
Abstract
Introduzione
I geni raramente funzionano in modo isolato per influenzare i fenotipi a livello cellulare o organismico. Molti studi hanno descritto come i geni agiscono in reti complesse per mantenere l’omeostasi attraverso la messa a punto di reazioni cellulari o organismiche a stimoli interni o esterni (Bergman & Siegal, 2003). Una perdita di buffering genetico può portare all’insorgenza di malattie come il cancro (Hartwell et al, 1997; Hartman et al , 2001). A loro volta, le mutazioni possono creare vulnerabilità genetiche nelle cellule tumorali, ad esempio, disattivando uno dei due percorsi del buffer genetico (Luo et al, 2009; Torti& Trusolino, 2011; Nagel et al, 2016). Gli approcci terapeutici tentano di sfruttare tali eventi inducendo selettivamente la morte cellulare nelle cellule tumorali e causando pochi danni alle cellule normali (Kaelin, 2005; Nijman, 2011).
Per identificare sistematicamente le interazioni genetiche, si possono effettuare esperimenti di knockout o knockdown genico a coppie (Mani et al, 2008) . Nei casi in cui un difetto di fitness misurato del doppio mutante è più forte del previsto sulla base dei due singoli fenotipi mutanti, l’interazione è chiamata aggravante o letale sintetica (Bridges, 1922). Al contrario, si osserva un’interazione tampone (o attenuante) quando il fenotipo misurato del doppio mutante è più debole del previsto. Schermi a matrice eseguiti mediante l’accoppiamento di ceppi di lieviti mutanti a perdita di funzione sono stati pionieri dello screening combinatorio (Tong et al, 2001; Davierwala et al, 2005; Baryshnikova et al, 2010; Costanzo et al, 2010, 2016). Metodi di perturbazione genica a coppie sono stati successivamente estesi utilizzando l’interferenza RNA combinatoria (RNAi) per mappare le interazioni genetiche nelle cellule metazoiche coltivate (Byrne et al, 2007; Horn et al, 2011; Laufer et al, 2013; Snijder et al, 2013; Fischer et al, 2015; Srivas et al, 2016) . Tuttavia, lo screening di tutte le combinazioni geniche a coppie è scarsamente scalabile con l’aumento delle dimensioni del genoma e nuovi approcci sono necessari per facilitare la generazione di grandi mappe di interazione genetica di organismi complessi, riducendo al minimo i costi e lo sforzo sperimentale.
Gli schermi di perturbazione su scala genomica possono ora essere eseguiti in modo efficiente in molte linee cellulari utilizzando CRISPR/Cas9 (Barrangou, 2014 ; Doudna & Charpentier, 2014; Wang et al, 2014; Shalem et al, 2015); Heigwer et al, 2016; Horlbeck et al , 2016) o RNAi (Brummelkamp et al, 2002; Sims et al, 2011; Kampmann et al , 2013) per la perturbazione mirata dei geni tramite knockout o knockdown. Poiché ogni linea cellulare ha un background genetico diverso, ciò consente di indagare le vulnerabilità specifiche del genotipo (Garnett et al, 2012; Hart et al, 2015); Iorio et al, 2016; Tzelepis et al , 2016; Martin et al, 2017; McDonald et al, 2017; Steinhart et al, 2017; Tsherniak et al, 2017; Wang et al, 2017 ). Per descrivere un’interazione genetica, gli studi precedenti si sono basati principalmente sulla definizione di “epistasi statistica” introdotta da R. A. Fisher (Fisher, 1930). Qui, un’interazione genetica è definita come una deviazione statistica dalla combinazione additiva di due loci nel modo in cui influenzano un fenotipo di interesse (Phillips, 2008). Questa definizione non presuppone necessariamente un background genetico standardizzato e fornisce quindi un quadro teorico applicabile per mappare le interazioni genetiche nelle linee cellulari tumorali nonostante la presenza di ulteriori mutazioni confondenti. Per sfruttare lo sforzo collettivo della comunità per caratterizzare funzionalmente le linee cellulari tumorali, è auspicabile combinare e analizzare gli schermi genetici di diversa origine in modo integrato. Questo, tuttavia, non è facile da mettere in pratica poiché varie fonti di variazione tecnica, come diverse biblioteche di sgRNA o protocolli sperimentali, possono influenzare i dati e confondere le analisi comparative.
Qui, proponiamo un quadro computazionale che integra gli schermi CRISPR/Cas9 di diversa origine per mappare le interazioni genetiche nelle cellule tumorali. Applichiamo questo approccio, che abbiamo chiamato MINGLE, ad un dataset curato composto da 85 schermi CRISPR/Cas9 a scala genomica in 60 diverse linee di cellule tumorali umane generate in diversi laboratori (Fig 1A). Per prima cosa, mostriamo che un approccio di normalizzazione in due fasi può essere applicato per consentire il confronto quantitativo di fenotipi derivati da diversi schermi (Fig EV1A). Dimostriamo poi come i concetti che sono stati applicati in precedenza per mappare le reti genetiche in organismi modello possono essere adattati e applicati a questo dataset per valutare le combinazioni gene-gene per le interazioni genetiche. Combinando il profilo intrinseco delle alterazioni genetiche di ogni linea cellulare presente nel dataset con i fenotipi di vitalità a livello genico, abbiamo testato 2,1 milioni di combinazioni geniche a coppie confrontando il tipo selvaggio con alleli alterati nelle linee cellulari (Fig 1B e C). Utilizzando queste previsioni, siamo stati in grado di identificare nuovi regolatori del percorso di segnalazione Wnt/β-catenina. I nostri risultati suggeriscono che i geni PRKCSH e GANAB, che insieme formano il complesso della glucosidasi II, regolano la secrezione dei ligandi Wnt attivi. Infine, abbiamo funzionalmente clusterizzato i geni dalla somiglianza dei loro profili di interazione e dimostrare che questi profili sono predittori informativi di somiglianza genica funzionale (Fig 1D). Abbiamo generato una mappa delle interazioni genetiche nelle cellule tumorali collegando i geni con profili simili e identificando moduli di rete con caratteristiche funzionali simili.
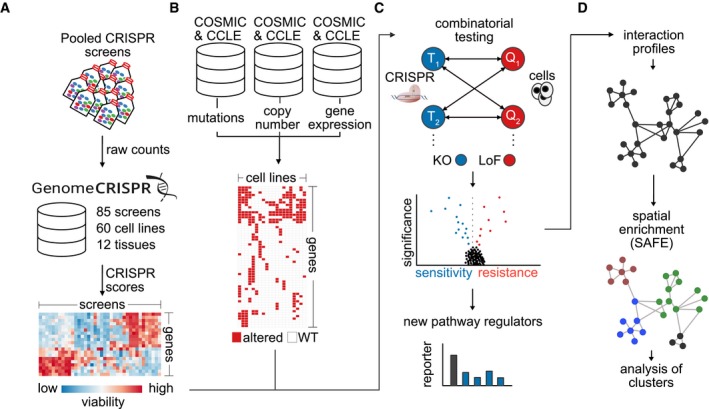
Figura 1.Un approccio di analisi integrata per identificare le interazioni genetiche nelle cellule tumorali
I dati degli schermi CRISPR/Cas9 in 60 linee cellulari tumorali sono stati rianalizzati e integrati. I risultati sono stati integrati in un profilo globale di risposta alla perturbazione. Mutazioni, numero di copie e dati di espressione di mRNA dai database COSMIC e CCLE sono stati combinati per creare una mappa delle alterazioni genetiche attraverso queste linee cellulari.Sono stati esaminati 1 milione di combinazioni gene-gene per dedurre le interazioni genetiche.i profili di interazione sono stati calcolati per le combinazioni geniche in base alla correlazione delle loro interazioni, come determinato dai punteggi di interazione (punteggi π). L’analisi dell’arricchimento spaziale è stata eseguita per identificare i moduli funzionali nella rete.I dati provenienti da schermi CRISPR/Cas9 in 60 linee cellulari tumorali sono stati rianalizzati e integrati. I risultati sono stati integrati in un profilo di risposta globale di perturbazione.Mutazioni, numero di copie e dati di espressione degli mRNA provenienti dai database COSMIC e CCLE sono stati combinati per creare una mappa delle alterazioni genetiche di queste linee cellulari.Per identificare le dipendenze genetiche tra combinazioni di geni che potrebbero far luce sul cablaggio genetico delle cellule tumorali, è stata esaminata una risposta di perturbazione di oltre 2,1 milioni di combinazioni gene-gene per dedurre le interazioni genetiche.I profili di interazione sono stati calcolati per le combinazioni di geni in base alla correlazione delle loro interazioni, come determinato dai punteggi di interazione (punteggi π). L’analisi dell’arricchimento spaziale è stata eseguita per identificare i moduli funzionali nella rete.
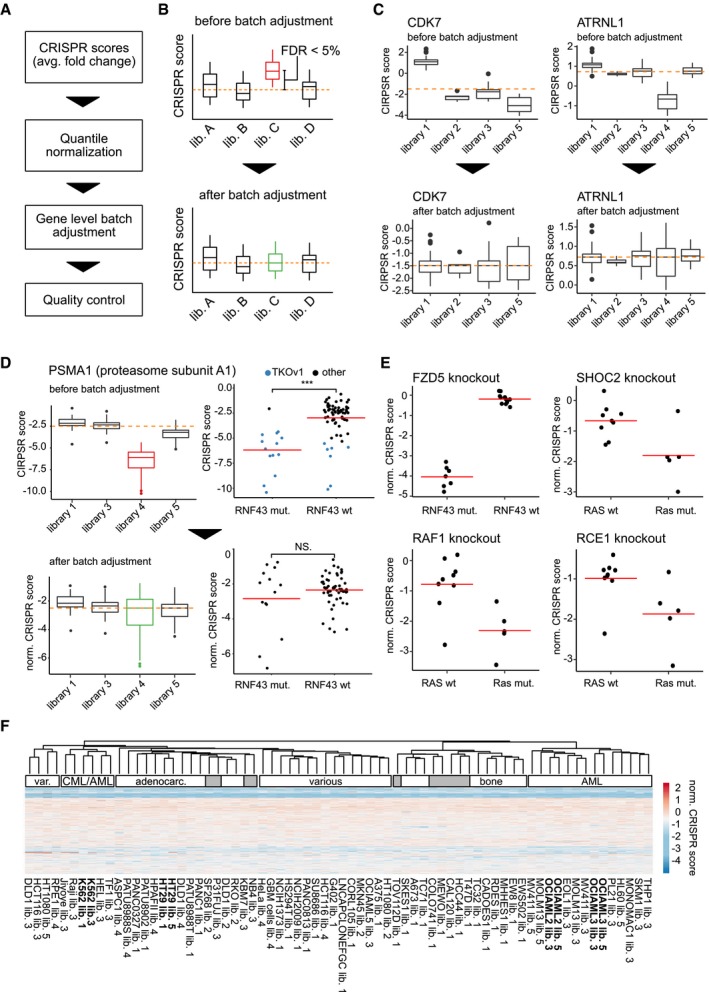
Figura EV1.Integrazione e normalizzazione dei fenotipi di vitalità in diversi schermi
Schema del flusso di lavoro del processo di normalizzazione.Illustrazione schematica della regolazione del lotto a livello genico per correggere le differenze tra le librerie. Ogni casella della trama corrisponde al fenotipo osservato nelle schermate che utilizzano la stessa libreria sgRNA quando si mira ad un gene specifico di interesse. Nel caso in cui i fenotipi di una libreria differiscano significativamente (FDR < 5%; Benjamini-Hochberg) dal previsto (mediana di tutti i fenotipi per lo stesso gene), la libreria viene regolata sottraendo la differenza stimata tra la media della libreria e il fenotipo previsto da ogni punto di dati corrispondente a quella libreria.Esempi di due geni in cui si osservano effetti batch che mostrano gli effetti della normalizzazione.Esempio in cui una libreria di sgRNA associata ad un effetto batch può, se non regolata, portare ad una interazione falso-positiva tra PSMA1 e RNF43. Ogni punto rappresenta un esperimento di screening. Il gruppo mutato RNF43 è composto da 14 punti di dati, il gruppo RNF43 wt è composto da 71 punti di dati.quattro esempi di interazioni genetiche riportate in precedenza dove i fenotipi sono conservati attraverso il processo di normalizzazione. Ogni punto rappresenta un esperimento di screening. Il numero di punti di dati corrispondenti a ciascun gruppo è di 7 (FZD5 konkcout, RNF43 wt), 12 (FZD5 knockout, RNF43 wt), 9 (tutti i gruppi RAS wt) e 5 (tutti i gruppi RAS wt). Le linee rosse orizzontali indicano il significato del gruppo.Clustering dei punteggi CRISPR normalizzati di tutti gli esperimenti usati nell’analisi basati su geni condivisi tra le biblioteche. La mappa termica è simile a quella mostrata in Fig 1A con tutte le colonne etichettate.informazioni sui dati: (B-D) Le linee rosse tratteggiate indicano il punteggio CRISPR mediano di tutte le librerie. Le linee orizzontali all’interno di ogni casella indicano la mediana. I confini superiore e inferiore di ogni casella rappresentano rispettivamente il 75° e il 25° percentile. Le barre di errore indicano il10° e il 90° percentile e gli outlier sono rappresentati come cerchi.Flusso di lavoro schematico del processo di normalizzazione.Illustrazione schematica della regolazione del lotto a livello genico per correggere le differenze tra le librerie. Ogni casella nel grafico corrisponde al fenotipo osservato nelle schermate che utilizzano la stessa libreria di sgRNA quando si mira ad uno specifico gene di interesse. Nel caso in cui i fenotipi di una libreria differiscano significativamente (FDR < 5%; Benjamini-Hochberg) dal previsto (mediana di tutti i fenotipi per lo stesso gene), la libreria viene regolata sottraendo la differenza stimata tra la media della libreria e il fenotipo previsto da ogni punto di dati corrispondente a quella libreria.Esempi di due geni in cui si osservano effetti batch che mostrano gli effetti della normalizzazione.Esempio in cui un effetto batch associato ad una libreria di sgRNA può, se non regolato, portare ad una interazione falso-positiva tra PSMA1 e RNF43. Ogni punto rappresenta un esperimento di screening. Il gruppo mutato RNF43 è composto da 14 punti dati, il gruppo RNF43 wt è composto da 71 punti dati.Quattro esempi di interazioni genetiche riportate in precedenza dove i fenotipi sono conservati attraverso il processo di normalizzazione. Ogni punto rappresenta un esperimento di screening. Il numero di punti di dati corrispondenti a ciascun gruppo è di 7 (FZD5 konkcout, RNF43 wt), 12 (FZD5 knockout, RNF43 wt), 9 (tutti i gruppi RAS wt) e 5 (tutti i gruppi RAS wt). Le linee rosse orizzontali indicano il significato del gruppo.Clustering dei punteggi CRISPR normalizzati di tutti gli esperimenti usati nell’analisi basati su geni condivisi tra le biblioteche. La mappa termica è simile a quella mostrata in Fig 1A con tutte le colonne etichettate.
Figura 1.Un approccio di analisi integrata per identificare le interazioni genetiche nelle cellule tumorali
I dati degli schermi CRISPR/Cas9 in 60 linee cellulari tumorali sono stati rianalizzati e integrati. I risultati sono stati integrati in un profilo globale di risposta alla perturbazione. Mutazioni, numero di copie e dati di espressione di mRNA dai database COSMIC e CCLE sono stati combinati per creare una mappa delle alterazioni genetiche attraverso queste linee cellulari.Sono stati esaminati 1 milione di combinazioni gene-gene per dedurre le interazioni genetiche.i profili di interazione sono stati calcolati per le combinazioni geniche in base alla correlazione delle loro interazioni, come determinato dai punteggi di interazione (punteggi π). L’analisi dell’arricchimento spaziale è stata eseguita per identificare i moduli funzionali nella rete.I dati provenienti da schermi CRISPR/Cas9 in 60 linee cellulari tumorali sono stati rianalizzati e integrati. I risultati sono stati integrati in un profilo di risposta globale di perturbazione.Mutazioni, numero di copie e dati di espressione degli mRNA provenienti dai database COSMIC e CCLE sono stati combinati per creare una mappa delle alterazioni genetiche di queste linee cellulari.Per identificare le dipendenze genetiche tra combinazioni di geni che potrebbero far luce sul cablaggio genetico delle cellule tumorali, è stata esaminata una risposta di perturbazione di oltre 2,1 milioni di combinazioni gene-gene per dedurre le interazioni genetiche.I profili di interazione sono stati calcolati per le combinazioni di geni in base alla correlazione delle loro interazioni, come determinato dai punteggi di interazione (punteggi π). L’analisi dell’arricchimento spaziale è stata eseguita per identificare i moduli funzionali nella rete.
