
Introduzione
I virus respiratori di origine umana hanno causato malattie nelle scimmie selvatiche in tutta l’Africa subsahariana e rappresentano una minaccia significativa e crescente per la salute e la conservazione delle scimmie selvatiche[1,2]. Per esempio, le malattie respiratorie sono la principale causa di morbilità e mortalità tra gli scimpanzé(Pan trogloditi) nel Gombe Stream National Park, Tanzania[3,4] e nel Kibale National Park, Uganda[5], due popolazioni che sono state studiate continuamente per decenni. La mortalità da pneumovirus respiratorio antroponotico (famiglia Pneumoviridae) e paramixovirus (famiglia Paramyxoviridae) è stata documentata negli scimpanzé occidentali (P. t. verus) in Costa d’Avorio[1,6], scimpanzé orientali(P.t. schweinfurthii) in Tanzania[7], gorilla di montagna(Gorilla beringei beringei) in Ruanda, gorilla di pianura(G. g. g. gorilla) nella Repubblica Centrafricana[8] e bonobos(P. paniscus) nella Repubblica Democratica del Congo [9]. Anche il rinovirus C[10] e il coronavirus OC43[11] di origine umana hanno causato la mortalità degli scimpanzé in Uganda e una lieve malattia respiratoria in Costa d’Avorio, rispettivamente.
Le somiglianze biologiche tra gli esseri umani e le scimmie li predispongono alla trasmissione incrociata di agenti patogeni[12], e le alterazioni dell’habitat possono esacerbare il contatto tra le specie e il rischio di trasmissione antropogenica[2]. Sebbene le infezioni simultanee di scimmie e di persone con lo stesso virus respiratorio siano state raramente confermate direttamente[8,11], i virus che sono relativamente benigni nell’uomo possono causare focolai letali nelle popolazioni di scimmie, indicando la mancanza di resistenza nelle scimmie. Strategie di prevenzione adeguate hanno incluso il miglioramento dell’igiene e dell’igiene[13], la riduzione delle visite umane[14] e la vaccinazione su larga scala delle scimmie (se un giorno saranno disponibili vaccini efficaci)[15]. Tali politiche potrebbero anche giovare alla salute pubblica umana riducendo il rischio di trasmissione zoonotica[16,17].
Qui, riportiamo focolai simultanei di malattie respiratorie in due comunità di scimpanzé vicine in Uganda, causate da due distinti virus RNA a senso negativo di origine umana. I focolai si sono verificati da dicembre 2016 a febbraio 2017 nelle comunità di scimpanzé Ngogo e Kanyawara nel Parco Nazionale di Kibale[18] (Figura S1). Solo 10 km a parte, le comunità sono interconnesse da una foresta umida sempreverde contigua, separate da una sola comunità di scimpanzé intervenuta che non è attualmente studiata, ed entrambe le comunità hanno generalmente un basso tasso di mortalità [19,20].
Sebbene i focolai di malattie respiratorie negli scimpanzé selvatici siano comuni, le loro cause rimangono spesso non diagnosticate[10]. Le carcasse fresche spesso non vengono recuperate e la lontananza dei siti sul campo complica la conservazione e l’analisi dei campioni. La diagnostica non invasiva (di solito da feci) si è dimostrata altamente informativa[6], ma i risultati negativi in questi casi sono inconcludenti[10]. Nel caso in questione, il rapido recupero delle carcasse, la presenza di veterinari addestrati e la disponibilità di capacità di base di laboratorio sul campo hanno fornito un’opportunità per le diagnosi eziologiche. Inoltre, la raccolta coordinata e prospettica di dati osservazionali tra i due siti ha offerto un’insolita possibilità di confrontare direttamente le caratteristiche cliniche ed epidemiologiche dei focolai.
