
Introduzione
I Coronavirus (CoV) sono una famiglia di virus RNA appartenenti alla famiglia dei Coronaviridae e alla sottofamiglia delle Coronavirine e costituiscono il più grande gruppo di virus RNA a singolo filamento a senso positivo. Da un punto di vista accademico, i CoV possono essere suddivisi in quattro generi: Alphacoronaviruses, Betacoronaviruses, Gammacoronaviruses e Deltacoronaviruses. Gli alfacoronavirus e i betacoronavirus si trovano di solito nei mammiferi, mentre i gammacoronavirus e i deltacoronavirus sono principalmente associati agli uccelli1,2. La SARS-CoV è l’agente causale dell’epidemia di sindrome respiratoria acuta grave (SARS) che si è verificata nel 2002-2003. Questa epidemia di SARS è stata la prima pandemia umana scoppiata dall’inizio del 21° secolo e ha causato quasi 8000 casi di infezione e 800 morti in tutto il mondo.3,4. SARS-CoV appartiene al genere Betacoronavirus, e la sua sequenza genomica mostra bassi livelli di somiglianza con i CoVs-OC43 e 229E umani precedentemente identificati. Abbiamo quindi ipotizzato che la SARS-CoV abbia subito un lungo e indipendente processo evolutivo. Il genoma della SARS-CoV di solito codifica quattro proteine strutturali: la proteina spike (S), la proteina dell’involucro (E), la proteina di membrana (M) e la proteina nucleocapside (N). Tra queste, la proteina S è una glicoproteina trimerica, cellula-superficie glicoproteica che consiste di due sottounità (S1 e S2), mentre la sottounità S1 è responsabile del legame del recettore. Le variazioni della proteina S, in larga misura, sono responsabili del tropismo dei tessuti e delle gamme di ospiti di diversi CoV5,6.
L’origine della SARS-CoV è sempre stata al centro della ricerca. Gli zibetti di palma sono stati inizialmente considerati il serbatoio naturale della SARS-CoV a causa dell’isolamento di diversi ceppi di SARS-CoV dagli zibetti di palma che sono stati scambiati nei mercati umidi della provincia cinese del Guangdong nel 2003.7. Tuttavia, studi successivi hanno dimostrato che il virus è stato rilevato solo negli zibetti di palma di origine commerciale che sono stati testati prima dell’abbattimento, ma non in quelli testati in seguito; anche gli zibetti di palma catturati dall’ambiente naturale sono risultati negativi al virus. Questo risultato ha suggerito che gli zibetti di palma servivano solo come serbatoio intermedio e non sono quindi un serbatoio naturale per la SARS-CoV8,9. Recentemente, i pipistrelli hanno catturato la nostra attenzione grazie alla loro capacità di agire come serbatoi naturali per una grande varietà di virus, tra cui molti importanti virus zoonotici che sono associati a diverse forme gravi di malattie infettive emergenti, come il virus Ebola, il virus Nipah, il virus Hendra e il virus Marburg.10,11. Nel 2005, squadre provenienti da Hong Kong e dalla Cina continentale hanno scoperto quasi contemporaneamente la presenza di SL-CoV in pipistrelli a ferro di cavallo cinesi selvatici (Rhinolophus sinicus) provenienti dalla Cina. Questi risultati hanno suggerito che i pipistrelli erano gli ospiti naturali della SARS-CoV12. In particolare, durante la sorveglianza longitudinale della colonia di Rhinolophus sinicus nella provincia cinese dello Yunnan negli ultimi anni, un gruppo di ricerca cinese ha isolato con successo un campione vivo SL-CoV da cellule Vero E6 che sono state incubate nelle feci dei pipistrelli nel 2013.13. Il virus isolato ha mostrato più del 95% dell’identità della sequenza genomica con le SARS-CoV umane e civili. Ulteriori studi su questi hanno indicato che la SL-CoV da pipistrelli può infettare direttamente gli esseri umani e non richiede un ospite intermedio. SL-CoV, simile alla SARS-CoVs, possiede la capacità di infiltrarsi nelle cellule utilizzando la sua proteina S per combinarsi con i recettori dell’enzima di conversione dell’angiotensina 2 (ACE2)14. Questa osservazione ha indicato che la SARS-CoV ha avuto origine dai pipistrelli cinesi a ferro di cavallo e che la SL-CoV isolata dai pipistrelli rappresenta quindi una potenziale minaccia per gli esseri umani.
Negli ultimi anni, molti nuovi SL-CoV sono stati identificati in una varietà di specie di pipistrelli in tutto il mondo, tra cui Asia, Europa, Africa e America. La maggior parte delle SL-CoV sono state scoperte in rinofidi provenienti da Cina, Slovenia, Bulgaria e Italia.15–17mentre nuovi beta-coronavirus correlati alla SARS-CoV sono stati rilevati in specie di Hipposideros e Chaerophon provenienti dal Kenya e dalla Nigeria.18,19. Tuttavia, l’analisi della sequenza di aminoacidi RNA polimerasi RNA-dipendente (RdRp) ha mostrato che le sequenze genomiche di questi campioni di pipistrello SL-CoV ottenuti da diverse parti del mondo condividevano tra loro l’80-90% di identità e mostravano un’identità dell’87-92% con i SARS-CoV estratti da fonti umane o da zibetti.20,21. Questi risultati indicano che la SARS-CoV si è probabilmente evoluta nei pipistrelli per periodi di tempo più lunghi. Una precedente ricerca condotta dal nostro gruppo ha rivelato che i pipistrelli trovati nel sud-est della Cina hanno un’elevata capacità di carico per i SL-CoV.22. Dopo aver condotto un’indagine epidemiologica sui pipistrelli portatori di CoV, sono stati identificati due nuovi SL-CoV nei campioni di Rhinolophus pusillus della città di Zhoushan, provincia di Zhejiang, Cina; successivamente, è stato utilizzato un modello di infezione da ratto per valutare il potenziale di trasmissione incrociata dei virus.