Figura EV1.Integrazione e normalizzazione dei fenotipi di vitalità in diversi schermi
Schema del flusso di lavoro del processo di normalizzazione.Illustrazione schematica della regolazione del lotto a livello genico per correggere le differenze tra le librerie. Ogni casella della trama corrisponde al fenotipo osservato nelle schermate che utilizzano la stessa libreria sgRNA quando si mira ad un gene specifico di interesse. Nel caso in cui i fenotipi di una libreria differiscano significativamente (FDR < 5%; Benjamini-Hochberg) dal previsto (mediana di tutti i fenotipi per lo stesso gene), la libreria viene regolata sottraendo la differenza stimata tra la media della libreria e il fenotipo previsto da ogni punto di dati corrispondente a quella libreria.Esempi di due geni in cui si osservano effetti batch che mostrano gli effetti della normalizzazione.Esempio in cui una libreria di sgRNA associata ad un effetto batch può, se non regolata, portare ad una interazione falso-positiva tra PSMA1 e RNF43. Ogni punto rappresenta un esperimento di screening. Il gruppo mutato RNF43 è composto da 14 punti di dati, il gruppo RNF43 wt è composto da 71 punti di dati.quattro esempi di interazioni genetiche riportate in precedenza dove i fenotipi sono conservati attraverso il processo di normalizzazione. Ogni punto rappresenta un esperimento di screening. Il numero di punti di dati corrispondenti a ciascun gruppo è di 7 (FZD5 konkcout, RNF43 wt), 12 (FZD5 knockout, RNF43 wt), 9 (tutti i gruppi RAS wt) e 5 (tutti i gruppi RAS wt). Le linee rosse orizzontali indicano il significato del gruppo.Clustering dei punteggi CRISPR normalizzati di tutti gli esperimenti usati nell’analisi basati su geni condivisi tra le biblioteche. La mappa termica è simile a quella mostrata in Fig 1A con tutte le colonne etichettate.informazioni sui dati: (B-D) Le linee rosse tratteggiate indicano il punteggio CRISPR mediano di tutte le librerie. Le linee orizzontali all’interno di ogni casella indicano la mediana. I confini superiore e inferiore di ogni casella rappresentano rispettivamente il 75° e il 25° percentile. Le barre di errore indicano il10° e il 90° percentile e gli outlier sono rappresentati come cerchi.Flusso di lavoro schematico del processo di normalizzazione.Illustrazione schematica della regolazione del lotto a livello genico per correggere le differenze tra le librerie. Ogni casella nel grafico corrisponde al fenotipo osservato nelle schermate che utilizzano la stessa libreria di sgRNA quando si mira ad uno specifico gene di interesse. Nel caso in cui i fenotipi di una libreria differiscano significativamente (FDR < 5%; Benjamini-Hochberg) dal previsto (mediana di tutti i fenotipi per lo stesso gene), la libreria viene regolata sottraendo la differenza stimata tra la media della libreria e il fenotipo previsto da ogni punto di dati corrispondente a quella libreria.Esempi di due geni in cui si osservano effetti batch che mostrano gli effetti della normalizzazione.Esempio in cui un effetto batch associato ad una libreria di sgRNA può, se non regolato, portare ad una interazione falso-positiva tra PSMA1 e RNF43. Ogni punto rappresenta un esperimento di screening. Il gruppo mutato RNF43 è composto da 14 punti dati, il gruppo RNF43 wt è composto da 71 punti dati.Quattro esempi di interazioni genetiche riportate in precedenza dove i fenotipi sono conservati attraverso il processo di normalizzazione. Ogni punto rappresenta un esperimento di screening. Il numero di punti di dati corrispondenti a ciascun gruppo è di 7 (FZD5 konkcout, RNF43 wt), 12 (FZD5 knockout, RNF43 wt), 9 (tutti i gruppi RAS wt) e 5 (tutti i gruppi RAS wt). Le linee rosse orizzontali indicano il significato del gruppo.Clustering dei punteggi CRISPR normalizzati di tutti gli esperimenti usati nell’analisi basati su geni condivisi tra le biblioteche. La mappa termica è simile a quella mostrata in Fig 1A con tutte le colonne etichettate.
Risultati
Integrazione dei fenotipi CRISPR/Cas9 di diversi studi
Al fine di prevedere sistematicamente le interazioni tra i geni messi fuori uso da CRISPR/Cas9 e i geni compromessi funzionalmente dalle mutazioni nelle cellule tumorali, abbiamo rianalizzato una serie di 85 schermi di vitalità CRISPR/Cas9 in 60 linee cellulari (Fig 1A, Dataset EV1). Questi schermi sono stati eseguiti in diversi laboratori e variano in termini di libreria e design vettoriale così come i protocolli di screening. Al fine di integrare questi dati (Fig EV1A),abbiamo prima calcolato i punteggi CRISPR a livello genico individualmente per ogni schermata (media log2 fold change of sgRNA abundance; Wang et al, 2017). Poiché, per esempio, i tempi di selezione variabili possono portare a differenze nella forza fenotipica, abbiamo poi quantile-normalizzato i dati per correggere i bias sistematici tra le schermate. L’esame del set di dati risultante ha rivelato considerevoli effetti di batch guidati principalmente dalla libreria sgRNA utilizzata per lo screening (Fig EV1B).Questi effetti di batch sembravano essere non sistematici e diversi da gene a gene. Per esempio, la chinasi 7 ciclo-dipendente (CDK7) è un gene noto per svolgere ruoli importanti in entrambi, nella progressione del ciclo cellulare e nella trascrizione (Fisher, 2005 ), e ci si aspetta che sia un gene ampiamente essenziale (Hart et al, 2017). Di conseguenza, il knockout del CDK7 ha costantemente portato ad una diminuzione della vitalità nella maggior parte degli esperimenti. Gli schermi in cui non è stato osservato alcun fenotipo di vitalità al knockout del CDK7 sono stati tutti condotti utilizzando la stessa libreria (Fig EV1C).Poiché le linee cellulari schermate con questa libreria sono derivate da vari tessuti diversi e tipi di cancro, una resistenza comune al knockout CDK7 sembra improbabile. Una spiegazione più probabile per l’effetto del lotto osservato potrebbe essere l’incapacità del CDK7 che colpisce gli sgRNA in questa libreria di generare un knockout in primo luogo. Se non considerati e corretti, tali effetti di batch possono introdurre false previsioni (Fig EV1D),sottolineando il requisito di una strategia efficiente per il loro aggiustamento. A tal fine, abbiamo ipotizzato che un knockout genico dovrebbe, in media, avere lo stesso effetto su tutti gli schermi, indipendentemente dalla libreria utilizzata. Abbiamo poi applicato un approccio basato su un modello per la scansione sistematica dei potenziali effetti di batch in cui i fenotipi generati da una libreria differivano significativamente (FDR < 5%) dal fenotipo mediano osservato in tutte le librerie. Al fine di proteggere gli effetti biologici reali, abbiamo utilizzato un modello lineare robusto per i test, che è robusto verso i forti effetti biologici presenti nei dati sotto forma di outlier. Nei casi in cui è stato possibile rilevare una differenza significativa tra i fenotipi generati da una biblioteca e il fenotipo mediano in tutte le biblioteche, abbiamo effettuato un aggiustamento sottraendo la differenza stimata tra la biblioteca interessata dall’effetto batch e le restanti biblioteche (Fig EV1B).E ‘importante sottolineare, che questo approccio può essere inappropriato quando vi è una correlazione tra una biblioteca sgRNA e una covariata biologica, per esempio, se la maggior parte delle linee cellulari schermate con questa specifica biblioteca sono derivati da tessuti simili. Questo non è il caso per la maggior parte delle biblioteche incluse in questa analisi. Ad esempio, le librerie GeCKOv2 e TKOv1 sono state utilizzate per lo screening di una grande varietà di linee cellulari derivate da diversi tessuti e tipi di cancro (Hart et al, 2015; Aguirre et al , 2016; Steinhart et al, 2017 ). Fanno eccezione, tuttavia, gli schermi eseguiti da Wang et al (2017 )e Tzelepis et al (2016 ). In questi studi, gli schermi sono stati eseguiti principalmente in linee cellulari di leucemia mieloide acuta (LAM). Al fine di preservare tali fenotipi specifici dei tessuti attraverso la correzione dei lotti, il nostro approccio basato su modelli permette di includere covariate biologiche come il tessuto di una linea cellulare o il tipo di cancro nella modellazione dei lotti, che può quindi distinguere tra variabilità tecnica e biologica.
Per convalidare il nostro approccio di integrazione dei dati, abbiamo eseguito una serie di analisi di controllo della qualità. In primo luogo, abbiamo raggruppato tutti gli schermi sulla base dei punteggi CRISPR normalizzati (figg. 2AedEV1F) .In molti casi, gli schermi che sono stati eseguiti in diversi laboratori con diverse biblioteche ma utilizzando la stessa linea di celle raggruppate insieme. Inoltre, abbiamo osservato una tendenza per le linee cellulari che condividono la stessa origine del tessuto per raggruppare insieme. Ad esempio, abbiamo potuto identificare distinti cluster di linee cellulari AML e linee cellulari di adenocarcinoma. Questi risultati suggeriscono un’adeguata correzione della polarizzazione tecnica, lasciando la variabilità biologica tra le linee cellulari come il driver principale del raggruppamento. Abbiamo poi valutato se i punteggi CRISPR normalizzati possono essere confrontati quantitativamente attraverso gli schermi. Qui, abbiamo selezionato in modo casuale nove polimerasi essenziali per il nucleo e abbiamo tracciato i punteggi CRISPR normalizzati per questi geni attraverso gli schermi (Fig 2B). I punteggi CRISPR per le polimerasi essenziali erano negativi e approssimativamente allo stesso livello, senza differenze evidenti tra gli schermi pubblicati in diversi studi, suggerendo che il confronto quantitativo dei punteggi è effettivamente fattibile e che i fenotipi di vitalità negativa prevista dei knockout di geni essenziali del nucleo sono conservati durante la normalizzazione. Ci siamo chiesti se la procedura di normalizzazione potrebbe potenzialmente introdurre falsi fenotipi. In generale, questo può essere escluso con l’aiuto di controlli non mirati, che, tuttavia, non erano disponibili per tutti gli esperimenti del nostro dataset. In sostituzione, abbiamo quindi selezionato tutte le schermate eseguite in linee cellulari femminili e abbiamo tracciato i punteggi CRISPR normalizzati per nove geni selezionati in modo casuale situati sul cromosoma Y (Fig 2C). Abbiamo osservato che i punteggi CRISPR sono approssimativamente 0, il che implica che nessun fenotipo falso viene introdotto artificialmente dalla normalizzazione. Successivamente, abbiamo determinato quanto bene i geni di riferimento essenziali e non essenziali (Hart et al, 2015, 2017) possano essere separati sulla base dei punteggi CRISPR normalizzati generando curve di precisione-richiamo (Fig 2D), sulla base delle quali abbiamo osservato buone prestazioni su tutti gli schermi. Abbiamo ulteriormente esaminato se i punteggi CRISPR normalizzati potessero catturare esempi ben studiati di dipendenza da oncogeno. La dipendenza da oncogeni descrive un fenomeno in cui le cellule tumorali, pur ospitando molte aberrazioni molecolari, diventano fortemente dipendenti da una sola di esse. L’inversione di questa anomalia porta all’inibizione della crescita e all’apoptosi (Weinstein & Joe, 2006 ). Abbiamo selezionato gli oncogeni ben studiati KRAS , NRAS, BRAF e PIK3CA e abbiamo confrontato i punteggi CRISPR delle linee cellulari che ospitano una mutazione di questi geni con il resto delle linee cellulari (Fig 2E-H). Come previsto, abbiamo osservato fenotipi notevolmente più forti nelle cellule mutate rispetto alle cellule wild-type. Infine, abbiamo determinato se le dipendenze genetiche precedentemente identificate negli schermi utilizzati per la nostra analisi potevano essere riprodotte (Fig EV1E).In tutti i casi, abbiamo potuto ottenere risultati comparabili a quelli pubblicati in precedenza, confermando l’uso di punteggi CRISPR normalizzati per analisi interscreen valide.
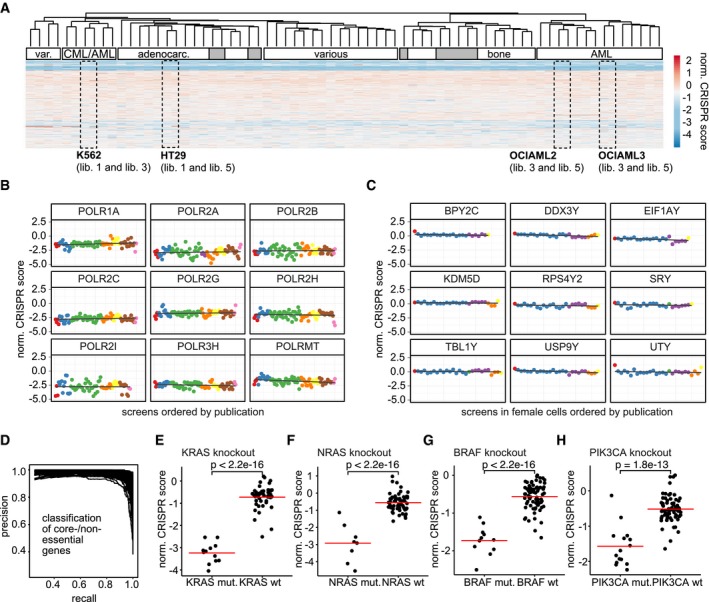
E-H.Risultati e controllo qualità dell’integrazione e della normalizzazione dei dati
La mappa termica AA mostra un clustering di punteggi CRISPR normalizzati (media log2 fold change degli sgRNA che prendono di mira un gene) per i geni presenti in ogni libreria di sgRNA utilizzata nelle schermate incluse nell’analisi. Le finestre rettangolari evidenziano gli esperimenti in cui gli schermi eseguiti nella stessa linea cellulare ma in diversi laboratori si raggruppano. Le barre bianche di annotazione indicano le proprietà biologiche condivise delle linee cellulari in ogni cluster. Le barre grigie indicano che la linea cellulare annotata non si adatta all’annotazione di altre linee cellulari nello stesso cluster.BNormalized CRISPR punteggi attraverso gli esperimenti sono visualizzati per un insieme selezionato in modo casuale di nove polimerasi essenziali del nucleo. Ogni punto corrisponde ad una schermata, e colori diversi evidenziano le pubblicazioni da cui sono stati derivati i dati. Punteggi CRISPR più negativi indicano una risposta di vitalità più negativa al knockout del gene.i punteggi CRISPR CNormalized attraverso gli esperimenti in linee cellulari femminili sono visualizzati per un insieme selezionato in modo casuale di nove geni situati sul cromosoma Y che servono come controlli non bersaglio. Colori raffigurano diverse pubblicazioni.DPrecisione-richiamo curve di richiamo che mostrano le prestazioni dei punteggi CRISPR normalizzati a distinguere il nucleo essenziale dai geni non essenziali. Ogni linea corrisponde ad un esperimento. L’elevato richiamo, pur mantenendo un’elevata precisione, indica una buona performance.E-HComparison dei punteggi CRISPR normalizzati in un diverso background genetico per quattro diverse dipendenze di controllo. Le linee rosse indicano i mezzi del gruppo. Significato statistico è stato determinato utilizzando un t-test a due facce di Studente. Ogni punto dati rappresenta un esperimento di screening. I gruppi sono composti da 12 (KRAS mut.), 44 (KRAS wt), 7 (NRAS mut.), 56 (NRAS wt), 11 (BRAF mut.), 63 (BRAF wt), 15 (PIK3CA mut.) e 59 (PIK3CA wt) punti dati.Una mappa termica mostra un clustering di punteggi CRISPR normalizzati (media log2 fold change degli sgRNA che prendono di mira un gene) per i geni presenti in ogni libreria di sgRNA utilizzata nelle schermate incluse nell’analisi. Le finestre rettangolari evidenziano gli esperimenti in cui gli schermi eseguiti nella stessa linea cellulare ma in diversi laboratori si raggruppano. Le barre bianche di annotazione indicano le proprietà biologiche condivise delle linee cellulari in ogni cluster. Le barre grigie indicano che la linea cellulare annotata non si adatta all’annotazione di altre linee cellulari nello stesso cluster.Punteggi CRISPR normalizzati attraverso gli esperimenti sono visualizzati per un insieme selezionato in modo casuale di nove polimerasi essenziali del nucleo. Ogni punto corrisponde ad una schermata, e colori diversi evidenziano le pubblicazioni da cui sono stati derivati i dati. Punteggi CRISPR più negativi indicano una risposta di vitalità più negativa al knockout del gene.Punteggi CRISPR normalizzati attraverso esperimenti in linee cellulari femminili sono visualizzati per un insieme selezionato in modo casuale di nove geni situati sul cromosoma Y che servono come controlli non bersaglio. I colori raffigurano diverse pubblicazioni.Precisione-richiamo le curve di precisione che mostrano le prestazioni dei punteggi CRISPR normalizzati a distinguere il nucleo essenziale dai geni non essenziali. Ogni linea corrisponde ad un esperimento. Un elevato richiamo, pur mantenendo un’elevata precisione, indica una buona prestazione.Confronto dei punteggi CRISPR normalizzati in un diverso background genetico per quattro diverse dipendenze di controllo. Le linee rosse indicano i mezzi del gruppo. La significatività statistica è stata determinata utilizzando un t-test a due facce. Ogni punto dati rappresenta un esperimento di screening. I gruppi sono composti da 12 (KRAS mut.), 44 (KRAS wt), 7 (NRAS mut.), 56 (NRAS wt), 11 (BRAF mut.), 63 (BRAF wt), 15 (PIK3CA mut.) e 59 (PIK3CA wt) punti dati.
