
Abstract
Introduzione
L’intricato ecosistema dei microbi intestinali, il microbiota intestinale, partecipa attivamente a diverse funzioni dell’ospite, oltre alla digestione (Shanahan, 2002; Turnbaugh et al, 2006; Backhed et al , 2007; Velagapudi et al, 2010; Reinhardt et al, 2012; Serino et al, 2012a). L’alterata proporzione e attività dei gruppi batterici dei microbioti intestinali, chiamati disbiosi, caratterizza patologie multiple (Tomasello et al, 2011; Haahtela et al , 2013; Serban , 2014), come il diabete di tipo 2 e l’obesità (Serino et al, 2009; Le Chatelier et al, 2013). Vi sono anche chiare prove che la disbiosi dei microbioti intestinali ha un impatto sul fegato promuovendo la steatosi epatica (Dumas et al, 2006; Le Roy et al, 2013), una caratteristica comune della sindrome metabolica. Abbiamo riferito che la suscettibilità alle malattie metaboliche indotte dalla dieta è caratterizzata da un particolare microbiota intestinale (Serino et al, 2012b). Da notare che il targeting dei microbioti intestinali attraverso il trattamento dietetico (Cani et al, 2007) , fibre (Serino et al, 2012b) o antibiotici (Cani et al, 2008; Membrez et al, 2008) può ripristinare l‘omeostasi del glucosio riducendo l’infiammazione metabolica (Shoelson et al, 2006).
La nostra comprensione dell’impatto dei microbioti intestinali sul metabolismo dell’ospite (Shanahan, 2002; Turnbaugh et al, 2006; Backhed et al , 2007; Reinhardt et al, 2012; Serino et al, 2012a) si basa sull ‘uso di topi assenici. Questi topi hanno permesso la scoperta di pochi meccanismi molecolari attraverso i quali il microbiota intestinale modula il metabolismo dell’ospite (Backhed et al, 2007) . È significativo che la colonizzazione di topi assenici con microbiota intestinale da modelli animali di patologia (ad esempio topi obesi; Turnbaugh et al, 2006) o feci umane (Chung et al, 2012; Atarashi et al, 2013) abbia trasferito il relativo fenotipo, suggerendo che il microbiota intestinale sia un fattore eziologico putativo di tale patologia.
La mancanza di microbiota nei topi assenici determina alterazioni sia strutturali che funzionali come l’iperpermeabilità intestinale e l’atrofia del sistema immunitario (Shanahan, 2002 ). Pertanto, abbiamo considerato se gli effetti dannosi dei microbioti intestinali disbiotici osservati nei topi assenici potrebbero essere osservati anche nei topi sani convenzionali. Per studiare il ruolo della disbiosi dei microbioti intestinali nell’eziologia delle malattie metaboliche, abbiamo inoculato topi sani e convenzionali con microbioti intestinali disbiotici provenienti da topi ob obesi e indotti dalla dieta o da topi obesi o con microbioti intestinali eubiotici provenienti da topi magri.
Abbiamo scoperto che il trasferimento di microbiota dell’intestino disbiotico ai topi convenzionali riduce acutamente i marcatori della gluconeogenesi epatica durante il pasto normale e protegge verso i marcatori ad alto contenuto di grassi della gluconeogenesi epatica e dell’adiposità, insieme ai cambiamenti sia nel microbiota che nel microbioma dell’intestino. Risultati metabolici simili sono stati ottenuti quando i topi sono stati inoculati con un microbiota intestinale disbiotico da topi ob/ob. Al contrario, il trasferimento di microbiota intestinale eubiotico ha leggermente influenzato sia la composizione dei microbiota intestinali che le relative funzioni metaboliche batteriche dei topi riceventi, che non hanno mostrato marcatori alterati di gluconeogenesi epatica su cibo normale.
I nostri risultati mostrano che il trasferimento di un microbiota intestinale disbiotico può essere vantaggioso per l’ospite, proponendo di riconsiderare il ruolo della disbiosi dei microbioti intestinali nell’eziologia delle malattie metaboliche.
Risultati
Per studiare gli effetti metabolici del trasferimento di microbiota intestinale, sono stati utilizzati topi riceventi mai precedentemente trattati con antibiotici, poiché è stato dimostrato che gli antibiotici smorzano il dismetabolismo indotto dalla disbiosi (Ellekilde et al, 2014) o addirittura limitano la formazione di microbiota esogeni (Manichanh et al, 2010).
Effetti metabolici del trasferimento di microbiota intestinale disbiotico vs. eubiotico in topi convenzionali alimentati con un normale chow (NC)
Per studiare il ruolo della disbiosi dei microbioti intestinali nell’eziologia delle malattie metaboliche, abbiamo trasferito il contenuto cecale da topi obesi ad alto contenuto di grassi indotti dalla dieta (HFD-microbiota) in topi convenzionali (Conv) alimentati con un NC (Conv +).OM (HFD); OM sta per “obese microbiota”) e abbiamo confrontato questo gruppo con topi inoculati con il veicolo (Conv + PBS) o con un microbiota eubiotico da topi magri (Conv + LM; LM sta per “lean microbiota”; Fig 1A). Sia per i topi donatori che per quelli riceventi, le caratteristiche metaboliche basali sono riportate nell’Appendice Fig S1A-E. In primo luogo, abbiamo verificato che i batteri di entrambi i trapianti erano vitali. Abbiamo trovato una quantità ridotta di batteri coltivabili nell’inoculo di topi obesi, principalmente nei batteri anaerobici(Appendice Fig S1F e G). Poiché la maggior parte dei microbi intestinali non è coltivabile, abbiamo ulteriormente quantificato il contenuto di DNA in entrambi i trapianti. Come previsto (Daniel et al, 2014) , i trapianti da topi obesi o magri erano altamente divergenti in termini di quantità e tassonomia (Appendice Fig S1H e I). Al contrario, i due trapianti dallo stesso donatore hanno mostrato una forte omogeneità dopo 1 settimana (AppendiceFig S1J e K).
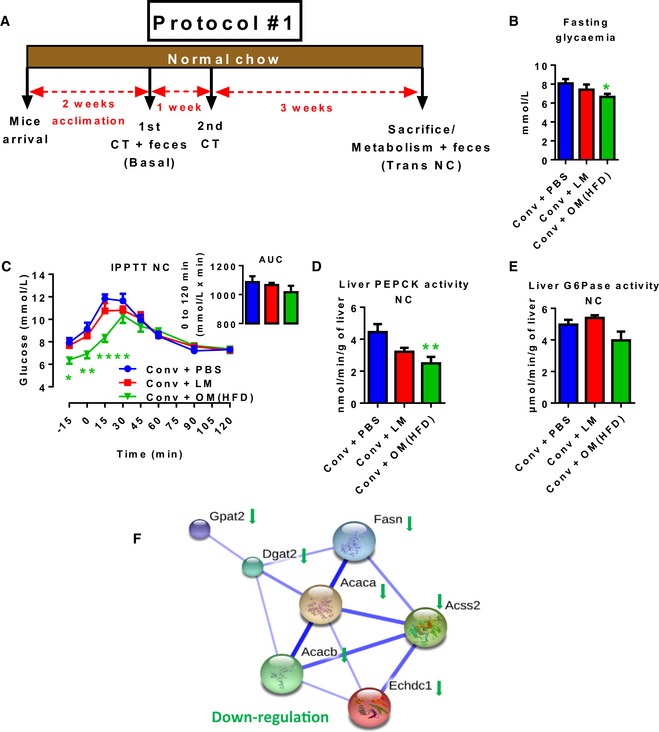
A-F.Il trasferimento del microbiota intestinale disbiotico vs. eubiotico in topi convenzionali alimentati a NC riduce la gluconeogenesi epatica
A-F(A) Timeline sperimentale:1 ° / 2 ° CT (trasferimento cecale); (B) 6 h glicemia a digiuno; (C) test di tolleranza intraperitoneale piruvato intraperitoneale e AUC come inset; epatica (D) PEPCK e (E) G6Pase attività enzimatica; (F) String analisi dei geni metabolici epatici significativamente modulata metabolica analizzata da microarray in antibiotico-topi convenzionali alimentati a NC liberi inoculati con il veicolo (PBS) o microbiota cecale da topi magri o da topi alimentati con HFD (rispettivamente Conv + PBS, Conv + LM, Conv + OM(HFD)). I dati sono indicati come media ± SEM; n = 6, *P < 0,05 ,**P < 0,01, ****P < 0.0001; t-test dello studente non accoppiato per (B), ANOVA a due vie e post-test di Sidak vs. Conv + PBS (C), ANOVA a una via e post-test di Dunnett vs. Conv + PBS (D). Basale, linea di base; Trans NC, trasferimento durante NC; Conv, convenzionale; OM, microbiota obeso; LM, microbiota magro.(A) Timeline sperimentale:1 ° / 2 ° CT (trasferimento cecale); (B) 6 h glicemia a digiuno; (C) test di tolleranza intraperitoneale piruvato intraperitoneale e AUC come inset; epatica (D) PEPCK e (E) G6Pase attività enzimatica; (F) String analisi dei geni metabolici epatici significativamente modulata metabolica analizzati da microarray in antibiotico-topi convenzionali alimentati a NC liberi inoculati con il veicolo (PBS) o microbiota cecale da topi magri o da topi alimentati con HFD (rispettivamente Conv + PBS, Conv + LM, Conv + OM(HFD)). I dati sono indicati come media ± SEM; n = 6, *P < 0,05 ,**P < 0,01, ****P < 0.0001; t-test dello studente non accoppiato per (B), ANOVA a due vie e post-test di Sidak vs. Conv + PBS (C), ANOVA a una via e post-test di Dunnett vs. Conv + PBS (D). Basale, linea di base; Trans NC, trasferimento durante NC; Conv, convenzionale; OM, microbiota obeso; LM, microbiota magro.
Per quanto riguarda l’impatto metabolico di entrambi i trapianti, i topi che ricevono il microbiota HFD-microbiota hanno mostrato una glicemia a digiuno inferiore di 6 ore rispetto ai topi di controllo (Fig 1B). La via del glucagone nel sangue non è stata influenzata in modo significativo, come dimostrato dall’analisi della fosforilazione epatica dei target di glucagone PKA, insieme a nessuna variazione del contenuto di glicogeno epatico(Appendice Fig S2A e B). Successivamente, abbiamo analizzato la gluconeogenesi epatica eseguendo un test di tolleranza al piruvato; i topi che hanno ricevuto il microbiota HFD-microbiota hanno mostrato una glicemia a digiuno significativamente più bassa e una concomitante gluconeogenesi epatica più bassa rispetto ai topi di controllo, mentre l’inoculazione con microbiota magro non ha indotto un effetto significativo (Fig 1C). Il livello proteico degli enzimi chiave della gluconeogenesi epatica PEPCK e G6Pase non è stato modificato in modo significativo(Appendice Fig S2C). Al contrario, i topi inoculati con il microbiota HFD-microbiota hanno mostrato una minore attività per la PEPCK (Fig 1D) ma non G6Pase (Fig 1E), senza alcuna variazione indotta dal microbiota magro. La minore attività per la PEPCK potrebbe offrire un meccanismo per spiegare la gluconeogenesi epatica inferiore. Inoltre, poiché l’area sotto la curva mostra un effetto HFD-microbiota non significativo (Fig 1C), anche la glicemia a digiuno rappresenta l’andamento osservato della gluconeogenesi epatica ridotta. Diversi parametri metabolici non sono stati influenzati nei topi inoculati, tra cui il peso corporeo ed epatico, il contenuto di trigliceridi epatici, l’infiammazione del fegato, il danno epatico, la tolleranza al glucosio per via orale(Appendice Fig S2D-J) o un indice di infiammazione sistemica analizzato attraverso l’enumerazione delle cellule immunitarie plasmatiche (AppendiceFig S3A-D). Questi dati mostrano che la riduzione della gluconeogenesi epatica non era dovuta a danni epatici.
