
Abstract
Introduzione
La nostra comprensione dei processi biologici a livello cellulare è stata sostenuta dalle discipline tradizionali della genetica, biochimica e biologia molecolare. Nell’ultimo decennio, l’attenzione si è spostata verso studi su larga scala di genomi e trascrittomi, questi ultimi come surrogati dei proteomi cellulari. Questi, combinati con studi di interazione proteica ad alto rendimento, hanno portato alla nuova disciplina della biologia dei sistemi, dove le proteine sono considerate nel contesto di reti di percorsi biochimici e di sviluppo. Nella visione di rete del comportamento delle proteine, ogni proteina o isoforma proteica può partecipare a molte interazioni proteine-proteine, ma mancano gli strumenti disponibili che permettono ai ricercatori di testare le ipotesi nel contesto biologico. Tecnologie come l’RNAi e CRISPR-Cas9 che abbassano o abbattono l’espressione delle proteine sono strumenti importanti, ma possono offuscare l’interpretazione di una relazione proposta tra un dato prodotto genico o dominio proteico e il fenotipo cellulare osservato. La prossima generazione di strumenti dovrebbe avere la capacità di bloccare sistematicamente le interazioni proteina-proteina senza influenzare i livelli di espressione.
Gli strumenti comunemente usati per studiare l’espressione e la funzione delle proteine includono gli anticorpi. Gli anticorpi hanno dimostrato di essere strumenti eccellenti in molte applicazioni, ma vi sono crescenti preoccupazioni circa la difficoltà di reperire anticorpi convalidati e rinnovabili(Bordeaux et al., 2010; Bradbury e Plückthun, 2015; Taussig et al., 2007). Mentre ci sono oltre 500.000 anticorpi diversi sul mercato, è stato riportato che fino al 75% non è stato convalidato, mostra un basso livello di convalida o semplicemente non si comporta in modo adeguato in alcune applicazioni(Berglund et al., 2008). Inoltre, l’uso di anticorpi per bloccare la funzione proteica all’interno delle cellule viventi viene comunemente eseguito, ma è limitato a causa dell’ambiente riducente delle cellule(Marschall et al., 2015). Anche se i frammenti di anticorpi, chiamati intracorpi(Marschall et al., 2015) o cromocorpi(Rothbauer et al., 2006) possono essere espressi nel citoplasma delle cellule dei mammiferi, solo una frazione del repertorio delle IgG è correttamente piegata nell’ambiente riducente del citoplasma(Biocca et al., 1995; Wörn e Plückthun, 2001), diminuendo la loro efficacia nelle applicazioni funzionali (Marschall et al.,2015).
Sono stati costituiti diversi consorzi(Stoevesandt e Taussig, 2012) per la generazione e la validazione degli anticorpi e dei loro derivati(Berglund et al., 2008; Renewable Protein Binder Working Group et al., 2011; Nilsson et al., 2005; Uhlén et al., 2005). Questi consorzi hanno generato anticorpi policlonali e monoclonali contro le proteine e i domini proteici. Pur dimostrandosi un successo, nel fornire un ampio catalogo di anticorpi convalidati, tali sforzi hanno richiesto grandi gruppi multidisciplinari in tutta Europa e negli Stati Uniti(Uhlén et al., 2015). Tuttavia, la capacità di generare rapidamente e a costi contenuti reagenti leganti rinnovabili per applicazioni come lo studio della funzione proteica sia in vitro che in vivo e per progetti proteomici rappresenterebbe un importante progresso. I reagenti leganti rinnovabili in questo contesto si riferiscono a reagenti che vengono prodotti ricombinatamente a partire da una sequenza nota.
Lo sviluppo di proteine leganti alternative ha fornito l’opportunità per tali progressi(Škrlec et al., 2015; Vazquez-Lombardi et al., 2015). Questi includono reagenti come DARPins(Binz et al., 2003), Monobodies(Koide et al., 1998), e Affibodies(Nord et al., 1995) e molti altri (per una recente revisione si veda[Škrlec et al., 2015]). Negli ultimi due decenni questi reagenti si sono dimostrati strumenti utili in molte applicazioni anticorpali, tra cui il rilevamento di proteine per la diagnostica(Theurillat et al., 2010), per lo studio della funzione proteica(Kummer et al., 2012), per il targeting intracellulare della funzione proteica(Spencer-Smith et al., 2017; Wojcik et al., 2010) e come chaperon di cristallizzazione(Sennhauser e Grütter, 2008). Nel 2010, l’Università di Leeds e il NHS Teaching Hospital Trust di Leeds hanno istituito il BioScreening Technology Group (BSTG), per consentire la rapida identificazione di proteine leganti alternative contro bersagli biologici, in particolare quelli di interesse clinico. Ora riportiamo alcuni dei risultati degli oltre 350 screening effettuati con successo dal BSTG fino ad oggi, e suggeriamo che l’accesso a questa e ad altre strutture simili (ad esempio la struttura di selezione dei leganti ad alta produttività dell’Università di Zurigo) dovrebbe fornire gli strumenti necessari per integrare gli anticorpi nella dissezione delle funzioni biologiche delle singole proteine e delle isoforme proteiche. Il nostro lavoro è sostenuto dallo sviluppo di una nuova impalcatura proteica ingegnerizzata per la visualizzazione dei peptidi(Figura 1). L’impalcatura Adhiron è una proteina sintetica originariamente basata su una sequenza di consenso cistatina e mostra una notevole stabilità termica (Tm = 101°C) (Tiede et al.,2014). La sua struttura è correlata a un’impalcatura precedentemente riportata, costruita con stefano umano A(Stadler et al., 2011). Le proteine leganti derivate da queste due impalcature sono ora indicate collettivamente come proteine Affimer, e usiamo questo termine successivamente.10.7554/eLife.24903.003Figure 1.Ribbon diagrams of three crystal structures for Affimer (Adhiron) reagents.(A) X-ray crystal structure of Affimer scaffold (PDB ID no. 4N6T) a 1,75 A resolution. Gli amminoacidi dei loop che collegano i quattro fogli beta antiparalleli sono evidenziati in rosa.(B) Struttura cristallina di un Affimer contro p300 (PDB ID n. 5A0O)(C) Struttura cristallina di un Affimer isolato contro le proteine umane SUMO (PDB ID n. 5ELJ). Le regioni variabili in B e C sono indicate in rosa.DOI:http://dx.doi.org/10.7554/eLife.24903.003
In precedenza abbiamo dimostrato l’uso di Affimers in una serie di saggi, compresi quelli di tipo immunitario (affinità), nei biosensori e abbiamo testato la loro capacità di essere espressi in cellule di mammifero per manipolare la segnalazione cellulare(Tiede et al., 2014; Kyle et al., 2015; Rawlings et al., 2015; Stadler et al., 2014; Sharma et al.,2016). Qui abbiamo vagliato la nostra consolidata libreria di fago Affimer(Tiede et al., 2014) rispetto a un’ampia gamma di obiettivi, compresi i membri della famiglia delle proteine omologhe, per isolare reagenti leganti altamente specifici e rinnovabili che possono essere utilizzati sia in vitro che in vivo. Per un’ampia applicabilità e per rimuovere il collo di bottiglia nella produzione di proteine target, abbiamo anche testato la nostra capacità di generare reagenti contro piccole quantità di proteine target provenienti da fonti commerciali. Dimostriamo la generazione di Affimers contro varie molecole target, tra cui una piccola molecola organica, e ne riportiamo l’uso in una serie di saggi biochimici e di biologia cellulare ampiamente utilizzati.
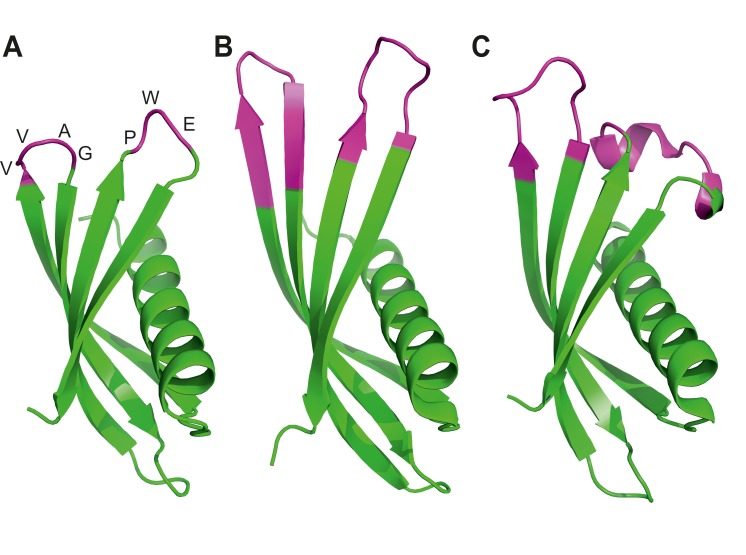
Figura 1.Diagrammi a nastro di tre strutture cristalline per i reagenti di Affimer (Adhiron).(A) Struttura a cristalli a raggi X dell’impalcatura di Affimer (PDB ID n. 4N6T) con risoluzione di 1,75 A. Gli amminoacidi dei loop che collegano i quattro fogli beta antiparalleli sono evidenziati in rosa.(B) Struttura cristallina di un Affimer contro p300 (PDB ID n. 5A0O)(C) Struttura cristallina di un Affimer isolato contro le proteine umane SUMO (PDB ID n. 5ELJ). Le regioni variabili in B e C sono indicate in rosa.DOI:
http://dx.doi.org/10.7554/eLife.24903.003
Risultati
Dissezionare i percorsi di segnalazione intracellulare
Una sfida nella biologia cellulare è quella di sviluppare strumenti altamente specifici per rilevare e modulare la funzione di un membro di una famiglia di proteine strutturalmente e funzionalmente simili. Reagenti biologici che mirano specificamente ad una singola proteina, o un sottoinsieme di una famiglia di proteine introdurrebbe una maggiore selettività agli studi in vivo.
Per dimostrare questa funzionalità, abbiamo isolato i leganti Affimer a vari domini Src-Homology 2 (SH2). SH2 domini sono brevi (~ 100 aminoacidi) domini proteici che si legano specificamente ai motivi contenenti fosfotirosina nelle proteine del partner, ma non alle isoforme de-fosforilate. Sono anche stati recentemente trovati per legarsi ai lipidi di segnalazione(Park et al., 2016) e sono coinvolti nella mediazione di molteplici aspetti della trasduzione del segnale cellulare e della comunicazione. Il genoma umano codifica circa 120 domini SH2 trovati in 111 proteine(Liu et al., 2011). La capacità di rilevare e disattivare specificamente ogni dominio SH2 è un passo avanti nella nostra comprensione di questi percorsi; l’uso di siRNA, ad esempio, può essere utilizzato per rimuovere un’intera proteina, come la proteina chinasi Syk o Zap70, da una cellula, ma non permetterà di determinare quale dei due domini SH2, trasportati da ciascuna di queste chinasi, mediano quale evento(i) di segnalazione. La capacità di sezionare questi eventi di segnalazione con reagenti leganti altamente specifici ha già identificato una nuova funzione biologica utilizzando i mono-corpi(Wojcik et al., 2010; Grebien et al., 2011; Sha et al., 2013). Abbiamo affrontato la questione se i leganti alternativi possono mirare a uno specifico dominio SH2 selezionando Affimers contro una serie di domini SH2.
Abbiamo scelto cinque domini SH2, alcuni dei quali erano stati precedentemente presi di mira utilizzando anticorpi(Renewable Protein Binder Working Group et al., 2011; Pershad et al., 2010). In questi rapporti precedenti, sono stati identificati reagenti leganti altamente specifici contro la proteina ricombinante, ma solo un numero limitato ha funzionato in modo efficiente nelle applicazioni testate(Renewable Protein Binder Working Group et al., 2011). In precedenza abbiamo dimostrato la capacità di isolare i reagenti contro il dominio Grb2 SH2(Tiede et al., 2014). Nel presente studio abbiamo adottato una diversa strategia di target capture producendo ogni dominio SH2 con un peptide accettore di biotina N-terminale per facilitare la semplice cattura diretta dal lisato cellulare e la presentazione per lo screening del display dei fagi. Ogni target è stato controllato per una biotinilazione efficiente da Western blot(Figura 2-figure supplement 1) ed è degno di nota che questa biotinilazione è stata ottenuta in un ceppo derivato da BL21(DE3) senza la necessità di un’ulteriore espressione della biotina ligasi. Da ogni schermata abbiamo selezionato in modo casuale i cloni fagemidici e con il fago ELISA abbiamo confermato che Affimers era stato selezionato contro ciascuno dei nove domini SH2. La proporzione di cloni legati a ciascun target, ma non al controllo, era compresa tra il 50% e il 100% con una media dell’87,6%. Successivamente, abbiamo valutato la specificità del target Affimer con l’ELISA dei fago(Figura 2A). Le proteine Grb sono proteine legate al recettore del fattore di crescita che contengono domini SH2. Inizialmente, gli Affimers del dominio Grb2, 7, 10 e 14 SH2 sono stati testati per la cross-reattività rispetto agli altri membri della famiglia Grb, e hanno mostrato un legame specifico al dominio Grb SH2, ad eccezione degli Affimers Grb14 che hanno mostrato una debole cross-reattività con le proteine Grb7 e Grb10 ma non Grb2. Il livello di omologia della sequenza a coppie tra Grb7, 10 e 14 è compreso tra il 65 e il 72% (Daly, 1998). È da notare che sono stati isolati gli Affimers che si legano specificamente a Grb7 e Grb10 senza la necessità di panning negativo per rimuovere i leganti cross-reattivi. Prevediamo che gli schermi che includono il pre-panning contro domini simili si tradurrebbero in isolamento di Affimers specifici che possono legare solo Grb14.10.7554/eLife.24903.004Figure 2.Isolamento e caratterizzazione di Affimers leganti del dominio SH2.(A) Phage ELISA da 24 reagenti monoclonali Affimer isolati contro i rispettivi domini SH2 della famiglia Grb. Specificità è stato testato attraverso l’estensione del legame con gli altri membri della famiglia SH2.(B) Western blot che mostra Affimer-mediata affinità-precipitazione di Affimer-mediata della proteina Grb2 endogena espressa da lisati cellulari U2OS utilizzando cinque Grb2 Affimers legati a perle magnetiche colbalto (n = 2). Un lievito SUMO legame Affimer è stato utilizzato come controllo negativo.(C) Fago ELISA da 24 reagenti monoclonali Affimer isolati contro p85 alpha N-terminal domain membro della famiglia di dominio SH2. La specificità è stata testata attraverso l’estensione del legame con gli altri membri della famiglia SH2 del dominio p85 alpha N.(D) Western blot di immunoprecipitazione utilizzando un anticorpo p110 su lisati cellulari da cellule che esprimono il dominio p85 SH2 affimeri di legame (n = 3).(E) Western blot e la quantificazione per densitometria di fosforilazione AKT in presenza di p85 SH2 dominio espresso legame Affimers p85 SH2 dominio vincolante (n = 2).DOI:http://dx.doi.org/10.7554/eLife.24903.00410.7554/eLife.24903.005Figure2-figure supplement 1.Western blot risultati di proteine del dominio Avi-Tag SH2 utilizzando un coniugato streptavidina-HRP per rilevare la presenza di biotina.DOI:http://dx.doi.org/10.7554/eLife.24903.005
Abbiamo poi esaminato la capacità dei reagenti di Affimer di legarsi alle proteine endogene. Cinque degli Affimers Grb2-binding Affimers sono stati purificati e legati a perle magnetiche a base di cobalto e la loro capacità di tirare giù Grb2 endogeno da lisati cellulari della linea cellulare umana U2OS è stata valutata(Figura 2B, n = 2). Tutti e cinque i reagenti hanno tirato giù con successo Grb2 mentre un lievito SUMO-legante di controllo Affimer(Tiede et al., 2014) non è stato in grado di tirare giù Grb2.
Per valutare ulteriormente la capacità di isolare gli Affimeri isoformi specifici di Affimer abbiamo studiato il fosfoinositide 3-chinasi (PI3K) una proteina eterodimerica che comprende una sottounità catalitica p110 e una sottounità regolatoria p85/p55. Abbiamo esaminato la specificità degli Affimers, sollevata contro il dominio N-terminale SH2 della variante p85α, per la cross-reattività con i domini p85β e p55γ della variante N-terminale SH2 e contro i domini C-terminale SH2 di tutte e tre le isoforme (Figura 2C). Nonostante un elevato grado di identità di sequenza (a coppie tra 83-90%) un certo numero di p85α specifici Affimers sono stati isolati (ad esempio, i cloni 1 e 2; Figura 2C) . Sono stati isolati anche gli Affimers che riconoscevano il dominio α e γ ma non il dominio β (cloni 3 e 23). Nessuno di questi Affimers legati a uno qualsiasi dei domini C-terminale p85/p55 SH2. Questi risultati dimostrano ulteriormente la capacità di isolare gli Affimers che mostrano un’elevata specificità di legame contro gli obiettivi correlati, anche all’interno di una singola proteina.
Gli Affimers p85 SH2 dominio-specifici Affimers sono stati espressi in cellule NIH 3T3 e la loro capacità di legarsi alla proteina p85 endogena è stata valutata da saggi di co-immunopreciptazione, in cui p85α è stato tirato giù. L’anticorpo p85α anche tirato giù sia p110α e il FLAG-tagged Affimers (Figura 2D). I diversi livelli di Affimer recuperati possono essere dovuti a differenze nei livelli di espressione di Affimer, come mostrato nella Figura 2E, e ad eventuali differenze di affinità di legame. È interessante notare che Affimer 1 sembra legarsi a p85 endogeno con alta affinità(Figura 2D) ma ha poco effetto sulla segnalazione(Figura 2E) suggerendo che si lega al di fuori della regione di interazione chiave SH2. Inoltre gli Affimers non hanno disturbato il complesso complessivo di p85/p110 in cui p85 interagisce con p110 attraverso tre domini, con l’attività di regolazione del dominio SH2 attraverso il binding di p110(Vivanco e Sawyers, 2002). Questi risultati dimostrano che l’Affimer sta specificamente vincolando l’interazione del dominio SH2 senza influenzare gli altri due domini di binding p85/p110.
Abbiamo quindi valutato la capacità degli Affimers di bloccare la funzione del dominio SH2 p85 N-terminale esaminando se gli Affimers hanno portato ad un aumento della proteina chinasi fosforilata B (AKT), un effettore a valle di p110. Cinque delle sei proteine Affimer hanno mediato un aumento della fosforilazione AKT(Figura 2E) dimostrando che inibiscono l’interazione tra il dominio N-terminale p85 SH2 e p110, ma soprattutto non bloccano la formazione di complessi p85-p110. Questo supporta un rapporto che l’inibizione siRNA di p85α da sola ha avuto poco effetto sulle cellule, ma che p85 e p110 entrambi dovevano essere eliminati per produrre un fenotipo e un effetto sulla fosforilazione AKT (Kimet al., 2005). Così i nostri dati evidenziano un vantaggio di Affimers, e potenzialmente altri reagenti alternativi, per studiare le interazioni proteina-proteina all’interno del contesto cellulare.
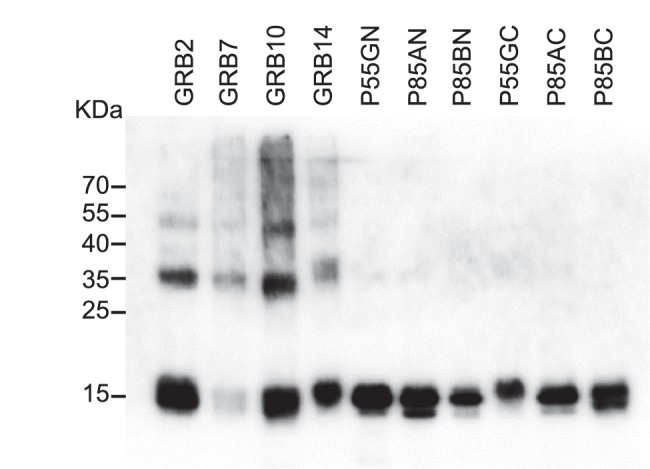
Figura 2-figure supplemento 1.Figura 2—figura 1. Isolamento e caratterizzazione del dominio SH2 che lega Affimers.Western blot risultati di proteine del dominio SH2 Avi-Tag utilizzando un coniugato streptavidina-HRP per rilevare la presenza di biotina.(A) Fago ELISA da 24 reagenti monoclonali Affimer isolati contro i rispettivi domini SH2 membri della famiglia Grb. La specificità è stata testata attraverso l’estensione del legame con gli altri membri della famiglia SH2.(B) Western blot che mostra Affimer-mediata affinità-precipitazione di Affimer-mediata della proteina Grb2 endogena espressa da lisati cellulari U2OS utilizzando cinque Grb2 Affimers legati a perle magnetiche colbalto (n = 2). Un lievito SUMO legame Affimer è stato utilizzato come controllo negativo.(C) Fago ELISA da 24 reagenti monoclonali Affimer isolati contro p85 alpha N-terminal domain membro della famiglia di dominio SH2. La specificità è stata testata attraverso l’estensione del legame con gli altri membri della famiglia SH2 del dominio p85 alpha N.(D) Western blot di immunoprecipitazione utilizzando un anticorpo p110 su lisati cellulari da cellule che esprimono il dominio p85 SH2 affimeri di legame (n = 3).(E) Western blot e la quantificazione per densitometria di fosforilazione AKT in presenza di p85 SH2 dominio espresso legame Affimers p85 SH2 dominio vincolante (n = 2).DOI:
http://dx.doi.org/10.7554/eLife.24903.004DOI:
http://dx.doi.org/10.7554/eLife.24903.005
Figura 2-figure supplement 1.2. Western blot risultati di proteine del dominio Avi-Tag SH2 utilizzando un coniugato streptavidina-HRP per rilevare la presenza di biotina.DOI:
http://dx.doi.org/10.7554/eLife.24903.005
Gli Affimers possono essere usati per inibire la funzione dei recettori extracellulari
I Vascular Endothelial Growth Factors (VEGFs) sono una famiglia di proteine secrete che regolano molti aspetti della biologia vascolare e linfatica, tra cui la vasculogenesi (formazione de novo del sistema vascolare), l’angiogenesi (formazione di nuovi capillari, ad esempio in risposta all’ipossia), la linfangiogenesi (formazione de novo del sistema linfatico) e l’arteriogenesi (formazione di nuove arterie, ad esempio a seguito di ischemia). Gli effetti biologici della famiglia VEGF sono mediati attraverso il legame con un recettore vascolare del fattore di crescita endoteliale legato alla membrana (VEGFR), sottofamiglia della tirosina chinasi che comprende VEGFR1, 2 e 3. Mentre il VEGFR1 è implicato come regolatore negativo dell’angiogenesi, il VEGFR2 è un importante regolatore della vasculogenesi, dell’angiogenesi e dell’arteriogenesi. L’attivazione del VEGFR3 è implicata nella specificazione della linfangiogenesi, ma il cross-talk tra i diversi VEGFR può modulare questi diversi processi(Aspelund et al., 2016). La dissezione dei ruoli dei diversi VEGFR è un obiettivo importante, soprattutto se si considera il successo degli agenti terapeutici che mirano al VEGF-A in malattie che vanno dal cancro metastatico alla degenerazione maculare. In questo contesto, il VEGFR2 è una molecola chiave che regola molti aspetti della fisiologia vascolare e della formazione dei vasi sanguigni, in particolare l’angiogenesi, ed è associato alla neovascolarizzazione tumorale(Kofler e Simons, 2015).
