
Abstract
Introduzione
La sovraespressione delle proteine è a volte dannosa per la crescita cellulare(Makanae et al., 2013; Sopko et al., 2006), e sono stati proposti alcuni meccanismi che potrebbero portare a difetti di crescita innescati dalla sovraespressione(Moriya, 2015). Il sovraccarico di risorse, lo squilibrio stechiometrico, l’interazione promiscua e la modulazione del percorso sono innescati dalla sovraespressione di, rispettivamente, (i) una proteina che ha una forte domanda di risorse cellulari(Dong et al., 1995; Kintaka et al., 2016; Stoebel et al., 2008), (ii) una proteina che fa parte di un complesso proteico(Kaizu et al., 2010; Makanae et al., 2013; Papp et al., 2003), (iii) una proteina con un dominio di interazione non specifico(Ma et al., 2010; Vavouri et al., 2009), e (iv) una proteina che catalizza un percorso(Prelich, 2012; Youn et al., 2017). Il meccanismo dei difetti di crescita innescati dalla sovraespressione della proteina dipende dalle caratteristiche strutturali e funzionali della proteina, che non sempre sono ancora completamente comprese. Pertanto, è ancora difficile prevedere se la sovraespressione di una particolare proteina sarà dannosa per la crescita cellulare e quali meccanismi causano l’effetto dannoso.
La sovraespressione finale di una proteina potrebbe essere dannosa per la crescita cellulare, perché monopolizza ed esaurisce le limitate risorse che sono coinvolte nella produzione di proteine, come i ribosomi e gli aminoacil-tRNA(Gong et al., 2006; Shachrai et al., 2010; Vind et al., 1993). Questo fenomeno è noto come effetto di carico proteico/costo(Kafri et al., 2016; Snoep et al., 1995). Le proteine che non hanno effetti dannosi sulle funzioni cellulari possono essere sovraespresse fino ad un livello che causa difetti di crescita dovuti al carico proteico. Al contrario, se una proteina non può essere sovraespressa fino a quel livello perché influisce negativamente sul funzionamento cellulare, allora la sovraespressione di quella proteina causerà difetti di crescita a livelli di espressione relativamente bassi, e dovremmo considerare i meccanismi che causano i difetti.
In precedenza abbiamo sviluppato un metodo di tiro alla fune genetico (gTOW) che può essere utilizzato per stimare il limite di espressione di una proteina target che innesca i difetti di crescita nel lievito Saccharomyces cerevisiae(Makanae et al., 2013; Moriyaet al., 2006, 2012 ). Abbiamo stimato che il limite di espressione di una proteina fluorescente verde (GFP) era circa il 15% della proteina cellulare totale in S. cerevisiae(Kintaka et al., 2016). Poiché la GFP è una proteina citoplasmatica altamente strutturata, non correlata alle funzioni cellulari del lievito e quindi innocua, questo livello potrebbe essere considerato il limite di espressione per qualsiasi proteina che causa difetti di crescita innescati dall’effetto proteine-carico.
Abbiamo previsto che i limiti di espressione di alcune proteine glicolitiche native altamente espresse sarebbero stati simili (>15% della proteina cellulare totale) (Moriya, 2015), suggerendo che la sovraespressione di queste proteine sarebbe innocua anche se hanno funzioni metaboliche nel lievito. La previsione è stata eseguita dal calcolo dei livelli di espressione nativa delle proteine (Kulaket al., 2014) e dei loro limiti di numero di copie geniche come determinato dall’analisi gTOW, e non è stata ancora convalidata sperimentalmente (Moriya,2015). In questo studio, quindi, abbiamo cercato di misurare i limiti di espressione di 29 proteine glicolitiche per valutare se sono espresse fino a livelli che causano difetti di crescita innescati dall’effetto proteine-carico. Ci sono cinque ragioni per cui abbiamo scelto le proteine glicolitiche: (1) perché sono generalmente altamente espresse e quindi considerate non nocive ad alti livelli di espressione, sono ottimi bersagli per esaminare se sono espresse fino al limite di proteina-cartuccia; (2) perché sono state studiate intensamente, abbiamo informazioni che possono permetterci di manipolare le loro attività catalitiche; (3) perché la via glicolitica è una delle vie metaboliche più conosciute, possiamo prevedere e misurare i cambiamenti metabolici in caso di sovraespressione di questeproteine; (4) includono un eteromero (Pfks), una proteina localizzata mitocondrialmente (Adh3) e proteine di membrana (Hxts), in modo da poter valutare come queste caratteristiche influenzano i limiti di espressione; e (5) includono paraloghi le cui espressioni sono regolate in modo diverso, in modo da poter testare come le loro differenze influenzano i loro limiti di espressione.
Abbiamo scoperto che i limiti di espressione della maggior parte delle 29 proteine erano paragonabili a quelli delle GFP ed erano determinati in modo indipendente dalle loro attività catalitiche, come suggerito da un modello cinetico di glicolisi del lievito, a conferma che la loro sovraespressione era innocua. Inoltre, alcune delle proteine avevano limiti di espressione molto più bassi di quelli che avrebbero creato un carico proteico (il limite di espressione delle GFP), e i loro effetti dannosi erano derivati dalla loro localizzazione e dalle perturbazioni metaboliche. A causa della loro ottimalità del codone, gli isoenzimi nativi mal espressi non sono stati prodotti a livelli sufficienti a causare difetti di crescita, anche quando sono stati espressi dal forte promotore TDH3 sui plasmidi multicopy. Alcune proteine glicolitiche formavano aggregati S-S-bond-mediate quando erano sovraespresse, e questa aggregazione sembrava anche limitare i loro limiti di espressione.
Risultati
La sovraespressione di proteine glicolitiche da un forte promotore su un plasmide multicopia causa difetti di crescita
La Figura 1A mostra il sistema sperimentale (plasmide) utilizzato per esprimere le proteine glicolitiche ai limiti che causano difetti di crescita. Le proteine glicolitiche target analizzate in questo studio e le loro caratteristiche sono riassunte nel file supplementare 1. Abbiamo clonato ogni gene target sul plasmide gTOW (pTOW40836)(Moriya et al., 2012), in modo tale che la proteina target è stata espressa sotto il controllo del forte promotore TDH3. PFK1 e PFK2 sono stati eccezionalmente espressi dal promotore meno forte PYK1/CDC19 perché la loro espressione dal promotore TDH3 era troppo forte e di conseguenza la crescita dei trasformanti era molto scarsa (dati non mostrati). I plasmidi sono stati utilizzati per trasformare il ceppo S. cerevisiae BY4741(ura3Δ leu2Δ ). Il numero di copie del plasmide all’interno della cellula è stato controllato attraverso il cambiamento delle condizioni di crescita: fino a 35 copie per cellula in condizioni di +leucina (-uracile) (condizioni di bassa copia) e fino a 150 copie per cellula in condizioni di -leucina (condizioni di alta copia) a causa delle distorsioni 2 µm ORI e leu2-89 (Moriya et al.,2012). In questo sistema sperimentale, i tassi di crescita massima delle cellule con il vettore in condizioni di +leucina sono molto maggiori di quelli in condizioni di -leucina (vedi Figura 1B-C) , probabilmente perché il numero di copie di leu2-89 non è sufficiente per supportare pienamente il requisito di leucina in condizioni di -leucina. Abbiamo misurato i limiti di espressione della maggior parte delle 29 proteine target in condizioni di bassa copia perché i livelli di espressione prodotti in queste condizioni erano già sufficienti a causare difetti di crescita.
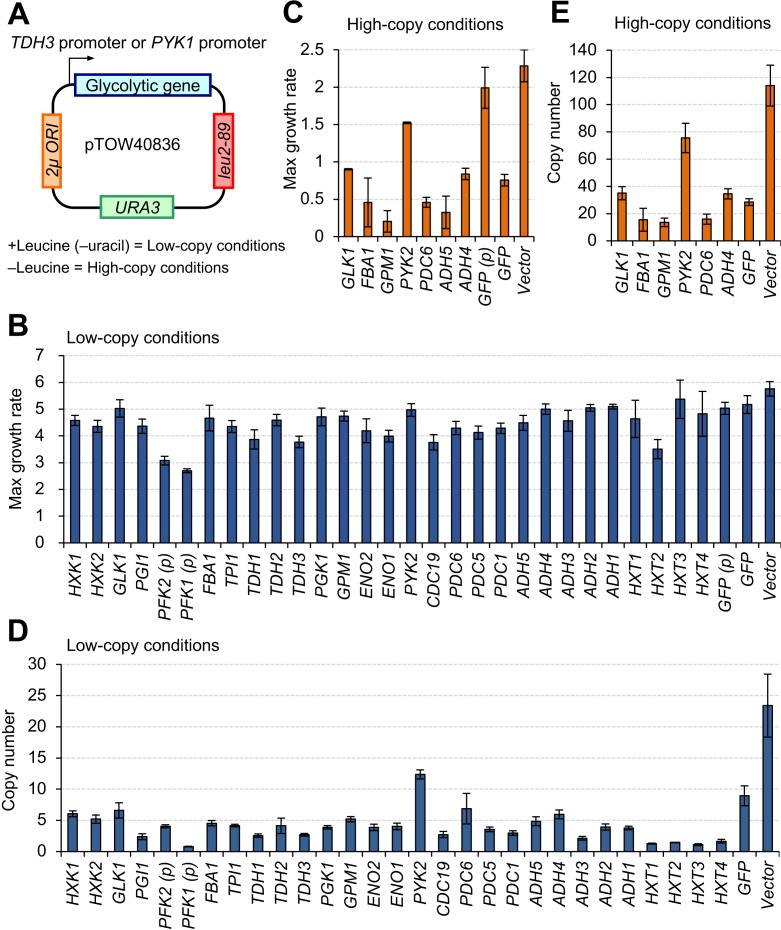
Figura 1 – Dati fonte 1.La sovraespressione della maggior parte delle proteine glicolitiche utilizzando un forte promotore e un plasmide multicopia causa difetti di crescita. Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura.(A) Il plasmide utilizzato in questo studio. Ogni gene glicolitico è stato clonato in un plasmide multicopia a base di 2 µm (pTOW40836) ed espresso dal promotore TDH3 (TDH3pro) (ad eccezione del PYK1 che è stato espresso dal promotore PYK1 [PYK1pro], rappresentato nella figura come (p)). In condizioni di +leucina, il numero di copie del plasmide è relativamente basso (~30). In condizioni di -leucina, il numero di copie arriva a 150 copie per cella a causa della polarizzazione di leu2-89 (Moriyaet al., 2012). Qui, designiamo queste condizioni di bassa e alta copia, rispettivamente.(B e C) Tasso di crescita massimo di cellule di lievito che ospitano il plasmide che sovraesprimono ogni proteina glicolitica nelle condizioni di crescita indicate. L’unità è min-1 × 10-4. (D ed E) Numero di copie del plasmide che sovraesprimono ogni proteina glicolitica nelle condizioni di crescita indicate. L’unità è il numero di copia per genoma aploide. Le barre di errore mostrano la deviazione standard di almeno tre misurazioni biologiche indipendenti.10.7554/eLife.34595.004Figure 1-source data 1.This foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura.
Abbiamo misurato per la prima volta i tassi di crescita delle cellule che ospitano i plasmidi gTOW. Come mostrato nella figura 1B, tutte le cellule che esprimono proteine glicolitiche, con l’eccezione di quelle che esprimono HXT1, HXT3 , e HXT4, hanno mostrato un significativo ritardo di crescita rispetto alle cellule di controllo vettoriale in condizioni di bassa copia (p<0,01, t-test di Welch, Figura 1-dati fonte 1), indicando che l’espressione della maggior parte delle proteine glicolitiche ha causato difetti di crescita. Questa osservazione è stata confermata dalla misurazione della crescita nelle condizioni di alta copia mostrate nella Figura 1C, poiché le cellule che esprimono la maggior parte delle proteine glicolitiche non sono cresciute in queste condizioni. Le cellule che esprimono GLK1, FBA1, GPM1, GPM1, PYK2, PDC6, ADH5, e ADH4 potrebbero crescere in condizioni di alta copia, anche se il loro tasso di crescita era significativamente inferiore a quello del controllo vettoriale (p<0,01, Welch t-test, Figura 1-source data 1).
Come precedentemente riportato, il numero di copie del plasmide gTOW all’interno della cellula riflette inversamente l’effetto deleterio dell’espressione della proteina da parte del plasmide a causa dell’effetto gTOW: il numero di copie del plasmide è basso se l’espressione della proteina target è dannosa per la crescita cellulare, e alto se l’espressione della proteina target è meno dannosa(Kintaka et al., 2016; Makanae et al., 2013; Moriya et al.,2006, 2012). La Figura 1D ed E mostra il numero di copie dei plasmidi gTOW in condizioni di bassa e alta copia. In condizioni di alta copia, i numeri di copia di plasmidi gTOW che esprimono solo GLK1, FBA1, GPM1, GPM1, PYK2, PDC6, e ADH4 sono stati determinati perché il lievito contenente plasmidi che esprimono gli altri geni codificanti le proteine non è riuscito a crescere. I numeri di copia di tutti i plasmidi gTOW contenenti i geni bersaglio erano significativamente inferiori a quelli contenenti il vettore vuoto (p<0,05, t-test di Welch, Figura 1-source data 1), confermando che sono stati espressi fino a livelli che hanno causato difetti di crescita in questo sistema sperimentale. Poiché il numero di copie di plasmidi che esprimono la maggior parte delle proteine glicolitiche testate qui, oltre a GLK1, PYK2 e ADH4, non era superiore al numero di copie di GFP, l’espressione della maggior parte delle proteine glicolitiche in questo sistema sperimentale non sembrava meno difettosa di quella delle GFP.
