
Abstract
Introduzione
L’obesità è molto diffusa nei paesi sviluppati[1] e contribuisce ad un sostanziale carico di morbilità e mortalità[2,3]. Nonostante i progressi nella comprensione delle varianti genetiche, dei fattori di stile di vita e delle interazioni gene-ambiente associate all’obesità [4- 7], gran parte della variazione interindividuale del peso corporeo rimane inspiegata da uno stile di vita misurabile e da fattori genetici. La metilazione del DNA, una delle modifiche epigenetiche più frequenti e ben caratterizzate, riflette a livello molecolare una vasta gamma di esposizioni ambientali e di influenze genetiche[8]. Stabilizzando la struttura della cromatina e alterando l’espressione genica, la metilazione del DNA ha il potenziale di influenzare la suscettibilità di un individuo all’obesità (vedi revisione in [9]). Inoltre, i cambiamenti nella metilazione del DNA possono verificarsi secondariamente all’obesità e possono di conseguenza influenzare lo sviluppo di malattie correlate all’adiposità come il diabete, la dislipidemia, l’ipertensione e le malattie cardiovascolari. Rimangono grandi lacune nelle conoscenze su come le modificazioni epigenetiche umane si riferiscono all’obesità e alle sue conseguenze.
I biomarcatori epigenetici rappresentano una risorsa ampiamente non sfruttata della medicina di precisione per guidare le decisioni terapeutiche utilizzando il profilo epigenetico di un individuo ottenuto da campioni di sangue [10]. L’identificazione di loci epigenetici clinicamente rilevanti nel sangue ha il potenziale di creare una base su cui basare futuri studi funzionali e sperimentazioni per testare il processo decisionale clinico epigeneticamente guidato per le malattie cardiometaboliche. Inoltre, possiamo acquisire nuove conoscenze sulle basi molecolari dell’obesità e delle malattie correlate alle adiposità attraverso lo studio dei loci di DNA metilato differenzialmente nel sangue. Ciò può portare all’identificazione di obiettivi terapeutici biologicamente rilevanti.
Il presente studio fornisce i risultati di uno studio di associazione a livello di epigenoma (EWAS) dell’indice di massa corporea (IMC) in oltre 3.700 partecipanti dello studio Framingham Heart Study (FHS) e delle coorti di nascita di Lothian (LBC) del 1921 e 1936 (LBC1921 e LBC1936). Abbiamo condotto la replicazione esterna indipendente in oltre 4.000 individui del Rischio di Aterosclerosi nelle Comunità (ARIC), della Genetica dei farmaci per l’abbassamento dei lipidi e della rete dietetica (GOLDN), e dell’Indagine prospettica sulla vascolarizzazione degli anziani di Uppsala (PIVUS). Abbiamo esaminato la rilevanza funzionale dei loci identificati interrogando le note funzioni di regolazione del transessuale e i cambiamenti concomitanti nell’espressione genica nel sangue. Inoltre, abbiamo esplorato la rilevanza clinica dei risultati per le malattie correlate all’adiposità con analisi delle variabili genetiche strumentali (IV) utilizzando approcci di randomizzazione Mendeliana bidirezionale e a due fasi trans-tissue Mendeliana (MR)[11- 13].
Metodi
Studio Design
Lo studio comprende due componenti principali. In primo luogo, abbiamo condotto un EWAS di BMI. In secondo luogo, i loci metilati differenzialmente legati all’IMC sono stati portati avanti per ulteriori analisi per comprendere meglio l’entità dell’associazione, l’annotazione normativa, le implicazioni funzionali e la rilevanza clinica(Fig 1). Il progetto di scoperta/replicazione e i modelli secondari per l’EWAS dell’IMC sono stati definiti a priori(S1 Text). Le analisi a valle per caratterizzare i loci scoperti sono state delineate a priori, ma l’approccio finale è stato principalmente guidato dai risultati e dai progressi simultanei sul campo.
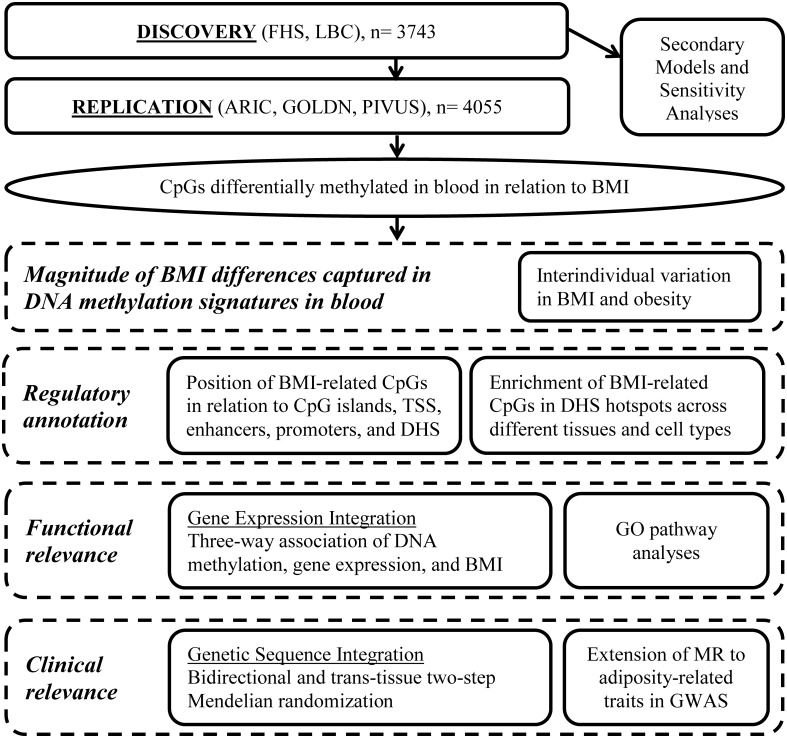
Fig. 1.Serie di analisi condotte per lo studio di associazione a livello epigenomico dell’indice di massa corporea.ARIC, Atherosclerosis Risk in Communities; BMI, indice di massa corporea; DHS, DNasi I sito ipersensibile; FHS, Framingham Heart Study; GO, Ontologia genica; GOLDN, Genetics of Lipid Lowering Drugs and Diet Network; GWAS, studio di associazione a livello genomico; LBC, Lothian Birth Cohorts; MR, randomizzazione mendeliana; PIVUS, Prospective Investigation of the Vasculature in Uppsala Seniors; TSS, sito di inizio trascrizione.
Fig. 1.Serie di analisi condotte per lo studio di associazione a livello di epigenoma dell’indice di massa corporea.ARIC, Atherosclerosis Risk in Communities; BMI, indice di massa corporea; DHS, DNasi I sito ipersensibile; FHS, Framingham Heart Study; GO, Ontologia genica; GOLDN, Genetics of Lipid Lowering Drugs and Diet Network; GWAS, studio di associazione a livello genomico; LBC, Lothian Birth Cohorts; MR, randomizzazione mendeliana; PIVUS, Prospective Investigation of the Vasculature in Uppsala Seniors; TSS, sito di partenza della trascrizione.
Etica
I protocolli FHS e i moduli di consenso dei partecipanti sono stati approvati dal comitato di revisione istituzionale della Boston University School of Medicine. Il permesso etico per la LBC1921 è stato ottenuto dal Lothian Research Ethics Committee (Wave 1: LREC/1998/4/183). Il permesso etico per la LBC1936 è stato ottenuto dal Multi-Centre Research Ethics Committee for Scotland (Wave 1: MREC/01/0/56) e dal Lothian Research Ethics Committee (Wave 1: LREC/2003/2/29). Il consenso informato scritto è stato ottenuto da tutti i partecipanti alla discovery coort (FHS e LBC) e alla replication coort (ARIC, GOLDN e PIVUS).
Partecipanti allo studio
I dati per la fase di scoperta di questa indagine sono stati ricavati dalla coorte di discendenti della FHS[14] e dai LBC del 1921 e del 1936[15- 17]. Come descritto in precedenza[14], la coorte di discendenti della FHS è stata inizialmente reclutata nel 1971 e comprendeva 5.124 discendenti (e i loro coniugi) della coorte originaria della FHS[18]. Il campione ammissibile per questa indagine era costituito dai 3.021 partecipanti alla coorte della prole della FHS che hanno partecipato all’ottavo ciclo di esami dal 2005 al 2008. I campioni LBC1921 e LBC1936 derivano dalle Indagini Mentali Scozzesi del 1932 e 1947, rispettivamente, quando quasi tutti i bambini di 11 anni in Scozia hanno completato un test di tipo IQ a scuola. Gli studi LBC hanno fornito un follow-up dei partecipanti sopravvissuti, la maggior parte dei quali vivevano nella regione di Lothian (città di Edimburgo e periferia) della Scozia. Lo studio attuale si basa sugli esami di base per gli anziani di 551 partecipanti a LBC1921 reclutati nel 1999-2001 e di 1.091 partecipanti a LBC1936 reclutati nel 2004-2007.
Misure antropometriche
L’altezza e il peso sono stati misurati in ogni studio utilizzando i protocolli stabiliti, come descritto in dettaglio nei metodi S1. L’IMC è stato calcolato come peso (in chilogrammi) diviso per l’altezza (in metri) al quadrato.
Genomica molecolare
Il DNA da campioni di sangue intero è stato raccolto alla stessa valutazione d’esame delle misure antropometriche e covariate in entrambi gli studi. La metilazione del DNA, saggiata con il BeadChip Infinium HumanMethylation450[19] (Illumina), era disponibile per 2.846 partecipanti FHS e 1.518 partecipanti LBC (514 da LBC1921 e 1.004 da LBC1936). I dettagli sui rigorosi controlli di qualità, le procedure di normalizzazione e le esclusioni di sonde non automatiche, di sonde a ibridazione incrociata e di sonde con polimorfismi a singolo nucleotide (SNP) sono descritti nei metodi S1. Ogni coorte di scoperta e di replicazione ha condotto condotte di pretrattamento specifiche per ogni coorte che hanno permesso ad ogni coorte di affrontare gli effetti tecnici e di lotto specifici dello studio. Questo progetto ha permesso la selezione di veri segnali biologici indipendenti dalla polarizzazione introdotta da metodi di elaborazione uniformi. Dopo il controllo di qualità nelle coorti di scoperta, c’erano 402.358 sonde di metilazione CpG (citosina-fosfato-guanina) condivise disponibili per le analisi in 2.377 FHS e 1.366 partecipanti LBC (446 da LBC1921 e 920 da LBC1936). La dimensione finale del campione è stata determinata dal numero di partecipanti delle coorti di scoperta che hanno acconsentito agli studi genomici e che avevano a disposizione test di DNA e di metilazione che hanno superato le misure di controllo della qualità. Nella FHS, i dati SNP sono stati ottenuti dall’array di Affymetrix 550K imputati al pannello di riferimento del Progetto 1000 Genomi, come precedentemente riportato[20]. I campioni LBC sono stati genotipizzati utilizzando la piattaforma di genotipizzazione Illumina Human610-Quad v1.0 e imputati anche al pannello di riferimento del 1000 Genomes Project. L’espressione genica nel sangue era disponibile nella FHS ed è stata misurata utilizzando l’Affymetrix Human Exon 1.0 ST GeneChip come descritto nei metodi S1.
Studio dell’associazione dell’epigenoma in tutto l’IMC
Nella FHS, sono stati condotti modelli di regressione lineare a effetti misti per testare l’associazione tra la metilazione del DNA specifico del sito e l’IMC. Il modello primario è stato regolato per l’età, il sesso, la parentela familiare (effetto casuale), e le variabili surrogate (per tener conto delle proporzioni cellulari differenziali e degli effetti tecnici)[21], con il BMI come variabile indipendente di interesse e la metilazione del DNA (inversa-normale trasformata) come variabile dipendente. Nel LBC, i modelli di regressione lineare sono stati condotti regolando per età, sesso e conta dei globuli bianchi, con ogni sonda di metilazione del DNA (residuo preso in avanti da un modello lineare generalizzato con una funzione di collegamento logistico che regola gli effetti tecnici e batch) come variabile dipendente e BMI come variabile indipendente di interesse. Ulteriori dettagli analitici per le coorti di scoperta sono descritti nei metodi S1. In entrambe le coorti, sono stati condotti modelli secondari: (1) regolazione aggiuntiva per lo stato di fumo, (2) limitata ai partecipanti con IMC 18-35 kg/m2 per evitare confusioni dovute a fragilità o obesità morbosa e malattie correlate all’obesità, e (3) test per l’età e le interazioni sessuali. I risultati di FHS e LBC sono stati meta-analizzati utilizzando metodi che pesano il valore pper dimensione del campione[22]. La coerenza direzionale degli effetti statisticamente significativi di coorte-specifici è stata confermata per tutti i risultati significativi a livello di metiloma a livello di meta-analisi della scoperta. Abbiamo concentrato le nostre analisi sulla statistica del test risultante e la direzione dell’effetto dalla variabile indipendente di interesse (BMI), poiché i coefficienti di regressione lineare coorte-specifici non erano direttamente comparabili a causa delle differenze nell’approccio di pre-elaborazione tra le coorti. La soglia di significatività statistica nella fase di scoperta è stata definita dalla correzione Bonferroni per test multipli di 0,05/405.000(p-valore< 1,2 × 10-7). Un diagramma di flusso delle analisi condotte è presentato in Fig 1.