Per valutare se le proteine Affimer che perturbano la funzione VEGFR2 possano essere selezionate abbiamo confrontato le proteine Affimer con il VEGFR2 e poi abbiamo testato gli Affimer per la loro capacità di legare la proteina ricombinante VEGFR2 in vitro(Figura 3A). In questo caso l’analisi della sequenza del DNA ha rivelato che i cloni positivi rappresentavano solo due sequenze distinte. Le affinità per il VEGFR2 delle proteine rappresentative di Affimer, A9 e B8, sono state determinate da SPR per essere rispettivamente 41 ± 17 nM e 240 ± 124 nM (Figura 3-figure supplement 1). Le proteine Affimer sono state poi etichettate alla cisteina del C-terminale con un unico gruppo di biotina e utilizzate per sondare vari tipi di tessuto per la colorazione specifica da confrontare con il modello prodotto da un anticorpo policlonale disponibile in commercio(Figura 3B). L’efficienza dell’etichettatura Affimer con la biotina è stata determinata per l’80-90% dalla spettrometria di massa (dati non mostrati). Per confrontare direttamente i modelli di anticorpi e di Affimer è stato utilizzato un anticorpo secondario biotinilato per rilevare il legame dell’anticorpo primario anti-VEGFR2. Successivamente, sia l’anticorpo che il legame dell’Affimer sono stati rilevati dall’attività della perossidasi di rafano accoppiata alla streptavidina. I reagenti di Affimer hanno mostrato esattamente lo stesso schema di colorazione degli anticorpi, con la colorazione VEGFR2 prevalentemente localizzata nelle cellule epiteliali e con una colorazione più intensa sulla membrana cellulare(Figura 3B; vedi frecce). In questo caso, la colorazione si è sviluppata più rapidamente per i leganti Affimer che per l’anticorpo che indica una maggiore sensibilità della colorazione.10.7554/eLife.24903.006Figure 3.Characterisation of VEGFR2 binding Affimers.(A) Phage ELISA per 32 reagenti monoclonali Affimer isolati contro il VEGFR2. Il controllo negativo conteneva solo streptavidina.(B) Immuno- e istochimica di affinità di un anticorpo policlonale anti-VEGFR2 e degli Affimer rappresentativi B8 e A9. La colorazione è mostrata come un colore marrone chiaro, controcolorazione con emotossilina (blu). Le frecce mostrano modelli di colorazione simili.(C) Saggio di tubulogenesi in presenza e assenza del fattore di crescita endoteliale vascolare A e dei due Affimers con quantificazione della lunghezza dei tubuli e del numero del punto di di diramazione mostrati a destra. Il controllo è in assenza di qualsiasi Affimer e il controllo Affimer è un legante contro il lievito SUMO (n = 3). L’analisi statistica è stata eseguita utilizzando un ANOVA bidirezionale seguito dal test di confronto multiplo Bonferroni utilizzando il software GraphPad Prism (La Jolla, USA). p valori p inferiori a 0,05 (*), 0,01 (**) sono indicati sui grafici. Le barre di errore nei grafici indicano ± errore standard della media.(D) I risultati del Western blot mostrano cambiamenti nella segnalazione a valle in HUVEC trattati per 0, 5 e 15 min in presenza del fattore di crescita endoteliale vascolare A e di concentrazioni crescenti del VEGFR2 che lega Affimer B8 (n = 3).DOI:http://dx.doi.org/10.7554/eLife.24903.00610.7554/eLife.24903.007FigureSupplemento a 3 cifre 1.SPR per gli Affimer leganti anti-VEGFR2, TNC e TNT.DOI:http://dx.doi.org/10.7554/eLife.24903.007
Anche se l’immunoistochimica è una tecnica qualitativa piuttosto che quantitativa, si tratta di un’osservazione interessante date le affinità leganti apparentemente modeste, la natura monomerica e lo stato mono-biotinilato dei leganti Affimer rispetto alla natura bivalente delle molecole di anticorpi policlonali biotinilati moltiplicati. Dimostra il valore degli Affimer come reagenti istochimici di affinità. La sensibilità differenziale della colorazione può essere dovuta alla differenza di dimensioni tra l’anticorpo e l’Affimer con quest’ultimo meglio in grado di penetrare nel tessuto fisso in modo più efficiente. L’Affimer può anche avere un sito di legame più esposto rispetto all’anticorpo. Una o più di queste proprietà possono consentire un maggior numero di eventi di legame al bersaglio con conseguente maggiore sensibilità della colorazione di Affimer.
Sono state segnalate impalcature alternative che inibiscono il VEGFR2, tra cui le proteine Nanobody(Behdani et al., 2012), Adnectin(Tolcher et al., 2011), Affibody (Fleetwood et al., 2014) e DARPin(Hyde et al., 2012), per cui ci siamo chiesti se le proteine di Affimer possano anche inibire la segnalazione del VEGFR2 nelle cellule endoteliali vascolari umane (HUVEC). Precedenti studi siRNA(Murga et al., 2005) hanno dimostrato che la segnalazione VEGFR2 è necessaria per la formazione di tubuli vascolari da parte degli HUVEC trasfettati, anche se questo effetto mediato da siRNA richiede 24-48 ore dopo la trasfezione. Al contrario, l’effetto inibitorio di Affimer B8 ha potuto essere misurato in soli 30 minuti di trattamento e ha anche portato ad una diminuzione della lunghezza dei tubuli dipendente dal VEGF e della formazione di punti di di diramazione in un test di tubulogenesi(Figura 3C). Coerentemente con gli effetti sulla tubulogenesi, Affimer B8 ha anche inibito la fosforilazione VEGF-dipendente del VEGFR2 e la segnalazione a valle, con una minore attivazione dei mediatori di segnalazione cellulare PLCg1, AKT, ERK, p38 ed eNOS (n = 3; Figura 3D) . Al contrario, il controllo Affimers non ha avuto alcun effetto sulla segnalazione. Nel complesso queste osservazioni dimostrano che gli Affimers rappresentano utili reagenti di ricerca che sono in grado di bloccare la funzione biologica di specifici recettori su tempi biologicamente rilevanti.
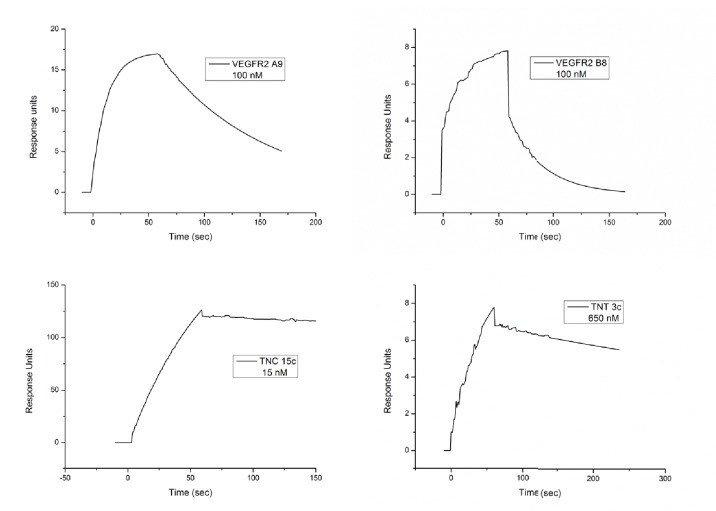
Figura 3-figure supplemento 1.Caratterizzazione dei grafici VEGFR2 che legano gli Affimers.SPR per gli Affimers anti-VEGFR2, TNC e TNT.(A) ELISA fago per 32 reattivi Affimer monoclonali isolati contro il VEGFR2. Il controllo negativo conteneva solo streptavidina.(B) Immuno- e istochimica di affinità di un anticorpo policlonale anti-VEGFR2 e degli Affimer rappresentativi B8 e A9. La colorazione è mostrata come un colore marrone chiaro, controcolorazione con emotossilina (blu). Le frecce mostrano modelli di colorazione simili.(C) Saggio di tubulogenesi in presenza e assenza del fattore di crescita endoteliale vascolare A e dei due Affimers con quantificazione della lunghezza dei tubuli e del numero del punto di di diramazione mostrati a destra. Il controllo è in assenza di qualsiasi Affimer e il controllo Affimer è un legante contro il lievito SUMO (n = 3). L’analisi statistica è stata eseguita utilizzando un ANOVA bidirezionale seguito dal test di confronto multiplo Bonferroni utilizzando il software GraphPad Prism (La Jolla, USA). p valori p inferiori a 0,05 (*), 0,01 (**) sono indicati sui grafici. Le barre di errore nei grafici indicano ± errore standard della media.(D) I risultati del Western blot mostrano cambiamenti nella segnalazione a valle in HUVEC trattati per 0, 5 e 15 min in presenza del fattore di crescita endoteliale vascolare A e di concentrazioni crescenti del VEGFR2 che lega Affimer B8 (n = 3).DOI:
http://dx.doi.org/10.7554/eLife.24903.006DOI:
http://dx.doi.org/10.7554/eLife.24903.007
Figura 3-figure supplement 1.2. Trame SPR per gli Affimers anti-VEGFR2, TNC e TNT vincolanti Affimers.DOI:
http://dx.doi.org/10.7554/eLife.24903.007
Leganti Affimer per la funzione di canale ionico modulante
I canali ionici sono coinvolti in una serie di processi fisiologici e sono importanti bersagli farmacologici(Overington et al., 2006). Tuttavia, rimane una mancanza di reagenti in grado di modulare i canali ionici con la selettività e la specificità necessarie per prevenire gli effetti fuori bersaglio(Skerratt e West, 2015). Gli anticorpi si sono dimostrati utili come reagenti per l’imaging dei canali ionici e hanno recentemente dimostrato di essere promettenti come terapeutiche(Lee et al., 2014; Sun e Li, 2013). A complemento del repertorio di anticorpi disponibili, si stanno utilizzando sempre più piccoli biologhi per studiare i canali ionici, ad esempio, fornendo chaperon di cristallizzazione(Stockbridge et al., 2015; Zhou et al., 2001). Inoltre, l’elevata selettività spesso associata a tali biologici, insieme alla loro capacità di accedere a fessure funzionali, può fornire ulteriori opportunità per modulare la funzione dei canali ionici. In effetti, il puntamento dei canali ionici sia ligandi che a tensione da parte di Nanobodies e scFv, rispettivamente, ha già dimostrato questo potenziale (Danquahet al., 2016; Harley et al., 2016).
Qui, ci siamo proposti di isolare gli Affimers in grado di legarsi e modulare l’attivazione del canale ionico del Transient Receptor Potential Vanilloid 1 (TRPV1) schermando contro un peptide derivato dal dominio dei pori esterni. Tredici cloni Affimer unici sono stati identificati da 24 cloni positivi identificati da un ELISA fago di 96 colonie selezionate in modo casuale dalla schermata della libreria dei fagi(Figura 4A). Nessuno dei 13 leganti ha mostrato reattività crociata ad un peptide distinto derivato dalla regione dei pori di un canale di sodio a tensione, Nav1.7. Affinità-fluorescenza studi sono stati eseguiti per esaminare la capacità delle proteine Affimer come reagenti di rilevamento. Solo Affimer 2 cellule U2-OS colorate che esprimono la lunghezza completa TRPV1(Figura 4B) che mostra la co-localizzazione con un anticorpo anti-TRPV1(Figura 4C). Affimer 2 non ha mostrato alcuna colorazione di cellule di controllo TRPV1-negative U2-OS. Nessuno degli altri 12 leganti lavorato in questo saggio.10.7554/eLife.24903.008Figure 4.Characterisation di Affimers TRPV1 vincolante.(A) Fago ELISA per 96 reagenti monoclonali Affimer isolato contro il peptide TRPV1. Il controllo negativo conteneva una diversa sequenza di peptidi idrofobici.(B) Affinità-citochimica su cellule U2-OS transitoriamente trasfettate con TRPV1 (TRPV1+) o controllo (TRPV1-) utilizzando Affimer 2. Il legame è stato rilevato utilizzando un anticorpo anti-HIS etichettato fluorescentemente con FITC. Il legame dell’Affimer è mostrato come un verde e DAPI (una macchia di DNA) mostrato come blu (n = 3), (C) Co-localizzazione della colorazione di Affimer con un anticorpo anti-TRPV1. La colorazione dell’anticorpo è mostrata in rosso.(D) Una Flexstation è stata utilizzata per misurare l’assorbimento di Fluo-4 AM, una piccola molecola fluorescente legante il calcio, per misurare i livelli di calcio nelle cellule stimolate con capsaicina in presenza di controllo Affimer e Affimeri leganti TPRV1 (n = 3).DOI:http://dx.doi.org/10.7554/eLife.24903.008
Successivamente abbiamo studiato la modulazione TRPV1 misurando i livelli di calcio intracellulare in risposta al trattamento con le proteine Affimer. Mentre non è stata osservata alcuna modulazione diretta sei Affimers hanno mostrato un significativo miglioramento dell’attivazione del TRPV1 al trattamento con la capsaicina agonista(Figura 4D) rispetto alle cellule trattate con la sola capsaicina. La ricerca precedente ha esplorato l’uso di composti di piccole molecole come modulatori positivi di TRPV1 per desensibilizzare e ridurre il dolore(Kaszas et al., 2012). Il composto (MRS1477) è stato ipotizzato per interagire con la regione di formazione dei pori del TRPV1, portando a un aumento di tre volte l’attivazione della capsaicina quando applicata a basse concentrazioni micromolari – un effetto simile a quello qui riportato per alcuni degli Affimers. Nel complesso, questo studio dimostra che le proteine di Affimer possono essere sollevate contro un surrogato peptidico per riconoscere e alterare la funzione del canale ionico attraverso la modulazione allosterica positiva, un meccanismo suggerito per il trattamento del dolore cronico indotto dal TRPV1(Lebovitz et al., 2012) e può rappresentare un nuovo approccio e una strategia terapeutica per il sollievo dal dolore cronico.
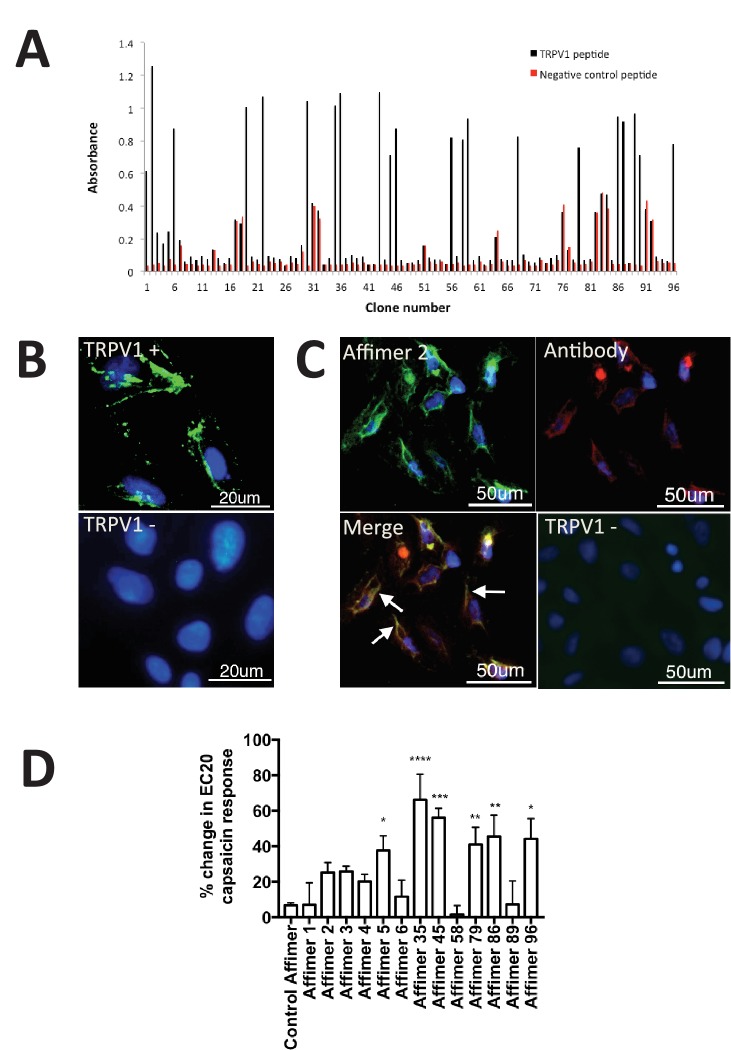
Figura 4.Caratterizzazione di TRPV1 che lega gli Affimers.(A) ELISA fago per 96 reagenti monoclonali Affimer isolati contro il peptide TRPV1. Il controllo negativo conteneva una diversa sequenza di peptidi idrofobici.(B) Affinità-citochimica su cellule U2-OS transitoriamente trasfettate con TRPV1 (TRPV1+) o controllo (TRPV1-) utilizzando Affimer 2. Il legame è stato rilevato utilizzando un anticorpo anti-HIS etichettato fluorescentemente con FITC. Il legame dell’Affimer è mostrato come un verde e DAPI (una macchia di DNA) mostrato come blu (n = 3), (C) Co-localizzazione della colorazione di Affimer con un anticorpo anti-TRPV1. La colorazione dell’anticorpo è mostrata in rosso.(D) Una Flexstation è stata utilizzata per misurare l’assorbimento di Fluo-4 AM, una piccola molecola fluorescente legante il calcio, per misurare i livelli di calcio nelle cellule stimolate con capsaicina in presenza di controllo Affimer e Affimeri leganti TPRV1 (n = 3).DOI:
http://dx.doi.org/10.7554/eLife.24903.008
Leganti Affimer per imaging in vivo
La tenascina C (TNC) è una proteina a matrice extracellulare che è abbondante durante lo sviluppo precoce, è espressa a bassi livelli nei tessuti adulti ed è spesso up-regolata nei tessuti tumorali ed è associata a metastasi (Minn et al., 2005; Oskarsson et al., 2011) e ad esiti poveri per i pazienti(Lowy e Oskarsson, 2015). Come tale, offre un potenziale come marker tumorale per l’imaging e/o il targeting terapeutico in vivo(Hicke et al., 2006). I leganti di Affimer al TNC sono stati isolati dalla libreria di visualizzazione dei fagi(Figura 5A). Una proteina Affimer con alta affinità per TNC (KD = 5,7 ± 2,8 nM da SPR – sup Figura 2) è stato utilizzato in saggi successivi. Per valutare la sua specificità per TNC abbiamo confrontato il modello di colorazione dell’Affimer con quello di un anticorpo anti-TNC in sezioni di tessuto xenotrapianto di cancro colorettale umano e glioblastoma. Modelli di colorazione con C-terminalmente biotinilato TNC Affimer erano simili a quelli ottenuti con l’anticorpo TNC(Figura 5B).10.7554/eLife.24903.009Figure 5.Characterisation di tenascina C (TNC) Affimer vincolante da affinità-istochimica e imaging ex vivo di xenotrapianti.(A) Fago ELISA per 48 Affimer monoclonali contro TNC. I due controlli sono la tenascina X (TNX) e la streptavidina.(B) Immunoistochimica di sezioni seriali di uno xenotrapianto di topo (linea cellulare SW620), mostrando la colorazione per TNC. La colorazione degli anticorpi e dell’effimero è mostrata come un colore marrone chiaro con controcolorazione di emotossilina (blu).(C) e(D) I topi sono stati iniettati attraverso la loro vena caudale con Affimer con legante TNC marcato con rodamina o Affimer con legante GFP di controllo. Dopo 24, 48, 72 e 96 ore, lo xenotrapianto e gli organi sono stati rimossi e visualizzati.(C) Immagini degli organi a 24 ore.(D) Quantificazione della fluorescenza della rodamina (efficienza radiante in p/s/cm2/sr/μW/cm2) ex vivo (n = 3). L’intensità media della fluorescenza di fondo è stata normalizzata per simulare tumori e organi di controllo iniettati.DOI:http://dx.doi.org/10.7554/eLife.24903.009
Nella clinica, l’imaging dei tumori con anticorpi etichettati può essere limitato da un elevato background di anticorpi etichettati in circolazione fino a quando questo non viene eliminato dal corpo. Questo porta a lunghe degenze ospedaliere o alla necessità di visite multiple dei pazienti per un singolo test. Al contrario, le dimensioni più piccole delle proteine leganti alternative significano che le molecole che non si legano al bersaglio, saranno cancellate più rapidamente dal sistema circolatorio, consentendo una visualizzazione più comoda poco dopo la somministrazione dell’agente imaging. Per dimostrare questo nei topi portatori di tumori abbiamo visualizzato la distribuzione di TNC Affimer rispetto ad un controllo GFP-binding Affimer, entrambi C-terminalmente etichettati con Rosso Rodamina(Figura 5C). Per massimizzare il rilevamento del segnale, abbiamo immaginato i tumori asportati e gli organi post-sacrificio e abbiamo quantificato il segnale come fold-change sopra lo sfondo. Come previsto, entrambe le sonde Affimer sono state rilevate nei reni che indicano la clearance renale. Tuttavia, rispetto al TNC Affimer questa clearance è stata più veloce per il GFP Affimer in quanto ha mostrato una significativa diminuzione (p = 0,04) in fluorescenza da 24 a 48 ore dopo l’iniezione (fold change 9,23 ± 3,10 a 24 ore a 2,98 ± 0,77 a 48 ore; Figura 5D). Il segnale del TNC Affimer a 24 ore dopo l’iniezione era significativamente più alto (p=0,02) nei tumori (6,26 ± 1,62) rispetto al gruppo di controllo GFP Affimer (2,32 ± 0,61), suggerendo che il TNC Affimer si è accumulato nel TNC esprimendo il tumore. La sonda TNC è stato anche rilevato nei tessuti del fegato sia a causa di clearance epatobiliare o per il fatto che il TNC mostra un basso livello di espressione in sinusoidi epatiche normali(Van Eyken et al., 1990). Inoltre, il rapporto dei leganti anti-TNC Affimer nel tumore rispetto alla milza, per esempio, era >6 a 24 ore (Figura 5D); al contrario, gli anticorpi anti-TNC hanno impiegato 2 giorni per raggiungere un rapporto tumore/pleena di 5, anche se questo è migliorato a 20-30 al giorno 10 (De Santis etal., 2006). Quindi il più rapido tasso di clearance delle proteine leganti alternative, come gli Affimers, rispetto agli anticorpi, ha il potenziale per consentire una più rapida diagnostica per immagini dei tumori. Sono in corso ulteriori lavori per migliorare la rilevazione del segnale in vivo con Affimers(Fisher et al., 2015).