Risultati
Dal 31 dicembre 2016 all’8 febbraio 2017, la comunità di scimpanzé Ngogo del Parco Nazionale di Kibale, Uganda (Figura S1), ha sperimentato un’epidemia di grave malattia respiratoria. Nello stesso periodo, anche la vicina comunità di Kanyawara (Figura S1) ha sperimentato un’epidemia di malattia respiratoria. All’inizio delle epidemie, la comunità di Ngogo era composta da 205 scimpanzé di età compresa tra <1 anno e 67 anni, e la comunità di Kanyawara era composta da 55 scimpanzé di età compresa tra <1 anno e 51 anni. Le curve delle epidemie(figura 1) mostrano che le epidemie di Ngogo e Kanyawara si sono verificate ciascuna in un’unica fase, con la maggior parte dei casi che si sono verificati nel gennaio 2017. A Ngogo, il 43,8% degli scimpanzé osservati tra il 3 dicembre 2016 e il 28 febbraio 2017 ha mostrato segni respiratori. A Kanyawara, il 69,1% degli scimpanzé osservati nello stesso periodo ha mostrato segni respiratori. A Ngogo, 25 scimpanzé (12,2%) sono morti durante il periodo dell’epidemia(Figura 1). Al contrario, a Kanyawara nessuno scimpanzé è morto durante il periodo dell’epidemia, ad eccezione di una femmina che si è ripresa dalla malattia ed è morta a seguito di un’aggressione conspecifica (vedi sotto). I segni respiratori consistevano in tosse, starnuti, dispnea ed essudato nasale; altri segni includevano letargia, immobilità e drammatica perdita delle condizioni corporee (Figura S2).Figura 1.Curve epidemiche per le epidemie di malattie respiratorie degli scimpanzé del 2016/2017 nelle comunità di Ngogo (in alto) e Kanyawara (in basso) nel Parco Nazionale di Kibale, Uganda. Le linee tratteggiate indicano i tassi medi dei segni respiratori del 2017 (linea inferiore) e 2 deviazioni standard sopra la media (linea superiore). Gli asterischi sopra le barre indicano la tempistica stimata dei singoli eventi di mortalità attribuiti alle malattie respiratorie. La casella grigia nel grafico inferiore rappresenta le date con dati mancanti (nessun segno clinico è stato osservato a Kanyawara prima di questo periodo).
La modellazione epidemiologica dei focolai di Ngogo e Kanyawara(Tabella 1 e Figura S3) ha prodotto stime del tasso di trasmissione giornaliera di 1,13 e 0,338, e durate di infettività di 1,12 e 4,55 giorni, rispettivamente. Questi parametri hanno prodotto numeri riproduttivi di base(R0) di 1,27 e 1,48 per Ngogo e Kanyawara, rispettivamente. Questi sono simili ai valori stimati da un’epidemia di rinovirus C a Kanyawara nel 2013(Tabella 1), durante la quale è morto l’8,9% degli scimpanzé, e ai valori pubblicati per il “raffreddore comune” umano [10]. Tuttavia, i limiti di fiducia del 95% intorno a queste stime non si sovrappongono per la velocità di trasmissione giornaliera di tutti e tre i focolai e la durata dell’infettività e R0in Ngogo (entrambi inferiori a quelli di Kanyawara nel 2013 o 2016/2017).Tabella 1.Parametri epidemiologici derivati da modelli matematici SIR del metapneumovirus e respirovirus umano 3 focolai negli scimpanzé delle comunità Ngogo e Kanyawara, rispettivamente. Parametri derivati in modo simile da un focolaio di rinovirus C umano del 2013 (solo Kanyawara) sono mostrati per confronto (intervalli di confidenza del 95% tra parentesi)[10]. Ngogo 2017Kanyawara 2017Kanyawara 2013Agente causativoMetapneumovirusMetapneumovirusHuman respirovirus 3Rhinovirus CEpidemic size78 individui38 individui31,7 individuiDurata dell’epidemia35 giorni>33 giorni20.7 giorniMortalità25 individui0 individui0 individui5 individuiTasso di trasmissione(β )1,13/giorno (1,12-1,15)0,326/giorno (0,312-0,340)0,68/giorno (0,44-0,85)Durata dell’infettività (1/γ )1.12 giorni (1,10-1,13)4,55 giorni (4,30-4,83)3,2 giorni (1,6-5,8)R0 (β /γ)1, 27 (1,23-1,30)1,48 (1,34-1,64)1,83 (1,38-2,56)
L’analisi dei fattori di rischio (Tabella S2) ha mostrato che l’età prevedeva in modo significativo la morbilità a entrambi i Ngogo (χ 2=10.097, DF=3, p=0 ,018) e Kanyawara (χ 2=12,154, DF=3, p=0,007). In entrambe le comunità, i segni respiratori sono stati osservati meno frequentemente tra i neonati e sono aumentati attraverso le categorie di età successive. Il sesso non ha influito sulla morbilità o sulla mortalità a Ngogo, ma le femmine hanno avuto una probabilità significativamente maggiore di mostrare segni respiratori a Kanyawara rispetto ai maschi (χ 2 = 6,310, DF = 1, p = 0,012). A Ngogo, l’età prevista per la mortalità (χ 2 = 19,153, DF = 3, p<0,001), con la mortalità più alta tra i neonati (OR 5,01, 95% CI: 1,53-19,56) e gli individui ≥30 anni (OR 3,86, 95% CI: 1,16-15,15), rispetto alle età intermedie.