Per spiegare la riduzione della glicemia a digiuno, abbiamo condotto un’analisi approfondita con microarray per cercare le variazioni complessive dell’espressione genica epatica. Della totalità dei geni significativamente(P< 0,05) modulato (1.021 dal HFD-microbiota contro Conv + PBS e 1.329 dal microbiota magro contro. Conv + PBS), abbiamo identificato una rete di geni metabolici epatici la cui espressione è stata ridotta dal HFD-microbiota (Fig 1F) e coinvolta nella lipogenesi de novo ( Appendice Fig S2K). Tra i 1.021 geni modulati in modo significativo da HFD-microbiota nessuno di essi è stato direttamente implicato nella gluconeogenesi, suggerendo che la diminuzione dei marcatori della produzione di glucosio epatico osservata sopra non è dovuta ad un cambiamento nell’espressione genica.
Nei topi inoculati con l’HFD-microbiota, abbiamo anche trovato una firma metabolomica sierica del fenotipo epatico da livelli più elevati di precursori glucogenici, come lattato e piruvato (Fig 2A-C), suggerendo la riduzione della gluconeogenesi epatica mostrata nella Fig 1C.
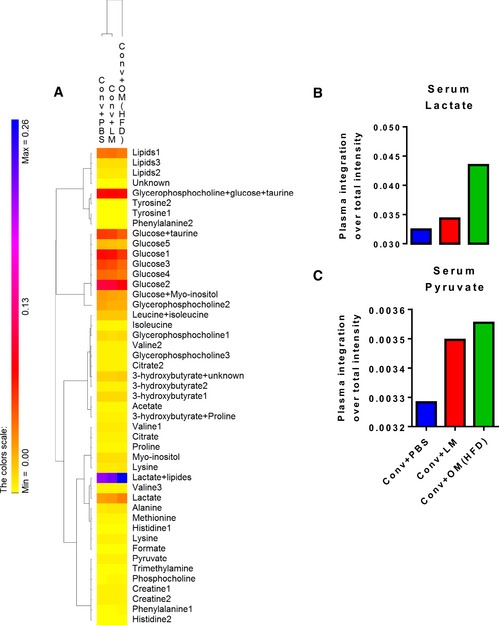
A-C.Il trasferimento del microbiota intestinale disbiotico vs. eubiotico nei topi convenzionali alimentati a NC influisce sul metabolismo sierico
A-C(A) Analisi della mappa termica del metaboloma del siero; istogrammi dettagliati per (B) lattato di siero e (C) piruvato di siero in NC-alimentato topi convenzionali inoculati con il veicolo (PBS) o microbiota cecale da topi magri o da topi alimentati con HFD (Conv + PBS, Conv + LM, Conv + OM(HFD), rispettivamente). È stato utilizzato un pool di campioni di siero per gruppo(n= 6).(A) Analisi della mappa termica del metaboloma del siero; istogrammi dettagliati per (B) lattato di siero e (C) piruvato di siero in NC-alimentato topi convenzionali inoculati con il veicolo (PBS) o microbiota cecale da topi magri o da topi alimentati con HFD (Conv + PBS, Conv + LM, Conv + OM(HFD), rispettivamente). È stato utilizzato un pool di campioni di siero per gruppo(n= 6).
Questi dati mostrano che il trasferimento di HFD-microbiota ha abbassato la glicemia a digiuno e i marcatori della gluconeogenesi epatica in associazione con una ridotta attività enzimatica gluconeogenica, senza influenzare né la segnalazione del glucagone né il contenuto di glicogeno epatico.
A-F.Il trasferimento del microbiota intestinale disbiotico vs. eubiotico nei topi convenzionali alimentati a NC riduce la gluconeogenesi epatica
A-F(A) Timeline sperimentale:1 ° / 2 ° CT (trasferimento cecale); (B) 6 h glicemia a digiuno; (C) test di tolleranza intraperitoneale piruvato intraperitoneale e AUC come inset; epatica (D) PEPCK e (E) G6Pase attività enzimatica; (F) String analisi dei geni metabolici epatici significativamente modulata metabolica analizzata da microarray in antibiotico-topi convenzionali alimentati a NC liberi inoculati con il veicolo (PBS) o microbiota cecale da topi magri o da topi alimentati con HFD (rispettivamente Conv + PBS, Conv + LM, Conv + OM(HFD)). I dati sono indicati come media ± SEM; n = 6, *P < 0,05 ,**P < 0,01, ****P < 0.0001; t-test dello studente non accoppiato per (B), ANOVA a due vie e post-test di Sidak vs. Conv + PBS (C), ANOVA a una via e post-test di Dunnett vs. Conv + PBS (D). Basale, linea di base; Trans NC, trasferimento durante NC; Conv, convenzionale; OM, microbiota obeso; LM, microbiota magro.(A) Timeline sperimentale:1 ° / 2 ° CT (trasferimento cecale); (B) 6 h glicemia a digiuno; (C) test di tolleranza intraperitoneale piruvato intraperitoneale e AUC come inset; epatica (D) PEPCK e (E) G6Pase attività enzimatica; (F) String analisi dei geni metabolici epatici significativamente modulata metabolica analizzati da microarray in antibiotico-topi convenzionali alimentati a NC liberi inoculati con il veicolo (PBS) o microbiota cecale da topi magri o da topi alimentati con HFD (rispettivamente Conv + PBS, Conv + LM, Conv + OM(HFD)). I dati sono indicati come media ± SEM; n = 6, *P < 0,05 ,**P < 0,01, ****P < 0.0001; t-test dello studente non accoppiato per (B), ANOVA a due vie e post-test di Sidak vs. Conv + PBS (C), ANOVA a una via e post-test di Dunnett vs. Conv + PBS (D). Basale, linea di base; Trans NC, trasferimento durante NC; Conv, convenzionale; OM, microbiota obeso; LM, microbiota magro.
A-C.Il trasferimento di microbiota intestinale disbiotico vs. eubiotico in topi convenzionali alimentati con NC influisce sul metabolismo del siero
A-C(A) Analisi della mappa termica del metaboloma del siero; istogrammi dettagliati per (B) lattato di siero e (C) piruvato di siero in NC-alimentato topi convenzionali inoculati con il veicolo (PBS) o microbiota cecale da topi magri o da topi alimentati con HFD (Conv + PBS, Conv + LM, Conv + OM(HFD), rispettivamente). È stato utilizzato un pool di campioni di siero per gruppo(n= 6).(A) Analisi della mappa termica del metaboloma del siero; istogrammi dettagliati per (B) lattato di siero e (C) piruvato di siero in NC-alimentato topi convenzionali inoculati con il veicolo (PBS) o microbiota cecale da topi magri o da topi alimentati con HFD (Conv + PBS, Conv + LM, Conv + OM(HFD), rispettivamente). È stato utilizzato un pool di campioni di siero per gruppo(n= 6).
Analisi della barriera intestinale nei topi convenzionali alimentati con un NC e inoculati con un microbiota intestinale disbiotico o eubiotico
Per spiegare se il fenotipo epatico può dipendere da alterazioni dell’asse intestinale (Szabo et al, 2010), abbiamo analizzato la barriera intestinale. In primo luogo, né il trasferimento dei microbioti dell’intestino disbiotico né quello dell’intestino eubiotico hanno influito in modo significativo sulla permeabilità in vivo dell’intestino (Fig 3A), secondo i livelli plasmatici LPS invariati (Fig 3B). Poi, poiché abbiamo già mostrato l’impatto della disbiosi dei microbioti intestinali sull’ileo (Amar et al, 2011; Serino et al , 2012b), ci siamo concentrati su questa regione intestinale. Le cellule del calice e l’espressione del Muc-2 (il principale gene produttore di muco) sono stati utilizzati come indici di produzione di muco e non sono stati influenzati in modo significativo (Fig 3C e D). Non è stata osservata alcuna modulazione significativa dell’espressione dei geni a giunzione stretta (Claudin-2/-7, Jam-A, Occludin o ZO-1) (Fig 3E). Per quanto riguarda l’infiammazione, non è stato osservato alcun cambiamento significativo per l’espressione del gene FoxP3, IL-17a, IFNγ e NF-kB nei topi inoculati con il microbiota HFD-microbiota; solo i topi che ricevono il microbiota magro hanno mostrato un aumento significativo dell’espressione IL-17a (Fig 3F). Queste osservazioni sono molto probabilmente dovute alla maggiore quantità batterica del trapianto dai topi magri rispetto a quella dei topi obesi(Appendice Fig S1F-I). Questi dati sono stati in accordo con la produzione di difese invariata di entrambi i gruppi di topi inoculati (Fig 3G). Nel complesso, nessuno dei trasferimenti ha influenzato in modo significativo l’architettura generale dell’ileo (Fig 3H).

A-H.Impatto intestinale del trasferimento del microbiota intestinale disbiotico vs. eubiotico nei topi convenzionali alimentati a NC
A-H(A) permeabilità intestinale in vivo; (B) siero LPS; (C) conteggio delle cellule del calice ileum/villo; espressione del gene dell’ileo per (D) Muc-2, (E) proteine a giunzione stretta, (F) marcatori infiammatori, (G) difensori e (H) istologia dell’ileo in antibiotico-topi convenzionali alimentati a NC liberi inoculati con il veicolo (PBS) o microbiota cecale da topi magri o da topi alimentati con HFD (Conv + PBS, Conv + LM, Conv + OM(HFD), rispettivamente). I dati sono indicati come media ± SEM; n = 6, ****P < 0 ,0001; ANOVA bidirezionale e post-test di Dunnett vs. Conv + PBS (F).(A) Permeabilità intestinale in vivo; (B) siero LPS; (C) conta delle cellule dell’ileo in calice; espressione del gene dell’ileo per (D) Muc-2, (E) proteine a giunzione stretta, (F) marcatori infiammatori, (G) difensori e (H) istologia dell’ileo in antibiotico-topi convenzionali alimentati a NC liberi inoculati con il veicolo (PBS) o microbiota cecale da topi magri o da topi alimentati con HFD (Conv + PBS, Conv + LM, Conv + OM(HFD), rispettivamente). I dati sono indicati come media ± SEM; n = 6, ****P < 0 ,0001; ANOVA bidirezionale e post-test di Dunnett vs. Conv + PBS (F).
Questi dati mostrano che il trasferimento di microbiota né HFD- né magro gioca un ruolo importante nella modulazione dell’infiammazione intestinale, sistemica ed epatica, escludendo il coinvolgimento di questi processi nella modulazione della gluconeogenesi epatica.