Replica esterna dei risultati dell’EWAS
I CpG significativi a livello di metilomio dalla meta-analisi FHS e LBC sono stati portati alla replicazione esterna in tre coorti indipendenti che utilizzavano lo stesso microarray di metilazione: lo studio ARIC, che utilizzava DNA derivato dal sangue intero di 2.096 partecipanti di origine africana; lo studio GOLDN, che utilizzava DNA derivato da cellule CD4+ di 992 partecipanti di origine europea; e lo studio PIVUS, che utilizzava DNA derivato dal sangue intero di 967 partecipanti di origine svedese. La descrizione e i metodi analitici delle coorti di replicazione sono forniti nei metodi S1. Le coorti di replicazione hanno anche condotto una pre-elaborazione specifica della coorte. Replicazione è stato esaminato all’interno di ogni coorte individualmente e poi in una meta-analisi di tutte e tre le coorti di replicazione (utilizzando p-valore-ponderatametodi e garantire la coerenza direzionale come descritto in precedenza). La soglia per la replicazione statisticamente significativa è stata determinata dalla correzione Bonferroni per essere 0,05 diviso per il numero di CpG portati avanti dalla scoperta.
Modelli di sensibilità che regolano i risultati dell’EWAS per potenziali confusioni per variazione genetica
Al fine di dimostrare se i risultati della metilazione del DNA e dell’associazione dell’IMC erano indipendenti dalle varianti genetiche che influenzano la metilazione (metilazione dei loci dei tratti quantitativi [meQTLs]), abbiamo condotto modelli di sensibilità nella FHS per le CpG replicate legate all’IMC condizionate al cis-meQTL superiore (selezionato in base al valore ppiù basso; ±500 kb dalla CpG) per ogni CpG replicata. L’approccio per identificare i cis-meQTL per i CpG legati all’IMC è descritto nei metodi S1.
Variazione interindividuale dell’IMC e distribuzione dell’obesità in relazione ai risultati dell’EWAS
Al fine di determinare l’entità della variazione dell’IMC contenuto all’interno delle firme epigenetiche studiate nel sangue, abbiamo esaminato la variazione catturata in tre modi. In primo luogo, abbiamo esaminato l’aumento del modello R2a partire dal modello di regressione lineare covariata di base, con il BMI come variabile dipendente, quando si aggiungono i CpG replicati non ridondanti (|r| < 0,7) come variabili indipendenti in ordine decrescente di significatività statistica. Abbiamo condotto questa analisi in due set di test di scoperta: (1) i CpG significativi a livello di metiloma nel FHS sono stati testati solo nei LBC e (2) i CpG replicati non ridondanti del BMI EWAS sono stati testati in una delle coorti di replicazione, PIVUS. A causa delle differenze rispetto alle coorti di scoperta nell’etnia (ascendenza africana in ARIC) e nella linea cellulare (cellule CD4+ in GOLDN), abbiamo condotto le analisi delle variazioni solo in PIVUS. In secondo luogo, abbiamo creato una misura additiva composita della stessa CpG replicata non ridondante statisticamente significativa ponderata per la dimensione dell’effetto. La misura composita di metilazione è stata generata per ogni individuo sommando il prodotto del valore beta della metilazione e la dimensione dell’effetto specifico della coorte (inclusa la direzione dell’effetto) per ognuno dei CpG replicati non ridondanti. La distribuzione dell’IMC e la prevalenza dell’obesità (IMC ≥ 30 kg/m2) è stata valutata in decili della misura composita ponderata dell’additivo nella coorte PIVUS. In terzo luogo, la variazione dell’IMC e le probabilità di sovrappeso (IMC 25-29,9 kg/m2) e di obesità sono state testate in modelli di regressione lineare e logistica, corretti in base all’età e al sesso, per ogni variazione della deviazione standard (SD) nella misura composita ponderata per additivo nella coorte PIVUS. La somma ponderata della misura composita di metilazione è stata convertita in unità SD (media = 0, SD = 1) per migliorare l’interpretabilità dei risultati. Poiché alcune delle variazioni della metilazione differenziale trasversale si prevedeva che fossero secondarie alle differenze dell’IMC, lo scopo di queste analisi non era quello di sviluppare un biomarcatore o un predittore di rischio per le misure dell’IMC trasversale, ma di determinare se una grande proporzione della variazione dell’IMC e dell’obesità, e quindi del rischio cardiometabolico correlato all’obesità, si riflette nei modelli di metilazione del DNA nel sangue. Ulteriori analisi esaminano i percorsi molecolari che sono interessati e cercano di dedurre quali cambiamenti di metilazione stanno influenzando causalmente l’IMC, che sono secondari rispetto alle differenze di IMC, e che hanno rilevanza per gli esiti clinici della malattia.
Analisi dell’espressione genica
Abbiamo analizzato i dati di espressione del gene del sangue intero nel FHS per identificare quale CpG differenzialmente metilato legato all’IMC ha dimostrato l’associazione con l’espressione genica alterata. I CpG replicati sono stati testati utilizzando modelli di effetti misti lineari per l’associazione, con il livello di espressione del gene corrispondente nel sangue intero (basato sull’annotazione del produttore) come variabile dipendente e la metilazione del DNA come variabile indipendente, regolata per l’età, il sesso e gli effetti tecnici e di lotto (ulteriori dettagli nei metodi S1).
Annotazione funzionale e regolamentare
Abbiamo studiato l’Ontologia Genetica (GO) processo biologico, funzione molecolare, e le vie della componente cellulare (rilascio 2016-08-22) dei geni identificati nel BMI EWAS utilizzando il PANTHER (annotazione della proteina attraverso la relazione evolutiva) test di sovrarappresentazione[23]. In secondo luogo, abbiamo limitato l’analisi ai geni di maggiore certezza che hanno dimostrato di aver alterato l’espressione del gene del sangue intero in associazione con la metilazione differenziale BMI-correlata, come descritto nella sezione precedente. Se più sonde sono state annotate allo stesso gene, allora il gene è stato incluso solo una volta (non pesato). Poiché la matrice di metilazione copre il 99% dei geni RefSeq, l’universo di fondo dei geni testati non è stato limitato. I risultati sono stati corretti per i test multipli all’interno di ogni categoria.
Inoltre, abbiamo usato eFORGE v1.2(http://eforge.cs.ucl.ac.uk/)[24] per identificare se i CpG replicati sono stati arricchiti in siti ipersensibili DNase I (DHS) (marcatori di regioni normative attive) e loci con modifiche dell’istone sovrapposte (H3Kme1, H3Kme4, H3K9me3, H3K27me3, e H3K36me3) attraverso le linee cellulari e i tessuti disponibili del progetto Roadmap Epigenomics Project, BLUEPRINT Epigenome, e ENCODE (Enciclopedia degli elementi del DNA) dati dei consorzi[25-27].
Randomizzazione bidirezionale e a due fasi del tessuto mendeliano trans-tissue
Analisi IV utilizzando SNP come IV per (1) la metilazione del DNA, (2) l’espressione genica e (3) l’IMC sono state condotte al fine di dedurre potenziali relazioni causali tra i risultati dell’EWAS, l’IMC e le malattie legate all’adiposità (la serie di analisi condotte è descritta nella Tabella 1). L’approccio dettagliato è fornito nei metodi S1. In breve, le differenze nella metilazione e nell’espressione sono state modellate utilizzando i loci dei tratti quantitativi (QTL), facendo così leva sul contributo della variazione genetica ai tratti epigenetici per dedurre le relazioni causali. Sangue QTL IVs sono stati selezionati come il singolo top SNP metilazione SNP singolo o associazione di espressione (dal più basso p-valore) nel FHS con la replicazione nelle coorti esterne o set di dati pubblici. Come QTLs variano in effetto in diversi tipi di tessuto, abbiamo selezionato metilazione specifica del tessuto e QTLs espressione per esaminare gli effetti specifici del tessuto (dettagli in S1 Metodi). Per modellare l’effetto del BMI sulla metilazione (causalità inversa), l’IV per BMI è stato assemblato come un punteggio di rischio genetico ponderato additivo dai 97 SNPs significativi a livello genomico del consorzio GIANT (Genetic Investigation of ANthropometric Traits) 2015 genome-wide association study (GWAS)[7]. Un’analisi di sensibilità utilizzando un singolo SNP nel locus FTO(massa grassa e obesità associata) come IV per l’IMC è stata condotta per esaminare un IV meno incline alla polarizzazione della pleiotropia, ma anche meno potente per rilevare potenziali relazioni causali.
| Metodo | Esposizione | IV | Fonte di IV | Selezione | Risultato | Impostazione |
|---|---|---|---|---|---|---|
| Avanti MR | Metilazione del DNA | meQTL | Coorti FHS/replicazione | Tutti i CpG replicati (come esposizione) | BMI | Consorzio FHS/GIANT |
| Due fasi MR-primo passo | Metilazione del DNA | meQTL | Coorti FHS/replicazione | Significativo in avanti MR | Espressione genica in più tessuti | Set di dati FHS/eQTL esterni provenienti da sangue, fegato e tessuto adiposo |
| Due passi MR-secondo passo | Espressione genica in più tessuti | eQTL | Set di dati FHS/GTEx/eQTL esterni | Significativo in avanti MR | BMI | Consorzio FHS/GIANT |
| Estensione delle relazioni causali ai tratti legati all’adiposità | Metilazione del DNA | meQTL | Coorti FHS/replicazione | Significativo in avanti MR | Tratti legati all’adiposità | Risultati GWAS |
| Espressione genica in più tessuti | eQTL | Set di dati FHS/GTEx/eQTL esterni | Significativo in due fasi MR | Tratti legati all’adiposità | Risultati GWAS | |
| MR inversa | BMI | BMI GRS | Risultati GWAS | Tutti i CpG replicati (come risultato) | Metilazione del DNA | FHS |
La RM in avanti, utilizzando il metodo dei minimi quadrati a due stadi, verifica la relazione causale della metilazione differenziale con l’IMC. SNP IVs che implicava un effetto causale della metilazione differenziale su BMI dalla MR avanti (Bonferroni-corretto e, secondariamente, nominale p-valorecausale nominale < 0,05) sono stati testati nel transessuale a due fasi MR. Il trans-tessuale a due fasi MR è stato implementato per rompere ulteriormente il rapporto tra la metilazione del DNA e BMI e per dedurre se il mediatore ipotizzato (espressione genica in tessuti multipli) è influenzato dall’esposizione (metilazione del DNA) e, in secondo luogo, se il mediatore (espressione genica in tessuti multipli) influisce sul risultato (BMI). SNP IVs che implicava un effetto causale della metilazione differenziale e l’espressione sul BMI sono stati testati per le associazioni con fenotipi legati all’adiposità dai risultati GWAS pubblicati. Infine, la risonanza magnetica inversa è stata condotta per testare la relazione causale dell’IMC con i cambiamenti a valle della metilazione del DNA.
Risultati
Caratteristiche della Coorte di Scoperta
Il campione di scoperta comprendeva 3.743 individui: 2.377 della FHS e 1.366 della LBC(n = 446 della LBC1921 e n = 920 della LBC1936). Le coorti FHS, LBC1921, e LBC1936 erano adulti più anziani (età media [SD] rispettivamente 67 [9], 79 [1], e 70 [1] y) e avevano una distribuzione sessuale simile (50%-60% femminile) e una proporzione di fumatori attuali (8%-11%) (Tabella 2).
| Caratteristica | FHS | LBC1936 | LBC1921 |
|---|---|---|---|
| N | 2,377 | 920 | 446 |
| Età (anni) | 67 ± 9 | 70 ± 1 | 79 ± 1 |
| Femmina | 55% | 40% | 61% |
| IMC (kg/m2) | 28.3 ± 5.4 | 27.8 ± 4.4 | 26.2 ± 4.0 |
| Fumo corrente | 8% | 11% | 7% |
Studio dell’associazione Epigenome-Wide Association di BMI
Scoperta
Nella meta-analisi FHS-LBC EWAS, 135 CpG sono stati significativamente associati al BMI dopo la correzione per test multipli nel modello primario aggiustato per età e sesso(p < 1,2 × 10-7; l’elenco completo e i coefficienti di regressione sono forniti nella tabella S1; grafici Q-Q in S1 e S2 Figs; grafico di Manhattan in S3 Fig; fattore di inflazione genomica della meta-analisi di scoperta, λ = 1,14). Risultati simili sono stati osservati dopo un ulteriore aggiustamento per lo stato di fumo e dopo aver escluso 313 individui con IMC al di fuori di 18-35 kg/m2 (Modelli 2-3 in S2 Table ; S4 Fig).
