
Abstract
Introduzione
Trecento anni fa, il semplice microscopio di Anton van Leeuwenhoek rivelò che le “animacole” cambiavano forma mentre si muovevano attraverso una goccia d’acqua (van Leewenhoeck,1677). Successive scoperte nella tecnologia del microscopio e nella biologia molecolare hanno rafforzato il legame tra morfologia cellulare e locomozione, e ora sappiamo che le cellule striscianti di molti phyla eucarioti creano una varietà di sporgenze dinamiche che proiettano in avanti, in direzione della migrazione.
Le sporgenze sottili e piatte prodotte da cellule fortemente aderenti sono generalmente chiamate lamellipodia. Precedenti lavori, principalmente su fibroblasti e cellule epiteliali, hanno stabilito che il movimento delle lamellipodia all’avanguardia è guidato dalla crescita di reti di actina ramificate nucleate e reticolate dal complesso Arp2/3. Man mano che avanzano, queste lamellipodia promuovono la creazione di nuove aderenze focali che forniscono trazione e sostengono il movimento continuo(Case and Waterman, 2015). Questa modalità di migrazione cellulare sembra essere limitata al lignaggio animale, dove si basa sulla presenza di specifici ligandi nell’ambiente extracellulare e gioca ruoli essenziali nello sviluppo embrionale, nella guarigione delle ferite e nell’omeostasi dei tessuti.
Una forma più ampiamente dispersa di cellule striscianti, probabilmente presente nell’ultimo antenato comune di tutti gli eucarioti (Fritz-Laylinet al., 2017), richiede solo una debole interazione non specifica con l’ambiente extracellulare e può raggiungere velocità di diversi ordini di grandezza più veloci rispetto al movimento basato sull’adesione (Loomiset al., 2012; Prestone King, 1978; Lämmermann et al., 2008; Buenemann et al., 2010; Butler et al., 2010; Loomis et al., 2012; Petrie e Yamada, 2015; Fritz-Laylin et al., 2017 ). Questa modalità di migrazione cellulare è associata alla formazione di pseudopodi complessi, tridimensionali, pieni di reti ramificate di filamenti di actina, nucleati e organizzati dal complesso Arp2/3. Questi pseudopodi tridimensionali possono contribuire al movimento in avanti delle cellule che strisciano e nuotano(Van Haastert, 2011), ma lavori recenti hanno rivelato che la formazione di pseudopodi perturbatori può aumentare la velocità e la persistenza della migrazione(Leithner et al., 2016; Vargas et al., 2016), il che implica che gli pseudopodi complessi potrebbero essere più importanti per esplorare l’ambiente esterno che per guidare il movimento in avanti.
Sappiamo meno della morfologia e della funzione degli pseudopodi complessi che dei lamellipod aderenti, in parte a causa dei limiti tecnici della microscopia a cellule vive. La microscopia a fluorescenza a riflessione interna confocale e totale (TIRF), ad esempio, fornisce una visione adeguata delle lamellipodia aderenti perché queste strutture lo sono: (i) lente, (ii) sottili, e (iii) strettamente aderenti al coprioggetto del microscopio. Al contrario, gli pseudopodi complessi creati da cellule in rapido movimento: (i) crescono rapidamente (10-100 um/min), (ii) adottano forme tridimensionali complesse, e (iii) spesso passano la loro vita lontano dalla superficie del coprioggetti. Inoltre, il photobleaching deve essere ridotto al minimo per osservare le cellule viventi per un tempo sufficiente a tracciare la loro complessa migrazione tridimensionale.
Anche se alcuni lavori hanno suggerito che i lamellipodici bidimensionali di cellule aderenti riflettono la planarità della superficie a cui sono attaccati(Burnette et al., 2014), non sono stati proposti meccanismi per spiegare le morfologie più complesse degli pseudopodi tridimensionali creati da cellule veloci e debolmente aderenti, soprattutto quando si strisciano in ambienti irregolari. Inoltre, non è chiaro se questi complessi pseudopodi siano completamente amorfi o se condividano caratteristiche strutturali comuni che potrebbero far luce sulle loro funzioni e sui meccanismi di assemblaggio.
Per affrontare questi problemi, abbiamo utilizzato il microscopio a foglio luminoso a reticolo recentemente sviluppato(Chen et al., 2014), che offre una combinazione unica di immagini tridimensionali ad alta risoluzione spaziale e temporale e bassa fototossicità. Per rendere e interpretare i grandi set di dati prodotti dalla microscopia a foglio luminoso a reticolo, abbiamo sviluppato un nuovo software di visualizzazione e lo abbiamo combinato con sonde molecolari biochimiche per rilevare e caratterizzare gli pseudopodi nelle cellule viventi.
Utilizzando questi nuovi strumenti, abbiamo scoperto che gli pseudopodi creati dai neutrofili in rapido movimento non sono completamente amorfi, ma rappresentano varie disposizioni di un unico motivo strutturale: un foglio lamellare autoportante. Così, gli pseudopodi complessi e tridimensionali che appaiono amorfi in microscopia a largo campo e confocale si rivelano essere rosette formate da lamelle multiple e interfogliate. A differenza dei lamellipodici aderenti, questi blocchi lamellari autoportanti non sono modelli da una superficie piana e la loro morfologia suggerisce fortemente che essi derivano da una disposizione lineare delle molecole di regolazione associate alla membrana plasmatica. Inoltre, i nostri metodi automatizzati per rilevare e quantificare le dimensioni di pseudopodi complessi rivelano che queste strutture cellulari dinamiche giocano un ruolo chiave nel pathfinding cellulare.
Risultati
La resa superficiale dei dati del foglio luminoso del reticolo rivela la dinamica tridimensionale delle membrane e delle reti di actina nei neutrofili in migrazione
Neutrofili come HL-60 cellule costruire pseudopodi che sono pieni di actina polimerizzata, facilmente visualizzabili in cellule fisse con falloidina(Figura 2-figure supplemento 1, Figura 3-figure supplemento 1). Per visualizzare la dinamica degli pseudopodi in cellule in migrazione attiva, abbiamo costruito HL-60 linee cellulari che esprimono stabilmente marcatori fluorescenti per i filamenti di actina e la membrana plasmatica(Figura 1). Nessuna sonda di actina si lega esclusivamente alla dinamica, reti di actina ramificata dello pseudopode, tra cui la sonda ampiamente utilizzato, Lifeact(Figura 1-figure supplement 1). Pertanto, abbiamo scelto la sonda di actina basata sull’utrofina, che si lega preferibilmente all’actina corticale che circonda il corpo cellulare ed è in gran parte esclusa dalle reti di actina più dinamiche e ramificate che guidano l’estensione degli pseudopodi(Belin et al., 2014). Quando combinato con un marcatore a membrana, la selettività della nostra sonda di actina basata sull’utrofina ci ha permesso di sviluppare metodi automatizzati per identificare e tracciare gli pseudopodi tridimensionali nelle cellule in movimento (vedi sotto). Questo approccio di ‘spazio negativo’ per identificare gli pseudopodi evita anche i problemi associati alla velocità con cui le molecole della sonda si diffondono attraverso le reti di actina densa (Belin etal., 2014).
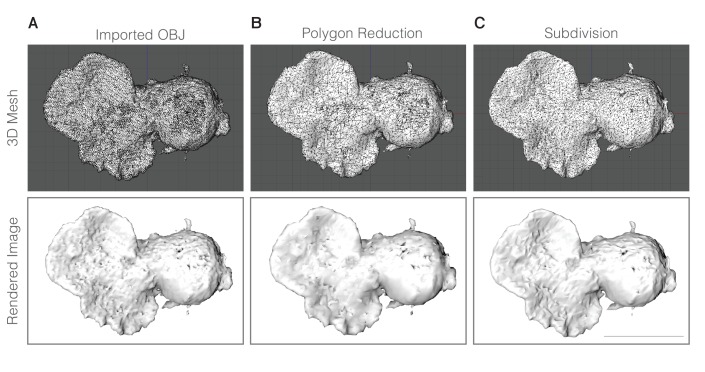
Figura 1-figure supplemento 3.Figura 1—supplemento di figura 3. Elaborazione dei dati e visualizzazione.A Lifeact-based sonda fluorescente basata su Lifeact-non etichetta actina filamentosa in HL-60 pseudopodi.confronto delle viste isosuperficie normale ai piani di imaging XY, YZ, e XZ, illustrando la risoluzione quasi isotropa di microscopia a foglio di luce reticolare.Mesh elaborazione per le figure, che mostra sia i conteggi poligonali (o ‘mesh tridimensionale’) e le immagini renderizzate.(A) Pipeline di elaborazione dati. A sinistra: esempio rappresentativo di dati grezzi LLSm che mostra un singolo piano di immagine che passa attraverso una cella di tipo neutrofilo HL-60 (pannello inferiore) migrando attraverso una rete di collagene (pannello superiore). La risoluzione è ~ 230 in X e Y, ~ 370 in Z. Il collagene è direttamente etichettato con fluoresceina e il citoscheletro di actina della cellula è evidenziato utilizzando una sonda a base di utrofene (vedi sotto). Medio: i dati grezzi sono deconvolti in una coppia di pile di immagini verticali a un colore (qui mostrato unito). A destra: gli stack di immagini verticali sono poi resi come isosuperfici tridimensionali in UCSF Chimera.(B) Rappresentante fluorescenza iso-superfici immagini rappresentative di una cella HL-60 che esprime marcatori per la membrana plasmatica (in alto) e citoscheletro di actina (al centro) e strisciando su un vetrino coprioggetti rivestito in fibronectina. In alto: fluorescenza della membrana plasmatica palmitoilata m-Emerald. Medio: fluorescenza dei filamenti di actina corticale nel corpo cellulare etichettato da Utrophin-mCherry. In basso: falso colore, sovrapposizione tridimensionale delle immagini membrana-mEmerald e Utrophin-mCherry.(C) Rappresentante fluorescenza iso-superficie immagini rappresentative di HL-60 cellule che migrano attraverso non etichettati (colonna di sinistra) e fluorescenti etichettati (colonna di destra) reti di collagene tridimensionale. Barre di scala = 10 µm. Gli assi indicano l’orientamento relativo attraverso le viste ruotate. Per le celle che strisciano su una superficie 2D, l’orientamento del coprioggetti è indicato da ombreggiature grigie. Se non diversamente specificato, le cellule sono state illuminate con luce 488 e 560 nm a 37C in 1 × HBSS integrato con 3% FBS, 1 × penna / stilo, e 40 nM del tripeptide formil-MLP (per la migrazione di stimolazione).In alto: UCSF Chimera rendering di una fluorescenza isosuperficie di una sonda a membrana al plasma (palmitoylated m-Emerald) espresso in una cella mobile HL-60. Medio: una resa superficiale simile di mCherry-Lifeact fluorescenza nella stessa cella. Questo costrutto di fusione Lifeact è identico alla fusione mCherry-Utrophin utilizzata in tutto questo lavoro, ad eccezione della sostituzione del peptide Lifeact F-act(Riedl et al., 2008) per il dominio Utrophin CH-ch. In basso: fusione tridimensionale della membrana-mEmerald e della fluorescenza mCherry-Lifeact. La sovrapposizione mostra che, analogamente alla sonda Utrophin, Lifeact non si accumula negli pseudopodi lamellari. Assi come indicato in Figura 1. Barra di scala = 10 µm.Pannello superiore: resa superficiale tridimensionale della fluorescenza del collagene coniugato con fluoresceina, orientato in modo che l’angolo di visualizzazione è normale al piano dell’immagine del microscopio (XY), e parallelo all’asse ottico del microscopio (asse Z). In basso a sinistra del pannello: la stessa matrice di collagene etichettata come sopra, ma con l’angolo di visualizzazione ruotato di 90 gradi in modo che l’asse ottico sia diventato l’asse verticale del rendering. Pannello in basso a destra: la stessa matrice di collagene degli altri due pannelli ma con l’angolo di visualizzazione ruotato di 90 gradi all’interno del piano dell’immagine del microscopio. La pila tridimensionale è stata resa in Cinema4D, con una vista superiore (X,Y), anteriore (X,Z) e laterale (Y,Z). I numeri sugli assi rappresentano le dimensioni dell’immagine in pixel. Barra di scala = 10 µm.(A) File Wavefront iniziale esportato da UCSF Chimera.(B) Risultati dell’applicazione dello strumento di riduzione del poligono in Cinema4D. (C) Versione finale che utilizza Polygon Subdivison, un metodo per calcolare una superficie levigata da una mesh poligonale ridotta. Barra di scala = 10 µm.
Abbiamo usato la microscopia a fogli di luce reticolare per creare sequenze di time-lapse ad alta risoluzione (230 × 230×370 nm [Chen et al., 2014]), quasi isotrope, immagini tridimensionali di cellule HL-60 che strisciano attraverso i coprioggetti rivestiti di fibronectina o che si muovono attraverso reti casuali di fibre di collagene (Figura 1e Figura 1-figure supplement 2). Manipolando i dati grezzi, ci siamo presto resi conto che la capacità della microscopia a foglio luminoso a reticolo di fornire nuove intuizioni sulla biologia cellulare è ostacolata, in parte, dalla mancanza di strumenti di calcolo per la visualizzazione e l’analisi dei dati tridimensionali di intensità di fluorescenza. Gli strumenti più utilizzati per l’analisi dei dati microscopici possono eseguire operazioni di base come la trebbiatura, la normalizzazione, l’allineamento e la correzione della deriva, ma questi strumenti sono stati progettati per gestire immagini bidimensionali. Invece di tentare di estendere le capacità di una piattaforma di calcolo principalmente bidimensionale, abbiamo invece adattato un software progettato fin dall’inizio per manipolare e analizzare set di dati tridimensionali. In particolare, abbiamo scelto il programma di visualizzazione molecolare UCSF Chimera(Pettersen et al., 2004) per questo scopo perché è liberamente disponibile e incorpora l’esperienza collettiva della comunità di biologia strutturale nel rendere e manipolare dati tridimensionali.
In breve, abbiamo aggiunto a UCSF Chimera la capacità di importare una serie temporale di serie di dati tridimensionali sull’intensità della fluorescenza. Una volta importati, questi dati possono essere resi e manipolati dalle funzioni native di UCSF Chimera, inclusi gli elementi del toolkit ‘vseries’, che eseguono versioni volumetriche di trebbiatura, normalizzazione, allineamento e adattamento. Inoltre, la capacità di UCSF Chimera di rendere superfici di uguale intensità di fluorescenza aumenta notevolmente la capacità di giudicare la prossimità e/o la co-localizzazione delle sonde fluorescenti in tre dimensioni. Infine, ispirandoci agli strumenti di rendering disponibili nei software di animazione professionale (ad esempio Cinema4D; www.maxon.net), abbiamo aggiunto una tecnica di ombreggiatura tonale, chiamata ambient occlusion(Tarini et al., 2006), a una recente release di UCSF Chimera(http://www.rbvi.ucsf.edu/chimera/data/ambient-jul2014/ambient.html). La resa delle superfici tonali con l’ambient occlusion rende le strutture cellulari più facilmente interpretabili, enfatizzando le relazioni spaziali delle superfici tridimensionali e le variazioni di texture delle superfici. La maggiore capacità di rilevare e interpretare le texture e le sporgenze può essere vista confrontando la proiezione tradizionale di massima intensità e le versioni renderizzate dei dati confocali(Figura 3-figure supplement 1) e rendering simili dei dati del foglio di luce reticolare (Figura 3-figure supplement 2).