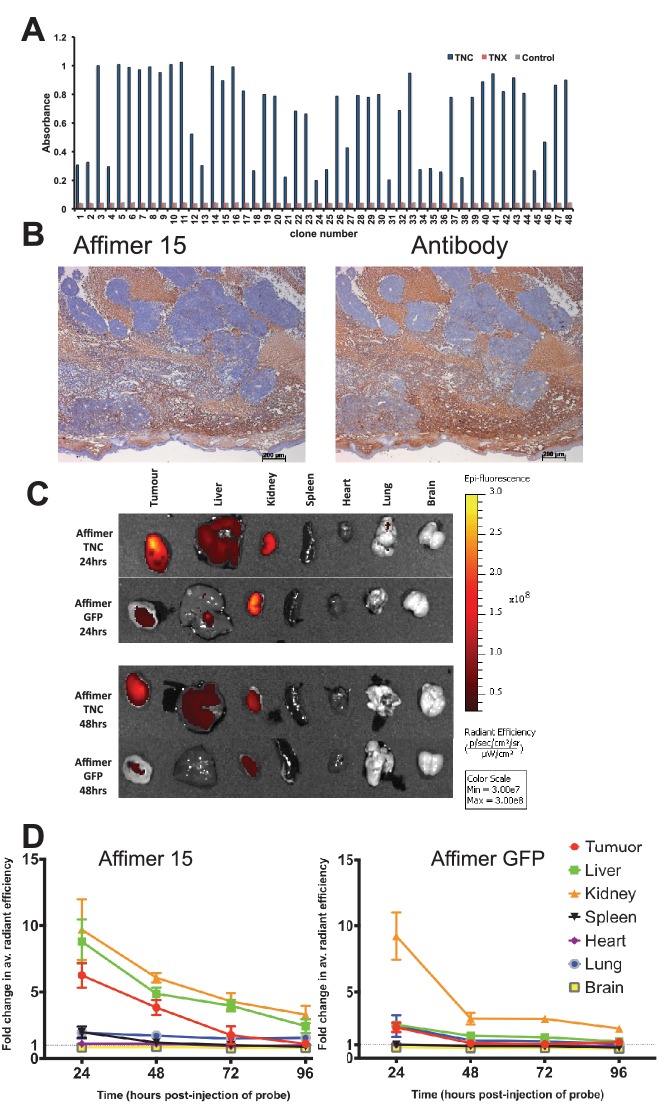
Figura 5.Caratterizzazione del tenascin C (TNC) che lega Affimer per affinità-istochimica e imaging ex vivo di xenotrapianti.(A) Fago ELISA per 48 Affimers monoclonali contro TNC. I due controlli sono la tenascina X (TNX) e la streptavidina.(B) Immunoistochimica di sezioni seriali di uno xenotrapianto di topo (linea cellulare SW620), mostrando la colorazione per TNC. La colorazione degli anticorpi e dell’effimero è mostrata come un colore marrone chiaro con controcolorazione di emotossilina (blu).(C) e(D) I topi sono stati iniettati attraverso la loro vena caudale con Affimer con legante TNC marcato con rodamina o Affimer con legante GFP di controllo. Dopo 24, 48, 72 e 96 ore, lo xenotrapianto e gli organi sono stati rimossi e visualizzati.(C) Immagini degli organi a 24 ore.(D) Quantificazione della fluorescenza della rodamina (efficienza radiante in p/s/cm2/sr/μW/cm2) ex vivo (n = 3). L’intensità media della fluorescenza di fondo è stata normalizzata per simulare tumori e organi di controllo iniettati.DOI:
http://dx.doi.org/10.7554/eLife.24903.009
Affinità-fluorescenza in celle fisse
La malattia di Marek, causata dal virus della malattia di Marek (MDV-1), è una malattia neoplastica dei polli significativa a livello globale ed economico, attualmente controllata mediante vaccinazione con il relativo Herpes Virus del tacchino (HVT). Nei campioni sul campo, i test per la malattia di Marek dovrebbero essere in grado di distinguere tra le proteine dell’HVT e i loro omologhi nell’MDV. A riprova del principio, abbiamo sottoposto a screening la libreria di fago contro la proteina derivata dall’HTV UL49, con controschermi contro le proteine dell’ospite così come le proteine correlate MDV (RB1B) e DEV UL49. L’ELISA dei fagi, l’affinità-fluorescenza e la Western in-cell hanno confermato che gli Affimers selezionati erano specifici per le proteine ricombinanti HTV e la loro capacità di colorare in modo specifico la proteina target nei fibroblasti embrionali di pollo primari (CEF) contenenti cloni di cromosomi artificiali batterici (BAC) di MDV-1 (RB1B), HVT o DEV (ceppo 2085)(Figura 6A,B e C).10.7554/eLife.24903.010Figure 6.Affimer detection of HVT UL49 in cellule infette tramite Western in-cell e affinity-fluorescence.(A) Phage ELISA per 24 Affimers monoclonali contro HVT schermato contro HVT, RB1B, DEV e lisati CEF per confermare la specificità per HVT.(B) L’infezione delle cellule è stata confermata utilizzando un anticorpo anti-GFP di capra e un anticorpo 680 (verde) di asino anti capra per rilevare GFP che è costitutivamente espresso dai virus derivati da BAC. I CEF infetti sono stati sottoposti a screening con candidati HVT UL49 Affimers a 1,5 µg/ml con successiva rilevazione (rosso) da parte di Streptavidina 800 coniugata (Licor). (n = 3) (D) I CEF infetti da HVT sono stati sottoposti a screening con Affimers HVT UL49 Affimers e visualizzati con Streptavidina-568 (rosso). I nuclei sono stati colorati con DAPI (blu). Il solo controllo con Streptavidina (nessun Affimer) non mostra alcuna etichettatura osservabile di CEF infetti. Barra = 10 µm.DOI:http://dx.doi.org/10.7554/eLife.24903.01010.7554/eLife.24903.011FigureSupplemento a 6 cifre 1.Bilayer Interferometria a 6 cifre per gli Affimer HVT vincolanti.DOI:http://dx.doi.org/10.7554/eLife.24903.011
L’affinità degli Affimers anti HVT UL49 Affimers era nel range basso nM, valori KD da 1,5 nM a 7,5 nM con una media di 3,3 nM, per gli otto cloni testati(Figura 6-figure supplement 1). L’elevata specificità e affinità dovrebbe essere vantaggiosa nello sviluppo dei test DIVA (differenziando gli animali infetti da quelli vaccinati) per la discriminazione dei virus del vaccino e dei ceppi di campo. Abbiamo testato le prestazioni degli Affimers anti-HVT UL49 Affimers in affinità-fluorescenza(Figura 6C). Le colture di CEF primarie infettate con clone BAC GFP BAC HVT sono state sottoposte a colorazione in affinità-fluorescenza utilizzando Affimers anti HVT UL49 biotinilati e sono state visualizzate con coniugato streptavidina-Alexa Fluor 568(Figura 6C). Rispetto alla colorazione a basso background con il controllo solo streptavidina (solo SA) ci sono pronunciati focolai citoplasmatici rilevati da Affimers nelle cellule infette. Questi focolai sono coerenti con i dati del relativo alphaherpesvirus MDV(Denesvre et al., 2007; Rémy et al., 2013) o del modello alphaherpesviruses Herpes Simplex tipo 1 (HSV-1) (Stylianou etal., 2009) e del virus Pseudorabies (PrV) (delRio et al., 2002)e indicano probabilmente i siti citoplasmatici dell’involucro virale secondario HVT. Questa distribuzione è coerente anche per i diversi cloni di Affimer testati ed è visibile solo all’interno delle cellule infette. Pertanto gli Affimer si dimostrano promettenti come alternative agli anticorpi tradizionali e sono probabilmente particolarmente preziosi laddove la disponibilità/prestazione dei reagenti anticorpali esistenti è scarsa.
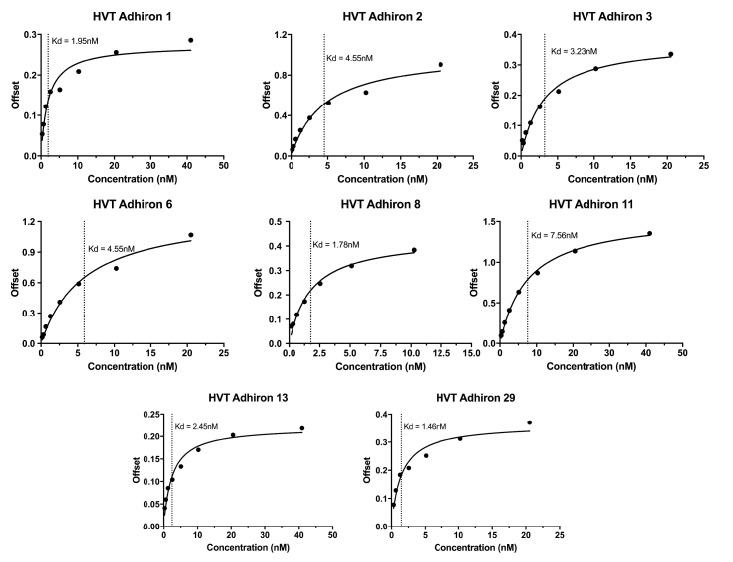
Figura 6-figure supplement 1.Figura 6—supplemento alla figura 1. Rilevamento dell’HVT UL49 nelle cellule infette da parte degli Affimer tramite la fluorescenza per affinità e la fluorescenza in cella.trame di interferometria a doppio strato per gli Affimer che legano l’HVT.(A) ELISA fago per 24 Affimers monoclonali contro l’HVT schermato contro l’HVT, RB1B, DEV e lisati CEF per confermare la specificità per l’HVT.(B) L’infezione delle cellule è stata confermata utilizzando un anticorpo anti-GFP di capra e un anticorpo 680 (verde) di asino anti capra per rilevare la GFP che è costitutivamente espressa dai virus derivati da BAC. I CEF infetti sono stati sottoposti a screening con candidati HVT UL49 Affimers a 1,5 µg/ml con successiva rilevazione (rosso) da parte di Streptavidina 800 coniugata (Licor). (n = 3) (D) I CEF infetti da HVT sono stati sottoposti a screening con Affimers HVT UL49 Affimers e visualizzati con Streptavidina-568 (rosso). I nuclei sono stati colorati con DAPI (blu). Il solo controllo con Streptavidina (nessun Affimer) non mostra alcuna etichettatura osservabile di CEF infetti. Barra = 10 µm.DOI:
http://dx.doi.org/10.7554/eLife.24903.010DOI:
http://dx.doi.org/10.7554/eLife.24903.011
Figura 6-figure supplement 1.2. Trame di interferometria a doppio strato per gli Affimers di rilegatura HVT.2. DOI:
http://dx.doi.org/10.7554/eLife.24903.011
Affimers come sonde per la microscopia a super risoluzione e l’inseguimento di particelle singole
La microscopia a super risoluzione fornisce la capacità di localizzare le proteine all’interno di una cella con una risoluzione di circa 20 nanometri. Una delle principali limitazioni di questo approccio è la mancanza di reagenti altamente specifici in grado di posizionare il fluoroforo in prossimità della proteina bersaglio endogena. Gli anticorpi sono grandi proteine multidominio che sono normalmente etichettate con fluorofori in siti casuali che limitano la risoluzione ottenibile. Al contrario, le proteine Affimer più piccole possono essere etichettate in modo specifico per il sito, fornendo un posizionamento spaziale più vicino del fluoroforo alla proteina bersaglio, facilitando così l’uso delle attuali tecniche di super-risoluzione. Questo approccio è stato recentemente dimostrato utilizzando i Nanobodies, dove la microscopia a super-risoluzione è stata utilizzata per l’immagine delle proteine marcate GFP e del complesso dei pori nucleari(Pleiner et al., 2015; Ries et al., 2012).
Il recettore 4 del fattore di crescita epidermico umano 4 (HER4), noto anche come c-erbB-4, è una proteina chinasi oncogena del recettore transmembrana(Lemmon e Schlessinger, 2010). Sebbene la funzione di questa proteina non sia ancora pienamente compresa, è noto che è associata a un aumento della sopravvivenza e a una minore proliferazione nelle pazienti affette da cancro al seno(Machleidt et al., 2013). Abbiamo esaminato la libreria di visualizzazione dei fagi contro HER4(Figura 7A) e due Affimers sono stati ricombinati con una cisteina C-terminale per l’etichettatura con il fluoroforo Alexa Fluor 647 o CF640R maleimmide. L’Affimer che mostra il segnale più alto da imaging fluorescente è stato utilizzato per ulteriori studi. I nostri risultati mostrano che l’Affimer HER4 può legarsi sia alle cellule CHO che esprimono transitoriamente HER4 che a MCF7, una linea cellulare del cancro al seno che esprime livelli fisiologici più bassi di HER4(Figura 7). Quando HER4 è sovra-espressa nelle cellule CHO l’Affimer ha mostrato un legame crescente a concentrazioni da 5 nM a 100 nM, come determinato dall’intensità del segnale di membrana dalle immagini della microscopia confocale, mentre nelle cellule MCF7 che esprimono livelli fisiologici di legame HER4 aumenta da 10 a 200 nM Affimer(Figura 7B).10.7554/eLife.24903.012Figure 7.Use of HER4 binding Affimers in super-resolution imaging e single molecule tracking.(A) Phage ELISA per HER4 binding Affimers. (B) Media fotoni conteggi / pixel per HER4-legante Affimer etichettati con CF640R e legato alle cellule CHO trasfettate con HER4 e alle cellule MCF7 che esprimono livelli endogeni di HER4.(C) Immagine ad ampio campo delle cellule CHO trasfettate con HER4-CYT-eGFP che mostra la localizzazione di HER4 attraverso la fluorescenza GFP (in alto) ed etichettate con HER Affimer-Alexa647 (al centro). La corrispondente immagine dSTORM di HER4 Affimer coniugata con Alexa647 (in basso) con una precisione di localizzazione di 25 nm. Barra di scala = 2 μm. Trame a destra per mostrare il numero di molecole e la dimensione dei cluster di cluster identificati da dSTORM.(D) Coefficienti di diffusione (pannello sinistro), e la curva MSD (pannello destro) di HER4 Affimers etichettati con CF640R e tracciati su cellule MCF7 che esprimono HER4.DOIendogeno:http://dx.doi.org/10.7554/eLife.24903.012
Le immagini di fluorescenza a doppio colore a campo ampio del recettore HER4 fuse all’estremità intracellulare del C-terminale con eGFP (HER4-CYT-eGFP) nelle cellule CHO (Figura 7C – in alto) ed etichettate nella regione extracellulare con Affimer-Alexa 647(Figura 7C al centro) mostrano che l’Affimer HER4 può essere utilizzato per etichettare specificamente l’HER4 associato alla membrana, attraverso la co-localizzazione della GFP e la fluorescenza Affimer etichettata. La corrispondente immagine diretta della Microscopia di ricostruzione ottica stocastica (dSTORM)(Figura 7C – in basso) ha una precisione di localizzazione di circa 25 nm. Una cluster analysis bayesiana(Griffié et al., 2016) dell’immagine dSTORM mostra che la dimensione dell’ammasso più prevalente di oligomeri HER4 è compresa tra 8,1 e 12 nm in raggio. Ciò corrisponde al numero più prevalente di molecole di HER4 in un cluster compreso tra 4 e 8. Questi dati mostrano che HER4 forma oligomeri grandi come quelli precedentemente trovati in EGFR.(Needham et al., 2016)
Questo Affimer è anche adatto all’immagine HER4 in modalità TIRF (Total Internal Reflection Fluorescence – Fluorescenza a riflessione interna totale) per effettuare il tracciamento a singola particella su cellule vive(Figura 7D). Per rilevare particelle singole, l’affinità di legame dell’Affimer deve essere nel range basso nM per evitare di saturare il campione e per ridurre il legame non specifico. Le particelle HER4 sono state tracciate con un algoritmo di tracciamento bayesiano(Rolfe et al., 2011) e il coefficiente di diffusione e lo Spostamento Quadrato Minimo (MSD) sono stati calcolati a partire dalle traiettorie risultanti. I dati mostrano che c’è una popolazione immobile, o scarsamente mobile, di recettori HER4 su cellule MCF7, associata ad una coda di molecole altamente mobili(Figura 7D, pannello a sinistra). La pendenza quasi rettilinea del grafico MSD indica che, a differenza di EGFR(Needham et al., 2016; Zanetti-Domingues et al., 2012), la diffusione di HER4 non è limitata sui tempi indagati(Figura 7D, pannello di destra).