E-H.Risultati e controllo qualità dell’integrazione e della normalizzazione dei dati
La mappa termica AA mostra un clustering di punteggi CRISPR normalizzati (media log2 fold change degli sgRNA che prendono di mira un gene) per i geni presenti in ogni libreria di sgRNA utilizzata nelle schermate incluse nell’analisi. Le finestre rettangolari evidenziano gli esperimenti in cui gli schermi eseguiti nella stessa linea cellulare ma in diversi laboratori si raggruppano. Le barre bianche di annotazione indicano le proprietà biologiche condivise delle linee cellulari in ogni cluster. Le barre grigie indicano che la linea cellulare annotata non si adatta all’annotazione di altre linee cellulari nello stesso cluster.BNormalized CRISPR punteggi attraverso gli esperimenti sono visualizzati per un insieme selezionato in modo casuale di nove polimerasi essenziali del nucleo. Ogni punto corrisponde ad una schermata, e colori diversi evidenziano le pubblicazioni da cui sono stati derivati i dati. Punteggi CRISPR più negativi indicano una risposta di vitalità più negativa al knockout del gene.i punteggi CRISPR CNormalized attraverso gli esperimenti in linee cellulari femminili sono visualizzati per un insieme selezionato in modo casuale di nove geni situati sul cromosoma Y che servono come controlli non bersaglio. Colori raffigurano diverse pubblicazioni.DPrecisione-richiamo curve di richiamo che mostrano le prestazioni dei punteggi CRISPR normalizzati a distinguere il nucleo essenziale dai geni non essenziali. Ogni linea corrisponde ad un esperimento. L’elevato richiamo, pur mantenendo un’elevata precisione, indica una buona performance.E-HComparison dei punteggi CRISPR normalizzati in un diverso background genetico per quattro diverse dipendenze di controllo. Le linee rosse indicano i mezzi del gruppo. Significato statistico è stato determinato utilizzando un t-test a due facce di Studente. Ogni punto dati rappresenta un esperimento di screening. I gruppi sono composti da 12 (KRAS mut.), 44 (KRAS wt), 7 (NRAS mut.), 56 (NRAS wt), 11 (BRAF mut.), 63 (BRAF wt), 15 (PIK3CA mut.) e 59 (PIK3CA wt) punti dati.Una mappa termica mostra un clustering di punteggi CRISPR normalizzati (media log2 fold change degli sgRNA che prendono di mira un gene) per i geni presenti in ogni libreria di sgRNA utilizzata nelle schermate incluse nell’analisi. Le finestre rettangolari evidenziano gli esperimenti in cui gli schermi eseguiti nella stessa linea cellulare ma in diversi laboratori si raggruppano. Le barre bianche di annotazione indicano le proprietà biologiche condivise delle linee cellulari in ogni cluster. Le barre grigie indicano che la linea cellulare annotata non si adatta all’annotazione di altre linee cellulari nello stesso cluster.Punteggi CRISPR normalizzati attraverso gli esperimenti sono visualizzati per un insieme selezionato in modo casuale di nove polimerasi essenziali del nucleo. Ogni punto corrisponde ad una schermata, e colori diversi evidenziano le pubblicazioni da cui sono stati derivati i dati. Punteggi CRISPR più negativi indicano una risposta di vitalità più negativa al knockout del gene.Punteggi CRISPR normalizzati attraverso esperimenti in linee cellulari femminili sono visualizzati per un insieme selezionato in modo casuale di nove geni situati sul cromosoma Y che servono come controlli non bersaglio. I colori raffigurano diverse pubblicazioni.Precisione-richiamo le curve di precisione che mostrano le prestazioni dei punteggi CRISPR normalizzati a distinguere il nucleo essenziale dai geni non essenziali. Ogni linea corrisponde ad un esperimento. Un elevato richiamo, pur mantenendo un’elevata precisione, indica una buona prestazione.Confronto dei punteggi CRISPR normalizzati in un diverso background genetico per quattro diverse dipendenze di controllo. Le linee rosse indicano i mezzi del gruppo. La significatività statistica è stata determinata utilizzando un t-test a due facce. Ogni punto dati rappresenta un esperimento di screening. I gruppi sono composti da 12 (KRAS mut.), 44 (KRAS wt), 7 (NRAS mut.), 56 (NRAS wt), 11 (BRAF mut.), 63 (BRAF wt), 15 (PIK3CA mut.) e 59 (PIK3CA wt) punti dati.
Le interazioni tra knockout genici e alterazioni tumorali rivelano mappe di cablaggio genetico
Per determinare le interazioni genetiche, abbiamo formato tutte le combinazioni a coppie tra i geni messi fuori uso da CRISPR/Cas9 negli schermi di vitalità in pool (geni bersaglio) e i geni alterati nelle cellule tumorali (geni di interrogazione) (Fig 1C). Abbiamo considerato i geni come query solo se contengono un’alterazione in almeno tre linee cellulari distinte(Dataset EV2). Un’alterazione del cancro è stata definita come una mutazione somatica, un’alterazione del numero di copie somatiche (SCNA) o espressione differenziale di un gene. Abbiamo messo in comune le alterazioni per ogni gene sulla base di tre ipotesi: Abbiamo ipotizzato che (i) una perdita di numero di copia del gene si comporti in modo simile a una mutazione somatica dirompente (ad esempio, una mutazione frame-shift o una mutazione senza senso), (ii) un guadagno di numero di copia si comporti in modo simile a un guadagno di espressione genica, e che (iii) le mutazioni somatiche dello stesso gene abbiano, in media, una conseguenza funzionale simile. Anche se queste ipotesi, specialmente la numero 3, in realtà non sempre sono vere, abbiamo trovato che siano un’approssimazione utile a giudicare dai risultati ottenuti nelle analisi di interazione genetica a valle. Inoltre, abbiamo ulteriormente perfezionato alcune delle alterazioni genetiche messe in comune mediante cura manuale, escludendo le linee cellulari con alterazioni note per essere funzionalmente dissimili da altre alterazioni dello stesso gene. Questo, tuttavia, è stato possibile solo per i geni ben caratterizzati. In totale, abbiamo formato 3,8 milioni di coppie di geni di 17.218 geni bersaglio e 221 geni di query.
Supponendo che due geni nella maggior parte dei casi non interagiscano tra loro, abbiamo prima eseguito un test statistico per ogni coppia di geni, confrontando i punteggi CRISPR normalizzati di cellule che contengono un’alterazione del gene di interrogazione con cellule che non contengono l’alterazione. In questo caso, abbiamo usato un modello multilivello che includeva la linea cellulare corrispondente ad ogni punto di dati come effetto casuale per tener conto dei pregiudizi che potenzialmente potevano essere introdotti quando una linea cellulare veniva schermata più volte. In alcuni casi, abbiamo osservato un’elevata correlazione tra diversi geni di interrogazione (Fig EV2A).Questa osservazione può, ad esempio, essere spiegata da una co-delezione di geni che si trovano uno vicino all’altro sul genoma. Per esempio, il CDKN2A, un gene soppressore di tumori (Liggett & Sidransky, 1998 ) localizzato sulla banda cromosomica 9p21, è spesso co-cancellato con i geni circostanti (Muller et al, 2015). In questi casi, non è possibile determinare con quale dei due potenziali geni interrogatori un gene target dovrebbe essere previsto interagire. Abbiamo affrontato questo problema aggregando geni di query identici, determinati dalla correlazione dei loro coefficienti del modello, in “meta-geni” che abbiamo poi utilizzato per le analisi a valle (Fig EV2B). Per quantificare la forza di interazione di ogni coppia di geni, abbiamo calcolato i punteggi π (Fig 3A e B)comedescritto in precedenza (Horn et al, 2011 ; Laufer et al, 2013; Fischer et al, 2015). Complessivamente, la nostra analisi ha previsto 17.545 interazioni gene-gene a FDR < 20% (0,8% del totale delle combinazioni testate dopo l’aggregazione di meta-gene).
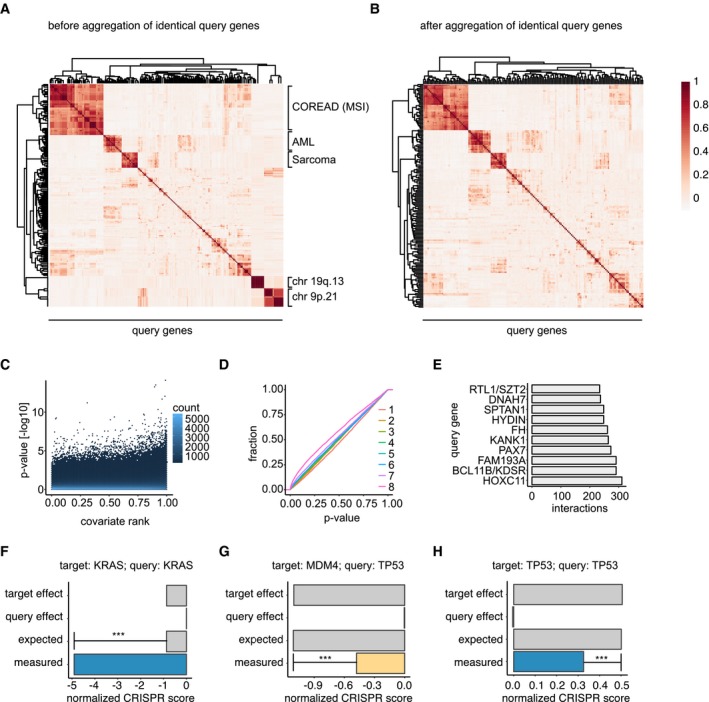
F-H.Previsione delle interazioni genetiche tra coppie di geni
Corrispondenze tra i geni dell’interrogazione. I cluster di geni di query altamente correlati sono etichettati indicando la fonte della correlazione.BQuery correlazioni dopo l’aggregazione di tutti i geni di query correlati (coefficiente di correlazione di Pearson = 1).CScatter plot di valori P in scala log10 contro il rango covariato di ogni coppia di geni come usato per la correzione di test multipli utilizzando IHW. La covariata è la varianza dei punteggi CRISPR del gruppo di linee cellulari mutate.DEmpirical cumulative distribution function plot of P-values of genetic interactions based on the covariate.ETen query genes with the highest predicted interaction count. Simboli di geni multipli separati da barre in avanti indicano i geni di query che sono stati aggregati a causa di alta correlazione.F-HEpistasis trame per tre esempi di interazioni genetiche conosciute. Il colore blu indica un’interazione negativa e il giallo indica un’interazione positiva. Per determinare un’interazione genetica, viene quantificata la differenza tra il fenotipo combinato misurato e quello atteso. La significatività statistica è stata determinata utilizzando un modello lineare di effetti misti con il metodo lmerTest. ***, FDR < 0,0001.Correlazioni a coppie tra i geni dell’interrogazione. I cluster di geni di query altamente correlati sono etichettati indicando la fonte della correlazione.Correlazioni di query dopo l’aggregazione di geni query completamente correlati (coefficiente di correlazione di Pearson = 1).Scatter plot di valori P in scala log10 contro il rango covariato di ogni coppia di geni, come usato per la correzione di test multipli usando IHW. La covariata è la varianza dei punteggi CRISPR del gruppo di linee cellulari mutate.Trama di funzione di distribuzione cumulativa empirica dei valori P delle interazioni genetiche basate sulla covariata.Dieci geni di query con il più alto numero di interazioni previste. Simboli di geni multipli separati da barre in avanti indicano geni di query che sono stati aggregati a causa di un’elevata correlazione.Trame di epistasi per tre esempi di interazioni genetiche conosciute. Il colore blu indica un’interazione negativa e il giallo indica un’interazione positiva. Per determinare un’interazione genetica, viene quantificata la differenza tra il fenotipo combinato misurato e quello atteso. La significatività statistica è stata determinata utilizzando un modello lineare di effetti misti con il metodo lmerTest. ***, FDR < 0,0001.
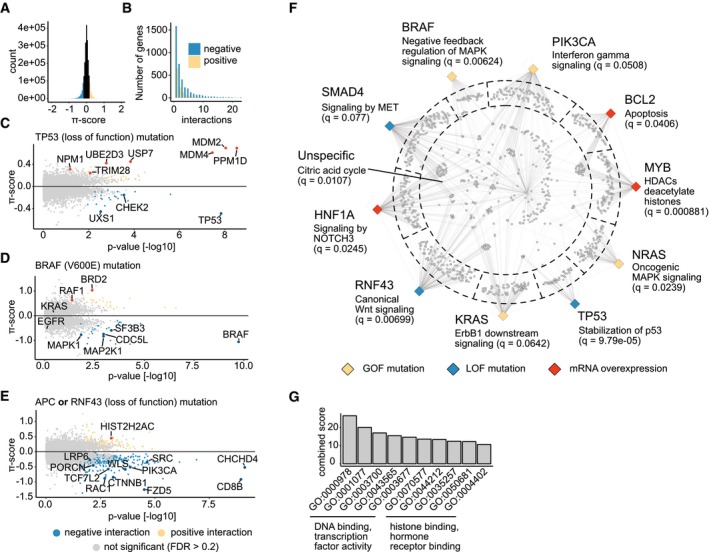
G.Risultati delle interazioni genetiche previste
ADistribuzione dei punti π calcolati per ogni interazione a coppie. I valori negativi indicano interazioni negative (interazioni aggravanti), e i valori positivi indicano interazioni positive (buffering). I valori > 0,2 e < -0,2 sono colorati rispettivamente in giallo e blu.BIl numero di interazioni positive e negative per gene. Le interazioni con un π-score > 0.2 sono considerate positive, e le interazioni con un π-score di < -0.2 sono considerate negative.I grafici C-EVolcano che mostrano i geni che interagiscono con le mutazioni a perdita di funzione (C) di TP53, le mutazioni BRAF V600E (D) e le mutazioni a perdita di funzione APC o RNF43 (E). Ogni punto corrisponde a un gene. Le interazioni che sono significative a FDR < 0,2 sono colorate in blu nel caso in cui l’interazione sia negativa o gialla se è positiva. I geni selezionati sono evidenziati ed etichettati.FA grafico di rete che mostra i risultati di arricchimento del set di geni per i gruppi di partner di interazione. Ciascuno dei diamanti colorati corrisponde a una delle 10 alterazioni della query selezionate. Il colore di ogni diamante indica il tipo di alterazione come descritto nella legenda in basso. Ogni punto grigio collegato a uno o più nodi del gene della query rappresenta un gene target che interagisce (FDR < 0.2) con la query. L’analisi di arricchimento del set di geni è stata eseguita per i geni che cadono nello stesso compartimento come indicato dalla linea tratteggiata. I geni nei compartimenti verso il bordo interagiscono con una query specifica. I geni posizionati al centro del cerchio hanno un profilo di interazione più promiscuo. I termini di percorso arricchiti selezionati sono usati per etichettare i nodi del gene della query.I termini GGO arricchiti tra i 40 geni della query con il maggior numero di interazioni (|π| > 0.2, FDR < 0.2).Distribuzione dei punti π calcolati per ogni interazione a coppie. I valori negativi indicano interazioni negative (interazioni aggravanti), e i valori positivi indicano interazioni positive (buffering). I valori > 0.2 e < -0.2 sono colorati rispettivamente in giallo e blu.Il numero di interazioni positive e negative per gene. Le interazioni con un π-score > 0,2 sono considerate positive, e le interazioni con un π-score di < -0,2 sono considerate negative.Trame di vulcano che mostrano geni che interagiscono con le mutazioni a perdita di funzione (C) del TP53, le mutazioni BRAF V600E (D) e le mutazioni a perdita di funzione APC o RNF43 (E). Ogni punto corrisponde a un gene. Le interazioni che sono significative a FDR < 0,2 sono colorate in blu nel caso in cui l’interazione sia negativa o gialla se è positiva. I geni selezionati sono evidenziati ed etichettati.Un grafico di rete che mostra i risultati dell’arricchimento dei set di geni per i set di partner di interazione. Ciascuno dei diamanti colorati corrisponde a una delle 10 alterazioni della query selezionate. Il colore di ogni diamante indica il tipo di alterazione come descritto nella legenda in basso. Ogni punto grigio collegato a uno o più nodi del gene della query rappresenta un gene target che interagisce (FDR < 0.2) con la query. L’analisi di arricchimento del set di geni è stata eseguita per i geni che cadono nello stesso compartimento come indicato dalla linea tratteggiata. I geni nei compartimenti verso il bordo interagiscono con una query specifica. I geni posizionati al centro del cerchio hanno un profilo di interazione più promiscuo. Per etichettare i nodi dei geni della query vengono utilizzati termini di percorso arricchiti selezionati.I termini GO arricchiti tra i 40 geni della query con il maggior numero di interazioni (|π| > 0.2, FDR < 0.2).
Esaminando le interazioni proposte, abbiamo scoperto che la nostra analisi è stata in grado di recuperare molte dipendenze precedentemente caratterizzate attraverso diversi percorsi che sono stati ampiamente studiati in passato (Figg. 3 e EV2F-H). Per esempio, abbiamo identificato molte interazioni positive (cioè, le cellule contenenti un’alterazione del gene di interrogazione sono più resistenti alla perturbazione del gene target) tra TP53 e diversi geni coinvolti nella stabilizzazione della proteina p53 (Fig 3C). Nelle cellule di tipo selvatico, la p53 è mantenuta a bassa abbondanza dalle ligasi ubiquitiniche E3/E4, tra cui, ad esempio, MDM2 e MDM4 ( Fig EV2G),chepossono mediarne la degradazione attraverso il proteasoma (Lavin & Gueven, 2006; Frum & Grossman, 2014). L’eliminazione di queste ligasi ubiquitiniche porta probabilmente ad un accumulo di p53, che potrebbe quindi mediare l’apoptosi e impedire la proliferazione con conseguente fenotipo di vitalità negativa. Nelle cellule tumorali, le mutazioni missenso del gene TP53 possono inibire la degradazione della p53 (Lavin & Gueven, 2006 ; Frum& Grossman, 2014) dove può accumularsi e agire come un oncogene (Oren & Rotter, 2010), che potrebbe spiegare la resistenza delle linee cellulari mutilate con TP53 alla perdita di E2/E3 ubiquitina ligasi. Un’interazione che a prima vista potrebbe sembrare sorprendente è un’interazione negativa del TP53 con se stesso (cioè, le cellule con una mutazione TP53 sono più sensibili al knockout del TP53 ). Nel contesto dell’epistasi, tuttavia, questo potrebbe essere spiegato dal fatto che nelle cellule di tipo selvaggio della TP53, dove la TP53 agisce come soppressore di tumori, il suo knockout porta ad un guadagno di fenotipo di vitalità, che non è il caso delle cellule tumorali che già ospitano mutazioni nella TP53 ( Fig EV2H). Successivamente, abbiamo esaminato le interazioni previste dell’oncogene BRAF. Non sorprende che abbiamo trovato interazioni negative con il BRAF stesso così come MAP2K1 (MEK1) e MAPK1 (ERK2), entrambi si trovano a valle del BRAF nella cascata di segnalazione MAPK (Seger & Krebs, 1995 ). Al contrario, non sono state trovate interazioni per le componenti a monte del percorso come KRAS o EGFR (Fig 3D), probabilmente perché l’attivazione costitutiva del BRAF causata dalla sua mutazione conferisce indipendenza alle componenti del percorso a monte. In seguito a studi precedenti (Brockmann et al, 2017) , abbiamo ragionato sul fatto che i geni che interagiscono specificamente con uno o pochi geni di query correlati dovrebbero essere funzionalmente correlati. Abbiamo quindi selezionato dieci geni di query, compresi i loro partner di interazione previsti a FDR < 20% ed eseguito un’analisi di sovrarappresentazione dei set di geni (Kamburov et al, 2013) per gruppi di geni target che interagiscono specificamente con una delle query selezionate (Fig 3F). Guardando i percorsi sovrarappresentati all’interno dell’insieme di geni analizzati, abbiamo trovato diverse relazioni ben caratterizzate che collegano, per esempio, mutazioni di KRAS, NRAS , o BRAF alla segnalazione MAPK, BCL2 all’apoptosi o TP53 alla stabilizzazione della stessa, suggerendo un elevato numero di vere previsioni. Inoltre, la nostra analisi propone interazioni genetiche per molti altri geni di query meno ben studiati (un elenco completo delle interazioni previste si trova nel Dataset EV3). Per trovare tratti condivisi tra i geni di query per i quali sono stati previsti elevati numeri di interazioni (Fig EV2E),abbiamo eseguito l’analisi di GO (Ashburner et al , 2000) per l’arricchimento delle funzioni molecolari(Kuleshov et al, 2016) . Non sorprende che abbiamo scoperto che i termini GO con i punteggi di arricchimento più alti sono stati correlati all’attività del fattore di trascrizione (Fig 3G). Altri termini GO di alto rango erano correlati al rimodellamento della cromatina e al legame dei recettori ormonali.
