
Abstract
Introduzione
Calcio / calmodulino-dipendente proteina chinasi II (CAMK2) è un calcio-attivati serina / treonina chinasi che è estremamente abbondante nel cervello, che comprende fino al 0,3% del contenuto totale di proteine cerebrali(Bennett et al., 1983). CAMK2 è altamente arricchito alle sinapsi ed è necessario per il processo di potenziamento a lungo termine (LTP), il rafforzamento dipendente dall’attività e la modulazione dell’attività sinaptica che si pensa sia la base molecolare di alcune forme di apprendimento e di memoria(Kandel et al., 2014; Lisman et al., 2002). Negli esseri umani, ci sono quattro geni che codificano isoenzimi distinti di CAMK2. CAMK2A e CAMK2B sono le isoforme predominanti nel sistema nervoso, con CAMK2A espresso 3-4 volte superiore a CAMK2B (Hansone Schulman, 1992). Ogni enzima comprende un dominio chinasi, un dominio di regolazione e un dominio di associazione. Strutturalmente gli oloenzimi CAMK2 sono omo- o etero-oligomeri, costituiti da 12 o 14 sottounità CAMK2(Hudmon e Schulman, 2002b). L’assemblaggio dell’oloenzima richiede il dominio di associazione carbossi-terminale, che forma coppie impilate di anelli esamerici o eptamerici con il dominio regolatorio e chinasi che proiettano radialmente per interagire con altre proteine essenziali per la funzione e la localizzazione di CAMK2(Bhattacharyya et al., 2016). In assenza di segnale di calcio, CAMK2 è inattivo, in quanto il dominio di regolazione inibisce la funzione chinasi(Yang e Schulman, 1999). Questa auto-inibizione è alleviata quando la calmodulina carica di calcio si lega al dominio di regolazione, esponendo così il dominio chinasi e permettendogli di fosforilare i substrati bersaglio(Meador et al., 1993). Ca2+-Calmodulina vincolante espone anche Thr286 sul dominio di regolazione di CAMK2A, che diventa fosforilato in trans dalle sottounità adiacenti. Una volta che questo residuo è fosforilato, l’enzima è costantemente attivo, anche in assenza di una continua segnalazione di Ca2+-Calmodulina(Rich e Schulman, 1998; Stratton et al., 2013). Si ritiene che questo passaggio dall’autoinibizione all’attività autonoma e persistente costituisca una forma biochimica della memoria, che segna il neurone per aver sperimentato un precedente afflusso di calcio(Bhattacharyya et al., 2016; Stratton et al., 2014; Stratton et al., 2013).
I topi che sono omozigoti null per CAMK2A sono vitali e mostrano una memoria spaziale compromessa e LTP ridotta nell’ippocampo(Silva et al., 1992a, 1992b). Il mutante eterozigote(Camk2a+/-) mostrano un deficit significativo nella memoria di lavoro spaziale e nella memoria della paura contestuale(Frankland et al., 2001; Matsuo et al., 2009). Più recentemente, è stato dimostrato che i topi con knock-out condizionale specifico per i neuroni di CAMK2A mostravano analogamente deficit di apprendimento e difetti di LTP paragonabili a quelli dei topi knockout completi(Achterberg et al., 2014). Questi risultati suggeriscono che la funzione neurone-intrinseca CAMK2A è indispensabile durante il periodo di apprendimento per la formazione della memoria. Il CAMK2 è conservato in invertebrati, come D. melanogaster e C. elegans, dove la chinasi gioca anche ruoli critici nei tratti comportamentali e cognitivi(Cho et al., 1991; Hudmon e Schulman, 2002a; Reiner et al., 1999; Rongo e Kaplan, 1999). In C. elegans, l’unico CAMK2 è codificato dal gene unc-43, essenziale per la funzione sinaptica(Rongo e Kaplan, 1999). La perdita di unc-43 fa sì che i vermi abbiano un tono muscolare flaccido, difetti di locomozione e contrazioni spontanee del corpo che assomigliano a crisi epilettiche(Reiner et al., 1999).
Nell’assistenza pediatrica, il ritardo globale dello sviluppo nei neonati è definito come un ritardo funzionale significativo in due o più domini dello sviluppo, tra cui la funzione motoria lorda e fine, il linguaggio, la parola e il linguaggio, la cognizione, lo sviluppo sociale e le abilità personali(Quality Standards Subcommittee dell’American Academy of Neurology et al., 2003). Questi difetti vengono rilevati in tenera età nei bambini di età inferiore ai cinque anni e possono persistere per tutta la vita(Shevell, 2008). Circa il 25-50% dei casi identificati sono causati da alterazioni genetiche della linea germinale, tra cui anomalie cromosomiche, varianti del numero di copie e mutazioni monogeniche (Sroure Shevell, 2014; van Bokhoven, 2011). Per molti pazienti con ritardo nello sviluppo neurologico globale, l’eziologia genetica rimane sconosciuta.
Risultati
Qui, riportiamo l’identificazione di una famiglia consanguinea proveniente dalla Giordania con due bambini affetti da ritardo nello sviluppo neurologico globale con frequenti crisi epilettiche e convulsioni. I due fratelli colpiti non avevano caratteristiche dismorfiche, ma non sono riusciti a sviluppare la capacità di camminare o parlare(Figura 1A,B, Figura 1-figure supplement 1A). Hanno mostrato un progressivo ritardo psicomotorio con muscoli ipotonici (Materiale supplementare, Video 1 e 2). Elettroencefalogramma (EEG) l’analisi ha rivelato anormale epilettiforme transitori(Figura 1C, Figura 1-figure supplemento 1B), coerente con frequenti crisi miocloniche. Risonanza magnetica per immagini (RM) la scansione non ha mostrato difetti strutturali importanti nel cervello di proband II: 4(Figura 1-figure supplemento 1C). I livelli di metaboliti nel siero erano normali, escludendo potenziali disturbi di stoccaggio lisosomiali.
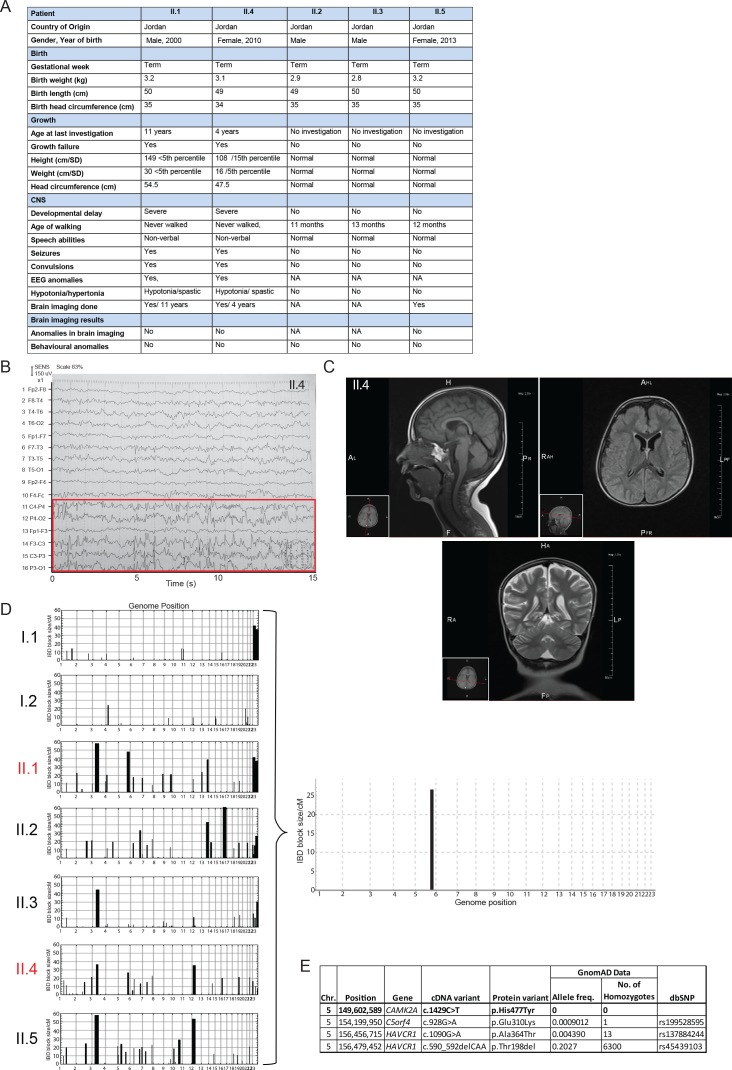
Figura 1-figure supplement 1.Una nuova sindrome di ritardo globale dello sviluppo neurologico con convulsioni causate da una mutazione biallelica in CAMK2A.Genetic e risultati clinici dei due pazienti con ritardo globale dello sviluppo.2.(A) Pedigree di una famiglia giordana consanguinea con due fratelli affetti da mutazioni omozigoti in CAMK2A. I genotipi di tutti gli individui sono stati verificati con il sequenziamento Sanger.(B) Fotografie dei due fratelli affetti con circonferenze craniche normali.(C) Grafico EEG del paziente II.I che mostra i transitori epilettici anormali (scatole rosse)(D) Mappatura omozigosi delinea un candidato locus sul cromosoma 5.(E) CAMK2A struttura esonica CAMK2A e domini proteici CAMK2A. I pazienti II:1 e II:4 portano la mutazione missensa biallelica p. H477Y situata nel dominio di associazione CAMK2A (AD). Il cambiamento del nucleotide c.1429 C > T si riferisce alla posizione sul cDNA CAMK2A.(A) Tabella clinica che illustra in dettaglio i parametri di crescita e i deficit di apprendimento dei due bambini colpiti.(B) EEG del paziente II.4 che mostra forme d’onda anomale (casella rossa).(C) Immagini di risonanza magnetica del paziente II.4 che non mostra anomalie strutturali grossolane nel cervello.(D) Grafici che mostrano le regioni omozigoti identificati attraverso la mappatura IBD per ogni membro della famiglia prima del filtraggio(E) Tabella di 4 geni omozigoti che si trovano all’interno della regione di Chr. 5 IBD.