Gli Affimers sollevati contro HER4 dimostrano la capacità di isolare i reagenti che possono essere utilizzati in una serie di tecniche di microscopia a super risoluzione. Tuttavia, non essendoci un confronto diretto con un anticorpo, questo esempio non evidenzia il vantaggio delle proteine alternative rispetto alle sonde anticorpali più grandi. Per fornire questa dimostrazione sono stati sollevati anche gli Affimers contro i microtubuli polimerizzati(Figura 8). L’Affimer abbiamo selezionato le etichette dei microtubuli interfasici in modo simile a un anticorpo ampiamente utilizzato(Figura 8A). Tuttavia, nelle cellule mitotiche, l’Affimer etichetta il fuso ma non i microtubuli astrali(Figura 8A) riflettendo probabilmente il fatto che l’anticorpo riconosce i microtubuli tirosinizzati a differenza dell’Affimer. È interessante notare che l’Affimer è in grado di etichettare la regione centrale del solco citocinetico(Figura 8A), in cui i microtubuli sono molto compressi. Gli anticorpi sono solitamente esclusi da questa regione, per cui l’analisi di questa caratteristica è stata problematica(Hu et al., 2012). Questo evidenzia un vantaggio dell’utilizzo di sonde più piccole, come le proteine leganti alternative, per la microscopia a super-risoluzione e per esempio in questo caso permetterà un’ulteriore chiarificazione del ruolo della tubulina nei solchi cito-cinetici.10.7554/eLife.24903.013Figure 8.Use of tubulin binding Affimer in super-resolution microscopy.(A) Immagini confocali di microtubuli in cellule HeLa, colorate con un anticorpo di ratto α-tubulina (YL1/2) che riconosce la tubulina tirosinata, e un Affimer per tubulina polimerizzata, coniugato con Alexa Fluor 647. Sono mostrate immagini di una cellula interfasica e metafasica, insieme ad un’immagine del solco cito cinetico. Frecce nella cellula metafasica puntano a microtubuli astrali che sono prevalentemente etichettati con l’anticorpo. Le frecce nel solco citochinetico indicano la regione centrale (corpo di Fleming). La barra di scala è di 10 μm.(B) 3D dSTORM immagini dSTORM di microtubuli in una cellula HeLa, etichettati con Alexa Fluor 647 coniugato con un anticorpo primario di ratto α-tubulina (a sinistra) e un Affimer per la tubulina polimerizzata (a destra). Queste immagini provengono da cellule separate. Le localizzazioni sono state aggregate in contenitori da 10 nm e proiettate su un unico piano, con lisciatura gaussiana. Barra di scala 1 µm.(C) Profilo di intensità attraverso l’immagine microtubula etichettata in(B) (scatola gialla), in media lungo 510 nm della sua lunghezza. La diminuzione centrale dell’intensità riflette la struttura cava del microtubule.(D) Confronto tra il profilo di intensità media dell’immagine del microtubule con la colorazione degli anticorpi (tratteggiata, media di 6 sezioni di microtubule), la colorazione Affimer (solida, media di 8 sezioni di microtubule) e la dimensione effettiva del microtubule (cerchio nero). L’FWHM di ogni profilo medio (come in(C)) è stato trovato per un adattamento gaussiano e una distribuzione gaussiana è qui tracciata usando la media FWHM per ogni metodo di colorazione.DOI:http://dx.doi.org/10.7554/eLife.24903.013
Le immagini 3D dSTORM dei microtubuli che utilizzano l’anticorpo e Affimer hanno un aspetto simile(Figura 8B) con analisi che mostrano che l’etichettatura Affimer ha la prevista diminuzione centrale della fluorescenza per il legame all’esterno del microtubule(Figura 8C). Tuttavia, la media dei profili per microtubuli multipli sia per l’Affimer che per l’anticorpo mostra una maggiore precisione di localizzazione con l’Affimer, rispetto agli anticorpi(Figura 8D). Mentre la densità di localizzazione potrebbe non essere completamente ottimizzata in questi campioni(Huang et al., 2008), i profili medi dei microtubuli erano sostanzialmente più ristretti con l’etichettatura con Affimer (47 ± 11 nm) rispetto all’etichettatura con anticorpi primari (73 ± 10 nm) (FWHM, media ± s.d.) e dovrebbero consentire un’ulteriore chiarificazione delle strutture tubuliniche che non sono state precedentemente risolte. In generale, gli Affimers, e presumibilmente altre proteine leganti alternative, hanno un vantaggio rispetto agli anticorpi nell’etichettatura per dSTORM.
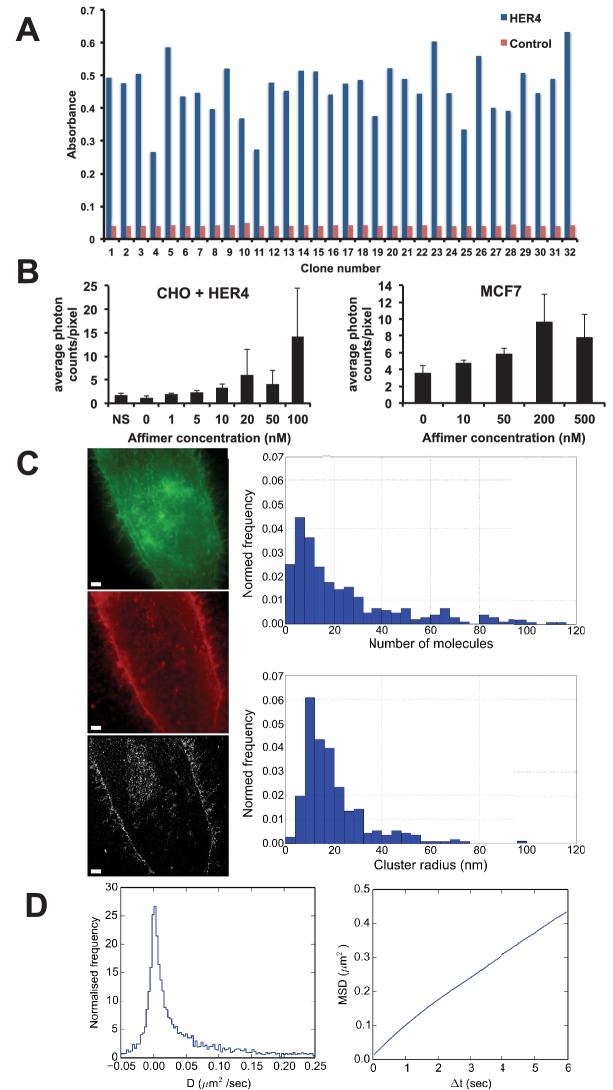
Figura 7.Figura 7. Uso di HER4 Affimers legante Affimers in super-risoluzione di imaging e tracciamento delle singole molecole.(A) Fago ELISA per HER4 Affimers binding Affimers.(B) Media fotoni conteggi / pixel per HER4-legante Affimer etichettati con CF640R e legato alle cellule CHO trasfettate con HER4 e alle cellule MCF7 che esprimono livelli endogeni di HER4.(C) Immagine ad ampio campo delle cellule CHO trasfettate con HER4-CYT-eGFP che mostra la localizzazione di HER4 attraverso la fluorescenza GFP (in alto) ed etichettate con HER Affimer-Alexa647 (al centro). La corrispondente immagine dSTORM di HER4 Affimer coniugata con Alexa647 (in basso) con una precisione di localizzazione di 25 nm. Barra di scala = 2 μm. Trame a destra per mostrare il numero di molecole e la dimensione dei cluster di cluster identificati da dSTORM.(D) Coefficienti di diffusione (pannello sinistro), e la curva MSD (pannello destro) di HER4 Affimers etichettati con CF640R e tracciati su cellule MCF7 che esprimono HER4 endogeno.DOI:
http://dx.doi.org/10.7554/eLife.24903.012
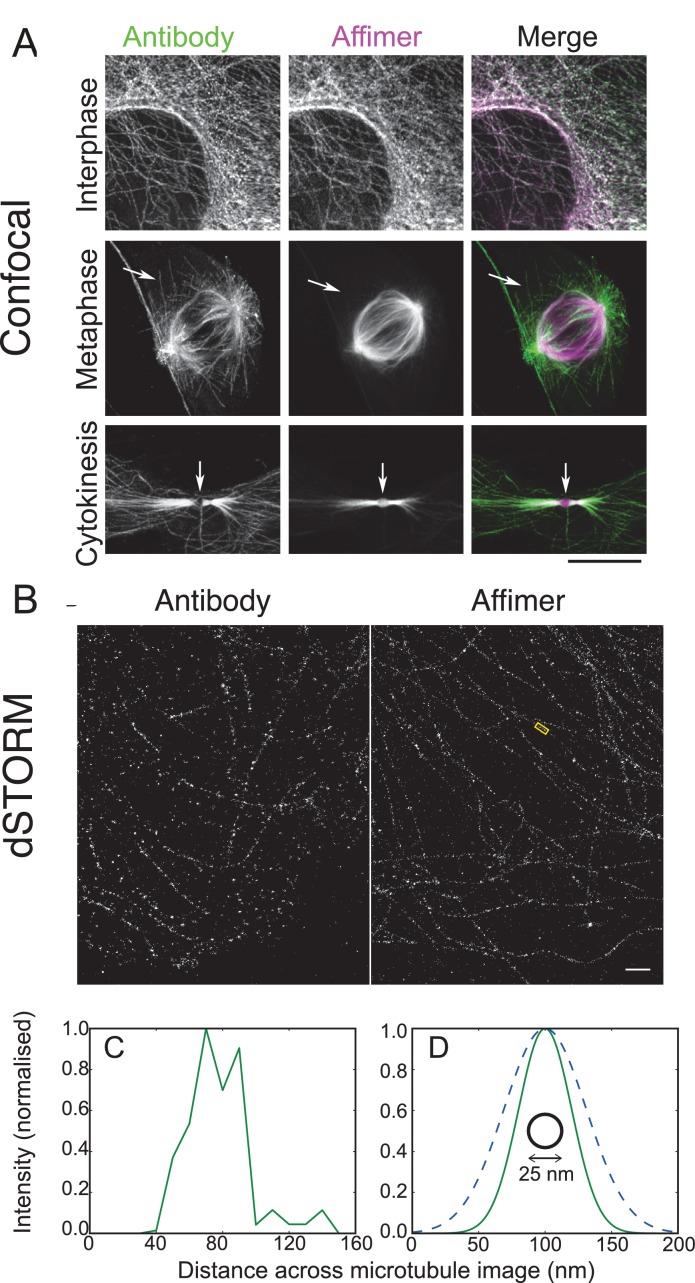
Figura 8.Uso di Affimer legante tubulina in microscopia a super-risoluzione.(A) Immagini confocali di microtubuli in cellule HeLa, colorate con un anticorpo di ratto α-tubulina (YL1/2) che riconosce la tubulina tirosinata, e un Affimer per la tubulina polimerizzata, coniugato con Alexa Fluor 647. Sono mostrate immagini di una cellula interfasica e metafasica, insieme ad un’immagine del solco cito cinetico. Frecce nella cellula metafasica puntano a microtubuli astrali che sono prevalentemente etichettati con l’anticorpo. Le frecce nel solco citochinetico indicano la regione centrale (corpo di Fleming). La barra di scala è di 10 μm.(B) 3D dSTORM immagini dSTORM di microtubuli in una cellula HeLa, etichettati con Alexa Fluor 647 coniugato con un anticorpo primario di ratto α-tubulina (a sinistra) e un Affimer per la tubulina polimerizzata (a destra). Queste immagini provengono da cellule separate. Le localizzazioni sono state aggregate in contenitori da 10 nm e proiettate su un unico piano, con lisciatura gaussiana. Barra di scala 1 µm.(C) Profilo di intensità attraverso l’immagine microtubula etichettata in(B) (scatola gialla), in media lungo 510 nm della sua lunghezza. La diminuzione centrale dell’intensità riflette la struttura cava del microtubule.(D) Confronto tra il profilo di intensità media dell’immagine del microtubule con la colorazione degli anticorpi (tratteggiata, media di 6 sezioni di microtubule), la colorazione Affimer (solida, media di 8 sezioni di microtubule) e la dimensione effettiva del microtubule (cerchio nero). L’FWHM di ogni profilo medio (come in(C)) è stato trovato per un adattamento gaussiano e una distribuzione gaussiana è qui tracciata usando la media FWHM per ogni metodo di colorazione.DOI:
http://dx.doi.org/10.7554/eLife.24903.013
Gli Affimers possono essere selezionati contro i piccoli composti organici
La generazione di reagenti efficaci che legano i composti organici a bassa massa molecolare è tecnicamente impegnativa. Le piccole molecole non mostrano un’immunogenicità innata e quindi sono tipicamente coniugate alle proteine portatrici per ottenere un’efficace risposta immunitaria. Anche così può essere un problema che solleva una risposta immunitaria alle molecole tossiche e a quelle che si coniugano male con le proteine portatrici. Per esaminare se potevamo isolare i reagenti di Affimer contro una piccola molecola organica abbiamo usato il 2,4,6-trinitrotoluene (TNT). Studi precedenti hanno dimostrato che la presentazione del TNT come hapten per la produzione di anticorpi è noto per essere vitale per il successo dell’isolamento degli anticorpi specifici del TNT(Ramin e Weller, 2012). L’analogo del TNT 2,4,6-trinitobenzene acido solfonico (TNBS)(Figura 9A) contiene gruppi di nitro (NO2) che si trovano nelle stesse posizioni del TNT sull’anello di benzene, mentre il metile (CH3) gruppo è sostituito da un acido solfonico (SO2OH) gruppo. Questo gruppo funzionale reagisce con le ammine primarie ed è stato utilizzato per preparare sia TNBS-ovalbumina e TNBS-IgG coniugati per lo screening di visualizzazione dei fagi, con schermi contatori eseguiti contro l’ovoalbumina e IgG per arricchire per il legame delle piccole molecole.10.7554/eLife.24903.014Figure 9.Affimer selezione e specificità contro TNT e DNT’s. (A) Strutture chimiche di TNBS, TNT, 2,3-DNT, 2,4-DNT e 2,6-DNT.(B) ELISA fago risultati da 32 reattivi monoclonali Affimer isolati contro TNBS legato all’ovoalbumina. La specificità del legame è stata anche testata contro TNBS legato a IgG e a ovoalbumina e IgG non coniugate.(C) ELISA da competizione di quattro TNT-Affimer per verificare la specificità contro una gamma di molecole in un profilo di concentrazione. Barre di errore = deviazione standard dalle ripetizioni tecniche di un ELISA rappresentativo.DOI:http://dx.doi.org/10.7554/eLife.24903.014
Per confermare la specificità di legame, i cloni selezionati sono stati testati sia contro i coniugati TNBS che contro la proteina non coniugata con il fago ELISA(Figura 9B). Il numero di cloni che hanno mostrato un forte legame con il TNBS coniugato sia con l’ovoalbumina che con le IgG è stato relativamente alto (22/32) con un ulteriore 8/32 che ha mostrato un ragionevole legame. Dei 32 cloni testati sono state identificate 14 sequenze distinte, alcune contenenti solo la regione variabile 1 che indica la selezione da una piccola sub-popolazione della biblioteca originale di visualizzazione dei fagi. All’interno del ciclo variabile 1 è stato possibile definire una breve sequenza di consenso che implica una modalità di binding comune. Quattro proteine Affimer sono state purificate e testate per il legame con TNT e vari dinitrotolueni (DNT; Figura 9A) dalla concorrenza ELISA(Figura 9C). Tutti e quattro gli Affimer hanno mostrato un legame con il coniugato originale TNBS e con il TNT, ma differivano per la loro specificità per il DNT. L’Affimer 4, un Affimer solo VR1, ha mostrato un livello di specificità per il TNT più alto di qualsiasi altro DNT. Al contrario, Affimer 3 si lega al TNT e fornisce anche una discriminazione tra il 2,4-DNT e le altre due DNT. Questa elevata selettività di riconoscimento del gruppo nitro sulla posizione 4, dimostra che i reagenti di Affimer possono mostrare una notevole specificità per tali piccole differenze molecolari. È molto probabile che modificare la strategia di panning per includere le fasi di competizione con gli analoghi permetterebbe di selezionare la specificità e la sensibilità degli Affimer per i target di piccole molecole organiche.
La capacità delle proteine Affimer di legarsi a piccole molecole aumenta la possibilità che esse possano essere utilizzate nelle cellule per spegnere gli effetti di molecole come Shield o doxorubicina che sono attualmente utilizzate per regolare il comportamento delle proteine, consentendo ai ricercatori di valutare gli effetti di spegnimento delle interazioni proteiche con la stessa velocità con cui sono attualmente attivate.
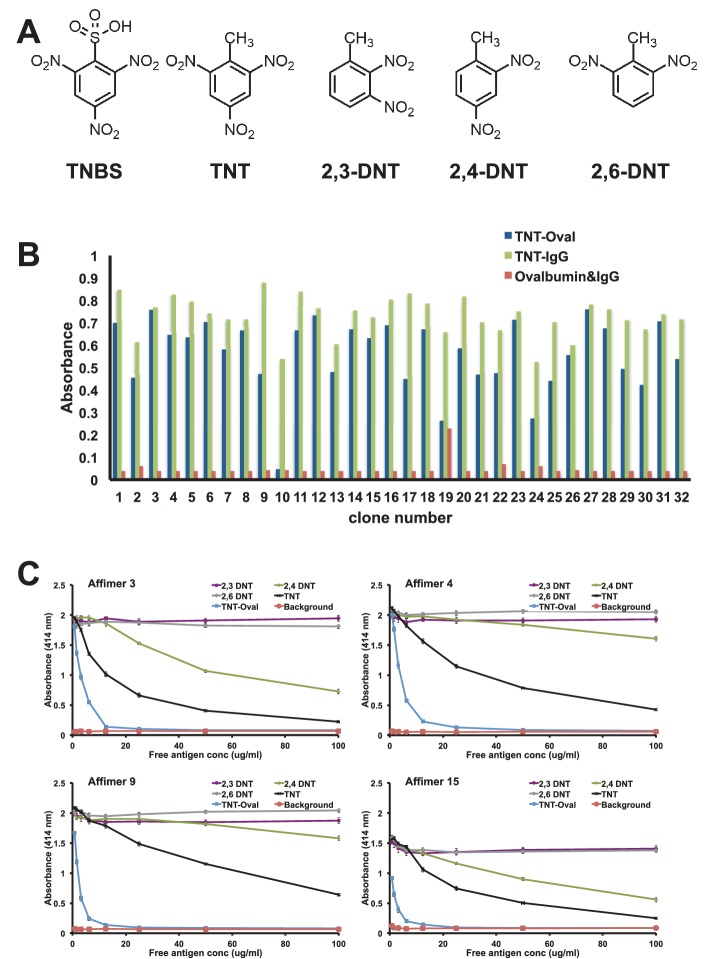
Figura 9.Selezione e specificità dell’affimer contro TNT e DNT.(A) Strutture chimiche di TNBS, TNT, 2,3-DNT, 2,4-DNT e 2,6-DNT.(B) ELISA fago risultati da 32 reattivi monoclonali Affimer isolati contro TNBS legato all’ovoalbumina. La specificità del legame è stata anche testata contro TNBS legato a IgG e a ovoalbumina e IgG non coniugate.(C) ELISA da competizione di quattro TNT-Affimer per verificare la specificità contro una gamma di molecole in un profilo di concentrazione. Barre di errore = deviazione standard dalle ripetizioni tecniche di un ELISA rappresentativo.DOI:
http://dx.doi.org/10.7554/eLife.24903.014
Discussione
La capacità di isolare rapidamente reagenti alternativi altamente specifici per l’affinità delle proteine leganti altamente specifici che si comportano in modo coerente in una vasta gamma di applicazioni scientifiche è il ‘Santo Graal’ per la produzione di reagenti leganti rinnovabili. Per un’impalcatura di proteine leganti artificiali recentemente sviluppata, conosciuta come Affimer, dimostriamo tale applicabilità in una serie di studi molecolari e cellulari. Abbiamo isolato le proteine Affimer contro più di 350 bersagli, ma qui abbiamo esemplificato il loro uso come strumenti molecolari e cellulari contro 12 diverse molecole bersaglio. Tipicamente ogni regime di screening, che consiste normalmente in tre cicli di panning di visualizzazione dei fagi e di ELISA dei fagi, è stato normalmente completato in 12 giorni. Così la selezione Affimer, come con altri approcci alternativi di selezione di proteine o frammenti di anticorpi che utilizzano la visualizzazione dei fagi o altre selezioni in vitro, è molto più veloce rispetto alle tecniche di produzione di anticorpi e nanobrodi che coinvolgono l’inoculazione animale. Essi permettono anche l’identificazione efficiente dei reagenti di legame contro gli epitopi conformazionali, poiché le proteine bersaglio sono lo screening nel loro stato di piegatura.
Ogni regione di codifica Affimer selezionata è stata subclonata in un vettore di espressione di E. coli e la proteina ricombinante è stata purificata per altri sette giorni. Senza automazione, la piattaforma di visualizzazione dei fago permette ad un individuo di schermare fino a 24 bersagli contemporaneamente. Il vantaggio di tale screening manuale è che è possibile esercitare un maggiore controllo per moderare i regimi di screening individuali all’interno di un insieme di campioni. Un ulteriore vantaggio delle proteine Affimer è la capacità di esprimere la proteina ricombinante ad alto rendimento in E. coli. Dei 36 Affimers qui riportati un rendimento medio di 83,3 mg/L (1,5-188 mg/L) coltura è stato raggiunto con una purezza superiore al 95% a seguito di una singola fase di affinità metallica immobilizzata. Non abbiamo cercato di ottimizzare il livello di produzione di proteine, ma in genere si coltivano solo colture da 50 mL per la purificazione delle proteine che forniscono quantità adeguate di proteine per la maggior parte delle applicazioni.
La chiave per il successo dell’isolamento di Affimers per gli studi cellulari è l’uso di antigeni di alta qualità normalmente presentati tramite biotina/streptavidina su piastre e perline. Le fonti ricombinanti di proteine sono normalmente di alta qualità in quanto il tempo e lo sforzo per purificare la proteina sono necessari. Per alcune proteine ricombinanti, in particolare quelle espresse in cellule di mammifero, questo può essere più impegnativo. Abbiamo espresso i piccoli domini SH2 con un peptide accettore di biotina N-terminale per facilitare la biotinilazione in vivo sito-specifica che consente l’immobilizzazione target su piastre di streptavidina, direttamente dal lisato cellulare(Figura 2-figure supplement 1). Questo approccio dovrebbe avere un’ampia applicabilità per la presentazione della proteina ricombinante e del dominio target per i protocolli di screening. Per gli antigeni proteici di origine commerciale (tenascina C, VEGFR2, tubulina e HER4) è diventato evidente che il successo dello screening dipende dalla fonte commerciale. Ad esempio, il tenascin C è stato ottenuto da diversi fornitori commerciali, ma solo uno di essi ha permesso di selezionare gli Affimers adatti (dati non mostrati). Fortunatamente la potenziale disponibilità di migliaia di proteine di alta qualità provenienti da consorzi di genomica strutturale, insieme alla capacità di esprimere in vivo domini proteici biotinilati per la cattura, senza purificazione, dovrebbe ridurre il rischio di fallimento dello schermo associato alla qualità e alla disponibilità dell’obiettivo.
