
Introduzione
Una migliore comprensione della trasmissione della tubercolosi (TBC) è fondamentale per il controllo della TBC nel 21° secolo. Le risorse economiche sono molto limitate in molti paesi ad alto carico, mentre nei paesi a basso carico il controllo della TBC è messo a rischio dalla diminuzione delle risorse, poiché la TBC non è percepita come un problema di salute pubblica[1]. I limitati finanziamenti vengono spesi per rintracciare i contatti di persone a cui è stata diagnosticata la TBC; molti di questi contatti risultano negativi al test per l’infezione da TBC, mentre altri contatti che hanno avuto un’esposizione sostanziale potrebbero non essere sottoposti a screening. Storicamente è stata utilizzata una dicotomia tra malattia attiva e latente a livello epidemiologico per differenziare i casi di TBC che possono trasmettere (malattia da TBC attiva) da quelli che non la trasmettono (latente). Tuttavia, prove più recenti suggeriscono che la transizione tra questi diversi stati è confusa e che lo sviluppo della TBC può essere meglio rappresentato come uno spettro di stati clinici e subclinici[2]. Il grado in cui la malattia subclinica contribuisce alla trasmissione è in gran parte sconosciuto, soprattutto perché gli strumenti per individuare la malattia subclinica sono disponibili solo di recente[3,4].
Il sequenziamento del genoma intero (WGS) degli isolati del paziente mostra una maggiore concordanza con le indagini di contatto rispetto ai marcatori precedenti[5]. È importante notare che il WGS è anche uno strumento superiore per delineare i cluster di trasmissione e può essere utilizzato per stimare l’onere della trasmissione[6]. Ma solo approcci molto limitati sono stati sviluppati utilizzando il WGS per identificare i singoli collegamenti di trasmissione. Le analisi filodinamiche e delle reti di trasmissione basate sull’uso combinato del WGS e dei dati epidemiologici si sono limitate principalmente all’analisi di grandi epidemie[7-10]. Tuttavia, i cluster di trasmissione che si estendono su decenni sono più un’eccezione che una regola nell’epidemiologia della TBC[11,12]. Per la maggior parte degli scenari epidemiologici, 2 limitazioni chiave impediscono l’uso di modelli filodinamici e di rete per prevedere i legami di trasmissione: la diversità dei batteri è estremamente bassa, e l’arco di tempo non permette una buona correlazione tra il tempo e l’accumulo di variazioni.
Le analisi basate sulla popolazione, in cui si possono identificare decine o centinaia di cluster di trasmissione, implicano tipicamente dimensioni di cluster di 1-15 casi di tubercolosi e tempi di campionamento di 2-5 anni. Nei paesi ad alto carico, le dimensioni dei cluster possono essere più grandi, ma i tempi sono ancora brevi. Abbiamo quindi sviluppato un approccio che ci ha permesso di analizzare simultaneamente i piccoli cluster da uno studio di 3 anni sulla popolazione nella regione di Valencia in Spagna. Il nostro approccio offre casi di indice e stima dei tempi di trasmissione.
Metodi
La nostra analisi complessiva ha proceduto come segue: raccolta di isolati, analisi di sequenziamento, identificazione di cluster di trasmissione che soddisfano determinati criteri, ricostruzione filogenetica dell’albero, calcolo del tempo dell’albero con diverse scelte di frequenza dell’orologio molecolare e, infine, analisi della trasmissione bayesiana.
Definizioni dei casi
Caso a grappolo
Un caso a grappolo è un caso genomicamente vicino ad un altro caso nella popolazione secondo una soglia genetica. In genere, per la trasmissione recente si usano 12 o 5 SNP, ma si veda sotto.
Caso indice
Il caso indice è il primo individuo documentato in un’epidemia di TBC, di solito quello che genera un’indagine epidemiologica. Nella maggior parte delle indagini epidemiologiche sulla TBC, questo coincide con (o si presume che sia) il primo individuo diagnosticato.
Molto probabilmente il genotipo ancestrale (MLAG)
Il MLAG è il genotipo ricostruito di un ipotetico caso ancestrale di un’epidemia. Può coincidere o meno con il caso indice dell’indagine epidemiologica. Una corrispondenza del MLAG con qualsiasi genotipo campionato suggerisce che il genotipo campionato è probabilmente un caso indice.
Dichiarazione etica
Questo studio è stato approvato dal Comitato Etico per la Ricerca Clinica dell’Agenzia Regionale di Sanità Pubblica di Valencia (Comité Ético de Investigación Clínica de la Dirección General de Salud Pública y Centro Superior de Investigación en Salud Pública). Si è rinunciato al consenso informato sulla base del fatto che la TBC fa parte del programma regionale di sorveglianza obbligatoria delle malattie trasmissibili. Tutte le informazioni personali sono state rese anonime e non sono stati conservati dati che consentano l’identificazione individuale.
Studiare la popolazione e isolare la raccolta
La regione di Valencia ha 4.974.475 milioni di abitanti ed è composta da 3 province, Castellón, Valencia e Alicante. Nel 2018, sono stati segnalati 315 individui affetti da tubercolosi in tutta la regione (tasso di incidenza di 6,4/100.000 abitanti); Valencia è considerata una regione a basso carico di tubercolosi. La ricerca di contatti è la procedura standard per individuare i cluster di trasmissione e viene effettuata nel 74,1% di tutti i casi di TBC notificati.
Abbiamo eseguito uno studio genomico basato sulla popolazione che ha coinvolto 785 casi positivi alla cultura della TBC nella regione di Valencia, Spagna, nel periodo 2014-2016, come parte di uno studio di epidemiologia genomica locale in corso. Utilizzando i dati WGS per delineare la trasmissione (sulla base delle distanze SNP, taglio di ≤15 SNP; vedi sotto), abbiamo identificato 121 cluster, la maggior parte dei quali ha coinvolto 2 casi per cluster (n = 325 casi di cluster; vedi S1 Text). Per la presente analisi abbiamo incluso tutti i cluster di trasmissione che coinvolgevano almeno 4 casi di TB e avevano più di 1 SNP (variante) tra i ceppi. Sulla base del feedback di un revisore, abbiamo eseguito un test chi-squared per confermare che i cluster selezionati per questo studio erano una buona rappresentazione del numero totale di casi di cluster nella popolazione.
Un totale di 21 cluster ha soddisfatto i criteri, coinvolgendo un totale di 117 persone affette da tubercolosi. Per 115 di questi abbiamo avuto dati epidemiologici, compresa la data di diagnosi e l’insorgenza dei sintomi diagnostici, oltre ad altri dati clinici e demografici. Per 2 individui abbiamo usato la data della positività della coltura con una correzione di 2 settimane per dedurre la data della diagnosi.
Analisi WGS e delineazione della trasmissione
È stato estratto il DNA dai tubi dell’indicatore di crescita dei micobatteri positivi alla coltura della TB (Becton Dickinson). Le librerie di sequenziamento sono state costruite con Nextera XT DNA Library Prep Kit (Illumina) e sequenziate sullo strumento Illumina MiSeq. Le letture di sequenziamento paired-end generate sono state tagliate e le probabili letture di contaminanti che potrebbero essere presenti nella cultura clinica sono state filtrate utilizzando il software KRAKEN[13]. L’analisi bioinformatica è stata eseguita seguendo una precedente pipeline[14]. In breve, le letture di sequenziamento sono state mappate e allineate ad un derivato del complesso di Mycobacterium tuberculosis complex (MTBC), molto probabilmente il genoma di un antenato comune. Successivamente, le varianti sono state separate in INDELS (piccole inserzioni e delezioni) e SNPs. Sono state selezionate varianti con almeno 10 letture in entrambi i ceppi e un punteggio di qualità di 20. Poiché volevamo rilevare la trasmissione genomica, ci siamo concentrati sugli SNPs che erano presenti con almeno il 90% di frequenza. Infine, gli SNPs annotati in regioni difficili da mappare come le sequenze ripetitive e i geni PPE/PE-PGRS sono stati rimossi dall’analisi, così come quelli rilevati in una finestra di 10 varianti vicino a INDELS. Inoltre, sono state rimosse le varianti che notoriamente conferiscono resistenza ai farmaci[15,16].
