
Introduzione
Al suo interno, un sarcomero è composto da filamenti di miosina II “spessi” e da filamenti di actina “sottili” (Figura 1A) (Au,2004). Un sarcomero è misurato da linea Z a linea Z, che contengono α-acttinina 2 (Figura 1A). La corretta costituzione dei sarcomeri cardiaci durante lo sviluppo e la loro successiva manutenzione è fondamentale per la funzione cardiaca. Precedenti studi sui miociti in coltura hanno dimostrato la presenza di fasci di actina chiamati ‘strutture simili a fibre da stress’ simili in apparenza alle classiche fibre da stress (Dlugoszet al., 1984). Queste fibre da stress sono state spesso trovate vicino al bordo del miocita con sarcomeri esistenti più lontano dal bordo(Rhee et al., 1994). Questi studi hanno proposto che le fibre da stress servissero da modello per la formazione di sarcomeri(Dlugosz et al., 1984; Rhee et al., 1994; Sanger et al., 2005). Il modello originale che lo propose si chiamava Templating Model(Dlugosz et al., 1984), e fu proposto prima che si sapesse che queste fibre da stress contenevano sia proteine non muscolari che sarcomeriche(Rhee et al., 1994). Oltre alla miosina non muscolare IIB (NMIIB), presente nelle cellule non muscolari, le fibre da stress nelle cellule muscolari contengono proteine specifiche del muscolo, come α-actinina, tropomiosina, troponine e tropomodulina (Almenar-Queralt et al.,1999; Rhee et al., 1994; Sanger et al., 2005). Ognuna di queste proteine ha paraloghi non muscolari, che probabilmente svolgono funzioni simili(Bryce et al., 2003; Colpan et al., 2013; Côté, 1983; Gunning et al., 2015; Lim et al., 1986; Sjöblom et al., 2008). In parte in risposta alla presenza di proteine specifiche del muscolo nelle fibre da sforzo, il Modello di Templating è stato modificato al “Modello Pre-Myofibril” (Rhee et al.,1994; Sanger et al., 2005). Anche se questi modelli hanno nomi diversi e sono spesso presentati come reciprocamente esclusivi, sono molto simili nelle loro previsioni. In particolare, entrambi i modelli pongono un fascio di actina che appare strutturalmente simile ad una fibra da stress acquisirà nel tempo una fila di sarcomeri per diventare una ‘miofibrilla’ (Dlugoszet al., 1984; Rhee et al. , 1994; Sanger et al., 2005) (Figura 1A). C’è una grande quantità di dati di localizzazione in cardiomiociti fissi per supportare questi modelli. Tuttavia, ci sono pochissimi dati dinamici nelle cellule vive che suggeriscono che le fibre da stress danno origine a sarcomeri. Il supporto dinamico più forte proviene da imaging fluorescente taggato α-actinina 2 nei miociti. I montaggi temporali dei miotobuli scheletrici dei pulcini hanno mostrato piccoli puncta di α-actinina 2 che si aggiungono alle linee Z preesistenti (McKennaet al., 1986). Successivamente, un montaggio temporale è stato utilizzato per mostrare un fenomeno simile che si verifica nei cardiomiociti dei pulcini(Dabiri et al., 1997).
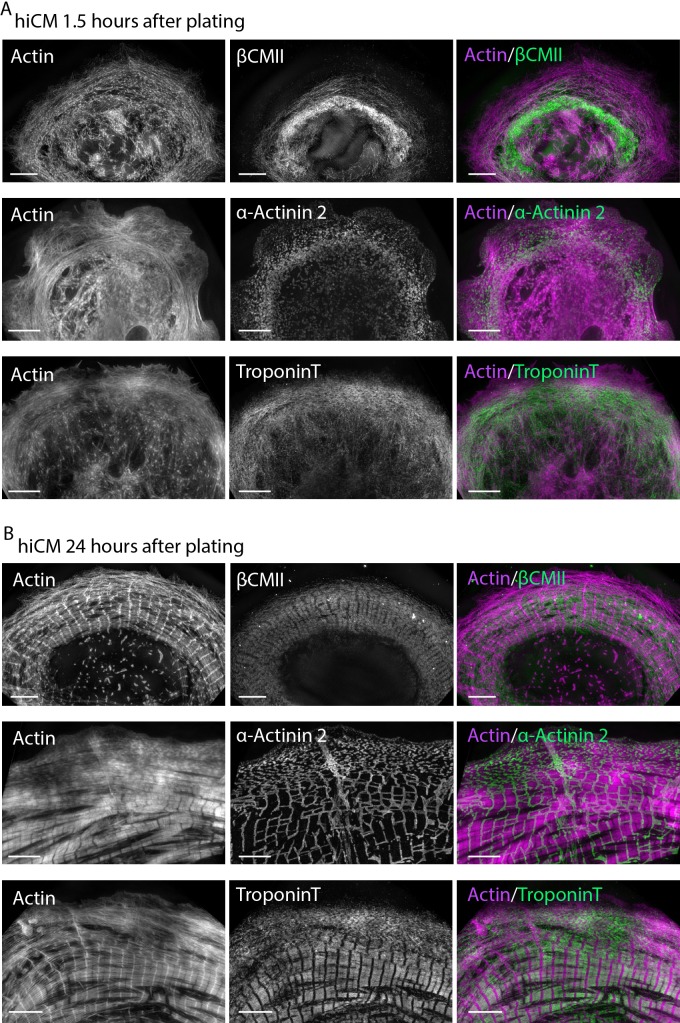
Figura 1-figure supplement 1.I sarcomeri derivano direttamente dai precursori delle fibre muscolari stressanti (MSF).hiCMs non contengono sarcomeri nei primi punti temporali dopo la placcatura.(A) Microscopia elettronica (EM) schema di un sarcomero cardiaco da topo adulto. regioni di elettroni densi sui bordi di un sarcomero sono Z-dischi (Z), mentre il nucleo del sarcomero è composto da sottili filamenti di actina e spessi filamenti di miosina II (A). Più sarcomeri allineati adiacenti formano una miofibrilla (mag EM inferiore, a destra).(B) hiCM permesso di diffondere per 24 ore dopo la placcatura e l’imaging con SIM. hiCM è stato colorato per l’actina e la codifica a colori è una rappresentazione di altezza (piano Z) all’interno della cella dopo l’imaging 3D (Z-altezza, a sinistra). Si noti la fibra di stress chiaro e l’organizzazione di actina come sarcomero nella parte anteriore e posteriore della cella nella casella 1 e 2, rispettivamente.(C) Diffondere U2OS colore cella U2OS codificato per Z come in Figura 1B, visualizzando le fibre di tensione ad arco di actina prominente dietro il bordo anteriore della cella e ripreso con SIM. Il riquadro 1 mostra gli archi di actina appena dietro il bordo di entrata della cellula, mentre il riquadro 2 mostra gli archi di actina sulla superficie dorsale del corpo della cellula (actina di colore verde e blu), mentre le fibre di stress ventrale (actina di colore rosso) sono sulla superficie inferiore della cellula.(D) Percentuale di hiCMs, U2OS, e cellule HeLa con le fibre di stress ad arco di actina. hiCMs; 1372 cellule su tre esperimenti. U2OS; 37 cellule su quattro esperimenti. HeLa; 186 cellule su quattro esperimenti.(E) Intervallo di tempo ad ampio raggio di hiCM trasfettato con Lifeact-mEmerald per visualizzare l’actina. MSF davanti a hiCM subisce il flusso retrogrado e acquisisce i sarcomeri (frecce gialle).(F) Laser-scanning microscopia confocale di hiCM che esprime Lifeact-mApple che mostra MSF a transizione sarcomero. hiCM manca di sarcomeri al primo punto di tempo, e MSF al bordo della cella subisce flusso retrogrado e acquisisce sarcomeri (frecce gialle).(G) 3D laser-scanning microscopia confocale di hiCM che esprime Lifeact-mApple formando sarcomeri. Si noti come la superficie ventrale (montaggio a sinistra) non contiene strutture di sarcomeri, mentre il montaggio dei sarcomeri avviene sulla superficie dorsale della cella (montaggio a destra). Barre di scala;(A) 500 nm alto mag (sinistra), 2 µm basso mag (destra); (B) 10 µm basso mag, 5 µm alto magnete inserti; (C) 10 µm basso mag, 5 µm alto magnete inserti; (E), (F), (G), 10 µm. I valori P indicati nei grafici.(A) hiCMs permesso di diffondere per 1,5 ore, fisso, e colorato per actina e βCMII (in alto), actina e α-acttinina 2 (al centro), actina e TroponinT (in basso), e ripreso con SIM. Si noti la mancanza di struttura del sarcomero in tutti i marcatori di sarcomero mostrato.(B) hiCMs permesso di diffondere per 24 ore, fisso, e colorato per actina e βCMII (in alto), actina e α-acttinina 2 (in mezzo), actina e TroponinT (in basso), e ripreso con SIM. Si noti come, a differenza di 1,5 ore, gli hiCM sparsi per 24 ore contengono robusti sarcomeri in tutti i marcatori mostrati. Barre di scala, 5 µm.
Alcuni dati in vivo supportano il modello Template/Pre-Myofibril, mentre altri non lo fanno. A forte sostegno del modello Template/Pre-Myofibril, le immagini statiche del tessuto cardiaco dei pulcini hanno essenzialmente rivelato ogni struttura descritta nei cardiomiociti primari dei pulcini in coltura(Du et al., 2008). La presenza di fibre da stress contenenti NMIIB nei cardiomiociti era particolarmente chiara(Du et al., 2008). Anche i topi NMIIB a eliminazione dei germi (KO) sono stati segnalati per avere un minor numero di sarcomeri disorganizzati via EM(Tullio et al., 1997). D’altra parte, diversi studi hanno messo in discussione il ruolo delle fibre di stress nell’assemblaggio dei sarcomeri. In primo luogo, diversi studi che esaminano i cardiomiociti all’interno del tessuto cardiaco di topo o di pulcino non hanno trovato fibre da stress contenenti NMIIB(Ehler et al., 1999; Kan-O et al., 2012; Ma et al., 2009). Inoltre, un topo KO condizionato che rimuove geneticamente l’NMIIB a P9 apparentemente aveva ancora strutture di sarcomeri striati(Ma et al., 2009). Infine, un KO condizionato del cuore dell’altro paralogio principale del NMII, il NMIIA, è stato anche segnalato come non abbia difetti apparenti nella formazione del cuore(Conti et al., 2004; Conti et al., 2015). Nel complesso, la mancanza di dati chiari che mostrino le fibre da stress nei cardiomiociti e le incongruenze per un ruolo della NMII nell’assemblaggio dei sarcomeri mette in dubbio se il Modello Modello Pre-Miofibrilla sia un costrutto valido per la comprensione dell’assemblaggio dei sarcomeri(Sanger et al., 2005; Sparrow e Schöck, 2009).
Ci sono ulteriori dati che suggeriscono che un meccanismo diverso da quello descritto nel modello Template/Pre-Myofibril potrebbe guidare l’assemblaggio dei sarcomeri. Questo modello alternativo chiamato “Modello di cucitura” si basa sull’idea che le parti di un sarcomero siano assemblate indipendentemente e poi riunite (cioè cucite) (Holtzer et al.,1997; Lu et al., 1992; Sangeret al., 2005). A sostegno del modello di cucitura, gli studi di Drosophila hanno dimostrato la presenza di piccoli filamenti di miosina dopo l’abbattimento (KD) di componenti separati della linea Z(Rui et al., 2010). Questi dati suggeriscono che i filamenti di miosina possono essere assemblati indipendentemente dalle linee Z. Infatti, ci sono anche micrografie elettroniche che sembrano mostrare pile di filamenti di miosina II (cioè, bande A) senza filamenti di actina rilevabili nel muscolo scheletrico(Holtzer et al., 1997; Lu et al., 1992; Sanger et al., 2005). L’esame di micrografie elettroniche supporta anche l’idea che corpi contenenti componenti della linea Z e filamenti di actina chiamati ‘I-Z-I’ possano esistere anche nel muscolo scheletrico senza filamenti di miosina II apparente (Holtzer et al.,1997; Lu et al., 1992; Sanger et al., 2005). Sulla base di questi dati, è stato proposto che la cucitura possa avvenire attraverso l’assemblaggio sequenziale aggiungendo nuovi corpi I-Z-I e filamenti di miosina II(Holtzer et al., 1997; Lu et al., 1992; Sanger et al., 2005).
Il Modello Modello di Template/Pre-Myofibril e il Modello di cucitura sono stati proposti come spiegazioni reciprocamente esclusive di come nascono i sarcomeri. Il Modello Modello Modello Pre-Miofibrilla prevede che più sarcomeri appariranno approssimativamente simultaneamente lungo la lunghezza di una fibra di tensione, mentre il Modello Stitching prevede che i sarcomeri appariranno adiacenti uno ad uno, in sequenza (vedi modelli originali in(Dlugosz et al., 1984; Holtzer et al., 1997; Rhee et al., 1994)). Qui, facciamo leva sulla nostra scoperta che i cardiomiociti pluripotenti indotti dall’uomo immaturo, derivati da cellule staminali pluripotenti (hiCM), indotti dall’uomo, si smontano completamente e poi rimontano i loro sarcomeri dopo la placcatura per testare queste possibilità. Utilizzando questo test, dimostriamo che i sarcomeri sono assemblati direttamente da modelli di fibre di actina da stress, e ci riferiamo a queste fibre da stress come Muscle Stress Fibers (MSFs). I nostri dati suggeriscono che l’assemblaggio dei sarcomeri dipende dal nucleatore del filamento di actina formina, FHOD3, dalla miosina non muscolare IIA e dalla miosina non muscolare IIB. Sorprendentemente, i nostri dati non supportano completamente né il Modello Modello di Template/Pre-Myofibril né il Modello di cucitura, ma piuttosto alcuni aspetti di ciascuno di essi. Come tale, proponiamo ora un modello unificato di assemblaggio dei sarcomeri basato sulla formazione di MSF e sulla loro successiva transizione in miofibrille contenenti sarcomeri.
Figura 1-figure supplement 1.I sarcomeri derivano direttamente dalla fibra di stress muscolare (MSF) precursori.hiCMs non contengono sarcomeri nei primi punti di tempo dopo la placcatura.(A) Microscopia elettronica (EM) schema di un sarcomero cardiaco da topo adulto. regioni di elettroni densi sui bordi di un sarcomero sono Z-dischi (Z), mentre il nucleo del sarcomero è composto da sottili filamenti di actina e spessi filamenti di miosina II (A). Più sarcomeri allineati adiacenti formano una miofibrilla (mag EM inferiore, a destra).(B) hiCM permesso di diffondere per 24 ore dopo la placcatura e l’imaging con SIM. hiCM è stato colorato per l’actina e la codifica a colori è una rappresentazione di altezza (piano Z) all’interno della cella dopo l’imaging 3D (Z-altezza, a sinistra). Si noti la fibra di stress chiaro e l’organizzazione di actina come sarcomero nella parte anteriore e posteriore della cella nella casella 1 e 2, rispettivamente.(C) Diffondere U2OS colore cella U2OS codificato per Z come in Figura 1B, visualizzando le fibre di tensione ad arco di actina prominente dietro il bordo anteriore della cella e ripreso con SIM. Il riquadro 1 mostra gli archi di actina appena dietro il bordo di entrata della cellula, mentre il riquadro 2 mostra gli archi di actina sulla superficie dorsale del corpo della cellula (actina di colore verde e blu), mentre le fibre di stress ventrale (actina di colore rosso) sono sulla superficie inferiore della cellula.(D) Percentuale di hiCMs, U2OS, e cellule HeLa con le fibre di stress ad arco di actina. hiCMs; 1372 cellule su tre esperimenti. U2OS; 37 cellule su quattro esperimenti. HeLa; 186 cellule su quattro esperimenti.(E) Intervallo di tempo ad ampio raggio di hiCM trasfettato con Lifeact-mEmerald per visualizzare l’actina. MSF davanti a hiCM subisce il flusso retrogrado e acquisisce i sarcomeri (frecce gialle).(F) Laser-scanning microscopia confocale di hiCM che esprime Lifeact-mApple che mostra MSF a transizione sarcomero. hiCM manca di sarcomeri al primo punto di tempo, e MSF al bordo della cella subisce flusso retrogrado e acquisisce sarcomeri (frecce gialle).(G) 3D laser-scanning microscopia confocale di hiCM che esprime Lifeact-mApple formando sarcomeri. Si noti come la superficie ventrale (montaggio a sinistra) non contiene strutture di sarcomeri, mentre il montaggio dei sarcomeri avviene sulla superficie dorsale della cella (montaggio a destra). Barre di scala;(A) 500 nm alto mag (sinistra), 2 µm basso mag (destra); (B) 10 µm basso mag, 5 µm alto magnete inserti; (C) 10 µm basso mag, 5 µm alto magnete inserti; (E), (F), (G), 10 µm. I valori P indicati nei grafici.(A) hiCMs permesso di diffondere per 1,5 ore, fisso, e colorato per actina e βCMII (in alto), actina e α-acttinina 2 (al centro), actina e TroponinT (in basso), e ripreso con SIM. Si noti la mancanza di struttura del sarcomero in tutti i marcatori di sarcomero mostrato.(B) hiCMs permesso di diffondere per 24 ore, fisso, e colorato per actina e βCMII (in alto), actina e α-acttinina 2 (in mezzo), actina e TroponinT (in basso), e ripreso con SIM. Si noti come, a differenza di 1,5 ore, gli hiCM sparsi per 24 ore contengono robusti sarcomeri in tutti i marcatori mostrati. Barre di scala, 5 µm.
Figura 1-figure supplement 1.hiCMs non contengono sarcomeri nei primi punti di tempo dopo la placcatura.(A) hiCMs permesso di diffondere per 1,5 ore e mezzo, fisso, e colorato per actina e βCMII (in alto), actina e α-acttinina 2 (al centro), actina e TroponinT (in basso), e ripreso con SIM. Si noti la mancanza di struttura sarcomero in tutti i marcatori di sarcomero mostrato.(B) hiCMs permesso di diffondere per 24 ore, fisso, e colorato per actina e βCMII (in alto), actina e α-acttinina 2 (in mezzo), actina e TroponinT (in basso), e ripreso con SIM. Si noti come, a differenza di 1,5 ore, gli hiCM sparsi per 24 ore contengono robusti sarcomeri in tutti i marcatori mostrati. Barre di scala, 5 µm.
Risultati
Sviluppo di un saggio per testare l’assemblaggio del sarcomero
Per affrontare le modalità di assemblaggio dei sarcomeri cardiaci, abbiamo utilizzato gli hiCM come sistema modello (vedi Materiali e metodi)(Takahashi et al., 2007). Abbiamo notato per la prima volta i filamenti di actina negli hiCMs, che si erano diffusi per 24 ore, avevano due organizzazioni distinte, le fibre di stress muscolare (MSF) e le miofibrille contenenti sarcomeri(Figura 1B). Diffondere hiCMs visualizzato MSFs al bordo anteriore e organizzato strutture sarcomeriche nel corpo cellulare(Figura 1B). Sorprendentemente, super-risoluzione di imaging ha rivelato la MSF in hiCMs assomigliava a una classica fibra di actina stress trovato in cellule non muscolari, indicato come archi di actina(Figura 1C e D)(Heath, 1983; Hotulainen e Lappalainen, 2006). Gli archi di actina sono fibre di stress sulla superficie dorsale (in alto) della cellula che sono parallele al bordo di entrata e si colorano continuamente con falloidina fluorescente(Figura 1C). Allo stesso modo, sia MSF e sarcomeri in hiCM sono sulla superficie dorsale(Figura 1B). Abbiamo poi cercato di testare il concetto che un MSF ottenuto sarcomeri come previsto dal modello Templating/Pre-Myofibril.
Per verificare se le MSF danno origine a sarcomeri, abbiamo dovuto sviluppare un test di assemblaggio dei sarcomeri. Abbiamo notato che hiCMs che era stato appena placcato (1,5-4 hr post placcatura) non conteneva sarcomeri sia al bordo della cellula o del corpo cellulare, come visualizzato da SIM (Figura 1-figure supplemento 1A). La perdita della struttura del sarcomero è stata confermata dalla visualizzazione di più proteine sarcomeriche, tra cui actina, beta miosina cardiaca II (βCMII), α-acttinina 2, e TroponinT (Figura 1-figuresupplement 1A). Anche se hiCMs non conteneva sarcomeri ai primi punti di tempo post placcatura, hiCMs ha fatto visualizzare MSFs al bordo della cella(Figura 1-figure supplemento 1A). hiCMs successivamente assemblato strutture sarcomeri nel corso di 24 ore (Figura 1-figure supplemento 1B). Abbiamo poi cercato di testare se MSFs template sarcomeri. Infatti, microscopia time-lapse di hiCMs che esprimono la sonda di actina Lifeact-mEmerald(Riedl et al., 2008) ha rivelato che MSFs acquisire sarcomeri nel tempo, con i primi sarcomeri che appaiono tra 4 e 16 ore(Figura 1E e F e Video 1). Non sorprende che questa transizione avvenga sulla superficie dorsale della cellula(Figura 1G). È importante notare che, per visualizzare l’assemblaggio dei sarcomeri in hiCMs dal vivo, abbiamo iniziato la nostra immagine nei primi punti di tempo post placcatura (cioè, 1,5-4 ore). In questi punti di tempo, hiCMs non contengono sarcomeri(Figura 1-figure supplemento 1A), e questo ci ha assicurato che stavamo visualizzando l’evento iniziale di assemblaggio sarcomero, e non sarcomeri riorganizzazione o riorganizzazione.

Video 1.Filamenti di actina in un hiCM che assembla i sarcomeri. hiCM trasfezionato con Lifeact-mApple e ripreso con SIM.Le MSF subiscono un flusso retrogrado e la transizione al sarcomero contenente miofibrille verso il corpo cellulare. Tabella di ricerca: arancione caldo. 30,5 per 20,9 µm. Lunghezza del video: 9,5 ore.
Per caratterizzare ulteriormente il nostro test di assemblaggio dei sarcomeri, abbiamo usato α-actinina 2, che è un classico marcatore di linee Z (Lutero, 2009). Endogena α-actinina 2 localizzata sia a MSF che a sarcomeri (Figura 2A). Piccoli punti di α-actinina 2 localizzati a MSF, mentre i sarcomeri avevano α-actinina 2 lineare che etichettava le linee Z (Figura 2A). Come è stato mostrato in altri sistemi(Dabiri et al., 1997; Du et al., 2008), la spaziatura tra α-acttinina 2 puncta aumenta durante la transizione da MSF a sarcomero, con la spaziatura di α-acttinina 2 ~ 0,5 µm in MSF e ~1,7 µm in sarcomeri (Figura 2B). In hiCMs, α-actinina 2 puncta in MSF si alterna con NMII, come è stato mostrato per altri sistemi (Figura 2-figure supplement 1) (Ehler etal., 1999; Hotulainen e Lappalainen, 2006; Rhee et al., 1994; Sanger et al., 2005). È interessante notare che la spaziatura di α-acttinina 2 puncta associata a MSF in hiCM era molto simile alla spaziatura di α-acttinina 4 (cioè, un paralogico non muscolare di α-acttinina) in archi di actina nelle cellule U2OS (Figura 2C). Se le MSF servivano come modello per i sarcomeri, abbiamo chiesto se le molecole di α-actinina 2 nelle MSF erano anche incorporate nelle strutture dei sarcomeri. I dati precedenti suggeriscono che α-actinina 2 puncta si uniscono alle linee Z esistenti (Dabiri etal., 1997; McKenna et al., 1986). Per testare questa ipotesi, abbiamo utilizzato una sonda fotoconvertibile, tdEOS, che converte la fluorescenza dal verde al rosso per marcare specificamente la α-actinina 2 puncta delle MSF (Nienhauset al., 2006; Wiedenmann et al., 2004). Abbiamo trovato che un sottoinsieme di fotoconvertito α-acttinina 2-tdEOS puncta sono stati effettivamente incorporati in linee Z (Figura 2D). Collettivamente, questi risultati suggeriscono fortemente che le MSF danno origine a sarcomeri in hiCM.
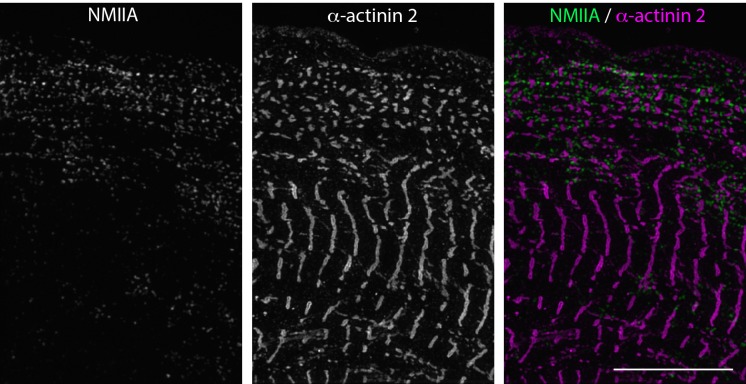
Figura 2-figure supplemento 1.α-Actininin 2 spaziatura e la dinamica in hiCMs.SIM di NMIIA e α-actinina 2localizzazione in hiCM(A) Rappresentazione codificata a colori per l’altezza Z dell’α-acttinina 2 endogena in MSF (riquadro 1) e dei sarcomeri (riquadro 2) di hiCM ripresi con SIM. Si noti la differenza nella struttura e nella spaziatura dell’α-acttinina 2 in MSF (riquadro 1) e dei sarcomeri (riquadro 2) (B) Distanza tra le strutture di α-acttinina 2 in MSF e i sarcomeri. MSF; 14 celle, tre esperimenti, 827 misure, sarcomeri; 15 celle, tre esperimenti, 527 misure. La distanza tra le strutture aumenta con il passaggio dei MSF ai sarcomeri.(C) Istogramma che rappresenta la distribuzione delle distanze tra α-acttinina 2 strutture in MSF in hiCMs (in alto) e α-acttinina 4 trovato in archi di actina di cellule non muscolari (in basso). La distribuzione è simile tra i tipi di cellule.(D) Ampio campo di montaggio di fotoconversione di α-acttinina 2-tdEOS in hiCM. MSF al bordo anteriore della cellula sono stati fotoconvertiti (da verde a rosso) e ripresi nel tempo. Montaggio (al centro) rappresenta α-acttinina 2-tdEOS puncta di MSF (punte di freccia gialla cava) transizione in strutture di sarcomeri (centro, a destra). Barre di scala;(A) 10 µm di magneti bassi, 5 µm di magneti alti.(B), 10 µM. Valori P indicati nei grafici.NMIIA localizza a MSF, ma è escluso dai sarcomeri (vedi Figura 7A e B). NMIIA si trova tra piccoli α-acttinina 2 puncta in MSF a bordo di hiCM (alternando verde e magenta nell’immagine a destra), ed è escluso da grandi α-acttinina 2 linee Z nel corpo hiCM. Barra di scala: 10 μm.
