
Abstract
Introduzione
La malaria è causata da parassiti eucariotici unicellulari del genere Plasmodium. Questi organismi hanno un ciclo vitale complesso che comprende molti stadi di sviluppo diversi. Nel sangue dei pazienti infetti, la replicazione asessuata intra-eritrocitica dei parassiti è l’unica responsabile della patogenesi, mentre gli stadi sessuali, chiamati gametociti, sono l’unico stadio in grado di trasmettersi all’ospite successivo attraverso il vettore zanzara. Questi stadi distinti della vita sono stati ampiamente investigati utilizzando approcci trascrittomici(Otto et al., 2010; Bozdech et al., 2003a; López-Barragán et al., 2011; Llinás et al., 2006; Hall et al., 2005; Lasonder et al., 2016; Otto et al., 2014), ma questo è stato in gran parte a livello di popolazione. Si sa poco di come le singole cellule variano all’interno delle varie fasi.
L’RNA-seq monocellulare (scRNA-seq) produce profili trascrittomici per più cellule individuali. Questo ha permesso la decomposizione di popolazioni cellulari(Haber et al., 2017), ha scoperto tipi di cellule precedentemente sconosciuti(Grün et al., 2015) e ha migliorato la nostra comprensione dei percorsi di sviluppo (Mohammed et al.,2017). Sono stati ora descritti diversi metodi scRNA-seq con diversi attributi(Ziegenhain et al., 2017), con alcuni che forniscono profondità – una buona rappresentazione di trascrizioni a lunghezza intera (Picelliet al., 2013) da decine o centinaia di cellule – e altri che forniscono un’ampiezza, con una rappresentazione più scarsa dei trascrittomi, ma da un numero molto maggiore di cellule (Macoskoet al., 2015). scRNA-seq promette nuovi potenti esami di organismi unicellulari, specialmente quelli che sono difficili da ottenere in gran numero o che non sono adatti alla coltivazione in vitro. Una serie di importanti questioni di biologia della malaria beneficeranno della tecnologia monocellulare. Per esempio, quali sono gli interruttori trascrizionali nei singoli parassiti che guidano i fenotipi come l’impegno nel percorso di sviluppo sessuale(Sinha et al., 2014; Kafsack et al., 2014), il sequestro dei parassiti(Tembo et al., 2014) e l’evasione immunitaria(Scherf et al., 2008).
Un recente studio(Poran et al., 2017) ha dimostrato l’uso di una tecnica scRNA-seq ad alto rendimento e bassa copertura (Drop-seq[Macosko et al., 2015]) per identificare una firma di impegno sessuale nel Plasmodium. Qui, usiamo un approccio più basso throughput (meno cellule), ma con una copertura più alta (sia più geni rilevati che più della lunghezza di ciascun gene rilevato attraverso il sequenziamento delle trascrizioni a tutta lunghezza) per esaminare le dinamiche trascrizionali del parassita durante le fasi del sangue sia nel più popolare parassita modello roditore (P.berghei) che nel più mortale parassita della malaria umana (P.falciparum). Dimostriamo che questo metodo è molto efficace nel catturare la variazione trascrizionale associata a diversi stadi parassitari e stati del ciclo cellulare, e scopriamo anche aspetti precedentemente sconosciuti della progressione del parassita attraverso il suo ciclo asessuato e nelle sue fasi sessuali.
Risultati
Ottimizzazione di un protocollo RNA-seq monocellulare per i parassiti del Plasmodio
La maggiore copertura di geni nelle cellule dei mammiferi che utilizzano lo scRNA-seq è stata raggiunta con il protocollo Smart-Seq2 (Picelli et al., 2013). In questo metodo, le cellule sono ordinate per FACS in singoli pozzetti, seguito da una generazione di cDNA a lunghezza piena utilizzando una trascrittasi inversa virale. Questo media l’aggiunta di una tripla sovrapposizione di citosina all’estremità 3′ al primo filamento cDNA che permette la ricottura di un oligonucleotide di commutazione del filamento per la sintesi del secondo filamento e l’amplificazione diretta del cDNA tramite PCR. Questo approccio basato su piastre tende a determinare il rilevamento di più trascrizioni da più geni rispetto ad altri approcci(Svensson et al., 2017). Inoltre, si tratta di un metodo di trascrizione a lunghezza completa, che fornisce informazioni sulla struttura dei trascritti, consentendo la deconvoluzione delle varianti di splicing e l’inferenza sul filamento di origine(Wu et al., 2015).
Inizialmente, abbiamo provato la versione standard del protocollo Smart-seq2(Picelli et al., 2013) su ordinati, Plasmodium falciparum-infectedsingoli globuli rossi(Figura 1A), regolando solo il numero di cicli di PCR (30 piuttosto che 18) per tenere conto del contenuto relativamente basso di RNA delle cellule protozoo. Tuttavia, in media, solo il 10% delle letture mappate sui geni del genoma parassita e più della metà di queste mappate sui geni rRNA(Figura 1B).
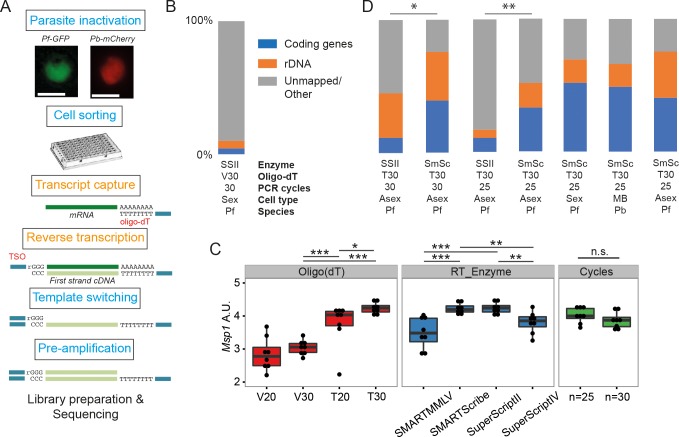
Figura 1.Figura 1. Definizione di un protocollo robusto per l’analisi trascrittomica monocellulare dei parassiti del Plasmodium.(A) Panoramica del protocollo RNAseq monocellulare. Passi nel protocollo originale Smart-seq2(Picelli et al., 2013) che ha portato a significativi guadagni sono evidenziati in arancione.(B) Il numero relativo di letture di mappatura per codificare RNA e rDNA per la nostra prova iniziale di sequenziamento, in media su tutte le cellule in quella prova (n = 5).(C) Il protocollo è stato valutato utilizzando qPCR della trascrizione msp-1 (PF3D7_09303000) su pool ordinati di 10 parassiti asessuali (n = 8) (significato dal test di Mann-Whitney, p≤0,05 *p≤0,01 **p≤0,001 ***). Sono stati testati i seguenti reagenti: Oligo(dT)s contenenti una base di ancoraggio terminale (A,G,C,V) o meno (T) e di lunghezza variabile (20 Ts vs. 30 Ts); quattro enzimi della trascrittasi inversa; 25 o 30 cicli di preamplificazione.(D) Numeri relativi di mappature di lettura per la codifica dell’RNA e dell’rDNA per le prove di ottimizzazione (6, 5, 6, 6 cellule, rispettivamente) e i principali gametociti di P. falciparum (n = 237), P. berghei sangue misto (n = 182) e P. falciparum asessuato (n = 189) set di dati (ultime tre barre). Gli asterischi indicano selezionate differenze significative tra le proporzioni della mappatura di lettura dei geni codificanti, calcolate utilizzando Mann-Whitney U (p≤0,05 *p≤0,01 **).
Per migliorare la resa, abbiamo testato l’impatto di: rimozione della base di ancoraggio dall’oligo(dT) e variazione della lunghezza del primer dell’oligo(dT) (20 vs 30); modifica degli enzimi di trascrizione inversa (SuperScriptII, SuperScriptIV, SMARTMMLV e SmartScribe); e variazione del numero di cicli di amplificazione (25 o 30). Abbiamo generato librerie per pool di 10 cellule P. falciparum di stadio avanzato ordinate e abbiamo testato l’abbondanza di trascrizioni dal gene msp-1 per RT-PCR quantitativa. Un più lungo, non ancorato oligo (dT) primer (T30) ha migliorato significativamente il rendimento e SuperScript II e SMARTScribe sono stati i più alti rendimento trascrizioni inverse trascrizioni(Figura 1C). L’amplificazione per 25 e 30 cicli sembrava dare risultati equivalenti(Figura 1C). Per capire l’impatto di queste permutazioni sulla complessità della sequenza di trascrittomi, abbiamo ordinato le singole cellule di P. falciparum e generato librerie di trascrittomi monocellulari utilizzando l’oligo dT30, sia gli enzimi SuperScript II o SmartScribe e sia 25 o 30 cicli di PCR(Figura 1D). Sono stati rilevati un numero significativamente maggiore di geni, con una contaminazione da rRNA drasticamente ridotta, utilizzando l’enzima SMARTScribe(Figura 1D; Tabella 1). Dati i risultati equivalenti per 25 o 30 cicli, abbiamo scelto di utilizzare il minor numero di cicli per tutti gli esperimenti successivi.
| Condizioni testate | Protocollo | SSII, V30, 30 cicli | SSII, T30, 30 cicli | SmSc, T30, 30 cicli | SSII, T30, 25 cicli | SmSc, T30, 25 cicli | SmSc, T30, 25 cicli | SmSc, T30, 25 cicli | SmSc, T30, 25 cicli |
|---|---|---|---|---|---|---|---|---|---|
| Celle | Sessuale | Asessuale | Asessuale | Asessuale | Asessuale | Sessuale | Sangue misto | Asessuale | |
| Specie | Pf | Pf | Pf | Pf | Pf | Pf | Pb | Pf | |
| Volume del tampone di lisi | 2 µl | ✓ | |||||||
| 4 µl | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| Oligo Dt (IDT) | Ancorato 30 bp | ✓ | |||||||
| Non-Anchored 30 bp | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ✓ | ||
| Trascrittasi inversa | Superscript II (Tecnologie per la vita) 10U | ✓ | ✓ | ✓ | |||||
| Smartscribe (Clontech) 5U | ✓ | ✓ | ✓ | ✓ | ✓ | ||||
| Numero di ciclo | 25 | ✓ | ✓ | ✓ | ✓ | ✓ | |||
| 30 | ✓ | ✓ | ✓ | ||||||
| Sequenziatore | HiSeq | ✓ | ✓ | ✓ | |||||
| MiSeq | ✓ | ✓ | ✓ | ✓ | ✓ | ||||
| Riepilogo dei risultati della sequenza | % rRNA | 5.7 | 33.5 | 36.2 | 6.4 | 18.4 | 17.8 | 16.7 | 34.8 |
| % di geni codificanti | 4.4 | 11.3 | 39.3 | 10.5 | 33 | 51.7 | 49 | 40.5 | |
| % altro | 90 | 55.2 | 24.4 | 83.1 | 48.6 | 30.5 | 34.2 | 24.6 | |
| Geni mediani rilevati per 50k di lettura | 25 | 84 | 145 | 174 | 181 | 502.5 | NA | NA | |
| Totale celle | 5 | 6 | 6 | 6 | 6 | 237 | 182 | 174 | |
| Filtri di passaggio delle celle | NA | NA | NA | NA | NA | 191 | 144 | 161 | |
| Conteggio dei geni mediano | NA | NA | NA | NA | NA | 2011 | 1922.5 | 1793 |
Due potenziali fonti di contaminazione sono importanti da considerare negli esperimenti di scRNA-seq. In primo luogo, le celle a singolo ordinamento potrebbero in realtà comprendere più celle, dando luogo ad un segnale ibrido che aggiunge rumore alle analisi a valle. In secondo luogo, l’RNA ambientale proveniente da cellule lisate nella sospensione cellulare potrebbe essere trasferito insieme alle cellule intatte in ogni pozzetto. Per valutare queste potenziali fonti di contaminazione, abbiamo fatto un flow-sorted di singoli parassiti da una miscela di GFP P. falciparum (Pf) e mCherry P. berghei (Pb), etichettati a fluorescenza, in una piastra a 96 pozzetti(Figura 2-figure supplement 1). Abbiamo quindi preparato e messo in sequenza le librerie di trascrittomi per ogni cella. Le letture sono state mappate su un riferimento combinato di entrambe le sequenze del genoma. Non è stata trovata alcuna prova di eventi doppi(Figura 2A) e, per ogni cellula, la stragrande maggioranza delle letture (98,1% per P.berghei, 99,4% per P. falciparum) mappate in modo univoco al genoma della specie prevista(Figura 2A). Le poche trascrizioni che hanno mappato il genoma sbagliato erano quelle più altamente espresse nelle altre specie e più probabilmente raccolte dalla soluzione(Figura 2-figure supplement 1B,C). È stato rilevato un numero molto basso di singole trascrizioni ambientali individuali(Figura 2-figure supplement 1D,E). Solo 15 delle 3566 trascrizioni rilevate nelle cellule di P. berghei erano di P. falciparum, e nessuno di questi è stato espresso in modo differenziato, suggerendo che non influenzerà la nostra analisi a valle.
Figura 2-figure supplemento 2.Figura 2— figura 2. Valutazione della purezza, della diversità e dell’accuratezza della sequenza del trascrittoma di una singola cellula.la doppia selezione delle cellule di P. berghei e P. falciparum mostra che la contaminazione da RNA ambientale è bassa.il contenuto di GC dei frammenti di trascritto è in accordo con il contenuto di GC dei geni.(A) Individualmente ordinato P . falciparum e cellule P. ber ghei da un pool misto non ha rivelato doppioni e poca contaminazione.(B) Distribuzioni di numeri di geni identificati come espressi nei nostri tre set di dati principali.(C) I geni espressi (quelli con almeno 10 letture in almeno cinque cellule) erano rappresentativi della lunghezza media del gene, suggerendo che anche se la trascrittasi inversa potrebbe non copiare l’insieme delle trascrizioni lunghe, frammenti di geni lunghi sono ancora rilevati.(D) Sequenziamento preparazione biblioteca spesso introduce polarizzazione fine, dove o la fine 5 ‘o 3′ di trascrizioni tendono ad essere meglio coperti. Il nostro protocollo ha introdotto un piccolo 5′-bias, che potrebbe essere attribuibile alla trascrizione inversa a volte iniziando all’interno di trascrizioni in regioni poliA interne, piuttosto che nella coda 3’ poly-A.(A) Purificato asessuato stadio di sangue asessuato tardivo di GFP P . falciparum e mCherry P. berghei sono stati mescolati ad un rapporto 1:1, inattivato in RNAlater, e ordinati singolarmente per citometria a flusso, gated sui rispettivi canali fluorescenti.(B) P. falciparum trascrizioni di P . ber ghei che contaminano le cellule di P. berghei e(C) P. berghei trascrizioni di P. berghei che contaminano le cellule di P. falciparum suggerendo che i contaminanti riflettono la perdita da cellule lisate (D) Istogramma del numero di letture (log-trasformato) da cellule di P . berghei che mappato ai genomi di P. berghei e P. falciparum. Una piccola percentuale dei geni rilevati proveniva dalla specie non corretta e questi tendevano ad essere espressi a bassi livelli.(E) Istogramma del numero di letture (log-trasformato) dalle cellule di P . falciparum che hanno mappato a P. falciparum e P. berghei.Non vi era alcuna apparente sovrarappresentazione o sottorappresentazione delle regioni ricche di GC.
Avendo stabilito l’affidabilità del protocollo, abbiamo generato 188 trascriptometri monocellulari di parassiti asessuali e sessuali (gametociti) a stadio ematico del modello di malaria dei roditori P. berghei. Dopo il filtraggio per rimuovere i trascrittomi con meno di 25.000 letture totali e meno di 1000 geni rilevati (con almeno una lettura), sono rimasti 144 trascrittomi di alta qualità. Abbiamo quindi rimosso i geni a meno che non avessero almeno dieci letture in ciascuna delle cinque o più cellule. In totale, abbiamo rilevato l’espressione di 4579 geni: oltre il 90% dei geni nel genoma di P. berghei. Da ogni cellula, abbiamo identificato l’espressione da, in media, 1981 geni (~ 33%), simile alla proporzione di trascrittomi rilevati in esperimenti su una singola cellula di mammifero(Treutlein et al., 2014)(Figura 2B).
Abbiamo anche generato trascrittomi monocellulari per il parassita della malaria umana P. falciparum e li abbiamo elaborati utilizzando la stessa procedura di filtraggio come per P. berghei. Ne sono risultati 191 trascrittomi monocellulari di alta qualità (su 237 totali) per gli stadi sessuali, con una media di 2090 geni rilevati, e 161 trascrittomi monocellulari di alta qualità (su 174 totali) per gli stadi asessuali, con una media di 1712 geni rilevati(Figura 2B).
Abbiamo usato il dataset di P. berghei per esplorare i pregiudizi nella rappresentazione delle trascrizioni sequenziate con il nostro protocollo. In primo luogo, abbiamo controllato se alcune regioni erano sovrarappresentate tra le nostre sequenze di trascrizione a causa dell’amplificazione preferenziale di sequenze meno ricche di AT mediante PCR. In secondo luogo, poiché la trascrittasi inversa dovrebbe elaborare un mRNA completo per produrre cDNA, abbiamo determinato se c’era un bias contro i geni lunghi. Infatti, né il contenuto di GC(Figura 2-figure supplement 2) né la lunghezza del gene (Figura 2C) hanno avuto un impatto sul rilevamento delle trascrizioni. Nel caso di molti geni lunghi, la mancanza di un bias di lunghezza potrebbe essere dovuta al sequenziamento di frammenti di mRNA, piuttosto che a sequenze a lunghezza intera. Questo suggerisce che il protocollo Smart-seq2 è suscettibile di priming interno da oligo-d (T) (come descritto in[Nam et al., 2002]) e modello di commutazione alle estremità esposte 5 ‘di frammenti di mRNA. Il vantaggio di questo è che siamo in grado di saggiare i livelli di trascrizione dei geni lunghi e corti con una precisione simile. Molti approcci RNA-seq mostrano un bias di segnale verso la fine 5′ o 3′ delle trascrizioni e nei nostri dati è stato rilevato un leggero bias di 5’ che potrebbe anche riflettere il legame di oligo(dT) alle regioni interne ricche di poliA delle trascrizioni (Figura 2D).
Figura 1.Figura 1. Definizione di un protocollo robusto per l’analisi trascrittomica monocellulare dei parassiti del Plasmodio.(A) Panoramica del protocollo RNAseq monocellulare. I passi del protocollo originale Smart-seq2(Picelli et al., 2013) che hanno portato a significativi guadagni sono evidenziati in arancione.(B) Il numero relativo di letture di mappatura per codificare RNA e rDNA per la nostra prova iniziale di sequenziamento, in media su tutte le cellule in quella prova (n = 5).(C) Il protocollo è stato valutato utilizzando qPCR della trascrizione msp-1 (PF3D7_09303000) su pool ordinati di 10 parassiti asessuali (n = 8) (significato dal test di Mann-Whitney, p≤0,05 *p≤0,01 **p≤0,001 ***). Sono stati testati i seguenti reagenti: Oligo(dT)s contenente una base di ancoraggio terminale (A,G,C,V) o meno (T) e di lunghezza variabile (20 Ts vs. 30 Ts); quattro enzimi della trascrittasi inversa; 25 o 30 cicli di preamplificazione.(D) Numeri relativi di mappature di lettura per la codifica dell’RNA e dell’rDNA per le prove di ottimizzazione (6, 5, 6, 6 cellule, rispettivamente) e i principali gametociti di P. falciparum (n = 237), P. berghei sangue misto (n = 182) e P. falciparum asessuato (n = 189) set di dati (ultime tre barre). Gli asterischi indicano selezionate differenze significative tra le proporzioni della mappatura di lettura dei geni codificanti, calcolate utilizzando Mann-Whitney U (p≤0,05 *p≤0,01 **).
