è stato coniugato con un linker di acido 6-aminocaproico (AHX) con due aggiunte doppie di 30 minuti come descritto sopra. Infine, il fluoroforo FITC è stato accoppiato al AHX-peptide-resina al buio sotto azoto con due aggiunte 30 minuti in un fango denso di 8 mg FITC in piridina / DMF / DCM (12:7:5) seguita da successiva scissione, purificazione e verifica.
Anisotropia di polarizzazione a fluorescenza (FPA)
Un saggio FPA è stato sviluppato e caratterizzato da esperimenti eseguiti in micropiastre nere, non trattate a 96 pozzetti (Nunc #12-566-23). Gli esperimenti sono stati eseguiti utilizzando triplicati tecnici con tre repliche biologiche raccolte in giorni separati e con preparati proteici separati. Le reazioni sono state condotte in 1x PBS a pH 7,4 in 100 µL volumi di reazione. Inizialmente, 100 nM His6-MCL1 proteina e variabile peptidi P18x senza etichetta o dimetilsolfossido di zolfo (DMSO) sono stati aggiunti al pozzo e incubati a temperatura ambiente (RT) per 10 minuti. Poi sono stati aggiunti 10 µL di 100 nM FITC-AHX-BAK (concentrazione finale 10 nM) e i campioni sono stati incubati per un ulteriore 1 ora, agitando a RT al buio. Le piastre sono state lette su un lettore di piastre Victor X5 (Perkin Elmer) usando l’impostazione FP-Fluoresceina (1.0s) (energia della lampada CW, 65535; Filtro della lampada CW, F485-slot A5; Filtro delle emissioni, F535-Slot A5; tempo di conteggio 1s; e fattore G, 1. L’adattamento della curva è stato eseguito utilizzando il software a prisma (software Graphpad, Inc) utilizzando l’equazione, Y=Bottom+(Top-bottom)/(1+10^((LogIC50-X)*HillSlope)).
Coltura cellulare
PC-3, WAC2, HELA, MDA-MB-231, e cellule DU-145 sono stati mantenuti in RPMI-1640 medium integrato con il 10% di siero bovino fetale, 2,05 mM l-glutammina, 100 unità per mL ciascuno di penicillina e streptomicina, e 0,25 mg/mL di antimicotica Fungizone (Life Technologies) in un’atmosfera umidificata con il 5% di CO2. La validazione delle linee cellulari è stata completata utilizzando un breve profilo di ripetizione in tandem contro le firme ATCC pubblicate. Il Silencer Select ha convalidato i piccoli RNA interferenti (siRNA) e i siRNA di controllo negativo sono stati acquistati da Ambion. Gli ID del prodotto siRNA sono: si-MCL1 (s8583), P18 (118622), e il controllo negativo, siGFP (AM4626) è stato utilizzato senza alcuna somiglianza significativa sequenza di sequenze di geni umani. cDNA per P18 (clone ID: 3907917) e MCL1 (clone ID: 3138465) è stato ottenuto da Life Technologies e la sequenza di codifica è stata clonata nel pcDNA3.1 plasmide per l’espressione esogena. le trasfezioni di siRNA e plasmide sono state effettuate utilizzando rispettivamente i reagenti Lipofectamina RNA iMAX e Lipofectamina 3000 (Life Technologies). Le cellule sono state raccolte per l’analisi dopo 48 o 72 trasfezioni come indicato nel testo. Per gli esperimenti CellTrace™ Violet stainings cells (1×106/mL) sono state colorate con 100 nm CTV, C34557, acquistate da Thermo Scientific e poi placcate e trasfettate dopo 24 ore.
Prodotti chimici
ABT-199 (S8048), leupeptina emisolfato (57830) e calcitriolo (S1466) sono stati acquistati da Selleck Chemicals. S63845 è stato acquistato da Chemietek.
Trascrizione quantitativa inversa PCR (RT-qPCR)
L’RNA totale è stato isolato dalle cellule con il reagente Trizol (Life Technologies) e purificato con il PureLink RNA Mini Kit (Ambion). Il DNA genomico è stato rimosso utilizzando il trattamento RNAse-free DNAse. Le concentrazioni finali di RNA sono state misurate per assorbanza a 260 nm e la qualità è stata confermata utilizzando un rapporto A260/280 di ~ 2,0. cDNA è stato preparato utilizzando 1 mg di RNA totale in 20 ml di reazione di trascrizione inversa con qScript cDNA SuperMix (Quanta Biosciences) secondo il protocollo del produttore. RT-qPCR reazioni sono state eseguite in una reazione di 10 μL contenente 4 μL di DNA complementare diluito (cDNA), 5 μL PerfeCta SYBR verde FastMix, Low ROX (Quanta Biosciences), e 0,5 μL ciascuno di primer avanti e indietro a concentrazioni finali di 250 nM. Tutte le reazioni qPCR sono state eseguite in quadruplicato su piastre ottiche MicroAmp a 384 pozzetti su uno strumento ViiA 7 (Applied Biosystems). Le condizioni di amplificazione consistevano nella fase iniziale di denaturazione a 95°C per 3 minuti, seguita da 40 cicli di 10 a 95°C e 30 a 60°C. Successivamente, sono state generate curve di fusione per confermare la presenza di un singolo picco uniforme. I risultati sono stati analizzati nel software ViiA 7 con il metodo comparativo CT (ΔΔCT) utilizzando GAPDH come controllo di normalizzazione ed esportati per l’analisi e la presentazione in GraphPad Prism (GraphPad Software, La Jolla, CA, USA). Sono stati utilizzati i seguenti primer (5′-3′):
MCL1: (F) GGACATCAAAAACGAAGAGACG e (R) GCAGCTCTCTTGGTTGTTTGTTATGGG; GAPDH: (F) CCACATCGCTCAGACACCAT e (R) CCAGGCGCCCAATACG; P18: (F) GGGGACCTAGAGCAACTTA e (R) CAGCGCAGTCCTTCCAAAT.
Estrazione di proteine e western blot
I lisati a cellule intere sono stati preparati lisando le cellule su ghiaccio con 1× RIPA buffer (Boston BioProducts): 50 mM Tris-HCl pH 7,4, 150 mM NaCl, 1% NP40, 0.5% sodio desossicolato e acqua (a riposo) integrato con inibitori della proteasi (1mM AEBSF, 0,8μM aprotinina, 0,05mM bestatina, 0,015mM E-64, 0,02mM leupeptina, 0,01mM pepstatina A). I lisati proteici sono stati risolti mediante elettroforesi in gel di sodio dodecil solfato di poliacrilammide e trasferiti alla membrana di fluoruro di polivinilidene in un sistema di trasferimento a umido per 1 ora a 100 V. Le membrane sono state incubate con Abs anti-MCL1 (D2W9E e D35A5, Cell Signaling), Abs anti-BAX (D2E11, Cell Signaling), Abs anti-P18, DCS-118 da Cell Signaling, Abs anti-β-actina, PA1-21167, da Pierce (Rockford, IL, USA), e anti-CDK6 Ab, (C-21): sc-177, da Santa Cruz ad una diluizione di 1:1000, a 4°C durante la notte. HRP-coniugato Abs secondario coniugato sono stati utilizzati per la rilevazione utilizzando il reagente ECL2 (Pierce). Immunoblot sono stati visualizzati su Bio-Rad ChemiDoc sistema di imaging MP.
Citometria a flusso
Le celle PC-3, WAC2, MDA-MB-231 o DU-145 (2×105) sono state trasfettate con il solo controllo siRNA o si-P18 o si-MCL1. Le cellule PC-3, WAC2, MDA-MB-231, o DU-145 (2×105) sono state trasfettate con il plasmide Vehicle o MCL1 per il solo per 48 ore o per 30 ore e poi trattate con S63845 o ABT-199, concentrazione finale DMSO 0,5% per 18 ore. Dopo 18&h per esperimenti di siRNA o 48&h per esperimenti di sovraespressione, sono state raccolte le cellule. Le cellule sono state raccolte per FACS mediante lavaggio in 1× dPBS (Corning) e trattamento con tripsina (Gibco) per 5 minuti a 37°C. La tripsina è stata inattivata con l’aggiunta di mezzi RPMI con FBS e le cellule raccolte sono state centrifugate a 500 giri al minuto per 5 minuti, mezzi e tripsina aspirata, e lavata in 1x PBS e centrifugata a 500 giri al minuto per 5 minuti, aspirata e il pellet cellulare risospeso in 1× PBS con 0,1% di formaldeide (Fisher) mediante pipettaggio. Le cellule sono stati fissati con l’aggiunta di ghiaccio freddo 70% di etanolo (Fisher) e incubazione a -20 ° C per 2 h per le cellule di colorazione PI sono stati pellet a 1500 giri al minuto per 10 minuti e lavato 2× con PBS, 1 mg / ml RNase A (Thermo Fisher) è stato aggiunto alle cellule e incubato a 37 ° C per 30 minuti. In tutto, 1×105 cellule sono statecolorate con 0,1 mg/mL di ioduro di propidio (BD Biosciences, San Jose, CA, USA) a temperatura ambiente per 15 minuti. Poi le cellule sono state sottoposte a citometria a flusso su BD LSRFortessa FACS, 50.000 eventi sono stati raccolti per ogni campione ei dati sono stati analizzati da FlowJo V10. Per gli esperimenti di Annexin V/PI, le cellule sono state raccolte in modo simile, lavate 2x in PBS, risospese in 1x Annexin V tampone legante (CBD Pharmingen), e colorato con FITC Annexin V (CBD Pharmingen) e PI. Poi le cellule sono state sottoposte a citometria a flusso su BD LSRFortessa FACS, 50.000 eventi sono stati raccolti per ogni campione e i dati sono stati analizzati da FlowJo V10. Per le cellule CellTrace™ Violet colorate, dopo 24, 48 o 72 cellule di trasfezione sono state raccolte in modo simile alle cellule PI colorate, ma fissate in etanolo al 70% di etanolo (Fisher) ghiacciato e incubate a -20°C per 20 minuti. Poi le cellule sono state sottoposte a citometria a flusso su BD LSRFortessa FACS, 50.000 eventi sono stati raccolti per ogni campione ei dati sono stati analizzati da FlowJo V10. Tutti i dati sono stati raccolti con tre repliche biologiche analizzate in giorni separati con duplicati tecnici per ogni analisi.