Figura 1-figure supplement 3.Figura 1—supplemento alla figura 3. Elaborazione e visualizzazione dei dati.una sonda fluorescente Lifeact-based non etichetta l’actina filamentosa in HL-60 pseudopods.Comparazione delle viste isosuperficiali normali con i piani di imaging XY, YZ, e XZ, illustrando la risoluzione quasi isotropa della microscopia dei fogli di luce del reticolo.elaborazione della mesh per le figure, mostrando sia i conteggi dei poligoni (o ‘mesh tridimensionale’) che le immagini renderizzate.(A) Pipeline di elaborazione dati. A sinistra: esempio rappresentativo di dati grezzi LLSm che mostrano un singolo piano di immagine che passa attraverso una cella di tipo neutrofilo HL-60 (pannello inferiore) migrando attraverso una rete di collagene (pannello superiore). La risoluzione è ~ 230 in X e Y, ~ 370 in Z. Il collagene è direttamente etichettato con fluoresceina e il citoscheletro di actina della cellula è evidenziato utilizzando una sonda a base di utrofene (vedi sotto). Medio: i dati grezzi sono deconvolti in una coppia di pile di immagini verticali a un colore (qui mostrato unito). A destra: gli stack di immagini verticali sono poi resi come isosuperfici tridimensionali in UCSF Chimera.(B) Rappresentante fluorescenza iso-superfici immagini rappresentative di una cella HL-60 che esprime marcatori per la membrana plasmatica (in alto) e citoscheletro di actina (al centro) e strisciando su un vetrino coprioggetti rivestito in fibronectina. In alto: fluorescenza della membrana plasmatica palmitoilata m-Emerald. Medio: fluorescenza dei filamenti di actina corticale nel corpo cellulare etichettato da Utrophin-mCherry. In basso: falso colore, sovrapposizione tridimensionale delle immagini membrana-mEmerald e Utrophin-mCherry.(C) Rappresentante fluorescenza iso-superficie immagini rappresentative di HL-60 cellule che migrano attraverso non etichettati (colonna di sinistra) e fluorescenti etichettati (colonna di destra) reti di collagene tridimensionale. Barre di scala = 10 µm. Gli assi indicano l’orientamento relativo attraverso le viste ruotate. Per le celle che strisciano su una superficie 2D, l’orientamento del coprioggetti è indicato da ombreggiature grigie. Se non diversamente specificato, le cellule sono state illuminate con luce 488 e 560 nm a 37C in 1 × HBSS integrato con 3% FBS, 1 × penna / stilo, e 40 nM del tripeptide formil-MLP (per la migrazione di stimolazione).In alto: UCSF Chimera rendering di una fluorescenza isosuperficie di una sonda a membrana al plasma (palmitoylated m-Emerald) espresso in una cella mobile HL-60. Medio: una resa superficiale simile di mCherry-Lifeact fluorescenza nella stessa cella. Questo costrutto di fusione Lifeact è identico alla fusione mCherry-Utrophin utilizzata in tutto questo lavoro, ad eccezione della sostituzione del peptide Lifeact F-act(Riedl et al., 2008) per il dominio Utrophin CH-ch. In basso: fusione tridimensionale della membrana-mEmerald e della fluorescenza mCherry-Lifeact. La sovrapposizione mostra che, analogamente alla sonda Utrophin, Lifeact non si accumula negli pseudopodi lamellari. Assi come indicato in Figura 1. Barra di scala = 10 µm.Pannello superiore: resa superficiale tridimensionale della fluorescenza del collagene coniugato con fluoresceina, orientato in modo che l’angolo di visualizzazione è normale al piano dell’immagine del microscopio (XY), e parallelo all’asse ottico del microscopio (asse Z). In basso a sinistra del pannello: la stessa matrice di collagene etichettata come sopra, ma con l’angolo di visualizzazione ruotato di 90 gradi in modo che l’asse ottico sia diventato l’asse verticale del rendering. Pannello in basso a destra: la stessa matrice di collagene degli altri due pannelli ma con l’angolo di visualizzazione ruotato di 90 gradi all’interno del piano dell’immagine del microscopio. La pila tridimensionale è stata resa in Cinema4D, con una vista superiore (X,Y), anteriore (X,Z) e laterale (Y,Z). I numeri sugli assi rappresentano le dimensioni dell’immagine in pixel. Barra di scala = 10 µm.(A) File Wavefront iniziale esportato da UCSF Chimera.(B) Risultati dell’applicazione dello strumento di riduzione del poligono in Cinema4D. (C) Versione finale che utilizza Polygon Subdivison, un metodo per calcolare una superficie levigata da una mesh poligonale ridotta. Barra di scala = 10 µm.

Figura 1-figure supplement 1.Una sonda fluorescente Lifeact-based non etichetta l’actina filamentosa negli pseudopodi HL-60.In alto: UCSF Chimera rendering di una fluorescenza isosuperficie di una sonda a membrana al plasma (palmitoylated m-Emerald) espresso in una cella mobile HL-60 HL-60. Medio: una resa superficiale simile di mCherry-Lifeact fluorescenza nella stessa cella. Questo costrutto di fusione Lifeact è identico alla fusione mCherry-Utrophin utilizzata in tutto questo lavoro, ad eccezione della sostituzione del peptide Lifeact F-act(Riedl et al., 2008) per il dominio Utrophin CH-ch. In basso: fusione tridimensionale della membrana-mEmerald e della fluorescenza mCherry-Lifeact. La sovrapposizione mostra che, analogamente alla sonda Utrophin, Lifeact non si accumula negli pseudopodi lamellari. Assi come indicato in Figura 1. Barra di scala = 10 µm.

Figura 1-figure supplement 2.Figura 1. Confronto delle viste isosuperficiali normali ai piani di imaging XY, YZ, e XZ, che illustra la risoluzione quasi isotropa della microscopia a foglio di luce reticolare.Pannello superiore: resa superficiale tridimensionale della fluorescenza del collagene coniugato con fluoresceina, orientato in modo che l’angolo di visualizzazione è normale al piano di immagine del microscopio (XY), e parallelo all’asse ottico del microscopio (asse Z). In basso a sinistra del pannello: la stessa matrice di collagene etichettata come sopra, ma con l’angolo di visualizzazione ruotato di 90 gradi in modo che l’asse ottico sia diventato l’asse verticale del rendering. Pannello in basso a destra: la stessa matrice di collagene degli altri due pannelli ma con l’angolo di visualizzazione ruotato di 90 gradi all’interno del piano dell’immagine del microscopio. La pila tridimensionale è stata resa in Cinema4D, con una vista superiore (X,Y), anteriore (X,Z) e laterale (Y,Z). I numeri sugli assi rappresentano le dimensioni dell’immagine in pixel. Barra di scala = 10 µm.
Figura 1-figure supplement 3.Elaborazione della mesh per le figure, che mostra sia il conteggio dei poligoni (o ‘mesh tridimensionale’) che le immagini renderizzate.(A) File Wavefront iniziale esportato da UCSF Chimera.(B) Risultati dell’applicazione dello strumento di riduzione dei poligoni in Cinema4D. (C) Versione finale che utilizza Polygon Subdivison, un metodo per calcolare una superficie levigata da una mesh poligonale ridotta. Barra di scala = 10 µm.
Gli pseudopodi dinamici sono composti da elementi intrinsecamente lamellari
Utilizzando questi strumenti di visualizzazione, abbiamo osservato due tipi distintivi di sporgenza dinamica della membrana sul bordo anteriore delle cellule HL-60 che strisciano su coprioggetti rivestiti in fibronectina: (i) lamina sottile, simile a un foglio (Figura 2 e Video 1- 6), e (ii) “rosette” formate dalla coincidenza di più lamine (Figura 3,Video 7- 10). La morfologia di queste sporgenze si differenzia dai blebs sferici prodotti quando la membrana plasmatica si stacca dalla corteccia sottostante(Charras e Paluch, 2008), invece assomigliano a movimenti di membrana guidati da un rapido assemblaggio di actina filamentosa. Utilizzando la microscopia confocale di cellule fisse etichettate con falloidina, abbiamo verificato che i fogli di bordo e le rosette sono pieni di fitte reti di actina(Figura 2-figure supplemento 1 e Figura 3-figure supplemento 1). I fogli e le rosette dinamiche sono facilmente visibili nei rendering di superficie dei dati di microscopia tridimensionale a reticolo di luce dei fogli, ma non sono facilmente identificabili in proiezioni bidimensionali(Figura 3-figure supplements 1 e 2), coerente con il fallimento della microscopia ottica convenzionale per identificare queste strutture. Utilizzando la microscopia elettronica a scansione, abbiamo osservato simili sporgenze lamellari in cellule fisse Jurkat T, un altro tipo di cellule ameboidi che impiega veloce, a bassa adesione strisciando attraverso ambienti complessi(Figura 3-figure supplement 3).

Video 1.Esempio di pseudopodi lamellari formati da cellule che strisciano su una superficie piana (coprioggetti in vetro rivestito di fibronectina). Il video gioca a 10,5 × in tempo reale.
Video 2.Un altro esempio di pseudopodi lamellari formati da cellule che strisciano su una superficie piana (coprivetrino in vetro rivestito di fibronectina).Il video viene riprodotto a 11 × tempo reale. Vedi anche Video 1.
Video 3.Esempio in cui i fogli non vengono mai a contatto con la superficie.Poiché il campione è tenuto verticalmente nella camera di imaging, i bordi delle celle che cadono dal coprioggetti possono passare attraverso il campo visivo, visto qui come oggetti che fluiscono da destra a sinistra. Il video viene riprodotto a 9,8 × tempo reale.
Video 4.Un altro esempio in cui i fogli non vengono mai a contatto con la superficie.Il video viene riprodotto a 11 × tempo reale. Vedi anche Video 3.
Video 5.Esempio di pseudopodi lamellari formati da cellule che strisciano attraverso reti di collagene tridimensionali. Il video viene riprodotto a 9,5 × tempo reale.
Video 6.Un altro esempio di pseudopodi lamellari formati da cellule che strisciano attraverso reti di collagene tridimensionali.Si prega di notare che lo stadio viene riposizionato più volte perché la cellula migra fuori dal campo visivo. Il video gioca a 10 × tempo reale. Vedi anche Video 5.
Video 7.Esempio di pseudopodi a ‘rosetta’ costruiti da cellule che strisciano su una superficie piana (coprivetrini in vetro rivestito di fibronectina). Il video 7 suonaa 11 × tempo reale.
Video 8.Un altro esempio di pseudopodi a ‘rosetta’ costruiti da cellule che strisciano su superficie piana (coprivetrini in vetro rivestito di fibronectina).Il video viene riprodotto a 11 × tempo reale. Vedi anche Video 7.
Video 9.Esempio di pseudopodi a ‘coccarda’ costruiti da cellule che strisciano attraverso reti di collagene polimerizzato.Il video gioca a 11 × tempo reale.
Video 10.Un altro esempio di pseudopodi a ‘coccarda’ costruiti da cellule che strisciano attraverso reti di collagene polimerizzato.Il video gioca a 10 × tempo reale. Vedi anche Video 9.
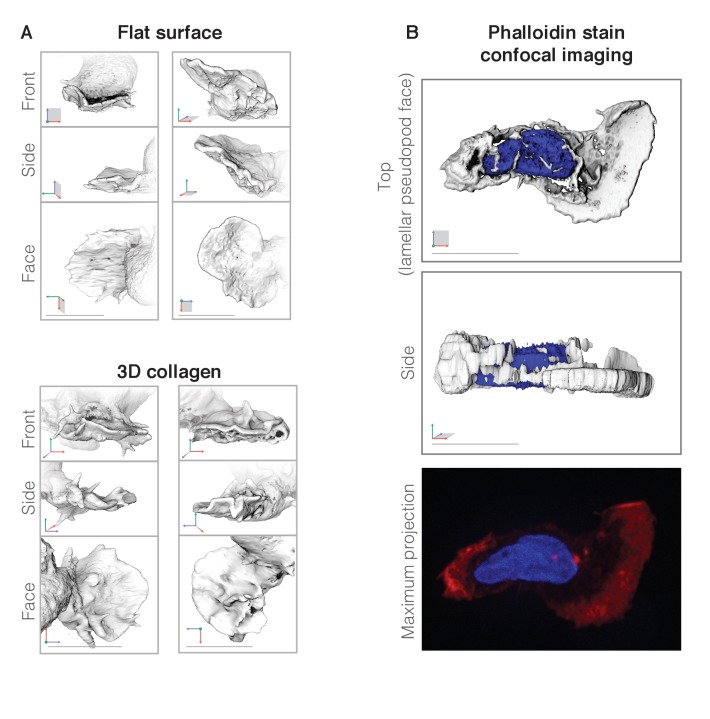
Figura 2-figure supplemento 1.I neutrofili formano pseudopodi lamellari, indipendentemente dal fatto che siano striscianti su superfici bidimensionali o in movimento attraverso complessi ambienti tridimensionali.i neutrofili formano pseudopodi lamellari.(A-B) Per illustrare la morfologia delle sporgenze di membrana in rapida crescita abbiamo sovrapposto più immagini iso-superficiali di fluorescenza multiple prese in diversi punti di tempo di singoli HL-60 cellule che esprimono una sonda a membrana (palmitoylated m-Emerald). I rendering delle singole superfici sono colorati in base al tempo. Gli inserti mostrano ogni cella nel punto temporale finale.(A) HL-60 cellule HL-60 strisciando attraverso un vetrino coprioggetti rivestito di fibronectina.(B) HL-60 cella HL-60 strisciando attraverso una rete di collagene polimerizzato.(C) rendering tridimensionale superficie di due pseudopodi di due cellule diverse, uno strisciando su vetro rivestito di fibronectina (a sinistra) e uno che si muove attraverso una matrice di fibre di collagene (a destra). Ogni set di dati LLSm è reso da tre diversi angoli di visualizzazione: en faccia (in alto), dal lato (al centro) e dall’alto (in basso). Per ulteriori esempi, vedere la Figura 2-figure supplement 1.(D) Box and whisker plot (con mediana, quartili e range) di errore verticale (stima della planarità) per ogni pseudopode, calcolato misurando la distanza media euclidea tra i punti originali sul bordo d’attacco e un piano di riferimento con una larghezza pari alla larghezza media misurata degli pseudopodi lamellari (banda grigia). I primi tre sono misure da cellule che strisciano su una superficie piana (marrone), e i tre successivi da cellule che strisciano attraverso una rete di collagene polimerizzato (blu).(E) Esempio di uno pseudopodo lamellare che mantiene uno spessore simile (cerchi verdi) durante l’estensione e la retrazione, come indicato dalla distanza dalla punta dello pseudopodo al corpo della cellula (quadrati neri). Assi come indicato nella Figura 1. Tutte le barre della scala = 10 µm.(A) Ulteriori visualizzazioni tridimensionali di singoli punti di tempo da quattro cellule indipendenti (due su vetro e due in matrice) che formano pseudopodi lamellari mostrate in faccia, dal lato, e dall’alto.(B) Spinning disco immagine confocale disco di una cella fissa con uno pseudopode lamellare. La pila tridimensionale risultante è stata resa in UCSF Chimera, con una vista superiore (X,Y) e laterale (Z,Y) vista mostrata. Il pannello inferiore è una proiezione di massima intensità di colorazione di falloidina (rosso) e DNA colorato con DAPI (blu), rivelando che gli pseudopodi planari sono pieni di actina polimerizzata. Assi come indicato in Figura 1. Barre di scala = 10 um.
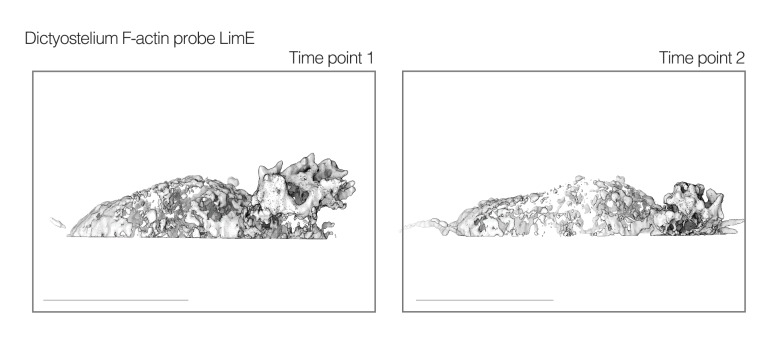
Figura 3-figure supplement 5.Figura 3. Neutrofili costruire complessi pseudopodi (‘rosette’) formato da fogli lamellari multipli.immagine disco di filatura confocale disco di cella fissa con una complessa rosetta pseudopod.proiezione di massima intensità di intensità di luce deconvolto reticolo foglio luce dati microscopia (a sinistra) rispetto alla resa superficiale (a destra).micrografie a scansione elettronica di cellule Jurkat T su vetrini coprioggetti.Box e baffi trama della vita di vita di semplice (a sinistra) e rosetta (a destra) pseudopodi costruito da cellule strisciando attraverso le superfici di vetro rivestito (blu) e attraverso le maglie di collagene (marrone).UCSF Chimera visualizzazione di due punti di tempo di due punti di dati pubblicati Dictyostelium cella cella pubblicato ripreso con microscopia a fascio Bessel(Gao et al., 2014) che mostra le sporgenze a rosetta.(A) Visualizzazioni tridimensionali di rosette costruite da cellule che strisciano su superfici bidimensionali (a sinistra) così come attraverso maglie di collagene senza etichetta (a destra). Singoli punti temporali di due celle indipendenti da ogni condizione sono mostrati di lato (pannelli superiori) e in faccia (pannelli inferiori).(B) Box e trama baffo che mostra che semplici pseudopodi lamellari hanno uno spessore simile a quello dei singoli petali di pseudopodi a rosetta complessi. (n = 18 pseudopodi, da 6 cellule di due repliche biologiche).(C) Viste ingrandite di tre punti temporali di una porzione di una singola cellula (intera cellula mostrata di seguito) che mostra le fasi iniziali della formazione di pseudopodi a rosetta visti in faccia (in alto) e dal lato (in basso). Si noti che, in questo caso, il nuovo pseudopodo emerge dal retro della cella, di fronte ad uno pseudopodo formatosi in precedenza.(D) Due punti temporali che mostrano la dissoluzione di uno pseudopode separato mostrato in faccia (in alto) e di lato (in basso). Per(A) e(C): Assi come indicato in Figura 1. Barre di scala = 10 µm.La pila tridimensionale risultante è stata resa in UCSF Chimera, con una vista in alto (X,Y) e una vista laterale (Z,Y). (In basso) La proiezione di massima intensità della colorazione di falloidina (rosso) rivela che gli pseudopodi a rosetta sono pieni di actina polimerizzata, con DNA colorato con DAPI (blu). Barra di scala = 10 µm. L’angolo in basso a destra della pila di dati è stato rimosso perché una seconda cella ha interferito con la visualizzazione tridimensionale dei dati. Assi come indicato in Figura 1.Questo particolare punto temporale è stato scelto per evidenziare la necessità di strumenti di visualizzazione appropriati per determinare le relazioni spaziali tra le strutture che sporgono dalla superficie della cella.Protrusioni planari su questi linfociti ricordano quelli che osserviamo nelle cellule HL-60. Barra di scala = 2 µm per il pannello in alto a destra e 5 µm per gli altri pannelli.Nove cellule sono state analizzate da tre esperimenti indipendenti.La cellula sta esprimendo il Ditostelio F-actina sonda LimE(Gao et al., 2014). Barra di scala = 10 µm.
Gli pseudopodi lamellari non erano limitati al piano del coprivetrino e, nei film tridimensionali, abbiamo osservato molti fogli pseudopodiali viaggiare attraverso la superficie dorsale della cellula, da davanti a dietro(Figura 2A e Video 1) o da lato a lato(Video 2). È importante notare che la maggior parte dei fogli pseudopodiali è emersa dal corpo della cellula e proiettata direttamente nel mezzo liquido senza mai toccare la superficie del coprioggetti(Video 3 e 4 e la sovrapposizione del tempo nella Figura 2A). Nonostante non siano supportati, questi pseudopodi hanno mantenuto una morfologia lamellare con uno spessore approssimativamente costante per tutta la loro vita(Figura 2E).