A-H.Impatto intestinale del trasferimento di microbiota intestinale disbiotico vs. eubiotico nei topi convenzionali alimentati a NC
A-H(A) permeabilità intestinale in vivo; (B) siero LPS; (C) conteggio delle cellule del calice ileum/villo; espressione del gene dell’ileo per (D) Muc-2, (E) proteine a giunzione stretta, (F) marcatori infiammatori, (G) difensori e (H) istologia dell’ileo in antibiotico-topi convenzionali alimentati a NC liberi inoculati con il veicolo (PBS) o microbiota cecale da topi magri o da topi alimentati con HFD (Conv + PBS, Conv + LM, Conv + OM(HFD), rispettivamente). I dati sono indicati come media ± SEM; n = 6, ****P < 0 ,0001; ANOVA bidirezionale e post-test di Dunnett vs. Conv + PBS (F).(A) Permeabilità intestinale in vivo; (B) siero LPS; (C) conta delle cellule dell’ileo in calice; espressione del gene dell’ileo per (D) Muc-2, (E) proteine a giunzione stretta, (F) marcatori infiammatori, (G) difensori e (H) istologia dell’ileo in antibiotico-topi convenzionali alimentati a NC liberi inoculati con il veicolo (PBS) o microbiota cecale da topi magri o da topi alimentati con HFD (Conv + PBS, Conv + LM, Conv + OM(HFD), rispettivamente). I dati sono indicati come media ± SEM; n = 6, ****P < 0 ,0001; ANOVA bidirezionale e post-test di Dunnett vs. Conv + PBS (F).
Analisi del microbiota intestinale e del microbioma in topi convenzionali alimentati con un NC e inoculati con un microbiota intestinale disbiotico o eubiotico
Alla luce del fenotipo epatico osservato sopra e del ruolo della disbiosi dei microbioti intestinali sul fegato (Dumas et al, 2006; Le Roy et al , 2013), abbiamo indagato i cambiamenti presunti dei microbioti intestinali dei topi riceventi indotti dal trasferimento. Abbiamo analizzato i microbiota delle feci sia a livello tassonomico che a livello funzionale correlato prima (Basal) e dopo il trasferimento (Trans NC).
Al basale (Basal), il microbiota dei diversi gruppi ha mostrato un certo grado di divergenza, soprattutto per il gruppo da inoculare con il microbiota HFD-microbiota, come mostrato dall’analisi delle coordinate principali (PCoA) (Fig 4A, pannello superiore). Infatti, i topi designati ad appartenere al gruppo di controllo hanno mostrato una maggiore quantità di ordine Parabacteroides; i topi da inoculare con il microbiota eubiotico hanno mostrato una maggiore quantità di Firmicutes e i topi da inoculare con il microbiota disbiotico hanno mostrato una maggiore quantità di Bacteroidetes (Fig 4A, pannello inferiore). Fondamentalmente, i cambiamenti metagenomici osservati al basale non hanno avuto un impatto sulla produzione di glucosio epatico basale(Appendice Fig S1C). Pertanto, è probabile che la gluconeogenesi epatica ridotta possa essere attribuita al trasferimento di microbiota intestinale.

A, B.Effetti del trasferimento di microbiota intestinale disbiotico ed eubiotico a topi convenzionali alimentati a NC sul microbiota intestinale
A, B(A) Analisi delle coordinate principali (PCoA) per il microbiota intestinale al basale (Basale) (pannello superiore) e relativo cladogramma che mostra il taxa batterico significativamente arricchito in ogni gruppo (pannello inferiore); (B) PCoA per il microbiota intestinale dopo il trasferimento su NC (Trans NC) (pannello superiore) e relativo cladogramma (pannello inferiore) in antibiotico-topi convenzionali alimentati a NC liberi inoculati con il veicolo o il microbiota cecale da topi magri o da topi alimentati con HFD (rispettivamente Conv + PBS, Conv + LM, Conv + OM(HFD)) (n = 6).(A) Analisi delle coordinate principali (PCoA) per il microbiota intestinale al basale (Basale) (pannello superiore) e relativo cladogramma che mostra il taxa batterico significativamente arricchito in ogni gruppo (pannello inferiore); (B) PCoA per il microbiota intestinale dopo il trasferimento su NC (Trans NC) (pannello superiore) e relativo cladogramma (pannello inferiore) in antibiotico-topi convenzionali alimentati a NC liberi inoculati con il veicolo o il microbiota cecale da topi magri o da topi alimentati con HFD (rispettivamente Conv + PBS, Conv + LM, Conv + OM(HFD)) (n = 6).
Dopo l’inoculazione, i tre profili dei tre microbioti intestinali presentavano alcune sovrapposizioni (Fig 4B, pannello superiore). Tuttavia, l’Actinobacteria taxon era significativamente più elevato nei topi che ricevevano il microbiota eubiotico, mentre i topi inoculati con microbiota disbiotico mostravano una maggiore quantità di Firmicuti (Fig 4B, pannello inferiore; l’elenco completo dei cladogrammi nella Fig 4 è riportato nell’Appendice Fig S4).
Per quanto riguarda il microbioma analizzato dal PICRUSt (Langille et al, 2013) , al basale (Basal), i topi hanno mostrato alcune divergenze funzionali (Fig 5A-C),inaccordo con i profili dei microbioti intestinali sopra riportati (Fig 4A). Dopo l’inoculazione, il microbiota disbiotico ha modificato il microbioma intestinale dei topi riceventi in modo diverso da quello del microbiota eubiotico, come riportato dall’analisi PCA rispetto ai topi di controllo (Fig 5D). Infatti, i topi inoculati con microbiota HFD-microbiota o con microbiota magro non condividevano alcuna via microbica (Fig 5E e F).
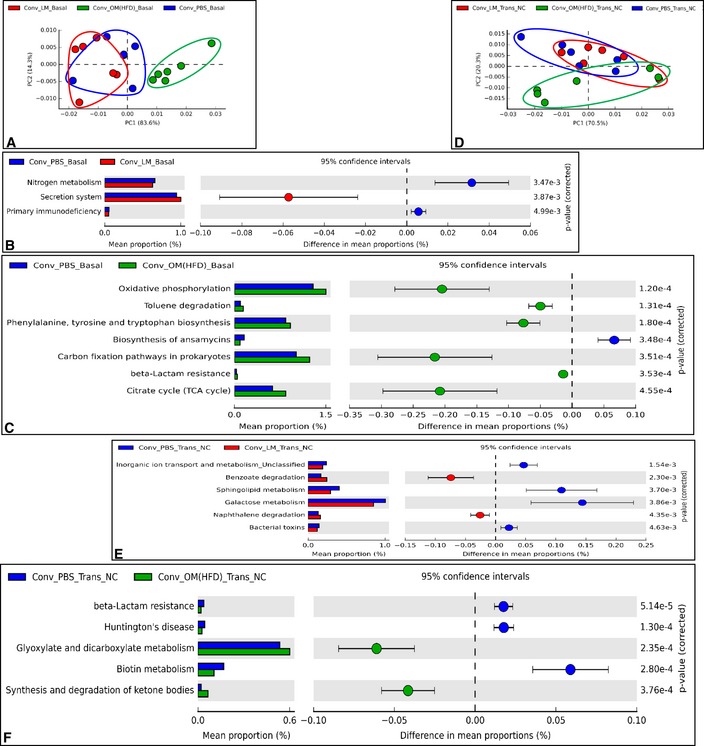
A-F.Effetti del trasferimento di microbiota intestinale disbiotico ed eubiotico a topi convenzionali alimentati a NC sul microbioma intestinale
Analisi dei componenti A-FPrincipal che mostra lo studio del microbioma intestinale basato sul PICRUSt-based gut a basale (Basal) (A) e dopo il trasferimento su NC (Trans NC) (D) e top modulato (basato sul t-test bifacciale di Welch) percorsi microbici in una coppia-saggio confronto (B, C, E, F) in topi convenzionali alimentati senza antibiotici NC inoculati con il veicolo o microbiota cecale da topi magri o da topi alimentati con HFD (rispettivamente Conv + PBS, Conv + LM, Conv + OM(HFD)) (n = 6).Analisi dei componenti principali che mostrano lo studio del microbioma intestinale basato su PICRUSt-based gut a basale (Basal) (A) e dopo il trasferimento su NC (Trans NC) (D) e top modulato (basato sul t-test bifacciale di Welch ) percorsi microbici in una coppia-saggio confronto (B, C, E, F) in topi convenzionali alimentati senza antibiotici NC inoculati con il veicolo o microbiota cecale da topi magri o da topi alimentati con HFD (rispettivamente Conv + PBS, Conv + LM, Conv + OM(HFD)) (n = 6).
Per valutare l’impatto netto del trasferimento microbico sul profilo metagenomico dei topi riceventi, abbiamo eseguito un’analisi intragruppo sia per il microbiota intestinale che per il microbioma. Il microbiota intestinale dei topi di controllo ha mostrato alcune divergenze rispetto alla linea di base(Appendice Fig S5A). Nei topi inoculati, le modifiche indotte dal microbiota magro erano simili a quelle indotte dal PBS, ad eccezione della modulazione del phylum Tenericutes(Appendice Fig S5B, pannello inferiore). Al contrario, le modifiche indotte dal microbiota HFD-microbiota erano più distinte dagli altri due gruppi (AppendiceFig S5C, pannello inferiore). Questa evidenza suggerisce che il trasferimento di microbiota HFD-microbiota ha modificato il microbiota intestinale dei topi riceventi in misura maggiore rispetto ai microbiota magri. Questo dato è in forte accordo con la maggiore modulazione metabolica indotta dal microbiota HFD-microbiota durante tutto lo studio.
L’analisi microbicrobica intragruppo rifletteva i cambiamenti sopra citati nei microbioti intestinali, con il trasferimento dei microbioti HFD-microbiota che mostra il maggiore impatto (AppendiceFig S5D-I); il gruppo di controllo e i topi inoculati con il microbiota magro hanno mostrato ciascuno una certa sovrapposizione (Appendice FigS5D e F) con solo tre vie microbiche significativamente influenzate (Appendice Fig S5Ee G). Al contrario, i topi inoculati con il microbiota HFD-microbiota hanno mostrato una separazione distinta (AppendiceFig S5H), suggerendo il maggiore impatto del microbiota HFD-microbiota rispetto al microbiota magro. Questo risultato è anche sostenuto dal maggior numero (venti) di vie microbiche significativamente modulate dal microbiota HFD (AppendiceFig S5I) rispetto alle tre vie microbiche trovate modulate sopra (AppendiceFig S5E e G).
Questi dati mostrano che il trasferimento di microbiota HFD-microbiota in topi convenzionali alimentati a NC senza antibiotici è in grado di influenzare sia i microbiota (tassonomia) che il microbioma (funzione), in misura maggiore rispetto ai microbiota magri.
A, B.Effetti del trasferimento dei microbioti intestinali disbiotici ed eubiotici ai topi convenzionali alimentati con NC sui microbioti intestinali
A, B(A) Analisi delle coordinate principali (PCoA) per il microbiota intestinale al basale (Basale) (pannello superiore) e relativo cladogramma che mostra il taxa batterico significativamente arricchito in ogni gruppo (pannello inferiore); (B) PCoA per il microbiota intestinale dopo il trasferimento su NC (Trans NC) (pannello superiore) e relativo cladogramma (pannello inferiore) in antibiotico-topi convenzionali alimentati a NC liberi inoculati con il veicolo o il microbiota cecale da topi magri o da topi alimentati con HFD (rispettivamente Conv + PBS, Conv + LM, Conv + OM(HFD)) (n = 6).(A) Analisi delle coordinate principali (PCoA) per il microbiota intestinale al basale (Basale) (pannello superiore) e relativo cladogramma che mostra il taxa batterico significativamente arricchito in ogni gruppo (pannello inferiore); (B) PCoA per il microbiota intestinale dopo il trasferimento su NC (Trans NC) (pannello superiore) e relativo cladogramma (pannello inferiore) in antibiotico-topi convenzionali alimentati a NC liberi inoculati con il veicolo o il microbiota cecale da topi magri o da topi alimentati con HFD (rispettivamente Conv + PBS, Conv + LM, Conv + OM(HFD)) (n = 6).