Video 1.Video del paziente II.1
Video 2.Video del paziente II.4
Ipotizzando un’eredità autosomica recessiva, abbiamo eseguito la mappatura dell’omozigosi identitaria per discendenza (IBD) utilizzando DNA genomico di entrambi i genitori, due probandi affetti e tre fratelli sani. Solo un locus candidato maggiore di 5 cM sul cromosoma 5 che abbraccia 28 Mb è stato delineato(Figura 1D, Figura 1-figure supplement 1D). Il sequenziamento dell’intero esoma è stato successivamente eseguito sul caso indice II:1. Dopo il filtraggio per le varianti con bassa qualità e bassa copertura di sequenziamento, sono state identificate 72 varianti omozigoti, di cui quattro si trovano all’interno della regione di Chr. 5 IBD(Tabella 1). Tre delle varianti omozigoti erano state precedentemente annotate come polimorfismi noti con frequenze alleliche minori > 0,0001. Inoltre, individui sani che sono omozigoti per gli alleli minori erano stati identificati in database di sequenziamento genomico come dbSNP e gnomAD(Figura 1-figure supplement 1E). Abbiamo quindi filtrato queste varianti come non patogene. La quarta variante omozigote all’interno della regione IBD mappato al gene CAMK2A (MIM: 114078), con conseguente mutazione missenso p.His477Tyr che non è mai stata osservata in precedenti database di sequenziamento su larga scala e la nostra coorte etnica interna(Figura 1E, Figura 1-figure supplement 1E). Utilizzando il sequenziamento Sanger, questa mutazione privata è stata confermata per segregarsi con la malattia in tutti i sette membri della famiglia(Figura 1A,D).
| Chr | Posizione | Gene | variante cDNA | Variante proteica |
|---|---|---|---|---|
| 1 | 33,476,435 | AK2 | c.*45-1G > T | |
| 1 | 36,752,343 | FRAGGIO3 | c.512C > T | p.Ser171Phe |
| 1 | 36,932,102 | CSF3R | c.2273C > T | p.Thr758Ile |
| 1 | 39,758,439 | MACF1 | c.1931G > T | p.Gly644Val |
| 1 | 145,365,316 | NBPF10 | c.9941G > A | p.Gly3314Glu |
| 2 | 11,758,842 | GREB1 | c.3841G > A | p.Ala1281Thr |
| 2 | 29,404,617 | CLIP4 | c.1976G > A | p.Arg659Gln |
| 2 | 64,779,195 | AFTPH | c.587G > A | p.Gly196Glu |
| 2 | 238,277,211 | COL6A3 | c.4895G > A | p.Arg1632Gln |
| 2 | 241,987,827 | SNED1 | c.1369G > A | p.Glu457Lys |
| 3 | 38,348,802 | SLC22A14 | c.574G > A | p.Ala192Thr |
| 3 | 44,672,687 | ZNF197 | c.524C > T | p.Ala175Val |
| 3 | 47,452,772 | PTPN23 | c.3484C > T | p.Arg1162Trp |
| 3 | 52,556,184 | STAB1 | c.6403C > G | p.Pro2135Ala |
| 3 | 67,426,232 | SUCLG2 | c.1235T > C | p.Ile412Thr |
| 3 | 197,422,844 | KIAA0226 | c.1366C > T | p.Arg456Trp |
| 4 | 9,174,981 | FAM90A26P | c.83T > G | p.Val28Gly |
| 4 | 9,175,603 | FAM90A26P | c.211C > G | p.Pro71Ala |
| 4 | 10,089,539 | WDR1 | c.743A > G | p.His248Arg |
| 4 | 15,529,151 | CC2D2A | c.1231T > G | p.Ser411Ala |
| 5 | 74,021,852 | GFM2 | c.1820_1825delTTGAGT | p.Glu608_Phe609del |
| 5 | 78,610,479 | JMY | c.2464C > A | p.Pro822Thr |
| 5 | 149,602,589 | CAMK2A | c.1429C > T | p.His477Tyr |
| 5 | 154,199,950 | C5orf4 | c.928G > A | p.Glu310Lys |
| 5 | 156,456,715 | HAVCR1 | c.1090G > A | p.Ala364Thr |
| 5 | 156,479,452 | HAVCR1 | c.590_592delCAA | p.Thr198del |
| 6 | 26,509,392 | BTN1A1 | c.1571G > A | p.Gly524Glu |
| 6 | 27,215,709 | PRSS16 | c.119G > A | p.Ser40Asn |
| 6 | 32,806,007 | TAP2 | c.4C > T | p.Arg2Trp |
| 6 | 33,260,924 | RGL2 | c.1876G > A | p.Gly626Arg |
| 6 | 38,704,952 | DNAH8 | c.221C > A | p.Ala74Asp |
| 6 | 43,412,643 | ABCC10 | c.2807C > T | p.Pro936Leu |
| 6 | 129,932,746 | ARHGAP18 | c.1054C > T | p.Arg352Ter |
| 6 | 131,946,054 | MED23 | c.235C > T | p.Leu79Phe |
| 6 | 151,674,121 | AKAP12 | c.4595_4596insGGGC | p.Asp1532delinsGluAla |
| 6 | 168,479,677 | FRMD1 | c.98A > C | p.Glu33Ala |
| 7 | 5,352,665 | TNRC18 | c.7851_7856dupCTCCTC | p.Ser2618_Ser2619dup |
| 7 | 45,123,857 | NACAD | c.1922T > C | p.Val641Ala |
| 7 | 143,884,437 | ARHGEF35 | c.1040C > T | p.Thr347Ile |
| 7 | 149,422,981 | KRBA1 | c.1304C > T | p.Ala435Val |
| 7 | 151,680,130 | GALNTL5 | c.428A > G | p.Tyr143Cys |
| 8 | 12,285,064 | FAM86B1|FAM86B2 | c.310T > C | p.Ser104Pro |
| 8 | 12,285,250 | FAM86B2 | c.808C > T | p.Arg270Trp |
| 8 | 86,574,132 | REXO1L1 | c.1595A > C | p.Asp532Ala |
| 9 | 12,775,863 | LURAP1L | c.149_150insTGGGCGG | p.Gly49_Gly50dup |
| 9 | 40,706,047 | FAM75A3 | c.3704A > G | p.His1235Arg |
| 9 | 41,323,425 | FAM75A4 | c.1908C > T | p.Arg637Trp |
| 9 | 41,323,469 | FAM75A4 | c.1864G > A | p.Gly622Asp |
| 9 | 43,822,668 | CNTNAP3B | c.1222C > T | p.Leu408Phe |
| 10 | 51,748,684 | AGAP6 | c.209G > A | p.Arg70Gln |
| 10 | 81,471,741 | FAM22B | c.2137T > C | p.Trp713Arg |
| 11 | 1,651,198 | KRTAP5-5 | c.129_137delAGGCTGTGG | p.Gly44_Gly46del |
| 11 | 12,316,388 | MICALCL | c.1408_1410dupCCT | p.Pro470dup |
| 12 | 7,045,917 | ATN1 | c.1488_1508delGCAGCAGCAGCAGCAGCAGCAGCAGCAGCA | p.Gln496_Gln502del |
| 12 | 7,045,920 | ATN1 | c.1491_1508delGCAGCAGCAGCAGCAGCAGCAGCA | p.Gln497_Gln502del |
| 13 | 99,461,564 | DOCK9 | c.1271_1272insA | p.Leu425LeufsTer? |
| 13 | 114,503,875 | FAM70B | c.500_509 + 72delCCTGGCGGGGGGGAGG TGAGGGGACCGGACGGGGACCCCCATATC TACACCTGGGGGGGGGGGGTGGGGGGC GCTGGGGGACCCGTATCTACA | |
| 14 | 105,411,514 | AHNAK2 | c.10274C > T | p.Ala3425Val |
| 14 | 106,994,118 | IGHV3-48 | c.47G > A | p.Gly16Asp |
| 16 | 29,496,359 | c.916T > C | p.Ser306Pro | |
| 16 | 30,772,988 | C16orf93 | c.82G > A | p.Ala28Thr |
| 16 | 70,215,817 | CLEC18C | c.521C > T | p.Ala174Val |
| 17 | 39,211,189 | KRTAP2-2 | c.275G > C | p.Cys92Ser |
| 19 | 1,026,716 | CNN2 | c.56A > C | p.Lys19Thr |
| 19 | 10,084,460 | COL5A3 | c.3584T > C | p.Val1195Ala |
| 19 | 14,517,213 | CD97 | c.1892G > A | p.Ser631Asn |
| 21 | 36,042,462 | CLIC6 | c.776_805delGCGTAGAGAGAGAGGGGGGGGGTCCCGGGGGGGGGGGACA | p.Val260_Ser269del |
| 22 | 18,834,773 | c.329C > T | p.Thr110Ile | |
| X | 48,920,059 | CCDC120 | c.110A > G | p.Asp37Gly |
| X | 55,116,478 | PAGINA2 | c.25T > A | p.Ser9Thr |
| X | 150,832,702 | PASD1 | c.954_971delCCCAATGGGACCAGCAGGA | p.Pro319_Asp324del |
| X | 153,050,158 | SRPK3 | c.1_5delGACAG | p.Thr2LeufsTer57 |
| X | 154,010,046 | MPP1 | c.978A > C | p.Glu326Asp |
CAMK2A è un neurone specifico, altamente abbondante serino/treonina chinasi che gioca ruoli critici nella plasticità sinaptica. Per capire come la funzione neuronale è influenzata a causa della mutazione in CAMK2A, abbiamo riprogrammato i fibroblasti dermici primari del paziente II.4 in iPSC, li abbiamo differenziati in neuroni e misurato l’attività neuronale a livello di popolazione utilizzando un sistema multi-elettrodo array (MEA)(Figura 2A). Rispetto ai neuroni derivati da cellule staminali embrionali H1 e ad un controllo iPSC non correlato CAMK2A wild-type, gli iPSC del paziente sono stati ugualmente efficienti nel differenziare in neuroni maturi che esprimono i marcatori neuronali TUJ1 e MAP2 dopo 21 giorni di differenziazione in vitro (Figura 2B). Quando questi neuroni differenziati sono stati placcati su piastre MEA per misurare i potenziali di azione spontanea, abbiamo osservato una significativa riduzione sia del numero totale di picchi spontanei(Figura 2C, a sinistra) e il tasso di cottura media(Figura 2C, a destra) nei neuroni derivati dal paziente che ospitano la mutazione p.H477Y rispetto ai controlli wild-type, suggerendo che CAMK2AH477Y provoca un profondo difetto funzionale nei neuroni in coltura.

Figura 2.CAMK2A neuroni mutanti di derivazione iPSC sono funzionalmente meno attivi.(A) Schema del hPSC-derivato neuronale saggio di attività neuronale con immagine rappresentativa di neuroni derivati iPSC placcati su un array multi-elettrodo(B) immagini rappresentative confocale di immunofluorescenza colorazione di marcatori di lignaggio neuronale TUJ1 (verde) e MAP2 (rosso) mostrano differenziazione efficiente di iPSC in neuroni. La barra di scala rappresenta 20 µm.(C) I grafici che mostrano il numero di picchi evocati dai neuroni e la velocità media di cottura rilevata da array multielettrodo. (n = 7 per linea per punto temporale; Valori indicati come media ±SEM; ANOVA bidirezionale con test post-hoc Tukey; *p<0,05 e ***p<0,001).
L’enzima CAMK2A è costituito da un dominio catalitico chinasi catalitica N-terminale, un dominio di regolazione vincolante Ca2+-calmodulina e un dominio di associazione C-terminale (AD) necessario per l’assemblaggio dell’oloenzima a 12 o 14 sottounità. La mutazione identificata, p.H477Y si trova nel dominio di associazione (Figura 1E) e colpisce un residuo di istidina che è invariante attraverso tutti i vertebrati e invertebrati omologhi CAMK2A. È anche conservato in altri paraloghi umani CAMK2 umani con un dominio di associazione simile come CAMK2B, CAMK2D e CAMK2G(Figura 3A,Figura 3-figure supplement 1A). Inoltre, questa mutazione è prevista essere deleteria sia dagli algoritmi SIFT, PolyPhen e MCAP. Strutturalmente, His477 si trova in corrispondenza dell’interfaccia dimerica che fa parte della superficie di interazione estesa al piano ‘equatoriale’ dell’oloenzima CAMK2A (Figura 3B). Sulla base di precedenti scoperte che l’oligomerizzazione CAMK2A attraverso il suo dominio di associazione è indispensabile per la fosforilazione del substrato e la localizzazione sinaptica(Bhattacharyya et al., 2016), si ipotizza che l’allele p.H477Y è ipomorfo e che la perdita biallelica di funzione in CAMK2A è la causa diretta per i fenotipi di sviluppo neurologico nei due probandi.