Risultati
Campionamento
Tra il 2015 e il 2017, sono stati prelevati 334 pipistrelli da Zhoushan, Cina. Questi pipistrelli appartenevano alla specie Rhinolophus pusillus come determinato dalle sequenze del gene del citocromo mitocondriale b nei loro tessuti muscolari23. Tutti i 334 campioni di pipistrello sono stati sottoposti a screening per l’RNA CoV utilizzando un test di trascrizione inversa pan-coronavirus (RT)-PCR. La prevalenza complessiva del virus è stata del 26,65% (89/334, pipistrelli; Tabella 1). Inoltre, è stata osservata una prevalenza più elevata nei campioni raccolti in luglio (66,7% nel 2015) rispetto a quelli raccolti in ottobre (21% nel 2016) o febbraio (13% nel 2017). È stato costruito un albero filogenetico secondo le sequenze parziali RdRp 440 bp e i campioni positivi sono stati classificati in Alphacoronavirus e Betacoronavirus. Come mostrato nella Fig. S1, 89 ampliconi sono stati raggruppati in cinque clades con il 66-100% di identità nucleotidiche tra loro, e hanno condiviso il 94-100% di identità con i virus che sono stati estratti da Hong Kong, Guangdong e Hainan in Cina e quelli provenienti dalla Spagna.Tabella 1Riepilogo del rilevamento dei bat-CoV nei pipistrelli della provincia cinese di ZhejiangTimeLocusSample numberBat speciesCoV positiveJuly, 15Dinghai, Zhoushan city (ZXC)45Rhinolophus sinicus66.7% (30/45)Gennaio, 16Dinghai, Zhoushan città (Z2)120Rhinolophus sinicus25% (30/120)Ottobre, 16Daishan, Zhoushan città (DXC)84Rhinolophus sinicus21% (18/84)Febbraio, 17Dinghai, Zhoushan città (ZC)85Rhinolophus sinicus13% (11/85)Totale33426,65 (89/334)
Confronto completo della sequenza genomica e analisi della ricombinazione
Per esplorare ulteriormente l’evoluzione di SL-CoV di Zhoushan, sono state generate due sequenze genomiche complete dei CoV rappresentativi derivati dai pipistrelli mediante il sequenziamento di diversi ampliconi sovrapposti. In particolare, le sequenze sono state generate dai seguenti campioni: SL-CoV ZXC21 (MG772934) pipistrello estratto da un campione prelevato nel luglio 2015 e SL-CoV ZC45 (MG772933) pipistrello estratto da un campione prelevato nel febbraio 2017. I genomi completi di ZXC21 e ZC45 consistevano rispettivamente di 29.732 nt e 29.802 nt. L’organizzazione genomica in entrambi i casi era simile a quella dei più noti bat-SL-CoV. Utilizzando il programma RDP, sono stati inizialmente previsti i potenziali eventi ricombinanti tra ZXC21, ZC45 e altri ceppi rappresentativi di 13 CoV umani/celluto e bat-SL-CoVs simili alla SARS. I risultati non hanno identificato alcun potenziale evento di ricombinazione. La somiglianza della sequenza genomica tra i cinque bat-SL-CoVs e il ceppo SARS-CoV SZ3 è stata esaminata con l’analisi Simplot (Fig. 1). I risultati hanno mostrato che i genomi avevano un contenuto di GC del 38,9% ed avevano 13 frame di lettura aperti (ORF) simili al ceppo HKU3-1. I due nuovi pipistrelli SL-CoVs condividevano tra loro il 97% di identità di sequenza genomica. L’identità complessiva della sequenza nucleotidica di questi due genomi con civetta SARS-CoV (ceppo SZ3) era dell’81%, che era inferiore alle osservazioni precedentemente riportate associate ai bat SL-CoV raccolti dalla Cina (88-92%). Dalle analisi di omologia di diversi ORF, i frammenti di ORF8 hanno mostrato l’omologia più bassa con i dati di omologia SL-CoV segnalati24Fig. 1A della mappa genica dei due nuovi SL-CoV e l’analisi di ricombinazione dei nuovi SL-CoV con altri SL-CoV.Le trame di somiglianza sono state condotte con la SARS CoV SZ3 come query e bat SL-CoV, tra cui Rs3367, Longquan-140, e HKU3-1, come potenziali sequenze parentali. L’analisi è stata eseguita utilizzando il modello Kimura, con una dimensione della finestra di 2000 coppie di base e una dimensione del passo di 200 coppie di base
La proteina S è responsabile dell’ingresso del virus ed è funzionalmente divisa in due domini, S1 e S2. Il pipistrello SL-CoV Rs3367 è il virus più strettamente correlato alla SARS-CoV umana e ha l’89,9% di identità di sequenza aminoacidica alla SARS-CoV rispetto all’intera proteina spike. In confronto, le proteine S di ZXC21 e ZC45 identificate in questo studio erano leggermente più diverse rispetto alla loro controparte nella SARS-CoV, che ha mostrato un’identità del 77% a livello di aminoacidi. Le analisi filogenetiche basate sulla proteina S hanno suggerito che le proteine S di ZXC21 e ZC45 rappresentavano un clade separato relativo al lignaggio B CoVs (Fig. 2b). La più alta identità di sequenza di aminoacidi condivisa con il ceppo Rs806 era solo dell’83%. Come altri bat-SL-CoVs, il dominio S1 dei CoVs simili alla SARS dei pipistrelli ha mostrato una somiglianza nucleotidica molto bassa con i CoV della SARS, e ci sono diverse delezioni e mutazioni chiave nella maggior parte delle regioni variabili all’interno del dominio legante del recettore (RBD) (Fig. 2a).Fig. 2Caratterizzazione dei domini S1 dei CoV della SARS e SL-CoVs. un confronto della sequenza di amminoacidi della sottounità S1. Il dominio legante del recettore (aa 318-510) della SARS-CoV. b Un’analisi filogenetica di tutte le sequenze di amminoacidi S1 basata sul metodo dell’unione adiacente. I ceppi SARS-CoV-GD01, BJ302 e GZ02 sono stati isolati dai pazienti dell’epidemia di SARS nel 2003. La SARS-CoV SZ3 è stata identificata dagli zibetti nel 2003. Le sequenze di SL-CoV in questo studio sono contrassegnate come triangoli pieni.