A-F.Effetti del trasferimento del microbiota intestinale disbiotico ed eubiotico a topi convenzionali alimentati a NC sul microbioma intestinale
Analisi dei componenti A-FPrincipal che mostra lo studio del microbioma intestinale basato sul PICRUSt-based gut a basale (Basal) (A) e dopo il trasferimento su NC (Trans NC) (D) e top modulato (basato sul t-test bifacciale di Welch) percorsi microbici in una coppia-saggio confronto (B, C, E, F) in topi convenzionali alimentati senza antibiotici NC inoculati con il veicolo o microbiota cecale da topi magri o da topi alimentati con HFD (rispettivamente Conv + PBS, Conv + LM, Conv + OM(HFD)) (n = 6).Analisi dei componenti principali che mostrano lo studio del microbioma intestinale basato su PICRUSt-based gut a basale (Basal) (A) e dopo il trasferimento su NC (Trans NC) (D) e top modulato (basato sul t-test bifacciale di Welch ) percorsi microbici in una coppia-saggio confronto (B, C, E, F) in topi convenzionali alimentati senza antibiotici NC inoculati con il veicolo o microbiota cecale da topi magri o da topi alimentati con HFD (rispettivamente Conv + PBS, Conv + LM, Conv + OM(HFD)) (n = 6).
Il trasferimento di due diversi microbiota intestinali disbiotici nei topi convenzionali riduce i marcatori della gluconeogenesi epatica su NC e previene l’alterazione epatica e l’adiposità su 72% di HFD
Per capire se l’origine della disbiosi (cioè nutrizionale vs. genetica) possa avere un ruolo nella modulazione metabolica osservata, abbiamo inoculato un altro gruppo di topi (Conv + OM(ob)) con il microbiota intestinale di topi geneticamente obesi (ob/ob, ob-microbiota di seguito). Su NC (Fig 6A), i marcatori della gluconeogenesi epatica erano di nuovo più bassi nei topi inoculati con l’ob-microbiota rispetto ai topi di controllo, anche se non nella stessa misura osservata nei topi che ricevono l’HFD-microbiota (Appendice FigS6A). Non è stato osservato alcun cambiamento significativo per il peso corporeo, indipendentemente dal gruppo(Appendice Fig S6C e D).

A-G.Il trasferimento di microbiota intestinale disbiotico nei topi convenzionali impedisce la gluconeogenesi epatica aumentata da HFD
A-G(A) Timeline sperimentale: 1°/2°CT (trasferimento cecale); dopo l’accensione 72% HFD: (B) glicemia alimentata; (C) test di tolleranza intraperitoneale del piruvato e AUC come inset; (D) analisi Western blot del fegato per la fosforilazione dei substrati PKA, PEPCK e G6Pase, tutti normalizzati su β-actina (controllo del carico, viene mostrato un singolo topo per corsia) e relativi istogrammi (E); attività enzimatica epatica (F) PEPCK e (G) G6Pase in topi convenzionali alimentati a NC senza antibiotici inoculati con il veicolo o microbiota cecale di C57Bl/6 ob/ob o topi alimentati con HFD (Conv + PBS, + OM(ob), + OM(HFD), rispettivamente). I dati sono indicati come media ± SEM; n = 5-6, *P < 0,05, **P < 0,01, ***P < 0,001, ****P < 0.0001; t-test dello studente non accoppiato vs. Conv + PBS (per B, C insetto C, E-G) e ANOVA bidirezionale con il post-test di Dunnett vs. Conv + PBS (C).(A) Timeline sperimentale:1°/2° CT (trasferimento cecale); dopo l’accensione 72% HFD: (B) glicemia alimentata; (C) test di tolleranza intraperitoneale del piruvato e AUC come inset; (D) analisi Western blot del fegato per la fosforilazione dei substrati PKA, PEPCK e G6Pase, tutti normalizzati su β-actina (controllo del carico, viene mostrato un singolo topo per corsia) e relativi istogrammi (E); attività enzimatica epatica (F) PEPCK e (G) G6Pase in topi convenzionali alimentati a NC senza antibiotici inoculati con il veicolo o microbiota cecale di C57Bl/6 ob/ob o topi alimentati con HFD (Conv + PBS, + OM(ob), + OM(HFD), rispettivamente). I dati sono indicati come media ± SEM; n = 5-6, *P < 0,05, **P < 0,01, ***P < 0,001, ****P < 0.0001; t-test dello studente non accoppiato vs. Conv + PBS (per B, C insetto C, E-G) e ANOVA bidirezionale con il post-test di Dunnett vs. Conv + PBS (C).
I dati di partenza sono disponibili online per questa cifra.
Un sottogruppo di topi inoculati è stato poi tenuto su NC (segnalato come “Normal chow long term”), e un altro sottogruppo è stato alimentato con un 72% di HFD (Fig 6A). La scelta di questa particolare dieta (Branchereau et al, 2016; Blasco-Baque et al, 2017) si è basata sul suo bassissimo livello di carboidrati (< 1%). Pertanto, in questo modello, la glicemia riflette la gluconeogenesi epatica. Nel gruppo di topi tenuti su NC, la gluconeogenesi epatica ridotta indotta dall’inoculazione non è stata più osservata(Appendice Fig S6N), suggerendo un impatto metabolico acuto.
Sul 72% HFD, la glicemia alimentata era più bassa nei topi inoculati con HFD-microbiota rispetto ai topi di controllo (Fig 6B). Questo dato non è stato associato ad un cambiamento nell’insulinemia alimentata(Appendice Fig S6B). Inoltre, i topi inoculati con il HFD-microbiota hanno mostrato ancora una volta una glicemia a digiuno più bassa e una gluconeogenesi epatica più bassa rispetto ai topi di controllo. Si noti anche che una gluconeogenesi epatica più bassa, ma non una glicemia a digiuno più bassa significativa, è stata osservata nel 72% dei topi alimentati con HFD inoculati con l’ob-microbiota (Fig 6C). Anche in questo caso, non abbiamo osservato cambiamenti significativi nella fosforilazione dei substrati PKA, mentre abbiamo trovato una significativa riduzione sia nella quantità che nell’attività di PEPCK e G6Pase (Fig 6DeG). Anche in questo caso, la modulazione dell’attività di questi enzimi gluconeogenici chiave insieme ad un cambiamento nella loro quantità di proteine forniscono un meccanismo per spiegare e corroborare la modulazione osservata della produzione di glucosio epatico, escludendo il mero impatto della riduzione della glicemia a digiuno. Né l’architettura epatica né il peso del fegato, né i trigliceridi epatici e i livelli plasmatici delle transaminasi sono stati significativamente influenzati(Appendice Fig S6E-I). Al contrario, su NC, abbiamo osservato un piccolo miglioramento della tolleranza al glucosio e un significativo miglioramento della tolleranza all’insulina(Appendice Fig S6J e K). La migliore tolleranza al glucosio, ma non all’insulina, è stata mantenuta sul 72% HFD(Appendice Fig S6L e M).
Abbiamo recentemente dimostrato che il sensore microbico NOD2 media l’insorgenza di malattie metaboliche nei topi (Denou et al, 2015) ; pertanto, per valutare se questo recettore può essere coinvolto nella regolazione del fenotipo epatico osservato, abbiamo inoculato topi convenzionali NOD2 KO. La mancanza del sensore microbico NOD2 ha attenuato la riduzione della gluconeogenesi epatica indotta nei topi WT dal trasferimento con HFD- e ob-microbiota (AppendiceFig S6O).
Poiché uno studio precedente aveva riportato un aumento dell’adiposità nel 72% dei topi alimentati con HFD (Serino et al, 2012b), abbiamo analizzato l’effetto del trasferimento dei microbioti intestinali nel tessuto adiposo bianco (WAT). Nonostante la mancanza di un cambiamento significativo nel peso corporeo, nel grasso e nella massa magra(Appendice Fig S7A-C), i topi inoculati con il microbiota HFD-microbiota hanno mostrato adipociti significativamente più piccoli (Appendice FigS7D) rispetto ai topi di controllo. Inoltre, questi topi hanno mostrato livelli plasmatici di acidi grassi liberi (FFA) significativamente più alti (AppendiceFig S7E) rispetto ai topi di controllo, mentre l’inoculazione non ha influenzato significativamente i livelli plasmatici di colesterolo totale, trigliceridi, lipoproteine HDL e LDL (Appendice Fig S7F-I). Al contrario, l’ob-microbiota non ha avuto un impatto significativo su questi parametri (Appendice Fig S7A-I). Questo risultato suggerisce che una disbiosi genetica vs. nutrizionale del microbiota intestinale può avere un impatto metabolico divergente sull’ACQUA.
Abbiamo anche indagato se le diverse origini dei microbioti intestinali disbiotici possono influenzare l’infiammazione intestinale e la permeabilità. L’inoculazione con HFD-microbiota ha indotto un aumento significativo dell’ileo di espressione dei geni iNOS, IFNγ e IL-6 e una tendenza ad aumentare la maggior parte dei marcatori infiammatori analizzati, anche nei linfonodi mesenterici, mentre l’ob-microbiota non ha influenzato significativamente questi parametri (Appendice Fig S8Ae B). Né il microbiota intestinale disbiotico né quello disbiotico hanno modificato in modo significativo l’espressione delle proteine della giunzione stretta, mentre entrambi i trasferimenti hanno indotto una tendenza ad aumentare la produzione di difese(Appendice Fig S8C e D).
Complessivamente, questi dati mostrano che i topi inoculati con HFD- o ob-microbiota avevano una gluconeogenesi epatica inferiore sia a seguito di dieta acuta NC e il 72% HFD, più piccole dimensioni delle cellule WAT e una minore infiammazione intestinale con nessun cambiamento nella permeabilità intestinale.
A-G.Il trasferimento di microbiota intestinale disbiotico nei topi convenzionali previene la gluconeogenesi epatica aumentata da HFD
A-G(A) Timeline sperimentale: 1°/2°CT (trasferimento cecale); dopo l’accensione 72% HFD: (B) glicemia alimentata; (C) test di tolleranza intraperitoneale del piruvato e AUC come inset; (D) analisi Western blot del fegato per la fosforilazione dei substrati PKA, PEPCK e G6Pase, tutti normalizzati su β-actina (controllo del carico, viene mostrato un singolo topo per corsia) e relativi istogrammi (E); attività enzimatica epatica (F) PEPCK e (G) G6Pase in topi convenzionali alimentati a NC senza antibiotici inoculati con il veicolo o microbiota cecale di C57Bl/6 ob/ob o topi alimentati con HFD (Conv + PBS, + OM(ob), + OM(HFD), rispettivamente). I dati sono indicati come media ± SEM; n = 5-6, *P < 0,05, **P < 0,01, ***P < 0,001, ****P < 0.0001; t-test dello studente non accoppiato vs. Conv + PBS (per B, C insetto C, E-G) e ANOVA bidirezionale con il post-test di Dunnett vs. Conv + PBS (C).(A) Timeline sperimentale:1°/2° CT (trasferimento cecale); dopo l’accensione 72% HFD: (B) glicemia alimentata; (C) test di tolleranza intraperitoneale del piruvato e AUC come inset; (D) analisi Western blot del fegato per la fosforilazione dei substrati PKA, PEPCK e G6Pase, tutti normalizzati su β-actina (controllo del carico, viene mostrato un singolo topo per corsia) e relativi istogrammi (E); attività enzimatica epatica (F) PEPCK e (G) G6Pase in topi convenzionali alimentati a NC senza antibiotici inoculati con il veicolo o microbiota cecale di C57Bl/6 ob/ob o topi alimentati con HFD (Conv + PBS, + OM(ob), + OM(HFD), rispettivamente). I dati sono indicati come media ± SEM; n = 5-6, *P < 0,05, **P < 0,01, ***P < 0,001, ****P < 0.0001; t-test dello studente non accoppiato vs. Conv + PBS (per B, C insetto C, E-G) e ANOVA bidirezionale con il post-test di Dunnett vs. Conv + PBS (C).