Figura 2-figure supplement 2.La valutazione della purezza, della diversità e dell’accuratezza della sequenza del trascrittoma di una singola cellula, la doppia selezione delle cellule di P. berghei e P. falciparum mostra che la contaminazione da RNA ambientale è bassa, il contenuto di GC dei frammenti di trascritto è in accordo con il contenuto di GC dei geni.(A) Individualmente ordinato P . falciparum e cellule P. ber ghei da un pool misto non ha rivelato doppioni e poca contaminazione.(B) Distribuzioni di numeri di geni identificati come espressi nei nostri tre set di dati principali.(C) I geni espressi (quelli con almeno 10 letture in almeno cinque cellule) erano rappresentativi della lunghezza media del gene, suggerendo che anche se la trascrittasi inversa potrebbe non copiare l’insieme delle trascrizioni lunghe, frammenti di geni lunghi sono ancora rilevati.(D) Sequenziamento preparazione biblioteca spesso introduce polarizzazione fine, dove o la fine 5 ‘o 3′ di trascrizioni tendono ad essere meglio coperti. Il nostro protocollo ha introdotto un piccolo 5′-bias, che potrebbe essere attribuibile alla trascrizione inversa a volte iniziando all’interno di trascrizioni in regioni poliA interne, piuttosto che nella coda 3’ poly-A.(A) Purificato asessuato stadio di sangue asessuato tardivo di GFP P . falciparum e mCherry P. berghei sono stati mescolati ad un rapporto 1:1, inattivato in RNAlater, e ordinati singolarmente per citometria a flusso, gated sui rispettivi canali fluorescenti.(B) P. falciparum trascrizioni di P . ber ghei che contaminano le cellule di P. berghei e(C) P. berghei trascrizioni di P. berghei che contaminano le cellule di P. falciparum suggerendo che i contaminanti riflettono la perdita da cellule lisate (D) Istogramma del numero di letture (log-trasformato) da cellule di P . berghei che mappato ai genomi di P. berghei e P. falciparum. Una piccola percentuale dei geni rilevati proveniva dalla specie non corretta e questi tendevano ad essere espressi a bassi livelli.(E) Istogramma del numero di letture (log-trasformato) dalle cellule di P . falciparum che hanno mappato a P. falciparum e P. berghei.Non vi era alcuna apparente sovrarappresentazione o sottorappresentazione delle regioni ricche di GC.
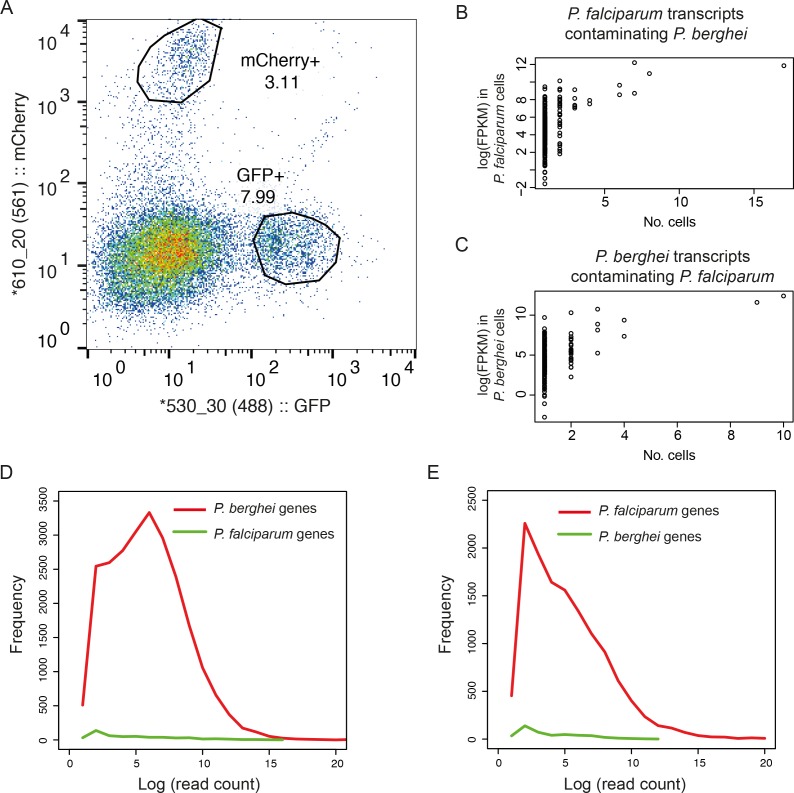
Figura 2-figure supplemento 1.La doppia selezione delle cellule di P. berghei e P. fal ciparum mostra che la contaminazione da RNA ambientale è bassa.(A) Purificato asessuato stadio di sangue asessuato tardivo di GFP P . falciparum e mCherry P. berghei sono stati mescolati ad un rapporto 1:1, inattivato in RNAlater, e ordinati singolarmente per citometria a flusso, gated sui rispettivi canali fluorescenti.(B) P. falciparum trascrizioni di P . ber ghei che contaminano le cellule di P. berghei e(C) P. berghei trascrizioni di P. berghei che contaminano le cellule di P. falciparum suggerendo che i contaminanti riflettono la perdita da cellule lisate (D) Istogramma del numero di letture (log-trasformato) da cellule di P . berghei che mappato ai genomi di P. berghei e P. falciparum. Una piccola percentuale dei geni rilevati proveniva dalla specie non corretta e questi tendevano ad essere espressi a bassi livelli.(E) Istogramma del numero di letture (log-trasformato) dalle cellule di P . falciparum che hanno mappato a P. falciparum e P. berghei.
Figura 2-figure supplement 2.Il contenuto di GC dei frammenti di trascrizione è in accordo con il contenuto di GC dei geni.Non c’è stata una apparente sovrarappresentazione o sotto-rappresentazione delle regioni ricche di GC.
Utilizzo di RNA-seq monocellulare per risolvere le popolazioni di parassiti
Dopo aver sviluppato e valutato il nostro protocollo per il sequenziamento dei trascrittomi monocellulari, abbiamo poi determinato se i diversi stadi parassitari potevano essere risolti tra i 144 P. berghei trascripttomi dello stadio di sangue misto. Utilizzando una combinazione di analisi dei componenti principali (PCA), k-mea clustering utilizzando SC3(Kiselev, 2016), e il confronto con i set di dati di trascrittomi di massa(Otto et al., 2014; Hoo et al., 2016), abbiamo classificato ogni cellula come maschio, femmina o asessuato(Figura 3A). La classificazione delle cellule è un passo importante nell’analisi dei dati del trascrittoma monocellulare, ma la classificazione di tutte le cellule in un particolare set di dati può essere una sfida. Per il Plasmodium, la disponibilità di una varietà di serie di dati pubblicati in massa di RNA-seq e microarray ci ha permesso di determinare lo stadio di vita approssimativo di ogni cellula. Per P. berghei, abbiamo utilizzato un dataset di microarray(Hoo et al., 2016) che ha esaminato il ciclo asessuato di 24 ore a intervalli di 2 ore e un dataset RNA-seq(Otto et al., 2014) che comprendeva campioni in tre punti temporali asessuali (anelli, trofozoiti e schizonti) e gametociti sessuali misti. Per ogni cellula, abbiamo confrontato l’elenco dei geni classificati per livello di espressione con quelli di ogni campione dei set di dati di cui sopra, scegliendo il punto temporale meglio correlato. I gametociti maschili e femminili sono stati differenziati esaminando i geni marcatori dei cluster di cellule realizzati con SC3(Kiselev et al., 2017). Abbiamo stabilito una classificazione di consenso annotata manualmente per ogni cellula sulla base delle analisi di cui sopra. Alcune cellule sembravano avere trascrittomi intermedi tra asessuali e gametociti e questi sono stati etichettati come outlier. Questi possono derivare da globuli rossi individuali co-infettati.
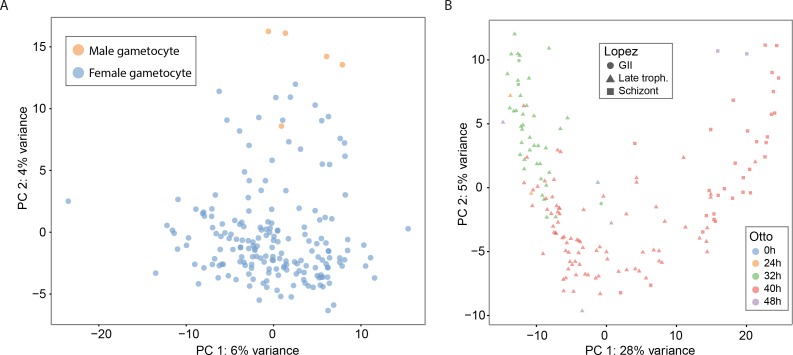
Figura 3-figure supplemento 2.Figura 3—figura 2. Diversi tipi di cellule sono stati risolti con successo utilizzando il trascrittoma monocellulare. Rileviamo trascrizioni specifiche per ogni fase a vari livelli di espressione.(A) Una combinazione di Analisi dei Componenti Principali (PCA), k-media clustering e confronto con i set di dati RNA-seq alla rinfusa è stato utilizzato per classificare 144 cellule singole di alta qualità P. berghei, e ha rivelato tre sottopopolazioni distinte. Gli outlier possono rappresentare eritrociti infettati con stadi sia sessuali che asessuali o stadi iniziali dello sviluppo dei gametociti.(B) Tre marcatori ben consolidati del lignaggio maschile, femminile e asessuato(Mair et al., 2006; Liu et al., 2008; Moss et al., 2012) sono concordi con la nostra classificazione.Geni specifici per gli stadi a diversi livelli di espressione, sono stati identificati dai dati RNA-seq di(Otto et al., 2014) per gli stadi asessuali (A),(B) gametociti maschili e(C) gametociti femminili. La media FPKM tra i campioni replicati viene visualizzata qui.(A) Per classificare 191 gametociti di alta qualità P . falciparum gametocytes di P. falciparum è stata utilizzata una combinazione di analisi dei componenti principali (PCA), clustering k-means e confronto con set di dati RNA-seq in massa. Il consenso sul clustering e il confronto con l’RNA-seq in massa ci ha permesso di distinguere i gametociti maschili e i gametociti femminili.(B) Un diagramma PCA che mostra i trascrittomi monocellulari di cellule asessuate di P. fal ciparum annotate utilizzando i set di dati dei trascrittomi di Lopez et al. e Otto et al.
L’accuratezza della nostra classificazione è stata fortemente supportata da marcatori stadi specifici stabiliti(Figura 3B; Figura 3-figure supplement 1). Inoltre, la confermata assenza di parassiti contaminanti di altri stadi del ciclo di vita ci ha permesso di determinare un nuovo e più lungo elenco di marcatori specifici per gli stadi(file supplementare 1). Abbiamo condotto analisi simili per due campioni di P. falciparum composti da stadi asessuali e sessuali. Poiché provenivano da due campioni puri distinti, la loro classificazione è stata più semplice ed entrambi i set di cellule (asessuate e sessuali) sono stati correlati come previsto con i set di dati di massa precedentemente pubblicati(Otto et al., 2010; López-Barragán et al., 2011; Lasonder et al., 2016).(Figura 3-figure supplement 2).
Figura 3-figure supplement 2.I diversi tipi di cellule sono stati risolti con successo utilizzando il trascrittoma monocellulare. Rileviamo trascrizioni specifiche per ogni fase a diversi livelli di espressione . (A) Una combinazione di Analisi dei Componenti Principali (PCA), k-media clustering e confronto con i set di dati RNA-seq alla rinfusa è stato utilizzato per classificare 144 cellule singole di alta qualità P. berghei, e ha rivelato tre sottopopolazioni distinte. Gli outlier possono rappresentare eritrociti infettati con stadi sia sessuali che asessuali o stadi iniziali dello sviluppo dei gametociti.(B) Tre marcatori ben consolidati del lignaggio maschile, femminile e asessuato(Mair et al., 2006; Liu et al., 2008; Moss et al., 2012) sono concordi con la nostra classificazione.Geni specifici per gli stadi a diversi livelli di espressione, sono stati identificati dai dati RNA-seq di(Otto et al., 2014) per gli stadi asessuali (A),(B) gametociti maschili e(C) gametociti femminili. La media FPKM tra i campioni replicati viene visualizzata qui.(A) Per classificare 191 gametociti di alta qualità P . falciparum gametocytes di P. falciparum è stata utilizzata una combinazione di analisi dei componenti principali (PCA), clustering k-means e confronto con set di dati RNA-seq in massa. Il consenso sul clustering e il confronto con l’RNA-seq in massa ci ha permesso di distinguere i gametociti maschili e i gametociti femminili.(B) Un diagramma PCA che mostra i trascrittomi monocellulari di cellule asessuate di P. fal ciparum annotate utilizzando i set di dati dei trascrittomi di Lopez et al. e Otto et al.
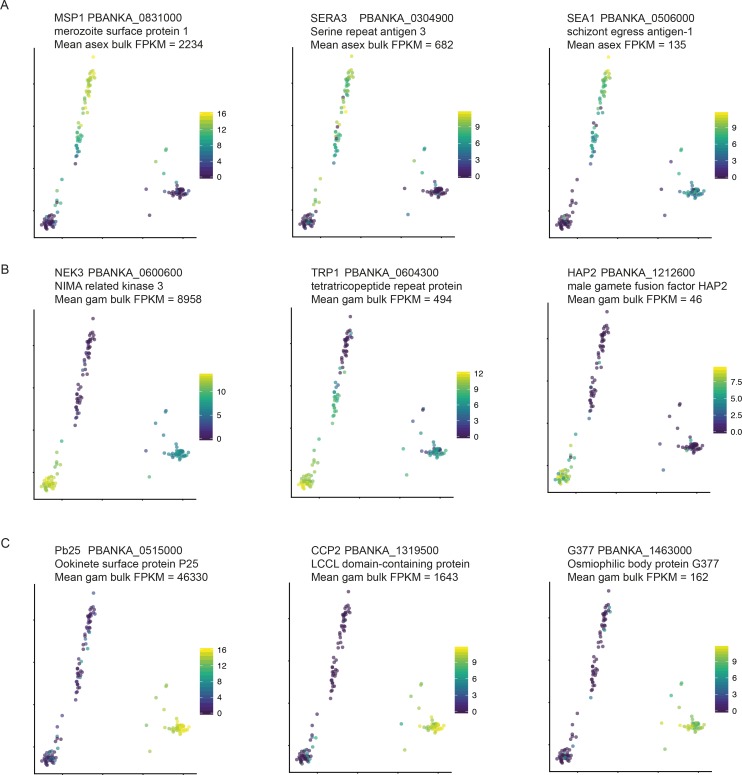
Figura 3-figure supplemento 1.Rileviamo trascrizioni specifiche per ogni fase a vari livelli di espressione.I geni specifici di uno stadio a diversi livelli di espressione, sono stati identificati dai dati RNA-seq di(Otto et al., 2014) per(A) stadi asessuali,(B) gametociti maschili e(C) gametociti femminili. La media FPKM tra i campioni replicati viene visualizzata qui.
Figura 3-figure supplement 2.Componenti principali Analisi e classificazione delle cellule di gametociti P. falciparum. (A) Per classificare 191 gametociti di P. falciparum falciparum di alta qualità è stata usata una combinazione di Analisi dei Componenti Principali (PCA), clustering k-means e confronto con i set di dati RNA-seq alla rinfusa, per classificare 191 gametociti di P . fal ciparum di alta qualità. Il consenso sul clustering e il confronto con l’RNA-seq in massa ci ha permesso di distinguere i gametociti maschili e i gametociti femminili.(B) Un diagramma PCA che mostra i trascrittomi monocellulari di cellule asessuate di P. fal ciparum annotate utilizzando i set di dati dei trascrittomi di Lopez et al. e Otto et al.
L’analisi dello pseudotempo rivela una brusca dinamica trascrizionale attraverso le fasi asessuate
Lo sviluppo asessuato delplasma è replicativo, ma non segue la progressione canonica del ciclo cellulare eucariotico e sebbene si ritenga che esistano dei punti di controllo, non sono stati caratterizzati(Gerald et al., 2011). Gli studi Bulk RNA-seq che monitorano gli schemi trascrizionali lungo il ciclo asessuato completo delle specie di parassiti della malaria umana e dei roditori hanno costantemente rivelato una cascata continua di iniziazione alla trascrizione(Hoo et al., 2016; Bozdech et al., 2003b) simile a quella vista in altri eucarioti(Spellman et al., 1998). Sebbene queste analisi abbiano utilizzato popolazioni di parassiti sincronizzate che permettono di saggiare finestre di espressione ragionevolmente strette, la loro risoluzione è stata limitata dal rilevamento di pool di cellule all’interno di ogni finestra di espressione che possono differire nella progressione dello sviluppo di diverse ore. L’RNA-seq monocellulare permette di campionare popolazioni non sincronizzate, da ampie parti del ciclo, e di identificare l’ordine delle cellule nel ciclo utilizzando analisi pseudotemporali(Trapnell et al., 2014). L’analisi pseudotemporale ordina le cellule nelle traiettorie di sviluppo identificando le cellule con i trascrittimi più simili tra loro e mettendo in ordine quelle più vicine tra loro. Per ricostruire l’ultima parte del ciclo di sviluppo asessuato, abbiamo usato per la prima volta M3Drop(Andrews e Hemberg, 2016) per identificare i geni che variano tra le cellule asessuate. Questo strumento tiene conto del gran numero di valori di zero (drop out) nei dati che sono dovuti al basso tasso di cattura insito negli approcci monocellulari. Abbiamo quindi utilizzato questi geni per confrontare ogni trascrittoma ed effettuare un’analisi pseudotemporale con Monocle 2(Trapnell et al., 2014). Questo ci ha permesso di posizionare ogni cellula asessuata di P. berghei e P. falciparum lungo una traiettoria di sviluppo. Gli ordinamenti delle cellule determinati dall’analisi pseudotemporale erano altamente concordanti con gli studi trascrizionali del corso del tempo di sviluppo pubblicati in precedenza(Otto et al., 2010; López-Barragán et al., 2011; Otto et al., 2014; Hoo et al., 2016) (Figura 4A,B,Figura 4-figure supplement 1A,B). Questo dimostra che singole cellule di Plasmodium da un pool non sincronizzato possono essere ordinate dalle loro firme trascrizionali per ricavare accuratamente una mappa trascrizionale dello sviluppo nel ciclo asessuato tardivo(Figura 4C, Figura 4-figure supplement 1C).