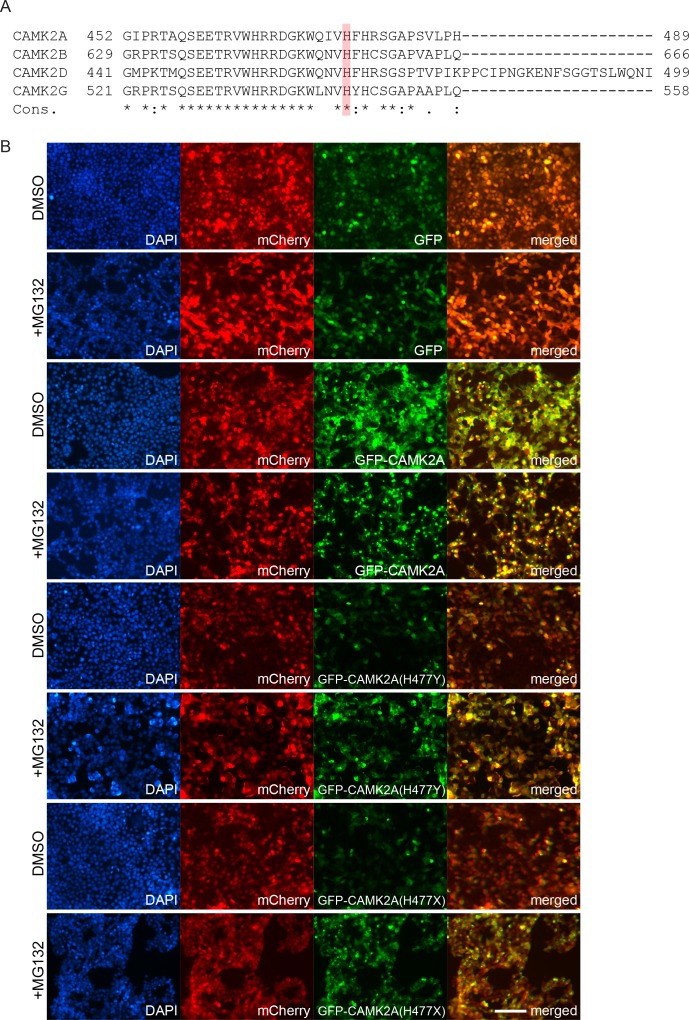
Figura 3-figure supplemento 1.p.H477Y colpisce l’oligomerizzazione CAMK2A e la stabilità delle proteine.la stabilità diminuita e la localizzazione citoplasmatica difettosa del mutante CAMK2AH477Y.(A) Conservazione della sequenza degli omologhi CAMK2A. L’istidina 477 (H477) è evidenziata in rosso.(B) Struttura a cristalli a raggi X del tetradecamer umano CAMK2A AD (PDB: 5IG3). H477 (rosso) si trova all’interfaccia del dimero equatoriale.(C) Oligomerizzazione difettosa del mutante p.H477Y. 293 cellule T sono state transitoriamente trasfettate con FLAG tagged wild-type CAMK2A e CAMK2AH477Y. Un terzo mutante, CAMK2AH477X che manca di parte dell’AD (a.a. 478-489) è stato utilizzato come controllo positivo.(D) Difettosa auto-associazione del mutante p.H477Y. L’indicato FLAG- e HA-tagged CAMK2A CAMK2A wild-type e proteine mutanti sono stati sintetizzati in vitro utilizzando coniglio lisato reticolocita lisato reticolo di coniglio. FLAG-GFP è stato utilizzato un controllo negativo. Le proteine con marchio FLAG sono state immunoprecipitate utilizzando la resina agarosio anti-FLAG M2 in presenza dell’1% di NP40. Le proteine immunoprecipitate sono state analizzate con SDS-PAGE. *, catena leggera IgG. ^, IgG catena pesante.(E) p.H477Y mutazione p.H477Y abbassa l’espressione di CAMK2A nelle cellule. 293 cellule T sono state trasfettate con plasmidi reporter che codificano i plasmidi GFP-tagging GFP wild-type CAMK2A, CAMK2AH477Y e CAMK2AH477X mutanti, seguito da peptide T2A e mCherry. Le immagini confocali rappresentative mostrano una minore espressione del mutante GFP- CAMK2AH477Y rispetto al wild-type. La barra di scala rappresenta 100 µm.(F). p.H477Y diminuisce la stabilità CAMK2A attraverso la degradazione proteasomica. 293 cellule T sono stati trasfettati come in(E) e trattati con 10 µM MG132 o DMSO per 16 ore 10 µg lisato cellulare totale è stato utilizzato per SDS-PAGE e Western blot.(A) Conservazione della sequenza dei paraloghi CAMK2. Il suo477 (rosso) è invariante in tutti i paraloghi umani CAMK2 con un dominio di associazione.(B) p.H477Y mutante è soggetto a degradazione proteasomica. 293 cellule T sono state trasfettate con plasmidi che codificano i plasmidi che codificano i mutanti CAMK2A, p.H477Y e p.H477X con marchio GFP wild-type. Le cellule sono state incubate con DMSO o 2,5 μM MG132 per 16 ore 1 giorno dopo la trasfezione. Intensità GFP nelle cellule è aumentata nel CAMK2A p.H477Y e p.H477X mutanti dopo il trattamento MG132. La barra di scala rappresenta 100 µm.
Per misurare il potenziale di oligomerizzazione del mutante CAMK2AH477Y nelle cellule, abbiamo espresso transitoriamente FLAG-tagged wild-type CAMK2A e CAMK2AH477Y mutante in 293 cellule T, che non esprimono CAMK2A endogeno. Un terzo mutante CAMK2AH477X, che manca di aminoacidi 477-489 e quindi codifica un dominio di associazione troncata, è stato utilizzato come controllo aggiuntivo. Utilizzando condizioni di lisi nativa che conservano le interazioni macromolecolari non covalenti, abbiamo trovato che wild-type CAMK2A forma un complesso prominente con un peso molecolare apparente ~ 1 MDa (Figura 3C, Native-PAGE, corsia 2), che è coerente con il 12 o 14 sottounità CAMK2A oloenzima (Bhattacharyyaet al., 2016). Rispetto al wild-type CAMK2A, il ~ 1 MDa, specie oligomerica presunta, è stato drasticamente ridotto per il mutante p.H477Y ed è stato impercettibile per il mutante p.H477X (Figura 3C, Native-PAGE, corsia 3 e 4 contro la corsia 2). Successivamente, abbiamo esaminato la capacità di CAMK2A tradotto in vitro per auto-oligomerizzare in un sistema senza cellule. In contrasto con la proteina di controllo negativo GFP, wild-type FLAG-CAMK2A efficacemente co-immunoprecipitato con HA-CAMK2A(Figura 3D, corsia 10 vs 11). Questa auto-associazione è stata preservata tra CAMK2AWT e CAMK2AH477Y(Figura 3D, corsie 12 e 15), ma è stata completamente persa tra CAMK2A wild-type e il mutante p.H477X(Figura 3D, corsia 13). Al contrario, non abbiamo potuto rilevare alcuna auto-associazione tra FLAG- CAMK2AH477Y e HA- CAMK2AH477Y(Figura 3D, corsia 16). Presi insieme, questi risultati suggeriscono che il missenso p.H477Y interrompe parzialmente l’auto-associazione tra molecole identiche di CAMK2A, che si era dimostrato necessario per l’assemblaggio dell’oloenzima. La perdita parziale di funzione del mutante p.H477Y, rispetto ad una mutazione più grave p.H477X, è coerente con l’ereditarietà autosomica recessiva osservata della malattia in famiglia, dove i portatori eterozigoti non mostrano sintomi apparenti di sviluppo neurologico.
Nel corso di questa analisi, abbiamo notato che sia p.H477Y che p.H477X mutanti avevano ridotto l’abbondanza di proteine. Questo effetto sul mutante p.H477Y era, tuttavia, sottile e non poteva facilmente spiegare la differenza nel CAMK2A oligomero osservato nel gel nativo(Figura 3C, SDS-PAGE). Come è noto, in generale i complessi oligomerici completamente assemblati hanno una maggiore stabilità in vivo rispetto ai complessi parzialmente assemblati con piegatura perturbata come CAMK2AH477Y(Lord, 1996; Oromendia et al., 2012), abbiamo ipotizzato che il mutante p.H477Y possa mostrare una stabilità ridotta e subire una degradazione proteasomica. Per testare questo, 293T cellule sono state trasfettate con i costrutti reporter che codificano GFP tagged wild-type e mutante CAMK2A seguito da un auto-clearing peptide T2A e mCherry(Figura 3E). L’intensità della fluorescenza GFP è stata utilizzata come misura quantitativa diretta della stabilità del CAMK2A con mCherry come controllo interno per la trasfezione e l’efficienza traslazionale. Abbiamo osservato una significativa riduzione dell’intensità delle GFP nelle cellule che esprimono CAMK2AH477Y e CAMK2AH477X rispetto al tipo selvaggio CAMK2A o GFP solo(Figura 3E), nonostante i livelli di fluorescenza mCherry comparabili. Questa riduzione è stata salvata quando abbiamo trattato le cellule con MG132, che ha bloccato la degradazione proteasomica. MG132 treamentled a maggiore accumulo del p.H477Y e p.H477X mutante, con un effetto maggiore su p.H477X(Figura 3F, corsia 3, 4 vs corsia 7, 8, Figura 3-figure supplemento 1B). Al contrario, il livello della proteina wild-type è stato ridotto, probabilmente a causa degli effetti tossici di MG132(Figura 3F, corsia 2 vs corsia 6). Questi risultati suggeriscono che, oltre a causare un ridotto assemblaggio di oloenzima, la mutazione p.H477Y potrebbe anche ridurre direttamente o indirettamente i livelli complessivi di CAMK2A compromettendo il suo tempo di dimezzamento.