Analisi statistiche
Tutti gli esperimenti sono stati ripetuti almeno tre repliche biologiche utilizzando due o tre repliche tecniche, come riportato, con i dati espressi come media±S.D. Nessun campione è stato escluso. Le differenze tra due serie di dati sono state calcolate utilizzando un t-test Studente non accoppiato a due code con P<0,05 considerato statisticamente significativo. L’analisi statistica è stata eseguita in Prism (Graphpad Inc.) o Microsoft Excel. *P<0.05, **P<0.01, ***P<0.001.
Risultati
P18 si lega a MCL1 in vitro
Per verificare se il motivo rBH3 funziona come un motivo di legame con MCL1 nelle proteine umane native, abbiamo seguito l’analisi BLAST eseguita sulla sequenza iniziale rBH3-115. Questa analisi ha identificato un rBH3 putativo nella c-terminale alfa-elica della proteina regolatrice del ciclo cellulare P18 (P18INK4C, CDKN2C). Come è stato osservato in rBH3-1 (Fig. 1a), la P18 rBH3 (residui 150-161) contiene una sostituzione omologa in cui il residuo di acido aspartico conservato nella sequenza di consenso BH3 viene sostituito con un residuo di acido glutammico (E151). Inoltre, i residui vicini di leucina (L155) e metionina (M156) si trovano in posizione idrofobica che fa contatto con la tasca P2 di MCL1 mentre un residuo di valina è posizionato in posizione idrofobica P3. Per confermare che la sequenza rBH3 in P18 è in grado di legarsi a MCL1 abbiamo utilizzato un test di polarizzazione fluorescente competitivo (FP) per valutare la capacità dei peptidi rBH3 derivati da P18 o della proteina P18 a tutta lunghezza di inibire il legame di un partner di legame nativo marcato con fluoresceina, in questo caso l’elica 23 dell’aminoacido BAK BH3 (F-BAK). Abbiamo osservato che la proteina P18 è in grado di inibire l’associazione F-BAK con MCL1 con un IC50 di 113,1±3,4 Nm. Per localizzare questo legame al motivo rBH3, abbiamo preparato due peptidi che sono composti dal c-terminale 21 o 12 residui di P18, residui 141-161 (21mer) o 160-161 (12mer), rispettivamente. Abbiamo osservato che sia il peptide 21-mer che il peptide 12-mer, sebbene in conformazioni non globali, hanno mantenuto un’inibizione competitiva di F-BAK con valori IC50 di 778,2±6,7±nM e 2502±15±nM, rispettivamente (Fig. 1b). Questa diminuzione dell’affinità è coerente con studi precedenti che hanno esaminato l’impatto che la stabilizzazione delle eliche alfa contenenti BH3 ha sull’associazione con le proteine BCL2 anti-apoptotiche16. In questo caso, la sequenza rBH3 nella proteina P18 a lunghezza intera è nativamente stabilizzata nella piega globulare, mentre i peptidi sintetizzati non lo sono. Abbiamo poi cercato di determinare l’importanza dei principali residui acidi e idrofobici che imitano la sequenza BH3 sintetizzando sostituzioni alanina di E151 e M156, rispettivamente. Abbiamo osservato che entrambi i mutanti hanno indotto una significativa riduzione del legame con valori IC50 di >30.000 nM (Fig. 1c). Questi studi dimostrano in vitro l’associazione in vitro di P18, attraverso la sua sequenza rBH3, con MCL1 e che tale associazione inibisce l’associazione F-BAK con MCL1 in modo biologicamente rilevante.Fig. 1P18 rBH3 sequenza inibisce l’associazione BAK con MCL1.a Tabella delle sequenze peptidiche e valori IC50 di FPA in 1B. b Saggio di anisotropia a polarizzazione di fluorescenza della concorrenza (FPA) della proteina P18 e dei peptidi concorrenti F-BAK23mer (10 nM) per il legame MCL1 (100 nM). c FPA di P1821mer e peptidimutanti concorrenti F-BAK23mer (10 nM) per il legame MCL1 (100 nM). Tutti i dati N=3.
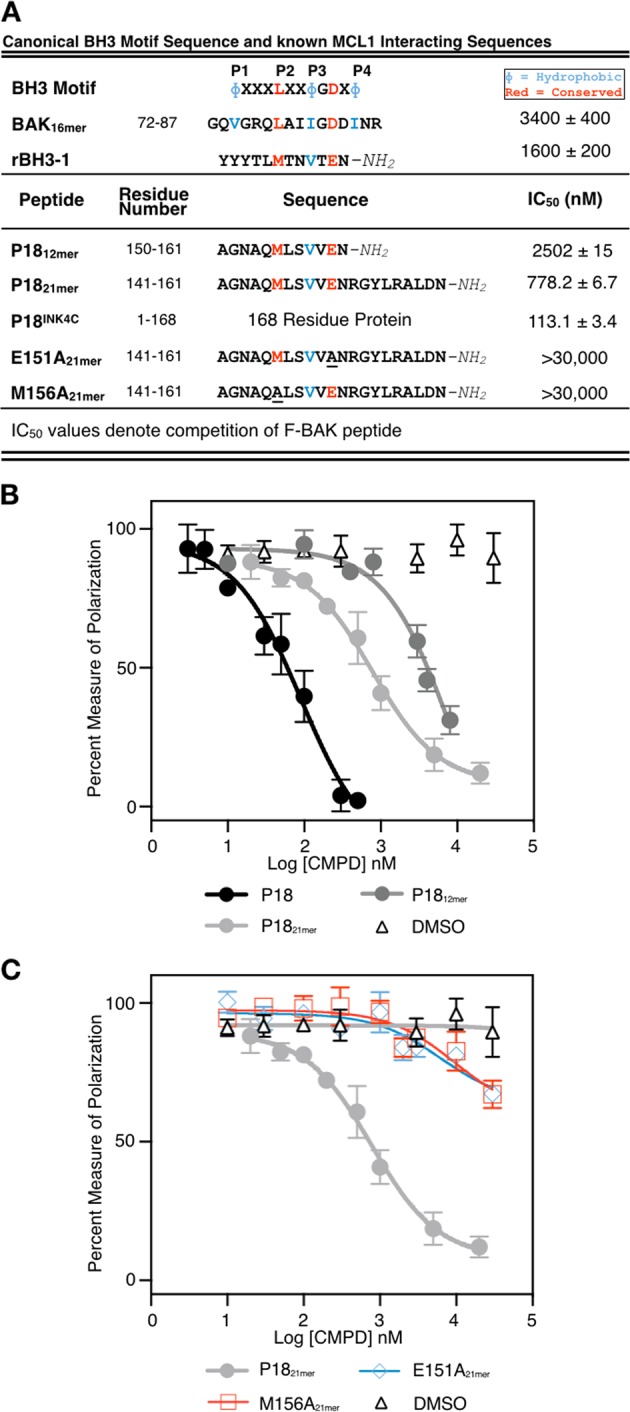
Fig. 1.La sequenza P18 rBH3 inibisce l’associazione del BAK con MCL1.a Tabella delle sequenze peptidiche e dei valori IC50 di FPA in 1B. b Saggio di anisotropia a polarizzazione di fluorescenza da competizione (FPA) della proteina P18 e dei peptidi concorrenti F-BAK23mer (10 nM) per il legame MCL1 (100 nM). c FPA di P1821mer e peptidimutanti concorrenti F-BAK23mer (10 nM) per il legame MCL1 (100 nM). Tutti i dati N=3.