Due lavori hanno descritto la generazione di anticorpi e frammenti di anticorpi contro i domini SH2(Renewable Protein Binder Working Group et al., 2011; Pershad et al., 2010). Il documento iniziale ha utilizzato il display fago per selezionare i reagenti leganti che mostravano una squisita specificità contro i domini SH2 ricombinanti in vitro(Pershad et al., 2010). Tuttavia, nessuno dei reagenti sono stati utilizzati nei saggi per dimostrare il legame alle proteine endogene delle cellule. Colwill et al. hanno valutato la capacità di isolare i reagenti leganti contro la stessa famiglia di bersagli utilizzando sia la visualizzazione fagica di librerie di frammenti di anticorpi che gli anticorpi monoclonali(Renewable Protein Binder Working Group et al., 2011). Hanno anche isolato con successo i reagenti di legame, anche se solo una bassa proporzione di proteine endogene legate nei saggi testati. Al contrario, con la stessa classe di target SH2, un’alta percentuale dei reagenti monoclonali Affimer qui riportati ha avuto successo nei saggi pull down e potrebbe bloccare la funzione proteica quando espressa in cellule. Le differenze tra questi risultati possono essere il risultato della qualità della biblioteca piuttosto che una caratteristica intrinseca dell’impalcatura. Tuttavia, lo consideriamo più probabile a causa delle differenze nella presentazione di strutture ad anello variabili tra gli anticorpi e l’impalcatura di Affimer.
La capacità di esprimere gli Affimers intracellulari in cellule di mammifero, come dimostrato dall’inibizione del dominio p85 SH2(Figura 2), rappresenta un’opportunità entusiasmante, con la mancanza di legami di disolfuro nell’impalcatura adatta all’ambiente riducente della cellula. Questa caratteristica è simile ad altre proteine leganti artificiali che possono essere espresse anche nel citoplasma delle cellule dei mammiferi(Kummer et al., 2012; Spencer-Smith et al., 2017; Wojcik et al., 2010). Ciò solleva l’intrigante possibilità di generare i reagenti necessari, basati su Affimers e altre proteine leganti artificiali, per mirare a specifici domini proteici dell'”interattomo” umano. Questi fornirebbero strumenti estremamente potenti per comprendere la funzione delle proteine e per identificare nuovi bersagli farmacologici nella malattia. La mancanza di legami disolfuro nel Affimer e molte altre proteine artificiali, come DARPins, Monobodies e Affibodies, permette anche l’introduzione diretta di residui di cisteina (s) per la modificazione chimica specifica del sito, compresa l’aggiunta di una singola biotina o fluoroforo.
Uno dei principali vantaggi dello screening dei fagi è la capacità di isolare reagenti altamente specifici eseguendo contro-schermi contro molecole target molto simili. È interessante notare che per i target SH2 non è stato effettuato alcun contro-schermatura, eppure con l’analisi ELISA sono stati recuperati Affimers specifici per i domini Grb2, 7 e 10 e p85 SH2. Ulteriori studi determineranno se questo livello di specificità viene osservato a livello cellulare. Questi risultati, tuttavia, forniscono una promessa per l’isolamento di reagenti altamente specifici di legame cellulare. Questo alto livello di specificità è stato dimostrato anche con gli Affimers che legano il piccolo composto organico TNT comunemente usato come composto organico modello. Gli Affimers hanno rivelato una notevole specificità considerando le piccole dimensioni della molecola (mol. massa <300 Da) e il numero limitato di giri di panning utilizzati. Sarebbe interessante determinare le strutture molecolari di Affimer legate al TNT e al 2,4-DNT per capire il meccanismo di riconoscimento e spiegare la discriminazione tra le diverse molecole di DNT. La possibilità di selezionare rapidamente gli Affimer che rilevano specificamente bersagli di piccole molecole rappresenta un utile approccio aggiuntivo alla generazione di reagenti per applicazioni diagnostiche e di monitoraggio di agenti chimici, ad esempio, in ambito sanitario, di sicurezza o ambientale. Ci sono molti esempi di anticorpi che si legano alle piccole molecole, anche se la natura in vivo dell’allevamento di tali reagenti può presentare delle sfide per alcuni composti, come le tossine e i prodotti farmaceutici, insieme ai tempi di inoculazione e isolamento e alla necessità di utilizzare animali. L’esposizione dei fagi è stata utilizzata anche per isolare i frammenti di anticorpi(Dörsam et al., 1997; Vaughan et al., 1996) e le lipocaline che riconoscono le piccole molecole (Beste et al., 1999; Schlehuber et al., 2000).
La selezione dei reagenti contro le piccole molecole solleva anche la prospettiva di riconoscere una serie di modifiche post-traslazionali, potenzialmente nel contesto di una specifica proteina. Ad esempio, sono state descritte le DARPine che discriminano tra proteine fosforilate e non fosforilate(Kummer et al., 2012). Queste riconoscono i cambiamenti conformazionali dovuti all’evento di fosforilazione piuttosto che all’amminoacido fosforilato. Solo il tempo ci dirà se i reagenti leganti alternativi sono in grado di segnalare direttamente le modifiche post-traslazionali delle proteine in modo simile agli anticorpi.
Le affinità di legame degli Affimers selezionati in questo lavoro erano tipicamente nella gamma nanomolare bassa, anche se alcuni dei leganti VEGFR2 avevano affinità più deboli. Anche così, questi leganti più deboli hanno comunque funzionato efficacemente e hanno mostrato specificità nei saggi istochimici di affinità e hanno inibito la funzione recettoriale nei saggi biologici. Poiché i nostri reagenti monoclonali sono stati identificati selezionando in modo casuale i cloni da tre cicli di panning, si prevede che gli inibitori con una maggiore affinità possano essere sviluppati, sia attraverso un’analisi più dettagliata del pool di fago utilizzando il sequenziamento di prossima generazione, sia attraverso la maturazione dell’affinità. Mentre la nostra strategia di screening identifica i reagenti monoclonali, questi possono anche essere combinati per generare reagenti policlonali che possono migliorare la sensibilità per alcune applicazioni in vitro.
In conclusione, abbiamo dimostrato la capacità di isolare rapidamente i reagenti Affimer, che sono strumenti efficaci in una serie di applicazioni di biologia molecolare e cellulare(Figura 10). Ciò evidenzia la possibilità di creare una pipeline per isolare i reagenti leganti rinnovabili coerenti contro un’ampia varietà di molecole target. Gli Affimers sono piccoli, termostabili e semplici da progettare e forniscono un sistema che completa piuttosto che sostituire gli anticorpi e altre impalcature proteiche alternative. Uno dei principali obiettivi del nostro laboratorio è quello di esplorare ulteriormente le capacità dei reagenti Affimer e di testare le loro potenziali proprietà modulanti delle proteine per l’uso nella dissezione di specifici percorsi di segnalazione delle cellule e nello studio delle interazioni proteine-proteine su scala proteomica. La tecnologia Affimer è disponibile in commercio attraverso Avacta Life Sciences o per collaborazioni accademiche attraverso l’Università di Leeds. In alternativa, la biblioteca può essere sintetizzata come descritto da Tiede et al. (2014) e vagliata nei singoli laboratori, rendendo questa tecnologia immediatamente accessibile alla comunità scientifica.10.7554/eLife.24903.015Figure 10.Overview schema di una serie di applicazioni che sono state testate con Affimers.DOI:http://dx.doi.org/10.7554/eLife.24903.015
Figura 10.Schema generale di una serie di applicazioni che sono state testate con Affimers.DOI:
http://dx.doi.org/10.7554/eLife.24903.015
Materiali e metodi
Espressione e purificazione degli obiettivi
Le sequenze di codifica del dominio umano SH2 erano in vettori AP SacB AP (Open Biosystems) resistenti alla kanamicina pET28, con un tag N-terminale istidina. Una biotina peptide accettore peptide (BAP) sequenza è stata clonata nel vettore per dare un N-terminale BAP-Histag-SH2 dominio sequenza e il DNA vettore modificato vettore introdotto in Rosetta 2 (DE3) cellule. Singole colonie sono state coltivate in 10 ml di Terrific Broth (TB) integrato con 100 µg/ml kanamicina e 34 µg/ml cloramfenicolo, durante la notte a 37 ° C e 2 ml è stato utilizzato per inoculare 400 ml TB/100 µg/ml kanamicina e colture coltivate fino a OD600 ~ 2. Dopo ilraffreddamento a 18 ° C, per 1 ora IPTG è stato aggiunto a 0,5 mM. Le cellule sono state raccolte per centrifugazione (800 g; 20 min, 4 ° C) e risospesi in 10 ml di tampone di lisi 1 (50 mM NaH2PO4; 300 mM NaCl; 30 mM imidazolo; 10% di glicerolo; Benzonase Nucleasi (Novagen); 1% Halt Protease Inhibitor Cocktail, senza EDTA; 1% Triton-X100; 1% lisozima) e lasciato dondolare per tutta la notte a 18°C prima che le proteine target fossero purificate con la resina Amintra Ni-NTA (Expedeon). Le proteine sono state eluite utilizzando tampone di eluizione (50 mM NaH2PO4; 500 mM NaCl; 300 mM imidazolo; 10% glicerolo). L’espressione e la biotinilazione in vivo dei bersagli è stata confermata da western blotting.
Il gene UL49 di Herpesvirus of Turkeys (HVT) è stato amplificato mediante PCR utilizzando la DNA polimerasi Q5 (NEB, UK) e clonato utilizzando il Gibson Assembly kit (NEB) in un pMT-V5/6His modificato (Invitrogen) in cui la cassetta V5/6His è stata sostituita con 6His-AviTag e un sito SmaI che è stato utilizzato per generare una fusione N-terminale al prodotto genetico UL49. pMT HVT UL49 è stato co-trasformato con pCoHygro (Invitrogen) in cellule di Drosophila S2 utilizzando precipitazione di fosfato di calcio e cellule stabilmente trasformate sono state selezionate con igromicina secondo le istruzioni del produttore (Invitrogen) prima del test di espressione da analisi western blot 24 ore dopo l’induzione con 500 µM di solfato di rame. Per la purificazione, la proteina è stata estratta da cellule trasformate stabilmente 36 ore dopo l’induzione con solfato di rame a 20 ° C con un tampone di lisi modificato (25 mM Tris (pH8), 1,5% Triton-X100, 50 mM arginina (pH8), 10 mM imidazolo, 7,5% glicerolo, 300 mM KCl). Le proteine sono state eluite da Ni-NTA (QIAGEN) in tampone di eluizione (25 mM Tris (pH8), 50 mM arginina (pH8), 200 mM imidazolo, 7,5% glicerolo, 300 mM KCl). Proteine purificate sono state sottoposte a biotinilazione in vitro con BirA purificato secondo le istruzioni del produttore (Avidity LLC).
Preparazione di 2,4,6-trinitrobenzene coniugato proteico
Un coniugato proteico di 2,4,6-trinitrobenzene è stato preparato miscelando l’ovoalbumina (frazione VI, Sigma) o la Gamma Globulina di Coniglio (RGG) (Sigma) ad una concentrazione di 1 mg ml-1 con lo 0,05% (p/v) di acido 2,4,6-trinitrobenzene sulfonico (TNBSA) (Thermo Scientific) in tampone bicarbonato di sodio 0,1 M (pH 8,5). La miscela è stata incubata a 37 ° C per 2 ore e il complesso risultante è stato poi tampone scambiato in PBS e concentrato utilizzando un Vivaspin sei colonne (MWCO: 10 kDa; Sartorius) per eliminare TNBSA non coniugato e tampone in eccesso. Il prodotto concentrato è stato quantificato utilizzando un test di Acido Bicinconinico Micro Pierce (BCA) (Thermo Scientific), in conformità con le linee guida dei produttori che utilizzano sieroalbumina bovina come proteina standard comparativa.
Visualizzazione dei fagi
La biotinilazione target e la selezione degli Affimers tramite visualizzazione dei fagi è stata eseguita come descritto in precedenza(Tiede et al., 2014) con alcune modifiche nel secondo e terzo turno di panning. I target biotinilati sono stati legati ai pozzi rivestiti di streptavidina (Pierce) per 1 ora, poi sono stati aggiunti1012 fago cfu pre-pannellati (fago preincubato nei pozzi rivestiti di streptavidina) per 2,5 ore con agitazione. I pozzi in pani sono stati lavati 10 volte e il fago è stato eluito con 50 mM glicina-HCl (pH 2,2) per 10 minuti, neutralizzato con 1 M Tris-HCL (pH 9,1), ulteriormente eluito con trietilammina 100 mM per 6 minuti, e neutralizzato con 1 M Tris-HCl (pH 7). Fago eluito sono stati utilizzati per infettare le cellule ER2738 per 1 ora a 37 ° C e 90 rpm poi placcato su piastre di agar LB agar con 100 µg / ml di carbenicillina e cresciuto durante la notte. Le colonie sono state raschiate in 5 ml di terreno 2TY, inoculate in 25 ml di terreno 2TY con carbenicillina (100 µg/ml) e infettate con circa 1 × 109 M13K07 fago aiutante. Dopo 1 ora a 90 rpm, la kanamicina è stata aggiunta a 25 μg/ml per una notte a 25°C e 170 rpm. Fago sono stati precipitati con il 4% di polietilenglicole 8000, 0,3 M NaCl e risospesi in 1 ml di 10 mM Tris, pH 8,0, 1 mM EDTA (TE buffer). Un’aliquota di 2 µl di sospensione dei fagi è stata utilizzata per il secondo turno di selezione con perle magnetiche di streptavidina (Invitrogen). Perle di destinazione etichettati sono stati lavati e incubati con pre-panned fago per 1 ora e poi lavato cinque volte utilizzando una piattaforma robotica KingFisher KingFisher (ThermoFisher), incubato durante la notte a RT in glicerolo 20% in PBS-T con almeno un ulteriore passo di lavaggio e eluito e amplificato come sopra. La padella finale utilizzato neutravidina piastre ad alta capacità di legame (Pierce), come descritto in precedenza per il panning round uno con l’aggiunta di una incubazione finale durante la notte a RT in glicerolo al 20% in PBS-T, e fago eluito utilizzando 100 µl di 100 mM di ditiotreitolo. Elutati di fagi sono stati recuperati da pozzi contenenti proteine bersaglio e pozzi di controllo per determinare il livello di amplificazione nei pozzi bersaglio. Per le selezioni di contatore sono stati aggiunti al fago lisati cellulari appropriati o proteine omologhe ad una concentrazione di almeno 10 µg/ml per 1 ora a temperatura ambiente prima di trasferire il fago alle perle di panning o ai pozzetti. Sono stati eseguiti dei controschermi per il bersaglio UL49 contro il Virus della Malattia di Marek purificato di tipo 1 (MDV-1, ceppo RB1B) e il virus dell’enterite dell’anatra (ceppo DEV 684), così come il lisato cellulare derivato dai fibroblasti embrionali di pollo (CEF). Il TNBS-Ovoalbumina è stato selezionato contro l’ovoalbumina.
Nella seconda e terza padella, dopo il lavaggio dei pozzi / perline post incubazione con la biblioteca fago, i pozzi / perline sono stati incubati durante la notte a RT in glicerolo al 20% in PBS-T con almeno un passo lavaggio supplementare prima dell’eluizione. I fagi sono stati eluiti in 100 µl 50 mM glicina-HCl (pH 2,2) per 10 minuti, neutralizzati con 15 µl 1 M Tris-HCl (pH 9,1), ulteriormente eluiti con 100 µl di trietilammina 100 mM per 6 minuti, e neutralizzati con 50 µl 1 M Tris-HCl (pH 7). Screening ELISA fago è stato eseguito, come descritto in precedenza, su cloni selezionati in modo casuale dal pan round finale come metodo per la selezione di cloni positivi per un’ulteriore valutazione(Tiede et al., 2014).
Produzione della proteina Affimer
Le regioni di codifica degli affetti selezionati sono state amplificate mediante l’amplificazione di PCR utilizzando uno dei due primer inversi che generano proteine con o senza cisteina del terminale C. A seguito della digestione NheI/NotI le regioni di codifica sono state legate in un vettore derivato da pET11a e successivamente espresse in cellule BL21 (DE3) come descritto in precedenza(Tiede et al., 2014). In breve, una singola colonia è stata utilizzata per inoculare una cultura 5 ml durante la notte in 2TY/100 µg/mL di carbenicillina. Poi 50 ml LB-carb media è stato inoculato con 1 ml di cultura notte e cresciuto per circa 2 ore a 37 ° C e 230 rpm a un OD600 tra 0,6-0,8, prima dell’aggiunta di IPTG a 0,1 mM e ulteriormente cresciuta per 6-8 ore o durante la notte a 25 ° C a 150 rpm. Le cellule sono state raccolte, lisate in 1 ml di tampone di lisi. Il lisato è stato poi incubato con 300 µl di liquame NiNTA lavato per 1 ora, lavato (50 mM NaH2PO4, 500 mM NaCl, 20 mM Imidazolo, pH 7,4) e eluito in 50 mM (NaH2PO4, 500 mM NaCl, 300 mM Imidazolo, 20% glicerolo, pH 7,4).
Biotinilazione degli Affimers
Gli affimers con cisteina C-terminale sono stati biotinilati direttamente dopo l’eluizione. Per ogni Affimer 150 µl di tris (2-carbossietil)fosfina (TCEP) di resina immobilizzata (ThermoFisher Scientific) sono stati lavati e incubati con 150 µl di soluzione di Affimer da 40 µM su un bilanciere per 1 ora. La soluzione è stata centrifugata per 1 minuto a 1500 g e 120 µl del surnatante è stato trasferito in una provetta fresca contenente 6 µl di 2 mM di biotina-maleimmide (Sigma) e incubato per 2 ore a temperatura ambiente. L’eccesso di biotina linker è stato rimosso utilizzando una colonna desalinizzante di spin Zeba (Thermo Scientific) secondo il protocollo del produttore o per dialisi.
Immunoprecipitazione e analisi western blot
Il Western blotting è stato eseguito su domini SH2 con marchio BAP espresso per controllare la biotinilazione in vivo dei bersagli. I campioni di proteine sono stati risospesi in 4 X tampone campione (8% (w / v) SDS, 0,2 M Tris-HCl (pH 7) 20% glicerolo, 1% blu di bromofenolo) e riscaldato a 95 ° C per 10 min. I campioni sono stati caricati su un gel risolvente al 15% di SDS-poliacrilammide con un gel impilabile al 5%. I campioni elettroforesi sono stati poi trasferiti su una membrana in PVDF con pori di 0,2 µm utilizzando un Trans-Blot Turbo Transfer System (Bio-Rad, Hercules, USA). Le membrane sono state bloccate al 3% di BSA in soluzione salina tampone Tris (TBS) contenente lo 0,1% di Tween-20 (TBS-T) durante la notte a 4°C, seguite da incubazione con Streptavidina ad alta sensibilità-HRP (ThermoFisher Scientific) e visualizzate utilizzando Luminata Forte Western HRP Substrate (Merck Millipore).
I lisati cellulari per il pull down di Grb2 sono stati isolati da cellule U-2 OS (acquistate da ATCC, STR profilate e negative al micoplasma – RRID:CVCL_0042). Le cellule sono state lavate con ghiaccio 1 X PBS e lisate in tampone di lisi (50 mM Tris, 150 mM NaCl, 1% (v/v) Nonidet P-40, 1 ml per ogni pallone da 75 cm2), su ghiaccio. L’esperimento di pull down è stato eseguito utilizzando la piattaforma automatizzata KingFisher. In breve, Affimers sono stati espressi in 50 ml di BL21 Star (DE3) IPTG indotta cultura notturna, le cellule BL21 Star (DE3) sono stati pellettato, lisato e chiarito per centrifugazione. Il lisato cellulare è stato incubato con perle magnetiche a base di cobalto (ThermoFisher) su piattaforma Kingfisher per 10 minuti prima di un singolo lavaggio, e incubato con lisato cellulare U-2 OS (circa 500 µg) per 90 minuti. Le perle sono state lavate altre tre volte sul KingFisher prima di essere aggiunte a 50 µl di tampone di eluizione. Un’aliquota di 15 µl di proteine eluite è stata mescolata con il tampone di carico prima di essere riscaldata a 95°C per 5 minuti e caricata su un gel SDS-PAGE. Western blot analisi è stata eseguita utilizzando un coniglio monoclonale anti-Grb2 anticorpo monoclonale, (Abcam; ab32037) come anticorpo primario e anticorpo anti-coniglio-HRP, capra policlonale (Signal Technology; 7074) come secondario (n = 2, replicato biologico; il numero di volte l’esperimento è stato ripetuto indipendentemente).
Per p85 tirare giù esperimenti NIH-3T3 cellule trasfettate con p85-nSH2 Affimers sono stati lisati in CellLytic M Cell Lysis Reagent (Sigma) con cocktail di inibitori della proteasi (Sigma) e lisati cancellati per centrifugazione. Per l’attivazione AKT analisi western blot, le cellule erano affamate di siero per 30 minuti prima della raccolta delle proteine. Immunoprecipitazione di p85α coinvolto incubazione 400 microgrammi di proteina totale con 3 microlitri di anticorpo anti-p85α (Abcam) a 4 ° C con rotazione durante la notte seguita da perle di sefarosio proteina G (Sigma) per 4 ore. Le perle sono stati lavati quattro volte con PBS e risospesi in 2 X SDS campione tampone SDS con β-mercaptoetanolo (Sigma), riscaldato a 95 ° C per 3 min e le proteine sono state risolte in 7% SDS-poliacrilammide gel. Per western blotting, 20 μg di proteine totali è stato denaturato con 5 X Laemmli campione tampone con β-mercaptoetanolo e risolto in 12% SDS-poliacrilamide gel di poliacrilamide. Le proteine sono state trasferite alle membrane di difluoruro di polivinilidene (Biorad), bloccate nel 5% di siero albumina bovina in PBS 0,1% Tween, e incubate con anti-pAKT (Ser473), anti-panAKT, anti-p110α (Cell Signaling Technology), anti-bandiera M2 (Sigma), anti-tubulina alfa (AbD Serotec), e anticorpi anti-p85α. L’anticorpo legato è stato rilevato utilizzando anticorpo antimouse/coniugato con perossidasi di rafano e chemiluminescenza (substrato Luminata Forte Western HRP, Millipore).