Video 1.2. Filamenti di actina in un hiCM che assembla i sarcomeri. hiCM trasformato con Lifeact-mApple e ripreso con SIM.MSFs subiscono il flusso retrogrado e la transizione al sarcomero contenente miofibrille verso il corpo cellulare. Tabella di ricerca: arancione caldo. 30,5 per 20,9 µm. Lunghezza del video: 9,5 ore.
Figura 2-figure supplemento 1.α-Actininin 2 spaziatura e la dinamica in hiCMs.SIM di NMIIA e α-actinina 2localizzazione in hiCM(A) Rappresentazione codificata a colori per l’altezza Z dell’α-acttinina 2 endogena in MSF (riquadro 1) e dei sarcomeri (riquadro 2) di hiCM ripresi con SIM. Si noti la differenza nella struttura e nella spaziatura dell’α-acttinina 2 in MSF (riquadro 1) e dei sarcomeri (riquadro 2) (B) Distanza tra le strutture di α-acttinina 2 in MSF e i sarcomeri. MSF; 14 celle, tre esperimenti, 827 misure, sarcomeri; 15 celle, tre esperimenti, 527 misure. La distanza tra le strutture aumenta con il passaggio dei MSF ai sarcomeri.(C) Istogramma che rappresenta la distribuzione delle distanze tra α-acttinina 2 strutture in MSF in hiCMs (in alto) e α-acttinina 4 trovato in archi di actina di cellule non muscolari (in basso). La distribuzione è simile tra i tipi di cellule.(D) Ampio campo di montaggio di fotoconversione di α-acttinina 2-tdEOS in hiCM. MSF al bordo anteriore della cellula sono stati fotoconvertiti (da verde a rosso) e ripresi nel tempo. Montaggio (al centro) rappresenta α-acttinina 2-tdEOS puncta di MSF (punte di freccia gialla cava) transizione in strutture di sarcomeri (centro, a destra). Barre di scala;(A) 10 µm di magneti bassi, 5 µm di magneti alti.(B), 10 µM. Valori P indicati nei grafici.NMIIA localizza a MSF, ma è escluso dai sarcomeri (vedi Figura 7A e B). NMIIA si trova tra piccoli α-acttinina 2 puncta in MSF a bordo di hiCM (alternando verde e magenta nell’immagine a destra), ed è escluso da grandi α-acttinina 2 linee Z nel corpo hiCM. Barra di scala: 10 μm.
Figura 2-figure supplemento 1.SIM di NMIIA e α-actinina 2localizzazione in hiCMNMIIA si localizza in MSF ma è escluso dai sarcomeri (vedi Figura 7A e B). NMIIA si trova tra la piccola α-acttinina 2 puncta in MSF al bordo dell’hiCM (alternando verde e magenta nell’immagine a destra), ed è escluso dalla grande α-acttinina 2 linee Z nel corpo dell’hiCM. Barra di scala: 10 μm.
Actina flusso retrogrado in hiCMs e cellule non muscolari
Abbiamo poi voluto indagare ulteriormente sulle somiglianze tra MSF e archi di actina. Le fibre di stress da arco di actina nelle cellule non muscolari subiscono un robusto ‘flusso retrogrado’ lontano dal bordo della cellula, come si può vedere nelle cellule U2OS, un classico modello di migrazione mesenchimale (Figura 3Ae C) (Hotulainene Lappalainen, 2006; Ponti et al., 2004). Le misurazioni chimografiche hanno rilevato che gli archi di actina nelle cellule U2OS si sono spostati a ~200 nm/min, in accordo con i risultati precedentemente pubblicati (Figura 3A e C)(Pontiet al., 2004). Abbiamo trovato MSFs anche sottoposti a flusso retrogrado(Figura 3B e C). Sorprendentemente, tuttavia, la chimografia ha rivelato MSF in hiCMs spostato significativamente più lento di archi di actina in cellule U2OS(Figura 3C). Questa è stata la prima indicazione che gli archi di actina in cellule non muscolari sono diversi da MSF in hiCMs. Abbiamo poi voluto definire i meccanismi che regolano le MSF e la loro acquisizione di sarcomeri. Poiché i meccanismi di formazione e mantenimento dell’arco di actina sono stati ben studiati(Burnette et al., 2014; Hotulainen e Lappalainen, 2006; Murugesan et al., 2016), eravamo interessati ad usare il nostro saggio per verificare se gli stessi meccanismi che guidano la dinamica dell’arco di actina stavano governando la dinamica dei MSF.
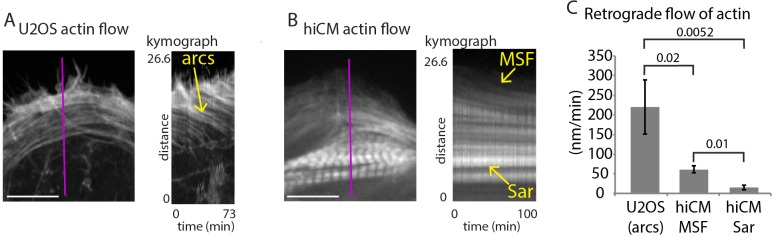
Figura 3.Figura 3. Flusso retrogrado di actina in cellule non muscolari e hiCMs.(A) Ancora di cella U2OS che esprime Lifeact-mEmerald (a sinistra) ripreso con disco in rotazione confocale. Kymograph (a destra) preso dalla linea viola dell’immagine di sinistra. Si noti il movimento robusto delle fibre di tensione dell’arco di actina (freccia gialla).(B) Ancora di hiCM che esprime Lifeact-mApple (sinistra) ripreso con disco rotante confocale. Kymograph (a destra) ripreso dalla linea viola dell’immagine di sinistra. Si noti il movimento più lento di MSF in hiCM rispetto agli archi di actina nella cella U2OS, e la natura stazionaria dei sarcomeri (Sar). La correzione dell’immagine gamma di 0,5 è stata utilizzata per visualizzare strutture relativamente luminose (cioè, sarcomeri) e fioche (cioè, MSF).(C) Quantificazione dei tassi di traslocazione delle fibre di actina stress in cellule U2OS e hiCM. U2OS; 3 cellule su tre esperimenti. hiCMs; 12 cellule su tre esperimenti. Barre di scala;(A),(B), 10 µM. P-valori indicati nel grafico.
Figura 3.Figura 3. Flusso retrogrado di actina in cellule non muscolari e hiCMs.(A) Ancora di cella U2OS che esprime Lifeact-mEmerald (a sinistra) ripreso con la filatura del disco confocale. Kymograph (a destra) preso dalla linea viola dell’immagine di sinistra. Si noti il movimento robusto delle fibre di tensione dell’arco di actina (freccia gialla).(B) Ancora di hiCM che esprime Lifeact-mApple (sinistra) ripreso con disco rotante confocale. Kymograph (a destra) ripreso dalla linea viola dell’immagine di sinistra. Si noti il movimento più lento di MSF in hiCM rispetto agli archi di actina nella cella U2OS, e la natura stazionaria dei sarcomeri (Sar). La correzione dell’immagine gamma di 0,5 è stata utilizzata per visualizzare strutture relativamente luminose (cioè, sarcomeri) e fioche (cioè, MSF).(C) Quantificazione dei tassi di traslocazione delle fibre di actina stress in cellule U2OS e hiCM. U2OS; 3 cellule su tre esperimenti. hiCMs; 12 cellule su tre esperimenti. Barre di scala;(A),(B), 10 µM. P-valori indicati nel grafico.
Formine, ma non il complesso Arp2/3, sono necessari per la formazione di sarcomeri basati su MSF
Il complesso Arp2/3 è ben noto per essere richiesto per la formazione di archi di actina in cellule non muscolari(Hotulainen e Lappalainen, 2006). Per testare il ruolo del complesso Arp2/3 durante l’assemblaggio dei sarcomeri, abbiamo permesso ad hiCMs di diffondersi in presenza di CK666, un inibitore del complesso Arp2/3(Nolen et al., 2009). Sorprendentemente, hiCMs permesso di diffondere in presenza di CK666 formato MSFs robusto e sarcomeri comparabili a cellule di controllo non trattati(Figura 4A, inset, e 4B). Le cellule sono state quantificate come contenenti sarcomeri se contenevano tre linee Z parallele in fila, ciascuna separata dalla linea Z adiacente di 1 µm – 2,5 µm. Una localizzazione α-actinina 2 era definita come una linea Z se era almeno 2 volte la lunghezza del limite di risoluzione del microscopio (vedi Materiali e metodi). Inoltre, la spaziatura tra le linee Z tra controllo e CK666 hiCMs era invariata(Figura 4C). Abbiamo anche trovato il flusso retrogrado di MSF è rimasto invariato tra il controllo e CK666 hiCMs trattati(Figura 4D ed E). Per confermare l’inibizione del complesso Arp2/3 da CK666, abbiamo esaminato la localizzazione endogena del complesso Arp2/3 con e senza trattamento CK666. La localizzazione forte del complesso Arp2/3 al bordo del controllo hiCMs era assente in CK666 hiCMs trattati(Figura 4F e G). Per confermare ulteriormente questa osservazione, abbiamo analizzato la perdita di p16b, una sottounità del complesso Arp2/3, dal bordo anteriore del hiCMs via CK666 in hiCMs vivo. Infatti, hiCMs ha mostrato una rapida perdita di p16b-mEGFP dal bordo di testa a seguito della somministrazione di CK666(Figura 4H e I). La delocalizzazione del complesso Arp2/3 dal bordo anteriore è coerente con l’inattivazione da CK666, come mostrato in precedenza in cellule non muscolari(Henson et al., 2015). Presi insieme, i nostri dati suggeriscono che il complesso Arp2/3 non ha bisogno di essere localizzato al bordo d’attacco per i sarcomeri da assemblare.
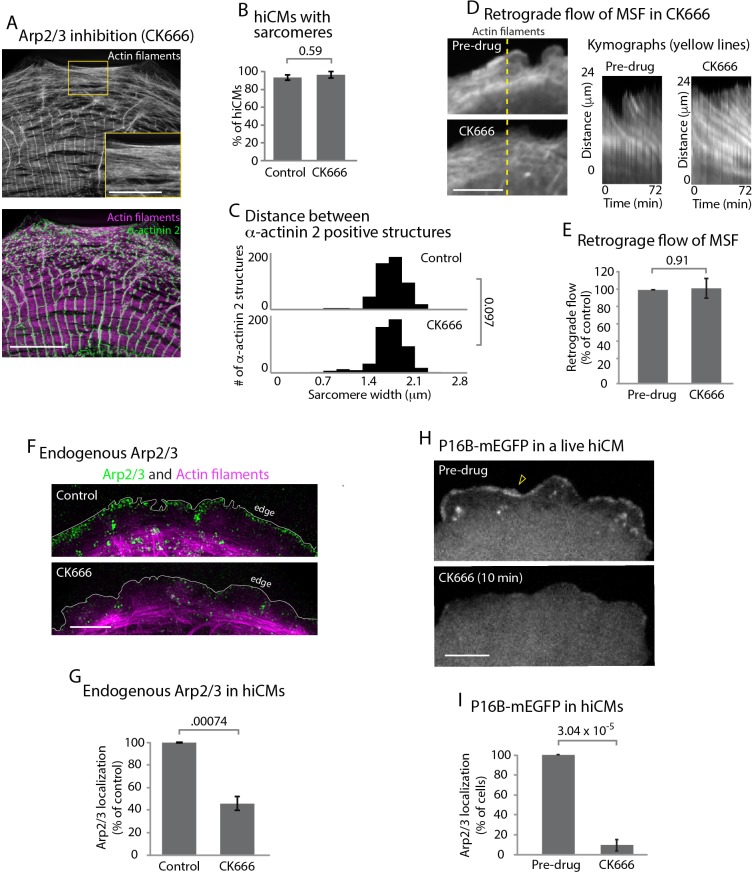
Figura 4.Il complesso Arp2/3 non è necessario per l’assemblaggio dei sarcomeri.(A) hiCM ha permesso di diffondere per 24 ore in presenza di 25 µM CK666, etichettato con actina e α-actinina 2 (cioè, linee Z) e ripreso con SIM. Il riquadro indica la presenza di MSF.B) Quantificazione della percentuale di cellule con sarcomeri a 24 ore dopo la placcatura in controllo e 25 µM CK666. Controllo: 76 cellule, 10 esperimenti; 25 µM CK666: 41 cellule, tre esperimenti.(C) Istogramma della distribuzione delle distanze tra α-acttinina 2 linee Z. Si noti la stretta distribuzione delle linee Z in entrambe le condizioni. Controllo: 14 cellule, tre esperimenti, 317 misure. 25 µM CK666: 16 cellule, tre esperimenti, 530 misure.(D) Alambicchi di hiCM che esprimono Lifeact-mApple pre (in alto) e post (in basso) aggiunta di 25 µM CK666 e ripresi con disco rotante confocale. Kymographs (a destra) presi da linea gialla tratteggiata (a sinistra).(E) Tassi di flusso retrogrado di hiCMs raffigurato come variazione percentuale in CK666 dalla condizione pre-droga. 8 cellule su tre esperimenti.(F) Localizzazione del complesso Arp2/3 in controllo (in alto) e 25 µM CK666 trattati (in basso) hiCMs ripresi con SIM. Nota perdita di Arp2/3 al bordo del CK666 hiCM trattati. Cellule diffuse per 24 ore in presenza di 25 µM CK666 come in Figura 4A (G) Quantificazione della perdita del complesso Arp2/3 dal bordo di testa degli hiCMs. Controllo; 36 cellule su tre esperimenti. 25 uM CK666; 29 cellule su tre esperimenti.(H) Live hiCM esprimendo P16B-mEGFP (un componente del complesso Arp2/3) e ripreso con la filatura disco confocale. La localizzazione di P16B-mEGFP all’avanguardia nel controllo pre-droga (in alto) è acutamente perso dopo l’aggiunta di 25 µM CK666 (in basso).(I) Quantificazione degli hiCM che visualizzano la localizzazione del complesso Arp2/3 (P16B-mEGFP) pre e post 25µM CK666 in hiCM dal vivo (come nella Figura 4H). 27 cellule su tre esperimenti. Barre di scala;(A) 10 µm basso magnete, 5 µm alto magnete inset.(D),(F), (H), (H), 10 µm. Valori P indicati nei grafici.
Oltre al complesso Arp2/3, la polimerizzazione dell’actina mediata dalla formina ha dimostrato di essere cruciale per la formazione di archi di actina e la dinamica in più tipi di cellule(Hotulainen e Lappalainen, 2006; Murugesan et al., 2016). Come punto di partenza per verificare se le formine sono necessarie per l’assemblaggio dei sarcomeri, abbiamo permesso ad hiCMs di diffondersi in presenza di un pan-inibitore della polimerizzazione dell’actina mediata dalla formina, piccolo inibitore molecolare dell’omologia della formina dominio 2, SMIFH2(Rizvi et al., 2009). SMIFH2 ha dimostrato di arrestare la polimerizzazione dell’actina mediata dalla formina, la formazione di archi di actina e il flusso retrogrado nelle cellule non muscolari(Henson et al., 2015; Murugesan et al., 2016; Rizvi et al., 2009). Abbiamo trovato hiCMs diffusione in presenza di SMIFH2 completamente non è riuscito a formare sarcomeri(Figura 5A e B, e Figura 5-figure supplemento 1). Questo effetto era reversibile, in quanto i sarcomeri si sono formati dopo la rimozione di SMIFH2(Figura 5-figure supplement 2). Distanze tra le strutture di α-acttinina 2 sono state anche significativamente diminuite in hiCM trattati con SMIFH2, con la distribuzione di α-acttinina 2 più simile a MSF rispetto ai sarcomeri (Figure 5Ce 2C). Tuttavia, gli allineamenti della α-actinina 2 puncta non erano simili alle MSF nelle hiCM di controllo, in quanto non erano periodici (Figure5A e 2A). Questo risultato ha fortemente suggerito che le formine sono necessarie per il montaggio del sarcomero.
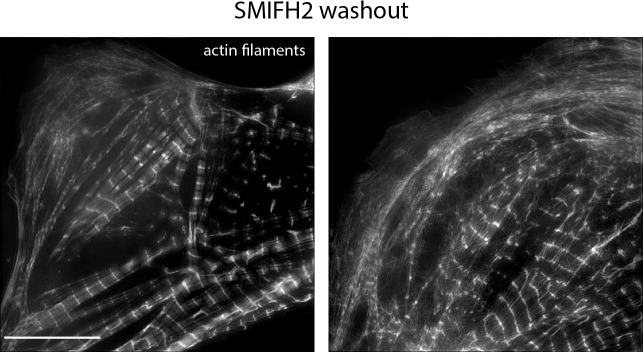
Figura 5-figure supplemento 2.Le formine sono necessarie per l’assemblaggio dei sarcomeri e la diffusione di MSF dynamics.hiCM in presenza di 25 µM SMIFH2 .hiCMs assemblare i sarcomeri dopo il lavaggio di SMIFH2.(A) hiCM permesso di diffondere in presenza di 25 µM SMIFH2 per 24 ore, etichettati con actina e α-actinina e ripresi con SIM. Box indica la perdita di MSF trasversale dietro il bordo d’attacco di hiCM.(B) Quantificazione della percentuale di cellule con sarcomeri a 24 ore dopo la placcatura. Controllo: 76 cellule, 10 esperimenti; 25 µM SMIFH2, 16 cellule, 16 cellule, tre esperimenti (C) Istogramma della distribuzione delle distanze tra α-acttinina 2 linee Z. Controllo: 14 cellule, tre esperimenti, 317 misure, 25 µM SMIFH2: 11 cellule, tre esperimenti, 468 misure.(D) Stills dal vivo hiCM esprimere Lifeact-mApple che sono stati sparsi per 24 ore e sarcomeri assemblati. hiCM prima (a sinistra) e 90 minuti dopo l’aggiunta di 25 µM SMIFH2 (a destra) (farmaco somministrato 24 ore dopo la diffusione) e ripreso con la microscopia a disco di filatura. Si noti come i sarcomeri e l’architettura complessiva dell’actina rimane imperturbata a 90 min dopo 25 µM SMIFH2.(E) I chimografi di MSF e il flusso retrogrado dei sarcomeri presi dalla linea viola in(D). Si noti l’immediata perdita di flusso retrogrado dopo l’aggiunta di 25 µM SMIFH2.(F) Quantificazione del flusso retrogrado dell’actina in hiCMs prima e dopo l’aggiunta di 25 µM SMIFH2. Controllo: 12 cellule, tre misure da ogni cella, tre esperimenti; 25 µM SMIFH2, 12 cellule, tre misure da ogni cella, tre esperimenti. Barre di scala:(A) 10 µm basso magnete, 5 µm alto magnete inset.(D), 10 µM. P valori indicati nei grafici.Spinning-disk microscopia confocale di hiCM che esprime Lifeact-mApple e diffusione in presenza di 25 µM SMIFH2 (aggiunto 1,5 hr post placcatura). Nota i sarcomeri non si formano in presenza di 25 µM SMIFH2. Barra di scala; 20 µm.Esempi di due hiCMs contenenti MSFs e sarcomeri a seguito di lavaggio di 25 µM SMIFH2 e ripresi con SIM. hiCMs sono stati sparsi in 25 µM SMIFH2 (come in Figura 5A) per 24 ore, e media freschi senza droga è stato scambiato. hiCMs sono stati successivamente permesso di diffondere per un ulteriore 24 ore prima della fissazione. Barra graduata; 10 µm.
Abbiamo poi chiesto se l’inibizione della formina stesse colpendo direttamente le MSF o i sarcomeri. Per testare questo, abbiamo permesso ad hiCMs di diffondersi per 24 ore (dopo che hanno stabilito sarcomeri) e imaged loro citoscheletro actina tramite microscopia a cellule vive prima e dopo la somministrazione di SMIFH2(Figura 5D). Dopo l’aggiunta di SMIFH2, la formazione di nuove MSF è stata immediatamente bloccata, insieme al flusso retrogrado delle MSF esistenti(Figura 5D-5F). Tuttavia, non abbiamo rilevato alcun cambiamento nella struttura del sarcomero nel breve periodo di tempo dell’esperimento, e gli hiCM hanno continuato a battere in presenza di SMIFH2 (si noti la struttura del sarcomero nella Figura 5D). Poiché ci sono 15 geni di formina di mammifero, abbiamo poi chiesto quale formina specifica era richiesta per l’assemblaggio del sarcomero.
Abbiamo eseguito l’analisi di sequenziamento RNA di mRNA isolato da hiCMs. Conta lettura normalizzata ha rivelato che una formina, FHOD3, è stato espresso superiore a tutte le altre formine(Figura 6A). Infatti, i dati precedenti da cardiomiociti di ratto isolati hanno mostrato FHOD3 come cruciale per la manutenzione del sarcomero(Iskratsch et al., 2010; Kan-O et al., 2012; Taniguchi et al., 2009). I cardiomiociti di ratto contenenti miofibrille hanno successivamente perso le loro miofibrille in seguito all’abbattimento dell’FHOD3(Iskratsch et al., 2010; Taniguchi et al., 2009). Tuttavia, il ruolo dell’FHOD3 durante l’assemblaggio del sarcomero de novo non è stato testato. Pertanto, abbiamo cercato di utilizzare il nostro test per verificare direttamente se il modulo FHOD3 era necessario per l’assemblaggio dei sarcomeri basato su MSF. Abbiamo abbattuto FHOD3 utilizzando siRNA, e gli hiCM non sono stati in grado di assemblare i sarcomeri dopo la placcatura(Figura 6B e C). È interessante notare che KD delle due formine più altamente espresse dopo FHOD3, DAAM1 e DIAPH1, non ha fermato il montaggio dei sarcomeri(Figura 6C e Figura 6-figure supplement 1). Tuttavia, ci sono evidenti difetti nell’organizzazione dell’actina al bordo della cellula e nei sarcomeri(Figura 6-figure supplement 1). Poiché FHOD3 aveva il fenotipo più prominente, abbiamo deciso di concentrare la nostra ulteriore analisi su questa condizione. In linea con l’inibizione pan-formin, l’organizzazione dell’actina e la spaziatura tra α-acttinina 2 in FHOD3 KD hiCMs molto simile hiCMs diffusa in presenza di SMIFH2 (Figura 5A-5C e Figura 6B-6D ).
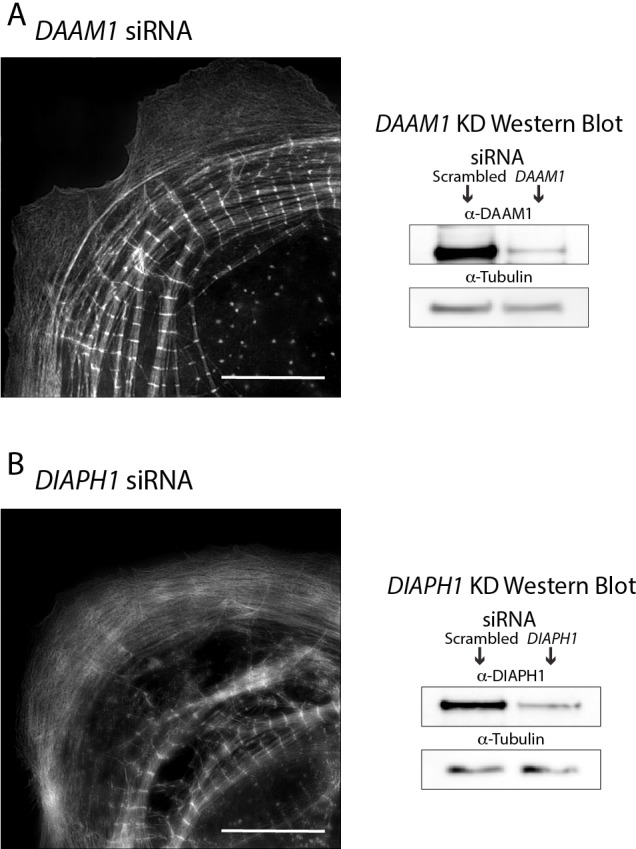
Figura 6-figure supplement 1.Formin FHOD3 è necessario per i sarcomeri assembly.hiCMs assemblare i sarcomeri a seguito di knockdown (KD) di DAAM1 e DIAPH1.(A) Frame normalizzati per milione di Kilobase (FPKM) di mRNA espressione dei primi tre formini espressi in hiCMs, tre esperimenti, tre corse separate.(B) Actin di siRNA scramble control e siRNA FHOD3 hiCMs permesso di diffondere per 24 ore e ripreso con SIM. Si noti la perdita di sarcomeri paragonabile al trattamento SMIFH2 in FHOD3 KD hiCMs(Figura 5A). Western blot (a destra) indica la perdita di proteine in FHOD3 KD.(C) Quantificazione della percentuale di cellule con sarcomeri a 24 ore dopo la placcatura nel controllo scramble, siFHOD3, siDAAM1, e siDIAHP1 hiCMs. Controllo: 76 cellule, 10 esperimenti; siRNA FHOD3: 33 cellule, tre esperimenti; siRNA DAAM1: 29 cellule, tre esperimenti; siRNA Dia1: 26 cellule, tre esperimenti.(D) Istogramma della distribuzione delle distanze tra α-acttinina 2 linee Z. Controllo: 14 cellule, tre esperimenti, 317 misure; siFHOD3: 15 cellule, tre esperimenti, 488 misure.(E) hiCM trasfettato con FHOD3-mEGFP, fissato a 24 ore, colorato per l’actina e ripreso con SIM. Le caselle 1-6 rappresentano la localizzazione di FHOD3-mEGFP lungo le MSF (caselle 1 e 2) e nei sarcomeri (caselle 4, 5 e 6).(F) Le caselle da (E) mostrano una maggiore organizzazione di FHOD3-mEGFP da MSF a sarcomeri. Si noti la struttura sempre più organizzata e la localizzazione tra le linee Z di FHOD3-mEGFP. Barre di scala;(B),(E), 10 µm.(F), 5 µm. Valori P indicati nei grafici.(A) siRNA DAAM1 hiCM siRNA diffusa per 24 ore colorata per l’actina, e ripreso con SIM (a sinistra). DAAM1 KD hiCMs ancora assemblare sarcomeri, e hanno esteso lamellipodia. Rappresentante western blot che mostra KD di proteina DAAM1 (a destra).(B) siRNA DIAPH1 hiCM siRNA diffusa per 24 ore, macchiato per l’actina e ripreso con SIM (a sinistra). DIAPH1 KD hiCMs sembrano avere meno sarcomeri di hiCMs di controllo scramble, ma ancora assemblare sarcomeri. Rappresentativo western blot che mostra KD di proteina DIAPH1 (a destra). Barre di scala;(A),(B), 10 µm.