Questa pipeline è stata convalidata dai laboratori internazionali di riferimento sulla tubercolosi per la salute pubblica(http://tgu.ibv.csic.es/?page_id=1794) e pubblicata[17,18]. I parametri utilizzati nella pipeline sono comuni tra la comunità di ricerca sulla TBC genomica[19].
Delineazione dei cluster di trasmissione basata sulle distanze SNP e sulla filogenesi
I cluster di trasmissione sono stati definiti utilizzando un taglio sciolto di ≤15 SNPs. Inoltre, tutti i gruppi rilevati sono stati confermati costruendo una filogenesi che comprendeva tutti gli isolati. Questa filogenesi è stata dedotta utilizzando l’approccio filogenetico di massima probabilità con RAxML v8.2[20], applicando il modello General Time Reversible di sostituzione nucleotidica con la distribuzione gamma (GTRGAMMA). I cluster di trasmissione con più di 1 SNP tra i ceppi e composti da almeno 4 individui di TB sono stati mantenuti per le analisi successive. I metodi descritti di seguito sono agnostici al valore di cutoff, ma con una soglia di 15 SNPs, siamo stati sicuri di incorporare eventi di trasmissione recenti e vecchi. In ogni caso, la maggior parte dei campioni era al di sotto del valore di cutoff di 12 SNP, e l’82% era al di sotto del cutoff di 5 varianti.
Ricostruzione delle reti di correlazione genetica
L’allineamento SNP risultante per ogni cluster è stato utilizzato per dedurre una rete di parentela genetica. A causa della natura monomorfa e non ricombinante della MTBC[21] e della possibilità che il genotipo ancestrale fosse presente nei campioni, abbiamo utilizzato un algoritmo basato sulla parsimonia per la ricostruzione della rete implementato nel software PopART[22]. Abbiamo scelto un approccio di giunzione mediana della rete (MJN) perché permette ai casi di occupare posizioni centrali nella rete; si ipotizza che i genotipi nei punti di ramificazione nell’albero della parsimonia siano stati presenti ma non sono stati campionati. Inoltre, un antenato recente ricostruito del cluster basato sulla topologia filogenetica è stato aggiunto alla rete in modo da poter (1) ipotizzare il MLAG e (2) dedurre la direzionalità di un SNP (wild-type versus mutant status) dato il MLAG. Nell’analisi della rete genetica, abbiamo considerato che qualsiasi ceppo che corrisponde al MLAG per il suo cluster di trasmissione era candidato ad essere il caso indice del cluster.
Ricostruzione ad albero temporizzato
Il valore accettato per il tasso di sostituzione in TB è di circa 0,3-0,5 sostituzioni per genoma all’anno [8,23], anche se i nostri dati sembrano suggerire che questo tasso può variare sia tra i cluster che a livello di singolo lignaggio all’interno dei cluster. Per la prima volta abbiamo stimato gli alberi a tempo per tutti i cluster utilizzando il pacchetto treedater in R[24] con 5 diversi valori di clock rate (che vanno da 0,327 a 1,103) campionati da una distribuzione log-normale in seguito a una meta-analisi. Anche se abbiamo generato previsioni per una gamma di tassi, per chiarezza, i risultati nel testo principale saranno basati su un tasso di clock di 0,363, che corrisponde strettamente al tasso medio identificato nella nostra meta-analisi e in una recente pubblicazione[25] per il lignaggio MTBC 4, che domina la nostra popolazione. I parametri utilizzati per ottenere i diversi valori di clock rate, così come la meta-analisi effettuata, sono descritti e mostrati nel testo S1 e nella tabella S1.
Inferenza della trasmissione
Abbiamo sviluppato un metodo di inferenza di trasmissione simultanea su molti cluster basato sul TransPhylo, un approccio di analisi bayesiana che utilizza il metodo della catena di Markov Monte Carlo (MCMC) per ricostruire gli alberi di trasmissione dalla filogenesi patogena[26]. La differenza principale tra il nostro metodo e le precedenti capacità del TransPhylo è che possiamo eseguire l’inferenza con più cluster di trasmissione contemporaneamente, scegliendo quali parametri devono essere condivisi tra i cluster.
L’albero di trasmissione risultante contiene informazioni su chi ha infettato chi e quando, e anche se un caso viene campionato o meno. Queste informazioni sono rappresentate da una matrice le cui colonne sono i tempi di infezione, i tempi di campionamento e i trasmettitori, e le cui righe corrispondono agli individui del cluster. Se un individuo nell’albero ricostruito non viene campionato, la voce corrispondente per il tempo di campionamento è vuota. TransPhylo produce un campione posteriore di tali alberi. Da questa raccolta, possiamo estrarre (1) la probabilità posteriore che il caso indice di un cluster venga campionato e (2) la probabilità posteriore che ogni host abbia trasmesso la TB nel suo cluster. Un protocollo dettagliato che include tutte le equazioni del metodo TransPhylo si trova nel testo S1.
Per testare e convalidare il nostro metodo, abbiamo eseguito simulazioni di 2 focolai. Abbiamo osservato larghezze più strette di intervalli credibili per tutti i parametri(S2 Text e S1 e S2 Figs) utilizzando l’approccio simultaneo. Questo metodo è stato incorporato nell’ultima versione del pacchetto TransPhylo[26].
Analisi statistica
Abbiamo selezionato i casi indice e i campioni con più di 0,6 probabilità posteriori di essere trasmettitori come previsto da TransPhylo (23 trasmettitori rispetto ai rimanenti 84 casi a grappolo), con analisi di sensibilità della soglia successiva in S1 Text e S1 Table. Poi, abbiamo calcolato il rapporto di probabilità (OR) e gli intervalli di confidenza del 95% (il test esatto di Fisher) per esplorare le variabili epidemiologiche associate all’essere un trasmettitore. Inoltre, abbiamo eseguito una regressione logistica multivariata per confermare il nostro risultato univario. Sulla base del feedback della peer review, abbiamo confrontato statisticamente le variabili epidemiologiche associate ai trasmettitori con quelle dei casi non raggruppati identificati nell’intero dataset.
Risultati
Le reti genetiche suggeriscono casi di indici mancanti
Utilizzando una soglia iniziale di 15 SNP, abbiamo identificato un totale di 21 cluster di trasmissione che coinvolgono 117 casi di TB(Tabella 1). Questa soglia di 15 SNP ci ha permesso di esaminare i vecchi eventi di trasmissione, anche se la maggior parte dei casi (81,2%) erano all’interno di 5 SNP di un altro caso, in linea con una trasmissione molto recente. La maggior parte dei cluster aveva più di 1 caso con un genotipo identico (0 SNP di differenza); 5 cluster non avevano coppie identiche(Tabella S2). Non è stata osservata alcuna differenza statistica per le variabili cliniche, epidemiologiche e demografiche disponibili tra i 21 cluster di trasmissione che soddisfacevano i nostri criteri di inclusione(n = 109) e i campioni totali di cluster nella popolazione(n = 325) (vedi S1 Text e S3 Table).