Replica esterna
I 135 CpG statisticamente significativi della meta-analisi BMI EWAS (modello primario) sono stati testati per la replicazione esterna nelle coorti ARIC (n = 2.096), GOLDN(n = 992), e PIVUS (n= 967). Sono stati replicati all’esterno 83 di 135 CpG su 135 CpG in almeno una coorte (73 in ARIC, 22 in GOLDN e 19 in PIVUS; S5 Fig) al valore p< 3,7 × 10-4 (valore p corretto da Bonferroni per 135 test), e 83 di 135 CpG replicati nelle meta-analisi delle tre coorti di replicazione e sono stati portati avanti per analisi successive (Tabella S3). Una maggiore metilazione è stata associata a un IMC più elevato a 49 (59%) degli 83 CpG replicati. La maggior parte dei CpG legati all’IMC (65%-85% dei CpG a seconda della coorte) aveva livelli medi di metilazione dei CpG del campione tra il 20% e l’80% (Tabella S4). Cinquanta degli 83 CpG replicati con metilazione differenziata non sono stati precedentemente riportati in EWAS di BMI basati su microarray[28- 36](Tabella 3).
| CpG | Gene | Scoperta | Replica | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| FHS-LBC(n = 3.743) | ARIC(n = 2.096) | ORO(n = 992) | PIVUS(n = 967) | Meta-analisi | ||||||
| p-Valore | Dir | p-Valore | Dir | p-Valore | Dir | p-Valore | Dir | p-Valore | ||
| cg17501210 | RPS6KA2 | 4.14E−21 | – − − − − | 1.72E−04 | − | 1.36E−06 | − | 4.15E−02 | − | 1.07E−06 |
| cg14870271 | LGALS3BP | 4.87E−16 | + + + + | 2.28E−04 | + | 3.62E−03 | + | 4.80E−02 | + | 1.51E−05 |
| cg11202345 | LGALS3BP | 6.22E−15 | + + + + | 7.70E−05 | + | 1.36E−02 | + | 1.83E−01 | + | 4.20E−05 |
| cg19750657 | UFM1 | 2.21E−13 | + + + + | 5.55E−06 | + | 6.92E−07 | + | 2.27E−08 | + | 5.60E−13 |
| cg17901584 | DHCR24 | 2.87E−12 | – − − − − | 6.26E−07 | − | 1.23E−04 | − | 7.99E−07 | − | 4.14E−12 |
| cg10179300 | TRIO | 5.71E−12 | + + + + | 7.37E−04 | + | 1.16E−02 | + | 8.82E−02 | + | 1.06E−04 |
| cg10508317 | SOCS3 | 2.90E−11 | – − − − − | 1.31E−03 | − | 3.49E−05 | − | 2.85E−02 | − | 1.12E−05 |
| cg17836612 | LGALS3BP | 1.05E−10 | + + + + | 4.73E−06 | + | 1.99E−03 | + | 7.40E−04 | + | 1.24E−08 |
| cg26950531 | DPF1 | 1.13E−10 | – − − − − | 6.28E−04 | − | 6.49E−03 | − | 9.90E−04 | − | 2.22E−06 |
| cg21429551 | GARS | 1.31E−10 | – − − − − | 3.95E−03 | − | 1.13E−05 | − | 8.58E−02 | − | 6.76E−05 |
| cg01881899 | ABCG1 | 1.37E−10 | + + + + | 4.90E−03 | + | 4.99E−04 | + | 8.25E−02 | + | 1.98E−04 |
| cg17782974 | TRIM8 | 1.79E−10 | + + + + | 7.54E−06 | + | 5.71E−02 | + | 1.39E−01 | + | 1.05E−05 |
| cg16611584 | AKAP10 | 2.40E−10 | + + + + | 2.08E−04 | + | 9.92E−01 | + | 1.98E−02 | + | 1.40E−04 |
| cg07730360 | 3.32E−10 | + + + + | 1.37E−06 | + | 3.31E−02 | + | 5.38E−02 | + | 8.00E−07 | |
| cg17738521 | HIVEP2 | 3.51E−10 | – − − − − | 2.36E−04 | − | 1.37E−01 | − | 7.56E−02 | − | 1.07E−04 |
| cg18098839 | GOLIM4 | 1.21E−09 | – − − − + | 1.48E−03 | − | 1.12E−03 | − | 3.66E−04 | − | 1.42E−06 |
| cg00108715 | NT5DC2 | 1.71E−09 | + + + + | 1.98E−07 | + | 4.60E−01 | + | 4.00E−04 | + | 1.61E−08 |
| cg24531955 | LOXL2 | 2.26E−09 | – − − − − | 3.85E−05 | − | 1.67E−02 | − | 9.20E−03 | − | 1.46E−06 |
| cg17058475 | CPT1A | 2.28E−09 | – − − − − | 3.18E−04 | − | 1.64E−05 | − | 1.29E−01 | − | 1.17E−05 |
| cg24678869 | DENND4B | 2.56E−09 | + + + + | 4.72E−08 | + | 6.92E−01 | − | 4.46E−01 | + | 2.64E−05 |
| cg1313134297 | 3.21E−09 | – − − − − | 1.10E−04 | − | 5.62E−03 | − | 2.06E−01 | − | 4.59E−05 | |
| cg10474597 | SERPINE3 | 3.82E−09 | + + + + | 3.51E−04 | + | 2.13E−01 | + | 1.39E−04 | + | 2.19E−06 |
| cg03725309 | SARS | 3.93E−09 | – − − − − | 2.67E−04 | − | 2.75E−04 | − | 2.43E−03 | − | 6.00E−07 |
| cg26894079 | ASAM | 4.06E−09 | – − − − − | 2.31E−06 | − | 4.60E−05 | − | 1.96E−03 | − | 3.52E−09 |
| cg22012981 | ACOX2 | 4.65E−09 | + + + + | 2.64E−04 | + | 2.27E−02 | + | 1.97E−03 | + | 2.71E−06 |
| cg2636361535 | ZC3H3 | 5.46E−09 | – − + + + | 1.56E−07 | + | 2.65E−02 | + | 1.37E−02 | + | 3.43E−08 |
| cg04286697 | B3GNT7 | 5.96E−09 | + + + + | 8.70E−06 | + | 4.51E−02 | + | 7.53E−01 | + | 1.22E−04 |
| cg26651978 | 6.24E−09 | – − − − − | 6.42E−03 | − | 3.07E−04 | − | 2.76E−03 | − | 1.68E−05 | |
| cg19017142 | 7.86E−09 | – − − − − | 1.48E−04 | − | 1.92E−02 | − | 1.80E−02 | − | 8.35E−06 | |
| cg10919522 | C14orf43 | 9.71E−09 | – − − − − | 3.98E−06 | − | 2.78E−04 | − | 8.58E−04 | − | 5.44E−09 |
| cg25649826 | USP22 | 1.05E−08 | + + + + | 9.69E−06 | + | 1.41E−03 | + | 6.62E−02 | + | 1.18E−06 |
| cg07037944 | DAPK2 | 1.19E−08 | – − − − − | 2.47E−04 | − | 1.36E−01 | − | 4.12E−09 | − | 4.32E−09 |
| cg24145109 | 1.23E−08 | + + + + | 2.69E−05 | + | 8.59E−02 | + | 2.28E−04 | + | 1.62E−07 | |
| cg01751802 | KANK2 | 1.32E−08 | + + + + | 6.33E−07 | + | 8.89E−01 | + | 3.31E−01 | + | 4.30E−05 |
| cg13274938 | RARA | 1.43E−08 | + + + + | 5.48E−07 | + | 3.47E−03 | + | 3.70E−02 | + | 9.57E−08 |
| cg11673687 | SLC9A1 | 2.52E−08 | + + + + | 7.80E−04 | + | 8.37E−01 | − | 7.96E−03 | + | 2.51E−04 |
| cg26800893 | ATPGD1 | 2.87E−08 | – − − − − | 5.04E−04 | − | 8.10E−04 | − | 3.80E−04 | − | 4.29E−07 |
| cg01368219 | CACNA2D3 | 3.20E−08 | + + + + | 3.90E−06 | + | 2.21E−02 | + | 2.62E−02 | + | 6.87E−07 |
| cg01130991 | 4.00E−08 | + + + + | 7.46E−06 | + | 2.49E−04 | + | 2.91E−02 | + | 2.19E−07 | |
| cg27470213 | LGALS3BP | 4.63E−08 | – − − − − | 3.88E−05 | − | 1.23E−03 | − | 4.18E−03 | − | 2.64E−07 |
| cg26955383 | CALHM1 | 6.04E−08 | + + + + | 2.02E−04 | + | 3.52E−02 | + | 1.70E−01 | + | 1.15E−04 |
| cg03500056 | ABAT | 6.19E−08 | + + + + | 4.21E−06 | + | 1.53E−02 | + | 8.29E−03 | + | 2.10E−07 |
| cg09182678 | 6.22E−08 | – − − − − | 1.03E−05 | − | 1.32E−03 | − | 5.88E−07 | − | 1.59E−10 | |
| cg02286155 | 6.39E−08 | + + + + | 2.99E−03 | + | 6.56E−03 | + | 7.40E−02 | + | 2.41E−04 | |
| cg12593793 | 7.12E−08 | – − − − − | 1.59E−04 | − | 8.31E−03 | − | 2.04E−08 | − | 1.06E−09 | |
| cg23172671 | 7.47E−08 | + + + + | 1.22E−04 | + | 7.70E−02 | + | 1.25E−01 | + | 8.01E−05 | |
| cg13139542 | 7.93E−08 | – − + + + | 1.23E−05 | + | 1.05E−02 | + | 3.78E−01 | + | 2.67E−05 | |
| cg02571142 | DKK4 | 9.91E−08 | + + + + | 6.33E−04 | + | 4.34E−03 | + | 6.97E−02 | + | 5.34E−05 |
| cg21766592 | SLC1A5 | 1.07E−07 | – − − − − | 7.69E−03 | − | 5.41E−01 | − | 3.33E−04 | − | 1.34E−04 |
| cg01526748 | FGF12 | 1.18E−07 | + + + + | 5.32E−04 | + | 4.17E−04 | + | 3.32E−02 | + | 1.11E−05 |
Età e interazioni sessuali tra i risultati dell’IMC EWAS
Tra i 135 CpG scoperti, una significativa interazione sessuale è stata dimostrata nelle coorti scoperte per un CpG non preannotato (cg26651978 sul cromosoma 17q25.3; <3 kbp dalla fine 3′ di LGALS3BP [lectinagalattoside-binding solubile 3-binding protein]), e una significativa interazione età per un CpG (cg24678869 cg24678869; DENND4B [DENNdominio 4B Rab GDP-GTP fattore di scambio DENN 4B]) a p-valore <3,7 × 10-4 (Bonferroni-corretto p-valore per 135 test) (S4 Tabella). L’interazione sessuale identificata a cg26651978(LGALS3BP) è modestamente replicata nelle coorti esterne (meta-analisi di replicazione p = 0,02), con coefficienti di regressione più grandi e valorip inferiori nei modelli stratificati tra gli uomini che tra le donne (meta-analisi di replicazione p = 1,73 × 10-6 e 0,002 negli uomini e nelle donne, rispettivamente; coefficienti di regressione complessivi e stratificati per sesso per ciascuna coorte nella Tabella S5) . L’interazione per età a cg24678869(DENND4B) non si è replicata nelle coorti esterne (meta-analisi di replicazione p = 0,9). A causa della ristretta fascia di età in PIVUS, tuttavia, questa interazione è stata testata solo in ARIC e GOLDN(n = 3.079).
Metilazione del locus HIF3A
Esaminando una metilazione differenziale legata all’IMC precedentemente identificata nel locus HIF3A[28], abbiamo dimostrato modeste associazioni con l’IMC nelle coorti di scoperta FHS-LBC per i tre CpG segnalati(p = 0,02 per cg22891070, p = 0,03 per cg16672562, e p = 0,04 per cg27146050; nessuna interazione sessuale significativa). I modelli di stratificazione all’età mediana di 66 anni nel FHS (fascia di età troppo ristretta nel LBC per la stratificazione) hanno rivelato associazioni più forti nel sottoinsieme più giovane e associazioni nulle nel sottoinsieme più vecchio (per cg22891070, cg16672562, e cg27146050, p = 0.003, p = 0,008, e p = 0,046, rispettivamente, tra i partecipanti ≤66 y di età, e p = 0,9, p = 0,6, e p = 0,4, rispettivamente, tra i partecipanti >66 y di età).
Modelli di sensibilità che condizionano i loci del tratto quantitativo della metilazione cis
I modelli di sensibilità condizionata sul cis-meQTL superiore (selezionato dal valore ppiù basso; ±500 kb dal CpG) nel FHS hanno dimostrato un’attenuazione minima della statistica di prova per l’associazione del BMI, con la metilazione differenziale alla maggior parte dei CpG (81/83 [98%]) attenuata di meno del 20% (Tabella S6).