A Ngogo, la carcassa di una femmina di scimpanzé di 20 anni è stata recuperata subito dopo l’insorgenza dei segni respiratori e la successiva morte. L’analisi post-mortem di questo individuo ha rivelato il consolidamento dei lobi dipendenti di entrambi i polmoni e un versamento pericardico sierosanguineo, ma non altre gravi anomalie patologiche (Figura S4). A Kanyawara, la carcassa di una femmina di scimpanzé di 22 anni è stata recuperata circa 10 giorni dopo aver recuperato i segni respiratori (ma ancora debole), subito dopo essere stata attaccata da conspecifici (per ragioni poco chiare). L’analisi post-mortem di questo individuo ha mostrato una pleuropolmonite grave e diffusa con aderenze fibrinose alla parete toracica e un grave consolidamento di tutti i lobi di entrambi i polmoni, nonché un versamento pericardico sierosanguineo (Figura S4).
L’analisi di campioni fecali accoppiati (prima e durante il periodo di epidemia) utilizzando un test Luminex che i test per una serie di agenti respiratori umani hanno rivelato diverse eziologie virali per ogni comunità(Tabella 2). Il metapneumovirus (MPV, Pneumoviridae: Metapneumovirus) è stato rilevato in 7 degli 11 individui (63,6%) di scimpanzé Ngogo che presentavano segni clinici durante, ma non prima del periodo di epidemia (P=0,0030 esatto di Fisher). Il respirovirus umano 3 (HRV3; Paramyxoviridae; Respirovirus, precedentemente noto come virus parainfluenzale 3) è stato rilevato in 5 dei 14 individui (35,7%) degli scimpanzé Kanyawara che mostravano segni clinici durante, ma non prima, il periodo di epidemia (P=0,0005 esatto di Fisher). Gli Adenovirus(Adenoviridae) erano presenti nei campioni sia di Ngogo (36,4%) che di Kanyawara (78,6%) ma non hanno mostrato alcuna associazione con il periodo dell’epidemia (Fisher’s esatto P=0,454545 e 0,5291, rispettivamente) e sono stati precedentemente caratterizzati in questa popolazione a frequenze comparabili [10]. Anche gli enterovirus(Picornaviridae) erano presenti a bassa frequenza in campioni di entrambe le comunità, analogamente non hanno mostrato alcuna associazione con il periodo dell’epidemia (P=1.000 esatto di Fisher in entrambi i casi), e sono stati anche precedentemente caratterizzati in questa popolazione a frequenze comparabili [10].
Tabella 2.Risultati dei test diagnostici effettuati dal saggio Luminex su campioni fecali accoppiati di scimpanzé delle comunità di Ngogo e Kanyawara prima (4° trimestre 2016) e durante (1° trimestre 2017) del periodo di epidemia, rispettivamente.Ngogo AdV EV MPV MPV HRV3ID Q4/16Q1/17 Q4/16Q1/17 Q4/16Q1/17 Q4/16Q1/17Q4/16Q1/17AB BABT CNDX GTMI PTWI WNZLKanyawara AdV EV MPV MPV HRV3ID Q4/16Q1/17 Q4/16Q1/17 Q4/16Q1/17 Q4/16Q1/17Q4/16Q1/17AL ANAT AZBT LKOG POQV RD UMWC WLWOI virus sono adenovirus (AdV), enterovirus (EV), metapneumovirus (MPV) e respirovirus umano 3 (HRV3). Le cellule sfumate indicano risultati positivi per gli individui elencati con l’abbreviazione di due lettere nella prima colonna, che si sono tutti ammalati clinicamente durante il periodo dell’epidemia. I risultati per altri agenti patogeni virali e batterici inclusi nel test sono stati negativi.
L’analisi metagenomica dei campioni di tampone del tratto respiratorio dello scimpanzé esaminati post mortem a Ngogo ha dato 16.107.924 letture dopo la rifilatura, di cui 27.393 assemblate per produrre un genoma MPV codificante-completo di 13.230 basi con una copertura media di 196, coerente con i risultati Luminex sopra descritti. Questo genoma (numero di adesione GenBank MH428626) è stato più simile (98,69%) a una variante di derivazione umana del 2010 proveniente dal Brasile (numero di adesione GenBank MG431250). Incredibilmente, il virus era quasi altrettanto simile (98,67%) a una variante di un gorilla di montagna del Ruanda nel 2008 (numero di adesione GenBank HM197719) rilevata durante un’epidemia letale[21]. L’RNA del MPV era presente in tutte le sezioni delle vie respiratorie, compreso il parenchima polmonare, con la proporzione di sequenza virale che si legge in diminuzione monotona dalla parte superiore a quella inferiore delle vie respiratorie (Figura S5). Sequenziamento di una porzione 480 nucleotide del gene F virale del gene F da campioni fecali ha avuto successo per altri tre scimpanzé Luminex-positivi a Ngogo (AB, MI e WI; Tabella 2), producendo sequenze identiche all’interno di questa regione genomica variabile (GenBank numeri di adesione GenBank MH428628- MH428630).