| Caratteristica | Tutti i pazienti(n = 109)* |
|---|---|
| Età (anni) | |
| <18 | 11 (10%) |
| 19–34 | 20 (18%) |
| 35–65 | 66 (61%) |
| >65 | 12 (11%) |
| Sesso | |
| Femmina | 33 (30%) |
| Maschio | 76 (70%) |
| Luogo di nascita | |
| Spagna | 80 (73%) |
| Altro paese | 29 (27%) |
| Striscio di espettorato | |
| Positivo | 66 (61%) |
| Negativo | 41 (38%) |
| Tipo di malattia | |
| Polmonare | 100 (92%) |
| Extrapolmonare | 9 (8%) |
| Alcolismo | 25 (23%) |
| Diabete | 13 (12%) |
| Infetto da HIV | 10 (9%) |
| Emarginazione sociale | 13 (12%) |
| Operatore sanitario | 5 (5%) |
| Detenzione | 8 (7%) |
| Ritardo diagnostico (giorni) | |
| ≤30 | 46 (42%) |
| 31–60 | 25 (23%) |
| 61–89 | 14 (13%) |
| ≥90 | 32 (29%) |
Le reti genetiche sono un approccio popolare per cercare di capire la trasmissione senza la necessità di ulteriori dati epidemiologici. Utilizzando i dati di allineamento SNP, abbiamo applicato l’algoritmo MJN per stabilire la correlazione genetica tra i ceppi. In totale sono stati previsti 22 collegamenti mancanti (che coinvolgono 14 su 21 reti genetiche). In 5 delle reti genetiche il genotipo mancante previsto corrispondeva al MLAG, il che suggerisce che il caso indice non è stato campionato. In altri cluster mancavano genotipi intermedi. Al contrario, in 7 reti (33%) non abbiamo previsto alcun anello mancante, indicando che il MLAG previsto era presente tra i casi di TBC analizzati.
Nell’approccio MJN è ragionevole stimare che il ceppo con lo stesso genotipo del MLAG sia anche il caso indice più probabile. Tuttavia, in diversi cluster (Figg. 1, S3 e S4), più di un ceppo corrisponde al MLAG, e quindi l’approccio, che si basa esclusivamente sui genotipi, non può prevedere quale dei casi corrispondenti sia il caso indice più probabile. Una caratteristica sorprendente delle reti in cui possiamo identificare un MLAG tra i casi di TBC campionati è che questo ipotetico caso indice non sempre coincide con il primo caso diagnosticato(Fig 1A). Questa situazione si è verificata in 2 delle 5 reti in cui si è verificato un caso con lo stesso genotipo del MLAG (cluster CL045 e CL078). Insieme al fatto che in altre 14 reti genetiche il MLAG non era presente, questo suggerisce che l’ipotesi comune che il primo caso diagnosticato sia il caso indice non è necessariamente corretta. Tutte le reti ricostruite con l’approccio della rete genetica si trovano in S3 e S4 Figs.
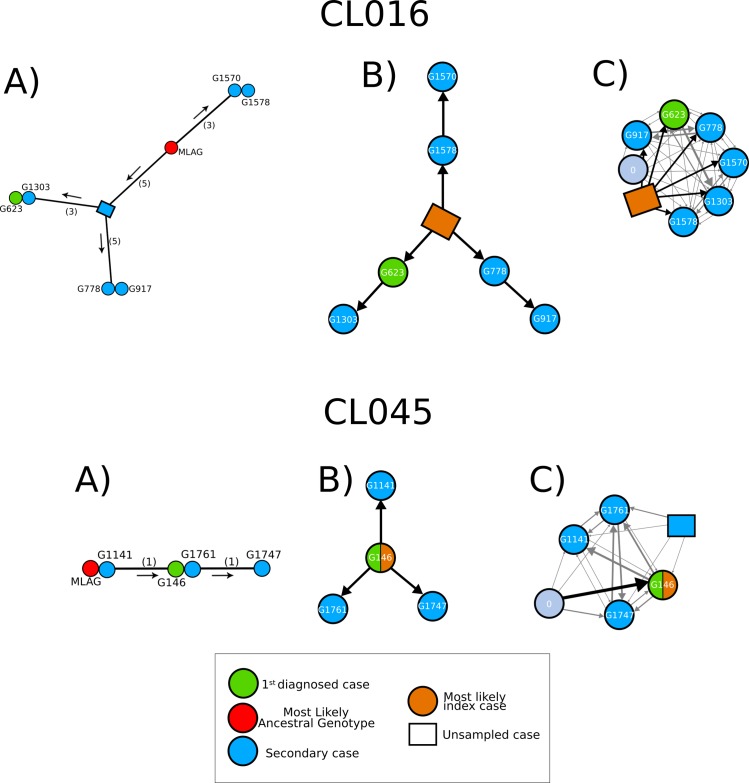
Fig 1.Confronto dei metodi di ricostruzione della trasmissione.La figura mostra per i cluster CL045 e CL016 la rete genetica dedotta (A) e l’albero di trasmissione del consenso dedotto da TransPhylo (B e C). Inoltre si mostra la forza della previsione TransPhylo (C). Quando il caso indice viene campionato, è rappresentato da una freccia nera diretta che collega il cerchio grigio “0” al rispettivo individuo. Questo è il caso di G146 in CL045. Quando manca la cassa dell’indice, questa è rappresentata da un quadrato arancione collegato a tutti i casi, come in CL016. Ogni altro caso di tubercolosi non campionato è rappresentato con un simbolo quadrato blu.
Le reti genetiche non riflettono necessariamente la trasmissione, in quanto non integrano le informazioni chiave. Ad esempio, il numero di sostituzioni osservate è influenzato dal tempo trascorso dall’infezione e dalla diversità all’interno dell’ospite; più cloni possono coesistere nello stesso individuo e possono essere trasmessi in modo differenziato. Pertanto, l’ipotesi che gli SNP siano ottenuti da un genotipo ancestrale ricostruito e che gli eventi di diversificazione rappresentino eventi di trasmissione può non essere corretta.
Fig. 1.Confronto dei metodi di ricostruzione della trasmissione.La figura mostra per i cluster CL045 e CL016 la rete genetica dedotta (A) e l’albero di trasmissione di consenso dedotto da TransPhylo (B e C). Inoltre si mostra la forza della previsione TransPhylo (C). Quando il caso indice viene campionato, è rappresentato da una freccia nera diretta che collega il cerchio grigio “0” al rispettivo individuo. Questo è il caso di G146 in CL045. Quando manca la cassa dell’indice, questa è rappresentata da un quadrato arancione collegato a tutti i casi, come in CL016. Ogni altro caso di tubercolosi non campionato è rappresentato con un simbolo quadrato blu.
TransPhylo identifica i casi indice non rilevati mediante il tracciamento dei contatti
L’approccio TransPhylo integra la temporizzazione del campione e la correlazione genetica e consente la diversità all’interno dell’ospite, evitando così il presupposto che la diversificazione rappresenti una trasmissione. TransPhylo produce eventi di trasmissione posteriori ricostruiti e tempi di trasmissione per ogni cluster, che possono essere visualizzati in molti modi, compresi gli alberi di consenso(Fig 1B) e la probabilità posteriore di infezione tra i casi(Fig 1C). Nel nostro studio, TransPhylo ha stimato che ci sono stati casi non campionati, con un numero diverso di casi non campionati in diversi cluster. Per i risultati principali, abbiamo selezionato un valore di clock rate di 0,363 SNP/genome/anno, che è il tasso ottenuto da altri[23,27]. I risultati mostrano che la maggior parte dei cluster di trasmissione ha avuto 2 o meno casi non campionati (62%). Solo 1 cluster (CL026) ha avuto un numero mediano di casi non campionati superiore a 5(Fig 2). Il numero stimato di casi non campionati è inferiore se si ipotizza un tasso di sostituzione più elevato, con pochissimi casi non campionati con un’ipotesi di clock veloce(S5 Fig). Questo effetto si verifica perché con un tasso di clock più veloce ipotizzato, i rami degli alberi a tempo sono più corti, e TransPhylo ha meno probabilità di posizionare i casi non campionati lungo i rami.
