
Abstract
Introduzione
Le piante sono organismi sessili che si sono adattati al loro habitat per ottimizzare il tempo di fioritura e garantire così il successo riproduttivo e la sopravvivenza. La temperatura è uno dei principali fattori di controllo del tempo di fioritura, in particolare prima dell’inverno, ma anche durante la primavera, quando le temperature fresche ambientali generalmente ritardano e le temperature calde favoriscono la fioritura. Diversi percorsi molecolari che controllano la fioritura in diversi intervalli di temperatura e ambienti sono stati geneticamente sezionati(Capovilla et al., 2015; Verhage et al., 2014).
In molte specie di piante e in molte accessioni della specie modello vegetale Arabidopsis thaliana (Arabidopsis), il percorso di vernalizzazione ben studiato previene la fioritura prematura prima dei lunghi periodi freddi dell’inverno(Song et al., 2012). Nell’Arabidopsis, la fioritura delle accessioni invernali senza vernalizzazione è fortemente ritardata dal fattore di trascrizione MADS-box FLOWERING LOCUS C(FLC)(Song et al., 2012; Michaels and Amasino, 1999, 2001; Johanson et al., 2000). FLC forma un complesso di repressori attraverso interazioni con il fattore di trascrizione MADS-box SVP (SHORT VEGETATIVE PHASE) per reprimere la trascrizione dei geni promotori della fioritura FLOWERING LOCUS T(FT) e SUPPRESSOR OF OVEREXPRESSION OF CO1 (SOC1)(Li et al., 2008; Lee et al., 2007). Come risultato dell’esposizione prolungata a temperature fredde durante l’inverno, l’abbondanza di FLC viene gradualmente ridotta, principalmente attraverso meccanismi epigenetici, e la repressione della fioritura viene gradualmente alleviata(Song et al., 2012). Sostanziali variazioni naturali nell’espressione che dipende dalla vernalizzazione della FLC sono già state descritte e caratterizzate in molte adesioni di Arabidopsis(Coustham et al., 2012; Li et al., 2014).
Durante la primavera, la temperatura ambiente è un importante fattore climatico. Le variazioni di temperatura di pochi gradi Celsius (°C) durante i periodi primaverili freddi o caldi spostano il tempo di fioritura in molte specie di piante. Poiché le variazioni di temperatura associate al riscaldamento globale potrebbero portare a simili cambiamenti di fioritura e minacciare i sistemi di produzione agricola, chiarire il percorso della temperatura ambiente ha recentemente ricevuto una maggiore attenzione(Moore e Lobell, 2015; Wheeler e von Braun, 2013; Jagadish et al., 2016).
Le complessità del percorso di fioritura a temperatura ambiente stanno appena iniziando a essere comprese(Capovilla et al., 2015; Verhage et al., 2014). Dopo la vernalizzazione o nelle adesioni non sensibili alla vernalizzazione, la fioritura a temperatura ambiente è in gran parte sotto il controllo della FLC relativa alla FIORITURA LOCUS M(FLM) e la FLC residua mantiene solo un ruolo minore(Gu et al., 2013; Balasubramanian et al., 2006; Blázquez et al., 2003; Lee et al., 2013). FLM controlla la fioritura nell’intervallo tra 5°C e 23°C e FLM può formare, proprio come FLC, un complesso repressivo della fioritura con SVP (Lee etal., 2013; Posé et al., 2013 ). Oltre all’FLM, come regolatore dominante, la fioritura a temperatura ambiente è regolata anche dagli omologhi FLM MAF2 – MAF4(Li et al., 2008; Gu et al., 2013; Lee et al., 2013; Ratcliffe et al., 2003; Airoldi et al., 2015). Al contrario, le mutazioni a perdita di funzione dell’SVP portano a una fioritura precoce e non sensibile alla temperatura nell’intervallo compreso tra 5°C e 27°C (Lee etal., 2007, 2013).
All’interno dell’intervallo di temperatura ambiente, l’FLM è giuntato in modo differenziato in funzione della temperatura attraverso l’uso alternativo degli esoni 2(FLM-ß ) e 3 (FLM-δ). Sulla base di esperimenti transgenici, è stato proposto un modello secondo il quale FLM-ß e FLM-δ funzionano in modo antagonistico con SVP. Di conseguenza, FLM-ß si impegna in interazioni repressive di fioritura con SVP a temperature più fredde. Al contrario, FLM-δ sarebbe superiore a FLM-ß in temperature più calde e forma eterodimeri con SVP incapace di legare il DNA e reprimere la fioritura (Lee et al., 2013; Posé et al., 2013). Dati recenti suggeriscono che questo attraente modello, basato esclusivamente su esperimenti transgenici, potrebbe non essere valido in contesti naturali(Sureshkumar et al., 2016; Lutz et al., 2015).
Finora non si sa ancora se le variazioni genetiche del locus del gene FLM svolgano un ruolo nella regolazione del tempo di fioritura attraverso l’adesione dell’Arabidopsis e, in caso affermativo, quali variazioni determinino l’espressione e lo splicing FLM basale e dipendente dalla temperatura. Sebbene due alleli di delezione FLM che conferiscono una fioritura precoce non sensibile alla temperatura siano stati identificati nelle accessioni dell’Arabidopsis Niederzenz-1 (Nd-1) e Eifel-6 (Ei-6), la loro limitata diffusione demografica e genetica ha indicato che le delezioni FLM possono essere svantaggiose(Werner et al., 2005; Balasubramanian e Weigel, 2006). Una prima variante di espressione FLM è stata determinata a partire dall’adesione alla prima fioritura Killian-0 (Kil-0). In Kil-0, un inserimento di LINE retrotransposon nell’introne FLM 1 ha causato la terminazione prematura della trascrizione, l’aberrante giunzione FLM, riducendo di conseguenza l’espressione FLM e la fioritura precedente. Questo fenotipo era particolarmente evidente ad una temperatura di 15°C , che è più vicina alla temperatura media nell’intervallo nativo della specie rispetto ai 21°C comunemente usati (Lutzet al., 2015; Hoffmann , 2002; Weigel, 2012). La successiva identificazione di nove ulteriori adesioni con un identico inserimento di LINE è stata suggestiva per un recente sweep selettivo adattativo(Lutz et al., 2015). Si è così concluso che gli alleli FLM che modulano l’espressione FLM sono vantaggiosi per l’adattamento del tempo di fioritura nell’intervallo di temperatura ambiente, soprattutto perché non ci sono altri cambiamenti di crescita pleiotropica osservati negli alleli mutanti FLM.
Il lavoro precedente aveva dimostrato che un cDNA FLM senza introni espresso da un frammento di promotore FLM non era in grado di salvare il fenotipo mutante flm. Poiché questo ha suggerito che informazioni importanti per l’espressione FLM possono risiedere in sequenze FLM non codificanti, abbiamo esaminato i polimorfismi di sequenza non codificanti con un ruolo potenziale nel controllo dell’espressione FLM e dello splicing. Utilizzando l’impronta filogenetica, abbiamo trovato che il promotore FLM conservato e le regioni dell’introne 1 sono essenziali per l’espressione FLM. Attraverso l’analisi delle associazioni utilizzando dati di polimorfismo provenienti da ≈800 adesioni di Arabidopsis, abbiamo identificato una piccola regione polimorfica nel promotore FLM e una terzina nucleotidica altamente polimorfica nell ‘introne 6 dell’FLM che controlla l’espressione FLM basale e dipendente dalla temperatura. Piccoli cambiamenti nella relativa abbondanza della variante FLM-ß della giunzione FLM-ß hanno modulato dinamicamente la fioritura a 15°C su un intervallo di 15 foglie. Quando testato in un background genetico omogeneo, l’abbondanza FLM correlato quasi perfettamente con il tempo di fioritura (R2 = 0,94) e contribuito (R2 = 0,21) alla variazione del tempo di fioritura in eterogenee popolazioni naturali Arabidopsis eterogenee. I nostri dati suggeriscono che FLM-ß è un importante fattore determinante della fioritura durante i periodi primaverili freddi o caldi.
Risultati
Le sequenze introniche sono necessarie per l’espressione FLM basale e sensibile alla temperatura
Le variazioni in funzione della temperatura nell’abbondanza di FLM e la giunzione alternativa sono fondamentali per il controllo del tempo di fioritura a temperatura ambiente in Arabidopsis. Come l’espressione FLM è regolata e se le adesioni dell’Arabidopsis hanno impiegato l’espressione FLM differenziale e lo splicing per adattarsi ai diversi climi di temperatura è rimasto da dimostrare. In precedenza abbiamo scoperto che le linee transgeniche dell’Arabidopsis che esprimono un FLM senza introni da un frammento di promotore FLM funzionale non possono esprimere FLM a livelli rilevabili e non riescono a salvare il fenotipo del tempo di fioritura di un mutante a perdita di funzione flm-3(Lutz et al., 2015). Abbiamo successivamente confrontato l’espressione FLM in linee transgeniche che esprimono un frammento FLM genomico, ottenuto dal Columbia-0 (Col-0) wild type, contenente tutti e sei gli introni (pFLM::gFLM; FLMCol-0) con linee che esprimono il FLM-ß ( pFLM::FLM-ß) o FLM-δ (pFLM ::FLM-δ ) varianti di giunzione che mantengono l’introne 1 ma mancano tutti gli altri introni (Figura 1A). La presenza dell’introne 1 era sufficiente a ripristinare l’espressione basale di FLM alle temperature ambiente di 15°C, 21°C e 27°C, ma gli introni 2-6 erano necessari per la regolazione sensibile alla temperatura delle varianti di giunzione FLM-ß e FLM-δ ( Figura 1B) . Abbiamo concluso che l’introne 1 può contenere informazioni critiche per l’espressione FLM e introni 2-6 possono contribuire alla temperatura-dipendente espressione FLM.10.7554/eLife.22114.002Figure 1.FLM sequenzeintroniche determinare basale e temperatura-dipendente espressione FLM. (A) Rappresentazione schematica del pFLM::gFLM (FLMCol-0), pFLM::FLM-ß, e pFLM::FLM-δ costruisce. La scala è impostata a 1 secondo la A del codice di partenza ATG. Le caselle nere rappresentano esoni, linee grigie 5′- o 3′- regioni non tradotte, linee grigio scuro-grigio introni. Le frecce indicano i punti di legame del primer; il primer in avanti F1 è stato usato per amplificare FLM-ß e FLM-δ con i primer inversi R1 e R2, rispettivamente.(B) Media e SD (tre repliche biologiche) delle analisi qRT-PCR di FLM-ß e FLM-δ in piante di dieci giorni che ospitano i transgeni mostrati in (A). Per la normalizzazione, i rispettivi valori misurati a 21°C sono stati impostati a 1.DOI:http://dx.doi.org/10.7554/eLife.22114.002

Figura 1.Le sequenze intronicheFLM determinano l’espressione FLM basale e dipendente dalla temperatura.(A) Rappresentazione schematica dei costrutti pFLM::gFLM (FLMCol-0), pFLM::FLM-ß, e pFLM::FLM-δ. La scala è impostata a 1 secondo la A del codice di partenza ATG. Le caselle nere rappresentano esoni, linee grigie 5′- o 3′- regioni non tradotte, linee grigio scuro-grigio introni. Le frecce indicano i punti di legame del primer; il primer in avanti F1 è stato usato per amplificare FLM-ß e FLM-δ con i primer inversi R1 e R2, rispettivamente.(B) Media e SD (tre repliche biologiche) delle analisi qRT-PCR di FLM-ß e FLM-δ in piante di dieci giorni che ospitano i transgeni mostrati in (A). Per la normalizzazione, i rispettivi valori misurati a 21°C sono stati impostati a 1.DOI:
http://dx.doi.org/10.7554/eLife.22114.002
L’impronta filogenetica individua le regioni essenziali per l’espressione basale nel promotore e nel primo introne
Per identificare le regioni non codificanti importanti per l’espressione FLM, abbiamo eseguito allineamenti di sequenze multiple di Arabidopsis FLM con il suo omologo di sequenza più vicino MAF3 e omologhi FLM di altre cinque specie di Brassicaceae(Figura 2-figure supplement 1A,B) (Martinez-Castilla e Alvarez-Buylla, 2003; Van de Velde et al., 2014). All’interno delle sequenze non codificanti, abbiamo rilevato una regione promotrice di 250 bp e due regioni dell’introne 1 di 373 e 101 bp con una maggiore conservazione della sequenza (>60%) (Figura 2A e Figura 2-figure supplement 1). Abbiamo poi generato PROΔ225bp, INT1Δ373bp, e INT1Δ101bp linee transgeniche che esprimono pFLM::gFLM varianti (FLMCol-0) condelezioni delle tre regioni nell’adesione di cancellazione FLM Nd-1 per misurare gli effetti su FLM-ß e FLM-δ espressione a 15 ° C e 23 ° C (Figura 2B e Figura 2-figure supplement 2) (Werner et al.,2005). Per normalizzare per la variabilità tra le linee transgeniche, abbiamo esaminato pool di linee indipendenti di segregamento T2 (n = 21-34). Abbiamo convalidato questa strategia di pooling dimostrando che il comportamento stabilito di FLM nelle adesioni a Col-0 e Kil-0 potrebbe essere ricapitolato fedelmente quando si eseguono analisi equivalenti con linee T2 che esprimono gFLMCol-0 e gFLMCol-0 che recano l’inserzione della Kil-0 LINE(Lutz et al., 2015)(Figura 2-figure supplement 2C,D). Mentre abbiamo rilevato un forte downregulation di FLM a 15°C e 23°C nelle linee PROΔ225bp e INT1Δ373bp, la cancellazione in INT1Δ101bp ha avuto effetti relativamente minori sull’espressione FLM ( Figura 2C,D). Ciò indicava che il promotore prossimale da 225 bp e le regioni dell’introne 1 da 337 bp contenevano sequenze importanti per l’espressione FLM basale. Esperimenti successivi hanno mostrato che il promotore 254 bp frammento promotore da solo non era, tuttavia, sufficiente a conferire espressione FLM di un frammento FLM genomico (PRO254bp; Figura 2B-D).10.7554/eLife.22114.003Figure 2.Phylogenetic footprinting identifica le regioni del promotore e dell’introne 1 necessarie per l’espressione FLM.(A) Impronta filogenetica del promotore e delle regioni genomiche di FLM e ortologie FLM putativi di sei specie di Brassicaceae. La copertura è indicata in blu, le identità sono indicate in ocre (≥30%) e in rosso (<30%). Gli esoni sono visualizzati in verde, le regioni ad alta conservazione della sequenza non codificata sono visualizzate in arancione.(B) Illustrazione schematica della regione genomica FLM e delle varianti transgeniche FLMCol-0 utilizzate per l’analisi dell’espressione.(C) e(D) Media e SD (quattro pool di replicazione con cinque a dieci linee transgeniche T2 indipendenti) dell’analisi qRT-PCR dell’espressione FLM-β (C) e FLM-δ (D ) a 15°C e 23°C in piantine di dieci giorni di 21-40 linee transgeniche T2 in massa. T-test degli studenti: *, p≤0,05; **, p≤0,01; ***, p≤0,001; n.s., non significativo.DOI:http://dx.doi.org/10.7554/eLife.22114.00310.7554/eLife.22114.004FigureSupplemento a 2 cifre 1.Impronta filogenetica dell’impronta del promotore e delle regioni genomiche di FLM e di ortologi putativi FLM di sei specie di Brassicaceae. (A) e (B) Allineamenti delle sequenze del promotore (A) e delle sequenze geniche (B) di FLM e MAF3, nonché ortologi putativi FLM di sei specie di Brassicaceae. La copertura è mostrata in blu, le identità sono basate su una parola a finestra scorrevole di dimensioni = 30 e visualizzate in ocre (≥30%) e rosso (<30%). Le regioni esoniche FLM sono mostrate in verde(B). La numerazione è indicata in base alla posizione nell’allineamento.DOI:http://dx.doi.org/10.7554/eLife.22114.00410.7554/eLife.22114.005Figuresupplemento a 2 cifre 2.I transgeni gFLMCol-0e gFLMCol-0+LINE ricapitolano fedelmente l’espressione FLM nelle adesioni di Col-0 e Kil-0.(A) Fotografie rappresentative di 42 piante di Col-0, flm-3 e Nd-1 di 42 giorni, cresciute a 15°C nel fotoperiodo di una lunga giornata. Le immagini delle piante sono state giuntate insieme, ma provengono dalla stessa fotografia, come indicato da una linea verticale bianca.(B) Analisi quantitativa del tempo di fioritura di Col-0, flm-3, e Nd-1 coltivate a 15°C e 23°C in fotoperiodo a lunga giornata.(C) e(D) qRT-PCR analisi qRT-PCR di FLM-β (C) e FLM-δ (D ) espressione a 15 ° C e 23 ° C in dieci giorni flm-3 mutanti flm-3 completati con un gFLMCol-0 o gFLMCol-0 + allele LINE(Lutz et al.,2015). Sono stati utilizzati pool di 35-40 linee transgeniche T2 indipendenti rispetto alle linee omozigote Col-0 e Kil-0. Sono mostrate la media e la SD di tre (Col-0, Kil-0) e quattro pool di replicazione che comprendono ciascuna da 8 a 10 linee transgeniche. Le percentuali si riferiscono alla riduzione dell’espressione FLM tra la linea transgenica gFLMCol-0+LINE rispetto alla linea gFLMCol-0 o tra Kil-0 e Col-0, rispettivamente. Lettere simili indicano che non vi sono differenze significative nel numero totale di foglie (Tukey HSD, p<0,05).DOI:http://dx.doi.org/10.7554/eLife.22114.005