Sulla base dei nostri risultati, se FHOD3 è coinvolto nel montaggio di sarcomeri, dovrebbe localizzare a MSF. FHOD3-mEGFP localizzato a entrambi i MSF al bordo di hiCMs, e poi diventa sempre più organizzato lontano dal bordo di testa della cella in cui si trovano i sarcomeri(Figura 6E). Questa localizzazione è coerente con un ruolo per FHOD3 nel mediare la transizione da MSF a sarcomeri. Presi insieme, i nostri dati mostrano che la formin FHOD3 si localizza sia a MSF che a sarcomeri, ed è necessaria per l’assemblaggio de novo dei sarcomeri. Abbiamo poi voluto indagare su altri potenziali meccanismi che regolano l’assemblaggio dei sarcomeri.
Figura 4.Il complesso Arp2/3 non è richiesto per l’assemblaggio dei sarcomeri.(A) hiCM ha permesso di diffondere per 24 ore in presenza di 25 µM CK666, etichettato con actina e α-actinina 2 (cioè, linee Z) e ripreso con SIM. Il riquadro indica la presenza di MSF.B) Quantificazione della percentuale di cellule con sarcomeri a 24 ore dopo la placcatura in controllo e 25 µM CK666. Controllo: 76 cellule, 10 esperimenti; 25 µM CK666: 41 cellule, tre esperimenti.(C) Istogramma della distribuzione delle distanze tra α-acttinina 2 linee Z. Si noti la stretta distribuzione delle linee Z in entrambe le condizioni. Controllo: 14 cellule, tre esperimenti, 317 misure. 25 µM CK666: 16 cellule, tre esperimenti, 530 misure.(D) Alambicchi di hiCM che esprimono Lifeact-mApple pre (in alto) e post (in basso) aggiunta di 25 µM CK666 e ripresi con disco rotante confocale. Kymographs (a destra) presi da linea gialla tratteggiata (a sinistra).(E) Tassi di flusso retrogrado di hiCMs raffigurato come variazione percentuale in CK666 dalla condizione pre-droga. 8 cellule su tre esperimenti.(F) Localizzazione del complesso Arp2/3 in controllo (in alto) e 25 µM CK666 trattati (in basso) hiCMs ripresi con SIM. Nota perdita di Arp2/3 al bordo del CK666 hiCM trattati. Cellule diffuse per 24 ore in presenza di 25 µM CK666 come in Figura 4A (G) Quantificazione della perdita del complesso Arp2/3 dal bordo di testa degli hiCMs. Controllo; 36 cellule su tre esperimenti. 25 uM CK666; 29 cellule su tre esperimenti.(H) Live hiCM esprimendo P16B-mEGFP (un componente del complesso Arp2/3) e ripreso con la filatura disco confocale. La localizzazione di P16B-mEGFP all’avanguardia nel controllo pre-droga (in alto) è acutamente perso dopo l’aggiunta di 25 µM CK666 (in basso).(I) Quantificazione degli hiCM che visualizzano la localizzazione del complesso Arp2/3 (P16B-mEGFP) pre e post 25µM CK666 in hiCM dal vivo (come nella Figura 4H). 27 cellule su tre esperimenti. Barre di scala;(A) 10 µm basso magnete, 5 µm alto magnete inset.(D),(F), (H), (H), 10 µm. Valori P indicati nei grafici.
Figura 5-figure supplement 2.Le formine sono necessarie per l’assemblaggio di sarcomeri e la diffusione di MSF dynamics.hiCM in presenza di 25 µM SMIFH2 .hiCMs assemblare sarcomeri dopo il lavaggio di SMIFH2.(A) hiCM permesso di diffondere in presenza di 25 µM SMIFH2 per 24 ore, etichettati con actina e α-actinina e ripresi con SIM. Box indica la perdita di MSF trasversale dietro il bordo d’attacco di hiCM.(B) Quantificazione della percentuale di cellule con sarcomeri a 24 ore dopo la placcatura. Controllo: 76 cellule, 10 esperimenti; 25 µM SMIFH2, 16 cellule, 16 cellule, tre esperimenti (C) Istogramma della distribuzione delle distanze tra α-acttinina 2 linee Z. Controllo: 14 cellule, tre esperimenti, 317 misure, 25 µM SMIFH2: 11 cellule, tre esperimenti, 468 misure.(D) Stills dal vivo hiCM esprimere Lifeact-mApple che sono stati sparsi per 24 ore e sarcomeri assemblati. hiCM prima (a sinistra) e 90 minuti dopo l’aggiunta di 25 µM SMIFH2 (a destra) (farmaco somministrato 24 ore dopo la diffusione) e ripreso con la microscopia a disco di filatura. Si noti come i sarcomeri e l’architettura complessiva dell’actina rimane imperturbata a 90 min dopo 25 µM SMIFH2.(E) I chimografi di MSF e il flusso retrogrado dei sarcomeri presi dalla linea viola in(D). Si noti l’immediata perdita di flusso retrogrado dopo l’aggiunta di 25 µM SMIFH2.(F) Quantificazione del flusso retrogrado dell’actina in hiCMs prima e dopo l’aggiunta di 25 µM SMIFH2. Controllo: 12 cellule, tre misure da ogni cella, tre esperimenti; 25 µM SMIFH2, 12 cellule, tre misure da ogni cella, tre esperimenti. Barre di scala:(A) 10 µm basso magnete, 5 µm alto magnete inset.(D), 10 µM. P valori indicati nei grafici.Spinning-disk microscopia confocale di hiCM che esprime Lifeact-mApple e diffusione in presenza di 25 µM SMIFH2 (aggiunto 1,5 hr post placcatura). Nota i sarcomeri non si formano in presenza di 25 µM SMIFH2. Barra di scala; 20 µm.Esempi di due hiCMs contenenti MSFs e sarcomeri a seguito di lavaggio di 25 µM SMIFH2 e ripresi con SIM. hiCMs sono stati sparsi in 25 µM SMIFH2 (come in Figura 5A) per 24 ore, e media freschi senza droga è stato scambiato. hiCMs sono stati successivamente permesso di diffondere per un ulteriore 24 ore prima della fissazione. Barra graduata; 10 µm.
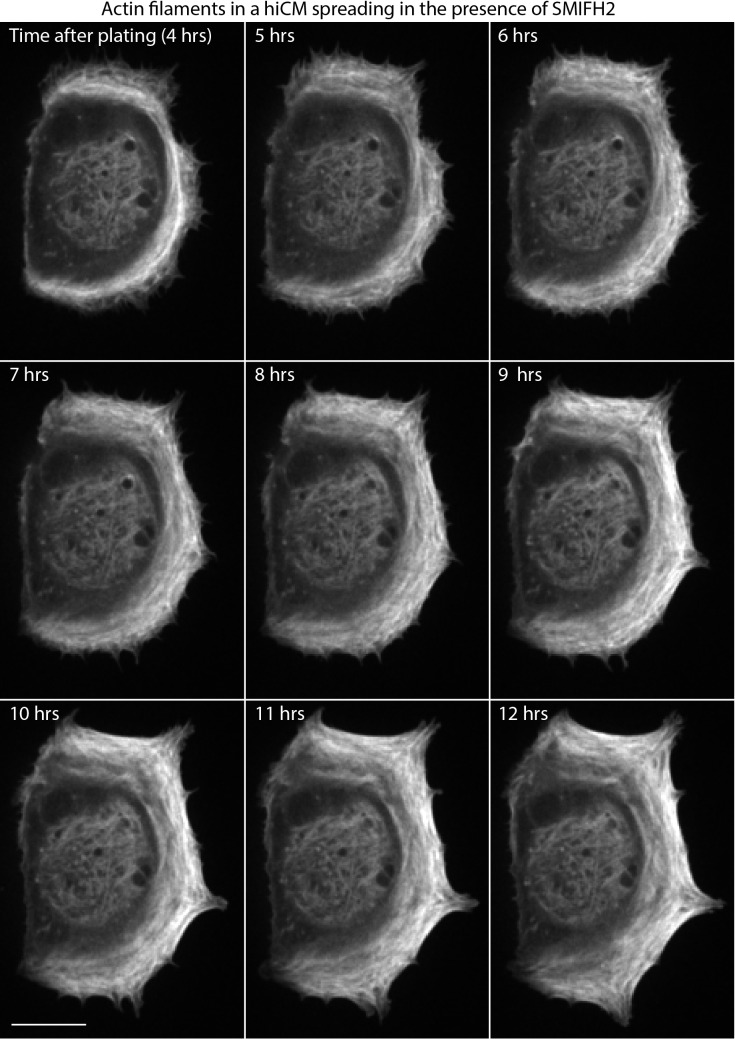
Figura 5-figure supplemento 1.hiCM diffusione in presenza di 25 µM SMIFH2 .Spinning-disk microscopia confocale di hiCM che esprime Lifeact-mApple e diffusione in presenza di 25 µM SMIFH2 (aggiunto 1,5 hr post placcatura). Nota i sarcomeri non si formano in presenza di 25 µM SMIFH2. Barra di scala; 20 µm.
Figura 5-figure supplemento 2.hiCMs assemblare i sarcomeri dopo il lavaggio di SMIFH2.Esempi di due hiCMs contenenti MSFs e sarcomeri dopo il lavaggio di 25 µM SMIFH2 e ripresi con SIM. hiCMs sono stati sparsi in 25 µM SMIFH2 (come in Figura 5A) per 24 ore, e media freschi senza droga è stato scambiato. hiCMs sono stati successivamente permesso di diffondere per un ulteriore 24 ore prima della fissazione. Barra graduata; 10 µm.
Figura 6-figure supplemento 1.Formin FHOD3 è necessario per il montaggio di sarcomeri assembly.hiCMs assemblare sarcomeri dopo knockdown (KD) di DAAM1 e DIAPH1.(A) Frame normalizzati per milione di Kilobase (FPKM) di mRNA espressione dei primi tre formini espressi in hiCMs, tre esperimenti, tre corse separate.(B) Actin di siRNA scramble control e siRNA FHOD3 hiCMs permesso di diffondere per 24 ore e ripreso con SIM. Si noti la perdita di sarcomeri paragonabile al trattamento SMIFH2 in FHOD3 KD hiCMs(Figura 5A). Western blot (a destra) indica la perdita di proteine in FHOD3 KD.(C) Quantificazione della percentuale di cellule con sarcomeri a 24 ore dopo la placcatura nel controllo scramble, siFHOD3, siDAAM1, e siDIAHP1 hiCMs. Controllo: 76 cellule, 10 esperimenti; siRNA FHOD3: 33 cellule, tre esperimenti; siRNA DAAM1: 29 cellule, tre esperimenti; siRNA Dia1: 26 cellule, tre esperimenti.(D) Istogramma della distribuzione delle distanze tra α-acttinina 2 linee Z. Controllo: 14 cellule, tre esperimenti, 317 misure; siFHOD3: 15 cellule, tre esperimenti, 488 misure.(E) hiCM trasfettato con FHOD3-mEGFP, fissato a 24 ore, colorato per l’actina e ripreso con SIM. Le caselle 1-6 rappresentano la localizzazione di FHOD3-mEGFP lungo le MSF (caselle 1 e 2) e nei sarcomeri (caselle 4, 5 e 6).(F) Le caselle da (E) mostrano una maggiore organizzazione di FHOD3-mEGFP da MSF a sarcomeri. Si noti la struttura sempre più organizzata e la localizzazione tra le linee Z di FHOD3-mEGFP. Barre di scala;(B),(E), 10 µm.(F), 5 µm. Valori P indicati nei grafici.(A) siRNA DAAM1 hiCM siRNA diffusa per 24 ore colorata per l’actina, e ripreso con SIM (a sinistra). DAAM1 KD hiCMs ancora assemblare sarcomeri, e hanno esteso lamellipodia. Rappresentante western blot che mostra KD di proteina DAAM1 (a destra).(B) siRNA DIAPH1 hiCM siRNA diffusa per 24 ore, macchiato per l’actina e ripreso con SIM (a sinistra). DIAPH1 KD hiCMs sembrano avere meno sarcomeri di hiCMs di controllo scramble, ma ancora assemblare sarcomeri. Rappresentativo western blot che mostra KD di proteina DIAPH1 (a destra). Barre di scala;(A),(B), 10 µm.
Figura 6-figure supplemento 1.hiCMs assemblare sarcomeri dopo l’abbattimento (KD) di DAAM1 e DIAPH1.(A) siRNA DAAM1 hiCM siRNA diffusa per 24 ore colorata per l’actina, e ripreso con SIM (a sinistra). hiCM DAAM1 KD hiCMs ancora assemblare sarcomeri, e hanno esteso lamellipodia. Rappresentante western blot che mostra KD di proteina DAAM1 (a destra).(B) siRNA DIAPH1 hiCM siRNA diffusa per 24 ore, macchiato per l’actina e ripreso con SIM (a sinistra). DIAPH1 KD hiCMs sembrano avere meno sarcomeri di hiCMs di controllo scramble, ma ancora assemblare sarcomeri. Rappresentativo western blot che mostra KD di proteina DIAPH1 (a destra). Barre di scala;(A),(B), 10 µm.
Non miosina muscolare II è richiesto per i filamenti di actina del sarcomero cardiaco
Oltre ai nucleatori di actina, l’attività della miosinanon muscolare II (NMII) si è dimostrata necessaria per la formazione di archi di actina e l’organizzazione in tipi di cellule non muscolari(Hotulainen e Lappalainen, 2006; Medeiros et al., 2006). Così, abbiamo chiesto se l’NMII era necessaria per la formazione di MSF e/o la transizione di MSF al sarcomero. Per prima cosa abbiamo localizzato i due principali paraloghi di NMII negli esseri umani, NMIIA e NMIIB, in hiCM di diffusione(Vicente-Manzanares et al., 2009). Sia l’NMIIA che l’NMIIB si localizzano in archi di actina in cellule non muscolari(Kolega, 1998). Coerentemente con questo, sia NMIIA e NMIIB localizzati in MSF, e sono stati limitati dal centro della cella in cui sono stati localizzati i sarcomeri(Figura 7A e B). Infatti, la microscopia time-lapse ha rivelato filamenti NMIIA formato al bordo di hiCMs e sono stati sottoposti a flusso retrogrado come nelle cellule non-muscolari(Figura 7C). Tuttavia, NMIIA è rimasto al bordo di hiCMs ed è stato limitato dal corpo cellulare dove si formano i sarcomeri(Figura 7C e Video 2). NMIIB anche rimasto al bordo di hiCMs ed è stato limitato dal corpo cellulare in cui si formano i sarcomeri(Video 3). La stragrande maggioranza dei filamenti NMIIA e NMIIB si sono sovrapposti, tranne che sul bordo di testa, dove NMIIA è localizzato leggermente più avanti di NMIIB nelle hiCM(Figura 7A e B). La microscopia a super-risoluzione ha rivelato che la maggior parte dei filamenti NMIIA conteneva NMIIA e NMIIB(Figura 7D). NMIIA e NMIIB co-filamenti sono stati precedentemente riportati in cellule non muscolari(Beach et al., 2014; Shutova et al., 2014). Le misurazioni delle lunghezze dei co-filamenti NMII in hiCM hanno mostrato lunghezze in accordo con le misurazioni precedentemente pubblicate in cellule non muscolari(Figura 7D ed E)(Beach et al., 2014; Shutova et al., 2014). Presi insieme, questi dati suggeriscono l’organizzazione e la dinamica NMII appaiono simili in MSF di hiCMs come in archi di actina di cellule non-muscolari.
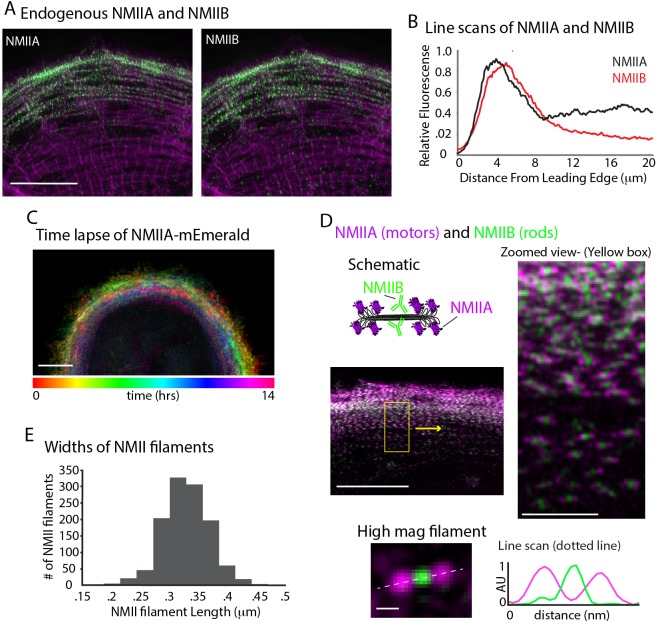
Figura 7.Figura 7. Localizzazione e dinamica NMII in hiCMs.(A) Localizzazione di NMIIA endogeno (a sinistra) e NMIIB (a destra) nella stessa hiCM e ripreso con SIM. Sia NMIIA e NMIIB localizzare a MSF al bordo anteriore di hiCMs.(B) Scansioni di linea a partire dal bordo degli hiCM che mostrano la localizzazione di NMIIA (nero) e NMIIB (rosso). Nota NMIIA è localizzato leggermente, ~1 µm davanti a NMIIB. NMIIA: 15 cellule, due esperimenti; NMIIB: 32 cellule, quattro esperimenti.(C) La proiezione a colori di time-lapse di hiCM che esprime NMIIA-mEmerald e ripreso con la scansione laser confocale. Si noti come NMIIA-mEmerald rimane al bordo di hiCMs.(D) hiCM trasfettato con NMIIA-mEmerald (N-terminal motors), colorato per il dominio endogeno NMIIB C-terminal rod dominio (cartone animato schematico e centrale a sinistra), e ripreso con SIM. Viste ad alta risoluzione dei co-filamenti NMIIA-NMIIB (a destra) dal riquadro giallo (in mezzo a sinistra). Vista ad alta risoluzione di un singolo co-filamento NMIIA-NMIIB (in basso) e scansione della linea attraverso la linea tratteggiata bianca, dai motori N-terminali (viola) e dai domini C-terminali ad asta (verde).(E) Quantificazione della lunghezza del co-filamento NMII. L’istogramma mostra la distribuzione delle lunghezze dei co-filamenti NMII (da dominio motore a dominio motore). Barre di scala;(A),(C), 10 µm, (D) 10 µm low mag (sinistra), 2 µm inserto ‘vista ingrandita’ (destra), 200 nm inserto ‘high mag filament’ (in basso).
Video 2.Dinamica dei filamenti NMIIA durante l’assemblaggio del sarcomero. hiCM trasfettato con NMIIA-mEmerald e Lifeact-mApple e ripreso con scansione laser 3D confocale.Si noti come i filamenti NMIIA-mEmerald si formano ai margini degli hiCM, sono localizzati in MSF, ma sono limitati dai sarcomeri durante l’assemblaggio dei sarcomeri. 28 per 35 μm. Lunghezza del video: 14 ore.
Video 3.Dinamica dei filamenti NMIIB durante l’assemblaggio dei sarcomeri. hiCM trasfettato con pHalo-NMIIB e α-acttinina 2-mEmerald e ripreso con microscopia ad ampio campo.Si noti come i filamenti NMIIB-Halo si formano ai margini degli hiCM, sono localizzati a MSF, ma sono limitati dai sarcomeri durante l’assemblaggio dei sarcomeri. 47 per 27 μm. Lunghezza del video: 7 ore.
Data la presenza di NMIIA e NMIIB in ogni filamento all’interno di MSF, abbiamo poi chiesto se NMIIA e/o NMIIB erano necessari per l’assemblaggio dei sarcomeri. NMIIA è stato precedentemente dimostrato di essere richiesto per l’assemblaggio dell’arco di actina in cellule non muscolari(Figura 8-figure supplement 1) (Burnetteet al., 2014; Fenix et al., 2016). Così, abbiamo ipotizzato che NMIIA sarebbe probabilmente la chiave paralogica richiesta per la formazione di MSF e il successivo assemblaggio del sarcomero. Sorprendentemente, KD di NMIIA non ha portato ad una completa inibizione di MSF o di assemblaggio di sarcomeri, anche se i sarcomeri in NMIIA KD hiCMs erano disorganizzati(Figura 8A-8E e Figura 8-figure supplementi 2 e 3). In particolare, gli hiCM NMIIA KD hanno mostrato una distribuzione simile delle distanze tra le strutture α-actinina 2 rispetto agli hiCM di controllo (Figura 8D). Questa misurazione mostra che, sebbene ci siano meno e più sarcomeri disorganizzati nel NMIIA KD, le loro larghezze misurate da linea Z a linea Z sono simili agli hiCM di controllo. Tuttavia, le cellule NMIIA KD aveva significativamente più breve Z-linee rispetto al controllo hiCMs(Figura 8E). Presi insieme, questi dati suggeriscono che NMIIA è coinvolto nel montaggio sarcomero e l’organizzazione.

Figura 8-figure supplemento 6.NMIIA e NMIIB sono necessari per l’assemblaggio del sarcomero in hiCMs.NMIIA ma non NMIIB è richiesto per la formazione di archi di actina in cellule HeLa.Knockdown di NMIIA e NMIIB in hiCMs.Montaggio live di diffusione NMIIA KD hiCM.montaggio live di diffusione NMIIA KD hiCM.montaggio live di diffusione NMIIB KD hiCM.montaggio live di diffusione hiCM in 100 µM blebbistatina.NMIIA KD e NMIIB KD hiCMs contengono sarcomeri prima della placcatura.(A) Actin del controllo rappresentativo scramble (in alto), NMIIA KD (siRNA MYH9, al centro), e NMIIB KD (siRNA MYH10, in basso) hiCMs permesso di diffondere per 24 ore e ripreso con SIM. NMIIA KD hiCMs (al centro) visualizzare sarcomeri disorganizzati, mentre NMIIB KD hiCMs (in basso) non visualizzare alcun sarcomero a base di actina.(B) Rappresentative macchie occidentali di 2 esperimenti separati che mostrano l’abbattimento di NMIIA (siRNA MYH9, in alto) e NMIIB (siRNA MYH10, in basso).(C) Percentuale di controllo scramble, NMIIA KD (siRNA MYH9), e NMIIB KD (siRNA MYH10) hiCMs con sarcomeri a base di actina a 24 ore di diffusione. Controllo: 49 cellule, sei esperimenti; NMIIA KD: 34 cellule, tre esperimenti; NMIIB KD: 59 cellule, quattro esperimenti.(D) Istogramma di distribuzione delle strutture α-actina 2 nel controllo scramble, NMIIA KD (siMYH9)e NMIIB KD (siMYH10) hiCMs. siScrambled: 554 misurazioni, 14 celle, tre esperimenti; siMYH9: 332 misurazioni, 15 celle, tre esperimenti; siMYH10: 772 misurazioni, 15 celle, tre esperimenti.(E) Quantificazione delle lunghezze della linea Z nel controllo scramble e NMIIA KD (siMYH9) hiCMs. Controllo: 22 celle, quattro esperimenti; NMIIA: 14 celle, tre esperimenti.(F) Quantificazione di hiCMs con sarcomeri nel controllo scramble, NMIIA KD (siMYH9), e NMIIB KD (siMYH10) hiCMs prima di ri-placcatura. siScrambled: 772 cellule, quattro esperimenti; siMYH9: 642 cellule, due esperimenti; siMYH10: 385 cellule, due esperimenti. Barra di scala;(A) 10 µm. I valori P indicati nei grafici.(A) cellule HeLa trattati con controllo, anti myh10, o anti myh9 (superiore, centrale, inferiore, rispettivamente) siRNA, colorato per l’actina, e ripreso con SIM. Si noti come le cellule KD di controllo e NMIIB KD visualizzano le fibre di tensione dell’arco di actina prominente, ma le cellule NMIIA KD non contengono archi di actina.(B) Quantificazione delle cellule HeLa con fibre di actina in condizioni indicate. Barre di scala: 5 µm.(A) Quantificazione da blot occidentali che mostrano KD di NMIIA (siMYH9) e NMIIB(siMYH10) in hiCMs rispetto al controllo scramble. N = 3 per ogni condizione.(B) Quantificazione dei livelli NMIIB negli hiCM siMYH9 (sinistra) e NMIIA negli hiCM siMYH10 (destra) rispetto al controllo scramble. N = 3 per ogni condizione.Laser-scanning confocale di diffusione NMIIA KD hiCM che esprime Lifeact-mApple. NMIIA KD hiCM è in grado di assemblare strutture di sarcomeri. Barra di scala; 10 µm.Scansione laser confocale di diffusione NMIIB KD hiCM che esprime Lifeact-mApple.NMIIB KD hiCM non forma sarcomeri. Barra graduata; 20 µm.Laser-scanning confocale di hiCM diffusione in presenza di 100 µM blebbistatina aggiunto 1,5 hr post placcatura. hiCM non assembla sarcomeri in presenza di 100 µM blebbistatina. Barra di scala; 20 µm.(A) siScrambled (in alto) siMYH9 (NMIIA KD, al centro), e siMYH10 (NMIIB KD, in basso) hiCMs trattati in 96 piastre pozzo, fisso, e localizzato per α-acttinina 2 imaged con microscopia ad ampio campo. hiCMs sono stati autorizzati ad attaccare per 4 giorni (secondo la raccomandazione del produttore, vedi Materiali e metodi), garantendo robusto assemblaggio sarcomero prima del trattamento siRNA iniziato. Si noti che tutte le condizioni contengono la colorazione sarcomerica di α-acttinina 2. Cercare tabella è mpl-magma nelle Fiji.
E ‘stato precedentemente mostrato NMIIB non è necessario per la formazione di archi di actina in cellule non muscolari(Kuragano et al., 2018; Shutova et al., 2017)(Figura 8-figure supplemento 1). Abbiamo ipotizzato che NMIIB non sarebbe necessario per MSF o assemblaggio di sarcomeri in hiCM. Sorprendentemente, NMIIB KD ha portato ad una completa incapacità di hiCMs di formare sarcomeri dopo la placcatura(Figura 8A-8D e Figura 8-figure supplementi 2 e 4). Inoltre, la larghezza tra le strutture di α-actinina 2 era significativamente più piccola delle hiCM di controllo (Figura 8D). Questi risultati sostengono che NMIIB è anche uno dei principali attori richiesti per l’assemblaggio di sarcomeri in hiCMs. Per confermare ulteriormente che la miosina II è necessaria per l’assemblaggio dei sarcomeri, abbiamo poi inibito farmacologicamente tutti i paraloghi della miosina II nelle hiCM con blebbistatina(Straight et al., 2003). le hiCM che si diffondevano in presenza di blebbistatina non erano in grado di assemblare le strutture dei sarcomeri(Figura 8-figure supplement 5). Mentre questi difetti nell’assemblaggio dei sarcomeri erano drammatici, abbiamo notato che gli hiCM trattati con siRNA contro NMIIA o NMIIB stavano ancora battendo prima della placcatura. Questo implicava che i sarcomeri preesistenti degli hiCM erano ancora intatti dopo il KD prima della placcatura. Pertanto, abbiamo immuno-localizzato α-actinina 2 per visualizzare i sarcomeri in hiCMs prima della placcatura. Sorprendentemente, abbiamo trovato che non ci sono state differenze tra il controllo, NMIIA, e NMIIB cellule KD prima della placcatura(Figura 8F e Figura 8-figure supplemento 6) Collettivamente, questi dati suggerirebbero NMIIA e NMIIB sono necessari per la formazione de novo sarcomeri, ma non omeostasi (cioè, il fatturato) di sarcomeri preesistenti.