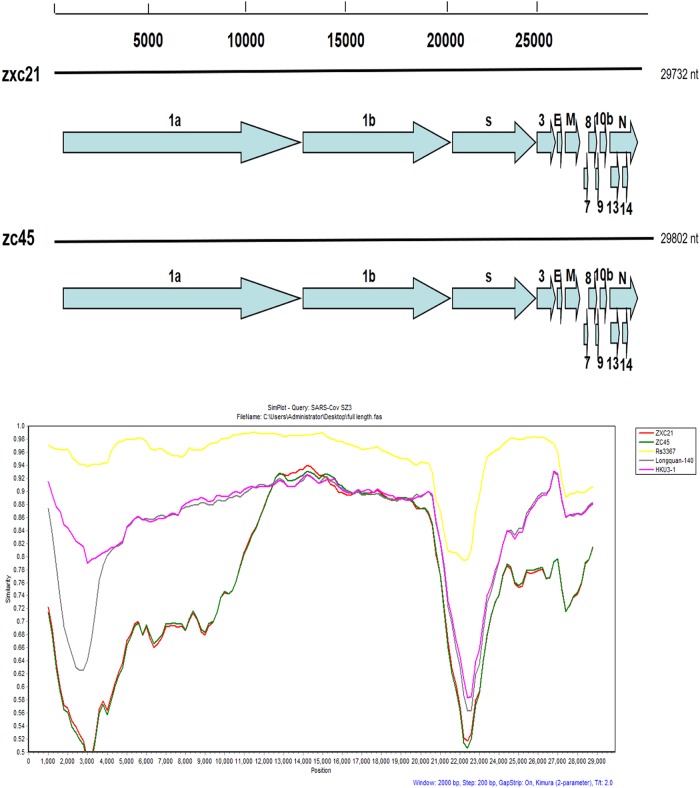
Fig. 1.Fig. 1. Una mappa genica dei due nuovi SL-CoV e l’analisi della ricombinazione dei nuovi SL-CoV con altri SL-CoV.Trame di somiglianza sono state condotte con la SARS CoV SZ3 come query e bat SL-CoV, tra cui Rs3367, Longquan-140, e HKU3-1, come potenziali sequenze parentali. L’analisi è stata eseguita utilizzando il modello Kimura, con una dimensione della finestra di 2000 coppie di base e una dimensione del passo di 200 coppie di base
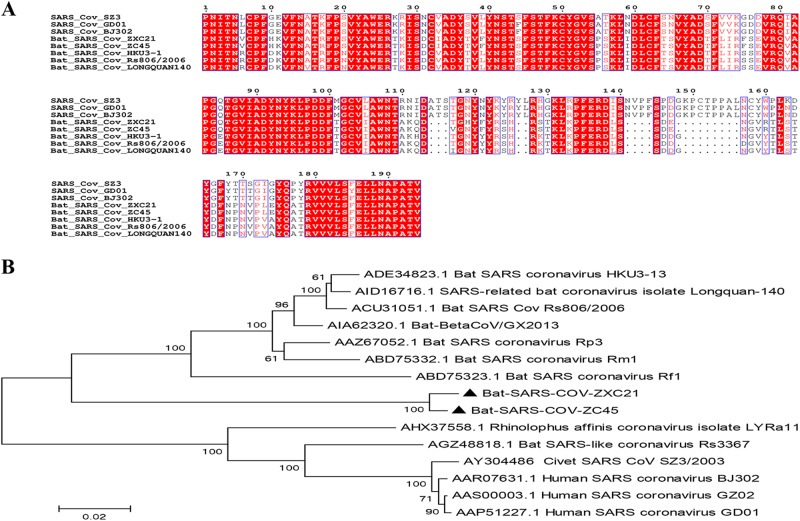
Fig. 2.Caratterizzazione dei domini S1 della SARS CoV e SL-CoV.un confronto della sequenza di aminoacidi della sottounità S1. Il dominio legante del recettore (aa 318-510) della SARS-CoV. b Un’analisi filogenetica di tutte le sequenze di amminoacidi S1 basata sul metodo dell’unione adiacente. I ceppi SARS-CoV-GD01, BJ302 e GZ02 sono stati isolati dai pazienti dell’epidemia di SARS nel 2003. La SARS-CoV SZ3 è stata identificata dagli zibetti nel 2003. Le sequenze di SL-CoV in questo studio sono contrassegnate come triangoli pieni.
Test per l’individuazione di infezioni da ratti e virus
Nonostante il fallito isolamento del virus infettivo da campioni positivi alla PCR nelle cellule Vero E6, abbiamo cercato di isolare il virus dai ratti da latte infettandoli con campioni di tessuto positivi al coronavirus. Dopo 15 giorni, l’analisi patologica ha mostrato che i tessuti e gli organi dei ratti infetti mostravano diversi gradi di infiammazione, e la reazione infiammatoria nei tessuti cerebrali era più evidente. Dei dieci ratti da latte, quattro hanno mostrato sintomi clinici, tra cui sonnolenza, azione lenta e depressione mentale. I nuovi ratti da latte infettati dal tessuto cerebrale malato avevano ancora un esordio irregolare, mentre cinque degli 11 ratti da latte in un nido avevano sintomi clinici. Numerosi neuroni apoptotici sono stati visti nelle aree focali del tessuto cerebrale, e la cromatina nei nuclei era condensata e poco chiara. I tessuti polmonari erano ben strutturati, ma le cavità alveolari erano parzialmente fuse insieme e mostravano chiari segni di lieve enfisema. L’analisi del tessuto intestinale ha mostrato una perdita nella struttura della mucosa intestinale; le membrane mucose erano sottili, le cripte erano poco profonde, le ghiandole intrinseche erano ridotte, e lo stroma mostrava un infiltrato infiammatorio disperso (Fig. 3). Successivamente, la carica virale di diversi tessuti è stata rilevata mediante PCR quantitativa, e le cariche virali dei tessuti polmonari sono rimaste le più alte, mostrando circa 104 copie del genoma virale per 1 μl di sospensione del tessuto (dati non mostrati).Fig. 3Light osservazioni microscopiche al microscopio dei tessuti di ratto infettati con bat-SL-CoVs: I tessuti cerebrali, intestinali, polmonari ed epatici sezionati sono stati prelevati da ratti infettati con bat-SL-CoV ZC45
Sospette particelle virali sono state osservate nei nuclei di neuroni denaturati nei tessuti cerebrali dei ratti utilizzando la microscopia elettronica a trasmissione (TEM). Queste particelle virali presentavano la tipica morfologia del coronavirus ed erano grandi circa 100 nm con picchi superficiali apparenti (Fig. 4). Contemporaneamente, vari test RT-PCR virali sono stati condotti sui tessuti per rilevare le particelle virali. I tessuti sono stati testati per la presenza di particelle virali associate a una grande varietà di virus, come CoV, enipavirus, respirovirus, avulavirus, rubulavirus, e il virus dell’influenza-A della famiglia Orthomyxoviridae, utilizzando metodi precedentemente pubblicati25,26. I risultati del test hanno rivelato che i tessuti erano positivi solo per CoV.Fig. 4Transmission micrografie elettroniche di tessuti cerebrali di ratto infetti. a, b particelle simili a CoV sono considerate SL-CoVs ZC45 in diverse posizioni dei tessuti cerebrali di ratto infetti