Abbiamo concluso che la maggior parte delle proteine glicolitiche sono state espresse vicino ai loro limiti superiori, anche in condizioni di bassa copia, e che un aumento del numero di copie in condizioni di alta copia era necessario per esprimere GLK1, FBA1, GPM1, GPM1, PYK2, PDC6, e ADH4 ai loro limiti.
Figura 1 – Dati sorgente 1.La sovraespressione della maggior parte delle proteine glicolitiche utilizzando un forte promotore e un plasmide multicopia causa difetti di crescita. Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura.(A) Il plasmide utilizzato in questo studio. Ogni gene glicolitico è stato clonato in un plasmide multicopia a base di 2 µm (pTOW40836) ed espresso dal promotore TDH3 (TDH3pro) (ad eccezione del PYK1 che è stato espresso dal promotore PYK1 [PYK1pro], rappresentato nella figura come (p)). In condizioni di +leucina, il numero di copie del plasmide è relativamente basso (~30). In condizioni di -leucina, il numero di copie arriva a 150 copie per cella a causa della polarizzazione di leu2-89 (Moriyaet al., 2012). Qui, designiamo queste condizioni di bassa e alta copia, rispettivamente.(B e C) Tasso di crescita massimo di cellule di lievito che ospitano il plasmide che sovraesprimono ogni proteina glicolitica nelle condizioni di crescita indicate. L’unità è min-1 × 10-4. (D ed E) Numero di copie del plasmide che sovraesprimono ogni proteina glicolitica nelle condizioni di crescita indicate. L’unità è il numero di copia per genoma aploide. Le barre di errore mostrano la deviazione standard di almeno tre misurazioni biologiche indipendenti.10.7554/eLife.34595.004Figure 1-source data 1.This foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura.
Misurazione dei limiti di espressione delle proteine glicolitiche
Successivamente, abbiamo misurato i livelli di espressione delle proteine all’interno delle cellule, sovraesprimendole dal plasmide gTOW. La figura 2A mostra come è stata stimata l’abbondanza di proteine. Come riportato in precedenza, quando la GFP è espressa fino al suo limite di espressione (e probabilmente al livello necessario per innescare l’effetto proteine-carico), la proteina è visibile all’interno di proteine cellulari intere separate da elettroforesi in gel di poliacrilamide di sodio dodecil solfato di sodio (SDS-PAGE) (Kintakaet al., 2016). Poiché la maggior parte delle proteine glicolitiche sono state espresse a livelli simili anche dal nostro sistema sperimentale, abbiamo misurato i livelli di espressione in unità arbitrarie (AU) come intensità relative delle bande di proteine target all’interno della proteina totale separata da SDS-PAGE. Si ritiene che l’AU rifletta il numero totale di amminoacidi all’interno della banda, e il numero relativo di molecole proteiche può essere stimato dividendo l’AU per la lunghezza della proteina. Quando due proteine di dimensioni diverse danno bande dello stesso numero di AU, il numero di molecole della proteina più grande nella banda dovrebbe essere inferiore a quello della proteina più piccola. Il rapporto tra l’AU e la percentuale di proteina totale che abbiamo precedentemente riportato(Kintaka et al., 2016) è stato stimato, come mostrato nella figura 2-figure supplement 1, come% proteina totale = 5,5 × AU. Immagini rappresentative di SDS-PAGE-separati proteine totali da cellule che ospitano plasmidi gTOW contenenti i geni della proteina glicolitica bersaglio sono mostrati in Figura 2-figure supplement 2. Come mostrato in Figura 2B, la maggior parte delle proteine sono state espresse a livelli abbastanza alti da renderle visibili all’interno della SDS-PAGE-separati proteine cellulari intere, ei livelli di espressione di Pgk1, Gmp1, Eno2, e Eno1 erano superiori a quella delle GFP. Al contrario, i livelli di espressione di Pfk1, Adh3, e Hxts erano quasi impercettibili con questo sistema sperimentale. L’aumento x-piega l’espressione di ogni proteina bersaglio oltre il suo livello nativo è mostrato in Figura 2-figure supplemento 3. L’espressione di alcune proteine è stata aumentata di oltre 10.000 volte in questo sistema sperimentale. L’espressione di Tdh3 e Gpm1 ulteriormente aumentato in condizioni di -leucina (Figura 2C), ma le cellule in questa condizione aveva arrestato la crescita (Figura 1C).
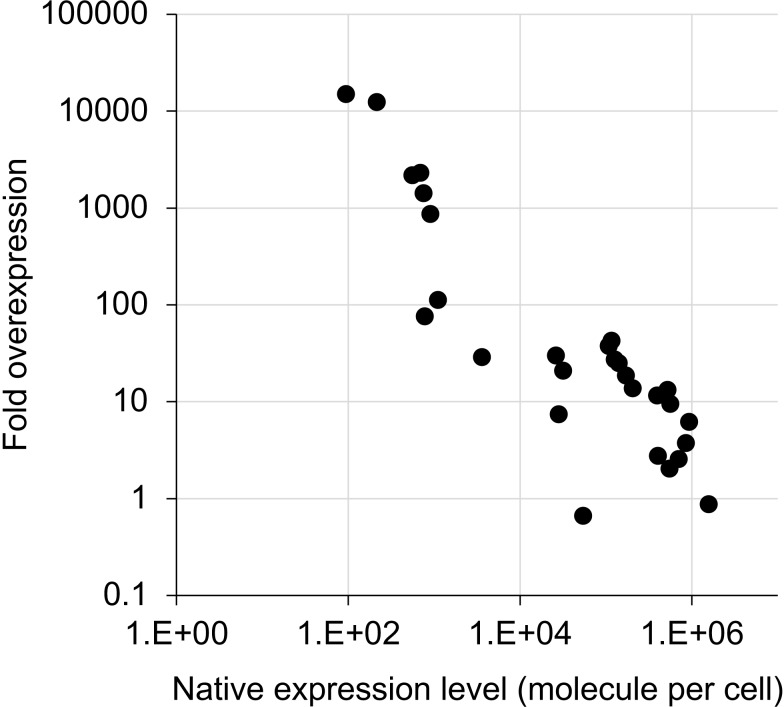
Figura 2-figure supplemento 3.Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura. Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura. relazione tra percentuale di proteina totale in(Kintaka et al., 2016) e livello proteico (AU) in questo studio. SDS-PAGE-separato proteine cellulari totali delle cellule sovraespressive proteine glicolitiche.(A) Misurazione del livello di espressione di una proteina glicolitica sovraespressa. Proteine cellulari intere sono state colorate da un colorante fluorescente e separate da SDS-PAGE. La sovraespressione della proteina bersaglio (TBS), stimata dall’intensità della sua banda sul gel e il suo peso molecolare, è stata confrontata con l’espressione corrispondente del controllo vettoriale (TBV), dopo la normalizzazione utilizzando bande di controllo (CBS e CBV) per calcolare l’espressione della proteina (AU). La misurazione del livello di espressione Gpm1 viene mostrata a titolo di esempio.(B e C) Il livello di espressione delle proteine glicolitiche sovraespresse utilizzando il sistema sperimentale mostrato in Figura 1 nelle condizioni indicate. Il promotore TDH3 è stato usato per l’espressione di tutti i geni, tranne dove (p) indica l’uso del promotore PYK1.(D) Relazione tra numero di copie e livello proteico in condizioni di bassa copia. I dati del numero di copie sono gli stessi della Figura 1D e i dati del livello proteico sono gli stessi della Figura 2B. Le barre di errore mostrano la deviazione standard di almeno tre misurazioni biologiche indipendenti.10.7554/eLife.34595.009Figure 2-source data 1.This spreadsheet contiene tutti i dati e i valori statistici associati alla figura.L’equazione derivata dalla regressione lineare dei dati e il valore R-squared sono mostrati. Ogni cerchio indica una misura indipendente dagli esperimenti Gpm1 (sei campioni), Pgk1 (tre campioni) e Adh4 (quattro campioni).Le proteine sono state analizzate in condizioni di bassa copia. I punti rossi indicano la dimensione prevista delle proteine target. Ilpromotore TDH3 è stato usato per l’espressione di tutti i geni, tranne dove (p) indica l’uso del promotore PYK1.Il livello di espressione nativa di ciascuna proteina (A) e la percentuale per la proteina S. cerevisiae totale sono stati ottenuti e calcolati utilizzando i dati pubblicati (Kulaket al., 2014). La percentuale di ciascuna proteina iperespressa (B) è stata calcolata utilizzando i dati della Figura 2B e l’equazione della Figura 2-figure supplement 1. La sovraespressione di ciascuna proteina target rispetto al livello nativo è stata poi calcolata come B/A.
Ci potrebbero essere due ragioni che spiegano perché il livello di espressione di una proteina è basso: (i) la sua forte sovraespressione è dannosa per la crescita cellulare e (ii) la sua espressione è repressa. Possiamo distinguere queste due possibilità confrontando i numeri di copia e l’abbondanza della proteina come mostrato nella Figura 2D perché il numero di copia del plasmide riflette inversamente l’effetto di deflessione dell’espressione della proteina come descritto sopra. La sovraespressione di Pfk1, Adh3 e Hxts sembrava dannosa perché il loro numero di copie era inferiore a quello delle altre proteine (cerchi rossi nella Figura 2D). Al contrario, l’espressione di Glk1, Pyk2 e Pdc6 sembrava essere repressa perché il loro numero di copie era più alto di quello delle altre proteine (cerchi blu nella Figura 2D). La relazione tra i livelli di espressione delle proteine e i numeri di copia (mostrati nella Figura 2D) suggerisce anche che i livelli di espressione non sono determinati esclusivamente dal promotore, perché non c’è una correlazione significativa tra i livelli di espressione e i numeri di copia del plasmide (Pearson r=0, 28, p=0,12).
Figura 2-figure supplement 3.Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura. Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura. Relazione tra la percentuale di proteine totali in(Kintaka et al., 2016) e il livello proteico (AU) in questo studio.(A) Misurazione del livello di espressione di una proteina glicolitica sovraespressa. Proteine cellulari intere sono state colorate da un colorante fluorescente e separate da SDS-PAGE. La sovraespressione della proteina bersaglio (TBS), stimata dall’intensità della sua banda sul gel e il suo peso molecolare, è stata confrontata con l’espressione corrispondente del controllo vettoriale (TBV), dopo la normalizzazione utilizzando bande di controllo (CBS e CBV) per calcolare l’espressione della proteina (AU). La misurazione del livello di espressione Gpm1 viene mostrata a titolo di esempio.(B e C) Il livello di espressione delle proteine glicolitiche sovraespresse utilizzando il sistema sperimentale mostrato in Figura 1 nelle condizioni indicate. Il promotore TDH3 è stato usato per l’espressione di tutti i geni, tranne dove (p) indica l’uso del promotore PYK1.(D) Relazione tra numero di copie e livello proteico in condizioni di bassa copia. I dati del numero di copie sono gli stessi della Figura 1D e i dati del livello proteico sono gli stessi della Figura 2B. Le barre di errore mostrano la deviazione standard di almeno tre misurazioni biologiche indipendenti.10.7554/eLife.34595.009Figure 2-source data 1.This spreadsheet contiene tutti i dati e i valori statistici associati alla figura.L’equazione derivata dalla regressione lineare dei dati e il valore R-squared sono mostrati. Ogni cerchio indica una misura indipendente dagli esperimenti Gpm1 (sei campioni), Pgk1 (tre campioni) e Adh4 (quattro campioni).Le proteine sono state analizzate in condizioni di bassa copia. I punti rossi indicano la dimensione prevista delle proteine target. Ilpromotore TDH3 è stato usato per l’espressione di tutti i geni, tranne dove (p) indica l’uso del promotore PYK1.Il livello di espressione nativa di ciascuna proteina (A) e la percentuale per la proteina S. cerevisiae totale sono stati ottenuti e calcolati utilizzando i dati pubblicati (Kulaket al., 2014). La percentuale di ciascuna proteina iperespressa (B) è stata calcolata utilizzando i dati della Figura 2B e l’equazione della Figura 2-figure supplement 1. La sovraespressione di ciascuna proteina target rispetto al livello nativo è stata poi calcolata come B/A.