Al contrario, né l’HRV3 né qualsiasi altro virus è stato rilevato nel tratto respiratorio dello scimpanzé morto a Kanyawara, probabilmente a causa di una precedente infezione e della clearance virale. Un genoma completo di codifica dell’HRV3 (15.407 basi) è stato quindi ricostruito da un campione fecale di questo stesso individuo raccolto quando tossiva circa 2 settimane prima, utilizzando la PCR con primer specifici per il virus e il sequenziamento Sanger (Tabella S1). Questo genoma (numero di adesione GenBank MH428627) era più simile (99,38%) a una variante di derivazione umana del 2009 proveniente dagli USA (numero di adesione GenBank KY674929). Sequenziamento di una variabile ed epidemiologicamente informativo 348-nucleotide porzione del gene F virale del gene F da campioni fecali ha avuto successo per altri tre scimpanzé Luminex-positivi a Kanyawara (AL, AN e AZ; Tabella 2), producendo sequenze identiche all’interno di questa regione genomica (GenBank numeri di adesione GenBank MH428631-MH428633). La rianalisi dei dati metagenomici delle vie respiratorie di questo individuo e dell’individuo di Ngogo ha rivelato che una piccola percentuale di letture nelle vie respiratorie superiori di entrambi gli animali (0,05% e 0.16%, rispettivamente) mappati al genoma di riferimento dello Staphylococcus pneumoniae (numero di adesione GenBank NC_003098), che può infettare secondariamente gli scimpanzé durante le epidemie di malattie respiratorie virali[9,22], indicando la presenza di questo o di un batterio correlato; tuttavia, nessuna lettura mappata a questo organismo nei polmoni di entrambi gli animali.
L’analisi filogenetica ha rivelato che il virus MPV dell’epidemia di Ngogo è stato classificato all’interno di un sottoclave di virus del sottotipo B2[23] provenienti da Brasile, Perù, Ruanda e Stati Uniti(Figura 2). Questo sottoclade contiene virus raccolti di recente (2009-2015), compresa la variante dei gorilla di montagna del Ruanda. La variante MPV di Ngogo appartiene a un sottotipo diverso rispetto a un virus B1 precedentemente segnalato da un focolaio di scimpanzé in Tanzania[7], ed è meno strettamente correlata a un sottotipo B2 precedentemente segnalato da un focolaio di scimpanzé in Costa d’Avorio[1] rispetto ad altre varianti all’interno del suo sottoclade, inclusa la variante derivata dai gorilla, basata sulla regione nucleotide 867 disponibile del gene virale P (numeri di adesione GenBank EU240454-EU240456; non mostrato).
Figura 2.Alberi filogenetici a massima probabilità di metapneumovirus (MPV) da Ngogo (in alto) e respirovirus umano 3 (HRV3) da Kibale (in basso) costruiti con allineamenti nucleotidici dei genomi virali completi di codifica. I nomi dei taxon indicano il paese di origine (AU = Australia; BR = Brasile; CA = Canada; CH = Canada.Cile; FR = Francia; JP = Giappone; MX = Messico; NL = Paesi Bassi; PE = PEPerù; RW = Ruanda; SA = Sud Africa; TA = Taiwan; TH = Thailandia; UG = Uganda; US = USA; ZA = Zambia), anno di raccolta (se specificato), e numero di adesione GenBank tra parentesi. Per l’HRV3, i clades principali sono stati collassati per la visualizzazione, e il numero di sequenze all’interno di ogni clade è indicato tra parentesi. Le sequenze generate in questo studio dagli scimpanzé sono in grassetto con le sagome in alto. Le ellissi riempite indicano valori di bootstrap del 100%; le ellissi grigie indicano valori di bootstrap ≥75%. Le barre di scala indicano sostituzioni nucleotidiche per sito. Gli asterischi indicano sequenze di altri primati non umani (vedi testo per i dettagli). I dettagli completi di tutte le sequenze incluse sono riportati nella Tabella S3.
L’analisi filogenetica ha rivelato che l’HRV3 dell’epidemia di Kanyawara è stato classificato all’interno di un sottoclave di virus di origine umana molto simili provenienti dal Perù e dagli Stati Uniti e raccolti tra il 2006 e il 2015 (non più recentemente di altri sottoclavi; Figura 2). In particolare, l’HRV3 di Kanyawara era diverso dai virus di altri primati non umani, compresi i virus dei babbuini selvatici dello Zambia(Papio cynocephalus)[24] e l’agente scimmiesco 10, originariamente isolato da una scimmia blu(Cercopithecus mitis) in Sud Africa[25].