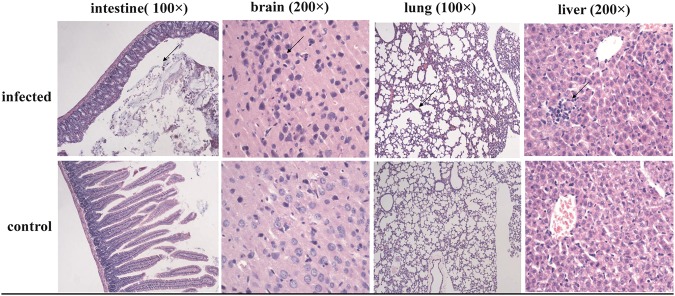
Fig. 3.Osservazioni al microscopio ottico di tessuti di ratto infettati con bat-SL-CoVs: I tessuti cerebrali, intestinali, polmonari ed epatici sezionati sono stati prelevati da ratti infettati con bat-SL-CoV ZC45
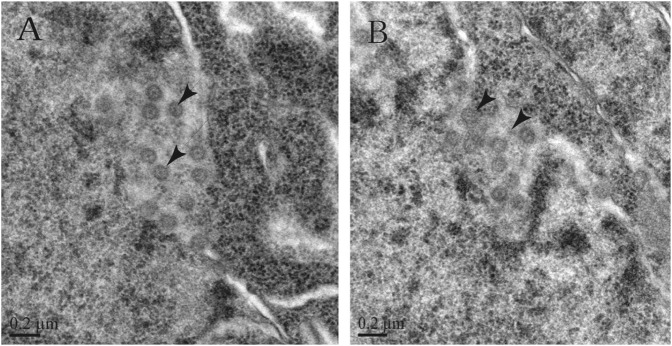
Fig. 4.Micrografie elettroniche a trasmissione di tessuti cerebrali di ratto infetti.a, b Le particelle simili a CoV sono considerate SL-CoVs ZC45 in diverse posizioni dei tessuti cerebrali infetti del ratto
Analisi dell’antigene della proteina N e western blotting
Simile ad altri CoV, la proteina nucleocapside è uno dei componenti fondamentali della SARS-CoV. La proteina N è una delle proteine maggiormente espresse durante le prime fasi dell’infezione da SARS-CoV ed è stata un interessante strumento diagnostico grazie all’avvio di una forte risposta immunitaria contro di essa. Le analisi evolutive hanno dimostrato che l’omologia tra la proteina N e le sue controparti nella ben nota SARS-CoV e bat SL-CoV variava dall’89 al 91%. L’analisi antigenica si è basata sulla sequenza di aminoacidi della proteina N (Fig. 5), e i risultati hanno suggerito che i due peptidi antigenici alternativi, tra cui KHD2016288-1:KDKKKKADELQALPQ e KHD2016288-2:QQQGQTVTKKSAAEA, sono stati selezionati per la sintesi peptidicaFig. 5Prediction dell’antigenicità della proteina N del pipistrello SL-CoV. a L’antigenicità prevista per la proteina N. b Sequenza di aminoacidi della proteina N. La porzione di antigenicità elevata è indicata nel cerchio rosso. I due polipeptidi sintetizzati sono indicati in rosso
Per caratterizzare ulteriormente la reattività antigenica del virus nei tessuti murini infetti con anticorpi specifici per ZC45 rispetto a quelli di ZC45, sono stati generati anticorpi policlonali contro i polipeptidi (KHD2016288-1:KDKKKKADELQALPQ) derivati dalle proteine ZC45 N e successivamente sottoposti ad analisi di Western Blotting (Fig. 6). Gli anti-polipeptidi sono stati derivati dagli anticorpi della proteina ZC45 N da sei diverse fonti di proteine N (50 kDa), compresi i tessuti intestinali, i tessuti cerebrali e i tessuti polmonari dei ratti infetti. I risultati hanno indicato che l’antigene polipeptide è stato sintetizzato correttamente, e gli anticorpi policlonali prodotti contro questo polipeptide potrebbero reagire con le proteine N del pipistrello SL-CoV. Gli anticorpi policlonali hanno reagito specificamente con i tessuti infetti di ratto, ma non con i tessuti di ratto derivati dai campioni di controllo. Questi risultati hanno indicato che il virus può circolare e proliferare nei ratti infetti. Fig. 6. Rilevamento dell’espressione della proteina N nei tessuti di ratto infetti da western blotting.sono state analizzate le proteine dei seguenti tessuti: cervello di ratto dal campione di controllo (corsia 1), tessuto intestinale del pipistrello ZC45 (corsia 2), tessuto intestinale del ratto infetto (corsia 3,6), tessuto polmonare del ratto infetto (corsia 4,7) e tessuto cerebrale del ratto infetto (corsia 5,8).
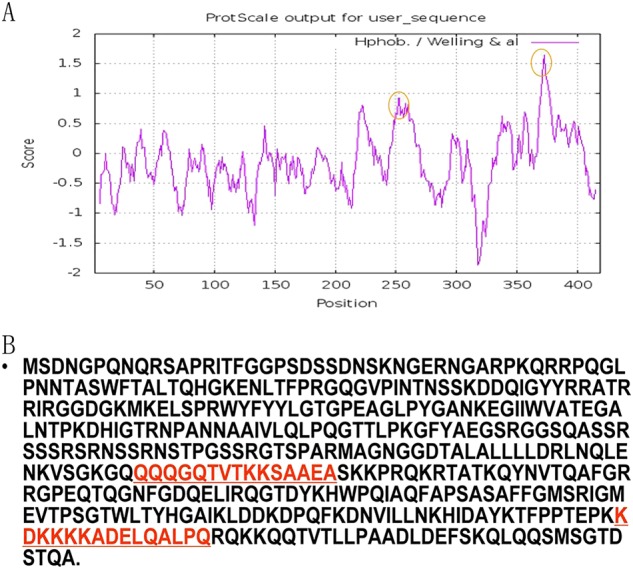
Fig. 5.Previsione dell’antigenicità della proteina SL-CoV N del pipistrello.a L’antigenicità prevista per la proteina N. b Sequenza di aminoacidi della proteina N. La porzione ad alta antigenicità è indicata nel cerchio rosso. I due polipeptidi sintetizzati sono indicati in rosso
Fig. 6.Fig. 6. Rilevazione dell’espressione della proteina N nei tessuti infetti di ratto tramite Western Blotting.Sono state analizzate le proteine dei seguenti tessuti: cervello di ratto dal campione di controllo (corsia 1), tessuto intestinale del pipistrello ZC45 (corsia 2), tessuto intestinale del ratto infetto (corsia 3,6), tessuto polmonare del ratto infetto (corsia 4,7) e tessuto cerebrale del ratto infetto (corsia 5,8).