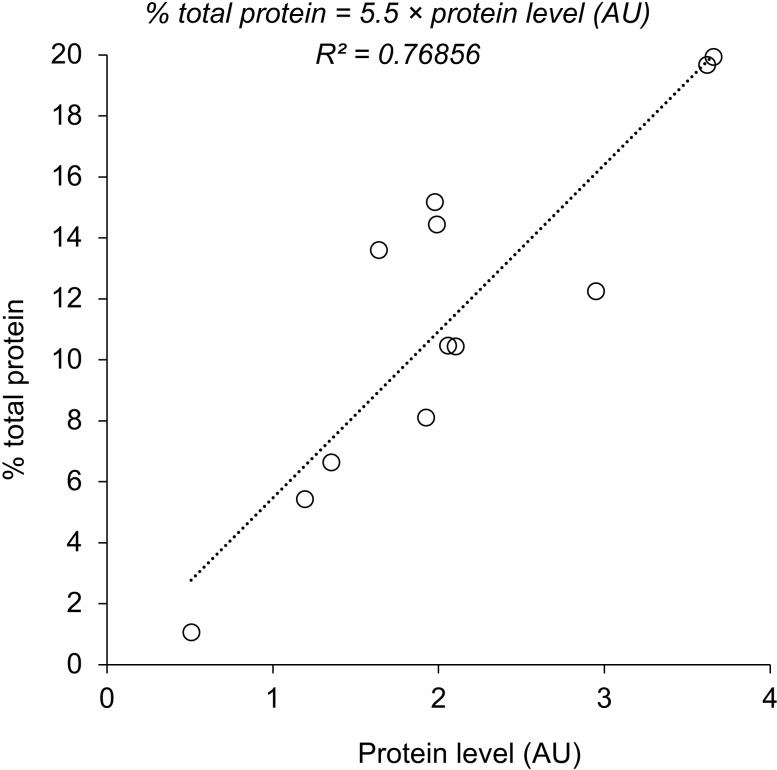
Figura 2-figure supplement 1.Relazione tra percentuale di proteina totale in(Kintaka et al., 2016) e livello proteico (AU) in questo studio.L’equazione derivata dalla regressione lineare dei dati e il valore R-squared sono mostrati. Ogni cerchio indica una misura indipendente dagli esperimenti Gpm1 (sei campioni), Pgk1 (tre campioni) e Adh4 (quattro campioni).
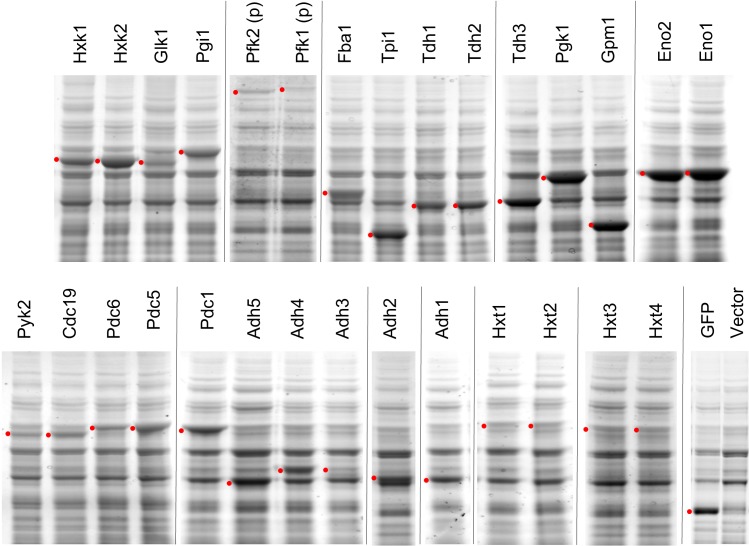
Figura 2-figure supplemento 2.SDS-PAGE-separato proteine cellulari totali delle cellule sovraespressive proteine glicolitiche.Le proteine sono state analizzate in condizioni di bassa copia. I punti rossi indicano la dimensione prevista delle proteine target. Ilpromotore TDH3 è stato utilizzato per l’espressione di tutti i geni, tranne dove (p) indica l’uso del promotore PYK1.
Figura 2-figure supplement 3.Stima della sovraespressione della piega delle proteine glicolitiche in questo studio.Il livello di espressione nativa di ciascuna proteina (A) e la percentuale per la proteina S. cerevisiae totale sono stati ottenuti e calcolati utilizzando i dati pubblicati (Kulaket al., 2014). La percentuale di ciascuna proteina iperespressa (B) è stata calcolata utilizzando i dati della Figura 2B e l’equazione della Figura 2-figure supplement 1. La sovraespressione di ciascuna proteina target rispetto al livello nativo è stata poi calcolata come B/A.
2. Le mutazioni nei centri catalitici non influenzano i limiti di espressione della maggior parte delle proteine glicolitiche
Successivamente, abbiamo cercato di rivelare i fattori che causano effetti dannosi che limitano i limiti di espressione, e i meccanismi che reprimono l’espressione delle proteine. La sovrapproduzione di proteine glicolitiche potrebbe causare perturbazioni metaboliche accelerando le reazioni che queste proteine catalizzano. Per verificare se l’inibizione della crescita causata dalle perturbazioni metaboliche limita i livelli di espressione delle proteine glicolitiche, abbiamo analizzato i limiti di espressione delle proteine mutanti con ridotta attività enzimatica introducendo mutazioni nei centri catalitici (qui chiamiamo il mutante “CC mutant”). Le mutazioni introdotte nelle proteine glicolitiche sono riassunte nel file supplementare 1. La Figura 3A mostra i livelli di espressione delle proteine wild-type e mutanti in condizioni di bassa copia. I livelli di espressione di tutte le proteine tranne Pfk1, Fba1, Tdh3 ed Eno1 non sono stati modificati in modo significativo introducendo mutazioni. I livelli di espressione di Pfk1 e Tdh3 mutanti erano significativamente più alti di quelli delle proteine wild-type e i livelli di espressione di Fba1 e Pgk1 mutanti erano significativamente più bassi di quelli delle proteine wild-type (p<0,05, test di Welch, Figura 3 – dati fonte 1). Per Pfk1, Tdh3 e Pfk2 (che catalizza la stessa reazione con Pfk1), abbiamo ulteriormente analizzato i livelli di espressione in condizioni di alta copia (Figura 3B). Il livello di espressione del Pfk2 mutante Pfk2 è aumentato significativamente rispetto a quello del Pfk2 wild-type (p=0,046, il t-test di Welch). I livelli di espressione sia del wild-type che del Pfk1 mutante erano quasi impercettibili in queste condizioni, probabilmente perché la loro espressione ad alto livello era troppo tossica per le cellule del lievito. Poiché il livello di espressione del wild-type Tdh3 era maggiore di quello del Tdh3 mutante, l’attività enzimatica del Tdh3 probabilmente non ha limitato il suo limite di espressione della proteina. Abbiamo concluso che i limiti di espressione della maggior parte delle proteine glicolitiche qui studiate non sono limitati da perturbazioni metaboliche innescate dalla loro sovrapproduzione, mentre i limiti di espressione di Pfk1 e Pfk2 sono eccezionalmente limitati da perturbazioni metaboliche. I livelli di espressione di Pfk1 e Pfk2 mutanti, tuttavia, sono rimasti nettamente inferiori a quelli di altre proteine glicolitiche, suggerendo che anche altri fattori influenzano i loro limiti di espressione.

Figura 3-figure supplement 1.Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura. Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura. Confronto delle sequenze di amminoacidi N-terminali di Adh1, Adn5, Adh3 e Adh3 mutanti senza la sequenza di targeting mitocondriale (ΔMTS-Adh3).(A e B) Livelli di espressione delle proteine glicolitiche mutanti di tipo selvatico e CC nelle condizioni indicate. Ogni CC mutante ha una mutazione nella posizione indicata nel file supplementare 1.(C) SDS-PAGE immagini gel SDS-PAGE di proteine cellulari intere che sovraesprimono Adh3 e ΔMTS-Adh3 in condizioni di bassa copia. I punti rossi indicano le dimensioni previste delle proteine target.(D) I livelli di espressione delle proteine Adh3 e ΔMTS-Adh3 in condizioni di bassa copia. Le barre di errore indicano la deviazione standard della media. *p<0,05; **p<0,01 nel t-test di Welch .10.7554/eLife.34595.012Figure 3-source data 1.Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura.Le lettere rosse indicano la sequenza di puntamento mitocondriale prevista.
Figura 3-figure supplemento 1.Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura. Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura. Confronto delle sequenze amminoacidiche N-terminali dei mutanti Adh1, Adn5, Adh3 e Adh3 senza la sequenza di targeting mitocondriale (ΔMTS-Adh3).(A e B) Livelli di espressione delle proteine glicolitiche mutanti di tipo selvatico e CC nelle condizioni indicate. Ogni CC mutante ha una mutazione nella posizione indicata nel file supplementare 1.(C) SDS-PAGE immagini gel SDS-PAGE di proteine cellulari intere che sovraesprimono Adh3 e ΔMTS-Adh3 in condizioni di bassa copia. I punti rossi indicano le dimensioni previste delle proteine target.(D) I livelli di espressione delle proteine Adh3 e ΔMTS-Adh3 in condizioni di bassa copia. Le barre di errore indicano la deviazione standard della media. *p<0,05; **p<0,01 nel t-test di Welch .10.7554/eLife.34595.012Figure 3-source data 1.Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura.Le lettere rosse indicano la sequenza di puntamento mitocondriale prevista.
Figura 3-figure supplemento 1.Figura 3—supplemento alla figura 1. Confronto delle sequenze amminoacidiche N-terminali di Adh1, Adn5, Adh3 e Adh3 mutanti senza la sequenza di targeting mitocondriale (ΔMTS-Adh3).Le lettere rosse indicano la sequenza di puntamento mitocondriale prevista.
La localizzazione mitocondriale limita il limite di espressione di Adh3
Successivamente, ci siamo concentrati su Adh3, il cui livello di espressione era inferiore a quello delle altre proteine glicolitiche, probabilmente perché l’espressione ad alto livello di Adh3 è dannosa(Figura 2D). Questo effetto dannoso, tuttavia, non è innescato da perturbazioni metaboliche, perché il livello di espressione del mutante Adh3 con ridotta attività enzimatica era quasi uguale a quello dell’Adh3 wild-type (Figura 3A). Tra le proteine glicolitiche testate in questo studio, Adh3 da solo è una proteina mitocondriale(Young and Pilgrim, 1985). Per verificare se la localizzazione mitocondriale di Adh3 limita il suo limite di espressione della proteina, abbiamo costruito un mutante senza la sequenza di targeting mitocondriale (ΔMTS-Adh3, Figura 3-figure supplement 1) e confrontato il suo livello di espressione con quello di wild-type Adh3. Come mostrato in Figura 3C e D, il livello di espressione di ΔMTS-Adh3 era circa tre volte superiore a quello di Adh3 wild-type. Abbiamo concluso che la localizzazione mitocondriale di Adh3 limita il suo limite di espressione, probabilmente perché l’espressione ad alto livello di questa proteina mitocondriale causa difetti di crescita dovuti al sovraccarico delle risorse di trasporto mitocondriale (Kintaka etal., 2016).
Perturbazioni metaboliche innescate dalla sovraespressione delle proteine glicolitiche
I risultati suggeriscono che la sovraespressione della maggior parte delle proteine glicolitiche non provoca gravi perturbazioni metaboliche. Per verificare se questa speculazione è teoricamente supportata, abbiamo usato un modello cinetico della via glicolitica del lievito(Smallbone et al., 2013); un diagramma schematico di cui è mostrato in Figura 4-figure supplement 1. La Figura 4A-D mostra il cambiamento x-fold dei metaboliti glicolitici nelle simulazioni in cui ogni proteina glicolitica è sovraespressa fino a 128 volte rispetto alla simulazione wild-type. La sovrapproduzione di 14 di 20 proteine glicolitiche non ha causato più di un doppio cambiamento metabolico (linee grigie nella Figura 4A), indicando che la sovraespressione della maggior parte delle proteine glicolitiche non causa gravi perturbazioni metaboliche. Al contrario, la sovrapproduzione di Hxk1 e Hxk2 ha interessato il metabolismo glicolitico in tutto il suo complesso, e la sovrapproduzione di Pdc1 e Cdc19 ha interessato localmente il metabolismo (Figura 4B-C). Poiché i risultati sperimentali utilizzando mutanti CC suggerito che la loro sovraespressione non ha innescato perturbazioni metaboliche che portano ai difetti di crescita, meccanismi sconosciuti per spiegare la discrepanza potrebbe esistere. La sovrapproduzione di Pfk1 o Pfk2 non ha causato un cambiamento metabolico, perché, nel modello, questi singoli enzimi non hanno catalizzato la reazione Pfk, mentre il complesso Pfk1-Pfk2 lo ha fatto. La sovrapproduzione simultanea di Pfk1 e Pfk2 ha causato gravi cambiamenti metabolici(Figura 4D), il cui modello era abbastanza simile ai cambiamenti causati dalla sovraespressione di Hxk1 e Hxk2(Figura 4 dati fonte 1) (tranne i livelli G6P e F6P non sono cambiati in quanto questi metaboliti sono a monte della reazione Pfk). Anche se i cambiamenti metabolici in seguito alla sovraespressione di Pfks e Hxks hanno mostrato un modello simile, la sovraespressione di Pfks ma non Hxks ha causato difetti di crescita (Figure1 e 2), e le mutazioni catalitiche di solo Pfks hanno aumentato il limite di espressione di questa proteina (Figura3). Quindi, i cambiamenti metabolici osservati nella simulazione non spiegano da soli i difetti di crescita innescati dalla sovraespressione di Pfks.