Per verificare che la caratteristica forma lamellare dei fogli pseudopodiali non richieda l’interazione con una superficie piana, abbiamo anche immaginato cellule HL-60 con membrana etichettata HL-60 che strisciano attraverso reti di collagene casuali(Figura 2B). Indipendentemente dal fatto che le cellule strisciano attraverso il vetro o attraverso una matrice di collagene, i loro pseudopodi lamellari condividono diverse caratteristiche comuni, tra cui un profilo simile(Figura 2C) e lo spessore (780 ± 137 nm deviazione standard, n = 18 cellule). Lo spessore misurato dai dati LLSm riferiti alla superficie concorda con le misurazioni effettuate su altri tipi di dati, tra cui: dati grezzi del foglio luminoso del reticolo (567 ± 64 nm su vetro e 689 ± 86 nm in collagene); immagini confocali su disco rotante dell’actina macchiata di falloidina (505± 49 nm per un foglio e 533 ± 123 nm per un petalo di rosetta); e micrografie elettroniche a scansione, che suggeriscono uno spessore di ~640 nm (Fleck et al., 2005). Data la risoluzione della microscopia a lamelle (~230 nm in XY[Chen et al., 2014]), si stima che lo spessore effettivo degli pseudopodi lamellari sia compreso tra 430 e 800 nm.
Gli pseudopodi lamellari non sono perfettamente piatti e presentano vari gradi di pucker e ondulazione. Per determinare se la planarità dei fogli lamellari è influenzata dalle interazioni della superficie cellulare, abbiamo prima quantificato la loro planarità: (i) scegliendo i punti distribuiti lungo il bordo d’attacco di ogni pseudopodo, (ii) utilizzandoli per definire un piano di riferimento che si adatti al meglio, e (iii) calcolando la distanza minima media euclidea, o l’errore verticale medio, tra i punti originali sul bordo d’attacco e il piano di riferimento. Le celle che strisciano attraverso i coprioggetti, così come quelle che strisciano attraverso le reti di collagene, costruiscono fogli con una planarità simile(Figura 2D), sollevando la questione di come le celle possano formare strutture piane senza il meccanismo di sagoma della superficie proposto per l’assemblaggio di lamellipodia piatti di cellule aderenti (vedi Discussione).
Video 1.Esempio di pseudopodi lamellari formati da cellule che strisciano su una superficie piana (coprivetrino in vetro rivestito di fibronectina). Il video riproduce a 10,5 × in tempo reale.
Video 2.Un altro esempio di pseudopodi lamellari formati da cellule che strisciano su una superficie piana (coprivetrino in vetro rivestito di fibronectina).Il video viene riprodotto a 11 × tempo reale. Vedi anche Video 1.
Video 3.Esempio in cui i fogli non vengono mai a contatto con la superficie.Poiché il campione è tenuto verticalmente nella camera di imaging, i bordi delle celle che cadono dal coprioggetti possono passare attraverso il campo visivo, visto qui come oggetti che fluiscono da destra a sinistra. Il video viene riprodotto a 9,8 × tempo reale.
Video 4.Un altro esempio in cui i fogli non vengono mai a contatto con la superficie.Il video viene riprodotto a 11 × tempo reale. Vedi anche Video 3.
Video 5.Esempio di pseudopodi lamellari formati da cellule che strisciano attraverso reti di collagene tridimensionali. Il video viene riprodotto a 9,5 × tempo reale.
Video 6.Un altro esempio di pseudopodi lamellari formati da cellule che strisciano attraverso reti di collagene tridimensionali.Si prega di notare che lo stadio viene riposizionato più volte perché la cellula migra fuori dal campo visivo. Il video gioca a 10 × tempo reale. Vedi anche Video 5.
Video 7.Esempio di pseudopodi a ‘rosetta’ costruiti da cellule che strisciano su una superficie piana (coprivetrini in vetro rivestito di fibronectina). Il video 7 suonaa 11 × tempo reale.
Video 8.Un altro esempio di pseudopodi a ‘rosetta’ costruiti da cellule che strisciano su superficie piana (coprivetrini in vetro rivestito di fibronectina).Il video viene riprodotto a 11 × tempo reale. Vedi anche Video 7.
Video 9.Esempio di pseudopodi a ‘coccarda’ costruiti da cellule che strisciano attraverso reti di collagene polimerizzato.Il video gioca a 11 × tempo reale.
Video 10.Un altro esempio di pseudopodi a ‘coccarda’ costruiti da cellule che strisciano attraverso reti di collagene polimerizzato.Il video gioca a 10 × tempo reale. Vedi anche Video 9.
Figura 2-figure supplemento 1.I neutrofili formano pseudopodi lamellari, indipendentemente dal fatto che siano striscianti su superfici bidimensionali o in movimento attraverso complessi ambienti tridimensionali.i neutrofili formano pseudopodi lamellari.(A-B) Per illustrare la morfologia delle sporgenze di membrana in rapida crescita abbiamo sovrapposto più immagini iso-superficiali di fluorescenza multiple prese in diversi punti di tempo di singoli HL-60 cellule che esprimono una sonda a membrana (palmitoylated m-Emerald). I rendering delle singole superfici sono colorati in base al tempo. Gli inserti mostrano ogni cella nel punto temporale finale.(A) HL-60 cellule HL-60 strisciando attraverso un vetrino coprioggetti rivestito di fibronectina.(B) HL-60 cella HL-60 strisciando attraverso una rete di collagene polimerizzato.(C) rendering tridimensionale superficie di due pseudopodi di due cellule diverse, uno strisciando su vetro rivestito di fibronectina (a sinistra) e uno che si muove attraverso una matrice di fibre di collagene (a destra). Ogni set di dati LLSm è reso da tre diversi angoli di visualizzazione: en faccia (in alto), dal lato (al centro) e dall’alto (in basso). Per ulteriori esempi, vedere la Figura 2-figure supplement 1.(D) Box and whisker plot (con mediana, quartili e range) di errore verticale (stima della planarità) per ogni pseudopode, calcolato misurando la distanza media euclidea tra i punti originali sul bordo d’attacco e un piano di riferimento con una larghezza pari alla larghezza media misurata degli pseudopodi lamellari (banda grigia). I primi tre sono misure da cellule che strisciano su una superficie piana (marrone), e i tre successivi da cellule che strisciano attraverso una rete di collagene polimerizzato (blu).(E) Esempio di uno pseudopodo lamellare che mantiene uno spessore simile (cerchi verdi) durante l’estensione e la retrazione, come indicato dalla distanza dalla punta dello pseudopodo al corpo della cellula (quadrati neri). Assi come indicato nella Figura 1. Tutte le barre della scala = 10 µm.(A) Ulteriori visualizzazioni tridimensionali di singoli punti di tempo da quattro cellule indipendenti (due su vetro e due in matrice) che formano pseudopodi lamellari mostrate in faccia, dal lato, e dall’alto.(B) Spinning disco immagine confocale disco di una cella fissa con uno pseudopode lamellare. La pila tridimensionale risultante è stata resa in UCSF Chimera, con una vista superiore (X,Y) e laterale (Z,Y) vista mostrata. Il pannello inferiore è una proiezione di massima intensità di colorazione di falloidina (rosso) e DNA colorato con DAPI (blu), rivelando che gli pseudopodi planari sono pieni di actina polimerizzata. Assi come indicato in Figura 1. Barre di scala = 10 um.
Figura 2-figure supplement 1.I neutrofili formano pseudopodi lamellari.(A) Ulteriori visualizzazioni tridimensionali di singoli punti di tempo da quattro cellule indipendenti (due su vetro e due in matrice) che formano pseudopodi lamellari visualizzati in faccia, dal lato, e dall’alto.(B) Spinning disco immagine confocale disco di una cella fissa con uno pseudopode lamellare. La pila tridimensionale risultante è stata resa in UCSF Chimera, con una vista superiore (X,Y) e laterale (Z,Y) vista mostrata. Il pannello inferiore è una proiezione di massima intensità di colorazione di falloidina (rosso) e DNA colorato con DAPI (blu), rivelando che gli pseudopodi planari sono pieni di actina polimerizzata. Assi come indicato in Figura 1. Barre di scala = 10 um.
Figura 3-figure supplement 5.Figura 3. Neutrofili costruire complessi pseudopodi (‘rosette’) formato da fogli lamellari multipli.immagine disco di filatura confocale disco di cella fissa con una complessa rosetta pseudopod.proiezione di massima intensità di intensità di luce deconvolto reticolo foglio luce dati microscopia (a sinistra) rispetto alla resa superficiale (a destra).micrografie a scansione elettronica di cellule Jurkat T su vetrini coprioggetti.Box e baffi trama della vita di vita di semplice (a sinistra) e rosetta (a destra) pseudopodi costruiti da cellule strisciando attraverso le superfici di vetro rivestito (blu) e attraverso le maglie di collagene (marrone).UCSF visualizzazione Chimera di due punti di tempo di due punti di dati pubblicati Dictyostelium cella cella pubblicato ripreso con microscopia a fascio Bessel(Gao et al., 2014) che mostra le sporgenze a rosetta.(A) Visualizzazioni tridimensionali di rosette costruite da cellule che strisciano su superfici bidimensionali (a sinistra) così come attraverso maglie di collagene senza etichetta (a destra). Singoli punti temporali di due celle indipendenti da ogni condizione sono mostrati di lato (pannelli superiori) e in faccia (pannelli inferiori).(B) Box e trama baffo che mostra che semplici pseudopodi lamellari hanno uno spessore simile a quello dei singoli petali di pseudopodi a rosetta complessi. (n = 18 pseudopodi, da 6 cellule di due repliche biologiche).(C) Viste ingrandite di tre punti temporali di una porzione di una singola cellula (intera cellula mostrata di seguito) che mostra le fasi iniziali della formazione di pseudopodi a rosetta visti in faccia (in alto) e dal lato (in basso). Si noti che, in questo caso, il nuovo pseudopodo emerge dal retro della cella, di fronte ad uno pseudopodo formatosi in precedenza.(D) Due punti temporali che mostrano la dissoluzione di uno pseudopode separato mostrato in faccia (in alto) e di lato (in basso). Per(A) e(C): Assi come indicato in Figura 1. Barre di scala = 10 µm.La pila tridimensionale risultante è stata resa in UCSF Chimera, con una vista in alto (X,Y) e una vista laterale (Z,Y). (In basso) La proiezione di massima intensità della colorazione di falloidina (rosso) rivela che gli pseudopodi a rosetta sono pieni di actina polimerizzata, con DNA colorato con DAPI (blu). Barra di scala = 10 µm. L’angolo in basso a destra della pila di dati è stato rimosso perché una seconda cella ha interferito con la visualizzazione tridimensionale dei dati. Assi come indicato in Figura 1.Questo particolare punto temporale è stato scelto per evidenziare la necessità di strumenti di visualizzazione appropriati per determinare le relazioni spaziali tra le strutture che sporgono dalla superficie della cella.Protrusioni planari su questi linfociti ricordano quelli che osserviamo nelle cellule HL-60. Barra di scala = 2 µm per il pannello in alto a destra e 5 µm per gli altri pannelli.Nove cellule sono state analizzate da tre esperimenti indipendenti.La cellula sta esprimendo il Ditostelio F-actina sonda LimE(Gao et al., 2014). Barra di scala = 10 µm.
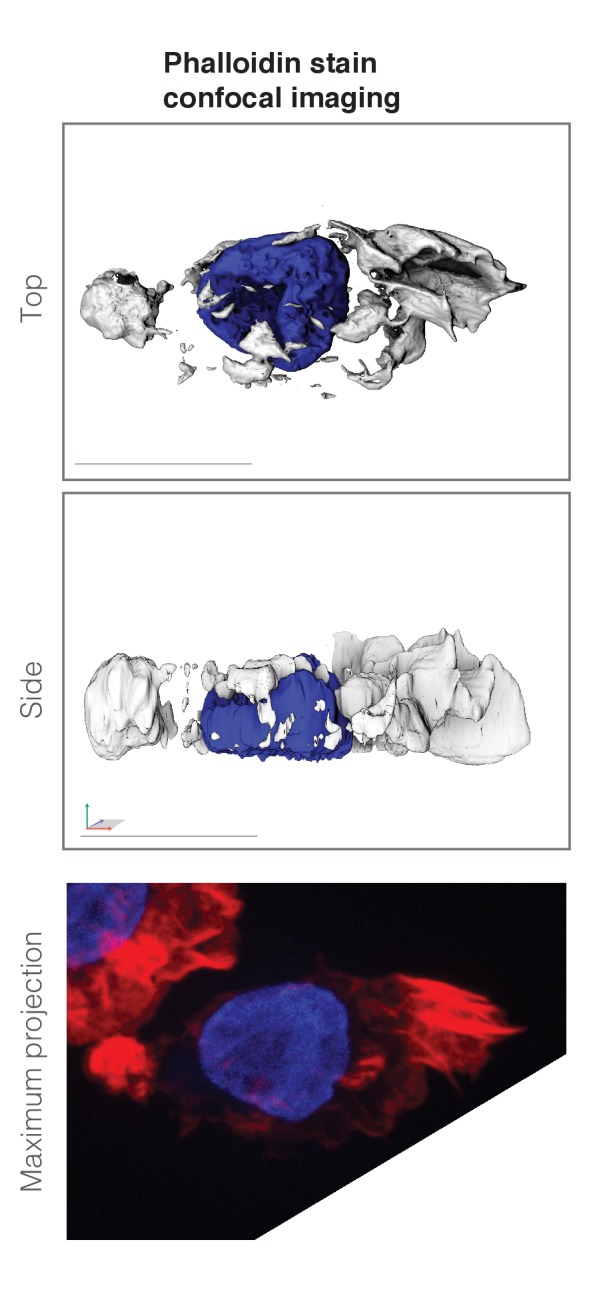
Figura 3-figure supplemento 1.Immagine confocale del disco rotante di una cella fissa con uno pseudopod a rosetta complesso.La pila tridimensionale risultante è stata resa in UCSF Chimera, con una vista dall’alto (X,Y) e una vista laterale (Z,Y). (In basso) La proiezione di massima intensità della colorazione di falloidina (rosso) rivela che gli pseudopodi a rosetta sono pieni di actina polimerizzata, con DNA colorato con DAPI (blu). Barra di scala = 10 µm. L’angolo in basso a destra della pila di dati è stato rimosso perché una seconda cella ha interferito con la visualizzazione tridimensionale dei dati. Assi come indicato in Figura 1.
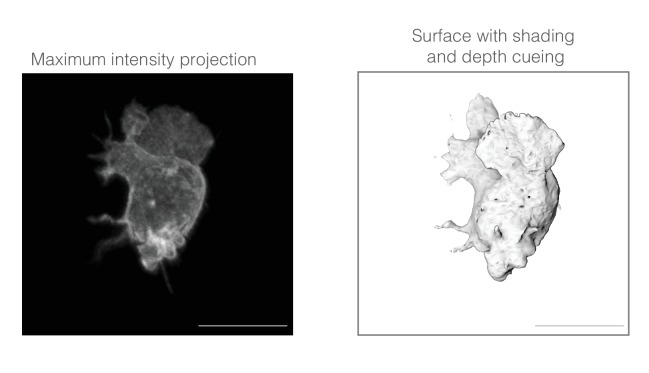
Figura 3-figure supplemento 2.Massima intensità di proiezione di intensità massima dei dati di microscopia a reticolo di luce deconvolto foglio luce microscopia (a sinistra) rispetto alla resa superficiale (a destra).Questo particolare punto temporale è stato scelto per evidenziare la necessità di strumenti di visualizzazione appropriati per determinare le relazioni spaziali tra le strutture che sporgono dalla superficie della cella.
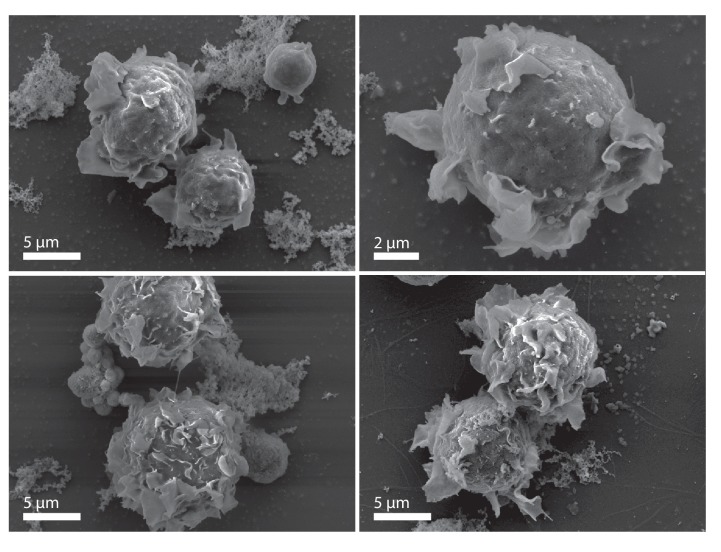
Figura 3-figure supplemento 3.Scansione micrografie elettroniche di cellule Jurkat T su vetrini coprioggetti.Protrusioni planari su questi linfociti ricordano quelli che osserviamo nelle cellule HL-60. Barra di scala = 2 µm per il pannello in alto a destra e 5 µm per gli altri pannelli.