Per dimostrare inequivocabilmente la patogenicità dell’allele p.H477Y in vivo, abbiamo effettuato esperimenti di salvataggio utilizzando C. elegans. Un unico ortologo di CAMK2, codificato da unc-43, è presente nel genoma del verme e le sue funzioni nel sistema nervoso sono ben documentate(Reiner et al., 1999). Per studiare i difetti neuronali causati dalla perdita di unc-43, ci siamo concentrati sul motoneurone, DA9, che ha il suo corpo cellulare situato nel ganglio preanale con un dendriti che si estende anteriormente e un assone orientato posteriormente che si estende attraverso una commissura nel cordone del nervo dorsale, dove procede anteriormente a formare circa 25 en sinapsi passanti sui muscoli della parete del corpo e neuroni inibitori reciproci(Figura 4A)(Klassen e Shen, 2007; Maeder et al., 2014). Utilizzando mCherry-tagging UNC-43, abbiamo osservato che UNC-43 potrebbe essere trovato lungo l’intero neurone DA9, ma concentrato in bouton sinaptici(Figura 4B). L’omologa mutazione paziente-specifica p.H466Y in UNC-43 (omologa a p.H477Y nel CAMK2A umano) ha perturbato significativamente la localizzazione sinaptica di UNC-43 e ha causato la dispersione della proteina in tutto l’assone(Figura 4B). Per testare la conseguenza della mutazione UNC-43 sulla funzione sinaptica DA9, abbiamo usato il promotore itr-1 pB per esprimere il marcatore della vescicola sinaptica fluorescente coniugato RAB-3 (RAB3::GFP) all’interno di DA9 sia nel wild-type che unc-43 mutante di sfondo. Negli animali wild-type, RAB-3::GFP accumulato in discreti puncta lungo l’assone di DA9 in posizioni sinaptiche stereotipate. Nel canonico unc-43(e408) mutante a perdita di funzione, abbiamo osservato una riduzione dell’intensità di fluorescenza individuale pre-sinaptica puncta rispetto agli animali wild-type. Questo fenotipo potrebbe essere salvato da un’espressione cellulare autonoma di unc-43 wild-type in DA9. Tuttavia, l’espressione di unc-43 che ospita la mutazione omologa del paziente UNC-43H466Y non è riuscita a salvare questo difetto(Figura 4C,D). Inoltre, l’espressione transgenica del CAMK2A umano wild-type CAMK2A ha salvato completamente questo difetto, confermando l’alto grado di conservazione funzionale tra gli omologhi CAMK2A, mentre il CAMK2AH477Y umano derivato dal paziente non è riuscito a farlo (Figura 4C, pannelli in basso). Questi risultati suggeriscono che la mutazione p.H477Y è difettosa nel supportare la funzione presinaptica in C. elegans.
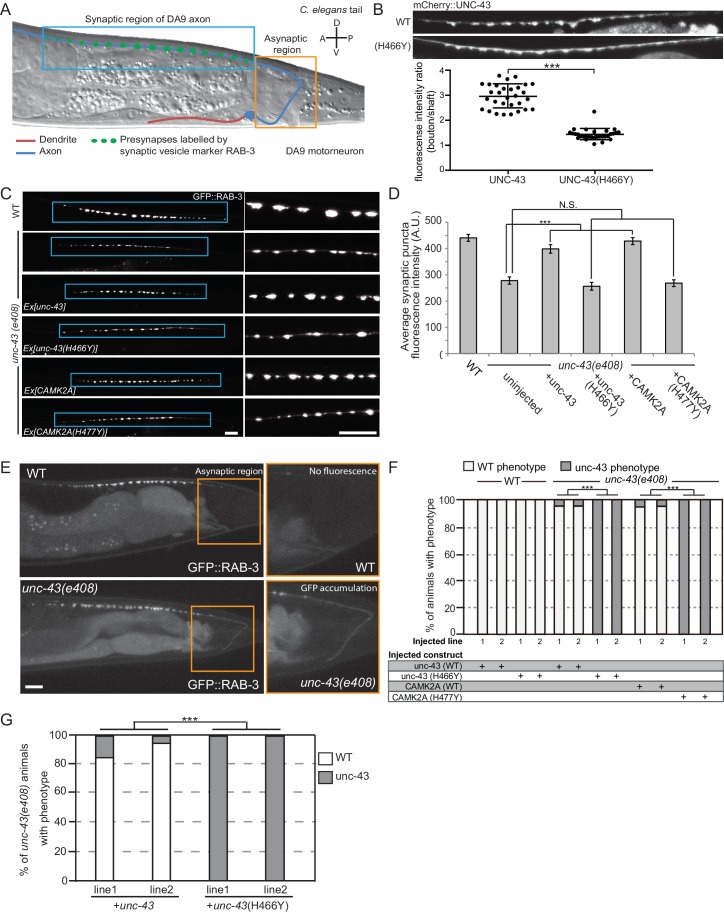
Figura 4.Il mutante CAMK2AH477Y non riesce a salvare i difetti sinaptici nei neuroni unc-43 C. eleg ans.(A) Disegno schematico del motoneurone C. elegans, DA9 nella regione della coda . DA9 estende un dendriti (rosso) anteriormente e un assone (blu) che si estende posteriormente attraversa la linea mediana dell’animale e anteriormente nel cordone del nervo dorsale (DNC). Forma circa 20 en sinapsi passanti all’interno di un tratto discreto lungo la DNC (scatola blu). DA9 vescicole presinaptiche sono stati contrassegnati con RAB-3 (GFP::RAB-3). La regione asinaptica (scatola gialla) è priva di qualsiasi accumulo di vescicole sinaptiche.(B) La localizzazione di mCherry::UNC-43 e mCherry::UNC-43(H466Y) in sinapsi DA9. Si noti che l’UNC-43 si accumula in corrispondenza dei bouton sinaptici mentre l’UNC-43(H466Y) è diffusamente localizzato. Intensità fluorescente di mCherry::UNC-43 è stata misurata in corrispondenza dei bouton sinaptici e lungo l’asse assonale. Grafico trama il rapporto di intensità di fluorescenza a bouton sinaptici rispetto all’albero assonale di 30 sinapsi da tre animali. Il grafico mostra la media e le barre di errore mostrano SEM, ***p-valore 6,32e-19, Student’s T-test.(C) Immagini rappresentative confocali che dimostrano i cambiamenti di dimensione dei puncta presinaptici tra WT e unc-43 (e408) mutanti. i mutanti unc-43 hanno puncta presinaptici più piccoli lungo il DNC. Questo difetto viene salvato con l’espressione di UNC-43 o CAMK2A in DA9 mentre i mutanti UNC-43H466Y e CAMK2AH477Y non riescono a salvare.(D) Quantificazione dell’intensità puncta media degli animali WT e unc-43(e408). Le barre di errore rappresentano il SEM con numero di puncta sinaptici quantificati n > 80, N.S. non è significativo, ***p-valore<0,001 (uninjected vs unc-43 p-valore 5 .0e8, uninjected vs unc-43H466Y p-valore 4,17, uninjected vs CAMK2A p-valore 4,25e-12, uninjected vs CAMK2AH477Y p-valore 9,40), ANOVA a senso unico con test post oculistico Bonferroni.(E) Immagini confocali rappresentative che mostrano la dislocazione delle PFP::RAB-3 nella regione asinaptica (scatola gialla) nel neurone unc-43 DA9.(F) Salvataggio del fenotipo unc-43(e408) da DA9 espressione cellulare specifica di UNC-43 o CAMK2A. UNC-43H466Y e CAMK2AH477Y non riescono a salvare il fenotipo unc-43. Il grafico mostra la percentuale di animali con il fenotipo mutante WT e unc-43. ***p<0.001 (unc-43vs unc-43H466Y p-valorep 2,13e-51, CAMK2A vs CAMK2AH477Yp-valore p 3,77e-50), Fisher Test esatto con n = 100 animali segnati per ogni riga.(G) Salvataggio comportamentale esprimendo UNC-43 o UNC-43H466Y di tipo selvaggio in unc-43(e408) mutanti. Il comportamento è stato segnato come wild-type o unc-43. Sono state analizzate due linee di vermi indipendenti per ogni condizione. *** p-valore 5.29e-41, Fisher Esatto test con n = 100 animali punteggio per ogni linea.
Immediatamente posteriore al tratto di puncta presinaptico è un dominio asinaptico all’interno dell’assone DA9 che è privo di qualsiasi fluorescenza RAB-3::GFP negli animali selvatici(Figura 4E, pannello superiore). La perdita di unc-43 comporta la dislocazione del marcatore sinaptico RAB3::GFP in questa regione asinaptica(Figura 4E, pannello inferiore). Questo difetto potrebbe anche essere salvato da espressione cellulare autonomo di unc-43 o umano CAMK2A. La mutazione derivata dal paziente CAMK2AH477Y o la mutazione omologa del verme UNC-43H466Y non sono riuscite a salvare questo fenotipo(Figura 4E). Inoltre, l’espressione di UNC-43H466Y in animali wild-type non ha causato alcun difetto sinaptico o mislocalizaton di RAB3::GFP, suggerendo che la mutazione non ha effetti negativi dominanti(Figura 4F).
Abbiamo inoltre testato se la mutazione derivata dal paziente in CAMK2A ha avuto un impatto sul comportamento locomotorio del verme. I mutanti nulli per unc-43 sono flaccidi in postura e si muovono con una forma d’onda appiattita non coordinata. Gli animali sono variabilmente convulsi, spesso si contraggono spontaneamente e rilassano i muscoli delle pareti del corpo in brevi raffiche ripetute che assomigliano a crisi epilettiche(Reiner et al., 1999). Abbiamo espresso sia il tipo selvaggio UNC-43 o UNC-43H466Y nei muscoli e nei neuroni dei vermi mutanti unc-43 (e408) e abbiamo segnato il comportamento dei giovani adulti in un esperimento in doppio cieco. Solo il wildtype UNC-43 è stato in grado di salvare i difetti di movimento in unc-43 (e408) animali, ma non UNC-43H466Y, suggerendo che UNC-43H466Y non è funzionale(Figura 4G). Insieme, i dati mostrano che CAMK2AH477Y è una mutazione a perdita di funzione che non riesce a supportare la funzione sinaptica in vivo.
In sintesi, abbiamo identificato una sindrome autosomica recessiva dello sviluppo neurologico caratterizzata da ritardo della crescita, frequenti crisi epilettiche e grave disabilità intellettuale che è causata da una mutazione biallelica a perdita di funzione in CAMK2A. Meccanicamente questa mutazione perturba meccanicamente l’autooligomerizzazione del CAMK2A e l’assemblaggio dell’oloenzima attraverso il suo dominio di associazione. I nostri risultati funzionali sono coerenti con l’elevato grado di conservazione evolutiva del residuo interessato H477 negli ortologi CAMK2A, così come i dati strutturali precedenti che dimostrano che His477 si trova nell’interfaccia tra due sottounità CAMK2A sovrapposte, che insieme formano l’unità di base di ripetizione dell’oloenzima ad anello(Bhattacharyya et al., 2016; Stratton et al., 2014). La patogenicità della mutazione biallelica p.H477Y è ulteriormente evidenziata dagli esperimenti di salvataggio in C. elegans, dove in contrasto con il wild-type umano CAMK2A il mutante p.H477Y non è riuscito a salvare il difetto neuronale e comportamentale in unc-43 (CAMK2 ortologia) vermi null. Insieme, questi dati dimostrano che la perdita di funzione di CAMK2A è la causa genetica più plausibile per i difetti dello sviluppo neurologico osservati nei due fratelli colpiti.
Figura 1-figure supplemento 1.Una nuova sindrome di ritardo globale dello sviluppo neurologico con crisi epilettiche causate da una mutazione biallelica in CAMK2A.Genetic e risultati clinici dei due pazienti con ritardo globale dello sviluppo.2.(A) Pedigree di una famiglia giordana consanguinea con due fratelli affetti da mutazioni omozigoti in CAMK2A. I genotipi di tutti gli individui sono stati verificati con il sequenziamento Sanger.(B) Fotografie dei due fratelli affetti con circonferenze craniche normali.(C) Grafico EEG del paziente II.I che mostra i transitori epilettiformi anormali (scatole rosse)(D) Mappatura omozigosi delinea un candidato locus sul cromosoma 5.(E) CAMK2A struttura esonica CAMK2A e domini proteici CAMK2A. I pazienti II:1 e II:4 portano la mutazione missensa biallelica p. H477Y situata nel dominio di associazione CAMK2A (AD). Il cambiamento del nucleotide c.1429 C > T si riferisce alla posizione sul cDNA CAMK2A.(A) Tabella clinica che illustra in dettaglio i parametri di crescita e i deficit di apprendimento dei due bambini colpiti.(B) EEG del paziente II.4 che mostra forme d’onda anomale (casella rossa).(C) Immagini di risonanza magnetica del paziente II.4 che non mostra anomalie strutturali grossolane nel cervello.(D) Grafici che mostrano le regioni omozigoti identificati attraverso la mappatura IBD per ogni membro della famiglia prima del filtraggio(E) Tabella di 4 geni omozigoti che si trovano all’interno della regione di Chr. 5 IBD.