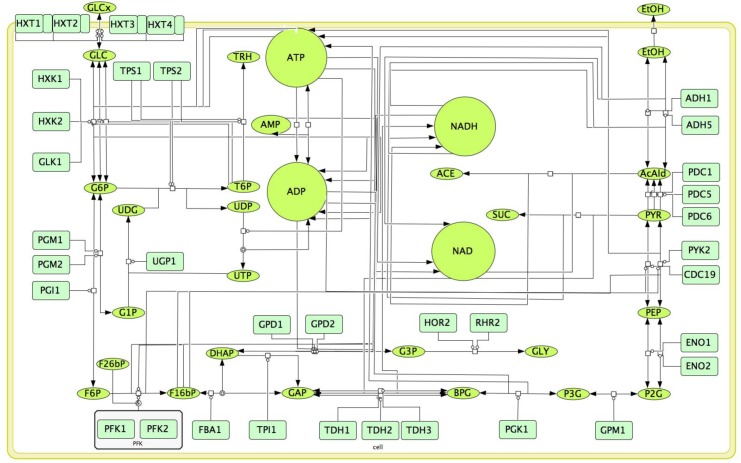
Figura 4-figure supplemento 1.Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura. Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura. Rappresentazione schematica del modello cinetico del metabolismo glicolitico utilizzato in questo studio(Smallbone et al., 2013).(A-D) Cambiamento metabolico innescato dalla sovraespressione della proteina glicolitica indicata in un modello cinetico del metabolismo glicolitico (Smallboneet al., 2013). Log10 fold-change in ogni livello metabolico in una simulazione con 128 volte la sovraespressione di ogni proteina glicolitica rispetto a quella del tipo wild-type. In (D), Pfk1 e Pfk2 sono contemporaneamente sovraespressi.10.7554/eLife.34595.015Figure 4-source data 1.This spreadsheet contiene tutti i dati e i valori statistici associati alla figura.Il diagramma è disegnato utilizzando la Notazione Grafica di Biologia dei Sistemi(Le Novère et al., 2009).
Per caratterizzare ulteriormente le condizioni fisiologiche che sono innescati dalla sovraespressione di Pfks, abbiamo poi analizzato i cambiamenti metabolici nelle cellule di lievito che sovraesprimono il tipo selvaggio e CC mutante Pfk2 sul controllo vettoriale misurando 35 metaboliti (Figura5-dati sorgente1), perché i mutanti CC hanno mostrato limiti di espressione aumentati (Figura 3B). La Figura 5A mostra i cambiamenti nei livelli di nove metaboliti glicolitici. La sovraespressione sia del wild-type che del mutante CC Pfk2 ha innescato riduzioni significative in alcuni metaboliti (p<0,05, test t di Welch, Figura 5 – dati fonte 1). Inoltre, i modelli dei cambiamenti metabolici non erano coerenti con quelli previsti dal modello(Figura 5-figure supplement 1). Queste riduzioni metaboliche non sono state quindi innescate dall’attività catalitica del Pfk2. Abbiamo notato, tuttavia, che il livello di F16bP nelle cellule sovraespressiva wild-type Pfk2 era >3 volte superiore a quello del mutante CC Pfk2 (Figura 5A, p<0,05, Welch t-test, Figura 5-source data 1). F16bP è il prodotto della catalisi di Pfk e la simulazione ha previsto un aumento del livello di F16bP in seguito alla sovraespressione di Pfks(Figura 4D), suggerendo che l’attività catalitica di Pfk2 innesca questa differenza metabolica.
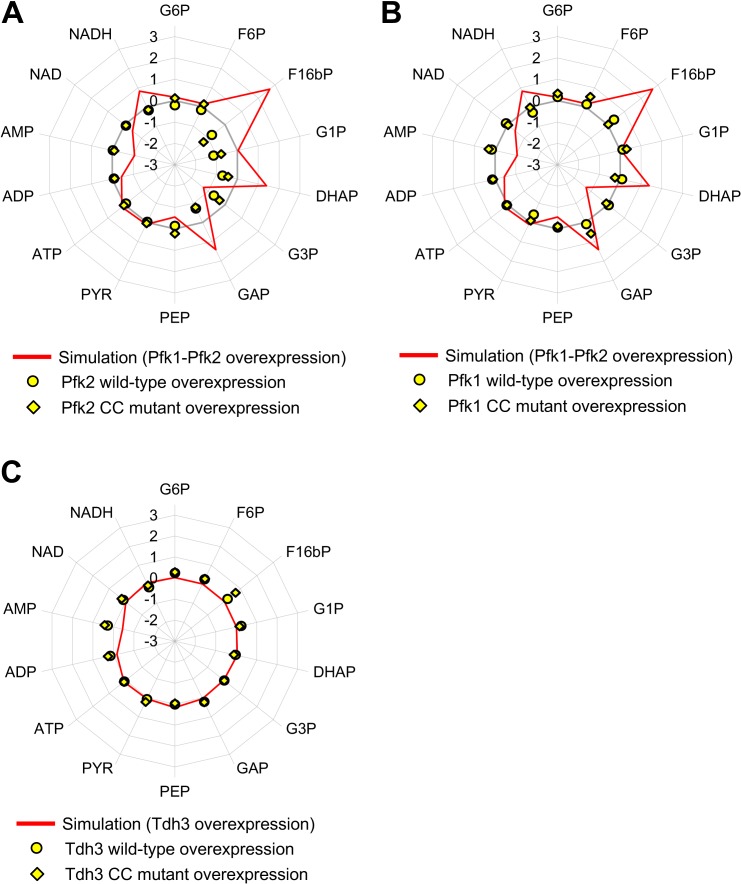
Figura 5-figure supplemento 1.Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura. Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura. Confronto dei cambiamenti metabolici innescati dalla sovraespressione delle proteine glicolitiche in silico e in vivo.(A-C) Il grafico a barre mostra il cambiamento log2-fold in ogni metabolita nelle cellule che sovraesprimono il tipo selvaggio e mutante Pfk2, Pfk1, e Tdh3 sui controlli vettoriali. Il cerchio rosso mostra la log2-differenza di piega in ogni metabolita tra il wild-type e le misure di mutante CC. I metaboliti sono stati misurati in cellule in crescita esponenziale coltivate in condizioni di bassa copia. Le barre di errore indicano le deviazioni standard della media per tre (Pfk2) e due (Pfk1, Tdh3) repliche biologiche. *p<0,05 nel t-test di Welch.10.7554/eLife.34595.018Figure 5-source data 1.Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura.Il grafico a linee mostra il risultato della simulazione e i dati sono gli stessi di quelli utilizzati nella Figura 4. I cerchi e i diamanti mostrano le misurazioni metaboliche in vivo, ed i dati sono gli stessi di quelli usati nella Figura 5.
Abbiamo poi misurato i cambiamenti metabolici in 29 metaboliti in cellule che esprimono in modo eccessivo il tipo selvaggio Pfk1 e Tdh3 e i loro mutanti CC, perché questi mutanti CC hanno anche mostrato limiti di espressione aumentati (Figura 3A). Come mostrato nella Figura 5B e C, i livelli di metaboliti glicolitici nelle cellule che sovraesprimono il wild-type Pfk1 e Tdh3 non sono stati modificati più di tre volte rispetto al controllo vettoriale. Non abbiamo osservato alcun aumento riproducibile del livello di F16bP nelle cellule che sovraesprimono il Pfk1 wild-type Pfk1 rispetto ai livelli nel suo mutante CC. Inoltre, i cambiamenti metabolici complessivi erano più elevati nelle cellule che esprimono una sovraespressione di CC mutante rispetto a quelle che esprimono il wild-type Pfk1(Figura 5B). Non abbiamo osservato alcuna differenza riproducibile nei cambiamenti metabolici tra le cellule che esprimono in modo eccessivo il wild-type Tdh3 e il suo mutante CC(Figura 5C). Abbiamo così concluso che la sovraespressione di Pfk1 e Tdh3 non ha innescato cambiamenti metabolici significativi attraverso le loro attività catalitiche, almeno nei metaboliti glicolitici rilevati.
Figura 4-figure supplement 1.Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura. Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura. Rappresentazione schematica del modello cinetico del metabolismo glicolitico utilizzato in questo studio(Smallbone et al., 2013).(A-D) Cambiamento metabolico innescato dalla sovraespressione della proteina glicolitica indicata in un modello cinetico del metabolismo glicolitico (Smallboneet al., 2013). Log10 fold-change in ogni livello metabolico in una simulazione con 128 volte la sovraespressione di ogni proteina glicolitica rispetto a quella del tipo wild-type. In (D), Pfk1 e Pfk2 sono contemporaneamente sovraespressi.10.7554/eLife.34595.015Figure 4-source data 1.This spreadsheet contiene tutti i dati e i valori statistici associati alla figura.Il diagramma è disegnato utilizzando la Notazione Grafica di Biologia dei Sistemi(Le Novère et al., 2009).
Figura 4-figure supplement 1.Rappresentazione schematica del modello cinetico del metabolismo glicolitico utilizzato in questo studio(Smallbone et al., 2013).Il diagramma è disegnato utilizzando la Notazione Grafica di Biologia dei Sistemi(Le Novère et al., 2009).
Figura 5-figure supplement 1.Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura. Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura.(A-C) Il grafico a barre mostra il cambiamento log2-fold in ogni metabolita nelle cellule che sovraesprimono il tipo selvaggio e mutante Pfk2, Pfk1, e Tdh3 sui controlli vettoriali. Il cerchio rosso mostra la log2-differenza di piega in ogni metabolita tra il wild-type e le misure di mutante CC. I metaboliti sono stati misurati in cellule in crescita esponenziale coltivate in condizioni di bassa copia. Le barre di errore indicano le deviazioni standard della media per tre (Pfk2) e due (Pfk1, Tdh3) repliche biologiche. *p<0,05 nel t-test di Welch.10.7554/eLife.34595.018Figure 5-source data 1.Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura.Il grafico a linee mostra il risultato della simulazione e i dati sono gli stessi di quelli utilizzati nella Figura 4. I cerchi e i diamanti mostrano le misurazioni metaboliche in vivo, ed i dati sono gli stessi di quelli usati nella Figura 5.
Figura 5-figure supplement 1.Confronto dei cambiamenti metabolici innescati dalla sovraespressione delle proteine glicolitiche in silico e in vivo.Il grafico a linee mostra il risultato della simulazione e i dati sono gli stessi di quelli utilizzati nella Figura 4. I cerchi e i diamanti mostrano le misurazioni metaboliche in vivo, e i dati sono gli stessi di quelli usati nella Figura 5.
L’ottimalità di Codon spiega la minore espressione delle proteine glicolitiche non nocive
Ci siamo poi concentrati su Glk1, Pyk2, e Pdc6, in quanto i loro livelli di espressione erano inferiori a quelli di altre proteine glicolitiche in condizioni di bassa copia, mentre non sembravano essere dannose(Figura 2D). Inoltre, i livelli di espressione di Glk1 e Pyk2 erano significativamente elevati in condizioni di alta copia (Figura 6A). Questi risultati hanno aumentato la possibilità che i livelli di proteine espresse per singola copia genica siano inferiori a quelli degli altri geni, sia perché i tassi di sintesi proteica sono bassi, sia perché i tassi di degradazione delle proteine sono elevati. L’ottimalità di Codon contribuisce fortemente al tasso di allungamento totale e alla stabilità dell’mRNA(Presnyak et al., 2015). Pertanto, abbiamo analizzato l’indice di adattamento tRNA di un gene (tAIg)(Tuller et al., 2010) per i geni glicolitici qui studiati (Figura 6B e Figura 6-figure supplement 1) e abbiamo notato che GLK1, PYK2 , e PDC6 aveva un tAIg molto più basso rispetto agli altri geni glicolitici. Per verificare se l’ottimalità del codone di GLK1 influisce sul livello di espressione della proteina, abbiamo costruito il codone ottimizzato GLK1(CoGLK1) e misurato il suo livello di espressione della proteina(Figura 6A). Glk1 espresso da CoGLK1 era presente a livelli 3,6 e 4,7 volte superiori a quello espresso dal GLK1 nativo in condizioni di bassa e alta copia, rispettivamente. Abbiamo concluso che l’espressione di Glk1 era bassa a causa della sua bassa ottimalità del codone.
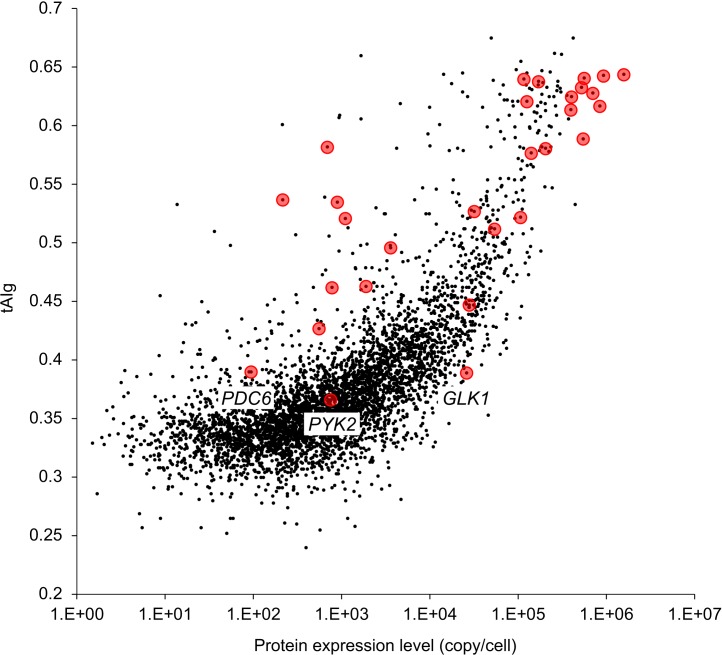
Figura 6-figure supplement 1.Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura . Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura.(A) Livelli di espressione di Glk1, Pyk2, Pdc6, e GLK1 (CoGlk1) ottimizzato per il codone nelle condizioni indicate.(B) Relazione tra il tAIg e il livello di espressione di ciascuna proteina glicolitica in condizioni di bassa copia. I dati relativi al livello di proteine sono gli stessi di quelli indicati nella Figura 2B.(C) Curve di crescita e fluorescenza GFP delle cellule che esprimono GFP ottimizzate per il codone. oG-GFP (tAIg = 0,40): un gene GFP i cui codoni sono stati ottimizzati per l’uso del codone GLK1. oT-GFP (tAIg = 0,64): un gene GFP i cui codoni sono stati ottimizzati per l’uso del codone TDH3.(D) Tempo di ritardo tra i tempi con la massima fluorescenza GFP e il massimo tasso di crescita. I tempi della massima fluorescenza GFP e del massimo tasso di crescita sono i punti temporali con i secondi massimi derivati della fluorescenza GFP e delle curve di crescita. Le barre di errore indicano le deviazioni standard dei mezzi. **p<0,01; ***p<0,001 nel t-test di Welch .10.7554/eLife.34595.021Figure 6-source data 1.Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura.Il tAIg è stato calcolato sulla base del metodo descritto da Tulleret al. (2010). Per il livello di espressione nativa della proteina S. cerevisiae è stato utilizzato un set di dati pubblicato(Kulak et al., 2014). I punti rossi indicano i geni/proteine glicolitiche che vengono analizzati in questo studio. I tAIgs di tutte le proteine del lievito sono elencati nella Figura 6-dati fonte 1.