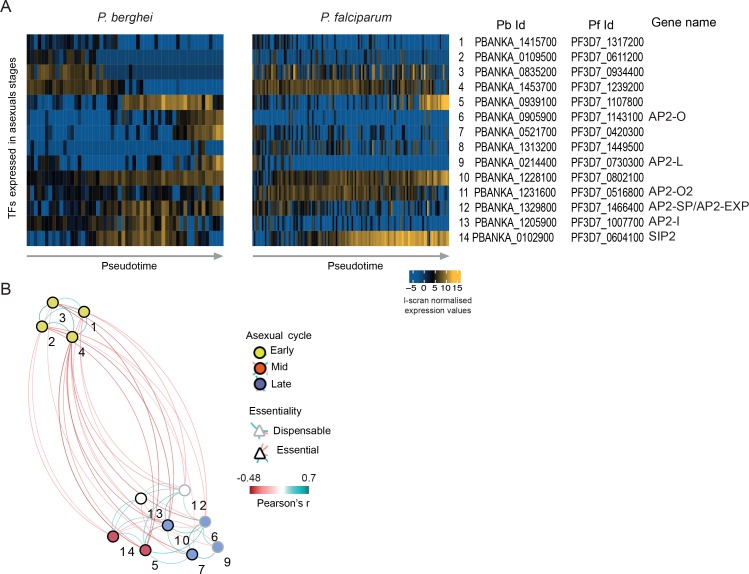
Figura 4-figure supplement 4.Gli stessi sottoinsiemi di trascrizioni mostrano diversi modelli di espressione intorno alla fine del ciclo cellulare asessuato in dati RNA-seq convenzionali in bulk RNA-seq e ricostruzioni pseudotime di dati RNAseq a singola cellula .I dati RNAseq a singola cellula a bassa copertura e ad alto rendimento, pubblicati di recente, supportano la nostra scoperta di cambiamenti di fase nell’espressione genica nel ciclo asessuato di P. falciparum.l’analisi del modello di co-espressione della famiglia ApiAP2 di fattori di trascrizione (TFs) nei parassiti asessuali.(A) Pseudotime ordinamento (utilizzando[Trapnell et al., 2014]) delle cellule asessuate in era in stretto accordo con la massa RNA-seq dataset di massa (fase prevista = consenso; vedi Materiali e metodi).(B) Pseudotime ordinamento (utilizzando[Trapnell et al., 2014]) del 125 P. falciparum cellule asessuate tardive era in stretto accordo con la massa RNA-seq dataset di massa (punto di tempo previsto da[Otto et al., 2010], fase prevista = consenso; vedi Materiali e metodi).(C) I geni espressi in modo diverso (identificati utilizzando M3Drop [Andrewse Hemberg, 2016]) sono stati raggruppati lungo gruppi di geni pseudotemporali rivelatori di geni con bruschi cambiamenti di profilo di espressione durante il ciclo asessuato tardivo. L’arricchimento funzionale nei cluster era in accordo con il previsto passaggio dal trofozoite in crescita allo schizonte in erba (IMC = Inner Membrane Complex; i micronemi e i rhoptries sono organelli secretori). Hoo è il punto temporale più simile in sviluppo nel dataset Hoo et al. (2016).PCA di 155 cellule P. falciparum colorate da pseudotempo(A) o stato Monocle(B); i rami di traiettoria identificati sono visualizzati come numeri cerchiati 1 e 2.(C) I geni espressi in modo diverso sono stati tracciati lungo lo pseudotempo per le cellule nella traiettoria principale (Stati 1, 3 e 4). Il numero di geni per cella è visualizzato sulla parte superiore della mappa termica, mentre lo pseudotempo e la previsione di fase(Otto et al., 2010) visualizzati sul lato della mappa termica. La transizione tra trofozoiti e schizonti è associata ad un duro spostamento trascrizionale, come si è visto per P. berghei.Un insieme condiviso di 651 geni identificati come segue un modello di espressione sigmoidale attraverso il ciclo di sviluppo intraeritrocitario (vedi Materiali e metodi) sono mostrati sia nei dati trascriptomi di massa(Hoo et al., 2016)(A) che nei dati monocellulari ordinati per pseudotempo(B) per P. berghei. Un cambiamento molto più drammatico nell’espressione genica si osserva nei dati del trascrittoma monocellulare. Un modello simile è osservato tra P. falciparum bulk(Otto et al., 2010)(C) e single-cell(D) RNA-seq. Nei pannelli b e d, i modelli di espressione genica sono valori l-scran media-normalizzati. Solo i parassiti di stadio avanzato (frecce grigie nelle serie di dati di riferimento bulk) dovrebbero essere presenti nelle serie di dati monocellulari.Una mappa termica che mostra i valori di espressione media-normalizzati registrati per i parassiti asessuali tardivi(Poran et al., 2017) ordinati per pseudotempo. I geni sono stati ordinati come per la Figura 4-figure supplement 2A che mostra che questi dati mostrano anche un modello di espressione discontinua, a differenza di bulk RNA-seq dati della stessa parte del ciclo.(A) Espressione dei geni Plasmodium ApiAP2 nei parassiti asessuali. I geni ortologi sono presentati sulle stesse righe.(B) Una rete di co-espressione per P. berghei è stata costruita utilizzando significative correlazioni positive e negative (p<0,05 dalla correlazione di Pearson) tra le TF e ponderate in base al loro coefficiente di correlazione. I numeri sulla rete si riferiscono alle TF così come presentate in A.
In netto contrasto con le transizioni lisce osservate in precedenza negli esperimenti di corso del tempo di massa(Bozdech et al., 2003a; Hoo et al., 2016), abbiamo osservato bruschi cambiamenti nell’espressione genica durante il ciclo cellulare sia di P. berghei e P. falciparum(Figura 4C, Figura 4-figure supplement 1). Mentre una cascata continua di iniziazione della trascrizione lungo il ciclo asessuato può essere visto in massa RNA-seq dati, dati monocellulari hanno chiaramente rivelato una brusca transizione in espressione per gli stessi geni(Figura 4-figure supplement 2). Abbiamo anche analizzato i dati di P. falciparum Drop-seq pubblicati di recente(Poran et al., 2017) e abbiamo osservato un modello simile(Figura 4-figure supplement 3). La progressione graduale nel ciclo rappresenta un allontanamento dalla visione comune e suggerisce un modello trascrizionale precedentemente nascosto, conservato attraverso i parassiti del Plasmodium. Nascente filamento bulk RNA-seq aveva già messo in discussione la natura a cascata dell’iniziazione della trascrizione nel ciclo asessuato(Lu et al., 2017).
Sospettiamo che la media attraverso le fasi del ciclo di vita leggermente asincrona negli studi di RNA-seq alla rinfusa abbia in precedenza mascherato la vera natura delle transizioni lungo il ciclo cellulare asessuato. I singoli parassiti non procedono lungo un percorso incrementale di cambiamento trascrizionale, ma sembrano invece generalmente subire spostamenti trascrizionali, accendendo o spegnendo l’espressione di un intero repertorio di geni contemporaneamente. Mentre questi moduli trascrizionali sembrano essere rapidamente accesi e spenti durante lo sviluppo, possono sovrapporsi e le cellule possono esprimere due moduli contemporaneamente. Un’analisi k-means in pseudotime ha identificato tre cluster di geni(Trapnell et al., 2014) per ogni specie(Figura 4C, Figura 4-figure supplement 1, Supplementary file 2). Cluster 1 in P. berghei (equivalente al cluster 2 in P. falciparum; Figura 4-figure supplement 1) è stato arricchito per la dinamica delle proteine e il metabolismo energetico, comprese molte sottounità ribosomiali, sottounità proteasome e ATPasi (Figura 4C). Cluster 2 in P. berghei (equivalente al cluster 3 in P. falciparum) è stato associato con l’organulo secretorio rhoptry, tra cui ron2, ron4, ron5, ron12, rop14, rap1 e rap2/3. Il gruppo 3 in P. berghei è stato arricchito per l’organulo secretorio microneme e il complesso di membrana interna, compresi sub2, ama1, ripr, imc1c, imc1e, imc1f, imc1g, imc1m e isp3. Quest’ultimo cluster non è stato catturato in P. falciparum. Questi cluster possono rappresentare moduli trascrizionali discreti che sono alla base dei punti di controllo del ciclo cellulare parassitario durante la transizione da un trofozoite metabolicamente attivo e in rapida crescita ad uno schizonte multinucleato in erba. Notiamo che due fattori di trascrizione essenziali ApiAP2(Figura 4-figure supplement 4) sono stati associati a cluster di espressione genica equivalente in entrambe le specie: PBANKA_1453700 (PF3D7_1239200) con il cluster precoce (1) e PBANKA_0939100 (PF3D7_1107800) con il cluster tardivo (2), implicandoli come potenziali regolatori di questi moduli.
Figura 4-figure supplement 4.Figura 4—supplemento di figura 4. RNA-seq a singola cella rivela la variazione trascrizionale nascosta nel ciclo asessuato delle cellule.Pseudotime ricostruzione della traiettoria asessuata tardiva di P. falciparum.gli stessi sottoinsiemi di trascrizioni mostrano diversi modelli di espressione intorno alla fine del ciclo asessuato delle cellule in massa convenzionale RNA-seq dati e ricostruzioni pseudotime di dati RNAseq a singola cella.I dati RNAseq a singola cellula a bassa copertura e ad alto rendimento, pubblicati di recente, supportano la nostra scoperta di cambiamenti di fase nell’espressione genica nel ciclo asessuato di P. falciparum.l’analisi del modello di co-espressione della famiglia ApiAP2 di fattori di trascrizione (TFs) nei parassiti asessuali.(A) Pseudotime ordinamento (utilizzando[Trapnell et al., 2014]) delle cellule asessuate in era in stretto accordo con la massa RNA-seq dataset di massa (fase prevista = consenso; vedi Materiali e metodi).(B) Pseudotime ordinamento (utilizzando[Trapnell et al., 2014]) del 125 P. falciparum cellule asessuate tardive era in stretto accordo con la massa RNA-seq dataset di massa (punto di tempo previsto da[Otto et al., 2010], fase prevista = consenso; vedi Materiali e metodi).(C) I geni espressi in modo diverso (identificati utilizzando M3Drop [Andrewse Hemberg, 2016]) sono stati raggruppati lungo gruppi di geni pseudotemporali rivelatori di geni con bruschi cambiamenti di profilo di espressione durante il ciclo asessuato tardivo. L’arricchimento funzionale nei cluster era in accordo con il previsto passaggio dal trofozoite in crescita allo schizonte in erba (IMC = Inner Membrane Complex; i micronemi e i rhoptries sono organelli secretori). Hoo è il punto temporale più simile in sviluppo nel dataset Hoo et al. (2016).PCA di 155 cellule P. falciparum colorate da pseudotempo(A) o stato Monocle(B); i rami di traiettoria identificati sono visualizzati come numeri cerchiati 1 e 2.(C) I geni espressi in modo diverso sono stati tracciati lungo lo pseudotempo per le cellule nella traiettoria principale (Stati 1, 3 e 4). Il numero di geni per cella è visualizzato sulla parte superiore della mappa termica, mentre lo pseudotempo e la previsione di fase(Otto et al., 2010) visualizzati sul lato della mappa termica. La transizione tra trofozoiti e schizonti è associata ad un duro spostamento trascrizionale, come si è visto per P. berghei.Un insieme condiviso di 651 geni identificati come segue un modello di espressione sigmoidale attraverso il ciclo di sviluppo intraeritrocitario (vedi Materiali e metodi) sono mostrati sia nei dati trascriptomi di massa(Hoo et al., 2016)(A) che nei dati monocellulari ordinati per pseudotempo(B) per P. berghei. Un cambiamento molto più drammatico nell’espressione genica si osserva nei dati del trascrittoma monocellulare. Un modello simile è osservato tra P. falciparum bulk(Otto et al., 2010)(C) e single-cell(D) RNA-seq. Nei pannelli b e d, i modelli di espressione genica sono valori l-scran media-normalizzati. Solo i parassiti di stadio avanzato (frecce grigie nelle serie di dati di riferimento bulk) dovrebbero essere presenti nelle serie di dati monocellulari.Una mappa termica che mostra i valori di espressione media-normalizzati registrati per i parassiti asessuali tardivi(Poran et al., 2017) ordinati per pseudotempo. I geni sono stati ordinati come per la Figura 4-figure supplement 2A che mostra che questi dati mostrano anche un modello di espressione discontinua, a differenza di bulk RNA-seq dati della stessa parte del ciclo.(A) Espressione dei geni Plasmodium ApiAP2 nei parassiti asessuali. I geni ortologi sono presentati sulle stesse righe.(B) Una rete di co-espressione per P. berghei è stata costruita utilizzando significative correlazioni positive e negative (p<0,05 dalla correlazione di Pearson) tra le TF e ponderate in base al loro coefficiente di correlazione. I numeri sulla rete si riferiscono alle TF così come presentate in A.
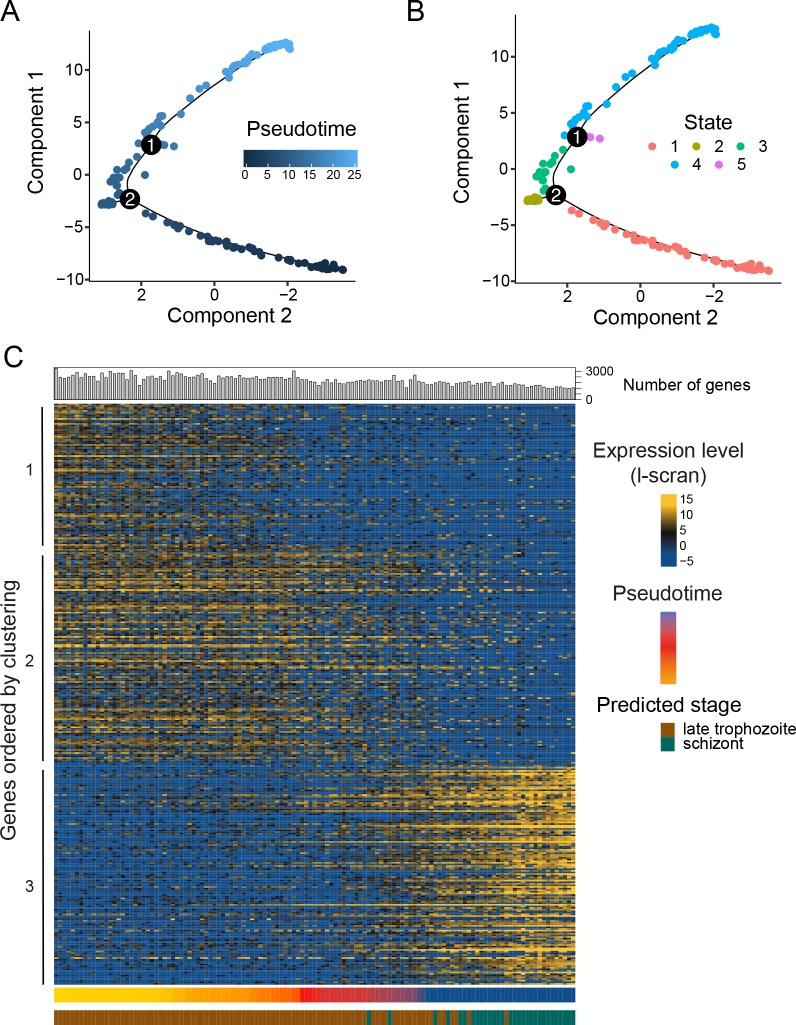
Figura 4-figure supplement 1.Ricostruzione pseudotempo della traiettoria asessuata tardiva di P. falciparum.PCA di 155 cellule di P . fal ciparum colorate da pseudotime(A) o stato Monocle(B); i rami della traiettoria identificati sono visualizzati come numeri 1 e 2 cerchiati.(C) I geni espressi in modo diverso sono stati tracciati lungo lo pseudotempo per le cellule nella traiettoria principale (Stati 1, 3 e 4). Il numero di geni per cella è visualizzato sulla parte superiore della mappa termica, mentre lo pseudotempo e la previsione di fase(Otto et al., 2010) visualizzati sul lato della mappa termica. La transizione tra trofozoiti e schizonti è associata ad un duro spostamento trascrizionale, come si è visto per P. berghei.
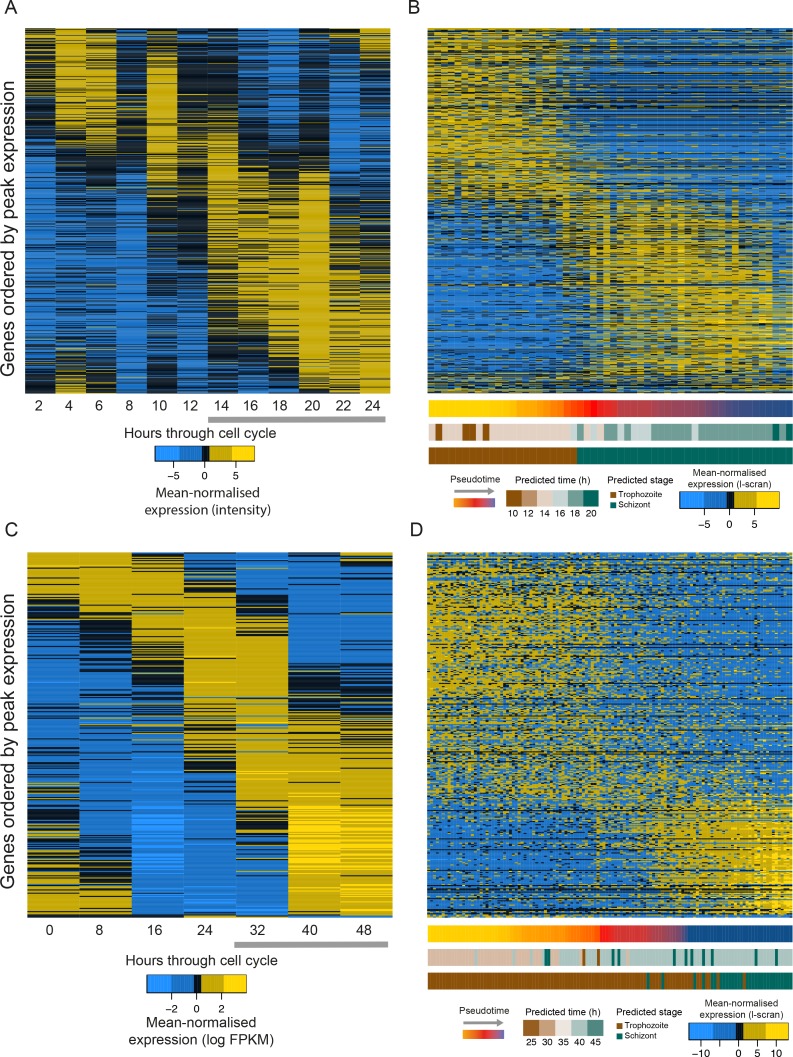
Figura 4-figure supplement 2.Gli stessi sottoinsiemi di trascrizioni mostrano diversi modelli di espressione intorno alla fine del ciclo cellulare asessuato in dati RNA-seq convenzionali alla rinfusa e ricostruzioni pseudotemporali di dati RNAseq a cella singola.Un insieme condiviso di 651 geni identificati come segue un modello di espressione sigmoidale attraverso il ciclo di sviluppo intraeritrocitario (vedi Materiali e metodi) sono mostrati sia in dati trascrittomatici in massa(Hoo et al., 2016)(A) e dati a singola cellula ordinati da pseudotime(B) per P. berghei. Un cambiamento molto più drammatico nell’espressione genica si osserva nei dati del trascrittoma monocellulare. Un modello simile è osservato tra P. falciparum bulk(Otto et al., 2010)(C) e single-cell(D) RNA-seq. Nei pannelli b e d, i modelli di espressione genica sono valori l-scran media-normalizzati. Solo i parassiti di stadio avanzato (frecce grigie nelle serie di dati di riferimento bulk) dovrebbero essere presenti nelle serie di dati monocellulari.
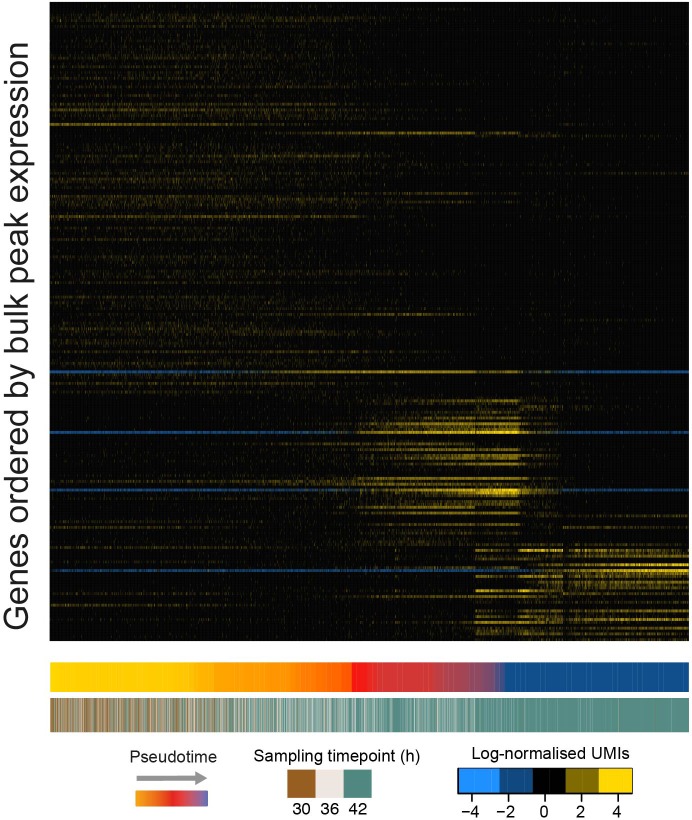
Figura 4-figure supplement 3.I dati RNA-seq monocellulari a bassa copertura e ad alto rendimento, pubblicati di recente, supportano la nostra scoperta di cambiamenti di fase nell’espressione genica nel ciclo asessuato di P. falciparum.Una mappa termica che mostra i valori di espressione media-normalizzati registrati per i parassiti asessuali tardivi(Poran et al., 2017) ordinati per pseudotempo. I geni sono stati ordinati come per la Figura 4-figure supplement 2A che mostra che questi dati mostrano anche un modello di espressione discontinua, a differenza di bulk RNA-seq dati della stessa parte del ciclo.