Discussione
Dal primo rapporto sull’origine di SL-CoVs da pipistrelli nel 2005, i CoVs sono stati trovati in dieci diverse specie di pipistrelli all’interno di sei famiglie di più di dieci paesi, tra cui Cina, Africa ed Europa.21,27. La nostra sorveglianza longitudinale di 2 anni dei pipistrelli a Zhoushan ha indicato che tutti i 334 pipistrelli che sono stati raccolti appartenevano alla specie Rhinolophus sinicus, suggerendo che era la specie di pipistrello dominante trovata nel nostro studio ed è stato dimostrato che è il serbatoio naturale della SARS-CoV. L’amplificazione PCR nidificata della regione conservata della RdRp ha mostrato che il tasso di trasporto CoV associato a questa specie di pipistrello era molto più alto di quello riportato in precedenza.28,29. Allo stesso tempo, il tasso di carico estivo è stato superiore a quello delle altre stagioni a causa dell’influenza della distribuzione stagionale. In questa regione sono stati identificati due cladi di Alphacoronavirus e tre cladi di Betacoronavirus, a indicare che una grande varietà di CoV circolano nei pipistrelli dell’area di Zhoushan, e questi CoV erano i più diffusi nelle colonie di pipistrelli che si trovavano in questa regione.
Per esplorare la possibilità di trasmissione di CoV dai pipistrelli in questa zona, sono stati prelevati due campioni a lunghezza intera di bat-SL-CoVs dai pipistrelli infetti da virus. Questi due pipistrelli SL-CoV sono stati ottenuti dalla stessa località ma in stagioni diverse; tra di essi è stata presentata un’identità di sequenza genomica dell’88-99%, indicando che i pipistrelli sono i serbatoi naturali di questi SL-CoV e che questi SL-CoV possono circolare all’interno di singole colonie. Nel frattempo, c’era una grande differenza tra i due virus descritti in questo studio e i virus descritti in studi precedenti, soprattutto per quanto riguarda l’ipervariabilità del dominio S130,31. Si è notato che il gene che codifica la proteina S ha mostrato un alto grado di variabilità. La proteina S è responsabile dell’ingresso virale ed è funzionalmente divisa in due domini, S1 e S2. Il dominio S1 è coinvolto nel legame del recettore, mentre il dominio S2 è coinvolto nella fusione della membrana cellulare. Il dominio S1 può essere funzionalmente suddiviso in due domini, un dominio N-terminale (S1-NTD) e un dominio C-terminale (S1-CTD), ed entrambi possono legarsi ai recettori dell’host, funzionando quindi come RBD32. ZXC21 e ZC45 hanno mostrato enormi diversità con i CoV di pipistrelli precedentemente segnalati associati alla regione S1, e il livello più alto di identità condivisa è stato solo dell’83%. Nel corso di questo studio si è cercato di effettuare un’analisi di ricombinazione. Nel nostro studio non è stato possibile identificare alcun potenziale evento di ricombinazione. Ciò potrebbe essere dovuto al fatto che i due ceppi hanno avuto origine da una discendenza SL-CoV non campionata che risiede in una specie di pipistrello filogeneticamente più vicina allo ZXC21 e allo ZC45 rispetto a tutti gli altri campioni noti di pipistrello SL-CoV. Quindi, abbiamo usato simplot per analizzare la somiglianza di sequenza di cinque bat-SL-CoV e della SARS-CoV SZ3. Il ceppo Longquan-140 è il più omologo allo ZC45 e allo ZXC21, l’Rs3367 è il ceppo di origine pipistrello più vicino al coronavirus umano patogeno della SARS, e l’SZ3 è il ceppo rappresentativo di origine civet.
In questo studio, un modello di ratto da latte è stato inizialmente utilizzato per studiare la possibilità di proliferazione di CoV di origine pipistrello in altri animali. In precedenza, solo un rapporto aveva mostrato risultati promettenti associati all’isolamento di SL-CoVs vivi dai campioni fecali di pipistrelli con cellule Vero E613. Il vivo SL-CoV coltivato in cellule Vero E6 ha presentato una morfologia tipica CoV e ha la capacità di utilizzare ACE2 da esseri umani, zibetti, e pipistrelli a ferro di cavallo cinese per l’ingresso delle cellule33. Un tentativo di isolare il virus con le cellule Vero E6 non ha avuto successo, probabilmente a causa di una bassa carica virale o della mancanza di compatibilità con le cellule Vero E6. Questo studio ha trovato che le SL-CoVs derivate da pipistrelli potrebbe replicare con successo nei ratti da latte, e l’esame patologico ha mostrato il verificarsi di reazioni infiammatorie negli organi esaminati dei ratti da latte. Questo risultato ha indicato che il virus può proliferare nei ratti e ha il potenziale di trasmissione tra specie incrociate. Quando le particelle di CoV procurate dai tessuti cerebrali infetti dei ratti sono state studiate al microscopio elettronico, la morfologia delle particelle è risultata identica alle tipiche particelle del coronavirus, come descritto negli studi precedenti34. Tuttavia, i tipici picchi non potevano essere visualizzati dalla microscopia elettronica. Questa osservazione può essere parzialmente spiegata dall’ipotesi che i domini S1 e S2 della proteina S (che non sono ben collegati) sono stati facilmente staccati dal virione utilizzando un eccessivo congelamento-scongelamento o ultracentrifugazione6. Così, c’è stata una perdita di domini S1, che probabilmente si è verificato durante la preparazione dei campioni per la microscopia elettronica. Nel frattempo, i tessuti infetti di ratto potrebbero reagire con gli anticorpi policlonali associati alla proteina ZC45 N, secondo i risultati del test western blotting, che indica che il virus può circolare nei ratti. Nonostante i risultati negativi del Western blotting negativo nei tessuti intestinali di ratto e i risultati positivi del Western blotting nel cervello e nei tessuti polmonari, abbiamo considerato che queste differenze possono essere causate da diverse cariche virali in diversi tessuti.