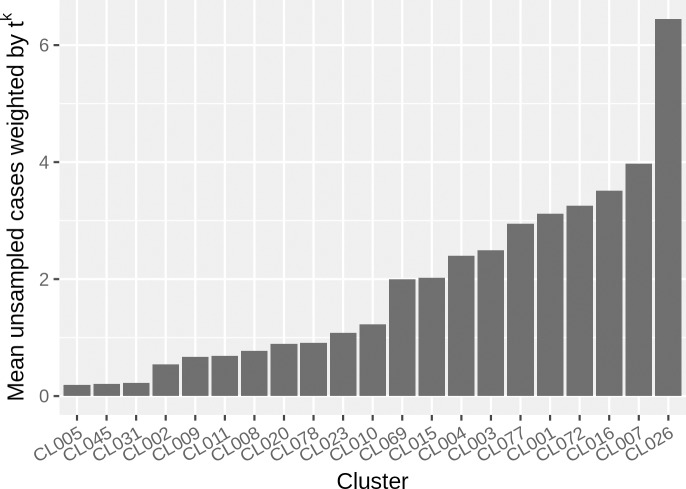
Fig 2.Numero medio ponderato di casi di tubercolosi non campionati.Per ogni albero di trasmissione posteriore associamo un fattore di ponderazione tkdove k è il numero di casi campionati per i quali la trasmissione è avvenuta dopo la diagnosi, e t = 0,1. Questo spiega il fatto che gli individui vengono trattati una volta diagnosticati e quindi hanno meno probabilità di trasmettere. Questa figura mostra il numero medio di casi non campionati per una delle frequenze di clock simulate (0,363). I risultati per tutte le frequenze di clock appaiono in S5 Fig.
L’approccio MCMC incrementato di TransPhylo ci permette di estrarre il caso indice dedotto per ogni albero posteriore. La figura 3 mostra per ogni cluster la probabilità che ogni individuo diagnosticato nel cluster fosse il caso indice, insieme ai tempi di diagnosi degli individui. Ci sono 6 cluster in cui il caso indice è stato molto probabilmente non campionato. Per i cluster in cui il caso indice è stato probabilmente campionato, il caso indice non è sempre il primo individuo diagnosticato (33%); la diagnosi del caso indice può avvenire molti mesi dopo la prima diagnosi (ad esempio, CL005). La maggior parte dei casi raggruppati non sono stati rilevati come contatti nelle indagini epidemiologiche di tracciamento dei contatti.
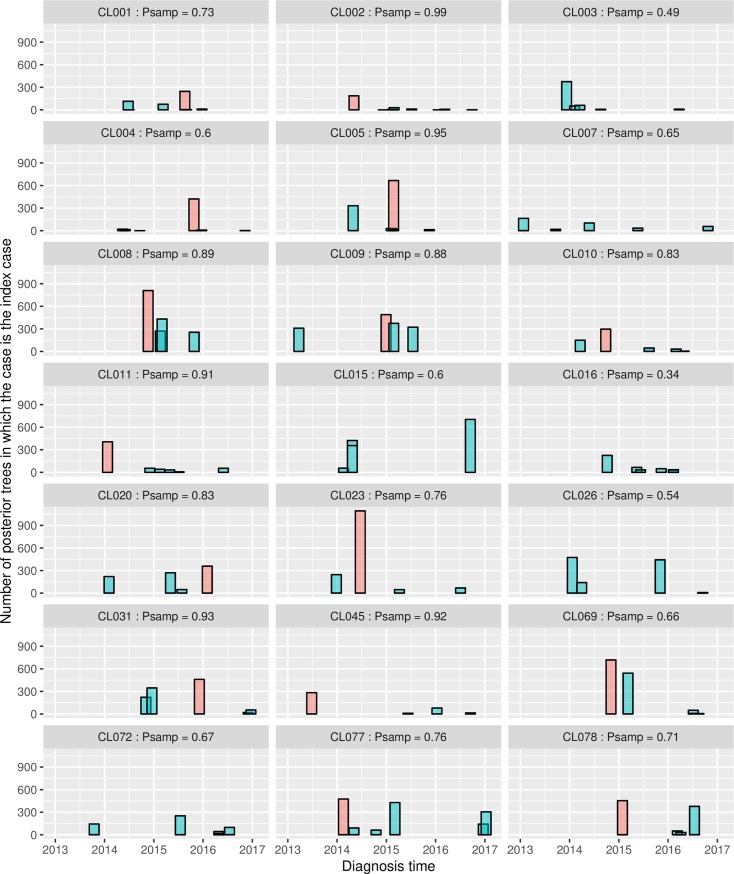
Fig. 3.La probabilità posteriore che ogni individuo sia il caso indice di un cluster rispetto al momento della diagnosi dell’individuo.L’individuo con la più alta probabilità posteriore di essere il caso indice è mostrato in rosso per ogni cluster. In alcuni cluster, il primo caso diagnosticato è stato il caso indice stimato, in quanto aveva la più alta probabilità di essere il caso indice (ad esempio, CL002). Al contrario, nella maggior parte dei cluster il caso indice più probabile non era il primo individuo diagnosticato (ad esempio, CL010 e CL023) o non è stato campionato (ad esempio, CL016 e CL003). I valori di Psamp sono la probabilità posteriore che il caso indice fosse uno qualsiasi degli individui campionati – in alcuni cluster (ad esempio, CL003) il caso indice era probabilmente un individuo non campionato.
Esiste un accordo generale tra TransPhylo e l’approccio della rete genetica nell’identificare i cluster in cui è probabile che il caso indice sia stato campionato. Per 7 cluster (33%), entrambi gli approcci hanno previsto che il caso indice fosse stato campionato. TransPhylo ha previsto la presenza di un caso indice in 8 cluster aggiuntivi in cui non si è verificato l’esatto genotipo MLAG, e di conseguenza l’approccio della rete genetica non ha previsto che il caso indice fosse stato campionato. Per gli altri cluster(n = 6), né TransPhylo né la rete genetica hanno identificato un probabile caso indice. Tuttavia, nonostante questo accordo generale, i metodi non sempre concordano su quale paziente fosse il caso indice probabile.
Le reti genetiche hanno previsto lo stesso caso indice di TransPhylo solo in 2 (13%) dei 15 cluster con un probabile caso indice campionato. Questo disaccordo è probabilmente associato al fatto che il tempo di campionamento e il tasso di cambiamento genetico non sono presi in considerazione nella previsione della rete genetica. Inoltre, l’approccio della rete genetica prevedeva più genotipi non campionati rispetto al TransPhylo, riflettendo il fatto che alcuni dei genotipi mancanti probabilmente esistevano, ma si sono evoluti all’interno di un ospite e non sono stati trasmessi(S3 e S4 Figs).
Fig 2.Fig. 2. Numero medio ponderato di casi di tubercolosi non campionati.Per ogni albero di trasmissione posteriore, associamo un fattore di ponderazione tkdove k è il numero di casi campionati per i quali la trasmissione è avvenuta dopo la diagnosi, e t = 0,1. Questo spiega il fatto che gli individui vengono trattati una volta diagnosticati e quindi hanno meno probabilità di trasmettere. Questa figura mostra il numero medio di casi non campionati per una delle frequenze di clock simulate (0,363). I risultati per tutte le frequenze di clock appaiono in S5 Fig.
Fig. 3.La probabilità posteriore che ogni individuo sia il caso indice di un cluster rispetto al momento della diagnosi dell’individuo.L’individuo con la più alta probabilità posteriore di essere il caso indice è mostrato in rosso per ogni cluster. In alcuni cluster, il primo caso diagnosticato è stato il caso indice stimato, in quanto aveva la più alta probabilità di essere il caso indice (ad esempio, CL002). Al contrario, nella maggior parte dei cluster il caso indice più probabile non era il primo individuo diagnosticato (ad esempio, CL010 e CL023) o non è stato campionato (ad esempio, CL016 e CL003). I valori di Psamp sono la probabilità posteriore che il caso indice fosse uno qualsiasi degli individui campionati – in alcuni cluster (ad esempio, CL003) il caso indice era probabilmente un individuo non campionato.