Figura 7.NMII Localizzazione e dinamica in hiCMs.(A) Localizzazione di NMIIA endogeno (a sinistra) e NMIIB (a destra) nello stesso hiCM e ripreso con SIM. Sia NMIIA che NMIIB si localizzano in MSF al bordo anteriore degli hiCM.(B) Scansioni di linea a partire dal bordo degli hiCM che mostrano la localizzazione di NMIIA (nero) e NMIIB (rosso). Nota NMIIA è localizzato leggermente, ~1 µm davanti a NMIIB. NMIIA: 15 cellule, due esperimenti; NMIIB: 32 cellule, quattro esperimenti.(C) La proiezione a colori di time-lapse di hiCM che esprime NMIIA-mEmerald e ripreso con la scansione laser confocale. Si noti come NMIIA-mEmerald rimane al bordo di hiCMs.(D) hiCM trasfettato con NMIIA-mEmerald (N-terminal motors), colorato per il dominio endogeno NMIIB C-terminal rod dominio (cartone animato schematico e centrale a sinistra), e ripreso con SIM. Viste ad alta risoluzione dei co-filamenti NMIIA-NMIIB (a destra) dal riquadro giallo (in mezzo a sinistra). Vista ad alta risoluzione di un singolo co-filamento NMIIA-NMIIB (in basso) e scansione della linea attraverso la linea tratteggiata bianca, dai motori N-terminali (viola) e dai domini C-terminali ad asta (verde).(E) Quantificazione della lunghezza del co-filamento NMII. L’istogramma mostra la distribuzione delle lunghezze dei co-filamenti NMII (da dominio motore a dominio motore). Barre di scala;(A),(C), 10 µm, (D) 10 µm low mag (sinistra), 2 µm ‘vista ingrandita’ inset (destra), 200 nm ‘high mag filament’ inset (in basso).
Video 2.Dinamica dei filamenti NMIIA durante l’assemblaggio del sarcomero. hiCM trasfettato con NMIIA-mEmerald e Lifeact-mApple e ripreso con scansione laser 3D confocale.Si noti come i filamenti NMIIA-mEmerald si formano ai margini degli hiCM, sono localizzati in MSF, ma sono limitati dai sarcomeri durante l’assemblaggio dei sarcomeri. 28 per 35 μm. Lunghezza del video: 14 ore.
Video 3.Dinamica dei filamenti NMIIB durante l’assemblaggio dei sarcomeri. hiCM trasfettato con pHalo-NMIIB e α-acttinina 2-mEmerald e ripreso con microscopia ad ampio campo.Si noti come i filamenti NMIIB-Halo si formano ai margini degli hiCM, sono localizzati a MSF, ma sono limitati dai sarcomeri durante l’assemblaggio dei sarcomeri. 47 per 27 μm. Lunghezza del video: 7 ore.
Figura 8-figure supplemento 6.NMIIA e NMIIB sono necessari per l’assemblaggio di sarcomeri in hiCMs.NMIIA ma non NMIIB è richiesto per la formazione di archi di actina in cellule HeLa.Knockdown di NMIIA e NMIIB in hiCMs.Montaggio live di diffusione NMIIA KD hiCM.montaggio live di diffusione NMIIA KD hiCM.montaggio live di diffusione NMIIB KD hiCM.montaggio live di diffusione hiCM in 100 µM blebbistatina.NMIIA KD e NMIIB KD hiCMs contengono sarcomeri prima della placcatura.(A) Actin del controllo rappresentativo scramble (in alto), NMIIA KD (siRNA MYH9, al centro), e NMIIB KD (siRNA MYH10, in basso) hiCMs permesso di diffondere per 24 ore e ripreso con SIM. NMIIA KD hiCMs (al centro) visualizzare sarcomeri disorganizzati, mentre NMIIB KD hiCMs (in basso) non visualizzare alcun sarcomero a base di actina.(B) Rappresentative macchie occidentali di 2 esperimenti separati che mostrano l’abbattimento di NMIIA (siRNA MYH9, in alto) e NMIIB (siRNA MYH10, in basso).(C) Percentuale di controllo scramble, NMIIA KD (siRNA MYH9), e NMIIB KD (siRNA MYH10) hiCMs con sarcomeri a base di actina a 24 ore di diffusione. Controllo: 49 cellule, sei esperimenti; NMIIA KD: 34 cellule, tre esperimenti; NMIIB KD: 59 cellule, quattro esperimenti.(D) Istogramma di distribuzione delle strutture α-actina 2 nel controllo scramble, NMIIA KD (siMYH9)e NMIIB KD (siMYH10) hiCMs. siScrambled: 554 misurazioni, 14 celle, tre esperimenti; siMYH9: 332 misurazioni, 15 celle, tre esperimenti; siMYH10: 772 misurazioni, 15 celle, tre esperimenti.(E) Quantificazione delle lunghezze della linea Z nel controllo scramble e NMIIA KD (siMYH9) hiCMs. Controllo: 22 celle, quattro esperimenti; NMIIA: 14 celle, tre esperimenti.(F) Quantificazione di hiCMs con sarcomeri nel controllo scramble, NMIIA KD (siMYH9), e NMIIB KD (siMYH10) hiCMs prima di ri-placcatura. siScrambled: 772 cellule, quattro esperimenti; siMYH9: 642 cellule, due esperimenti; siMYH10: 385 cellule, due esperimenti. Barra di scala;(A) 10 µm. I valori P indicati nei grafici.(A) cellule HeLa trattati con controllo, anti myh10, o anti myh9 (superiore, centrale, inferiore, rispettivamente) siRNA, colorato per l’actina, e ripreso con SIM. Si noti come le cellule KD di controllo e NMIIB KD visualizzano le fibre di tensione dell’arco di actina prominente, ma le cellule NMIIA KD non contengono archi di actina.(B) Quantificazione delle cellule HeLa con fibre di actina in condizioni indicate. Barre di scala: 5 µm.(A) Quantificazione da blot occidentali che mostrano KD di NMIIA (siMYH9) e NMIIB(siMYH10) in hiCMs rispetto al controllo scramble. N = 3 per ogni condizione.(B) Quantificazione dei livelli NMIIB negli hiCM siMYH9 (sinistra) e NMIIA negli hiCM siMYH10 (destra) rispetto al controllo scramble. N = 3 per ogni condizione.Laser-scanning confocale di diffusione NMIIA KD hiCM che esprime Lifeact-mApple. NMIIA KD hiCM è in grado di assemblare strutture di sarcomeri. Barra di scala; 10 µm.Scansione laser confocale di diffusione NMIIB KD hiCM che esprime Lifeact-mApple.NMIIB KD hiCM non forma sarcomeri. Barra graduata; 20 µm.Laser-scanning confocale di hiCM diffusione in presenza di 100 µM blebbistatina aggiunto 1,5 hr post placcatura. hiCM non assembla sarcomeri in presenza di 100 µM blebbistatina. Barra di scala; 20 µm.(A) siScrambled (in alto) siMYH9 (NMIIA KD, al centro), e siMYH10 (NMIIB KD, in basso) hiCMs trattati in 96 piastre pozzo, fisso, e localizzato per α-acttinina 2 imaged con microscopia ad ampio campo. hiCMs sono stati autorizzati ad attaccare per 4 giorni (secondo la raccomandazione del produttore, vedi Materiali e metodi), garantendo robusto assemblaggio sarcomero prima del trattamento siRNA iniziato. Si noti che tutte le condizioni contengono la colorazione sarcomerica di α-acttinina 2. Cercare tabella è mpl-magma nelle Fiji.
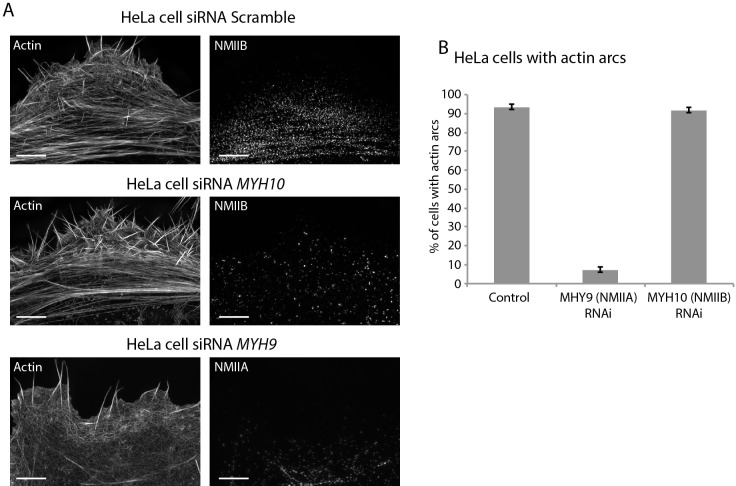
Figura 8-figure supplemento 1.NMIIA ma non NMIIB è richiesto per la formazione di archi di actina nelle cellule HeLa.(A) Cellule HeLa trattate con controllo, anti myh10, o anti myh9 (superiore, centrale, inferiore, rispettivamente) siRNA, colorato per l’actina, e ripreso con SIM. Si noti come le cellule KD di controllo e NMIIB KD visualizzano le fibre di tensione dell’arco di actina prominente, ma le cellule NMIIA KD non contengono archi di actina.(B) Quantificazione delle cellule HeLa con fibre di actina in condizioni indicate. Barre di scala: 5 µm.
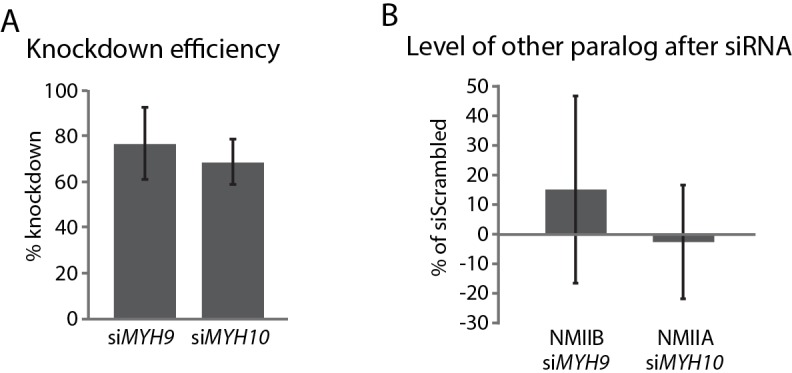
Figura 8-figure supplement 2.Abbattimento di NMIIA e NMIIB in hiCMs.(A) Quantificazione da blot occidentale che mostra KD di NMIIA (siMYH9) e NMIIB(siMYH10) in hiCMs rispetto al controllo scramble. N = 3 per ogni condizione.(B) Quantificazione dei livelli NMIIB negli hiCM siMYH9 (sinistra) e NMIIA negli hiCM siMYH10 (destra) rispetto al controllo scramble. N = 3 per ogni condizione.
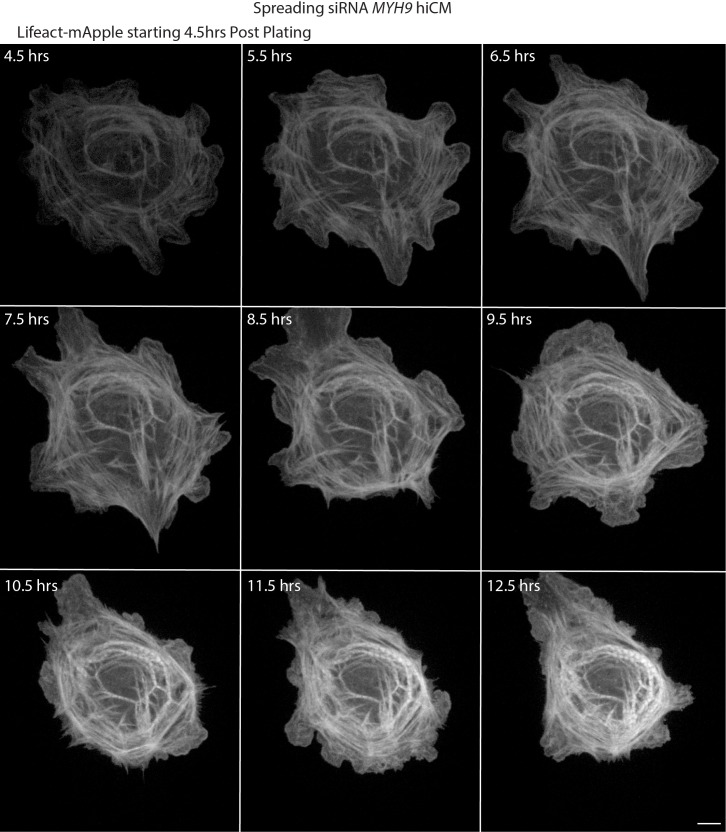
Figura 8-figure supplement 3.Figura 8 = 3. Montaggio dal vivo della diffusione NMIIA KD hiCM.Laser-scanning confocale di diffusione NMIIA KD hiCM che esprime Lifeact-mApple. NMIIA KD hiCM è in grado di assemblare strutture di sarcomeri. Barra di scala; 10 µm.
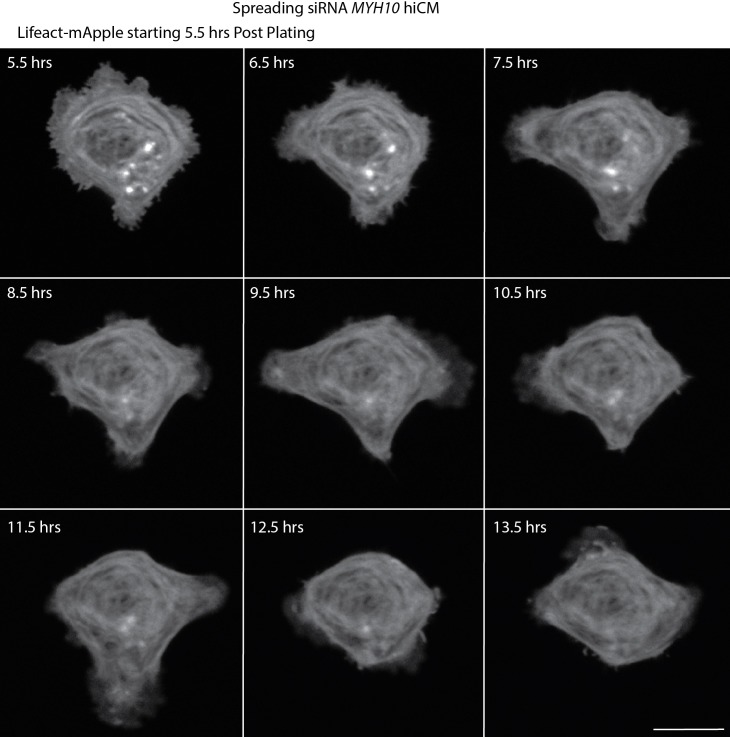
Figura 8-figure supplemento 4.Montaggio dal vivo di diffusione NMIIB KD hiCM.Laser-scanning confocale di diffusione NMIIB KD hiCM che esprime Lifeact-mApple.NMIIB KD hiCM non forma sarcomeri. Barra graduata; 20 µm.
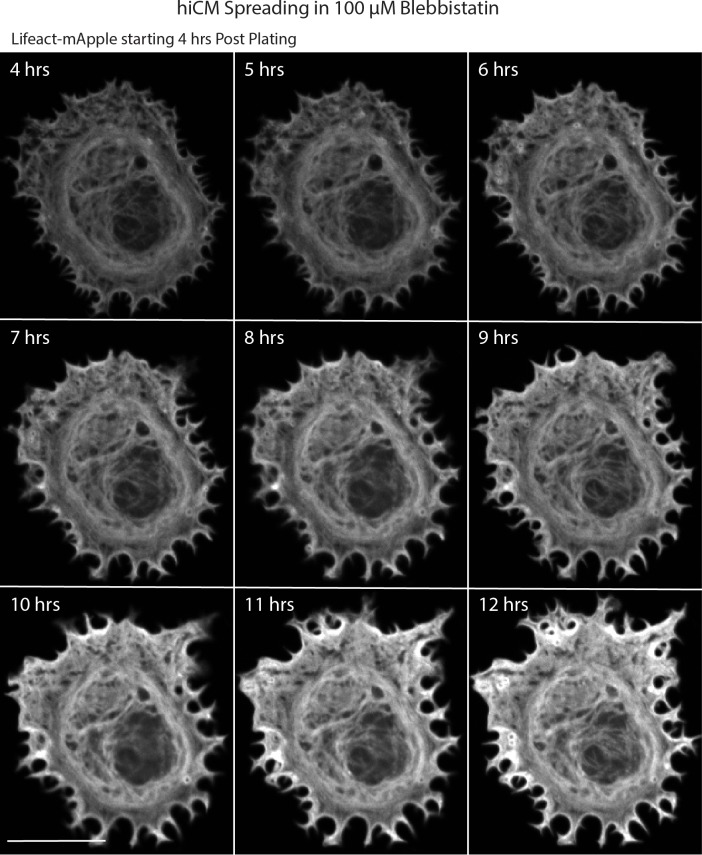
Figura 8-figure supplemento 5.Figura 8—figura supplemento 5. Montaggio dal vivo di hiCM che si diffonde in 100 µM blebbistatina.Laser-scanning confocale di hiCM diffusione in presenza di 100 µM blebbistatina aggiunto 1,5 ore e mezzo post placcatura. hiCM non assemblare sarcomeri in presenza di 100 µM blebbistatina. Barra di scala; 20 µm.
Figura 8-figure supplemento 6.NMIIA KD e NMIIB KD hiCMs contengono sarcomeri prima della placcatura.(A) siScrambled (in alto) siMYH9 (NMIIA KD, al centro), e siMYH10 (NMIIB KD, in basso) hiCMs trattati in 96 piastre pozzo, fisso, e localizzato per α-acttinina 2 imaged con microscopia ad ampio campo. hiCMs sono stati autorizzati ad attaccare per 4 giorni (secondo la raccomandazione del produttore, vedi Materiali e metodi), garantendo robusto assemblaggio sarcomero prima del trattamento siRNA iniziato. Si noti che tutte le condizioni contengono la colorazione sarcomerica di α-acttinina 2. Cercare tabella è mpl-magma nelle Fiji.
NMIIB e FHOD3 sono necessari per la formazione organizzata della banda A
Finora i nostri risultati evidenziano l’importanza della polimerizzazione dell’actina mediata dalla forma e dell’NMII per una corretta architettura dei filamenti di actina durante l’assemblaggio dei sarcomeri. Abbiamo poi voluto affrontare il modo in cui i filamenti spessi della miosina II β cardiaca (βCMII ) al centro del sarcomero (cioè, banda A, Figura 1A ) si assemblano. Pertanto, abbiamo iniziato con la localizzazione di βCMII endogeno e filamenti NMIIB (Figura 9A). βCMII prevalentemente localizzato dietro NMIIB in strutture sarcomeriche organizzate e ha mostrato una localizzazione di picco ~ 15 micron dietro il bordo anteriore della cella, con una leggera area di sovrapposizione con NMIIB (Figura 9A eB). Abbiamo notato che l’area di sovrapposizione conteneva co-filamenti NMIIB-βCMII (Figura 9C e D, e Figura 9-figure supplement 1). Inoltre, abbiamo trovato anche co-filamenti NMIIA-βCMII in hiCMs (Figura 9-figure supplement 2). A nostra conoscenza, questa è la prima volta che è stata segnalata una specie di filamento di miosina II che contiene un paralogame non muscolare e muscolare all’interno delle cellule. Inoltre, abbiamo anche trovato co-filamenti di NMIIB-βCMII nei tessuti del topo e del cuore umano, indicando che i co-filamenti di NMIIB-βCMII sono presenti in vivo (Figura 9Ce Figura 9-figure supplement 1). I co-filamenti contenenti NMIIB e βCMII erano di lunghezza simile ai filamenti NMIIA/B (Figura 9D). Infatti, abbiamo notato che vicino al bordo di entrata della cellula, i filamenti βCMII sono tipicamente più piccoli e non organizzati in pile che assomigliano alle bande A (Figura 9A e C-E). Questo suggerisce che i filamenti βCMII sono polimerizzati sul bordo e successivamente diventano più grandi man mano che si allontanano dal bordo d’attacco (Figura 9E). La presenza di NMII prima che i filamenti βCMII crescano in filamenti più grandi ci ha portato a testare l’ipotesi che NMII avrebbe avuto un ruolo nella formazione dei filamenti βCMII.
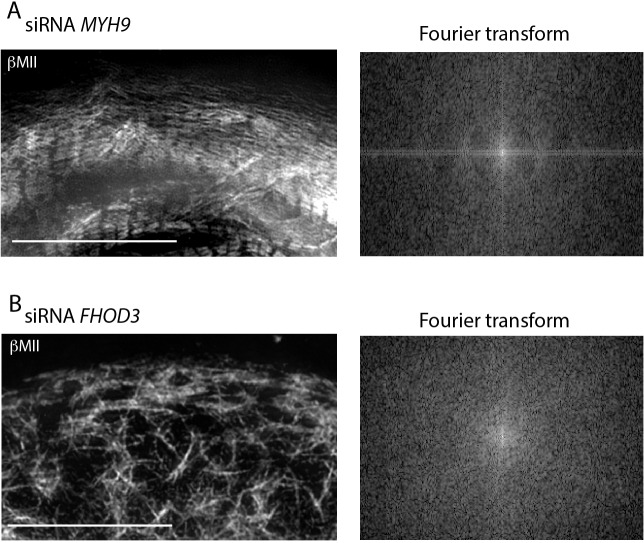
Figura 9-figure supplemento 4.Figura 9—supplemento alla figura 4. β Miosina II Cardiaca (βCMII) Assemblaggio del filamento di miosina II (βCMII) nelle hiCM.La localizzazione endogena di NMIIA e βCMII nelle hiCM.Actina, βCMII, e la localizzazione del DNA nelle hiCM trattate con Latrunculina A.βCMII assemblaggio del filamento è perturbato in hiCM NMIIA e FHOD3 KD hiCM.(A) Localizzazione endogena di NMIIB (a sinistra) e βCMII nella stessa hiCM e ripreso con SIM. (B) Media line-scan di NMIIB (Figura 7B) e la localizzazione βCMII in hiCMs diffusione per 24 ore. Si noti la fluorescenza di picco di βCMII è più verso il corpo cellulare di fluorescenza di picco di NMIIB. 23 cellule da quattro esperimenti sono stati utilizzati per la localizzazione βCMII.(C) Schema (in alto) di NMIIB-βCMII co-filamenti. Viste ad alta risoluzione dei co-filamenti βCMII-NMIIB (in basso). Colorazione endogena di hiCM (in basso, a sinistra) per βCMII (motori a N-terminali) e NMIIB (dominio dell’asta) e ripresa con SIM. Topo e tessuto umano (in basso al centro e in basso a destra, rispettivamente) colorati per βCMII (motori) e NMIIB (dominio dell’asta) e ripresi con SIM e Zeiss 880 con Airyscan, rispettivamente.(D) Istogrammi che mostrano la larghezza dei filamenti NMII (in alto), βCMII filamenti (in mezzo), e NMIIB-βCMII co-filamenti in hiCMs. Misure effettuate dal dominio del motore al dominio del motore come in Figura 7D e E.(E) Istogrammi che mostrano la distribuzione della larghezza dei filamenti βCMII rispetto alla loro posizione in hiCM. Nota βCMII filamenti tendono a crescere più grande come si muovono verso il centro della cella. Le misure non sono state prese da strutture di sarcomeri ‘maturi’ in bande A altamente organizzate.(F) Actin e βCMII di controllo scramble hiCM (in alto) e NMIIB KD (siMYH10) hiCM(in basso) si è diffuso per 24 ore. Si noti la perdita di bande A organizzate ma la presenza di filamenti βCMII in NMIIB KD hiCM.(G) Fourier trasforma il segnale βCMII da scatole bianche in Figura 11F da controllo scramble e NMIIB KD hiCMs (rispettivamente sopra e sotto). Le frecce gialle indicano la periodicità sarcomerica in hiCMs di controllo scramble, che manca nelle celle NMIIB KD.(H) Actin e α-actinina 2 localizzata in un hiCM dopo NMIIB KD.(I) viste ad alta mag di actina e βCMII in NMIIB KD (siRNA MYH10 ) hiCM ripreso con SIM. βCMII filamenti localizzati a filamenti residui di actina.(J) βCMII in hiCM diffuso per un totale di 24 ore, con le ultime 6 ore in 5 µM di Latrunculina B e ripreso con SIM. Si noti la mancanza di bande βCMII A nella periferia di hiCM, e grandi aggregati di filamenti βCMII (scatola gialla).(K) Percentuale di controllo scramble, NMIIB KD (siRNA MYH10), e 5 µM Latrunculin B hiCMs con bande βCMII A. Controllo: 26 cellule, tre esperimenti; NMIIB KD: 26 cellule, due esperimenti; Latrunculin B: 11 cellule, tre esperimenti. I corpi delle cellule non sono stati analizzati nell’esperimento latrunculina a causa della densità di localizzazione βCMII. Barre di scala;(A) 10 µm, (C)200 nm, (F), (G) 5µm, (H) 1 µm.(I) 10 µm basso magnete, 5 µm alto magnete inserti.(J) 10 µm a basso magnete, 5 µm ad alto magnete. Valori P indicati nei grafici.(A) hiCM trasfettato con βCMII-mEGFP (motori a terminali N), colorato per le aste NMIIB endogene e ripreso con SIM. Lo schema illustra la strategia di visualizzazione. Basso mag di hiCM sinistra, e alto mag esempi in basso a destra.(B) Vista mag bassa del tessuto cardiaco del mouse P3 macchiato per βCMII (motori) e NMIIB (aste), e ripreso con la scansione laser confocale (in alto). Come negli hiCM, NMIIB è limitato da strutture di sarcomeri, ma è localizzato adiacente ai sarcomeri. Le frecce indicano aree di possibili co-filamenti. Esempio di magnete alto (in basso) preso dal tessuto del mouse P3 ripreso con SIM da un’area simile indicata dalla freccia sull’immagine a basso magnete. Lo schema indica la strategia di visualizzazione.(C) Localizzazione di βCMII e NMIIB nel muscolo settale umano da un paziente con cardiomiopatia ipertrofica. Low-mag immagine (a sinistra) di βCMII (magenta) e NMIIB (verde) ripreso su Zeiss 880 con AiryScan. Gli esempi ad alta immagine (a destra) da Boxes in low-mag image (a sinistra) indicano i co-filamenti βCMII-NMIIB. Lo schema indica la strategia di visualizzazione.(D) High-mag esempi di NMIIA (sinistra) e NMIIB-βCMII (destra) co-filamenti da un paziente con cardiomiopatia ipertrofica umana ripreso su Zeiss 880 con AiryScan. Schema del cartone animato indica la strategia di imaging. Barre di scala: (A, sinistra), 10 μm; (A, destra), 200 nm, (B, in alto), 20 µm: (B, in basso), 200 nm: (C, sinistra), 5 µm: (C, destra), 1 µm:(D), 200 nm.(A) SIM di localizzazione endogena di NMIIA (verde) e βCMII (magenta) in hiCM. Le caselle gialle indicano aree di sovrapposizione tra NMIIA e βCMII.(B) Viste ad alta risoluzione dei co-filamenti NMIIA-βCMII presi dai riquadri gialli in (A).(A) hiCM spread per un totale di 24 ore e trattati con 5 μM Latrunculina A nelle ultime 6 ore di diffusione prima della fissazione e ripreso con SIM. Si noti la perdita di actina sarcomerica e βCMII. Dopo il trattamento con Latrunculina A, βCMII si trova adiacente al nucleo (DNA) e in grandi aggregati in periferia cellulare (box 1 e 2).(B) Viste ad alta immaginazione da(A), che mostra βCMII aggregati filamento βCMII localizzati ad aggregati di actina nella periferia cellulare dopo il trattamento con 5 microlitri di Latrunculina A. Barre di scala: 10 μm in (A), 5 μm in (B).(A) Endogeno βCMII localizzato in NMIIA KD (siRNA MYH9 ) hiCMs(a sinistra) ripreso con SIM. trasformata di Fourier (a destra) di hiCM mostra una certa periodicità di βCMII che non è così facilmente evidente rispetto al controllo scramble hiCMs (Figura 9G).(B) βCMII endogeno localizzato in FHOD3 KD (siRNA FHOD3 ) hiCM(a sinistra) ripreso con SIM. Si noti la perdita di βCMII bande A. Le trasformazioni di Fourier (a destra) mostrano perdita di periodicità sarcomerica. Barre di scala: 10 μm.