In conclusione, sulla base del rilevamento precoce di un alto tasso di trasporto di SL-CoV, che ha avuto origine dai pipistrelli di Zhoushan, in Cina, questo studio ha comportato una sorveglianza continua degli SL-CoV che hanno avuto origine dai pipistrelli di questa regione. Diversi pipistrelli SL-CoV sono stati identificati in questa regione, e gli SL-CoV di questa regione sono rimasti stabili e possono essere trasmessi l’uno all’altro. Anche se ci sono state diverse differenze tra le SARS-CoVs e i bat-SL-CoVs ottenuti da questa regione sulla base dei due campioni di lunghezza completa ottenuti in questo studio, in particolare per quanto riguarda la regione della proteina S, questo ceppo potrebbe ancora causare l’infezione nei ratti neonatali. Questa osservazione evidenzia la possibilità di trasmissione incrociata di questi virus. Questi risultati suggeriscono fortemente la necessità di una sorveglianza continua dei virus provenienti da animali selvatici e promuovono ulteriori ricerche per studiare la possibilità di trasmissione incrociata di questi virus.
Materiali e metodi
Dichiarazione etica
Le procedure di campionamento dei chirotteri sono state esaminate e approvate dal Comitato amministrativo per il benessere degli animali dell’Istituto di Zhejiang CDC Veterinario (Autorizzazione del Comitato per la cura e l’uso degli animali da laboratorio). Tutti i chirotteri vivi sono stati mantenuti e gestiti secondo i principi e le linee guida per la medicina degli animali da laboratorio (2006), Ministero della Scienza e della Tecnologia, Cina. Tutti gli esperimenti sugli animali sono stati approvati dal Comitato Etico dell’Istituto di Ricerca per la Medicina, Comando di Nanchino. Tutti i metodi sono stati eseguiti in conformità alle linee guida e ai regolamenti pertinenti (numero di approvazione: 2015011).
Campionamento
In totale, 334 pipistrelli adulti sono stati catturati dal vivo nella grotta di montagna con reti a nebbia in quattro momenti diversi, da luglio 2015 a febbraio 2017, nella città di Zhoushan (compresi Dinghai e Daishan), provincia di Zhejiang, Cina. Tutti i pipistrelli sono apparsi sani e non presentavano segni clinici evidenti al momento della cattura. Dopo il completamento della raccolta da ogni sito di campionamento, tutti i pipistrelli sono stati immediatamente sezionati, e i dettagli dei pipistrelli sono mostrati nella Tabella 1. Ogni campione (circa 1 g di tessuto intestinale) è stato immediatamente trasferito nel mezzo di trasporto virale (soluzione salina bilanciata di Earle, 0.2% di bicarbonato di sodio, 0,5% di siero albumina bovina, 18 g/l di amicacina, 200 g/l di vancomicina, 160 U/l di nistatina), conservato in azoto liquido prima del trasporto al laboratorio, e infine conservato a -80°C.
Estrazione dell’RNA e screening RT-PCR
Tutti i campioni sono stati raggruppati e sottoposti ad analisi RT-PCR nidificate come riportato nello studio precedente22. In breve, ogni campione intestinale (circa 0,1 g) è stato omogeneizzato in un macinino di vetro con dieci volumi di tampone SM (50 mM Tris, 10 mM MgSO4, 0,1 m NaCl, pH 7,5). L’omogeneizzato è stato centrifugato a 12.000 g per 10 minuti a 4°C, ma è stato utilizzato solo il supernatante. Il supernatante di ogni campione è stato passato attraverso 0,22 μm filtri Pellicon II (Millipore, Billerica, MA) per filtrare i tessuti rotti, batteri e altre impurità. L’RNA virale è stato estratto con un Kit Mini RNA Virale (Qiagen, Hilden, Germania) secondo le raccomandazioni del produttore. L’RNA è stato eluito in 35 ml di RNase-free H2O e conservato a -80 ° C. Trascrizione inversa è stata effettuata utilizzando il primo kit di sintesi di cDNA (TaKaRa, Dalian, Cina) secondo il protocollo del produttore con acqua bidistillata (ddH2O) come controllo negativo. Tutti i campioni sono stati amplificati da una PCR nidificata che ha preso di mira un frammento 440-nt nel gene RdRp di tutti gli alfa e betacoronavirus noti35,36. Per la prima PCR rotonda, la miscela di reazione da 20 μl conteneva 18 μl di soluzione di reazione PCR (Takara), 10 pmol di ogni primer e 1 μl di DNA template. L’amplificazione è stata eseguita nelle seguenti condizioni: 94°C per 3 minuti; 40 cicli a 94°C per 30 s, 52°C per 30 s e 72°C per 1 minuto per 40 cicli di reazione interna; ed estensione a 72°C per 10 minuti. Per la seconda fase di PCR, la miscela di reazione da 20 μl conteneva 18 μl di tampone di reazione PCR, 10 pmol di ogni primer e 1 μl di prodotto della prima fase di PCR. L’amplificazione è stata eseguita nelle seguenti condizioni: 94°C per 3 minuti, seguiti da 30 cicli composti da 94°C per 30 s, 52°C per 30 s, 72°C per 30 s, e un’estensione finale di 72°C per 10 minuti con ddH2O come controllo negativo. I prodotti PCR positivi sono stati sequenziati in entrambe le direzioni da un ABI 3730 DNA Analyzer (Invitrogen, Beijing, China).
Sequenziamento dei genomi a lunghezza intera
Per ottenere le sequenze genomiche complete di ZXC21 e ZC45, 19 coppie di primer PCR degenerate sono state progettate mediante l’allineamento multiplo delle sequenze SARS-CoV e SL-CoV disponibili depositate in GenBank, mirando quasi a tutta la lunghezza del genoma. Le sequenze di primer sono disponibili su richiesta. Sequenze di 5′ e 3′ estremità genomiche sono state ottenute rispettivamente da 5′ e 3′ RACE (Takara). I prodotti PCR con dimensioni previste sono stati purificati con gel e direttamente sottoposti a sequenziamento. Le sequenze di frammenti genomici sovrapposti sono state assemblate per ottenere le sequenze genomiche a lunghezza intera, con ogni sequenza sovrapposta più lunga di 600 bp.
Analisi filogenetica degli ampliconi
Tutti gli ampliconi da 440 bp sono stati allineati con i loro vicini filogenetici più vicini in GenBank utilizzando ClustalW v.2.0. Nell’allineamento sono stati inclusi rappresentanti di diverse specie dei generi Alphacoronavirus e Betacoronavirus, nonché alcune specie non approvate. Gli alberi filogenetici basati su sequenze nucleotidiche sono stati costruiti utilizzando il metodo di congiunzione dei vicini utilizzando il MEGA v.7 con il modello Maximum Composite Likelihood e un valore di bootstrap di 100037.