Abbiamo ipotizzato che dovrebbe essere possibile combinare i geni dell’interrogazione funzionalmente correlati per migliorare la previsione dei regolatori dei percorsi di segnalazione. Di conseguenza, abbiamo combinato la perdita di mutazioni funzionali dei geni APC e RNF43(Dataset EV3) in un metagene di query “Wnt mutation”. Entrambi, APC e RNF43, sono regolatori negativi frequentemente mutati del percorso di segnalazione Wnt/β-catenina (Polakis, 2012; de Lau et al, 2014; Tsukiyama et al, 2015; Zhan et al, 2017 )-un percorso che è aberrantemente regolato in vari tipi di cancro (Polakis, 2012; Giannakis et al, 2014; Zhan et al, 2017). In assenza di ligandi Wnt, l’APC regola l’attività della β-catenina attraverso la formazione di un complesso di distruzione con GSK3β e Axin1, che media la fosforilazione della β-catenina. Fosforilato β-catenina è poi mirata per la degradazione da parte del proteasoma. Il legame dei Wnts canonici ai recettori frizzled e ai co-recettori LRP5/6 sulla superficie cellulare inibisce la formazione del complesso di distruzione, che si traduce in stabilizzazione della β-catenina e la sua traslocazione al nucleo. All’interno del nucleo, la β-catenina interagisce con i fattori di trascrizione TCF/LEF e attiva la trascrizione dei geni target Wnt, che mediano la crescita cellulare e la sopravvivenza (MacDonald et al, 2009). RNF43 è una ligasi ubiquitinica E3 che può indurre l’ubiquitinazione e la conseguente degradazione del complesso Wnt-Frizzled (MacDonald et al, 2009; Clevers & Nusse, 2012), inibendo così la segnalazione della β-catenina. Di conseguenza, le mutazioni dirompenti in APC o RNF43 possono promuovere l’attivazione del percorso. Esaminando i geni che si prevede interagiscano con le mutazioni a perdita di funzione di APC o RNF43, abbiamo osservato molti regolatori noti della segnalazione di Wnt/β-catenina (Fig 3E). Tra questi, abbiamo identificato, per esempio, i regolatori della secrezione del ligando Wnt, TCF7L2 e CTNNB1 che insieme formano il complesso del fattore di trascrizione TCF/β-catenina, e altri geni, che sono stati precedentemente collegati al percorso Wnt/β-catenina (Chen et al, 2014; Ormanns et al, 2014).
F-H.Previsione delle interazioni genetiche tra coppie di geni
Corrispondenze tra i geni dell’interrogazione. I cluster di geni di query altamente correlati sono etichettati indicando la fonte della correlazione.BQuery correlazioni dopo l’aggregazione di tutti i geni di query correlati (coefficiente di correlazione di Pearson = 1).CScatter plot di valori P in scala log10 contro il rango covariato di ogni coppia di geni come usato per la correzione di test multipli utilizzando IHW. La covariata è la varianza dei punteggi CRISPR del gruppo di linee cellulari mutate.DEmpirical cumulative distribution function plot of P-values of genetic interactions based on the covariate.ETen query genes with the highest predicted interaction count. Simboli di geni multipli separati da barre in avanti indicano i geni di query che sono stati aggregati a causa di alta correlazione.F-HEpistasis trame per tre esempi di interazioni genetiche conosciute. Il colore blu indica un’interazione negativa e il giallo indica un’interazione positiva. Per determinare un’interazione genetica, viene quantificata la differenza tra il fenotipo combinato misurato e quello atteso. La significatività statistica è stata determinata utilizzando un modello lineare di effetti misti con il metodo lmerTest. ***, FDR < 0,0001.Correlazioni a coppie tra i geni dell’interrogazione. I cluster di geni di query altamente correlati sono etichettati indicando la fonte della correlazione.Correlazioni di query dopo l’aggregazione di geni query completamente correlati (coefficiente di correlazione di Pearson = 1).Scatter plot dei valori P in scala log10 contro il rango covariato di ogni coppia di geni, come usato per la correzione di test multipli usando IHW. La covariata è la varianza dei punteggi CRISPR del gruppo di linee cellulari mutate.Trama di funzione di distribuzione cumulativa empirica dei valori P delle interazioni genetiche basate sulla covariata.Dieci geni di query con il più alto numero di interazioni previste. Simboli di geni multipli separati da barre in avanti indicano geni di query che sono stati aggregati a causa di un’elevata correlazione.Trame di epistasi per tre esempi di interazioni genetiche conosciute. Il colore blu indica un’interazione negativa e il giallo indica un’interazione positiva. Per determinare un’interazione genetica, viene quantificata la differenza tra il fenotipo combinato misurato e quello atteso. La significatività statistica è stata determinata utilizzando un modello lineare di effetti misti con il metodo lmerTest. ***, FDR < 0,0001.
G.Risultati delle interazioni genetiche previste
ADistribuzione dei punti π calcolati per ogni interazione a coppie. I valori negativi indicano interazioni negative (interazioni aggravanti), e i valori positivi indicano interazioni positive (buffering). I valori > 0,2 e < -0,2 sono colorati rispettivamente in giallo e blu.BIl numero di interazioni positive e negative per gene. Le interazioni con un π-score > 0.2 sono considerate positive, e le interazioni con un π-score di < -0.2 sono considerate negative.I grafici C-EVolcano che mostrano i geni che interagiscono con le mutazioni a perdita di funzione (C) di TP53, le mutazioni BRAF V600E (D) e le mutazioni a perdita di funzione APC o RNF43 (E). Ogni punto corrisponde a un gene. Le interazioni che sono significative a FDR < 0,2 sono colorate in blu nel caso in cui l’interazione sia negativa o gialla se è positiva. I geni selezionati sono evidenziati ed etichettati.FA grafico di rete che mostra i risultati di arricchimento del set di geni per i gruppi di partner di interazione. Ciascuno dei diamanti colorati corrisponde a una delle 10 alterazioni della query selezionate. Il colore di ogni diamante indica il tipo di alterazione come descritto nella legenda in basso. Ogni punto grigio collegato a uno o più nodi del gene della query rappresenta un gene target che interagisce (FDR < 0.2) con la query. L’analisi di arricchimento del set di geni è stata eseguita per i geni che cadono nello stesso compartimento come indicato dalla linea tratteggiata. I geni nei compartimenti verso il bordo interagiscono con una query specifica. I geni posizionati al centro del cerchio hanno un profilo di interazione più promiscuo. I termini di percorso arricchiti selezionati sono usati per etichettare i nodi del gene della query.I termini GGO arricchiti tra i 40 geni della query con il maggior numero di interazioni (|π| > 0.2, FDR < 0.2).Distribuzione dei punti π calcolati per ogni interazione a coppie. I valori negativi indicano interazioni negative (interazioni aggravanti), e i valori positivi indicano interazioni positive (buffering). I valori > 0.2 e < -0.2 sono colorati rispettivamente in giallo e blu.Il numero di interazioni positive e negative per gene. Le interazioni con un π-score > 0,2 sono considerate positive, e le interazioni con un π-score di < -0,2 sono considerate negative.Trame di vulcano che mostrano geni che interagiscono con mutazioni a perdita di funzione (C) di TP53, mutazioni BRAF V600E (D) e mutazioni a perdita di funzione APC o RNF43 (E). Ogni punto corrisponde a un gene. Le interazioni che sono significative a FDR < 0,2 sono colorate in blu nel caso in cui l’interazione sia negativa o gialla se è positiva. I geni selezionati sono evidenziati ed etichettati.Un grafico di rete che mostra i risultati dell’arricchimento dei set di geni per i set di partner di interazione. Ciascuno dei diamanti colorati corrisponde a una delle 10 alterazioni della query selezionate. Il colore di ogni diamante indica il tipo di alterazione come descritto nella legenda in basso. Ogni punto grigio collegato a uno o più nodi del gene della query rappresenta un gene target che interagisce (FDR < 0.2) con la query. L’analisi di arricchimento del set di geni è stata eseguita per i geni che cadono nello stesso compartimento come indicato dalla linea tratteggiata. I geni nei compartimenti verso il bordo interagiscono con una query specifica. I geni posizionati al centro del cerchio hanno un profilo di interazione più promiscuo. Per etichettare i nodi dei geni della query vengono utilizzati termini di percorso arricchiti selezionati.I termini GO arricchiti tra i 40 geni della query con il maggior numero di interazioni (|π| > 0.2, FDR < 0.2).
L’analisi di dipendenza delle alterazioni del percorso Wnt rivela nuovi regolatori di segnalazione Wnt/β-catenina
Abbiamo ipotizzato che tra i modulatori noti della segnalazione Wnt/β-catenina, la nostra analisi dovrebbe anche identificare i regolatori di percorso finora sconosciuti. Mutazioni inattivanti del gene RNF43, per esempio, hanno dimostrato in precedenza di conferire dipendenza dalla segnalazione di Wnt/β-catenina (Jiang et al, 2013; Steinhart et al, 2017), per cui abbiamo ragionato che le interazioni negative del gene RNF43 potrebbero indicare regolatori di percorso positivi. Oltre ai noti regolatori di percorso Wnt, la nostra analisi ha rivelato interazioni negative tra RNF43 e diversi geni sconosciuti(Dataset EV3). Abbiamo cercato di convalidare sperimentalmente queste previsioni e abbiamo proceduto selezionando tre geni candidati ad alto punteggio di cui è stato segnalato il coinvolgimento nella glicosilazione delle proteine (D’Alessio & Dahms, 2015) per il follow-up (Fig 4A). Due di questi geni, PRKCSH e GANAB, insieme formano la glucosidasi eterodimerica II. Il terzo candidato, UGP2, è coinvolto nella sintesi dei carboidrati (Wang et al, 2016) . Abbiamo abbattuto ciascuno dei geni candidati utilizzando almeno tre diversi siRNA (Figg. 4BeEV3B, Materiali e Metodi) o un pool costituito dagli stessi reagenti nelle cellule HEK293T (Fig. 4B) (Thomas& Smart, 2005). Le cellule HEK293T sono state scelte come un modello consolidato per l’attivazione canonica di segnalazione Wnt, che non ospitano mutazioni note nel percorso Wnt. Inoltre, le cellule HEK293T sono dotate di uno stato inattivo di segnalazione Wnt canonica, motivo per cui il percorso può essere attivato da una sovraespressione di diversi componenti chiave (Wnt3, Dvl3, e β-catenina).
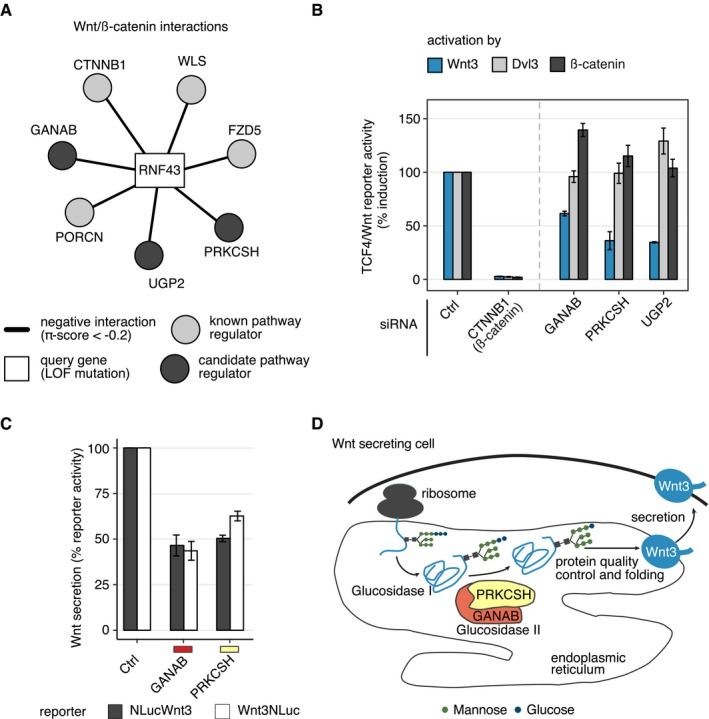
Figura 4.I geni candidati GANAB e PRKCSH regolano la secrezione Wnt
Tre geni candidati (cerchi grigio scuro) interagiscono con il gene di interrogazione RNF43 (rettangolo), simile a componenti di percorso ben caratterizzati (cerchi grigi chiari).cellule HEK293T sono stati invertiti trasfettati con pool di siRNA di targeting per i geni etichettati sull’asse x. 24 ore dopo la trasfezione, la segnalazione Wnt è stata attivata dalla sovraespressione di Wnt3, Dvl3, o β-catenina plasmidi. Il segnale TCF4/Wnt Firefly luciferasi è stato normalizzato al segnale actina-Renilla. I risultati sono mostrati come medie di 3-4 esperimenti indipendenti ± s.e.m.HEK293T cellule sono stati invertiti trasfettati con siRNA in pool di targeting GANAB o PRKCSH. Dopo 24 ore, i costrutti indicati Wnt3 NanoLuciferase Wnt3 sono stati trasfettati insieme con un reporter CMV Firefly luciferasi. 48 ore dopo, i segnali di luciferasi sono stati misurati nel mezzo e lisato. Attività % reporter denota il segnale Wnt3 NanoLuciferase nel medio normalizzato a NanoLuciferase e Firefly segnali luciferasi nel lisato. I risultati sono mostrati come medie di tre esperimenti indipendenti ± s.e.m.Rappresentazione schematica di un ipotetico meccanismo in cui la secrezione Wnt3 è controllata dalla glucosidasi II.Tre geni candidati (cerchi grigio scuro) interagiscono con il gene della query RNF43 (rettangolo), simile a componenti di percorso ben caratterizzati (cerchi grigio chiaro).Le cellule HEK293T sono state sottoposte a trasfezione inversa con pool di siRNA che prendono di mira i geni etichettati sull’asse x. 24 ore dopo la trasfezione, la segnalazione Wnt è stata attivata dalla sovraespressione di Wnt3, Dvl3, o β-catenina plasmidi. Il segnale TCF4/Wnt Firefly luciferasi è stato normalizzato al segnale actina-Renilla. I risultati sono mostrati come medie di 3-4 esperimenti indipendenti ± s.e.m.HEK293T cellule HEK293T sono stati invertiti trasfettati con siRNA in pool di targeting GANAB o PRKCSH. Dopo 24 ore, i costrutti indicati Wnt3 NanoLuciferase Wnt3 sono stati trasfettati insieme con un reporter CMV Firefly luciferasi. 48 ore dopo, i segnali di luciferasi sono stati misurati nel mezzo e lisato. Attività % reporter denota il segnale Wnt3 NanoLuciferase nel medio normalizzato a NanoLuciferase e Firefly segnali luciferasi nel lisato. I risultati sono mostrati come medie di tre esperimenti indipendenti ± s.e.m.Rappresentazione schematica di un ipotetico meccanismo in cui la secrezione Wnt3 è controllata dalla glucosidasi II.
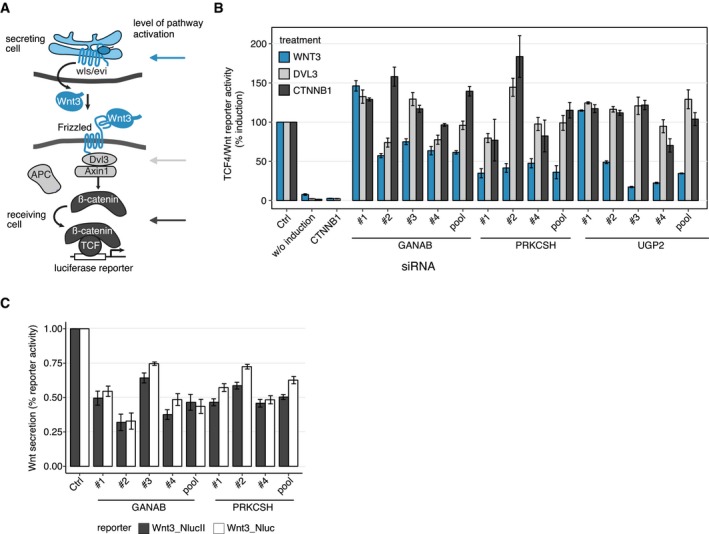
Figura EV3.Analisi dei regolatori candidati di segnalazione Wnt/β-catenina
Schema che spiega il saggio Wnt/TCF4 utilizzato per determinare l’influenza dei geni candidati sulla segnalazione di Wnt/β-catenina. Le frecce indicano i livelli di attivazione del percorso tramite sovraespressione di WNT3 (blu), DVL3 (grigio), o CTNNB1/β-catenina (nero).dati completi per il saggio di attività Wnt presentato nella Fig 4B. I gruppi di barre indicano gli effetti dei singoli siRNA che colpiscono i geni candidati. I risultati sono mostrati come medie di 3-4 esperimenti indipendenti ± s.e.m.Dati completi per il saggio sulla secrezione di Wnt presentato in Fig 4C. Gruppi di barre illustrano l’effetto delle singole perturbazioni di siRNA. I risultati sono mostrati come medie di tre esperimenti indipendenti ± s.e.m.Schema che spiega il saggio Wnt/TCF4 reporter utilizzato per determinare l’influenza dei geni candidati sulla segnalazione Wnt/β-catenina. Le frecce indicano i livelli di attivazione del percorso tramite sovraespressione di WNT3 (blu), DVL3 (grigio) o CTNNB1/β-catenina (nero).Dati completi per il saggio di attività Wnt presentato in Fig 4B. I gruppi di barre indicano gli effetti dei singoli siRNA che colpiscono i geni candidati. I risultati sono mostrati come medie di 3-4 esperimenti indipendenti ± s.e.m.I dati completi per il saggio sulla secrezione Wnt presentati nella Fig 4C. Gruppi di barre illustrano l’effetto delle singole perturbazioni di siRNA. I risultati sono mostrati come medie di tre esperimenti indipendenti ± s.e.m.