Per verificare se il NMIIB era richiesto anche per la formazione del filamento βCMII e della banda A, abbiamo esaurito gli hiCMs di NMIIB e localizzato il βCMII 24 ore dopo la placcatura. Rispetto agli hiCMs di controllo, gli hiCMs KD-hiCMs NMIIB hanno mostrato una significativa diminuzione della capacità di formare strutture simili alla banda A (come definito nei Materiali e metodi) e un numero complessivo ridotto di filamenti βCMII (Figure 9F, G,I e K). Anche se si sono formati filamenti βCMII, erano altamente disorganizzati rispetto alle cellule di controllo, come valutato da Fourier transform (Pasqualiniet al., 2015) (Figura 9G). Come l’actina puncta rimasto dopo NMIIB KD conteneva α-acttinina 2 (Figura 9H), abbiamo ipotizzato che questi puncta potrebbe anche essere legato a filamenti βCMII. In effetti, abbiamo scoperto che i filamenti βCMII si estendevano alla distanza tra i puncta strettamente distanziati (Figura 9I). Questi dati suggeriscono che anche quando il citoscheletro dell’actina è gravemente perturbato, esistono ancora meccanismi che portano all’associazione tra esso e i filamenti βCMII. Abbiamo poi cercato di approfondire questa osservazione e di verificare se i difetti osservati nell’assemblaggio dei filamenti βCMII nel NMIIB KD hiCMs sono stati causati dalla perturbazione del citoscheletro dell’actina. Per testare questo, abbiamo permesso alle hiCM di formare sarcomeri per 18 ore, poi abbiamo trattato le hiCM con l’agente di sequestro dell’actina monomero Latrunculina B per 6 ore(Spector et al., 1983; Wakatsuki et al., 2001). Studi precedenti in cellule non muscolari hanno dimostrato che il trattamento con latrunculina elimina l’actina dalla maggior parte delle cellule e lascia aggregati di filamenti di actina sparsi nel citoplasma(Ayscough et al., 1997; Gronewold et al., 1999). Abbiamo anche trovato aggregati di filamenti di actina nella periferia di hiCM trattati con latrunculina(Figura 9-figure supplement 3). βCMII filamenti sembravano localizzare esclusivamente a questi aggregati, ma non erano in organizzato in bande A (Figura11I J). Presi insieme, questi risultati sostengono che l’organizzazione dei filamenti di actina è un fattore importante nell’organizzazione dei filamenti βCMII. Poiché FHOD3 KD ha anche portato ad una grave disorganizzazione dell’architettura dei filamenti di actina, abbiamo localizzato βCMII in questa condizione. Infatti, FHOD3 KD hiCM aveva anche disorganizzato i filamenti βCMII rispetto agli hiCM di controllo (Figura 9-figure supplement 4).
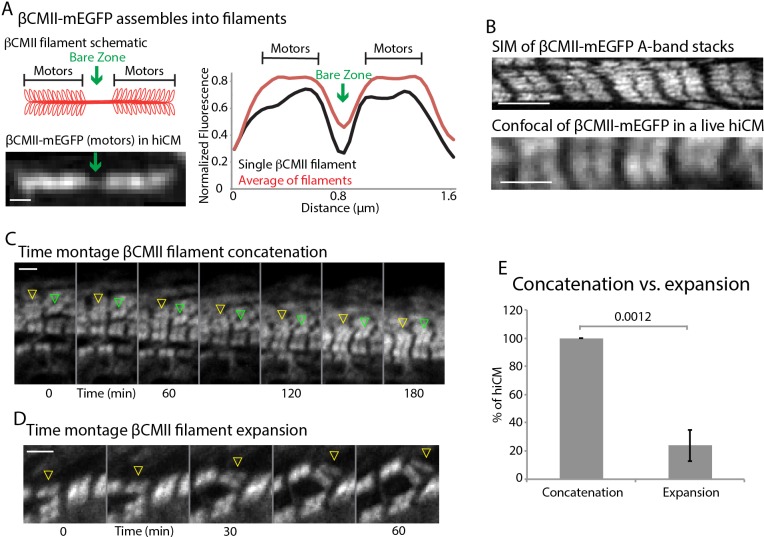
Figura 11.βCMII filamenti concatenati per formare strutture più grandi in banda A.(A) Cartone animato del filamento βCMII (a sinistra, in alto). N-terminale tagged umano βCMII-mEGFP filamento umano espresso in hiCM (a sinistra, in basso). Gap in segnale rappresenta la mancanza di motori a zona nuda (frecce verdi). βCMII filamento singolo e filamento in banda A (filamenti βCMII trovati all’interno di bande A organizzate) larghezze misurate da scansioni di linea (a destra). βCMII Filamenti βCMII: 16 filamenti, tre esperimenti. βCMII miofibrille: 28 miofibrille, tre esperimenti. Si noti più ‘plateau’ di livello del segnale dei motori in filamenti βCMII in banda A.(B) SIM di rappresentante βCMII-mEGFP miofibrille in hiCM (in alto) e la scansione laser confocale (in basso).(C) montaggio rappresentativo che mostra due eventi di concatenazione separati. La punta della freccia gialla denota una grande pila di filamenti βCMII-mEGFP concatenazione con una pila più piccola di filamenti βCMII-mEGFP mentre subiscono il flusso retrogrado. La punta della freccia verde denota un filamento βCMII-mEGFP più piccolo concatenato con una pila più grande di βCMII-mEGFP mentre subiscono il flusso retrogrado. Entrambi gli eventi danno come risultato uno stack di filamenti βCMII-mEGFP più grande e più organizzato (cioè la banda A).(D) Esempio di evento di scissione dei filamenti βCMII-mEGFP. Si noti come la piccola pila di filamenti βCMII-mEGFP si divide per creare due filamenti βCMII-mEGFP più piccoli e non risulta in pile di filamenti βCMII-mEGFP più grandi o più organizzati.(E) Quantificazione della % di hiCMs che mostrano eventi di concatenazione o di espansione dei filamenti βCMII. Barre di scala;(A) 200 nm, (B),(C) e(D) 2 µm. Valori P indicati nel grafico.
Per indagare ulteriormente i meccanismi di A-band assembly, abbiamo creato una lunghezza completa, umano βCMII costruire βCMII contenente un tag mEGFP sul dominio del motore (cioè, N-terminale) (Figura 11A). Questo costrutto correttamente integrato sia nei singoli filamenti che nelle miofibrille più mature(Figura 11A B). Nelle cellule U2OS non muscolari, le pile di filamenti NMIIA in banda A simili a pile di NMIIA sono spesso formate attraverso un processo chiamato ‘Espansione’ (Fenix etal., 2016). Durante l’Espansione, i filamenti NMIIA che sono vicini l’uno all’altro (cioè in un fascio stretto) si allontanano l’uno dall’altro nello spazio, ma rimangono parte dello stesso insieme, dove possono essere allineati in una pila simile alla miosina muscolare II nella banda A. Oltre all’espansione, i filamenti NMIIA sono anche, ma più raramente, “concatenati” (Fenix etal., 2016). La concatenazione è definita da filamenti NMIIA separati spazialmente che si muovono l’uno verso l’altro per creare una pila. Per testare come si formano le pile di filamenti βCMII, abbiamo ripetuto il nostro test di formazione di sarcomeri a cellule vive usando il nostro costrutto βCMII-mEGFP. In contrasto con i nostri precedenti risultati con NMIIA, abbiamo trovato che il principale meccanismo fisico della formazione di stack di filamenti βCMII è la concatenazione, dove i filamenti βCMII preesistenti si sono incrociati e cuciti insieme per formare la banda A (Figura11C E e Video 4) . Una piccola percentuale di hiCMs ha mostrato un evento di espansione di βCMII-mEGFP, tuttavia questo è stato significativamente meno frequente che nelle cellule non muscolari e non sembrano risultare in una più organizzata A-band (Figura 11D E). Infatti, ciascuno degli hiCMs quantificati nella Figura 11E ha mostrato un solo evento di espansione.
Video 4.βCMII filamenti in un hiCM assemblando sarcomeri. hiCM trasfettato con βCMII-mEGFP e ripreso con SIM.Si noti che i filamenti si concatenano. Tabella di ricerca: arancione caldo. 30,7 per 29,7 μm. Lunghezza del video: 7,5 ore.
Figura 9-figure supplement 4.Figura 9—supplemento figura 4. β Miosina II Cardiaca (βCMII) Assemblaggio del filamento in hiCMs.miosina II co-filamenti in hiCMs e in vivo.localizzazione endogena di NMIIA e βCMII in hiCMs.Actin, βCMII, e la localizzazione del DNA in hiCMs trattati con Latrunculin A.βCMII montaggio filamento è perturbato in hiCMs NMIIA e FHOD3 KD hiCMs.(A) Localizzazione endogena di NMIIB (a sinistra) e βCMII nella stessa hiCM e ripreso con SIM. (B) Media line-scan di NMIIB (Figura 7B) e la localizzazione βCMII in hiCMs diffusione per 24 ore. Si noti la fluorescenza di picco di βCMII è più verso il corpo cellulare di fluorescenza di picco di NMIIB. 23 cellule da quattro esperimenti sono stati utilizzati per la localizzazione βCMII.(C) Schema (in alto) di NMIIB-βCMII co-filamenti. Viste ad alta risoluzione dei co-filamenti βCMII-NMIIB (in basso). Colorazione endogena di hiCM (in basso, a sinistra) per βCMII (motori a N-terminali) e NMIIB (dominio dell’asta) e ripresa con SIM. Topo e tessuto umano (in basso al centro e in basso a destra, rispettivamente) colorati per βCMII (motori) e NMIIB (dominio dell’asta) e ripresi con SIM e Zeiss 880 con Airyscan, rispettivamente.(D) Istogrammi che mostrano la larghezza dei filamenti NMII (in alto), βCMII filamenti (in mezzo), e NMIIB-βCMII co-filamenti in hiCMs. Misure effettuate dal dominio del motore al dominio del motore come in Figura 7D e E.(E) Istogrammi che mostrano la distribuzione della larghezza dei filamenti βCMII rispetto alla loro posizione in hiCM. Nota βCMII filamenti tendono a crescere più grande come si muovono verso il centro della cellula. Le misure non sono state prese da strutture di sarcomeri ‘maturi’ in bande A altamente organizzate.(F) Actin e βCMII di controllo scramble hiCM (in alto) e NMIIB KD (siMYH10) hiCM(in basso) si è diffuso per 24 ore. Si noti la perdita di bande A organizzate ma la presenza di filamenti βCMII in NMIIB KD hiCM.(G) Fourier trasforma il segnale βCMII da scatole bianche in Figura 11F da controllo scramble e NMIIB KD hiCMs (rispettivamente sopra e sotto). Le frecce gialle indicano la periodicità sarcomerica in hiCMs di controllo scramble, che manca nelle celle NMIIB KD.(H) Actin e α-actinina 2 localizzata in un hiCM dopo NMIIB KD.(I) viste ad alta mag di actina e βCMII in NMIIB KD (siRNA MYH10 ) hiCM ripreso con SIM. βCMII filamenti localizzati a filamenti residui di actina.(J) βCMII in hiCM diffuso per un totale di 24 ore, con le ultime 6 ore in 5 µM di Latrunculina B e ripreso con SIM. Si noti la mancanza di bande βCMII A nella periferia di hiCM, e grandi aggregati di filamenti βCMII (scatola gialla).(K) Percentuale di controllo scramble, NMIIB KD (siRNA MYH10), e 5 µM Latrunculin B hiCMs con bande βCMII A. Controllo: 26 cellule, tre esperimenti; NMIIB KD: 26 cellule, due esperimenti; Latrunculin B: 11 cellule, tre esperimenti. I corpi delle cellule non sono stati analizzati nell’esperimento latrunculina a causa della densità di localizzazione βCMII. Barre di scala;(A) 10 µm, (C)200 nm, (F), (G) 5µm, (H) 1 µm.(I) 10 µm basso magnete, 5 µm alto magnete inserti.(J) 10 µm a basso magnete, 5 µm ad alto magnete. Valori P indicati nei grafici.(A) hiCM trasfettato con βCMII-mEGFP (motori a terminali N), colorato per le aste NMIIB endogene e ripreso con SIM. Lo schema illustra la strategia di visualizzazione. Basso mag di hiCM sinistra, e alto mag esempi in basso a destra.(B) Vista mag bassa del tessuto cardiaco del mouse P3 macchiato per βCMII (motori) e NMIIB (aste), e ripreso con la scansione laser confocale (in alto). Come negli hiCM, NMIIB è limitato da strutture di sarcomeri, ma è localizzato adiacente ai sarcomeri. Le frecce indicano aree di possibili co-filamenti. Esempio di magnete alto (in basso) preso dal tessuto del mouse P3 ripreso con SIM da un’area simile indicata dalla freccia sull’immagine a basso magnete. Lo schema indica la strategia di visualizzazione.(C) Localizzazione di βCMII e NMIIB nel muscolo settale umano da un paziente con cardiomiopatia ipertrofica. Low-mag immagine (a sinistra) di βCMII (magenta) e NMIIB (verde) ripreso su Zeiss 880 con AiryScan. Gli esempi ad alta immagine (a destra) da Boxes in low-mag image (a sinistra) indicano i co-filamenti βCMII-NMIIB. Lo schema indica la strategia di visualizzazione.(D) High-mag esempi di NMIIA (sinistra) e NMIIB-βCMII (destra) co-filamenti da un paziente con cardiomiopatia ipertrofica umana ripreso su Zeiss 880 con AiryScan. Schema del cartone animato indica la strategia di imaging. Barre di scala: (A, sinistra), 10 μm; (A, destra), 200 nm, (B, in alto), 20 µm: (B, in basso), 200 nm: (C, sinistra), 5 µm: (C, destra), 1 µm:(D), 200 nm.(A) SIM di localizzazione endogena di NMIIA (verde) e βCMII (magenta) in hiCM. Le caselle gialle indicano aree di sovrapposizione tra NMIIA e βCMII.(B) Viste ad alta risoluzione dei co-filamenti NMIIA-βCMII presi dai riquadri gialli in (A).(A) hiCM spread per un totale di 24 ore e trattati con 5 μM Latrunculina A nelle ultime 6 ore di diffusione prima della fissazione e ripreso con SIM. Si noti la perdita di actina sarcomerica e βCMII. Dopo il trattamento con Latrunculina A, βCMII si trova adiacente al nucleo (DNA) e in grandi aggregati in periferia cellulare (box 1 e 2).(B) Viste ad alta immaginazione da(A), che mostra βCMII aggregati filamento βCMII localizzati ad aggregati di actina nella periferia cellulare dopo il trattamento con 5 microlitri di Latrunculina A. Barre di scala: 10 μm in (A), 5 μm in (B).(A) Endogeno βCMII localizzato in NMIIA KD (siRNA MYH9 ) hiCMs(a sinistra) ripreso con SIM. trasformata di Fourier (a destra) di hiCM mostra una certa periodicità di βCMII che non è così facilmente evidente rispetto al controllo scramble hiCMs (Figura 9G).(B) βCMII endogeno localizzato in FHOD3 KD (siRNA FHOD3 ) hiCM(a sinistra) ripreso con SIM. Si noti la perdita di βCMII bande A. Le trasformazioni di Fourier (a destra) mostrano perdita di periodicità sarcomerica. Barre di scala: 10 μm.

Figura 9-figure supplement 1.Co-filamenti di miosina II in hiCMs e in vivo.(A) hiCM trasfettato con βCMII-mEGFP (N-terminal motors), colorato per le aste NMIIB endogene e ripreso con SIM. schema illustra la strategia di visualizzazione. Basso mag di hiCM sinistra, e alto mag esempi in basso a destra.(B) Vista mag bassa del tessuto cardiaco del mouse P3 macchiato per βCMII (motori) e NMIIB (aste), e ripreso con la scansione laser confocale (in alto). Come negli hiCM, NMIIB è limitato da strutture di sarcomeri, ma è localizzato adiacente ai sarcomeri. Le frecce indicano aree di possibili co-filamenti. Esempio di magnete alto (in basso) preso dal tessuto del mouse P3 ripreso con SIM da un’area simile indicata dalla freccia sull’immagine a basso magnete. Lo schema indica la strategia di visualizzazione.(C) Localizzazione di βCMII e NMIIB nel muscolo settale umano da un paziente con cardiomiopatia ipertrofica. Low-mag immagine (a sinistra) di βCMII (magenta) e NMIIB (verde) ripreso su Zeiss 880 con AiryScan. Gli esempi ad alta immagine (a destra) da Boxes in low-mag image (a sinistra) indicano i co-filamenti βCMII-NMIIB. Lo schema indica la strategia di visualizzazione.(D) High-mag esempi di NMIIA (sinistra) e NMIIB-βCMII (destra) co-filamenti da un paziente con cardiomiopatia ipertrofica umana ripreso su Zeiss 880 con AiryScan. Schema del cartone animato indica la strategia di imaging. Barre di scala: (A, sinistra), 10 μm; (A, destra), 200 nm, (B, in alto), 20 µm: (B, in basso), 200 nm: (C, sinistra), 5 µm: (C, destra), 1 µm:(D), 200 nm.

Figura 9-figure supplement 2.Figura 9. Localizzazione endogena di NMIIA e βCMII in hiCMs.(A) SIM di localizzazione endogena di NMIIA (verde) e βCMII (magenta) in hiCM. scatole gialle indicano aree di sovrapposizione tra NMIIA e βCMII.(B) Viste ad alta risoluzione dei co-filamenti NMIIA-βCMII presi dai riquadri gialli in (A).
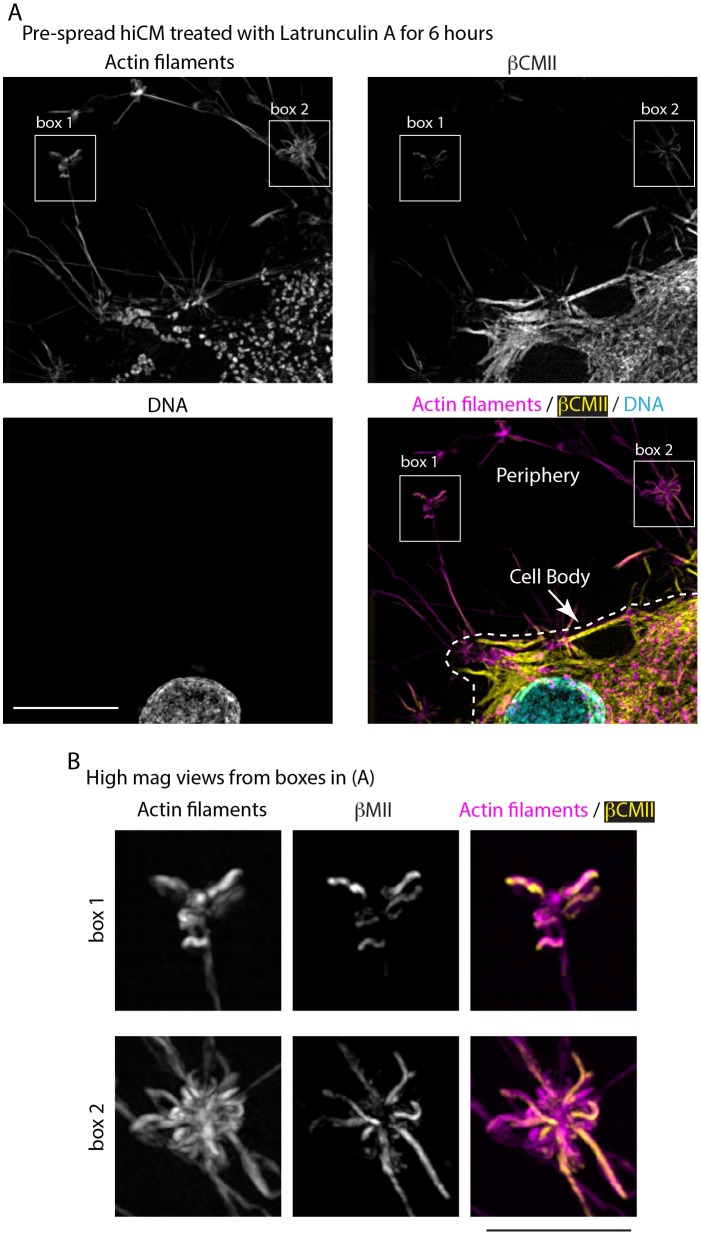
Figura 9-figure supplemento 3.Figura 9. Actina, βCMII, e la localizzazione del DNA in hiCMs trattati con Latrunculin A.(A) hiCM diffuso per un totale di 24 ore e trattati con 5 μM Latrunculina A nelle ultime 6 ore di diffusione prima della fissazione e ripreso con SIM. Si noti la perdita di actina sarcomerica e βCMII. Dopo il trattamento con Latrunculina A, βCMII si trova adiacente al nucleo (DNA) e in grandi aggregati in periferia cellulare (box 1 e 2).(B) Viste ad alta immaginazione da(A), che mostra βCMII aggregati filamento βCMII localizzati ad aggregati di actina nella periferia cellulare dopo il trattamento con 5 microlitri di Latrunculina A. Barre di scala: 10 μm in (A), 5 μm in (B).
Figura 9-figure supplemento 4.βCMII montaggio filamento è perturbato in NMIIA e FHOD3 KD hiCMs.(A) Endogeno βCMII localizzato in NMIIA KD (siRNA MYH9 ) hiCMs(a sinistra) ripreso con SIM. trasformata di Fourier (a destra) di hiCM mostra una certa periodicità di βCMII che non è così facilmente evidente rispetto ai hiCMs di controllo scramble (Figura 9G).(B) βCMII endogeno localizzato in FHOD3 KD (siRNA FHOD3 ) hiCM(a sinistra) ripreso con SIM. Si noti la perdita di βCMII bande A. Le trasformazioni di Fourier (a destra) mostrano perdita di periodicità sarcomerica. Barre di scala: 10 μm.
Figura 11.I filamenti βCMII si concatenano a formare strutture più grandi della banda A.(A) Cartone animato del filamento βCMII (a sinistra, sopra). N-terminale tagged umano βCMII-mEGFP filamento umano espresso in hiCM (a sinistra, sotto). Gap in segnale rappresenta la mancanza di motori a zona nuda (frecce verdi). βCMII filamento singolo e filamento in banda A (filamenti βCMII trovati all’interno di bande A organizzate) larghezze misurate da scansioni di linea (a destra). βCMII Filamenti βCMII: 16 filamenti, tre esperimenti. βCMII miofibrille: 28 miofibrille, tre esperimenti. Si noti più ‘plateau’ di livello del segnale dei motori in filamenti βCMII in banda A.(B) SIM di rappresentante βCMII-mEGFP miofibrille in hiCM (in alto) e la scansione laser confocale (in basso).(C) montaggio rappresentativo che mostra due eventi di concatenazione separati. La punta della freccia gialla denota una grande pila di filamenti βCMII-mEGFP concatenazione con una pila più piccola di filamenti βCMII-mEGFP mentre subiscono il flusso retrogrado. La punta della freccia verde denota un filamento βCMII-mEGFP più piccolo concatenato con una pila più grande di βCMII-mEGFP mentre subiscono il flusso retrogrado. Entrambi gli eventi danno come risultato uno stack di filamenti βCMII-mEGFP più grande e più organizzato (cioè la banda A).(D) Esempio di evento di scissione dei filamenti βCMII-mEGFP. Si noti come la piccola pila di filamenti βCMII-mEGFP si divide per creare due filamenti βCMII-mEGFP più piccoli e non risulta in pile di filamenti βCMII-mEGFP più grandi o più organizzati.(E) Quantificazione della % di hiCMs che mostrano eventi di concatenazione o di espansione dei filamenti βCMII. Barre di scala;(A) 200 nm, (B),(C) e(D) 2 µm. Valori P indicati nel grafico.
Video 4.βCMII filamenti in un hiCM assemblando sarcomeri. hiCM trasfezionato con βCMII-mEGFP e ripreso con SIM.Nota i filamenti concatenati. Tabella di ricerca: arancione caldo. 30,7 per 29,7 μm. Lunghezza del video: 7,5 ore.
Discussione
L’obiettivo di questo studio è stato quello di affrontare una questione decennale riguardante l’origine dei sarcomeri che si assemblano in cardiomiociti. Alcuni laboratori hanno proposto che i sarcomeri derivino da un precursore simile a una fibra di stress, mentre altri propongono modelli in cui componenti di sarcomeri separati si uniscono in sequenza per formare sarcomeri(Holtzer et al., 1997; Lin et al., 1994; Lu et al., 1992; Rhee et al., 1994; Rui et al., 2010; Sanger et al., 2005). Sulla base della precedente colorazione dei filamenti di actina e della localizzazione NMII a queste fibre da stress, la nostra ipotesi iniziale era che i meccanismi alla base della polimerizzazione e dell’organizzazione dei filamenti di actina sarebbero stati correlati a quelli che governano le fibre non da stress muscolare note come archi di actina (vedi modello in Figura 12). A sostegno di questa ipotesi, sia i precedenti rapporti sui cardiomiociti non umani che il nostro lavoro con gli hiCM mostrano che l’organizzazione dell’actina di questi precursori appare identica agli archi di actina(Heath, 1983; Rhee et al., 1994; Sanger et al., 2005; Tojkander et al., 2012). Infatti, sia i MSF che gli archi di actina contengono NMII, si colorano continuamente di falloidina, si trovano sulla superficie dorsale della cellula e mostrano un flusso retrogrado in cui si allontanano dal bordo della cellula (Figura 12). Tuttavia, il nostro studio ha anche rivelato differenze distinte tra la regolazione e la dinamica delle MSF e gli archi di actina.