Figura 4-figure supplement 4.Analisi del modello di co-espressione della famiglia di fattori di trascrizione (TF) ApiAP2 nei parassiti asessuali.(A) Espressione dei geni Plasmodium ApiAP2 nei parassiti asessuali. I geni ortologi sono presentati sulle stesse righe.(B) Una rete di co-espressione per P. berghei è stata costruita utilizzando significative correlazioni positive e negative (p<0,05 dalla correlazione di Pearson) tra le TF e ponderate in base al loro coefficiente di correlazione. I numeri sulla rete si riferiscono alle TF così come presentate in A.
Alcuni tipi di trascrizioni variano indipendentemente dal ciclo cellulare asessuato e sono conservate tra gli stadi e tra le specie
Come molti altri tipi di cellule(Spellman et al., 1998; Kowalczyk et al., 2015), il punto in cui i parassiti del Plasmodium sono all’interno del loro ciclo cellulare domina la variazione trascrizionale osservata all’interno di una popolazione geneticamente clonale. Tuttavia, ci sono anche geni che variano indipendentemente dal ciclo cellulare, comprese le famiglie di geni clonali varianti, che si trovano in gran parte nelle regioni subtelomeriche del genoma(Rovira-Graells et al., 2012). Si pensa che un ambiente unico della cromatina permetta di passare da un’espressione all’altra dei diversi membri delle famiglie di geni e questo meccanismo permette alle popolazioni di parassiti di adattarsi al sistema immunitario dell’ospite( genivar ) (Scherf et al., 2008), di stabilire un’infezione cronica (genipir pirici )(Scherf et al., 2008) e di variare le vie di invasione dei globuli rossi (p235)(Preiser et al., 1999). Poiché consentono al parassita di adattarsi ad ambienti inaspettati, i membri di queste famiglie multigene sono stati definiti geni di emergenza(Reid, 2015). Ci sono anche prove di variazione nell’espressione in risposta al rilevamento dei nutrienti(Mancio-Silva et al., 2017) e a una varietà di interventi chimici(Hu et al., 2010). Abbiamo utilizzato un approccio di regressione per identificare i geni che variano indipendentemente dal ciclo cellulare (scLVM)(Buettner et al., 2015) rimuovendo la variazione dipendente dal ciclo cellulare dalle cellule asessuate di P. falciparum. Per addestrare questo metodo, abbiamo usato geni che variano in pseudotempo (cioè il ciclo cellulare). Abbiamo scoperto che i primi due fattori latenti dei dati di espressione sono stati guidati dal ciclo cellulare, ognuno dei quali spiega almeno il 5% della variazione dei geni del ciclo cellulare(Figura 5-figure supplement 1). Dopo aver regolato per questi, abbiamo identificato 56 geni in P. falciparum cellule asessuate che hanno mostrato una variazione residua(Figura 5A; file supplementare 2). A differenza dei geni della variante clonale identificati nel lavoro precedente(Rovira-Graells et al., 2012), questi 56 geni non erano localizzati in regioni subtelomeriche. I prodotti di questi geni sono stati coinvolti nell’assemblaggio del nucleosoma, nel proteasoma e nell’acidificazione vacuolare, suggerendo un ruolo nel controllo dell’espressione genica attraverso l’iniziazione della trascrizione, la stabilità delle proteine e la localizzazione delle proteine. I modelli di espressione dei 56 geni non erano correlati, come ci si sarebbe potuto aspettare se fossero stati parte di una risposta trascrizionale coordinata, come la risposta allo stress. Abbiamo quindi indagato se il modello di espressione osservato è risultato da variazioni dei livelli di mRNA stazionari a causa dell’espressione intermittente di questi geni, seguita da un rapido decadimento dell’mRNA. Da un dataset pubblicato di emivite di mRNA nel ciclo asessuato, abbiamo scoperto che questi geni hanno in realtà una durata media di emivita moderatamente superiore alla media(Figura 5B). Ciò suggerisce che la variabilità di questi geni era più probabile che la variabilità di questi geni fosse guidata dall’inizio di trascrizione variabile piuttosto che dal decadimento rapido. Abbiamo scoperto che questi geni sono più conservati in evoluzione di quanto ci si aspettasse per caso (p=2,2e-16), e che questo non è semplicemente perché tendono ad essere altamente espressi(Figura 5C). È interessante notare che 22 di questi 56 geni sono anche geni espressi in modo variabile negli stadi sessuali, il che suggerisce una variabilità intrinseca lungo tutto il ciclo vitale(file supplementare 3E). Inoltre, tipi simili di geni erano variabili negli stadi sessuali di P. berghei(file supplementare 1A,C), ma non siamo stati in grado di identificare molti geni variabili indipendenti dal ciclo cellulare nelle cellule asessuate di P. berghei, forse a causa di troppo poche cellule esaminate. È ancora da vedere se l’espressione volatile di questi geni si riflette anche nell’abbondanza di proteine.
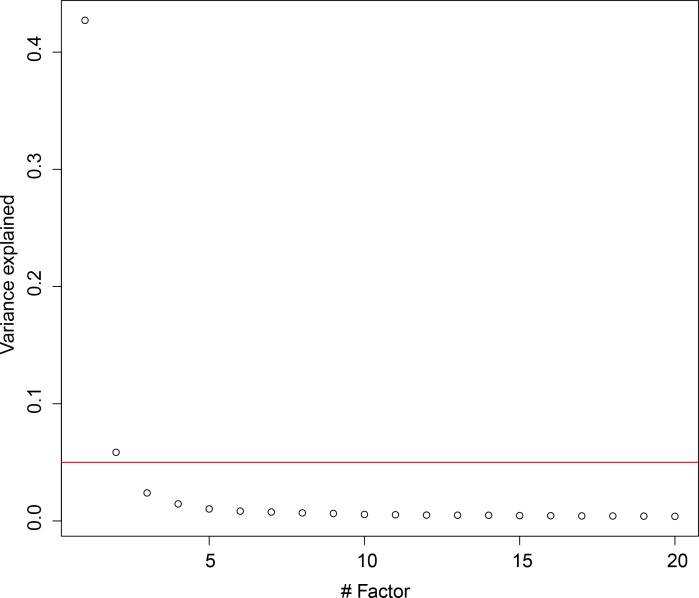
Figura 5-figure supplemento 1.Dopo aver rimosso il segnale di progressione del ciclo cellulare, identifichiamo una nuova classe di geni variabili indipendenti dal ciclo cellulare.analisi del fattore latente della variazione di espressione nei geni del ciclo cellulare.(A) P. falciparum geni con >= 50% della loro varianza attribuita alla variabile latente associata al ciclo cellulare associato variabile latente uno variano in pseudotempo. Dopo aver rimosso la variazione associata al ciclo cellulare, 56 geni con >= 50% della loro varianza è rimasto. Altamente arricchito termini funzionali associati ai due gruppi di geni sono mostrati.(B) Qui, mostriamo che trascrizioni variabili indipendenti dal ciclo cellulare trascrizioni variabili hanno emivita simili ai geni in generale durante le fasi di anello e trofozoiti. Tuttavia, durante la fase di schizofrenia e più tardi, sono significativamente più lunghe. I dati sono stati ricavati da(Shock et al., 2007).(C) Un punteggio di conservazione, calcolato sulla base della sostituzione media degli aminoacidi tra le proteine di P. berghei e P. falciparum, è stato tracciato contro il livello di espressione (scran-l) per ogni cellula-ciclo-dipendente e per ogni gene cellula-ciclo-indipendente in P. falciparum. I grafici di densità mostrano le distribuzioni di ciascuno di questi parametri, evidenziando che i geni indipendenti dal ciclo cellulare tendono ad avere punteggi di conservazione più alti, ma livelli di espressione simili.Abbiamo scoperto che solo le prime due variabili latenti spiegano almeno il 5% della variazione dei geni del ciclo cellulare (linea rossa).
Figura 5-figure supplement 1.Dopo aver rimosso il segnale della progressione del ciclo cellulare, abbiamo identificato una nuova classe di geni indipendenti dal ciclo cellulare.analisi del fattore latente della variazione di espressione nei geni del ciclo cellulare.(A) P. falciparum geni con >= 50% della loro varianza attribuita alla variabile latente associata al ciclo cellulare associato variabile latente uno variano in pseudotempo. Dopo aver rimosso la variazione associata al ciclo cellulare, 56 geni con >= 50% della loro varianza è rimasto. Altamente arricchito termini funzionali associati ai due gruppi di geni sono mostrati.(B) Qui, mostriamo che trascrizioni variabili indipendenti dal ciclo cellulare trascrizioni variabili hanno emivita simili ai geni in generale durante le fasi di anello e trofozoiti. Tuttavia, durante la fase di schizofrenia e più tardi, sono significativamente più lunghe. I dati sono stati ricavati da(Shock et al., 2007).(C) Un punteggio di conservazione, calcolato sulla base della sostituzione media degli aminoacidi tra le proteine di P. berghei e P. falciparum, è stato tracciato contro il livello di espressione (scran-l) per ogni cellula-ciclo-dipendente e per ogni gene cellula-ciclo-indipendente in P. falciparum. I grafici di densità mostrano le distribuzioni di ciascuno di questi parametri, evidenziando che i geni indipendenti dal ciclo cellulare tendono ad avere punteggi di conservazione più alti, ma livelli di espressione simili.Abbiamo scoperto che solo le prime due variabili latenti spiegano almeno il 5% della variazione dei geni del ciclo cellulare (linea rossa).
Figura 5-figure supplement 1.Analisi del fattore latente della variazione di espressione nei geni del ciclo cellulare.Abbiamo trovato che solo le prime due variabili latenti spiegato almeno il 5% della variazione dei geni del ciclo cellulare (linea rossa).
I gametociti mostrano l’espressione di una variabile specifica del sesso dei geni di contingenza
Sorprendentemente, i geni espressi in modo più variabile nelle fasi sessuali erano quelli delle famiglie di geni di contingenza: var in P. falciparum e pir in P. berghei(Figura 6; File supplementare 4). Le famiglie di geni di contingenza sono estremamente labili dal punto di vista evolutivo e diverse specie hanno repertori diversi(Reid, 2015). Tra P. falciparum e P. berghei, non vi è alcuna evidenza di omologia tra queste famiglie e mentre molte sono note o si suppone che svolgano un ruolo nelle interazioni ospite-parassita, non è chiaro fino a che punto possano svolgere funzioni sovrapposte nelle due specie. Poco si sa sul ruolo di queste famiglie negli stadi sessuali e sebbene non siano state osservate variazioni trascrizionali, l’espressione lo ha fatto(Florens et al., 2002) e suggerisce un ruolo per i geni di emergenza nella trasmissione. Diverse parti importanti della trasmissione potrebbero richiedere geni di emergenza che codificano le proteine di superficie delle cellule. In primo luogo, i gametociti maturi si trovano nel sangue e sono quindi suscettibili di essere attaccati dal sistema immunitario adattivo dell’ospite in modo molto simile agli anelli di P. falciparum o agli anelli di P. berghei e ai trofozoiti. In secondo luogo, è stato suggerito che i gametociti possono raggrupparsi per rendere la trasmissione più affidabile e questo potrebbe richiedere proteine di superficie cellulare antigenicamente variabili(Pichon et al., 2000). Infine, dopo la trasmissione, i gameti devono affrontare un ambiente complesso e ostile nel midgut della zanzara, dove i gameti maschi devono trovare rapidamente le femmine, cosa che fanno a ritmi difficili da spiegare senza invocare movimenti non casuali come la chemiotassi(Lawniczak e Eckhoff, 2016). I nostri dati hanno rivelato che i maschi e le femmine sono molto diversi nella loro espressione delle famiglie di geni di contingenza. In P. berghei gametociti maschili, abbiamo osservato una significativa variabilità di un insieme di geni pir pir(Otto et al., 2014) (p=0,014; Figura 6-figure supplement 1A; Supplementary file 4), i cui prodotti proteici sono stati precedentemente identificati nei gameti maschili (Talmanet al., 2014), indicando un potenziale ruolo nella fecondazione. Ciò solleva l’intrigante possibilità che la variazione nell’espressione di questi geni possa avere un impatto sulle interazioni maschio/femmina durante la fecondazione. Abbiamo trovato nessun gene pir pir specifico femminile, invece, le femmine hanno mostrato una variazione trascrizionale nei membri delle famiglie subtelomeriche multigene fam-a e fam-b(Figura 6A; File supplementare 4).
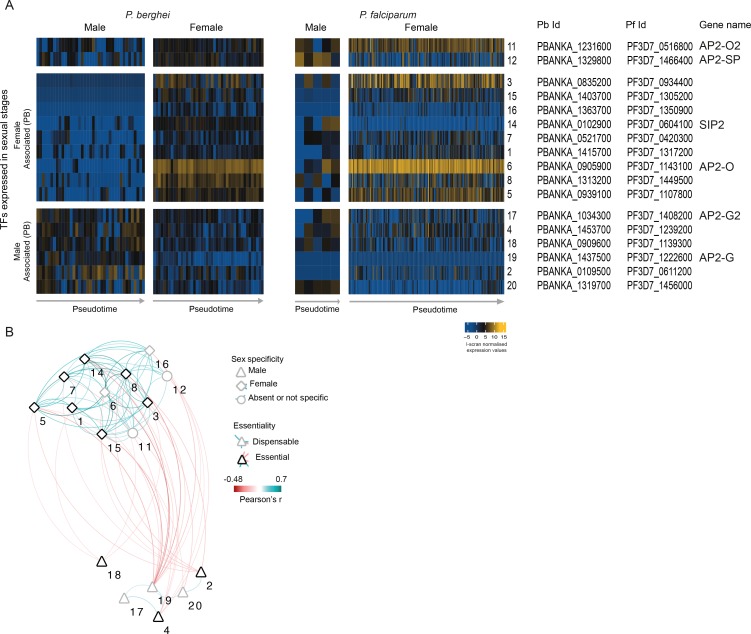
Figura 6-figure supplemento 2.Le famiglie multigene mostrano un’espressione variabile all’interno e tra gli stadi sessuali di entrambe le P.Le famiglie multigene mostrano un’espressione variabile negli stadi sessuali di entrambe le P.Analisi del modello di co-espressione della famiglia ApiAP2 dei fattori di trascrizione (TF) nei parassiti sessuali.berghei e P. falciparum.a) La heatmap mostra i livelli di espressione genica per i membri della famiglia multigenica espressi in modo differenziato tra gametociti maschili e femminili di P. berghei. * gene espresso in modo variabile all’interno del maschio (arancione) o della femmina (verde), Lpl= lisofosfolipasi, ema1 = antigene di membrana dell’eritrocita 1, (b) I conteggi di lettura per gli mRNA var in singole cellule di gametociti femminili di P. falciparum e nelle popolazioni di gametociti femminili e maschili da dati RNA-seq in massa. Sono state incluse solo le letture che hanno riguardato gli introni del var e solo i geni con almeno due di queste letture. Le singole cellule maschili non erano sufficienti per l’analisi.Berghei e P. Falciparum, rispettivamente.(A) L’espressione del gene Pir era altamente variabile tra i gametociti maschili. Inoltre, sono stati espressi più geni pir nei maschi che nelle femmine. Si tratta di sottofamiglie distinte di geni pir da quelli espressi in modo variabile in fasi asessuate.(B) Leggere i conteggi per i var mRNA in P. falciparum gametociti femminili a cellule singole e popolazioni di gametociti femminili e maschili a partire da dati RNA-seq di massa. Sono state incluse solo le letture che hanno riguardato gli introni del var e solo i geni con almeno due di queste letture. Le singole cellule maschili non erano sufficienti per l’analisi. Per i geni di var, evidenziamo il tipo (ad esempio upsB, upsC o var2csa) e se abbiamo trovato prove di espressione dell’mRNA, piuttosto che letture che potrebbero rappresentare ncRNA noti per essere espressi da questi geni. Epf1= famiglia proteica esportata 1, gbp = proteina legante della glicoforina, emp3 = proteina di membrana eritrocita 3.(A) Espressione dei geni Plasmodium ApiAP2 nei parassiti sessuali. I geni ortologi sono presentati sulle stesse righe.(B) Una rete di co-espressione per P. berghei è stato costruito utilizzando significative correlazioni positive e negative (p<0,05 dalla correlazione di Pearson) tra le TF e ponderate in base al loro coefficiente di correlazione. I numeri sulla rete si riferiscono alle TF così come presentate in A.
In P. falciparum, i geni var sono fondamentali per stabilire le infezioni croniche attraverso la citoaderenza e la variazione antigenica(Scherf et al., 2008). Invece di trovare una variazione significativa nei maschi, come ci si aspettava dai nostri risultati in P. berghei, sono state le femmine a mostrare una variazione trascrizionale all’interno dei geni var (p=0,0006; Figura 6B). Nei parassiti asessuali, l’espressione di due diverse trascrizioni var non codificanti è comune ed è coinvolta nel mantenimento dell’espressione del gene var reciprocamente esclusivo che è essenziale per il loro ruolo di evasione immunitaria(Amit-Avraham et al., 2015; Guizetti e Scherf, 2013). Entrambi sono trascritti da un promotore bidirezionale all’interno del singolo introne var. Ciò significa che la presenza di trascrizioni var codificanti nei gametociti può essere valutata identificando le letture intron-spanning. Abbiamo scoperto che all’interno di ogni singola cellula femminile, solo un singolo gene var aveva letto le letture che supportavano il corretto splicing, suggerendo che l’espressione mutuamente esclusiva dei geni var si verifica in fasi sessuali, come avviene nei parassiti asessuali(Guizetti e Scherf, 2013). Le trascrizioni dei codici var erano sempre da cluster di geni var interni, spesso con la classe di promotori upsC, distinti dai geni var subtelomerici visti negli stadi asessuali, con promotori upsB e upsA(Figura 6B; Figura 6-figure supplement 1B). I singoli gametociti maschili non erano ben rappresentati in questo studio, così abbiamo invece esaminato i dati RNAseq dei gametociti maschili e femminili RNAseq(Lasonder et al., 2016) per l’espressione del gene var maschile. I gametociti maschili hanno mostrato solo l’mRNA di un singolo gene var, var2csa, noto per la sua importanza nella malaria correlata alla gravidanza(Figura 6B; File supplementare 4). Questo gene è stato proposto anche come importante regolatore della commutazione dell’espressione del gene var(Mok et al., 2008). La nostra nuova osservazione che i gametociti mostrano una significativa variazione sessuale specifica nell’espressione di grandi famiglie multigene, finora note per la loro importanza negli stadi asessuali, suggerisce che la loro evoluzione e funzione può essere guidata anche dalla biologia degli stadi sessuali.
IlPlasmodium non ha cromosomi sessuali e il fondamento genetico del dimorfismo sessuale è molto poco compreso. Per esplorare la regolazione del dimorfismo sessuale, abbiamo esaminato l’espressione sessuale specifica dei fattori di trascrizione in entrambe le specie e abbiamo condotto un’analisi di co-espressione nei maschi e nelle femmine. Abbiamo osservato un marcato e conservato modello sessuale specifico dell’espressione TF(Figura 6B, Figura 6-figure supplement 2). È interessante notare che una TF femmina-specifica in particolare (ap2-o) ha dimostrato in precedenza di avere una funzione femminile ed è probabile che abbia un ruolo nella differenziazione delle forme maschili e femminili(Modrzynska et al., 2017).