P18 si associa endogicamente con MCL1
Dopo aver confermato la capacità della P18 di legarsi a MCL1, abbiamo poi cercato di sondare l’interazione endogena della P18 con MCL1. Abbiamo testato l’espressione della P18 in una selezione di linee cellulari tumorali solide e ne abbiamo scelte due, PC-3 (cancro alla prostata) e WAC2 (neuroblastoma), per studiare ulteriormente l’interazione basata sui livelli di espressione della P18. In entrambe le linee cellulari, abbiamo sondato e osservato la co-immunoprecipitazione (coIP) di entrambi P18 e la nota proteina legante MCL1 BAX a seguito di immunoprecipitazione (IP) di MCL1 endogeno o IgG di controllo (Fig. 2a e Supplementay Fig. S1A). Nelle cellule PC-3, siamo stati anche in grado di eseguire l’IP inverso della P18 endogena utilizzando un anticorpo monoclonale specifico per la P18 o IgG di controllo e abbiamo sondato e osservato la coIP sia della MCL1 che della nota proteina di legame P18 CDK6 (Supplementay Fig. S1B). Questi dati dimostrano l’interazione endogena tra MCL1 e P18 in più linee cellulari di derivazione umana.Fig. 2MCL1 si lega specificamente alla P18 attraverso il C-terminale rBH3.un Western blot di coIP endogeno di MCL1 (IP) con P18 (IB), BAX (IB), e P18 (IB) in WAC2. b Western blot di pulldown in vitro di proteine ricombinanti (IP), ANK1-4, ANK4-5, P18, p16, e chimera, con MCL1 ricombinante (rMCL1) (IB). Il cartone animato delle proteine p16, P18 e chimera è mostrato sotto il blot. L’elica contenente rBH3 è evidenziata in giallo (Residui: 150-161). c Western blot di pulldown in vitro di P18 ricombinante (IP) con rMCL1 (IB) con e senza inibitore MCL1 S63845.
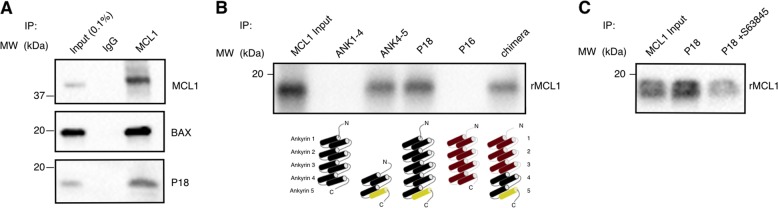
Fig. 2.MCL1 si lega specificamente a P18 attraverso il terminale C rBH3.un Western blot di coIP endogeno di MCL1 (IP) con P18 (IB), BAX (IB), e P18 (IB) in WAC2. b Western blot di pulldown in vitro di proteine ricombinanti (IP), ANK1-4, ANK4-5, P18, p16, e chimera, con MCL1 ricombinante (rMCL1) (IB). Il cartone animato delle proteine p16, P18 e chimera è mostrato sotto il blot. L’elica contenente rBH3 è evidenziata in giallo (Residui: 150-161). c Western blot di pulldown in vitro di P18 ricombinante (IP) con rMCL1 (IB) con e senza inibitore MCL1 S63845.
Il motivo P18 rBH3 è necessario e sufficiente per mediare il legame
P18 è un membro della famiglia delle proteine INK4 [P16INK4A (P16, CDKN2A), P15INK4B (P15, CDKN2B), P18INK4C (P18, CDKN2C) e P19INK4D (P19, CDKN2D)] che regolano la progressione del ciclo cellulare G1/S con tutte le proteine INK4 che mantengono la funzione inibitoria omologa CDK4/6. Le proteine della famiglia INK4 hanno strutture omologhe contenenti quattro o cinque ripetizioni di anchilina (ANK), un’unità strutturale elicoidale di rotazione dell’elica nota per mediare le interazioni proteine-proteine15,17. In particolare, P15 e P16 sono composti da quattro ripetizioni di achirin, mentre P18 e P19 hanno cinque unità di ripetizione di achirin. L’rBH3 in P18 risiede nella sua quinta ripetizione di una certa quantità di colore (residui 141-168). Per determinare se il motivo rBH3 della P18 è sufficiente a mediare il legame con MCL1 e che i due non interagiscono semplicemente attraverso un complesso cellulare condiviso, abbiamo testato la capacità delle proteine ricombinanti e chimeriche P18 e P16 di ricombinare esogeneamente MCL1. Sulla base di precedenti studi di stabilità delle ripetizioni dell’achilina all’interno delle proteine della famiglia INK418,19abbiamo progettato due tronconi di P18 (Tabella supplementare S1). La prima, P18-ANK1-4 (ANK1-4), rimuove la quinta ripetizione della rBH3 contenente la quinta anchilina. Il secondo, P18-ANK4-5 (ANK4-5), mantiene il dominio rBH3 e la ripetizione 4 dell’anchilina per facilitare la piegatura. Come controllo negativo, abbiamo espresso la ricombinante P16 che contiene nativamente quattro ripetizioni di una certa acne e non contiene un motivo putativo di rBH317,20. Abbiamo osservato che solo i costrutti delle proteine ANK4-5 e P18 (Fig. 2b, corsie 3 e 4), entrambi contenenti il motivo rBH3 trovato in ANK5, hanno tirato giù con successo MCL1 mentre sia le proteine ANK1-4 che P16 (Fig. 2b, corsie 2 e 5) non hanno mostrato alcuna interazione (Fig. 2b). Per dimostrare ulteriormente che la rBH3 contenente ANK5 media il legame, abbiamo progettato una proteina chimerica a cinque ripetizione di cherica composta da P16-ANK1-3 e P18-ANK4-5 (chimera) (Tabella supplementare S1). Questa proteina chimerica ha acquisito la capacità di pulldown ricombinante MCL1 (Fig. 2b, corsia 6). Infine, per dimostrare che l’interazione tra P18 e MCL1 utilizza la tasca legante BH3 per mediare l’interazione, abbiamo impiegato una piccola molecola MCL1 specifica per MCL1 mimetica BH3, S6384521e ha testato la sua capacità di interrompere il pulldown. Coerentemente con la sequenza rBH3 in P18 che si lega alla tasca BH3 di MCL1, l’aggiunta di S63845 alla miscela ha soppresso la capacità di P18 di abbattere MCL1 (Fig. 2c). Questi dati dimostrano che la porzione rBH3 contenente ANK5 di P18 è necessaria e sufficiente per mediare l’associazione diretta di P18 con MCL1 e l’inibizione della tasca BH3 in MCL1 blocca questa interazione.
MCL1 induce la perdita di P18
Quando è attivo, le proteine CDK4 e CDK6 fosforilano il soppressore tumorale, RB1, che poi rilascia il fattore di trascrizione chiave dell’ingresso in fase S, E2F122,23. I tumori umani eliminano comunemente P16 e/o P15 per superare l’inibizione della crescita, ma il membro contenente rBH3, P18, la cui espressione ha dimostrato di compensare la perdita di P16, viene raramente eliminato.24 (Fig. supplementare S2). Per confermare la capacità del motivo rBH3 di mediare l’interazione diretta proteina-proteina tra MCL1 e P18 e chiarire l’impatto che questo legame ha sulla crescita cellulare e la vitalità abbiamo scelto la linea cellulare del neuroblastoma, WAC2, che contiene una delezione omozigote di P16, come il modello cellulare principale per questi studi25. Abbiamo confermato i nostri risultati nelle cellule PC-3, un modello di cancro alla prostata resistente alla castrazione spesso utilizzato negli studi della famiglia BCL2.
Tipicamente, l’interferenza del BH3 che si lega ai membri della famiglia BCL2 antiapoptotica è stata implicata nella segnalazione a favore della morte e questo modello serve come base per il recente sviluppo di composti antitumorali destinati alla famiglia BCL2.26. Come tale, abbiamo innanzitutto cercato di determinare se l’aumento della P18 avrebbe indotto una risposta apoptotica. In ciascuna delle linee cellulari testate, abbiamo osservato che la sovraespressione della P18 non ha mostrato alcuna evidenza di stress cellulare o di induzione dell’apoptosi (Fig. 3 e Supplementare Fig. S3).La trasfezione della Fig. 3P18 non induce apoptosi.a Trasfezione del controllo del veicolo (sinistra) o della P18 (destra) (48 h) in WAC2 con l’analisi delle FACs di colorazione Annexin V e PI. b Western blot che conferma la trasfezione dell’espressione della P18 e la sovraespressione. Tutti i dati sono presentati come media±±S.D., N==3. La significatività statistica è stata determinata dal test t dello studente non accoppiato dove *P>0.05 ;**P>0.01; ***P>0.001.