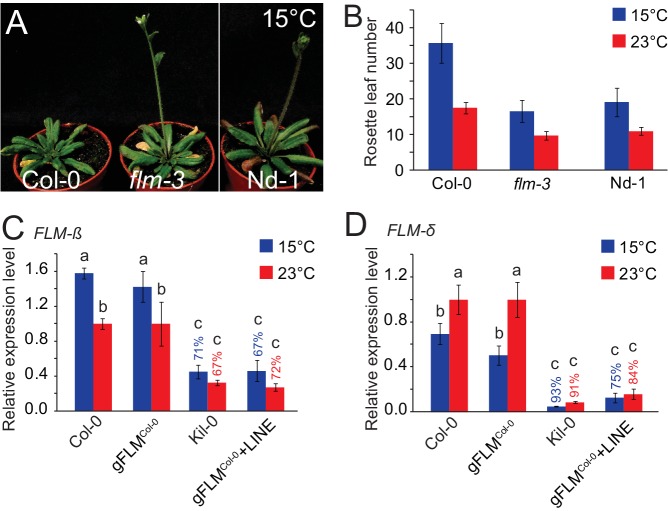
Figura 2-figure supplement 2.L’impronta filogenetica identifica le regioni del promotore e dell’introne 1 necessarie per l’espressione FLM.L’impronta filogenetica del promotore e delle regioni genomiche di FLM e degli ortologi FLM putativi di sei specie di Brassicaceae.I transgeni gFLMCol-0 e gFLMCol-0+LINE ricapitolano fedelmente l’espressione FLM nelle adesioni di Col-0 e Kil-0.(A) Impronta filogenetica dell’impronta del promotore e delle regioni genomiche di FLM e degli ortologi FLM putativi di sei specie di Brassicaceae. La copertura è indicata in blu, le identità sono indicate in ocre (≥30%) e in rosso (<30%). Gli esoni sono visualizzati in verde, le regioni ad alta conservazione della sequenza non codificata sono visualizzate in arancione.(B) Illustrazione schematica della regione genomica FLM e delle varianti transgeniche FLMCol-0 utilizzate per l’analisi dell’espressione.(C) e(D) Media e SD (quattro pool di replicazione con cinque a dieci linee transgeniche T2 indipendenti) dell’analisi qRT-PCR dell’espressione FLM-β (C) e FLM-δ (D ) a 15°C e 23°C in piantine di dieci giorni di 21-40 linee transgeniche T2 in massa. T-test degli studenti: *, p≤0,05; **, p≤0,01; ***, p≤0,001; n.s., non significativo.DOI:
http://dx.doi.org/10.7554/eLife.22114.003(A) e (B) Allineamenti delle sequenze del promotore (A)e delle sequenze geniche (B) di FLM e MAF3 , nonché ortologie FLM putativi di sei specie di Brassicaceae. La copertura è mostrata in blu, le identità sono basate su una parola a finestra scorrevole di dimensioni = 30 e visualizzate in ocre (≥30%) e rosso (<30%). Le regioni esoniche FLM sono mostrate in verde(B). La numerazione è indicata in base alla posizione nell’allineamento.DOI:
http://dx.doi.org/10.7554/eLife.22114.004(A) Fotografie rappresentative di 42 piante di 42 giorni di Col-0, flm-3 e Nd-1 cresciute a 15°C nel fotoperiodo di una lunga giornata. Le immagini delle piante sono state unite insieme, ma provengono dalla stessa fotografia, come indicato da una linea verticale bianca.(B) Analisi quantitativa del tempo di fioritura di Col-0, flm-3, e Nd-1 coltivate a 15°C e 23°C in fotoperiodo a lunga giornata.(C) e(D) qRT-PCR analisi qRT-PCR di FLM-β (C) e FLM-δ (D ) espressione a 15 ° C e 23 ° C in dieci giorni flm-3 mutanti flm-3 completati con un gFLMCol-0 o gFLMCol-0 + allele LINE(Lutz et al.,2015). Sono stati utilizzati pool di 35-40 linee transgeniche T2 indipendenti rispetto alle linee omozigote Col-0 e Kil-0. Sono mostrate la media e la SD di tre (Col-0, Kil-0) e quattro pool di replicazione che comprendono ciascuna da 8 a 10 linee transgeniche. Le percentuali si riferiscono alla riduzione dell’espressione FLM tra la linea transgenica gFLMCol-0+LINE rispetto alla linea gFLMCol-0 o tra Kil-0 e Col-0, rispettivamente. Lettere simili indicano che non vi sono differenze significative nel numero totale di foglie (Tukey HSD, p<0,05).DOI:
http://dx.doi.org/10.7554/eLife.22114.005

Figura 2-figure supplement 1.2. Impronta filogenetica dell’impronta del promotore e delle regioni genomiche di FLM e di ortologi FLM putativi di sei specie di Brassicaceae.2.(A) e (B) Allineamenti delle sequenze del promotore (A)e delle sequenze genomiche (B) di FLM e MAF3 , nonché ortologie FLM putativi di sei specie di Brassicaceae. La copertura è mostrata in blu, le identità sono basate su una parola a finestra scorrevole di dimensioni = 30 e visualizzate in ocre (≥30%) e rosso (<30%). Le regioni esoniche FLM sono mostrate in verde(B). La numerazione è indicata in base alla posizione nell’allineamento.DOI:
http://dx.doi.org/10.7554/eLife.22114.004
Figura 2-figure supplement 2.I transgeni gFLMCol-0 e gFLMCol-0+LINE ricapitolano fedelmente l’espressione FLM nelle adesioni a Col-0 e Kil-0.(A) Fotografie rappresentative di 42 piante di 42 giorni di Col-0, flm-3 , e Nd-1 cresciute a 15°C nel fotoperiodo di una lunga giornata. Le immagini delle piante sono state unite insieme, ma provengono dalla stessa fotografia, come indicato da una linea verticale bianca.(B) Analisi quantitativa del tempo di fioritura di Col-0, flm-3, e Nd-1 coltivate a 15°C e 23°C in fotoperiodo a lunga giornata.(C) e(D) qRT-PCR analisi qRT-PCR di FLM-β (C) e FLM-δ (D ) espressione a 15 ° C e 23 ° C in dieci giorni flm-3 mutanti flm-3 completati con un gFLMCol-0 o gFLMCol-0 + allele LINE(Lutz et al.,2015). Sono stati utilizzati pool di 35-40 linee transgeniche T2 indipendenti rispetto alle linee omozigote Col-0 e Kil-0. Sono mostrate la media e la SD di tre (Col-0, Kil-0) e quattro pool di replicazione che comprendono ciascuna da 8 a 10 linee transgeniche. Le percentuali si riferiscono alla riduzione dell’espressione FLM tra la linea transgenica gFLMCol-0+LINE rispetto alla linea gFLMCol-0 o tra Kil-0 e Col-0, rispettivamente. Lettere simili indicano che non vi sono differenze significative nel numero totale di foglie (Tukey HSD, p<0,05).DOI:
http://dx.doi.org/10.7554/eLife.22114.005
La variazione di sequenza non codificata dei principali aplotipi FLM influenza l’espressione FLM
Per trovare ulteriori determinanti non codificanti per l’espressione FLM, abbiamo analizzato la variazione nucleotidica FLM in 776 adesioni sequenziate di Arabidopsis(The 1001 Genomes Consortium, 2016). Abbiamo identificato 45 SNPs promotori e intronici con una frequenza di allele minore (MAF)≥5% e li abbiamo utilizzati per definire dieci aplotipi principali (H1–H10) che rappresentano 379 (49%) adesioni (Figura 3-figure supplement 1A,B; File supplementare 1). Abbiamo definito un set iniziale con 41 adesioni selezionando da cinque a dodici adesioni da sei dei dieci gruppi di aplotipi(Figura 3A). Poiché gli introni 2-6 sembravano importanti per l’espressione FLM sensibile alla temperatura, abbiamo aggiunto 11 adesioni con diversi aplotipi di introni 2-6 (HI2-6) ma identici aplotipi di introni 1 (HI1; Figura 3A e Figura 3-figure supplement 1D-G). Infine, abbiamo aggiunto Col-0 (H3) e Kil-0 (H1) grazie alla loro regolazione FLM ben caratterizzata per ottenere un set di aplotipi FLM rappresentativo con 54 adesioni(file supplementare 2). Abbiamo assicurato, mediante PCR analitica, che nessuna di queste adesioni, ad eccezione di Kil-0, portava l’inserimento della LINEA dell’introne 1 precedentemente descritto(Lutz et al., 2015).10.7554/eLife.22114.006Figure 3.FLM haplotype analysis of 776 accessions identifica i principali aplotipi determinati da variazioni non codificate.(A) Affiliazione del gruppo di aplotipi di 54 adesioni dell’insieme di aplotipi FLM basata su 45 SNPs per il locus FLM 7 kb (H7kb), l’introne 1 (HI1) o gli introni 2-6 (HI2-6). La numerazione e la colorazione del gruppo dipendono dalle dimensioni del gruppo.(B) Valori di espressione riassunti dell’espressione totale FLM (esone 1), FLM-ß, e FLM-δ dell’insieme di aplotipi FLM. I valori erratici sono stati determinati sulla base di 1,5 x IQR (intervallo interquartile). Il coefficiente di variazione (CV) è mostrato nel grafico in basso.DOI:http://dx.doi.org/10.7554/eLife.22114.00610.7554/eLife.22114.007Figuresupplemento a 3 cifre 1.Haplotype analysis basato su 45 SNPs da 776 adesioni.(A) Rappresentazione della posizione, della frequenza e del tipo di 45 SNPs da 776 adesioni utilizzate per il clustering di aplotipi.(B),(D) e(E) Analisi della rete di aplotipi dei dieci(B) gruppi più frequenti di una regione genomica FLM a 7 kb basati su 45 SNP o dei dieci (D) o undici(E) gruppi di aplotipi più grandi quando si considerano solo gli SNP dell’introne 1 (HI1; D) o degli introni 2-6 (HI2-6; E). Le dimensioni del cerchio e il codice colore illustrano il numero di adesioni in ogni gruppo con il gruppo 1 che rappresenta rispettivamente 174(B), 220(D) e 356 (E) adesioni. In(B), i numeri dei bulloni indicano i gruppi da cui sono state scelte le adesioni rappresentative.(C) Distribuzione geografica dei dieci gruppi più frequenti di una regione genomica FLM a 7 kb.(F) Diagramma di Venn che indica la sovrapposizione delle adesioni incluse nei dieci maggiori gruppi aplotipi HI1 e HI2-6.(G) Clustering dei gruppi di aplotipi delle 589 adesioni incluse nei dieci maggiori gruppi di aplotipi HI1 e HI2-6. Le adesioni sono state ordinate in base ai numeri dei gruppi di aplotipi HI1 e HI2-6. I numeri a sinistra indicano il numero del gruppo di aplotipi HI1, come indicato anche in(D). A destra è mostrato un ingrandimento di un sottoinsieme di adesioni evidenziato dalle caselle tratteggiate. La codifica a colori per i numeri dei gruppi di aplotipi, che illustra il numero del gruppo e la dimensione del gruppo, come mostrato in(D) e(E), è specificata sotto il grafico.DOI:http://dx.doi.org/10.7554/eLife.22114.00710.7554/eLife.22114.008Figuresupplemento a 3 cifre 2.Tempo di fioritura delle adesioni del set di aplotipi FLM suggeriscono un requisito di vernalizzazione per molte adesioni.(A) Analisi del tempo di fioritura delle adesioni del set di aplotipi FLM. Le adesioni a fioritura estremamente tardiva o le adesioni che non avevano fiorito sono rappresentate al margine delle foglie a 60 rosette. I valori anomali sono stati determinati sulla base di 1,5 x IQR (intervallo interquartile).(B) Media e SD (tre repliche biologiche) nelle analisi qRT-PCR di FLM-β e FLM-δ in dodici giorni Col-0, flc-3 e Col-0 con un modulo divernalizzazioneFRISf-2FLC (Michaelse Amasino, 1999).DOI:http://dx.doi.org/10.7554/eLife.22114.00810.7554/eLife.22114.009Figure 3-figure supplement 3.Correlation analysis identifica FLM-β come la trascrizione principale FLM a 15°C e 23°C nelle adesioni all’insieme di aplotipi FLM.(A) e (B) Correlazione (regressione lineare semplice) tra l’espressione FLM come determinata da qRT-PCR sull’esone 1 FLM con misure di abbondanza della variante di giunzione FLM-β e FLM-δ a 15°C (A) e 23°C (B) nelle 54 adesioni dell’insieme di aplotipi FLM. FLM-ß , 15°C, p<0.0001; FLM-δ , 15°C , p=0.009; FLM-ß , 23°C, p<0. 0001; FLM-δ, 23°C, p>0.05.DOI:http://dx.doi.org/10.7554/eLife.22114.00910.7554/eLife.22114.010Figure 3-figure supplement 4.119 siti polimorfici tra le adesioni dell’insieme di aplotipi FLM.(A) Posizione, frequenza e tipo di ciascuno dei 119 siti polimorfici tra le 54 adesioni dell’insieme di aplotipi FLM utilizzati per semplici test di associazione di singoli locus.(B) Albero di congiunzione tra i vicini che mostra la relazione genetica tra 54 adesioni dell’insieme di aplotipi FLM basato sui 119 siti polimorfici mostrati in(A). I valori dell’aplotipo sono indicati in ogni ramo.DOI:http://dx.doi.org/10.7554/eLife.22114.01010.7554/eLife.22114.011FigureSupplemento a 3 cifre 5.Risultati del test di associazione a singolo locus semplice tra i 119 siti polimorfi con valori di espressione FLM.(A) e (B) Rappresentazione dei valori p -log(10)-trasformati dei test di associazione a singolo locus semplice di ciascuno dei 119 siti che sono stati inclusi nell’analisi. La linea orizzontale nera corrisponde ad una soglia di significatività di p=0,05. I punti neri rappresentano i siti polimorfici che si associano con p>0,05, i punti rossi quelli con p<0,05. I punti verdi rappresentano i siti con p<0,05 che sono stati scelti per un’analisi sperimentale dettagliata come mostrato nella Figura 4A. Il modello genomico del gene FLM è quello introdotto nella Figura 1. Ogni grafico rappresenta l’analisi del tratto di espressione indicato.(A) mostra tutti i confronti con i valori di espressione relativi e(B) mostra tutti i confronti dei cambiamenti relativi dei livelli di trascrizione tra FLM-β e FLM-δ in risposta a variazioni di temperatura (15 ° C a 23 ° C) o rapporti tra i livelli di trascrizione (FLM-δ /FLM-β) a 15° C e 23 ° C come lettura per l’abbondanza relativa delle due forme di giunzione con funzioni antagoniste ipotizzate. I valori quantitativi sono riassunti nel file supplementare 5.DOI:http://dx.doi.org/10.7554/eLife.22114.01110.7554/eLife.22114.012Figuresupplemento a 3 cifre 6.Linkage analysis for the simple single locus association test.Pairwise linkage analysis (R2)dei siti utilizzati per il semplice test di associazione a singolo locus tra le 54 adesioni del set di aplotipi FLM. Sono mostrati solo i 109 siti biallelici dei 119 siti. I cerchi rossi mostrano i siti PRO1, PRO2 e due SNP biallelici della tripletta nucleotidica INT6 (a bp +3975 e +3976). La posizione è impostata a 1 secondo la A del codice di partenza ATG.DOI:http://dx.doi.org/10.7554/eLife.22114.01210.7554/eLife.22114.013FigureSupplemento a 3 cifre 7.Effetti dei polimorfismi PRO1, PRO2 e INT6 sui polimorfismi del gene FLM.Effetti degli alleli PRO1, PRO2 e INT6 sui livelli di espressione relativi e sui rapporti di questi valori di espressione tra le 54 adesioni del set di aplotipi FLM. Il colore di sfondo di ogni grafico indica che l’allele minore si associa rispettivamente con l’upregulation (verde) e downregulation (rosso). vengono mostrati i valori p dei test di associazione: *, p<0,05; **, p≤0,01; ***, p<0,001 come mostrato in ogni grafico. I confronti non significativi (p>0,05) sono mostrati con uno sfondo grigio. I singoli valori sono rappresentati come punti jitterati. Scala e numerazione sono impostate a 1 secondo la A del codice di partenza ATG. I valori quantitativi sono riassunti nel file supplementare 5.DOI:http://dx.doi.org/10.7554/eLife.22114.013
Per trovare i polimorfismi che regolano l’espressione FLM e lo splicing, abbiamo eseguito un’analisi di associazione genotipo-fenotipo. Poiché il percorso di vernalizzazione ha fortemente ritardato la fioritura in 24 delle 54 adesioni e di conseguenza ha soppresso gli effetti FLM, abbiamo usato l’espressione FLM come fenotipo per l’analisi di associazione(Figura 3-figure supplement 2A). Per accertare che l’espressione FLM non è stata influenzata da FLC, abbiamo esaminato FLM in mutanti non virtualizzati di tipo selvaggio Col-0 e flc-3, così come in Col-0 con un modulo di vernalizzazione funzionale(Michaels e Amasino, 1999). In concomitanza con i rapporti precedenti, non abbiamo rilevato un’influenza di FLC sull’abbondanza di trascrizioni FLM nelle nostre condizioni(Figura 3-figure supplement 2B) (Scortecciet al., 2001; Ratcliffe et al., 2001). Abbiamo quindi ottenuto i dati di espressione FLM dal set di aplotipi FLM e misurato i livelli totali di trascrizione FLM, nonché i livelli FLM-ß e FLM-δ a 15°C o 23°C (file supplementare3). In linea con il comportamento riportato di FLM in Col-0, abbiamo osservato che l’espressione FLM-ß è diminuita (in media 0,6 volte) e FLM-δ è aumentata (in media 1,4 volte) con l’aumento della temperatura nella maggior parte delle adesioni (Figura 3Be file supplementare 3) (Lee et al.,2013; Posé et al. , 2013; Lutz et al., 2015). Allo stesso tempo, abbiamo anche osservato una variazione sostanziale nell’espressione FLM-ß e FLM-δ tra le adesioni dell’insieme di aplotipi FLM, invitando a concludere che la variazione di sequenza non codificata modula l’espressione FLM (Figura 3Be file supplementare 3). Poiché i livelli totali di trascrizione FLM sono fortemente correlati con FLM-ß ma non con l’abbondanza FLM-δ a 15°C e a 23°C, si potrebbe suggerire che FLM-ß rappresenta la forma FLM principale tra le trascrizioni FLM ( Figura 3-figuresupplement 3A,B).