Figura 6-figure supplement 2.Le famiglie multigene mostrano un’espressione variabile all’interno e tra gli stadi sessuali di entrambe le P.Le famiglie multigene mostrano un’espressione variabile negli stadi sessuali di entrambe le P.Analisi del modello di co-espressione della famiglia ApiAP2 dei fattori di trascrizione (TF) nei parassiti sessuali.berghei e P. falciparum.a) La heatmap mostra i livelli di espressione genica per i membri della famiglia multigene, espressi in modo differenziato tra gametociti maschili e femminili di P. berghei. * gene espresso in modo variabile all’interno del maschio (arancione) o della femmina (verde), Lpl= lisofosfolipasi, ema1 = antigene di membrana dell’eritrocita 1, (b) I conteggi di lettura per gli mRNA var in singole cellule di gametociti femminili di P. falciparum e nelle popolazioni di gametociti femminili e maschili da dati RNA-seq in massa. Sono state incluse solo le letture che hanno riguardato gli introni del var e solo i geni con almeno due di queste letture. Le singole cellule maschili non erano sufficienti per l’analisi.Berghei e P. Falciparum, rispettivamente.(A) L’espressione del gene Pir era altamente variabile tra i gametociti maschili. Inoltre, sono stati espressi più geni pir nei maschi che nelle femmine. Si tratta di sottofamiglie distinte di geni pir da quelli espressi in modo variabile in fasi asessuate.(B) Leggere i conteggi per i var mRNA in P. falciparum gametociti femminili a cellule singole e popolazioni di gametociti femminili e maschili da dati RNA-seq di massa. Sono state incluse solo le letture che hanno riguardato gli introni del var e solo i geni con almeno due di queste letture. Le singole cellule maschili non erano sufficienti per l’analisi. Per i geni di var, evidenziamo il tipo (ad esempio upsB, upsC o var2csa) e se abbiamo trovato prove di espressione dell’mRNA, piuttosto che letture che potrebbero rappresentare ncRNA noti per essere espressi da questi geni. Epf1= famiglia proteica esportata 1, gbp = proteina legante della glicoforina, emp3 = proteina di membrana eritrocita 3.(A) Espressione dei geni Plasmodium ApiAP2 nei parassiti sessuali. I geni ortologi sono presentati sulle stesse righe.(B) Una rete di co-espressione per P. berghei è stato costruito utilizzando significative correlazioni positive e negative (p<0,05 dalla correlazione di Pearson) tra le TF e ponderate in base al loro coefficiente di correlazione. I numeri sulla rete si riferiscono alle TF così come presentate in A.
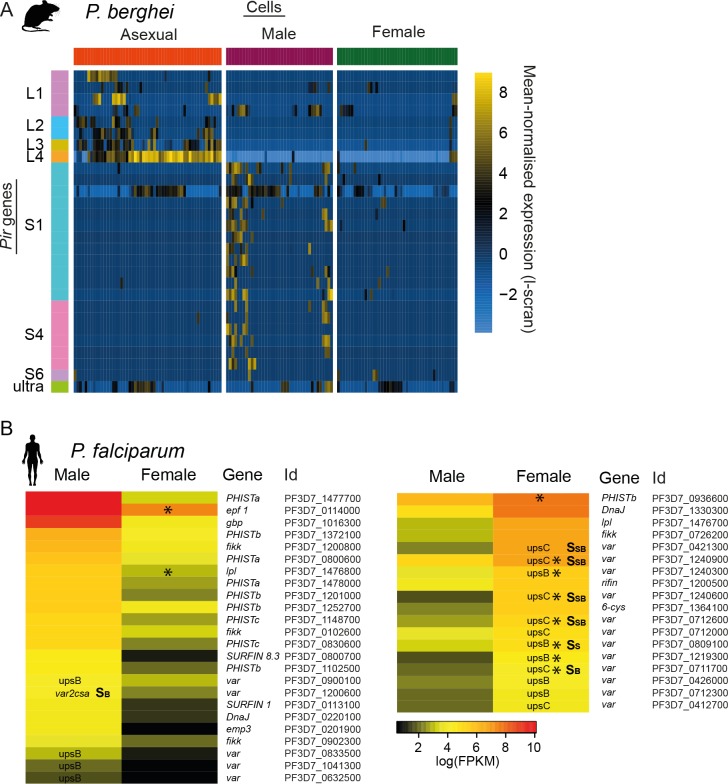
Figura 6-figure supplement 1.Le famiglie multigene mostrano un’espressione variabile nelle fasi sessuali di entrambe le P.berghei e P. falciparum, rispettivamente.(A) L’espressione del gene Pir era altamente variabile tra i gametociti maschili. Inoltre, più geni pir sono stati espressi nei maschi che nelle femmine. Si tratta di sottofamiglie di geni pir distinte da quelle espresse in modo variabile in fasi asessuate.(B) Leggere i conteggi per i var mRNA in P. falciparum gametociti femminili a cellule singole e popolazioni di gametociti femminili e maschili da dati RNA-seq di massa. Sono state incluse solo le letture che hanno riguardato gli introni del var e solo i geni con almeno due di queste letture. Le singole cellule maschili non erano sufficienti per l’analisi. Per i geni di var, evidenziamo il tipo (ad esempio upsB, upsC o var2csa) e se abbiamo trovato prove di espressione dell’mRNA, piuttosto che letture che potrebbero rappresentare ncRNA noti per essere espressi da questi geni. Epf1= famiglia proteica esportata 1, gbp = proteina legante della glicoforina, emp3 = proteina di membrana eritrocita 3.
Figura 6-figure supplemento 2.Analisi del modello di co-espressione della famiglia ApiAP2 dei fattori di trascrizione (TF) nei parassiti sessuali.(A) Espressione dei geni Plasmodium ApiAP2 nei parassiti sessuali. I geni ortologi sono presentati sulle stesse righe.(B) Una rete di co-espressione per P. berghei è stata costruita utilizzando significative correlazioni positive e negative (p<0,05 dalla correlazione di Pearson) tra le TF e ponderate in base al loro coefficiente di correlazione. I numeri sulla rete si riferiscono alle TF così come presentate in A.
Discussione
Abbiamo stabilito un protocollo ottimizzato per la generazione di sequenze di trascrittomi monocellulari di parassiti del Plasmodio con il potere di identificare non solo i diversi tipi di cellule, ma anche di esplorare le potenziali variazioni funzionali da una cellula all’altra. Questo protocollo permette la valutazione di trascrizioni a lunghezza completa, cosa necessaria per valutare i complessi schemi trascrizionali che abbiamo osservato per i geni var ma che attualmente non è possibile con approcci basati su tag 3′ (Poranet al., 2017). Inoltre, questo metodo ha anche il vantaggio di fornire informazioni su un numero di geni per cellula quasi tre volte maggiore rispetto alle valutazioni Drop-seq della stessa specie (~1900 in media qui contro ~650 in media per Drop-seq) (Poran et al.,2017).
I futuri studi sulla malaria trarranno grande beneficio dalla disponibilità di entrambi (i) metodi basati su goccioline a bassa copertura che permettono di analizzare un gran numero di cellule e (ii) metodi di trascrizione a tutta lunghezza ad alta copertura, che permettono un’analisi ad alta definizione e focalizzata delle cellule ordinate con citometria a flusso. Durante l’ottimizzazione del nostro protocollo per i parassiti del Plasmodium, abbiamo identificato diversi passaggi decisivi e reagenti permutabili che, se modificati, sono stati determinanti per la qualità del trascrittoma. Ci auguriamo che questo quadro di ottimizzazione possa aiutare ad estendere lo scRNA-seq del trascritto a una gamma molto più ampia di diversi tipi di cellule eucariotiche.
Oltre a stabilire un nuovo strumento, il nostro studio ha fatto diverse nuove osservazioni sulla biologia del Plasmodio. In primo luogo, abbiamo utilizzato dati monocellulari per produrre indagini ad alta risoluzione della schizogonia e abbiamo osservato transizioni trascrizionali nette durante il ciclo di vita asessuato, che in precedenza si pensava fosse un processo continuo. Il ciclo intracellulare del Plasmodium è complesso, composto da diversi cicli di replicazione del DNA endomitotico seguiti da una citochinesi finale sincronizzata. Sebbene i punti di controllo siano molto probabilmente necessari per garantire la tempestività di eventi cellulari complessi, come l’assemblaggio del macchinario di invasione dei globuli rossi, non sono ancora stati identificati(Gerald et al., 2011). Si ipotizza che le transizioni nette che abbiamo osservato corrispondano a tali punti di controllo. Anche se abbiamo trovato indizi sulla possibile architettura normativa sottostante, i veri regolatori restano ancora da confermare.
Una seconda importante scoperta del nostro studio è stata la variazione inaspettata da cellula a cellula nell’espressione genica. La maggior parte dei geni sono noti per variare durante il ciclo cellulare asessuato, stadio del sangue con un singolo picco di espressione(Bozdech et al., 2003a). Alcuni geni nelle regioni subtelomeriche sono noti per variare indipendentemente dal ciclo cellulare, accendendo e spegnendo i singoli parassiti. Tra questi vi sono le famiglie multigene di geni di emergenza noti per essere coinvolti nel sequestro e nell’infezione cronica(var e pir). Ma inaspettatamente abbiamo trovato un’altra classe di geni che variano indipendentemente dal ciclo cellulare sia nel ciclo che nelle cellule arrestate. Abbiamo scoperto che, a differenza dei geni di contingenza, essi erano altamente conservati tra le specie e gli stessi tipi di geni erano variabili nelle specie parassite che infettavano sia gli esseri umani che i roditori. Si potrebbe ipotizzare che ciò sia dovuto a segnali più rumorosi associati specificamente ad alcune funzioni cellulari per le quali è utile rilassarsi nel controllo trascrizionale. La generazione di variazioni in una popolazione di molti milioni di parassiti strettamente imparentati che occupano un ambiente ospite sempre diverso può essere una strategia di scommessa che favorisce il successo di almeno alcuni dei membri di questa popolazione.
Infine, poiché il nostro approccio è stato in grado di sezionare le trascrizioni dei gametociti maschili e femminili e di valutare l’espressione delle famiglie multigene, siamo stati in grado di scoprire un’inaspettata specificità sessuale nell’espressione di diverse famiglie multigene. Particolarmente intrigante è che queste famiglie sono note per codificare le proteine extracellulari coinvolte nelle interazioni ospite-parassita nelle fasi asessuate del sangue. Potrebbero avere funzioni interattive host simili non ancora descritte per gli stadi sessuali o avere ruoli inesplorati nel comportamento sessuale del parassita. Nell’ospite mammifero, potrebbero essere coinvolti nel sequestro di gametociti maturi nella vascolarizzazione periferica, come strategia di evasione immunitaria o per aiutare la trasmissione attraverso una puntura di zanzara. La natura sessuale specifica dell’espressione dei geni var e pirica potrebbe anche indicare un possibile ruolo nella fecondazione nel midgut della zanzara.
L’RNA-seq monocellulare avrà molte applicazioni per i parassiti della malaria. La rilevazione dei parassiti direttamente da campioni di pazienti in infezioni naturali porterà senza dubbio a nuove conoscenze dei geni alla base di importanti fenotipi. Oltre agli aspetti qui trattati, può essere particolarmente potente per affrontare i seguenti problemi: (i) analisi di piccoli campioni provenienti da stadi del ciclo di vita non coltivabili o da specie di Plasmodium che non possono ancora essere coltivate, come il parassita umano prevalente P. vivax, (ii) scoperta di stati cellulari rari/indescrivibili, (iii) caratterizzazione dell’effetto delle alterazioni genetiche per generare fenotipi ad alta dimensione per molti mutanti in parallelo(Bushell et al., 2017), e (iv) esame della variabilità da cellula a cellula di fronte a farmaci e vaccini.
Materiali e metodi
Isolamento dei parassiti di P. berghei
La linea ANKA mCherry-expressing P. berghei, clone RMgm-928(Khan et al., 2013), è stata propagata in un topo femmina di Theiler di 6-8 settimane fornito da Envigo UK. I parassiti sono stati purificati da una coltura notturna (20 ore) di 50 ml di 1 ml di sangue infetto utilizzando un cuscino Histodenz al 55% (SIGMA), a seguito di un protocollo di purificazione dello schizonte stabilito e dettagliato altrove(Gomes et al., 2015). Gli stadi tardivi purificati (asessuato e sessuale) sono stati pellettizzati a 450 g per 3 minuti e incubati con 500 µL di RNALater (ThermoFisher) per 5 minuti, e ulteriormente diluiti in 3 mL di 1x PBS prima della selezione delle cellule. Tutte le ricerche sugli animali sono state condotte con licenze del Ministero dell’Interno del Regno Unito e hanno utilizzato protocolli approvati dal comitato etico del Wellcome Sanger Institute.
Cultura in vitro P. falciparum
Un passaggio precoce di 3D7-HTGFP (ceppo MR4-1029), un ceppo trasmissibile GFP-espressivo P. falciparum, (non più di tre espansioni da stock congelati dalla clonazione iniziale),(Talman et al., 2010), è stato mantenuto in globuli rossi O-negativi ottenuti dal NHSBT, utilizzando un terreno di coltura RPMI 1640 (GIBCO) integrato con 25 mM HEPES (SIGMA), 10 mM D-Glucosio (SIGMA), 50 mg/L ipoxantina (SIGMA), 10% di siero umano (ottenuto localmente in conformità con i protocolli eticamente approvati), e gassato utilizzando una miscela contenente 5% O2, 5%CO2 e 90% N2. I parassiti sono stati altamente sincronizzati utilizzando due cicli consecutivi di trattamento con Percoll-Sorbitolo(Kutner et al., 1985). I parassiti asessuali tardivi (trofozoiti e schizonti) sono stati purificati su un cuscino di Percoll al 63% (GE Healthcare). I gametociti di stadio V sono stati ottenuti utilizzando una coltura standard di gametociti(Fivelman et al., 2007) e purificati magneticamente con una colonna LS(Ribaut et al., 2008) (Miltenyi Biotec). Dopo la purificazione di ogni stadio, tutti i parassiti di P. falciparum sono stati pellettizzati a 800 g per 5 minuti, incubati con 500 µL di RNALater (ThermoFisher) per 5 minuti, e ulteriormente diluiti in 3 mL di 1x PBS prima della selezione delle cellule. Parasitemia è stata determinata da Giemsa macchia di Giemsa striscio di sangue sottile.
Selezione delle cellule
Quattro microlitri di tampone di lisi (0,8% di Triton-X privo di RNAse (Fisher) in acqua priva di nucleasi (Ambion)), trattato UV per 30 minuti con un reticolante UV Stratalinker 2400 a 200, 000 µJ/cm2, 2,5 mM dNTPs (Life Technologies), 2,5 µM di oligo(dT) (Non-Anchored 30 bp: 5′-AAGCAGGTGGTGTATCAACGCAGGAGTACT(x30)-3′; Ancorato 30 bp: 5′-AAGCAGTGGTGTGTATCAACGCAGGAGTACT(x30)VN-3′;Non-Ancorporato 20 bp: 5′-AAGCAGTGGTGTGTATCAACGCAGGAGTACT(x20)-3′;;Ancorato 20 bp: 5′-AAGCAGTGGGTGTATCAACGCAGGAGTACT(x20)VN-3’IDT; vedi Tabella 1 per i dettagli) e 2U di SuperRNAsin (Life Technologies) sono stati dispensati in ogni pozzetto della piastra a 96 pozzetti priva di RNAse (Abgene) immediatamente prima della selezione e conservati in ghiaccio. Nel primo esperimento sono stati utilizzati solo 2 µL di tampone di lisi, ma l’efficienza di cattura delle cellule osservata era molto scarsa e quindi il volume è stato aumentato. La selezione delle cellule è stata condotta su un selezionatore cellulare Influx (BD Biosciences) con un ugello di 70 µm. I parassiti sono stati ordinati per gating su eventi monocellulari e su GFP(P. falciparum) o mCherry(P. berghei) fluorescenza. Un pozzo di controllo negativo non ordinato e un pozzo di controllo positivo a 100 celle sono stati inclusi in ogni piastra accanto alle singole celle. Piastre ordinate sono state girate a 200 G per 10 s e immediatamente posizionate su ghiaccio secco.
Primo e secondo filamento cDNA sintesi e preamplificazione del cDNA
Le cellule in piastre sono state incubate a 72°C per 3 min. Un master mix di trascrizione inversa è stato aggiunto ai campioni contenenti 1 µM di LNA-oligonucleotide (5′-AGCAGTGGTGTATCAACGCAACGCAGGGTACATrGrG+G-3′; Exiqon), 6 µM MgCl2, 1M Betaina (Affymetrix), 1X buffer di trascrizione inversa, 50 µM DTT, 0.5 U di SuperRNAsin, e 0,5 µL di trascrittasi inversa (Tabella1). Il volume totale della reazione è stato di 10 µL. La piastra è stata incubata utilizzando il seguente programma: 1 ciclo di 42°C per 90 min; 10 cicli (42°C/2 min, 50°C/2 min); 1 ciclo di 70°C per 15 min. I campioni sono stati poi integrati con 1X KAPA Hotstart HiFi Readymix e 2,5 µM del primer ISO SMART (Picelliet al., 2013) e incubati utilizzando il seguente programma di cicli 1 ciclo di 98°C per 3 min; 25 o 30 cicli (98°C/20 s, 67°C/15 s, 72°C/6 min); 1 ciclo di 72°C per 5 min (Tabella 1). I campioni sono stati poi purificati con perle di Ampure 1X Agencourt (Beckman Coulter) in una stazione di lavoro Zephyr G3 SPE (Perkin Elmer) secondo la raccomandazione del produttore. Il cDNA amplificato è stato eluito in 10 µL di acqua priva di nucleasi. I dettagli delle diverse permutazioni del protocollo testato durante il processo di ottimizzazione sono riportati nella tabella 1.
Controllo di qualità dei campioni di cDNA
La qualità di un sottoinsieme di campioni di cDNA amplificati è stata monitorata con il chip di DNA ad alta sensibilità su un Bioanalizzatore Agilent 2100. I campioni sono stati verificati con qPCR utilizzando LightCycler 480 SYBR Green I Master e primer MSP-1 ad una concentrazione di 0,4 µM (Forward: 5′-TCCCAATCAGGGAGAAACAGAACAG-3′; Reverse: 5′-GATGGGGTTGTGGTGGGTGGTGGTAATG-3′), su un Roche Lightcycler 480 II. Le reazioni sono state incubate secondo il seguente programma ciclico: un ciclo, 95°C/10 min; 45 cicli (98°C/20 s, 58°C/10 s, 68°C/30 s). Le trascrizioni sono state quantificate con il metodo della quantificazione assoluta utilizzando una diluizione standard.
Preparazione della biblioteca e sequenziamento
Le biblioteche sono state preparate utilizzando il kit Nextera XT (Illumina) secondo le raccomandazioni del produttore. Sono state utilizzate 96 o 384 diverse combinazioni di indici per consentire il multiplexing durante il sequenziamento. Dopo l’indicizzazione, le biblioteche sono state messe in comune per la pulizia con un rapporto 4:5 di perle di Ampure di Agencourt (Beckman Coulter). La qualità delle biblioteche è stata monitorata con il chip di DNA ad alta sensibilità su un Bioanalizzatore Agilent 2100. I controlli a pozzetto vuoto e le singole cellule sono stati raggruppati separatamente dai controlli a 100 cellule e caricati in proporzione al loro contenuto cellulare previsto per il sequenziamento su un Illumina MiSeq o HISeq 4000.
Sequenziamento di biblioteche a cella singola
Il protocollo originale Smart-seq2 con l’enzima Superscript II e l’oligo(dT) originale con una base di ancoraggio è stato eseguito con 30 cicli di PCR di preamplificazione su 10 campioni. I campioni comprendevano un singolo controllo senza cellule, cinque singoli gametociti P. falciparum, due controlli a 10 cellule e due controlli a 100 cellule. Questi sono stati multiplexati, insieme a tre campioni ciascuno di cellule carcinoma polmonare umano individuale (A549) e sequenziati su un singolo MiSeq eseguire con 150 bp accoppiato fine lettura.
Per testare l’effetto di diversi enzimi della trascrittasi inversa e diversi numeri di cicli di PCR, abbiamo sequenziato P. falciparum schizont librerie P. preparato utilizzando l’enzima SmartScribe (Clontech) o l’enzima SuperScript II (Thermofisher) per ciascuna delle sei singole cellule, un 100-cellula di controllo e due controlli no-cellula, utilizzando 25 cicli di PCR in ogni caso. I campioni sono stati multiplexati su un singolo MiSeq eseguire e sequenziato come 150 bp fine accoppiato legge.
Per determinare se i campioni monocellulari potrebbero essere contaminati con cellule aggiuntive o RNA da cellule lisate, singoli mCherry P. berghei (RMgm-928[Khan et al., 2013]) e GFP P. falciparum(Talman et al., 2010) schizonti sono stati mescolati in un rapporto 1:1, inattivato con fissazione RNAlatere poi ordinati. È stata preparata una libreria multiplex composta da 32 singoli schizonti P. berghei, due controlli di schizonti P. berghei a 100 celle, un controllo no-cell, 40 singoli schizonti P. falciparum e due controlli di schizonti P. falciparum a 100 celle. Queste librerie sono state sequenziate come un pool multiplex su un singolo MiSeq eseguito con 150 bp lettura finale accoppiato.