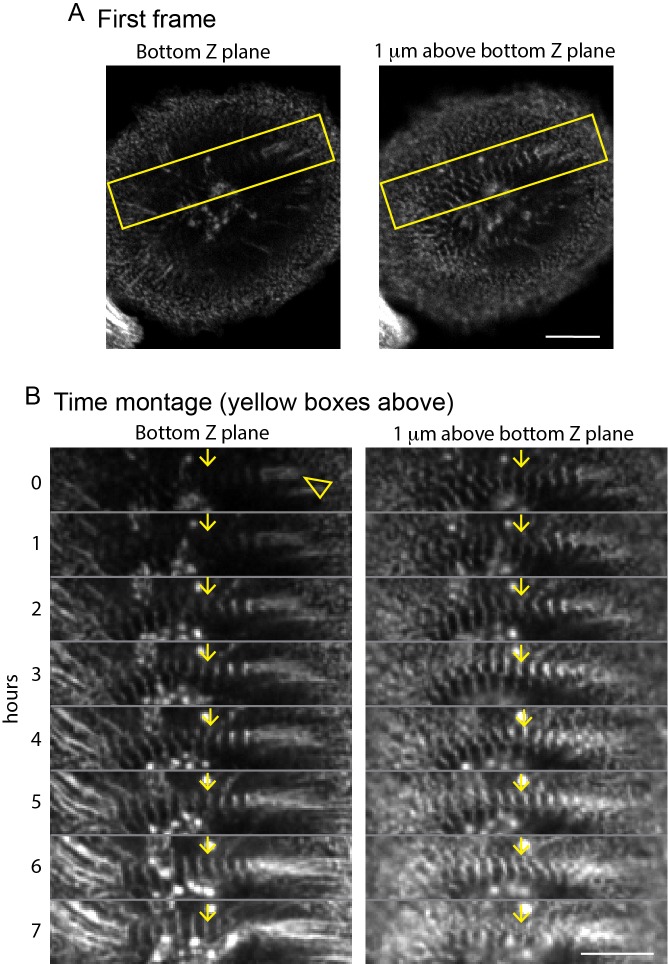
Figura 12-figure supplemento 1.Modello di actomiosina stress formazione di fibre in cellule non muscolari e cardiomiociti umani.sarcomeri forma sulla superficie dorsale di hiCMs e successivamente si muovono verso la superficie ventrale.(A) Actina e miosina II stress formazione di fibre di stress in cellule non-muscolari. fibre di stress Actin sono formate attraverso il complesso Arp2/3 e la formina mDia1. NMIIA è l’isoforma predominante al bordo anteriore delle cellule non muscolari, e la formazione di fibre da sforzo è dipendente NMIIA. Le cellule non muscolari mostrano un robusto flusso retrogrado di fibre di actina da stress e mostrano un rapido turnover. Le grandi pile di NMIIA si formano attraverso la crescita e l’espansione di filamenti NMIIA più piccoli. Le citazioni che portano a questo modello sono presentate nel cartone animato.(B) Modello della formazione di fibre di stress di actina e miosina II nei cardiomiociti umani. I sarcomeri sono modelli di fibre da stress muscolare (MSF). MSFs non richiedono il complesso Arp2/3, e richiedono la formina FHOD3. MSFs visualizzare il flusso lento retrogrado rispetto alle fibre di stress non muscolare. Sia NMIIA che NMIIB sono localizzate ai margini delle hiCM, e visualizzano i co-filamenti NMII prominenti. Anche i co-filamenti NMIIB-βCMII sono presenti con le MSF. Grandi pile di filamenti βCMII si formano attraverso la concatenazione e la cucitura dei singoli filamenti βCMII.(A) hiCM che esprime α-actinina 2-mCherry placcato per 24 ore e ripreso con la scansione laser confocale. Si noti che quando si visualizza la superficie ventrale (in basso) (immagine a sinistra), nessun modello sarcomerico di α-actinina 2 può essere visto. Tuttavia, quando si visualizza la superficie dorsale (in alto) della cellula (immagine a destra), i sarcomeri sono chiaramente presenti.(B) Tempo di montaggio da scatola gialla in(A). Myofibrilla dalla parte superiore della cella (montaggio a destra, freccia gialla) si muove verso il fondo della cella (immagine a sinistra, freccia gialla). La testa della freccia (montaggio a sinistra) indica α-acttinina 2 contenente adesione.
E ‘stato ben stabilito che i filamenti di actina di archi di actina in cellule non muscolari richiedono sia la nucleazione mediata dal complesso Arp2/3 e formine(Henson et al., 2015; Hotulainen e Lappalainen, 2006; Murugesan et al., 2016). I nostri dati suggeriscono che l’Arp2/3 non è formalmente richiesto per l’assemblaggio di MSF o di sarcomeri in hiCM. Tuttavia, il complesso Arp2/3 è localizzato ai margini degli hiCM, e sarà necessario un lavoro futuro per chiarire il suo ruolo in biologia cardiaca. A differenza del complesso Arp2/3, i nostri dati suggeriscono che le formine sono necessarie per l’assemblaggio di MSF e sarcomeri. In particolare, abbiamo trovato che la formina FHOD3 ha un ruolo importante nell’assemblaggio di MSF e sarcomeri(Figura 12). Questo si aggiunge al ruolo già consolidato di FHOD3 nell’omeostasi dei sarcomeri (cioè il turn-over)(Iskratsch et al., 2010; Kan-O et al., 2012; Taniguchi et al., 2009). In futuro sarà necessario un lavoro per chiarire il meccanismo preciso di come e dove l’FHOD3 potenzialmente nuclea i filamenti di actina nei sarcomeri. I filamenti di actina sarcomerica canonica hanno le loro estremità spinate incorporate all’interno di una linea Z (vedi schema in Figura 1A). Come tale, è qui che avremmo previsto la localizzazione dell’FHOD3. Tuttavia, i dati della nostra SIM mostrano che FHOD3 non localizza le linee Z(Figura 6F, riquadri 4, 5 e 6). Invece, FHOD3 sembra localizzare su entrambi i lati di ogni linea Z. Ciò potrebbe indicare che FHOD3 è solo transitoriamente associato alle estremità spinate dei filamenti di actina sarcomerica canonica. In alternativa, ci potrebbero essere estremità spinate dei filamenti di actina in questa regione, il che potrebbe indicare che l’organizzazione e/o la dinamica dei filamenti di actina all’interno di un sarcomero è più complessa. Infine, mentre FHOD3 KD mostrava il fenotipo più severo, KD di DAAM1 e DIAPH1 in hiCM ha portato a difetti sarcomerici e non sarcomerici nell’architettura dell’actina. Questo concorda con la letteratura precedente che mostra i ruoli per le formine multiple in cardiomiociti di topo spiegati nella manutenzione della struttura del sarcomero(Rosado et al., 2014). I diversi ruoli delle formine durante l’assemblaggio dei sarcomeri e la successiva manutenzione dovrebbero essere esplorati nei lavori futuri.
Dimostriamo anche che NMIIA e NMIIB sono necessari per il corretto assemblaggio dei sarcomeri nel nostro sistema di modelli hiCM. È interessante notare che i nostri dati suggeriscono che ogni motore può svolgere un ruolo diverso nell’assemblaggio dei sarcomeri. NMIIB KD ha portato a non rilevare il montaggio del sarcomero(Figura 8 e Figura 8-figure supplemento 4), suggerendo che NMIIB potrebbe essere richiesto per le fasi iniziali ed eventualmente successivi del montaggio. D’altra parte, NMIIA KD hiCMs sono stati in grado di assemblare i sarcomeri, anche se questi erano wispy con significativamente più breve Z-lines(Figura 8 e Figura 8-figure supplemento 3). Questo fenotipo NMIIA KD potrebbe essere il risultato di diverse possibilità, tra cui un ruolo di NMIIA nella maturazione, nell’allineamento o nella stabilità dei sarcomeri. Ovviamente, questo non è un elenco esaustivo dei potenziali meccanismi. Oltre a mostrare NMIIA e NMIIB erano necessari per l’assemblaggio dei sarcomeri, mostriamo che NMIIA e NMIIB formano co-filamenti di miosina II con βCMII. I filamenti βCMII trovati nei co-filamenti erano relativamente piccoli, (~300 nm), e successivamente sono diventati più grandi man mano che passavano ai filamenti più grandi della banda A (~1,6 micron), e hanno perso NMII. Singoli filamenti βCMII individuali (o un piccolo fascio al di sotto del limite di risoluzione della nostra modalità di imaging) concatenati per formare le pile di filamenti βCMII della banda A. Questo processo richiede la presenza di tracce di actina(Figura 12).
Collettivamente, i nostri dati forniscono una nuova visione meccanicistica e dinamica dell’assemblaggio dei sarcomeri e mettono in evidenza le differenze chiave tra le fibre classiche, ben studiate, non muscolari da stress, e le MSF nei miociti cardiaci(Figura 12). Inoltre, il nostro modello unifica alcuni aspetti dei modelli di assemblaggio dei sarcomeri precedentemente proposti(Figura 12). Questi modelli precedentemente proposti, pur essendo presentati come reciprocamente esclusivi l’uno dall’altro, sono in realtà abbastanza simili sotto certi aspetti. Evidenziando questo, i nostri dati supportano una caratteristica importante condivisa tra il modello di modello e il modello di Pre-Myofibril. In particolare, le fibre stressanti sono precursori dei sarcomeri contenenti miofibrille. Queste fibre da sforzo sono state originariamente chiamate “strutture simili alle fibre da sforzo” quando è stato proposto il Modello di Modello (Dlugoszet al., 1984; Sanger et al., 2005). Più tardi furono rinominate Pre-Myofibrils al posto di una caratterizzazione più approfondita(Rhee et al., 1994; Sanger et al., 2005). È stato dimostrato che queste fibre da stress non contenevano le stesse proteine delle fibre non da stress muscolare(Rhee et al., 1994). Tuttavia, la maggior parte delle proteine note nelle fibre da stress che si trovano nei cardiomiociti hanno paraloghi nelle fibre non da stress muscolare. Come tali, troviamo che le “strutture simili alle fibre da stress” siano una descrizione appropriata. Pertanto, suggeriamo di chiamare semplicemente queste strutture ‘fibre da sforzo’ è preferibile. In questo studio, stiamo confrontando i meccanismi di regolazione della fibra non muscolare stress fibra di cui si fa riferimento come archi di actina, ai precursori di sarcomeri nei miociti cardiaci. Così, abbiamo deciso di chiamarle fibre da stress muscolare (MSF) per evitare confusione.
I nostri dati supportano il concetto che le MSF si trasformano nel tempo in miofibrille contenenti sarcomeri. Questo è il modo in cui vengono presentati i modelli a cartoni animati sia del Modello che dei Modelli di Pre-Miofibrilla (vedi modelli originali(Dlugosz et al., 1984; Rhee et al., 1994)). Rimane aperta la questione se ci sia un vero modello durante questo processo. Il modello originale del modello presupponeva che le “strutture simili alle fibre da sforzo” [MSF] sarebbero scomparse dopo essere state usate come modello per costruire una miofibrilla (Dlugoszet al., 1984). Mentre alcuni componenti di MSF (ad esempio, α-actinina 2) sembrano persistere durante la transizione da MSF a sarcomero, altri chiaramente non lo fanno (ad esempio, NMIIA/B). Inoltre, i nostri dati suggeriscono che i filamenti NMIIA/B potrebbero essere essi stessi un modello per l’aggiunta di βCMII, poiché tutti e tre i paraloghi possono essere trovati in co-filamenti insieme nella regione in cui NMIIA/B e βCMII si sovrappongono. Oltre ai modelli Templating/Pre-myofibril, i nostri dati supportano anche gli aspetti del modello di cucitura. Il modello di cucitura originariamente proponeva che i componenti preassemblati del sarcomero (cioè, le bande A e i corpi I-Z-I) sarebbero stati cuciti insieme in sequenza per formare una miofibrilla(Holtzer et al., 1997). Non abbiamo rilevato corpi I-Z-I preformati o bande A nel nostro saggio(Figura 1-figure supplement 1) o assemblaggio sequenziale di sarcomeri per formare una miofibrilla (Figura 1). Tuttavia, alcuni aspetti della dinamica βCMII giustificano il confronto con il modello di cucitura. Abbiamo trovato filamenti βCMII separati concatenati e “cuciti” insieme per formare pile di filamenti βCMII più grandi (cioè la banda A) (Figura 11C).
I nostri dati mostrano la transizione di un MSF ad un sarcomero contenente miofibrilla si verifica sulla superficie dorsale (in alto) della cellula. Tuttavia, un recente studio anche imaging iPSC derivati miociti cardiaci sostiene che i sarcomeri si formano sulla superficie ventrale (in basso) della cellula vicino adesioni matrice extracellulare(Chopra et al., 2018). Questo gruppo riferisce anche che i primi sarcomeri si formano tra le 24 e le 48 ore dopo la placcatura, ben dopo che abbiamo rilevato la comparsa dei primi sarcomeri (Chopra et al., 2018). Questo ci ha portato a chiederci cosa potrebbe portare a questi due risultati apparentemente opposti. È importante notare che questo gruppo sembra essere stato immaginato come la superficie ventrale (in basso) dei loro miociti, dato che il focus del loro studio era sulle aderenze focali. Un attento esame dei loro filmati time-lapse ha rivelato strutture sbiadite e sfocate che corrispondono a modelli sarcomerici che compaiono nell’inquadratura proprio prima della comparsa dei sarcomeri. Questo supporta l’idea che si tratta di sarcomeri di imaging che stanno entrando a fuoco, e non di assemblaggi “de novo”. Per testare questa idea, abbiamo immaginato gli hiCM con la microscopia confocale 3D dopo che erano stati placcati per 24 ore(Figura 12-figure supplement 1). Mentre vediamo anche modelli simili di sarcomeri che appaiono sulla superficie ventrale come(Chopra et al., 2018), i nostri dati hanno rivelato che questi sarcomeri si muovono verso il basso dalla superficie dorsale e non si assemblano sulla superficie ventrale(Figura 12-figure supplement 1 e Video 5). Di interesse, il fenomeno degli archi di actina che si spostano verso la superficie ventrale delle cellule non muscolari è stato riportato anche in precedenza(Gao et al., 2012; Hotulainen e Lappalainen, 2006). Infine,(Chopra et al., 2018) sostengono anche che né la NMIIA né la NMIIB sono necessarie per l’assemblaggio dei sarcomeri. Anche questo ci sembra strano. Mentre il loro doppio NMIIA/NMIIB knockout (KO) linea di cellule cardiomiocitarie ha strutture positive α-actinina 2, non contengono linee Z continue etichettate allineate parallelamente l’una all’altra paragonabile alla linea delle cellule di controllo. Gli autori non hanno misurato le lunghezze delle linee Z, la spaziatura o altri criteri necessari per definire i sarcomeri. Queste discrepanze tra il nostro studio e il loro, compreso il ruolo della NMII, devono essere armonizzate, in quanto influiranno direttamente sulla nostra interpretazione dei futuri dati in vivo relativi all’assemblaggio dei sarcomeri.
Video 5.Sarcomeri che viaggiano dalla superficie dorsale a quella ventrale di hiCM hiCM placcati per 24 ore ed esprimono α-actinina 2-mCherry.Quattro viste da quattro piani Z ripresi con microscopia confocale a scansione laser. Nel primo fotogramma, nessun sarcomero può essere visualizzato sul fondo della cella (a sinistra la maggior parte dei fotogrammi), ma può essere chiaramente visto nel piano Z di 2 μm (fotogramma centrale destro). Mentre il video continua, i sarcomeri nel piano 2 μm Z viaggiano verso la superficie ventrale (piano Z in basso) dell’hiCM, che può essere facilmente visto come la miofibrilla entra a fuoco prima nel piano 1 μm Z (metà sinistra), e poi nel piano Z in basso (sinistra). Altezza: 38,2 μm. Lunghezza del video: 17,5 ore.
I dati degli studi in vivo che cercano di rispondere alla domanda del contributo NMII all’assemblaggio dei sarcomeri nei topi sono stati difficili da interpretare(Sparrow e Schöck, 2009). I topi NMIIA KO di Germline non riescono a gastrurare e quindi l’assemblaggio dei sarcomeri è impossibile da esaminare(Conti et al., 2004). Un topo NMIIA KO condizionale è stato generato utilizzando un promotore che si attiva dopo l’inizio della formazione del cuore, e non sono stati rilevati difetti cardiaci(Conti et al., 2015). Un topo NMIIB KO germline ha formato un cuore funzionale, anche se la maggior parte dei cuccioli è morta prima della nascita a causa di un’insufficienza cardiaca(Tullio et al., 1997). È stata mostrata solo un’immagine ad alto ingrandimento dell’animale KO, che ha dimostrato una grave disorganizzazione del sarcomero(Tullio et al., 1997). Di interesse, gli animali NMIIB KO hanno anche mostrato livelli di proteina NMIIA altamente aumentati rispetto ai controlli(Tullio et al., 1997). Così, la capacità osservata di questo animale di formare strutture simili a quelle dei sarcomeri potrebbe essere dovuta alla compensazione genetica da parte di NMIIA. Un topo KO NMIIB condizionale è stato anche fatto(Ma et al., 2009). Mentre gli autori hanno mostrato un impressionante KD NMIIB nel cervelletto attraverso un driver neuronale specifico, c’erano alti livelli di proteina NMIIB nel cuore al momento dell’analisi nel KO specifico del cuore(Ma et al., 2009). Inoltre, il KO condizionale specifico del cuore è spinto fuori dal promotore della catena pesante della miosina alfa, che si accende dopo l’inizio dell’assemblaggio del sarcomero e in effetti dopo che i campi del cuore hanno iniziato a battere(Ma et al., 2009; Ng et al., 1991). A complicare ulteriormente la questione, l’NMIIB è stato difficile da localizzare nel tessuto. Riteniamo che questo sia il risultato dell’incorporazione della paraffina e dei successivi protocolli di rimozione e reidratazione della paraffina. Per esempio, abbiamo localizzato con successo l’NMIIB nel tessuto umano e nel topo fissato in formalina(Figura 9 e Figura 9-figure supplement 1), ma non siamo riusciti a localizzare l’NMIIB nel tessuto paraffinico incorporato. I futuri confronti tra i set di dati in vivo e in vitro dovranno essere affrontati.
Rimangono da testare anche domande intriganti e senza risposta. Ad esempio, come vengono stabiliti e mantenuti i gradienti osservati di NMII e βCMII nei miociti cardiaci? Tali domande sono state in precedenza da difficili a impossibili da testare a causa di sistemi modello complicati e tecnicamente impegnativi. Qui presentiamo un sistema modello relativamente facile da usare per testare queste domande. Gli hiCM che utilizziamo in questo studio sono disponibili in commercio. I nostri metodi delineano protocolli per la trasfezione con DNA per l’espressione delle proteine e siRNA per il knockdown delle proteine, e protocolli di tripsinizzazione per la ri-placcatura. Andando avanti, crediamo che gli studi che utilizzano l’impostazione sperimentale che descriviamo qui non solo continuerà a chiarire i modelli precedenti, ma anche rivelare nuove intuizioni sia l’assemblaggio di sarcomeri, e la biologia delle cellule cardiache.
Figura 12-figure supplemento 1.Figura 12—figura supplemento 1. Modello della formazione di fibre di tensione actomiosina in cellule non muscolari e cardiomiociti umani.i sarcomeri si formano sulla superficie dorsale di hiCMs e successivamente si muovono verso la superficie ventrale.(A) Actina e miosina II stress formazione di fibre di stress in cellule non-muscolari. fibre di stress Actin sono formate attraverso il complesso Arp2/3 e la formina mDia1. NMIIA è l’isoforma predominante al bordo anteriore delle cellule non muscolari, e la formazione di fibre da sforzo è dipendente NMIIA. Le cellule non muscolari mostrano un robusto flusso retrogrado di fibre di actina da stress e mostrano un rapido turnover. Le grandi pile di NMIIA si formano attraverso la crescita e l’espansione di filamenti NMIIA più piccoli. Le citazioni che portano a questo modello sono presentate nel cartone animato.(B) Modello della formazione di fibre di stress di actina e miosina II nei cardiomiociti umani. I sarcomeri sono modelli di fibre da stress muscolare (MSF). MSFs non richiedono il complesso Arp2/3, e richiedono la formina FHOD3. MSFs visualizzare il flusso lento retrogrado rispetto alle fibre di stress non muscolare. Sia NMIIA che NMIIB sono localizzate ai margini delle hiCM, e visualizzano i co-filamenti NMII prominenti. Anche i co-filamenti NMIIB-βCMII sono presenti con le MSF. Grandi pile di filamenti βCMII si formano attraverso la concatenazione e la cucitura dei singoli filamenti βCMII.(A) hiCM che esprime α-actinina 2-mCherry placcato per 24 ore e ripreso con la scansione laser confocale. Si noti che quando si visualizza la superficie ventrale (in basso) (immagine a sinistra), nessun modello sarcomerico di α-actinina 2 può essere visto. Tuttavia, quando si visualizza la superficie dorsale (in alto) della cellula (immagine a destra), i sarcomeri sono chiaramente presenti.(B) Tempo di montaggio da scatola gialla in(A). Myofibrilla dalla parte superiore della cella (montaggio a destra, freccia gialla) si muove verso il fondo della cella (immagine a sinistra, freccia gialla). La testa della freccia (montaggio a sinistra) indica α-acttinina 2 contenente adesione.
Figura 12-figure supplemento 1.Sarcomeri forma sulla superficie dorsale di hiCMs e successivamente si muovono verso la superficie ventrale.(A) hiCM che esprime α-acttinina 2-mCherry placcato per 24 ore e ripreso con laser-scansione confocale. Si noti che quando si visualizza la superficie ventrale (in basso) superficie (immagine a sinistra), nessun modello sarcomerico di α-acttinina 2 può essere visto. Tuttavia, quando si visualizza la superficie dorsale (in alto) della cellula (immagine a destra), i sarcomeri sono chiaramente presenti.(B) Tempo di montaggio da scatola gialla in(A). Myofibrilla dalla parte superiore della cella (montaggio a destra, freccia gialla) si muove verso il fondo della cella (immagine a sinistra, freccia gialla). La testa della freccia (montaggio a sinistra) indica α-acttinina 2 contenente adesione.
Video 5.Sarcomeri che viaggiano da dorsale a superficie ventrale di hiCM hiCM placcato per 24 ore e che esprime α-actinina 2-mCherry.6. Quattro viste da quattro piani Z ripresi con microscopia confocale a scansione laser. Nel primo fotogramma, nessun sarcomero può essere visualizzato sul fondo della cella (a sinistra la maggior parte dei fotogrammi), ma può essere chiaramente visto nel piano Z di 2 μm (fotogramma centrale destro). Mentre il video continua, i sarcomeri nel piano 2 μm Z viaggiano verso la superficie ventrale (piano Z in basso) dell’hiCM, che può essere facilmente visto come la miofibrilla entra a fuoco prima nel piano 1 μm Z (metà sinistra), e poi nel piano Z in basso (sinistra). Altezza: 38,2 μm. Lunghezza del video: 17,5 ore.