Le sequenze complete allineate sono state inizialmente scansionate per gli eventi di ricombinazione utilizzando il Recombination Detection Program (RDP)38. I potenziali eventi di ricombinazione tra ZXC21, ZC45, Rs3367 (KC881006), Longquan-140 (KF294457.1) e HKU3-1 (DQ022305.2), come suggerito da RDP con forti valori di P (<10-20), sono stati ulteriormente analizzati mediante analisi di similarità plot e bootscan utilizzando SimPlot v.3.5.139.
Saggio infettante per ratti che succhiano
Per testare la patogenicità dell’agente ZC45, sono stati effettuati esperimenti di infezione nei ratti da latte. 3 giorni di allattamento BALB / c ratti (SLAC, Cina) sono stati inoculati intracerebrali con 20 ml di volume supernatante macinazione del tessuto intestinale ZC45. Cura degli animali e tutti gli esperimenti sugli animali sono stati eseguiti in una struttura di livello di biosicurezza 3 (BSL-3) e sono stati approvati dal comitato etico locale. Dopo 14 giorni, il cervello, i polmoni, l’intestino, e tessuti del fegato da ratti infetti sono stati selezionati per preparare sezioni patologiche. In breve, i tessuti sono stati fissati nel 10% (vol / vol) neutrale-buffered formalina. Dopo l’elaborazione dei tessuti di routine, compresa la disidratazione da soluzioni alcoliche graduate, lavaggio, e l’incubazione in paraffina, 4 sezioni di spessore µm sono stati tagliati e colorati con ematossilina ed eosina (H & E). Circa 2 ore più tardi, le sezioni di tessuto preparato sono stati ripresi con la microscopia ottica (Olympus, Giappone).
La TEM è stata utilizzata per ottenere informazioni patologiche più dettagliate responsabili dei sintomi principali. I campioni di tessuto sono stati fissati in dialdeide al 2,5% (vol/vol) per 2 ore, postfissati in tetraossido di osmio all’1% (vol/vol) per 1 ora, disidratati in etanolo graduato e incorporati in resina epossidica Epon-812. Successivamente, sono state prodotte sezioni ultrasottili da 70 nm e rapidamente colorate in acetato di uranile acquoso e citrato di piombo di Reynolds. Infine, le sezioni di tessuto generate sono state esaminate utilizzando un JEM-1200 TEM (Jeol Ltd. Tokyo, Giappone).
Quantitativa RT-PCR è stata eseguita utilizzando sospensioni tissutali di ratti positivi per SL-CoV da RT-PCR. cDNA è stato amplificato in reazioni di fluorescenza SYBR Green I (Roche) utilizzando primer specifici (5′-TGTGACAGAGAGCCATGCCCCTAA-3′ e 5′-ATCTTATTACCATCAGTTGAAAGA-3′)12. È stato utilizzato un plasmide con la sequenza target per la generazione della curva standard. Alla fine del test, i prodotti di PCR (frammento di pol a 280 bp) sono stati sottoposti all’analisi della curva di fusione (65-95°C, 0,1°C/s) per confermare la specificità del test.
Preparazione di antisiero di coniglio contro due peptidi
Per ottenere l’anticorpo policlonale della proteina N del pipistrello SL-CoV ZC45, sono stati sintetizzati due peptidi parziali con residui di 15 amminoacidi della proteina N (Sangon Biotech, Shanghai, Cina) dopo una ricerca di omologia secondo l’analisi bioinformatica e la previsione del peptide di segnale (SignalIP-4.1), idrofilia e antigenicità della proteina N. I conigli bianchi della Nuova Zelanda (2-2,3 kg) sono stati iniettati per via sottocutanea utilizzando 0,6 mg di due peptidi in 1 ml di soluzione salina tamponata con fosfato (PBS) emulsionata con 1 ml di coadiuvante completo di Freund (Sigma). Gli animali sono stati stimolati due volte per lo stesso percorso a intervalli di 2 settimane con circa 0,3 mg di due peptidi in 1 ml di PBS emulsionato con 1 ml di adiuvante incompleto (Sigma) di Freund. Una settimana dopo l’ultima immunizzazione del booster, sono stati raccolti campioni di sangue, e sieri sono stati isolati per i saggi di attività biologica. Il titolo anticorpale è stato testato con un test di immunosorbimento indiretto legato ad enzimi. Il siero di coniglio preimmune è stato raccolto prima della prima iniezione.
Determinazione dell’infettività del virus con il saggio western blotting
Western blotting è stato eseguito per caratterizzare la reattività antigenica del tessuto di ratto infetto con anticorpo della proteina N di pipistrello SL-CoV-ZC45. I campioni di tessuto intestinale, polmonare e cerebrale infetto sono stati omogeneizzati e lisati in tampone RIPA integrato con inibitori della proteinasi. Quantità uguali di proteine (40 mg) sono state caricate e separate su gel SDS-PAGE (elettroforesi su gel di sodio dodecil solfato poliacrilamide) all’8%. A seguito di elettroforesi, le proteine sono state trasferite su una membrana di PVDF (polivinilidene difluoruro), bloccata con il 5% (p/v) del latte, e incubate con anticorpi primari e secondari. I blot sono stati sviluppati e rilevati da una chemiluminescenza potenziata (GE Healthcare, Little Chalfont, UK). Tessuti di ratto dai campioni di controllo e tessuti intestinali da pipistrello ZC45 sono stati utilizzati come controlli negativi e positivi, rispettivamente.
Numeri di adesione della sequenza nucleotidica
Tutte le sequenze di ampliconi e i genomi completi di ZXC21 e ZC45 generati in questo studio sono stati depositati in GenBank con i numeri di adesione da MG772844 a MG772934.