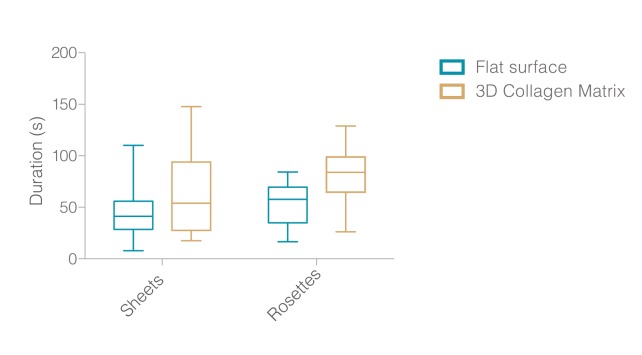
Figura 3-figure supplemento 4.Scatola e baffo trama di vita di semplice (a sinistra) e rosetta (a destra) pseudopodi costruiti da cellule che strisciano attraverso le superfici di vetro rivestito (blu) e attraverso le maglie di collagene (marrone).Nove cellule sono state analizzate da tre esperimenti indipendenti.
Figura 3-figure supplemento 5.UCSF Chimera visualizzazione UCSF di due punti temporali di dati pubblicati Dictyostelium cella dataset pubblicato ripreso con microscopia a fascio di Bessel(Gao et al., 2014) che mostra rosetta come sporgenze.La cellula sta esprimendo il Ditostelio F-actina sonda LimE(Gao et al., 2014). Barra di scala = 10 µm.
I fogli pseudopodiali si intrecciano a formare rosette complesse
Oltre a semplici fogli, le celle HL-60 producono anche pseudopodi più complessi. Quando sono state riprese al microscopio convenzionale a campo largo o confocale, queste sporgenze sono apparse come striature irregolari e tridimensionali(Figura 3-figure supplement 1, proiezione di massima intensità). Tuttavia, quando sono state riprese ad alta risoluzione con la microscopia a reticolo e visualizzate con la resa tridimensionale della superficie, queste strutture apparentemente amorfe si rivelano essere “rosette”, composte da molteplici componenti lamellari intrecciati in disegni simili a rose. Le rosette sono assemblate da cellule che strisciano su una superficie bidimensionale(Figura 3, Video 7 e 8), e da cellule che strisciano attraverso maglie di collagene tridimensionali(Figura 3, Video 9, e Video 10). In campi uniformi di chemioattrattore, sia lamellare e pseudopodi a rosetta sono altamente dinamici ed effimeri(Figura 4A).
Diverse linee di evidenza sostengono che gli elementi lamellari che formano queste rosette sono identici agli pseudopodi lamellari descritti sopra. In primo luogo, sia gli pseudopodi a foglio che quelli a rosetta hanno in gran parte escluso le sonde di actina basate sull’utrofina (Figura 1B) ed entrambi i tipi di protrusione sono scomparsi in presenza di CK-666 (Figura 4B,Video 11, e Video 12), un inibitore delle piccole molecole del complesso Arp2/3 (Nolen et al.,2009), sostenendo che i fogli e le rosette sono formati da reti di actina ramificate che si assemblano rapidamente. In secondo luogo, le lamelle all’interno delle rosette hanno lo stesso spessore (690 ± 20 nm, n = 18, Figura 3B) dei semplici pseudopodi lamellari. In terzo luogo, sequenze di immagini time-lapse spesso rivelato pseudopodiali fogli convergenti o ramificazione per formare rosette (ad esempio, Figura 4A, Video 4- 5). In quarto luogo, in un campo uniforme di chemioattrattore, i fogli pseudopodiali e le rosette hanno entrambi una durata di vita di circa 60 s, dalla comparsa iniziale alla scomparsa finale(Figura 3-figure supplement 4). Questa durata di vita rimane simile sia che le cellule stiano migrando attraverso superfici piane o attraverso reti di collagene(Figura 3-figure supplement 4), indicando che la durata di vita della protrusione non dipende dal supporto meccanico esterno. Infine, circa il 75% degli pseudopodi sia a foglio che a rosetta scompaiono in modo identico, collassando in un groviglio di strutture lineari che assomigliano a filopodi(Figura 3E (rosetta) e Video 1 (foglio), vedi anche Figura 4A).
Video 11.Esempio di cellule trattate con CK-666 che migrano attraverso una rete di collagene polimerizzato.Il video viene riprodotto a 11 × tempo reale.
Video 12.Un altro esempio di cellule trattate con CK-666 che migrano attraverso una rete di collagene polimerizzato.Il video riproduce a 11 × tempo reale. Vedi anche Video 11.
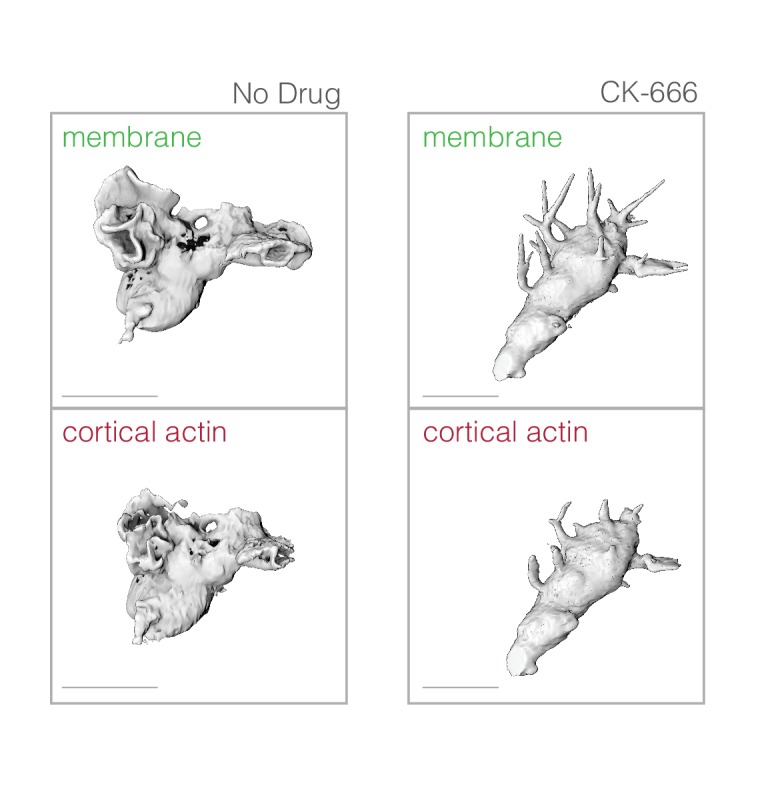
Figura 4-figure supplemento 1.Figura 4—figura supplemento 1. Sia lamellare semplice e pseudopodi a rosetta sono formati de novo dal corpo cellulare e richiedono Arp2/3 attività per il montaggio.confronto di sporgenze formate da controllo (colonna di sinistra) e CK-666 trattati (colonna di destra) le cellule che esprimono sia membrana (riga superiore) e actina corticale actina Utrophin-sonde a base di actina (riga inferiore).(A) Rendering superficiale della membrana di controllo HL-60 cella di controllo ad intervalli di ~ 19 s. Frecce verdi evidenziano la crescita di pseudopodi, mentre restringendo pseudopodi sono indicati da frecce rosse. Inserto (in alto a sinistra): sovrapposizione del contorno delle cellule in ogni punto di tempo per evidenziare l’estensione degli pseudopodi e il movimento delle cellule.(B) Una cella simile trattata con l’inibitore Arp2/3 CK-666 per dieci minuti prima dell’imaging. Un singolo piano dell’immagine tridimensionale è oscurato e indicato con una freccia nera per evidenziare l’estensione delle sporgenze tubolari aberranti. Barra di scala = 10 µm.
Video 11.Esempio di cellule trattate con CK-666 che migrano attraverso una rete di collagene polimerizzato.Il video viene riprodotto a 11 × tempo reale.
Video 12.Un altro esempio di cellule trattate con CK-666 che migrano attraverso una rete di collagene polimerizzato.Il video riproduce a 11 × tempo reale. Vedi anche Video 11.
Figura 4-figure supplemento 1.Figura 4—figura supplemento 1. Sia lamellare semplice e pseudopodi a rosetta sono formati de novo dal corpo cellulare e richiedono Arp2/3 attività per il montaggio.confronto di sporgenze formate da controllo (colonna di sinistra) e CK-666 trattati (colonna di destra) le cellule che esprimono sia membrana (riga superiore) e actina corticale actina Utrophin-sonde a base di actina (riga inferiore).(A) Rendering superficiale della membrana di controllo HL-60 cella di controllo ad intervalli di ~ 19 s. Frecce verdi evidenziano la crescita di pseudopodi, mentre restringendo pseudopodi sono indicati da frecce rosse. Inserto (in alto a sinistra): sovrapposizione del contorno delle cellule in ogni punto di tempo per evidenziare l’estensione degli pseudopodi e il movimento delle cellule.(B) Una cella simile trattata con l’inibitore Arp2/3 CK-666 per dieci minuti prima dell’imaging. Un singolo piano dell’immagine tridimensionale è oscurato e indicato con una freccia nera per evidenziare l’estensione delle sporgenze tubolari aberranti. Barra di scala = 10 µm.
Figura 4-figure supplement 1.Confronto delle sporgenze formate da controllo (colonna sinistra) e CK-666 trattati (colonna destra) cellule che esprimono sia membrana (riga superiore) e actina corticale actina Utrofina a base di actina sonde (riga inferiore).
Gli pseudopodi lamellari crescono da creste lineari
Gli elementi lamellari che formano pseudopodi – sia fogli che rosette – sono apparsi per la prima volta come deformazioni lineari che attraversano la superficie della membrana al plasma (Figura 3C). Queste basse ‘creste’ hanno formato pseudopodi maturi crescendo rapidamente lontano dal corpo cellulare, ad un tasso caratteristico di 11,9 ± 5,4 µm/min (n = 4). Sorprendentemente, man mano che ogni protrusione nascente cresceva, il suo bordo distale rimaneva lineare, lasciando uno pseudopod piatto nella sua scia. Fogli pseudopodiali isolati a volte producevano rosette riproducendo fogli secondari, ma anche questi fogli secondari emergevano come creste lineari dal lato del foglio originale (ad esempio Video 5).
Gli pseudopodi lamellari a contatto con il coprioggetti appaiono come ‘onde’ dinamiche di attivazione Arp2/3
L’assemblaggio di reti di actine ramificate è in gran parte controllato attraverso l’attivazione del complesso Arp2/3, che negli pseudopodi è mediato dal complesso normativo WAVE(Bear et al., 1998; Weiner et al., 2006). Uno studio precedente(Weiner et al., 2007) ha impiegato la microscopia TIRF per la localizzazione di immagini e la dinamica del complesso regolatorio WAVE in cellule HL-60 che strisciano su vetrini coprioggetti. Quando viene ripreso nella regione prossimale (entro ~ 100 nm) al coprioggetti, il complesso normativo WAVE forma una serie di archi sottili, o ‘onde’, che hanno origine da qualche parte nel corpo della cellula e poi avanzare verso l’esterno lungo la superficie ventrale della cellula. La nostra osservazione che lo pseudopod di una cellula HL-60 è composto da più fogli lamellari suggerisce che ogni onda in movimento verso l’esterno è in realtà il bordo di un singolo foglio premuto nel piano TIRF mentre si estende in avanti.
Per testare questa ipotesi, abbiamo condotto la microscopia TIRF quasi simultanea e la microscopia a contrasto con interferenza a riflessione (RICM) sia per visualizzare la localizzazione complessa dell’onda che per sondare la distanza tra la superficie ventrale della cella e la superficie di vetro su cui migra(Figura 5, Video 13). RICM ha rivelato che la superficie ventrale della cellula non è liscia e non fa uniformemente stretto contatto con il coprioggetti rivestito di fibronectina a cui aderisce, coerente con le osservazioni precedenti (Sullivane Mandell, 1983). Invece, la superficie ventrale forma modelli dinamici e complessi di contatto ravvicinato, comprese le linee che viaggiano verso la parte anteriore della cellula strisciante(Figura 5, Video 13). Coerentemente con la nostra interpretazione, vediamo anche l’arricchimento del complesso delle Onde in corrispondenza di queste linee di contatto ravvicinate in movimento(Figura 5, Video 13).
Video 13.I video di fluorescenza WAVE in TIRF (a sinistra) e la distanza della superficie della cella ventrale dal coprioggetti in RICM (al centro), e overlay (a destra).Per il RICM, le aree scure rappresentano distanze maggiori tra il coprioggetti e la superficie della cellula ventrale; il bianco brillante (o magenta, nella sovrapposizione) indica lo stretto contatto della cellula sul vetro. Sono mostrate due diverse celle di esempio. I grafici mostrano l’intensità TIRF e RICM lungo le linee disegnate sulle sovrapposizioni delle immagini (all’estrema destra). Il video viene riprodotto a 5 × tempo reale.
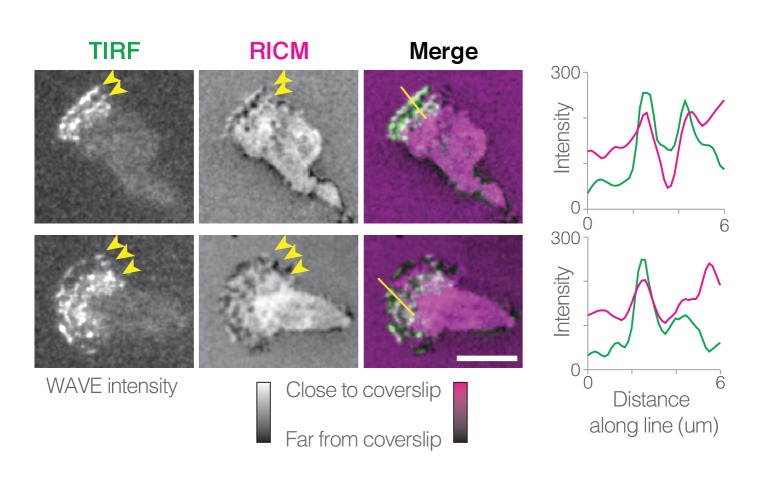
Figura 5.Il complesso WAVE si localizza su sporgenze ventrali.Time-lapse immagini di fluorescenza complesso WAVE in TIRF (a sinistra) e la distanza della superficie delle cellule ventrali dalla superficie del vetro coprioggetti in microscopia a contrasto di interferenza riflettente (RICM, al centro), e overlay (a destra). Per il RICM, la colormap è stata invertita rispetto ai dati grezzi: le aree scure rappresentano distanze maggiori tra il coprioggetti e la superficie della cellula ventrale; il bianco brillante (o magenta, nella sovrapposizione) indica lo stretto contatto della cellula sul vetro. Le scansioni di linea dell’intensità TIRF e RICM lungo le linee mostrate sulle sovrapposizioni dell’immagine (all’estrema destra). Barre di scala = 5 µm. Vedere anche il Video 13 per la localizzazione dinamica e le linescans.
Video 13.Video della fluorescenza WAVE in TIRF (a sinistra) e della distanza della superficie della cellula ventrale dal coprioggetti in RICM (al centro), e overlay (a destra).Per il RICM, le aree scure rappresentano distanze maggiori tra il coprioggetti e la superficie della cellula ventrale; il bianco brillante (o magenta, nella sovrapposizione) indica lo stretto contatto della cellula sul vetro. Sono mostrate due diverse celle di esempio. I grafici mostrano l’intensità TIRF e RICM lungo le linee disegnate sulle sovrapposizioni delle immagini (all’estrema destra). Il video viene riprodotto a 5 × tempo reale.
Figura 5.Il complesso WAVE si localizza su sporgenze ventrali.Time-lapse immagini di fluorescenza complesso WAVE in TIRF (a sinistra) e la distanza della superficie delle cellule ventrali dalla superficie del vetro coprioggetti in microscopia a contrasto di interferenza riflettente (RICM, al centro), e overlay (a destra). Per il RICM, la colormap è stata invertita rispetto ai dati grezzi: le aree scure rappresentano distanze maggiori tra il coprioggetti e la superficie della cellula ventrale; il bianco brillante (o magenta, nella sovrapposizione) indica lo stretto contatto della cellula sul vetro. Le scansioni di linea dell’intensità TIRF e RICM lungo le linee mostrate sulle sovrapposizioni dell’immagine (all’estrema destra). Barre di scala = 5 µm. Vedere anche il Video 13 per la localizzazione dinamica e le linescans.
Gli pseudopodi a foglio e a rosetta promuovono cambiamenti nella direzione della migrazione ma non sono necessari per la locomozione delle cellule
Per indagare il ruolo degli pseudopodi dinamici – sia fogli e rosette – nella locomozione cellulare, abbiamo confrontato la morfologia e il movimento delle cellule di controllo non trattate con le cellule trattate con l’inibitore Arp2/3 CK-666 per 10 minuti. Questa analisi è stata limitata alle cellule che si muovono attraverso le matrici tridimensionali di collagene perché abbiamo trovato che la perdita di attività Arp2/3 ha ridotto drasticamente l’adesione delle cellule HL-60 a fibronectina rivestita coprivetrini. Questa perdita di adesione ha posto problemi particolari per i coprioggetti orientati verticalmente utilizzati nel nostro microscopio a foglio luminoso a reticolo. Mentre le cellule di controllo costruite pseudopodi lamellari, CK-666 cellule trattate formavano lunghe sporgenze tubolari(Figura 4A e B).