Durante gli studi iniziali per ottenere il coIP di P18 e MCL1, abbiamo testato un certo numero di condizioni di sovraespressione MCL1 prima dell’immunoprecipitazione con gli anticorpi monoclonali MCL1-specifici o P18-specifici. Tuttavia, l’analisi delle macchie occidentali risultanti da questi test ci ha portato a sospettare che gli elevati livelli di proteina MCL1 avevano un effetto deleterio sui livelli di proteina P18. A seguito di questa osservazione, abbiamo confermato che la transfezione transitoria e la successiva sovraespressione di MCL1 si traduce in una diminuzione della proteina P18 (Fig. 4a). Abbiamo inoltre confermato che questa diminuzione della proteina P18 non coincide con una diminuzione dell’espressione di mRNA P18 (Fig. 4b). In particolare, bortezomib (Velcade, BTZ), un inibitore del proteasoma (che agisce sulle modalità enzimatiche del proteasoma simili alla chimotripsina e alla caspasi)27,28 non salva questo effetto. Infatti, il trattamento con bortezomib ha causato un aumento della proteina MCL1 e una conseguente diminuzione della proteina P18 (Fig. 5 e Supplementare Fig. S4). Abbiamo quindi cercato di determinare se una manipolazione alternativa del proteasoma potesse salvare la perdita di P18. Abbiamo osservato che il trattamento con l’inibitore cisteina-proteasi-specifico, la leupeptina (LeuP), ha avuto un impatto marginale a nessun impatto su MCL1 e P18 come singolo agente. Tuttavia, il co-trattamento di BTZ con la LeuP ha aumentato significativamente l’espressione sia di MCL1 che di P18 e ha salvato l’effetto negativo che il singolo agente BTZ ha sulla P18 (Fig. 5). Questi dati suggeriscono che MCL1 ha un effetto negativo trascrizionalmente indipendente sulla proteina P18 che è mediata attraverso un processo di degradazione della cisteina-proteasi.Fig. 4MCL1 influenza negativamente la proteina P18.un Western blot di transfezione transitoria MCL1. MCL1, P18 e actina IB in PC-3 e WAC2 mostrano che l’aumento di MCL1 corrisponde alla diminuzione della proteina P18. Quantificazione dell’intensità della banda mostrata di seguito blot. b RT-qPCR analisi RT-qPCR in PC-3 e WAC2 di MCL1 transfezione transitoria o di controllo del veicolo. Espressione normalizzata a GAPDH. Tutti i dati sono presentati come media±S.D., N==3. La significatività statistica è stata determinata dal t-test dello studente non accoppiato dove *P> 0.05; **P>0.01; ***P>0.001.Fig. 5MCL1 impatto sulla degradazione del P18.Western blot di controllo DMSO (0,3%), 10 µM di leupeptina (LeuP), 10 µM di leupeptina (LeuP), 10 µM di bortezomib (BTZ), o 30 µM di bortezomib trattato con BTZ (a) cellule PC3 o (b) cellule WAC2 per 4 ore. Quantificazione dell’intensità della banda indicata di seguito. Tutti i dati sono presentati come media±±S.D., N==3. La significatività statistica è stata determinata dal test t dello studente non accoppiato dove *P>0,05 ;**P>0,01; ***P>0,001.
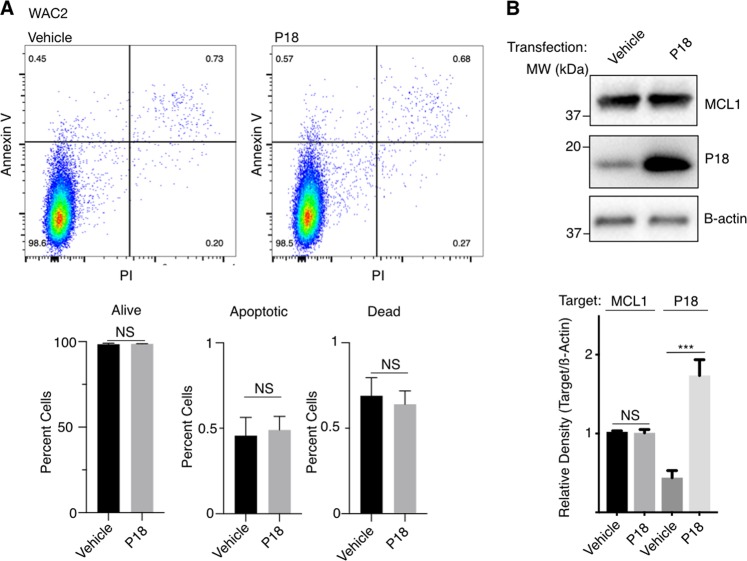
Fig. 3.La trasfezione P18 non induce apoptosi.a Trasfezione del controllo del veicolo (sinistra) o P18 (destra) (48 ore) in WAC2 con l’analisi dei FACs di colorazione di Annexin V e PI. b Western blot che conferma la trasfezione dell’espressione P18 e la sovraespressione. Tutti i dati sono presentati come media±±S.D., N==3. La significatività statistica è stata determinata dal test t dello studente non accoppiato dove *P>0.05 ;**P>0.01; ***P>0.001.
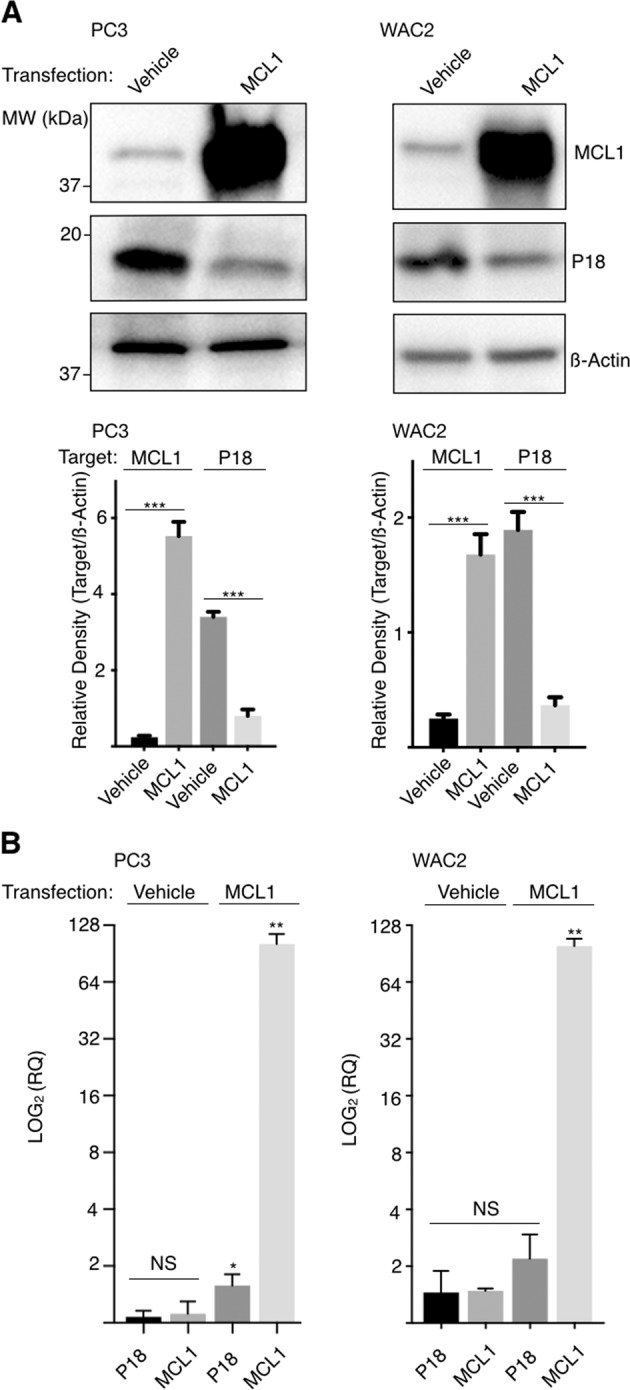
Fig. 4.MCL1 influenza negativamente la proteina P18.una Western blot di transfezione transitoria MCL1. MCL1, P18, e actina IB in PC-3 e WAC2 mostrano aumento in MCL1 corrisponde con la diminuzione della proteina P18. Quantificazione dell’intensità della banda mostrata di seguito blot. b RT-qPCR analisi RT-qPCR in PC-3 e WAC2 di MCL1 transfezione transitoria o di controllo del veicolo. Espressione normalizzata a GAPDH. Tutti i dati sono presentati come media±±S.D., N==3. La significatività statistica è stata determinata dal t-test dello studente non accoppiato dove *P>0,05 ;**P>0,01; ***P>0,001.