Figura 1-figure supplemento 1.1. Risultati genetici e clinici dei due pazienti con ritardo di sviluppo globale.(A) Tabella clinica che descrive in dettaglio i parametri di crescita e i deficit di apprendimento dei due bambini affetti.(B) EEG del paziente II.4 che mostra forme d’onda anomale (scatola rossa).(C) Immagini di risonanza magnetica del paziente II.4 che non mostra anomalie strutturali grossolane nel cervello.(D) Grafici che mostrano le regioni omozigoti identificati attraverso la mappatura IBD per ogni membro della famiglia prima del filtraggio(E) Tabella di 4 geni omozigoti che si trovano all’interno della regione di Chr. 5 IBD.
Video 1.Video del paziente II.1
Video 2.Video del paziente II.4
Figura 2.I neuroni mutantiCAMK2A di derivazione iPSC sono funzionalmente meno attivi.(A) Schema del test di attività neuronale derivata da hPSC con immagine rappresentativa dei neuroni derivati da iPSC placcati su un array multi-elettrodo(B) Immagini rappresentative confocali di colorazione in immunofluorescenza dei marcatori di lignaggio neuronale TUJ1 (verde) e MAP2 (rosso) mostrano una differenziazione efficiente degli iPSC in neuroni. La barra di scala rappresenta 20 µm.(C) I grafici che mostrano il numero di picchi evocati dai neuroni e la velocità media di cottura rilevata da array multielettrodo. (n = 7 per linea per punto temporale; Valori indicati come media ±SEM; ANOVA bidirezionale con test post-hoc Tukey; *p<0,05 e ***p<0,001).
Figura 3-figure supplement 1.p.H477Y colpisce l’oligomerizzazione CAMK2A e la stabilità delle proteine.la stabilità diminuita e la localizzazione citoplasmatica difettosa del mutante CAMK2AH477Y.(A) Conservazione della sequenza degli omologhi CAMK2A. L’istidina 477 (H477) è evidenziata in rosso.(B) Struttura a cristalli a raggi X del tetradecamer umano CAMK2A AD (PDB: 5IG3). H477 (rosso) si trova all’interfaccia del dimero equatoriale.(C) Oligomerizzazione difettosa del mutante p.H477Y. 293 cellule T sono state transitoriamente trasfettate con FLAG tagged wild-type CAMK2A e CAMK2AH477Y. Un terzo mutante, CAMK2AH477X che manca di parte dell’AD (a.a. 478-489) è stato utilizzato come controllo positivo.(D) Difettosa auto-associazione del mutante p.H477Y. L’indicato FLAG- e HA-tagged CAMK2A CAMK2A wild-type e proteine mutanti sono stati sintetizzati in vitro utilizzando coniglio lisato reticolocita lisato reticolo di coniglio. FLAG-GFP è stato utilizzato un controllo negativo. Le proteine con marchio FLAG sono state immunoprecipitate utilizzando la resina agarosio anti-FLAG M2 in presenza dell’1% di NP40. Le proteine immunoprecipitate sono state analizzate con SDS-PAGE. *, catena leggera IgG. ^, IgG catena pesante.(E) p.H477Y mutazione p.H477Y abbassa l’espressione di CAMK2A nelle cellule. 293 cellule T sono state trasfettate con plasmidi reporter che codificano i plasmidi GFP-tagging GFP wild-type CAMK2A, CAMK2AH477Y e CAMK2AH477X mutanti, seguito da peptide T2A e mCherry. Le immagini confocali rappresentative mostrano una minore espressione del mutante GFP- CAMK2AH477Y rispetto al wild-type. La barra di scala rappresenta 100 µm.(F). p.H477Y diminuisce la stabilità CAMK2A attraverso la degradazione proteasomica. 293 cellule T sono stati trasfettati come in(E) e trattati con 10 µM MG132 o DMSO per 16 ore 10 µg lisato cellulare totale è stato utilizzato per SDS-PAGE e Western blot.(A) Conservazione della sequenza dei paraloghi CAMK2. Il suo477 (rosso) è invariante in tutti i paraloghi umani CAMK2 con un dominio di associazione.(B) p.H477Y mutante è soggetto a degradazione proteasomica. 293 cellule T sono state trasfettate con plasmidi che codificano i plasmidi che codificano i mutanti CAMK2A, p.H477Y e p.H477X con marchio GFP wild-type. Le cellule sono state incubate con DMSO o 2,5 μM MG132 per 16 ore 1 giorno dopo la trasfezione. Intensità GFP nelle cellule è aumentata nel CAMK2A p.H477Y e p.H477X mutanti dopo il trattamento MG132. La barra di scala rappresenta 100 µm.
Figura 3-figure supplemento 1.2. Diminuzione della stabilità e localizzazione citoplasmatica difettosa del mutante CAMK2AH477Y.2.(A) Conservazione della sequenza dei paraloghi CAMK2. Il suo477 (rosso) è invariante in tutti i paraloghi umani CAMK2 con un dominio di associazione.(B) p.H477Y mutante è soggetto a degradazione proteasomica. 293 cellule T sono state trasfettate con plasmidi che codificano i plasmidi che codificano i mutanti CAMK2A, p.H477Y e p.H477X con marchio GFP wild-type. Le cellule sono state incubate con DMSO o 2,5 μM MG132 per 16 ore 1 giorno dopo la trasfezione. Intensità GFP nelle cellule è aumentata nel CAMK2A p.H477Y e p.H477X mutanti dopo il trattamento MG132. La barra di scala rappresenta 100 µm.
Figura 4.CAMK2AH477Y mutante CAMK2AH477Y non riesce a salvare i difetti sinaptici in unc-43 C. elegans neuroni.(A) Disegno schematico del motoneurone C. elegans, DA9 nella regione della coda . DA9 estende un dendriti (rosso) anteriormente e un assone (blu) che si estende posteriormente attraversa la linea mediana dell’animale e anteriormente nel cordone del nervo dorsale (DNC). Forma circa 20 en sinapsi passanti all’interno di un tratto discreto lungo la DNC (scatola blu). DA9 vescicole presinaptiche sono stati contrassegnati con RAB-3 (GFP::RAB-3). La regione asinaptica (scatola gialla) è priva di qualsiasi accumulo di vescicole sinaptiche.(B) La localizzazione di mCherry::UNC-43 e mCherry::UNC-43(H466Y) in sinapsi DA9. Si noti che l’UNC-43 si accumula in corrispondenza dei bouton sinaptici mentre l’UNC-43(H466Y) è diffusamente localizzato. Intensità fluorescente di mCherry::UNC-43 è stata misurata in corrispondenza dei bouton sinaptici e lungo l’asse assonale. Grafico trama il rapporto di intensità di fluorescenza a bouton sinaptici rispetto all’albero assonale di 30 sinapsi da tre animali. Il grafico mostra la media e le barre di errore mostrano SEM, ***p-valore 6,32e-19, Student’s T-test.(C) Immagini rappresentative confocali che dimostrano i cambiamenti di dimensione dei puncta presinaptici tra WT e unc-43 (e408) mutanti. i mutanti unc-43 hanno puncta presinaptici più piccoli lungo il DNC. Questo difetto viene salvato con l’espressione di UNC-43 o CAMK2A in DA9 mentre i mutanti UNC-43H466Y e CAMK2AH477Y non riescono a salvare.(D) Quantificazione dell’intensità puncta media degli animali WT e unc-43(e408). Le barre di errore rappresentano il SEM con numero di puncta sinaptici quantificati n > 80, N.S. non è significativo, ***p-valore<0,001 (uninjected vs unc-43 p-valore 5 .0e8, uninjected vs unc-43H466Y p-valore 4,17, uninjected vs CAMK2A p-valore 4,25e-12, uninjected vs CAMK2AH477Y p-valore 9,40), ANOVA a senso unico con test post oculistico Bonferroni.(E) Immagini confocali rappresentative che mostrano la dislocazione delle PFP::RAB-3 nella regione asinaptica (scatola gialla) nel neurone unc-43 DA9.(F) Salvataggio del fenotipo unc-43(e408) da DA9 espressione cellulare specifica di UNC-43 o CAMK2A. UNC-43H466Y e CAMK2AH477Y non riescono a salvare il fenotipo unc-43. Il grafico mostra la percentuale di animali con il fenotipo mutante WT e unc-43. ***p<0.001 (unc-43vs unc-43H466Y p-valorep 2,13e-51, CAMK2A vs CAMK2AH477Yp-valore p 3,77e-50), Fisher Test esatto con n = 100 animali segnati per ogni riga.(G) Salvataggio comportamentale esprimendo UNC-43 o UNC-43H466Y di tipo selvaggio in unc-43(e408) mutanti. Il comportamento è stato segnato come wild-type o unc-43. Sono state analizzate due linee di vermi indipendenti per ogni condizione. *** p-valore 5.29e-41, Fisher Esatto test con n = 100 animali punteggio per ogni linea.
Discussione
Il CAMK2 svolge ruoli importanti ed evolutivi conservati nella plasticità sinaptica, nella trasmissione neuronale e nella cognizione in quasi tutti gli organismi modello esaminati, e diversi gruppi hanno dimostrato che le mutazioni somatiche nelle isoforme umane CAMK2 possono contribuire a disturbi neurologici(Ghosh e Giese, 2015; Robison, 2014; Takemoto-Kimura et al., 2017). In particolare, è stato dimostrato che una mutazione de novo p.E183V nel dominio catalitico CAMK2A causa disturbi dello spettro autistico(Stephenson et al., 2017). Questa mutazione ha dimostrato di agire in modo dominante-negativo per ridurre l’autofosforilazione e la localizzazione di tipo selvatico CAMK2A a spine dendritiche. Mentre questo manoscritto era in fase di revisione, S. Küry et al. hanno riportato diverse famiglie con disabilità intellettiva causata da de novo, mutazioni eterozigoti sia in CAMK2A che in CAMK2B chinasi e domini auto-regolatori, che hanno perturbato la fosforilazione CAMK2A e causato difetti migratori neuronali nei modelli murini (Küryet al., 2017).