La sovraespressione di Wnt3 imita l’attivazione automatica e paracrina della segnalazione Wnt canonica a livello della cellula di secrezione Wnt che ha dimostrato di dipendere dai componenti Wnt-secretariali Porcn e Evi/Wls (Bänziger et al, 2006; Bartscherer et al, 2006; Bartscherer & Boutros, 2008; Herr & Basler, 2012) . Al contrario, la sovraespressione del Dvl3 induce il percorso a valle del complesso recettoriale nelle cellule riceventi. Sovraespressione di β-catenina porta all’attivazione della via a valle di APC (Figg. 4B e EV3A). Abbiamo osservato che l’abbattimento di ciascuno dei geni candidati testati seguito da un’attivazione della via indotta dall’espressione Wnt3 ha portato ad una forte riduzione dell’attivazione di un reporter TCF4/Wnt, che imita l’attivazione della trascrizione dei geni regolati da β-catenina (Fig 4B). È interessante notare che l’abbattimento di GANAB, PRKCSH, o UGP2 non ha mostrato un forte effetto sull’attività del reporter o anche una maggiore induzione alla trasfezione con plasmidi di espressione Dvl3 o β-catenina (Fig 4B). Questi risultati permettono di concludere un’interferenza dei candidati indagati a livello di secrezione Wnt o a livello dei recettori, poiché l’effetto negativo sull’attività Wnt viene abolito in caso di ulteriore attivazione della via a valle con Dvl3 o β-catenina.
Per indagare ulteriormente il ruolo del complesso della glucosidasi II e da questa glicosilazione proteica, la secrezione e il controllo di qualità della glicoproteina che si ripiega in ER nel contesto della segnalazione Wnt, abbiamo eseguito un test di secrezione Wnt al momento dell’abbattimento di PRKCSH e GANAB (Fig 4D; D‘Alessio & Dahms, 2015). Per questo, abbiamo accoppiato Wnt3 a una sequenza di NanoLuciferase (Hall et al, 2012) all’interno di un plasmide di espressione Wnt3. La sequenza NanoLuciferasi è stata integrata o dopo il peptide di segnale (NLucWnt3) o al C-termino di Wnt3 (Wnt3NLuc) per escludere un effetto di accoppiamento NanoLuciferasi sulla secrezione Wnt3. Una lettura della NanoLuciferasi ha successivamente permesso di rilevare le proteine Wnt3 secrete nel supernatante della coltura cellulare e di normalizzarla alla quantità di Wnt3 nel lisato cellulare. Dopo l’abbattimento di GANAB o PRKCSH, la secrezione di Wnt3 è stata ridotta di circa il 40-50% utilizzando i costrutti NLucWnt3 o Wnt3NLuc (figg. 4C e EV3C). Questi dati confermano una necessità già pubblicata della glicosilazione del ligando Wnt per la secrezione delle proteine Wnt (Fig 4D; Komekado et al , 2007).
Figura 4.I geni candidati GANAB e PRKCSH regolano la secrezione di Wnt
Tre geni candidati (cerchi grigio scuro) interagiscono con il gene di interrogazione RNF43 (rettangolo), simile a componenti di percorso ben caratterizzati (cerchi grigi chiari).cellule HEK293T sono stati invertiti trasfettati con pool di siRNA di targeting per i geni etichettati sull’asse x. 24 ore dopo la trasfezione, la segnalazione Wnt è stata attivata dalla sovraespressione di Wnt3, Dvl3, o β-catenina plasmidi. Il segnale TCF4/Wnt Firefly luciferasi è stato normalizzato al segnale actina-Renilla. I risultati sono mostrati come medie di 3-4 esperimenti indipendenti ± s.e.m.HEK293T cellule sono stati invertiti trasfettati con siRNA in pool di targeting GANAB o PRKCSH. Dopo 24 ore, i costrutti indicati Wnt3 NanoLuciferase Wnt3 sono stati trasfettati insieme con un reporter CMV Firefly luciferasi. 48 ore dopo, i segnali di luciferasi sono stati misurati nel mezzo e lisato. Attività % reporter denota il segnale Wnt3 NanoLuciferase nel medio normalizzato a NanoLuciferase e Firefly segnali luciferasi nel lisato. I risultati sono mostrati come medie di tre esperimenti indipendenti ± s.e.m.Rappresentazione schematica di un ipotetico meccanismo in cui la secrezione Wnt3 è controllata dalla glucosidasi II.Tre geni candidati (cerchi grigio scuro) interagiscono con il gene di interrogazione RNF43 (rettangolo), simile a componenti di percorso ben caratterizzati (cerchi grigio chiaro).Le cellule HEK293T sono state sottoposte a trasfezione inversa con pool di siRNA che prendono di mira i geni etichettati sull’asse x. 24 ore dopo la trasfezione, la segnalazione Wnt è stata attivata dalla sovraespressione di Wnt3, Dvl3, o β-catenina plasmidi. Il segnale TCF4/Wnt Firefly luciferasi è stato normalizzato al segnale actina-Renilla. I risultati sono mostrati come medie di 3-4 esperimenti indipendenti ± s.e.m.HEK293T cellule HEK293T sono stati invertiti trasfettati con siRNA in pool di targeting GANAB o PRKCSH. Dopo 24 ore, i costrutti indicati Wnt3 NanoLuciferase Wnt3 sono stati trasfettati insieme con un reporter CMV Firefly luciferasi. 48 ore dopo, i segnali di luciferasi sono stati misurati nel mezzo e lisato. Attività % reporter denota il segnale Wnt3 NanoLuciferase nel medio normalizzato a NanoLuciferase e Firefly segnali luciferasi nel lisato. I risultati sono mostrati come medie di tre esperimenti indipendenti ± s.e.m.Rappresentazione schematica di un ipotetico meccanismo in cui la secrezione Wnt3 è controllata dalla glucosidasi II.
Figura EV3.Analisi dei regolatori candidati di segnalazione Wnt/β-catenina
Schema che spiega il saggio Wnt/TCF4 utilizzato per determinare l’influenza dei geni candidati sulla segnalazione di Wnt/β-catenina. Le frecce indicano i livelli di attivazione del percorso tramite sovraespressione di WNT3 (blu), DVL3 (grigio), o CTNNB1/β-catenina (nero).dati completi per il saggio di attività Wnt presentato nella Fig 4B. I gruppi di barre indicano gli effetti dei singoli siRNA che colpiscono i geni candidati. I risultati sono mostrati come medie di 3-4 esperimenti indipendenti ± s.e.m.Dati completi per il saggio sulla secrezione di Wnt presentato in Fig 4C. Gruppi di barre illustrano l’effetto delle singole perturbazioni di siRNA. I risultati sono mostrati come medie di tre esperimenti indipendenti ± s.e.m.Schema che spiega il saggio Wnt/TCF4 reporter utilizzato per determinare l’influenza dei geni candidati sulla segnalazione Wnt/β-catenina. Le frecce indicano i livelli di attivazione del percorso tramite sovraespressione di WNT3 (blu), DVL3 (grigio) o CTNNB1/β-catenina (nero).Dati completi per il saggio di attività Wnt presentato in Fig 4B. I gruppi di barre indicano gli effetti dei singoli siRNA che colpiscono i geni candidati. I risultati sono mostrati come medie di 3-4 esperimenti indipendenti ± s.e.m.I dati completi per il saggio sulla secrezione Wnt presentati nella Fig 4C. Gruppi di barre illustrano l’effetto delle singole perturbazioni di siRNA. I risultati sono mostrati come medie di tre esperimenti indipendenti ± s.e.m.
La somiglianza dei profili di interazione predice le relazioni funzionali dei geni
Diversi studi hanno precedentemente dimostrato che geni funzionalmente simili possono essere identificati confrontando i loro profili di interazione. Qui, i vettori dei punteggi di interazione tra i geni della query vengono confrontati per tutte le possibili coppie di geni target utilizzando una misura di somiglianza – più comunemente la loro correlazione. Due geni target con profili di interazione altamente correlativi sono quindi previsti per condividere la funzione biologica attraverso il senso di colpa per associazione (Fig 1D). Incoraggiati dall’osservazione dell’arricchimento del percorso tra i geni target che si prevede interagiscano con la stessa query, abbiamo ragionato che un’analisi della somiglianza dei profili di interazione dovrebbe essere possibile anche sulla base dei nostri risultati, nonostante un numero relativamente basso di geni query (167 dopo l’aggregazione di geni query altamente simili). Di conseguenza, abbiamo correlato i coefficienti di correlazione di Pearson dei profili di interazione π-score per tutte le combinazioni a coppie di geni target. Abbiamo ragionato sul fatto che i dati sulla co-membership di complessi proteici conosciuti dovrebbero essere in grado di servire come riferimento per stimare il potere predittivo del nostro approccio. Quindi, abbiamo scaricato tutti i dati sul complesso proteico umano dal database CORUM (Ruepp et al, 2010) e abbiamo confrontato le nostre associazioni previste con i dati noti sul complesso proteico per analisi delle caratteristiche dell’operatore ricevente (ROC). Inizialmente, questa analisi ha rivelato che le nostre previsioni di co-associazione del complesso proteico non erano soddisfacenti. Dopo un’attenta ispezione delle relazioni previste, abbiamo notato che il coefficiente di correlazione è stato nella maggior parte dei casi notevolmente influenzato da piccolissimi π-score. Tali punti di dati non contengono molte informazioni biologiche in quanto indicano semplicemente che potrebbe non esserci alcuna connessione tra un target e un gene di interrogazione basato su un fenotipo di vitalità. Quindi, abbiamo ipotizzato che escludendo le interazioni con punteggi π molto bassi, si dovrebbe spostare più peso su punti di dati più informativi e dovrebbe quindi portare a previsioni più significative di co-funzionalità. Di conseguenza abbiamo escluso tutte le interazioni con π-score < 0.2 e abbiamo ripetuto l’analisi di cui sopra. Poiché l’esclusione delle interazioni con un basso π-score viola l’assunto di normalità della correlazione di Pearson, abbiamo usato invece la correlazione non parametrica di Spearman. Abbiamo calcolato questa correlazione per tutte le coppie di geni bersaglio in cui erano disponibili almeno cinque punti di dati completi per coppia. Ripetendo l’analisi ROC come descritto in precedenza, è emerso un notevole miglioramento delle previsioni risultanti che ha portato a risultati superiori all’assegnazione casuale (Fig 5A). Al fine di identificare le soglie di parametro più adatte, abbiamo ripetuto sistematicamente questa analisi utilizzando diverse combinazioni dei parametri πmin ( minimo π-score da considerare) e nmin (numero minimo di punti dati completi a coppie). Abbiamo notato che soglie di parametro più conservative portano a prestazioni più elevate nella previsione dei complessi proteici. Tuttavia, più queste soglie diventano conservative e più i geni devono essere esclusi dall’analisi a causa di dati insufficienti. Pertanto, abbiamo deciso di selezionare πmin = 0,2 e nmin = 15 comeparametri per le analisi a valle, supponendo che questi cutoff presentino un buon compromesso tra il potere predittivo dell’analisi e il numero di geni che possono essere considerati. Sulla base di questi parametri, abbiamo scoperto che la nostra analisi ha il potere di associare correttamente molti geni che interagiscono strettamente, come CTNNB1 e TCF7L2, che insieme formano il complesso del fattore di trascrizione TCF/β-catenina (Morin et al, 1997) o il complesso del recettore del ligando WNT10A/FZD5(Voloshanenko et al, 2017; Fig 5B). Profili di interazione simili si possono trovare anche per diversi membri del complesso del mediatore, un complesso multisubunitario importante per la regolazione trascrizionale della RNA polimerasi II (Fig 5C).
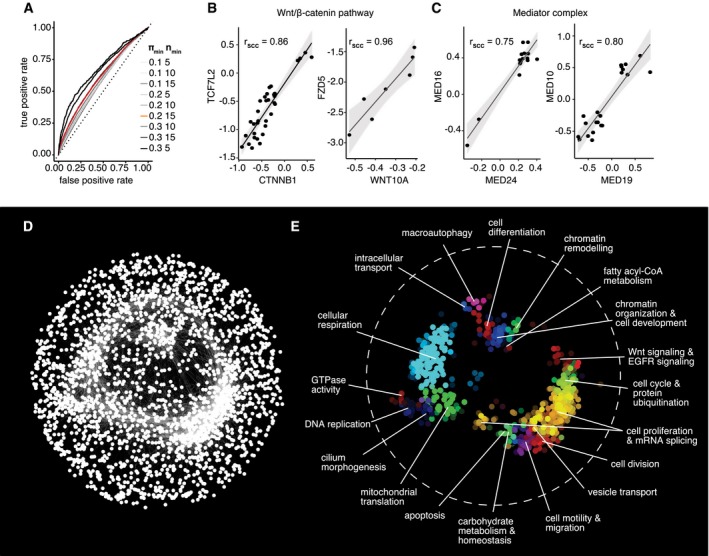
E.Profili di interazione altamente correlati possono prevedere la somiglianza funzionale
Curva AROC che mostra le prestazioni della somiglianza del profilo di interazione a prevedere la co-membership del complesso proteico. Le curve sono mostrate per diverse combinazioni di parametri di filtraggio. La curva corrispondente alla combinazione di parametri utilizzata per l’analisi a valle (πmin = 0,2; nmin = 15) è evidenziata in rosso. Una linea tratteggiata grigia indica la performance attesa dall’assegnazione casuale.B, CExamples di complessi proteici in cui i membri complessi mostrano profili di interazione altamente correlati(rSCC= Spearman’s correlation coefficient).DNetwork di geni con profili di interazione altamente correlati. In totale, 2.497 nodi (geni) sono collegati da 19.044 link (FDR di singoli link < 1,5e-05). Per posizionare i nodi è stato utilizzato un layout edge-weighted spring-embedded per posizionare i nodi.L’analisi dell’arricchimento spaziale con l’algoritmo SAFE evidenzia i moduli di rete costituiti da geni con annotazioni funzionali simili basate su processi biologici di ontologia genica. Le etichette in figura riassumono i termini GO associati a ciascun modulo.Curva ROC che mostra le prestazioni della similarità del profilo di interazione a prevedere la co-membership di proteine complesse. Le curve sono mostrate per diverse combinazioni di parametri di filtraggio. La curva corrispondente alla combinazione di parametri utilizzata per l’analisi a valle (πmin = 0,2; nmin = 15) è evidenziata in rosso. Una linea tratteggiata grigia indica la performance attesa dall’assegnazione casuale.Esempi di complessi proteici in cui i membri complessi mostrano profili di interazione altamente correlati(rSCC= coefficiente di correlazione di Spearman).Rete di geni con profili di interazione altamente correlati. In totale, 2.497 nodi (geni) sono collegati da 19.044 link (FDR di singoli link < 1,5e-05). Per il posizionamento dei nodi è stato utilizzato un layout a molla ponderato a bordo.L’analisi dell’arricchimento spaziale con l’algoritmo SAFE evidenzia moduli di rete costituiti da geni con annotazioni funzionali simili basate su processi biologici di ontologia genica. Le etichette nella figura riassumono i termini GO associati a ciascun modulo.
I dati di origine sono disponibili online per questa figura.
Abbiamo usato un rigoroso cutoff per selezionare tutte le coppie di geni target per le quali il valore P asintotico regolato della loro somiglianza di profilo (correlazione di Spearman) era più piccolo di 1,5e-05 e le abbiamo collegate ad una rete. La rete risultante mostrava un rapporto bordo/nodo paragonabile alle reti di lieviti precedentemente riportate (Costanzo et al, 2016) con un bordo che rappresentava in media una correlazione di profilo di interazione di 0,85 (Fig EV4D). Abbiamo visualizzato la rete applicando un layout integrato a molla diretto a forza che può posizionare geni molto simili tra loro in modo prossimale (Fig 5D). Abbiamo poi utilizzato l’analisi spaziale dell’arricchimento funzionale (SAFE; Baryshnikova, 2016a,b) per identificare le regioni della rete arricchite per specifici processi biologici come annotato dall’ontologia genica (GO; Ashburner et al, 2000; Fig 5E). L’analisi SAFE ha rivelato il raggruppamento di 19 sottoreti, che sono state associate a 217 diversi termini GO e che comprendono in totale 2.479 geni.
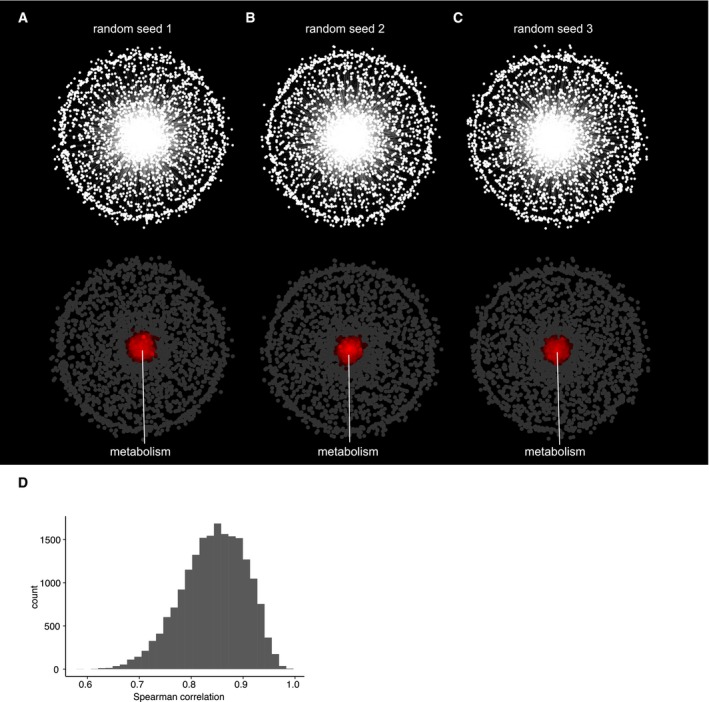
D.Validazione computazionale della rete genetica prevista
Rete A-CGenetica dopo la permutazione casuale dei bordi utilizzando tre diversi semi casuali. È stata applicata una disposizione a molla con permutazione forzata per i nodi di posizione simile alla rete mostrata in Fig 5. Ogni punto corrisponde ad un gene. Sono stati mantenuti i geni nella rete e il numero di bordi.Il DHistogramma dei coefficienti di correlazione di Spearman che rappresentano i bordi della rete genetica mostrata in Fig 5D.Rete genetica dopo permutazione casuale dei bordi utilizzando tre diversi semi casuali. È stata applicata una disposizione a molla con permutazione forzata dei nodi di posizione simile alla rete mostrata in Fig 5. Ogni punto corrisponde ad un gene. I geni nella rete così come il numero di bordi sono stati mantenuti.Istogramma dei coefficienti di correlazione di Spearman che rappresentano i bordi della rete genetica mostrata in Fig 5D.