I dati di partenza sono disponibili online per questa cifra.
Analisi del microbiota intestinale e del microbioma in topi convenzionali inoculati con un microbiota intestinale disbiotico e alimentati con un NC e un HFD al 72%.
Abbiamo analizzato sia i taxa che le relative funzioni metaboliche tre volte: prima dell’inoculazione (Basal); dopo l’inoculazione mentre i topi riceventi erano su NC (Trans NC) e dopo che i topi inoculati sono stati alimentati con un 72% di HFD (Trans 72% HFD).
Al basale (Basal), i topi hanno mostrato nuovamente alcune divergenze nella tassonomia microbica, come riportato da PCoA (Fig 7A, pannello superiore). Infatti, i topi designati ad appartenere al gruppo di controllo (blu) presentavano una quantità maggiore di Batteroidi e di generi Clostridium; i topi designati a ricevere l’ob-microbiota (verde) avevano una quantità maggiore di Proteobatteri e Batteroidi ; i topi designati a ricevere l’HFD-microbiota (rosso) avevano una quantità maggiore di Firmicutes ( Fig 7A, pannello inferiore). Ancora una volta, le differenze metagenomiche osservate al basale non hanno influito sulla produzione di glucosio epatico basale(Appendice Fig S1C).

A-C.Il trasferimento di microbiota intestinale disbiotico nei topi convenzionali cambia il microbiota intestinale a seconda dell’origine della disbiosi sia sul NC che sul 72% di HFD
A-C(A) Analisi delle coordinate principali (PCoA) per il microbiota intestinale al basale (Basale) (pannello superiore) e relativo cladogramma che mostra i taxa batterici significativamente arricchiti in ogni gruppo (pannello inferiore); (B) PCoA per il microbiota intestinale dopo il trasferimento su NC (Trans NC) (pannello superiore) e relativo cladogramma (pannello inferiore); (C) PCoA per microbiota intestinale dopo trasferimento su 72% HFD (Trans 72% HFD) (pannello superiore) e relativo cladogramma (pannello inferiore) in NC-alimentato topi convenzionali inoculati con il veicolo o il microbiota cecale di C57Bl/6 ob/ob o topi alimentati con HFD (Conv + PBS, + OM(ob), + OM(HFD), rispettivamente) e poi alimentati con un 72% di HFD (n = 5-6).(A) Analisi delle coordinate principali (PCoA) per il microbiota intestinale al basale (Basal) (pannello superiore) e relativo cladogramma che mostra il taxa batterico significativamente arricchito in ogni gruppo (pannello inferiore); (B) PCoA per il microbiota intestinale dopo il trasferimento su NC (Trans NC) (pannello superiore) e relativo cladogramma (pannello inferiore); (C) PCoA per microbiota intestinale dopo trasferimento su 72% HFD (Trans 72% HFD) (pannello superiore) e relativo cladogramma (pannello inferiore) in NC-alimentato topi convenzionali inoculati con il veicolo o il microbiota cecale di C57Bl/6 ob/ob o topi alimentati con HFD (Conv + PBS, + OM(ob), + OM(HFD), rispettivamente) e poi alimentati con un 72% di HFD (n = 5-6).
Dopo il trasferimento, i topi inoculati con il microbiota HFD-microbiota e alimentati con un NC mostravano un microbiota intestinale profondamente diverso dai topi di controllo (Fig 7B, in verde nel pannello superiore e rosso in quello inferiore). Al contrario, il microbiota intestinale dei topi inoculati con l’ob-microbiota era quasi simile a quello dei topi di controllo (Fig 7B, inrossonel pannello superiore e verde in quello inferiore). Tuttavia, i topi inoculati con l’ob-microbiota avevano Actinobatteri più alti , mentre i topi di controllo mostravano classi di Bacilli e Batteroidia più alte (Fig 7B, pannello inferiore).
Quando i topi inoculati sono stati alimentati con un 72% di HFD, il microbiota intestinale di entrambi i gruppi inoculati è apparso più simile a quello del gruppo di controllo (Fig 7C, pannello superiore). Questo dato è in accordo con la forte capacità della dieta di influenzare il microbiota intestinale (Carmody et al, 2015) . Tuttavia, alcuni taxa batterici erano ancora significativamente diversi in ogni gruppo di topi inoculati (Fig 7C, pannello inferiore; L’elenco completo dei cladogrammi nella Fig 7 è riportato nell’Appendice Fig S9).
Per quanto riguarda il microbioma, alla linea di base (Basale), i topi hanno mostrato un alto grado di sovrapposizione, ad eccezione di due topi (Fig 8A), anche se alcuni percorsi microbici sono stati trovati significativamente modulati (Fig 8B e C). Dopo il trasferimento, su NC, la separazione dei tre profili microbici intestinali (Fig 8D) è stata simile alla separazione dei profili microbici intestinali (Fig 7B). Si noti che le vie microbiche significativamente modulate rispetto ai topi di controllo sono state identificate solo nei topi inoculati con il microbiota HFD (Fig 8E).
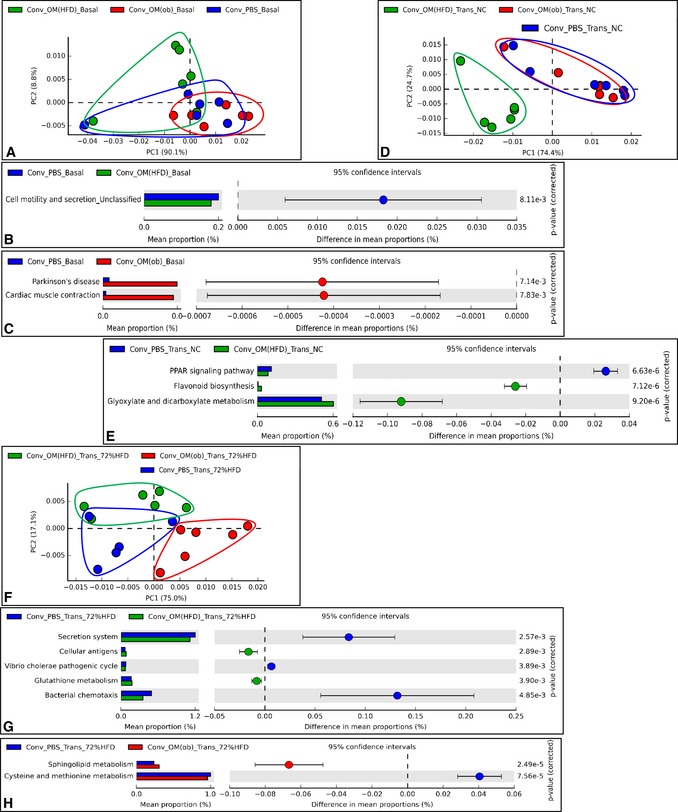
A-H.Il trasferimento di microbiota intestinale disbiotico nei topi convenzionali cambia il microbiota intestinale e il microbioma secondo l’origine della disbiosi sia sul NC che sul 72% di HFD
Analisi del componente A-HPrincipal che mostra lo studio del microbioma intestinale basato sul PICRUSt-based gut a basale (Basal) (A), dopo il trasferimento su NC (Trans NC) (D) e su 72% HFD (Trans 72% HFD) (F) e top modulato (basato sul t-test di Welch su due lati) percorsi microbici in una coppia-saggio confronto (B, C, E, G, H) in topi convenzionali alimentati a NC senza antibiotici inoculati con il veicolo o microbiota cecale di C57Bl/6 ob/ob o topi alimentati con HFD (Conv + PBS, + OM(ob), + OM(HFD), rispettivamente) e poi alimentati con un 72% di HFD (n = 5-6).Analisi dei componenti principali che mostrano lo studio del microbioma intestinale basato su PICRUSt-based gut a basale (Basal) (A), dopo il trasferimento su NC (Trans NC) (D) e su 72% HFD (Trans 72% HFD) (F) e top modulato (basato sui due lati del t-test di Welch ) percorsi microbici in una coppia-saggio confronto (B, C, E, G, H) in topi convenzionali alimentati a NC senza antibiotici inoculati con il veicolo o microbiota cecale di C57Bl/6 ob/ob o topi alimentati con HFD (Conv + PBS, + OM(ob), + OM(HFD), rispettivamente) e poi alimentati con un 72% di HFD (n = 5-6).
Abbiamo osservato un cambiamento nei marcatori della gluconeogenesi epatica in entrambi i protocolli (#1 e #2), che ci ha spinto a cercare una via microbica comune associata a questo fenotipo epatico confrontando le due analisi microbiche. La via microbica gliossilato e dicarbossilato è stata l’unica che è stata trovata ad essere significativamente modulata in entrambi i protocolli e aumentata dal HFD-microbiota (figg. 5F e 8E). Questa via microbica ha mostrato una forte correlazione negativa e significativa con l’area IPPTT sotto la curva (AUC)(Appendice Fig S10), suggerendo un ruolo putativo per il metabolismo microbico di gliossilato e dicarbossilato nella modulazione della produzione di glucosio epatico.
Per quanto riguarda l’analisi microbica di topi inoculati una volta alimentati con un 72% di HFD, i topi inoculati con l’ob-microbiota hanno mostrato alcune divergenze rispetto ai topi di controllo (Fig 8F). Tuttavia, in entrambi i gruppi di topi inoculati, abbiamo potuto identificare alcune vie microbiche significativamente modulate (Fig 8GeH).
Per valutare l’impatto netto del trasferimento sul profilo metagenomico dei topi riceventi, abbiamo eseguito un’analisi intragruppo sia per il microbiota intestinale che per il microbioma. Il microbiota intestinale dei topi di controllo ha mostrato alcune differenze rispetto alla linea di base(Appendice Fig S11A). Entrambi i trasferimenti hanno cambiato il microbiota intestinale dei topi riceventi(Appendice Fig S11B e C) come osservato anche per il microbioma (Appendice Fig S11D-I). Inoltre, il microbiota HFD-microbiota ha interessato un numero maggiore (dieci) di vie microbiche (AppendiceFig S11I) rispetto al microbiota ob (uno) (Appendice FigS11G). Abbiamo anche eseguito un’analisi intragruppo sia per i microbioti intestinali che per il microbioma confrontando il NC e il 72% degli stati nutrizionali HFD. Per quanto riguarda il microbiota intestinale, questa analisi ha mostrato che l’impatto del 72% di HFD era importante in tutti i gruppi, ma in misura maggiore nei topi inoculati con il HFD-microbiota (Appendice Fig S12A-C). In termini di microbioma intestinale, i percorsi microbici modulati erano altamente specifici per ogni gruppo. Il percorso microbico gliossilato e dicarbossilato è stato aumentato solo nei topi inoculati con HFD-microbiota quando alimentati con un NC. Questo suggerisce che questa via non spiegherebbe totalmente la riduzione della gluconeogenesi ancora osservata una volta che i topi inoculati sono stati alimentati con un 72% di HFD.