L’espressione Glk1 aumenta dopo uno spostamento diauxico – uno spostamento di fase di crescita innescato dall’alterazione della fonte di carbonio dal glucosio all’etanolo (Zamparet al., 2013). Abbiamo ipotizzato che GLK1 potrebbe avere un uso di codone ottimizzato per il pool di tRNA dopo uno spostamento diauxico e il suo tasso di traslazione potrebbe essere più alto dopo lo spostamento. Per investigare questa possibilità, abbiamo monitorato i livelli di espressione delle GFP con diversi usi del codone in diverse condizioni di crescita. Abbiamo costruito due geni GFP i cui codoni sono stati ottimizzati in modo diverso: (i) oG-GFP, i cui codoni sono stati selezionati a caso con probabilità ottenute dalla tabella di utilizzo del codone della GLK1, e (ii) oT-GFP, i cui codoni sono stati sostituiti dal sinonimo di codone usato più frequentemente in TDH3. Abbiamo aggiunto l’ornitina decarbossilasi degron(Jungbluth et al., 2010) al C-termino di questi geni GFP per consentire un accurato monitoraggio dei tempi delle loro sintesi. La Figura 6C mostra la fluorescenza delle GFP e la crescita delle cellule che esprimono i geni GFP. La fluorescenza delle GFP di entrambi i geni ha raggiunto il picco durante le loro fasi di crescita esponenziale. Successivamente, abbiamo misurato il tempo di ritardo tra i punti di inflessione della curva di fluorescenza GFP e la curva di crescita (dove dovrebbe avvenire lo spostamento diauxico), come mostrato nella Figura 6D. Poiché i tempi di ritardo non erano significativamente diversi (p=0,44), abbiamo concluso che l’utilizzo del codone di GLK1 non era ottimizzato per massimizzare la loro traslazione dopo lo spostamento diauxico.
Figura 6-figure supplement 1.Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura . Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura.(A) Livelli di espressione di Glk1, Pyk2, Pdc6, e GLK1 (CoGlk1) ottimizzato per il codone nelle condizioni indicate.(B) Relazione tra il tAIg e il livello di espressione di ciascuna proteina glicolitica in condizioni di bassa copia. I dati relativi al livello di proteine sono gli stessi di quelli indicati nella Figura 2B.(C) Curve di crescita e fluorescenza GFP delle cellule che esprimono GFP ottimizzate per il codone. oG-GFP (tAIg = 0,40): un gene GFP i cui codoni sono stati ottimizzati per l’uso del codone GLK1. oT-GFP (tAIg = 0,64): un gene GFP i cui codoni sono stati ottimizzati per l’uso del codone TDH3.(D) Tempo di ritardo tra i tempi con la massima fluorescenza GFP e il massimo tasso di crescita. I tempi della massima fluorescenza GFP e del massimo tasso di crescita sono i punti temporali con i secondi massimi derivati della fluorescenza GFP e delle curve di crescita. Le barre di errore indicano le deviazioni standard dei mezzi. **p<0,01; ***p<0,001 nel t-test di Welch .10.7554/eLife.34595.021Figure 6-source data 1.Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura.Il tAIg è stato calcolato sulla base del metodo descritto da Tulleret al. (2010). Per il livello di espressione nativa della proteina S. cerevisiae è stato utilizzato un set di dati pubblicato(Kulak et al., 2014). I punti rossi indicano i geni/proteine glicolitiche che vengono analizzati in questo studio. I tAIgs di tutte le proteine del lievito sono elencati nella Figura 6-dati fonte 1.
Figura 6-figure supplement 1.2. Trama di dispersione che mostra il tAIg e il livello di espressione nativa della proteina S. cerevisiae.Il tAIg è stato calcolato sulla base del metodo descritto da Tulleret al. (2010). Per il livello di espressione nativa della proteina S. cer evisiae è stato utilizzato un dataset pubblicato(Kulak et al., 2014). I punti rossi indicano i geni/proteine glicolitiche che vengono analizzati in questo studio. I tAIgs di tutte le proteine del lievito sono elencati nella Figura 6-dati fonte 1.
La sovraespressione – l’aggregazione di proteine attraverso i legami S-S limita i limiti di espressione del Tpi1
Quando abbiamo misurato i livelli di espressione delle proteine Eno2 e Pgk1, abbiamo inaspettatamente osservato bande ad alto peso molecolare le cui dimensioni (~125 e 100 kDa) erano diverse dalle dimensioni dei monomeri o dimeri di Eno2 e Pgk1 (45 e 90 kDa, rispettivamente)(Figura 7A). La formazione delle bande era indipendente dalle attività catalitiche di Eno2 perché le bande sono state osservate anche nell’esperimento con Eno2 CC mutante (Figura 7A). La banda nell’esperimento Eno2 sembrava essere S-S-bond-connesso aggregati proteici perché è scomparso dopo il trattamento con l’agente riducente ditiotreitolo (DTT) (Figura 7B). Abbiamo confermato che le cisteine erano responsabili della creazione di queste bande perché sono scomparsi quando i residui di cisteina sono stati rimossi da Pgk1 e Eno2(Figura 7-figure supplemento 1). Per identificare le specie proteiche nelle bande, le abbiamo analizzate con la cromatografia liquida – spettrometria di massa in tandem (LC-MS/MS). Come mostrato nella Figura 7C e D, abbiamo rilevato principalmente proteine glicolitiche, fattori di allungamento traslazionale, e fattori di iniziazione di traduzione, oltre a ciascuna proteina sovraespressa. La maggior parte delle proteine rilevate sono state rilevate anche nell’esperimento CC mutante(Figura 7C). Questa aggregazione non sembrava influenzare i limiti di espressione di Eno3 e Pgk1 perché i limiti di espressione delle proteine wild-type e dei mutanti senza cisteina (Eno2-C248S e Pgk1-C98S) erano indistinguibili (Figura 7-figuresupplement 2).
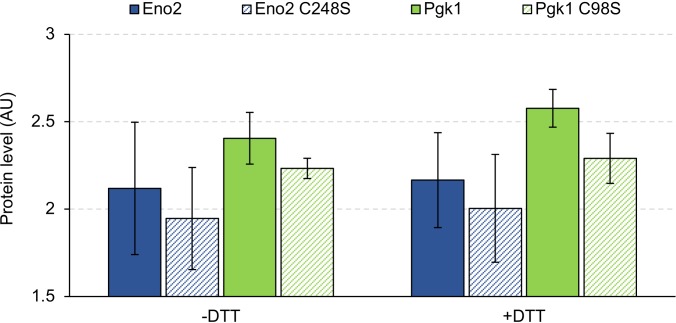
Figura 7-figure supplement 2.Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura. L’aggregazione di Eno2 e Pgk1 richiede residui di cisteina in Eno2 e Pgk1.Effetto delle sostituzioni di cisteina sui livelli di espressione di Eno2 e Pgk1.(A) SDS-PAGE-separato proteine cellulari totali dalle cellule che sovraesprimono le proteine indicate.(B) SDS-PAGE-separate proteine cellulari totali separate da cellule che sovraesprimono l’Eno2 dopo il trattamento con (+) o senza (-) l’agente riducente DTT.(C e D) Proteine arricchite nelle bande ad alto contenuto molecolare da cellule che sovraesprimono le proteine indicate. Le proteine arricchite nelle bande ad alto peso molecolare > 1,5 volte il controllo vettoriale sono mostrati. Le proteine sono state analizzate in condizioni di bassa copia. Il punto rosso indica il peso molecolare previsto delle proteine sovraespresse. L’asterisco indica la banda ad alto peso molecolare specificamente osservata in caso di sovraespressione di ogni proteina glicolitica. Le immagini del gel sono state contrastate in modo che le bande ad alto peso molecolare erano visibili. CC mut.: centro catalitico mutante.10.7554/eLife.34595.025Figure 7-source data 1.This foglio di calcolo contiene tutti i dati e valori statistici associati alla figura.(A e C) SDS-PAGE-separati proteine totali delle cellule che sovraesprimono le proteine indicate. Tre repliche biologiche per ogni esperimento sono mostrati. C248S: sostituzione di cisteina 248 a serina; C98S: sostituzione di cisteina 98 a serina. La freccia da A a B indica la direzione dell’analisi dell’intensità dei pixel mostrata in (B) e (D).(B e D) L’analisi dell’intensità dei pixel del gel corrispondente alla posizione contenente l’aggregato proteico mostrato come la freccia tra A e B. Il software Image Quant TL è stato utilizzato per l’analisi dell’immagine. L’asterisco indica la banda ad alto peso molecolare specificamente osservata in caso di sovraespressione di ogni proteina glicolitica.C248S: sostituzione della cisteina 248 con serina; C98S: sostituzione della cisteina 98 con serina. Le proteine sono state analizzate in condizioni di bassa copia.
Successivamente, ci siamo concentrati sul Tpi1 perché è stato rilevato in entrambi gli aggregati Pgk1 ed Eno2 (Figura 7C-D) e perché il suo limite di espressione (1,7 U) era inferiore a quello delle proteine a più alto limite come Pgk1 e Gpm1 (>2,0 U) (Figura 2B). Come mostrato nella Figura 8A, il Tpi1 costituiva molte bande di aggregazione al momento della sua sovraespressione. La maggior parte di queste bande sono scomparse quando i residui di cisteina sono stati rimossi dal Tpi1 (C41S, C126S), o dopo il trattamento DTT. Questi risultati suggeriscono che l’aggregazione non specifica S-S-collegato si è verificata in seguito alla sovraespressione del Tpi1. Per verificare se l’aggregazione limita il limite di espressione del Tpi1, abbiamo misurato i limiti di espressione del Tpi1 senza cisteina. Come mostrato in Figura 8B, i livelli di espressione di Tpi1 senza cisteina sono aumentati significativamente al di sopra di quelli di Tpi1 wild-type. Poiché i livelli di Tpi1 mutanti erano superiori a quelli del wild-type Tpi1, anche in condizioni +DTT, la rimozione dei residui di cisteina non solo impedirebbe la formazione di aggregati, ma aumenterebbe anche il limite di espressione del Tpi1.
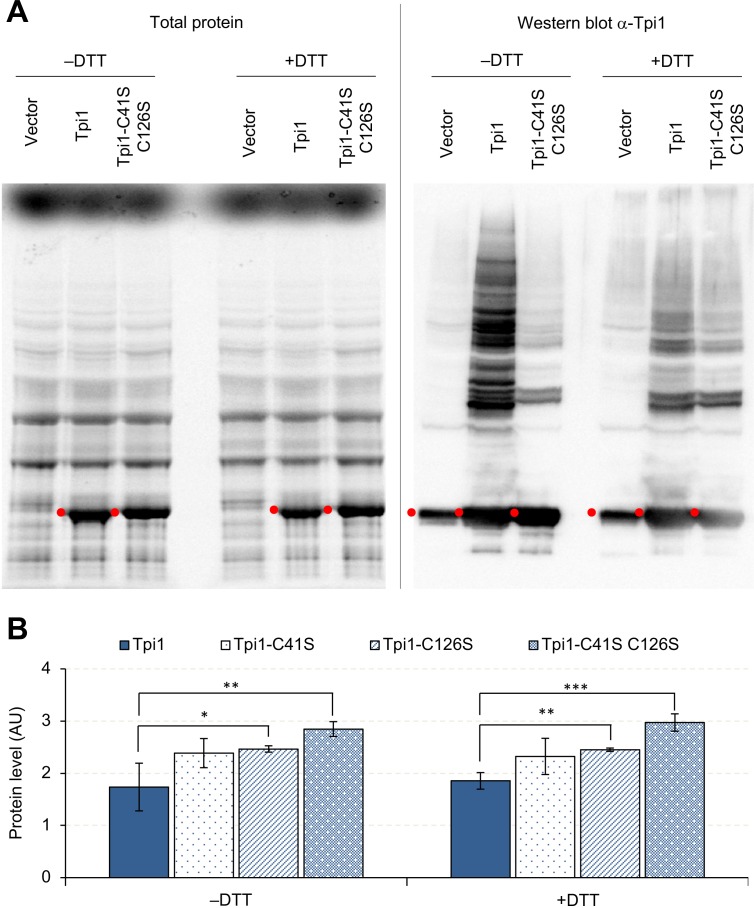
Figura 8 – Dati sorgente 1.Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura.(A) SDS-PAGE-separati proteine totali da cellule che sovraespressano le proteine indicate, e la loro immagine Western blot utilizzando anticorpi anti-Tpi1. I punti rossi indicano il peso molecolare previsto delle proteine sovraespresse.(B) Effetto delle sostituzioni di cisteina sul livello di espressione di Tpi1. *p<0,05; **p<0,01; ***p<0,001 nel t-test di Welch. C41S: sostituzione della cisteina 41 alla serina; C126S: sostituzione della cisteina 126 alla serina. Le proteine sono state analizzate in condizioni di bassa copia.10.7554/eLife.34595.027Figure 8-source data 1.This foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura.