I campioni di sangue misto P. berghei fase di P. berghei sangue misto fase comprendeva 182 singole cellule di P. berghei, più quattro controlli no-cellulare e sei controlli a 100 cellule. Questi sono stati multiplexati con altri 192 campioni non analizzati in questo lavoro e sequenziati su una singola corsia di HiSeq 4000 utilizzando HiSeq v4 con 75 bp lettura di estremità accoppiata. I campioni di gametociti di P. falciparum sono stati sequenziati come tre pool multiplexati di 84, utilizzando la stessa chimica. Tre campioni tecnici duplicati sono stati esclusi dall’analisi. I campioni asessuati di P. falciparum sono stati sequenziati come due pool di 96, ciascuno su una corsia di illuminazione HiSeq 2500 utilizzando la chimica HiSeq v4 con 75 bp paired-end. Ogni lotto di 96 campioni conteneva tre controlli a 100 celle. Il secondo lotto (corsia 7) conteneva sei campioni di gametociti di stadio I e sei campioni di gametociti di stadio II, ciascuno con un singolo controllo a 100 celle. Questi non sono stati inclusi nell’analisi, lasciando 176 campioni monocellulari.
Mappatura delle letture e calcolo dei conteggi delle letture
Tutti gli esperimenti di sequenziamento sono stati elaborati nel modo seguente. I file CRAM di lettura sono stati acquisiti dalla pipeline del core pipeline WTSI, convertiti in BAM utilizzando samtools-1.2 view -b, ordinati utilizzando samtools sort -n, convertiti in fastq utilizzando samtools-1 .2 bam2fq e poi deinterleaved (Liet al., 2009). Le sequenze di adattatori Nextera sono state tagliate usando trim_galore -q 20 -a CTGTCTCTTATACACACATCT –paired –stringency 3 –lunghezza 50 -e 0.1 (v0.4.1). Gli indici HISAT2 (v2.0.0-beta)(Kim et al., 2015) sono stati prodotti per le sequenzegenomiche P. falciparum v3 (http://www.genedb.org/Homepage/Pfalciparum) o P. berghei v3(Fougère et al., 2016), scaricate da GeneDB (Logan-Klumpleret al., 2012), utilizzando parametri predefiniti (ottobre 2016). Le letture tagliate e accoppiate sono state mappate su una delle due sequenze del genoma utilizzando hisat2 –max-intronlen 5000 p. 12. Per l’esperimento del doppio ordinamento, abbiamo mappato rispetto a un riferimento combinato, permettendoci di escludere le letture che mappano entrambi i genomi. I file SAM sono stati convertiti in BAM usando la vista -b di samtools-1.2 e ordinati con l’ordinamento samtools-1 .2. I file GFF sono stati scaricati da GeneDB (ottobre 2016) e convertiti in file GTF utilizzando uno script interno. Tutti i tipi di funzionalità (mRNA, rRNA, tRNA, tRNA, snRNA, snoRNA, pseudogenic_transcript e ncRNA) sono stati conservati, con le loro singole regioni di ‘codifica’ etichettate come CDS in ogni caso per comodità. Dove sono state annotate più trascrizioni per un singolo gene, è stata considerata solo la trascrizione primaria. Le letture sono state sommate contro i geni utilizzando HTSeq: htseq-count -f bam -r pos -s no -t CDS (v0.6.0 [Anderset al., 2015];). HTSeq esclude le letture multimappatura per impostazione predefinita (-a 10). Ciò significa che le letture di mappature ambigue con geni simili della stessa famiglia non sono considerate nella nostra analisi. Per l’analisi a valle (escluso l’esame dei conteggi degli rRNA), sono state escluse le trascrizioni non incluse nei file di sequenza cDNA del GeneDB. I conteggi di lettura grezzi per gli stadi di sangue misto P. berghei, P. falciparum gametocytes e P. falciparum asessuato sono presentati nel file supplementare 5.
Classificazione delle letture per il controllo di qualità
Per determinare la resa utile dei diversi protocolli di amplificazione dell’RNA (riassunti nella Tabella 1), abbiamo classificato le letture risultanti in quelle mappature di geni rRNA, altri geni, non mappati o ambigui (che rientrano in più di una categoria). Ci siamo concentrati qui sull’rRNA perché avevamo osservato che questo era un problema particolare. Per fare questo abbiamo iniziato con i file HISAT2 BAM prodotti come descritto sopra. Le coppie di lettura totali erano tutti gli identificatori di coppia di lettura unici. Le letture dell’RNA ribosomiale sono state contate utilizzando l’intersezione di bedtools (v2.17.0[Quinlan e Hall, 2010];) per trovare la sovrapposizione degli identificativi di coppia di lettura unici con le caratteristiche dell’rRNA. Altre letture di codifica sono state contate allo stesso modo, ma cercando la sovrapposizione con tutte le altre caratteristiche. Le letture non mappate sono state identificate utilizzando la vista samtools -f 0 × 8 (v1. 2) ed estraendo gli identificatori univoci della coppia di lettura. Quando una coppia di lettura si verificava in più di una di queste liste, veniva considerata ambigua.
Abbiamo confrontato la complessità della libreria di diverse iterazioni del nostro protocollo per determinare se più letture hanno portato ad una maggiore complessità, o semplicemente più letture dagli stessi geni, forse a causa di un gran numero di cicli di PCR. I diversi cicli di sequenziamento avevano dimensioni di libreria molto diverse e quindi abbiamo effettuato un downsampling dei dati. Per massimizzare il numero di celle incluse, pur consentendo un numero ragionevole di letture per cella, abbiamo scelto di effettuare un downsample a 50.000 letture per cella. Per fare questo, sono stati campionati a caso 50.000 conteggi da HTSeq per ogni cella. I conteggi associati ai geni della codifica delle proteine sono stati enumerati e i geni sono stati chiamati come rilevati se c’erano almeno 10 letture mappate su di essi.
Valutazione del bias nelle librerie di sequenziamento a singola cellula
Diversi protocolli di preparazione della biblioteca e di sequenziamento mostrano diversi pregiudizi nella rappresentazione di sequenze ricche di GC/AT e di trascrizioni di 5 o 3 finali. Al fine di valutare tali distorsioni, abbiamo adottato un approccio di utilizzare i dati mappati RNA-seq per identificare i frammenti di geni che sono stati espressi ed esaminato la copertura dei geni da questi frammenti. La ragione per fare questo, piuttosto che guardare alla profondità di copertura, era che avevamo notato che i geni spesso non avevano una copertura completa, in particolare quando erano molto lunghi o espressi a basso livello. Questo suggerisce che, anche se ci aspetteremmo che Smartseq-2 amplificasse le trascrizioni a piena lunghezza, in alcuni casi solo trascrizioni parziali sono sopravvissute al protocollo completo. Abbiamo usato Stringtie (v1.2; opzioni predefinite[Pertea et al., 2016];) per richiamare frammenti espressi dai nostri file HISAT2 BAM. Abbiamo poi cercato le caratteristiche di trascrizione di Stringtie che si sovrappongono ad ogni caratteristica mRNA nella nostra annotazione di riferimento. Dove più trascrizioni di Stringtie si sovrappongono, queste sono state fuse. Abbiamo poi determinato, per ogni gene, la sequenza esonica coperta dalle trascrizioni fuse di Stringtie. Sono stati calcolati la lunghezza, il contenuto GC e l’inizio e la fine relativa di queste regioni. Il contenuto di GC osservato è stato confrontato con il contenuto di GC per l’intera regione di codifica. Ogni posizione relativa lungo una sequenza di codifica (0-100) coperta da un frammento è stata incrementata per ogni frammento che la copre. La copertura di ogni posizione relativa per ogni gene è stata poi normalizzata tra 0 e 1 sulla base della copertura più alta in quella sequenza di codifica. Per esaminare l’effetto della lunghezza del gene, abbiamo confrontato la distribuzione della lunghezza di tutti i 4943 geni P. berghei utilizzati nella nostra analisi iniziale con i 4579 che hanno superato i nostri criteri di filtraggio (avendo almeno 10 letture in almeno cinque cellule).
Analisi della contaminazione con un doppio tipo di P. falciparum e P. berghei schizonts
Le letture per i campioni del doppio tipo sono state mappate come sopra, ma con un riferimento combinato di entrambi i parassiti, consentendo di mappare ugualmente bene i due genomi, poiché non è stato possibile determinare la loro origine. I conteggi delle letture sono stati convertiti in FPKM e le trascrizioni con un FPKM >= 10 sono state contate come espresse. Abbiamo usato questi dati per dimostrare che nessun pozzo conteneva più di una cella, cioè pozzi con buoni dati (un gran numero di geni espressi) non hanno mai avuto un numero simile di geni di entrambe le specie. Inoltre, nessun pozzetto buono conteneva un gran numero di geni della specie sbagliata. Per esplorare se i geni contaminanti erano simili in pozzi diversi, abbiamo confrontato i geni di P. falciparum identificati nei pozzi con una cellula di P. berghei ordinata in essi e viceversa tra i pozzi. La somiglianza è stata calcolata come il numero di geni contaminanti comuni con un FPKM >= 10, diviso per il numero medio di geni contaminanti tra i due pozzi. Ogni cellula conteneva pochissimi geni contaminanti. Questo è stato più alto per la contaminazione di P. falciparum P . berghei rispetto alla contaminazione di P. berghei P. falciparum, suggerendo che le cellule di P. falciparum contribuiscono maggiormente all’RNA extracellulare nel mezzo. Diverse cellule hanno condiviso pochissime trascrizioni contaminanti, ma i contaminanti più comunemente presenti erano anche più espressi nelle loro cellule di origine.
Per esaminare l’effetto della contaminazione sugli esperimenti a valle, abbiamo filtrato i trascrittomi di P. berghei per escludere quelli con meno di 10.000 letture e meno di 500 geni, lasciando 16 cellule su 32. Abbiamo poi escluso i geni che non erano presenti con almeno cinque conteggi di lettura in almeno due cellule. Di 3566 trascrizioni rilevate in cellule di P. berghei (con almeno cinque conteggi di lettura in almeno due cellule), 15 provenivano da P. falciparum. Il più altamente espresso di questi erano due geni RNA ribosomiali – PF3D7_0532000 e PF3D7_0726000. C’erano anche altri geni di RNA ribosomiale, istoni e diversi noti geni altamente espressi come MSP1, S-antigene, 60S proteina ribosomiale L6-2 e ETRAMP2. Abbiamo poi usato M3Drop (min.geni = 500, soglia MT FDR = 0,01) per determinare se c’erano geni variabili attraverso i campioni. Ne abbiamo trovati solo quattro, nessuno dei quali era di P. falciparum.
Filtraggio e normalizzazione dei dati di conteggio a lettura monocellulare
I tre set di dati principali (Pb mixed, Pf asex, Pf sex) sono stati elaborati utilizzando Scater v1.0.4(McCarthy et al., 2016). In primo luogo abbiamo rimosso i geni senza conteggio in ogni cellula, e le cellule di controllo (100 pool di cellule). Abbiamo poi rimosso le cellule con un totale di meno o uguale a 25000 conteggi di lettura e/o meno di 1000 geni con almeno una lettura. Successivamente abbiamo rimosso i geni che non avevano almeno 10 letture in 5 cellule. Per il set di dati di P. berghei, questo ha portato a 144/183 cellule e 4579 geni unici rilevati in tutte le cellule. Per il set di dati dei gametociti di P. falciparum, dopo il filtraggio, c’erano 191/238 cellule e 4454 geni unici e per il set di dati asessuato di P. falciparum 161/180 cellule e 4387 geni unici. I conteggi sono stati poi normalizzati utilizzando scran(Lun et al., 2016) (v1.0.3). La normalizzazione è richiesta a causa della variazione tecnica tra i campioni dovuta, ad esempio, alla profondità di sequenziamento variabile e all’efficienza di cattura. I dati di lettura dei conteggi di RNA-seq a cella singola contengono molti zeri rispetto ai dati di RNA-seq alla rinfusa. Questi sono causati dal drop out di geni a bassa espressione o dalla variazione tra le cellule e riducono l’accuratezza dei metodi di normalizzazione progettati per i dati RNA-seq alla rinfusa. Scran utilizza un approccio di pooling per ridurre questi zeri. Inoltre, consente un raggruppamento iniziale dei dati e la normalizzazione all’interno di questi cluster (ad esempio, i tipi di cellule), prima di una fase finale di normalizzazione attraverso l’intero set di dati. Questo è particolarmente utile per i nostri dati di P. berghei, dove le cellule asessuate, maschili e femminili dei gametociti differiscono molto nei loro modelli di espressione. La fase iniziale di clustering è stata eseguita con la funzione scran quickCluster (dimensione minima = 30). Ne sono risultati tre cluster che rappresentano le popolazioni asessuate, maschile e femminile di gametociti. La funzione computeSumFactors è stata eseguita utilizzando questi cluster, con dimensioni = 20 e positivo = VERO. Tutte le analisi a valle sono state eseguite con i dati normalizzati dello scran, tranne dove indicato. Per i gametociti di P. falciparum, la funzione computeSumFactors è stata eseguita con dimensioni = 15. Per le fasi asessuate di P. fal ciparum, abbiamo impostato min.size = 20 per quickCluster e la funzione computeSumFactors è stata eseguita con sizes = 15.
Per alcune applicazioni, è necessario normalizzare i dati per lunghezza di trascrizione. Ad esempio, quando si confrontano i valori di espressione genica classificati con i dati di riferimento per determinare lo stadio del ciclo di vita di una cellula. Abbiamo quindi normalizzato i valori di scran prendendo l’esponente (2x), moltiplicando per 1000 e dividendo per la lunghezza del cDNA, determinata dal file GeneDB cDNA FASTA (solo sequenza di codifica, senza UTR). Questo è simile al calcolo di FPKM, tranne che la normalizzazione delle dimensioni della libreria è già realizzata. Ci si riferisce a questi valori come l-scran, per i valori di scran normalizzati in lunghezza.
Determinazione delle fasi del ciclo di vita dei parassiti utilizzando i dati di riferimento alla rinfusa e il clustering
Abbiamo usato diversi set di dati RNA-seq di massa per assegnare uno stadio del ciclo di vita ad ogni cella. Per gli stadi asessuali di P. berghei, abbiamo usato sia i dati microarray di Hoo et al. (2016) che cattura il ciclo di sviluppo asessuato di 24 ore con risoluzione di 2 ore, sia i dati RNA-seq di Otto et al., 2014, che cattura diversi stadi distinti (Otto et al., 2014). Nell’esperimento Hoo microarray, Cy5 è stato usato per etichettare ogni punto temporale, mentre Cy3 è stato usato per etichettare un pool di tutti i campioni. I valori “F635 Median – B635” sono la differenza di intensità di Cy5 tra il primo piano mediano e lo sfondo mediano. Questo valore di intensità è correlato al livello di espressione reale e questi sono i valori che abbiamo usato. I loro dati sono stati generati utilizzando l’assieme del genoma P. berghei v2, quindi abbiamo rimappato le loro sequenze di sonda contro v3 utilizzando HISAT2 (parametri predefiniti;[Kim et al., 2015]). Abbiamo poi usato htseq-count -a 200 f sam -r name -s no per identificare i geni su cui le sonde mappate (cut -f1,21 probes_berghei_htseq.sam | grep PBANKA | grep -v ambiguo >probes_berghei.map). Abbiamo quindi utilizzato i file GPR forniti da ArrayExpress(Parkinson et al., 2005) (adesione GSE80015) e la mappa delle sonde per produrre una tabella dei ranghi percentili per ogni gene in ogni condizione. Le letture RNA-seq dal dataset di Otto et al. sono state scaricate dall’ENA (PRJNA212241). Sono stati mappati su P. berghei ANKA v3 sequenze di trascrizione utilizzando Bowtie2 v2.2.9 (-a -X 800;[Langmead e Salzberg, 2012]) ed eXpress v1.5.1(RobertsePachter, 2013). I conteggi di lettura risultanti sono stati convertiti in FPKM.
I valori di espressione genica monocellulare sono stati convertiti in valori di scran normalizzati in lunghezza (l-scran), come descritto sopra, al fine di produrre livelli di espressione del rango più accurati per i nostri dati scRNA-seq. Abbiamo confrontato ogni profilo di espressione monocellulare con ogni set di dati di riferimento. Per ridurre il rumore, sono stati rimossi i geni che non variano molto tra le condizioni dei dati di riferimento. Per i dati di riferimento del ciclo di sviluppo intraeritrocitario di P. berghei 24 ore(Hoo et al., 2016), i geni sono stati inclusi solo se il loro profilo di espressione aveva un rango medio superiore a 30 e inferiore a 70 e una deviazione standard nel rango tra campioni superiori a 3. Sono stati anche rimossi i geni dal set di dati della query con l-scran <3. Per calcolare una correlazione tra di loro sono stati necessari almeno 100 geni rimanenti comuni sia al profilo di riferimento che a quello di interrogazione. La correlazione di rango Spearman è stata utilizzata per rendere il microarray e gli insiemi di dati RNA-seq più comparabili. La migliore correlazione di un profilo di espressione monocellulare con un profilo di espressione di riferimento è stata presa come previsione di fase per quella singola cellula. Man mano che diventano disponibili nuovi dati (ad esempio, l’analisi monocellulare dei punti temporali attraverso l’intero ciclo di sviluppo eritrocitario sincronizzato), gli algoritmi di stadiazione di benchmarking diventeranno fattibili. Non erano disponibili dati RNA-seq di massa per classificare direttamente i maschi e le femmine di P. berghei. Pertanto, abbiamo utilizzato dati RNA-seq in massa(Otto et al., 2014) che includono campioni di gametociti di sesso misto, dopo aver convertito i profili in v3 utilizzando l’annotazione id precedente di PlasmoDB(Aurrecoechea et al., 2017).
Per determinare gruppi distinti di singole cellule in base ai loro modelli di espressione, abbiamo utilizzato lo strumento di clustering SC3(Kiselev, 2016). Abbiamo usato la distanza combinata Euclidea, Pearson e Spearman, più la PCA combinata e la trasformazione spettrale. Per il set di dati P. berghei il k ottimale era 3 (larghezza media della silhouette = 0,99), con quattro che erano quasi altrettanto buoni (larghezza media della silhouette = 0,97). Abbiamo scoperto che il cluster aggiuntivo divideva i parassiti asessuali in trofozoiti e schizonti, mentre entrambi i valori k mantenevano i gametociti maschili e femminili come cluster separati. Tuttavia, c’era ancora un’ampia variazione all’interno di questi cluster, così abbiamo ulteriormente indagato questo escludendo le cellule asessuate e il clustering. Con questo set di dati ridotto siamo stati in grado di ottenere un nuovo, robusto clustering con k = 3 (larghezza = 0,99). In questo caso, si tratta di valori erratici sia del presunto cluster maschile che di quello femminile raggruppati insieme, escludendo il nucleo dei cluster maschile e femminile. I marcatori suggeriscono che sei di queste cellule anomale possedevano entrambi i geni maschili e geni asessuali, mentre una singola cellula possedeva entrambi i geni femminili e geni asessuali. È possibile che queste cellule siano gametociti precoci, schizonti commessi o cellule doppiamente infettate da parassiti sia asessuali che sessuali. Questi sono stati esclusi da ulteriori analisi. La funzione dei marcatori di SC3 (soglia AUROC 0,85, soglia p-valore 0,01) è stata utilizzata sul clustering iniziale, con k = 3, per identificare nuovi marcatori per asessuali, maschi e femmine (file supplementare1).
I dati RNA-seq di massa di Otto et al. (2010) e López-Barragán et al. (2011) sono stati utilizzati per classificare 161 P. falciparum cellule asessuate in fase asessuata. RNA-seq legge da Otto et al. (2010) solo per le biblioteche Illumina 36 bp, sono stati scaricati dall’Archivio Europeo dei Nucleotidi (adesione ERX001048). Sono stati mappati sulla sequenza del genoma di P. falciparum 3D7 utilizzando HISAT2 v2.0.0-beta(Kim et al., 2015) e le letture sono state contate utilizzando HTSeq v0.6.0(Anders et al., 2015). I conteggi delle letture sono stati poi convertiti in FPKM per una successiva analisi. Le letture RNA-seq da López-Barragán et al. (2011) sono state scaricate dall’Archivio Europeo dei Nucleotidi (adesione SRX105940) e mappate sulle sequenze di trascrizione di P. falciparum 3D7 utilizzando Bowtie2 v2.2.9 (-a -X 800; [Langmead eSalzberg, 2012]) ed eXpress v1.5.1 (Roberts ePachter, 2013). I conteggi di lettura risultanti sono stati convertiti in FPKM. Il López-Barragán et al. (2011) è stato utilizzato come previsione di consenso, la previsione comprendeva sei gametociti di stadio II che sono stati rimossi da ulteriori analisi pseudotemporali (n = 155).