Figura 3-figure supplemento 7.L’analisi degli aplotipiFLM di 776 adesioni identifica i principali aplotipi determinati da variazioni non codificate.l’analisi degli aplotipi basata su 45 SNP di 776 adesioni.il tempo di fioritura delle adesioni dell’insieme di aplotipi FLM suggerisce un requisito di vernalizzazione per molte adesioni.l’analisi di correlazione identifica FLM-β come la trascrizione FLM principale a 15°C e 23°C nelle adesioni dell’insieme di aplotipi FLM.119 Risultati del semplice test di associazione a singolo locus tra i 119 siti polimorfici con valori di espressione FLM.Analisi dei collegamenti per il semplice test di associazione a singolo locus.Effetti dei polimorfismi PRO1, PRO2 e INT6 sull’espressione genica FLM.(A) Affiliazione del gruppo aplotipo di 54 adesioni dell’insieme aplotipo FLM basato su 45 SNP per il locus FLM a 7 kb (H7kb), l’introne 1 (HI1) o gli introni 2-6 (HI2-6). La numerazione e la colorazione del gruppo dipendono dalle dimensioni del gruppo.(B) Valori di espressione riassunti dell’espressione totale FLM (esone 1), FLM-ß, e FLM-δ dell’insieme di aplotipi FLM. I valori erratici sono stati determinati sulla base di 1,5 x IQR (intervallo interquartile). Il coefficiente di variazione (CV) è mostrato nel grafico in basso.DOI:
http://dx.doi.org/10.7554/eLife.22114.006(A) Rappresentazione della posizione, della frequenza e del tipo di 45 SNPs da 776 adesioni utilizzate per il clustering di aplotipi.(B),(D) e(E) Analisi della rete di aplotipi dei dieci(B) gruppi più frequenti di una regione genomica FLM a 7 kb basati su 45 SNP o dei dieci (D) o undici(E) gruppi di aplotipi più grandi quando si considerano solo gli SNP dell’introne 1 (HI1; D) o degli introni 2-6 (HI2-6; E). Le dimensioni del cerchio e il codice colore illustrano il numero di adesioni in ogni gruppo con il gruppo 1 che rappresenta rispettivamente 174(B), 220(D) e 356 (E) adesioni. In(B), i numeri dei bulloni indicano i gruppi da cui sono state scelte le adesioni rappresentative.(C) Distribuzione geografica dei dieci gruppi più frequenti di una regione genomica FLM a 7 kb.(F) Diagramma di Venn che indica la sovrapposizione delle adesioni incluse nei dieci maggiori gruppi aplotipi HI1 e HI2-6.(G) Clustering dei gruppi di aplotipi delle 589 adesioni incluse nei dieci maggiori gruppi di aplotipi HI1 e HI2-6. Le adesioni sono state ordinate in base ai numeri dei gruppi di aplotipi HI1 e HI2-6. I numeri a sinistra indicano il numero del gruppo di aplotipi HI1, come indicato anche in(D). A destra è mostrato un ingrandimento di un sottoinsieme di adesioni evidenziato dalle caselle tratteggiate. La codifica a colori per i numeri dei gruppi di aplotipi, che illustra il numero del gruppo e la dimensione del gruppo, come mostrato in(D) e(E), è specificata sotto il grafico.DOI:
http://dx.doi.org/10.7554/eLife.22114.007(A) Analisi del tempo di fioritura delle adesioni dell’insieme aplotipo FLM. Le adesioni a fioritura estremamente tardiva o le adesioni che non avevano fiorito sono rappresentate al margine delle foglie a 60 rosette. I valori anomali sono stati determinati sulla base di 1,5 x IQR (intervallo interquartile).(B) Media e SD (tre repliche biologiche) nelle analisi qRT-PCR di FLM-β e FLM-δ in dodici giorni Col-0, flc-3 e Col-0 con un modulo divernalizzazioneFRISf-2FLC (Michaelse Amasino, 1999).DOI:
http://dx.doi.org/10.7554/eLife.22114.008(A) e (B) Correlazione (regressione lineare semplice) tra l’espressione FLM come determinata da qRT-PCR sull’esone 1 FLM con misure dell’abbondanza della variante di giunzione FLM-β e FLM-δ a 15°C (A) e 23°C (B) nelle 54 adesioni dell’insieme aplotipo FLM. FLM-ß , 15°C, p<0,0001; FLM-δ , 15°C , p=0,009; FLM-ß , 23°C, p<0, 0001; FLM-δ, 23°C, p>0,05.DOI:
http://dx.doi.org/10.7554/eLife.22114.009(A) Posizione, frequenza e tipo di ciascuno dei 119 siti polimorfici tra 54 adesioni del set di aplotipi FLM utilizzati per semplici prove di associazione di singoli locus.(B) Albero di congiunzione tra i vicini che mostra la relazione genetica tra 54 adesioni dell’insieme di aplotipi FLM basato sui 119 siti polimorfici mostrati in(A). I valori dell’aplotipo sono indicati in ogni ramo.DOI:
http://dx.doi.org/10.7554/eLife.22114.010(A) e (B)Rappresentazione dei valori p -log(10)-trasformati dei semplici test di associazione a singolo locus di ciascuno dei 119 siti che sono stati inclusi nell’analisi. La linea orizzontale nera corrisponde ad una soglia di significatività di p=0,05. I punti neri rappresentano i siti polimorfici che si associano con p>0,05, i punti rossi quelli con p<0,05. I punti verdi rappresentano i siti con p<0,05 che sono stati scelti per un’analisi sperimentale dettagliata come mostrato nella Figura 4A. Il modello genomico del gene FLM è quello introdotto nella Figura 1. Ogni grafico rappresenta l’analisi del tratto di espressione indicato.(A) mostra tutti i confronti con i valori di espressione relativi e(B) mostra tutti i confronti dei cambiamenti relativi dei livelli di trascrizione tra FLM-β e FLM-δ in risposta a variazioni di temperatura (15 ° C a 23 ° C) o rapporti tra i livelli di trascrizione (FLM-δ /FLM-β) a 15° C e 23 ° C come lettura per l’abbondanza relativa delle due forme di giunzione con funzioni antagoniste ipotizzate. I valori quantitativi sono riassunti nel file supplementare 5.DOI:
http://dx.doi.org/10.7554/eLife.22114.011Analisi del collegamento a coppie (R2) dei siti utilizzati per il semplice test di associazione a singolo locus tra le 54 adesioni del set di aplotipi FLM. Sono mostrati solo i 109 siti biallelici dei 119 siti. I cerchi rossi mostrano i siti PRO1, PRO2 e due SNP biallelici della tripletta nucleotidica INT6 (a bp +3975 e +3976). La posizione è impostata a 1 secondo la A del codice di partenza ATG.DOI:
http://dx.doi.org/10.7554/eLife.22114.012Effetti degli alleli PRO1, PRO2 e INT6 sui livelli di espressione relativi e sui rapporti di questi valori di espressione tra le 54 adesioni dell’insieme di aplotipi FLM. Il colore di sfondo di ogni grafico indica che l’allele minore si associa rispettivamente con l’upregulation (verde) e downregulation (rosso). sono mostrati i valori p dei test di associazione: *, p<0,05; **, p≤0,01; ***, p<0,001 come mostrato in ogni grafico. I confronti non significativi (p>0,05) sono mostrati con uno sfondo grigio. I singoli valori sono rappresentati come punti jitterati. Scala e numerazione sono impostate a 1 secondo la A del codice di partenza ATG. I valori quantitativi sono riassunti nel file supplementare 5.DOI:
http://dx.doi.org/10.7554/eLife.22114.013
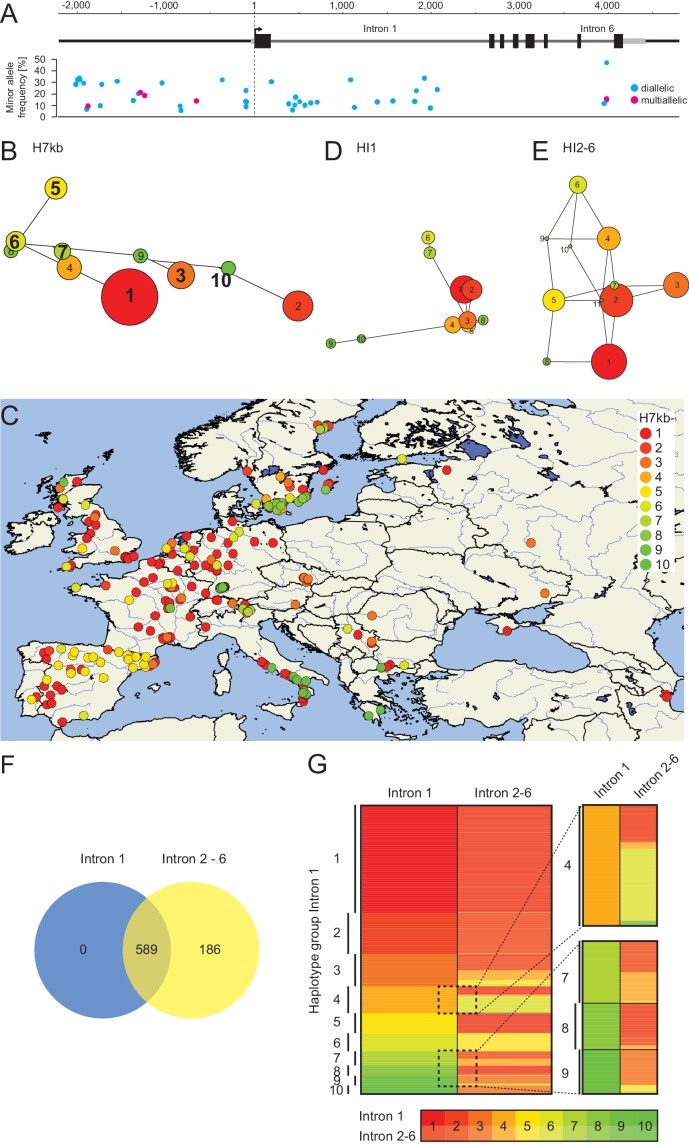
Figura 3-figure supplement 1.Analisi aplotipo basata su 45 SNPs da 776 adesioni.A) Rappresentazione della posizione, della frequenza e del tipo di 45 SNPs da 776 adesioni utilizzate per il clustering degli aplotipi.(B),(D) e(E) Analisi della rete di aplotipi dei dieci(B) gruppi più frequenti di una regione genomica FLM a 7 kb basati su 45 SNP o dei dieci (D) o undici(E) gruppi di aplotipi più grandi quando si considerano solo gli SNP dell’introne 1 (HI1; D) o degli introni 2-6 (HI2-6; E). Le dimensioni del cerchio e il codice colore illustrano il numero di adesioni in ogni gruppo con il gruppo 1 che rappresenta rispettivamente 174(B), 220(D) e 356 (E) adesioni. In(B), i numeri dei bulloni indicano i gruppi da cui sono state scelte le adesioni rappresentative.(C) Distribuzione geografica dei dieci gruppi più frequenti di una regione genomica FLM a 7 kb.(F) Diagramma di Venn che indica la sovrapposizione delle adesioni incluse nei dieci maggiori gruppi aplotipi HI1 e HI2-6.(G) Clustering dei gruppi di aplotipi delle 589 adesioni incluse nei dieci maggiori gruppi di aplotipi HI1 e HI2-6. Le adesioni sono state ordinate in base ai numeri dei gruppi di aplotipi HI1 e HI2-6. I numeri a sinistra indicano il numero del gruppo di aplotipi HI1, come indicato anche in(D). A destra è mostrato un ingrandimento di un sottoinsieme di adesioni evidenziato dalle caselle tratteggiate. La codifica a colori per i numeri dei gruppi di aplotipi, che illustra il numero del gruppo e la dimensione del gruppo, come mostrato in(D) e(E), è specificata sotto il grafico.DOI:
http://dx.doi.org/10.7554/eLife.22114.007
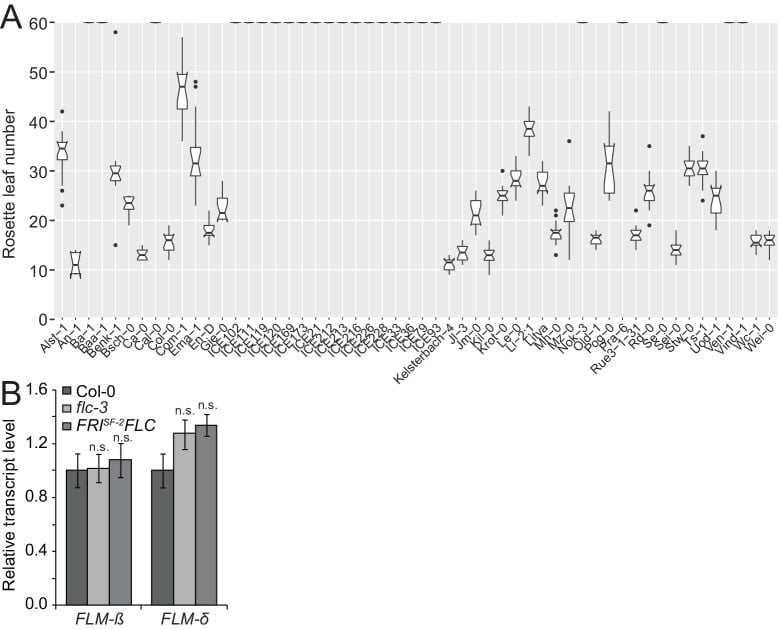
Figura 3-figure supplement 2.Il tempo di fioritura delle adesioni del set di aplotipi FLM suggerisce un requisito di vernalizzazione per molte adesioni.(A) Analisi del tempo di fioritura delle adesioni dell’insieme di aplotipi FLM. Le adesioni a fioritura estremamente tardiva o le adesioni che non avevano fiorito sono rappresentate al margine delle foglie a 60 rosette. I valori anomali sono stati determinati sulla base di 1,5 x IQR (intervallo interquartile).(B) Media e SD (tre repliche biologiche) nelle analisi qRT-PCR di FLM-β e FLM-δ in dodici giorni Col-0, flc-3 e Col-0 con un modulo divernalizzazioneFRISf-2FLC (Michaelse Amasino, 1999).DOI:
http://dx.doi.org/10.7554/eLife.22114.008

Figura 3-figure supplement 3.L’analisi di correlazione identifica FLM-β come la trascrizione principale FLM a 15°C e 23°C nelle adesioni degli aplotipi FLM.4.(A) e (B) Correlazione (regressione lineare semplice) tra l’espressione FLM come determinata da qRT-PCR sull’esone 1 di FLM con misurazioni dell’abbondanza della variante di giunzione FLM-β e FLM-δ a 15°C (A) e 23°C (B) nelle 54 adesioni dell’insieme di aplotipi FLM. FLM-ß , 15°C, p<0,0001; FLM-δ , 15°C , p=0,009; FLM-ß , 23°C, p<0, 0001; FLM-δ, 23°C, p>0,05.DOI:
http://dx.doi.org/10.7554/eLife.22114.009

Figura 3-figure supplement 4.119 siti polimorfici tra le adesioni del set di aplotipi FLM.(A) Posizione, frequenza e tipo di ciascuno dei 119 siti polimorfici tra le 54 adesioni dell’insieme di aplotipi FLM utilizzati per semplici test di associazione di singoli locus.(B) Albero di congiunzione tra i vicini che mostra la relazione genetica tra 54 adesioni dell’insieme di aplotipi FLM basato sui 119 siti polimorfici mostrati in(A). I valori dell’aplotipo sono indicati in ogni ramo.DOI:
http://dx.doi.org/10.7554/eLife.22114.010

Figura 3-figure supplement 5.Risultati del semplice test di associazione a singolo locus tra i 119 siti polimorfici con valori di espressione FLM.(A) e (B)Rappresentazione dei valori p -log(10)-trasformati dei test di associazione a singolo locus semplice di ciascuno dei 119 siti che sono stati inclusi nell’analisi. La linea orizzontale nera corrisponde ad una soglia di significatività di p=0,05. I punti neri rappresentano i siti polimorfici che si associano con p>0,05, i punti rossi quelli con p<0,05. I punti verdi rappresentano i siti con p<0,05 che sono stati scelti per un’analisi sperimentale dettagliata come mostrato nella Figura 4A. Il modello genomico del gene FLM è quello introdotto nella Figura 1. Ogni grafico rappresenta l’analisi del tratto di espressione indicato.(A) mostra tutti i confronti con i valori di espressione relativi e(B) mostra tutti i confronti dei cambiamenti relativi dei livelli di trascrizione tra FLM-β e FLM-δ in risposta a variazioni di temperatura (15 ° C a 23 ° C) o rapporti tra i livelli di trascrizione (FLM-δ /FLM-β) a 15° C e 23 ° C come lettura per l’abbondanza relativa delle due forme di giunzione con funzioni antagoniste ipotizzate. I valori quantitativi sono riassunti nel file supplementare 5.DOI:
http://dx.doi.org/10.7554/eLife.22114.011

Figura 3-figure supplement 6.Analisi dei collegamenti per il semplice test di associazione di un singolo locus.Analisi del collegamento a coppie (R2) dei siti utilizzati per il semplice test di associazione a singolo locus tra le 54 adesioni del set di aplotipi FLM. Sono mostrati solo i 109 siti biallelici dei 119 siti. I cerchi rossi mostrano i siti PRO1, PRO2 e due SNP biallelici della tripletta nucleotidica INT6 (a bp +3975 e +3976). La posizione è impostata a 1 secondo la A del codice di partenza ATG.DOI:
http://dx.doi.org/10.7554/eLife.22114.012
Figura 3 cifre supplemento 7.Effetti dei polimorfismi PRO1, PRO2 e INT6 sull’espressione del gene FLM.Effetti degli alleli PRO1, PRO2 e INT6 sui livelli di espressione relativi e sui rapporti di questi valori di espressione tra le 54 adesioni dell’insieme di aplotipi FLM. Il colore di sfondo di ogni grafico indica che l’allele minore si associa rispettivamente con l’upregulation (verde) e downregulation (rosso). sono mostrati i valori p dei test di associazione: *, p<0,05; **, p≤0,01; ***, p<0,001 come mostrato in ogni grafico. I confronti non significativi (p>0,05) sono mostrati con uno sfondo grigio. I singoli valori sono rappresentati come punti jitterati. Scala e numerazione sono impostate a 1 secondo la A del codice di partenza ATG. I valori quantitativi sono riassunti nel file supplementare 5.DOI:
http://dx.doi.org/10.7554/eLife.22114.013
L’analisi dell’associazione identifica i siti polimorfici con potenziali funzioni normative FLM
Utilizzando il set di aplotipi FLM, abbiamo poi eseguito test di associazione tra l’espressione FLM a 15°C e 23°C o rapporti di espressione derivati da questi valori e un set esteso di 119 polimorfismi lungo la regione FLM a 7 kb, che comprendeva i 45 SNP, ma anche SNP poco frequenti (MAF>1%) e indici (≤2 bp; MAF>1%) (Figura 3-figuresupplement 4). L’analisi delle associazioni ha rilevato polimorfismi con associazioni significative (p<0,001) (Figura 3-figure supplement 5 e Supplementary file 4). Abbiamo deciso di indagare il ruolo potenziale della delezione di una singola coppia di base (PRO1T/-; bp -215) e di un SNP geneticamente leggermente legato (PRO2A/C;bp-93), perché entrambi si trovavano nella parte prossimale del promotore FLM. Inoltre, abbiamo studiato tre nucleotidi geneticamente non collegati perché erano posizionati come una tripletta nucleotidica altamente diversificata nell’introne 6 (INT6A/C-A/C-A/T/C; bp +3975-+ 3977) in un contesto di sequenza altrimenti conservato (Figura 4A,Figura 3-figure supplements 5- 7, Supplementary file5) .10.7554/eLife.22114.014Figure 4.Polymorphisms in PRO2+ e INT6+ sites influenza l’espressione FLM basale e dipendente dalla temperatura.(A) Rappresentazione schematica del locus genomico FLM come mostrato in Figura 1. I punti verdi indicano le posizioni dei siti PRO1, PRO2 e INT6 scelti per ulteriori indagini. I loghi di sequenza mostrano le frequenze degli alleli di questi siti tra le 54 adesioni del set di aplotipi FLM.(B) Loghi di sequenza e distribuzione delle frequenze degli alleli dei polimorfismi PRO2+ e INT6+ tra 840 adesioni di Arabidopsis. Tutti i residui polimorfici sono contrassegnati da asterischi e le rispettive frequenze alleliche sono indicate dal logo della sequenza. L’aplotipo di riferimento Col-0 è contrassegnato in giallo, gli aplotipi scelti per ulteriori indagini sono contrassegnati in verde.(C) e(D) Allineamento dei polimorfismi PRO2+ e INT6+(C) e rappresentazione schematica del costrutto di riferimento FLMCol-0 (pFLM::gFLM) nonché delle varianti FLMCol-0 selezionate per l’analisi transgenica nell’adesione alla cancellazione FLM Nd-1. Le basi che differiscono tra FLMCol-0 e le varianti sono colorate. Le cancellazioni nei costrutti di cancellazione dei siti PRO2+ e INT6+ e i motivi PolyA vengono visualizzati con linee rosse (non disegnate in scala).(E) e(F) Media e SD (quattro pool di replicazione con quattro a undici linee transgeniche T2 indipendenti) delle analisi qRT-PCR di FLM-β (E) e FLM-δ (F ). Per un confronto più semplice, i valori ottenuti con FLMCol-0 sono visualizzati come linee tratteggiate rosse e blu. T-test degli studenti: *p≤0,05; **p≤0,01; ***p≤0,001; n.s., non significativo.DOI:http://dx.doi.org/10.7554/eLife.22114.01410.7554/eLife.22114.015FigureSupplemento a 4 cifre 1.Aumentando il numero di cancellazioni di motivi PolyA si ottiene una graduale riduzione dell’espressione FLM-ß.(A) Rappresentazione schematica del costrutto diriferimento FLMCol-0 (pFLM::gFLM) e delle varianti di cancellazione FLM PolyA selezionate per l’analisi transgenica in Nd-1. Le delezioni dei motivi PolyA vengono visualizzate con linee rosse (non disegnate in scala).(B) e(C) Media e SD (quattro pool di replicazione che comprendono da cinque a dieci linee transgeniche T2 indipendenti) da analisi qRT-PCR di FLM-β (B) e FLM-δ (C ) espressione in piantine di dieci giorni. T-test dello studente: *p≤0,05; **p≤0,01; ***p≤0,001; altrimenti non significativo.DOI:http://dx.doi.org/10.7554/eLife.22114.015
Sorprendentemente, i siti PRO2 e INT6 erano direttamente affiancati o in prossimità di un totale di quattro motivi PolyA di lunghezza variabile ([A]7-11), uno dei quali rappresentava una cosiddetta CArG-box, un potenziale sito vincolante per i fattori di trascrizione MADS-box(Zhang et al., 2016). Altri due motivi PolyA risiedevano negli introni 3 e 5(Figura 4D e Figura 4-figure supplement 1A). Dal momento che i motivi PolyA erano stati segnalati come importanti per l’espressione genica, abbiamo ragionato che questi motivi potevano essere rilevanti per la regolazione FLM, in isolamento o in combinazione con le variazioni non codificanti altamente significative(O’Malley et al., 2016; Horton et al., 2012).