Variazione interindividuale dell’IMC e distribuzione dell’obesità
La variazione interindividuale dell’IMC e la distribuzione dell’obesità catturata nei risultati dell’IMC EWAS è stata valutata. La regressione dell’IMC sui 77 CpG non ridondanti (correlazione inter-sonda |r| < 0.7) dai 83 CpG replicati identificati nel BMI EWAS ha rivelato che il 18% della variazione interindividuale (R2 corretto) nel BMI è catturato dalla metilazione differenziale oltre l’età e il sesso nella coorte di replicazione esterna PIVUS (S6Fig). Questa proporzione è simile a quella osservata durante l’esame di un set di test di scoperta completamente indipendente utilizzando i 75 CpG che erano significativi a livello di metiloma nella coorte di scoperta FHS (nessuna replicazione), che rappresentava il 17,5% della variazione interindividuale dell’IMC (rettificato R2) oltre l’età e il sesso nei LBC. La creazione di una misura composita additiva ponderata dei 77 CpG replicati non ridondanti ed esaminando la distribuzione dell’IMC e dell’obesità (IMC ≥ 30 kg/m2) attraverso i decili della misura ha dimostrato che l’IMC mediano è aumentato in modo graduato da 22 a 34 kg/m2 e la prevalenza dell’obesità è aumentata dallo 0% al 50% (figg. 2 e S7 ). Per ogni aumento di SD nella misura della metilazione del DNA composito nella coorte di replicazione PIVUS, l’IMC è aumentato di 1,63 (errore standard 0,13) kg/m2(p = 3,7 × 10-34). I rapporti di probabilità per l’obesità (IMC ≥ 30 kg/m2) e il sovrappeso (IMC 25-29,9 kg/m2) rispetto al gruppo di riferimento (IMC < 25 kg/m2) erano2,8 (95% CI 2,3-3,5; p = 1.6 × 10-25) e 1,9(95% CI 1,6-2,2; p = 2,5 × 10-18), rispettivamente, per ogni aumento di SD nella misura della metilazione nei modelli corretti per età e sesso.
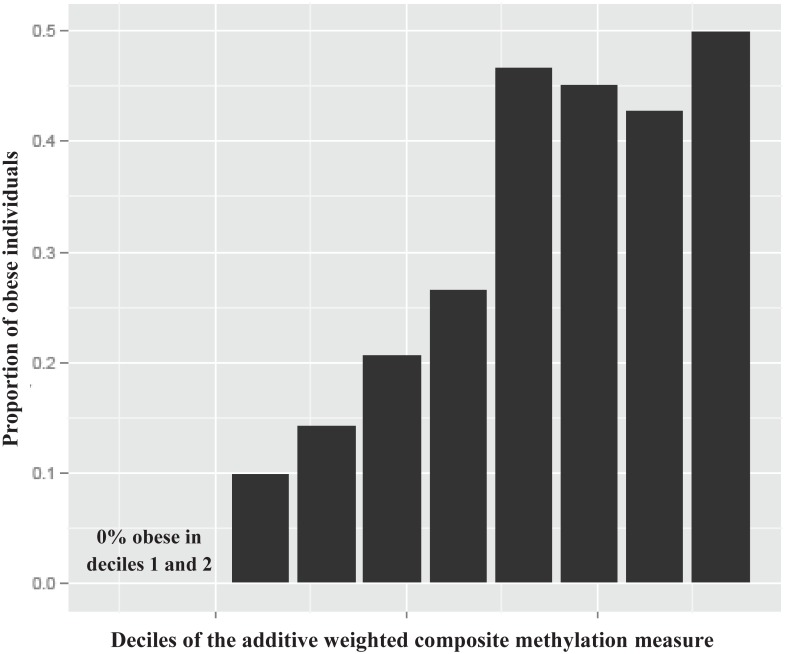
Fig. 2.Fig. 2. Istogramma della proporzione di individui obesi (IMC ≥ 30 kg/m2) nella coorte PIVUS attraverso decili della misura di metilazione composita ponderata dell’additivo dei 77 CpG replicati non ridondanti (|r| < 0,7) dallo studio di associazione a livello di epigenoma dell’IMC.BMI, indice di massa corporea; PIVUS, Prospective Investigation of the Vasculature in Uppsala Seniors.
Fig 2.Fig. 2. Istogramma della proporzione di individui obesi (IMC ≥ 30 kg/m2) nella coorte PIVUS attraverso decili della misura di metilazione composita additiva ponderata dei 77 CpG replicati non ridondanti (|r| < 0.7) dallo studio di associazione a livello di epigenoma dell’IMC.BMI, indice di massa corporea; PIVUS, Prospective Investigation of the Vasculature in Uppsala Seniors.
Associazione a tre vie di metilazione del DNA, espressione genica e IMC
Abbiamo esaminato l’associazione della metilazione del DNA presso gli 83 CpG correlati all’IMC replicati con l’espressione genica tra 2.246 partecipanti al FHS, al fine di determinare quali geni nel sangue possono essere influenzati dalla metilazione differenziale dei BMI EWAS CpG. Degli 83 CpG replicati, l’espressione genica annotata dal sangue intero era disponibile per 62 coppie di espressione genica CpG (tre risultati di trascrizione non erano disponibili sul microarray, e 18 CpG erano intergenici). Ci sono state associazioni significative(p-valore< 8 × 10-4; 0.05/62) tra la metilazione differenziale del DNA e l’espressione genica nel sangue intero per 19 coppie di espressione genica CpG, che rappresentano dieci trascrizioni geniche uniche (ABCG1, CPT1A , SREBF1, LGALS3BP, DHCR24 , PHGDH , SARS , NOD2, CACNA2D3 , e SLC1A5 ), con quasi tutte le coppie di espressione genica CpG (18/19; 95%) che dimostrano un’associazione inversa della metilazione con l’espressione (tabella S7). Ci sono state significative associazioni a tre vie (CpG contro BMI; CpG contro espressione genica; espressione genica contro BMI) per 11 CpG con sette geni annotati unici (Tabella 4). Cinque dei sette geni (71%) con significative associazioni a tre vie tra espressione genica CpG e BMI sono noti per mostrare fenotipi cardiometabolici nei modelli murini di knockout genico [37-44].
| Gene (GEx Probe Number) | CpG | CpG contro IMC | CpG contro GEx | GEx contro BMI | Funzione proteica e fenotipo cardiometabolico del topo transgenico | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Dir | p-Valore | Dir | R2 | p-Valore | Dir | R2 | p-Valore | |||
| ABCG1 (#3922444) | cg06500161 | + | 7.6 × 10−43 | − | 0.112 | 1.2 × 10−59 | − | 0.023 | 6.2 × 10−73 | Funzione: membrana trasportatore per il colesterolo e fosfolipidi; topo transgenico: diminuita suscettibilità all’obesità indotta dalla dieta, adipociti più piccoli, peso inferiore[37]. |
| cg2724243685 | + | 2.3 × 10−15 | − | 0.054 | 4.4 × 10−29 | − | 0.023 | 6.2 × 10−73 | ||
| cg01881899 | + | 1.4 × 10−10 | − | 0.035 | 3.8 × 10−19 | − | 0.023 | 6.2 × 10−73 | ||
| cg10192877 | + | 1.7 × 10−08 | − | 0.014 | 1.7 × 10−08 | − | 0.023 | 6.2 × 10−73 | ||
| CACNA2D3 (#2624639) | cg01368219 | − | 3.2 × 10−08 | − | 0.014 | 2.9 × 10−08 | − | 0.006 | 1.1 × 10−08 | Funzione: complesso dei canali del calcio dipendente dalla tensione; topo transgenico: diminuzione degli acidi grassi liberi del siero[38]. |
| CPT1A (#3379644) | cg00574958 | − | 2.2 × 10−29 | − | 0.025 | 7.7 × 10−14 | + | 0.017 | 4.5 × 10−24 | Funzione: trasportatore attraverso la membrana interna mitocondriale per l’ossidazione beta degli acidi grassi; topo transgenico: diminuzione del glucosio nel siero e aumento dei livelli di acidi grassi liberi nel siero dopo il digiuno[39]. |
| cg17058475 | − | 2.3 × 10−09 | − | 0.014 | 2.2 × 10−08 | + | 0.017 | 4.5 × 10−24 | ||
| DHCR24 (#2413907) | cg17901584 | − | 2.9 × 10−12 | − | 0.017 | 8.9 × 10−10 | + | 0.003 | 1.4 × 10−07 | Funzione: catalizza la riduzione degli intermedi di sterolo durante la biosintesi del colesterolo; topo transgenico: diminuzione dei depositi adiposi sottocutanei e mesenterici, diminuzione delle dimensioni del corpo, diminuzione del colesterolo in circolazione[40]. |
| SARS (#2350551) | cg03725309 | − | 3.9 × 10−09 | − | 0.011 | 6.6 × 10−07 | + | 0.011 | 4.7 × 10−10 | Funzione: catalizza il trasferimento di L-serina al tRNA; topo transgenico: nessun fenotipo cardiometabolico riportato |
| SLC1A5 (#3866276) | cg02711608 | + | 6.3 × 10−08 | − | 0.013 | 5.1 × 10−08 | + | 0.023 | 1.7 × 10−69 | Funzione: trasportatore di aminoacidi; topo transgenico: nessun fenotipo cardiometabolico riportato |
| SREBF1 (#374747966) | cg11024682 | + | 4.8 × 10−22 | − | 0.009 | 8.0 × 10−06 | − | 0.003 | 5.2 × 10−05 | Funzione: fattore di trascrizione per la biosintesi dello sterolo; topo transgenico: cellula grassa anormale e morfologia del cuscinetto di grasso, omeostasi lipidica anormale, resistenza all’insulina, fegato ingrossato[41- 44]. |
Annotazione funzionale e regolamentare dei risultati dell’IMC EWAS
Analisi del percorso di ontologia genica
Le analisi GO del processo biologico, della funzione molecolare e dei percorsi dei componenti cellulari dei 55 geni unici annotati ai 83 CpG replicati (dieci CpG sono stati annotati ai geni annotati ad altri CpG replicati, e 18 CpG erano intergenici) non hanno identificato alcun percorso statisticamente significativo dopo l’aggiustamento per test multipli. In secondo luogo, al fine di perfezionare ulteriormente la selezione dei geni per le analisi GO ai geni che hanno dimostrato un’espressione alterata, abbiamo limitato le analisi GO ai dieci geni unici per i quali la variazione nell’espressione è stata associata alla metilazione differenziale, come descritto nella sezione precedente. Abbiamo identificato una significativa sovrarappresentazione di un percorso di processo biologico nella regolazione positiva dei processi metabolici lipidici (GO:0045834; valore pcorretto = 0.002; arricchimento 64 volte; quattro geni sovrapposti[ABCG1, SREBF1, CPT1A e NOD2] di 130 geni totali nel percorso) e due processi correlati (regolazione positiva del colesterolo biosintetico [GO:0045542] e dei processi metabolici del colesterolo [GO:0090205]; valorep regolato = 0,02-0,03).
Annotazione regolamentare dei CpG associati all’espressione genica nel sangue
La maggior parte dei CpG legati all’IMC associati all’espressione genica alterata si trovavano entro 50 kb dal sito di inizio della trascrizione ed erano all’interno di enhancer o DHS conosciuti(S8 e S9 Figs). I CpG associati al BMI erano più probabili negli stimolatori e DHS ( valore p diarricchimento p = 4,5 × 10-7 e 9,4 × 10-4, rispettivamente) e meno probabili di risiedere nelle isole CpG (valore p di esaurimento p = 3,2 × 10-11) rispetto all’insieme completo dei CpG misurati sul microarray (tabella S8).
DNase I test di sito ipersensibile di tutti i CpG identificati
I test di arricchimento DHS specifici per i tessuti e le cellule utilizzando lo strumento eFORGE v1.2 hanno dimostrato che i CpG correlati all’IMC sono arricchiti in DHS in quasi tutti i tessuti e i tipi di cellule analizzati nei set di dati del progetto ENCODE, BLUEPRINT Epigenome e Roadmap Epigenomics(S10 e S11 Figs), sostenendo così l’idea che i CpG identificati nel sangue si trovano anche in regioni regolatorie note e attive non solo nel sangue, ma anche in altri tessuti metabolicamente attivi. Un’ulteriore stratificazione in base al fatto che le CpG legate all’IMC avevano una sovrapposizione dell’istonio metilazione H3 ha rivelato che le CpG legate all’IMC si sovrapponevano prevalentemente a regioni con la mono metilazione e, in misura minore, la trimetilazione della lisina 4 sull’istone H3K4 (H3K4me1 e H3K4me3) attraverso numerosi tessuti dei dati consolidati del Progetto Roadmap Epigenomics(S12- S14Figs ). I marcatori H3K4me1 sono indicativi degli esaltatori, i marcatori H3K4me3 sono indicativi dei promotori, ed entrambi sono noti marcatori di attivazione trascrizionale.
Analisi genetiche strumentali variabili (randomizzazione mendeliana)
Successive analisi genetiche IV sono state condotte per dedurre le relazioni causali tra metilazione differenziale, espressione genica e IMC, seguite da una valutazione dei cambiamenti epigenetici modellati sui tratti legati all’adiposità utilizzando i risultati del GWAS(Tabella 1).
Randomizzazione mendeliana in avanti
L’analisi dell’associazione causale della metilazione del DNA con l’IMC ha rivelato che la metilazione differenziale a due CpG aveva associazioni causali nominalmente significative(p-valore< 0,05) con l’IMC: (1) cg11024682(SREBF1; cis-meQTL SNP IV rs752579) e (2) cg07730360 (un CpG non annotato sul cromosoma 3q21.3; trans-meQTL SNP IV rs13437553), con valore pcausale = 0,02 e 0,04, rispettivamente(S15 Fig; S9 Table). Portando avanti le due CpG causali in scoperta per la validazione esterna, abbiamo trovato che la metilazione differenziale modellata a uno dei due CpG (cis-meQTL SNP IV rs752579 per la metilazione differenziale a cg11024682[SREBF1]) è stata associata al BMI nei risultati del consorzio GIANT 2015(p = 0,0003; tutte le antenate).