Figura 7-figure supplemento 2.Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura. Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura. L’aggregazione di Eno2 e Pgk1 richiede residui di cisteina in Eno2 e Pgk1.Effetto delle sostituzioni di cisteina sui livelli di espressione di Eno2 e Pgk1.(A) SDS-PAGE-separato proteine cellulari totali dalle cellule che sovraesprimono le proteine indicate.(B) SDS-PAGE-separate proteine cellulari totali separate da cellule che sovraesprimono l’Eno2 dopo il trattamento con (+) o senza (-) l’agente riducente DTT.(C e D) Proteine arricchite nelle bande ad alto contenuto molecolare da cellule che sovraesprimono le proteine indicate. Le proteine arricchite nelle bande ad alto peso molecolare > 1,5 volte il controllo vettoriale sono mostrati. Le proteine sono state analizzate in condizioni di bassa copia. Il punto rosso indica il peso molecolare previsto delle proteine sovraespresse. L’asterisco indica la banda ad alto peso molecolare specificamente osservata in caso di sovraespressione di ogni proteina glicolitica. Le immagini del gel sono state contrastate in modo che le bande ad alto peso molecolare erano visibili. CC mut.: centro catalitico mutante.10.7554/eLife.34595.025Figure 7-source data 1.This foglio di calcolo contiene tutti i dati e valori statistici associati alla figura.(A e C) SDS-PAGE-separati proteine totali delle cellule che sovraesprimono le proteine indicate. Tre repliche biologiche per ogni esperimento sono mostrati. C248S: sostituzione di cisteina 248 a serina; C98S: sostituzione di cisteina 98 a serina. La freccia da A a B indica la direzione dell’analisi dell’intensità dei pixel mostrata in (B) e (D).(B e D) L’analisi dell’intensità dei pixel del gel corrispondente alla posizione contenente l’aggregato proteico mostrato come la freccia tra A e B. Il software Image Quant TL è stato utilizzato per l’analisi dell’immagine. L’asterisco indica la banda ad alto peso molecolare specificamente osservata in caso di sovraespressione di ogni proteina glicolitica.C248S: sostituzione della cisteina 248 con serina; C98S: sostituzione della cisteina 98 con serina. Le proteine sono state analizzate in condizioni di bassa copia.
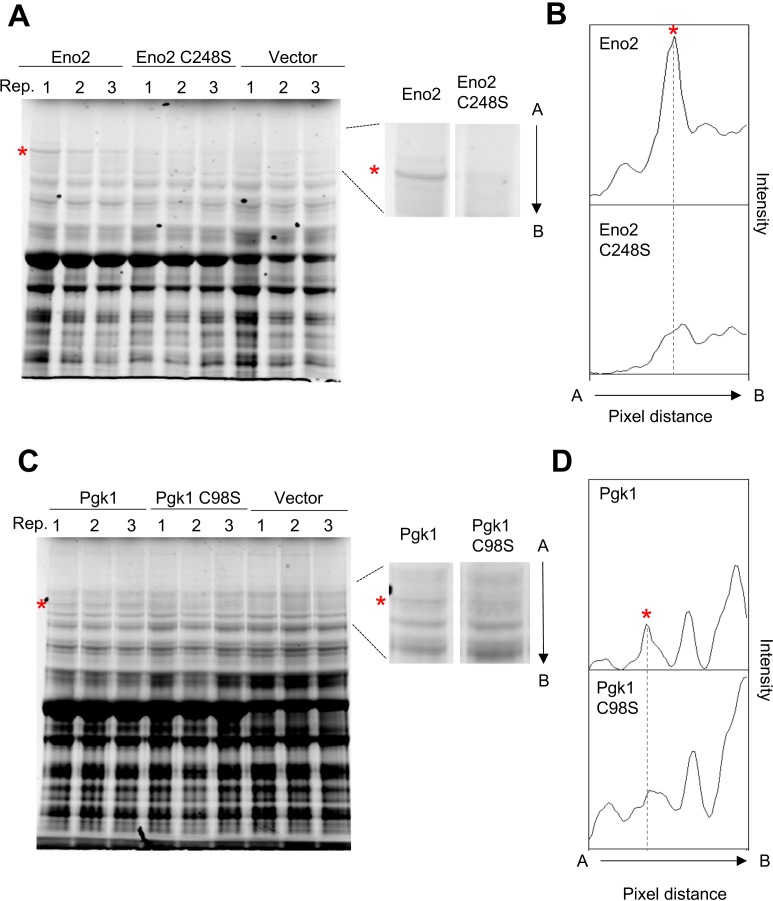
Figura 7-figure supplemento 1.L’aggregazione di Eno2 e Pgk1 richiede residui di cisteina in Eno2 e Pgk1.(A e C) SDS-PAGE-separati proteine totali delle cellule che sovraesprimono le proteine indicate. Tre repliche biologiche per ogni esperimento sono mostrati. C248S: sostituzione di cisteina 248 a serina; C98S: sostituzione di cisteina 98 a serina. La freccia da A a B indica la direzione dell’analisi dell’intensità dei pixel mostrata in (B) e (D).(B e D) L’analisi dell’intensità dei pixel del gel corrispondente alla posizione contenente l’aggregato proteico mostrato come la freccia tra A e B. Il software Image Quant TL è stato utilizzato per l’analisi dell’immagine. L’asterisco indica la banda ad alto peso molecolare specificamente osservata in caso di sovraespressione di ogni proteina glicolitica.
Figura 7-figure supplemento 2.Effetto delle sostituzioni di cisteina sui livelli di espressione di Eno2 e Pgk1.C248S: sostituzione della cisteina 248 con serina; C98S: sostituzione della cisteina 98 con serina. Le proteine sono state analizzate in condizioni di bassa copia.
Figura 8-dati fonte 1.Questo foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura.(A) SDS-PAGE-separati proteine totali da cellule che sovraespressano le proteine indicate, e la loro immagine Western blot utilizzando anticorpi anti-Tpi1. I punti rossi indicano il peso molecolare previsto delle proteine sovraespresse.(B) Effetto delle sostituzioni di cisteina sul livello di espressione di Tpi1. *p<0,05; **p<0,01; ***p<0,001 nel t-test di Welch. C41S: sostituzione della cisteina 41 alla serina; C126S: sostituzione della cisteina 126 alla serina. Le proteine sono state analizzate in condizioni di bassa copia.10.7554/eLife.34595.027Figure 8-source data 1.This foglio di calcolo contiene tutti i dati e i valori statistici associati alla figura.
Discussione
Secondo il concetto di proteine-carico(Dong et al., 1995; Kafri et al., 2016; Shah et al., 2013; Snoep et al., 1995; Stoebel et al., 2008), la sovraespressione finale di qualsiasi proteina potrebbe causare difetti di crescita sovraccaricando le risorse di produzione di proteine di base. Ma solo le proteine non nocive possono essere sovraespresse fino al livello finale, o il limite di proteina-burden, perché il limite di espressione delle proteine nocive dovrebbe essere limitato dai loro effetti dannosi. Conoscere il limite proteico-carico in sé è quindi essenziale quando si cerca di determinare se la sovraespressione di una proteina è dannosa per le funzioni cellulari. In precedenza abbiamo stimato il limite di proteina-cartuccia delle cellule di S. cerevisiae misurando il livello di espressione della GFP che causa difetti di crescita, pari al 15% della proteina cellulare totale (Kintaka et al., 2016).
In questo studio, abbiamo prima cercato di misurare i limiti di espressione delle proteine glicolitiche del lievito per confermare se il limite di proteina-burden misurato utilizzando GFP si applica alle proteine endogene. La maggior parte delle proteine glicolitiche qui studiate ha causato difetti di crescita quando sono state espresse da un forte promotore su un plasmide multicopia(Figura 1). I livelli di espressione di alcune proteine glicolitiche in queste condizioni erano, infatti, comparativi o addirittura superiori a quelli delle GFP(Figura 2). Inoltre, i loro livelli di espressione non sono aumentati a causa di mutazioni nei loro centri catalitici(Figura 3A). Questi risultati suggeriscono fortemente che l’effetto proteine-carico determina in gran parte il limite di espressione, e che il limite è di circa il 15% della proteina cellulare totale. Tra le proteine glicolitiche qui studiate, Pgk1, Gpm1, e Eno2 aveva i limiti di espressione più alti. Anche se Pgk1 (44,7 kDa) e Eno2 (46,9 kDa) sono 1,5 volte più grande di Gpm1 (27,6 kDa), i loro limiti di espressione erano simili a quelli di Gpm1 (Figura 2B). Questi risultati suggeriscono che la dimensione di una proteina non influenza il suo limite di espressione, almeno per le proteine in questo intervallo di peso molecolare. Questi dati suggeriscono anche che i limiti di espressione delle proteine non sono determinati dalle concentrazioni molari di tali proteine, ma dal costo di produzione della proteina.
Alcune altre proteine glicolitiche, come Pfk1, Pfk2, Adh3 e Hxts, hanno mostrato limiti di espressione molto al di sotto del limite di carico proteico del 15%(Figura 2), suggerendo che la sovraespressione di queste proteine è dannosa. Delle 18 proteine glicolitiche studiate, Pfk1 e Pfk2 sono state le uniche i cui limiti di espressione sono stati significativamente aumentati da mutazioni nei loro centri catalitici (Figura 3A-B),suggerendo che le loro funzioni metaboliche limitano i loro limiti di espressione. Pensiamo, tuttavia, che le perturbazioni metaboliche che hanno innescato la sovraespressione influenzino solo parzialmente i limiti di espressione perché i limiti di espressione delle proteine mutanti erano ancora molto inferiori a quelli delle altre proteine glicolitiche (Figura 3A-B). Pfk1 e Pfk2 formano un complesso etero-ottamerico e il loro squilibrio stechiometrico porta alla formazione di strutture filamentose di Pfk1 nel citosol (Schwocket al., 2004). Questo aggregato proteico stechiometrico-sbilanciamento-triggered stoichiometrico potrebbe causare difetti di crescita in seguito alla sovraespressione di Pfk1 (e Pfk2), anche se non abbiamo potuto confermare questa ipotesi perché la sovraespressione simultanea di Pfk1 e Pfk2 non ha aumentato i limiti di espressione di queste proteine (la nostra osservazione non pubblicata).
I mutanti CC di Fba1 e Pgk1 hanno mostrato limiti di espressione più bassi rispetto alle loro proteine wild-type (Figura 3A). Attualmente non abbiamo alcuna spiegazione sostanziale e coerente del perché questi mutanti CC abbiano limiti di espressione più bassi. Possiamo ipotizzare alcuni meccanismi generali: Le proteine CC mutanti sequestrano gli enzimi wild-type in complessi inattivi; le proteine CC mutanti sequestrano le molecole del substrato per gli enzimi wild-type; o la mutazione nel centro catalitico destabilizza la struttura dell’enzima. Ad esempio, Fba1 è un enzima omodimerico essenziale (UniProtKB: P14540). La sovraespressione delle molecole CC mutanti Fba1 potrebbe sequestrare le molecole Fba1 attive di tipo selvatico in complessi inattivi. Il limite di CC mutante Tdh3 era superiore a quello del wild-type in condizioni di bassa copia, mentre era inferiore in condizioni di alta copia (Figura 3A-B). Questo strano comportamento potrebbe essere correlato alla sua funzione lunare. L’attività catalitica di Tdh3 non sembra spiegare la differenza nei limiti di espressione del wild-type e del CC mutante Tdh3 (Figura 5C). Oltre alla sua funzione metabolica, Tdh3 si lega direttamente alla proteina Sir2 per promuovere il silenziamento trascrizionale, e una mutazione nel centro catalitico (C150G) riduce il silenziamento(Ringel et al., 2013). È quindi possibile che il mutante CC Tdh3 (C150S) causi il silenziamento in modo dose-dipendente facendo concorrenza al Tdh3 wild-type per il legame con Sir2.
Abbiamo ipotizzato che la localizzazione di Adh3 ai mitocondri e di Hxts alla membrana plasmatica abbia limitato i loro limiti di espressione perché le proteine localizzate sovraccaricano risorse di localizzazione più limitate (Kintakaet al., 2016). Questa ipotesi è stata confermata perché la rimozione del segnale mitocondriale da Adh3 ha aumentato il suo limite di espressione(Figura 3C, D). Abbiamo anche ipotizzato che i limiti di espressione delle proteine di membrana come Hxts dovrebbero essere limitati dalla loro localizzazione, anche se non ci sono ancora prove sperimentali a sostegno di questa ipotesi.