Questi dati mostrano l’impatto divergente dei due microbioti intestinali disbiotici su entrambi i microbioti intestinali e sul microbiota e sul microbiota del 72% di HFD.
A-C.Il trasferimento di microbiota intestinale disbiotico nei topi convenzionali cambia il microbiota intestinale a seconda dell’origine della disbiosi sia sul NC che sul 72% di HFD.
A-C(A) Analisi delle coordinate principali (PCoA) per il microbiota intestinale al basale (Basale) (pannello superiore) e relativo cladogramma che mostra i taxa batterici significativamente arricchiti in ogni gruppo (pannello inferiore); (B) PCoA per il microbiota intestinale dopo il trasferimento su NC (Trans NC) (pannello superiore) e relativo cladogramma (pannello inferiore); (C) PCoA per microbiota intestinale dopo trasferimento su 72% HFD (Trans 72% HFD) (pannello superiore) e relativo cladogramma (pannello inferiore) in NC-alimentato topi convenzionali inoculati con il veicolo o il microbiota cecale di C57Bl/6 ob/ob o topi alimentati con HFD (Conv + PBS, + OM(ob), + OM(HFD), rispettivamente) e poi alimentati con un 72% di HFD (n = 5-6).(A) Analisi delle coordinate principali (PCoA) per il microbiota intestinale al basale (Basal) (pannello superiore) e relativo cladogramma che mostra il taxa batterico significativamente arricchito in ogni gruppo (pannello inferiore); (B) PCoA per il microbiota intestinale dopo il trasferimento su NC (Trans NC) (pannello superiore) e relativo cladogramma (pannello inferiore); (C) PCoA per microbiota intestinale dopo trasferimento su 72% HFD (Trans 72% HFD) (pannello superiore) e relativo cladogramma (pannello inferiore) in NC-alimentato topi convenzionali inoculati con il veicolo o il microbiota cecale di C57Bl/6 ob/ob o topi alimentati con HFD (Conv + PBS, + OM(ob), + OM(HFD), rispettivamente) e poi alimentati con un 72% di HFD (n = 5-6).
A-H.Il trasferimento del microbiota intestinale disbiotico nei topi convenzionali cambia il microbiota intestinale e il microbioma in base all’origine della disbiosi sia sul NC che sul 72% di HFD
Analisi del componente A-HPrincipal che mostra lo studio del microbioma intestinale basato sul PICRUSt-based gut a basale (Basal) (A), dopo il trasferimento su NC (Trans NC) (D) e su 72% HFD (Trans 72% HFD) (F) e top modulato (basato sul t-test di Welch su due lati) percorsi microbici in una coppia-saggio confronto (B, C, E, G, H) in topi convenzionali alimentati a NC senza antibiotici inoculati con il veicolo o microbiota cecale di C57Bl/6 ob/ob o topi alimentati con HFD (Conv + PBS, + OM(ob), + OM(HFD), rispettivamente) e poi alimentati con un 72% di HFD (n = 5-6).Analisi dei componenti principali che mostrano lo studio del microbioma intestinale basato su PICRUSt-based gut a basale (Basal) (A), dopo il trasferimento su NC (Trans NC) (D) e su 72% HFD (Trans 72% HFD) (F) e top modulato (basato sui due lati del t-test di Welch ) percorsi microbici in una coppia-saggio confronto (B, C, E, G, H) in topi convenzionali alimentati a NC senza antibiotici inoculati con il veicolo o microbiota cecale di C57Bl/6 ob/ob o topi alimentati con HFD (Conv + PBS, + OM(ob), + OM(HFD), rispettivamente) e poi alimentati con un 72% di HFD (n = 5-6).
Discussione
In questo studio, riportiamo che i topi convenzionali privi di antibiotici inoculati con un microbiota intestinale disbiotico da topi ob obesi indotti da HFD o ob/ob obesi mostrano inaspettatamente una gluconeogenesi epatica inferiore acuta su NC e una protezione dal 72% di gluconeogenesi epatica aumentata da HFD e adiposità. Questi tratti fenotipici sono stati associati a cambiamenti sia nel microbiota intestinale che nel microbioma. I topi inoculati con HFD-microbiota hanno mostrato in entrambi i protocolli ridotto (i) glicemia a digiuno e (ii) marcatori della gluconeogenesi epatica, e più alto (iii) Firmicutes e (iv) percorso microbico gliossilato e dicarbossilato su NC. Al contrario, il trasferimento di microbiota intestinale da topi magri non ha influito sul metabolismo epatico, nonostante alcuni cambiamenti sia nel microbiota intestinale che nel microbioma. Infatti, la modulazione dei marcatori della gluconeogenesi epatica era in accordo con la diminuzione della quantità e/o dell’attività degli enzimi gluconeogenici chiave, a seconda della dieta dei topi riceventi. L’HFD al 72% può spiegare le discrepanze osservate per la regolazione della PEPCK durante il trasferimento disbiotico, principalmente influenzando il microbiota intestinale del ricevente, con conseguenti effetti sistemici.
I topi inoculati con HFD-microbiota hanno mostrato livelli plasmatici più elevati di lattato e piruvato, in accordo con la ridotta produzione di glucosio epatico (Madiraju et al, 2014). Questa riduzione non era dovuta a danni epatici, poiché il fegato dei topi inoculati non mostrava alcuna infiammazione né un aumento delle transaminasi e dei trigliceridi. Questo fenotipo epatico è stato smussato nei topi NOD2 KO, suggerendo il coinvolgimento del sensore microbico NOD2 nella gestione degli effetti metabolici indotti dall’inoculazione di microbiota intestinale nei topi riceventi. In entrambi i protocolli utilizzati, i topi inoculati con HFD-microbiota hanno mostrato una maggiore abbondanza relativa di Firmicutes , batteri Gram-positivi che ospitano un peptidoglicano più sviluppato di quelli Gram-negativi. Dato che il peptidoglicano è un ligando NOD2, si può ipotizzare che l’attivazione del NOD2 possa essere implicata nel fenotipo epatico osservato. I diversi microbioti intestinali ospitati dai topi NOD2 KO (Mondot et al, 2012; Denou et al , 2015) possono limitare la già citata riduzione della produzione di glucosio epatico indotta dall’inoculazione.
In generale, abbiamo osservato risultati contrastanti rispetto all’impatto metabolico del trasferimento di microbiota intestinale eubiotico nei pazienti affetti da sindrome metabolica (Vrieze et al, 2012) e di microbiota intestinale disbiotico nei topi assenici (Turnbaugh et al , 2006). La spiegazione che proponiamo è che la barriera funzionale intestinale e il sistema immunitario maturo di un topo convenzionale possono consentire una migliore gestione dei microbioti intestinali disbiotici rispetto ai topi assenici. Questo può portare ad una risposta immunitaria più efficiente, come suggerito dall’aumento dell’espressione del gene IL-17a nell’intestino di topi inoculati con microbiota magri. Al contrario, il microbiota HFD-microbiota non ha indotto questo aumento, in conformità con il nostro precedente rapporto (Garidou et al, 2015). Inoltre, il deterioramento della barriera intestinale è essenziale per le alterazioni metaboliche indotte dalla disbiosi (Serino et al, 2014). Pertanto, l’iperpermeabilità e l’alterazione dell’architettura dei villi che caratterizza l’intestino assenico (Reinhardt et al, 2012) possono favorire una diffusione incontrollata di entrambi i batteri e dei loro antigeni. Questo può scatenare un’infiammazione metabolica (Amar et al, 2011) responsabile delle alterazioni metaboliche indotte dalla disbiosi riportate nei topi assenici. Questa logica è sostenuta dai nostri risultati che dimostrano che la barriera intestinale non è influenzata dal trasferimento del microbiota intestinale in un topo convenzionale.
La risposta immunitaria efficiente può avere effetti benefici sistemici come quelli osservati sul fegato e sull’ACQUA. Per quanto riguarda quest’ultimo, nei topi inoculati con il microbiota HFD-microbiota la ridotta espressione dei geni lipogenici de novo è conforme ai più piccoli adipociti. Infatti, Eissing et al ( 2013) hanno dimostrato che gli enzimi lipogenici sono upregolati nel fegato dei pazienti obesi, e nel nostro studio, gli enzimi lipogenici sono downregolati nel fegato dei topi in associazione con adipociti più piccoli. L’aumento della FFA sierica è stato associato a piccoli adipociti anche nei topi assenici (Backhed et al, 2004), in accordo con i nostri dati.
Nel complesso, per spiegare i nostri risultati controintuitivi, abbiamo analizzato l’infiammazione dei topi riceventi. Notiamo che l’inoculazione di microbiota intestinale disbiotico nei topi convenzionali ha indotto l’up-regolazione della risposta dell’effettore (IFNγ) e l’down-regolazione della risposta regolatoria (FoxP3), nell’ileo e nel linfonodo mesenterico, in modo simile ai topi assenici colonizzati con microbiota intestinale normale (Naik et al, 2012). Questi risultati possono dipendere dalla capacità di un topo convenzionale di sviluppare una risposta efficace verso gli antigeni “obesi” a causa di un sistema immunitario maturo e di una barriera intestinale funzionale. La significativa correlazione negativa tra la via microbica gliossilato e dicarbossilato e l’IPPTT AUC suggerisce un collegamento tra questa attività microbica e la regolazione della produzione di glucosio epatico. La nostra ipotesi è supportata da una recente pubblicazione che mostra che la via microbica del gliossilato e del dicarbossilato è tra le più colpite nel modello dei ratti diabetici diabetici Zucker (Dong et al, 2016) . Pertanto, il targeting dei geni microbici coinvolti in questa via può essere efficace per il controllo della funzione epatica su un’alimentazione NC, ma non più sul 72% HFD.
In conclusione, i nostri risultati potrebbero aprire un nuovo dibattito sull’impatto della disbiosi dei microbioti intestinali sul metabolismo dell’ospite, descrivendo gli effetti benefici del trasferimento dei microbioti intestinali disbiotici, principalmente sul fegato. Così, la nostra nuova osservazione può incoraggiare a riesaminare il ruolo causale della disbiosi dei microbioti intestinali sulle malattie metaboliche.
Materiali e metodi
Modello animale e dieta
I topi maschi di sei settimane C57Bl/6 (WT o NOD2 KO) (Charles River, L’Arbresle, Francia) sono stati alimentati con un normale chow (NC) per 4 settimane (protocollo n. 1) o con un NC e poi con un alto livello di nutrizione.dieta dei grassi (HFD) (~72% di grassi (olio di mais e strutto), 28% di proteine e < 1% di carboidrati; SAFE, Augy, Francia) (Serino et al, 2012b ) per 6 settimane (protocollo #2). I topi sono stati ospitati in gruppo (5 o 6 topi per gabbia) in un ambiente controllato senza agenti patogeni specifici (invertito 12-h ciclo di luce diurna, luce spenta alle 10:00 del mattino). I topi con una velocità di sei ore sono stati sacrificati dalla dislocazione cervicale. Poi, i tessuti sono stati raccolti e congelati a scatto in azoto liquido. Tutte le procedure sperimentali su animali sono state approvate dal comitato etico locale dell’ospedale universitario di Rangueil (Tolosa, Francia).
Trasferimento dei microbioti intestinali
Sono stati eseguiti due protocolli: i topi riceventi erano topi maschi C57Bl/6 di 6 settimane alimentati a NC (Charles River, L’Arbresle, Francia), inoculati in una condizione di alimentazione e mai trattati precedentemente con antibiotici per entrambi i protocolli.