Al fine di garantire che i moduli osservati assomigliano in realtà a cluster funzionali biologicamente significativi e non sono solo artefatti casuali dell’analisi, abbiamo eseguito un’analisi di permutazione casuale (Fig EV4A-C). Come previsto, abbiamo osservato che in seguito al rimescolamento casuale dei collegamenti, mantenendo i geni e il numero di bordi uguali, la rete perde la sua struttura modulare, dando luogo ad un unico grande cluster di geni al centro della rete. L’analisi SAFE rivela che questo cluster si arricchisce per i geni del metabolismo, indicando che c’è una sovrarappresentazione generale dei geni del metabolismo tra i geni che si trovano a comportarsi in modo differenziato nelle cellule tumorali.
I cluster funzionalmente arricchiti non coprono solo i processi biologici che si trovano comunemente implicati nel cancro (ad es, “divisione cellulare”, “segnalazione Wnt & EGFR”, o “differenziazione cellulare”) ma anche processi di importanza generale nello sviluppo e nel comportamento cellulare (ad esempio, “morfogenesi del cilium”, “trasporto intracellulare” e “macroautofagia”). Ciò implica che l’approccio qui presentato è effettivamente in grado di identificare nuovi regolatori delle assemblee di percorso noto e dei membri precedentemente sconosciuti dei processi biologici funzionali noti. In questo modo, abbiamo creato una risorsa senza precedenti di cluster di geni funzionali da sfruttare in studi futuri per una più profonda comprensione dei nuovi meccanismi che influenzano i bioprocessi conosciuti, non solo importanti nel cancro, ma che coprono una vasta gamma di biologia. Questa risorsa può anche essere utilizzata per convalidare l’assunzione preventiva di funzioni geniche in qualsiasi studio funzionale. Prevediamo che, man mano che i dati in più linee cellulari e fenotipi diventano disponibili, questa mappa funzionale di una cellula continuerà a crescere e a migliorare.
E.Profili di interazione altamente correlati possono prevedere la somiglianza funzionale
Curva AROC che mostra le prestazioni della somiglianza del profilo di interazione a prevedere la co-membership del complesso proteico. Le curve sono mostrate per diverse combinazioni di parametri di filtraggio. La curva corrispondente alla combinazione di parametri utilizzata per l’analisi a valle (πmin = 0,2; nmin = 15) è evidenziata in rosso. Una linea tratteggiata grigia indica la performance attesa dall’assegnazione casuale.B, CExamples di complessi proteici in cui i membri complessi mostrano profili di interazione altamente correlati(rSCC= Spearman’s correlation coefficient).DNetwork di geni con profili di interazione altamente correlati. In totale, 2.497 nodi (geni) sono collegati da 19.044 link (FDR di singoli link < 1,5e-05). Per posizionare i nodi è stato utilizzato un layout edge-weighted spring-embedded per posizionare i nodi.L’analisi dell’arricchimento spaziale con l’algoritmo SAFE evidenzia i moduli di rete costituiti da geni con annotazioni funzionali simili basate su processi biologici di ontologia genica. Le etichette in figura riassumono i termini GO associati a ciascun modulo.Curva ROC che mostra le prestazioni della somiglianza del profilo di interazione a prevedere la co-membership di proteine complesse. Le curve sono mostrate per diverse combinazioni di parametri di filtraggio. La curva corrispondente alla combinazione di parametri utilizzata per l’analisi a valle (πmin = 0,2; nmin = 15) è evidenziata in rosso. Una linea tratteggiata grigia indica la performance attesa dall’assegnazione casuale.Esempi di complessi proteici in cui i membri complessi mostrano profili di interazione altamente correlati(rSCC= coefficiente di correlazione di Spearman).Rete di geni con profili di interazione altamente correlati. In totale, 2.497 nodi (geni) sono collegati da 19.044 link (FDR di singoli link < 1,5e-05). Per il posizionamento dei nodi è stato utilizzato un layout a molla ponderato a bordo.L’analisi dell’arricchimento spaziale con l’algoritmo SAFE evidenzia moduli di rete costituiti da geni con annotazioni funzionali simili basate su processi biologici di ontologia genica. Le etichette nella figura riassumono i termini GO associati a ciascun modulo.
I dati di origine sono disponibili online per questa figura.
D.Convalida computazionale della rete genetica prevista
Rete A-CGenetica dopo la permutazione casuale dei bordi utilizzando tre diversi semi casuali. È stata applicata una disposizione a molla con permutazione forzata per i nodi di posizione simile alla rete mostrata in Fig 5. Ogni punto corrisponde ad un gene. Sono stati mantenuti i geni nella rete e il numero di bordi.Il DHistogramma dei coefficienti di correlazione di Spearman che rappresentano i bordi della rete genetica mostrata in Fig 5D.Rete genetica dopo permutazione casuale dei bordi utilizzando tre diversi semi casuali. È stata applicata una disposizione a molla con permutazione forzata dei nodi di posizione simile alla rete mostrata in Fig 5. Ogni punto corrisponde ad un gene. I geni nella rete così come il numero di bordi sono stati mantenuti.Istogramma dei coefficienti di correlazione di Spearman che rappresentano i bordi della rete genetica mostrata in Fig 5D.
Discussione
Per identificare nuove funzioni di geni conosciuti o per assegnare funzioni cellulari a geni sconosciuti, sono stati condotti degli schermi genetici in avanti in molti sistemi modello che vanno dai batteri alle cellule umane (Boutros & Ahringer, 2008 ). La combinazione di metodi di screening ad alto rendimento con la capacità di mettere fuori combattimento in modo affidabile ogni gene del genoma umano mediante nucleasi programmabili apre ora la possibilità di studiare le conseguenze di mutazioni a perdita totale o parziale di funzione con un’accuratezza senza precedenti in vari contesti mutazionali. Sono stati eseguiti schermi a livello genomico, prevalentemente per l’essenzialità del gene, che hanno identificato un gran numero di geni essenziali noti, nuovi e specifici del contesto (Wang et al, 2014, 2015; Hart et al, 2015; Evers et al, 2016; Morgens et al, 2016; Zhan & Boutros , 2016; Rauscher et al, 2017). Abbiamo sviluppato un approccio computazionale per integrare decine di schermi ad alta produttività CRISPR/Cas9, indipendentemente dalle dimensioni dello schermo, dalla libreria, dal tipo Cas9 e dal protocollo di screening. Poiché, rispetto ad altre tecniche, gli schermi CRISPR/Cas9 hanno dimostrato di essere un metodo più sensibile con cui è possibile scoprire fenotipi indotti dalla perturbazione nelle cellule umane (Hart et al, 2015; Wang et al, 2015), un tale approccio mostra grandi promesse per la scoperta sistematica delle vulnerabilità al cancro. Abbiamo sviluppato MINGLE, un framework computazionale che integra schermi CRISPR/Cas9 di diversa origine per mappare le interazioni genetiche nelle cellule tumorali. Abbiamo applicato questo approccio per integrare i dati di 85 schermi in linee cellulari tumorali umane e abbiamo analizzato gli effetti di vitalità delle perturbazioni di CRISPR/Cas9 nel contesto del background genetico delle linee cellulari. Valutando sistematicamente 2,1 milioni di combinazioni di geni, abbiamo scoperto mappe di cablaggio genetico che includono molte dipendenze note e nuove tra i geni implicati nella tumorigenesi e nella resistenza alla terapia. Abbiamo inoltre dimostrato che queste mappe possono identificare nuovi regolatori di percorsi che giocano ruoli importanti in specifici tipi di cancro, per esempio, la segnalazione Wnt dipendente dalla β-catenina.
Qui, dimostriamo che i membri del complesso di controllo della glucosidasi II controllano l’attività di segnalazione mediante la regolazione della secrezione di ligando Wnt3, probabilmente mediata dalla proteina N-glicosilazione. La glicosilazione legata a N è un processo basato su ER essenziale per la secrezione e il ripiegamento delle proteine (Xu & Ng, 2015; Fig 4D ). Mentre la glicosilazione legata a N di Wnt3a è già stata descritta in passato (Smolich et al, 1993), l’importanza della glicosilazione del ligando Wnt per la secrezione e l’attivazione del percorso è controversa. Mentre alcuni autori affermano una chiara correlazione tra glicosilazione del ligando Wnt e la secrezione in una linea cellulare umana (Komekado et al, 2007) , altri non hanno potuto osservare la perdita di secrezione di proteine per la soppressione della proteina N-glicosilazione in Drosophila (Herr & Basler, 2012; Tang et al, 2012). I nostri risultati supportano un ruolo di tre geni coinvolti nella glicosilazione della proteina sull’attivazione della via Wnt, che potrebbe essere ulteriormente supportata da una riduzione della secrezione di ligando Wnt al momento dell’abbattimento di GANAB e PRKCSH.
Tradizionalmente, le interazioni genetiche sono stati esaminati da perturbazione simultanea di due geni. La nostra analisi si basa sull’idea che una di queste perturbazioni può essere imitata da alterazioni genetiche che si verificano naturalmente nelle cellule tumorali. Anche se troviamo che questo concetto può essere applicato per identificare efficacemente le vere interazioni, esso pone una serie di sfide. Prima di tutto, le alterazioni genetiche di ogni gene devono essere messe in comune, richiedendo alcune ipotesi sulla somiglianza delle loro conseguenze funzionali. In natura, tuttavia, questi presupposti non sempre sono veri, il che può confondere l’analisi. In questo studio, abbiamo cercato di affrontare questo problema dividendo le alterazioni in gruppi logici, per esempio, mettendo insieme mutazioni senza senso e mutazioni frame-shift come varianti a perdita di funzione. Abbiamo ulteriormente perfezionato queste annotazioni con la cura manuale, escludendo le linee cellulari con varianti note per essere funzionalmente distinte dalle altre. Anche se questo è attualmente possibile solo per i geni ben caratterizzati, siamo fiduciosi che i futuri progressi nella caratterizzazione funzionale delle varianti tumorali andranno a beneficio del nostro approccio. È importante sottolineare che, sebbene l’assenza di espressione genica dovrebbe essere funzionalmente simile a una perdita completa della funzione genica dovuta alla mutazione, non abbiamo preso in considerazione le informazioni sui geni non espressi. Ciò è dovuto al fatto che i profili trascrittomici delle linee cellulari tumorali sono stati per lo più derivati da esperimenti di microarray. Pertanto, è difficile distinguere tra geni non espressi e geni che sono espressi a basso livello (Mirnics et al, 2001). Noi crediamo che una volta che i dati RNA-seq diventano ampiamente disponibili per le linee cellulari tumorali, questo problema può essere superato. Un’altra sfida è posta dal fatto che alcune alterazioni genetiche sono correlate perché coesistono nelle stesse linee cellulari o negli stessi tipi di cancro. Un esempio è la delezione del locus del cromosoma 9p21 dove si trova il soppressore tumorale CDKN2A. Il CDKN2A è spesso co-cancellato con i suoi geni vicini (Muller et al, 2015), e non è quindi facilmente possibile capire quale di essi sia il vero motore di una proposta di interazione. Questo può ulteriormente introdurre un pregiudizio nella rete di somiglianze genetiche. Nel nostro studio, affrontiamo questo problema aggregando i geni della query completamente correlati in “meta-geni” che poi procediamo a utilizzare per calcolare le interazioni e generare la rete di somiglianza genetica. Per evitare distorsioni, calcoliamo ulteriormente le correlazioni dei profili di interazione genetica basati solo su un sottoinsieme di geni di query in modo che non ci siano due geni di query più simili del 70% in termini di composizione della loro linea cellulare.
In questo studio abbiamo richiesto che un gene venga alterato in almeno tre diverse linee cellulari per essere considerato come un gene di interrogazione per l’analisi delle interazioni. Con l’aumentare dei dati disponibili, tuttavia, ci aspettiamo che il numero di possibili geni da interrogare a questa soglia cresca rapidamente, il che può imporre un notevole onere di test multipli sul nostro approccio. Pertanto, riteniamo che questo taglio debba essere rivalutato quando l’analisi viene ripetuta con un set di dati più ampio, al fine di trovare il miglior compromesso tra la copertura genica e la potenza statistica.
È stato precedentemente dimostrato che i profili delle interazioni genetiche sintetiche possono raggruppare i geni funzionalmente correlati attraverso il “senso di colpa per associazione”. Gli studi sulle cellule umane si sono precedentemente basati sull’interferenza dell’RNA. Tuttavia, è stato dimostrato che questo metodo ha dei limiti, come off-targeting e gli effetti di compensazione del dosaggio, che possono essere superati da CRISPR / Cas9. I nostri approcci ci hanno permesso di analizzare i profili di interazione utilizzando i dati di molti esperimenti CRISPR/Cas9 ad alto rendimento. Questi profili hanno il potere di prevedere le relazioni funzionali dei geni, come dimostriamo attraverso il benchmarking con il database del complesso proteico CORUM. Poiché le interazioni fisiche delle proteine come si verificano nei complessi proteici rappresentano solo un sottoinsieme di possibili relazioni funzionali, crediamo che questo benchmarking possa essere interpretato come un limite inferiore per il potere predittivo dell’analisi. Abbiamo creato una rete che raggruppa i geni in cluster con profili funzionali arricchiti. I risultati di questa analisi possono essere importanti per due motivi: In primo luogo, le ipotesi sulla funzione di geni debolmente caratterizzati che sono spesso cancellati nelle cellule tumorali possono essere generate osservando i partner di interazione comuni all’interno dei moduli di rete funzionale; e in secondo luogo, tale rete può servire come un potente strumento per dedurre la funzione di geni del tutto sconosciuti basati sulla funzione dei geni connessi. Ad esempio, oltre il 10% dei geni della nostra rete non è annotato con i processi biologici GO.
Allo stato attuale, un fattore limitante di questo tipo di analisi è la quantità di dati disponibili. Attualmente, ci sono circa 200 geni che sono stati trovati frequentemente alterati nelle linee cellulari incluse nei nostri dati e per i quali possono essere testate interazioni genetiche sintetiche. Pertanto, attualmente è possibile esaminare solo i geni che interagiscono con questi geni. Tuttavia, questo numero aumenterà rapidamente con la pubblicazione di nuovi dati, che consentiranno la creazione di reti di interazione sempre più complesse. La messa in comune di alterazioni funzionalmente correlate di diversi geni, come dimostriamo sull’esempio di RNF43 e APC, può ampliare ulteriormente l’insieme dei possibili geni di interrogazione. Nel complesso, riteniamo che l’approccio presentato possa essere un modo efficace per scoprire sistematicamente le interazioni genetiche sintetiche che possono essere di interesse clinico. Inoltre, crediamo che possa servire come un’importante risorsa per la ricerca di una comprensione più completa del funzionamento dei geni umani. Le scale del flusso di lavoro presentate e la crescente quantità di dati sono sempre più disponibili.
Ci aspettiamo che in futuro vengano effettuate molte altre schermate CRISPR/Cas9 in varie linee cellulari. Amplieremo la nostra analisi una volta che questi dati saranno disponibili per migliorare e diversificare le nostre scoperte. Infine, puntiamo ad estendere la nostra analisi per includere anche i dati di altri tipi di esperimenti, come le interazioni fisiche derivate da studi di interazione proteina-proteina. La maggior parte delle interazioni genetiche sintetiche, per esempio, non collegano geni che sono membri degli stessi percorsi, ma collegano invece membri di due percorsi che interagiscono tra loro (Kelley & Ideker, 2005 ). Pertanto, l’integrazione di interazioni sintetiche e di interazioni fisiche derivate da esperimenti di interazione proteina-proteina potrebbe fornire nuove importanti intuizioni su come i percorsi biologici interagiscono tra loro.
Il nostro obiettivo è inoltre quello di rendere disponibili le interazioni previste per la navigazione e il download attraverso il database GenomeCRISPR, in quanto riteniamo che possano essere una risorsa utile per informare la selezione dei geni candidati per esperimenti che non possono essere effettuati su scala genomica. Questi includono, ad esempio, schermi in vivo in modelli di topo geneticamente modificati che sono spesso limitati dal numero di cellule che possono essere trasfettate o da esperimenti di perturbazione a coppie, in quanto sono ora condotti su cellule umane utilizzando CRISPR/Cas9 (Du et al , 2017; Shen et al, 2017 ), che sono limitati dal numero di possibili combinazioni di geni.
Materiali e metodi
Profili genetici delle linee cellulari tumorali
Per generare profili di alterazioni genetiche nelle linee cellulari tumorali di GenomeCRISPR (Rauscher et al, 2017), ci siamo basati su dati pubblicamente disponibili nel progetto COSMIC Cell Lines (Forbes et al. al, 2017), l’Enciclopedia delle linee cellulari cancerogene (CCLE; Barretina et al, 2012) e i dati aggiuntivi pubblicati in precedenza da Bürckstümmer et al (2013) per la linea cellulare KBM7 e Klijn et al (2014) (Fig 1B). Presi insieme, questi dati possono caratterizzare tutti tranne due (una linea cellulare di glioblastoma derivata dal paziente e la linea cellulare RPE1) linee cellulari attualmente incluse in GenomeCRISPR. In totale, sono state incluse nell’analisi 60 diverse linee cellulari. Per ognuna di queste linee cellulari è stato generato un elenco di geni alterati, tenendo in considerazione i seguenti tipi di alterazioni: (i) guadagno di eventi del numero di copie, (ii) perdita di eventi del numero di copie, (iii) mutazioni somatiche, escluse le mutazioni silenziose e le inserzioni o cancellazioni in-frame, e (iv) sovraespressione dell’mRNA.