Il fatto che i limiti di espressione della maggior parte delle proteine glicolitiche non sono stati influenzati da mutazioni nei loro centri catalitici (Figura 3A) suggerisce che la loro sovraespressione non provoca perturbazioni metaboliche. Questo risultato è stato teoricamente confermato da simulazioni che utilizzano un modello cinetico del metabolismo glicolitico(Figura 4). La ragione per cui la loro sovraespressione non causa perturbazioni metaboliche è probabilmente dovuta al fatto che si tratta di enzimi bidirezionali: il flusso metabolico dovrebbe essere determinato solo dalla disponibilità di substrati quando le concentrazioni di questi enzimi sono superiori ad un certo livello. A sostegno di questa idea, la sovraespressione di 14 enzimi bidirezionali ha mostrato cambiamenti metabolici minori, mentre la sovraespressione di 6 enzimi unidirezionali (tra cui Hxks, Pfks, Cdc19 e Pdc1) ha mostrato forti cambiamenti metabolici nella simulazione (Figura 4). I limiti di espressione di Hxks nelle cellule, tuttavia, erano vicini al limite di carico proteico(Figura 2B) e non sono stati colpiti da mutazioni nel centro catalitico(Figura 3A). Questi risultati suggeriscono un meccanismo aggiuntivo che non è implementato nel modello che permette alle cellule di evitare gli effetti di grandi cambiamenti metabolici sulla sovraespressione di Hxks: un meccanismo che impedisce che queste perturbazioni metaboliche si verifichino, o un meccanismo che impedisce a queste perturbazioni metaboliche di causare difetti di crescita.
Attraverso l’analisi metabolica, ci siamo resi conto che attualmente non abbiamo alcun modo sistematico per identificare i cambiamenti metabolici che sono direttamente innescati dalla sovraespressione di un enzima, perché il metabolismo è interconnesso e la sovraespressione di una proteina potrebbe causare perturbazioni non specifiche che alla fine influenzano il metabolismo. Inoltre, sappiamo molto poco su quanto cambiamento in cui il metabolita innesca un difetto di crescita. Il confronto dei cambiamenti metabolici nelle cellule che sovraesprimono gli enzimi wild-type e gli enzimi CC-mutanti potrebbe essere una soluzione per questo. Infatti, abbiamo osservato una differenza di tre volte tra le cellule che esprimono il wild-type e CC mutante Pfk2 (Figura 5A). Tuttavia, ancora una volta, non possiamo concludere, sulla base delle nostre attuali conoscenze, che questa differenza causi la differenza nei limiti di espressione di queste due forme di Pfk2. Utilizzando un modello matematico, abbiamo cercato di prevedere i potenziali cambiamenti metabolici che sarebbero stati innescati dalla sovraespressione di un enzima senza considerare effetti sconosciuti diversi dall’attività metabolica dell’enzima. Nelle simulazioni, la sovraespressione di Pfks e Hxks ha innescato cambiamenti metabolici divergenti e quasi catastrofici (aumento di circa 1000 volte in alcuni metaboliti, Figura 4B,D), suggerendo che la loro sovraespressione causerebbe difetti di crescita a causa di queste forti perturbazioni metaboliche. Ci aspettavamo quindi di ottenere cambiamenti metabolici simili su sovraespressione di Pfks, i cui mutanti CC avevano limiti di espressione più elevati. Non abbiamo, tuttavia, osservato tali grandi cambiamenti (Figura 5A-B e Figura 5-figure supplement 1). Per rispondere a questi problemi con precisione, abbiamo bisogno di una comprensione molto più profonda delle connessioni tra i livelli dei metaboliti e la crescita cellulare.
Il tasso traslazionale di alcune proteine glicolitiche, tra cui il Glk1, sembrava basso a causa della loro bassa ottimalità del codone (Figura 6). In realtà, l’ottimalità codone di Glk1 (tAIg = 0,38) è vicino alla media per tutti i geni del lievito (tAIg = 0,37), e l’ottimalità codone di altre proteine glicolitiche qui studiate è eccezionalmente elevata (Figura 6-figuresupplement 1). Queste osservazioni suggeriscono che l’ottimalità del codone della maggior parte dei geni del lievito non è abbastanza alta da permettere l’espressione delle loro proteine fino al limite della proteina-burden, anche se sono espresse da un promotore forte su un plasmide multicopia.
La sovraespressione di Eno2, Pgk1, e Tpi1 ha innescato l’aggregazione S-S-collegato (Figure7 e 8), e gli aggregati che si formano contengono altre proteine glicolitiche e fattori traslazionali (Figura 7C-D). Pensiamo che questa aggregazione sia innescata dalla formazione spontanea non specifica del legame S-S tra le proteine esistenti in alte concentrazioni. È interessante notare che abbiamo anche rilevato le stesse proteine all’interno del gel del peso molecolare corrispondente nel controllo vettoriale, anche se le quantità stimate da LC-MS/MS erano inferiori e non possono essere identificati come bande proteiche visibili(Figura 7-dati fonte 1). Pertanto, abbiamo ipotizzato che l’aggregazione della proteina S-S-bond-mediata si verifica anche in condizioni fisiologiche normali, ma è accelerata da un aumento della concentrazione di proteine citoplasmatiche su sovraespressione di proteine glicolitiche. Questa aggregazione potrebbe influenzare i limiti di espressione delle proteine glicolitiche contenenti cisteina, perché la modifica dei residui di cisteina di Tpi1 in residui di serina aumenta il limite di espressione della proteina (Figura 8B). Poiché la quantità di proteina corrispondente al monomero Tpi1 non è stata modificata dal trattamento DTT, il livello di espressione di Tpi1 non dovrebbe essere ridotto semplicemente per aggregazione, ma per l’effetto nocivo della formazione spontanea del legame S-S. Questa ipotesi è supportata dal fatto che la proteina glicolitica più altamente espressa Gpm1, che ha un peso molecolare simile a quello del Tpi1, non ha un residuo di cisteina. L’effetto deleterio di questa aggregazione, tuttavia, sembra proteine-specifico perché i limiti di espressione di Pgk1 e Eno1 erano tra i più alti misurati (Figura 2A), e la rimozione della loro cisteina non ha aumentato i loro limiti di espressione (Figura 7-figuresupplemento 1).
Come descritto sopra, abbiamo rivelato meccanismi che limitano i limiti di espressione di alcune proteine glicolitiche. Non pensiamo, tuttavia, che questi meccanismi siano gli unici fattori che limitano i limiti di espressione di queste proteine. I limiti di espressione di ΔMTS-Adh3 (0,45 AU, Figura 3D) e CoGlkl (1,07 AU, Figura 6A) sono ancora inferiori a quelli di altre proteine ad alto limite come Pgk1 e Gpm1 (2,26 AU e 2,63 AU, rispettivamente, Figura 2B). È quindi probabile che più meccanismi limitino i limiti di espressione di queste proteine.
Si ritiene che il misfolding delle proteine o la loro cattiva interazione causino tossicità in caso di espressione ad alto livello di una proteina con bassa robustezza traslazionale, bassa stabilità al ripiegamento, o un’alta propensione alla cattiva interazione(Drummond e Wilke ,2009; Zhang e Yang, 2015). In generale, le proteine altamente espresse come le proteine glicolitiche sono quindi evolute per evitare queste caratteristiche (Zhang eYang, 2015), e questo dovrebbe essere un requisito per una proteina da esprimere fino al limite di proteina-burden. La Cdc19, una delle proteine glicolitiche qui studiate, si aggrega in maniera indotta da stress e reversibile attraverso una regione a bassa complessità compositiva (Saad et al., 2017). Questa capacità di aggregazione di Cdc19 potrebbe spiegare perché il suo limite di espressione (0,42 AU) è inferiore al limite di carico proteico (>2,0 AU) (Figura 2B). La nostra scoperta nella Figura 8 ha suggerito che l’espressione ad alto livello di una proteina contenente cisteina potrebbe anche causare un effetto tossico indotto da un’interazione errata; quindi le cisteine non importanti dovrebbero essere evitate nelle proteine altamente espresse. Concentrazione-dipendente fase liquida separazione dipendente dalla concentrazione è anche considerato per causare la tossicità in caso di sovraespressione di proteine strutturalmente disordinato e nucleico-acido-legante (Bolognesiet al., 2016). Non pensiamo che questo meccanismo abbia causato difetti di crescita in caso di sovraespressione delle proteine glicolitiche qui studiate perché sono meno strutturalmente disordinate (Moriya, 2015)e non proteinenucleo-acido-leganti.
Riassumiamo la nostra analisi nel file supplementare 1. In conclusione, abbiamo stabilito il livello di espressione finale che causa difetti di crescita cellulare a causa dell’effetto proteine-carico come circa il 15% della proteina cellulare totale. Il prossimo tema interessante è quello di identificare le caratteristiche delle proteine che possono essere sovraespresse fino al limite della proteina-cartuccia perché tali proteine sono considerate non dannose per le funzioni cellulari. Queste caratteristiche dovrebbero invece implicare le proprietà delle proteine che sono dannose quando sono sovraespresse.
Materiali e metodi
Ceppi, condizioni di crescita e trasformazione del lievito
BY4741(MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0) (Brachmann et al.,1998) è stato usato come ceppo ospite per gli esperimenti. La coltura e la trasformazione del lievito sono state effettuate come descritto in precedenza(Amberg et al., 2005). Per la coltura del lievito è stato utilizzato un mezzo sintetico completo (SC) senza uracile (Ura) o leucina (Leu), come indicato.
Plasmidi utilizzati nello studio
I plasmidi utilizzati nello studio sono elencati nella tabella delle risorse chiave (file supplementare2). I plasmidi sono stati costruiti dall’attività omologa di ricombinazione delle cellule di lievito(Oldenburg et al., 1997), e le loro sequenze sono state verificate con il sequenziamento del DNA.
Misurazione del numero di copie di plasmidi
Il numero di copie di plasmide è stato misurato con la reazione a catena della polimerasi in tempo reale, come descritto in precedenza(Moriya et al., 2006), utilizzando un sistema LightCycler480 (Roche). I set di primer LEU2 (LEU2-2F e LEU2-2R) e LEU3 (LEU3-3F e LEU3-3R) sono stati utilizzati per amplificare i frammenti di DNA del plasmide pTOW40836 e dei DNA genomici, rispettivamente. I valori medi, le deviazioni standard (SD) e i valori p del t-test di Welch sono stati calcolati da triplicati biologici.
Analisi delle proteine
La proteina totale è stata estratta da cellule in fase di log con un tampone NuPAGE LDS (ThermoFisher) dopo un trattamento NaOH 0,2N(Kushnirov, 2000). Per ogni analisi è stata utilizzata la proteina totale estratta da due unità di cellule a densità ottica (OD) con OD600. Per la visualizzazione della proteina totale, la proteina totale estratta è stata etichettata con Ezlabel FluoroNeo (ATTO), come descritto nel protocollo del produttore, e separati da 4-12% SDS-PAGE. Le proteine sono state rilevate e misurate utilizzando l’analizzatore di immagini LAS-4000 (GE Healthcare) in modalità di rilevamento della fluorescenza SYBR-verde e il software Image Quant TL (GE Healthcare). L’espressione di ogni proteina target (AU) è stata calcolata, come mostrato in Figura 2. Valori medi, SD, e p-valori di Welch t-testsono stati calcolati da triplici biologici. Per il rilevamento di Tpi1, le proteine SDS-PAGE-separate sono state trasferite su una membrana in PVDF (ThermoFisher). Tpi1 è stato rilevato utilizzando un anticorpo anti-Tpi1 (RRID:AB_11130951), un anticorpo secondario coniugato con perossidasi (Nichirei Biosciences), e un reagente chemiluminescente (ThermoFisher). L’immagine chemiluminescente è stata acquisita con un analizzatore di immagini LAS-4000 in modalità di rilevamento della chemiluminescenza.
Misurazione del tasso di crescita e della fluorescenza GFP
La crescita cellulare e la fluorescenza GFP sono state misurate monitorando rispettivamente OD595 e Ex485 nm/Em 535 nm, ogni 30 minuti, utilizzando un lettore di micropiastre Infinite F200 (Tecan). Il tasso di crescita massimo (MGR) è stato calcolato come descritto in precedenza(Moriya et al., 2006). I valori medi, SD, e i valori p del t-testdi Welch sono stati calcolati a partire da triplici biologici. Definiamo difetto di crescita sulla base di una significativa riduzione del tasso di crescita massimo delle cellule che sovraesprimono una proteina bersaglio rispetto a quella delle cellule che sovraesprimono il vettore di controllo (p<0,01, t-test di Welch).
In silico analisi della sovraespressione delle proteine glicolitiche
Abbiamo usato un modello cinetico del percorso glicolitico del lievito sviluppato in precedenza (Smallboneet al., 2013). Per prevedere i cambiamenti metabolici in caso di sovraespressione delle proteine glicolitiche, abbiamo cambiato la concentrazione iniziale di ogni proteina bersaglio 128 volte rispetto alla concentrazione originale, e calcolato la concentrazione di ogni metabolita allo stato stazionario. Non abbiamo analizzato il metabolismo per la sovrapproduzione di Pyk2, Adh2, Adh2, Adh3, Adh4 e Adh5, perché non sono stati inclusi o perché i loro rapporti di turnover sono stati impostati a 0 nel modello. Non abbiamo nemmeno analizzato la sovraespressione di Hxts, perché la sua concentrazione non era modificabile nel modello.