La temporizzazione degli eventi rivela casi di TBC che trasmettono prima della diagnosi o dell’insorgenza del sintomo
Poiché integra le informazioni sulla tempistica del caso e l’orologio molecolare insieme alla correlazione genetica degli isolati, TransPhylo può stimare la tempistica della trasmissione, che può essere confrontata con i tempi di diagnosi e i tempi dei sintomi riportati. Quindi, la triangolazione delle date e dei tempi rilevanti dovrebbe permetterci di usare TransPhylo per valutare quanto la trasmissione potrebbe essere evitata identificando prima gli individui affetti da TBC o isolando i pazienti durante le prime fasi del trattamento.
In primo luogo, abbiamo estratto gli alberi di trasmissione corrispondenti a uno dei tassi di clock molecolari (0,363 SNP/genome/anno) e abbiamo selezionato tutti gli individui per i quali la probabilità di trasmissione era maggiore di 0,6. Abbiamo poi confrontato i tempi di trasmissione dedotti con i tempi di diagnosi e con i tempi di insorgenza dei sintomi. Un totale di 14 individui aveva un’alta probabilità di essere trasmettitori(Fig 4). Abbiamo ragionato che se la nostra previsione era accurata, molti eventi di trasmissione dovrebbero verificarsi tra l’insorgenza dei sintomi e la diagnosi; questo è il caso di 9 dei 14 individui affetti da TBC. Tuttavia, quando abbiamo esaminato il momento della trasmissione negli altri 5 casi, la trasmissione è avvenuta prima dell’insorgenza dei sintomi o della diagnosi (G815, G258, G201, G1775 e G1449). In particolare, 3 dei 5 individui su 5 erano negativi all’espettorato al momento della diagnosi, il che suggerisce che erano infettivi prima, ma non al momento della diagnosi. L’ora del primo evento di trasmissione per tutti i casi in ogni cluster è riportata in S6- S12Figs, comprese le combinazioni di diverse probabilità e velocità di clock.
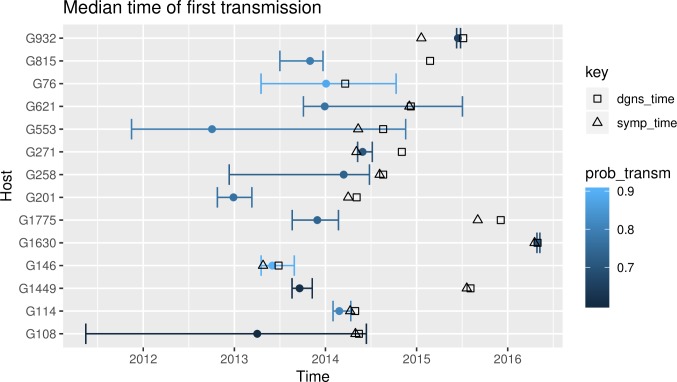
Fig 4.Tempo mediano ricampionato della prima trasmissione.Il grafico rappresenta il tempo mediano della prima trasmissione altamente probabile per gli individui per i quali la probabilità posteriore di trasmissione (prob_transm) era maggiore di 0,6, sotto un valore di clock rate di 0,363 SNP/genome/anno. Per ogni caso, vengono aggiunti il tempo di diagnosi (dgns_time; quadrati) e, dove noto, il tempo di insorgenza del sintomo (symp_time; triangoli). Grafici analoghi per i diversi cutoff di probabilità di trasmissione, e senza cutoff, sono mostrati in S6- S12Figs.
Per valutare la fattibilità che la trasmissione sia avvenuta prima dei sintomi, abbiamo analizzato il tracciato dei contatti e i dati epidemiologici disponibili per 1 dei casi. G1449 era un trasmettitore credibile prima dell’insorgenza dei sintomi(Fig 4). Il G1449 si è raggruppato con un altro caso, il G1011, che era la figlia diciottenne del G1449. Entrambi sono stati identificati quasi simultaneamente, ma la figlia è stata la prima a cercare assistenza. Così, è stata considerata il caso indice e sono stati controllati i contatti. Il G1449 è stato identificato durante lo screening pochi giorni dopo. Stimiamo che G1449 abbia infettato G1011 meno di 2 anni prima, il che è compatibile con il tempo di incubazione della TBC latente in persone senza fattori di rischio noti. Al contrario, se G1011 ha infettato G1449 dopo l’insorgenza dei sintomi, allora G1449 ha dovuto sviluppare i sintomi in meno di 1 mese dall’infezione, il che è meno probabile dell’altro scenario.
Abbiamo anche ragionato che la probabilità di trasmissione dovrebbe essere compatibile con le caratteristiche epidemiologiche note dei pazienti. Abbiamo utilizzato il tempo di arrivo dei cittadini stranieri per valutare la fattibilità che la trasmissione sia avvenuta quando abbiamo previsto. In tutti gli individui con un’alta probabilità di trasmissione della TBC, la trasmissione è avvenuta dopo l’arrivo nel Paese. Al contrario, ci sono stati 5 individui per i quali è stato previsto che la trasmissione sia avvenuta prima dell’arrivo, quindi per questi individui c’è una contraddizione tra la previsione (se si trattava di trasmettitori) e la storia epidemiologica. In tutti e 5 i casi, il nostro approccio non li ha identificati come trasmettitori credibili (probabilità di trasmissione < 0,3; tabella S4 ).
Infine, abbiamo esaminato se gli individui con tempi stimati più lunghi tra l’infezione e la diagnosi avevano un numero maggiore di casi di TBC secondaria. Questo ci si aspetterebbe, poiché una diagnosi tardiva dà a un individuo l’opportunità di esporre gli altri e di diventare il caso indice di un cluster. Abbiamo scoperto che il tempo stimato per la diagnosi era più lungo per quegli individui che si prevedeva avessero infettato 2 o più casi secondari, ma i risultati sono variabili, come previsto, dato che molti altri fattori influenzano le probabilità di trasmissione e di infezione (vedi S13 Fig).
Fig 4.Tempo mediano ricampionato della prima trasmissione.Il grafico rappresenta il tempo mediano della prima trasmissione altamente probabile per gli individui per i quali la probabilità posteriore di trasmissione (prob_transm) era maggiore di 0,6, sotto un valore di clock rate di 0,363 SNP/genome/anno. Per ogni caso, vengono aggiunti il tempo di diagnosi (dgns_time; quadrati) e, dove noto, il tempo di insorgenza del sintomo (symp_time; triangoli). Grafici analoghi per i diversi cutoff di probabilità di trasmissione, e senza cutoff, sono mostrati in S6- S12Figs.
L’identificazione dei trasmettitori permette l’associazione dei fattori di rischio alla trasmissione
Per il 66% dei cluster analizzati, il caso indice identificato da TransPhylo non è stato sottoposto a campionamento o non è stato il primo caso diagnosticato (14 su 21). Ciò suggerisce che i casi indice basati sulle date diagnostiche possono essere fuorvianti. Inoltre, le analisi dei fattori di rischio associati alla trasmissione utilizzando i dati di epidemiologia molecolare sono state tradizionalmente eseguite su misure di gruppo di clustering (clustering rispetto a casi unici, associazione con le dimensioni dei cluster). Questo approccio evita il fatto che non tutti gli individui affetti da TBC sono trasmettitori, e quindi i fattori di rischio associati alla trasmissione sono difficili da distinguere da quelli associati all’infezione. La nostra identificazione dei casi di indice e dei trasmettitori probabili ci permette di esplorare se i fattori di rischio hanno una diversa distribuzione specifica tra i probabili trasmettitori. Abbiamo combinato i casi di probabili trasmettitori con i casi di indice previsti da TransPhylo(n = 23) e li abbiamo confrontati con gli altri casi a grappolo(n = 61). La nostra analisi statistica è limitata dal basso numero di cluster e dal basso numero di trasmettitori che sono stati inequivocabilmente identificati. Inoltre, i casi di cluster sono un insieme di trasmettitori, non trasmettitori, e quei casi che non possono essere assegnati in modo sicuro ad entrambe le categorie. Tuttavia, è possibile identificare differenze rilevanti tra i probabili trasmettitori e il resto dei casi di cluster(Fig 5).