La nostra scoperta di una nuova sindrome neuro-sviluppo neurologico causata da mutazioni bialleliche CAMK2A amplia ulteriormente lo spettro dei disturbi neurologici umani causati dalla famiglia delle chinasi CAMK2. Per quanto ne sappiamo, questa rappresenta la prima malattia umana mendeliana causata da mutazioni CAMK2A bialleliche. La nostra caratterizzazione funzionale della nuova mutazione p.H477Y in vitro e in vivo rivela anche nuove intuizioni su come l’oloenzima CAMK2A regola la funzione neuronale. A differenza di tutte le mutazioni precedentemente segnalate in CAMK2A nelle sindromi di disabilità intellettiva, il p.H477Y si trova all’interno del dominio dell’associazione C-terminale e si traduce in una parziale ma significativa interruzione dell’autooligomerizzazione, suggerendo che l’assemblaggio degli oligomeri CAMK2A, oltre alla sua funzione chinasi, è necessario per la funzione neuronale. È interessante notare che il mutante CAMK2AH477Y mantiene la capacità di interagire con il CAMK2A wild-type ma non con se stesso(Figura 3D). CAMK2A mostra modelli di espressione cellulare e subcellulare molto specifici che è importante per regolare la fosforilazione del substrato nelle cellule(Liu e Murray, 2012; Tsui et al., 2005). Il missenso p.H477Y influenza la localizzazione subcellulare nei neuroni e questo può influenzare la sua capacità di funzionare in modo efficiente. Questi risultati biochimici forniscono una base meccanicistica per la natura autosomica recessiva della malattia nella nostra famiglia: l’allele p.H477Y è ipomorfo e diventa patogeno quando viene ereditato in modo recessivo nello stato omozigote. Si ipotizza che i portatori eterozigoti conservino un’attività CAMK2A sufficiente per una corretta funzione neuronale. Rispetto ai fenotipi di malattia più blandi riportati da Kury et. al., i sintomi che affliggono i pazienti p.H477Y rappresentano probabilmente la manifestazione più grave della disfunzione CAMK2A nell’uomo.
A causa dell’elevato grado di conservazione del CAMK2A attraverso l’evoluzione, abbiamo impiegato il mutante C. elegans unc-43 stabilito per dimostrare la patogenicità della mutazione p.H477Y. UNC-43 è l’omologo verme del vertebrato CAMK2. Abbiamo dimostrato che il CAMK2A umano wild-type CAMK2A potrebbe salvare i difetti locomotori dei mutanti unc-43, mentre il mutante p.H477Y non è riuscito a farlo, probabilmente a causa della sua incapacità di localizzarsi in sinapsi neuronali(Figura 4). Prevediamo che questo test funzionale in vivo in C. elegans sia ampiamente applicabile per valutare la patogenicità degli alleli CAMK2 appena scoperti che si trovano nelle malattie umane.
Materiali e metodi
| Tipo di reagente (specie) o risorsa | Designazione | Fonte o riferimento | Identificatori | Ulteriori informazioni |
|---|---|---|---|---|
| Gene (umano) | CAMK2A Calcio/Calmodulina Dipendente da Calcio/Calmodulina Proteina Kinasi II Alfa | Uniprot: Isoform B (identificatore: Q9UQM7-2) | Q9UQM7-2 | |
| Gene(C. elegans) | unc-43 Calcio/calmodulina-proteina chinasi dipendente tipo II | Proteina UNC-43 isoform d (Wormbase CDS K11E8.1d) | K11E8.1d | |
| Ceppo, fondo di ceppo(C. elegans) | Verme (C. elegans) N2 Bristol Strain;unc-43(e408) | Caenorhabditis Genetics Center (CGC) PMID: 17941711 | 17941711 | |
| DNA ricombinante | pCDH-CMV-MCS-EF1α-Neo | Sistemi Bioscienze (SBI) | CD514B-1 | |
| DNA ricombinante | vettore pSM (un derivato di pPD49.26 con ulteriori siti di clonazione) | Modificato per questo articolo | NA | AGGIUNGERE https://media.addgene.org/cms/files/Vec95.pdf |
| DNA ricombinante | pCDH-CMV-CAMK2A-T2A-mCherry | Questo documento; basato su pCDH-CMV-MCS-EF1α-Neo | NA | |
| DNA ricombinante | pMIG-hOCT4 | Addgene | Plasmide #17225 | |
| DNA ricombinante | MSCV h c-MYC IRES PFP | Addgene | Plasmide #18119 | |
| DNA ricombinante | pMIG-hKLF4 | Addgene | Plasmide #17227 | |
| DNA ricombinante | pMIG-hSOX2 | Addgene | Plasmide #17226 | |
| Linea cellulare (umana) | Neuroni iPS derivati dal paziente | Questo documento | NA | |
| Linea cellulare (umana) | 293T | Stock di laboratorio | NA | |
| Composto chimico, farmaco | MG132 | Sigma-Aldrich | M7449 | |
| Kit commerciale | Pagina dei marchi nativi | ThermoFisher | BN1002BOX | |
| Kit commerciale | TnT Sistema di trascrizione/traslazione ad accoppiamento rapido | Promega | L1170 | |
| Anticorpo | Clone Anti-FLAG M2 | Sigma-Aldrich | F3165 | |
| Anticorpo | Anti-HA Clone Y-11 | Santa Cruz Biotecnologia | sc-7392 | |
| Anticorpo | Anti-GADPH | Santa Cruz Biotecnologia | sc-47724 | |
| Anticorpo | Anti-Tuj1 | Ricerca sulla covanza | MMS-435P | |
| Anticorpo | Anti-MAP2 | Sistemi sinaptici | 188 004 | |
| Reagente di coltura cellulare | 20% di sostituzione del siero knock out | Thermo Fisher | 10828–028 | |
| Reagente di coltura cellulare | bFGF | Stemgent | 37316 | |
| Reagente di coltura cellulare | Matrice a membrana Matrigel Basement Membrane | Corning | 354234 | |
| Reagente di coltura cellulare | mTeSR1 | Tecnologie STEMCELL | 85850 | |
| Reagente di coltura cellulare | Kit di riprogrammazione CytoTune-iPS 2.0 Sendai | ThermoFisher | A16517 | |
| Linea cellulare (umana) | Cellule staminali embrionali H1 | Regalo del Dr. Lawrence W. Stanton, WiCell | RRID:CVCL_C813 | |
| Anticorpo | Alexa Fluor 594 secondario Ab | ThermoFisher | Cat# A-11076, RRID:AB_2534120 | |
| Anticorpo | Alexa Fluor 488 secondario Ab | ThermoFisher | Cat# A-11001, RRID:AB_2534069 | |
| Sistema di dosaggio/kit | Sistema Maestro MEA | Biosistema Axion | – |
Diagnosi delle malattie e consenso informato
I pazienti sono stati identificati e diagnosticati da genetisti clinici presso il King Hussein Medical Centre, Amman, Giordania. È stato ottenuto il consenso informato delle famiglie per i test genetici in conformità con le linee guida etiche istituzionali approvate (Institute of Medical Biology, Singapore, A*STAR, Singapore, NUS-IRB codice di riferimento 10-051). Per i pazienti della Figura 1, il consenso informato alla pubblicazione di fotografie e video è stato ottenuto dai genitori. I campioni di DNA genomico sono stati isolati dalla saliva utilizzando il kit di raccolta del DNA di Oragene (OG-500, DNAGenotek) ed è stata effettuata una biopsia cutanea punch dal paziente II.4.
Sequenziamento dell’intero esoma
L’intero sequenziamento dell’exome di proband II-1 è stato eseguito utilizzando il kit di arricchimento dell’exome Illumina TruSeq Exome Enrichment Kit per la cattura dell’exome utilizzando 1 ug di DNA genomico. La modalità ad alto rendimento Illumina HiSeq2000 è stata utilizzata per il sequenziamento come 100 bp paired-end eseguito presso l’UCLA Clinical Genomics Centre e presso l’UCLA Broad Stem Cell Research Centre come descritto in precedenza(Hu et al., 2014). Una copertura media di 34X è stata raggiunta in tutto l’exome con l’87% di queste basi coperte a ≥10X. Dopo il filtraggio, un totale di 73 varianti omozigoti, 125 eterozigoti composti e 493 eterozigoti erano varianti che cambiavano le proteine con frequenze alleliche minori della popolazione < 1%.
Mappatura dell’omozigosi
Entrambi i genitori e i loro cinque figli sono stati genotipizzati utilizzando Illumina Humancore-12v1 BeadChips seguendo le istruzioni del produttore. I tassi di chiamata erano superiori al 99%. Sesso e relazioni sono stati verificati utilizzando Illumina BeadStudio. La mappatura è stata eseguita cercando le regioni condivise che sono omozigoti e identiche per discendenza (IBD) nei due bambini colpiti utilizzando programmi personalizzati scritti nel software di analisi dei dati Mathematica (Wofram Research, Inc.) (File di codice sorgente 1 – programma di collegamento IBD). Le regioni candidate sono state ulteriormente perfezionate escludendo i segmenti omozigoti comuni con i membri della famiglia non affetti. I criteri di fiducia per identificare i blocchi IBD erano di almeno 5 cM. Abbiamo identificato un loci candidato condiviso sul cromosoma 5.
Coltura cellulare e trasfezione di plasmidi
HEK 293 cellule T (ATCC Cat# CRL-3216, RRID:CVCL_0063, da Lab Stock) sono state coltivate in media DMEM (Gibco) integrate con il 10% di FBS. L’identità della linea cellulare è stata autenticata da profilazione umana commerciale STR con ATCC (ATCC, #135-XV). Tutte le linee cellulari utilizzate in questo documento sono risultate negative al micoplasma utilizzando Lonza MycoAlert (Lonza LT07). Per la trasfezione transitoria, 6 × 105 cellule per pozzo sono state seminate in 6 piastre di pozzo 24 ore prima di essere trasfettati con Lipofectamina 2000 (ThermoFisher) – plasmidi complessi in OPTIMEM (ThermoFisher). Per costruire i plasmidi di espressione CAMK2A, il cDNA CAMK2A umano è stato amplificato in PCR da ImageClone con siti di restrizione AscI e PacI e clonato in pCDNA3.1 con un N-terminale 3xFLAG o 3xHA tag. Tutti i mutanti CAMK2A sono stati generati utilizzando QuikChange XL (Agilent). Le cellule sono state trattate con MG132 (Sigma, M8699) a 5 µM per bloccare la degradazione del proteasoma.
Generazione di iPSC
Controllare le iPSC da un individuo non correlato ma etnicamente e sessualmente compatibile
I fibroblasti sono stati trasdotti con OCT4, SOX2, KLF4 e c-MYC (i plasmidi Addgene #17225, #17226, #17227 erano regali di George Daley, e #18119 un regalo di John Cleveland) come precedentemente descritto(Park et al., 2008). Dopo 4 giorni, le cellule trasdotte sono state riseminate su fibroblasti embrionali di topo irradiato in medium cellulare umano ES (DMEM/F12 (Sigma, D6421) integrato con il 20% di Knock Out Serum Replacement (Thermo Fisher Scientific, 10828-028), 0,1 mM 2-mercaptoetanolo (Thermo Fisher Scientific, 21985-023), 2 mM L-glutammina (Thermo Fisher Scientific, 25030), 0.2 mM NEAA (Thermo Fisher Scientific, 11140-050) e 5 ng/mL bFGF (Stemgent, 03-0002). Le colonie iPSC sono state raccolte tra i giorni 17-28 e mantenute in matrigel (Corning, 354234) e mTeSR1 (STEMCELL Technologies, 85850) per l’espansione.
iPSC di proband, II.4
I fibroblasti primari dermici sono stati riprogrammati utilizzando il kit di riprogrammazione CytoTune-iPS 2.0 Sendai (Thermo Fisher Scientific, A16517) secondo le istruzioni del produttore. Le cellule sono state passate e placcate su alimentatori embrionali di topo irradiato 7 giorni dopo la trasfezione virale in mezzo cellulare umano ES (DMEM/F12 (Sigma, D6421) integrato con il 20% di Knock Out Serum Replacement (Thermo Fisher Scientific, 10828-028), 0,1 mM 2-mercaptoetanolo (Thermo Fisher Scientific, 21985-023), 2 mM L-glutammina (Thermo Fisher Scientific, 25030), 0.2 mM NEAA (Thermo Fisher Scientific, 11140-050) e 5 ng/mL bFGF (Stemgent, 03-0002). Le colonie iPSC sono state raccolte tra i giorni 17-28 e mantenute in Matrigel Basement Membrane Matrix (Corning, 354234) e mTeSR1 (STEMCELL Technologies, 85850) per l’espansione.