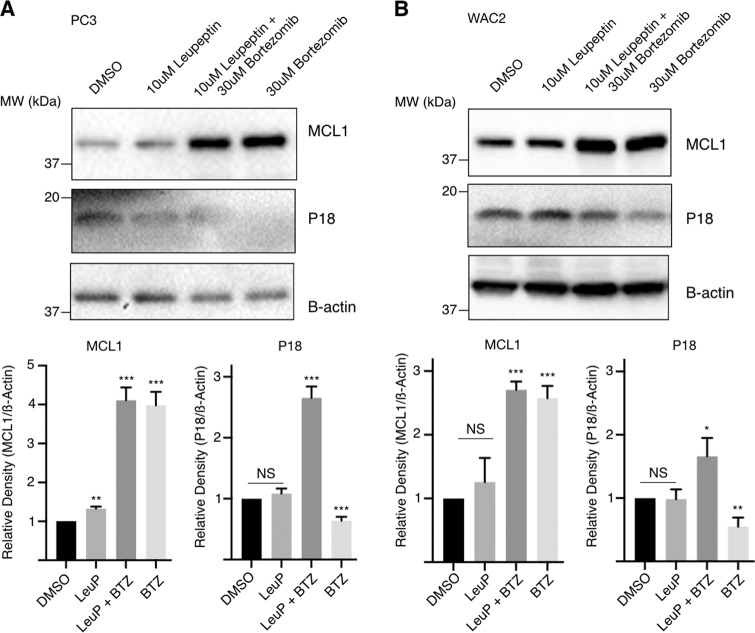
Fig. 5.MCL1 impatto sulla degradazione della P18.Western blot di controllo DMSO (0,3%), 10 µM di leupeptina (LeuP), 10 µM di leupeptina (LeuP), 10 µM di bortezomib (BTZ), o 30 µM di bortezomib trattato con BTZ (a) cellule PC3 o (b) cellule WAC2 per 4 ore. Quantificazione dell’intensità della banda indicata di seguito. Tutti i dati sono presentati come media±±S.D., N==3. La significatività statistica è stata determinata dal test t dello studente non accoppiato dove *P>0,05 ;**P>0,01; ***P>0,001.
MCL1 regola la P18 per promuovere la progressione G1/S
Il ruolo stabilito della P18 come regolatore negativo del ciclo cellulare in combinazione con la nostra osservazione che MCL1 media l’esaurimento della proteina P18, ci ha portato a valutare come la sovraespressione MCL1 influisce sul ciclo cellulare. Utilizzando la colorazione del contenuto di DNA dello ioduro di propidio (PI) e l’analisi FACS, abbiamo osservato che nelle linee cellulari non sincronizzate WAC2 e PC-3, la sovraespressione di MCL1 si traduce in una diminuzione della popolazione di cellule G1 (Fig. 6a e Supplementare S5) con un corrispondente aumento delle popolazioni S (PC-3) e G2/M (WAC2). Per mettere la modulazione MCL1 del ciclo cellulare in prospettiva di P18, abbiamo eseguito l’inibizione del siRNA per misurare l’impatto che la perdita di P18 ha sulle popolazioni del ciclo cellulare. Abbiamo osservato che si-P18 ha mostrato una diminuzione del G1 e aumento della proliferazione cellulare in entrambe le linee cellulari WAC2 e PC-3 (Supplementare Figs. S6 e S7, rispettivamente).Fig. 6MCL1 modulazione del ciclo cellulare è RB dipendente.un cromatogrammi PI (a sinistra) di veicolo o MCL1 sovraespressione nella linea cellulare RB1-positivo, WAC2. Analisi della popolazione del ciclo cellulare dei cromatogrammi e studi corrispondenti di MCL1 sovraespressi e trattati con mimetici BH3 S63845 o ABT-199 (a destra). b Cromatogrammi PI (a sinistra), controllo del veicolo e transitorio MCL1 sovraespressione nella linea cellulare RB1-mutante, DU-145 con l’analisi della popolazione del ciclo cellulare corrispondente a destra. Tutti i dati sono presentati come media±±S.D., N==3. La significatività statistica è stata determinata dal t-test dello studente non accoppiato dove *P> 0,05 ;**P>0,01; ***P>0,001.
Sulla base degli studi di cui sopra, abbiamo ipotizzato che la tasca BH3 di MCL1 interagisse con il motivo rBH3 in P18 per indurre il degrado e quindi promuovere la transizione G1/S. Per confermare che le interazioni con la tasca BH3 di MCL1 modulano direttamente la progressione G1 osservata, abbiamo impiegato il MCL1-specifico (S63845) e BCL2-specifico (ABT-199) inibitori delle piccole molecole21,29,30. Entrambi questi inibitori agiscono come mimetici BH3 e si legano nelle tasche BH3 di MCL1 o BCL2, rispettivamente, per sopprimere l’interazione con le proteine leganti delle tasche BH3, come l’interazione P18 con MCL1. Abbiamo osservato che l’inibitore specifico per MCL1 ha salvato la diminuzione indotta dalla sovraespressione di MCL1 nella popolazione G1 di nuovo a livelli di controllo (Fig. 6a), mentre l’inibitore specifico per BCL2 non ha avuto alcun impatto sul cambiamento nella popolazione G1. Questi dati suggeriscono che le interazioni BH3 tra MCL1 e proteine bersaglio forniscono un nuovo meccanismo per MCL1 libero per modulare la progressione cellulare attraverso il punto di controllo INK4 G1/S precoce. Inoltre, la mancanza di impatto dell’inibitore BCL2 sulla progressione G1/S, anche se con un impatto più significativo sulla popolazione cellulare sub-G1, dimostra che questo è un effetto specifico MCL1 e non un meccanismo generale di proteine leganti BH3.
Al fine di contestualizzare l’effetto di MCL1 sulla transizione cellulare G1/S all’interno del percorso P18 regolato CDK4/6-RB1, abbiamo valutato l’impatto di tale sovraespressione MCL1 ha sulle cellule DU-145, un P18 che esprime ma RB1-mutante (proteina non funzionale attraverso l’esone 21 delezione) linea cellulare del cancro alla prostata31. Con la cancellazione di RB1, ci aspettiamo che le modifiche in P18 non abbiano alcun effetto sulla transizione G1/S. Abbiamo quindi confermato per la prima volta che il si-P18 non ha alcun effetto sulla popolazione del ciclo cellulare in DU-145 (Supplemento Fig. S6). Abbiamo poi determinato che la sovraespressione MCL1 mantiene la capacità di influenzare negativamente l’espressione della proteina P18 nelle cellule DU-145 (Supplemento Fig. S8). Infine, abbiamo valutato l’impatto della sovraespressione MCL1 nelle cellule DU-145 e abbiamo scoperto che la sovraespressione MCL1 non induce cambiamenti nella popolazione G1 rispetto al controllo (Fig. 6b). Questi dati dimostrano che la regolazione MCL1 dei livelli di proteina P18 non è un effetto secondario del suo impatto sul ciclo cellulare. Inoltre, che la regolazione MCL1 del ciclo cellulare è RB-1 dipendente.
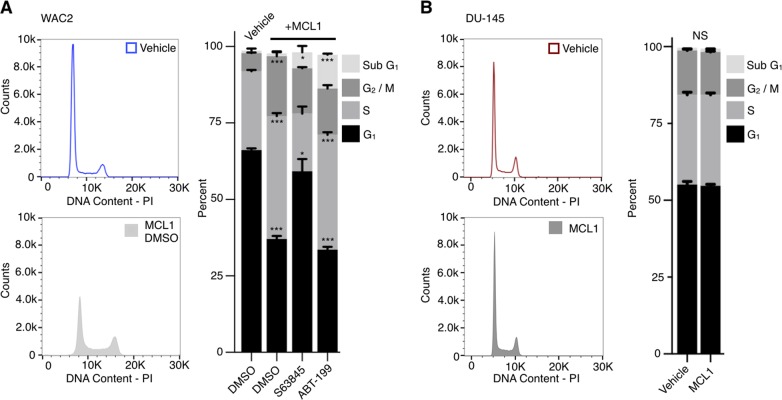
Fig. 6.La modulazione MCL1 del ciclo cellulare dipende da RB.un cromatogramma PI (a sinistra) del veicolo o sovraespressione MCL1 nella linea cellulare RB1-positiva, WAC2. 7. Analisi della popolazione del ciclo cellulare di popolazione di cromatografi così come gli studi corrispondenti di MCL1 sovraespressi e trattati con mimetici BH3 S63845 o ABT-199 (a destra). b Cromatogrammi PI (a sinistra), il controllo del veicolo e transitorio MCL1 sovraespressione MCL1 nella linea cellulare RB1-mutante, DU-145 con l’analisi della popolazione del ciclo cellulare corrispondente a destra. Tutti i dati sono presentati come media±±S.D., N==3. La significatività statistica è stata determinata dal t-test dello studente non accoppiato dove *P> 0,05 ;**P>0,01; ***P>0,001.