Randomizzazione mendeliana in due fasi (prima fase)
Nel primo passo (metilazione del DNA che colpisce il mediatore, espressione genica), il SNP IV (rs752579) utilizzato nelle analisi MR forward per modellare la metilazione differenziale del locus SREBF1 (cg11024682) è stato anche trovato essere fortemente associato con l’espressione alterata del gene SREBF1 nel sangue nel FHS(p = 3 × 10-12; espressione diminuita in relazione all’allele C), un set di dati pubblicato [45] locus di espressione quantitativa del tratto del sangue (eQTL) (p = 3).2 × 10-6; direzione dell’effetto nel sangue coerente con quella vista nella FHS), e nel fegato (p = 1 ×10-15; nella stessa direzione osservata nel sangue in una rianalisi di 958 campioni [46,47]).
Randomizzazione mendeliana in due fasi (seconda fase)
Nella seconda fase (espressione genica nel sangue e nei tessuti alternativi che influenzano l’IMC), abbiamo identificato adeguati eQTL per l’espressione SREBF1 nel sangue intero (rs1889018; p = 1,7 × 10-15) dalla FHS; nella ghiandola surrenale (rs4925138; p = 1.1 × 10-6) e nel fegato (rs11078366; p = 1,8 × 10-6) dal progetto Genotype-Tissue Expression (GTEx); e nel tessuto adiposo (rs4985779; p = 8,4 × 10-4) dal più grande dataset MuTHER[48]. I SREBF1 eQTL multi-tessuto sono stati selezionati per essere ampiamente indipendenti dal locus di metilazione SREBF1 SNP IV (dettagli nei metodi S1). Abbiamo identificato associazioni significative con il BMI (corretto per i quattro test, p < 0,013) nei risultati del consorzio GIANT per due dei quattro tipi di tessuto; in particolare, il BMI è stato associato al SNP IV per l’espressione SREBF1 nel sangue intero (rs1889018, p = 0.002) e della ghiandola surrenale (rs4925138, p = 0,0098), ma non del fegato (rs11078366, p = 0,89) o del tessuto adiposo (rs4985779, p = 0,80).
Tratti legati all’adiposità nei GWAS
Valutando altre associazioni di malattie cardiometaboliche da GWAS pubblicati, il SNP IV (rs752579) per l’esposizione alla metilazione differenziale al locus SREBF1 (cg11024682) è stato anche trovato per essere associato con (1) tratti legati all’adiposità[49- 51](rapporto vita-fianco aggiustato per BMI [p =2,0 × 10-4], adiponectina[p = 0.007], peso alla nascita[p = 0,046]), (2) tratti del diabete[52-55](diabete di tipo 2 [p =0, 002], insulina a digiuno regolata per l’IMC [p = 0, 001], HbA1C [p = 0, 003], HOMA-B [p =0.007]), (3) livelli di lipidi [56] (trigliceridi [p = 0,001], colesterolo lipoproteico ad alta densità [p = 0,03]) e (4) malattia coronarica[57] (p= 1,7 × 10-6). Inoltre, il SNP IV per l’aumento dell’espressione di SREBF1 nel sangue intero (rs1889018) è stato anche associato al rapporto vita-cintura (p = 0,0002), adiponectina( p = 0,003), e trigliceridi( p = 0,02) sulla base dei risultati GWAS[49,50,56]. Il SNP IV per l’aumento dell’espressione di SREBF1 nella ghiandola surrenale (rs4925138) è stato anche nominalmente associato all’adiponectina (p= 0,02), ai trigliceridi( p = 0,022) e alla modificazione delle lipoproteine a bassa densità in risposta al trattamento con statina(p = 0,04)[50,56,58].
Stime dell’effetto causale
Ogni aumento di SD nella metilazione del DNA al locus SREBF1 (cg11024682) è stato previsto per determinare una diminuzione di 2,8-kg/m2 dell’IMC nell’FHS (modellando l’effetto dell’allele C per rs752579). Al contrario, la relazione osservata tra la metilazione nel sangue e l’IMC nella FHS era nella direzione opposta: un aumento di 1,0-kg/m2 dell’IMC per ogni aumento di SD nella metilazione del DNA a cg11024682. La direzione di effetto prevista tra metilazione e IMC è in parte derivata dalla direzione di effetto osservata tra il SNP IV e la metilazione nel sangue. La letteratura precedente ha riportato QTLs cellulari dipendenti dal tipo di cellula con direzioni opposte di effetto tra un SNP e la metilazione o l’espressione a seconda del tipo di cellula o tessuto esaminato[59]. Poiché non sono disponibili estese banche dati di metilazione trans-tissuale non sono disponibili, abbiamo esaminato gli eQTL trans-tissuale per SREBF1 dal portale GTEx[60]. Una serie di eQTL per SREBF1 (tasso di falsa scoperta ≤ 0,05) dimostrano una direzione opposta dell’effetto tra sangue e ghiandola surrenale (valore p < 10-6) e tessuti aggiuntivi (al valore p <10-5) come il muscolo scheletrico, l’esofago, il tessuto dell’aorta e il nervo tibiale (http://www.gtexportal.org/home/bubbleHeatmapPage/SREBF1). Forte eQTLs per SREBF1 sono probabilmente presenti nel tessuto surrenale come SREBF1 è altamente espresso nella ghiandola surrenale rispetto ad altri tessuti(http://www.proteinatlas.org/ENSG00000072310-SREBF1/tissue). Ad esempio, rs854764 è un eQTL forte per SREBF1 sia nel sangue che nel tessuto surrenale ma in direzioni opposte(p = 3,8 × 10-12 e p = 4 × 10-6, rispettivamente, nel catalogo GTEx) ed è associato al BMI in GIANT (p = 0.001) e al rapporto vita-fianchi(p = 9,2 × 10-4), adiponectina(p = 0,02),HbA1C (p = 0,02), diabete di tipo 2 (p = 0,03), trigliceridi (p = 0,04)e malattie coronariche (p = 1,1× 10-5) nei risultati GWAS [4,7,50,52,54,57,61]. Questo SNP, rs854764, è anche un meQTL per SREBF1 locus metilazione a cg11024682 nel FHS(p = 2,8 × 10-18), ma l’associazione con SREBF1 locus metilazione nella ghiandola surrenale, il tessuto potenziale di effetto, è sconosciuta. Si veda la tabella S10 per le stime dell’effetto causale e gli intervalli di confidenza per la seconda fase delle analisi MR a due fasi.
Randomizzazione mendeliana inversa
Per verificare se l’IMC influisce sulla metilazione ai CpG identificati, è stato utilizzato come IV per l’IMC il punteggio di rischio genetico ponderato dell’additivo di 97 SNPs IMC noti[7](F-test statistic = 26). Sedici CpGs sono stati trovati per essere differenzialmente metilato come conseguenza di BMI utilizzando un valorenominale causale p-valore< 0,05 cutoff (elenco completo nella tabella S11 ). I 16 CpG a valle sono stati annotati a 12 geni(ABCG1, USP22, DPF1, RARA, KDM2B, KANK2, RALB, NT5DC2, DENND4B, B3GNT7, DKK4 e ABAT). Un’analisi di sensibilità che utilizza un singolo SNP nel locus FTO come BMI IV(Tabella S12) ha supportato ulteriormente le associazioni causali a valle del BMI a due dei 16 CpG ( valorenominale pcausale < 0,05 per cg06500161 e cg04286697, rispettivamente ai loci ABCG1 e B3GNT7). I geni annotati con metilazione differenziale correlata al BMI sono caratterizzati in Fig 3.
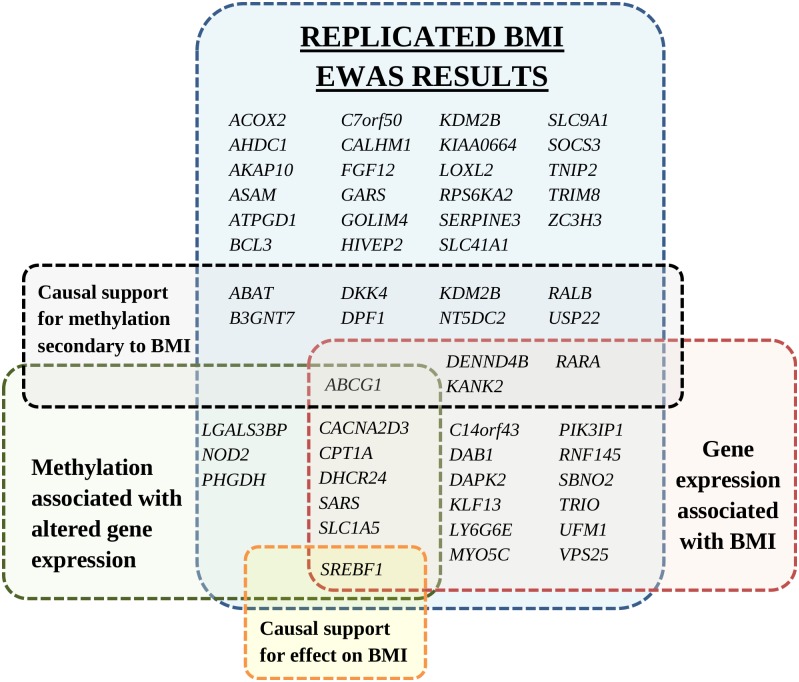
Fig 3.I geni annotati di CpG replicati con metilazione differenziale replicata e metilati identificati nello studio di associazione a livello di epigenoma BMI.I geni sono raggruppati per associazione con l’espressione genica, l’associazione dell’espressione genica con l’IMC e le analisi di randomizzazione mendeliana per il supporto causale. I nomi dei geni duplicati all’interno dello stesso gruppo non sono mostrati. La figura non include 18 CpG intergenici senza un’annotazione genica. BMI, indice di massa corporea; EWAS, studio di associazione a livello di epigenoma.
Fig 3.Fig. 3. Geni annotati di CpG replicati differenzialmente metilati identificati nello studio di associazione a livello di epigenome-wide BMI.I geni sono raggruppati per associazione con l’espressione genica, l’associazione dell’espressione genica con l’IMC e le analisi di randomizzazione mendeliana per il supporto causale. I nomi dei geni duplicati all’interno dello stesso gruppo non sono mostrati. La figura non include 18 CpG intergenici senza un’annotazione genica. BMI, indice di massa corporea; EWAS, studio di associazione a livello di epigenoma.
Discussione
In questa analisi dell’associazione dell’IMC con la metilazione differenziale del DNA derivato dal sangue, forniamo una solida evidenza di una connessione tra la segnalazione epigenetica replicabile a 83 CpG e l’IMC. Dimostriamo anche la correlazione della metilazione differenziale BMI-correlata con l’espressione alterata di dieci geni nel sangue intero che sono sovrarappresentati nelle vie del metabolismo lipidico. Tra gli 83 CpGs replicati BMI-correlati, un locus differenzialmente metilato (cg11024682) al fattore di trascrizione del metabolismo lipidico SREBF1 ha dimostrato la prova di un effetto causale sul BMI, l’esposizione geneticamente prevista alla metilazione differenziale e l’espressione di SREBF1 è stato trovato per essere associato con BMI e altri tratti di adiposità, tratti glicemici, dislipidemia, e malattia coronarica. Al contrario, abbiamo trovato che una parte sostanziale (16 su 83 [19%]) dei CpG differenzialmente metilati legati al BMI identificati in questo EWAS sono probabilmente una conseguenza del BMI (cioè, segnali a valle).
La variazione dell’IMC è riflessa nelle firme di metilazione del DNA nel sangue
Una parte sostanziale (~ 18%) della variazione interindividuale dell’IMC viene catturata dai CpG replicati con metilato differenziale nel sangue. L’entità della differenza di IMC (~12 kg/m2 tra il decile più alto e quello più basso) equivale a rischi sostanziali per la salute; per esempio, ogni aumento di 5-kg/m2 di IMC nella popolazione generale è associato ad un aumento del 30% della mortalità[62].
I nostri risultati suggeriscono che i biomarcatori epigenetici hanno il potenziale per migliorare la previsione del rischio e aiutare a personalizzare le scelte terapeutiche per prevenire o trattare le malattie cardiometaboliche. Ad esempio, a livello di popolazione, l’IMC è una misura efficace del rischio medio futuro di malattia cardiometabolica[63], ma non è sufficientemente predittivo a livello individuale. Indipendentemente dalla causalità, i biomarcatori ematici possono essere utili a fini prognostici o diagnostici. Sono necessarie ulteriori ricerche per determinare se perfezionare il rischio legato all’IMC incorporando biomarcatori epigenetici può migliorare la previsione del rischio e aiutare a guidare le decisioni di trattamento.