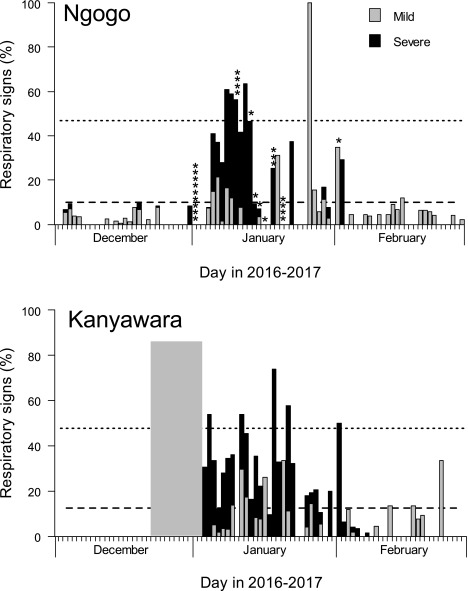
Figura 1.Curve epidemiche per le epidemie di malattie respiratorie degli scimpanzé del 2016/2017 nelle comunità di Ngogo (in alto) e Kanyawara (in basso) nel Parco Nazionale di Kibale, Uganda. Le linee tratteggiate indicano i tassi medi dei segni respiratori del 2017 (linea inferiore) e 2 deviazioni standard al di sopra della media (linea superiore). Gli asterischi sopra le barre indicano la tempistica stimata dei singoli eventi di mortalità attribuiti alle malattie respiratorie. La casella grigia nel grafico inferiore rappresenta le date con dati mancanti (nessun segno clinico è stato osservato a Kanyawara prima di questo periodo).

Figura 2.Figura 2. Alberi filogenetici di massima probabilità di metapneumovirus (MPV) da Ngogo (in alto) e respirovirus umano 3 (HRV3) da Kibale (in basso) costruiti con allineamenti nucleotidici dei genomi virali completi di codifica. I nomi dei taxon indicano il paese di origine (AU = Australia; BR = Brasile; CA = Canada; CH = Canada.Cile; FR = Francia; JP = Giappone; MX = Messico; NL = Paesi Bassi; PE = PEPerù; RW = Ruanda; SA = Sud Africa; TA = Taiwan; TH = Thailandia; UG = Uganda; US = USA; ZA = Zambia), anno di raccolta (se specificato), e numero di adesione GenBank tra parentesi. Per l’HRV3, i clades principali sono stati collassati per la visualizzazione, e il numero di sequenze all’interno di ogni clade è indicato tra parentesi. Le sequenze generate in questo studio dagli scimpanzé sono in grassetto con le sagome in alto. Le ellissi riempite indicano valori di bootstrap del 100%; le ellissi grigie indicano valori di bootstrap ≥75%. Le barre di scala indicano sostituzioni nucleotidiche per sito. Gli asterischi indicano sequenze di altri primati non umani (vedi testo per i dettagli). I dettagli completi di tutte le sequenze incluse sono riportati nella Tabella S3.
Discussione
Focolai di malattie respiratorie gravi e talvolta letali sono comuni negli scimpanzé in tutta l’Africa subsahariana, ma le loro cause rimangono spesso indeterminate[2,10]. Di conseguenza, e a causa delle differenze nei protocolli sul campo tra i siti di ricerca, si è rivelato difficile confrontare direttamente questi focolai. La simultaneità dei due focolai del 2016/2017 a Ngogo e Kanyawara, insieme al coordinamento dei dati e degli sforzi di raccolta dei campioni tra i due siti, ha permesso di effettuare un raro confronto diretto. Le analisi dimostrano che i due virus hanno mostrato diverse patogenicità e diverse dinamiche epidemiologiche nelle due comunità di scimpanzé.
Il virus MPV a Ngogo ha causato una mortalità sostanzialmente più elevata rispetto all’HRV3 a Kanyawara, nonostante abbia causato una morbilità apparentemente inferiore. Tuttavia, la velocità di trasmissione giornaliera stimata e la durata dell’infettività del virus MPV a Ngogo differivano dai valori corrispondenti per l’HRV3 a Kanyawara(Tabella 1), suggerendo che, nonostante valori R0comparabili, i virus mostravano dinamiche epidemiologiche diverse. La maggiore mortalità dovuta al MPV nei neonati e negli adulti più anziani rispetto alle categorie di età intermedie è congruente con i dati dell’uomo, dove i neonati, i bambini e gli adulti più anziani sono più suscettibili a gravi manifestazioni cliniche di infezione da MPV[26,27], così come con i dati demografici di precedenti focolai di MPV negli scimpanzé[1,6].