MCL1 promuove la crescita, non il blocco G2/M
Negli studi precedenti, ci siamo concentrati sull’effetto che MCL1 ha sulla popolazione cellulare G1, ma, negli studi sul ciclo cellulare, un cambiamento in un gruppo deve portare alla ridistribuzione in un’altra porzione del ciclo cellulare. Abbiamo osservato che la sovraespressione di MCL1 e la successiva diminuzione di G1 ha portato ad un aumento simultaneo della percentuale di cellule in G2/M. Studi precedenti hanno osservato questo cambiamento negli istogrammi PI e lo ha attribuito a MCL1 inducendo un blocco nella progressione G2/M. Data la nostra osservazione che MCL1 influenza la proteina P18 abbiamo supposto che un tale aumento potrebbe essere causato da un blocco G2/M o, in alternativa, attraverso un aumento della proliferazione cellulare. Per valutare quale si sta verificando e con conseguente cambiamento negli istogrammi PI, abbiamo tracciato la proliferazione cellulare in due modi. In primo luogo, abbiamo monitorato la crescita cellulare in un periodo di tempo di 64 ore. Abbiamo osservato che le cellule RB1-positive hanno tutte mostrato un aumento dei tassi di crescita a seguito di una sovraespressione di MCL1, mentre le cellule RB1-negative non mostrano alcun cambiamento (Fig. 7a, c, rispettivamente). In secondo luogo, abbiamo usato il marcatore di diluizione del colorante cellulare, CellTrace™ Violet (CTV), per tracciare la proliferazione32–34 (Fig. supplementare S9). In questo saggio, mentre le cellule dividono il colorante viene diviso in cellule figlie, causando una diminuzione della misurazione del colorante e un aumento del numero di cellule. Abbiamo osservato che la sovraespressione MCL1 nelle cellule RB1-positive ha indotto sia un aumento del numero di cellule che una diminuzione della colorazione CTV, coerente con l’aumento della proliferazione rispetto ai controlli (Fig. 7be Supplementare Figs. S7, S5B, S10). Al contrario, le cellule RB1-negative non hanno mostrato tale cambiamento in seguito alla sovraespressione MCL1 (Fig. 7d). Questo suggerisce fortemente che la diminuzione osservata in G1 e l’aumento delle popolazioni di cellule S o G2/M dalla sovraespressione MCL1 non è la prova di un blocco del ciclo cellulare, ma è piuttosto dovuto ad un aumento della proliferazione cellulare.Fig. 7MCL1 promuove la proliferazione in modo RB-dipendente.la proliferazione cellulare è stata misurata utilizzando il conteggio delle cellule in WAC2(a) e DU-145(c) linee cellulari oltre 64 ore dopo la trasfezione del veicolo o MCL1. Inoltre, il test di diluizione del colorante, Cell Trace Violet, è stato utilizzato per visualizzare la proliferazione in WAC2(b) e DU-145(d) linee a 48 e 72h dopo la trasfezione del veicolo o MCL1. L’analisi dell’intensità massima di picco per i saggi CTV è mostrata a destra di ogni coppia. Tutti i dati sono presentati come media±±S.D., N==3. La significatività statistica è stata determinata dal t-test dello studente non accoppiato dove *P>0.05 ;**P>0.01; ***P>0.001.
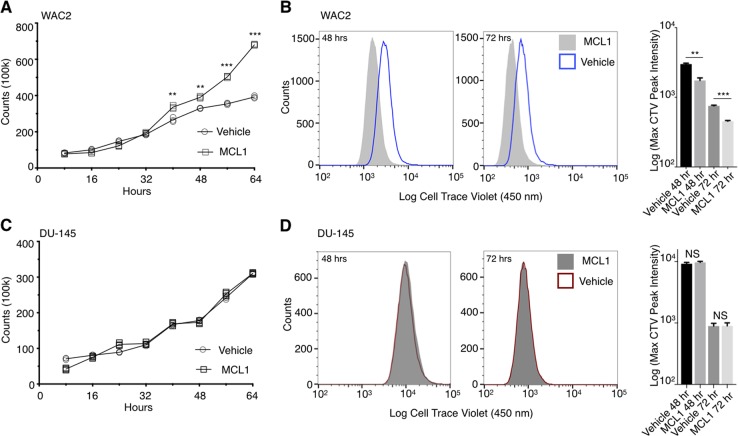
Fig. 7.MCL1 promuove la proliferazione in modo dipendente dalla RB.La proliferazione cellulare è stata misurata utilizzando il conteggio delle cellule in WAC2(a) e DU-145(c) linee cellulari oltre 64 ore dopo la trasfezione del veicolo o MCL1. Inoltre, il test di diluizione del colorante, Cell Trace Violet, è stato utilizzato per visualizzare la proliferazione in WAC2(b) e DU-145(d) linee a 48 e 72h dopo la trasfezione del veicolo o MCL1. L’analisi dell’intensità massima di picco per i saggi CTV è mostrata a destra di ogni coppia. Tutti i dati sono presentati come media±±S.D., N==3. La significatività statistica è stata determinata dal t-test dello studente non accoppiato dove *P>0.05 ;**P>0.01; ***P>0.001.
Discussione
Dalla scoperta dell’importanza del BCL2, prima nella tumorigenesi e poi per la sua regolazione dell’apoptosi, sono stati compiuti notevoli sforzi per comprendere la complessa rete di interazioni proteina-proteina che comprende la regolazione della famiglia BCL2 e la coordinazione della morte cellulare.2,35. In questi studi della famiglia BCL2, diversi lavori hanno evidenziato come le proteine della famiglia non-BCL2 possano influenzare i membri della famiglia BCL2 anti-apoptotici.36–38. Mentre la maggior parte di queste interazioni hanno dimostrato di agire in modo pro-apoptotico, alcune interazioni hanno suggerito una connessione tra la famiglia BCL2 e altri meccanismi omeostatici cellulari, compreso il ciclo cellulare39–44.
Infatti, per tutto il tempo in cui la famiglia BCL2 è stata studiata e a causa del coinvolgimento diretto di questa famiglia di proteine nella regolazione della vitalità cellulare, il ruolo della famiglia BCL2 nella proliferazione cellulare è stato ipotizzato essere come minimo un effetto secondario della vitalità mantenuta.45. Tuttavia, è diventato sempre più evidente che la famiglia BCL2 può avere un ruolo molto più diretto nella proliferazione attraverso le interazioni con i macchinari del ciclo cellulare e la presenza della famiglia BCL2 durante tutto il ciclo cellulare.46. In effetti, il lato anti-apoptotico della famiglia BCL2 sembra rinunciare al suo ruolo pro-sorveglianza durante tutto il ciclo cellulare tra i suoi membri. A partire da G1, sia BCL2 che BCLxL hanno dimostrato di mantenere la vitalità cellulare in G0 e G140,47,48. Oltre a sostenere la vitalità cellulare durante l’interfase, queste due proteine hanno anche dimostrato di prolungare G1, ritardando la transizione alla fase S in presenza di stress intracellulare49. Ciò è confermato dalle osservazioni di un recente articolo50 dove nel killer naturale specifico (NK) tipo di cellula; c’è una differenza nell’espressione delle proteine dei membri della famiglia BCL2 nel ciclismo rispetto alle cellule non ciclistiche. In particolare, BCL2 è stato osservato più in cellule non cicliche e MCL1 è stato osservato più in cellule NK ciclismo. Passando oltre G1 in fase S, la chiave G1/S fattore di transizione G1/S trascrizione, E2F1, è noto per sopprimere direttamente il promotore MCL1 51 e sopprime anche i livelli di RNA BCL2 e di proteine52. Al contrario, BCL2 ritarda la transizione G1/S attraverso l’inibizione di E2F140. Tuttavia, il ruolo di MCL1 è tutt’altro che chiaro in quanto l’espressione MCL1 è correlata ad un aumento del noto inibitore della proteina di transizione G1/S, p27, nei progenitori neurali53. Inoltre, è stato dimostrato che MCL1 interagisce con il PCNA, un morsetto scorrevole del DNA coinvolto nella processualità durante la fase S39. PCNA è una proteina promiscua con molti partner leganti, tra cui p21, CDK2/4/5/6, ciclina D1 e altri54. Fujise e colleghi hanno osservato che MCL1 è in grado di legare il PCNA utilizzando lievito due costrutti ibridi e di sovraespressione e che la sovraespressione MCL1 induce una diminuzione della percentuale di cellule in fase S come mostrato attraverso l’assorbimento di BRDU39. Al contrario, Jamil et al.55 hanno osservato che in seguito al blocco del ciclo cellulare, il CDK1 si lega ad una forma alternativa di MCL1 murino (snMcl-1) e che la sovraespressione di questa forma rallenta la crescita cellulare. Nonostante queste osservazioni contraddittorie sul ruolo di MCL1 nella transizione G1/S, il suo coinvolgimento non può essere ignorato, e sono necessari ulteriori studi per distinguere MCL1 nel contesto della regolazione proliferativa al checkpoint G1/S.