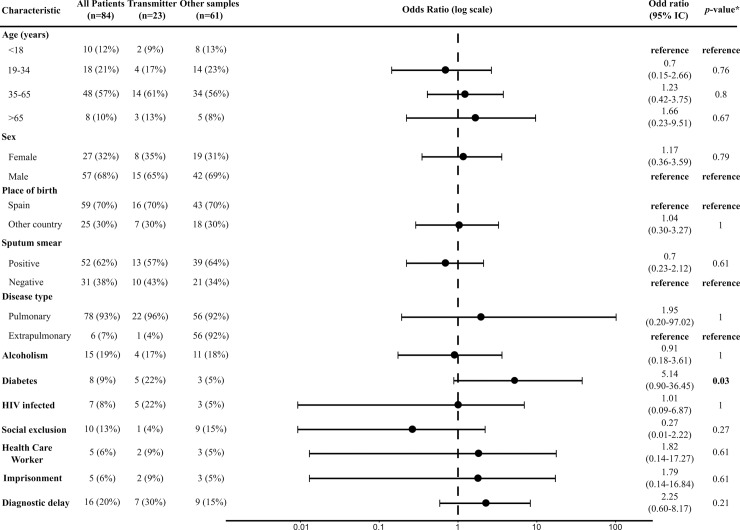
Fig 5.Caratteristiche epidemiologiche dei casi utilizzati per identificare i fattori di rischio di trasmissione.Si noti che i dati non includono tutti i campioni dello studio: per 5 cluster non siamo stati in grado di identificare un probabile evento di trasmissione, e questi cluster sono stati esclusi da questa analisi. I trasmettitori sono definiti come quegli individui che si stima siano probabili trasmettitori e/o probabili casi di indice rilevati da TransPhylo. La figura mostra i rapporti dispari stimati per ogni fattore di rischio testato. *Il test esatto del pescatore. Sono stati effettuati confronti tra i casi di trasmettitori e il resto dei campioni raggruppati.
Come prova del concetto, i trasmettitori tendevano ad essere diagnosticati in un secondo momento (ritardo diagnostico medio di 85 giorni contro 54 giorni), anche se questa differenza non è statisticamente significativa. Anche altre variabili suggeriscono importanti differenze tra l’essere un trasmettitore e il semplice fatto di far parte di un cluster. I trasmettitori sono stati significativamente arricchiti nei pazienti diabetici sia in analisi statistiche univariate (test esatto di Fisher; OR 0,19 [95% CI 0,02-1,10], p < 0,003) che multivariate (regressione logistica; OR 23,77 [95% CI 2,53-339,69], p < 0,009). È stato suggerito in precedenza che i pazienti diabetici tendono ad avere cavità di tubercolosi più grandi, un fattore noto per essere associato alla trasmissione[28]. Infine, confermiamo i rapporti precedenti che mostrano che gli individui che sono negativi allo striscio al momento della diagnosi possono essere trasmettitori (37% nel nostro dataset). Tuttavia, prendiamo questi risultati con cautela. Abbiamo ripetuto l’analisi confrontando i trasmettitori con i casi non raggruppati e il diabete era ancora arricchito (27% contro il 10%), ma non in modo significativo(p = 0,06). Anche se le piccole dimensioni del campione non ci permettono di trarre ulteriori conclusioni, questi risultati preliminari mostrano l’importanza di distinguere tra l’essere un trasmettitore e l’essere infetti.
Fig. 5.Caratteristiche epidemiologiche dei casi utilizzati per identificare i fattori di rischio di trasmissione.Si noti che i dati non includono tutti i campioni dello studio: per 5 cluster non siamo stati in grado di identificare un probabile evento di trasmissione, e questi cluster sono stati esclusi da questa analisi. I trasmettitori sono definiti come quegli individui che si stima siano probabili trasmettitori e/o probabili casi di indice rilevati da TransPhylo. La figura mostra i rapporti dispari stimati per ogni fattore di rischio testato. *Il test esatto del pescatore. Sono stati effettuati confronti tra i casi di trasmettitori e il resto dei campioni raggruppati.
Discussione
Presentiamo un approccio basato sulla genomica per svelare i singoli collegamenti di trasmissione della TBC tra i pazienti all’interno dei cluster di trasmissione. È importante notare che il nostro metodo ci permette di identificare, o di dedurre l’assenza del caso indice più probabile, nonché di stimare il numero di casi non campionati all’interno di un cluster. Questi risultati possono contribuire a riorientare le strategie di indagine dei contatti in termini di chi e dove deve essere eseguito il test per la TBC. Inoltre, identifichiamo i potenziali eventi di trasmissione durante la fase di malattia subclinica, suggerendo la necessità di incorporare le fasi iniziali della malattia nei modelli epidemiologici e nei programmi di controllo della TBC.
Il WGS ha dimostrato di essere superiore ai precedenti strumenti di genotipizzazione nell’identificare i casi di TBC che potrebbero essere di recente trasmissione[29]. Ciononostante, c’è solo un accordo del 30%-50% tra quelli identificati dal WGS come casi di TBC di recente trasmissione e quelli identificati dal contact tracing [30]. Questo scenario indica che mancano i casi di indice probabili e che sono necessarie migliori strategie di indagine sui contatti per individuare tali individui. Un recente studio clinico[31] ha mostrato che i contatti ravvicinati dei casi indice identificati dal reperimento di casi attivi hanno tassi di guarigione da TB migliori di quelli identificati dal reperimento passivo dei casi. Pertanto, l’identificazione dei casi indice ha implicazioni a livello di popolazione e di cura individuale. In questo studio, abbiamo dimostrato che fino al 28% dei cluster non vi sono prove che il caso indice sia incluso tra gli individui del cluster. Per quei cluster in cui è stato rilevato un caso indice, il 60% delle volte il caso indice non è stato il primo individuo a cui è stata diagnosticata la tubercolosi, il che suggerisce che gli sforzi per identificare la trasmissione sono imperfetti.
Le ragioni per cui i casi indice non vengono campionati in uno studio possono essere molteplici e probabilmente variano a seconda del contesto clinico. In primo luogo, la trasmissione dei casi indice potrebbe essere avvenuta prima del tempo di campionamento. Questo è molto probabile nelle nostre analisi, dove potenzialmente includiamo eventi di trasmissione più vecchi, anche se i cutoff SNP fissi possono non delineare perfettamente i cluster di trasmissione[32]. Inoltre, ci sono sfuggite quelle persone con uno stato di cultura negativo al momento della diagnosi, che potrebbero aver contribuito alla trasmissione. Tuttavia, è preoccupante che gli individui affetti da tubercolosi possano essere stati trascurati dai programmi di controllo e possano continuare a trasmettere attivamente nella popolazione. A Valencia, ogni anno vengono investigati circa 3.000 contatti seguendo le linee guida del Centro Europeo per la Prevenzione e il Controllo delle Malattie. Tuttavia, un’ampia percentuale dei casi raggruppati non è stata identificata come contatti, in linea con studi pubblicati simili[30,33-35], compresi i casi di indice previsti nella nostra analisi.