Protocollo n. 1
Topi donatori: Topi maschi di otto settimane C57Bl/6 (Charles River, L’Arbresle, Francia) sono stati alimentati con un 60% di HFD (60% di grassi, 20% di carboidrati, 20% di proteine) (Serino et al, 2007) o un NC per 3 mesi. Poi, il contenuto di cieco di questi topi è servito come trapianto ed è stato sospeso in PBS ridotto sterile (N2 gas e acido tioglicolico, Sigma Aldrich, St. Louis, MO) alla concentrazione di 200 mg / ml. Non-antibiotici trattati con C57Bl 6 settimane di età convenzionale C57Bl / 6 topi maschi (Charles River, L’Arbresle, Francia) servito come topi riceventi e sono stati gavaged con 200 microlitri di PBS sterile ridotto (Conv + PBS) o 200 microlitri a 200mg/ml sospensione ciecale di microbiota intestinale eubiotico di topi magri (Conv + LM) o di microbiota intestinale disbiotico di topi obesi indotti da HFD (Conv + OM(HFD)) una volta alla settimana, per 2 settimane (“LM” sta per microbiota magro e “OM” sta per microbiota obeso). Il contenuto di cieco da 3 a 6 topi per gruppo di donatori è stato messo in comune e fornito ai topi riceventi alla stessa concentrazione di 200 mg/ml.
Protocollo n. 2
Topi donatori: Undici – dodici settimane C57Bl/6 topi ob/ob maschi ( Charles River, L’Arbresle, Francia) o 20 settimane C57Bl/6 topi maschi (Charles River, L’Arbresle, Francia) alimentati con un 60% di HFD (Serino et al, 2007) sono serviti come topi donatori . Poi, il contenuto di cieco di questi topi è servito come trapianto ed è stato sospeso in PBS ridotto sterile (gas N2 e acido tioglicolico, Sigma Aldrich, St.) I topi maschi convenzionali C57Bl/6 non trattati con antibiotici di 6 settimane (Charles River, L’Arbresle, Francia) sono serviti come topi riceventi e sono stati sottoposti a gavage con 200 μl di PBS ridotto sterile (Conv + PBS) o 200 μl a 200 mg/ml di sospensione intestinale cieca di microbiota intestinale disbiotico da topi ob/ob (Conv + OM(ob)) o da topi obesi indotti da HFD (Conv + OM(HFD)) una volta alla settimana, per 2 settimane (“OM” sta per microbiota obeso). Il contenuto di cieco da 3 a 6 topi per gruppo di donatori è stato messo in comune e fornito ai topi riceventi alla stessa concentrazione di 200 mg/ml. Si noti che l’età ineguagliata per i topi donatori nel protocollo n. 2 è legata al fatto che il punto principale che abbiamo voluto indagare in questa sede è l’effetto metabolico putativo del trasferimento di un microbiota intestinale disbiotico; pertanto, non abbiamo intenzione di confrontare i donatori del protocollo n. 2 l’uno contro l’altro.
Criteri per la definizione di microbiota intestinale eubiotico vs. disbiotico
I microbioti intestinali eubiotici vs. disbiotici sono stati definiti in base alla quantità di batteri, più bassi nei microbioti intestinali disbiotici e alla loro elevata diversità a seconda del donatore (NC vs. topo nutrito con HFD; Appendice Fig S1F-K).
Analisi Western blot
L’analisi Western blot negli estratti di fegato è stata eseguita come descritto in precedenza (Serino et al, 2012b ). Sono stati utilizzati i seguenti anticorpi: substrati PKA, β-actina, PEPCK, tutti di Cell Signalling Technology. L’anticorpo G6Pase (De Vadder et al, 2014) è stato gentilmente fornito dal Dr. Gilles Mithieux e dal Dr. Fabienne Rajas (vedi Riconoscimenti).
Dosaggio del glicogeno epatico
50-100 mg di fegato di topi 6-h-fasted è stato sciolto in 200 μl di 1 M NaOH a 55 ° C per 1 h. I campioni sono stati neutralizzati con 200 μl 1 N HCl e poi centrifugati a 7.000 g per 5 minuti a 4°C. Poi, per idrolizzare il contenuto di glicogeno epatico, 10 microlitri di supernatante sono stati incubati in 40 microlitri di una soluzione di 50 U / ml di amiloglucosidasi (Sigma) diluito in 0,2 M tampone di acetato di sodio a pH 7,4. Come controllo, 10 μl sono stati incubati in 40 μl di tampone di acetato di sodio solo. Le provette sono state incubate per 1 h a 55°C. Poi, la concentrazione di glucosio è stata misurata con il reagente Glucose GOD FS (DiaSys Diagnostic Systems GmbH) secondo le istruzioni del produttore. La differenza di concentrazione di glucosio tra le due condizioni con e senza amiloglucosidasi rappresentava il contenuto di glicogeno epatico per campione. Il glicogeno è stato espresso come microgrammi di glucosio risultanti dall’idrolisi del glicogeno per milligrammi di fegato.
Test di tolleranza al piruvato intraperitoneale (IP), test di tolleranza al glucosio (IPPTT), test di tolleranza al glucosio (IPGTT) e test di tolleranza all’insulina (IPITT) o test di tolleranza al glucosio per via orale (OGTT).
Poiché i topi erano a ciclo di luce invertito, l’IPPTT è stato eseguito iniettando piruvato (2 g/kg) in topi a 6 h (Ribeiro et al, 2016). La glicemia è stata misurata come descritto in precedenza (Cani et al, 2007) a-15, 0, 15, 30, 45, 45, 60, 90 e 120 min. OGTT è stata eseguita come descritto altrove (Cani et al, 2007) . Per l’IPITT, ai topi a digiuno 3 ore sono state iniettate 0,75 U/kg di insulina (Serino et al, 2007). L’area sotto la curva (AUC) è anche mostrata come inset per IPPTT e IPGTTs/OGTT. AUC è stata calcolata con la regola trapezoidale (Le Floch et al, 1990) utilizzando GraphPad Prism versione 7.00 per Windows Vista (GraphPad Software, San Diego, CA) e mostrata come mmol/l × min.
Misurazione dei trigliceridi del fegato
I trigliceridi del fegato sono stati misurati utilizzando il reagente di glicerolo libero e il reagente trigliceride, entrambi della Sigma (Sigma Aldrich, St. Louis, MO).
Determinazione della dimensione degli adipociti
L’acqua epididimale è stata raccolta e fissata in etanolo al 70%. Il tessuto è stato trattato sul STP 120 Spin Tissue Processor mediante disidratazione dell’etanolo (aumentando il bagno dal 70% al 100%), sostituzione dello xilene e infiltrazione di paraffina. 5-μm sezioni di paraffina sono stati ottenuti utilizzando un Microtomo Microm HM 340E e colorato da ematossilina / eosina. Le sezioni colorate sono state riprese su un sistema Zeiss PALM MicroBeam con obiettivi ad aria Plan-Neofluar 10× (0,3 NA) e telecamera AxioCam MRm in bianco e nero. Le immagini sono state analizzate utilizzando il software MotionTracking (Collinet et al, 2010) a seguito di una pipeline sviluppata dal Dr Giovanni Marsico. In primo luogo, il centro degli adipociti è stato localizzato manualmente. Poi, il confine degli adipociti è stato segmentato automaticamente da un algoritmo di crescita regionale basato sulla trasformazione dello spartiacque. Poi, la dimensione degli adipociti è stata tracciata come distribuzione cumulativa.
Misurazione della massa grassa/perdita
La massa grassa/lean mass (%) è stata misurata tramite il sistema EchoMRI-100 TM 3 in 1 (EchoMRI LLC, Houston, TX, USA).
Saggi biochimici
Le transaminasi plasma aspartato (AST) e alanina (ALT), il colesterolo totale, le lipoproteine ad alta/bassa densità (HDL e LDL, rispettivamente), i trigliceridi e gli acidi grassi liberi (FFA) sono stati misurati con test multiplex dalla Piattaforma Fenotipage-ANEXPLO (US06-CREFRE).
Analisi metabolomica
I campioni di plasma (100 μl, su un pool di n = 6 topi per gruppo) sono stati diluiti con 600 μl di ossido di deuterio (D2O) e centrifugati a 5.000 g per 10 minuti prima di essere inseriti in tubi NMR da 5 mm. 1Hspettri NMR sono stati ottenuti su uno spettrometro Bruker DRX-600 Avance NMR che funziona a 600,13 MHz per la frequenza di risonanza 1H utilizzando una rivelazione inversa 5 mm 1H-13C-15N crioprobo collegato ad un CryoPlatform (l’unità di raffreddamento del preamplificatore). Gli spettri 1HNMR sono stati acquisiti a 300 K utilizzando la sequenza di impulsi di spin-eco Carr-Purcell-Meiboom_Gill (CPMG) con pre-saturazione, con un ritardo totale di spin-eco (2 nt) di 64 ms per attenuare ampi segnali da proteine e lipoproteine. Un totale di 128 transitori sono stati raccolti in 32 k punti di dati utilizzando un’ampiezza spettrale di 12 ppm, un ritardo di rilassamento di 5 s ed un tempo di acquisizione di 2,28 s. Prima della trasformazione di Fourier, al FID è stata applicata una funzione di allargamento esponenziale della linea di 0,3 Hz. Gli spettri NMR sono stati corretti in fase e la linea di base, e poi, i segnali dei metaboliti sono stati integrati, e normalizzati all’area spettrale totale.
Attività enzimatiche
L’attività della fosfatasi epatica glucosio-6 è stata determinata come descritto in precedenza (Rajas et al, 1999). L’attività della fosfoenolpiruvato carbossichinasi epatica è stata determinata con il metodo di Pogson e Smith (Pogson & Smith, 1975 ).
Studio dell’espressione genica di Microarray e analisi delle stringhe
L’analisi dell’espressione genica è stata eseguita presso lo stabilimento GeT-TRiX (GénoToul, Génopole Toulouse Midi-Pyrénées) utilizzando il mouse Agilent SurePrint G3 GE v2 8x60K microarray 8x60K (design ID 074809) seguendo le istruzioni del produttore (Agilent Technologies, Santa Clara, California). Per ciascuno dei sei campioni, il cRNA con etichettatura Cyanine-3 (Cy3) è stato preparato a partire da 200 ng di RNA totale utilizzando il kit di etichettatura One-Color Quick Amp (Agilent) secondo le istruzioni del produttore, seguito da Agencourt RNAClean XP (Agencourt Bioscience Corporation, Beverly, Massachusetts). 600 ng di cRNA marcato Cy3 sono stati ibridati sui vetrini per microarray seguendo le istruzioni del produttore. Subito dopo il lavaggio, i vetrini sono stati scansionati su Agilent G2505C Microarray Scanner utilizzando il software Agilent Scan Control A.8.5.1 e un segnale di fluorescenza estratto utilizzando il software Agilent Feature Extraction v10.10.1.1 con parametri predefiniti (griglia 074809_D_F_20150624 e protocollo GE1_1010_Sep10). I geni sono stati considerati espressi in modo diverso tra i gruppi Conv + OM(HFD) vs. Conv + PBS e tra i gruppi Conv + LM vs. Conv + PBS quando P < 0,05. Abbiamo anche considerato un logaritmo del cambiamento di piega contro Conv + PBS tra -2,2 (per la regolazione a valle) e 2,5 (per la regolazione a monte).