Per comprendere la variazione di sequenza che circonda i siti PRO1, PRO2 e INT6, abbiamo rianalizzato le sequenze di genoma Arabidopsis disponibili(The 1001 Genomes Consortium, 2016). Oltre alla delezione del PRO1 1 bp, la regione circostante il PRO1 è stata conservata e ci siamo quindi concentrati sul polimorfismo di delezione del PRO1T/-(Figura 4A). Poche basi a monte e a valle del PRO2 (bp -102 a bp -92), abbiamo rilevato quattro SNP aggiuntivi altamente diversificati e abbiamo designato questa regione PRO2+. Abbiamo anche identificato ulteriori aplotipi frequenti e frequenti della terzina INT6 (INT6+). Oltre ai due motivi PolyA situati a PRO2+ e INT6+, i quattro motivi PolyA rimanenti sono stati conservati in tutte le adesioni esaminate(Figura 4B).

Figura 4-figure supplement 1.I polimorfismi nei siti PRO2+ e INT6+ influenzano l’espressione FLM basale e dipendente dalla temperatura.aumentando il numero di delezioni di motivi PolyA si ottiene una graduale riduzione dell’espressione FLM-ß.(A) Rappresentazione schematica del locus genomico FLM come mostrato in Figura 1. I punti verdi indicano le posizioni dei siti PRO1, PRO2, e INT6 che sono stati scelti per ulteriori indagini. I loghi di sequenza mostrano le frequenze degli alleli di questi siti tra le 54 adesioni del set di aplotipi FLM.(B) Loghi di sequenza e distribuzione delle frequenze degli alleli dei polimorfismi PRO2+ e INT6+ tra 840 adesioni di Arabidopsis. Tutti i residui polimorfici sono contrassegnati da asterischi e le rispettive frequenze alleliche sono indicate dal logo della sequenza. L’aplotipo di riferimento Col-0 è contrassegnato in giallo, gli aplotipi scelti per ulteriori indagini sono contrassegnati in verde.(C) e(D) Allineamento dei polimorfismi PRO2+ e INT6+(C) e rappresentazione schematica del costrutto di riferimento FLMCol-0 (pFLM::gFLM) nonché delle varianti FLMCol-0 selezionate per l’analisi transgenica nell’adesione alla cancellazione FLM Nd-1. Le basi che differiscono tra FLMCol-0 e le varianti sono colorate. Le cancellazioni nei costrutti di cancellazione dei siti PRO2+ e INT6+ e i motivi PolyA sono visualizzati con linee rosse (non disegnate in scala).(E) e(F) Media e SD (quattro pool di replicazione con quattro a undici linee transgeniche T2 indipendenti) delle analisi qRT-PCR di FLM-β (E) e FLM-δ (F ). Per un confronto più semplice, i valori ottenuti con FLMCol-0 sono visualizzati come linee tratteggiate rosse e blu. T-test degli studenti: *p≤0,05; **p≤0,01; ***p≤0,001; n.s., non significativo.DOI:
http://dx.doi.org/10.7554/eLife.22114.014(A) Rappresentazione schematica del costrutto di riferimento FLMCol-0 (pFLM::gFLM) e delle varianti di cancellazione FLM PolyA selezionate per l’analisi transgenica in Nd-1. Le delezioni dei motivi PolyA vengono visualizzate con linee rosse (non disegnate in scala).(B) e(C) Media e SD (quattro pool di replicazione che comprendono da cinque a dieci linee transgeniche T2 indipendenti) da analisi qRT-PCR di FLM-β (B) e FLM-δ (C ) espressione in piantine di dieci giorni. T-test dello studente: *p≤0,05; **p≤0,01; ***p≤0,001; altrimenti non significativo.DOI:
http://dx.doi.org/10.7554/eLife.22114.015
Figura 4-figure supplement 1.Aumentando il numero di cancellazioni di motivi PolyA si ottiene una graduale riduzione dell’espressione FLM-ß.(A) Rappresentazione schematica del costrutto di riferimento FLMCol-0 (pFLM::gFLM) così come le varianti di cancellazione FLM PolyA selezionati per l’analisi transgenica in Nd-1. Le delezioni dei motivi PolyA vengono visualizzate con linee rosse (non disegnate in scala).(B) e(C) Media e SD (quattro pool di replicazione che comprendono da cinque a dieci linee transgeniche T2 indipendenti) da analisi qRT-PCR di FLM-β (B) e FLM-δ (C ) espressione in piantine di dieci giorni. T-test dello studente: *p≤0,05; **p≤0,01; ***p≤0,001; altrimenti non significativo.DOI:
http://dx.doi.org/10.7554/eLife.22114.015
La variazione di sequenza non codificata FLM regola l’abbondanza FLM in un fondo transgenico omogeneo
Per esaminare gli effetti delle variazioni di sequenza non codificate in uno sfondo omogeneo, abbiamo trasformato l’adesione di cancellazione Nd-1 con le varianti FLMCol-0 che portano i polimorfismi PRO1, PRO2+ e INT6+ o le delezioni di PRO2+ (PRO2+Δ16bp) e INT6+ (INT6+Δ17bp)(Figura 4C,D). Inoltre, abbiamo trasformato varianti con delezioni tra due e sei tratti PolyA (PolyA_2xΔa, PolyA_2xΔb, PolyA_3xΔ, PolyA_4xΔ, PolyA_4xΔ, PolyA_5xΔ, PolyA_6xΔ; Figura 4D e Figura 4-figure supplement 1A ). Utilizzando le linee di segregatura T2 raggruppate (n = 18-45) per ciascuno dei transgeni, abbiamo analizzato gli effetti della variazione di sequenza sull’espressione FLM-ß e FLM-δ a 15°C e 23°C. La cancellazione PRO1-, come presente nel riferimento FLMCol-0, non ha rivelato differenze nei livelli di trascrizione FLM rispetto al PRO1T (FLMCol-0) (Fig. Figura 4-figure supplement 1B,C). Tuttavia, due PRO2+ (PRO2+GGAAC, PRO2+AAACC) e tre varianti INT6+ (INT6+ACA, INT6+CAC e INT6+AAAA) hanno mostrato un upregulation di FLM-ß a 15°C rispetto a FLMCol-0 (Figura 4E). Gli aumenti e le diminuzioni dei livelli di FLM-δ hanno seguito in gran parte quelli dei livelli di FLM-ß ad eccezione di INT6+AAAA, che ha mostrato un upregulation esclusivamente di FLM-ß ( Figura 4E,F). La variante di cancellazione PRO2+Δ16bp ha mostrato un upregulation di FLM-ß ma INT6+Δ17bp non ha alterato significativamente i livelli di FLM-ß e FLM-δ rispetto a FLMCol-0 (Figura 4E,F). Abbiamo concluso che l’identità di queste regioni piuttosto che la loro presenza controllava l’espressione FLM e la regolazione FLM sensibile alla temperatura. Le varianti di cancellazione del motivo PolyA hanno mostrato una graduale diminuzione dei livelli FLM-ß e FLM-δ con un numero crescente di cancellazioni, mentre la cancellazione di tutti e sei i motivi PolyA (PolyA_6xΔ) ha avuto un effetto particolarmente forte sull’espressione FLM-ß e la sua espressione sensibile alla temperatura (Figura 4E,F eFigura 4-figure supplement 1B,C). Pertanto, i motivi PolyA hanno un ruolo regolatore, ma possono funzionare in modo ridondante.
I livelli FLM-ß ed il tempo di fioritura sono fortemente correlati in modo lineare
Le analisi dell’impronta filogenetica e delle associazioni hanno portato all’identificazione di polimorfismi e regioni che hanno alterato significativamente l’espressione FLM quando presenti in un contesto molecolare e genomico omogeneo. Per correlare l’espressione FLM con la fioritura, abbiamo misurato il tempo di fioritura in linee transgeniche T2 di otto varianti con abbondanza FLM significativamente diverse(Figura 5A e Figura 5-figure supplement 1A-C). Abbiamo rilevato una correlazione molto forte tra i livelli di trascrizione FLM-ß e il tempo di fioritura (R2 = 0,94) in piante cresciute a 15°C. Tempo di fioritura ha risposto in modo lineare ai livelli FLM-ß con 17 a 30 foglie a rosetta fino alla fioritura nella gamma dei livelli di trascrizione relativi da 0,6 a 2,4 (Figura 5B). Inoltre, le varianti a bassa espressione PolyA_6xΔ, INT1Δ373bp, e PROΔ225bp fiorirono già all’adesione di cancellazione Nd-1 suggerendo che ci deve essere una soglia critica inferiore per FLM-ß per essere efficace ( Figura 5A,B). La correlazione era molto più bassa per FLM-δ (R2 = 0,70) suggerendo che FLM-ß è il principale determinante per il tempo di fioritura in queste condizioni (Figura 5-figuresupplement 1D). All’interno della gamma di linee esaminate, non abbiamo rilevato un effetto di saturazione al livello di espressione superiore.10.7554/eLife.22114.016Figure 5.FLM-ß espressione mostra un’elevata correlazione con il tempo di fioritura delle piante transgeniche. (A)Box plot di analisi quantitativa del tempo di fioritura (numero di foglie a rosetta) di linee transgeniche indipendenti T2 a 15 ° C e fotoperiodo giorno lungo. Sono state analizzate dieci piante di tre pool di replicazione per ogni costruzione. I dati sono stati corretti per l’atteso 25% di segregants Nd-1 non transgenici (vedi Figura 5-figure supplement 1D per l’analisi non corretta). I singoli valori sono mostrati come punti jitterati, il colore rappresenta il tipo di variante introdotto nella Figura 4D, la mediana del riferimento FLMCol-0 è indicata come linea tratteggiata. Test del rango Wilcoxon: *p≤0,05; **p≤0,01; ***p≤0,001; n.s., non significativo.(B) Correlazione (semplice regressione lineare) tra FLM-ß e qRT-PCR dati di espressione come presentato in Figura 5-figure supplemento 1A,B e l’analisi del tempo di fioritura come mostrato in (A). Sono designati i datapoint delle varianti INT6+ con espressione FLM a contrasto e FLMCol-0. Il codice colore corrisponde a(A). Le varianti PolyA_6xΔ, INT1Δ373bp, e PROΔ225bp, che non rispondono in modo lineare, sono indicate come cerchi puntinati. Le aree ombreggiate indicano gli intervalli di confidenza del 95%; p<0,0001. Si noti che i dati sul tempo di fioritura mostrati in(A) sono stati corretti eliminando i valori di segregants T2 non transgenici. Un’analisi utilizzando i dati non corretti è mostrata in Figura 5-figure supplement 1E,F.DOI:http://dx.doi.org/10.7554/eLife.22114.01610.7554/eLife.22114.017Figure5-figure supplement 1.FLM-ß espressione supplemento 1.FLM-ß correla con il tempo di fioritura in linee transgeniche che trasportano diversi alleli FLM . (A) e (B) Risultatidelle analisi qRT-PCR di FLM-β (A) e FLM-δ (B ) espressione in piantine di dieci giorni di piscine (n = 18-40) di linee transgeniche T2 indipendenti. Le barre di errore indicano SD di quattro pool di replicazione che comprendono da quattro a dieci linee transgeniche o tre repliche di Col-0.C) Fotografie di piante rappresentative di 57 giorni che esprimono le varianti FLMCol-0 e PRO2+ e INT6+ coltivate a 15°C nel fotoperiodo di una giornata lunga. Col-0 e Nd-1, che sono serviti come sfondo genetico per l’analisi transgenica, sono mostrati in ogni fotografia per il confronto. Le piante di entrambe le foto provengono dallo stesso esperimento, ma sono state fotografate separatamente.(D) Correlazione (semplice regressione lineare) tra i dati di espressione FLM-δ come presentato in (B) e il tempo di fioritura, come mostrato in Figura 5A. Punti di dati di INT6 + varianti con contrasto FLM espressione FLM e FLMCol-0 sono designati. p = 0,005. Le aree ombreggiate indicano gli intervalli di confidenza al 95%.(E) Box plot di analisi quantitativa del tempo di fioritura (numero di foglie a rosetta) di linee transgeniche indipendenti T2 a 15°C e fotoperiodo del giorno lungo. Sono state analizzate dieci piante di tre pool di replicazione per ogni costruzione (dati non corretti). I singoli valori sono mostrati come punti jitterati, il colore rappresenta il tipo di variante come introdotto nella Figura 4D, la mediana del riferimento FLMCol-0 è indicata come linea tratteggiata. Il 25% delle piante che mostrano una fioritura molto precoce che molto probabilmente rappresentano il tipo selvatico Nd-1 segregants sono indicati con un cerchio rosso e questi sono stati estratti per la correzione come presentato in Figura 5A. Wilcoxon rango test: *p≤0,05; **p≤0,01; ***p≤0,001; n.s., non significativo.(F) Correlazione (semplice regressione lineare) tra FLM-ß e FLM-δ . qRT-PCR dati di espressione come presentato in (A) e (B) e l’analisi del tempo difioritura come mostrato in (D). FLM-ß , p<0,0001; FLM-δ , p=0,002. Le aree ombreggiate indicano gli intervalli di confidenza al 95%.DOI:http://dx.doi.org/10.7554/eLife.22114.017

Figura 5-figure supplement 1.L’espressioneFLM-ß mostra un’elevata correlazione con il tempo di fioritura delle piante transgeniche. L’espressioneFLM-ß è correlata al tempo di fioritura nelle linee transgeniche che portano diversi alleli FLM.(A) Box plot di analisi quantitativa del tempo di fioritura (numero di foglie a rosetta) di linee transgeniche indipendenti T2 a 15°C e fotoperiodo di un giorno lungo. Sono state analizzate dieci piante di tre pool di replicazione per ogni costruzione. I dati sono stati corretti per l’atteso 25% di segregants Nd-1 non transgenici (vedi Figura 5-figure supplement 1D per l’analisi non corretta). I singoli valori sono mostrati come punti jitterati, il colore rappresenta il tipo di variante introdotto nella Figura 4D, la mediana del riferimento FLMCol-0 è indicata come linea tratteggiata. Test del rango Wilcoxon: *p≤0,05; **p≤0,01; ***p≤0,001; n.s., non significativo.(B) Correlazione (semplice regressione lineare) tra FLM-ß e qRT-PCR dati di espressione come presentato in Figura 5-figure supplemento 1A,B e l’analisi del tempo di fioritura come mostrato in (A). Sono designati i datapoint delle varianti INT6+ con espressione FLM a contrasto e FLMCol-0. Il codice colore corrisponde a(A). Le varianti PolyA_6xΔ, INT1Δ373bp, e PROΔ225bp, che non rispondono in modo lineare, sono indicate come cerchi puntinati. Le aree ombreggiate indicano gli intervalli di confidenza del 95%; p<0,0001. Si noti che i dati sul tempo di fioritura mostrati in(A) sono stati corretti eliminando i valori di segregants T2 non transgenici. Un’analisi utilizzando i dati non corretti è mostrata in Figura 5-figure supplement 1E,F.DOI:
http://dx.doi.org/10.7554/eLife.22114.016(A) e (B) Risultati delle analisi qRT-PCR di FLM-β (A) e FLM-δ (B ) espressione in semenzali di dieci giorni di vita di pool (n = 18-40) di linee transgeniche T2 indipendenti. Le barre di errore indicano SD di quattro pool di replicazione che comprendono da quattro a dieci linee transgeniche o tre repliche di Col-0.C) Fotografie di piante rappresentative di 57 giorni che esprimono le varianti FLMCol-0 e PRO2+ e INT6+ coltivate a 15°C nel fotoperiodo di una giornata lunga. Col-0 e Nd-1, che sono serviti come sfondo genetico per l’analisi transgenica, sono mostrati in ogni fotografia per il confronto. Le piante di entrambe le foto provengono dallo stesso esperimento, ma sono state fotografate separatamente.(D) Correlazione (semplice regressione lineare) tra i dati di espressione FLM-δ come presentato in (B) e il tempo di fioritura, come mostrato in Figura 5A. Punti di dati di INT6 + varianti con contrasto FLM espressione FLM e FLMCol-0 sono designati. p = 0,005. Le aree ombreggiate indicano gli intervalli di confidenza al 95%.(E) Box plot di analisi quantitativa del tempo di fioritura (numero di foglie a rosetta) di linee transgeniche indipendenti T2 a 15°C e fotoperiodo del giorno lungo. Sono state analizzate dieci piante di tre pool di replicazione per ogni costruzione (dati non corretti). I singoli valori sono mostrati come punti jitterati, il colore rappresenta il tipo di variante come introdotto nella Figura 4D, la mediana del riferimento FLMCol-0 è indicata come linea tratteggiata. Il 25% delle piante che mostrano una fioritura molto precoce che molto probabilmente rappresentano il tipo selvatico Nd-1 segregants sono indicati con un cerchio rosso e questi sono stati estratti per la correzione come presentato in Figura 5A. Wilcoxon rango test: *p≤0,05; **p≤0,01; ***p≤0,001; n.s., non significativo.(F) Correlazione (semplice regressione lineare) tra FLM-ß e FLM-δ . qRT-PCR dati di espressione come presentato in (A) e (B) e l’analisi del tempo difioritura come mostrato in (D). FLM-ß , p<0,0001; FLM-δ , p=0,002. Le aree ombreggiate indicano gli intervalli di confidenza al 95%.DOI:
http://dx.doi.org/10.7554/eLife.22114.017
Figura 5-figure supplement 1.L’espressioneFLM-ß è correlata al tempo di fioritura nelle linee transgeniche che portano diversi alleli FLM.(A) e (B) Risultati delle analisi qRT-PCR di FLM-β (A) e FLM-δ (B ) espressione in semenzali di dieci giorni di vita di pool (n = 18-40) di linee transgeniche indipendenti T2. Le barre di errore indicano SD di quattro pool di replicazione che comprendono da quattro a dieci linee transgeniche o tre repliche di Col-0.C) Fotografie di piante rappresentative di 57 giorni che esprimono le varianti FLMCol-0 e PRO2+ e INT6+ coltivate a 15°C nel fotoperiodo di una giornata lunga. Col-0 e Nd-1, che sono serviti come sfondo genetico per l’analisi transgenica, sono mostrati in ogni fotografia per il confronto. Le piante di entrambe le foto provengono dallo stesso esperimento, ma sono state fotografate separatamente.(D) Correlazione (semplice regressione lineare) tra i dati di espressione FLM-δ come presentato in (B) e il tempo di fioritura, come mostrato in Figura 5A. Punti di dati di INT6 + varianti con contrasto FLM espressione FLM e FLMCol-0 sono designati. p = 0,005. Le aree ombreggiate indicano gli intervalli di confidenza al 95%.(E) Box plot di analisi quantitativa del tempo di fioritura (numero di foglie a rosetta) di linee transgeniche indipendenti T2 a 15°C e fotoperiodo del giorno lungo. Sono state analizzate dieci piante di tre pool di replicazione per ogni costruzione (dati non corretti). I singoli valori sono mostrati come punti jitterati, il colore rappresenta il tipo di variante come introdotto nella Figura 4D, la mediana del riferimento FLMCol-0 è indicata come linea tratteggiata. Il 25% delle piante che mostrano una fioritura molto precoce che molto probabilmente rappresentano il tipo selvatico Nd-1 segregants sono indicati con un cerchio rosso e questi sono stati estratti per la correzione come presentato in Figura 5A. Wilcoxon rango test: *p≤0,05; **p≤0,01; ***p≤0,001; n.s., non significativo.(F) Correlazione (semplice regressione lineare) tra FLM-ß e FLM-δ . qRT-PCR dati di espressione come presentato in (A) e (B) e l’analisi del tempo difioritura come mostrato in (D). FLM-ß , p<0,0001; FLM-δ , p=0,002. Le aree ombreggiate indicano gli intervalli di confidenza al 95%.DOI:
http://dx.doi.org/10.7554/eLife.22114.017
Il polimorfismo INT6+CAA conferisce un’espressione e una fioritura FLM sensibile alla temperatura
Per l’espressione FLM sensibile alla temperatura erano necessari gli introni 2-6 (Figura 1). Quando abbiamo testato statisticamente l’interazione tra genotipo e temperatura con un modello lineare multiplo, abbiamo trovato che la regolazione FLM-ß sensibile alla temperatura è stata significativamente ridotta in INT6+CAA tra 15°C e 23°C e rispetto alla variante dicontrollo FLMCol-0 (p=0,012) (Figure 4Ee 6A,B). In linea con la previsione, la fioritura di questa variante era infatti meno sensibile alle variazioni di temperatura quando testato a 15 ° C e 23 ° C in piante omozigote T3 progenie e rispetto al riferimento FLMCol-0 (Figura6C,D). Pertanto, le variazioni di espressione FLM-ß indipendenti dalla temperatura sono correlate con la fioritura non sensibile alla temperatura nell’intervallo di temperatura selezionato.10.7554/eLife.22114.018Figure 6.Il polimorfismo INT6+CAA riduce la sensibilità alla temperatura dell’espressione FLM e della fioritura.(A) analisi qRT-PCR e (B) rapporti di espressione FLM-β di FLMCol-0 (INT6+AAT ) eINT6+CAA cresciutia 15°C e 23°C. Le prove statistiche di sensibilità alla temperatura sono descritte nel testo.(C) Mezzi e SD di analisi quantitativa del tempo di fioritura (numero di foglie a rosetta). n = 5 (15°C) e 8 (23°C) si replica da cinque linee transgeniche indipendenti omozigote T3 omozigote T3 coltivate a 15°C e 23°C in fotoperiodo di lunga giornata.(D) Media e SD dei rapporti dei numeri di foglie a rosetta dall’analisi mostrata in(C). T-test dello studente: *p<0.05; **p≤0.01.DOI:http://dx.doi.org/10.7554/eLife.22114.018

Figura 6.Il polimorfismo INT6+CAA riduce la sensibilità alla temperatura dell’espressione FLM e della fioritura.(A) qRT-PCR analisi e (B) rapporti di espressione di FLM-β di FLMCol-0 (INT6+AAT ) eINT6+CAA cresciutoa 15 ° C e 23 ° C. Le prove statistiche di sensibilità alla temperatura sono descritte nel testo.(C) Mezzi e SD di analisi quantitativa del tempo di fioritura (numero di foglie a rosetta). n = 5 (15°C) e 8 (23°C) si replica da cinque linee transgeniche indipendenti omozigote T3 omozigote T3 coltivate a 15°C e 23°C in fotoperiodo di lunga giornata.(D) Media e SD dei rapporti dei numeri di foglie a rosetta dall’analisi mostrata in(C). T-test dello studente: *p<0,05; **p≤0,01.DOI:
http://dx.doi.org/10.7554/eLife.22114.018
L’abbondanza differenziale delle forme di giunzione FLM può essere correlata a cambiamenti nella trascrizione FLM o nella giunzione alternativa
Per capire se lo stesso o diversi meccanismi molecolari sono alla base dell’espressione FLM alterata nelle varianti FLM, abbiamo stimato la trascrizione FLM di varianti con abbondanza fortemente alterata dell’abbondanza FLM elaborata misurando i livelli di FLM non elaborati pre-mRNA da piante coltivate a 15°C. Rispetto ai livelli di mRNA FLM trasformati, le varianti PROΔ225bp e INT1Δ373bp hanno mostrato riduzioni altrettanto forti di pre-mRNA non trasformati, suggerendo che i polimorfismi di eliminazione rispettivi polimorfismi influenzano direttamente la trascrizione FLM (Figure 4E, F e 7A). A sua volta, le linee INT6+CAA e PolyA_6xΔ avevano ridotto i livelli di mRNA FLM ma, se confrontate con FLMCol-0, nonhanno mostrato cambiamenti sostanziali nei livelli di pre-mRNA non elaborati (Figure4E,F e e 7B). Dal momento che questo indicava che gli eventi post-trascrizionali possono essere influenzati in queste varianti, abbiamo testato per l’abbondanza di varianti differenziali di giunzione poliadenilati dopo semiquantitativa 3′-RACE-PCR e sequenziamento dei prodotti PCR clonati (Figura 7-figure supplemento 1A). Lì, abbiamo rilevato una riduzione relativa di trascrizioni FLM-ß in INT6+CAAe PolyA_6xΔ che è stata accompagnata da un aumento dell’abbondanza di due trascrizioni poliadenilate contenenti l’esone 1 e l’introne 1 (E1I1p) che era già stato notato in una pubblicazione precedente (Figura 7C,D) (Lutzet al.,2015). È importante notare che non abbiamo identificato un singolo clone FLM-δ tra i 163 cDNA sequenziati. L’aumento relativo di trascrizioni E1I1p potrebbe anche essere confermato in modo indipendente da E1I1p specifici qRT-PCR e suggerito in sintesi che la scelta del sito di giunzione alla giunzione esone 1 – introne 1 è cambiato nel INT6 +CAA e PolyA_6xΔ alleli (Figura 7-figure supplemento 1B,C). In relazione a tutte le trascrizioni contenenti l’esone 1, l’abbondanza complessiva di queste trascrizioni contenenti l’introne 1 era relativamente bassa(Figura 7-figure supplement 1C).10.7554/eLife.22114.019Figure 7.FLMpolimorfismi influenzano la trascrizione o la giunzione FLM a spese di FLM-ß .(A) e(B) Media e SD (n = 3) dei livelli FLM pre-mRNA . T-test dello studente: ***p≤0,001; n.s. = non significativo.(C) Rappresentazione schematica dei cDNA FLM rilevati più di una volta (n = 163). Le trascrizioni FLM-δ non sono state rilevate e sono mostrate solo per completezza. Le aree grigie corrispondono agli esoni FLM dei modelli dei geni FLM-ß e FLM-δ come specificato nella Figura 1A. La freccia indica la posizione del primer 3′ RACE utilizzato in combinazione con un primer invertito oligo(dT) per rilevare trascrizioni poliadenilate.(D) Distribuzione di frequenza delle trascrizioni visualizzate in(B) con il numero totale di sequenze rappresentate nel grafico.DOI:http://dx.doi.org/10.7554/eLife.22114.01910.7554/eLife.22114.020Figuresupplemento a 7 cifre 1.Linee transgeniche che esprimono diversi alleli FLM visualizzano l’espressione differenziale FLM e modelli di giunzione. (A) Immagine di un gel di agarosio con i risultati di un 3′-RACE PCR. Il primer utilizzato per l’amplificazione 3′ è indicato nella Figura 7C. Le porzioni di gel dal FLMCol-0, INT6 +CAA e PolyA_6xΔ prodotti di amplificazione incorniciato con una linea tratteggiata sono stati purificati, clonati e sottoposti a sequenziamento del DNA.(B) Rappresentazione schematica dei primer utilizzati per la quantificazione qRT-PCR dei livelli totali FLM (esone 1) e la sequenza E1I1p dell’introne 1 contenente trascrizioni come indicato dalle frecce sotto il modello genico. La scatola nera corrisponde all’esone 1 FLM, la scatola grigia alla porzione prossimale dell’introne 1. La numerazione è rispettiva alla A del codice di partenza ATG.(C) Media e SD delle analisi qRT-PCR di analisi qRT-PCR di trascrizioni contenenti la sequenza dell’introne 1 da quattro pool di trascrizioni replicate di linee T2 indipendenti. La diminuzione relativa dei valori di espressione E1I1p rispetto al trascritto dell’esone 1 è indicata per ogni riga del grafico. Si noti che il confronto dei valori di espressione tra i frammenti è approssimativo, poiché le efficienze del primer potrebbero non essere identiche. Le sequenze di primer sono elencate nel file supplementare 7.DOI:http://dx.doi.org/10.7554/eLife.22114.020

Figura 7-figure supplement 1.I polimorfismiFLM influenzano la trascrizione FLM o la giunzione FLM a scapito delle linee FLM-ß.Transgeniche che esprimono diversi alleli FLM mostrano l’espressione differenziale FLM e i modelli di giunzione.(A) e (B) Media e SD (n = 3) dei livelli FLM pre-mRNA. T-test dello studente: ***p≤0,001; n.s. = non significativo.(C) Rappresentazione schematica dei cDNA FLM rilevati più di una volta (n = 163). Le trascrizioni FLM-δ non sono state rilevate e sono mostrate solo per completezza. Le aree grigie corrispondono agli esoni FLM dei modelli dei geni FLM-ß e FLM-δ come specificato nella Figura 1A. La freccia indica la posizione del primer 3′ RACE utilizzato in combinazione con un primer invertito oligo(dT) per rilevare trascrizioni poliadenilate.(D) Distribuzione di frequenza delle trascrizioni visualizzate in(B) con il numero totale di sequenze rappresentate nel grafico.DOI:
http://dx.doi.org/10.7554/eLife.22114.019(A) Immagine di un gel di agarosio con i risultati di una PCR 3′-RACE. Il primer utilizzato per l’amplificazione a 3′ è indicato nella Figura 7C. Le porzioni di gel dal FLMCol-0, INT6 +CAA e PolyA_6xΔ prodotti di amplificazione incorniciato con una linea tratteggiata sono stati purificati, clonati e sottoposti a sequenziamento del DNA.(B) Rappresentazione schematica dei primer utilizzati per la quantificazione qRT-PCR dei livelli totali FLM (esone 1) e la sequenza E1I1p dell’introne 1 contenente trascrizioni come indicato dalle frecce sotto il modello genico. La scatola nera corrisponde all’esone 1 FLM, la scatola grigia alla porzione prossimale dell’introne 1. La numerazione è rispettiva alla A del codice di partenza ATG.(C) Media e SD delle analisi qRT-PCR di analisi qRT-PCR di trascrizioni contenenti la sequenza dell’introne 1 da quattro pool di trascrizioni replicate di linee T2 indipendenti. La diminuzione relativa dei valori di espressione E1I1p rispetto al trascritto dell’esone 1 è indicata per ogni riga del grafico. Si noti che il confronto dei valori di espressione tra i frammenti è approssimativo, poiché le efficienze del primer potrebbero non essere identiche. Le sequenze di primer sono elencate nel file supplementare 7.DOI:
http://dx.doi.org/10.7554/eLife.22114.020
Figura 7-figure supplement 1.Le linee transgeniche che esprimono diversi alleli FLM mostrano l’espressione differenziale FLM e i modelli di giunzione.(A) Immagine di un gel di agarosio con i risultati di un 3′-RACE PCR. Il primer utilizzato per l’amplificazione 3′ è indicato nella Figura 7C. Le porzioni di gel dal FLMCol-0, INT6 +CAA e PolyA_6xΔ prodotti di amplificazione incorniciato con una linea tratteggiata sono stati purificati, clonati e sottoposti a sequenziamento del DNA.(B) Rappresentazione schematica dei primer utilizzati per la quantificazione qRT-PCR dei livelli totali FLM (esone 1) e la sequenza E1I1p dell’introne 1 contenente trascrizioni come indicato dalle frecce sotto il modello genetico. La scatola nera corrisponde all’esone 1 FLM, la scatola grigia alla porzione prossimale dell’introne 1. La numerazione è rispettiva alla A del codice di partenza ATG.(C) Media e SD delle analisi qRT-PCR di analisi qRT-PCR di trascrizioni contenenti la sequenza dell’introne 1 da quattro pool di trascrizioni replicate di linee T2 indipendenti. La diminuzione relativa dei valori di espressione E1I1p rispetto al trascritto dell’esone 1 è indicata per ogni riga del grafico. Si noti che il confronto dei valori di espressione tra i frammenti è approssimativo, poiché le efficienze del primer potrebbero non essere identiche. Le sequenze di primer sono elencate nel file supplementare 7.DOI:
http://dx.doi.org/10.7554/eLife.22114.020
I polimorfismi PRO2+ e INT6+ contribuiscono alla variazione globale dei livelli FLM
I nove aplotipi PRO2+ e INT6+ testati in esperimenti transgenici erano presenti in 579 (69%) di tutte le 840 adesioni con informazioni disponibili sulla sequenza genomica(Figura 8-figure supplement 1A). Per esaminare se questi aplotipi spiegano la variazione naturale dei livelli di FLM-ß nelle adesioni naturali, abbiamo selezionato casualmente una popolazione sperimentale di 94 adesioni (da 2 a 14 adesioni per gruppo PRO2+/INT6+, in media 10) con un’ampia distribuzione genetica e geografica (Figura 8-figure supplement 1B,C). Abbiamo poi determinato l’espressione FLM-ß e, a seguito di un filtraggio di qualità, abbiamo raggruppato 85 adesioni secondo i loro aplotipi PRO2+ o INT6+ (file supplementare6A).
Abbiamo scoperto che l’aplotipo PRO2+/INT6+ ha influenzato significativamente i livelli di trascrizione FLM-ß (Figura 8-figure supplement 1D). Quando abbiamo integrato i valori medi di queste adesioni naturali con i rispettivi valori dell’analisi transgenica, abbiamo rilevato una correlazione positiva, anche se non significativa, una probabile conseguenza del numero ridotto di punti di dati(FLM-ß , R2= 0,13) (Figura 8-figuresupplement 1E).