Analisi dei metaboliti
Le cellule di lievito sono state coltivate aerobicamente a 30°C per 24-48 ore in un terreno SC-Ura. Le cellule sono state inoculate in 200 mL del mezzo a un OD600di 0,5 e poi coltivate aerobicamente a 30 ° C per 3 ore 1,0 mL di coltura contenente cellule con un di OD600 di 50 è statomescolato con 1,4 mL di soluzione di metanolo pre-raffreddato a -80 ° C. Il campione è stato centrifugato a 5.000 g a -20°C per 5 min. Dopo la rimozione del surnatante, 1,0 mL di etanolo al 75% preriscaldato a 95 ° C è stato aggiunto al campione, che è stato poi incubato per 3 minuti a 95 ° C. 10 µL di 17 µM di acido D-camforo solfonico µM è stato aggiunto al campione come standard interno per la cromatografia liquida triplo stadio quadrupolo-spettrometria di massa (LC-QqQ-MS) analisi. Dopo averlo messo in ghiaccio per 5 minuti, il campione è stato centrifugato a 5.000 g a 4°C per 5 minuti per rimuovere i detriti delle cellule. 950 µL del supernatante è stato trasferito in una nuova provetta e centrifugato a 15.000 giri al minuto a 4°C per 5 min. 300 µL del supernatante raccolto come estratto cellulare è stato essiccato sotto vuoto, e poi conservato a -80°C fino all’analisi in spettrometria di massa. Tutti i metaboliti sono stati misurati usando LC-QqQ-MS. L’analisi LC-QqQ-MS è stata eseguita secondo il metodo fornito da Kato et al. (2012). Abbiamo calcolato le aree di picco standard interne normalizzate per ogni metabolita. Sono stati analizzati campioni provenienti da tre colture indipendenti per le cellule sovraespressive Pfk2, Pfk2 CC mutante, e il controllo vettoriale. Campioni da due colture indipendenti sono stati analizzati per le cellule sovraespressiva Pfk1, Pfk1 CC mutante, Tdh3, Tdh3 CC mutante, e il controllo vettoriale.
Identificazione di specie proteiche aggregate
Gli estratti proteici totali nella sovraespressione di Eno2, Eno2 CC mutante, e Pgk1 sono stati separati da SDS-PAGE e colorati con la soluzione di colorazione Coomassie (ThermoFisher). Le proteine di interesse sono state asportate dai gel e digerite con la tripsina. I peptidi triptici sono stati analizzati con LC-MS/MS costituiti da uno spettrometro di massa LTQ-Orbitrap (ThermoFisher) e un sistema DiNa nano LC (KYA Technologies) secondo il metodo descritto in precedenza(Kito et al., 2016). La miscela peptidica è stata separata con cromatografia a fase inversa. La fase mobile A conteneva lo 0,1% di acido formico, e la fase mobile B conteneva lo 0,1% di acido formico/80% di acetonitrile. I peptidi sono stati eluiti ad una velocità di flusso di 200 nL / minuto con un gradiente di 55 minuti come segue: dallo 0% al 32% solvente B oltre 45 minuti, dal 32% al 40% solvente B oltre 5 minuti, e dal 40% all’80% solvente B oltre 5 minuti. Gli spettri MS/MS acquisiti sono stati sottoposti a una ricerca nel database contro le sequenze proteiche di S. cerevisiae. Le specie proteiche aggreganti in figura 7 sono quelle per le quali il numero di colpi peptidici nella ricerca nel database è stato di cinque o più ed è stato 1,5 volte superiore a quello del controllo vettoriale.
References
- Amberg DC, Burke D, Strathern JN, Cold Spring Harbor, L. Methods in Yeast Genetics: A Cold Spring Harbor Laboratory Course Manual. Cold Spring Harbor Laboratory Press; 2005.
- Bolognesi B, Lorenzo Gotor N, Dhar R, Cirillo D, Baldrighi M, Tartaglia GG, Lehner B. A Concentration-Dependent liquid phase separation can cause toxicity upon increased protein expression. Cell Reports. 2016; 16:222-231. DOI | PubMed
- Brachmann CB, Davies A, Cost GJ, Caputo E, Li J, Hieter P, Boeke JD. Designer deletion strains derived from Saccharomyces cerevisiae S288C: a useful set of strains and plasmids for PCR-mediated gene disruption and other applications. Yeast. 1998; 14:115-132. DOI | PubMed
- Dong H, Nilsson L, Kurland CG. Gratuitous overexpression of genes in Escherichia coli leads to growth inhibition and ribosome destruction. Journal of Bacteriology. 1995; 177:1497-1504. DOI | PubMed
- Drummond DA, Wilke CO. The evolutionary consequences of erroneous protein synthesis. Nature Reviews Genetics. 2009; 10:715-724. DOI | PubMed
- Gong M, Gong F, Yanofsky C. Overexpression of tnaC of Escherichia coli inhibits growth by depleting tRNA2Pro availability. Journal of Bacteriology. 2006; 188:1892-1898. DOI | PubMed
- Jungbluth M, Renicke C, Taxis C. Targeted protein depletion in Saccharomyces cerevisiae by activation of a bidirectional degron. BMC Systems Biology. 2010; 4DOI | PubMed
- Kafri M, Metzl-Raz E, Jona G, Barkai N. The cost of protein production. Cell Reports. 2016; 14:22-31. DOI | PubMed
- Kaizu K, Moriya H, Kitano H. Fragilities caused by dosage imbalance in regulation of the budding yeast cell cycle. PLoS Genetics. 2010; 6DOI | PubMed
- Kato H, Izumi Y, Hasunuma T, Matsuda F, Kondo A. Widely targeted metabolic profiling analysis of yeast central metabolites. Journal of Bioscience and Bioengineering. 2012; 113:665-673. DOI | PubMed
- Kintaka R, Makanae K, Moriya H. Cellular growth defects triggered by an overload of protein localization processes. Scientific Reports. 2016; 6DOI | PubMed
- Kito K, Ito H, Nohara T, Ohnishi M, Ishibashi Y, Takeda D. Yeast interspecies comparative proteomics reveals divergence in expression profiles and provides insights into proteome resource allocation and evolutionary roles of gene duplication. Molecular & Cellular Proteomics. 2016; 15:218-235. DOI | PubMed
- Kulak NA, Pichler G, Paron I, Nagaraj N, Mann M. Minimal, encapsulated proteomic-sample processing applied to copy-number estimation in eukaryotic cells. Nature Methods. 2014; 11:319-324. DOI | PubMed
- Kushnirov VV. Rapid and reliable protein extraction from yeast. Yeast. 2000; 16:857-860. DOI | PubMed
- Le Novère N, Hucka M, Mi H, Moodie S, Schreiber F, Sorokin A, Demir E, Wegner K, Aladjem MI, Wimalaratne SM, Bergman FT, Gauges R, Ghazal P, Kawaji H, Li L, Matsuoka Y, Villéger A, Boyd SE, Calzone L, Courtot M, Dogrusoz U, Freeman TC, Funahashi A, Ghosh S, Jouraku A, Kim S, Kolpakov F, Luna A, Sahle S, Schmidt E, Watterson S, Wu G, Goryanin I, Kell DB, Sander C, Sauro H, Snoep JL, Kohn K, Kitano H. The systems biology graphical notation. Nature Biotechnology. 2009; 27:735-741. DOI | PubMed
- Ma L, Pang CN, Li SS, Wilkins MR. Proteins deleterious on overexpression are associated with high intrinsic disorder, specific interaction domains, and low abundance. Journal of Proteome Research. 2010; 9:1218-1225. DOI | PubMed
- Makanae K, Kintaka R, Makino T, Kitano H, Moriya H. Identification of dosage-sensitive genes in Saccharomyces cerevisiae using the genetic tug-of-war method. Genome Research. 2013; 23:300-311. DOI | PubMed
- Moriya H, Makanae K, Watanabe K, Chino A, Shimizu-Yoshida Y. Robustness analysis of cellular systems using the genetic tug-of-war method. Molecular BioSystems. 2012; 8:2513-2522. DOI | PubMed
- Moriya H, Shimizu-Yoshida Y, Kitano H. In vivo robustness analysis of cell division cycle genes in Saccharomyces cerevisiae. PLoS Genetics. 2006; 2DOI | PubMed
- Moriya H. Quantitative nature of overexpression experiments. Molecular Biology of the Cell. 2015; 26:3932-3939. DOI | PubMed
- Oldenburg KR, Vo KT, Michaelis S, Paddon C. Recombination-mediated PCR-directed plasmid construction in vivo in yeast. Nucleic Acids Research. 1997; 25:451-452. DOI | PubMed
- Papp B, Pál C, Hurst LD. Dosage sensitivity and the evolution of gene families in yeast. Nature. 2003; 424:194-197. DOI | PubMed
- Prelich G. Gene overexpression: uses, mechanisms, and interpretation. Genetics. 2012; 190:841-854. DOI | PubMed
- Presnyak V, Alhusaini N, Chen YH, Martin S, Morris N, Kline N, Olson S, Weinberg D, Baker KE, Graveley BR, Coller J. Codon optimality is a major determinant of mRNA stability. Cell. 2015; 160:1111-1124. DOI | PubMed
- Ringel AE, Ryznar R, Picariello H, Huang KL, Lazarus AG, Holmes SG. Yeast Tdh3 (glyceraldehyde 3-phosphate dehydrogenase) is a Sir2-interacting factor that regulates transcriptional silencing and rDNA recombination. PLoS Genetics. 2013; 9DOI | PubMed
- Saad S, Cereghetti G, Feng Y, Picotti P, Peter M, Dechant R. Reversible protein aggregation is a protective mechanism to ensure cell cycle restart after stress. Nature Cell Biology. 2017; 19:1202-1213. DOI | PubMed
- Schwock J, Kirchberger J, Edelmann A, Kriegel TM, Kopperschläger G. Interaction of 6-phosphofructokinase with cytosolic proteins of Saccharomyces cerevisiae. Yeast. 2004; 21:483-494. DOI | PubMed
- Shachrai I, Zaslaver A, Alon U, Dekel E. Cost of unneeded proteins in E. coli is reduced after several generations in exponential growth. Molecular Cell. 2010; 38:758-767. DOI | PubMed
- Shah P, Ding Y, Niemczyk M, Kudla G, Plotkin JB. Rate-limiting steps in yeast protein translation. Cell. 2013; 153:1589-1601. DOI | PubMed
- Smallbone K, Messiha HL, Carroll KM, Winder CL, Malys N, Dunn WB, Murabito E, Swainston N, Dada JO, Khan F, Pir P, Simeonidis E, Spasić I, Wishart J, Weichart D, Hayes NW, Jameson D, Broomhead DS, Oliver SG, Gaskell SJ, McCarthy JE, Paton NW, Westerhoff HV, Kell DB, Mendes P. A model of yeast glycolysis based on a consistent kinetic characterisation of all its enzymes. FEBS Letters. 2013; 587:2832-2841. DOI | PubMed
- Snoep JL, Yomano LP, Westerhoff HV, Ingram LO. Protein burden in Zymomonas mobilis: negative flux and growth control due to overproduction of glycolytic enzymes. Microbiology. 1995; 141:2329-2337. DOI
- Sopko R, Huang D, Preston N, Chua G, Papp B, Kafadar K, Snyder M, Oliver SG, Cyert M, Hughes TR, Boone C, Andrews B. Mapping pathways and phenotypes by systematic gene overexpression. Molecular Cell. 2006; 21:319-330. DOI | PubMed
- Stoebel DM, Dean AM, Dykhuizen DE. The cost of expression of Escherichia coli lac operon proteins is in the process, not in the products. Genetics. 2008; 178:1653-1660. DOI | PubMed
- Tuller T, Carmi A, Vestsigian K, Navon S, Dorfan Y, Zaborske J, Pan T, Dahan O, Furman I, Pilpel Y. An evolutionarily conserved mechanism for controlling the efficiency of protein translation. Cell. 2010; 141:344-354. DOI | PubMed
- Vavouri T, Semple JI, Garcia-Verdugo R, Lehner B. Intrinsic protein disorder and interaction promiscuity are widely associated with dosage sensitivity. Cell. 2009; 138:198-208. DOI | PubMed
- Vind J, Sørensen MA, Rasmussen MD, Pedersen S. Synthesis of proteins in Escherichia coli is limited by the concentration of free ribosomes. Expression from reporter genes does not always reflect functional mRNA levels. Journal of Molecular Biology. 1993; 231:678-688. DOI | PubMed
- Youn J-Y, Friesen H, Nguyen Ba AN, Liang W, Messier V, Cox MJ, Moses AM, Andrews B. Functional analysis of kinases and transcription factors in Saccharomyces cerevisiae using an integrated overexpression library. G3: Genes|Genomes|Genetics. 2017; 7:911-921. DOI | PubMed
- Young ET, Pilgrim D. Isolation and DNA sequence of ADH3, a nuclear gene encoding the mitochondrial isozyme of alcohol dehydrogenase in Saccharomyces cerevisiae. Molecular and Cellular Biology. 1985; 5:3024-3034. DOI | PubMed
- Zampar GG, Kümmel A, Ewald J, Jol S, Niebel B, Picotti P, Aebersold R, Sauer U, Zamboni N, Heinemann M. Temporal system-level organization of the switch from glycolytic to gluconeogenic operation in yeast. Molecular Systems Biology. 2013; 9DOI | PubMed
- Zhang J, Yang JR. Determinants of the rate of protein sequence evolution. Nature Reviews Genetics. 2015; 16:409-420. DOI | PubMed
Fonte
Eguchi Y, Makanae K, Hasunuma T, Ishibashi Y, Kito K, et al. () Estimating the protein burden limit of yeast cells by measuring the expression limits of glycolytic proteins. eLife 7e34595. https://doi.org/10.7554/eLife.34595