Per il rilevamento di VEGFR2 segnalazione HUVECs (cellule endoteliali vascolari ombelicali umane primarie) sono stati lisati nel 2% (w / v) SDS contenente 1 mM PMSF, in PBS). Per l’analisi immunoblot, lisati sono stati incubati a 95 ° C per 5 min e sonicati per 3 s. 25 mg di lisato cellulare è stato risospeso in un volume uguale di 2 X SDS campione tampone (1 M Tris-HCl pH 6.8, 20% (v / v) glicerolo, 4% (w / v) SDS e 0,1% (w / v) blu di bromofenolo, 4% (v / v) mercaptoetanolo) e incubato a 95 ° C per 5 min. I lisati sono stati caricati su un 10% (v / v) SDS-poliacrilammide gel resolving con un 5% (w / v) SDS-poliacrilammide gel impilamento e separati a 130 V per 90 min in SDS-running buffer (192 mM glicina, 25 mM Tris, 0,1% (w / v) SDS). Le proteine sottoposte a SDS-PAGE sono state trasferite sulla membrana di nitrocellulosa (0,2 μm dimensione dei pori) (Schleicher e Schuell) in buffer di trasferimento (106 mM glicina, 25 mM Tris, 0,1% (p/v) SDS, 20% (v/v) metanolo) a 300 mA per 3 ore a 4°C. Le membrane sono state incubate nel 5% (p/v) di latte scremato (in TBS-T) per 30-60 min su un bilanciere. Le membrane sono state risciacquate in TBS-T, incubate con anticorpi primari (capra anti-VEGFR2 (R&D Systems), coniglio anti-phospho-VEGFR2 (Y1175), coniglio anticorpi contro il PLCγ nativo e fosforilato.1 (Y783), c-Akt (S473), p38 MAPK (T180/Y182) ed eNOS (S117), coniglio anti-ERK1/2, mouse anti-phospho-ERK1/2 (T202, Y204) (Cell Signalling Technologies), mouse anti-α-tubulina (Santa Cruz Biotechnology)) per una notte a 4°C e lavata 3 volte per 10 minuti in TBS-T prima dell’incubazione con anticorpo secondario coniugato con HRP (PerBio Sciences, Cramlington, UK) per 1 ora a temperatura ambiente, seguita da un secondo ciclo di lavaggio TBS-T e rilevamento con una soluzione chemiluminescente, EZ-ECL (Geneflow, Nottingham, UK).
Immuno- e Affinità-fluorescenza per TRPV1
Le cellule U2-OS sono state coltivate in mezzo DMEM integrato con il 10% (v/v) di siero bovino fetale (ThermoFisher Scientific), 2 mM L-glutammina, 1% penicillina-streptomicina, 10 mM piruvato di sodio al 5% diCO2 in aria a 37 ° C. Le cellule sono state seminate su vetrini coprioggetto in piastre a 24 pozzetti per raggiungere una densità di ~60% al momento della trasfezione. Le cellule sono state trasfettate con ratto TRPV1- codifica del DNA di ratto 48 ore prima dell’uso in esperimenti ICC utilizzando FuGENE Reagente Trasfezione Reagente (Promega) secondo le istruzioni dei produttori. A 48 ore dopo la trasfezione, Affimers sono stati incubati su cellule vive ad una concentrazione finale di 5 μg / ml in tampone di analisi (130 mM NaCl, 10 mM di glucosio, 5 mM KCl, 2 mM CaCl2, 1,2 mM MgCl2, 10mM HEPES, pH 7,4) per 20 min. Le cellule sono state poi lavate con lo stesso tampone tre volte con lo stesso tampone per 5 min a lavaggio prima della fissazione con il 4% di PFA per 10 min. Le cellule fisse sono stati lavati tre volte in PBS e poi permeabilizzato con 0,1% Triton X100 in PBS. Le cellule sono state bloccate per 30 min in 1% BSA in PBS a temperatura ambiente. Mouse monoclonale anti-6X Il suo anticorpo (Abcam: Ab18184) è stato incubato sulle cellule per un’ora a temperatura ambiente. Le cellule sono state poi lavate tre volte con PBS e incubate per un’ora al buio con l’anticorpo anti-topo 488 (Life Technologies: A11001) per 1 ora. Altri due lavaggi in PBS-T, due lavaggi in PBS e un lavaggio in ddH20 sono stati seguiti dal montaggio su vetrini di vetro con ProLong Diamond Antifade Mountant con DAPI (ThermoFisher Scientific). Il giorno successivo, i campioni sono stati ripresi utilizzando un sistema di imaging EVOS FL (ThermoFisher Scientific).
Per la colorazione di co-localizzazione con anticorpo anti-TRPV1, lo stesso protocollo è stato seguito con un ulteriore passaggio in cui è stata condotta un’ulteriore incubazione di 1 ora con anticorpo anti-TRPV1 (Abcam: ab10295) post-fissazione. Capra anti-guinea pig 647 (Abcam: ab150187) è stato poi applicato per la rilevazione di anticorpo anti-TRPV1 anticorpo per un’ora a temperatura ambiente al buio.
Saggio di modulazione TRPV1
Le cellule U2-OS sono state coltivate utilizzando le condizioni di cui sopra. Le cellule sono state seminate in fiaschi T75cm in modo che la confluenza avrebbe raggiunto ~ 60% al momento della trasfezione. 24 ore post-trasfezione con TRPV1 DNA, le cellule sono state tripsinizzate in a pareti nere 96 ben piastre (Greiner Bio-One) ad una confluenza di ~ 100.000 cellule per pozzo e incubato per un ulteriore 24 ore in condizioni di coltura cellulare precedentemente descritto.
Il giorno del test di modulazione, le cellule sono state lavate con tampone di analisi e caricate con 50 μL Fluo-4 AM (1 μM) (ThermoFisher Scientific: F14201) per 1 ora a 37 ° C. Le cellule sono state lavate di nuovo con 200 μL di tampone di analisi prima dell’aggiunta di Affimer ad una concentrazione di 1 μM in 50 μL di tampone di analisi. Dopo 30 minuti di incubazione a temperatura ambiente, l’aumento di Ca2+ intracellulare è stato misurato utilizzando una Flexstation 3 (Dispositivi Molecolari; Sunnyvale, CA, USA). La fluorescenza è stata rilevata per 60 sec a 485 nm di eccitazione e 525 nm di emissione, ma il picco di risposta Ca2 + (circa 5 sec dopo l’aggiunta dell’agonista ortoosterico TRPV1, capsaicina) è stato utilizzato per la successiva determinazione della risposta dell’agonista. Inizialmente, l’effetto di Affimers sulla risposta di Ca2+ è stato testato ad una concentrazione di capsaicina EC20, mentre gli esperimenti successivi hanno testato una gamma di concentrazioni di capsaicina in un cosiddetto test di spostamento della curva. Le unità di fluorescenza di picco relativo sono state normalizzate alla risposta osservata in assenza di Affimer. L’analisi dei dati – l’ANOVA ordinario a senso unico con confronti multipli è stata condotta per i saggi di modulazione contro una concentrazione di capsaicina EC20. Sono indicati valori P inferiori a 0,05 (*) 0,01 (**) e 0,001 (***) e 0,0001 (****).
Caratterizzazione del legame con gli analoghi del TNT e del DNT da parte della concorrenza ELISA
Immulon2 HB 96-pozzetti di microtitolazione a 96 pozzetti (Nunc) sono stati rivestiti con coniugato TNBS-ovalbumina ad una concentrazione di 10 µg ml-1 in PBS ( 100 µl per pozzetto) e incubati durante la notte a 5°C. Ogni pozzetto è stato lavato tre volte con PBS contenente 0,05 % v/v Tween20 (PBST) prima del blocco utilizzando il 2% (w / v) latte scremato in polvere (Marvel) in PBST (tampone di blocco), incubando per 60 minuti a temperatura ambiente. Ogni pozzetto è stato poi lavato e l’analogo TNBS-Ovalbumina, TNT o DNT libero è stato diluito in tampone di blocco e aggiunto a 6 pozzetti di replicazione per ogni Affimer ad una concentrazione di 100 µg/ml. Ogni hapten è stato poi diluito in serie lungo la piastra ad una concentrazione finale di 0,78 µg/ml in tampone di blocco. Successivamente, 50 µl di biotinilati Affimers TNT3, 4 e 9 (0,5 µg/ml) e TNT15 (1,0 µg/ml) in tampone di blocco è stato aggiunto in replicare a ciascuno dei pozzetti contenenti hapten libero per consentire sei curve di diluizione replica tecnica di ogni molecola hapten libero con ogni Affimer da generare (replica tecnica; il numero di volte l’esperimento è stato ripetuto all’interno di un esperimento). Per il controllo negativo, l’Affimer è stato sostituito con il buffer di blocco. Ogni piastra è stata incubata a temperatura ambiente per 1 ora, lavata tre volte con PBST e l’Affimer che è rimasto legato al coniugato TNBS-Ovalbumina è stato rilevato utilizzando un coniugato ad alta sensibilità streptavidina-HRP (ThermoFisher Scientific), diluito 1:2000 in tampone di blocco. La presenza di HRP è stata rilevata utilizzando perossido di idrogeno (Sigma) e substrato ABTS (Sigma) nel tampone del substrato (0,1 M di acido citrico, 0,2 M di Na2HPO4 a pH 4,37) con la risposta quantificata sulla base di letture a 414 nm in un lettore automatico di piastre (Anthos 2001, Anthos Labtec Instruments).
Immuno- e Affinità-istochimica per tenascina C
SW620 (micoplasma testato – RRID:CVCL_0547) sono stati sacrificati topi xenotrapianti; i tumori sono stati raccolti e incorporati nella cera di paraffina. Tessuto xenotrapianto è stato poi elaborato per l’immunoistochimica tenascina C come segue. In breve, 4 sezioni di paraffina μm sono stati tagliati e raccolti su vetrini rivestiti in polilisina. Le sezioni sono state decerate in soluzioni di xilene e reidratate in alcool graduato seguito da acqua distillata. Il recupero dell’antigene è stato effettuato mediante cottura a pressione in tampone di acido citrico 0,01 M, pH 6,0. A seguito di recupero dell’antigene, sezioni di tessuto sono stati lavati in acqua distillata e perossidasi endogena è stata bloccata con Bloxall bloccando reagente (10 min, SP-6000; Vector Laboratories Ltd, Peterborough, Regno Unito). Dopo il lavaggio con TBS-T, l’avidina endogena e la biotina sono state bloccate con il kit di blocco di avidina e biotina (SP-2001 Vector Laboratories Ltd, Peterborough, UK). Le sezioni di tessuto sono state lavate in TBS-T e i siti di legame proteico non specifici sono stati bloccati utilizzando 1 X caseina (20 min, SP-5020; Vector Laboratories Ltd, Peterborough, UK) preparato in diluente anticorpale (Sigma, Poole, UK). Le sezioni sono state poi incubate durante la notte (4°C) in anticorpo monoclonale anti-TNC di topo (1:25, 4F10TT, IBL, USA) e l’anticorpo legato è stato rilevato utilizzando il kit IHC polimero di topo sul topo (ab127055, Abcam, Cambridge, UK) secondo le istruzioni del produttore. Le sezioni di tessuto sono state contornate con ematossilina, disidratate, eliminate e montate in DPX. Le immagini sono state catturate con un microscopio Axioplan Zeiss e il software AxioVision 4.8 (Carl Zeiss Inc. Germania).
Colorazione dei tessuti utilizzando il TNC Affimer è stata eseguita in modo simile in ogni caso, dopo aver bloccato i siti non specifici di legame proteico con la caseina, le sezioni sono state incubate in TNC-Affimer biotinilato durante la notte a 4 ° C. Le sezioni sono state poi lavate in TBS-T e legato Affimer è stato visualizzato utilizzando Streptavidina / HRP (1:300, 30 min, SA-5004;, Vector Laboratories Ltd, Peterborough, UK) con 3,3′-diamminobenzidina (DAB) come substrato (SK41-05; ImmPACT DAB, Vector Laboratories Ltd, Peterborough, UK). Le sezioni sono state contornate e riprodotte come descritto in precedenza.
Immuno- e Affinità-istochimica per VEGFR2
Cera incorporato sezioni di tessuto di tessuto pancreatico umano sono stati raccolti e trattati per l’immunocolorazione in modo simile a quello descritto in precedenza. Dopo la decerizzazione e la reidratazione delle sezioni di tessuto, il recupero dell’antigene per l’epitopo VEGFR-2 è stato effettuato utilizzando il tampone Tris EDTA (pH 9,0). Perossidasi endogena, Avidina/Biotina e proteine sono state bloccate come descritto e le sezioni tissutali sono state incubate durante la notte a 4°C in coniglio monoclonale anticorpo anti VEGFR2 (1:25, clone 55B11, Cell Signaling Technology, Danvers, USA) o VEGFR2 Affimers (1-11 µg/ml). L’anticorpo legato è stato visualizzato utilizzando l’anticorpo policlonale di capra anti-coniglio biotinilato (1:200, 30 min, clone E0432, DAKO, UK) e Streptavidina/HRP con DAB come substrato. Gli Affimers sono stati visualizzati utilizzando Streptavidin/HRP con DAB come substrato. Le sezioni sono state contornate e visualizzate come descritto in precedenza (n = 3).
Coltura cellulare e trasfezione
Le cellule MCF7 (acquistate da ECACC, STR profilate e micoplasma negative – RRID:CVCL_0031) sono state coltivate in RPMI medium integrato con il 10% (v/v) di siero bovino fetale (ThermoFisher Scientific), 2 mM L-glutammina, 1% penicillina-streptomicina, 10 mM piruvato di sodio al 5% diCO2 in aria a 37°C. I fibroblasti di topo, NIH-3T3, sono stati coltivati in Dulbecco Modified Eagle’s Medium (Sigma) con 10% FCS e 2 mM L-glutammina in atmosfera umidificata a 37°C al 5% di CO2. Gli affimers specifici per il dominio N-terminale SH2 di p85 sono stati trasferiti in NIH-3T3 utilizzando TransIT 293 (Mirus, Madison, USA) secondo le istruzioni dei produttori.
Le cellule endoteliali della vena ombelicale umana (HUVEC) sono state isolate e coltivate in mezzo di crescita delle cellule endoteliali (ECGM). Cordoni ombelicali umani utilizzati per l’isolamento delle cellule endoteliali primarie sono stati forniti con il consenso informato scritto in conformità con le linee guida etiche e sotto l’approvazione etica (riferimento CA03/020) del Leeds NHS Hospitals Local Ethics Committee (UK). Gli HUVEC sono stati seminati in piastre a 6 pozzetti e coltivati (per almeno 24 ore) in ECGM fino a ~80% confluente, lavati due volte in PBS e poi affamati in MCDB131 più 0,2% (p/v) BSA per 2-3 ore. HUVECS sono stati trattati con 0, 50, 100 o 150 μg/ml Affimer per 30 min prima della stimolazione con 25 ng/ml VEGF-A (Genentech Inc., San Francisco, USA) per 0, 5 o 15 min.
I fibroblasti embrionali di pollo embrionale (CEF) sono stati derivati da embrioni di 10 giorni e mantenuti in mezzo E199 (Sigma), integrati con 10% di brodo di triptosio fosfato (BD) e 5% di siero fetale di vitello (Sigma), a 38,5°C. Le infezioni virali sono state generate dalla trasfezione di lipofectamina di cloni BAC di MDV-1 (ceppo RB1B), HVT o DEV (ceppo 2085). In breve, 2 µg di DNA BAC sono stati diluiti in 100 µl di reagente Opti-MEM, e mescolati con 10 µl di lipofectamina (Invitrogen) diluita in 100 µl Opti-MEM (Invitrogen). Dopo la formazione complessa per 30 minuti un ulteriore 800 µl di Opti-MEM è stato aggiunto e il campione di 1 ml trasferito su un pozzo di una piastra a sei pozzetti contenente CEF che era stato risciacquato due volte con Opti-MEM. Complessi di DNA sono stati lasciati su CEF per 6 ore in condizioni di coltura normali, dopo di che 2 ml di terreno di crescita è stato aggiunto per ogni pozzetto e restituito all’incubatrice. Una volta che la replicazione del virus è stato stabilito cellule infette sono state passate su CEF freschi come richiesto per mantenere l’infezione.
Affinità-fluorescenza e analisi occidentale in cella
Per gli esperimenti di immunofluorescenza e gli esperimenti occidentali in cella i CEF primari sono stati infettati con MDV-1 (ceppo RB1B), HVT, o DEV (ceppo 2085). In tutti i casi il virus è stato derivato da cloni BAC per trasfezione in CEF, questi virus esprimono costitutivamente un marcatore GFP sotto il controllo del promotore della timidina chinasi presente all’interno dell’elemento BAC. Per gli studi in-cellulari occidentali, le cellule infette sono state seminate in 96 piastre di pozzo e sono state lasciate aderire durante la notte. Le cellule sono state poi fissate con il 4% di paraformaldeide in PBS, lavate con PBS, permeabilizzate con lo 0,1% di TritonX100 in PBS e bloccate con lo 0,5% di BSA in PBS per 30 minuti a temperatura ambiente. Affimers sono stati poi aggiunti a 1,5 µg/ml o un anticorpo policlonale di capra anti-GFP (Sicgen) a 1:2000 diluizione in tampone di blocco e incubato a temperatura ambiente per 1 ora. I campioni sono stati poi ampiamente lavati con PBSa prima che l’anticorpo secondario in tampone di blocco è stato aggiunto. Per il Western in-cellula, l’anti capra asino 680 è stato utilizzato a diluizione 1:5000, con coniugato Streptavidina 800 coniugato a diluizione 1:5000 (Licor), e incubato per 1 ora a temperatura ambiente, poi ampiamente lavato con PBS. I western in cella sono stati ripresi con il sistema Licor Odyssey utilizzando sia i canali 700 nm che 800 nm secondo le raccomandazioni del produttore. Le immagini sono state esportate dal software proprietario del produttore ed elaborate utilizzando Adobe Illustrator. Per l’immunofluorescenza lo stesso approccio è stato seguito dopo la semina delle cellule per i coprioggetti. I coprioggetti sono stati incubati con Affimers come unico reagente di rilevamento primario, con successiva etichettatura con streptavidina-568 coniugato (Invitrogen) alla diluizione 1:1000. Dopo aver lavato via l’eccesso di streptavidina-568, le cellule sono state colorate con DAPI, seguite da tre lavaggi ad acqua deionizzata prima del montaggio su un mezzo di montaggio Vectashield (Vector Laboratories). Le immagini in immunofluorescenza sono state acquisite utilizzando un sistema Leica SP5 e il software del produttore, dal canale 488 nm, 568 nm e 405 nm utilizzando l’obiettivo 63 x e trattate come per le immagini occidentali in cella.
Determinazione delle costanti di equilibrio di dissociazione
I chip di accoppiamento amminico (chip sensore CM5, GE Healthcare) sono stati innescati in 0,1 M di acetato di sodio a pH 5,6 e funzionalizzati con EDC/NHS35 µl a 5 µL/min. Proteina target (1 mg / ml) è stato immobilizzato in una cella di flusso (300-600 unità di risposta), ad una velocità di flusso di 5 µL / min e la cella di flusso è stato tappato con 1 M etanolamina HCl (35 µL a 5 µL / min). La cella di flusso non funzionalizzata (che agisce come un vuoto) è stato trattato con EDC / NHS (35 µL a 5 µL / min) e 1 M etanolamina HCl (35 µL a 5 µL / min. Il sistema è stato innescato in PBS integrato con 0,1% TritonX 100. Cinque concentrazioni di Affimer (5-500 nM) sono stati testati. Ogni concentrazione è stata fatta scorrere sia sulle cellule di flusso funzionalizzate che su quelle non funzionalizzate a 40 μL/min e le costanti di associazione e di dissociazione ka e kd ,rispettivamente sono state calcolate utilizzando il software Biacore che permette di determinare la costante di dissociazione dell’equilibrio, KD come below.d[AB]/-dt=ka[A][B]∙d[AB]/dt=kd[AB]KD=kd/ka
I valori di KD sono stati determinati anche con l’interferometro Octet Red (Pall Fortebio) utilizzando i biosensori rivestiti di streptavidina (AMC, 18-5019) come descritto in precedenza (Kumaraswamye Tobias, 2015). Tutti gli esperimenti sono stati effettuati in tampone HBS-EP (10 mM HEPES (pH 7,4), 150 mM NaCl, 3 mM EDTA, 0,005% (v/v) Tween 20). I valori di KD sono stati determinati legando ogni Affimer biotinilato ad una fila di biosensori AMC ad una concentrazione costante di 50 nM. Successivamente una diluizione di 2 volte di proteina purificata non etichettata a partire da 41 nM è stata legata agli Affimer. I valori di offset grezzi sono stati tracciati contro la concentrazione di proteina purificata HVT UL49, determinata dalla densitometria, e modellati in un’equazione di legame specifica del sito utilizzando Graphpad Prism 6.