Per esaminare la correlazione tra l’espressione FLM-ß e il tempo di fioritura, abbiamo determinato il tempo di fioritura delle adesioni a 15°C. Per evitare forti interferenze dal percorso di vernalizzazione, abbiamo selezionato 27 adesioni estive-annuali geneticamente diversificate, che iniziano la fioritura senza la necessità di vernalizzazione(Figura 8-figure supplemento 2A, file supplementare 6B). Abbiamo misurato i livelli di trascrizione FLM-ß e il tempo di fioritura a 15°C. Poiché i residui di trascrizione FLC in queste adesioni estive annuali possono ancora influenzare il tempo di fioritura, abbiamo anche determinato i livelli di trascrizione FLC(Coustham et al., 2012; Li et al., 2014; Duncan et al., 2015). Utilizzando un approccio di regressione lineare multipla, abbiamo scoperto che FLM-ß e FLC spiegavano il 32,9% (R2 = 0,329, p=0,0083) della variazione del tempo di fioritura, con FLM-ß che spiegava significativamente una sottofrazione di 21.0% (R2 = 0,210, p=0,011) e FLC solo 11, 9%, ma non in modo significativo (R2 = 0,119, p>0,05) (Figura 8B e Figura 8-figure supplement 2B). Inoltre, grazie all’integrazione dei dati di espressione e dei dati sul tempo di fioritura in un modello lineare multiplo e al confronto delle pendenze, abbiamo scoperto che il tempo di fioritura ha risposto più forte ai livelli FLM-ß nelle adesioni che nelle varianti transgeniche (p=0,0321) (Figura 5B e Figura 8). Presi nel loro insieme, abbiamo concluso che le variazioni dei livelli di FLM-ß tengono conto del tempo di fioritura nelle temperature ambiente fresche della primavera in una popolazione diversificata di adesioni estive-annuali di Arabidopsis (Figura 9).10.7554/eLife.22114.021Figure 8.FLM-ß expression shows high correlation with flowering time in Arabidopsis accessions.(A) Correlation (simple linear regression) of FLM-ß transcript levels detected in transgenic plants and natural accessions with identical PRO2+ and INT6+ haplotypes. Quando è stato valutato il contributo dei singoli aplotipi, PRO2+AAAAC INT6+CAAAC ha mostrato una risposta eccezionale nelle adesioni ed è stato escluso. La figura 8-figure supplement 1E mostra l’analisi completa. Le barre di errore orizzontali e verticali rappresentano le SD di quattro pool di linee transgeniche replicate o le SD delle adesioni. I dati delle linee transgeniche sono stati presi dalla Figura 4E. L’area grigia indica l’intervallo di confidenza del 95%.(B) Correlazione dei livelli di trascrizione FLM-ß con il tempo di fioritura (n = 8-10) misurato in numero di foglie a rosetta delle adesioni estive-annuali. p=0.011.DOI:http://dx.doi.org/10.7554/eLife.22114.02110.7554/eLife.22114.022Figure 8-figure supplement 1.Distribuzione geografica e genetica degli aplotipi PRO2+/INT6+ tra 840 adesioni.(A) Grafico a torta con la distribuzione relativa degli aplotipi PRO2+/INT6+ tra 840 adesioni. L’aplotipo Col-0 è contrassegnato in giallo, gli aplotipi analizzati nell’ambito di questo studio sono contrassegnati in verde. Gli aplotipi rappresentati solo da una singola adesione non sono inclusi.(B) Numero di adesioni selezionate da ciascuno dei nove gruppi PRO2+/INT6+ (in media 10, in totale 94 adesioni). Il codice colore indica l’appartenenza al gruppo genetico (k = 9) di ogni adesione come recuperato dallo strumento 1001 Genomi ADMIXTURE(http://tools.1001genomes.org/) (IlConsorzio1001Genomi, 2016).(C) Distribuzione geografica delle adesioni mostrate in(B).(D) Livelli di trascrizione FLM-β delle adesioni mostrate in (B) e (C) raggruppati per l’aplotipo PRO2+/INT6+. Lettere simili non indicano alcuna differenza significativa del numero totale di foglie (Tukey HSD, p< 0,05).(E) Correlazione (regressione lineare semplice) tra i livelli di trascrizione FLM-ß rilevati nell’analisi transgenica e nelle accessioni con aplotipi PRO2+/INT6+ identici provenienti da piante coltivate a 15°C. L’aplotipo PRO2+AAAAC INT6+CAA, che è stato rimosso nella Figura 8A, è mostrato ed evidenziato con una freccia rossa. Le dimensioni del cerchio rappresentano il numero di adesioni analizzate di ciascun gruppo, come specificato nella legenda. Le barre di errore orizzontali e verticali rappresentano SD di quattro pool di linee transgeniche replicate o SD delle adesioni di ciascun gruppo. I dati per l’analisi delle linee transgeniche sono stati presi dalla Figura 4E. L’area grigia indica l’intervallo di confidenza del 95%.DOI:http://dx.doi.org/10.7554/eLife.22114.02210.7554/eLife.22114.023FigureSupplemento a 8 cifre 2.Distribuzione geografica delle adesioni estive annuali.(A) Distribuzionegeografica delle 27 adesioni estive annuali come descritto nella Figura 8B .(B) Correlazione dei livelli di trascrizione FLC con i dati del tempo di fioritura (n = 8-10) come misurato in numero di foglie a rosetta delle adesioni estive annuali. p>0.05.DOI:http://dx.doi.org/10.7554/eLife.22114.02310.7554/eLife.22114.024Figure 9.Model of the proposed role of PRO2+ and INT6+ haplotypes and temperature on FLM-ß abundance and flowering.The abundance of the flowering repressor FLM-ß decreases in response to higher temperature and flowering is consequently accelerated (Figures 1Band 4E) ( Lee et al., 2013; Posé et al., 2013; Lutz et al., 2015). Si noti che gli studi precedenti hanno mostrato un effetto particolarmente evidente dell’FLM in un intervallo compreso tra 9°C e 21°C (fotoperiodo di una lunga giornata) (Lee et al., 2013; Posé et al., 2013; Lutz et al., 2015). A livello genetico, l’abbondanza FLM-ß è innescata dall’aplotipo PRO2+ (viola) e/o dall’aplotipo INT6+ (rosa) e dal tempo di fioritura correlato all’abbondanza FLM-ß (Figura 4E). Tra le combinazioni PRO2+/INT6+ testate, l’allele di riferimento Col-0 (grigio) (PRO2+AAAAC/INT6+AAT) ha mostrato livelli intermedi FLM-ß (Figura 4E). Suggeriamo, che le variazioni del tempo di fioritura dovute al cambiamento della temperatura ambiente possono essere precisamente tamponate modificando le regioni PRO2+ e INT6+, come illustrato da simboli vegetali simili.DOI:http://dx.doi.org/10.7554/eLife.22114.024

Figura 8-figure supplement 2.L’espressioneFLM-ß mostra un’elevata correlazione con il tempo di fioritura nelle adesioni dell’Arabidopsis.distribuzione geografica e genetica degli aplotipi PRO2+/INT6+ tra 840 adesioni.distribuzione geografica delle adesioni estive-annuali.(A) Correlazione (regressione lineare semplice) dei livelli di trascrizione FLM-ß rilevati nelle piante transgeniche e nelle adesioni naturali con aplotipi PRO2+ e INT6+ identici. Quando è stato valutato il contributo dei singoli aplotipi, PRO2+AAAAC INT6+CAAAC ha mostrato una risposta eccezionale nelle adesioni ed è stato escluso. La figura 8-figure supplement 1E mostra l’analisi completa. Le barre di errore orizzontali e verticali rappresentano le SD di quattro pool di linee transgeniche replicate o le SD delle adesioni. I dati delle linee transgeniche sono stati presi dalla Figura 4E. L’area grigia indica l’intervallo di confidenza del 95%.(B) Correlazione dei livelli di trascrizione FLM-ß con il tempo di fioritura (n = 8-10) misurato in numero di foglie a rosetta delle adesioni estive-annuali. p=0,011.DOI:
http://dx.doi.org/10.7554/eLife.22114.021(A) Grafico a torta con la relativa distribuzione degli aplotipi PRO2+/INT6+ tra 840 adesioni. L’aplotipo Col-0 è contrassegnato in giallo, gli aplotipi analizzati nell’ambito di questo studio sono contrassegnati in verde. Gli aplotipi rappresentati solo da una singola adesione non sono inclusi.(B) Numero di adesioni selezionate da ciascuno dei nove gruppi PRO2+/INT6+ (in media 10, in totale 94 adesioni). Il codice colore indica l’appartenenza al gruppo genetico (k = 9) di ogni adesione come recuperato dallo strumento 1001 Genomi ADMIXTURE(http://tools.1001genomes.org/) (IlConsorzio1001Genomi, 2016).(C) Distribuzione geografica delle adesioni mostrate in(B).(D) Livelli di trascrizione FLM-β delle adesioni mostrate in (B) e (C) raggruppati per l’aplotipo PRO2+/INT6+. Lettere simili non indicano alcuna differenza significativa del numero totale di foglie (Tukey HSD, p< 0,05).(E) Correlazione (regressione lineare semplice) tra i livelli di trascrizione FLM-ß rilevati nell’analisi transgenica e nelle accessioni con aplotipi PRO2+/INT6+ identici provenienti da piante coltivate a 15°C. L’aplotipo PRO2+AAAAC INT6+CAA, che è stato rimosso nella Figura 8A, è mostrato ed evidenziato con una freccia rossa. Le dimensioni del cerchio rappresentano il numero di adesioni analizzate di ciascun gruppo, come specificato nella legenda. Le barre di errore orizzontali e verticali rappresentano SD di quattro pool di linee transgeniche replicate o SD delle adesioni di ciascun gruppo. I dati per l’analisi delle linee transgeniche sono stati presi dalla Figura 4E. L’area grigia indica l’intervallo di confidenza del 95%.DOI:
http://dx.doi.org/10.7554/eLife.22114.022(A) Distribuzione geografica delle 27 adesioni estive annuali come descritto nella Figura 8B.(B) Correlazione dei livelli di trascrizione FLC con i dati del tempo di fioritura (n = 8-10) come misurato in numero di foglie a rosetta delle adesioni estive-annuali. p> 0,05.DOI:
http://dx.doi.org/10.7554/eLife.22114.023

Figura 8-figure supplement 1.Distribuzione geografica e genetica degli aplotipi PRO2+/INT6+ tra 840 adesioni.(A) Grafico a torta con la distribuzione relativa degli aplotipi PRO2+/INT6+ tra 840 adesioni. L’aplotipo Col-0 è contrassegnato in giallo, gli aplotipi analizzati nell’ambito di questo studio sono contrassegnati in verde. Gli aplotipi rappresentati solo da una singola adesione non sono inclusi.(B) Numero di adesioni selezionate da ciascuno dei nove gruppi PRO2+/INT6+ (in media 10, in totale 94 adesioni). Il codice colore indica l’appartenenza al gruppo genetico (k = 9) di ogni adesione come recuperato dallo strumento 1001 Genomi ADMIXTURE(http://tools.1001genomes.org/) (IlConsorzio1001Genomi, 2016).(C) Distribuzione geografica delle adesioni mostrate in(B).(D) Livelli di trascrizione FLM-β delle adesioni mostrate in (B) e (C) raggruppati per l’aplotipo PRO2+/INT6+. Lettere simili non indicano alcuna differenza significativa del numero totale di foglie (Tukey HSD, p< 0,05).(E) Correlazione (regressione lineare semplice) tra i livelli di trascrizione FLM-ß rilevati nell’analisi transgenica e nelle accessioni con aplotipi PRO2+/INT6+ identici provenienti da piante coltivate a 15°C. L’aplotipo PRO2+AAAAC INT6+CAA, che è stato rimosso nella Figura 8A, è mostrato ed evidenziato con una freccia rossa. Le dimensioni del cerchio rappresentano il numero di adesioni analizzate di ciascun gruppo, come specificato nella legenda. Le barre di errore orizzontali e verticali rappresentano SD di quattro pool di linee transgeniche replicate o SD delle adesioni di ciascun gruppo. I dati per l’analisi delle linee transgeniche sono stati presi dalla Figura 4E. L’area grigia indica l’intervallo di confidenza del 95%.DOI:
http://dx.doi.org/10.7554/eLife.22114.022
Figura 8-figure supplement 2.Distribuzione geografica delle adesioni estive-annuali.(A) Distribuzione geografica delle 27 adesioni estive annuali come descritto nella Figura 8B.(B) Correlazione dei livelli di trascrizione FLC con i dati del tempo di fioritura (n = 8-10) come misurato in numero di foglie a rosetta delle adesioni estive-annuali. p> 0,05.DOI:
http://dx.doi.org/10.7554/eLife.22114.023

Figura 9.Modello del ruolo proposto degli aplotipi PRO2+ e INT6+ e della temperatura sull’abbondanza e sulla fioritura FLM-ß.L’abbondanza del repressore di fioritura FLM-ß diminuisce in risposta all’aumento della temperatura e la fioritura è di conseguenza accelerata (Figure1B e 4E) (Leeet al., 2013; Posé et al. , 2013; Lutz et al., 2015). Si noti che gli studi precedenti hanno mostrato un effetto particolarmente evidente dell’FLM in un intervallo compreso tra 9°C e 21°C (fotoperiodo di una lunga giornata) (Lee et al., 2013; Posé et al., 2013; Lutz et al., 2015). A livello genetico, l’abbondanza FLM-ß è innescata dall’aplotipo PRO2+ (viola) e/o dall’aplotipo INT6+ (rosa) e dal tempo di fioritura correlato all’abbondanza FLM-ß (Figura 4E). Tra le combinazioni PRO2+/INT6+ testate, l’allele di riferimento Col-0 (grigio) (PRO2+AAAAC/INT6+AAT) ha mostrato livelli intermedi FLM-ß (Figura 4E). Suggeriamo, che i cambiamenti nel tempo di fioritura a causa del cambiamento della temperatura ambiente può essere esattamente tamponato modificando le regioni PRO2 + e INT6 +, come illustrato da simboli vegetali simili.DOI:
http://dx.doi.org/10.7554/eLife.22114.024
Discussione
La temperatura ambiente durante la primavera è uno dei principali fattori che determinano il tempo di fioritura. Le temperature fredde in genere ritardano e le temperature calde favoriscono il tempo di fioritura dell’Arabidopsis. Il locus FLM spiega la variazione del tempo di fioritura in diverse temperature ambientali, ma le basi genetiche sottostanti della fioritura dipendente dall’FLM sono rimaste ampiamente poco chiare(Salomé et al., 2011; el-Lithy et al., 2006; O’Neill et al., 2008).
Abbiamo scoperto che, oltre al promotore FLM, anche le sequenze dell’introne 1 erano essenziali per l’espressione basale FLM e successivamente identificate dall’impronta filogenomica di una regione dell’introne 1 conservata a 373 bp essenziale per l’espressione basale FLM(Figura 2). Inoltre, attraverso analisi di associazione che utilizzano le informazioni della sequenza genomica, abbiamo scoperto le regioni di regolazione FLM (PRO2+ e INT6+) che controllano l’espressione FLM dipendente dalla temperatura in modo specifico per l’aplotipo(Figura 4). Mentre la maggior parte degli aplotipi PRO2+ e INT6+ mostravano un’espressione FLM differenziale basale ma sensibile alla temperatura, correlando fortemente con le variazioni del tempo di fioritura, una variante INT6+, INT6+CAA, era fortemente compromessa nella regolazione FLM sensibile alla temperatura e nella fioritura, indicando l’importanza di questa regione intronica per l’adattamento del tempo di fioritura(Figure 4 e 6).
In tutti i nostri esperimenti, FLM-ß altamente correlato (R2 = 0,94) con il tempo di fioritura in un ampio intervallo vegetativo (15-30 foglie a rosetta) se determinato a 15 ° C e testato in un fondo omozigote, indipendentemente dal tipo di variante (Figura 5). La nostra scoperta che FLM-ß ha avuto un effetto più forte nel controllo del tempo di fioritura rispetto a FLM-δ indica che i modelli funzionali precedentemente formulati proponendo che FLM-δ aveva un’attività antagonista a FLM-ß devono essere corretti (Posé et al., 2013;Sureshkumar et al., 2016). Questo è ulteriormente supportato dal fatto che i nostri risultati stimano che i livelli di FLM-δ sono complessivamente molto bassi e che non abbiamo identificato un singolo clone FLM-δ in un approccio di sequenziamento diretto che ha identificato 53 cloni FLM-ß . I nostri risultati supportano quindi studi più recenti che suggeriscono che la variante di giunzione FLM-δ può essere biologicamente irrilevante (Sureshkumaret al., 2016; Lutz et al., 2015). Uno di questi studi ha anche identificato un gran numero di trascrizioni biologicamente irrilevanti che potrebbero essere identificate con la combinazione di primer qui utilizzata per il rilevamento di FLM-δ (Sureshkumaret al., 2016). I nostri dati supportano quindi la conclusione che nessuno di questi prodotti di amplificazione ha una funzione biologicamente importante(Sureshkumar et al., 2016).
Mentre i polimorfismi PRO2+ e INT6+ testati hanno influito sull’abbondanza di trascrizioni FLM, le cause molecolari di questi cambiamenti di trascrizione variano tra i diversi polimorfismi(Figure 4 e 7). La cancellazione di una regione promotrice di 225 bp che include il sito polimorfico PRO2+ è stata associata a livelli decuplicati di livelli ridotti di FLM pre-mRNA, indicando che questa regione promotrice era essenziale per l’espressione FLM(Figura 7). Una doppia differenza relativa di livelli FLM-ß è stata trovata confrontando levarianti PRO2+GATACe PRO2+AAAACC con l’espressione FLM-ß più bassa e più alta, rispettivamente, e il nostro modello lineare prevede una differenza di tempo di fioritura di 14,4 foglie a 15°C (Figure 4 e5). La regione PRO2 + ospita un predetto sito di legame del fattore di trascrizione MADS-box e sono stati descritti diversi casi di auto-regolazione o di regolazione incrociata dei fattori MADS-box(de Folter et al., 2005; Kaufmann et al., 2009; Smaczniak et al., 2012). Si può quindi immaginare che l’espressione FLM sia regolata da fattori di trascrizione MADS-box e che i polimorfismi PRO2+ modulino l’efficienza o la specificità di questi eventi di legame e quindi modulino l’espressione FLM basale.
Allo stesso modo, la cancellazione di un frammento di 373 bp nell’introne 1 FLM ha portato ad una riduzione di undici volte l’espressione FLM e la fioritura precedente di 11,5 foglie come determinato sperimentalmente. Questa dimensione dell’effetto è molto più piccola di quanto previsto dal modello lineare. Tuttavia, come proposto in precedenza, questo può essere probabilmente dovuto ad una soglia critica inferiore per FLM-ß per diventare efficace (Figure4 e 5). Intronic cis-regulatory transcription factor binding sites sono stati identificati in altri fattori di trascrizione MADS-box e sono state segnalate interazioni di elementi potenziatori e silenziatori che risiedono nella sequenza del promotore o alla fine del 3′-end del primo introne (Honget al., 2003; Schauer et al., 2009). Quindi, meccanismi simili possono governare l’espressione di FLM nei siti dell’introne 1 che possono agire in modo isolato o insieme ad eventi vincolanti presso il promotore FLM.
È interessante notare che i polimorfismi naturali nell’introne 6 (INT6+) hanno portato a circa quattro volte le differenze nell’abbondanza di FLM-ß , che potrebbero, come previsto dal modello lineare, riferirsi ad un ritardo di fioritura di 28,8 foglie, suggerendo che l’INT6+ racchiude un esteso potenziale per la messa a punto della fioritura (Figure4 e 5). È importante notare che questo effetto molecolare sembra essere mediato, nel caso di INT6+CAA, dagli effetti sull’efficienza di giunzione e sulla specificità dell’introne distale 1. Lì, INT6+CAA promuove la formazione di brevi trascrizioni contenenti l’introne 1, a scapito di FLM-ß , che sono probabilmente soggette a degradazione per decadimento non mediato da un nonsense-mediato (Figura 7) (Lutzet al., 2015).
Il sito INT6+ è direttamente affiancato da un breve motivo PolyA e tali siti rappresentano potenziali siti di riconoscimento dei fattori di giunzione hnRNP (ribonucleoproteina nucleare eterogenea). Il modello di legame hnRNP previsto a INT6+ dipendeva infatti dall’aplotipo INT6+, quando previsto da algoritmi basati sul web, e la loro preferenza e attività di legame, di concerto con altri regolatori di splicing o trascrizionali, può essere alla fine la base dei cambiamenti di splicing qui osservati(Piva et al., 2012; Carrillo Oesterreich et al., 2011; Reddy et al., 2013).
La ricorrenza dei motivi PolyA ([A]7-11) con i siti PRO2+ e INT6+ aveva attirato la nostra attenzione(Figura 4-figure supplement 1). Le delezioni combinatorie di tutti e sei i motivi FLM PolyA hanno portato a una graduale diminuzione dell’abbondanza di FLM-ß, che potrebbe essere spiegata con la modifica dello splicing dell’introne 1 (Figura 7). Questo ha portato alla fine a livelli di FLM-ß al di sotto di una soglia effettiva più bassa nelle linee PolyA_6xΔ e di conseguenza una fioritura molto precoce (Figura 4-figuresupplement 1). Questo suggerisce che i motivi PolyA possono modulare l’espressione FLM modificando la giunzione FLM. A sostegno di questa conclusione, abbiamo trovato che un motivo PolyA esteso, come è presente nella variante INT6+AAAAA rispetto alla variante di riferimento Col-0 INT6+AAT, è correlato con forti aumenti in FLM-ß ma non in FLM-δ abbondanza (Figura 4). I motivi PolyA sono noti per prevenire il legame dei nucleosomi e i cambiamenti dell’architettura della cromatina possono influenzare lo splicing(Reddy et al., 2013; Suter et al., 2000; Wijnker et al., 2013). La conoscenza dei meccanismi molecolari sottostanti e dell’identità e specificità dei regolatori di giunzione nelle piante è ancora molto limitata. Abbiamo notato con interesse, tuttavia, che diversi hnRNP hanno un ruolo segnalato nella regolazione del tempo di fioritura in Arabidopsis e grano(Kippes et al., 2015; Fusaro et al., 2007; Streitner et al., 2012; Xiao et al., 2015).