Selezione delle alterazioni del numero di copie
In primo luogo, i dati relativi al numero di copie sono stati scaricati dal COSMIC Cell Lines Project v81, dal CCLE (file datato 27 maggio-2017) e dallapubblicazione Klijn et al ( 2014). Lo stato di guadagno e perdita del numero di copie è stato determinato per ogni gene come segue: COSMIC fornisce un’etichetta per ogni evento del numero di copie che indica se l’evento può essere classificato come evento di guadagno o perdita del numero di copie. Abbiamo adottato questa classificazione per la nostra analisi. Nell’articolo di Klijn e colleghi, l’amplificazione e la cancellazione di un gene sono state definite come > 1 o < -0,75 del numero di copia corretto per ploidia (Mermel et al, 2011; Klijn et al, 2014). Di conseguenza, nel nostro approccio sono state utilizzate le stesse soglie. Infine, il CCLE fornisce log2-transformato il numero di copia piegato tra campioni sani e linee di cellule tumorali a livello genico. Il numero assoluto di copie di ogni gene per linea cellulare è stato stimato a partire dai dati del cambiamento di piega come C=[2x×2]dove C è il numero assoluto di copie e x è il cambiamento di piega log2 tra linea cellulare e campione sano. Al fine di valutare se questo fornisce una stima realistica del numero totale di copie, abbiamo analizzato il numero di copie derivato per tutti i geni del cromosoma Y nelle linee cellulari femminili, dove sono stati stimati in modo robusto i numeri di copie di 0. Infine, abbiamo scaricato i dati pre-processati del numero di copie a livello genico da COSMIC. Tutti i geni in cui è stato stimato un numero di copie di 0 in una linea cellulare sono stati contrassegnati come geni a perdita di funzione. Gli eventi di alterazione del numero di copie che sono stati osservati in modo robusto su almeno 2 diverse fonti di dati sono stati mantenuti per l’analisi a valle dopo aver escluso le alterazioni sui cromosomi X e Y.
Selezione di mutazioni somatiche
I dati sulle mutazioni somatiche sono stati scaricati dal COSMIC Cell Lines Project (versione 81), dalle mutazioni CCLE (mutazioni Oncomap3 del 10 aprile 2012 e mutazioni Hybrid Capture del 05 maggio 2015) e dalle pubblicazioni Klijn et al e Bürckstümmer. Sono state selezionate le mutazioni missenso e le mutazioni frame-shift e sono state escluse le mutazioni segnalate in disaccordo tra le singole fonti di dati. Successivamente, le mutazioni missenso sono state classificate in conducente e passeggero e conducente come proposto da Anoosha et al (2016 ). Le mutazioni passeggeri presunte sono state escluse e le restanti mutazioni sono state conservate per l’analisi a valle. Dopo aver messo in comune le alterazioni del numero di copie e le mutazioni somatiche, abbiamo mantenuto tutti i geni come geni di query in cui è stata osservata un’alterazione in almeno tre diverse linee cellulari GenomeCRISPR.
Selezione dei geni sovraespressi
Al fine di definire i geni che sono sovraespressi nelle linee cellulari incluse in GenomeCRISPR, sono stati scaricati da CCLE (CCLE_Expression_2012-09-29.res del 17 ottobre 2012) e dal COSMIC Cell Lines Project (v81). ComBat (Leek et al, 2012) è stato utilizzato per rimuovere gli effetti batch tra le due diverse fonti di dati, e i livelli di espressione per le linee cellulari presenti in entrambe le fonti sono stati aggregati calcolando la media. Successivamente, sono stati calcolati i punti Z dell‘espressione genica per ogni gene in ogni linea cellulare. I geni dell’elenco COSMIC dei geni del censimento dei tumori per i quali è stato osservato uno Z-score> 2 in almeno cinque diverse linee cellulari del GenomeCRISPR sono stati mantenuti per l’analisi a valle.
Analisi degli schermi CRISPR/Cas9
Per confrontare i fenotipi di vitalità degli schermi CRISPR/Cas9 ad alta produttività, sono stati calcolati i punteggi CRISPR a livello genico aggregati per ogni esperimento. In primo luogo, tutte le schermate di selezione negativa per la vitalità cellulare sono state scaricate dal database GenomeCRISPR (Rauscher et al, 2017) . In primo luogo, tutti i geni bersaglio di meno di tre sgRNA e tutti gli sgRNA in cui < 30 conteggi sono stati osservati nel punto di tempo 0 (T0) campione, sono stati rimossi da ogni schermata singolarmente. Inoltre, abbiamo escluso tutti gli sgRNA della libreria GeCKOv2 (Sanjana et al, 2014) che erano contrassegnati come “isUsed = FALSE” nel sito web “Achilles_v3.3.8.reagent.table.txt”(https://portals.broadinstitute.org/achilles/datasets/7/download) del Progetto Achilles(Aguirre et al, 2016). Dopo il filtraggio, i conteggi delle letture grezze sono stati corretti per le differenze nella profondità di sequenziamento dividendo il conteggio di ogni lettura per la mediana di tutti i conteggi delle letture dei campioni sia a T0 che al punto temporale finale. Sulla base di questi valori, sono stati calcolati i cambiamenti di piega per le repliche tecniche, dopo aver aggiunto 1 a ogni conteggio per evitare log di 0, asfcsgRNA=log2rcsamplercT0where rccampione è il conteggio normalizzato della lettura misurato nella popolazione di cellule campione e rcT0 è il conteggio normalizzato letto misurato al punto di tempo 0. In alcuni casi, l’abbondanza di conteggio letto nel pool di DNA plasmide è stato dato invece di tempo punto 0 dati di sequenziamento delle cellule. In questi casi, i conteggi di lettura del DNA plasmidico sono stati utilizzati per calcolare i cambiamenti di piega per tutti i campioni replicati di quelle schermate. Inoltre, in due casi (Doench et al, 2016; Munoz et al , 2016), non erano disponibili dati di conteggio letti. Qui, abbiamo usato i valori originali di cambio di piega forniti dagli autori degli esperimenti.
Per valutare la qualità di ogni schermata è stata utilizzata l’Analisi Bayesiana dell’Essenzialità Genica (BAGEL; Hart & Moffat, 2016 ) per prevedere l’essenzialità genica. Utilizzando le curve di precisione-richiamo è stata esaminata la capacità di separare i geni essenziali e non essenziali sulla base dei dati di cambiamento della piega. Tutte le schermate in cui è stata osservata un’area sotto la curva di precisione-richiamo inferiore a 0,85 sono state escluse da ulteriori analisi. Dopo aver selezionato le schermate per l’analisi a valle(Dataset EV4), i punteggi CRISPR a livello genico sono stati calcolati come il cambiamento di piega media di tutti gli sgRNA che prendono di mira un gene. Abbiamo poi usato la normalizzazione quantile per normalizzare i punteggi CRISPR tra gli esperimenti.
Correzione a livello genico dell’effetto batch della libreria
Al fine di stimare gli effetti di batch introdotti dall’uso di diverse librerie, un robusto modello lineare della forma yi= β0 + β1xi1+ …+βn
xin+ εi con β0 =0 e yi= yCRISPR,i– Median(yCRISPR) è stato montato per ogni gene individualmente dove n è il numero di librerie incluso il gene, i è l’indice di un punto di dati, e yCRISPRsono punteggi CRISPR quantile-normalizzati. I coefficienti β1…βn sono quindi la differenza stimata tra i punteggi CRISPR visualizzati in una biblioteca e i punteggi CRISPR mediani di tutte le biblioteche. Un robusto test F come implementato nel pacchetto R “sfsmisc” è stato usato per testare l’ipotesi nulla che il punteggio CRISPR mediano osservato per un gene sia lo stesso in tutte le biblioteche. Il metodo Benjamini-Hochberg (Benjamini & Hochberg, 1995) è stato usato per stimare il tasso di falsificazione (FDR) per ogni test. Nel caso in cui l’ipotesi nulla potesse essere respinta al 5% di FDR, è stato ipotizzato un effetto batch specifico per la biblioteca e i punteggi CRISPR osservati usando quella biblioteca sono stati centrati sottraendo la sua distanza dalla mediana dei punteggi CRISPR di tutte le biblioteche. Una libreria è stata segnalata dalla correzione dei lotti nei casi in cui è stato previsto un effetto batch simile (stesso segno dei coefficienti del modello) per le librerie utilizzate nelle schermate di Wang et al (2017 )e Tzelepis et al (2016 ). Entrambe queste librerie sono state utilizzate per schermare principalmente le linee cellulari della leucemia mieloide acuta (LAM), e quindi, l’ipotesi nulla descritta sopra potrebbe non essere vera nel caso di geni specifici della LAM. Pertanto, in tali casi, non è stato effettuato alcun aggiustamento del lotto.
Controllo qualità dei punteggi CRISPR normalizzati
Per valutare l’adeguatezza delle fasi di normalizzazione sopra descritte, il controllo di qualità è stato effettuato esaminando diverse proprietà dei dati normalizzati. Prima di tutto, i campioni sono stati raggruppati per valutare se i campioni biologicamente correlati si sono raggruppati più strettamente rispetto ai campioni biologicamente distanti. Qui, l’insieme dei geni condivisi in tutte le biblioteche è stato determinato e Ward clustering (come implementato nel metodo “ward.D2” di R per il clustering gerarchico) è stato eseguito. Il pacchetto “pheatmap” R è stato usato per visualizzare la mappa termica mostrata in Fig 2A. Successivamente, le differenze nei punteggi CRISPR normalizzati tra i campioni sono state osservate sugli esempi di nove polimerasi essenziali del nucleo e nove geni situati sul cromosoma Y, tutti campionati in modo casuale dall’insieme dei geni della polimerasi essenziale del nucleo (Hart et al, 2017) e l’insieme dei geni del cromosoma Y, rispettivamente. Solo gli schermi nelle linee cellulari femminili sono stati tracciati nella Fig 2C. Per esaminare se i punteggi CRISPR normalizzati potevano distinguere i geni essenziali del nucleo (Hart et al, 2017) dai geni non essenziali (Hart et al, 2015), sono state generate curve di precisione-richiamo per ogni schermo usando il pacchetto ROCR R/Bioconduttore (Gentleman et al, 2004; Sing et al, 2005). Inoltre, sono stati selezionati alcuni oncogeni di controllo (KRAS, NRAS, BRAF e PIK3CA) per vedere se è possibile osservare una differenza prevista nella risposta al knockout del gene a seconda dello stato di mutazione del gene. I valori di P mostrati in Fig 2E-H sono stati calcolati utilizzando un t-test di Student’s t su due lati come implementato in R. Infine abbiamo verificato che i potenziali effetti indesiderati introdotti dalla correzione del lotto non distorcessero i risultati pubblicati negli articoli in cui i dati sono stati inclusi nella nostra pipeline. Per questi confronti, sono stati utilizzati punteggi CRISPR normalizzati per le linee cellulari presenti nelle pubblicazioni originali.
Test combinatoriali delle interazioni gene-gene
Per testare le differenze nella risposta di fitness in base ai genotipi a perdita di funzione, sono stati selezionati i punteggi di fitness per tutti gli schermi CRISPR/Cas9 nelle linee cellulari in cui erano disponibili informazioni sul genotipo. Abbiamo selezionato tutti i geni che sono stati contrassegnati come alterati da mutazioni somatiche o da modifiche del numero di copie in almeno tre o contrassegnati come sovraesposti in almeno cinque linee cellulari distinte come geni di interrogazione. In totale sono stati selezionati 221 geni. Di conseguenza, abbiamo identificato tutte le combinazioni tra questi geni di query e i geni perturbati negli schermi (geni target). I geni target sono stati selezionati in modo tale che i punteggi di fitness fossero disponibili per almeno tre linee cellulari distinte con e senza perdita di funzione della query. Nel complesso, abbiamo identificato circa 3,8 milioni di combinazioni di questo tipo. Come dati di input per il test, abbiamo usato punteggi CRISPR normalizzati come descritto sopra. Abbiamo montato un modello lineare ad effetti misti per ogni combinazione, modellando il genotipo a perdita di funzione come effetto fisso e la linea cellulare come effetto casuale per tenere conto dei pregiudizi specifici della linea cellulare. Per la modellazione è stato utilizzato il pacchetto R “lme4” (Bates et al, 2014). Il pacchetto R “lmerTest” (Kuznetsova et al, 2016) è stato utilizzato per calcolare una stima della significatività (P-value)per i coefficienti di ogni modello. Dopo i test, query simili sono state identificate calcolando la correlazione di Pearson dei coefficienti di modello stimati per ogni coppia di geni di query. Coppie di geni di query con una correlazione del 100% sono state fuse insieme in un gene “meta” query. Per controllare la frazione attesa di false scoperte effettuate durante i test multipli, sono stati utilizzati test di ipotesi indipendenti (IHW; Ignatiadis et al, 2016) utilizzando la varianza dei punteggi normalizzati CRIPSR del gruppo alterato (mutato o sovraespresso) come covariata per la ponderazione delle ipotesi (Fig EV2C e D).
Quantificazione delle interazioni genetiche
Le interazioni tra i geni sono state quantificate utilizzando la statistica π-score (Horn et al, 2011; Laufer et al, 2013 ; Fischer et al, 2015). I valori π-score sono stati calcolati utilizzando la funzione “HD2013SGImaineffects” implementata nel pacchetto R/Bioconductor “HD2013SGI” (Laufer et al, 2013). Per generare l’input per la funzione “HD2013SGImaineffects”, i punteggi CRISPR normalizzati sono stati inseriti sottraendo i mezzi delle colonne e scalati dividendo le colonne per la loro deviazione standard.
Gene set di arricchimento della rete di arricchimento
Per generare la rete di arricchimento dei set di geni mostrata in Fig 3F,abbiamoselezionato 10 geni di query e tutti i geni target che interagiscono con queste query a FDR < 20%. L’elenco dei bordi risultante è stato visualizzato in Cytoscape (Shannon et al, 2003) utilizzando un algoritmo di rete a molla a forza diretta. I nodi del gene della query sono stati disposti manualmente. ConsensusPathDB (Kamburov et al, 2013) è stato utilizzato per eseguire l’analisi della sovrarappresentazione dei set di geni, e per ogni gene di query è stato selezionato un termine di percorso dall’elenco dei risultati. I valori q visualizzati nella Fig 3F sono quelli forniti da ConsensusPathDB. Vorremmo ricordare che la Fig 3Fè stataispirata da un precedente studio di M. Brockmann e colleghi (Brockmann et al , 2017).
TCF4/Wnt-luciferase reporter assay
Le cellule HEK293T sono state coltivate nel MEM di Dulbecco (GIBCO) integrato con il 10% di siero bovino fetale (Biochrom GmbH, Berlino, Germania) senza antibiotici. Gli esperimenti sono stati eseguiti in un formato a 384 pozzetti utilizzando piastre di polistirolo bianche a fondo piatto (Greiner, Mannheim, Germania). Le cellule HEK293T sono state sottoposte a trasfezione inversa con 20 nM siRNA indicati con l’aiuto dell’1% di Lipofectamina RNAiMAX Reagente di Trasfezione RNAiMAX (# 13778150; Thermo Fisher Scientific Waltham, MA, USA). 24 ore dopo, le cellule sono state trasfettate con 0.2% del reagente di trasfezione TransIT-LT1 (731-0029; Mirus/VWR, Madison, USA), 20 ng di TCF4/Wnt Firefly luciferase reporter (Demir et al, 2013), e 10 ng di actina.Renilla luciferase reporter (Nickles et al, 2012), e la segnalazione Wnt canonica è stata indotta dall’aggiunta del Wnt3(20 ng)-, β-catenina (20 ng)-, o Dvl3 (5 ng)-espressione di plasmidi o lasciata senza induzione dall’aggiunta del Ctrl plasmide pcDNA3. La luminescenza è stata misurata con il lettore di plasmidi Mithras LB940 (Berthold Technologies, Bad Wildbad, Germania). Il segnale TCF4/Wnt-luciferasi è stato normalizzato al segnale di actina-Renillaluciferasi reporter. Tutte le sequenze e i costrutti siRNA utilizzati per il test TCF4/Wnt-luciferase reporter sono elencati nel Dataset EV5.
NanoLuciferase Wnt3 saggio di secrezione Wnt3
Simile al saggio TCF4/Wnt-luciferase reporter, le cellule HEK293T sono state sottoposte a trasfezione inversa con siRNA indicati e seminate in piastre di polistirolo bianco a fondo piatto in formato 384 pozzetti (Greiner, Mannheim, Germania). 24 ore dopo, le cellule sono state trasfettate con 20 ng di NLucWnt3 o Wnt3NLuc, insieme a 5 ng di CMV Firefly luciferase reporter plasmidi (Campeau et al,2009). Il costrutto NLucWnt3 è stato generato clonando la sequenza NanoLuciferasi (Hall et al, 2012) dopo il peptide di segnale di Wnt3 nel plasmide di espressione pcDNA Wnt3 (Najdi et al, 2012), mentre è stato clonato al C-termino di Wnt3 per il costrutto Wnt3NLuc. 48 ore dopo, le piastre sono state centrifugate e 20 μl di terreno di coltura sono stati trasferiti in una nuova piastra. Il segnale di NanoLuciferasi nel lisato e nel mezzo è stato rilevato con l’aiuto di un test di NanoLuciferasi-Glo Luciferase (#N1110) di Promega (USA) secondo le istruzioni del produttore. La luminescenza è stata misurata con il lettore di piastre Mithras LB940 (Berthold Technologies, Bad Wildbad, Germania). Nel caso del lisato, è stato misurato prima il segnale per Firefly luciferasi e poi per NanoLuciferasi. Il segnale della NanoLuciferasi nel mezzo di coltura è stato normalizzato al segnale della NanoLuciferasi nel lisato normalizzato al segnale della Firefly luciferasi. Tutte le sequenze di siRNA e i costrutti utilizzati per il saggio di secrezione Wnt3 sono elencati nel Dataset EV5.