Con il nostro approccio potremmo separare i probabili trasmettitori da altri casi di cluster, piuttosto che trattare ogni cluster come una singola unità, e quindi potremmo associare alla trasmissione variabili biologiche, epidemiologiche e demografiche. Il nostro set di dati presenta 2 grandi carenze – vale a dire il basso numero di collegamenti di trasmissione con un supporto statistico sufficiente e il fatto che solo 21 cluster soddisfano i criteri per l’analisi – e quindi i nostri cluster non sono necessariamente rappresentativi dell’intera popolazione. Tuttavia, i nostri dati suggeriscono che alcuni fattori di rischio ed epidemiologici si sono arricchiti tra i trasmettitori, mentre altri si sono esauriti. Inoltre, confermiamo che gli individui con uno stato di espettorato negativo possono contribuire alla trasmissione (40% dei casi di indice), come è stato discusso in precedenza[36,37]. Set di dati più ampi basati sulla popolazione, che includono un numero maggiore di cluster che soddisfano i criteri, contribuiranno a definire meglio il ruolo esatto di questi fattori.
La nostra scelta di TransPhylo come strumento per tracciare la trasmissione è stata guidata dalla necessità di considerare i potenziali casi non campionati. Ci sono altri approcci simili che non tengono conto dei casi non campionati[38] o che utilizzano un modello più adatto ai serbatoi ambientali[39,40]. Inoltre, non abbiamo potuto fare previsioni per alcuni cluster di trasmissione a causa della limitata diversità osservata all’interno dei cluster, come anticipato in precedenza[41]. Così, la nostra analisi si è concentrata su quegli eventi che abbiamo potuto stimare in modo robusto. È importante notare che le previsioni possono essere sensibili alle variazioni di velocità molecolare. Abbiamo focalizzato la nostra discussione sulle analisi che utilizzano un tasso molecolare appropriato per i ceppi del lignaggio MTBC 4, che dominano l’ambiente locale. Tuttavia, altre impostazioni dovranno calibrare il modello con una frequenza diversa, poiché sta diventando evidente che la frequenza per i diversi lignaggi può variare[25].
Il fatto che stimiamo che circa il 35% degli eventi di trasmissione si sia verificato prima dell’insorgenza dei sintomi potrebbe avere diverse spiegazioni. I tempi di insorgenza dei sintomi dichiarati dai pazienti sono soggettivi, e se i sintomi erano lievi, la malattia potrebbe non essere stata riconosciuta per un certo periodo di tempo. Tuttavia, nella maggior parte dei casi la differenza di tempo tra l’insorgenza dei sintomi e la trasmissione si estende per diverse settimane o addirittura mesi. Recentemente si è ipotizzato che la trasmissione subclinica possa esistere ed essere facilitata da una tosse non correlata[42]. Qui mostriamo le prove della trasmissione durante la fase asintomatica della malattia, in cui la probabilità di trasmissione è più bassa che durante la malattia esacerbata, ma non è trascurabile[42,43].
Ci sono prove da studi clinici di individui positivi allo striscio di espettorato che sono altrimenti sani e che sono potenziali trasmettitori[3]. Ciò è in linea con le recenti evidenze che mostrano uno spettro di diversi stati di malattia (da quasi sano a malato[4]) e la possibilità che una percentuale di quelli tradizionalmente considerati casi di TBC tradizionalmente infetti latenti in realtà siano casi di TBC attiva con malattia subclinica[3,44]. La nostra analisi della trasmissione suggerisce che la malattia subclinica può mettere a rischio le attuali strategie di controllo della TBC, in linea con i risultati dei modelli epidemiologici[44].
Un limite del nostro metodo è che non abbiamo potuto testarlo su altre serie di dati genomici pubblicamente disponibili. Una delle ragioni è che è difficile ottenere dati epidemiologici associati ai casi, specialmente quelli relativi all’insorgenza dei sintomi (che è una variabile chiave del nostro studio). Nonostante ciò, abbiamo convalidato il nostro metodo (1) conducendo analisi di sensibilità utilizzando diversi parametri TransPhylo e (2) confrontando il tempo di trasmissione previsto per i casi di TBC di origine straniera con il tempo di immigrazione. Tuttavia, la mancanza di set di dati pubblicati con i dati epidemiologici rilevanti evidenzia la necessità di incorporare queste variabili negli studi epidemiologici sulla TBC.
In conclusione, il nostro metodo di inferenza di trasmissione basato sull’individuo dimostra che molti probabili trasmettitori, compresi i casi di indice, non vengono presi in considerazione dalle indagini di contatto. Sorprendentemente, una parte sostanziale di questi trasmettitori probabilmente diffonde la TBC durante la malattia subclinica. Il lavoro futuro di allineamento dei biomarcatori e della ricerca epidemiologica aiuterà a chiarire i biomarcatori ospiti della trasmissione durante lo spettro dell’infezione da TBC, per progettare migliori strategie di controllo della TBC.
Informazioni di supporto
References
- Lönnroth K, Migliori GB, Abubakar I, D’Ambrosio L, de Vries G, Diel R. Towards tuberculosis elimination: an action framework for low-incidence countries. Eur Respir J. 2015; 45:928-52. DOI | PubMed
- Barry CE 3rd, Boshoff HI, Dartois V, Dick T, Ehrt S, Flynn J. The spectrum of latent tuberculosis: rethinking the biology and intervention strategies. Nat Rev Microbiol. 2009; 7:845. DOI | PubMed
- Drain PK, Bajema KL, Dowdy D, Dheda K, Naidoo K, Schumacher SG. Incipient and subclinical tuberculosis: a clinical review of early stages and progression of infection. Clin Microbiol Rev. 2018; 31:e00021-18. DOI | PubMed
- Lin PL, Flynn JL. The end of the binary era: revisiting the spectrum of tuberculosis. J Immunol. 2018; 201:2541-8. DOI | PubMed
- Wyllie DH, Davidson JA, Grace Smith E, Rathod P, Crook DW, Peto TEA. A quantitative evaluation of MIRU-VNTR typing against whole-genome sequencing for identifying Mycobacterium tuberculosis transmission: a prospective observational cohort study. EBioMedicine. 2018; 34:122-30. DOI | PubMed
- Hatherell H-A, Colijn C, Stagg HR, Jackson C, Winter JR, Abubakar I. Interpreting whole genome sequencing for investigating tuberculosis transmission: a systematic review. BMC Med. 2016; 14:21. DOI | PubMed
- Gardy JL, Johnston JC, Sui SJH, Cook VJ, Shah L, Brodkin E. Whole-genome sequencing and social-network analysis of a tuberculosis outbreak. N Engl J Med. 2011; 364:730-9. DOI | PubMed
- Roetzer A, Diel R, Kohl TA, Rückert C, Nübel U, Blom J. Whole genome sequencing versus traditional genotyping for investigation of a Mycobacterium tuberculosis outbreak: a longitudinal molecular epidemiological study. PLoS Med. 2013; 10(2):e1001387. DOI | PubMed
- Stucki D, Ballif M, Bodmer T, Coscolla M, Maurer A-M, Droz S. Tracking a tuberculosis outbreak over 21 years: strain-specific single-nucleotide polymorphism typing combined with targeted whole-genome sequencing. J Infect Dis. 2015; 211:1306-16. DOI | PubMed
- Folkvardsen DB, Norman A, Andersen ÅB, Rasmussen EM, Jelsbak L, Lillebaek T. Genomic epidemiology of a major Mycobacterium tuberculosis outbreak: retrospective cohort study in a low-incidence setting using sparse time-series sampling. J Infect Dis. 2017; 216:366-74. DOI | PubMed
- van Soolingen D.. Whole-genome sequencing of Mycobacterium tuberculosis as an epidemiological marker. Lancet Respir Med. 2014; 2:251-2. DOI | PubMed
- Guerra-Assunção J, Crampin A, Houben R, Mzembe T, Mallard K, Coll F. Large-scale whole genome sequencing of M. tuberculosis provides insights into transmission in a high prevalence area. Elife. 2015; 4:e05166.