Le nove combinazioni di aplotipi PRO2+/INT6+ incluse nei nostri esperimenti transgenici rappresentano il 69% della variazione mondiale PRO2+/INT6+(Figura 8-figure supplement 1A,B). Abbiamo scoperto che FLM-ß ha spiegato circa il 21% del tempo di fioritura in una popolazione geneticamente eterogenea di adesioni estive-annuali di Arabidopsis (Figura 8). Quindi, gli aplotipi PRO2+ e INT6+ regolano l’abbondanza di FLM-ß e, a loro volta, contribuiscono alla regolazione del tempo di fioritura nelle adesioni dell’Arabidopsis (Figure8 e 9). Non ci sorprende che la correlazione del tempo di fioritura con i livelli di FLM-ß (21% contro il 94%) e la reattività del tempo di fioritura ai livelli di FLM-ß differiscano tra una popolazione naturale geneticamente eterogenea e la popolazione transgenica omogenea. In primo luogo, abbiamo mostrato che la trascrizione residua di FLC può anche contribuire leggermente alla variazione del tempo di fioritura delle adesioni estive-annuali, interferendo eventualmente con gli effetti di FLM-ß (Balasubramanianet al., 2006; Li et al., 2006). Inoltre, un’ulteriore variazione della sequenza di FLM nelle adesioni naturali può contribuire a cambiamenti di espressione rilevanti, ad esempio nella parte dell’introne 1 identificata come essenziale per l’espressione FLM, ma non analizzata in modo più dettagliato in questa sede(Figura 2). Quindi, è possibile che tali adesioni portino a variazioni nel MAF2 – 4 relativo a FLM, e questa variazione può influenzare in modo differenziato il tempo di fioritura, in modo isolato o in combinazione con la variazione del loro partner di interazione comune SVP(Ratcliffe et al., 2003; Airoldi et al., 2015; Scortecci et al., 2003). Tenendo conto di queste possibili interferenze genetiche e ambientali, consideriamo considerevole l’effetto rilevato di FLM-ß sulla fioritura (21%) a temperature fresche (15°C) e suggeriamo che si tratta piuttosto di una sottovalutazione.
Abbiamo trovato una distribuzione uniforme di nove combinazioni di aplotipi PRO2+/INT6+ tra i cluster genetici che sono stati recentemente stabiliti sulla base dell’analisi delle sequenze genomiche da 1135 adesioni di Arabidopsis(The 1001 Genomes Consortium, 2016). È interessante notare che l’aplotipo PRO2+GGAAC/INT6+AAT era sovrarappresentato tra le adesioni relitte e l’aplotipo PRO2+AAAAC/INT6+AAAA era sovrarappresentato tra il gruppo asiatico(The 1001 Genomes Consortium, 2016)(Figura 8-figure supplement 1). Nei nostri esperimenti, entrambi questi aplotipi sono stati associati ad un aumento dei livelli di trascrizione FLM-ß e di conseguenza alla fioritura tardiva (Figura 4). Dal momento che i relitti e le adesioni asiatiche sono state proposte per originare da refugia glaciale da dove l’Europa centrale è stata ricolonizzata dopo l’ultima era glaciale, questi aplotipi PRO2+/INT6+ a fioritura tardiva possono rappresentare aplotipi originali(Sharbel et al., 2000; Schmid et al., 2006). Il fenotipo a fioritura tardiva associato a questi aplotipi sarebbe in linea con l’ipotesi che gli alleli a fioritura tardiva siano più antichi e che gli alleli a fioritura precoce siano stati derivati solo durante la più recente evoluzione dell’Arabidopsis(Toomajian et al., 2006). Inoltre, il legame genetico tra i siti polimorfici PRO2+ e INT6+ era complessivamente basso (R2 = 0,11-0,55) e le sequenze che circondano PRO2+ e INT6+ erano relativamente conservate. Presi insieme, questo suggerisce che le mutazioni nei siti PRO2+ e INT6+ sono emerse in modo indipendente più volte e che questi siti sono stati selezionati preferibilmente durante l’adattamento della fioritura dell’Arabidopsis. Tutti gli alleli PRO2+/INT6+ hanno mostrato un’ampia distribuzione geografica, eccetto PRO2+GGAAC/INT6+AAT, che è stato sovrarappresentato in Spagna(Figura 8-figure supplement 1). Probabilmente, i microclimi in specifiche località geografiche piuttosto che le condizioni climatiche generali in ampie regioni geografiche possono essere importanti per guidare l’adattamento della fioritura al cambiamento della temperatura ambiente, come è stato suggerito in precedenza per gli aplotipi non codificanti di FLC ampiamente distribuiti che spiegano la variazione nella vernalizzazione(Li et al., 2014; Weigel, 2012; Shindo et al., 2006).
Le mutazioni nelle regioni regolatorie cis sono importanti per l’adattamento e l’evoluzione fenotipica e hanno una bassa probabilità di generare effetti pleiotropici negativi(Swinnen et al., 2016; Meyer e Puruggananan, 2013; Wray, 2007). FLM può essere un candidato ideale per l’adattamento del tempo di fioritura attraverso la variazione di sequenza non codificata nei siti PRO2+ e INT6+, poiché le variazioni dell’abbondanza FLM-ß modulano con precisione la fioritura mantenendo la plasticità fenotipica e senza generare effetti pleiotropici negativi (Figura 9). I cambiamenti nei siti PRO2+ e INT6+ dovrebbero permettere di adattare il tempo di fioritura in risposta alle alterazioni della distribuzione geografica e di conseguenza delle condizioni climatiche, nonché durante i cambiamenti degli ambienti globali(Figura 9). Il ruolo degli ortologi FLM in altre Brassicacee, incluse alcune specie importanti dal punto di vista agronomico, non è ancora stato compreso. Non è quindi attualmente possibile prevedere il ruolo dei polimorfismi FLM dell’Arabidopsis per l’adattamento al tempo di fioritura e la riproduzione delle piante in questa famiglia di piante. Tuttavia, in considerazione della disponibilità di nuove metodologie di modifica del genoma, la conoscenza di importanti regioni non codificanti può essere utile per la messa a punto del tempo di fioritura(Shan et al., 2013). Questo può consentire di tamponare gli effetti negativi previsti sui sistemi agricoli che si verificano come conseguenza di piccoli cambiamenti di temperatura durante il cambiamento climatico(Moore e Lobell, 2015; Wheeler e von Braun, 2013).
Materiali e metodi
Materiale biologico
Tutte le accessioni di Arabidopsis thaliana utilizzate in questo studio e flm-3 (Salk_141971; Col-0) e Nd-1 (N1636) sono state fornite dal Nottingham Arabidopsis Stock Centre (NASC; Nottingham, Regno Unito). flc-3 e FRISF-2 FLC(Michaels and Amasino, 1999) sono stati un regalo di Franziska Turck e George Coupland (Max-Planck Institute of Plant Breeding Research, Colonia, Germania). pFLM::gFLM (pDP34), pFLM::FLM-ß (pDP96), e pFLM::FLM-δ (pDP97) sono stati precedentemente riportati (Posé etal., 2013). Primer per vagliare le adesioni per l’FLMLINEA sono stati utilizzati come descritto in precedenza(Lutz et al., 2015).
Esperimenti fisiologici
Per le analisi del tempo di fioritura, le piante sono state coltivate in condizioni di giorno lungo con 16 ore di luce bianca (110-130 μmol m-2 s-1)/8 ore di buio nelle camere di crescita MLR-351 SANYO (Ewald, Bad Nenndorf, Germania). Le piante sono state disposte a caso in vassoi e i vassoi sono stati riordinati ogni giorno. L’acqua veniva fornita mediante subirrigazione. Il tempo di fioritura è stato quantificato contando i numeri delle foglie a rosetta (RLN). Coerentemente con i rapporti precedenti, abbiamo osservato una forte correlazione tra i giorni di bullonatura e il numero di foglie a rosetta delle accessioni di Arabidopsis(Figura 8-figure supplement 2C) (Atwellet al., 2010). I test t degli studenti (valori normalmente distribuiti), i test di rango Wilcoxon (valori normalmente non distribuiti) e i modelli di regressione multipla sono stati calcolati con R(http://www.r-project.org/).
Procedura di clonazione
Un costrutto precedentemente descritto con il frammento di FLM genomico Col-0 pFLM::gFLM template (pDP34)(Posé et al., 2013) è stato ricombinato in un vettore di destinazione pDONR201 utilizzando il sistema Gateway (Life Technologies, Carlsbad, CA). Questo vettore è stato utilizzato come template (FLMCol-0) per generare mutazioni utilizzando un singolo primer fosforilato o una combinazione di primer forward e reverse(Sawano e Miyawaki, 2000; Hansson et al., 2008). Nel caso di varianti con modifiche multiple, le singole mutazioni sono state introdotte una alla volta. Gli inserti mutati sono stati ricombinati al vettore di espressione pFAST-R07 utilizzando il sistema Gateway (Life Technologies, Carlsbad, CA)(Shimada et al., 2010). Tutti i costrutti di espressione sono stati verificati mediante sequenziamento e trasformati nel ceppo GV3101 di Agrobacterium tumefaciens. Le piante Nd-1 sono state trasformate utilizzando il metodo dell’immersione floreale(Clough e Bent, 1998) e sono state identificate piante transgeniche sulla base della fluorescenza dei semi(Shimada et al., 2010). È stata esaminata la separazione delle linee T2 e sono state selezionate linee con singoli eventi di inserimento per ulteriori analisi basate su rapporti di separazione(Shimada et al., 2010). Un elenco dei primer e dei costrutti di espressione è elencato nel file supplementare 7.
PCR quantitativa in tempo reale
Per le analisi qRT-PCR delle linee omozigoti, l’RNA totale è stato isolato da tre repliche biologiche utilizzando il kit RNA NucleoSpin (Machery-Nagel, Düren, Germania). Il DNA è stato rimosso con un trattamento su colonna con rDNase (Machery-Nagel, Düren, Germania). 2-3 μg di RNA totale sono stati trascritti al contrario con M-MuLV Transcriptasi inversa (Thermo Fisher Scientific, Waltham, USA) utilizzando un primer oligo(dT). L’equivalente cDNA di 30-50 ng di RNA totale è stato utilizzato in una reazione PCR di 12 μl con SsoAdvancedUniversal SYBR Green Supermix (BioRad, München, Germania) in un CFX96 Real-Time System Cycler (BioRad, München, Germania). La relativa quantificazione è stata calcolata con il metodo ΔΔCt utilizzando ACT8 (AT1G49240) come standard (Pfaffl,2001). Per l’analisi delle linee transgeniche indipendenti T2 messe in comune, è stato campionato un quarto di tutte le linee indipendenti disponibili per ogni costrutto (da 18 a 45), ottenendo così quattro pool di replicazione che comprendono da quattro a undici linee. Circa 1000 semi sono stati utilizzati per il pooling per linea. Un campione di RNA è stato estratto per ogni pool di replicati ed elaborato come descritto sopra.
Gli esperimenti di espressione su larga scala sono stati eseguiti utilizzando un formato a 96 pozzetti precedentemente descritto(Figura 8 e file supplementare 3)(Box et al., 2011). Il DNA è stato digerito con DNaseI (Thermo Fisher Scientific, Waltham, USA) e trascrizione inversa e reazioni qPCR sono state eseguite come descritto in precedenza utilizzando un CFX384 Real-Time System Cycler (BioRad, München, Germania). ACT8 (AT1G49240 ) e BETA-TUBULIN-4 (AT5G44340) sono stati utilizzati come geni di riferimento. I t-test degli studenti sono stati calcolati con Excel (Microsoft). I primer qRT-PCR sono elencati nel file supplementare 7.
Recupero dati dal progetto del genoma 1001 Arabidopsis thaliana
Sono disponibili informazioni sulla sequenza dei dati di 776 e 840 adesioni, rispettivamente, che sono stati utilizzati in questo studio. Le sequenze genomiche di un set completo di 1135 adesioni di Arabidopsis sono state appena rilasciate e sono state utilizzate per le analisi conclusive(The 1001 Genomes Consortium, 2016). Le sequenze genomiche FLM (7 kb; Chr1: 28953637-28960296; inclusa la sequenza 2 kb a monte e 0,2 kb a valle) sono state estratte da 776 adesioni dal portale 1001 Genomes (http://1001genomes.org/datacenter/) utilizzando il dataset Wang (343 adesioni), il dataset GMI (180 adesioni), il dataset Salk (171 adesioni) e il dataset MPI (80 adesioni) (The1001 Genomes Consortium, 2016; Schmitz et al., 2013; Long et al., 2013; Cao et al., 2011). 45 SNP con un MAF>5% sono stati estratti e utilizzati per l’analisi dell’aplotipo del locus FLM (Figura 3A e Figura 3-figure supplement 1). 31 SNP erano polimorfici tra le 54 adesioni selezionate rappresentate nel set di aplotipi FLM(file supplementare 1).
Per ottenere un set esteso di sequenze che includesse non solo SNPs ma anche informazioni su inserimenti e cancellazioni, abbiamo estratto sequenze genomiche FLM (incluse 2 kb a monte e 0.38 kb a valle della sequenza) dalla risorsa GEBrowser 3.0 (http://signal.salk.edu/atg1001/3.0/gebrowser.php). Un set di 850 sequenze è stato curato manualmente e allineato usando MEGA7.0.14(Tamura et al., 2011). Alcune sequenze sono state escluse in quanto possedevano un elevato numero di basi ambigue. Il set di sequenze di base consisteva di 840 sequenze. Questo set di sequenze è stato utilizzato per l’analisi di associazione(Figura 3-figure supplements 4- 7) e tutte le analisi successive sulla variazione FLM PRO2+ e INT6+ (Figura 8e Figura 8-figure supplement 1). Gli identificatori di adesione, i dati geografici e l’appartenenza al gruppo ADMIXTURE (k = 9) sono stati ottenuti dalla raccolta di strumenti per i genomi 1001(http://tools.1001genomes.org/) (The 1001 GenomesConsortium, 2016).
Analisi degli aplotipi
Le analisi degli aplotipi sono state eseguite con DNaSP 5.10(Rozas e Rozas, 1995) utilizzando il set di dati SNP del set di 776 sequenze sopra descritto. I siti invariabili sono stati rimossi e le reti sono state generate utilizzando il software di rete FLUXUS(Bandelt et al., 1999).
Albero filogenetico e allineamenti
Gli allineamenti sono stati calcolati utilizzando ClustalW2 o MUSCLE e gli alberi vicini (metodo della massima probabilità composita, 1000 repliche di bootstrap) sono stati costruiti con Geneious vR7.0.5 (Biomatters Limited, Auckland, Nuova Zelanda) e MEGA7.0.14(Tamura et al., 2011).
Analisi LD
Il collegamento (R2) tra i siti polimorfici che sono stati utilizzati come input per il semplice test di associazione a singolo locus è stato calcolato utilizzando la funzione LD in GGT v2.0(van Berloo, 2008). Sono stati considerati solo gli SNP diallelici (109 su 119).
Test di Kruskal-Wallis
Per rilevare varianti con effetti significativi sui livelli di trascrizione FLM (semplice test di associazione di un singolo locus), 119 varianti di un locus FLM da 7 kb sono state estratte da un set di 840 sequenze recuperate dal GEBrowser 3.0, come descritto sopra. Per esaminare se una variante ha mostrato un effetto significativo sull’espressione FLM, i valori di espressione qRT-PCR delle 54 adesioni dal set di aplotipi FLM sono stati utilizzati come dati di input(file supplementare 4) per eseguire i test di Kruskal-Wallis utilizzando R(http://www.r-project.org/). p-valori di tutti i confronti sono stati corretti a seguito della correzione dei test multipli Benjamini-Hochberg e i valori risultanti sono stati -log(10) trasformati e tracciate lungo il modello genico (Figura 3-figure supplemento 5 e file supplementare 5).
Impronta filogenetica
Gli ortologiFLM sono stati identificati per primi da OrthoMCL (V2.0) clustering (PMID: 12952885; parametri standard, valore di inflazione = 1,5, BLAST e-value-cutoff = e-05), incorporando sequenze proteiche previste da Arabidopsis thaliana (TAIR10; AT1G77080.4, AT5G65060.1), Arabidopsis lyrata (V1.0; fgenesh2_kg.2__2000__AT1G77080, fgenesh2_kg.8__2543__AT5G65050, fgenesh2_kg.233__4__AT5G6505050), Boechera stricta (V1.2; Bostr.4104s0001.1), Brassica rapa (V1.3; Brara.B03928.1.p, Brara.F02378.1.p), Brassica oleracea (V2.1; Bo2g166500.1, Bo2g166560.1), Capsella grandiflora (V1.1; Cagra.0450s0030.1, Cagra.0917s0081.1), Capsella rosolia (V1.0; Carubv10020979m, Carubv10027327m), Gossypium raimondii (V2.1), Medicago truncatula (V4.0), Oryza sativa (MSU7), Populus trichocarpa (V3.0) e Solanum lycopersicum (ITAG2.3). I set di dati sono stati scaricati da Phytozome(Goodstein et al., 2012), PGSB PlantsDB(Spannagl et al., 2016) e Ensembl Plants(Kersey et al., 2016). I cluster contenenti FLM sono stati estratti e, in caso di presenza di più membri del gruppo provenienti da singole specie, sono stati ulteriormente filtrati utilizzando il miglior criterio bi-direzionale BLAST hit. Le sequenze genomiche e la regione a monte di 2 kb sono state allineate con ClustalW2(Larkin et al., 2007). Le regioni conservate nell’allineamento sono state annotate manualmente.
Analisi molecolari FLM
qRT-PCR per l’abbondanza di FLM pre-mRNA e 3′ RACE PCR sono state eseguite come descritto in precedenza(Lutz et al., 2015). Le sequenze di primer sono elencate nel file supplementare 7.