Analisi dei dati microarray basati su stringhe
Gli elenchi dei geni epatici espressi in modo diverso tra i gruppi Conv + OM (HFD) vs. Conv + PBS e tra i gruppi Conv + LM vs. Conv + PBS sono stati mappati utilizzando il database STRING (http://string-db.org/). Ogni gene è rappresentato da un nodo, e lo spessore delle linee tra i nodi illustra la forza delle interazioni basate sulla letteratura e sui database.
Estrazione dell’RNA e qPCR in fegato, ileo e MLN
L’RNA totale è stato estratto da tessuti congelati utilizzando il mini kit miRNeasy (Qiagen, Courtaboeuf, Francia). Per l’mRNA, la qPCR è stata eseguita come descritto in precedenza (Serino et al, 2012b ), ad eccezione dell’ileo e del linfonodo mesenterico (MLN), dove 500 ng di cDNA sono stati amplificati utilizzando il sistema ViiA7 (Applied Biosystems). I risultati sono stati espressi come 2-∆∆Ct come già descritto (Serino et al, 2012b ) e mostrato dopo la normalizzazione con la media dei valori di controllo (Conv + PBS). Il gene utilizzato in questo studio è la proteina Ribosomal L19 (RPL19).
Tutti i primer utilizzati in questo studio sono elencati nell’Appendice Tabella S1.
Analisi tassonomica dei microbioti intestinali mediante pirosequenziamento
Seguendo il protocollo n. 1 o n. 2, il DNA totale fecale è stato estratto come descritto in precedenza (Serino et al, 2012b ). L’intera regione del DNA batterico 16S V2 è stata presa di mira dai primer 28F-519R e pirosequenziata dalle 454 tecnologie FLX Roche presso il Research&Testing Laboratory(http://www.researchandtesting.com/, Texas, USA). Sono state generate in media 3.000 sequenze per campione.
Una descrizione completa dei filtri bioinformatici è disponibile sul sito http://www.rtlgenomics.com/docs/Data_Analysis_Methodology.pdf. I pannelli superiori delle figure 4AeB e 7A-C sono stati disegnati da XLSTAT per Windows Excel; i cladogrammi dei pannelli inferiori delle figure 4A e B e7A-Csono stati disegnati dalsito web dell’applicazione Huttenhower Galaxy(http://huttenhower.sph.harvard.edu/galaxy/) tramite l’algoritmo LEfSe (Segata et al, 2011).
Analisi funzionale del microbiota intestinale attraverso l’analisi del microbioma
L’analisi funzionale dei microbioti intestinali è stata effettuata tramite PICRUSt (Langille et al, 2013) . Le analisi dei componenti principali e le analisi delle barre di errore estese con un intervallo di confidenza del 95% per le figg. 5 e 8 sono state effettuate tramite il software Statistical Analysis of Metagenomic Profiles (STAMP) (Parks et al , 2014).
Analisi statistica
I risultati sono presentati come mezzi ± SEM. Le analisi statistiche sono state eseguite con ANOVA a senso unico o bidirezionale seguito dal post-test di Sidak o Dunnett, come riportato o dal t-test dello studente non accoppiato, utilizzando GraphPad Prism versione 7.00 per Windows Vista (GraphPad Software, San Diego, CA). Valori significativi considerati a P< 0.05 o come riportato. Le figure 5 e 8 sono state analizzate da un t-test di Welch su due lati; i pannelli superiori dell’appendice Le figure S1J/S5A-C/S11A-C/S11A-C/S12A-C sono state disegnate da XLSTAT per Windows Excel.
Disponibilità dei dati
I dati microarray di questa pubblicazione sono stati depositati nella banca dati Gene Expression Omnibus (GEO) https://www.ncbi.nlm.nih.gov/geo/ ed è stato assegnato l’identificatore (adesione GSE81318).
I dati metabolomici di questa pubblicazione sono disponibili nella versione elettronica Dataset EV1.
I dati metagenomici di questa pubblicazione sono stati depositati nella banca dati ENA http://www.ebi.ac.uk/ena e gli è stato assegnato l’identificatore PRJEB19465.
Contributi degli autori
SN ha eseguito e analizzato esperimenti; VB-B, AF, JG, PK, AW, FC, AM hanno eseguito esperimenti; RP, JSI ha eseguito analisi bioinformatiche su dati PICRUSt e microarray; FT ha interpretato i dati e rivisto il manoscritto; PDC ha eseguito il dosaggio plasma LPS; J-FT, RB ha letto il manoscritto; CK, MC ha analizzato i dati; MS ha progettato, eseguito esperimenti, analizzato e interpretato i dati e ha scritto il manoscritto. Tutti gli autori hanno approvato la versione finale da pubblicare.
Conflitto di interessi
Gli autori dichiarano di non avere alcun conflitto di interessi.
Informazioni di supporto
References
- Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: molecular mechanisms and probiotic treatment. EMBO Mol Med. 2011; 3:559-572. PubMed
- Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature. 2013; 500:232-236. PubMed
- The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci USA. 2004; 101:15718-15723. PubMed
- Mechanisms underlying the resistance to diet‐induced obesity in germ‐free mice. Proc Natl Acad Sci USA. 2007; 104:979-984. PubMed
- Associations between hepatic miRNA expression, liver triacylglycerols and gut microbiota during metabolic adaptation to high‐fat diet in mice. Diabetologia. 2017; 60:690. PubMed
- Periodontal dysbiosis linked to periodontitis is associated with cardiometabolic adaptation to high‐fat diet in mice. Am J Physiol Gastrointest Liver Physiol. 2016; 310:G1091-G1101. PubMed
- Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes. 2007; 56:1761-1772. PubMed
- Changes in gut microbiota control metabolic endotoxemia‐induced inflammation in high‐fat diet‐induced obesity and diabetes in mice. Diabetes. 2008; 57:1470-1481. PubMed
- Diet dominates host genotype in shaping the murine gut microbiota. Cell Host Microbe. 2015; 17:72-84. PubMed
- Gut immune maturation depends on colonization with a host‐specific microbiota. Cell. 2012; 149:1578-1593. PubMed
- Systems survey of endocytosis by multiparametric image analysis. Nature. 2010; 464:243-249. PubMed
- High‐fat diet alters gut microbiota physiology in mice. ISME J. 2014; 8:295-308. PubMed
- Microbiota‐generated metabolites promote metabolic benefits via gut‐brain neural circuits. Cell. 2014; 156:84-96. PubMed
- Defective NOD2 peptidoglycan sensing promotes diet‐induced inflammation, dysbiosis, and insulin resistance. EMBO Mol Med. 2015; 7:259-274. PubMed
- Metabolomics study of type 2 diabetes mellitus and the antidiabetic effect of berberine in zucker diabetic fatty rats using Uplc‐ESI‐Hdms. Phytother Res. 2016; 30:823-828. PubMed
- Metabolic profiling reveals a contribution of gut microbiota to fatty liver phenotype in insulin‐resistant mice. Proc Natl Acad Sci USA. 2006; 103:12511-12516. PubMed
- De novo lipogenesis in human fat and liver is linked to ChREBP‐beta and metabolic health. Nat Commun. 2013; 4:1528. PubMed
- Transfer of gut microbiota from lean and obese mice to antibiotic‐treated mice. Sci Rep. 2014; 4:5922. PubMed
- The gut microbiota regulates intestinal CD4 T cells expressing RORgammat and controls metabolic disease. Cell Metab. 2015; 22:100-112. PubMed
- WAO Special Committee on Climate Change and Biodiversity. The biodiversity hypothesis and allergic disease: world allergy organization position statement. World Allergy Organ J. 2013; 6:3. PubMed
- Predictive functional profiling of microbial communities using 16S rRNA marker gene sequences. Nat Biotechnol. 2013; 31:814-821. PubMed
- Richness of human gut microbiome correlates with metabolic markers. Nature. 2013; 500:541-546. PubMed
- Blood glucose area under the curve. Methodological aspects. Diabetes Care. 1990; 13:172-175. PubMed
- Intestinal microbiota determines development of non‐alcoholic fatty liver disease in mice. Gut. 2013; 62:1787-1794. PubMed
- Metformin suppresses gluconeogenesis by inhibiting mitochondrial glycerophosphate dehydrogenase. Nature. 2014; 510:542-546. PubMed
- Reshaping the gut microbiome with bacterial transplantation and antibiotic intake. Genome Res. 2010; 20:1411-1419. PubMed
- Gut microbiota modulation with norfloxacin and ampicillin enhances glucose tolerance in mice. Faseb J. 2008; 22:2416-2426. PubMed
- Altered gut microbiota composition in immune‐impaired Nod2(−/−) mice. Gut. 2012; 61:634-635. PubMed
- Compartmentalized control of skin immunity by resident commensals. Science. 2012; 337:1115-1119. PubMed
- STAMP: statistical analysis of taxonomic and functional profiles. Bioinformatics. 2014; 30:3123-3124. PubMed
- The activity of phosphoenolpyruvate carboxykinase in rat tissues. Assay techniques and effects of dietary and hormonal changes. Biochem J. 1975; 152:401-408. PubMed
- The glucose‐6 phosphatase gene is expressed in human and rat small intestine: regulation of expression in fasted and diabetic rats. Gastroenterology. 1999; 117:132-139. PubMed
- Tissue factor and PAR1 promote microbiota‐induced intestinal vascular remodelling. Nature. 2012; 483:627-631. PubMed
- Acephate exposure during a perinatal life program to type 2 diabetes. Toxicology. 2016; 372:12-21. PubMed
- Metagenomic biomarker discovery and explanation. Genome Biol. 2011; 12:R60. PubMed
- Gastrointestinal cancers: influence of gut microbiota, probiotics and prebiotics. Cancer Lett. 2014; 345:258-270. PubMed
- Mice heterozygous for tumor necrosis factor‐alpha converting enzyme are protected from obesity‐induced insulin resistance and diabetes. Diabetes. 2007; 56:2541-2546. PubMed
- Intestinal microflora and metabolic diseases. Diabetes Metab. 2009; 35:262-272. PubMed
- Intestinal MicrobiOMICS to define health and disease in human and mice. Curr Pharm Biotechnol. 2012a; 13:746-758. PubMed
- Metabolic adaptation to a high‐fat diet is associated with a change in the gut microbiota. Gut. 2012b; 61:543-553. PubMed
- Managing the manager: gut microbes, stem cells and metabolism. Diabetes Metab. 2014; 40:186-190. PubMed
- The host‐microbe interface within the gut. Best Pract Res Clin Gastroenterol. 2002; 16:915-931. PubMed
- Inflammation and insulin resistance. J Clin Invest. 2006; 116:1793-1801. PubMed
- Gut‐liver axis and sensing microbes. Dig Dis. 2010; 28:737-744. PubMed
- From gut microflora imbalance to mycobacteria infection: is there a relationship with chronic intestinal inflammatory diseases?. Ann Ital Chir. 2011; 82:361-368. PubMed
- An obesity‐associated gut microbiome with increased capacity for energy harvest. Nature. 2006; 444:1027-1031. PubMed
- The gut microbiota modulates host energy and lipid metabolism in mice. J Lipid Res. 2010; 51:1101-1112. PubMed
- Transfer of intestinal microbiota from lean donors increases insulin sensitivity in individuals with metabolic syndrome. Gastroenterology. 2012; 143:913-916. PubMed
Fonte
Nicolas S, Blasco‐Baque V, Fournel A, Gilleron J, Klopp P, et al. (2017) Transfer of dysbiotic gut microbiota has beneficial effects on host liver metabolism. Molecular Systems Biology 13(3): 921. https://doi.org/10.15252/msb.20167356