Per analizzare la complessa dinamica degli pseudopodi, abbiamo sviluppato un metodo automatizzato per il rilevamento degli pseudopodi e la stima del volume nei nostri set di dati tridimensionali(Figura 6A). Questo metodo sfrutta il legame preferenziale della sonda utrofina-actina a strutture stabili, come le reti di actina corticale, sulle reti più dinamiche e ramificate che guidano il movimento delle membrane(Belin et al., 2014). Come previsto, la nostra microscopia a foglio luminoso a reticolo ha rivelato una chiara colocalizzazione della sonda utrofina con il marcatore di membrana ovunque sulla superficie della cellula, tranne che all’interno dei fogli dinamici e delle rosette, che hanno in gran parte escluso la sonda(Figura 1B), nonostante contenga dense reti di actina(Figura 2-figure supplement 1 e Figura 3-figure supplement 1). Abbiamo utilizzato l’assenza di utrofina fluorescente dalle regioni membrano-prossimali della cellula come un comodo marker per identificare le sporgenze guidate dalla crescita di reti di actina ramificate, e abbiamo automatizzato questo processo sottraendo il volume racchiuso da una sonda di actina a base di utrofina dal volume racchiuso dal marker della membrana plasmatica(Figura 6A, Video 14). Questo metodo di identificazione e quantificazione automatizzata rivela che l’inibizione dell’attività complessa Arp2/3 diminuisce il volume dello pseudopodo più grande prodotto dalle cellule striscianti di una media del 40%(Figura 6-figure supplement 1C e Video 14- 15), una diminuzione significativa nonostante la variabilità all’interno delle cellule del volume degli pseudopodi (Figura 6-figuresupplement 2D-E). Allo stesso modo, l’ispezione visiva delle sequenze time-lapse ha rivelato che il tasso di formazione di pseudopodi è diminuito da 1,9 pseudopodi / min a 0,8 pseudopodi / min su Arp2/3 inibizione (Controllo: n = 9 cellule oltre 40,7 min; CK-666 trattati: n = 7 cellule oltre 22,1 min). Gli pseudopodi rimanenti possono essere dovuti ad inibizione incompleta di Arp2/3.
Video 14.Video che evidenzia l’automazione del rilevamento della protrusione nelle celle di controllo. Il video viene riprodotto a 10 × in tempo reale.
Video 15.Video15. Video 15. Evidenziazione video automazione della rilevazione delle protrusioni nelle celle CK666. Il video viene riprodotto a 11 × tempo reale.
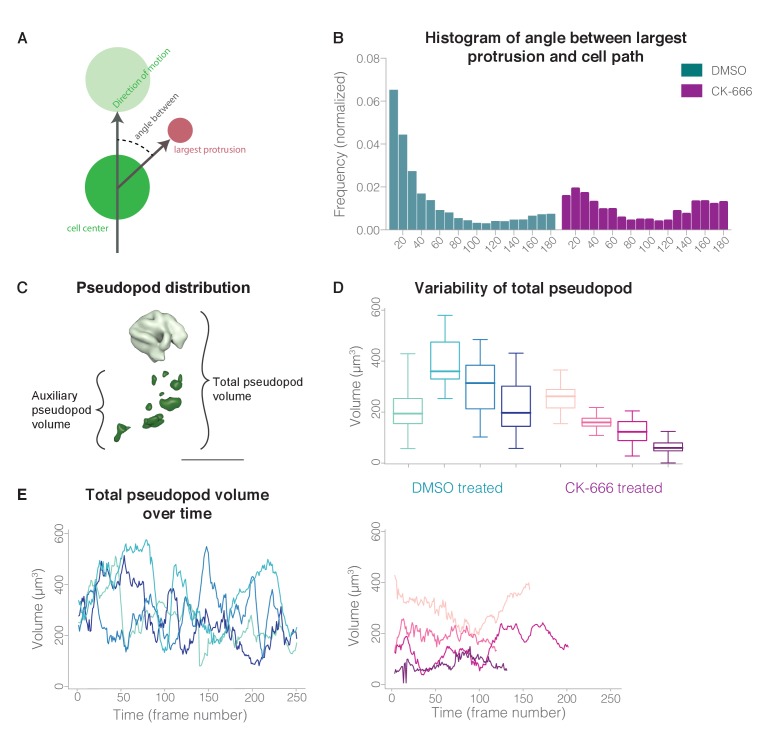
Figura 6-figure supplement 2.Gli pseudopodi lamellari sono associati alla rotazione delle cellule, ma non sono necessari per la locomozione.’protrusioni_chimera_script.py’ script Python utilizzato per calcolare i volumi di protrusione con le funzioni di analisi delle protrusioni delle cellule UCSF chimera.R’ script di analisi per l’uso in R per calcolare e tracciare le caratteristiche del percorso e le relazioni con l’attività degli pseudopod.’protrusioni_chimera_script.py’ script Python usato per calcolare i volumi di protrusione con le funzioni di analisi delle protrusioni delle cellule UCSF chimera.R’ script di analisi per l’uso in R per calcolare e tracciare le caratteristiche del percorso, e le relazioni con l’attività pseudopod.Calcolo dei punti di svolta (A) Velocità di svolta (calcolato con due metodi distinti) delle celle di controllo (n = 4 celle su un totale di 20.7 min, da almeno due esperimenti indipendenti) e CK-666 cellule trattate (n = 5 cellule su un totale di 13,7 min, da almeno due esperimenti indipendenti).Effetti dell’inibitore Arp2/3 sul comportamento degli pseudopodi.(A) Il rilevamento automatico pseudopod si basa sulla relativa esclusione della sonda di actina Utrophin-based actina dalle reti di actina dinamica all’interno di pseudopodi; pseudopodi sono identificati sottraendo prima il segnale di actina corticale (in alto a destra) dal segnale di membrana (in alto a sinistra), con conseguente mappa differenza (in basso a sinistra). Gli pseudopodi (in basso a destra) sono poi definiti come volumi chiusi che soddisfano tutti i seguenti tre criteri: più grandi di 1 µm3, entro 15 µm dal centro della cellula, e al di fuori del corpo cellulare (o più grandi di 15 um3). Il corpo cellulare è definito da una sfera con un volume pari al volume della mappa dell’actina; se il centro del volume chiuso era all’interno di questa sfera, non era considerato uno pseudopod a meno che il suo volume non superasse i 15 um3. Nei Video 14,15, il corpo cellulare è indicato da una sfera gialla.(B) Trame risultanti da un’analisi dei componenti principali della posizione della cellula. In vari punti lungo la traccia di una cella, la saturazione del colore indica il numero di pseudopodi e lo spessore della linea indica la distribuzione degli pseudopodi, o il rapporto tra il volume degli pseudopodi ausiliari e il volume totale degli pseudopodi. Due esempi ciascuno di cellule di controllo e CK-666 cellule trattate sono mostrati.(C) I risultati di un’analisi dei componenti principali della posizione delle cellule nello spazio tridimensionale per ogni punto temporale(punti blu). Curvatura bassa, dove la lunghezza del percorso è vicino alla distanza percorsa dalla cella in dieci punti temporali, e curvatura alta, dove la lunghezza del percorso è 2,5 volte più lungo della distanza percorsa, sono entrambi indicati da frecce rosse.(D) Curvatura del percorso di una cella di controllo rappresentativa e di una cella trattata CK-666.(E) Velocità del centroide delle celle di controllo e delle celle trattate CK-666 (n = 5 celle di almeno due esperimenti indipendenti, valore p = 0,024). Le barre di errore rappresentano la deviazione standard.(F) Velocità di rotazione delle cellule di controllo (n = 4 cellule su un totale di 20,7 min, da almeno due esperimenti indipendenti) e CK-666 cellule trattate (n = 5 cellule su un totale di 13,7 min, da almeno due esperimenti indipendenti). Le barre di errore rappresentano la deviazione standard.10.7554/eLife.26990.035Figure 6-source code 1.’protrusions_chimera_script.py’ script Python utilizzato per calcolare i volumi di protrusione con UCSF chimera.Questo produce due file di testo: uno contenente i dati per i volumi di pseudopod in ogni punto di tempo e uno degli angoli tra lo pseudopod principale e il percorso della cella.Questo produce due file di testo: uno contenente i dati per i volumi degli pseudopod in ogni punto temporale e uno degli angoli tra lo pseudopod principale e il percorso della cella.10.7554/eLife.26990.036Figure 6-source code 2.’cell protrusions analysis functions.R’ Script di analisi per l’uso in R per calcolare e tracciare le caratteristiche del percorso e le relazioni con l’attività degli pseudopod.Questo produce due file di testo: uno contenente i dati per i volumi degli pseudopod in ogni punto di tempo e uno degli angoli tra lo pseudopod principale e il percorso della cella.Le barre di errore rappresentano la deviazione standard.(B) Confronto dell’attività di svolta, calcolata come l’area sotto la curvatura del percorso delle celle mostrate in(A). Le barre di errore rappresentano la deviazione standard.(C) Trama a scatola e baffi del volume totale dello pseudopod più grande. Controllo (media ±SD, N): 184,0 ± 97,9 µm3, 980 punti di tempo; CK666 (media, SD, N): 110,5 ± 64,6 µm3, 600 punti di tempo.(A) Schema che mostra come viene calcolato l’angolo tra lo pseudopod principale e il percorso della cella.(B) Confronto di angolo tra il più grande pseudopod e la traiettoria di movimento delle cellule per il controllo e CK-666 cellule trattate mostrato in Figura 6-figure supplement 1. Ogni istogramma è normalizzato ad una curva sinusoidale per compensare la normale distribuzione prevista dell’angolo tra due vettori casuali nello spazio tridimensionale.(C) La distribuzione degli pseudopodi è determinato come il rapporto tra il volume ausiliario pseudopodi ausiliario e il volume totale.(D) Box-and-whisker trama di variabilità di volume totale pseudopod per il controllo e CK-666 cellule trattate.(E) Variabilità del volume totale degli pseudopodi nel tempo, con le stesse 8 cellule come in(D) mostrato.
Per valutare l’effetto di inibizione del complesso Arp2/3 sulla locomozione delle cellule, abbiamo confrontato il movimento dei centroidi delle cellule in assenza e presenza dell’inibitore Arp2/3 CK-666. Nonostante la perdita di pseudopodi dinamici, le cellule trattate con CK-666 hanno mantenuto la capacità di migrare attraverso le reti di collagene in un campo uniforme di chemioattrattore. Le cellule trattate, tuttavia, si muoveva più lentamente (5,42 vs 8,57 um/min, Figura 6E) e sembrava viaggiare lungo traiettorie molto più dritte rispetto alle cellule di controllo(Figura 6B). Abbiamo usato due metodi per quantificare e analizzare la rotazione delle cellule in tre dimensioni. In un approccio, abbiamo rilevato le svolte stimando la prima derivata della traiettoria della cella per identificare i punti di inflessione nel percorso della cella. Il secondo metodo calcola il rapporto tra la lunghezza del percorso percorso del centroide della cella su dieci punti temporali e la distanza euclidea tra il primo e l’ultimo punto, e definisce le svolte come regioni in cui questo rapporto è maggiore di 2,5(Figura 6C). Secondo entrambe le metriche, le cellule di controllo hanno cambiato direzione da 2 a 3 volte più spesso delle cellule trattate CK-666 (metodo 1: 2,73 vs 1,15 giri/min, Figura 6-figure supplement 1A e B; metodo 2: 3,41 vs 1,09 giri/min, Figura 6D-F), e hanno mostrato una maggiore attività di giro (Figura 6-figuresupplement 1B ).
La ridotta frequenza di rotazione in CK-666 cellule trattate suggeriscono che la dinamica, Arp2/3-generato pseudopods pseudopodi svolgono un ruolo nell’avviare o facilitare i cambiamenti di direzione. Per esplorare ulteriormente questa possibilità, abbiamo visualizzato le relazioni tra la formazione di pseudopodi e la rotazione delle cellule sovrapponendo il numero e il volume di pseudopodi dinamici sulle traiettorie tridimensionali delle cellule che strisciano attraverso una matrice di collagene(Figura 6B). In queste trame, la linea è una proiezione bidimensionale della traiettoria tridimensionale del centroide della cellula proiettata su un piano che massimizza la coincidenza parallela, mentre la larghezza della linea e la saturazione del colore indicano, rispettivamente, il numero e il volume degli pseudopodi dinamici in ogni punto. Le traiettorie delle cellule di controllo rivelano frequenti svolte, ognuna delle quali appare correlata ad un drammatico aumento della dinamica degli pseudopodi. Al contrario, CK-666 cellule trattate CK-666 fatto meno della metà del numero di giri(Figura 6F) e ha creato meno e più piccoli pseudopodi dinamici(Figura 6-figure supplemento 1C e Figura 6-figure supplemento 2D-E). Inoltre, mentre le cellule di controllo spesso costruiscono pseudopodi in direzione del movimento delle cellule, le sporgenze costruite da CK-666 cellule trattate sono più uniformemente distribuiti(Figura 6-figure supplement 2A e B).
Video 14.Video che evidenzia l’automazione del rilevamento delle protrusioni nelle celle di controllo. Il video viene riprodotto a 10 × in tempo reale.
Video 15.Video15 Video 15. Evidenziazione video automazione della rilevazione delle protrusioni nelle celle CK666. Il video viene riprodotto a 11 × tempo reale.
Figura 6-figure supplement 2.Gli pseudopodi lamellari sono associati alla rotazione delle cellule, ma non sono necessari per la locomozione.’protrusioni_chimera_script.py’ script Python utilizzato per calcolare i volumi di protrusione con le funzioni di analisi delle protrusioni delle cellule UCSF chimera.R’ script di analisi per l’uso in R per calcolare e tracciare le caratteristiche del percorso e le relazioni con l’attività degli pseudopod.’protrusioni_chimera_script.py’ script Python usato per calcolare i volumi di protrusione con le funzioni di analisi delle protrusioni delle cellule UCSF chimera.R’ script di analisi per l’uso in R per calcolare e tracciare le caratteristiche del percorso, e le relazioni con l’attività pseudopod.Calcolo dei punti di svolta (A) Velocità di svolta (calcolato con due metodi distinti) delle celle di controllo (n = 4 celle su un totale di 20.7 min, da almeno due esperimenti indipendenti) e CK-666 cellule trattate (n = 5 cellule su un totale di 13,7 min, da almeno due esperimenti indipendenti).Effetti dell’inibitore Arp2/3 sul comportamento degli pseudopodi.(A) Il rilevamento automatico pseudopod si basa sulla relativa esclusione della sonda di actina Utrophin-based actina dalle reti di actina dinamica all’interno di pseudopodi; pseudopodi sono identificati sottraendo prima il segnale di actina corticale (in alto a destra) dal segnale di membrana (in alto a sinistra), con conseguente mappa differenza (in basso a sinistra). Gli pseudopodi (in basso a destra) sono poi definiti come volumi chiusi che soddisfano tutti i seguenti tre criteri: più grandi di 1 µm3, entro 15 µm dal centro della cellula, e al di fuori del corpo cellulare (o più grandi di 15 um3). Il corpo cellulare è definito da una sfera con un volume pari al volume della mappa dell’actina; se il centro del volume chiuso era all’interno di questa sfera, non era considerato uno pseudopod a meno che il suo volume non superasse i 15 um3. Nei Video 14,15, il corpo cellulare è indicato da una sfera gialla.(B) Trame risultanti da un’analisi dei componenti principali della posizione della cellula. In vari punti lungo la traccia di una cella, la saturazione del colore indica il numero di pseudopodi e lo spessore della linea indica la distribuzione degli pseudopodi, o il rapporto tra il volume degli pseudopodi ausiliari e il volume totale degli pseudopodi. Due esempi ciascuno di cellule di controllo e CK-666 cellule trattate sono mostrati.(C) I risultati di un’analisi dei componenti principali della posizione delle cellule nello spazio tridimensionale per ogni punto temporale(punti blu). Curvatura bassa, dove la lunghezza del percorso è vicino alla distanza percorsa dalla cella in dieci punti temporali, e curvatura alta, dove la lunghezza del percorso è 2,5 volte più lungo della distanza percorsa, sono entrambi indicati da frecce rosse.(D) Curvatura del percorso di una cella di controllo rappresentativa e di una cella trattata CK-666.(E) Velocità del centroide delle celle di controllo e delle celle trattate CK-666 (n = 5 celle di almeno due esperimenti indipendenti, valore p = 0,024). Le barre di errore rappresentano la deviazione standard.(F) Velocità di rotazione delle cellule di controllo (n = 4 cellule su un totale di 20,7 min, da almeno due esperimenti indipendenti) e CK-666 cellule trattate (n = 5 cellule su un totale di 13,7 min, da almeno due esperimenti indipendenti). Le barre di errore rappresentano la deviazione standard.10.7554/eLife.26990.035Figure 6-source code 1.’protrusions_chimera_script.py’ script Python utilizzato per calcolare i volumi di protrusione con UCSF chimera.Questo produce due file di testo: uno contenente i dati per i volumi di pseudopod in ogni punto di tempo e uno degli angoli tra lo pseudopod principale e il percorso della cella.Questo produce due file di testo: uno contenente i dati per i volumi degli pseudopod in ogni punto temporale e uno degli angoli tra lo pseudopod principale e il percorso della cella.10.7554/eLife.26990.036Figure 6-source code 2.’cell protrusions analysis functions.R’ Script di analisi per l’uso in R per calcolare e tracciare le caratteristiche del percorso e le relazioni con l’attività degli pseudopod.Questo produce due file di testo: uno contenente i dati per i volumi degli pseudopod in ogni punto di tempo e uno degli angoli tra lo pseudopod principale e il percorso della cella.Le barre di errore rappresentano la deviazione standard.(B) Confronto dell’attività di svolta, calcolata come l’area sotto la curvatura del percorso per le celle mostrate in(A). Le barre di errore rappresentano la deviazione standard.(C) Trama a scatola e baffi del volume totale dello pseudopod più grande. Controllo (media ±SD, N): 184,0 ± 97,9 µm3, 980 punti di tempo; CK666 (media, SD, N): 110,5 ± 64,6 µm3, 600 punti di tempo.(A) Schema che mostra come viene calcolato l’angolo tra lo pseudopod principale e il percorso della cella.(B) Confronto di angolo tra il più grande pseudopod e la traiettoria di movimento delle cellule per il controllo e CK-666 cellule trattate mostrato in Figura 6-figure supplement 1. Ogni istogramma è normalizzato ad una curva sinusoidale per compensare la normale distribuzione prevista dell’angolo tra due vettori casuali nello spazio tridimensionale.(C) La distribuzione degli pseudopodi è determinato come il rapporto tra il volume ausiliario pseudopodi ausiliario e il volume totale.(D) Box-and-whisker trama di variabilità di volume totale pseudopod per il controllo e CK-666 cellule trattate.(E) Variabilità del volume totale degli pseudopodi nel tempo, con le stesse 8 cellule come in(D) mostrato.