Differenziazione neuronale e registrazioni di array multielettrodo
I PNG derivati dalle iPSC sono stati differenziati in neuroni per 21 giorni utilizzando un protocollo pubblicato in precedenza(Xu et al., 2017a). In breve, le PNG sono stati placcati ad una densità di 50.000 cellule / cm2 in un poli-L-ornitina e laminina rivestita piastre, coltivate in N2B27 mezzo integrato BDNF (20 ng / ml), GDNF (20 ng / ml), cAMP (N6,2′-O-dibutiladenosina 3′,5′ – monofosfato ciclico; Sigma; 0,3 mM) e acido ascorbico (0,2 mM). H1 cellule staminali embrionali (dono di Lawrence W. Stanton, WiCell, RRID:CVCL_C813) sono stati anche differenziati in neuroni da utilizzare come controlli. Le cellule staminali embrionali H1 sono state verificate mediante cariotipizzazione (Cytogenetics Laboratory, KK Women’s and Children’s Hospital, Singapore) e controllate per la pluripotenza mediante differenziazione nei tre strati germinali contrassegnati da Nestin (ectoderma), AFP (endoderma) e ASM-1 (mesoderma). Per la colorazione di immunofluorescenza, i neuroni sono stati fissati per 15 minuti in ghiaccio freddo 4% (w / v) paraformaldeide. La permeabilizzazione con 0,3% (v/v) Triton-X in 1X PBS è stata eseguita per 10 minuti e poi incubata con 1:1000 topo anti-Tuj1 (Covance Research Products Inc Cat# MMS-435P, RRID:AB_2313773), 1:1500 cavia anti-MAP2 (Synaptic Systems Cat# 188 004, RRID:AB_2138181) per una notte a 4°C in 5% (w/v) BSA diluito con 1X PBS. Per la visualizzazione, è stato applicato l’anticorpo secondario 1:1000 coniugato con Alexa Fluor 594 (Thermo Scientific Cat# A-11076, RRID:AB_2534120) o Alexa Fluor 488 (Thermo Scientific Cat# A-11001, RRID:AB_2534069). La controcolorazione dei nuclei è stata eseguita utilizzando Dapi. Le immagini sono state catturate utilizzando l’FV3000 Olympus confocal.
Per le registrazioni multi-array di elettrodi (MEA), i neuroni il giorno 21 sono stati dissociati e replicati su piastre MEA a 0,1 polietilenimmina (Sigma) rivestite di 48 pozzetti (Axion Biosystems) in media BrainPhys integrati con BDNF, GDNF, cAMP e acido ascorbico come descritto in precedenza(Xu et al., 2017b). L’attività neuronale spontanea è stata osservata e registrata a 37°C per 5 minuti ogni 2-3 giorni utilizzando il sistema Maestro MEA (Axion Biosystem). Misure indipendenti sono state prese da sette pozzi per ogni condizione (repliche tecniche).
Trascrizione in vitro / traduzione e co-immunoprecipitazione
Le proteine CAMK2A sono state sintetizzate in vitro utilizzando TNT T7 Quick Coupled Transcription/Translation (Promega) con 1 μg di plasmidi in 20 μl di volumi di reazione per 90 minuti a 30°C. Per la co-immunoprecipitazione, le reazioni sono state diluite 10x in TBS (100 mM Tris-HCl, pH = 8, 150 mM NaCl) con 1% Nonidet P40 (NP40) e incubato con 10 microlitri anti-FLAG M2 perle di agarosio (Sigma) a 4 ° C durante la notte. Le proteine sono state eluite con 1x Laemmli buffer dopo 3 lavaggi nel 1xTBS con 1% NP40.
Elettroforesi delle proteine e immunoblotting
Il lisato proteico totale è stato quantificato utilizzando un test standard di Bradford e 10 μg di lisato è stato utilizzato per esperimenti di immunoblotting. Per Blue Native PAGE, le cellule sono state lisate in 1x tampone di preparazione del campione (ThermoFisher) contenente l’1% di digitonina. 1% SDS è stato integrato per SDS-PAGE. Tutte le proteine sono state trasferite su membrane in PVDF utilizzando TurboBlot (Bio-rad) a 2,5 mA per sette minuti. Gli anticorpi primari utilizzati erano anti-FLAG (M2, Sigma-Aldrich Cat# F3165, RRID:AB_259529), anti-GAPDH (Santa Cruz Biotechnology Cat# sc-47724, RRID:AB_627678) e anti-HA (Y-11, Santa Cruz Biotechnology Cat# sc-805, RRID:AB_631618). Gli anticorpi secondari erano IgG-HRP anti-coniglio (Jackson ImmunoResearch Labs Cat# 111-035-003, RRID:AB_2313567), IgG-HRP anti-topo (specifici per catene leggere) (Jackson ImmunoResearch Labs Cat# 205-032-176, RRID:AB_2339056) e IgG-HRP anti-topo (Jackson ImmunoResearch Labs Cat# 115-035-003, RRID:AB_10015289).
Ceppi di vermi
Tutti i ceppi sono stati mantenuti a 20°C sulle piastre di crescita dei nematodi di E. coli OP50 come descritto (Brenner, 1974). I ceppi N2 Bristol (WB Cat# CB4852, RRID:WB-STRAIN:CB4852) sono stati usati come riferimento WT, e il mutante unc-43(e408) è stato usato. Per visualizzare le vescicole sinaptiche nel neurone DA9, è stato utilizzato wyIs85 [Pitr-1::GPF::RAB-3](Klassen e Shen, 2007).
Linee transgeniche
OTL70 wyIs85[Podr-1::dsred, Pitr-1::gfp::rab-3]; jpnEx40[Podr-1::gfp, Pmig-13::unc-43]
OTL71 wyIs85; jpnEx41[Podr-1::gfp, Pmig-13::unc-43]
OTL72 unc-43(e408); wyIs85; jpnEx44[Podr-1::gfp, Pmig-13::unc-43(H466Y)].
OTL73 unc-43(e408); wyIs85; jpnEx45[Podr-1::gfp, Pmig-13::unc-43(H466Y)].
OTL74 unc-43(e408); wyIs85; jpnEx42[Podr-1::gfp, Pmig-13::unc-43].
OTL75 unc-43(e408); wyIs85; jpnEx43[Podr-1::gfp, Pmig-13::unc-43].
OTL76 unc-43(e408); wyIs85; jpnEx47[Podr-1::gfp, Pmig-13::CAMK2A].
OTL77 unc-43(e408); wyIs85; jpnEx48[Podr-1::gfp, Pmig-13::CAMK2A]
OTL78 unc-43(e408); wyIs85; jpnEx49[Podr-1::gfp, Pmig-13::CAMK2AH477Y].
OTL79 unc-43(e408); wyIs85; jpnEx50[Podr-1::gfp, Pmig-13::CAMK2AH477Y]].
OTL82: jpnEx54[Podr-1::gfp, Pmig-13::mcheery::unc-43]
OTL83: jpnEx55[Podr-1::gfp, Pmig-13::mcherry::unc-43]
OTL84: jpnEx56[Podr-1::gfp, Pmig-13::mcherry::unc-43(H466Y)]
OTL85: jpnEx57[Podr-1::gfp, Pmig-13::mcherry::unc-43(H466Y)]
OTL86: unc-43(e408); jpnEx58[Punc-122::dsred, Punc-104::mcherry::unc-43, Phlh-1::mcherry::unc-43]
OTL87: unc-43(e408); jpnEx59[Punc-122::dsred, Punc-104::mcherry::unc-43, Phlh-1::mcherry::unc-43].
OTL88: unc-43(e408); jpnEx60[Punc-122::dsred, Punc-104::mcherry::unc-43(H466Y), Phlh-1::mcherry::unc-43(H466Y)].
OTL89: unc-43(e408); jpnEx61[Punc-122::dsred, Punc-104::mcherry::unc-43(H466Y), Phlh-1::mcherry::unc-43(H466Y)].
Plasmidi per l’espressione transgenica nei vermi
I plasmidi di espressione per le linee di vermi transgenici sono stati realizzati utilizzando il vettore pSM (C. Bargmann), un derivato di pPD49.26 (A. Fire). Il promotore mig-13 è stato clonato tra SphI/AscI, e C.elegans unc-43 isoform d o umano CAMK2α è stato clonato tra NheI/KpnI o AscI/NheI, rispettivamente. Le mutazioni P.H466Y e p.H477Y sono state introdotte dalla mutagenesi basata sulla PCR utilizzando la polimerasi del DNA polimerasi ad alta fedeltà KOD-plus- high fidelity (TOYOBO, Tokyo, Giappone). I vermi transgenici sono stati generati come descritto(Mello, 1995). I plasmidi sono stati iniettati negli animali a 10 ng/μl (nel caso di Pmig-13::unc-43) e 50 ng/μl (nel caso di Pmig-13::CAMK2α )insieme a marcatori di coiniezione a 90 ng/μl.
Quantificazione in fluorescenza e immagini confocali
Tutte le immagini a fluorescenza di DA9 sono state scattate in vermi vivi immobilizzati con il 5% di agar pad, 10 μM di levamisolo (Sigma) e perle di polistirolo da 0,1 mm (Polysciences, Inc., Warrington, PA, USA) con un obiettivo 63×/1,4 NA su un sistema di imaging Zeiss Axioplan 2 o un obiettivo Plan-Apochromat 63×/1,3 su un microscopio confocale Zeiss LSM710 utilizzando parametri di imaging simili per lo stesso marcatore attraverso genotipi diversi. La quantificazione in fluorescenza è stata determinata utilizzando il software Image J (ImageJ, RRID:SCR_003070).
Analisi comportamentale
mcherry::unc-43 o mcherry::unc-43(H466Y) sono stati espressi in vermi mutanti unc-43(e403) utilizzando sia il promotore unc-104 (promotore neuronale specifico) che il promotore hlh-1 (promotore muscolare specifico). Due linee indipendenti sono state stabilite e analizzate. Il fenotipo comportamentale dei vermi transgenici è stato segnato in doppio cieco con un microscopio stereo (SZX16, Olumpus, Tokyo, Giappone). Dal comportamento di movimento sulle piastre NGM dell’alimentatore OP50, ogni verme è stato classificato per comportarsi come ‘wild-type‘ o ‘unc-43’. Sono stati osservati 100 vermi per ogni genotipo nello stesso giorno da due linee transgeniche di derivazione indipendente.