Materiale elettronico supplementare
Figura supplementareLegenda della figura supplementare
References
- Cui J. Evolutionary relationships between bat coronaviruses and their hosts. Emerg. Infect. Dis.. 2007; 13:1526-1532. DOI | PubMed
- Perlman S, Netland J. Coronaviruses post-SARS: update on replication and pathogenesis. Nat. Rev. Microbiol.. 2009; 7:439-450. DOI | PubMed
- Chinese SMEC. Molecular evolution of the SARS coronavirus during the course of the SARS epidemic in China. Science. 2004; 303:1666-1669. DOI | PubMed
- Zhong NS. Epidemiology and cause of severe acute respiratory syndrome (SARS) in Guangdong, People’s Republic of China, in February, 2003. Lancet. 2003; 362:1353-1358. DOI | PubMed
- Marra MA. The genome sequence of the SARS-associated coronavirus. Science. 2003; 300:1399-1404. DOI | PubMed
- Xu Y. Crystal structure of severe acute respiratory syndrome coronavirus spike protein fusion core. J. Biol. Chem.. 2004; 279:49414-49419. DOI | PubMed
- Song HD. Cross-host evolution of severe acute respiratory syndrome coronavirus in palm civet and human. Proc. Natl. Acad. Sci. U. S. A.. 2005; 102:2430-2435. DOI | PubMed
- Kan B. Molecular evolution analysis and geographic investigation of severe acute respiratory syndrome coronavirus-like virus in palm civets at an animal market and on farms. J. Virol.. 2005; 79:11892-11900. DOI | PubMed
- Poon LL. Identification of a novel coronavirus in bats. J. Virol.. 2005; 79:2001-2009. DOI | PubMed
- Wynne JW, Wang LF. Bats and viruses: friend or foe?. PLoS. Pathog.. 2013; 9:e1003651. DOI | PubMed
- Wong S, Lau S, Woo P, Yuen KY. Bats as a continuing source of emerging infections in humans. Rev. Med. Virol.. 2007; 17:67-91. DOI | PubMed
- Lau SK. Severe acute respiratory syndrome coronavirus-like virus in Chinese horseshoe bats. Proc. Natl. Acad. Sci. U. S. A.. 2005; 102:14040-14045. DOI | PubMed
- Ge XY. Isolation and characterization of a bat SARS-like coronavirus that uses the ACE2 receptor. Nature. 2013; 503:535-538. DOI | PubMed
- Yang XL. Isolation and characterization of a novel bat coronavirus closely related to the direct progenitor of severe acute respiratory syndrome coronavirus. J. Virol.. 2015; 90:3253-3256. DOI | PubMed
- Drexler JF. Genomic characterization of severe acute respiratory syndrome-related coronavirus in European bats and classification of coronaviruses based on partial RNA-dependent RNA polymerase gene sequences. J. Virol.. 2010; 84:11336-11349. DOI | PubMed
- Rihtaric D, Hostnik P, Steyer A, Grom J, Toplak I. Identification of SARS-like coronaviruses in horseshoe bats (Rhinolophus hipposideros) in Slovenia. Arch. Virol.. 2010; 155:507-514. DOI | PubMed
- Balboni A, Palladini A, Bogliani G, Battilani M. Detection of a virus related to betacoronaviruses in Italian greater horseshoe bats. Epidemiol. Infect.. 2011; 139:216-219. DOI | PubMed
- Tong S. Detection of novel SARS-like and other coronaviruses in bats from Kenya. Emerg. Infect. Dis.. 2009; 15:482-485. DOI | PubMed
- Quan PL. Identification of a severe acute respiratory syndrome coronavirus-like virus in a leaf-nosed bat in Nigeria. mBio. 2010; 1:pii: e00208-pii: e00210. DOI
- Woo PC. Molecular diversity of coronaviruses in bats. Virology. 2006; 351:180-187. DOI | PubMed
- Hu B, Ge X, Wang LF, Shi Z. Bat origin of human coronaviruses. Virol. J.. 2015; 12:221. DOI | PubMed
- Hu D. Virome analysis for identification of novel mammalian viruses in bats from Southeast China. Sci. Rep.. 2017; 7:10917. DOI | PubMed
- Irwin DM, Kocher TD, Wilson AC. Evolution of the cytochrome b gene of mammals. J. Mol. Evol.. 1991; 32:128-144. DOI | PubMed
- Lau SK. Severe acute respiratory syndrome (SARS) coronavirus ORF8 protein is acquired from SARS-related coronavirus from greater horseshoe bats through recombination. J. Virol.. 2015; 89:10532-10547. DOI | PubMed
- Tong S, Chern SW, Li Y, Pallansch MA, Anderson LJ. Sensitive and broadly reactive reverse transcription-PCR assays to detect novel paramyxoviruses. J. Clin. Microbiol.. 2008; 46:2652-2658. DOI | PubMed
- Fouchier RA. Detection of influenza A viruses from different species by PCR amplification of conserved sequences in the matrix gene. J. Clin. Microbiol.. 2000; 38:4096-4101. PubMed
- Drexler JF, Corman VM, Drosten C. Ecology, evolution and classification of bat coronaviruses in the aftermath of SARS. Antivir. Res.. 2014; 101:45-56. DOI | PubMed
- Hu B. Discovery of a rich gene pool of bat SARS-related coronaviruses provides new insights into the origin of SARS coronavirus. PLoS. Pathog.. 2017; 13:e1006698. DOI | PubMed
- Wu Z. Deciphering the bat virome catalog to better understand the ecological diversity of bat viruses and the bat origin of emerging infectious diseases. Isme. J.. 2016; 10:609-620. DOI | PubMed
- Lau SK. Ecoepidemiology and complete genome comparison of different strains of severe acute respiratory syndrome-related Rhinolophus bat coronavirus in China reveal bats as a reservoir for acute, self-limiting infection that allows recombination events. J. Virol.. 2010; 84:2808-2819. DOI | PubMed
- Li W. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J.. 2005; 24:1634-1643. DOI | PubMed
- Li F. Evidence for a common evolutionary origin of coronavirus spike protein receptor-binding subunits. J. Virol.. 2012; 86:2856-2858. DOI | PubMed
- Menachery VD. SARS-like WIV1-CoV poised for human emergence. Proc. Natl. Acad. Sci. USA. 2016; 113:3048-3053. DOI | PubMed
- Ksiazek TG. A novel coronavirus associated with severe acute respiratory syndrome. N. Engl. J. Med.. 2003; 348:1953-1966. DOI | PubMed
- Lau SK. Complete genome sequence of bat coronavirus HKU2 from Chinese horseshoe bats revealed a much smaller spike gene with a different evolutionary lineage from the rest of the genome. Virology. 2007; 367:428-439. DOI | PubMed
- Falcon A. Detection of alpha and betacoronaviruses in multiple Iberian bat species. Arch. Virol.. 2011; 156:1883-1890. DOI | PubMed
- Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol.. 2016; 33:1870-1874. DOI | PubMed
- Martin DP. RDP3: a flexible and fast computer program for analyzing recombination. Bioinformatics. 2010; 26:2462-2463. DOI | PubMed
- Lole KS. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J. Virol.. 1999; 73:152-160. PubMed
Fonte
Hu D, Zhu C, Ai L, He T, Wang Y, et al. (2018) Genomic characterization and infectivity of a novel SARS-like coronavirus in Chinese bats. Emerging Microbes & Infections 7154. https://doi.org/10.1038/s41426-018-0155-5