La metilazione differenziale è identificata nei loci noti per essere coinvolti nell’adiposità
Metabolismo lipidico
Esperimenti condotti in precedenza sostengono un ruolo causale del SREBF1 nell’adiposità[64]. SREBF1 (noto anche come SREBP1) svolge un ruolo centrale nell’omeostasi energetica promuovendo la glicolisi, la lipogenesi e l’adipogenesi attraverso l’induzione della conversione dell’acetilcoA in trigliceridi(S16 Fig). SREBF1 promuove la conversione degli acidi grassi liberi in trigliceridi nel fegato e in lipoproteine ricche di trigliceridi nel sangue. In situazioni di eccesso calorico, SREBF1 è un mediatore chiave dell’induzione della lipogenesi nell’uomo[64]. Nei topi con insulino-resistenza indotta dalla dieta, l’inibizione del SREBF1 attenua l’aterosclerosi accelerata, sostenendo un collegamento con l’aterosclerosi e la malattia coronarica[65]. La connessione causale tra l’aumento delle lipoproteine ricche di trigliceridi e la malattia coronarica è supportata da studi genetici umani[66]. Evidenziamo il ruolo potenziale dell’espressione SREBF1 nella ghiandola surrenale nella regolazione del peso e nelle malattie correlate alle adiposità sulla base dei risultati delle analisi MR. Le malattie della ghiandola surrenale sono note per essere legate all’obesità grave, e l’adrenalectomia in modelli murini può invertire l’obesità indotta geneticamente[67,68]. I nostri risultati suggeriscono che l’alterazione della regolazione genomica del SREBF1 è causalmente correlata al BMI; tuttavia, la mancanza di grandi serie di dati di meQTL in numerosi tessuti e in varie condizioni, in combinazione con l’incapacità di condurre l’editing epigenetico mirato ai tessuti nei modelli sperimentali pertinenti, limita la nostra capacità di fare una definitiva inferenza causale. La regolazione del SREBF1 è un obiettivo poco esplorato per la prevenzione delle malattie coronariche.
Un altro dei nostri geni principali, CPT1A(carnitina palmitoyltransferase 1A), è un enzima esterno di membrana mitocondriale coinvolto nell’utilizzo di acetilcoA, che funziona come un enzima chiave nella beta-ossidazione degli acidi grassi a catena lunga nel metabolismo energetico mitocondriale. L’acetilcoA è stato recentemente identificato come un legame centrale tra la lipolisi alterata a causa di adiposità o infiammazione e i conseguenti cambiamenti nella resistenza all’insulina epatica con la comunicazione incrociata tra fegato e tessuto adiposo[69]. L’ABCG1(ATP binding cassette G1), un trasportatore di lipidi a membrana cellulare, ha un ruolo consolidato nel trasporto del colesterolo inverso; il suo ruolo nell’obesità è sostenuto in precedenti studi su animali[37] e sull’uomo[70]. DHCR24(24-deidrocolesterolo reduttasi) catalizza la riduzione degli intermedi dello sterolo durante la sintesi del colesterolo. La metilazione differenziale di SREBF1, CPT1A, ABCG1 e DHCR24 è stata riportata in precedenti EWAS di adiposità, tratti glicemici e lipidi[29- 31,71- 76]. Aggiungiamo alla letteratura pubblicata e forniamo la prova che la metilazione differenziale al locus ABCG1 è probabilmente un effetto a valle del BMI. Da questi risultati presi insieme, la disregolazione epigenetica sta emergendo come un collegamento comune tra l’obesità e le comorbidità correlate all’obesità. Anche se sono necessarie ulteriori ricerche funzionali, si ipotizza che l’obesità e le malattie correlate all’obesità siano in parte causate da cambiamenti nella metilazione del DNA, con conseguente disregolazione del bilancio energetico attraverso effetti sull’espressione dei geni del metabolismo lipidico. I meccanismi di regolazione coinvolti nell’omeostasi energetica sono stati proposti come obiettivi interessanti per il trattamento dell’obesità, della sindrome metabolica e delle malattie cardiache[77,78]. I nostri risultati dimostrano che queste connessioni sono evidenti negli esseri umani, aggiungendo alle precedenti prove di modelli animali[78].
Vie infiammatorie
Oltre ai geni del metabolismo lipidico, il nostro EWAS ha identificato una serie di loci coinvolti in percorsi infiammatori. Gli adipociti allargati negli individui obesi sono noti per promuovere l’infiammazione. Un BMI-correlato CpG differenzialmente metilato è stato identificato al locus NOD2(nucleotide binding oligomerizzazione dominio 2). NOD2, un recettore immunitario innato, è coinvolto nella risposta immunitaria ai lipopolisaccaridi batterici (LPS) attivando la segnalazione NF-κB. L’assorbimento di LPS dal microbiota intestinale ha dimostrato di provocare una maggiore internalizzazione delle lipoproteine ricche di LPS in adipociti e promuovere la conversione dei macrofagi dalla forma M2 alla forma infiammatoria M1[79]. NOD2 è anche incluso nel percorso GO per la regolazione del metabolismo lipidico (0045834) in quanto è un regolatore positivo dell’attività fosfatidilinositolo 3-chinasi ed è stato dimostrato di promuovere l’infiammazione vascolare e la formazione di lesioni aterosclerotiche ricche di lipidi in topi ipercolesterolemici LDLR-/-[ 80,81]. NOD2 interagisce con un altro BMI correlato con un altro locus genetico infiammatorio differenzialmente metilato a SOCS3(soppressore della segnalazione delle citochine 3), un regolatore negativo della segnalazione delle citochine. Inoltre, LGALS3BP(lectina, galattoside che lega la proteina 3 solubile legante 3), nota anche come MAC2BP(Mac-2-binding protein), è coinvolto nella risposta immunitaria associata alla citotossicità delle cellule killer attivate dalla linfocina e l’attivazione, la segnalazione e l’aggregazione delle piastrine. LGALS3BP è stato trovato per stimolare le difese dell’ospite ed è elevato in individui con vari tipi di cancro come il cancro al seno, ai polmoni, al colon-retto, alle ovaie e all’endometrio, molti dei quali sono correlati all’obesità. Inoltre, LGALS3BP è stato recentemente identificato come un promettente biomarcatore per la steatoepatite non alcolica e la pancreatite[82,83], malattie note legate all’obesità. La metilazione al locus LGALS3BP ha dimostrato una significativa interazione sessuale, con un effetto più forte negli uomini. Ciò può essere correlato a fattori ambientali più comuni negli uomini (come modelli dietetici specifici) o alla fisiologia maschile specifica.
La metilazione differenziale è identificata nei loci non precedentemente collegati all’adiposità
Metabolismo serinico
Due dei dieci geni espressi in modo differenziato in associazione con la metilazione legata all’IMC(PHGDH e SARS) sono coinvolti nel metabolismo della L-serina. PHGDH(fosfoglicerato deidrogenasi) è coinvolto nelle prime fasi della sintesi dell’aminoacido L-serina, che svolge un ruolo nell’ossidoreduttasi come accettore NADP nel ciclo dell’acido tricarbossilico. La SARS(sieril-TRNA sintetasi) catalizza il trasferimento della L-serina al tRNA. Inoltre, RPS6KA2(proteina ribosomica S6 chinasi A2), un locus non precedentemente segnalato come BMI-correlato, è un serino/treonina chinasi che agisce a valle della segnalazione MAPK ed è coinvolto nella proliferazione cellulare. L-serina è necessaria per funzioni specifiche nel sistema nervoso centrale, tuttavia, il legame tra l’adiposità e le conseguenze funzionali per la salute attraverso gli effetti sul metabolismo della serina è attualmente sconosciuto.
Cell-membrana trasportatori
Oltre ai trasportatori di cellule-membrane discussi, sono stati identificati due ulteriori trasportatori a membrana tra i dieci geni associati alla metilazione differenziale. SLC1A5(soluto vettore famiglia 1 membro 5), che è stato trovato per avere significative associazioni a tre vie con l’espressione genica alterata nel sangue e BMI, è un trasportatore sodio-dipendente di aminoacidi. È attivato dalla concentrazione di insulina, che è spesso elevata negli individui con obesità. La metilazione differenziale legata al BMI è stata identificata anche al locus CACNA2D3(canale del calcio, dipendente dalla tensione, alfa 2 / delta subunità 3). CACNA2D3 è coinvolto nella trasmissione del segnale nervoso e la conduzione cardiaca.
Risultati BMI EWAS nel contesto degli studi pubblicati di epidemiologia epigenetica
In precedenza nei metodi in silico per identificare i geni dell’obesità presunta regolata epigeneticamente hanno evidenziato SOCS3(soppressore della segnalazione delle citochine 3) e RARA(recettore dell’acido retinoico alfa)[84], entrambi identificati nella meta-analisi FHS-LBC(p = 2,7 × 10-11 per cg27637521 in SOCS3 e p = 1,3 × 10-8 per cg13274938 in RARA). Uno studio di associazione della metilazione del DNA e del BMI in 459 individui del Cardiogenics Consortium ha identificato un’associazione di metilazione a tre CpG intronic a HIF3A(fattore ipossia inducibile 3A) nel sangue e nelle cellule adipose con BMI[28]. Nel nostro studio abbiamo trovato modeste associazioni di metilazione differenziale e di espressione al locus HIF3A con BMI. Tuttavia, le associazioni erano più forti in individui più giovani nella FHS, suggerendo che la connessione può essere meno evidente in età più avanzata.
Ad un valorenominale causale p-valore< 0,05, abbiamo trovato che molti (16 [19%]) dei CpG replicati sono a valle del BMI. Questo è coerente con i recenti risultati dei dati di metilazione longitudinale e MR bidirezionale nello studio Avon Longitudinal Study of Parents and Children[85] che la metilazione HIF3A legata al BMI è probabilmente secondaria alle differenze di BMI.
C’è una sostanziale sovrapposizione tra i CpG correlati all’IMC identificati e le associazioni di CpG-metabolite nel sangue segnalate da 1.814 partecipanti alla coorte KORA (Kooperative Gesundheitsforschung in der Region Augsburg) [86](TabellaS13). In particolare, sono stati identificati ceramidi e sfingolipidi – noti per avere livelli alterati tra gli individui obesi e implicati nello sviluppo della sindrome metabolica [87-89]. Inoltre, il BMI-correlato CpG differenzialmente metilato CpG (cg03725309) presso il locus SARS, come discusso in precedenza nella sezione del metabolismo della serina, è stato trovato per essere associato con i livelli ematici di serina.
Risultati BMI EWAS nel contesto dei risultati BMI GWAS e varianti genetiche vicine
Da notare che nessuno dei CpG associati all’IMC era vicino ai geni precedentemente identificati nei GWAS dei tratti legati all’obesità, come FTO(massa grassa e obesità associata) o MC4R(recettore della melanocortina 4). Ipotizziamo che molti dei loci replicati differenzialmente metilati riflettono nuovi percorsi coinvolti nella regolazione delle adiposità o malattie correlate all’adiposità. Tuttavia, possono esistere interazioni a lungo raggio della metilazione del DNA con loci noti legati all’obesità[90]. È inoltre necessario un ulteriore lavoro per comprendere il ruolo dei nuovi loci in relazione all’adiposità. Inoltre, la combinazione di informazioni provenienti dalla metilazione del DNA con marcatori genetici identificati dalla variazione della sequenza del DNA può consentire miglioramenti nella previsione del rischio precedentemente non possibili con le sole varianti della sequenza[91].
Molti dei loci significativi della fase di scoperta (73 su 135) sono stati replicati negli afroamericani dallo studio ARIC[30]. Allo stesso modo, molti dei CpG a metile differenziato legati all’IMC identificati in questo studio sono stati riportati anche in relazione all’IMC in persone di origine araba[34]. Nei GWAS, la mancata replicazione tra gruppi razziali/etnici può essere dovuta a differenze nelle frequenze alleliche e nei modelli di disequilibrio di collegamento. Al contrario, l’alto tasso di replicazione dei risultati di replicazione della metilazione del DNA per l’IMC in individui di origine europea e africana e di altre origini suggerisce che le esposizioni ambientali condivise o i cambiamenti secondari alle differenze nell’IMC, e non la variazione genetica, possono essere alla base di molte delle associazioni. È necessario un ulteriore lavoro per identificare i fattori ambientali che promuovono o mitigano la disregolazione epigenetica correlata all’obesità. Le nostre analisi che hanno condizionato i meQTL superiori hanno mostrato un’attenuazione minima, suggerendo che l’associazione tra metilazione differenziale e IMC è in gran parte indipendente dalle varianti genetiche vicino ai CpG segnalati.