Etichettatura degli Affimers con rosso rodamina
I residui di cisteina C-terminale di Affimers TNC15C e GFP32C sono stati etichettati con Rhodamine Red C2 maleimide (Thermo Fisher Scientific). Campioni di Affimer (TNC15C o GFP32C) (80-200 µM) in tampone di eluizione (50 mM NaH2PO4, 500 mM NaCl, 300 mM imidazolo, 10% glicerolo, pH 7,4) sono stati dializzati (2 X con una diluizione di 1000x) in tampone di etichettatura (PBS contenente il 20% di glicerolo e 0,05% Tween-20; pH 7,4). I campioni sono stati poi trattati con TCEP inH2Oa 2,5 mM e Rhodamine Red C2 maleimide (20 mM in DMSO; 5 eq.) e cullati per 6 ore. Al termine della valutazione con la spettrometria di massa, le reazioni sono state estinte con β-mercaptoetanolo (100 eq.) e la miscela è stata concentrata in spin (3 kDa cut-off). La miscela concentrata è stata passata attraverso una colonna di scambio tampone (PD-10, GE Healthcare), eluizione di 0,5 mL frazioni con tampone di etichettatura. Le frazioni contenenti proteine sono state identificate con il saggio colorimetrico BioRad e messe in comune facendo attenzione a non includere le frazioni contenenti il colorante libero Rosso Rodamina che eluisce in seguito. Gli Affimers etichettati sono stati poi concentrati a 250-300 μM in un concentratore di spin (3 kDa cut-off). Le concentrazioni sono state stimate da analisi SDS-PAGE contro quantità note di BSA come standard. Le identità degli Affimers etichettati sono state confermate dalla spettrometria di massa. I campioni sono stati utilizzati immediatamente o flash congelati in azoto liquido e conservati a -80°C fino a quando richiesto.
Lavoro in vivo
Tutte le procedure sono state eseguite in conformità con la legge sugli animali (procedure scientifiche) del 1986 sotto l’approvazione della licenza di progetto (PPL 70/7965). L’esame e il monitoraggio etico sono stati effettuati dal Comitato per l’esame etico e il benessere degli animali (AWERC) dell’Università di Leeds. Ventiquattro topi femmina nudi BALB/c di 6-10 settimane (originariamente ottenuti da Charles River, Regno Unito, poi mantenuti in casa) sono stati iniettati per via sottocutanea nel fianco destro con 1 × 107 cellule SW620 (ottenute dall’ECACC e verificate con un’analisi di ripetizione tandem singola). Dopo 10-14 giorni di crescita tumorale, gli animali sono stati randomizzati per ricevere o il tenascin C Affimer o un controllo GFP Affimer coniugato con Rhodamine Red C2 maleimide, tramite iniezione della vena caudale (volume tumorale medio tenascin C Affimer gruppo 316,9 ± 192,0 mm3 vs 360,8 ± 216,6 mm3 nel controllo GFP Affimer gruppo; p=0, 64). Circa 300 µM etichettati Affimer in 100 μL PBS con il 20% di glicerolo e lo 0,05% di Tween-20 è stato iniettato per via endovenosa in ogni animale. Immagini in fluorescenza di tessuti raccolti (tumore, fegato, rene, milza, cuore, polmone e cervello) sono stati catturati ex vivo utilizzando IVIS Spectrum (eccitazione 570 nm, emissione 620 nm; Perkin Elmer, USA) Intensità di fluorescenza (efficienza radiante in p/s/cm2/sr/μW/cm2) per ogni tessuto è stato determinato per una regione di interesse di area unitaria definita utilizzando il software Living Image (v4.3.1, Perkin Elmer). L’intensità media della fluorescenza di fondo è stata normalizzata per simulare tumori e organi di controllo iniettati.
Metodi di microscopia a super-risoluzione
HER4: a) Etichettatura delle celle per le curve di legame
Chinese Hamster Ovary (CHO) cellule (STR profilato e micoplasma testato) sono stati seminati ad una densità di 0,75 × 105 cellule piatto su piatto non rivestito 35 mm n. 1,5 piatti a fondo di vetro (MatTek Corporation, USA). Le cellule sono state trasfettate con HER4-CYT-eGFP il giorno successivo e sono state affamate di siero durante la notte il secondo giorno dopo la trasfezione. Le MCF7 sono state seminate e coltivate per due giorni, prima di morire di fame durante la notte.
Le cellule sono state risciacquate e raffreddate a 4°C per 10 minuti, poi etichettate con HER4 Affimer etichettato con CF640R (Biotium) in concentrazioni che vanno da 1 a 100 nM (CHO/HER4-CYT-eGFP) o da 10 a 500 nM (MCF7). L’etichettatura è stata effettuata a 4°C per 1 ora. Le cellule sono state risciacquate e fissate con il 3% di paraformaldeide più lo 0,5% di glutaraldeide per 15 minuti a 4°C e poi 15 minuti a temperatura ambiente. L’imaging è stato eseguito in PBS utilizzando la modalità confocale standard in una Leica TCS sp8 (vedere l’analisi dei dati delle curve di legame qui sotto).
HER4: (b) Analisi dei dati delle curve di legame
Le immagini in fluorescenza acquisite in modalità conteggio fotoni sono state analizzate utilizzando le Fiji (ImageJ). I segnali della membrana sono stati isolati utilizzando la segmentazione della soglia di intensità seguita dalla dilatazione della maschera binaria risultante. Per ogni concentrazione di Affimer, i valori dei pixel delle aree di membrana segmentate di cinque regioni immaginarie sono stati combinati per produrre un istogramma dei valori dei pixel. È stata tracciata la deviazione media e standard delle distribuzioni dei valori dei pixel.
HER4: c) Etichettatura delle celle per l’imaging a super risoluzione
Le cellule CHO (1 × 105) sono state seminate in piatti a fondo di vetro non trattati da 35 mm ad alta precisione (MatTek Corporation, USA) e sottoposte a trasfezione inversa con HER4-CYT-eGFP in un rapporto di 4 ul ViaFect (Promega) a 1 ug DNA.due giorni dopo le cellule sono state etichettate con 100 nM HER4 Affimer5 coniugate con Alexa 647 per 1 ora a 4°C. Le cellule sono state risciacquate e fissate con il 3% di paraformaldeide più lo 0,5% di glutaraldeide per 15 minuti a 4°C e poi 15 minuti a temperatura ambiente prima del risciacquo.
HER4: (d) dSTORM imaging
Le immagini in super risoluzione dSTORM sono state scattate in un sistema Zeiss Elyra PS.1. Il fluoroforo Alexa Fluor 647 è stato foto-commutato utilizzando laser ad eccitazione 642 nm e 405 nm contemporaneamente, con 100 mM ditiotreitolo in PBS come buffer di commutazione. La densità di potenza dell’illuminazione a 642 nm e 405 nm sul piano del campione era di circa 4,6 kW/cm2 e 0,4 kW/cm2. Per l’imaging sono stati utilizzati un obiettivo ad immersione ad olio 100× NA 1,46 (Zeiss alpha Plan-Apochromat) e un filtro dicroico multi-banda (BP 420-480+ LP 650). Le immagini fluorescenti finali sono state proiettate su una telecamera EMCCD Andor iXon 897. Le immagini dSTORM a super risoluzione sono state ricostruite nel software ZEISS ZEN. Una precisione di localizzazione di 25 nm si ottiene secondo l’istogramma della precisione di localizzazione calcolata da ogni fluoroforo.
HER4: e) Etichettatura delle celle per esperimenti di tracciamento di singole molecole
Le cellule MCF7 sono state seminate alla densità di 3 × 105 celle/piatto su piatti a fondo di vetro da 35 mm. Ogni piatto contiene un fondo di vetro ad alta precisione n. 1,5 ± 0,170 di 14 mm di diametro (MatTek Corporation, USA). I piatti sono stati puliti con la soluzione Piranha e rivestiti con 1% di BSA, secondo il protocollo precedentemente descritto(Zanetti-Domingues et al., 2012). Prima dell’imaging, le cellule sono state affamate per 2 ore a 37°C in mezzo libero di siero integrato con 25 mM HEPES. Dopo la fame, le cellule sono state risciacquate due volte con mezzo libero di siero preriscaldato a 37 ° C. L’etichettatura con Affimer fluorescente è stata effettuata per 10 minuti a 37°C. HER4 Affimer 5 (HER4-5) è stato coniugato internamente con CF640R (Biotium) tintura maleimmide seguendo le istruzioni del produttore. Le cellule sono state risciacquate due volte con un mezzo libero da siero preriscaldato a 37°C e prontamente ripreso come descritto di seguito.
HER4: (f) Acquisizione di una singola molecola
Le immagini a singola molecola sono state acquisite utilizzando un microscopio Axiovert 200M con un illuminatore TIRF (Zeiss, UK) e incorporando un obiettivo a immersione in olio 100 x (α-Plan-Fluar, NA = 1,45; Zeiss, UK) e un EMCCD (iXon X3; Andor, UK). I campioni sono stati illuminati con un laser a 638 nm (100 mW, Vortran) alimentato al microscopio attraverso una polarizzazione mantenendo un combinatore laser triplo (Oz Optics) . In alternativa, sono state utilizzate le linee da 640 nm di un combinatore Vortran o di un combinatore laser Andor Revolution. Un incubatore avvolgente (Pecon XL S1) è stato utilizzato per mantenere una temperatura costante di 37°C. Il campo visivo di ogni canale era di 80 × 30 μm. I dati sono stati acquisiti a 20 Hz per 30 s. Le immagini sono state salvate in formato HDF5 per la successiva elaborazione con un software progettato su misura. Tutti i dati delle serie temporali a singola molecola sono stati analizzati utilizzando il software di analisi multidimensionale descritto in precedenza(Rolfe et al., 2011).
Microscopia di ricostruzione ottica stocastica diretta della tubulina 3D (3D dSTORM)
a) Preparazione del campione
L’anticorpo monoclonale purificato di ratto antitubulina IgG2a, clone YL1/2, (BioRad Antibodies) descritto in precedenza(Kilmartin et al., 1982; Wehland et al., 1983) è stato direttamente etichettato con Alexa Fluor 647. In breve, 20 μg di anticorpo è stato incubato con PBS contenente 120 mM NaHCO3 e 0,4 μg di estere di succinimidile dell’acido carbossilico Alexa Fluor 647 (A37573, Life Technologies Inc.) per 30 minuti a temperatura ambiente. Il colorante non incorporato è stato rimosso mediante filtrazione su gel utilizzando colonne NAP-5 (17-0853-02, GE Healthcare) seguendo il protocollo dei produttori. Rapporti di etichettatura anticorpo:colorante di circa 1:1 sono stati confermati da assorbanza misurata in uno spettrofotometro, con una concentrazione finale di 0,2 μg/μl. La cisteina C-terminale di Affimer 32, sollevata contro la tubulina, è stata etichettata immediatamente dopo la purificazione con il derivato maleimmide di Alexa Fluor 647 (Thermo Fisher Scientific). In breve, 150 μl di gel disolfuro-riducente TCEP immobilizzato è stato lavato tre volte con PBS contenente 1 mM EDTA prima di essere risospeso in 4 μl di PBS contenente 50 mM EDTA. Il gel è stato incubato con 150 μl di Affimer preparato a 0,5 mg/ml in PBS per 1 ora a temperatura ambiente per ridurre la cisteina pronta per l’etichettatura. L’Affimer ridotto è stato centrifugato a 1000 giri al minuto per 1 min per pellet il gel e 130 microlitri di supernatante contenente l’Affimer è stato mescolato con 6 microlitri di un 2 mM Alexa-647 maleimmide stock e incubato a temperatura ambiente per 2 ore. Unbound Alexa Fluor 647 è stato rimosso passando l’Affimer etichettato attraverso una colonna di desalinizzazione Zeba Spin Desalting Column, 7K MWCO (Thermo Scientific) secondo le istruzioni del produttore. L’Affimer etichettato è stato conservato a 4°C ad una concentrazione finale di 0,5 mg/ml.
Coprivetrini (# 1,5, 25 mm di diametro; Forniture Laboratorio Scientifico, MIC3350) sono stati puliti come descritto in precedenza(Shroff et al., 2007). Le cellule HeLa sono state seminate a 2 × 105 cellule per coprioggetto in piatti di coltura di 30 mm di diametro in DMEM (Gibco) integrato con 10% FCS, 1% P / S e incubato a 37 ° C, 5% di CO2 per 24 ore. Le cellule sono state fissate in paraformaldeide 2% (PFA) disciolto in tampone PEM (80 mM PIPES pH 6,8, 5 mM EGTA, 2 mM MgCl2) integrato con 0,1% di glutaraldeide per 20 minuti a temperatura ambiente prima di essere elaborato per la colorazione di immunofluorescenza. Le cellule sono state permeabilizzate con lo 0,5% di Triton X-100 per 5 minuti, lavate tre volte con PBS prima del blocco con il 5% di BSA in PBS per 1 ora. Le cellule sono state incubate con anticorpi anti-tubulina direttamente etichettati (1:20) o etichettati Affimer (1:2000) preparati in PBS integrato con 0,25% BSA per 1 ora a temperatura ambiente. I coprivetrini sono stati lavati tre volte prima dell’imaging. Le diluizioni dell’anticorpo e dell’Affimer sono state precedentemente ottimizzate per dare le migliori prestazioni in dSTORM. Per l’imaging confocale standard più piccolo (13 mm di diametro) sono stati utilizzati coprivetrini, la diluizione degli anticorpi e Affimer è stata di 1/500 e 1/100 rispettivamente, e un anticorpo secondario (etichettato con Alexa Fluor 488, Life Technologies) per l’anticorpo anti-tubulina di ratto è stato utilizzato a 1:400. La procedura di fissazione e colorazione era la stessa, e i coprioggetti sono stati montati in Antifade Pro-long prima dell’imaging su uno Zeiss LSM880 confocale, equipaggiato con un Airyscan.
Per l’imaging dSTORM, i coprioggetti sono stati montati nelle camere di imaging e posizionati sul microscopio. Coprivetrini sono stati poi incubati con 0,01% poli-L-lisina (Sigma-Aldrich, P4707) per 10 minuti, seguita da una sospensione di nanoparticelle d’oro 150 nm (Sigma-Aldrich, 742058, 1:10 in PBS), che servono come emettitori di riferimento stabile per le immagini di calibrazione e la correzione della deriva. dati dSTORM è stato acquisito in presenza di fluorescenza quenching buffer(Heilemann et al., 2009, 2008) (PBS (pH 8), 5 mg/ml di glucosio, 114 mM β-mercaptoetanolo, 0,5 mg/ml di glucosio ossidasi e 40 μg/ml di catalisi).
b) Acquisizione e ricostruzione di immagini
Il sistema 3D dSTORM era basato su un microscopio invertito (Olympus, IX81) come descritto in precedenza(Lambacher et al., 2016), con una lente 60x, 1.2 NA, obiettivo a immersione (Olympus, UPLSAPO60XW), e una lente cilindrica con f= 150 mm (Thorlabs, LJ1629RM-A) per generare astigmatismo. I laser a 642 nm e 405 nm (Omicron, LuxX) hanno fornito l’eccitazione a largo campo e la fotoattivazione di Alexa Fluor 647, insieme ad un espansore di fascio 2x prima della porta di illuminazione posteriore del microscopio. Le immagini sono state catturate da una telecamera CCD (EMCCD) retrodiluita ed elettroni moltiplicatori, raffreddata a -80°C (Andor Technology, iXON Ultra, modello DU-897U-CSO-#BV), utilizzando script pubblicati (York etal., 2011) richiamati dall’interfaccia della telecamera (Andor Technology, SOLIS).
Il flusso di lavoro di acquisizione è stato descritto in precedenza(York et al., 2011) (vedi https://github.com/AndrewGYork/palm3d per ulteriori dettagli), compresa l’acquisizione di immagini di calibrazione di una nanoparticella d’oro a passi di 50 nm in z su un intervallo di 4 µm. Le etichette del colorante fluorescente nel campione sono state eccitate utilizzando un laser a 642 nm che emetteva 100 mW, fino a quando un numero sufficiente è stato estinto(Heilemann et al., 2009, 2008) per separare nel tempo gli eventi di emissione spazialmente vicini, in seguito ai quali abbiamo iniziato la raccolta dei dati (tempo di esposizione 50 ms, EMCCD gain 150). Quando gli eventi di emissione sono diventati scarsi (dopo circa 10.000 s di fotogrammi), le etichette sono state riattivate stocasticamente (vande Linde et al., 2011) utilizzando un laser da 405 nm, con potenza crescente da 2 a 20 μW. La raccolta dei dati è terminata quando il numero di eventi di emissione per fotogramma è diventato trascurabile. Gli eventi di emissione che durano per più di un fotogramma sono stati collegati in localizzazioni mediate, che sono state infine raccolte in un istogramma per la visualizzazione, tenendo conto della distorsione da parte della lente cilindrica.
References
- Aspelund A, Robciuc MR, Karaman S, Makinen T, Alitalo K. Lymphatic system in Cardiovascular Medicine. Circulation Research. 2016; 118:515-530. DOI | PubMed
- Behdani M, Zeinali S, Khanahmad H, Karimipour M, Asadzadeh N, Azadmanesh K, Khabiri A, Schoonooghe S, Habibi Anbouhi M, Hassanzadeh-Ghassabeh G, Muyldermans S. Generation and characterization of a functional nanobody against the vascular endothelial growth factor receptor-2; angiogenesis cell receptor. Molecular Immunology. 2012; 50:35-41. DOI | PubMed
- Berglund L, Björling E, Oksvold P, Fagerberg L, Asplund A, Szigyarto CA, Persson A, Ottosson J, Wernérus H, Nilsson P, Lundberg E, Sivertsson A, Navani S, Wester K, Kampf C, Hober S, Pontén F, Uhlén M. A genecentric human protein atlas for expression profiles based on antibodies. Molecular & Cellular Proteomics. 2008; 7:2019-2027. DOI | PubMed
- Beste G, Schmidt FS, Stibora T, Skerra A. Small antibody-like proteins with prescribed ligand specificities derived from the lipocalin fold. PNAS. 1999; 96:1898-1903. DOI | PubMed
- Binz HK, Stumpp MT, Forrer P, Amstutz P, Plückthun A. Designing repeat proteins: well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. Journal of Molecular Biology. 2003; 332:489-503. DOI | PubMed
- Biocca S, Ruberti F, Tafani M, Pierandrei-Amaldi P, Cattaneo A. Redox state of single chain fv fragments targeted to the endoplasmic reticulum, cytosol and mitochondria. Bio/Technology. 1995; 13:1110-1115. DOI | PubMed
- Bordeaux J, Welsh A, Agarwal S, Killiam E, Baquero M, Hanna J, Anagnostou V, Rimm D. Antibody validation. BioTechniques. 2010; 48:197-209. DOI | PubMed
- Bradbury A, Plückthun A. Reproducibility: standardize antibodies used in research. Nature. 2015; 518:27-29. DOI | PubMed
- Daly RJ. The Grb7 family of signalling proteins. Cellular Signalling. 1998; 10:613-618. DOI | PubMed
- Danquah W, Meyer-Schwesinger C, Rissiek B, Pinto C, Serracant-Prat A, Amadi M, Iacenda D, Knop JH, Hammel A, Bergmann P, Schwarz N, Assunção J, Rotthier W, Haag F, Tolosa E, Bannas P, Boué-Grabot E, Magnus T, Laeremans T, Stortelers C, Koch-Nolte F. Nanobodies that block gating of the P2X7 ion channel ameliorate inflammation. Science Translational Medicine. 2016; 8DOI | PubMed
- De Santis R, Albertoni C, Petronzelli F, Campo S, D’Alessio V, Rosi A, Anastasi AM, Lindstedt R, Caroni N, Arseni B, Chiodi P, Verdoliva A, Cassani G, Chinol M, Paganelli G, Carminati P. Low and high tenascin-expressing tumors are efficiently targeted by ST2146 monoclonal antibody. Clinical Cancer Research. 2006; 12:2191-2196. DOI | PubMed
- del Rio T, Werner HC, Enquist LW. The pseudorabies virus VP22 homologue (UL49) is dispensable for virus growth in vitro and has no effect on virulence and neuronal spread in rodents. Journal of Virology. 2002; 76:774-782. DOI | PubMed
- Denesvre C, Blondeau C, Lemesle M, Le Vern Y, Vautherot D, Roingeard P, Vautherot JF. Morphogenesis of a highly replicative EGFPVP22 recombinant Marek’s disease virus in cell culture. Journal of Virology. 2007; 81:12348-12359. DOI | PubMed
- Dörsam H, Rohrbach P, Kürschner T, Kipriyanov S, Renner S, Braunagel M, Welschof M, Little M. Antibodies to steroids from a small human naive IgM library. FEBS Letters. 1997; 414:7-13. DOI | PubMed
- Fisher MJ, Williamson DJ, Burslem GM, Plante JP, Manfield IW, Tiede C, Ault JR, Stockley PG, Plein S, Maqbool A, Tomlinson DC, Foster R, Warriner SL, Bon RS. Trivalent Gd-DOTA reagents for modification of proteins. RSC Adv.. 2015; 5:96194-96200. DOI | PubMed
- Fleetwood F, Klint S, Hanze M, Gunneriusson E, Frejd FY, Ståhl S, Löfblom J. Simultaneous targeting of two ligand-binding sites on VEGFR2 using biparatopic affibody molecules results in dramatically improved affinity. Scientific Reports. 2014; 4DOI | PubMed
- Grebien F, Hantschel O, Wojcik J, Kaupe I, Kovacic B, Wyrzucki AM, Gish GD, Cerny-Reiterer S, Koide A, Beug H, Pawson T, Valent P, Koide S, Superti-Furga G. Targeting the SH2-kinase interface in Bcr-Abl inhibits leukemogenesis. Cell. 2011; 147:306-319. DOI | PubMed
- Griffié J, Shannon M, Bromley CL, Boelen L, Burn GL, Williamson DJ, Heard NA, Cope AP, Owen DM, Rubin-Delanchy P. A bayesian cluster analysis method for single-molecule localization microscopy data. Nature Protocols. 2016; 11:2499-2514. DOI | PubMed
- Harley CA, Starek G, Jones DK, Fernandes AS, Robertson GA, Morais-Cabral JH. Enhancement of hERG channel activity by scFv antibody fragments targeted to the PAS domain. PNAS. 2016; 113:9916-9921. DOI | PubMed
- Heilemann M, van de Linde S, Mukherjee A, Sauer M. Super-resolution imaging with small organic fluorophores. Angewandte Chemie International Edition. 2009; 48:6903-6908. DOI | PubMed
- Heilemann M, van de Linde S, Schüttpelz M, Kasper R, Seefeldt B, Mukherjee A, Tinnefeld P, Sauer M. Subdiffraction-resolution fluorescence imaging with conventional fluorescent probes. Angewandte Chemie International Edition. 2008; 47:6172-6176. DOI | PubMed
- Hicke BJ, Stephens AW, Gould T, Chang YF, Lynott CK, Heil J, Borkowski S, Hilger CS, Cook G, Warren S, Schmidt PG. Tumor targeting by an aptamer. Journal of Nuclear Medicine : Official Publication, Society of Nuclear Medicine. 2006; 47:668-678. PubMed
- Hu CK, Coughlin M, Mitchison TJ. Midbody assembly and its regulation during cytokinesis. Molecular Biology of the Cell. 2012; 23:1024-1034. DOI | PubMed
- Huang B, Jones SA, Brandenburg B, Zhuang X. Whole-cell 3D STORM reveals interactions between cellular structures with nanometer-scale resolution. Nature Methods. 2008; 5:1047-1052. DOI | PubMed
- Hyde CA, Giese A, Stuttfeld E, Abram Saliba J, Villemagne D, Schleier T, Binz HK, Ballmer-Hofer K. Targeting extracellular domains D4 and D7 of vascular endothelial growth factor receptor 2 reveals allosteric receptor regulatory sites. Molecular and Cellular Biology. 2012; 32:3802-3813. DOI | PubMed
- Kaszas K, Keller JM, Coddou C, Mishra SK, Hoon MA, Stojilkovic S, Jacobson KA, Iadarola MJ. Small molecule positive allosteric modulation of TRPV1 activation by vanilloids and acidic pH. Journal of Pharmacology and Experimental Therapeutics. 2012; 340:152-160. DOI | PubMed
- Kilmartin JV, Wright B, Milstein C. Rat monoclonal antitubulin antibodies derived by using a new nonsecreting rat cell line. The Journal of Cell Biology. 1982; 93:576-582. DOI | PubMed
- Kim IA, Bae SS, Fernandes A, Wu J, Muschel RJ, McKenna WG, Birnbaum MJ, Bernhard EJ. Selective inhibition of ras, phosphoinositide 3 kinase, and akt isoforms increases the radiosensitivity of human carcinoma cell lines. Cancer Research. 2005; 65:7902-7910. DOI | PubMed
- Kofler NM, Simons M. Angiogenesis versus arteriogenesis: neuropilin 1 modulation of VEGF signaling. F1000Prime Reports. 2015; 7DOI | PubMed
- Koide A, Bailey CW, Huang X, Koide S. The fibronectin type III domain as a scaffold for novel binding proteins. Journal of Molecular Biology. 1998; 284:1141-1151. DOI | PubMed
- Kumaraswamy S, Tobias R. Protein-Protein Interactions: Methods and Applications.. Springer New York: New York; 2015.