Materiali e metodi
| Tipo di reagente (specie) o risorsa | Designazione | Fonte o riferimento | Identificatori | Ulteriori informazioni |
|---|---|---|---|---|
| Linea cellulare (umana) | iCell Cardiomiociti^2 | Dinamica cellulare internazionale | miociti cardiaci derivati dall’ipsc | |
| Linea cellulare (umana) | Cardiomiociti Cor.4U | Ncardia (ex Axiogenesi) | miociti cardiaci derivati dall’ipsc | |
| Linea cellulare (umana) | HeLa | Collezione americana della cultura del tipo (ATCC) | CCL-2 | |
| Linea cellulare (umana) | U2-OS | Collezione americana della cultura del tipo (ATCC) | HTB-96 | |
| Reagente DNA ricombinante | pHalo-NMHC IIA-C18 | questo documento | vedi Materiali e metodi per i dettagli sulla creazione | |
| Reagente DNA ricombinante | pHalo-NMHC IIB-C18 | questo documento | vedi Materias e metodi per i dettagli sulla creazione | |
| Reagente DNA ricombinante | α-acttinina-2-mEmeraldo | Addgene | 53988 | |
| Reagente DNA ricombinante | α-acttinina-2-tdEOS | Addgene | 57577 | |
| Reagente DNA ricombinante | NMIIA-(N-terminale)-mEGFP | Addgene | 11347 | |
| Reagente DNA ricombinante | Lifeact-mEmerald | Regalo di Michael Davidson | ||
| Reagente DNA ricombinante | Lifeact-mApple | Regalo di Michael Davidson | ||
| Reagente DNA ricombinante | NMIIB-(N-terminale)-mEmerald | Addgene | 54192 | |
| Reagente DNA ricombinante | FHOD-mEGFP (manca l’esone T(D/E)5XE) | Regalo della Dott.ssa Elizabeth Ehler | Iskratsch et al. (2010) | |
| Reagente DNA ricombinante | βCMII-mEGFP | Genescript (Piscataway, NJ, USA) questo documento | vedi Materiali e metodi per i dettagli sulla creazione | |
| Reagente DNA ricombinante | p16b-eGFP | Regalo della Dr. Jenniffer Lippincott-Schwartz e della Dr. Pekka Lappalainen | Lai, F. P., et al 2008.Koestler et al., 2013. | |
| Anticorpo | coniglio IgG policlonale IgG anti-NMIIA | Biolegend | P909801 | Usato a 1:1000 |
| Anticorpo | coniglio IgG policlonale IgG anti-NMIIB | Segnalazione cellulare | P3404S | Usato a 1:200 |
| Anticorpo | coniglio IgG policlonale IgG anti-NMIIB | Biolegend | 909901 | Usato a 1:200 |
| Anticorpo | topo monoclonale IgG anti-βCMII | Banca Ibridioma Iowa | A4.1025 | Usato a 1:2 |
| Anticorpo | coniglio IgG policlonale IgG anti-α-actinina-2 | Sigma-Aldrich | clone EA-53 | Usato a 1:200 |
| Anticorpo | coniglio policlonale IgG anti-p34-Arc/ArpC2 (complesso Arp2/3) | Millipore Sigma | 07–227 | Usato a 1:100 |
| Anticorpo | coniglio IgG policlonale IgG anti-DAAM1 | Laboratori Bethyl | HPA026605 | Usato a 1:100 |
| Anticorpo | coniglio IgG policlonale IgG anti-DIAPH1 | Sigma-Aldrich | A300-078A | Usato a 1:100 |
| Anticorpo | topo monoclonale IgGanti-cardiaco troponina T | Santa Cruz Biotecnologia | CT3, sc-20025 | Usato a 1:50 |
| Anticorpo | capra anti-topo IgG Alexa Fluor 568 | ThermoFisher Scientific | A-11004 | Usato a 1:100 |
| Anticorpo | capra anti coniglio IgG Alexa Fluor 568 | ThermoFisher Scientific | A-11011 | Usato a 1:100 |
| Anticorpo | capra anti-topo IgG Alexa Fluor 488 | ThermoFisher Scientific | AA28175 | Usato a 1:100 |
| Anticorpo | capra anti coniglio IgG Alexa Fluor 647 | ThermoFisher Scientific | A-21244 | Usato a 1:100 |
| Anticorpo | capra anti-topo IgM Alexa Fluor 488 | ThermoFisher Scientific | A-21042 | Usato a 1:100 |
| Anticorpo | mouse monoclonale IgG anti-FHOD3 | Santa Cruz Biotecnologia | G-5, sc-374601 | Usato a 1:100 |
| Reagente a base di sequenza | SmartPool siRNA DIAPH1 | GE Dharmacon | E-010347-00-0005 | |
| Reagente a base di sequenza | SmartPool siRNA DAAM1 | GE Dharmacon | E-012925-00-0005 | |
| Reagente a base di sequenza | SmartPool siRNA FHOD3 | GE Dharmacon | E-023411-00-0005 | |
| Reagente a base di sequenza | SmartPool siRNA MYH10 (NMIIB) | GE Dharmacon | E-023017-00-0010 | |
| Reagente a base di sequenza | SmartPool siRNA MYH9 (NMIIA) | GE Dharmacon | E-007668-00-0005 | |
| Saggio commerciale o kit | Rneasy Mini Kit | Qiagen | 74104 | |
| Saggio commerciale o kit | QIAprep spin Kit Miniprep (250) | Qiagen | 27106 | |
| Composto chimico, farmaco | Blebbistatina | Sigma-Aldrich | B0560 | |
| Composto chimico, farmaco | LatrunculinB | Sigma-Aldrich | L5288 | |
| Composto chimico, farmaco | LatrucluinA | Sigma-Aldrich | L5163 | |
| Composto chimico, farmaco | CK666 | Sigma-Aldrich | SML0006 | |
| Composto chimico, farmaco | SMIFH2 | Sigma-Aldrich | S4826 | |
| Composto chimico, farmaco | TransIT-TKO | Mirus Bio | MIR 2150 | |
| Composto chimico, farmaco | FuGENE HD | Promega | E2311 | |
| Composto chimico, farmaco | Alexa Fluor 488 Falloidina | ThermoFisher Scientific | A12379 | |
| Composto chimico, farmaco | Viafect | promega | E4981 | |
| Software, algoritmo | (Fiji è solo) ImageJ | NIH (open source) |
Contatto per la condivisione di reagenti e risorse
Ulteriori informazioni e richieste di risorse e reagenti devono essere indirizzate e saranno soddisfatte dal Lead Contact, D.T.B. (dylan.burnette@vanderbilt.edu)
Modello sperimentale e dettagli del soggetto
Crescita della linea cellulare e condizioni sperimentali
I cardiomiociti staminali pluripotenti indotti dall’uomo (hiCM) sono stati acquistati da Axiogenesis (Cor.4u, Ncardia Cologne, Germania) o da Cellular Dynamics International (iCell caridomyocytes2, Madison, WI). Le cellule sono state coltivate secondo le istruzioni del produttore. hiCMs sono stati coltivati in 96 piastre pozzo e mantenuto in produttore proprietario fornito supporti fino a quando non è pronto per l’uso sperimentale. Esperimenti Knockdown in hiCMs sono stati avviati 4 giorni dopo il disgelo iniziale. Vedere il test di placcatura per un protocollo più dettagliato per la placcatura delle cellule. Vedi sotto per informazioni dettagliate sulla trasfezione di proteine e knockdown mediato da siRNA
Le cellule U-2 OS e HeLa (HTB-96; American Type Culture Collection, Manassas, VA) sono state coltivate in flaconi da 25 cm2 (25-207; Genessee Scientific Corporation, San Diego, CA) con un mezzo di crescita comprendente DMEM (10-013-CV; Mediatech, Manassas, VA) contenente 4.5 g/L di L-glutammina, D-glucosio e piruvato di sodio e integrato con il 10% di siero bovino fetale (F2442; Sigma-Aldrich, St. Louis, MO). Per gli esperimenti di espressione delle proteine nelle cellule U-2 OS, le cellule sono state transitoriamente trasfettate con FuGENE 6 (E2691; Promega, Madison, WI) secondo le istruzioni del produttore. Knockdown di NMIIA e NMIIB è stata eseguita come in precedenza, con il siRNA Accell SMARTpool siRNA umano MYH9, MYH10 o controllo Accell strapazzato acquistato da Thermo Fisher Scientific (Waltham, MA) in combinazione con Lipofectamina 2000 Reagente Trasfezione (cat # 11668027, Thermo Fisher Scientific, Waltham, MA) per le cellule HeLa. Sia per la microscopia a cellule vive che per la microscopia a cellule fisse, le cellule sono state placcate e riprese su piatti con fondo in vetro da 35 mm con un micropozzetto da 10 mm #1,5 di vetro di copertura (Cellvis, Mountain View, CA) rivestito con 25 µg/mL di laminina (114956-81-9, Sigma-Aldrich).
Plasmidi
I plasmidi che codificano NMIIA-(N-terminal)-mEGFP (11347; Addgene, Cambridge, MA) con mEGFP sul N-terminale della catena pesante NMIIA sono stati utilizzati come descritto in precedenza(Chua et al., 2009). La codifica Plasmid Lifeact-mEmerald e Lifeact-mApple sono stati doni di Michael Davidson. La codifica Plasmide NMIIB-(N-terminal)-mEmerald è stata acquistata da Addgene (54192; Addgene, Cambridge, MA). La codifica plasmidica α-actinina 2-tdEOS è stata acquistata da Addgene (57577; Addgene, Cambridge, MA). La codifica del plasmide βCMII umano è stata sintetizzata da Genscript (Piscataway, NJ, USA). In breve, la wild-type human MYH7 (βCMII) sequenza di tipo umano MYH7 (βCMII) dal National Center for Biotechnology Information (NCBI, Bethesda, MD, USA) è stata clonata in un pUC57 insieme a Gateway sequenze di ricombinazione del DNA al fine di facilitare la rapida integrazione e lo scambio di proteine fluorescenti (Gateway Technology, ThermoFisher Scientific, Waltham, MA). mEGFP contenente una sequenza linker precedentemente pubblicata è stata aggiunta al plasmide βCMII utilizzando il sistema di conversione vettoriale Gateway Vector Conversion System con cellule di sopravvivenza ccdB One Shot (ThermoFisher Scientific, Waltham, MA) per il costrutto βCMII-(N-terminal)-mEGFP utilizzato in questo studio. Il plasmide FHOD3-mEGFP privo dell’esone T(D/E)5XE è stato un regalo di Elizabeth Ehler. Questo costrutto è stato precedentemente mostrato per localizzare i sarcomeri in cardiomiociti di ratto neonatale(Iskratsch et al., 2010) (vedi pannello in alto a sinistra della Figura 2A in riferimento). L’assemblaggio di pHalo-NMHC IIB-C18: pEmerald-NMHC IIB-C18 (Addgene, #54192) è stato il gentile dono di Michael Davidson. Halo tag cDNA è stato PCR amplificato da pHalo-N1 e subclonato nel sito di Age I/BspEI mEmerald di pEmerald-NMHC IIB-C18, sostituendo mEmerald, per generare la sequenza verificata di pHalo-NMHC IIB-C18. Montaggio di pHalo-NMHC IIA-C18 (NMIIA-HaLo): Il frammento di cDNA NheI/BspEI Halo Halo dal costrutto pHalo-NMHC IIB-C18 è stato subclonato nel sito NheI/BspEI mEmerald di pEmerald-NMHC IIA-C18 (Addgene, #54190)(Burnette et al., 2014), sostituendo mEmerald, per generare la sequenza verificata del costrutto pHalo-NMHC IIA-C18. Il plasmide pEGFP-p16b (per visualizzare il complesso Arp2/3) è stato un regalo del Dr. Pekka Lappalainen e precedentemente pubblicato(Koestler et al., 2013; Lai et al., 2008).
Etichettatura Halo-tag
hiCM esprimendo costrutti Halo-tag sono stati etichettati con JF Dye 585 a diluizione 1:100 in terreno di coltura preriscaldato per 1 ora. La piastra è stata poi lavata scambiando delicatamente con il terreno di coltura fresco per 3 volte(Grimm et al., 2017; Grimm et al., 2015)
Autenticazione della linea cellulare
La linea cellulare HeLa utilizzata in questo studio è stata un regalo del Dr. DA Weitz (Università di Harvard). Il laboratorio Burnette ha fatto autenticare questa linea da Promega e ATCC utilizzando il loro ‘Cell Line Authentication Service’ nel 2015. I metodi e i risultati dei test ricevuti da Promega e ATCC sono i seguenti:
“Metodologia: Diciassette loci a breve ripetizione tandem (STR) più il locus che determina il genere, l’Amelogenina, sono stati amplificati utilizzando il kit PowerPlex 18D di Promega disponibile in commercio. Il campione della linea cellulare è stato elaborato utilizzando l’analizzatore genetico ABI Prism 3500xl. I dati sono stati analizzati utilizzando il software GeneMapper ID-X v1.2 (Applied Biosystems). Sono stati eseguiti e confermati adeguati controlli positivi e negativi per ogni campione presentato”.
“Interpretazione dei dati”: Le linee cellulari sono state autenticate utilizzando l’analisi Short Tandem Repeat (STR) come descritto nel 2012 in ANSI Standard (ASN-0002) Authentication of Human Cell Lines: Standardizzazione del profilo STR da parte dell’Organizzazione per lo sviluppo degli standard ATCC (SDO)”.
“Risultati dei test”: Il profilo presentato corrisponde esattamente alle seguenti linee cellulari umane ATCC nel database ATCC STR (otto loci principali più Amelogenina): CCL-2 (HeLa)”.
La linea cellulare U2 OS utilizzata in questo studio è stata acquistata direttamente da ATCC dal laboratorio Burnette nel 2014 (ATCC, HTB-96). Monitoraggio dei micoplasmi: Sia HeLa e U2 linee cellulari OS sono stati controllati per potenziale infezione da micoplasma utilizzando sia DAPI o Heoscht durante tutto il corso di questo studio.
Live-Cell test di visualizzazione del montaggio del sarcomero
Per visualizzare la formazione di sarcomeri, abbiamo sviluppato un metodo ripetibile, che può essere utilizzato per visualizzare qualsiasi proteina marcata fluorescentemente durante la formazione di sarcomeri in hiCM. Prima di eseguire questo saggio, le cellule sono mantenute nel vaso di coltura desiderato (i nostri hiCMs sono stati mantenuti in 96 piastre di pozzo). In questo saggio, le cellule sono transitoriamente trasfettate con la proteina desiderata fluorescente taggato, tripsinizzato, placcato sul piatto di imaging desiderato, e ripreso per osservare la formazione di sarcomeri. Un test di assemblaggio sarcomero procede come segue. In primo luogo, hiCMs sono trasfettati con la proteina desiderata fluorescente taggato come descritto di seguito (Viafect, trasfezione durante la notte). La miscela di trasfezione viene lavata via con i mezzi di coltura. Le cellule vengono poi staccate dal vaso di coltura utilizzando il metodo di tripsinizzazione descritto di seguito. Le cellule sono poi placcati su un vaso di coltura desiderato (in questo studio, 10 mm # 1,5 piatti di vetro di fondo sono stati utilizzati, CellVis, Mountain View, CA) per l’imaging cellulare vivo. I vasi di imaging sono stati pre-rivestiti con 25 µg/mL di lamina per 1 ora a 37° C e lavati con 1x PBS contenente Mg2+/Ca2+. Le cellule sono stati autorizzati ad attaccare per ~ 1,5 ore e media è stato aggiunto per riempire il piatto fondo di vetro. Le cellule sono stati poi ripresi utilizzando la modalità di imaging desiderato a 3-6 ore post placcatura. La finestra temporale di 3-6 ore era ottimale, in quanto le cellule non avevano ancora stabilito sarcomeri, ma erano abbastanza sani per tollerare la fluorescenza di imaging. Questo potente test può essere adattato per visualizzare la cinetica durante la formazione di sarcomeri di qualsiasi proteina marcata con fluorescenza. Abbiamo usato questo test per visualizzare l’actina (Lifeact), FHOD3, βCMII, α-actina 2, NMIIA e NMIIB. Queste ultime due proteine sono grandi (cioè, >250 KD).
Tripsinizzazione degli hiCM
Per tentare la tripsinizzazione degli hiCM Cellular Dynamics, sono state utilizzate le raccomandazioni dei produttori, come segue. Tutti i volumi applicati sono stati modificati dal formato a 24 pozzetti per 96 piastre a pozzetto. hiCMs sono stati lavati 2x con 100 uL 1x PBS senza Ca2+/Mg2+ (PBS*). PBS * è stato completamente rimosso da hiCMs e 40 uL 0,1% Trypsin-EDTA senza rosso fenolo (Invitrogen) è stato aggiunto a hiCMs e posto a 37 ° C per 2 min. Dopo l’incubazione, piatto di coltura è stato lavato 3x con tripsina all’interno del pozzo, ruotato di 180 gradi, e lavato un altro 3x. Tripsinizzazione è stato poi estinto con l’aggiunta di 160 µL di terreno di coltura e la miscela cellulare totale è stato posto in una provetta Eppendorf 1,5 mL. Le cellule sono state filate a 1000 g per 5 minuti, e il surnatante è stato aspirato. Le cellule sono state poi risospese in 200 uL di terreno di coltura e placcato su un piatto di vetro di 10 mm di fondo pre-rivestito con 25 µg / ml di lamina per 1 ora. Le cellule sono stati poi permesso di attaccare per almeno 1 ora, e 2-3 mLs di mezzi di coltura con o senza farmaco è stato aggiunto alle cellule.
Per trypsinize Axiogenesis hiCMs, le raccomandazioni dei produttori sono stati utilizzati, come segue. Le cellule sono state lavate 2x con 500 µL PBS*. Le cellule sono state poste in incubatrice a 37° C per 7 minuti in PBS*. Dopo 7 minuti, PBS * è stato aspirato e 40 µL 0,5% tripsina 0,5% (Invitrogen) è stato posto sulle cellule per 3 minuti in 37 ° C incubatrice. Dopo 3 min, 160 µL pieno Cor.4u media è stato utilizzato per estinguere tripsinizzazione e risospendere le cellule. Le cellule sono stati poi placcati su un piatto di vetro pre-rivestito fondo piatto e media aggiunto 1,5 ore dopo per diluire tripsinatura e riempire la camera. Nota * questo protocollo di tripsinizzazione è stato poi modificato da Axiogenesis (ora Ncardia). Vedi produttore per il nuovo protocollo.
È importante notare che il protocollo di tripsinizzazione si basa sui protocolli dei produttori di hiCMs per la placcatura delle cellule, e questi hiCMs sono stati tripsinizzati prima di saggi funzionali delle prestazioni cardiomiocitarie e la caratterizzazione (ad esempio, la risposta al farmaco, elettrofisiologia, maturazione, ecc …) (Fineet al., 2013; Ivashchenko et al., 2013; Mioulane et al., 2012).
Trasfezione transitoria di hiCM
Gli hiCM Cellular Dynamics sono stati trasfettati modificando le raccomandazioni del produttore come segue. I volumi utilizzati sono per la trasfezione in 96 piastre a pozzetto. A 6 µL Opti-MEM sono stati aggiunti 2 µL di plasmide totale 200 ng (contenente la proteina di interesse marcata con fluorescenza, diluita in Opti-MEM) e 2 µL di Viafect diluito 1:5 (Promega, E4981, in Opti-MEM). L’intera miscela di 10 µL è stata aggiunta a 96 pozzetti singoli di hiCM contenenti 100 µL di terreno di coltura completo appena scambiato. La trasfezione è stato permesso di andare durante la notte (~ 15 ore), e lavato 2x con mezzi di coltura completa. Per la trasfezione di sonde multiple, 2 µL di 200 ng di plasmide ng è stato utilizzato per ogni sonda insieme a 4 µL 1:5 diluito Viafect, in 2 µL Opti-MEM e la miscela è stata applicata alle cellule come sopra.
Axiogenesis hiCMs sono stati trasfettati attraverso la modifica delle raccomandazioni del produttore come segue. Un rapporto 3,5:1 Fugene a DNA è stato utilizzato per trasfettare Axiogenesis hiCMs. 1,2 µL di fuugenio +0,33 µg di DNA per 96 pozzetti in 5 µL di siero libero Cor.4u media è stato incubato per 15 minuti a temperatura ambiente. 95 µL media Cor.4u pieno è stato aggiunto alla miscela e l’intera miscela è stata aggiunta a hiCMs (in cima a 100 µL già in pozzo). Per la trasfezione di 2 plasmidi separati, rapporto 3,5:1 è stato utilizzato per entrambi i plasmidi e volume aggiuntivo è stato sottratto da 95 µL diluizione. Nota* questo protocollo di trasfezione è stato modificato dall’assiogenesi. Vedere il produttore per il nuovo protocollo.
Abbattimento delle proteine
Gli hiCM Cellular Dynamics sono stati utilizzati per esperimenti di knockdown attraverso la modifica delle raccomandazioni dei produttori. I volumi utilizzati sono per l’applicazione di siRNA in 96 piastre a pozzo. Dharmacon SmartPool siRNA (GE Dharmacon, Lafayette, CO) destinati a MYH9 (NMIIA), MYH10 (NMIIB), FHOD3, DAAM1, e DIAPH1 sono stati utilizzati (E-007668-00-00-00-0005, E-023017-00-0010, E-023411-00-00-00-0005, E-012925-00-00-00-0005, e E-010347-00-00-0005, rispettivamente). Per ottenere KD, una miscela master di 100 μl Opti-MEM (ThermoFisher, Waltham, MA)+4 μl Transkit-TKO (Mirus Bio, Madison WI)+5,5 μl 10 μM siRNA è stato incubato per 30 minuti a temperatura ambiente. 80 μl di media freschi, pre-riscaldati è stato aggiunto a hiCMs. Dopo l’incubazione di miscela siRNA, 8,3 microlitri di miscela è stato aggiunto ad ogni singolo pozzo di 96 piastra pozzo. hiCMs sono stati poi incubati per 2 giorni a 37 ° C. hiCMs sono stati poi lavati 2x con fresco, pre-riscaldato media. Per ottenere KD di NMIIA, NMIIB, FHOD3, DAAM1, e DIAPH1, 3 turni di siRNA mediato KD descritto sopra sono stati necessari. A seguito di 3 turni di trattamento siRNA di controllo scramble siRNA, hiCMs ancora battere e mantenuto la struttura del sarcomero (vediFigura 8-figure supplemento 6).
Esperimenti farmacologici
Per tutti gli esperimenti farmacologici, i farmaci sono stati aggiunti ai mezzi di comunicazione preriscaldati ed equilibrati prima di aggiungerli alle hiCM. Per i saggi di assemblaggio del sarcomero, 25 μM SMIFH2 (Sigma-Aldrich, S4826), 25 μM CK666 (Sigma-Aldrich, SML0006), e 100 μM Blebbistatina (Sigma-Aldrich, B0560) (separatamente) sono stati aggiunti 1,5 ore dopo la placcatura per consentire hiCMs di attaccare al substrato. hiCMs sono stati successivamente fissati (vedi sotto) a 24 ore post placcatura. Per gli esperimenti di cellule vive (come in Figura 5D), media in contenitore di imaging è stato aspirato utilizzando un sistema di vuoto, e media contenenti droga è stato aggiunto al hiCMs sul microscopio fase. Questo ha permesso la stessa hiCM per essere imaged stesso hiCM pre e post-applicazione di droga. Per l’esperimento LatrunculinB(Figura 9J), le cellule sono stati autorizzati a diffondersi per 18 ore in media normali, e media contenenti 5 μM LatrunculinB (Sigma-Aldrich, L5288) è stato aggiunto per 6 ore e hiCM sono stati fissati (per un tempo totale di diffusione di 24 ore).
Macchia occidentale
I campioni di gel sono stati preparati miscelando i lisati delle cellule con il buffer di campioni LDS (Life Technologies, #NP0007) e il buffer di riduzione del campione (Life Technologies, #NP00009) e bolliti a 95°C per 5 min. I campioni sono stati risolti su gel di Bis-Tris a gradiente Bolt 4-12% (Life Technologies, #NW04120BOX). Le bande di proteine sono state tamponate su una membrana di nylon (Millipore). Le macchie sono state bloccate usando 5% NFDM (Research Products International Corp, Mt. Prospect, IL, #33368) in TBST. Incubazioni di anticorpi sono state eseguite anche in 5% NFDM in TBST. I blot sono stati sviluppati utilizzando il kit di chemiluminescenza Immobilon (Millipore, #WBKLS0500).
Fissazione e immunoistochimica
hiCMs e cellule HeLa sono stati fissati con il 4% di paraformaldeide (PFA) in PBS a temperatura ambiente per 20 minuti e poi estratti per 5 minuti con 1% Tritone X-100% e 4% PFA in PBS come descritto in precedenza(Burnette et al., 2014). Le cellule sono state lavate tre volte in 1 × PBS. Per visualizzare βCMII endogeno e proteine trasfettate in cellule fisse, hiCMs sono stati estratti cellule vive prima della fissazione come descritto in precedenza per ridurre i filamenti di fondo e non citoscheletrico miosina II filamenti (cioè, il pool solubile). In breve, un citoscheletro-stabilizzante tampone di estrazione a cellule vive è stato fatto fresco contenente 2 ml di soluzione madre (500 mM acido 1,4-piperazinedietansolfonico, 25 mM acido etilenglicole tetraacetico, 25 mM MgCl2), 4 ml di 10% di poliossietilene glicole (PEG; 35.000 peso molecolare), 4 ml diH2O, e 100 μl di Triton X-100, 10 μM di paclitaxel e 10 μM di falloidina. Le cellule sono state trattate con questo tampone di estrazione per 1 minuto, seguito da un lavaggio di 1 minuto con tampone di lavaggio (tampone di estrazione senza PEG o Triton X-100). Le cellule sono state poi fissate con il 4% di PFA per 20 minuti. Dopo la fissazione, sono state utilizzate le seguenti procedure di etichettatura: per la visualizzazione dell’actina, la falloidina-488 in 1 × PBS (15 μl di falloidina di riserva per 200 μl di PBS) è stata utilizzata per 3 ore a temperatura ambiente. Per gli esperimenti di immunofluorescenza, le cellule sono state bloccate nel 10% di sieroalbumina bovina (BSA) in PBS. Gli anticorpi primari sono stati diluiti in BSA al 10%. L’anticorpo NMIIA (BioLegend, P909801) è stato utilizzato a 1:1000. L’anticorpo NMIIB (Cell Signaling, 3404S e BioLegend 909901) è stato usato a 1:200. L’anticorpo βCMII (Iowa Hybridoma Bank, A4.1025) è stato usato non diluito dal siero. L’anticorpo α-actinina 2 (Sigma-Aldrich, clone EA-53) è stato usato a 1:200. L’anticorpo FHOD3 (Santa Cruz Biotechnology, G-5, sc-374601) è stato utilizzato a 1:100. L’anticorpo Arp2/3 (Anti-p34-Arc/ARPC2, Millipore Sigma, 07-227) è stato usato a 1:100. L’anticorpo DAAM1 (Bethyl Laboratories, A300-078A) è stato usato a 1:100. L’anticorpo DIAPH1 (Sigma-Aldrich, HPA026605) è stato usato a 1:100. L’anticorpo della troponina T cardiaca (Santa Cruz Biotechnology, CT3, sc-20025) è stato utilizzato a 200 µg/ml. Gli anticorpi secondari sono stati diluiti al 10% di BSA a 1:100 e centrifugati a 13.000 rpm per 10 minuti prima dell’uso. Le cellule sono state riprese in VECTASHIELD Antifade Mounting Media con DAPI (H-1200, VECTOR LABORATORIES, Burlingame, CA, USA). Per etichettare sia NMIIA che NMIIB nello stesso campione (come nella Figura 7A), l’anticorpo primario NMIIA è stato etichettato direttamente usando un kit di etichettatura dell’anticorpo primario di Biotium (Mix-n-Stain CF Antibody Labeling Kits, Biotium, Inc. Fremont, CA). Seguendo il protocollo dei produttori, l’NMIIA è stato etichettato come anticorpo primario e la colorazione è stata confrontata visivamente con il protocollo standard di immunofluorescenza (vedi sopra) per convalidare il modello di localizzazione.
Analisi dei dati RNA-seq
Le letture RNA-seq sono state allineate al genoma umano di riferimento hg19 utilizzando STAR(Dobin et al., 2013) e quantificate da featureCounts(Liao et al., 2014). I conteggi delle letture sono stati normalizzati con il metodo Relative Log Expression (RLE) e i valori FPKM per ogni campione sono stati generati da DESeq2(Love et al., 2014).
Microscopia ad illuminazione strutturata
In questo studio sono stati utilizzati due microscopi SIM (gli esperimenti individuali sono stati sempre ripresi con lo stesso microscopio). L’imaging e l’elaborazione delle immagini SIM è stata effettuata su una GE Healthcare DeltaVision OMX equipaggiata con un obiettivo a olio 60×/1,42 NA e una telecamera sCMOS a temperatura ambiente. L’imaging e l’elaborazione delle immagini SIM è stata eseguita anche su una piattaforma di illuminazione strutturata Nikon N-SIM dotata di una telecamera EMCCD Andor DU-897 e di un obiettivo SR Apo TIRF (olio) 100 × 1,49 NA WD 0,12 a temperatura ambiente.
Microscopia a disco rotante
Le immagini confocali dei dischi rotanti sono state scattate su un disco rotante Nikon Spinning Disk equipaggiato con una testa a disco rotante Yokogawa CSU-X1, una fotocamera Andor DU-897 EMCCD e un obiettivo 100x Apo TIRF (olio) 1,49 NA WD 0,12 mm a 37 gradi C e 5%CO2.
Microscopia confocale a scansione laser
Le immagini confocali a scansione laser sono state scattate su un laser Nikon A1R dotato di un obiettivo 60x/1,40 Plan Apo Oil a 37 gradi C e 5%CO2. Le immagini confocali per laFigura 9C (in vivo, a destra) e la Figura 9-figuresupplement 1C-D sono state scattate su uno Zeiss 880 con AiryScan equipaggiato con un obiettivo Plan-Apochromat Oil 63x/1.40 a temperatura ambiente.