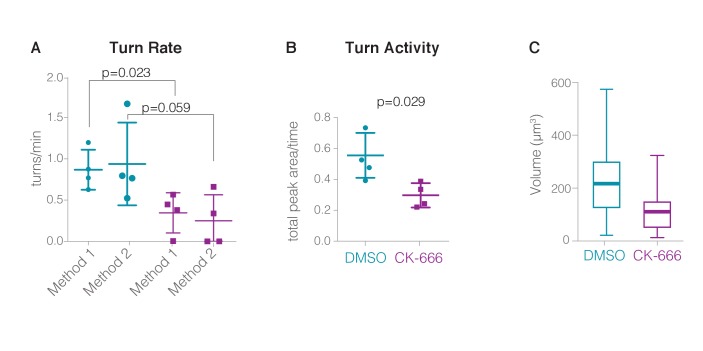
Figura 6-figure supplemento 1.Calcolo dei punti di svolta(A) Tassi di svolta (calcolato con due metodi distinti) di cellule di controllo (n = 4 cellule su un totale di 20,7 min, da almeno due esperimenti indipendenti) e CK-666 cellule trattate (n = 5 cellule su un totale di 13,7 min, da almeno due esperimenti indipendenti).Le barre di errore rappresentano la deviazione standard.(B) Confronto dell’attività di svolta, calcolato come l’area sotto la curvatura di trame di percorso per le cellule mostrate in(A). Le barre di errore rappresentano la deviazione standard.(C) Trama a scatola e baffi del volume totale dello pseudopod più grande. Controllo (media ±SD, N): 184,0 ± 97,9 µm3, 980 punti di tempo; CK666 (media, SD, N): 110,5 ± 64,6 µm3, 600 punti di tempo.
Figura 6-figure supplement 2.Effetti dell’inibitore Arp2/3 sul comportamento degli pseudopodi.(A) Schema che mostra come viene calcolato l’angolo tra pseudopod principale e il percorso delle cellule.(B) Confronto di angolo tra il più grande pseudopod e la traiettoria di movimento delle cellule per il controllo e CK-666 cellule trattate mostrato in Figura 6-figure supplement 1. Ogni istogramma è normalizzato ad una curva sinusoidale per compensare la normale distribuzione prevista dell’angolo tra due vettori casuali nello spazio tridimensionale.(C) La distribuzione degli pseudopodi è determinato come il rapporto tra il volume ausiliario pseudopodi ausiliario e il volume totale.(D) Box-and-whisker trama di variabilità di volume totale pseudopod per il controllo e CK-666 cellule trattate.(E) Variabilità del volume totale degli pseudopodi nel tempo, con le stesse 8 cellule come in(D) mostrato.
Discussione
Le lamelle libere si combinano per formare complessi pseudopodi
La microscopia a fogli luminosi a traliccio rivela che gli pseudopodi complessi formati da cellule HL-60 in rapido movimento sono in realtà composti da elementi lamellari dinamici. Alcuni pseudopodi sono isolati, lamelle autoportanti, mentre altri sono rosette composte da più elementi lamellari, spesso collegati da giunzioni a T e a Y. Questi blocchi lamellari hanno caratteristiche strutturali comuni, tra cui uno spessore caratteristico, ma la loro morfologia a lamelle non dipende dall’interazione con una superficie piana.
Abbiamo identificato simili sporgenze lamellari e a rosetta in micrografie elettroniche a scansione pubblicate di diversi tipi di cellule diverse, tra cui cellule differenziate HL-60(Fleck et al., 2005), cellule T Jurkat(Nicholson-Dykstra e Higgs, 2008)(Figura 3-figure supplement 3), neutrofili umani che migrano attraverso un gradiente chemioattrattore (Zigmonde Sullivan, 1979), celluledendritiche(Coateset al., 2003; Fisher et al., 2008; Felts et al., 2010), cellule T (Brown et al., 2003) e monociti(Majstoravich et al., 2004) da sangue umano, così come Dictyostelium amoebae(Gerisch et al., 2013). La microscopia ottica ha anche accennato all’esistenza di pseudopodi lamellari e a rosetta indipendenti, in particolare nelle immagini di cellule striscianti inclinate di 90 gradi rispetto all’angolo di visione convenzionale(Sullivan e Mandell, 1983). Utilizzando la microscopia confocale veloce su cellule dendritiche confinate, il laboratorio Sixt ha recentemente osservato quelle che sembravano essere sporgenze dinamiche a forma di foglio che non richiedono l’adesione ad alcun substrato(Leithner et al., 2016). Le nostre rosette assomigliano anche alle strutture protrusive osservate nelle amebe di Dictyostelium utilizzando l’illuminazione del piano del fascio di Bessel(Gao et al., 2012). Questa somiglianza è accresciuta dal surface-rendering dei dati pubblicati sul Dictostelium utilizzando UCSF Chimera, e le tecniche di visualizzazione sopra descritte(Figura 3-figure supplement 5). Dal nostro lavoro attuale e dalle osservazioni precedenti, proponiamo due principi di base che definiscono la morfologia degli pseudopodi in queste e altre cellule che costruiscono pseudopodi lamellari. In primo luogo, i leucociti in rapido movimento possono assemblare pseudopodi lamellari indipendenti senza l’aiuto di un ‘modello’ di superficie. In secondo luogo, gli pseudopodi tridimensionali più complessi possono essere risolti in componenti lamellari. Anche se suggerito dal lavoro precedente, stabilire questi fatti richiesti a seguito dell’intero ‘ciclo di vita’ delle sporgenze della membrana, dall’emergenza iniziale alla retrazione finale, utilizzando ad alta risoluzione, tridimensionale, time-lapse imaging ad alta risoluzione, tridimensionale, time-lapse. Il microscopio a foglio luminoso a reticolo ci ha permesso di vedere la nascita di pseudopodi lamellari come creste lineari, nonché la loro maturazione e coalescenza in forme più complesse.
Qual è il rapporto tra gli pseudopodi lamellari indipendenti di leucociti in rapido movimento e i lamellipod di cellule più lente? Diversi studi precedenti hanno descritto le “increspature” lamellipodiali libere che si staccano dal bordo anteriore dei fibroblasti o delle cellule epiteliali (Ingram,1969; Abercrombie et al., 1970; Abercrombie et al., 1971), ma la loro morfologia è stata generalmente assunta per riflettere l’attaccamento transitorio a una superficie (Burnette et al., 2014). È interessante notare che Ingram ha descritto le protrusioni ‘a lamina’ che crescono sul lato dorsale delle cellule di fibrosarcoma aderenti (Ingram,1969), e Gauthier e collaboratori hanno scoperto protrusioni ‘a pinna’ che viaggiano lungo la superficie dorsale dei fibroblasti aderenti mentre si avvolgono e strisciano lungo sottili nanofibre (Guetta-Terrieret al., 2015). Questi fogli e pinne non sembrano formarsi su un substrato esterno, ma la loro morfologia e la composizione molecolare suggeriscono che le cellule aderenti possono anche essere in grado di fare protrusioni lamellari libere, indipendenti dall’interazione superficiale. Anche se assomigliano a lamellipodia attaccati, i fogli pseudopodiali che descriviamo sono più spessi (~430-800 nm contro <200 nm (Abercrombieet al., 1971)) e crescono a ritmi più veloci (Ingram, 1969; Abercrombie et al., 1971). Inoltre, l’assemblaggio e la regolazione di fogli pseudopodiali indipendenti può richiedere diverse molecole regolatrici di actina(Fritz-Laylin et al., 2017). È necessario un ulteriore lavoro per comprendere le relazioni tra gli pseudopodi indipendenti dall’adesione e i lamellipodici delle cellule adesive.
Le sporgenze lamellari implicano l’organizzazione lineare delle molecole regolatrici
In che modo i leucociti in rapido movimento producono sporgenze simili a fogli, anche quando si muovono in ambienti tridimensionali complessi? La ricostituzione dell’assemblaggio di una rete di actina ramificata dipendente da Arp2/3 fornisce una semplice spiegazione: in vitro, la morfologia di una rete di actina ramificata è determinata dalla disposizione spaziale delle molecole regolatrici che attivano il complesso Arp2/3. La disposizione unidimensionale dell’attivatore Arp2/3 lungo una sottile fibra di vetro, ad esempio, produce una rete di actina piatta, simile a una lastra(Achard et al., 2010; Hu e Kuhn, 2012), mentre un quadrato bidimensionale dell’attivatore Arp2/3 produce un solido rettangolare più tridimensionale(Bieling et al., 2016). I modelli matematici della formazione di reti di actina ramificate concordano con questi risultati di ricostituzione e suggeriscono che, una volta formata, una rete di actina sottile e piatta dovrebbe rimanere stabile durante la sua crescita(Schmeiser e Winkler, 2015). Generalizzando, proponiamo che la dimensionalità approssimativa della nucleazione e dell’attività della polimerasi sulla membrana sia una in meno rispetto alla dimensionalità approssimativa della rete di actina (e della sporgenza) generata da questa attività. In altre parole, un filopodio (dimensione ~1) cresce da una sorgente puntiforme (dimensione ~0) di attività della nucleazione/polimerasi mentre uno pseudopode lamellare (dimensione ~2) cresce da una sorgente lineare (dimensione ~1). Proponiamo quindi che la natura bidimensionale delle sporgenze lamellari segua come diretta conseguenza dell’organizzazione unidimensionale della macchina di attivazione Arp2/3(Figura 7). Attualmente stiamo esplorando i possibili meccanismi che possono dare origine a questa organizzazione lineare degli attivatori Arp2/3.

Figura 7.Modello che mostra come la localizzazione dell’attivatore Arp2/3 (complesso WAVE attivato, verde) potrebbe portare alla formazione di una sporgenza lamellare indipendente dalle interazioni di superficie.Una volta formata, una sporgenza piatta continuerebbe a crescere come proiezione planare come descritto(Schmeiser e Winkler, 2015).
Questo modello per l’assemblaggio di pseudopodi piatti è supportato da precedenti osservazioni che gli attivatori dell’assemblaggio di actina Arp2/3-dipendente appaiono in modelli lineari sulla membrana. Quando osservati alla superficie della cellula dalla microscopia TIRF, gli attivatori diretti di Arp2/3 attività-WASP e l’onda di regolazione complessa forma dinamica, archi lineari che si propagano come onde viaggianti (Weineret al., 2007; Fritz-Laylin et al., 2017). Ad oggi, solo questi due attivatori Arp2/3 hanno dimostrato di formare linee, mentre ulteriori componenti a monte della cascata di attivazione, come il Rac, mostrano modelli di localizzazione più diffusi(Weiner et al., 2007; Yang et al., 2016).
Qui, mostriamo che queste “onde” WAVE si co-localizzano con regioni lineari di pseudopodi che premono vicino alla superficie del vetro, indicando le regioni di assemblaggio dell’actina e la generazione di forza. Proponiamo che queste zone di attivazione dinamica multipla Arp2/3 producano pseudopodi lamellari multipli che possono interagire per formare rosette complesse. Allo stesso modo, la recente microscopia del foglio di luce reticolare delle cellule di Ditostelio ha rivelato cerchi legati alla membrana del complesso WAVE (noto come SCAR in Ditostelio) iniziando la formazione di sporgenze cilindriche a forma di coppa durante la macropinocitosi(Veltman et al., 2016). Al contrario, le cellule che producono solo una singola linea stabile di attivatori Arp2/3 sul bordo anteriore (ad esempio cellule Drosophila S2[Kunda et al., 2003] e cellule epiteliali[Gautier et al., 2011]) producono solo una singola protrusione lamellare.
Figura 7.Modello che mostra come la localizzazione dell’attivatore Arp2/3 (complesso WAVE attivato, verde) potrebbe portare alla formazione di una protrusione lamellare indipendente dalle interazioni superficiali.Una volta formata, una sporgenza piatta continuerebbe a crescere come proiezione planare come descritto(Schmeiser e Winkler, 2015).
Gli pseudopodi lamellari come dispositivi efficienti per la ricerca dello spazio
Troviamo che, sebbene gli pseudopodi complessi non siano necessari per la migrazione delle cellule HL-60, essi giocano un ruolo nel cambiamento di direzione. Utilizzando metodi automatizzati per rilevare e misurare gli pseudopodi troviamo che le dimensioni e il numero di queste sporgenze della membrana aumentano drasticamente quando le cellule cambiano direzione e che il blocco della formazione di pseudopodi riduce la frequenza dei cambiamenti di direzione(Figura 6). Questi risultati forniscono una semplice spiegazione per il recente lavoro del laboratorio Sixt, che ha trovato che la riduzione dell’attività delle onde nelle cellule dendritiche abolisce la formazione di pseudopodi, ma accelera la strisciata delle cellule in semplici spazi ristretti(Leithner et al., 2016). È interessante notare che le cellule dendritiche pseudopod carenti di cellule dendritiche non riescono a riorientare in gradienti mutevoli di chemioattrattore, e migrano più lentamente verso una fonte chemiotattica all’interno di ambienti tridimensionali complessi rispetto alle cellule di controllo. Allo stesso modo, in un altro recente articolo, i laboratori Piel e Lennon-Dumenil hanno scoperto che l’attività Arp2/3 nella parte anteriore delle cellule dendritiche in migrazione è stata responsabile della velocità-fluttuante comportamento(Vargas et al., 2016). Insieme a questi risultati nelle cellule dendritiche, i nostri risultati suggeriscono che gli pseudopodi tridimensionali prodotti dai leucociti in rapido movimento promuovono la rotazione delle cellule necessaria per l’esplorazione dell’ambiente locale.
Oltre agli pseudopodi lamellari di leucociti e simili cellule in rapido movimento, altri tipi di protrusione avanzano il bordo anteriore di altri tipi di cellule striscianti, tra cui blebs sferiche(Charras e Paluch, 2008; Maugis et al., 2010) e filopodi lineari(Dent et al., 2007). Ci sono anche esempi di cellule individuali che passano rapidamente da un tipo di protrusione all’altro(Yoshida e Soldati, 2006; Bergert et al., 2012; Diz-Muñoz et al., 2016). Queste morfologie forniscono vantaggi funzionali e selettivi o rappresentano un elemento di casualità nell’evoluzione cellulare(Bonner, 2013)? Noi ipotizziamo che gli pseudopodia ampi e lamellari e le rosette estese possano essere strutture uniche per sondare l’ambiente di una cellula. Un foglio pseudopodiale è molto più sottile di una vescichetta sferica, ma può spazzare via un volume comparabile. Filopodia lineare, al contrario, dovrebbe muoversi sia avanti e indietro e da un lato all’altro per cercare lo stesso volume, e fornirebbe un contatto superficiale limitato.
La microscopia tridimensionale ad alta risoluzione richiede nuovi strumenti di visualizzazione
L’imaging tridimensionale time-lapse ad alta risoluzione spaziale e temporale pone sfide significative agli strumenti di analisi e al flusso di lavoro sviluppati per l’imaging bidimensionale. In particolare, la microscopia a foglio luminoso a reticolo richiede metodi efficaci per: (i) la gestione di grandi set di dati (~ 20 GByte per sequenza time-lapse); (ii) la normalizzazione, l’allineamento e la correzione della deriva di più canali di fluorescenza in tre dimensioni; (iii) l’identificazione di caratteristiche tridimensionali di interesse; e (iv) la quantificazione della forma e del movimento di superfici complesse. Il software open-source più utilizzato per l’analisi delle immagini non è adatto a questi compiti, e il software commerciale per il rendering di dati tridimensionali è costoso (~$10.000 per licenza, più tasse annuali) e difficile da modificare. Per affrontare questi problemi pratici abbiamo modificato UCSF Chimera per accettare set di dati tridimensionali di microscopia a foglio luminoso a reticolo. UCSF Chimera è una piattaforma software open-source liberamente disponibile, sviluppata per rendere e analizzare strutture atomiche tridimensionali(Pettersen et al., 2004) e mappe di densità(Goddard et al., 2007). La maggior parte degli strumenti che descriviamo qui sono disponibili nella recente release e incoraggiamo altri ad utilizzarli (Vedi Materiali e Metodi per l’elenco delle risorse online).