References
- Airoldi CA, McKay M, Davies B. MAF2 is regulated by temperature-dependent splicing and represses flowering at low temperatures in parallel with FLM. PLoS One. 2015; 10DOI | PubMed
- Atwell S, Huang YS, Vilhjálmsson BJ, Willems G, Horton M, Li Y, Meng D, Platt A, Tarone AM, Hu TT, Jiang R, Muliyati NW, Zhang X, Amer MA, Baxter I, Brachi B, Chory J, Dean C, Debieu M, de Meaux J, Ecker JR, Faure N, Kniskern JM, Jones JD, Michael T, Nemri A, Roux F, Salt DE, Tang C, Todesco M, Traw MB, Weigel D, Marjoram P, Borevitz JO, Bergelson J, Nordborg M. Genome-wide association study of 107 phenotypes in Arabidopsis thaliana inbred lines. Nature. 2010; 465:627-631. DOI | PubMed
- Balasubramanian S, Sureshkumar S, Lempe J, Weigel D. Potent induction of Arabidopsis thaliana flowering by elevated growth temperature. PLoS Genetics. 2006; 2DOI | PubMed
- Balasubramanian S, Weigel D. Temperature induced flowering in Arabidopsis thaliana. Plant Signaling & Behavior. 2006; 1:227-228. DOI | PubMed
- Bandelt HJ, Forster P, Röhl A. Median-joining networks for inferring intraspecific phylogenies. Molecular Biology and Evolution. 1999; 16:37-48. DOI | PubMed
- Blázquez MA, Ahn JH, Weigel D. A thermosensory pathway controlling flowering time in Arabidopsis thaliana. Nature Genetics. 2003; 33:168-171. DOI | PubMed
- Box MS, Coustham V, Dean C, Mylne JS. Protocol: a simple phenol-based method for 96-well extraction of high quality RNA from Arabidopsis. Plant Methods. 2011; 7:7. DOI | PubMed
- Cao J, Schneeberger K, Ossowski S, Günther T, Bender S, Fitz J, Koenig D, Lanz C, Stegle O, Lippert C, Wang X, Ott F, Müller J, Alonso-Blanco C, Borgwardt K, Schmid KJ, Weigel D. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nature Genetics. 2011; 43:956-963. DOI | PubMed
- Capovilla G, Schmid M, Posé D. Control of flowering by ambient temperature. Journal of Experimental Botany. 2015; 66:59-69. DOI | PubMed
- Carrillo Oesterreich F, Bieberstein N, Neugebauer KM. Pause locally, splice globally. Trends in Cell Biology. 2011; 21:328-335. DOI | PubMed
- Clough SJ, Bent AF. Floral dip: a simplified method for Agrobacterium-mediated transformation of Arabidopsis thaliana. The Plant Journal. 1998; 16:735-743. DOI | PubMed
- The 1001 Genomes Consortium. 1,135 genomes reveal the global pattern of polymorphism in Arabidopsis thaliana. Cell. 2016; 166:481-491. DOI | PubMed
- Coustham V, Li P, Strange A, Lister C, Song J, Dean C. Quantitative modulation of polycomb silencing underlies natural variation in vernalization. Science. 2012; 337:584-587. DOI | PubMed
- de Folter S. Comprehensive interaction map of the Arabidopsis MADS box transcription factors. The Plant Cell Online. 2005; 17:1424-1433. DOI
- Duncan S, Holm S, Questa J, Irwin J, Grant A, Dean C. Seasonal shift in timing of vernalization as an adaptation to extreme winter. eLife. 2015; 4DOI
- el-Lithy ME, Bentsink L, Hanhart CJ, Ruys GJ, Rovito D, Broekhof JL, van der Poel HJ, van Eijk MJ, Vreugdenhil D, Koornneef M. New Arabidopsis recombinant inbred line populations genotyped using SNPWave and their use for mapping flowering-time quantitative trait loci. Genetics. 2006; 172:1867-1876. DOI | PubMed
- Fusaro AF, Bocca SN, Ramos RL, Barrôco RM, Magioli C, Jorge VC, Coutinho TC, Rangel-Lima CM, De Rycke R, Inzé D, Engler G, Sachetto-Martins G. AtGRP2, a cold-induced nucleo-cytoplasmic RNA-binding protein, has a role in flower and seed development. Planta. 2007; 225:1339-1351. DOI | PubMed
- Goodstein DM, Shu S, Howson R, Neupane R, Hayes RD, Fazo J, Mitros T, Dirks W, Hellsten U, Putnam N, Rokhsar DS. Phytozome: a comparative platform for green plant genomics. Nucleic Acids Research. 2012; 40:D1178-D1186. DOI | PubMed
- Gu X, Le C, Wang Y, Li Z, Jiang D, Wang Y, He Y. Arabidopsis FLC clade members form flowering-repressor complexes coordinating responses to endogenous and environmental cues. Nature Communications. 2013; 4DOI | PubMed
- Hansson MD, Rzeznicka K, Rosenbäck M, Hansson M, Sirijovski N. PCR-mediated deletion of plasmid DNA. Analytical Biochemistry. 2008; 375:373-375. DOI | PubMed
- Hoffmann MH. Biogeography of arabidopsis thaliana (L.) Heynh. (Brassicaceae). Journal of Biogeography. 2002; 29:125-134. DOI
- Hong RL, Hamaguchi L, Busch MA, Weigel D. Regulatory elements of the floral homeotic gene AGAMOUS identified by phylogenetic footprinting and shadowing. The Plant Cell Online. 2003; 15:1296-1309. DOI | PubMed
- Horton MW, Hancock AM, Huang YS, Toomajian C, Atwell S, Auton A, Muliyati NW, Platt A, Sperone FG, Vilhjálmsson BJ, Nordborg M, Borevitz JO, Bergelson J. Genome-wide patterns of genetic variation in worldwide Arabidopsis thaliana accessions from the RegMap panel. Nature Genetics. 2012; 44:212-216. DOI | PubMed
- Jagadish SV, Bahuguna RN, Djanaguiraman M, Gamuyao R, Prasad PV, Craufurd PQ. Implications of high temperature and elevated CO2 on flowering time in plants. Frontiers in Plant Science. 2016; 7DOI | PubMed
- Johanson U, West J, Lister C, Michaels S, Amasino R, Dean C. Molecular analysis of FRIGIDA, a major determinant of natural variation in Arabidopsis flowering time. Science. 2000; 290:344-347. DOI | PubMed
- Kaufmann K, Muiño JM, Jauregui R, Airoldi CA, Smaczniak C, Krajewski P, Angenent GC. Target genes of the MADS transcription factor SEPALLATA3: integration of developmental and hormonal pathways in the Arabidopsis flower. PLoS Biology. 2009; 7DOI | PubMed
- Kersey PJ, Allen JE, Armean I, Boddu S, Bolt BJ, Carvalho-Silva D, Christensen M, Davis P, Falin LJ, Grabmueller C, Humphrey J, Kerhornou A, Khobova J, Aranganathan NK, Langridge N, Lowy E, McDowall MD, Maheswari U, Nuhn M, Ong CK, Overduin B, Paulini M, Pedro H, Perry E, Spudich G, Tapanari E, Walts B, Williams G, Tello-Ruiz M, Stein J, Wei S, Ware D, Bolser DM, Howe KL, Kulesha E, Lawson D, Maslen G, Staines DM. Ensembl genomes 2016: more genomes, more complexity. Nucleic Acids Research. 2016; 44:D574-D580. DOI | PubMed
- Kippes N, Debernardi JM, Vasquez-Gross HA, Akpinar BA, Budak H, Kato K, Chao S, Akhunov E, Dubcovsky J. Identification of the VERNALIZATION 4 gene reveals the origin of spring growth habit in ancient wheats from South Asia. PNAS. 2015; 112:E5401-E5410. DOI | PubMed
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG. Clustal W and clustal X version 2.0. Bioinformatics. 2007; 23:2947-2948. DOI | PubMed
- Lee JH, Ryu HS, Chung KS, Posé D, Kim S, Schmid M, Ahn JH. Regulation of temperature-responsive flowering by MADS-box transcription factor repressors. Science. 2013; 342:628-632. DOI | PubMed
- Lee JH, Yoo SJ, Park SH, Hwang I, Lee JS, Ahn JH. Role of SVP in the control of flowering time by ambient temperature in Arabidopsis. Genes & Development. 2007; 21:397-402. DOI | PubMed
- Li D, Liu C, Shen L, Wu Y, Chen H, Robertson M, Helliwell CA, Ito T, Meyerowitz E, Yu H. A repressor complex governs the integration of flowering signals in Arabidopsis. Developmental Cell. 2008; 15:110-120. DOI | PubMed
- Li P, Filiault D, Box MS, Kerdaffrec E, van Oosterhout C, Wilczek AM, Schmitt J, McMullan M, Bergelson J, Nordborg M, Dean C. Multiple FLC haplotypes defined by independent cis-regulatory variation underpin life history diversity in Arabidopsis thaliana. Genes & Development. 2014; 28:1635-1640. DOI | PubMed
- Li Y, Roycewicz P, Smith E, Borevitz JO. Genetics of local adaptation in the laboratory: flowering time quantitative trait loci under geographic and seasonal conditions in Arabidopsis. PLoS One. 2006; 1DOI | PubMed
- Long Q, Rabanal FA, Meng D, Huber CD, Farlow A, Platzer A, Zhang Q, Vilhjálmsson BJ, Korte A, Nizhynska V, Voronin V, Korte P, Sedman L, Mandáková T, Lysak MA, Seren Ü, Hellmann I, Nordborg M. Massive genomic variation and strong selection in Arabidopsis thaliana lines from Sweden. Nature Genetics. 2013; 45:884-890. DOI | PubMed
- Lutz U, Posé D, Pfeifer M, Gundlach H, Hagmann J, Wang C, Weigel D, Mayer KF, Schmid M, Schwechheimer C. Modulation of ambient Temperature-Dependent flowering in Arabidopsis thaliana by natural variation of FLOWERING LOCUS M. PLoS Genetics. 2015; 11DOI | PubMed
- Martinez-Castilla LP, Alvarez-Buylla ER. Adaptive evolution in the Arabidopsis MADS-box gene family inferred from its complete resolved phylogeny. PNAS. 2003; 100:13407-13412. DOI | PubMed
- Meyer RS, Purugganan MD. Evolution of crop species: genetics of domestication and diversification. Nature Reviews Genetics. 2013; 14:840-852. DOI | PubMed
- Michaels SD, Amasino RM. FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering. The Plant Cell Online. 1999; 11:949-956. DOI | PubMed
- Michaels SD, Amasino RM. Loss of FLOWERING LOCUS C activity eliminates the late-flowering phenotype of FRIGIDA and autonomous pathway mutations but not responsiveness to vernalization. The Plant Cell Online. 2001; 13:935-942. DOI | PubMed
- Moore FC, Lobell DB. The fingerprint of climate trends on European crop yields. PNAS. 2015; 112:2670-2675. DOI | PubMed
- O’Malley RC, Huang SS, Song L, Lewsey MG, Bartlett A, Nery JR, Galli M, Gallavotti A, Ecker JR. Cistrome and epicistrome features shape the regulatory DNA landscape. Cell. 2016; 165:1280-1292. DOI | PubMed
- O’Neill CM, Morgan C, Kirby J, Tschoep H, Deng PX, Brennan M, Rosas U, Fraser F, Hall C, Gill S, Bancroft I. Six new recombinant inbred populations for the study of quantitative traits in Arabidopsis thaliana. Theoretical and Applied Genetics. 2008; 116:623-634. DOI | PubMed
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Research. 2001; 29:45. DOI | PubMed
- Piva F, Giulietti M, Burini AB, Principato G. SpliceAid 2: a database of human splicing factors expression data and RNA target motifs. Human Mutation. 2012; 33:81-85. DOI | PubMed
- Posé D, Verhage L, Ott F, Yant L, Mathieu J, Angenent GC, Immink RG, Schmid M. Temperature-dependent regulation of flowering by antagonistic FLM variants. Nature. 2013; 503:414-417. DOI | PubMed
- Ratcliffe OJ, Nadzan GC, Reuber TL, Riechmann JL. Regulation of flowering in Arabidopsis by an FLC homologue. Plant Physiology. 2001; 126:122-132. DOI | PubMed
- Ratcliffe OJ. Analysis of the Arabidopsis MADS AFFECTING FLOWERING gene family: maf2 prevents vernalization by short periods of cold. The Plant Cell Online. 2003; 15:1159-1169. DOI
- Reddy AS, Marquez Y, Kalyna M, Barta A. Complexity of the alternative splicing landscape in plants. The Plant Cell. 2013; 25:3657-3683. DOI | PubMed
- Rozas J, Rozas R. DnaSP, DNA sequence polymorphism: an interactive program for estimating population genetics parameters from DNA sequence data. Computer Applications in the Biosciences. 1995; 11:621-625. DOI | PubMed
- Salomé PA, Bomblies K, Laitinen RA, Yant L, Mott R, Weigel D. Genetic architecture of flowering-time variation in Arabidopsis thaliana. Genetics. 2011; 188:421-433. DOI | PubMed
- Sawano A, Miyawaki A. Directed evolution of green fluorescent protein by a new versatile PCR strategy for site-directed and semi-random mutagenesis. Nucleic Acids Research. 2000; 28:E78-78. DOI | PubMed
- Schauer SE, Schlüter PM, Baskar R, Gheyselinck J, Bolaños A, Curtis MD, Grossniklaus U. Intronic regulatory elements determine the divergent expression patterns of AGAMOUS-LIKE6 subfamily members in Arabidopsis. The Plant Journal. 2009; 59:987-1000. DOI | PubMed
- Schmid KJ, Törjék O, Meyer R, Schmuths H, Hoffmann MH, Altmann T. Evidence for a large-scale population structure of Arabidopsis thaliana from genome-wide single nucleotide polymorphism markers. Theoretical and Applied Genetics. 2006; 112:1104-1114. DOI | PubMed
- Schmitz RJ, Schultz MD, Urich MA, Nery JR, Pelizzola M, Libiger O, Alix A, McCosh RB, Chen H, Schork NJ, Ecker JR. Patterns of population epigenomic diversity. Nature. 2013; 495:193-198. DOI | PubMed
- Scortecci K, Michaels SD, Amasino RM. Genetic interactions between FLM and other flowering-time genes in Arabidopsis thaliana. Plant Molecular Biology. 2003; 52:915-922. DOI | PubMed
- Scortecci KC, Michaels SD, Amasino RM. Identification of a MADS-box gene, FLOWERING LOCUS M, that represses flowering. The Plant Journal. 2001; 26:229-236. DOI | PubMed
- Shan Q, Wang Y, Li J, Zhang Y, Chen K, Liang Z, Zhang K, Liu J, Xi JJ, Qiu JL, Gao C. Targeted genome modification of crop plants using a CRISPR-Cas system. Nature Biotechnology. 2013; 31:686-688. DOI | PubMed
- Sharbel TF, Haubold B, Mitchell-Olds T. Genetic isolation by distance in Arabidopsis thaliana: biogeography and postglacial colonization of Europe. Molecular Ecology. 2000; 9:2109-2118. DOI | PubMed
- Shimada TL, Shimada T, Hara-Nishimura I. A rapid and non-destructive screenable marker, FAST, for identifying transformed seeds of Arabidopsis thaliana. The Plant Journal. 2010; 61:519-528. DOI | PubMed
- Shindo C, Lister C, Crevillen P, Nordborg M, Dean C. Variation in the epigenetic silencing of FLC contributes to natural variation in Arabidopsis vernalization response. Genes & Development. 2006; 20:3079-3083. DOI | PubMed
- Smaczniak C, Immink RG, Muiño JM, Blanvillain R, Busscher M, Busscher-Lange J, Dinh QD, Liu S, Westphal AH, Boeren S, Parcy F, Xu L, Carles CC, Angenent GC, Kaufmann K. Characterization of MADS-domain transcription factor complexes in Arabidopsis flower development. PNAS. 2012; 109:1560-1565. DOI | PubMed
- Song J, Angel A, Howard M, Dean C. Vernalization – a cold-induced epigenetic switch. Journal of Cell Science. 2012; 125:3723-3731. DOI | PubMed
- Spannagl M, Nussbaumer T, Bader KC, Martis MM, Seidel M, Kugler KG, Gundlach H, Mayer KF. PGSB PlantsDB: updates to the database framework for comparative plant genome research. Nucleic Acids Research. 2016; 44:D1141-D1147. DOI | PubMed
- Streitner C, Köster T, Simpson CG, Shaw P, Danisman S, Brown JW, Staiger D. An hnRNP-like RNA-binding protein affects alternative splicing by in vivo interaction with transcripts in Arabidopsis thaliana. Nucleic Acids Research. 2012; 40:11240-11255. DOI | PubMed
- Sureshkumar S, Dent C, Seleznev A, Tasset C, Balasubramanian S. Nonsense-mediated mRNA decay modulates FLM-dependent thermosensory flowering response in Arabidopsis. Nature Plants. 2016; 2:16055. DOI | PubMed
- Suter B, Schnappauf G, Thoma F. Poly(dA.dT) sequences exist as rigid DNA structures in nucleosome-free yeast promoters in vivo. Nucleic Acids Research. 2000; 28:4083-4089. DOI | PubMed
- Swinnen G, Goossens A, Pauwels L. Lessons from domestication: targeting Cis-Regulatory elements for crop improvement. Trends in Plant Science. 2016; 21:506-515. DOI | PubMed
- Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution. 2011; 28:2731-2739. DOI | PubMed
- Toomajian C, Hu TT, Aranzana MJ, Lister C, Tang C, Zheng H, Zhao K, Calabrese P, Dean C, Nordborg M. A nonparametric test reveals selection for rapid flowering in the Arabidopsis genome. PLoS Biology. 2006; 4DOI | PubMed
- van Berloo R. GGT 2.0: versatile software for visualization and analysis of genetic data. Journal of Heredity. 2008; 99:232-236. DOI | PubMed
- Van de Velde J, Heyndrickx KS, Vandepoele K. Inference of transcriptional networks in Arabidopsis through conserved noncoding sequence analysis. The Plant Cell. 2014; 26:2729-2745. DOI | PubMed
- Verhage L, Angenent GC, Immink RG. Research on floral timing by ambient temperature comes into blossom. Trends in Plant Science. 2014; 19:583-591. DOI | PubMed
- Weigel D. Natural variation in Arabidopsis: from molecular genetics to ecological genomics. Plant Physiology. 2012; 158:2-22. DOI | PubMed
- Werner JD, Borevitz JO, Warthmann N, Trainer GT, Ecker JR, Chory J, Weigel D. Quantitative trait locus mapping and DNA array hybridization identify an FLM deletion as a cause for natural flowering-time variation. PNAS. 2005; 102:2460-2465. DOI | PubMed
- Wheeler T, von Braun J. Climate change impacts on global food security. Science. 2013; 341:508-513. DOI | PubMed
- Wijnker E, Velikkakam James G, Ding J, Becker F, Klasen JR, Rawat V, Rowan BA, de Jong DF, de Snoo CB, Zapata L, Huettel B, de Jong H, Ossowski S, Weigel D, Koornneef M, Keurentjes JJ, Schneeberger K. The genomic landscape of meiotic crossovers and gene conversions in Arabidopsis thaliana. eLife. 2013; 2DOI | PubMed
- Wray GA. The evolutionary significance of cis-regulatory mutations. Nature Reviews Genetics. 2007; 8:206-216. DOI | PubMed
- Xiao J, Li C, Xu S, Xing L, Xu Y, Chong K. JACALIN-LECTIN LIKE1 regulates the nuclear accumulation of GLYCINE-RICH RNA-BINDING PROTEIN7, influencing the RNA processing of FLOWERING LOCUS C antisense transcripts and flowering time in Arabidopsis. Plant Physiology. 2015; 169:2102-2117. DOI | PubMed
- Zhang T, Marand AP, Jiang J. PlantDHS: a database for DNase I hypersensitive sites in plants. Nucleic Acids Research. 2016; 44:D1148-D1153. DOI | PubMed
Fonte
Lutz U, Nussbaumer T, Spannagl M, Diener J, Mayer KF, et al. () Natural haplotypes of . eLife 6e22114. https://doi.org/10.7554/eLife.22114