I dati di Lasonder et al. (2016) sono stati utilizzati per classificare le cellule di gametociti P. falciparum per sesso. I dati del conteggio grezzo sono stati scaricati dal Gene Expression Omnibus(Barrett et al., 2013) (adesione GSE75795) e convertiti in FPKM. I dati di Young e colleghi(Young et al., 2005), sono stati utilizzati per classificare le cellule di P. falciparum lungo il corso del tempo di sviluppo dei gametociti (giorni 1, 2, 3, 6, 8, 12). Per questo dataset i profili dei ranghi sono stati scaricati da PlasmoDB. I dati di Lasonder(Lasonder et al., 2016) hanno evidenziato cinque cellule maschili, mentre il resto è stato chiamato femmina. I dati di Young(Young et al., 2005) suggerivano che tutte le cellule erano in uno stadio di sviluppo coerente (otto giorni), anche se manca la risoluzione nei punti temporali più rilevanti, tra otto e dodici giorni.
La migliore classificazione di ogni cellula in base a ciascuno dei set di dati di massa usati sopra è elencata nel file supplementare 5.
Valutazione della variazione dell’espressione genica durante la maturazione asessuata
All’interno delle 54 cellule berghei P. identificate come asessuate, 275 geni sono stati trovati per essere variabile utilizzando M3Drop(Andrews e Hemberg, 2016) (ingresso conteggio grezzo, False Discovery Rate <= 0,01). I valori di espressione L-scran per questo sottoinsieme di geni sono stati utilizzati per ordinare le cellule in pseudotempo utilizzando Monocle 2(Trapnell et al., 2014); in particolare le funzioni reduceDimension() e orderCells (num_path = 2) sono state utilizzate per ricavare l’ordine delle cellule. Monocle 2 ha identificato uno stato di singola cella e le celle sono state ordinate in un’unica traiettoria(Figura 2a). Il pacchetto Monocle 2 è stato ulteriormente utilizzato per cluster di geni in pseudotime (k = 3) con la funzione clusterGenes(); il pacchetto Nbclust è stato utilizzato per definire il numero ottimale di cluster. Abbiamo cercato l’arricchimento dei termini dell’Ontologia dei geni all’interno dei tre cluster identificati, utilizzando topGO(Alexa et al., 2006) (riassunti nella Figura 2c).
Per il set di dati P. falciparum 155-cellule, 360 geni sono stati trovati per essere geni variabili con M3Drop (raw count input, False Discovery Rate <= 0.01) (Andrews e Hemberg, 2016), Monocle 2 ha identificato due rami che definiscono tre possibili traiettorie, anche se 2 di questi erano minori (Stati 2 e 5 nella Figura 4-figure supplement 1B). Le cellule ordinate in queste traiettorie minori non sembravano correlate con marcatori biologici noti, come i marcatori di impegno sessuale(ap2-g e gdv-1) e queste cellule sono state rimosse da tutte le ulteriori analisi. L’analisi pseudotempo è stata ripetuta sulla traiettoria principale delle cellule (125 cellule). Il pacchetto Monocle 2 è stato ulteriormente utilizzato per raggruppare i geni in pseudotime (k = 3) con la funzione clusterGenes(); il pacchetto Nbclust è stato utilizzato per definire il numero ottimale di cluster. Abbiamo cercato di arricchire i termini di Gene Ontology all’interno dei tre cluster identificati, utilizzando topGO(Alexa et al., 2006).
Confronto diretto di singole cellule in pseudotempo con dati RNA-seq alla rinfusa
Per determinare se lo stesso insieme di geni ha mostrato modelli diversi attraverso lo sviluppo in esperimenti di bulk e RNA-seq a singola cellula abbiamo fatto confronti diretti tra questi due approcci. Dopo aver ordinato le cellule asessuate di P. berghei per pseudotempo, i geni sono stati ordinati in base al loro picco di espressione basato su valori di espressione lineare (cioè non registrati), lunghezza-normalizzata scran. Per fare questo, i dati dei valori di espressione, ordinati per pseudotempo, sono stati normalizzati, poi trasformati a Fourier, ordinando le trascrizioni secondo la fase della frequenza più prominente. I rapporti segnale/rumore (S/N) sono stati calcolati per ogni segnale trasformato e normalizzati rispetto al valore massimo ottenibile per il set di dati. Le trasformazioni con un S/N normalizzato inferiore a 0,1 sono state escluse dai risultati in quanto prive di prove di periodicità. I dataset di Hoo et al.(Hoo et al., 2016) sono stati trattati allo stesso modo, ma inizialmente ordinati per punto di raccolta temporale piuttosto che per pseudotempo e utilizzando i valori di intensità come descritto sopra. Ciò ha portato a 1141 geni ordinati per i nostri dati a singola cellula e 2612 geni per i dati Hoo(Hoo et al., 2016). C’erano 651 geni condivisi, che sono stati utilizzati per confrontare i due set di dati(Figura 4-figure supplement 2A,B)
Le cellule asessuate di P. falciparum erano ordinate in modo diverso. Abbiamo usato come riferimento i dati del ciclo di sviluppo asessuato di Otto P. falciparum(Otto et al., 2010). Questi dati sono stati elaborati come descritto sopra. Abbiamo usato l’approccio di trasformazione di Fourier descritto sopra, con un rapporto S/N normalizzato di 0,5 per identificare 4517 geni dal set di dati di Otto et al.(Otto et al., 2010). Abbiamo poi identificato 336 geni comuni a questo elenco e l’elenco di 361 geni espressi in modo differenziato identificati attraverso le 155 singole cellule. Questo diverso approccio per P. falciparum è stato adottato perché la finestra di tempo catturata dalle nostre singole cellule era troppo stretta per identificare i geni ciclici usando l’approccio di Fourier (tutti i rapporti S/N normalizzati erano molto bassi, ad esempio <0,05). Abbiamo poi generato delle mappe termiche per il set di dati di massa e per le singole cellule, con i geni ordinati in base al loro tempo di picco nel set di dati di Otto etal. (Otto et al., 2010) in entrambi i casi(Figura 4-figure supplement 2C,D).
I dati Plasmodium Drop-seq (Poranet al., 2017) sono stati caricati con il pacchetto Seurat versione 2.1 (Butlere Satija, 2017), i passaggiQC e la normalizzazione sono stati eseguiti con gli stessi parametri del codice rilasciato(https://github.com/KafsackLab/scRNAseq-Malaria). 8581 celle AP2-DD (on e off) da tre punti temporali (30, 36, 42 ore dopo l’invasione) sono state sottratte e ordinate in pseudotime con Monocole 2(Trapnell et al., 2014).
Analisi AP2
I fattori di trascrizione ortografici P. falciparum e P. berghei ApiAP2 sono stati mappati all’interno dei dati delle singole cellule e ordinati secondo il modello di espressione in entrambe le specie. L’insieme di ApiAP2 espresso in ogni tipo di cellula P. berghei (>10 cellule) è stato isolato e utilizzato per generare una rete di correlazione di co-espressione utilizzando il pacchetto Hmisc (clustfunc – metodo = competere; distfunc – metodo = ‘euclidea’). I bordi con un coefficiente di Pearson superiore a 0,4 sono stati usati per rappresentare la rete con Cytoscape (impostazione del layout diretto a forza prefuse). I dati di essenzialità sono stati ricavati da Modrzynska et al., 2017. È stata calcolata una correlazione media di co-espressione delle 14 TF espresse in asessuato(Figura 4-figure supplement 4B) con ogni gene nei cluster identificati, e le TF erano ciascuna associata al cluster con cui avevano il maggiore coefficiente di correlazione se era maggiore di 0,4.
Correzione per il ciclo cellulare con scLVM
P. berghei trofozoite e schizont singole cellule sono state elaborate utilizzando scLVM v0.99.2(Buettner et al., 2015). I conteggi sono stati normalizzati in base a fattori dimensionali. Il rumore tecnico è stato stimato utilizzando un log fit e 1485 geni variabili sono stati chiamati utilizzando getVariableGenes con parametri di default. Per adattare il fattore del ciclo cellulare latente, abbiamo usato i 2104 geni identificati come differenzialmente espressi in pseudotempo utilizzando Monocle con valore p corretto<=0,01. Eravamo consapevoli dalla nostra analisi PCA che i primi due componenti principali erano probabilmente guidati dal ciclo cellulare, e abbiamo trovato utilizzando l’analisi del fattore di latenza che solo i primi due fattori spiegavano almeno il 5% della variazione dei geni del ciclo cellulare. Pertanto, abbiamo riadattato il modello con due fattori latenti. Abbiamo identificato solo quattro geni che avevano >= 50% della loro varianza attribuibile al rumore biologico dopo la correzione, per esempio la variazione nella loro espressione non è in gran parte guidata dal ciclo cellulare o dal rumore tecnico. Questi sembrano essere guidati da un piccolo numero di potenziali cellule anomale, che non erano state identificate nelle analisi precedenti. Un campione più grande di cellule asessuate di P. berghei sarebbe necessario per affrontare meglio la variazione di espressione genica indipendente dal ciclo cellulare in questa specie.
I dati della fase asessuata di P. falciparum asexual P. a single-cell RNA-seq sono stati filtrati come in precedenza ed elaborati con scLVM come sopra. Abbiamo usato 416 geni identificati come differenzialmente espressi in pseudotempo utilizzando Monocle con corretto p-valore<=0,01. Eravamo consapevoli dalla nostra analisi PCA che i primi due fattori latenti erano probabilmente guidati dal ciclo cellulare, così abbiamo riadattato il modello con due fattori latenti. Abbiamo identificato 56 geni che avevano >= 50% della loro varianza attribuibile al rumore biologico dopo la correzione, ad esempio la variazione nella loro espressione non è in gran parte guidata dal ciclo cellulare o dal rumore tecnico.
Abbiamo usato TopGO(Alexa et al., 2006) per identificare i termini GO arricchiti tra questi geni, con parametri come altrove. Abbiamo cercato correlazioni tra i geni indipendenti dal ciclo delle cellule inserendo un modello lineare misto (LMM) come suggerito nella vignetta scLVM(https://github.com/PMBio/scLVM/blob/master/R/tutorials/scLVM_vignette.Rmd). Non abbiamo trovato correlazioni significative con un coefficiente di correlazione >= 0,5 o <=-0,5.
Abbiamo ipotizzato che queste trascrizioni variabili potrebbero essere degradate più rapidamente di altre, facendole apparire più sporadicamente espresse in mRNA allo stato stazionario. Abbiamo esaminato le distribuzioni dell’emivita per le 56 trascrizioni variabili in P. falciparum. Abbiamo preso le stime dell’emivita per oligoelemento dal materiale supplementare di(Shock et al., 2007). Abbiamo poi usato l’oligo per la mappatura genica di(Bozdech et al., 2003b) e abbiamo convertito gli ids nel nuovo ids del gene PF3D7_ prefisso PF3D7_ usando ortologi one-to-one di PlasmoDB(Aurrecoechea et al., 2017). Per ogni stadio di vita, abbiamo confrontato la distribuzione dei valori di emivita nell’intero campione con quelli dei 56 geni variabili. Abbiamo avuto misurazioni dell’emivita per un numero di trascrizioni compreso tra 34 e 39, a seconda dello stadio. Abbiamo usato il test di Kolmogorov-Smirnov (a due facce) per determinare le differenze tra le distribuzioni(Figura 5B)
Determinazione del punteggio di conservazione del gene
Le sequenze proteiche per 4290 geni ortologi 1:1 in P. falciparum e P. berghei sono state recuperate da PlasmoDB(Aurrecoechea et al., 2017) e allineate con Muscle(Edgar, 2004). All’interno degli allineamenti, è stato calcolato un punteggio di sostituzione per ogni posizione di aminoacidi basato sulla matrice di sostituzione BLOSUM62(Henikoff e Henikoff, 1992). Il punteggio di conservazione per ogni gene corrisponde alla media dei punteggi di tutti gli aminoacidi della proteina che i geni codificano(MalariaGEN Plasmodium falciparum Community Project, 2016).
Determinazione della variabilità dell’espressione genica all’interno di diversi tipi di cellule
Per esaminare la variazione dell’espressione genica all’interno delle fasi della vita, abbiamo usato i set di dati filtrati e considerato le cellule asessuate, maschili e femminili separatamente per ogni specie. Per P. berghei, M3Drop(Andrews e Hemberg, 2016) è stato utilizzato per determinare la variabilità dell’espressione genica tra le cellule con FDR <= 0,01. Abbiamo identificato 115 geni variabili nelle femmine di P. berghei, 73 nei maschi e 275 negli asessuali. In P. falciparum, abbiamo trovato 360 geni variabili in asessuali, e 448 geni variabili nelle femmine. Non siamo stati in grado di analizzare la variabilità nei maschi di P. fal ciparum perché ne abbiamo trovati solo cinque.
Per esaminare le classi funzionali arricchite tra i geni variabili, abbiamo usato topGO con l’algoritmo weight01, la statistica di Fisher, la dimensione del nodo = 5 e il False Discovery Rate >= 0,05 (Alexa et al., 2006). I termini dell’ontologia genica per i geni P. berghei e P. falciparum sono stati estratti dai file EMBL di GeneDB. Le famiglie multigene in P. berghei non hanno termini associati all’ontologia genica (GO) e quindi abbiamo usato test ipergeometrici ad hoc per esaminare il loro arricchimento. Abbiamo scoperto che i geni pir pir pir sono stati arricchiti tra i geni variabili nei maschi (5 su 135, test ipergeometrico, p=0,014), ma non ce n’erano nelle femmine. Abbiamo scoperto che per le femmine di P. falciparum, i termini GO arricchiti includevano la modulazione per simbionte dell’eritrocita dell’ospite ela citoaderenza alla microvasculatura, mediata dalla proteina del simbionte. Questi termini si riferiscono ai 14 geni var che si trovano tra i geni variabili (14 di 60 geni, test ipergeometrico, p=0,0006).
I geni che mostrano una significativa variazione di espressione in ciascuna di queste categorie si trovano nel file supplementare 2 e 3, insieme con le loro analisi di arricchimento funzionale.
Identificazione delle trascrizioni dei var funzionali putativi
È noto che le trascrizioni antisenso sono espresse da un promotore bidirezionale nell’introne di ogni gene var(Guizetti e Scherf, 2013). Il nostro protocollo non conserva informazioni su quale filamento viene trascritto. Pertanto, la scoperta che legge la mappa di uno dei due esoni di un gene var non fornisce la prova che esso sia espresso funzionalmente. Al fine di identificare le trascrizioni dei sensi, abbiamo cercato la mappatura delle letture sul singolo introne di ogni gene var. Queste letture, che includono entrambi gli esoni, devono provenire da trascrizioni mRNA e quindi non da trascrizioni antisenso che iniziano all’interno dell’introne. Inizialmente abbiamo identificato le letture dalle mappature HISAT2 che si sono sovrapposte ai geni var annotati usando gli strumenti da letto che si intersecano (Quinlan e Hall, 2010). Dal file BAM risultante abbiamo selezionato quelle letture che includevano una N nella stringa CIGAR, che indicava una lettura suddivisa. Abbiamo poi identificato il gene var che ogni lettura si sovrapponeva e se era diviso esattamente sopra l’introne. Abbiamo chiamato espressione per un gene var dove c’erano almeno due letture mappate sopra l’introne.
Elaborazione di dati di gametociti RNA-seq in massa per esplorare l’espressione della famiglia multigene
I file SRA per Lasonder et al. (2016) sono stati scaricati da GEO (SRR2981459, SRR2981460, SRR2981461). Questi sono stati convertiti in formato FASTQ utilizzando il fastq-dump (v 2.3.5). Le letture sono state mappate sul genoma di riferimento P. falciparum 3D7 utilizzando HISAT2 (–rna-strandness R –max-intronlen 5000 p 12[Kim et al., 2015]). Le letture sono state confrontate con le caratteristiche utilizzando il conteggio htseq (-f bam -r pos -s reverse -t CDS;[Anders et al., 2015]).
Abbiamo voluto guardare più in generale a come l’espressione della famiglia multigene e la variazione tra le cellule differiscono tra maschi e femmine e tra le specie. Per determinare i geni DE tra maschi e femmine nei dati di Lasonder P. falciparum bulk RNA-seq, abbiamo usato EdgeR(Robinson et al., 2010). Questo strumento non è tuttavia appropriato per i dati monocellulari in quanto non tiene conto dei drop out. Pertanto, per i dati monocellulari di P. berghei abbiamo raccolto i dati monocellulari per formare tre repliche di trascrizioni pseudo-bulk per i maschi e tre per le femmine per ridurre i drop out e simulare i dati sfusi. Questo è stato fatto sommando ogni profilo di espressione genica monocellulare per replicare 1, 2 o 3 fino a quando non avevamo esaurito le singole cellule. Abbiamo continuato a cercare i geni che erano più altamente espressi nei maschi o nelle femmine usando EdgeR e che provenivano da famiglie multigene.
Per i dati di massa femminili, abbiamo visto l’evidenza di una trascrizione di senso per un insieme di geni var simile a quello delle singole cellule femminili. Quelli che non abbiamo osservato nei dati delle singole cellule erano anche tipi upsC1. Non abbiamo visto alcuna prova di espressione del senso dei geni var nelle nostre cinque cellule singole maschili. Tuttavia, nel Lasonder bulk RNA-seq maschio campione MG2, il gene var var var2csa (PF3D7_1200600) aveva 13 letture sovrapposte al PF3D7_1200600 var2csa introne.
Codice e disponibilità dei dati
I codici Perl, R e C+++ per le varie analisi sono disponibili suhttps://github.com/adamjamesreid/Plasmodium-single-cell-RNA-seq (Reid, 2018; copia archiviata suhttps://github.com/elifesciences-publications/Plasmodium-single-cell-RNA-seq). Le letture RNA-seq monocellulari sono disponibili presso l’Archivio Europeo dei Nucleotidi (adesione ERP021229) e ArrayExpress (adesione E-ERAD-611). I conteggi delle letture grezze e i metadati, comprese le classificazioni per ogni cella, sono presentati anche nei file supplementari 5 e 6.