Il nostro presente studio dimostra l’esistenza di un’interazione diretta proteina-proteina tra la famiglia BCL2, attraverso MCL1, e il checkpoint G1/S CDK4/6-RB1, attraverso P18. Questa interazione è mediata da un motivo rBH3 che si trova in P18. Al di là della specifica interazione di MCL1 e P18, questo convalida la rBH3 come un motivo proteico nativo in grado di mediare le interazioni proteina-proteina con la famiglia BCL2 anti-apoptotico. Inoltre, si dimostra che questa interazione si verifica con affinità biologicamente rilevante in quanto può sopprimere l’associazione del bersaglio nativo MCL1, il motivo BH3 di BAK.
Il risultato di questa interazione diretta proteina-proteina è stato imprevisto. Inizialmente abbiamo ipotizzato che l’associazione P18 con MCL1 si sarebbe comportata in modo pro apoptotico come un segnale di soppressione della crescita parallela. Questo rispecchierebbe studi recenti che hanno trovato altre proteine di risposta allo stress che sono in grado di interagire con le proteine della famiglia BCL2 anti-apoptotico per promuovere direttamente la morte cellulare56. Abbiamo osservato che la sovraespressione della P18 non ha alcun impatto sulla vitalità cellulare. Piuttosto, l’associazione di MCL1 con la P18 induce la degradazione della P18 attraverso un percorso dipendente dalla cisteina-proteasi.
Con l’impatto osservato sull’espressione della proteina P18, abbiamo cercato di determinare come la perdita di P18 potrebbe influenzare la progressione cellulare attraverso il checkpoint G1/S. In genere, P15 e P16 servono come i principali regolatori negativi di CDK4/6, ma nei tumori umani, la sovraespressione di P18 ha dimostrato di compensare la perdita di P15 e / o P1624. Sono stati effettuati studi limitati per caratterizzare l’effetto della sola perdita di P18, in quanto non è comunemente osservato. Abbiamo quindi effettuato studi ad eliminazione diretta che dimostrano che la perdita di P18 è in grado di promuovere la proliferazione cellulare. Osserviamo inoltre che la perdita della proteina P18 indotta dalla sovraespressione MCL1 può imitare questo effetto. Questo ci ha portato a determinare come MCL1 sta influenzando il ciclo cellulare e la proliferazione cellulare. Abbiamo osservato, in accordo con una serie di studi precedenti, che l’aumento dell’espressione di MCL1 porta ad un aumento della popolazione cellulare in G2/M. Mentre gli studi precedenti avevano suggerito che l’upregulation MCL1 induce un blocco G2/M sulla base di questi risultati, abbiamo sospettato che questo fosse piuttosto il risultato di MCL1 che guida la cellula attraverso il checkpoint G1/S e promuove la proliferazione. Abbiamo confermato questa ipotesi utilizzando sia gli studi sulla crescita cellulare che il test di diluizione del colorante CellTrace™ Violet. Questa osservazione sinergica con studi precedenti che hanno evidenziato il ruolo di MCL1 nel regolare l’apoptosi durante il ciclo cellulare, soprattutto durante la mitosi51,53,57–59. I nostri risultati suggeriscono che avere una quantità sufficiente di MCL1 libero, indicativo di uno stato non apoptotico, può promuovere la crescita cellulare e può aiutare ad assicurare che ci sia un MCL1 sufficiente per consentire l’uscita dalla mitosi una volta che il ciclo cellulare è completo. Significativamente, questo studio è la prima dimostrazione che MCL1 può avviare direttamente la proliferazione cellulare.
Mentre ci aspettiamo che l’interazione proteina-proteina tra P18 e MCL1 possa avvenire in qualsiasi linea o tipo di cellula in cui queste due proteine sono presenti, l’effetto sul controllo del ciclo cellulare sarà probabilmente specifico del tipo di cellula. Per esempio, abbiamo osservato un impatto molto più grande sulla popolazione G1 nelle cellule WAC2 a seguito di upregulation MCL1 rispetto a quanto osservato dopo il trattamento con si-P18. Ciò suggerisce che MCL1 può mirare a più proteine di regolazione del ciclo cellulare. Eppure, il trattamento con una piccola molecola che mira la tasca BH3 di MCL1 BH3 ha salvato con successo questo effetto completamente. Questo suggerisce che altre interazioni BH3 o rBH3 possono essere presenti che mediano il controllo MCL1 sul ciclo cellulare. Al contrario, abbiamo osservato che nelle cellule mutanti RB la degradazione mediata MCL1 di P18 si verifica, ma non induce la proliferazione cellulare. In modo simile a BH3 soppressione mimetica BH3 della proliferazione esogena MCL1 indotta, ci aspetteremmo che upregulation di pro-morte BH3 solo proteine che bersaglio MCL1 per sopprimere questo effetto proliferativo. Questo ha probabilmente implicazioni significative nell’impiego di mimetici MCL1 mirati al BH3, sviluppati di recente, che stanno attualmente entrando nella clinica.
Infine, ci aspettiamo che questa comunicazione tra la famiglia BCL2 e il percorso CDK4/6-RB esista al di là del regno del cancro e possa avere un impatto significativo nella normale proliferazione cellulare, nella crescita delle cellule staminali e nella differenziazione. In particolare, è di particolare interesse l’indagine su come questa interazione influisce sulla speciazione delle cellule progenitrici ematopoietiche e neuronali, dove MCL1 è stato precedentemente identificato come un mediatore chiave della differenziazione. In conclusione, abbiamo stabilito un’interazione proteina-proteina che crea un meccanismo di comunicazione diretta per accoppiare la morte cellulare e la proliferazione cellulare senza la necessità di intervenire sull’attivazione del fattore di trascrizione o sulla traduzione delle proteine.
Informazioni supplementari
Didascalie della figura supplementareCifre S1Cifre S2Cifre S3Cifre S4Cifre S5Cifre S6Cifre S7Cifre S8Cifre S9Cifre S10Tabella S1
References
- Reed JC. Dysregulation of apoptosis in cancer. J. Clin. Oncol.. 1999; 17:2941-2953. DOI | PubMed
- Chipuk JE, Moldoveanu T, Llambi F, Parsons MJ, Green DR. The BCL-2 family reunion. Mol. Cell. 2010; 37:299-310. DOI | PubMed
- Delbridge AR, Grabow S, Strasser A, Vaux DL. Thirty years of BCL-2: translating cell death discoveries into novel cancer therapies. Nat. Rev. Cancer. 2016; 16:99-109. DOI | PubMed
- Letai A. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002; 2:183-192. DOI | PubMed
- Kutuk O, Letai A. Alteration of the mitochondrial apoptotic pathway is key to acquired paclitaxel resistance and can be reversed by ABT-737. Cancer Res.. 2008; 68:7985-7994. DOI | PubMed
- Placzek WJ. A survey of the anti-apoptotic Bcl-2 subfamily expression in cancer types provides a platform to predict the efficacy of Bcl-2 antagonists in cancer therapy. Cell Death Dis.. 2010; 1:e40. DOI | PubMed
- Jackson RS. Sabutoclax, a Mcl-1 antagonist, inhibits tumorigenesis in transgenic mouse and human xenograft models of prostate cancer. Neoplasia. 2012; 14:656-665. DOI | PubMed
- Cui J, Placzek WJ. PTBP1 modulation of MCL1 expression regulates cellular apoptosis induced by antitubulin chemotherapeutics. Cell Death Differ.. 2016; 23:1681-1690. DOI | PubMed
- Bates D, Eastman A. Microtubule destabilising agents: far more than just antimitotic anticancer drugs. Br. J. Clin. Pharm.. 2017; 83:255-268. DOI
- Cree IA, Charlton P. Molecular chess? Hallmarks of anti-cancer drug resistance. BMC Cancer. 2017; 17:10. DOI | PubMed
- Whitaker RH, Placzek WJ. Regulating the BCL2 family to improve sensitivity to microtubule targeting agents. Cells. 2019; 8:346. DOI | PubMed
- Wei J. BI-97C1, an optically pure Apogossypol derivative as pan-active inhibitor of antiapoptotic B-cell lymphoma/leukemia-2 (Bcl-2) family proteins. J. Med. Chem.. 2010; 53:4166-4176. DOI | PubMed
- Lieber J. The BH3 mimetic ABT-737 increases treatment efficiency of paclitaxel against hepatoblastoma. BMC Cancer. 2011; 11:362. DOI | PubMed