Nessuno dei due virus è stato rilevato negli scimpanzé immediatamente prima dei focolai(Tabella 2). Il virus MPV è stato rilevato nel 63,6% degli scimpanzé Ngogo colpiti campionati durante l’epidemia e l’HRV3 è stato rilevato nel 35,7% degli scimpanzé Kanyawara colpiti campionati durante l’epidemia. Le sequenze virali degli individui colpiti erano identiche all’interno della regione variabile sequenziata. MPV e HRV3 erano quindi probabili cause di origine singola delle rispettive epidemie. Adenovirus ed enterovirus erano presenti sia prima che durante le epidemie a frequenze statisticamente indistinguibili (AdV) e basse (EV). Gli adenovirus sono presenti sia nelle scimmie selvatiche sane e malate[28-31] che negli scimpanzé durante le epidemie respiratorie [32], anche a Kanyawara [10], il che ha portato al dibattito sul loro ruolo eziologico; tuttavia, i nostri risultati confermano le precedenti conclusioni secondo cui questi virus sono molto probabilmente benigni. I nostri risultati supportano una conclusione simile per gli enterovirus, con le notevoli eccezioni del virus della poliomielite[3,33] e dei rinovirus[10], che possono causare malattie mortali negli scimpanzé.
Notiamo che, al momento delle epidemie, si presumeva che le epidemie di Ngogo e Kanyawara avessero la stessa causa, con il timore concomitante che un agente infettivo si stesse diffondendo rapidamente nel Parco nazionale di Kibale. Inoltre, l’influenza aviaria era stata recentemente segnalata negli uccelli acquatici del lago Vittoria, dall’altra parte dell’Uganda[34]. I nostri risultati evidenziano come una maggiore vigilanza sulle malattie infettive emergenti possa talvolta portare a conclusioni errate. I comuni fattori ambientali (ad esempio il tempo, i modelli di visita dell’uomo) possono far precipitare contemporaneamente focolai di virus antroponotici distinti nelle scimmie selvatiche, creando la comparsa di un’unica malattia con un modello stagionale. I nostri risultati suggeriscono anche che la trasmissione di virus respiratori tra comunità di scimpanzé è probabilmente non comune, nonostante gli alti tassi di trasmissione all’interno delle comunità di scimpanzé. Gli scimpanzé delle comunità vicine interagiscono raramente[33], il che può limitare le opportunità di trasmissione intercomunitaria anche di microbi altamente contagiosi.
La MPV e la HRV3 rappresentano entrambe un onere significativo per la salute pubblica globale[35]. L’infezione da MPV nell’uomo causa tosse, secrezioni nasali e perdita di peso[36]; nei modelli non umani i segni sono spesso più gravi che per altri virus correlati[37]. L’MPV causa anche malattie cliniche negli scimpanzé infettati sperimentalmente[38] e focolai gravi e talvolta letali di malattie respiratorie negli scimpanzé selvatici e nei gorilla, spesso con modelli demografici di infezione simili a quelli descritti nel presente documento[1,6,7,21], così come un virus correlato, il virus sinciziale respiratorio(Pneumoviridae, Orthopneumovirus)[1]. I nostri risultati mostrano che questa virulenza relativamente elevata di MPV è ricapitolata negli scimpanzé infetti in natura.
L’HRV3 causa malattie respiratorie e l’esacerbazione dell’asma nelle persone in tutto il mondo, soprattutto nei bambini e negli anziani, ma è meno virulento dell’MPV[39]. L’HRV3 ha un’ampia gamma di ospiti, tra cui primati, ruminanti e roditori[40]. L’agente scimmiesco 10, isolato per la prima volta nel 1963 da una scimmia blu apparentemente sana[41], è stato successivamente identificato come HRV3 e attribuito alla trasmissione antroponotica[25]. L’HRV3 è stato rilevato nelle vie respiratorie di babbuini gialli selvatici apparentemente sani in Zambia[24]. L’infezione sperimentale di uistitì(Saguinus mystax) con la HRV3 causa una malattia acuta e trasmissibile[42], ma l’infezione sperimentale di altri primati, tra cui i macachi del Rhesus(Macaca mulatta) e gli scimpanzé[43], è più lieve. I nostri risultati rispecchiano questi risultati e, più in generale, mostrano che alcuni virus respiratori RNA a senso negativo di origine umana causano solo infezioni lievi e autolimitanti negli scimpanzé selvatici.