References
- Alexa A, Rahnenführer J, Lengauer T. Improved scoring of functional groups from gene expression data by decorrelating GO graph structure. Bioinformatics. 2006; 22:1600-1607. DOI | PubMed
- Amit-Avraham I, Pozner G, Eshar S, Fastman Y, Kolevzon N, Yavin E, Dzikowski R. Antisense long noncoding RNAs regulate var gene activation in the malaria parasite Plasmodium falciparum. PNAS. 2015; 112:E982-E991. DOI | PubMed
- Anders S, Pyl PT, Huber W. HTSeq–a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015; 31:166-169. DOI | PubMed
- Andrews TS, Hemberg M. Modelling dropouts allows for unbiased identification of marker genes in scRNASeq experiments. biorxiv. 2016. DOI
- Aurrecoechea C, Barreto A, Basenko EY, Brestelli J, Brunk BP, Cade S, Crouch K, Doherty R, Falke D, Fischer S, Gajria B, Harb OS, Heiges M, Hertz-Fowler C, Hu S, Iodice J, Kissinger JC, Lawrence C, Li W, Pinney DF, Pulman JA, Roos DS, Shanmugasundram A, Silva-Franco F, Steinbiss S, Stoeckert CJ, Spruill D, Wang H, Warrenfeltz S, Zheng J. EuPathDB: the eukaryotic pathogen genomics database resource. Nucleic Acids Research. 2017; 45:D581-D591. DOI | PubMed
- Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, Robertson CL, Serova N, Davis S, Soboleva A. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Research. 2013; 41:D991-D995. DOI | PubMed
- Bozdech Z, Llinás M, Pulliam BL, Wong ED, Zhu J, DeRisi JL. The transcriptome of the intraerythrocytic developmental cycle of Plasmodium falciparum. PLoS Biology. 2003a; 1DOI | PubMed
- Bozdech Z, Zhu J, Joachimiak MP, Cohen FE, Pulliam B, DeRisi JL. Expression profiling of the schizont and trophozoite stages of Plasmodium falciparum with a long-oligonucleotide microarray. Genome Biology. 2003b; 4DOI | PubMed
- Buettner F, Natarajan KN, Casale FP, Proserpio V, Scialdone A, Theis FJ, Teichmann SA, Marioni JC, Stegle O. Computational analysis of cell-to-cell heterogeneity in single-cell RNA-sequencing data reveals hidden subpopulations of cells. Nature Biotechnology. 2015; 33:155-160. DOI | PubMed
- Bushell E, Gomes AR, Sanderson T, Anar B, Girling G, Herd C, Metcalf T, Modrzynska K, Schwach F, Martin RE, Mather MW, McFadden GI, Parts L, Rutledge GG, Vaidya AB, Wengelnik K, Rayner JC, Billker O. Functional profiling of a plasmodium genome reveals an abundance of essential genes. Cell. 2017; 170:260-272. DOI | PubMed
- Butler A, Satija R. Integrated analysis of single cell transcriptomic data across conditions, technologies, and species. bioRxiv. 2017. DOI
- Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Research. 2004; 32:1792-1797. DOI | PubMed
- Fivelman QL, McRobert L, Sharp S, Taylor CJ, Saeed M, Swales CA, Sutherland CJ, Baker DA. Improved synchronous production of Plasmodium falciparum gametocytes in vitro. Molecular and Biochemical Parasitology. 2007; 154:119-123. DOI | PubMed
- Florens L, Washburn MP, Raine JD, Anthony RM, Grainger M, Haynes JD, Moch JK, Muster N, Sacci JB, Tabb DL, Witney AA, Wolters D, Wu Y, Gardner MJ, Holder AA, Sinden RE, Yates JR, Carucci DJ. A proteomic view of the Plasmodium falciparum life cycle. Nature. 2002; 419:520-526. DOI | PubMed
- Fougère A, Jackson AP, Bechtsi DP, Braks JA, Annoura T, Fonager J, Spaccapelo R, Ramesar J, Chevalley-Maurel S, Klop O, van der Laan AM, Tanke HJ, Kocken CH, Pasini EM, Khan SM, Böhme U, van Ooij C, Otto TD, Janse CJ, Franke-Fayard B. Variant exported blood-stage proteins encoded by plasmodium multigene families are expressed in liver stages where they are exported into the parasitophorous vacuole. PLOS Pathogens. 2016; 12DOI | PubMed
- Gerald N, Mahajan B, Kumar S. Mitosis in the human malaria parasite Plasmodium falciparum. Eukaryotic Cell. 2011; 10:474-482. DOI | PubMed
- Gomes AR, Bushell E, Schwach F, Girling G, Anar B, Quail MA, Herd C, Pfander C, Modrzynska K, Rayner JC, Billker O. A genome-scale vector resource enables high-throughput reverse genetic screening in a malaria parasite. Cell Host & Microbe. 2015; 17:404-413. DOI | PubMed
- Grün D, Lyubimova A, Kester L, Wiebrands K, Basak O, Sasaki N, Clevers H, van Oudenaarden A. Single-cell messenger RNA sequencing reveals rare intestinal cell types. Nature. 2015; 525:251-255. DOI | PubMed
- Guizetti J, Scherf A. Silence, activate, poise and switch! Mechanisms of antigenic variation in Plasmodium falciparum. Cellular Microbiology. 2013; 15:718-726. DOI | PubMed
- Haber AL, Biton M, Rogel N, Herbst RH, Shekhar K, Smillie C, Burgin G, Delorey TM, Howitt MR, Katz Y, Tirosh I, Beyaz S, Dionne D, Zhang M, Raychowdhury R, Garrett WS, Rozenblatt-Rosen O, Shi HN, Yilmaz O, Xavier RJ, Regev A. A single-cell survey of the small intestinal epithelium. Nature. 2017; 551:333-339. DOI | PubMed
- Hall N, Karras M, Raine JD, Carlton JM, Kooij TW, Berriman M, Florens L, Janssen CS, Pain A, Christophides GK, James K, Rutherford K, Harris B, Harris D, Churcher C, Quail MA, Ormond D, Doggett J, Trueman HE, Mendoza J, Bidwell SL, Rajandream MA, Carucci DJ, Yates JR, Kafatos FC, Janse CJ, Barrell B, Turner CM, Waters AP, Sinden RE. A comprehensive survey of the Plasmodium life cycle by genomic, transcriptomic, and proteomic analyses. Science. 2005; 307:82-86. DOI | PubMed
- Henikoff S, Henikoff JG. Amino acid substitution matrices from protein blocks. PNAS. 1992; 89:10915-10919. DOI | PubMed
- Hoo R, Zhu L, Amaladoss A, Mok S, Natalang O, Lapp SA, Hu G, Liew K, Galinski MR, Bozdech Z, Preiser PR. Integrated analysis of the Plasmodium species transcriptome. EBioMedicine. 2016; 7:255-266. DOI | PubMed
- Hu G, Cabrera A, Kono M, Mok S, Chaal BK, Haase S, Engelberg K, Cheemadan S, Spielmann T, Preiser PR, Gilberger TW, Bozdech Z. Transcriptional profiling of growth perturbations of the human malaria parasite Plasmodium falciparum. Nature Biotechnology. 2010; 28:91-98. DOI | PubMed
- Kafsack BF, Rovira-Graells N, Clark TG, Bancells C, Crowley VM, Campino SG, Williams AE, Drought LG, Kwiatkowski DP, Baker DA, Cortés A, Llinás M. A transcriptional switch underlies commitment to sexual development in malaria parasites. Nature. 2014; 507:248-252. DOI | PubMed
- Khan SM, Kroeze H, Franke-Fayard B, Janse CJ. Standardization in generating and reporting genetically modified rodent malaria parasites: the RMgmDB database. Methods in Molecular Biology. 2013; 923:139-150. DOI | PubMed
- Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nature Methods. 2015; 12:357-360. DOI | PubMed
- Kiselev VY, Kirschner K, Schaub MT, Andrews T, Yiu A, Chandra T, Natarajan KN, Reik W, Barahona M, Green AR, Hemberg M. SC3: consensus clustering of single-cell RNA-seq data. Nature Methods. 2017; 14:483-486. DOI | PubMed
- Kiselev VY. SC3 – consensus clustering of single-cell RNA-Seq data. bioRxiv. 2016. DOI
- Kowalczyk MS, Tirosh I, Heckl D, Rao TN, Dixit A, Haas BJ, Schneider RK, Wagers AJ, Ebert BL, Regev A. Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Research. 2015; 25:1860-1872. DOI | PubMed
- Kutner S, Breuer WV, Ginsburg H, Aley SB, Cabantchik ZI. Characterization of permeation pathways in the plasma membrane of human erythrocytes infected with early stages of Plasmodium falciparum: association with parasite development. Journal of Cellular Physiology. 1985; 125:521-527. DOI | PubMed
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012; 9:357-359. DOI | PubMed
- Lasonder E, Rijpma SR, van Schaijk BC, Hoeijmakers WA, Kensche PR, Gresnigt MS, Italiaander A, Vos MW, Woestenenk R, Bousema T, Mair GR, Khan SM, Janse CJ, Bártfai R, Sauerwein RW. Integrated transcriptomic and proteomic analyses of P. falciparum gametocytes: molecular insight into sex-specific processes and translational repression. Nucleic Acids Research. 2016; 44:6087-6101. DOI | PubMed
- Lawniczak MK, Eckhoff PA. A computational lens for sexual-stage transmission, reproduction, fitness and kinetics in Plasmodium falciparum. Malaria Journal. 2016; 15DOI | PubMed
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078-2079. DOI | PubMed
- Liu Y, Tewari R, Ning J, Blagborough AM, Garbom S, Pei J, Grishin NV, Steele RE, Sinden RE, Snell WJ, Billker O. The conserved plant sterility gene HAP2 functions after attachment of fusogenic membranes in Chlamydomonas and Plasmodium gametes. Genes & Development. 2008; 22:1051-1068. DOI | PubMed
- Llinás M, Bozdech Z, Wong ED, Adai AT, DeRisi JL. Comparative whole genome transcriptome analysis of three Plasmodium falciparum strains. Nucleic Acids Research. 2006; 34:1166-1173. DOI | PubMed
- Logan-Klumpler FJ, De Silva N, Boehme U, Rogers MB, Velarde G, McQuillan JA, Carver T, Aslett M, Olsen C, Subramanian S, Phan I, Farris C, Mitra S, Ramasamy G, Wang H, Tivey A, Jackson A, Houston R, Parkhill J, Holden M, Harb OS, Brunk BP, Myler PJ, Roos D, Carrington M, Smith DF, Hertz-Fowler C, Berriman M. GeneDB–an annotation database for pathogens. Nucleic Acids Research. 2012; 40:D98-D108. DOI | PubMed
- Lu XM, Batugedara G, Lee M, Prudhomme J, Bunnik EM, Le Roch KG. Nascent RNA sequencing reveals mechanisms of gene regulation in the human malaria parasite Plasmodium falciparum. Nucleic Acids Research. 2017; 45:7825-7840. DOI | PubMed
- Lun AT, Bach K, Marioni JC. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biology. 2016; 17DOI | PubMed
- López-Barragán MJ, Lemieux J, Quiñones M, Williamson KC, Molina-Cruz A, Cui K, Barillas-Mury C, Zhao K, Su XZ. Directional gene expression and antisense transcripts in sexual and asexual stages of Plasmodium falciparum. BMC Genomics. 2011; 12DOI | PubMed
- Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, Tirosh I, Bialas AR, Kamitaki N, Martersteck EM, Trombetta JJ, Weitz DA, Sanes JR, Shalek AK, Regev A, McCarroll SA. Highly parallel genome-wide expression profiling of individual cells using nanoliter droplets. Cell. 2015; 161:1202-1214. DOI | PubMed
- Mair GR, Braks JA, Garver LS, Wiegant JC, Hall N, Dirks RW, Khan SM, Dimopoulos G, Janse CJ, Waters AP. Regulation of sexual development of Plasmodium by translational repression. Science. 2006; 313:667-669. DOI | PubMed
- MalariaGEN Plasmodium falciparum Community Project. Genomic epidemiology of artemisinin resistant malaria. eLife. 2016; 5DOI | PubMed
- Mancio-Silva L, Slavic K, Grilo Ruivo MT, Grosso AR, Modrzynska KK, Vera IM, Sales-Dias J, Gomes AR, MacPherson CR, Crozet P, Adamo M, Baena-Gonzalez E, Tewari R, Llinás M, Billker O, Mota MM. Nutrient sensing modulates malaria parasite virulence. Nature. 2017; 547:213-216. DOI | PubMed
- McCarthy DJ, Campbell KR, Lun ATL, Wills QF. scater: pre-processing, quality control, normalisation and visualisation of single-cell RNA-seq data in R. bioRxiv. 2016. DOI
- Modrzynska K, Pfander C, Chappell L, Yu L, Suarez C, Dundas K, Gomes AR, Goulding D, Rayner JC, Choudhary J, Billker O. A knockout screen of ApiAP2 genes reveals networks of interacting transcriptional regulators controlling the Plasmodium life cycle. Cell Host & Microbe. 2017; 21:11-22. DOI | PubMed
- Mohammed H, Hernando-Herraez I, Savino A, Scialdone A, Macaulay I, Mulas C, Chandra T, Voet T, Dean W, Nichols J, Marioni JC, Reik W. Single-cell landscape of transcriptional heterogeneity and cell fate decisions during mouse early gastrulation. Cell Reports. 2017; 20:1215-1228. DOI | PubMed
- Mok BW, Ribacke U, Rasti N, Kironde F, Chen Q, Nilsson P, Wahlgren M. Default Pathway of var2csa switching and translational repression in Plasmodium falciparum. PLoS One. 2008; 3DOI | PubMed
- Moss DK, Remarque EJ, Faber BW, Cavanagh DR, Arnot DE, Thomas AW, Holder AA. Plasmodium falciparum 19-kilodalton merozoite surface protein 1 (MSP1)-specific antibodies that interfere with parasite growth in vitro can inhibit MSP1 processing, merozoite invasion, and intracellular parasite development. Infection and Immunity. 2012; 80:1280-1287. DOI | PubMed
- Nam DK, Lee S, Zhou G, Cao X, Wang C, Clark T, Chen J, Rowley JD, Wang SM. Oligo(dT) primer generates a high frequency of truncated cDNAs through internal poly(A) priming during reverse transcription. PNAS. 2002; 99:6152-6156. DOI | PubMed
- Otto TD, Böhme U, Jackson AP, Hunt M, Franke-Fayard B, Hoeijmakers WA, Religa AA, Robertson L, Sanders M, Ogun SA, Cunningham D, Erhart A, Billker O, Khan SM, Stunnenberg HG, Langhorne J, Holder AA, Waters AP, Newbold CI, Pain A, Berriman M, Janse CJ. A comprehensive evaluation of rodent malaria parasite genomes and gene expression. BMC Biology. 2014; 12DOI | PubMed
- Otto TD, Wilinski D, Assefa S, Keane TM, Sarry LR, Böhme U, Lemieux J, Barrell B, Pain A, Berriman M, Newbold C, Llinás M. New insights into the blood-stage transcriptome of Plasmodium falciparum using RNA-Seq. Molecular Microbiology. 2010; 76:12-24. DOI | PubMed
- Parkinson H, Sarkans U, Shojatalab M, Abeygunawardena N, Contrino S, Coulson R, Farne A, Lara GG, Holloway E, Kapushesky M, Lilja P, Mukherjee G, Oezcimen A, Rayner T, Rocca-Serra P, Sharma A, Sansone S, Brazma A. ArrayExpress–a public repository for microarray gene expression data at the EBI. Nucleic Acids Research. 2005; 33:D553-D555. DOI | PubMed
- Pertea M, Kim D, Pertea GM, Leek JT, Salzberg SL. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nature Protocols. 2016; 11:1650-1667. DOI | PubMed
- Picelli S, Björklund ÅK, Faridani OR, Sagasser S, Winberg G, Sandberg R. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nature Methods. 2013; 10:1096-1098. DOI | PubMed
- Pichon G, Awono-Ambene HP, Robert V. High heterogeneity in the number of Plasmodium falciparum gametocytes in the bloodmeal of mosquitoes fed on the same host. Parasitology. 2000; 121 ( Pt 2:115-120. DOI | PubMed
- Poran A, Nötzel C, Aly O, Mencia-Trinchant N, Harris CT, Guzman ML, Hassane DC, Elemento O, Kafsack BFC. Single-cell RNA sequencing reveals a signature of sexual commitment in malaria parasites. Nature. 2017; 551:95-99. DOI | PubMed
- Preiser PR, Jarra W, Capiod T, Snounou G. A rhoptry-protein-associated mechanism of clonal phenotypic variation in rodent malaria. Nature. 1999; 398:618-622. DOI | PubMed
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841-842. DOI | PubMed
- Reid A. GitHub. 2018. Publisher Full Text
- Reid AJ. Large, rapidly evolving gene families are at the forefront of host-parasite interactions in Apicomplexa. Parasitology. 2015; 142:S57-S70. DOI | PubMed
- Ribaut C, Berry A, Chevalley S, Reybier K, Morlais I, Parzy D, Nepveu F, Benoit-Vical F, Valentin A. Concentration and purification by magnetic separation of the erythrocytic stages of all human Plasmodium species. Malaria Journal. 2008; 7DOI | PubMed
- Roberts A, Pachter L. Streaming fragment assignment for real-time analysis of sequencing experiments. Nature Methods. 2013; 10:71-73. DOI | PubMed
- Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139-140. DOI | PubMed
- Rovira-Graells N, Gupta AP, Planet E, Crowley VM, Mok S, Ribas de Pouplana L, Preiser PR, Bozdech Z, Cortés A. Transcriptional variation in the malaria parasite Plasmodium falciparum. Genome Research. 2012; 22:925-938. DOI | PubMed
- Scherf A, Lopez-Rubio JJ, Riviere L. Antigenic variation in Plasmodium falciparum. Annual Review of Microbiology. 2008; 62:445-470. DOI | PubMed
- Shock JL, Fischer KF, DeRisi JL. Whole-genome analysis of mRNA decay in Plasmodium falciparum reveals a global lengthening of mRNA half-life during the intra-erythrocytic development cycle. Genome Biology. 2007; 8DOI | PubMed
- Sinha A, Hughes KR, Modrzynska KK, Otto TD, Pfander C, Dickens NJ, Religa AA, Bushell E, Graham AL, Cameron R, Kafsack BFC, Williams AE, Llinas M, Berriman M, Billker O, Waters AP. A cascade of DNA-binding proteins for sexual commitment and development in Plasmodium. Nature. 2014; 507:253-257. DOI | PubMed
- Spellman PT, Sherlock G, Zhang MQ, Iyer VR, Anders K, Eisen MB, Brown PO, Botstein D, Futcher B. Comprehensive identification of cell cycle-regulated genes of the yeast Saccharomyces cerevisiae by microarray hybridization. Molecular Biology of the Cell. 1998; 9:3273-3297. DOI | PubMed
- Svensson V, Natarajan KN, Ly LH, Miragaia RJ, Labalette C, Macaulay IC, Cvejic A, Teichmann SA. Power analysis of single-cell RNA-sequencing experiments. Nature Methods. 2017; 14:381-387. DOI | PubMed
- Talman AM, Blagborough AM, Sinden RE. A Plasmodium falciparum strain expressing GFP throughout the parasite’s life-cycle. PLoS One. 2010; 5DOI | PubMed
- Talman AM, Prieto JH, Marques S, Ubaida-Mohien C, Lawniczak M, Wass MN, Xu T, Frank R, Ecker A, Stanway RS, Krishna S, Sternberg MJ, Christophides GK, Graham DR, Dinglasan RR, Yates JR, Sinden RE. Proteomic analysis of the Plasmodium male gamete reveals the key role for glycolysis in flagellar motility. Malaria Journal. 2014; 13DOI | PubMed
- Tembo DL, Nyoni B, Murikoli RV, Mukaka M, Milner DA, Berriman M, Rogerson SJ, Taylor TE, Molyneux ME, Mandala WL, Craig AG, Montgomery J. Differential PfEMP1 expression is associated with cerebral malaria pathology. PLoS Pathogens. 2014; 10DOI | PubMed
- Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, Lennon NJ, Livak KJ, Mikkelsen TS, Rinn JL. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nature Biotechnology. 2014; 32:381-386. DOI | PubMed
- Treutlein B, Brownfield DG, Wu AR, Neff NF, Mantalas GL, Espinoza FH, Desai TJ, Krasnow MA, Quake SR. Reconstructing lineage hierarchies of the distal lung epithelium using single-cell RNA-seq. Nature. 2014; 509:371-375. DOI | PubMed
- Wu L, Zhang X, Zhao Z, Wang L, Li B, Li G, Dean M, Yu Q, Wang Y, Lin X, Rao W, Mei Z, Li Y, Jiang R, Yang H, Li F, Xie G, Xu L, Wu K, Zhang J, Chen J, Wang T, Kristiansen K, Zhang X, Li Y, Yang H, Wang J, Hou Y, Xu X. Full-length single-cell RNA-seq applied to a viral human cancer: applications to HPV expression and splicing analysis in HeLa S3 cells. GigaScience. 2015; 4DOI | PubMed
- Young JA, Fivelman QL, Blair PL, de la Vega P, Le Roch KG, Zhou Y, Carucci DJ, Baker DA, Winzeler EA. The Plasmodium falciparum sexual development transcriptome: a microarray analysis using ontology-based pattern identification. Molecular and Biochemical Parasitology. 2005; 143:67-79. DOI | PubMed
- Ziegenhain C, Vieth B, Parekh S, Reinius B, Guillaumet-Adkins A, Smets M, Leonhardt H, Heyn H, Hellmann I, Enard W. Comparative Analysis of Single-Cell RNA Sequencing Methods. Molecular Cell. 2017; 65:631-643. DOI | PubMed
Fonte
Reid AJ, Talman AM, Bennett HM, Gomes AR, Sanders MJ, et al. () Single-cell RNA-seq reveals hidden transcriptional variation in malaria parasites. eLife 7e33105. https://doi.org/10.7554/eLife.33105