Nei film tridimensionali time-lapse, gli pseudopodi cambiano rapidamente forma, rendendoli difficili da identificare solo per morfologia. Abbiamo quindi sviluppato un nuovo metodo per il rilevamento automatizzato degli pseudopodi, che sfrutta una peculiarità biochimica del dominio dell’actina utrofina legante. L’utrofina si localizza nella rete di actina che forma la corteccia cellulare, ma è in gran parte esclusa dal rapido assemblaggio dei filamenti di actina ramificati creati dal complesso Arp2/3(Belin et al., 2014). La nostra sonda basata sull’utrofina localizza le reti di actina corticale in prossimità della membrana plasmatica ovunque, tranne che all’interno di reti di assemblaggio rapido che guidano l’estensione degli pseudopodi. Quando viene riprodotta in tre dimensioni e combinata con un marcatore per la membrana plasmatica, l’assenza di una sonda di actina basata sull’utrofina fornisce un metodo semplice per identificare gli pseudopodi dinamici e tracciare la loro dimensione e il loro movimento. Studi precedenti hanno descritto metodi per identificare e tracciare le sporgenze delle cellule in immagini bidimensionali(Bosgraaf e Van Haastert, 2010), ma, a nostra conoscenza, questo rappresenta il primo metodo automatizzato per identificare e misurare gli pseudopodi in tre dimensioni. Approcci simili si riveleranno probabilmente utili per definire altre strutture cellulari irregolari.
La moderna visualizzazione dei dati fornisce molto di più di semplici e graziose immagini; può facilitare l’analisi dei risultati sperimentali e la formulazione di domande scientifiche. Qui, la visualizzazione tridimensionale ha permesso il rilevamento automatico degli pseudopodi e l’analisi della relazione tra la formazione degli pseudopodi e la rotazione delle cellule. Inoltre, la rappresentazione della superficie ha risolto le complesse e apparentemente amorfe sporgenze nei fogli lamellari, un’osservazione che ha immediatamente suggerito un modello di sistema di segnalazione unidimensionale. Confidiamo che questi strumenti di visualizzazione e analisi forniranno progressi simili ad altre aree di indagine biologica.
Materiali e metodi
Linee cellulari
Filamento di actina corticale polimerizzata sono stati etichettati con un dominio di legame ad alta affinità di actina dall’utrofina fusa alla proteina fluorescente rossa mCherry(Belin et al., 2014), e membranaal plasma etichettata fondendo una sequenza di palmitilazione da Lyn chinasi(Inoue et al., 2005) alla proteina fluorescente verde mEmerald. Utrophin-mCherry HL-60 cellule sono state derivate da trasduzione lentivirale della linea cellulare #CCL-240 ottenuto dall’American Type Culture Center (ATCC), dove l’identità della linea cellulare è stata confermata da STR. HL-60 linee cellulari sono state coltivate in RPMI 1640 medio integrato con 15% FBS, 25 mM Hepes, e 2,0 g / L NaHCO3, e a 37C con il 5% di CO2. HL-60 linee cellulari HL-60 testato negativo per micoplasma sia da PCR e colorazione del DNA. Lentivirus è stato prodotto in HEK293T coltivato in piastre a 6 pozzetti e trasfettato con quantità uguali del vettore della spina dorsale lentivirale (un vettore di espressione proteica derivato da pHRSIN-CSGW(Demaison et al., 2002), clonando il dominio legante dell’actina dell’utrofano(Burkel et al., 2007) seguito da un linker flessibile (sequenza di aminoacidi: GDLELSRILTR) al N-termine di mCherry), pCMV∆8.91 (codifica dei geni essenziali del packaging) e pMD2.G (codifica del gene VSV-G in virus pseudotype). Dopo 48 ore, il supernatante da ogni pozzo è stato rimosso, centrifugato a 14.000 g per 5 minuti per rimuovere i detriti e poi incubato con ~ 1×10∧ 6 cellule HL-60 sospese in 1 mL RPMI completo per 5-12 ore. Terreno fresco è stato poi aggiunto e le cellule sono state recuperate per 3 giorni per consentire l’espressione della proteina bersaglio, e le cellule che esprimono sono stati selezionati da fluorescenza-attivati selezione delle cellule (FACS). Membrane-mEmerald e Lifeact-mCherry linee cellulari sono state derivate come sopra e fondendo la sequenza di palmitilazione da Lyn chinasi(Inoue et al., 2005) alla proteina verde fluorescente mEmerald, e il peptide Lifeact(Riedl et al., 2008) a mCherry, rispettivamente.
Prima di imaging, HL-60 cellule sono stati differenziati da un trattamento con 1,3% DMSO per 5 giorni. Per la migrazione bidimensionale, differenziato HL-60 cellule HL-60 sono stati autorizzati ad aderire a fibronectina rivestita coprivetrini per 30 minuti prima che coprivetrino è stato spostato nella camera di imaging. Per la migrazione tridimensionale, le cellule sono state sovrapposte su coprioggetti contenenti 1,7% di collagene preformato a matrice di collagene polimerizzato da una miscela 1:1 di collagene bovino non etichettato (catalogo Advanced Biomatrix n. 5005) e coniugato FITC (catalogo Sigma n. C4361), utilizzando protocolli standard(Sixt e Lämmermann, 2011). Le cellule sono state lasciate migrare nella rete per un’ora prima di rimuovere il coprioggetto nella camera di imaging. Le cellule sono stati ripresi in 1 × HBSS integrato con il 3% di FBS, 1 × penna / stelo, e 40 nM del tripeptide formil-MLP (per la migrazione di stimolazione). Le cellule trattate sono state esposte a 10 µm CK-666 (Sigma) o al solo vettore DMSO (controllo) per dieci minuti prima dell’imaging.
Lattice luce microscopia foglio foglio di luce di cellule viventi
Le sequenze time-lapse generate dal microscopio a fogli luminosi a reticolo comprendevano pile di immagini a due colori, raccolte attraverso interi volumi di cellule a intervalli di 1,3 s con un fotobleaching minimo e nessuna evidenza di fototossicità. La risoluzione spaziale dei dati risultanti era di circa 230 nm in XY e 370 nm in Z(Chen et al., 2014).
L’imaging delle cellule HL-60 è stato effettuato in microscopia a foglio luminoso a reticolo utilizzando fasci di Bessel disposti in una configurazione a reticolo quadrato in modalità dithered, come in(Chen et al., 2014). Per la migrazione 2D, le cellule differenziate HL-60 sono state lasciate aderire ai coprioggetti rivestiti in fibronectina per 30 minuti prima che il coprioggetti venisse spostato nella camera di imaging a 37°C. I dati sono stati acquisiti su una telecamera Hamamatsu ORCA-Flash 4.0 sCMOS, dove la cella in movimento è stata ripresa eccitando ogni piano con un laser a 488 nm a ~ 10 µW (all’apertura posteriore dell’obiettivo di eccitazione) per 5 ms, con un’eccitazione interna / esterna apertura numerica di 0,55/0,48 rispettivamente e una corrispondente lunghezza del foglio di luce di 15 µm. In ogni punto temporale, le celle sono state riprese con la modalità di scansione del campione e il foglio di luce dithered a passo di 400 nm, catturando così un volume di ~ 70 µm x 90 µm x 40 µm (corrispondente a 672 × 908 × 201 pixel in dati deskewed) ogni 1,2 s (che include 1 s di tempo di acquisizione e 0,2 s di pausa tra ogni punto temporale). Ci sono 143 punti di tempo per i periodi di imaging continuo di 2,86 min di durata. Per la migrazione tridimensionale, mCherry-utrofina HL-60 cellule sono stati sovrapposti su coprioggetti contenenti pre-formati 1,7% collagene a matrice polimerizzata come descritto (14) utilizzando il 50% FITC-collagene (Sigma) e ha permesso di migrare nella rete per un’ora prima di rimuovere il coprioggetti alla camera di imaging. La cella in movimento all’interno del collagene è stato ripreso da eccitare ogni piano con un laser 488 nm (collagene) e 568 nm laser (HL-60) a ~ 20 µW e 10 µW (all’apertura posteriore dell’obiettivo di eccitazione) per 5 ms separatamente prima di passare al piano z successivo, con un’eccitazione interna / esterna apertura numerica di 0,4 / 0,325 rispettivamente e una corrispondente lunghezza del foglio di luce di 20 µm. In ogni punto di tempo, le celle sono state immaginate in modalità di scansione obiettivo e il foglio di luce dithered a passo di 250 nm, catturando così un volume di ~ 70 µm x 36 µm x 35 µm (corrispondente a 672 × 352 × 141 pixel in dati grezzi) ogni 1,4 s (che include 1,3 s di tempo di acquisizione e 0,1 s di pausa tra ogni punto di tempo). Ci sono 250 punti di tempo per i periodi di imaging continuo di 5,83 min di durata.
Allineamento, compressione e normalizzazione dell’immagine
Con i set di dati che abbiamo raccolto, il volume dell’immagine era in genere cinque volte più grande della cella lungo ogni asse, in modo che la cella non sfuggisse alla vista. Per consentire la riproduzione in tempo reale da qualsiasi punto di vista abbiamo usato la compressione e la trebbiatura dei dati dell’immagine per ridurne le dimensioni di un fattore 100, utilizzando il fatto che la cella in qualsiasi istante occupava solo una piccola porzione della scatola. Per compensare la deriva del telescopio abbiamo allineato le caratteristiche statiche (filamenti di collagene extracellulare) di ogni immagine tridimensionale al tempo precedente massimizzando la correlazione incrociata per correggere il jitter del microscopio. Abbiamo normalizzato i livelli di intensità spostando la media a 0 e scalando l’intensità per ogni immagine tridimensionale in modo che il volume della cella chiusa fosse costante ad un valore di intensità standard (scelto arbitrariamente pari a 100). Questo ha corretto le variazioni di normalizzazione introdotte dal software di deconvoluzione del microscopio e ha anche compensato il fotobleaching che ha ridotto i livelli di intensità di circa un fattore di 2 per i periodi di tempo ripresi.
Rendering delle immagini
Le sporgenze delle cellule osservate subiscono continui cambiamenti di forma e possono emergere da tutti i lati della cellula. Per comunicare queste caratteristiche in immagini statiche bidimensionali abbiamo esportato la mesh di superficie da UCSF Chimera come file Wavefront (.obj), e le abbiamo importate in Cinema4D, un pacchetto software di animazione tridimensionale. Abbiamo usato la tecnica di ombreggiatura dell’occlusione ambientale (le aree rientranti appaiono scure) e lo Sketch e il Toon Shader entrambi con impostazioni predefinite per visualizzare chiaramente il comportamento dinamico tridimensionale (Figura 1-figure supplement 3).
Visualizzazione in tempo reale dei dati con il toolkit ‘vseries’.
Abbiamo sviluppato il software ‘vseries’ all’interno di UCSF Chimera per analizzare i grandi set di dati delle celle di scansione. Il toolkit ‘vseries’ include l’implementazione a riga di comando e una GUI della serie Volume. Esso consente agli utenti di visualizzare e manipolare una sequenza ordinata di serie di dati volumetrici e di applicare i comandi di analisi ad alcune o a tutte le mappe volumetriche in una sola volta. Abbiamo utilizzato la visualizzazione interattiva tridimensionale per vedere tutti gli aspetti del movimento della cella, consentendo di esaminare qualsiasi caratteristica in qualsiasi momento del movimento della cella. Le nuove capacità di visualizzazione e analisi del software sono state distribuite come comando ‘vseries’ del programma di visualizzazione UCSF Chimera. Per contrassegnare le caratteristiche di interesse abbiamo anche sviluppato visualizzazioni tridimensionali interattive di superficie per la visualizzazione in un browser web con la possibilità di posizionare a mano i marcatori (piccole sfere) sulle caratteristiche della cella desiderata, e annotarli con descrizioni di testo e scelta del colore. Questo software consente l’annotazione collaborativa online dei dati.
Quantificazione dello spessore della sporgenza e della planarità
Per misurare lo spessore delle celle tridimensionali renderizzate, sono state importate in Cinema4D delle maglie di superficie e sono stati creati dei marcatori per misurare la distanza dall’alto verso il basso lungo il bordo di un foglio pseudopodiale. La planarità è stata misurata in UCSF Chimera posizionando prima i marcatori lungo il bordo del foglio usando la funzione ‘Markers’(http://www.rbvi.ucsf.edu/chimera/current/docs/ContributedSoftware/volumepathtracer/framevolpath.html) e poi eseguendo uno script Python che calcola il piano più adatto e misura le distanze dal piano ai marcatori. Abbiamo anche misurato il bordo sottile dei fogli lamellari che intersecano le immagini del foglio di luce reticolare grezzo a 90° (567 ± 64 nm su vetro e 689 ± 86 nm in collagene) a metà dell’intensità massima di fluorescenza). Abbiamo anche misurato l’actina macchiata di falloidina, riprodotta con la microscopia confocale (505 ± 49 nm per un foglio e 533 ± 123 nm per un petalo di rosetta) che interseca il piano confocale a 90°, misurata a metà dell’intensità massima di fluorescenza.
Quantificazione del movimento delle cellule e dell’attività di protrusione
Per quantificare i movimenti abbiamo misurato la posizione del centroide della cella in funzione del tempo per ottenere velocità, direzione e cambi di direzione. Questa capacità è ora inclusa come comando ‘vseries measure’. Per ogni volume della serie di volumi, il comando calcola le coordinate del centroide (x,y,z), la distanza dal centroide precedente (‘passo’), la distanza cumulativa lungo il percorso lineare del pezzo dal primo centroide, il volume chiuso in superficie e la superficie. I risultati sono salvati in un file di testo. Il centroide è il centro di massa della mappa di densità in base alle regioni della mappa al di sopra della soglia; abbiamo impostato la soglia al nostro già stabilito livello normalizzato di 100.
Quantificazione dell’attività di sporgenza
Per automatizzare l’identificazione delle protuberanze di forma irregolare, abbiamo approfittato del fatto che l’etichettatura dell’actina corticale a base di utrofine entrava lentamente nelle protuberanze nascenti, mentre l’etichettatura a membrana era sempre presente nelle protuberanze. Abbiamo calcolato le mappe delle differenze tra i canali della membrana e dell’actina, abbiamo applicato una levigatura spaziale utilizzando un filtro gaussiano con una deviazione standard di 0,5 µm per ridurre il rumore. I volumi chiusi risultanti sono stati marcati come sporgenze individuali se erano maggiori di 1 µm3, meno di 15 µm dal centro, e il raggio del centro della cella esterna o maggiore di 15 µm3. Per ogni volume della serie di volumi abbiamo calcolato il volume totale di tutte le sporgenze, la distanza di ogni sporgenza dal centro della cella, l’angolo tra la linea che collega il centroide della cella al centroide della sporgenza e il percorso del centroide della cella, e il numero di sporgenze. Questo calcolo è stato implementato come script Python(Figura 6-codice sorgente 1).
Quantificazione dei punti di svolta
Cambiamenti di direzione delle cellule è apparso correlato con l’emergere di sporgenze multiple in diverse direzioni. Per esaminare questa correlazione abbiamo usato l’analisi dei componenti principali per trovare il piano primario di movimento per la rappresentazione grafica di un percorso del centroide cellulare in due dimensioni. Il primo metodo che abbiamo usato per quantificare i cambiamenti di direzione è stato quello di calcolare il rapporto della finestra temporale di scorrimento della lunghezza del percorso sulla distanza euclidea tra i punti finali utilizzando un intervallo di 10 punti temporali. Un rapporto maggiore di 2,5 è stato considerato di virata. Il nostro secondo approccio è stato un filtro Savitzky-Golay per calcolare una stima della derivata. I punti temporali erano considerati punti di svolta se la derivata era pari a zero e la cella aveva percorso almeno 1 µm dall’ultimo punto di svolta. Entrambi questi metodi sono stati scritti in R Studio(Figura 6-codice sorgente 2).
Microscopia convenzionale
La microscopia confocale RICM, TIRF e la microscopia a disco rotante sono state eseguite su un microscopio invertito Nikon Ti-E dotato di Spectral Diskovery e di un Andor iXon Ultra EMCCD. L’illuminazione RICM (chiamata anche ‘microscopia a riflessione di interferenza’) è stata prodotta con un beamsplitter 50/50 (Chroma 21000) nel cubo dicroico e un filtro di eccitazione a 550 nm (Thorlabs FB550-10) dopo una lampada alogena ad epi. Sufficienti filtri di densità neutra sono stati posizionati dopo la lampada alogena e il diaframma di apertura è stato regolato per massimizzare il contrasto delle immagini RICM. Le immagini RICM e TIRF sono state interlacciate a meno di 500 ms di distanza l’una dall’altra ruotando il cubo RICM dentro e fuori dal percorso ogni altro fotogramma. Tutto l’hardware è stato controllato utilizzando il software Micro-Manager(Edelstein et al., 2010). L’analisi delle immagini di RICM, TIRF e dei dati confocali del disco rotante è stata eseguita utilizzando ImageJ, se non diversamente specificato.
Microscopia elettronica a scansione
I linfociti Jurkat T sono stati ottenuti dalla American Type Culture Collection (ATCC) che conferma l’identità della linea cellulare tramite STR, e sono stati coltivati in sospensione in RPMI 1640 (Gibco) integrato con il 5% di siero bovino fetale (Atlanta Biologicals Inc), 2 mg/mL di glucosio, 1 mM di piruvato di sodio (Gibco) e 50 uM di beta-mercaptoetanolo (Gibco), e sono stati mantenuti a densità <1 milione di cellule/mL. Le cellule sono state fissate mescolando 1 milione di cellule con 1 vol di glutaraldeide al 7% di glutaraldeide (Electron Microscopy Sciences Inc) in 100 mM di fosfato di sodio a pH 7,4 mM e incubato 1 ora a 23 gradi C. Le cellule fisse sono stati lavati 5x con PBS (centrifugazione a 300xg per 5 minuti per pellet le cellule), poi risospeso in 100 uL PBS e ha permesso di stabilirsi per 20 minuti a 23 gradi C su 12 mm round coprivetrini (Fisher) pre-rivestito con 0,1% poli-L-lisina (Sigma-Aldrich). Le cellule sono state ri-fissate sul coprioggetti con 3,5% di glutaraldeide in PBS per 15 minuti, seguito da tre lavaggi con 20 mM di fosfato di sodio a pH 7,5, e la colorazione con 1% di tetrossido di osmio (Electron Microscopy Sciences Inc) in 100 mM di fosfato di sodio a pH 7,5. I coprioggetti sono stati lavati una volta con acqua, poi disidratati in sequenza con 30, 50, 70, 95, e 100% di etanolo. I coprivetrini sono stati lavati 2 × 5 min con diluizioni 1:1 e 1:2 di etanolo: esametildisilazano (HMDS; Sigma), poi 3 × 20 min con HMDS al 100%. I coperchi sono stati essiccati durante la notte sotto vuoto, poi hanno aderito a stub SEM (Electron Microscopy Sciences), rivestiti con uno strato di 3 nm di tetraossido di osmio utilizzando un OPC-60 osmio plasma coater (SPI Supplies), e ripresi in modalità SE con un FEI XL-30 FEG-ESEM FEI utilizzando una tensione di accelerazione di 5 kV, spot size 3.