- Kummer L, Parizek P, Rube P, Millgramm B, Prinz A, Mittl PR, Kaufholz M, Zimmermann B, Herberg FW, Plückthun A. Structural and functional analysis of phosphorylation-specific binders of the kinase ERK from designed ankyrin repeat protein libraries. PNAS. 2012; 109:E2248-E2257. DOI | PubMed
- Kyle HF, Wickson KF, Stott J, Burslem GM, Breeze AL, Tiede C, Tomlinson DC, Warriner SL, Nelson A, Wilson AJ, Edwards TA. Exploration of the HIF-1α/p300 interface using peptide and adhiron phage display technologies. Mol. BioSyst.. 2015; 11:2738-2749. DOI | PubMed
- Lambacher NJ, Bruel AL, van Dam TJ, Szymańska K, Slaats GG, Kuhns S, McManus GJ, Kennedy JE, Gaff K, Wu KM, van der Lee R, Burglen L, Doummar D, Rivière JB, Faivre L, Attié-Bitach T, Saunier S, Curd A, Peckham M, Giles RH, Johnson CA, Huynen MA, Thauvin-Robinet C, Blacque OE. TMEM107 recruits ciliopathy proteins to subdomains of the ciliary transition zone and causes Joubert syndrome. Nature Cell Biology. 2016; 18:122-131. DOI | PubMed
- Lebovitz EE, Keller JM, Kominsky H, Kaszas K, Maric D, Iadarola MJ. Positive allosteric modulation of TRPV1 as a novel analgesic mechanism. Molecular Pain. 2012; 8DOI | PubMed
- Lee JH, Park CK, Chen G, Han Q, Xie RG, Liu T, Ji RR, Lee SY. A monoclonal antibody that targets a NaV1.7 channel voltage sensor for pain and itch relief. Cell. 2014; 157:1393-1404. DOI | PubMed
- Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010; 141:1117-1134. DOI | PubMed
- Liu BA, Shah E, Jablonowski K, Stergachis A, Engelmann B, Nash PD. The SH2 domain-containing proteins in 21 species establish the provenance and scope of phosphotyrosine signaling in eukaryotes. Science Signaling. 2011; 4DOI | PubMed
- Lowy CM, Oskarsson T. Tenascin C in metastasis: a view from the invasive front. Cell Adhesion & Migration. 2015; 9:112-124. DOI | PubMed
- Machleidt A, Buchholz S, Diermeier-Daucher S, Zeman F, Ortmann O, Brockhoff G. The prognostic value of Her4 receptor isoform expression in triple-negative and Her2 positive breast Cancer patients. BMC Cancer. 2013; 13DOI | PubMed
- Marschall AL, Dübel S, Böldicke T. Specific in vivo knockdown of protein function by intrabodies. mAbs. 2015; 7:1010-1035. DOI | PubMed
- Minn AJ, Gupta GP, Siegel PM, Bos PD, Shu W, Giri DD, Viale A, Olshen AB, Gerald WL, Massagué J. Genes that mediate breast Cancer metastasis to lung. Nature. 2005; 436:518-524. DOI | PubMed
- Murga M, Fernandez-Capetillo O, Tosato G. Neuropilin-1 regulates attachment in human endothelial cells independently of vascular endothelial growth factor receptor-2. Blood. 2005; 105:1992-1999. DOI | PubMed
- Needham SR, Roberts SK, Arkhipov A, Mysore VP, Tynan CJ, Zanetti-Domingues LC, Kim ET, Losasso V, Korovesis D, Hirsch M, Rolfe DJ, Clarke DT, Winn MD, Lajevardipour A, Clayton AH, Pike LJ, Perani M, Parker PJ, Shan Y, Shaw DE, Martin-Fernandez ML. EGFR Oligomerization organizes kinase-active dimers into competent signalling platforms. Nature Communications. 2016; 7DOI | PubMed
- Nilsson P, Paavilainen L, Larsson K, Odling J, Sundberg M, Andersson AC, Kampf C, Persson A, Al-Khalili Szigyarto C, Ottosson J, Björling E, Hober S, Wernérus H, Wester K, Pontén F, Uhlen M. Towards a human proteome atlas: high-throughput generation of mono-specific antibodies for tissue profiling. Proteomics. 2005; 5:4327-4337. DOI | PubMed
- Nord K, Nilsson J, Nilsson B, Uhlén M, Nygren PA. A combinatorial library of an alpha-helical bacterial receptor domain. “Protein Engineering, Design and Selection”. 1995; 8:601-608. DOI | PubMed
- Oskarsson T, Acharyya S, Zhang XH, Vanharanta S, Tavazoie SF, Morris PG, Downey RJ, Manova-Todorova K, Brogi E, Massagué J. Breast Cancer cells produce tenascin C as a metastatic niche component to colonize the lungs. Nature Medicine. 2011; 17:867-874. DOI | PubMed
- Overington JP, Al-Lazikani B, Hopkins AL. How many drug targets are there?. Nature Reviews Drug Discovery. 2006; 5:993-996. DOI | PubMed
- Park MJ, Sheng R, Silkov A, Jung DJ, Wang ZG, Xin Y, Kim H, Thiagarajan-Rosenkranz P, Song S, Yoon Y, Nam W, Kim I, Kim E, Lee DG, Chen Y, Singaram I, Wang L, Jang MH, Hwang CS, Honig B, Ryu S, Lorieau J, Kim YM, Cho W. SH2 domains serve as Lipid-Binding modules for pTyr-Signaling proteins. Molecular Cell. 2016; 62:7-20. DOI | PubMed
- Pershad K, Pavlovic JD, Gräslund S, Nilsson P, Colwill K, Karatt-Vellatt A, Schofield DJ, Dyson MR, Pawson T, Kay BK, McCafferty J. Generating a panel of highly specific antibodies to 20 human SH2 domains by phage display. Protein Engineering, Design and Selection. 2010; 23:279-288. DOI | PubMed
- Pleiner T, Bates M, Trakhanov S, Lee CT, Schliep JE, Chug H, Böhning M, Stark H, Urlaub H, Görlich D. Nanobodies: site-specific labeling for super-resolution imaging, rapid epitope-mapping and native protein complex isolation. eLife. 2015; 4DOI | PubMed
- Ramin S, Weller MG. Extremely sensitive and selective antibodies against the explosive 2,4,6-trinitrotoluene by rational design of a structurally optimized hapten. Journal of Molecular Recognition. 2012; 25:89-97. DOI | PubMed
- Rawlings AE, Bramble JP, Tang AAS, Somner LA, Monnington AE, Cooke DJ, McPherson MJ, Tomlinson DC, Staniland SS. Phage display selected magnetite interacting Adhirons for shape controlled nanoparticle synthesis. Chemical Science. 2015; 6:5586-5594. DOI
- Renewable Protein Binder Working Group, Colwill K, Gräslund S. A roadmap to generate renewable protein binders to the human proteome. Nature Methods. 2011; 8:551-558. DOI | PubMed
- Rémy S, Blondeau C, Le Vern Y, Lemesle M, Vautherot JF, Denesvre C. Fluorescent tagging of VP22 in N-terminus reveals that VP22 favors Marek’s disease virus (MDV) virulence in chickens and allows morphogenesis study in MD tumor cells. Veterinary Research. 2013; 44DOI | PubMed
- Ries J, Kaplan C, Platonova E, Eghlidi H, Ewers H, A simple EH. A simple, versatile method for GFP-based super-resolution microscopy via nanobodies. Nature Methods. 2012; 9:582-584. DOI | PubMed
- Rolfe DJ, McLachlan CI, Hirsch M, Needham SR, Tynan CJ, Webb SE, Martin-Fernandez ML, Hobson MP. Automated multidimensional single molecule fluorescence microscopy feature detection and tracking. European Biophysics Journal. 2011; 40:1167-1186. DOI | PubMed
- Rothbauer U, Zolghadr K, Tillib S, Nowak D, Schermelleh L, Gahl A, Backmann N, Conrath K, Muyldermans S, Cardoso MC, Leonhardt H. Targeting and tracing antigens in live cells with fluorescent nanobodies. Nature Methods. 2006; 3:887-889. DOI | PubMed
- Schlehuber S, Beste G, Skerra A. A novel type of receptor protein, based on the lipocalin scaffold, with specificity for digoxigenin. Journal of Molecular Biology. 2000; 297:1105-1120. DOI | PubMed
- Sennhauser G, Grütter MG. Chaperone-assisted crystallography with DARPins. Structure. 2008; 16:1443-1453. DOI | PubMed
- Sha F, Gencer EB, Georgeon S, Koide A, Yasui N, Koide S, Hantschel O. Dissection of the BCR-ABL signaling network using highly specific monobody inhibitors to the SHP2 SH2 domains. PNAS. 2013; 110:14924-14929. DOI | PubMed
- Sharma R, Deacon SE, Nowak D, George SE, Szymonik MP, Tang AA, Tomlinson DC, Davies AG, McPherson MJ, Wälti C. Label-free electrochemical impedance biosensor to detect human interleukin-8 in serum with sub-pg/ml sensitivity. Biosensors and Bioelectronics. 2016; 80:607-613. DOI | PubMed
- Shroff H, Galbraith CG, Galbraith JA, White H, Gillette J, Olenych S, Davidson MW, Betzig E. Dual-color superresolution imaging of genetically expressed probes within individual adhesion complexes. PNAS. 2007; 104:20308-20313. DOI | PubMed
- Skerratt SE, West CW. Ion channel therapeutics for pain. Channels. 2015; 9:344-351. DOI | PubMed
- Škrlec K, Štrukelj B, Berlec A. Non-immunoglobulin scaffolds: a focus on their targets. Trends in Biotechnology. 2015; 33:408-418. DOI | PubMed
- Spencer-Smith R, Koide A, Zhou Y, Eguchi RR, Sha F, Gajwani P, Santana D, Gupta A, Jacobs M, Herrero-Garcia E, Cobbert J, Lavoie H, Smith M, Rajakulendran T, Dowdell E, Okur MN, Dementieva I, Sicheri F, Therrien M, Hancock JF, Ikura M, Koide S, O’Bryan JP. Inhibition of RAS function through targeting an allosteric regulatory site. Nature Chemical Biology. 2017; 13:62-68. DOI | PubMed
- Stadler LK, Hoffmann T, Tomlinson DC, Song Q, Lee T, Busby M, Nyathi Y, Gendra E, Tiede C, Flanagan K, Cockell SJ, Wipat A, Harwood C, Wagner SD, Knowles MA, Davis JJ, Keegan N, Ferrigno PK. Structure-function studies of an engineered scaffold protein derived from Stefin A. II: development and applications of the SQT variant. Protein Engineering, Design and Selection. 2011; 24:751-763. DOI | PubMed
- Stadler LK, Tomlinson DC, Lee T, Knowles MA, Ko Ferrigno P. The use of a neutral peptide aptamer scaffold to anchor BH3 peptides constitutes a viable approach to studying their function. Cell Death and Disease. 2014; 5DOI | PubMed
- Stockbridge RB, Kolmakova-Partensky L, Shane T, Koide A, Koide S, Miller C, Newstead S. Crystal structures of a double-barrelled fluoride ion channel. Nature. 2015; 525:548-551. DOI | PubMed
- Stoevesandt O, Taussig MJ. European and international collaboration in affinity proteomics. New Biotechnology. 2012; 29:511-514. DOI | PubMed
- Stylianou J, Maringer K, Cook R, Bernard E, Elliott G. Virion incorporation of the herpes simplex virus type 1 tegument protein VP22 occurs via glycoprotein E-specific recruitment to the late secretory pathway. Journal of Virology. 2009; 83:5204-5218. DOI | PubMed
- Sun H, Li M. Antibody therapeutics targeting ion channels: are we there yet?. Acta Pharmacologica Sinica. 2013; 34:199-204. DOI | PubMed
- Taussig MJ, Stoevesandt O, Borrebaeck CA, Bradbury AR, Cahill D, Cambillau C, de Daruvar A, Dübel S, Eichler J, Frank R, Gibson TJ, Gloriam D, Gold L, Herberg FW, Hermjakob H, Hoheisel JD, Joos TO, Kallioniemi O, Koegl M, Koegll M, Konthur Z, Korn B, Kremmer E, Krobitsch S, Landegren U, van der Maarel S, McCafferty J, Muyldermans S, Nygren PA, Palcy S, Plückthun A, Polic B, Przybylski M, Saviranta P, Sawyer A, Sherman DJ, Skerra A, Templin M, Ueffing M, Uhlén M. ProteomeBinders: planning a european resource of affinity reagents for analysis of the human proteome. Nature Methods. 2007; 4:13-17. DOI | PubMed
- Theurillat JP, Dreier B, Nagy-Davidescu G, Seifert B, Behnke S, Zürrer-Härdi U, Ingold F, Plückthun A, Moch H. Designed ankyrin repeat proteins: a novel tool for testing epidermal growth factor receptor 2 expression in breast Cancer. Modern Pathology. 2010; 23:1289-1297. DOI | PubMed
- Tiede C, Tang AA, Deacon SE, Mandal U, Nettleship JE, Owen RL, George SE, Harrison DJ, Owens RJ, Tomlinson DC, McPherson MJ. Adhiron: a stable and versatile peptide display scaffold for molecular recognition applications. Protein Engineering, Design and Selection. 2014; 27:145-155. DOI | PubMed
- Tolcher AW, Sweeney CJ, Papadopoulos K, Patnaik A, Chiorean EG, Mita AC, Sankhala K, Furfine E, Gokemeijer J, Iacono L, Eaton C, Silver BA, Mita M. Phase I and pharmacokinetic study of CT-322 (BMS-844203), a targeted adnectin inhibitor of VEGFR-2 based on a domain of human fibronectin. Clinical Cancer Research. 2011; 17:363-371. DOI | PubMed
- Uhlén M, Björling E, Agaton C, Szigyarto CA, Amini B, Andersen E, Andersson AC, Angelidou P, Asplund A, Asplund C, Berglund L, Bergström K, Brumer H, Cerjan D, Ekström M, Elobeid A, Eriksson C, Fagerberg L, Falk R, Fall J, Forsberg M, Björklund MG, Gumbel K, Halimi A, Hallin I, Hamsten C, Hansson M, Hedhammar M, Hercules G, Kampf C, Larsson K, Lindskog M, Lodewyckx W, Lund J, Lundeberg J, Magnusson K, Malm E, Nilsson P, Odling J, Oksvold P, Olsson I, Oster E, Ottosson J, Paavilainen L, Persson A, Rimini R, Rockberg J, Runeson M, Sivertsson A, Sköllermo A, Steen J, Stenvall M, Sterky F, Strömberg S, Sundberg M, Tegel H, Tourle S, Wahlund E, Waldén A, Wan J, Wernérus H, Westberg J, Wester K, Wrethagen U, Xu LL, Hober S, Pontén F. A human protein atlas for normal and Cancer tissues based on antibody proteomics. Molecular & Cellular Proteomics. 2005; 4:1920-1932. DOI | PubMed
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Pontén F, Proteomics PF. Proteomics. Tissue-based map of the human proteome. Science. 2015; 347DOI | PubMed
- van de Linde S, Löschberger A, Klein T, Heidbreder M, Wolter S, Heilemann M, Sauer M. Direct stochastic optical reconstruction microscopy with standard fluorescent probes. Nature Protocols. 2011; 6:991-1009. DOI | PubMed
- Van Eyken P, Sciot R, Desmet VJ. Expression of the novel extracellular matrix component tenascin in normal and diseased human liver. an immunohistochemical study. Journal of Hepatology. 1990; 11:43-52. DOI | PubMed
- Vaughan TJ, Williams AJ, Pritchard K, Osbourn JK, Pope AR, Earnshaw JC, McCafferty J, Hodits RA, Wilton J, Johnson KS. Human antibodies with sub-nanomolar affinities isolated from a large non-immunized phage display library. Nature Biotechnology. 1996; 14:309-314. DOI | PubMed
- Vazquez-Lombardi R, Phan TG, Zimmermann C, Lowe D, Jermutus L, Christ D. Challenges and opportunities for non-antibody scaffold drugs. Drug Discovery Today. 2015; 20:1271-1283. DOI | PubMed
- Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human Cancer. Nature Reviews Cancer. 2002; 2:489-501. DOI | PubMed
- Wehland J, Willingham MC, Sandoval IV. A rat monoclonal antibody reacting specifically with the tyrosylated form of alpha-tubulin. I. biochemical characterization, effects on microtubule polymerization in vitro, and microtubule polymerization and organization in vivo. The Journal of Cell Biology. 1983; 97:1467-1475. DOI | PubMed
- Wojcik J, Hantschel O, Grebien F, Kaupe I, Bennett KL, Barkinge J, Jones RB, Koide A, Superti-Furga G, Koide S. A potent and highly specific FN3 monobody inhibitor of the abl SH2 domain. Nature Structural & Molecular Biology. 2010; 17:519-527. DOI | PubMed
- Wörn A, Plückthun A. Stability engineering of antibody single-chain fv fragments. Journal of Molecular Biology. 2001; 305:989-1010. DOI | PubMed
- York AG, Ghitani A, Vaziri A, Davidson MW, Shroff H. Confined activation and subdiffractive localization enables whole-cell PALM with genetically expressed probes. Nature Methods. 2011; 8:327-333. DOI | PubMed
- Zanetti-Domingues LC, Martin-Fernandez ML, Needham SR, Rolfe DJ, Clarke DT. A systematic investigation of differential effects of cell culture substrates on the extent of artifacts in single-molecule tracking. PLoS One. 2012; 7DOI | PubMed
- Zhou Y, Morais-Cabral JH, Kaufman A, MacKinnon R. Chemistry of ion coordination and hydration revealed by a K+ channel-Fab complex at 2.0 A resolution. Nature. 2001; 414:43-48. DOI | PubMed
Fonte
Tiede C, Bedford R, Heseltine SJ, Smith G, Wijetunga I, et al. () Affimer proteins are versatile and renewable affinity reagents. eLife 6e24903. https://doi.org/10.7554/eLife.24903