Microscopia ad ampio campo
Le immagini ad ampio campo sono state scattate su una Nikon Eclipse Ti ad alta risoluzione ad ampio campo equipaggiata con un obiettivo ad olio Nikon 100x Plan Apo 1.45 NA e una fotocamera CMOS Nikon DS-Qi2. I dati presentati inFigura 8-figure supplement 6 sono stati acquisiti su un sistema di Live-Cell Analysis di Essen Incucyte S3 (Ann Arbor, MI) con l’obiettivo 20x. Foto-conversione esperimenti sono stati eseguiti come descritto in precedenza(Fenix et al., 2016).
Quantificazione e analisi statistica
Quantificazione del montaggio del sarcomero
Per quantificare la percentuale di hiCM con sarcomeri (cioè.., Figura 4B), il citoscheletro di actina (tramite falloidina fluorescente etichettato) è stato ripreso con microscopia a illuminazione strutturata. hiCMs sono stati quantificati come contenenti strutture sarcomeri se contenevano almeno una miofibrilla contenente almeno 3 Z-dischi (colorazione falloidina brillante che si sovrappone con α-actinina 2 colorazione come in Figura 4A) distanziati ~ 1,5-2 micron di distanza. Con queste metriche la nostra quantificazione della formazione del sarcomero non è una misura della maturità o dell’allineamento del sarcomero, ma una misura della capacità degli hiCM di assemblare i blocchi di costruzione del sarcomero (cioè i sottili filamenti di actina) in risposta ad una perturbazione. Così, mentre gli hiCMs NMIIA KD formano chiaramente degli hiCMs non allineati, disorganizzati, e meno sarcomeri e miofibrille rispetto agli hiCMs di controllo, essi mantengono ancora la capacità di assemblare le strutture dei sarcomeri, che si riflette nella nostra quantificazione(Figura 8A e C). Nella stessa ottica, ci rendiamo conto che la nostra quantificazione è una quantificazione molto liberale dell’assemblaggio dei sarcomeri. Mentre non ci aspettiamo che una piccola serie di sarcomeri rappresenti un cardiomiocita funzionalmente capace, stiamo investigando i primi passi dell’assemblaggio dei sarcomeri e la capacità degli hiCM di formare la struttura di base dell’actina-miosina del sarcomero.
β miosina cardiaca II (βCMII) filamento e quantificazione della pila
Una metodologia simile è stata utilizzata per quantificare le pile di filamenti in banda A della βCMII utilizzando la colorazione endogena della βCMII e la SIM al posto del citoscheletro dell’actina. Un filamento βCMII è stato quantificato come la minima unità risolvibile SIM βCMII risolvibile che aveva un’organizzazione bipolare (un filamento con domini motori su ogni lato), come rappresentato nella Figura 11A. hiCMs sono stati quantificati come contenenti stack di filamenti in banda A βCMII se contenevano anche un solo stack di filamenti βCMII nella cella. Una pila di filamenti βCMII è stata definita come più spessa del filamento βCMII risolvibile minimo della SIM, indicando più filamenti βCMII risolvibili della SIM. Infatti, con questa metrica, le pile di filamenti βCMII hanno più “domini motori” risolvibili dei filamenti βCMII.
Per l’arco di actina e le velocità di flusso retrogrado MSF (come nella Figura 3), sono state utilizzate 3 regioni di interesse (ROI) per cella. ROI sono stati disegnati utilizzando lo strumento linea in FIJI a partire da davanti al bordo anteriore (per garantire la formazione di nuovi MSF è stato catturato) al corpo della cellula in cui sono state localizzate le strutture di sarcomeri. Le ROI sono state poi utilizzate per misurare i tassi di traslocazione di MSF utilizzando lo strumento chimografo (larghezza della linea = 3) su hiCM che erano stati allineati utilizzando la funzione StackReg. I chimografi generati in questo modo sono stati poi misurati manualmente contando i pixel sull’asse X (distanza) e sull’asse Y (tempo) per una misurazione della distanza/tempo che ha portato a tassi di traslocazione. Questo metodo è simile ai metodi precedentemente descritti per la traslocazione dell’arco di actina in cellule non muscolari.
Per misurare la distanza tra le strutture di α-actina 2, sono stati utilizzati lo strumento linea e lo strumento di misura in FIJI (Fiji is Just ImageJ). Le linee sono state disegnate, le posizioni registrate (utilizzando lo strumento ROI), e le distanze misurate tra α-acttinina 2 strutture. Sono state utilizzate più regioni per cella per α-actinina 2 in MSF, e celle intere per le distanze tra i sarcomeri.
Per misurare le dimensioni della linea Z in hiCMs, lo strumento di linea e strumento di misura in FIJI è stato utilizzato. Le linee sono state disegnate su singole linee Z e le loro posizioni sono state registrate utilizzando lo strumento ROI. Questo ha dato una misura delle dimensioni della linea Z e della posizione all’interno della cella. Per le misurazioni della linea Z, come la Figura 8E, sono state misurate tutte le linee Z che potevano essere misurate in modo affidabile.
Quantificazione della localizzazione in immunofluorescenza
Per quantificare la localizzazione di NMIIA, NMIIB e βCMII, per ogni cella sono state effettuate scansioni di linea a partire dal bordo della cella e le localizzazioni medie normalizzate sono state utilizzate per calcolare la media del numero di esperimenti indicati per i modelli di localizzazione finale rappresentati nel grafico (come nella Figura 7B).
Per quantificare l’intensità di Arp2/3, è stata adottata una strategia simile ma modificata. quattro scatole separate per ogni cella sono state posizionate lungo il bordo della cella (cioè il lamellipodio) utilizzando il canale di actina come guida. Queste scatole sono state poi utilizzate per misurare l’intensità media del canale anti-p34 (cioè il complesso Arp2/3). Sfondo sottratto medie per ogni cellula in controllo e CK666 hiCM trattati sono stati utilizzati per quantificare la diminuzione percentuale come illustrato nella figura 4F.
Cellule utilizzate per questo studio
Per avere risultati comparabili, le cellule utilizzate per questo studio sono state standardizzate in base alla morfologia. In particolare, le cellule non muscolari diffuse e le hiCM con un ampio bordo d’attacco e la lamella sono state scelte come precedentemente mostrato negli studi sulla formazione del sistema contrattile sia non-muscolare che muscolare(Burnette et al., 2014; Rhee et al., 1994). Questo ha anche facilitato la capacità di osservare la MSF per la transizione del sarcomero in hiCMs vivo, ed è raccomandato per studi futuri che indagano l’assemblaggio del sarcomero. Tutti gli esperimenti di misurazione dell’assemblaggio dei sarcomeri sono stati condotti almeno 3 volte, separatamente, e le cellule sono state riprese utilizzando la SIM. Anche se questo si traduce in un numero relativamente piccolo di cellule per alcuni degli esperimenti, crediamo che le modalità di imaging a super-risoluzione come la SIM offrono una visione inestimabile sull’assemblaggio dei sarcomeri(Gustafsson, 2005). Infatti, l’imaging a subdiffrazione è necessario per localizzare in modo affidabile i co-filamenti di miosina II sia in vitro che in vivo. Inoltre, come si è visto in Drosophila, anche le modalità di imaging ad alta risoluzione come la microscopia confocale a scansione laser non sono sufficienti per rilevare sottili, ma importanti, cambiamenti strutturali in risposta alle perturbazioni(Fernandes e Schöck, 2014).
Statistiche
La significatività statistica è stata calcolata utilizzando i test T-test degli studenti a due code e non accoppiati eseguiti in Excel. Tutti gli esperimenti indipendenti rappresentano repliche biologiche. Le barre di errore in tutti i grafici rappresentano l’errore standard della media (SEM). Per tutti i grafici che rappresentano la % di celle (ad esempio, Figura 1D), il numero di celle e di esperimenti è indicato nella legenda delle figure. Le percentuali visualizzate rappresentano la media delle medie di tutti gli esperimenti eseguiti. Ad esempio, se le celle di controllo mostrano il 95%, 90% e 100% di tutte le celle che mostrano gli archi di actina in tre esperimenti separati, la percentuale rappresentata nel grafico sarebbe del 95%. Il SEM è stato calcolato dividendo la deviazione standard per la radice quadrata del numero di esperimenti separati. Per i tassi di traslocazione dell’actina sono state utilizzate almeno tre misurazioni per cella per calcolare i tassi medi di traslocazione per cella. I tassi di traslocazione sono stati poi calcolati nello stesso modo descritto sopra. I grafici a scansione di linea rappresentano la fluorescenza relativa normalizzata.
References
- Almenar-Queralt A, Gregorio CC, Fowler VM. Tropomodulin assembles early in myofibrillogenesis in chick skeletal muscle: evidence that thin filaments rearrange to form striated myofibrils. Journal of Cell Science. 1999; 112:1111-1123. PubMed
- Au Y. The muscle ultrastructure: a structural perspective of the sarcomere. Cellular and Molecular Life Sciences. 2004; 61:3016-3033. DOI | PubMed
- Ayscough KR, Stryker J, Pokala N, Sanders M, Crews P, Drubin DG. High rates of actin filament turnover in budding yeast and roles for actin in establishment and maintenance of cell polarity revealed using the actin inhibitor latrunculin-A. The Journal of Cell Biology. 1997; 137:399-416. DOI | PubMed
- Beach JR, Shao L, Remmert K, Li D, Betzig E, Hammer JA. Nonmuscle myosin II isoforms coassemble in living cells. Current Biology. 2014; 24:1160-1166. DOI | PubMed
- Bryce NS, Schevzov G, Ferguson V, Percival JM, Lin JJ, Matsumura F, Bamburg JR, Jeffrey PL, Hardeman EC, Gunning P, Weinberger RP. Specification of actin filament function and molecular composition by tropomyosin isoforms. Molecular Biology of the Cell. 2003; 14:1002-1016. DOI | PubMed
- Burnette DT, Shao L, Ott C, Pasapera AM, Fischer RS, Baird MA, Der Loughian C, Delanoe-Ayari H, Paszek MJ, Davidson MW, Betzig E, Lippincott-Schwartz J. A contractile and counterbalancing adhesion system controls the 3D shape of crawling cells. The Journal of Cell Biology. 2014; 205:83-96. DOI | PubMed
- Chopra A, Kutys ML, Zhang K, Polacheck WJ, Sheng CC, Luu RJ, Eyckmans J, Hinson JT, Seidman JG, Seidman CE, Chen CS. Force Generation via β-Cardiac Myosin, Titin, and α-Actinin Drives Cardiac Sarcomere Assembly from Cell-Matrix Adhesions. Developmental Cell. 2018; 44:87-96. DOI | PubMed
- Chua J, Rikhy R, Lippincott-Schwartz J. Dynamin 2 orchestrates the global actomyosin cytoskeleton for epithelial maintenance and apical constriction. PNAS. 2009; 106:20770-20775. DOI | PubMed
- Colpan M, Moroz NA, Kostyukova AS. Tropomodulins and tropomyosins: working as a team. Journal of Muscle Research and Cell Motility. 2013; 34:247-260. DOI | PubMed
- Conti MA, Even-Ram S, Liu C, Yamada KM, Adelstein RS. Defects in cell adhesion and the visceral endoderm following ablation of nonmuscle myosin heavy chain II-A in mice. Journal of Biological Chemistry. 2004; 279:41263-41266. DOI | PubMed
- Conti MA, Saleh AD, Brinster LR, Cheng H, Chen Z, Cornelius S, Liu C, Ma X, Van Waes C, Adelstein RS. Conditional deletion of nonmuscle myosin II-A in mouse tongue epithelium results in squamous cell carcinoma. Scientific Reports. 2015; 5DOI | PubMed
- Côté GP. Structural and functional properties of the non-muscle tropomyosins. Molecular and Cellular Biochemistry. 1983; 57:127-146. DOI | PubMed
- Dabiri GA, Turnacioglu KK, Sanger JM, Sanger JW. Myofibrillogenesis visualized in living embryonic cardiomyocytes. PNAS. 1997; 94:9493-9498. DOI | PubMed
- Dlugosz AA, Antin PB, Nachmias VT, Holtzer H. The relationship between stress fiber-like structures and nascent myofibrils in cultured cardiac myocytes. The Journal of Cell Biology. 1984; 99:2268-2278. DOI | PubMed
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15-21. DOI | PubMed
- Du A, Sanger JM, Sanger JW. Cardiac myofibrillogenesis inside intact embryonic hearts. Developmental Biology. 2008; 318:236-246. DOI | PubMed
- Ehler E, Rothen BM, Hämmerle SP, Komiyama M, Perriard JC. Myofibrillogenesis in the developing chicken heart: assembly of Z-disk, M-line and the thick filaments. Journal of Cell Science. 1999; 112:1529-1539. PubMed
- Fenix AM, Taneja N, Buttler CA, Lewis J, Van Engelenburg SB, Ohi R, Burnette DT. Expansion and concatenation of nonmuscle myosin IIA filaments drive cellular contractile system formation during interphase and mitosis. Molecular Biology of the Cell. 2016; 27:1465-1478. DOI
- Fernandes I, Schöck F. The nebulin repeat protein Lasp regulates I-band architecture and filament spacing in myofibrils. The Journal of Cell Biology. 2014; 206:559-572. DOI | PubMed
- Fine M, Lu FM, Lin MJ, Moe O, Wang HR, Hilgemann DW. Human-induced pluripotent stem cell-derived cardiomyocytes for studies of cardiac ion transporters. American Journal of Physiology-Cell Physiology. 2013; 305:C481-C491. DOI | PubMed
- Gao L, Shao L, Higgins CD, Poulton JS, Peifer M, Davidson MW, Wu X, Goldstein B, Betzig E. Noninvasive imaging beyond the diffraction limit of 3D dynamics in thickly fluorescent specimens. Cell. 2012; 151:1370-1385. DOI | PubMed
- Grimm JB, English BP, Chen J, Slaughter JP, Zhang Z, Revyakin A, Patel R, Macklin JJ, Normanno D, Singer RH, Lionnet T, Lavis LD. A general method to improve fluorophores for live-cell and single-molecule microscopy. Nature Methods. 2015; 12:244-250. DOI | PubMed
- Grimm JB, Brown TA, English BP, Lionnet T, Lavis LD. Synthesis of Janelia Fluor HaloTag and SNAP-Tag Ligands and Their Use in Cellular Imaging Experiments. Methods in Molecular Biology. 2017; 1663:179-188. DOI | PubMed
- Gronewold TM, Sasse F, Lünsdorf H, Reichenbach H. Effects of rhizopodin and latrunculin B on the morphology and on the actin cytoskeleton of mammalian cells. Cell and Tissue Research. 1999; 295:121-129. DOI | PubMed
- Gunning PW, Hardeman EC, Lappalainen P, Mulvihill DP. Tropomyosin – master regulator of actin filament function in the cytoskeleton. Journal of Cell Science. 2015; 128:2965-2974. DOI | PubMed
- Gustafsson MG. Nonlinear structured-illumination microscopy: wide-field fluorescence imaging with theoretically unlimited resolution. PNAS. 2005; 102:13081-13086. DOI | PubMed
- Heath JP. Behaviour and structure of the leading lamella in moving fibroblasts. I. Occurrence and centripetal movement of arc-shaped microfilament bundles beneath the dorsal cell surface. Journal of Cell Science. 1983; 60:331-354. PubMed
- Henson JH, Yeterian M, Weeks RM, Medrano AE, Brown BL, Geist HL, Pais MD, Oldenbourg R, Shuster CB. Arp2/3 complex inhibition radically alters lamellipodial actin architecture, suspended cell shape, and the cell spreading process. Molecular Biology of the Cell. 2015; 26:887-900. DOI | PubMed
- Holtzer H, Hijikata T, Lin ZX, Zhang ZQ, Holtzer S, Protasi F, Franzini-Armstrong C, Sweeney HL. Independent assembly of 1.6 microns long bipolar MHC filaments and I-Z-I bodies. Cell Structure and Function. 1997; 22:83-93. DOI | PubMed
- Hotulainen P, Lappalainen P. Stress fibers are generated by two distinct actin assembly mechanisms in motile cells. The Journal of Cell Biology. 2006; 173:383-394. DOI | PubMed
- Iskratsch T, Lange S, Dwyer J, Kho AL, dos Remedios C, Ehler E. Formin follows function: a muscle-specific isoform of FHOD3 is regulated by CK2 phosphorylation and promotes myofibril maintenance. The Journal of Cell Biology. 2010; 191:1159-1172. DOI | PubMed
- Ivashchenko CY, Pipes GC, Lozinskaya IM, Lin Z, Xiaoping X, Needle S, Grygielko ET, Hu E, Toomey JR, Lepore JJ, Willette RN. Human-induced pluripotent stem cell-derived cardiomyocytes exhibit temporal changes in phenotype. American Journal of Physiology-Heart and Circulatory Physiology. 2013; 305:H913-H922. DOI | PubMed
- Kan-O M, Takeya R, Abe T, Kitajima N, Nishida M, Tominaga R, Kurose H, Sumimoto H. Mammalian formin Fhod3 plays an essential role in cardiogenesis by organizing myofibrillogenesis. Biology Open. 2012; 1:889-896. DOI | PubMed
- Koestler SA, Steffen A, Nemethova M, Winterhoff M, Luo N, Holleboom JM, Krupp J, Jacob S, Vinzenz M, Schur F, Schlüter K, Gunning PW, Winkler C, Schmeiser C, Faix J, Stradal TE, Small JV, Rottner K. Arp2/3 complex is essential for actin network treadmilling as well as for targeting of capping protein and cofilin. Molecular Biology of the Cell. 2013; 24:2861-2875. DOI | PubMed
- Kolega J. Cytoplasmic dynamics of myosin IIA and IIB: spatial ‘sorting’ of isoforms in locomoting cells. Journal of Cell Science. 1998; 111:2085-2095. PubMed
- Kuragano M, Uyeda TQP, Kamijo K, Murakami Y, Takahashi M. Different contributions of nonmuscle myosin IIA and IIB to the organization of stress fiber subtypes in fibroblasts. Molecular Biology of the Cell. 2018; 29:911-922. DOI | PubMed
- Lai FP, Szczodrak M, Block J, Faix J, Breitsprecher D, Mannherz HG, Stradal TE, Dunn GA, Small JV, Rottner K. Arp2/3 complex interactions and actin network turnover in lamellipodia. The EMBO Journal. 2008; 27:982-992. DOI | PubMed
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014; 30:923-930. DOI | PubMed
- Lim SS, Hering GE, Borisy GG. Widespread occurrence of anti-troponin T crossreactive components in non-muscle cells. Journal of Cell Science. 1986; 85:1-19. PubMed
- Lin Z, Lu MH, Schultheiss T, Choi J, Holtzer S, DiLullo C, Fischman DA, Holtzer H. Sequential appearance of muscle-specific proteins in myoblasts as a function of time after cell division: evidence for a conserved myoblast differentiation program in skeletal muscle. Cell Motility and the Cytoskeleton. 1994; 29:1-19. DOI | PubMed
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology. 2014; 15DOI | PubMed
- Lu MH, DiLullo C, Schultheiss T, Holtzer S, Murray JM, Choi J, Fischman DA, Holtzer H. The vinculin/sarcomeric-alpha-actinin/alpha-actin nexus in cultured cardiac myocytes. The Journal of Cell Biology. 1992; 117:1007-1022. DOI | PubMed
- Luther PK. The vertebrate muscle Z-disc: sarcomere anchor for structure and signalling. Journal of Muscle Research and Cell Motility. 2009; 30:171-185. DOI | PubMed
- Ma X, Takeda K, Singh A, Yu ZX, Zerfas P, Blount A, Liu C, Towbin JA, Schneider MD, Adelstein RS, Wei Q. Conditional ablation of nonmuscle myosin II-B delineates heart defects in adult mice. Circulation Research. 2009; 105:1102-1109. DOI | PubMed
- McKenna NM, Johnson CS, Wang YL. Formation and alignment of Z lines in living chick myotubes microinjected with rhodamine-labeled alpha-actinin. The Journal of Cell Biology. 1986; 103:2163-2171. DOI | PubMed
- Medeiros NA, Burnette DT, Forscher P. Myosin II functions in actin-bundle turnover in neuronal growth cones. Nature Cell Biology. 2006; 8:216-226. DOI
- Mioulane M, Foldes G, Ali NN, Schneider MD, Harding SE. Development of high content imaging methods for cell death detection in human pluripotent stem cell-derived cardiomyocytes. Journal of Cardiovascular Translational Research. 2012; 5:593-604. DOI | PubMed
- Murugesan S, Hong J, Yi J, Li D, Beach JR, Shao L, Meinhardt J, Madison G, Wu X, Betzig E, Hammer JA. Formin-generated actomyosin arcs propel T cell receptor microcluster movement at the immune synapse. The Journal of Cell Biology. 2016; 215:383-399. DOI | PubMed
- Ng WA, Grupp IL, Subramaniam A, Robbins J. Cardiac myosin heavy chain mRNA expression and myocardial function in the mouse heart. Circulation Research. 1991; 68:1742-1750. DOI | PubMed
- Nienhaus GU, Nienhaus K, Hölzle A, Ivanchenko S, Renzi F, Oswald F, Wolff M, Schmitt F, Röcker C, Vallone B, Weidemann W, Heilker R, Nar H, Wiedenmann J. Photoconvertible fluorescent protein EosFP: biophysical properties and cell biology applications. Photochemistry and Photobiology. 2006; 82:351-358. DOI | PubMed
- Nolen BJ, Tomasevic N, Russell A, Pierce DW, Jia Z, McCormick CD, Hartman J, Sakowicz R, Pollard TD. Characterization of two classes of small molecule inhibitors of Arp2/3 complex. Nature. 2009; 460:1031-1034. DOI | PubMed
- Pasqualini FS, Sheehy SP, Agarwal A, Aratyn-Schaus Y, Parker KK. Structural phenotyping of stem cell-derived cardiomyocytes. Stem Cell Reports. 2015; 4:340-347. DOI | PubMed
- Ponti A, Machacek M, Gupton SL, Waterman-Storer CM, Danuser G. Two distinct actin networks drive the protrusion of migrating cells. Science. 2004; 305:1782-1786. DOI | PubMed
- Rhee D, Sanger JM, Sanger JW. The premyofibril: evidence for its role in myofibrillogenesis. Cell Motility and the Cytoskeleton. 1994; 28:1-24. DOI | PubMed
- Riedl J, Crevenna AH, Kessenbrock K, Yu JH, Neukirchen D, Bista M, Bradke F, Jenne D, Holak TA, Werb Z, Sixt M, Wedlich-Soldner R. Lifeact: a versatile marker to visualize F-actin. Nature Methods. 2008; 5:605-607. DOI | PubMed
- Rizvi SA, Neidt EM, Cui J, Feiger Z, Skau CT, Gardel ML, Kozmin SA, Kovar DR. Identification and characterization of a small molecule inhibitor of formin-mediated actin assembly. Chemistry & Biology. 2009; 16:1158-1168. DOI | PubMed
- Rosado M, Barber CF, Berciu C, Feldman S, Birren SJ, Nicastro D, Goode BL. Critical roles for multiple formins during cardiac myofibril development and repair. Molecular Biology of the Cell. 2014; 25:811-827. DOI | PubMed
- Rui Y, Bai J, Perrimon N. Sarcomere formation occurs by the assembly of multiple latent protein complexes. PLOS Genetics. 2010; 6DOI | PubMed
- Sanger JW, Kang S, Siebrands CC, Freeman N, Du A, Wang J, Stout AL, Sanger JM. How to build a myofibril. Journal of Muscle Research and Cell Motility. 2005; 26:343-354. DOI | PubMed
- Shutova MS, Spessott WA, Giraudo CG, Svitkina T. Endogenous species of mammalian nonmuscle myosin IIA and IIB include activated monomers and heteropolymers. Current Biology. 2014; 24:1958-1968. DOI | PubMed
- Shutova MS, Asokan SB, Talwar S, Assoian RK, Bear JE, Svitkina TM. Self-sorting of nonmuscle myosins IIA and IIB polarizes the cytoskeleton and modulates cell motility. The Journal of Cell Biology. 2017; 111DOI
- Sjöblom B, Salmazo A, Djinović-Carugo K. Alpha-actinin structure and regulation. Cellular and Molecular Life Sciences. 2008; 65:2688-2701. DOI | PubMed
- Sparrow JC, Schöck F. The initial steps of myofibril assembly: integrins pave the way. Nature Reviews Molecular Cell Biology. 2009; 10:293-298. DOI | PubMed
- Spector I, Shochet NR, Kashman Y, Groweiss A. Latrunculins: novel marine toxins that disrupt microfilament organization in cultured cells. Science. 1983; 219:493-495. DOI | PubMed
- Straight AF, Cheung A, Limouze J, Chen I, Westwood NJ, Sellers JR, Mitchison TJ. Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science. 2003; 299:1743-1747. DOI | PubMed
- Takahashi K, Tanabe K, Ohnuki M, Narita M, Ichisaka T, Tomoda K, Yamanaka S. Induction of pluripotent stem cells from adult human fibroblasts by defined factors. Cell. 2007; 131:861-872. DOI | PubMed
- Taniguchi K, Takeya R, Suetsugu S, Kan-O M, Narusawa M, Shiose A, Tominaga R, Sumimoto H. Mammalian formin fhod3 regulates actin assembly and sarcomere organization in striated muscles. Journal of Biological Chemistry. 2009; 284:29873-29881. DOI | PubMed
- Tojkander S, Gateva G, Lappalainen P. Actin stress fibers–assembly, dynamics and biological roles. Journal of Cell Science. 2012; 125:1855-1864. DOI | PubMed
- Tullio AN, Accili D, Ferrans VJ, Yu ZX, Takeda K, Grinberg A, Westphal H, Preston YA, Adelstein RS. Nonmuscle myosin II-B is required for normal development of the mouse heart. PNAS. 1997; 94:12407-12412. DOI | PubMed
- Vicente-Manzanares M, Ma X, Adelstein RS, Horwitz AR. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nature Reviews Molecular Cell Biology. 2009; 10:778-790. DOI | PubMed
- Wakatsuki T, Schwab B, Thompson NC, Elson EL. Effects of cytochalasin D and latrunculin B on mechanical properties of cells. Journal of Cell Science. 2001; 114:1025-1036. PubMed
- Wiedenmann J, Ivanchenko S, Oswald F, Schmitt F, Röcker C, Salih A, Spindler KD, Nienhaus GU. EosFP, a fluorescent marker protein with UV-inducible green-to-red fluorescence conversion. PNAS. 2004; 101:15905-15910. DOI | PubMed
Fonte
Fenix AM, Neininger AC, Taneja N, Hyde K, Visetsouk MR, et al. () Muscle-specific stress fibers give rise to sarcomeres in cardiomyocytes. eLife 7e42144. https://doi.org/10.7554/eLife.42144