- Wood DE, Salzberg SL. Kraken: ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014; 15:R46. DOI | PubMed
- Comas I, Coscolla M, Luo T, Borrell S, Holt KE, Kato-Maeda M. Out-of-Africa migration and Neolithic coexpansion of Mycobacterium tuberculosis with modern humans. Nat Genet. 2013; 45:1176-82. DOI | PubMed
- Feuerriegel S, Schleusener V, Beckert P, Kohl TA, Miotto P, Cirillo DM. PhyResSE: a web tool delineating Mycobacterium tuberculosis antibiotic resistance and lineage from whole-genome sequencing data. J Clin Microbiol. 2015; 53:1908-14. DOI | PubMed
- Miotto P, Tessema B, Tagliani E, Chindelevitch L, Starks AM, Emerson C. A standardised method for interpreting the association between mutations and phenotypic drug resistance in Mycobacterium tuberculosis. Eur Respir J. 2017; 50:1701354. DOI | PubMed
- Cancino-Muñoz I, Moreno-Molina M, Furió V, Goig GA, Torres-Puente M, Chiner-Oms Á. Cryptic resistance mutations associated with misdiagnoses of multidrug-resistant tuberculosis. J Infect Dis. 2019; 220:316-20. DOI | PubMed
- Chiner-Oms Á, Sánchez-Busó L, Corander J, Gagneux S, Harris SR, Young D. Genomic determinants of speciation and spread of the Mycobacterium tuberculosis complex. Sci Adv. 2019; 5:eaaw3307. DOI | PubMed
- Meehan CJ, Goig GA, Kohl TA, Verboven L, Dippenaar A, Ezewudo M. Whole genome sequencing of Mycobacterium tuberculosis: current standards and open issues. Nat Rev Microbiol. 2019; 17:533-45. DOI | PubMed
- Stamatakis A.. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics. 2014; 30:1312-3. DOI | PubMed
- Comas I, Homolka S, Niemann S, Gagneux S. Genotyping of genetically monomorphic bacteria: DNA sequencing in Mycobacterium tuberculosis highlights the limitations of current methodologies. PLoS ONE. 2009; 4(11):e7815. DOI | PubMed
- Leigh JW, Bryant D. POPART: full-feature software for haplotype network reconstruction. Methods Ecol Evol. 2015; 6:1110-6.
- Duchêne S, Holt KE, Weill F-X, Le Hello S, Hawkey J, Edwards DJ. Genome-scale rates of evolutionary change in bacteria. Microb Genomics. 2016; 2:e000094.
- Volz EM, Frost SDW. Scalable relaxed clock phylogenetic dating. Virus Evol. 2017; 3:vex025. DOI
- Menardo F, Duchêne S, Brites D, Gagneux S. The molecular clock of Mycobacterium tuberculosis. PLoS Pathog. 2019; 15(9):e1008067. DOI | PubMed
- Didelot X, Fraser C, Gardy J, Colijn C. Genomic infectious disease epidemiology in partially sampled and ongoing outbreaks. Mol Biol Evol. 2017; 34:997-1007. DOI | PubMed
- Merker M, Barbier M, Cox H, Rasigade J-P, Feuerriegel S, Kohl TA. Compensatory evolution drives multidrug-resistant tuberculosis in Central Asia. Elife. 2018; 7:e38200. DOI | PubMed
- Rodrigo T, Caylà JA, García de Olalla P, Galdós-Tangüis H, Jansà JM, Miranda P. Characteristics of tuberculosis patients who generate secondary cases. Int J Tuberc Lung Dis. 1997; 1:352-7. PubMed
- Comas I, Gardy JL. TB Transmission: closing the gaps. EBioMedicine. 2018; 34:4-5. DOI | PubMed
- Jajou R, Neeling A de, Hunen R van, Vries G de, Schimmel H, Mulder A. Epidemiological links between tuberculosis cases identified twice as efficiently by whole genome sequencing than conventional molecular typing: a population-based study. PLoS ONE. 2018; 13(4):e0195413. DOI | PubMed
- Fox GJ, Nhung NV, Sy DN, Hoa NLP, Anh LTN, Anh NT. Household-contact investigation for detection of tuberculosis in Vietnam. N Engl J Med. 2018; 378:221-9. DOI | PubMed
- Stimson J, Gardy J, Mathema B, Crudu V, Cohen T, Colijn C. Beyond the SNP threshold: identifying outbreak clusters using inferred transmissions. Mol Biol Evol. 2019; 36:587-603. DOI | PubMed
- Walker TM, Lalor MK, Broda A, Ortega LS, Morgan M, Parker L. Assessment of Mycobacterium tuberculosis transmission in Oxfordshire, UK, 2007–12, with whole pathogen genome sequences: an observational study. Lancet Respir Med. 2014; 2:285-92. DOI | PubMed
- Glynn JR, Guerra-Assunção JA, Houben RMGJ, Sichali L, Mzembe T, Mwaungulu LK. Whole genome sequencing shows a low proportion of tuberculosis disease is attributable to known close contacts in rural Malawi. PLoS ONE. 2015; 10(7):e0132840. DOI | PubMed
- Yang C, Lu L, Warren JL, Wu J, Jiang Q, Zuo T. Internal migration and transmission dynamics of tuberculosis in Shanghai, China: an epidemiological, spatial, genomic analysis. Lancet Infect Dis. 2018; 18:788-95. DOI | PubMed
- Behr M, Warren S, Salamon H, Hopewell P, de Leon AP, Daley C. Transmission of Mycobacterium tuberculosis from patients smear-negative for acid-fast bacilli. Lancet. 1999; 353:444-9. DOI | PubMed
- Tostmann A, Kik SV, Kalisvaart NA, Sebek MM, Verver S, Boeree MJ. Tuberculosis transmission by patients with smear-negative pulmonary tuberculosis in a large cohort in the Netherlands. Clin Infect Dis. 2008; 47:1135-42. DOI | PubMed
- Klinkenberg D, Backer JA, Didelot X, Colijn C, Wallinga J. Simultaneous inference of phylogenetic and transmission trees in infectious disease outbreaks. PLoS Comput Biol. 2017; 13(5):e1005495. DOI | PubMed
- Hall M, Woolhouse M, Rambaut A. Epidemic reconstruction in a phylogenetics framework: transmission trees as partitions of the node set. PLoS Comput Biol. 2016; 11:e1004613. DOI | PubMed
- De Maio N, Wu C-H, Wilson DJ. SCOTTI: efficient reconstruction of transmission within outbreaks with the structured coalescent. PLoS Comput Biol. 2016; 12(12):e1005130. DOI | PubMed
- Campbell F, Strang C, Ferguson N, Cori A, Jombart T. When are pathogen genome sequences informative of transmission events?. PLoS Pathog. 2018; 14(2):e1006885. DOI | PubMed
- Esmail H, Dodd PJ, Houben RMGJ. Tuberculosis transmission during the subclinical period: could unrelated cough play a part?. Lancet Respir Med. 2018; 6:244-6. DOI | PubMed
- Houben RMGJ, Esmail H, Emery JC, Joslyn LR, McQuaid CF, Menzies NA. Spotting the old foe—revisiting the case definition for TB. Lancet Respir Med. 2019; 7:199-201. DOI | PubMed
- Dowdy DW, Basu S, Andrews JR. Is passive diagnosis enough?: The impact of subclinical disease on diagnostic strategies for tuberculosis. Am J Respir Crit Care Med. 2013; 187:543-51. DOI | PubMed
Fonte
Xu Y, Cancino-Muñoz I, Torres-Puente M, Villamayor LM, Borrás R, et al. (2019) High-resolution mapping of tuberculosis transmission: Whole genome sequencing and phylogenetic modelling of a cohort from Valencia Region, Spain. PLoS Medicine 16(10): e1002961. https://doi.org/10.1371/journal.pmed.1002961