Limitazioni dello studio
Il nostro studio ha diversi limiti. I risultati delle analisi di risonanza magnetica che utilizzano la metilazione e i livelli di espressione geneticamente previsti non provano la causalità, ma forniscono una prova di supporto. I risultati delle analisi MR si basano su numerose ipotesi, ad esempio, che non esistono vie alternative attraverso le quali il SNP IV possa agire sul BMI (cioè gli effetti pleiotropici). Le ipotesi di MR non possono essere testate direttamente e possono influenzare i risultati. I risultati di MR in avanti non hanno raggiunto soglie di significatività Bonferroni-regolate per i test multipli; tuttavia, la convalida dei risultati nominalmente significativi nel più grande consorzio GIANT supporta i nostri risultati. Abbiamo evitato l’uso di multi-SNP score IVs, in quanto avevamo già identificato adeguati singoli SNP meQTLs e l’uso di multi-SNP score IVs avrebbe ulteriormente rischiato di introdurre bias a causa della pleiotropia. I meQTL per le analisi MR sono stati ricavati nella FHS e il risultato è stato testato all’interno della stessa coorte, il che può potenzialmente determinare un bias verso la significatività. Le analisi MR, utilizzando il meQTL IV del sangue, suggeriscono una relazione inversa tra la metilazione prevista del locus SREBF1 e l’IMC, l’inverso della relazione osservata, che può essere interpretata come un risultato nullo. Questo risultato è potenzialmente spiegato da diverse direzioni di effetto dei QTL in tessuti alternativi, che è stato supportato dall’esame dell’associazione di varianti genetiche nel sangue rispetto ad altri tessuti metabolicamente attivi nella risorsa del Progetto GTEx. Purtroppo, ci sono limitate serie di dati di meQTLs in vari tessuti per esplorare ulteriormente questo aspetto. L’osservazione di associazioni di IMC con metilazione alla stessa CpG in diverse direzioni di effetto nel sangue rispetto al DNA derivato dalle adiposità è stata precedentemente riportata in siti CpG correlati all’IMC[30]. Per SREBF1, presumiamo che le conseguenze metaboliche della metilazione alterata e l’effetto sul BMI si verifichino in tessuti diversi dal sangue, come la ghiandola surrenale, con le variazioni di metilazione nel sangue che siamo stati in grado di rilevare rappresentando un biomarcatore della metilazione differenziale transessuale[92]. Inoltre, è possibile che gli anelli di feedback positivi e negativi possano portare alla regolazione dello stesso gene ad essere sia un effetto causale che un effetto a valle dell’adiposità. Non saremmo in grado di discernere questo scenario dai dati osservazionali trasversali di questo studio.
Un test di metilazione alternativo sarebbe necessario per scopi clinici in quanto gli attuali microarray non sono adatti in un ambiente clinico. La ricerca futura sarebbe necessaria per la validazione tecnica a fini clinici. Il nostro studio supporta le cellule del sangue come un utile tessuto accessibile per la scoperta di biomarcatori epigenetici in studi su grandi popolazioni. Tuttavia, il nostro studio non sarebbe in grado di rilevare i cambiamenti di metilazione specifici del tessuto che si verificano nelle linee cellulari non sanguigne (ad esempio, modifiche epigenetiche neurone-specifiche in relazione all’IMC). Molti dei nostri migliori CpG replicati nello studio GOLDN, che ha valutato la metilazione del DNA in un singolo tipo di cellule del sangue (CD4+), suggerendo che le associazioni che abbiamo rilevato non sono probabilmente dovute a confondere per eterogeneità delle cellule del sangue. Molti dei geni associati alla metilazione differenziale legata al BMI erano noti per avere un ruolo nell’adiposità e nei tratti cardiometabolici dei modelli murini knockout; tuttavia, l’universo dei modelli knockout è probabilmente arricchito per lo studio dell’adiposità e dei tratti cardiometabolici, e non abbiamo potuto verificare direttamente se i nostri risultati hanno identificato più del previsto. Il nostro studio è stato condotto tra gli adulti in età avanzata, e i risultati potrebbero non essere generalizzabili alle età più giovani.
Conclusioni
Forniamo i risultati di un grande EWAS di IMC in quasi 8.000 individui che hanno identificato 83 loci replicabili di metilazione del DNA e prove di differenze trascriptomiche complementari che sono state arricchite per i prodotti genici coinvolti nel metabolismo lipidico. Le analisi genetiche IV danno priorità al locus SREBF1 per futuri studi funzionali per definire ulteriormente la relazione causale con l’adiposità, la resistenza all’insulina, le dislipidemie correlate all’obesità e le malattie coronariche. I nostri risultati forniscono una base per ulteriori ricerche per determinare se i profili epigenetici individualizzati possono essere utilizzati per guidare il processo decisionale clinico e migliorare i risultati di salute. I nostri risultati possono avere un’ulteriore rilevanza clinica e terapeutica se altri loci che sono differenziatamente metilati in relazione all’IMC rappresentano obiettivi interessanti per il trattamento o la prevenzione dell’obesità e delle malattie correlate all’adiposità.
Informazioni di supporto
References
- Ogden CL, Carroll MD, Kit BK, Flegal KM. Prevalence of childhood and adult obesity in the United States, 2011–2012. JAMA. 2014; 311(8):806-14. DOI | PubMed
- Masters RK, Reither EN, Powers DA, Yang YC, Burger AE, Link BG. The impact of obesity on US mortality levels: the importance of age and cohort factors in population estimates. Am J Public Health. 2013; 103(10):1895-901. DOI | PubMed
- Lu Y, Hajifathalian K, Ezzati M, Woodward M, Rimm EB, Danaei G. Metabolic mediators of the effects of body-mass index, overweight, and obesity on coronary heart disease and stroke: a pooled analysis of 97 prospective cohorts with 1.8 million participants. Lancet. 2014; 383(9921):970-83. DOI | PubMed
- Berndt SI, Gustafsson S, Magi R, Ganna A, Wheeler E, Feitosa MF. Genome-wide meta-analysis identifies 11 new loci for anthropometric traits and provides insights into genetic architecture. Nat Genet. 2013; 45(5):501-12. DOI | PubMed
- Speliotes EK, Willer CJ, Berndt SI, Monda KL, Thorleifsson G, Jackson AU. Association analyses of 249,796 individuals reveal 18 new loci associated with body mass index. Nat Genet. 2010; 42(11):937-48. DOI | PubMed
- van Vliet-Ostaptchouk JV, Snieder H, Lagou V. Gene-lifestyle interactions in obesity. Curr Nutr Rep. 2012; 1:184-96. DOI | PubMed
- Locke AE, Kahali B, Berndt SI, Justice AE, Pers TH, Day FR. Genetic studies of body mass index yield new insights for obesity biology. Nature. 2015; 518(7538):197-206. DOI | PubMed
- Relton CL, Davey Smith G. Epigenetic epidemiology of common complex disease: prospects for prediction, prevention, and treatment. PLoS Med. 2010; 7(10):e1000356. DOI | PubMed
- van Dijk SJ, Molloy PL, Varinli H, Morrison JL, Muhlhausler BS. Epigenetics and human obesity. Int J Obes (Lond). 2014; 39(1):85-97. DOI | PubMed
- Bray MS, Loos RJ, McCaffery JM, Ling C, Franks PW, Weinstock GM. NIH working group report-using genomic information to guide weight management: From universal to precision treatment. Obesity (Silver Spring). 2016; 24(1):14-22. DOI | PubMed
- Relton CL, Davey Smith G. Mendelian randomization: applications and limitations in epigenetic studies. Epigenomics. 2015; 7(8):1239-43. DOI | PubMed
- Relton CL, Davey Smith G. Two-step epigenetic Mendelian randomization: a strategy for establishing the causal role of epigenetic processes in pathways to disease. Int J Epidemiol. 2012; 41(1):161-76. DOI | PubMed
- Burgess S, Small DS, Thompson SG. A review of instrumental variable estimators for Mendelian randomization. Stat Methods Med Res. 2015. DOI | PubMed
- Kannel WB, Feinleib M, McNamara PM, Garrison RJ, Castelli WP. An investigation of coronary heart disease in families. The Framingham offspring study. Am J Epidemiol. 1979; 110(3):281-90. PubMed
- Deary IJ, Whiteman MC, Starr JM, Whalley LJ, Fox HC. The impact of childhood intelligence on later life: following up the Scottish mental surveys of 1932 and 1947. J Pers Soc Psychol. 2004; 86(1):130-47. DOI | PubMed
- Deary IJ, Gow AJ, Taylor MD, Corley J, Brett C, Wilson V. The Lothian Birth Cohort 1936: a study to examine influences on cognitive ageing from age 11 to age 70 and beyond. BMC Geriatr. 2007; 7:28. DOI | PubMed
- Deary IJ, Gow AJ, Pattie A, Starr JM. Cohort profile: the Lothian Birth Cohorts of 1921 and 1936. Int J Epidemiol. 2012; 41(6):1576-84. DOI | PubMed
- Dawber TR, Meadors GF, Moore FE. Epidemiological approaches to heart disease: the Framingham Study. Am J Public Health Nations Health. 1951; 41(3):279-81. PubMed
- Bibikova M, Barnes B, Tsan C, Ho V, Klotzle B, Le JM. High density DNA methylation array with single CpG site resolution. Genomics. 2011; 98(4):288-95. DOI | PubMed
- Zhang X, Johnson AD, Hendricks AE, Hwang SJ, Tanriverdi K, Ganesh SK. Genetic associations with expression for genes implicated in GWAS studies for atherosclerotic cardiovascular disease and blood phenotypes. Hum Mol Genet. 2014; 23(3):782-95. DOI | PubMed
- Leek JT, Johnson WE, Parker HS, Jaffe AE, Storey JD. The sva package for removing batch effects and other unwanted variation in high-throughput experiments. Bioinformatics. 2012; 28(6):882-3. DOI | PubMed
- Zaykin DV. Optimally weighted Z-test is a powerful method for combining probabilities in meta-analysis. J Evol Biol. 2011; 24(8):1836-41. DOI | PubMed
- Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large-scale gene function analysis with the PANTHER classification system. Nat Protoc. 2013; 8(8):1551-66. DOI | PubMed
- Breeze CE, Paul DS, van Dongen J, Butcher LM, Ambrose JC, Barrett JE. eFORGE: a tool for identifying cell type-specific signal in epigenomic data. Cell Rep. 2016; 17(8):2137-50. DOI | PubMed
- Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A. Integrative analysis of 111 reference human epigenomes. Nature. 2015; 518(7539):317-30. DOI | PubMed
- Adams D, Altucci L, Antonarakis SE, Ballesteros J, Beck S, Bird A. BLUEPRINT to decode the epigenetic signature written in blood. Nat Biotechnol. 2012; 30(3):224-6. DOI | PubMed
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489(7414):57-74. DOI | PubMed
- Dick KJ, Nelson CP, Tsaprouni L, Sandling JK, Aissi D, Wahl S. DNA methylation and body-mass index: a genome-wide analysis. Lancet. 2014; 383(9933):1990-8. DOI | PubMed
- Aslibekyan S, Demerath EW, Mendelson M, Zhi D, Guan W, Liang L. Epigenome-wide study identifies novel methylation loci associated with body mass index and waist circumference. Obesity (Silver Spring). 2015; 23(7):1493-501. DOI | PubMed
- Demerath EW, Guan W, Grove ML, Aslibekyan S, Mendelson M, Zhou YH. Epigenome-wide association study (EWAS) of BMI, BMI change and waist circumference in African American adults identifies multiple replicated loci. Hum Mol Genet. 2015; 24(15):4464-79. DOI | PubMed
- Ding J, Reynolds LM, Zeller T, Muller C, Lohman K, Nicklas BJ. Alterations of a cellular cholesterol metabolism network are a molecular feature of obesity-related type 2 diabetes and cardiovascular disease. Diabetes. 2015; 64(10):3464-74. DOI | PubMed
- Huang RC, Garratt ES, Pan H, Wu Y, Davis EA, Barton SJ. Genome-wide methylation analysis identifies differentially methylated CpG loci associated with severe obesity in childhood. Epigenetics. 2015; 10(11):995-1005. DOI | PubMed
- Ronn T, Volkov P, Gillberg L, Kokosar M, Perfilyev A, Jacobsen AL. Impact of age, BMI and HbA1c levels on the genome-wide DNA methylation and mRNA expression patterns in human adipose tissue and identification of epigenetic biomarkers in blood. Hum Mol Genet. 2015; 24(13):3792-813. DOI | PubMed
- Al Muftah WA, Al-Shafai M, Zaghlool SB, Visconti A, Tsai PC, Kumar P. Epigenetic associations of type 2 diabetes and BMI in an Arab population. Clin Epigenetics. 2016; 8:13. DOI | PubMed
- Mansego ML, Milagro FI, Zulet MA, Moreno-Aliaga MJ, Martinez JA. Differential DNA methylation in relation to age and health risks of obesity. Int J Mol Sci. 2015; 16(8):16816-32. DOI | PubMed