Benchmarking e modellazione della rete di somiglianza genica
Al fine di valutare se le reti di similitudine di interazione possono prevedere la co-membership del complesso proteico, sono state scaricate annotazioni del complesso proteico dai database CORUM (Ruepp et al, 2010) e sono stati selezionati i geni target inclusi nei dati CORUM. Abbiamo rimosso tutte le interazioni a coppie πtq con |πtq| < πmin dove πtq è il punteggio di interazione tra il gene target t e il gene q e πminè una soglia scelta. Successivamente, la correlazione di Spearman è stata calcolata come implementata nel pacchetto “Hmisc” R per ogni possibile coppia di geni target utilizzando osservazioni complete a coppie. Coppie di geni bersaglio in cui meno di nminpunti di dati sono stati utilizzati per calcolare la correlazione sono stati esclusi. Questa analisi è stata eseguita per sei diverse combinazioni dei parametri πmine nmin, e le curve ROC sono stati disegnati per visualizzare quanto bene le correlazioni risultanti potrebbero prevedere la co-membership complesso proteico come annotato in CORUM. Sulla base di questi risultati, e πmin= 0,2 e nmin = 15sono stati selezionati come soglie per calcolare le correlazioni Spearman tra tutte le possibili coppie di geni target come descritto sopra. Per tener conto del fatto che ogni correlazione si basa su un diverso numero di punti di dati, le coppie di geni sono state classificate in base al valore P invece delle correlazioni di Spearman grezze. Quindi, per ogni correlazione, il valore P asintotico P è stato calcolato utilizzando il pacchetto “Hmisc” R testando l’ipotesi nulla che la correlazione tra una coppia di geni sia 0. Per selezionare le coppie di geni come bordi per la rete di somiglianza genica mostrata in Fig 5D, il tasso di falsificazione (FDR) è stato controllato usando il metodo Benjamini-Hochberg alla soglia rigorosa di FDR < 1.5e-05. La rete è stata visualizzata utilizzando Cytoscape (Shannon et al, 2003) . Per posizionare i nodi della rete è stato utilizzato un layout a molla forzata per posizionare i nodi della rete senza ponderazione dei bordi. La rappresentazione visiva della rete è stata ispirata da studi precedenti in lievito (Costanzo et al, 2010, 2016) . L’analisi spaziale dell’arricchimento funzionale (SAFE; Baryshnikova, 2016a,b) Il plugin Cytoscape è stato utilizzato per identificare i moduli funzionali della rete. Per l’analisi SAFE è stata scelta la metrica di distanza basata sulla mappa con una soglia di distanza massima di 0,6 (percentile). Per costruire la mappa composita, è stata scelta una dimensione minima del paesaggio di 7 e la distanza Jaccard è stata usata come metrica di similitudine per gli attributi di gruppo con una soglia di similitudine di 0.75. Come sfondo per l’arricchimento, sono stati scelti tutti i nodi dello standard di annotazione. In SAFE, lo standard di annotazione è una matrice binaria di geni (righe) e termini di annotazione (colonne). Un valore di 1 indica che un gene è annotato con un termine di annotazione specifico. Per la nostra analisi, abbiamo generato un tale standard di annotazione contenente l’ontologia dei geni (GO; Ashburner et al, 2000) annotazioni di processo biologico per tutti i geni target testati. Le annotazioni GO sono state scaricate dalla sezione dati di esempio della pagina GitHub dell’algoritmo SAFE(https://github.com/baryshnikova-lab/safe-data/blob/master/attributes/go_Hs_P_160509.txt.gz; accessibile il 13.09.2017) e filtrate per contenere solo i geni testati nella nostra analisi di interazione.
Disponibilità di dati e software
Il codice informatico documentato per riprodurre le analisi descritte in questo studio può essere scaricato come pacchetto R da GitHub all’indirizzo https://github.com/boutroslab/Supplemental-Material/tree/master/Rauscher_2017.
Contributi degli autori
BR, FH e MB hanno progettato lo studio. BR ha scritto il codice di analisi. TH ha consultato l’analisi statistica. LH e OV hanno progettato ed eseguito gli esperimenti. Tutti gli autori hanno discusso e analizzato i risultati. BR, FH, LH, OV e MB hanno scritto il manoscritto. Tutti gli autori hanno letto e approvato il manoscritto finale.
Conflitto di interessi
Gli autori dichiarano di non avere alcun conflitto di interessi.
Informazioni di supporto
References
- Genomic copy number dictates a gene‐independent cell response to CRISPR/Cas9 targeting. Cancer Discov. 2016; 6:914-929. PubMed
- Exploring preferred amino acid mutations in cancer genes: applications to identify potential drug targets. Biochim Biophys Acta Biomembranes. 2016; 1862:155-165.
- Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet. 2000; 25:25-29. PubMed
- Wntless, a conserved membrane protein dedicated to the secretion of Wnt proteins from signaling cells. Cell. 2006; 125:509-522. PubMed
- RNA events. Cas9 targeting and the CRISPR revolution. Science. 2014; 344:707-708. PubMed
- The cancer cell line encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 2012; 483:603-607. PubMed
- Secretion of Wnt ligands requires Evi, a conserved transmembrane protein. Cell. 2006; 125:523-533. PubMed
- Regulation of Wnt protein secretion and its role in gradient formation. EMBO Rep. 2008; 9:977-982. PubMed
- Quantitative analysis of fitness and genetic interactions in yeast on a genome scale. Nat Methods. 2010; 7:1017-1024. PubMed
- Systematic functional annotation and visualization of biological networks. Cell Syst. 2016a; 2:412-421. PubMed
- Exploratory analysis of biological networks through visualization, clustering, and functional annotation in cytoscape. Cold Spring Harb Protoc. 2016b; 2016:pdb.prot077644. PubMed
- Fitting linear mixed effects models using lme4. J Stat Soft. 2014; 67:1-48.
- Controlling the false discovery rate: a practical and powerful approach to multiple testing on JSTOR. J R Stat Soc. 1995; 57:289-300.
- Evolutionary capacitance as a general feature of complex gene networks. Nature. 2003; 424:549-552. PubMed
- The art and design of genetic screens: RNA interference. Nat Rev Genet. 2008; 9:554-566. PubMed
- The origin of variations in sexual and sex‐limited characters. Am Nat. 1922; 56:51-63.
- Genetic wiring maps of single‐cell protein states reveal an off‐switch for GPCR signalling. Nature. 2017; 546:307-311. PubMed
- A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002; 296:550-553. PubMed
- A reversible gene trap collection empowers haploid genetics in human cells. Nat Methods. 2013; 10:965-971. PubMed
- A global analysis of genetic interactions in Caenorhabditis elegans. J Biol. 2007; 6:8. PubMed
- A versatile viral system for expression and depletion of proteins in mammalian cells. PLoS ONE. 2009; 4:e6529. PubMed
- Tyrosine phosphorylation of LRP6 by Src and Fer inhibits Wnt/‐catenin signalling. EMBO Rep. 2014; 15:1254-1267. PubMed
- Wnt/β‐catenin signaling and disease. Cell. 2012; 149:1192-1205. PubMed
- The genetic landscape of a cell. Science. 2010; 327:425-431. PubMed
- A global genetic interaction network maps a wiring diagram of cellular function. Science. 2016; 353:aaf1420. PubMed
- Glucosidase II and MRH‐domain containing proteins in the secretory pathway. Curr Protein Pept Sci. 2015; 16:31-48. PubMed
- The synthetic genetic interaction spectrum of essential genes. Nat Genet. 2005; 37:1147-1152. PubMed
- RAB8B is required for activity and caveolar endocytosis of LRP6. Cell Rep. 2013; 4:1224-1234. PubMed
- Optimized sgRNA design to maximize activity and minimize off‐target effects of CRISPR‐Cas9. Nat Biotechnol. 2016; 34:184-191. PubMed
- Genome editing. The new frontier of genome engineering with CRISPR‐Cas9. Science. 2014; 346:1258096. PubMed
- Genetic interaction mapping in mammalian cells using CRISPR interference. Nat Methods. 2017; 14:577-580. PubMed
- CRISPR knockout screening outperforms shRNA and CRISPRi in identifying essential genes. Nat Biotechnol. 2016; 34:631-633. PubMed
- A map of directional genetic interactions in a metazoan cell. Elife. 2015; 4:e05464.
- The genetical theory of natural selection. The Clarendon Press: Oxford; 1930.
- Secrets of a double agent: CDK7 in cell‐cycle control and transcription. J Cell Sci. 2005; 118:5171-5180. PubMed
- COSMIC: somatic cancer genetics at high‐resolution. Nucleic Acids Res. 2017; 45:D777-D783. PubMed
- Mechanisms of mutant p53 stabilization in cancer. Subcell Biochem. 2014; 85:187-197. PubMed
- Systematic identification of genomic markers of drug sensitivity in cancer cells. Nature. 2012; 483:570-575. PubMed
- Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004; 5:R80. PubMed
- RNF43 is frequently mutated in colorectal and endometrial cancers. Nat Genet. 2014; 46:1264-1266. PubMed
- Engineered luciferase reporter from a deep sea shrimp utilizing a novel imidazopyrazinone substrate. ACS Chem Biol. 2012; 7:1848-1857. PubMed
- High‐resolution CRISPR screens reveal fitness genes and genotype‐specific cancer liabilities. Cell. 2015; 163:1515-1526. PubMed
- BAGEL: a computational framework for identifying essential genes from pooled library screens. BMC Bioinformatics. 2016; 17:164. PubMed
- Evaluation and design of genome‐wide CRISPR/SpCas9 knockout screens. G3. 2017; 7:2719-2727. PubMed
- Principles for the buffering of genetic variation. Science. 2001; 291:1001-1004. PubMed
- Integrating genetic approaches into the discovery of anticancer drugs. Science. 1997; 278:1064-1068. PubMed
- CRISPR library designer (CLD): software for multispecies design of single guide RNA libraries. Genome Biol. 2016; 17:55. PubMed
- Porcupine‐mediated lipidation is required for Wnt recognition by Wls. Dev Biol. 2012; 361:392-402. PubMed
- Compact and highly active next‐generation libraries for CRISPR‐mediated gene repression and activation. Elife. 2016; 5:e19760. PubMed
- Mapping of signaling networks through synthetic genetic interaction analysis by RNAi. Nat Methods. 2011; 8:341-346. PubMed
- Data‐driven hypothesis weighting increases detection power in genome‐scale multiple testing. Nat Methods. 2016; 13:577-580. PubMed
- A landscape of pharmacogenomic interactions in cancer. Cell. 2016; 166:740-754. PubMed
- Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003; 4:249-264. PubMed
- Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma. Proc Natl Acad Sci USA. 2013; 110:12649-12654. PubMed
- The concept of synthetic lethality in the context of anticancer therapy. Nat Rev Cancer. 2005; 5:689-698. PubMed
- The ConsensusPathDB interaction database: 2013 update. Nucleic Acids Res. 2013; 41:D793-D800. PubMed
- Integrated platform for genome‐wide screening and construction of high‐density genetic interaction maps in mammalian cells. Proc Natl Acad Sci USA. 2013; 110:E2317-E2326. PubMed
- Systematic interpretation of genetic interactions using protein networks. Nat Biotechnol. 2005; 23:561-566. PubMed
- A comprehensive transcriptional portrait of human cancer cell lines. Nat Biotechnol. 2014; 33:306-312. PubMed
- Glycosylation and palmitoylation of Wnt‐3a are coupled to produce an active form of Wnt‐3a. Genes Cells. 2007; 12:521-534. PubMed
- Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016; 44:W90-W97. PubMed
- lmerTest: tests in linear mixed effects models. J Stat Soft. 2016; 13:1-16.
- The R‐spondin/Lgr5/Rnf43 module: regulator of Wnt signal strength. Genes Dev. 2014; 28:305-316. PubMed
- Mapping genetic interactions in human cancer cells with RNAi and multiparametric phenotyping. Nat Methods. 2013; 10:427-431. PubMed
- The complexity of p53 stabilization and activation. Cell Death Differ. 2006; 13:941-950. PubMed
- The sva package for removing batch effects and other unwanted variation in high‐throughput experiments. Bioinformatics. 2012; 28:882-883. PubMed
- Role of the p16 tumor suppressor gene in cancer. J Clin Oncol. 1998; 16:1197-1206. PubMed
- Principles of cancer therapy: oncogene and non‐oncogene addiction. Cell. 2009; 136:823-837. PubMed
- Wnt/beta‐catenin signaling: components, mechanisms, and diseases. Dev Cell. 2009; 17:9-26. PubMed
- Defining genetic interaction. Proc Natl Acad Sci USA. 2008; 105:3461-3466. PubMed
- A role for mitochondrial translation in promotion of viability in K‐Ras mutant cells. Cell Rep. 2017; 20:427-438. PubMed
- Project DRIVE: a compendium of cancer dependencies and synthetic lethal relationships uncovered by large‐scale, deep RNAi screening. Cell. 2017; 170:577-592. PubMed
- GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy‐number alteration in human cancers. Genome Biol. 2011; 12:R41. PubMed
- Analysis of complex brain disorders with gene expression microarrays: schizophrenia as a disease of the synapse. Trends Neurosci. 2001; 24:479-486. PubMed
- Systematic comparison of CRISPR/Cas9 and RNAi screens for essential genes. Nat Biotechnol. 2016; 34:634-636. PubMed
- Activation of beta‐catenin‐Tcf signaling in colon cancer by mutations in beta‐catenin or APC. Science. 1997; 275:1787-1790. PubMed
- Collateral lethality: a new therapeutic strategy in oncology. Trends Cancer. 2015; 1:161-173. PubMed
- CRISPR screens provide a comprehensive assessment of cancer vulnerabilities but generate false‐positive hits for highly amplified genomic regions. Cancer Discov. 2016; 6:900-913. PubMed
- Drugging the addict: non‐oncogene addiction as a target for cancer therapy. EMBO Rep. 2016; 17:1516-1531. PubMed
- A uniform human Wnt expression library reveals a shared secretory pathway and unique signaling activities. Differentiation. 2012; 84:203-213. PubMed
- A genome‐wide RNA interference screen identifies caspase 4 as a factor required for tumor necrosis factor alpha signaling. Mol Cell Biol. 2012; 32:3372-3381. PubMed
- Synthetic lethality: general principles, utility and detection using genetic screens in human cells. FEBS Lett. 2011; 585:1-6. PubMed
- Mutant p53 gain‐of‐function in cancer. Cold Spring Harb Perspect Biol. 2010; 2:a001107. PubMed
- WNT signaling and distant metastasis in colon cancer through transcriptional activity of nuclear β‐Catenin depend on active PI3K signaling. Oncotarget. 2014; 5:2999-3011. PubMed
- Epistasis–the essential role of gene interactions in the structure and evolution of genetic systems. Nat Rev Genet. 2008; 9:855-867. PubMed
- Wnt signaling in cancer. Cold Spring Harb Perspect Biol. 2012; 4:a008052. PubMed
- GenomeCRISPR ‐ a database for high‐throughput CRISPR/Cas9 screens. Nucleic Acids Res. 2017; 45:D679-D686. PubMed
- CORUM: the comprehensive resource of mammalian protein complexes—2009. Nucleic Acids Res. 2010; 38:D497-D501. PubMed
- Improved vectors and genome‐wide libraries for CRISPR screening. Nat Methods. 2014; 11:783-784. PubMed
- The MAPK signaling cascade. FASEB J. 1995; 9:726-735. PubMed
- High‐throughput functional genomics using CRISPR‐Cas9. Nat Rev Genet. 2015; 16:299-311. PubMed
- Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 2003; 13:2498-2504. PubMed
- Combinatorial CRISPR‐Cas9 screens for de novo mapping of genetic interactions. Nat Methods. 2017; 14:573-576. PubMed
- High‐throughput RNA interference screening using pooled shRNA libraries and next generation sequencing. Genome Biol. 2011; 12:R104. PubMed
- ROCR: visualizing classifier performance in R. Bioinformatics. 2005; 21:3940-3941. PubMed
- Wnt family proteins are secreted and associated with the cell surface. Mol Biol Cell. 1993; 4:1267-1275. PubMed
- Predicting functional gene interactions with the hierarchical interaction score. Nat Methods. 2013; 10:1089-1092. PubMed
- A network of conserved synthetic lethal interactions for exploration of precision cancer therapy. Mol Cell. 2016; 63:514-525. PubMed
- Genome‐wide CRISPR screens reveal a Wnt‐FZD5 signaling circuit as a druggable vulnerability of RNF43‐mutant pancreatic tumors. Nat Med. 2017; 23:60-68. PubMed
- Roles of N‐glycosylation and lipidation in Wg secretion and signaling. Dev Biol. 2012; 364:32-41. PubMed
- HEK293 cell line: a vehicle for the expression of recombinant proteins. J Pharmacol Toxicol Methods. 2005; 51:187-200. PubMed
- Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001; 294:2364-2368. PubMed
- Oncogene addiction as a foundational rationale for targeted anti‐cancer therapy: promises and perils. EMBO Mol Med. 2011; 3:623-636. PubMed
- Defining a cancer dependency map. Cell. 2017; 170:564-576. PubMed
- Molecular role of RNF43 in canonical and noncanonical Wnt signaling. Mol Cell Biol. 2015; 35:2007-2023. PubMed
- A CRISPR dropout screen identifies genetic vulnerabilities and therapeutic targets in acute myeloid leukemia. Cell Rep. 2016; 17:1193-1205. PubMed
- Mapping of Wnt‐Frizzled interactions by multiplex CRISPR targeting of receptor gene families. FASEB J. 2017; 31:4832-4844. PubMed
- Identification and characterization of essential genes in the human genome. Science. 2015; 350:1096-1101. PubMed
- SHP2 and UGP2 are biomarkers for progression and poor prognosis of gallbladder cancer. Cancer Invest. 2016; 34:255-264. PubMed
- Genetic screens in human cells using the CRISPR‐Cas9 system. Science. 2014; 343:80-84. PubMed
- Gene essentiality profiling reveals gene networks and synthetic lethal interactions with oncogenic ras. Cell. 2017; 168:890-903. PubMed
- Mechanisms of disease: oncogene addiction—a rationale for molecular targeting in cancer therapy. Nat Clin Pract Oncol. 2006; 3:448-457. PubMed
- Glycosylation‐directed quality control of protein folding. Nat Rev Mol Cell Biol. 2015; 16:742-752. PubMed
- Towards a compendium of essential genes ‐ From model organisms to synthetic lethality in cancer cells. Crit Rev Biochem Mol Biol. 2016; 51:74-85. PubMed
- Wnt signaling in cancer. Oncogene. 2017; 36:1461-1473. PubMed
Fonte
Rauscher B, Heigwer F, Henkel L, Hielscher T, Voloshanenko O, et al. (2018) Toward an integrated map of genetic interactions in cancer cells. Molecular Systems Biology 14(2): e7656. https://doi.org/10.15252/msb.20177656