- Billard C. BH3 mimetics: status of the field and new developments. Mol. Cancer Ther.. 2013; 12:1691-1700. DOI | PubMed
- Placzek WJ. Identification of a novel Mcl-1 protein binding motif. J. Biol. Chem.. 2011; 286:39829-39835. DOI | PubMed
- Walensky LD. Activation of apoptosis in vivo by a hydrocarbon-stapled BH3 helix. Science. 2004; 305:1466-1470. DOI | PubMed
- Venkataramani R, Swaminathan K, Marmorstein R. Crystal structure of the CDK4/6 inhibitory protein p18INK4c provides insights into ankyrin-like repeat structure/function and tumor-derived p16INK4 mutations. Nat. Struct. Biol.. 1998; 5:74-81. DOI | PubMed
- Kohl A. Designed to be stable: crystal structure of a consensus ankyrin repeat protein. Proc. Natl Acad. Sci. U.S.A.. 2003; 100:1700-1705. DOI | PubMed
- Sklenovsky P, Banas P, Otyepka M. Two C-terminal ankyrin repeats form the minimal stable unit of the ankyrin repeat protein p18INK4c. J. Mol. Model.. 2008; 14:747-759. DOI | PubMed
- Lilischkis R, Sarcevic B, Kennedy C, Warlters A, Sutherland RL. Cancer-associated mis-sense and deletion mutations impair p16INK4 CDK inhibitory activity. Int. J. Cancer. 1996; 66:249-254. DOI | PubMed
- Kotschy A. The MCL1 inhibitor S63845 is tolerable and effective in diverse cancer models. Nature. 2016; 538:477-482. DOI | PubMed
- Canepa ET. INK4 proteins, a family of mammalian CDK inhibitors with novel biological functions. IUBMB Life. 2007; 59:419-426. DOI | PubMed
- Grant GD, Cook JG. The temporal regulation of S phase proteins during G1. Adv. Exp. Med. Biol.. 2017; 1042:335-369. DOI | PubMed
- Wiedemeyer R. Feedback circuit among INK4 tumor suppressors constrains human glioblastoma development. Cancer Cell. 2008; 13:355-364. DOI | PubMed
- Dreidax D. Low p14ARF expression in neuroblastoma cells is associated with repressed histone mark status, and enforced expression induces growth arrest and apoptosis. Hum. Mol. Genet.. 2013; 22:1735-1745. DOI | PubMed
- Oltersdorf T. An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature. 2005; 435:677-681. DOI | PubMed
- Teicher BA, Ara G, Herbst R, Palombella VJ, Adams J. The proteasome inhibitor PS-341 in cancer therapy. Clin. Cancer Res.. 1999; 5:2638-2645. PubMed
- Groll M, Berkers CR, Ploegh HL, Ovaa H. Crystal structure of the boronic acid-based proteasome inhibitor bortezomib in complex with the yeast 20S proteasome. Structure. 2006; 14:451-456. DOI | PubMed
- Souers AJ. ABT-199, a potent and selective BCL-2 inhibitor, achieves antitumor activity while sparing platelets. Nat. Med.. 2013; 19:202-208. DOI | PubMed
- Leverson JD. Potent and selective small-molecule MCL-1 inhibitors demonstrate on-target cancer cell killing activity as single agents and in combination with ABT-263 (navitoclax). Cell Death Dis.. 2015; 6:e1590. DOI | PubMed
- Bookstein R. Tumor suppressor genes in prostatic oncogenesis. J. Cell Biochem. Suppl.. 1994; 19:217-223. PubMed
- Kueh HY, Champhekar A, Nutt SL, Elowitz MB, Rothenberg EV. Positive feedback between PU.1 and the cell cycle controls myeloid differentiation. Science. 2013; 341:670-673. DOI | PubMed
- Filby A, Day W, Purewal S, Martinez-Martin N. The analysis of cell cycle, proliferation, and asymmetric cell division by imaging flow cytometry. Methods Mol. Biol.. 2016; 1389:71-95. DOI | PubMed
- Martinez J. Corrigendum: Noncanonical autophagy inhibits the autoinflammatory, lupus-like response to dying cells. Nature. 2016; 539:124. DOI | PubMed
- Reed JC, Cuddy M, Slabiak T, Croce CM, Nowell PC. Oncogenic potential of bcl-2 demonstrated by gene transfer. Nature. 1988; 336:259-261. DOI | PubMed
- Beverly LJ. Regulation of anti-apoptotic BCL2-proteins by non-canonical interactions: the next step forward or two steps back?. J. Cell Biochem.. 2012; 113:3-12. DOI | PubMed
- Moldoveanu T, Follis AV, Kriwacki RW, Green DR. Many players in BCL-2 family affairs. Trends Biochem. Sci.. 2014; 39:101-111. DOI | PubMed
- Gabellini C, Trisciuoglio D, Del Bufalo D. Non-canonical roles of Bcl-2 and Bcl-xL proteins: relevance of BH4 domain. Carcinogenesis. 2017; 38:579-587. DOI | PubMed
- Fujise K, Zhang D, Liu J, Yeh ET. Regulation of apoptosis and cell cycle progression by MCL1. Differential role of proliferating cell nuclear antigen. J. Biol. Chem.. 2000; 275:39458-39465. DOI | PubMed
- Vairo G. Bcl-2 retards cell cycle entry through p27(Kip1), pRB relative p130, and altered E2F regulation. Mol. Cell. Biol.. 2000; 20:4745-4753. DOI | PubMed
- Hershko T, Ginsberg D. Up-regulation of Bcl-2 homology 3 (BH3)-only proteins by E2F1 mediates apoptosis. J. Biol. Chem.. 2004; 279:8627-8634. DOI | PubMed
- Antonucci LA, Egger JV, Krucher NA. Phosphorylation of the retinoblastoma protein (Rb) on serine-807 is required for association with Bax. Cell Cycle. 2014; 13:3611-3617. DOI | PubMed
- Wang P. Phosphorylation of the proapoptotic BH3-only protein bid primes mitochondria for apoptosis during mitotic arrest. Cell Rep.. 2014; 7:661-671. DOI | PubMed
- Sloss O, Topham C, Diez M, Taylor S. Mcl-1 dynamics influence mitotic slippage and death in mitosis. Oncotarget. 2016; 7:5176-5192. DOI | PubMed
- Craig RW. MCL1 provides a window on the role of the BCL2 family in cell proliferation, differentiation and tumorigenesis. Leukemia. 2002; 16:444-454. DOI | PubMed
- Maddika S. Cell survival, cell death and cell cycle pathways are interconnected: implications for cancer therapy. Drug Resist. Update. 2007; 10:13-29. DOI
- Greider C, Chattopadhyay A, Parkhurst C, Yang E. BCL-x(L) and BCL2 delay Myc-induced cell cycle entry through elevation of p27 and inhibition of G1 cyclin-dependent kinases. Oncogene. 2002; 21:7765-7775. DOI | PubMed
- Janumyan YM. Bcl-xL/Bcl-2 coordinately regulates apoptosis, cell cycle arrest and cell cycle entry. EMBO J.. 2003; 22:5459-5470. DOI | PubMed
- Deng X, Gao F, May WS. Bcl2 retards G1/S cell cycle transition by regulating intracellular ROS. Blood. 2003; 102:3179-3185. DOI | PubMed
- Haschka MD. The NOXA-MCL1-BIM axis defines lifespan on extended mitotic arrest. Nat. Commun.. 2015; 6:6891. DOI | PubMed
- Croxton R, Ma Y, Song L, Haura EB, Cress WD. Direct repression of the Mcl-1 promoter by E2F1. Oncogene. 2002; 21:1359-1369. DOI | PubMed
- Eischen CM. Bcl-2 is an apoptotic target suppressed by both c-Myc and E2F-1. Oncogene. 2001; 20:6983-6993. DOI | PubMed
- Hasan SM. Mcl1 regulates the terminal mitosis of neural precursor cells in the mammalian brain through p27Kip1. Development. 2013; 140:3118-3127. DOI | PubMed
- Maga G, Hubscher U. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J. Cell Sci.. 2003; 116:3051-3060. DOI | PubMed
- Jamil S. A proteolytic fragment of Mcl-1 exhibits nuclear localization and regulates cell growth by interaction with Cdk1. Biochem. J.. 2005; 387:659-667. DOI | PubMed
- Pihan P, Carreras-Sureda A, Hetz C. BCL-2 family: integrating stress responses at the ER to control cell demise. Cell Death Differ.. 2017; 24:1478-1487. DOI | PubMed
- Pawlikowska P. ATM-dependent expression of IEX-1 controls nuclear accumulation of Mcl-1 and the DNA damage response. Cell Death Differ.. 2010; 17:1739-1750. DOI | PubMed
- Zhou T. Downregulation of Mcl-1 through inhibition of translation contributes to benzyl isothiocyanate-induced cell cycle arrest and apoptosis in human leukemia cells. Cell Death Dis.. 2013; 4:e515. DOI | PubMed
- Wang S. Revisiting the role of MCL1 in tumorigenesis of solid cancer: gene expression correlates with antiproliferative phenotype in breast cancer cells and its functional regulatory variants are associated with reduced cancer susceptibility. Tumour Biol.. 2014; 35:8289-8299. DOI | PubMed
Fonte
Whitaker RH, Placzek WJ (2020) MCL1 binding to the reverse BH3 motif of P18INK4C couples cell survival to cell proliferation. Cell Death & Disease 11(2): 156. https://doi.org/10.1038/s41419-020-2351-1