Risorse per iniziare a utilizzare Chimera per i dati di microscopia ottica 3D
Sito web UCSF Chimera e download. https://www.cgl.ucsf.edu/chimera/
Vedi anche ChimeraX, la prossima generazione di software Chimera, che include il comando vseries. Supporta l’illuminazione dell’occlusione ambientale che non è disponibile in Chimera.http://www.cgl.ucsf.edu/chimerax/
Tutorial per l’uso generale di Chimera (altamente raccomandato per i nuovi utenti) www.rbvi.ucsf.edu/chimera/current/docs/UsersGuide/
Chimera Volume serie di documentazione dell’interfaccia grafica utente della serie Chimera, per la riproduzione di dati di microscopia ottica 5d. https://www.cgl.ucsf.edu/chimera/docs/ContributedSoftware/volseries/volseries.html
Documentazione dei comandi di Chimera vseries. Comando per riprodurre ed elaborare i dati delle immagini 5d (normalizzare le intensità, rimuovere il jitter, estrarre le sottoregioni, la soglia, la maschera, la compressione).https://www.cgl.ucsf.edu/chimera/docs/UsersGuide/midas/vseries.html
References
- Abercrombie M, Heaysman JE, Pegrum SM. The locomotion of fibroblasts in culture. II. "RRuffling". Experimental cell research. 1970; 60:437-444. DOI | PubMed
- Abercrombie M, Heaysman JE, Pegrum SM. The locomotion of fibroblasts in culture. IV. Electron microscopy of the leading lamella. Experimental cell research. 1971; 67:359-367. DOI | PubMed
- Achard V, Martiel JL, Michelot A, Guérin C, Reymann AC, Blanchoin L, Boujemaa-Paterski R. A "primer"-based mechanism underlies branched actin filament network formation and motility. Current Biology. 2010; 20:423-428. DOI | PubMed
- Bear JE, Rawls JF, Saxe CL. SCAR, a WASP-related protein, isolated as a suppressor of receptor defects in late Dictyostelium development. The Journal of Cell Biology. 1998; 142:1325-1335. DOI | PubMed
- Belin BJ, Goins LM, Mullins RD. Comparative analysis of tools for live cell imaging of actin network architecture. BioArchitecture. 2014; 4:189-202. DOI | PubMed
- Bergert M, Chandradoss SD, Desai RA, Paluch E. Cell mechanics control rapid transitions between blebs and lamellipodia during migration. PNAS. 2012; 109:14434-14439. DOI | PubMed
- Bieling P, Li TD, Weichsel J, McGorty R, Jreij P, Huang B, Fletcher DA, Mullins RD. Force Feedback Controls Motor Activity and Mechanical Properties of Self-Assembling Branched Actin Networks. Cell. 2016; 164:115-127. DOI | PubMed
- Bonner JT. Randomness in Evolution. Princeton University Press; 2013.
- Bosgraaf L, Van Haastert PJ. Quimp3, an automated pseudopod-tracking algorithm. Cell Adhesion & Migration. 2010; 4:46-55. DOI | PubMed
- Brown MJ, Nijhara R, Hallam JA, Gignac M, Yamada KM, Erlandsen SL, Delon J, Kruhlak M, Shaw S. Chemokine stimulation of human peripheral blood T lymphocytes induces rapid dephosphorylation of ERM proteins, which facilitates loss of microvilli and polarization. Blood. 2003; 102:3890-3899. DOI | PubMed
- Buenemann M, Levine H, Rappel WJ, Sander LM. The role of cell contraction and adhesion in dictyostelium motility. Biophysical Journal. 2010; 99:50-58. DOI | PubMed
- Burkel BM, von Dassow G, Bement WM. Versatile fluorescent probes for actin filaments based on the actin-binding domain of utrophin. Cell Motility and the Cytoskeleton. 2007; 64:822-832. DOI | PubMed
- Burnette DT, Shao L, Ott C, Pasapera AM, Fischer RS, Baird MA, Der Loughian C, Delanoe-Ayari H, Paszek MJ, Davidson MW, Betzig E, Lippincott-Schwartz J. A contractile and counterbalancing adhesion system controls the 3D shape of crawling cells. The Journal of Cell Biology. 2014; 205:83-96. DOI | PubMed
- Butler KL, Ambravaneswaran V, Agrawal N, Bilodeau M, Toner M, Tompkins RG, Fagan S, Irimia D. Burn injury reduces neutrophil directional migration speed in microfluidic devices. PLoS One. 2010; 5DOI | PubMed
- Case LB, Waterman CM. Integration of actin dynamics and cell adhesion by a three-dimensional, mechanosensitive molecular clutch. Nature Cell Biology. 2015; 17:955-963. DOI | PubMed
- Charras G, Paluch E. Blebs lead the way: how to migrate without lamellipodia. Nature Reviews Molecular Cell Biology. 2008; 9:730-736. DOI | PubMed
- Chen BC, Legant WR, Wang K, Shao L, Milkie DE, Davidson MW, Janetopoulos C, Wu XS, Hammer JA, Liu Z, English BP, Mimori-Kiyosue Y, Romero DP, Ritter AT, Lippincott-Schwartz J, Fritz-Laylin L, Mullins RD, Mitchell DM, Bembenek JN, Reymann AC, Böhme R, Grill SW, Wang JT, Seydoux G, Tulu US, Kiehart DP, Betzig E. Lattice light-sheet microscopy: imaging molecules to embryos at high spatiotemporal resolution. Science. 2014; 346DOI | PubMed
- Coates PT, Barratt-Boyes SM, Zhang L, Donnenberg VS, O’Connell PJ, Logar AJ, Duncan FJ, Murphey-Corb M, Donnenberg AD, Morelli AE, Maliszewski CR, Thomson AW. Dendritic cell subsets in blood and lymphoid tissue of rhesus monkeys and their mobilization with Flt3 ligand. Blood. 2003; 102:2513-2521. DOI | PubMed
- Demaison C, Parsley K, Brouns G, Scherr M, Battmer K, Kinnon C, Grez M, Thrasher AJ. High-level transduction and gene expression in hematopoietic repopulating cells using a human immunodeficiency [correction of imunodeficiency] virus type 1-based lentiviral vector containing an internal spleen focus forming virus promoter. Human Gene Therapy. 2002; 13:803-813. DOI | PubMed
- Dent EW, Kwiatkowski AV, Mebane LM, Philippar U, Barzik M, Rubinson DA, Gupton S, Van Veen JE, Furman C, Zhang J, Alberts AS, Mori S, Gertler FB. Filopodia are required for cortical neurite initiation. Nature Cell Biology. 2007; 9:1347-1359. DOI | PubMed
- Diz-Muñoz A, Romanczuk P, Yu W, Bergert M, Ivanovitch K, Salbreux G, Heisenberg CP, Paluch EK. Steering cell migration by alternating blebs and actin-rich protrusions. BMC Biology. 2016; 14DOI | PubMed
- Edelstein A, Amodaj N, Hoover K, Vale R, Stuurman N. Computer control of microscopes using µManager. Current protocols in molecular biology. 2010; Chapter 14DOI | PubMed
- Felts RL, Narayan K, Estes JD, Shi D, Trubey CM, Fu J, Hartnell LM, Ruthel GT, Schneider DK, Nagashima K, Bess JW, Bavari S, Lowekamp BC, Bliss D, Lifson JD, Subramaniam S. 3D visualization of HIV transfer at the virological synapse between dendritic cells and T cells. PNAS. 2010; 107:13336-13341. DOI | PubMed
- Fisher PJ, Bulur PA, Vuk-Pavlovic S, Prendergast FG, Dietz AB. Dendritic cell microvilli: a novel membrane structure associated with the multifocal synapse and T-cell clustering. Blood. 2008; 112:5037-5045. DOI | PubMed
- Fleck RA, Romero-Steiner S, Nahm MH. Use of HL-60 cell line to measure opsonic capacity of pneumococcal antibodies. Clinical and Vaccine Immunology. 2005; 12:19-27. DOI | PubMed
- Fritz-Laylin LK, Lord SJ, Mullins RD. WASP and SCAR are evolutionarily conserved in actin-filled pseudopod-based motility. The Journal of Cell Biology. 2017; 216:1673-1688. DOI | PubMed
- Gao L, Shao L, Chen BC, Betzig E. 3D live fluorescence imaging of cellular dynamics using Bessel beam plane illumination microscopy. Nature Protocols. 2014; 9:1083-1101. DOI | PubMed
- Gao L, Shao L, Higgins CD, Poulton JS, Peifer M, Davidson MW, Wu X, Goldstein B, Betzig E. Noninvasive imaging beyond the diffraction limit of 3D dynamics in thickly fluorescent specimens. Cell. 2012; 151:1370-1385. DOI | PubMed
- Gautier JJ, Lomakina ME, Bouslama-Oueghlani L, Derivery E, Beilinson H, Faigle W, Loew D, Louvard D, Echard A, Alexandrova AY, Baum B, Gautreau A. Clathrin is required for Scar/Wave-mediated lamellipodium formation. Journal of Cell Science. 2011; 124:3414-3427. DOI | PubMed
- Gerisch G, Ecke M, Neujahr R, Prassler J, Stengl A, Hoffmann M, Schwarz US, Neumann E. Membrane and actin reorganization in electropulse-induced cell fusion. Journal of Cell Science. 2013; 126:2069-2078. DOI | PubMed
- Goddard TD, Huang CC, Ferrin TE. Visualizing density maps with UCSF Chimera. Journal of Structural Biology. 2007; 157:281-287. DOI | PubMed
- Guetta-Terrier C, Monzo P, Zhu J, Long H, Venkatraman L, Zhou Y, Wang P, Chew SY, Mogilner A, Ladoux B, Gauthier NC. Protrusive waves guide 3D cell migration along nanofibers. The Journal of Cell Biology. 2015; 211:683-701. DOI | PubMed
- Hu X, Kuhn JR. Actin filament attachments for sustained motility in vitro are maintained by filament bundling. PLoS One. 2012; 7DOI | PubMed
- Ingram VM. A side view of moving fibroblasts. Nature. 1969; 222:641-644. DOI | PubMed
- Inoue T, Heo WD, Grimley JS, Wandless TJ, Meyer T. An inducible translocation strategy to rapidly activate and inhibit small GTPase signaling pathways. Nature Methods. 2005; 2:415-418. DOI | PubMed
- Kunda P, Craig G, Dominguez V, Baum B. Abi, Sra1, and Kette control the stability and localization of SCAR/WAVE to regulate the formation of actin-based protrusions. Current Biology. 2003; 13:1867-1875. DOI | PubMed
- Leithner A, Eichner A, Müller J, Reversat A, Brown M, Schwarz J, Merrin J, de Gorter DJ, Schur F, Bayerl J, de Vries I, Wieser S, Hauschild R, Lai FP, Moser M, Kerjaschki D, Rottner K, Small JV, Stradal TE, Sixt M. Diversified actin protrusions promote environmental exploration but are dispensable for locomotion of leukocytes. Nature Cell Biology. 2016; 18:1253-1259. DOI | PubMed
- Loomis WF, Fuller D, Gutierrez E, Groisman A, Rappel WJ. Innate non-specific cell substratum adhesion. PLoS ONE. 2012; 7DOI | PubMed
- Lämmermann T, Bader BL, Monkley SJ, Worbs T, Wedlich-Söldner R, Hirsch K, Keller M, Förster R, Critchley DR, Fässler R, Sixt M. Rapid leukocyte migration by integrin-independent flowing and squeezing. Nature. 2008; 453:51-55. DOI | PubMed
- Majstoravich S, Zhang J, Nicholson-Dykstra S, Linder S, Friedrich W, Siminovitch KA, Higgs HN. Lymphocyte microvilli are dynamic, actin-dependent structures that do not require Wiskott-Aldrich syndrome protein (WASp) for their morphology. Blood. 2004; 104:1396-1403. DOI | PubMed
- Maugis B, Brugués J, Nassoy P, Guillen N, Sens P, Amblard F. Dynamic instability of the intracellular pressure drives bleb-based motility. Journal of Cell Science. 2010; 123:3884-3892. DOI | PubMed
- Nicholson-Dykstra SM, Higgs HN. Arp2 depletion inhibits sheet-like protrusions but not linear protrusions of fibroblasts and lymphocytes. Cell Motility and the Cytoskeleton. 2008; 65:904-922. DOI | PubMed
- Nolen BJ, Tomasevic N, Russell A, Pierce DW, Jia Z, McCormick CD, Hartman J, Sakowicz R, Pollard TD. Characterization of two classes of small molecule inhibitors of Arp2/3 complex. Nature. 2009; 460:1031-1034. DOI | PubMed
- Petrie RJ, Yamada KM. Fibroblasts Lead the Way: A Unified View of 3D Cell Motility. Trends in Cell Biology. 2015; 25:666-674. DOI | PubMed
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera–a visualization system for exploratory research and analysis. Journal of Computational Chemistry. 2004; 25:1605-1612. DOI | PubMed
- Preston TM, King CA. An experimental study of the interaction between the soil amoeba Naegleria gruberi and a glass substrate during amoeboid locomotion. Journal of cell science. 1978; 34:145-158. PubMed
- Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, Sixt M, Wedlich-Soldner R. Lifeact: a versatile marker to visualize F-actin. Nature Methods. 2008; 5:605-607. DOI | PubMed
- Schmeiser C, Winkler C. The flatness of Lamellipodia explained by the interaction between actin dynamics and membrane deformation. Journal of Theoretical Biology. 2015; 380:144-155. DOI | PubMed
- Sixt M, Lämmermann T. In vitro analysis of chemotactic leukocyte migration in 3D environments. Methods in molecular biology. 2011; 769:149-165. DOI | PubMed
- Sullivan JA, Mandell GL. Motility of human polymorphonuclear neutrophils: microscopic analysis of substrate adhesion and distribution of F-actin. Cell Motility. 1983; 3:31-46. DOI | PubMed
- Tarini M, Cignoni P, Montani C. Ambient occlusion and edge cueing to enhance real time molecular visualization. IEEE Transactions on Visualization and Computer Graphics. 2006; 12:1237-1244. DOI | PubMed
- Van Haastert PJ. Amoeboid cells use protrusions for walking, gliding and swimming. PLoS One. 2011; 6DOI | PubMed
- van Leewenhoeck A. Observations, Communicated to the Publisher by Mr. Antony van Leewenhoeck, in a Dutch Letter of the 9th of Octob. 1676. Here English’d: concerning Little Animals by Him Observed in Rain-Well-Sea. and Snow Water; as Also in Water Wherein Pepper Had Lain Infused. Philosophical Transactions of the Royal Society of London. 1677; 12:821-831. DOI
- Vargas P, Maiuri P, Bretou M, Sáez PJ, Pierobon P, Maurin M, Chabaud M, Lankar D, Obino D, Terriac E, Raab M, Thiam HR, Brocker T, Kitchen-Goosen SM, Alberts AS, Sunareni P, Xia S, Li R, Voituriez R, Piel M, Lennon-Duménil AM. Innate control of actin nucleation determines two distinct migration behaviours in dendritic cells. Nature Cell Biology. 2016; 18:43-53. DOI | PubMed
- Veltman DM, Williams TD, Bloomfield G, Chen BC, Betzig E, Insall RH, Kay RR. A plasma membrane template for macropinocytic cups. eLife. 2016; 5DOI | PubMed
- Weiner OD, Marganski WA, Wu LF, Altschuler SJ, Kirschner MW. An actin-based wave generator organizes cell motility. PLoS Biology. 2007; 5DOI | PubMed
- Weiner OD, Rentel MC, Ott A, Brown GE, Jedrychowski M, Yaffe MB, Gygi SP, Cantley LC, Bourne HR, Kirschner MW. Hem-1 complexes are essential for Rac activation, actin polymerization, and myosin regulation during neutrophil chemotaxis. PLoS Biology. 2006; 4DOI | PubMed
- Yang HW, Collins S, Meyer T. Locally excitable Cdc42 signals steer cells during chemotaxis. Nature Cell Biology. 2016; 18:191-201. DOI | PubMed
- Yoshida K, Soldati T. Dissection of amoeboid movement into two mechanically distinct modes. Journal of Cell Science. 2006; 119:3833-3844. DOI | PubMed
- Zigmond SH, Sullivan SJ. Sensory adaptation of leukocytes to chemotactic peptides. The Journal of Cell Biology. 1979; 82:517-527. DOI | PubMed
Fonte
Fritz-Laylin LK, Riel-Mehan M, Chen B, Lord SJ, Goddard TD, et al. () Actin-based protrusions of migrating neutrophils are intrinsically lamellar and facilitate direction changes. eLife 6e26990. https://doi.org/10.7554/eLife.26990