Non possiamo escludere il ruolo di altri agenti patogeni co-infiammatori nel modulare la patogenicità di MPV e HRV3 nelle popolazioni oggetto del nostro studio. Ad esempio, l’HRV3 può causare malattie delle vie respiratorie superiori e predisporre gli scimpanzé a infezioni invasive da pneumococco[44], e il batterio Streptococcus pneumoniae coesiste con i metapneumovirus umani e i virus sinciziali respiratori sia in natura che nelle scimmie in cattività[9,22]. Il nostro riscontro di una bassa percentuale di sequenza metagenomica indica la presenza di S. pneumoniae (o di un batterio correlato) nelle vie respiratorie superiori di entrambi gli individui necropsiati, ma non nei loro polmoni, supporta l’idea che gli scimpanzé selvatici ospitano microbi che potrebbero facilmente invadere le vie respiratorie inferiori a seguito di un’infezione virale primaria. Indagini sulla popolazione di microbi endemici e valutazioni sierologiche di una precedente esposizione ad agenti che potrebbero predisporre gli scimpanzé ad un’infezione o migliorare la patogenesi potrebbero rivelarsi utili.
I segni clinici negli scimpanzé Ngogo erano congruenti con quelli degli esseri umani gravemente infetti[36]. Una femmina adulta di Ngogo (“Kidman”) ha mostrato un ansimare persistente dopo l’epidemia; nei bambini umani che soffrono di gravi infezioni da monovolume, i successivi episodi di ansimazione non sono rari [45]. L’analisi post-mortem di un individuo a Ngogo ha rivelato una patologia delle vie respiratorie sorprendentemente poco grave, coerente con l’infezione acuta e la morte rapida, e l’MPV è stato recuperato da tutte le sezioni delle vie respiratorie di quell’individuo, compreso il parenchima polmonare (Figure S4 e S5). Al contrario, l’analisi post-mortem di un individuo a Kanyawara ha rivelato una patologia avanzata delle vie respiratorie, coerente con l’infezione a lungo termine (Figura S5), ma non abbiamo recuperato l’HRV3 dalle vie respiratorie di questo individuo, nonostante il recupero di un genoma completo HRV3 dalle feci di questo stesso individuo raccolte circa 2 settimane prima. Questo individuo aveva probabilmente recuperato dalla sfida virale iniziale, ha eliminato il virus e forse ha acquisito un’infezione batterica secondaria[9,22]. In particolare, entrambi gli individui hanno avuto effusioni pericardiche, che negli esseri umani sono rare ma gravi sequele di alcune infezioni virali[46].
Le analisi filogenetiche hanno rivelato che i virus MPV di Ngogo e HRV3 di Kanyawara sono strettamente correlati ai virus umani circolanti a livello globale, indicando fonti antroponetiche indipendenti per entrambi i virus. Le associazioni geografiche apparenti in queste filogenie rappresentano probabilmente una distorsione nei dati sequenziali disponibili (ad esempio, un’elevata rappresentazione dei virus provenienti dal Perù e dagli USA; tabella S3). La variante Ngogo MPV cluster di varianti di Ngogo all’interno di un sottoclave di virus che è più recente (2008-2017) rispetto ad altri sottocladi, riflettendo forse la sostituzione antigenica dei ceppi di MPV nel tempo. In particolare, la variante Ngogo è molto simile a un virus derivante da un’infezione mortale di un gorilla di montagna in Ruanda nel 2009[21]. Questa particolare stirpe virale può quindi avere una propensione a infettare le scimmie, o può semplicemente circolare ampiamente in Africa orientale. Il virus è diverso, tuttavia, dai virus letali precedentemente descritti, virus letali degli scimpanzé selvatici in Tanzania[7] e in Costa d’Avorio[1], indicando che molteplici varianti di MPV possono infettare e uccidere gli scimpanzé selvatici.
Le vie attraverso le quali questi virus sono entrati negli scimpanzé dagli esseri umani rimangono fastidiosamente oscure. Le persone frequentano gli habitat di entrambe le comunità di scimpanzé per la ricerca e le attività associate (non solo per gli scimpanzé) e per viaggiare o raccogliere prodotti della foresta[47]. Il turismo è talvolta citato come un rischio per la trasmissione antroponotica dei virus alle grandi scimmie e contribuisce a informare le attuali raccomandazioni dell’Unione Internazionale per la Conservazione della Natura (IUCN) per prevenire tale trasmissione, come i requisiti di vaccinazione dei visitatori, le distanze minime di osservazione, il gel disinfettante a base di alcol e le maschere facciali[13]. Tuttavia, né Ngogo né Kanyawara sono un sito turistico. Ngogo confina con un sito turistico (Kanyanchu) che riceve migliaia di visitatori ogni anno, ma non ci sono state segnalazioni di una concomitante epidemia di malattie respiratorie negli scimpanzé di Kanyanchu, anche se eventi simili si sono verificati in altri periodi. Nel Parco nazionale di Gombe in Tanzania, il rifornimento di banane agli scimpanzé (una pratica abbandonata) è stato, insieme alla stagione, il più forte predittore dei segni respiratori degli scimpanzé[48], che ha colpito tra lo 0% e il 9% degli scimpanzé al mese tra il 2005 e il 2012[4]. Tuttavia, né lo scimpanzé Ngogo né lo scimpanzé Kanyawara sono forniti, e sono in vigore misure di biosicurezza basate sulle raccomandazioni dell’IUCN[13] per i visitatori di entrambi i siti. A Kanyawara, i dati relativi a 22 anni mostrano un andamento stagionale delle malattie respiratorie negli scimpanzé, che di solito raggiunge il suo apice a marzo, ma non vi è alcuna correlazione tra i segni clinici negli scimpanzé e l’andamento stagionale delle malattie respiratorie negli esseri umani che vivono nelle vicinanze[5]. Anche il ruolo di altre specie nella trasmissione di questi virus dall’uomo agli scimpanzé non è chiaro. Il Parco Nazionale di Kibale contiene una diversità di specie di primati e gruppi sociali, nessuno dei quali è stato osservato con segni respiratori al momento dell’epidemia, ma non si possono escludere modelli complessi di trasmissione incrociata tra specie dall’uomo agli scimpanzé. Tali possibilità potrebbero essere studiate attraverso un ampio campionamento di persone (specialmente il personale di ricerca) e di fauna selvatica durante le future epidemie, comprese le valutazioni sierologiche dell’esposizione precedente.