References
- Achterberg KG, Buitendijk GH, Kool MJ, Goorden SM, Post L, Slump DE, Silva AJ, van Woerden GM, Kushner SA, Elgersma Y. Temporal and region-specific requirements of αCaMKII in spatial and contextual learning. Journal of Neuroscience. 2014; 34:11180. DOI | PubMed
- Bennett MK, Erondu NE, Kennedy MB. Purification and characterization of a calmodulin-dependent protein kinase that is highly concentrated in brain. The Journal of Biological Chemistry. 1983; 258:12735-12744. PubMed
- Bhattacharyya M, Stratton MM, Going CC, McSpadden ED, Huang Y, Susa AC, Elleman A, Cao YM, Pappireddi N, Burkhardt P, Gee CL, Barros T, Schulman H, Williams ER, Kuriyan J. Molecular mechanism of activation-triggered subunit exchange in Ca(2+)/calmodulin-dependent protein kinase II. eLife. 2016; 5DOI | PubMed
- Brenner S. The Genetics of CAENORHABDITIS ELEGANS. Genetics. 1974; 77PubMed
- Cho KO, Wall JB, Pugh PC, Ito M, Mueller SA, Kennedy MB. The alpha subunit of type II Ca2+/calmodulin-dependent protein kinase is highly conserved in Drosophila. Neuron. 1991; 7:439-450. DOI | PubMed
- Frankland PW, O’Brien C, Ohno M, Kirkwood A, Silva AJ. Alpha-CaMKII-dependent plasticity in the cortex is required for permanent memory. Nature. 2001; 411:309-313. DOI | PubMed
- Ghosh A, Giese KP. Calcium/calmodulin-dependent kinase II and Alzheimer’s disease. Molecular Brain. 2015; 8DOI | PubMed
- Hanson PI, Schulman H. Neuronal Ca2+/calmodulin-dependent protein kinases. Annual Review of Biochemistry. 1992; 61:559-601. DOI | PubMed
- Hu WF, Pomp O, Ben-Omran T, Kodani A, Henke K, Mochida GH, Yu TW, Woodworth MB, Bonnard C, Raj GS, Tan TT, Hamamy H, Masri A, Shboul M, Al Saffar M, Partlow JN, Al-Dosari M, Alazami A, Alowain M, Alkuraya FS, Reiter JF, Harris MP, Reversade B, Walsh CA. Katanin p80 regulates human cortical development by limiting centriole and cilia number. Neuron. 2014; 84:1240-1257. DOI | PubMed
- Hudmon A, Schulman H. Neuronal CA2+/calmodulin-dependent protein kinase II: the role of structure and autoregulation in cellular function. Annual Review of Biochemistry. 2002a; 71:473-510. DOI | PubMed
- Hudmon A, Schulman H. Structure-function of the multifunctional Ca2+/calmodulin-dependent protein kinase II. Biochemical Journal. 2002b; 364:611. DOI | PubMed
- Kandel ER, Dudai Y, Mayford MR. The molecular and systems biology of memory. Cell. 2014; 157:163-186. DOI | PubMed
- Klassen MP, Shen K. Wnt signaling positions neuromuscular connectivity by inhibiting synapse formation in C. elegans. Cell. 2007; 130:704-716. DOI | PubMed
- Küry S, van Woerden GM, Besnard T, Proietti Onori M, Latypova X, Towne MC, Cho MT, Prescott TE, Ploeg MA, Sanders S, Stessman HAF, Pujol A, Distel B, Robak LA, Bernstein JA, Denommé-Pichon AS, Lesca G, Sellars EA, Berg J, Carré W, Busk ØL, van Bon BWM, Waugh JL, Deardorff M, Hoganson GE, Bosanko KB, Johnson DS, Dabir T, Holla ØL, Sarkar A, Tveten K, de Bellescize J, Braathen GJ, Terhal PA, Grange DK, van Haeringen A, Lam C, Mirzaa G, Burton J, Bhoj EJ, Douglas J, Santani AB, Nesbitt AI, Helbig KL, Andrews MV, Begtrup A, Tang S, van Gassen KLI, Juusola J, Foss K, Enns GM, Moog U, Hinderhofer K, Paramasivam N, Lincoln S, Kusako BH, Lindenbaum P, Charpentier E, Nowak CB, Cherot E, Simonet T, Ruivenkamp CAL, Hahn S, Brownstein CA, Xia F, Schmitt S, Deb W, Bonneau D, Nizon M, Quinquis D, Chelly J, Rudolf G, Sanlaville D, Parent P, Gilbert-Dussardier B, Toutain A, Sutton VR, Thies J, Peart-Vissers L, Boisseau P, Vincent M, Grabrucker AM, Dubourg C, Tan WH, Verbeek NE, Granzow M, Santen GWE, Shendure J, Isidor B, Pasquier L, Redon R, Yang Y, State MW, Kleefstra T, Cogné B, Petrovski S, Retterer K, Eichler EE, Rosenfeld JA, Agrawal PB, Bézieau S, Odent S, Elgersma Y, Mercier S, Undiagnosed Diseases Network, GEM HUGO, Deciphering Developmental Disorders Study. De novo mutations in protein kinase genes camk2a and CAMK2B cause intellectual disability. American Journal of Human Genetics. 2017; 101:768-788. DOI | PubMed
- Lisman J, Schulman H, Cline H. The molecular basis of CaMKII function in synaptic and behavioural memory. Nature reviews. Neuroscience. 2002; 3:175-190. DOI | PubMed
- Liu XB, Murray KD. Neuronal excitability and calcium/calmodulin-dependent protein kinase type II: location, location, location. Epilepsia. 2012; 53:45-52. DOI | PubMed
- Lord JM. Protein degradation: Go outside and see the proteasome. Current Biology. 1996; 6:1067-1069. DOI | PubMed
- Maeder CI, San-Miguel A, Wu EY, Lu H, Shen K. In vivo neuron-wide analysis of synaptic vesicle precursor trafficking. Traffic. 2014; 15:273-291. DOI | PubMed
- Matsuo N, Yamasaki N, Ohira K, Takao K, Toyama K, Eguchi M, Yamaguchi S, Miyakawa T. Neural activity changes underlying the working memory deficit in alpha-CaMKII heterozygous knockout mice. Frontiers in Behavioral Neuroscience. 2009; 3DOI | PubMed
- Meador WE, Means AR, Quiocho FA. Modulation of calmodulin plasticity in molecular recognition on the basis of x-ray structures. Science. 1993; 262:1718-1721. DOI | PubMed
- Mello C. DNA transformation. Methods Cell Biology. 1995; 48:451-482. DOI
- Oromendia AB, Dodgson SE, Amon A. Aneuploidy causes proteotoxic stress in yeast. Genes & Development. 2012; 26:2696-2708. DOI | PubMed
- Park IH, Zhao R, West JA, Yabuuchi A, Huo H, Ince TA, Lerou PH, Lensch MW, Daley GQ. Reprogramming of human somatic cells to pluripotency with defined factors. Nature. 2008; 451DOI | PubMed
- Shevell M, Ashwal S, Donley D, Flint J, Gingold M, Hirtz D, Majnemer A, Noetzel M, Sheth RD, Quality Standards Subcommittee of the American Academy of Neurology, Practice Committee of the Child Neurology Society. practice parameter: evaluation of the child with global developmental delay: report of the quality standards subcommittee of the american academy of neurology and the practice committee of the child neurology society. Neurology. 2003; 60:367-380. DOI | PubMed
- Reiner DJ, Newton EM, Tian H, Thomas JH. Diverse behavioural defects caused by mutations in Caenorhabditis elegans unc-43 CaM kinase II. Nature. 1999; 402:199-203. DOI | PubMed
- Rich RC, Schulman H. Substrate-directed function of calmodulin in autophosphorylation of Ca2+/calmodulin-dependent protein kinase II. Journal of Biological Chemistry. 1998; 273:28424-28429. DOI | PubMed
- Robison AJ. Emerging role of CaMKII in neuropsychiatric disease. Trends in Neurosciences. 2014; 37:653-662. DOI | PubMed
- Rongo C, Kaplan JM. CaMKII regulates the density of central glutamatergic synapses in vivo. Nature. 1999; 402:195-199. DOI | PubMed
- Shevell M. Global developmental delay and mental retardation or intellectual disability: conceptualization, evaluation, and etiology. Pediatric clinics of North America. 2008; 55:1071-1084. DOI | PubMed
- Silva AJ, Paylor R, Wehner JM, Tonegawa S. Impaired spatial learning in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992a; 257:206-211. DOI | PubMed
- Silva AJ, Stevens CF, Tonegawa S, Wang Y. Deficient hippocampal long-term potentiation in alpha-calcium-calmodulin kinase II mutant mice. Science. 1992b; 257:201-206. DOI | PubMed
- Srour M, Shevell M. Genetics and the investigation of developmental delay/intellectual disability. Archives of Disease in Childhood. 2014; 99:386-389. DOI | PubMed
- Stephenson JR, Wang X, Perfitt TL, Parrish WP, Shonesy BC, Marks CR, Mortlock DP, Nakagawa T, Sutcliffe JS, Colbran RJ. A novel human CAMK2A mutation disrupts dendritic morphology and synaptic transmission, and causes ASD-related behaviors. Journal of Neuroscience. 2017; 37:2216-2233. DOI | PubMed
- Stratton M, Lee IH, Bhattacharyya M, Christensen SM, Chao LH, Schulman H, Groves JT, Kuriyan J. Activation-triggered subunit exchange between CaMKII holoenzymes facilitates the spread of kinase activity. eLife. 2014; 3DOI | PubMed
- Stratton MM, Chao LH, Schulman H, Kuriyan J. Structural studies on the regulation of Ca2+/calmodulin dependent protein kinase II. Current Opinion in Structural Biology. 2013; 23:292-301. DOI | PubMed
- Takemoto-Kimura S, Suzuki K, Horigane SI, Kamijo S, Inoue M, Sakamoto M, Fujii H, Bito H. Calmodulin kinases: essential regulators in health and disease. Journal of Neurochemistry. 2017; 141:808-818. DOI | PubMed
- Tsui J, Inagaki M, Schulman H. Calcium/calmodulin-dependent protein kinase II (CaMKII) localization acts in concert with substrate targeting to create spatial restriction for phosphorylation. Journal of Biological Chemistry. 2005; 280:9210-9216. DOI | PubMed
- van Bokhoven H. Genetic and epigenetic networks in intellectual disabilities. Annual Review of Genetics. 2011; 45:81-104. DOI | PubMed
- Xu X, Radulescu C, Utami K, Pouladi M. Obtaining multi-electrode array recordings from human induced pluripotent stem cell–derived neurons. Bio-Protocol. 2017a; 7DOI
- Xu X, Tay Y, Sim B, Yoon SI, Huang Y, Ooi J, Utami KH, Ziaei A, Ng B, Radulescu C, Low D, Ng AYJ, Loh M, Venkatesh B, Ginhoux F, Augustine GJ, Pouladi MA. Reversal of phenotypic abnormalities by crispr/cas9-mediated gene correction in huntington disease patient-derived induced pluripotent stem cells. Stem Cell Reports. 2017b; 8:619-633. DOI | PubMed
- Yang E, Schulman H. Structural examination of autoregulation of multifunctional calcium/calmodulin-dependent protein kinase II. Journal of Biological Chemistry. 1999; 274:26199-26208. DOI | PubMed
Fonte
Chia PH, Zhong FL, Niwa S, Bonnard C, Utami KH, et al. () A homozygous loss-of-function . eLife 7e32451. https://doi.org/10.7554/eLife.32451