- Ali O, Cerjak D, Kent JW, James R, Blangero J, Carless MA. Methylation of SOCS3 is inversely associated with metabolic syndrome in an epigenome-wide association study of obesity. Epigenetics. 2016. DOI | PubMed
- Buchmann J, Meyer C, Neschen S, Augustin R, Schmolz K, Kluge R. Ablation of the cholesterol transporter adenosine triphosphate-binding cassette transporter G1 reduces adipose cell size and protects against diet-induced obesity. Endocrinology. 2007; 148(4):1561-73. DOI | PubMed
- Skarnes WC, Rosen B, West AP, Koutsourakis M, Bushell W, Iyer V. A conditional knockout resource for the genome-wide study of mouse gene function. Nature. 2011; 474(7351):337-42. DOI | PubMed
- Nyman LR, Cox KB, Hoppel CL, Kerner J, Barnoski BL, Hamm DA. Homozygous carnitine palmitoyltransferase 1a (liver isoform) deficiency is lethal in the mouse. Mol Genet Metab. 2005; 86(1–2):179-87. DOI | PubMed
- Wechsler A, Brafman A, Shafir M, Heverin M, Gottlieb H, Damari G. Generation of viable cholesterol-free mice. Science. 2003; 302(5653):2087. DOI | PubMed
- Shimano H, Horton JD, Hammer RE, Shimomura I, Brown MS, Goldstein JL. Overproduction of cholesterol and fatty acids causes massive liver enlargement in transgenic mice expressing truncated SREBP-1a. J Clin Invest. 1996; 98(7):1575-84. DOI | PubMed
- Kim S, Huang LW, Snow KJ, Ablamunits V, Hasham MG, Young TH. A mouse model of conditional lipodystrophy. Proc Natl Acad Sci U S A. 2007; 104(42):16627-32. DOI | PubMed
- Shimomura I, Hammer RE, Richardson JA, Ikemoto S, Bashmakov Y, Goldstein JL. Insulin resistance and diabetes mellitus in transgenic mice expressing nuclear SREBP-1c in adipose tissue: model for congenital generalized lipodystrophy. Genes Dev. 1998; 12(20):3182-94. PubMed
- Takahashi A, Shimano H, Nakagawa Y, Yamamoto T, Motomura K, Matsuzaka T. Transgenic mice overexpressing SREBP-1a under the control of the PEPCK promoter exhibit insulin resistance, but not diabetes. Biochim Biophys Acta. 2005; 1740(3):427-33. DOI | PubMed
- Fehrmann RS, Jansen RC, Veldink JH, Westra HJ, Arends D, Bonder MJ. Trans-eQTLs reveal that independent genetic variants associated with a complex phenotype converge on intermediate genes, with a major role for the HLA. PLoS Genet. 2011; 7(8):e1002197. DOI | PubMed
- Schadt EE, Molony C, Chudin E, Hao K, Yang X, Lum PY. Mapping the genetic architecture of gene expression in human liver. PLoS Biol. 2008; 6(5):e107. DOI | PubMed
- Greenawalt DM, Dobrin R, Chudin E, Hatoum IJ, Suver C, Beaulaurier J. A survey of the genetics of stomach, liver, and adipose gene expression from a morbidly obese cohort. Genome Res. 2011; 21(7):1008-16. DOI | PubMed
- Grundberg E, Meduri E, Sandling JK, Hedman AK, Keildson S, Buil A. Global analysis of DNA methylation variation in adipose tissue from twins reveals links to disease-associated variants in distal regulatory elements. Am J Hum Genet. 2013; 93(5):876-90. DOI | PubMed
- Heid IM, Jackson AU, Randall JC, Winkler TW, Qi L, Steinthorsdottir V. Meta-analysis identifies 13 new loci associated with waist-hip ratio and reveals sexual dimorphism in the genetic basis of fat distribution. Nat Genet. 2010; 42(11):949-60. DOI | PubMed
- Dastani Z, Hivert MF, Timpson N, Perry JR, Yuan X, Scott RA. Novel loci for adiponectin levels and their influence on type 2 diabetes and metabolic traits: a multi-ethnic meta-analysis of 45,891 individuals. PLoS Genet. 2012; 8(3):e1002607. DOI | PubMed
- Horikoshi M, Yaghootkar H, Mook-Kanamori DO, Sovio U, Taal HR, Hennig BJ. New loci associated with birth weight identify genetic links between intrauterine growth and adult height and metabolism. Nat Genet. 2013; 45(1):76-82. DOI | PubMed
- Morris AP, Voight BF, Teslovich TM, Ferreira T, Segre AV, Steinthorsdottir V. Large-scale association analysis provides insights into the genetic architecture and pathophysiology of type 2 diabetes. Nat Genet. 2012; 44(9):981-90. DOI | PubMed
- Manning AK, Hivert MF, Scott RA, Grimsby JL, Bouatia-Naji N, Chen H. A genome-wide approach accounting for body mass index identifies genetic variants influencing fasting glycemic traits and insulin resistance. Nat Genet. 2012; 44(6):659-69. DOI | PubMed
- Soranzo N, Sanna S, Wheeler E, Gieger C, Radke D, Dupuis J. Common variants at 10 genomic loci influence hemoglobin A(1)(C) levels via glycemic and nonglycemic pathways. Diabetes. 2010; 59(12):3229-39. DOI | PubMed
- Dupuis J, Langenberg C, Prokopenko I, Saxena R, Soranzo N, Jackson AU. New genetic loci implicated in fasting glucose homeostasis and their impact on type 2 diabetes risk. Nat Genet. 2010; 42(2):105-16. DOI | PubMed
- Teslovich TM, Musunuru K, Smith AV, Edmondson AC, Stylianou IM, Koseki M. Biological, clinical and population relevance of 95 loci for blood lipids. Nature. 2010; 466(7307):707-13. DOI | PubMed
- CARDIoGRAMplusC4D Consortium, Deloukas P, Kanoni S, Willenborg C, Farrall M, Assimes TL. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013; 45(1):25-33. DOI | PubMed
- Barber MJ, Mangravite LM, Hyde CL, Chasman DI, Smith JD, McCarty CA. Genome-wide association of lipid-lowering response to statins in combined study populations. PLoS ONE. 2010; 5(3):e9763. DOI | PubMed
- Dimas AS, Deutsch S, Stranger BE, Montgomery SB, Borel C, Attar-Cohen H. Common regulatory variation impacts gene expression in a cell type-dependent manner. Science. 2009; 325(5945):1246-50. DOI | PubMed
- Consortium GTEx. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015; 348(6235):648-60. DOI | PubMed
- Willer CJ, Schmidt EM, Sengupta S, Peloso GM, Gustafsson S, Kanoni S. Discovery and refinement of loci associated with lipid levels. Nat Genet. 2013; 45(11):1274-83. DOI | PubMed
- Whitlock G, Lewington S, Sherliker P, Clarke R, Emberson J, Halsey J. Body-mass index and cause-specific mortality in 900 000 adults: collaborative analyses of 57 prospective studies. Lancet. 2009; 373(9669):1083-96. DOI | PubMed
- Mongraw-Chaffin ML, Peters SA, Huxley RR, Woodward M. The sex-specific association between BMI and coronary heart disease: a systematic review and meta-analysis of 95 cohorts with 1.2 million participants. Lancet Diabetes Endocrinol. 2015; 3(6):437-49. DOI | PubMed
- Strable MS, Ntambi JM. Genetic control of de novo lipogenesis: role in diet-induced obesity. Crit Rev Biochem Mol Biol. 2010; 45(3):199-214. DOI | PubMed
- Li Y, Xu S, Mihaylova MM, Zheng B, Hou X, Jiang B. AMPK phosphorylates and inhibits SREBP activity to attenuate hepatic steatosis and atherosclerosis in diet-induced insulin-resistant mice. Cell Metab. 2011; 13(4):376-88. DOI | PubMed
- Do R, Willer CJ, Schmidt EM, Sengupta S, Gao C, Peloso GM. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013; 45(11):1345-52. DOI | PubMed
- Feldkircher KM, Mistry AM, Romsos DR. Adrenalectomy reverses pre-existing obesity in adult genetically obese (ob/ob) mice. Int J Obes Relat Metab Disord. 1996; 20(3):232-5. PubMed
- Solomon J, Mayer J. The effect of adrenalectomy on the development of the obese-hyperglycemic syndrome in ob-ob mice. Endocrinology. 1973; 93(2):510-2. DOI | PubMed
- Perry RJ, Camporez JP, Kursawe R, Titchenell PM, Zhang D, Perry CJ. Hepatic acetyl CoA links adipose tissue inflammation to hepatic insulin resistance and type 2 diabetes. Cell. 2015; 160(4):745-58. DOI | PubMed
- Frisdal E, Le Lay S, Hooton H, Poupel L, Olivier M, Alili R. Adipocyte Atp-binding cassette G1 promotes triglyceride storage, fat mass growth and human obesity. Diabetes. 2015; 64(3):840-55. DOI | PubMed
- Hidalgo B, Irvin MR, Sha J, Zhi D, Aslibekyan S, Absher D. Epigenome-wide association study of fasting measures of glucose, insulin, and HOMA-IR in the Genetics of Lipid Lowering Drugs and Diet Network Study. Diabetes. 2014; 63(2):801-7. DOI | PubMed
- Irvin MR, Zhi D, Joehanes R, Mendelson M, Aslibekyan S, Claas SA. Epigenome-wide association study of fasting blood lipids in the Genetics of Lipid-lowering Drugs and Diet Network study. Circulation. 2014; 130(7):565-72. DOI | PubMed
- Pfeiffer L, Wahl S, Pilling LC, Reischl E, Sandling JK, Kunze S. DNA methylation of lipid-related genes affects blood lipid levels. Circ Cardiovasc Genet. 2015; 8(2):334-42. DOI | PubMed
- Chambers JC, Loh M, Lehne B, Drong A, Kriebel J, Motta V. Epigenome-wide association of DNA methylation markers in peripheral blood from Indian Asians and Europeans with incident type 2 diabetes: a nested case-control study. Lancet Diabetes Endocrinol. 2015; 3(7):526-34. DOI | PubMed
- Sayols-Baixeras S, Subirana I, Lluis-Ganella C, Civeira F, Roquer J, Do AN. Identification and validation of seven new loci showing differential DNA methylation related to serum lipid profile: an epigenome-wide approach. The REGICOR study. Hum Mol Genet. 2016. DOI
- Dekkers KF, van Iterson M, Slieker RC, Moed MH, Bonder MJ, van Galen M. Blood lipids influence DNA methylation in circulating cells. Genome Biol. 2016; 17(1):138. DOI | PubMed
- Folmes CD, Lopaschuk GD. Role of malonyl-CoA in heart disease and the hypothalamic control of obesity. Cardiovasc Res. 2007; 73(2):278-87. DOI | PubMed
- Schreurs M, Kuipers F, van der Leij FR. Regulatory enzymes of mitochondrial beta-oxidation as targets for treatment of the metabolic syndrome. Obes Rev. 2010; 11(5):380-8. DOI | PubMed
- Hersoug LG, Moller P, Loft S. Gut microbiota-derived lipopolysaccharide uptake and trafficking to adipose tissue: implications for inflammation and obesity. Obes Rev. 2016; 17(4):297-312. DOI | PubMed
- Bansal K, Balaji KN. Intracellular pathogen sensor NOD2 programs macrophages to trigger Notch1 activation. J Biol Chem. 2011; 286(7):5823-35. DOI | PubMed
- Yuan H, Zelkha S, Burkatovskaya M, Gupte R, Leeman SE, Amar S. Pivotal role of NOD2 in inflammatory processes affecting atherosclerosis and periodontal bone loss. Proc Natl Acad Sci U S A. 2013; 110(52):E5059-68. DOI | PubMed
- Kamada Y, Ono M, Hyogo H, Fujii H, Sumida Y, Mori K. A novel noninvasive diagnostic method for nonalcoholic steatohepatitis using two glycobiomarkers. Hepatology. 2015; 62(5):1433-43. DOI | PubMed
- Maekawa T, Kamada Y, Ebisutani Y, Ueda M, Hata T, Kawamoto K. Serum Mac-2 binding protein is a novel biomarker for chronic pancreatitis. World J Gastroenterol. 2016; 22(17):4403-10. DOI | PubMed
- Campion J, Milagro FI, Martinez JA. Individuality and epigenetics in obesity. Obes Rev. 2009; 10(4):383-92. DOI | PubMed
- Richmond RC, Sharp GC, Ward ME, Fraser A, Lyttleton O, McArdle WL. DNA methylation and body mass index: investigating identified methylation sites at HIF3A in a causal framework. Diabetes. 2016; 65(5):1231-44. DOI | PubMed
- Petersen AK, Zeilinger S, Kastenmuller G, Romisch-Margl W, Brugger M, Peters A. Epigenetics meets metabolomics: an epigenome-wide association study with blood serum metabolic traits. Hum Mol Genet. 2014; 23(2):534-45. DOI | PubMed
- Bikman BT, Summers SA. Ceramides as modulators of cellular and whole-body metabolism. J Clin Invest. 2011; 121(11):4222-30. DOI | PubMed
- Borodzicz S, Czarzasta K, Kuch M, Cudnoch-Jedrzejewska A. Sphingolipids in cardiovascular diseases and metabolic disorders. Lipids Health Dis. 2015; 14:55. DOI | PubMed
- Bellini L, Campana M, Mahfouz R, Carlier A, Veret J, Magnan C. Targeting sphingolipid metabolism in the treatment of obesity/type 2 diabetes. Expert Opin Ther Targets. 2015; 19(8):1037-50. DOI | PubMed
- Smemo S, Tena JJ, Kim KH, Gamazon ER, Sakabe NJ, Gomez-Marin C. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014; 507(7492):371-5. DOI | PubMed
- Shah S, Bonder MJ, Marioni RE, Zhu Z, McRae AF, Zhernakova A. Improving phenotypic prediction by combining genetic and epigenetic associations. Am J Hum Genet. 2015; 97(1):75-85. DOI | PubMed
- Ma B, Wilker EH, Willis-Owen SA, Byun HM, Wong KC, Motta V. Predicting DNA methylation level across human tissues. Nucleic Acids Res. 2014; 42(6):3515-28. DOI | PubMed
Fonte
Mendelson MM, Marioni RE, Joehanes R, Liu C, Hedman K, et al. (2017) Association of Body Mass Index with DNA Methylation and Gene Expression in Blood Cells and Relations to Cardiometabolic Disease: A Mendelian Randomization Approach. PLoS Medicine 14(1): e1002215. https://doi.org/10.1371/journal.pmed.1002215