Nel complesso, questi risultati ampliano la nostra comprensione dei virus umani che possono causare malattie negli scimpanzé selvatici e mostrano che le loro manifestazioni cliniche possono variare notevolmente. Il nostro studio mostra anche che le analisi epidemiologiche, rese possibili da una raccolta di dati osservazionali prospettici e coordinati, possono essere in grado di distinguere i virus in base ai modelli di infezione in fase iniziale. In tal caso, le analisi epidemiologiche “in tempo reale” sarebbero un prezioso complemento ai test diagnostici molecolari, in quanto potrebbero aiutare a guidare le strategie di risposta al progredire delle epidemie. Le analisi epidemiologiche potrebbero anche informare i futuri sforzi di monitoraggio per individuare e ridurre al minimo l’impatto degli eventi di trasmissione antropogenica sulle popolazioni di scimmie selvatiche.
Materiali e metodi
Dichiarazione etica
Lo studio è stato osservativo e non invasivo. I protocolli sono stati approvati dai comitati istituzionali per la cura e l’uso degli animali (IACUC) dell’Università di Harvard (protocollo 96-03) e dell’Università del New Mexico (protocollo 14-101186-MCC) e sono stati esentati dalla IACUC dell’Università di Boston e dall’Università del Michigan. I protocolli hanno seguito il Weatherall Report, NIH Guide for the Care and Use of Laboratory Animals, USDA Animal Welfare Act, Institute for Laboratory Animal Research Guide for the Care and Use of Laboratory Animals, US Public Health Service e National Academies of Sciences National Research Council, e US Centers for Disease Control and Prevention.
Dati osservazionali
Durante le epidemie, assistenti sul campo addestrati hanno raccolto quotidianamente dati di osservazione a livello individuale sugli scimpanzé. Gli scimpanzé delle comunità Ngogo e Kanyawara sono identificabili individualmente, rendendo possibile la raccolta di tali dati. Abbiamo compilato questi dati in misure di segni clinici (tosse o starnuti, ulteriormente classificati come lievi o gravi) e tempo di osservazione per scimpanzé. Abbiamo stimato le date degli eventi di mortalità come il punto centrale dell’ultima data in cui uno scimpanzé è stato visto vivo e la prima data in cui è stato assente da un gruppo che comprendeva soci frequenti, compresa la prole dipendente.
Analisi epidemiologiche
Per dedurre i parametri di trasmissione epidemiologica, abbiamo costruito due modelli matematici SIR (Susceptible-Infectious-Removed) – uno per ogni comunità – seguendo Althaus [49], adattando le curve ai dati di incidenza cumulativa utilizzando l’algoritmo di ottimizzazione Nelder e Mead nel pacchetto optim in R, versione 3.3.2 [50]. Per entrambe le comunità, abbiamo assunto la libera commistione e non abbiamo incorporato l’eterogeneità dell’interazione sociale per minimizzare la complessità del modello, come abbiamo fatto in precedenza per un’epidemia di rinovirus C a Kanyawara[10].
Abbiamo analizzato i fattori di rischio per la morbilità e la mortalità utilizzando un modello lineare generalizzato (GLM) con distribuzione binomiale e funzione log-link g in R[51]. L’età, il sesso e la loro interazione sono stati inclusi come variabili predittive e le ore di osservazione sono state incluse come variabile di controllo. L’età è stata modellata come variabile categoriale con quattro livelli: neonati (<5 anni), giovani (5-14,9 anni), adulti (15-29,9 anni) e più anziani (≥30 anni). A Ngogo, dove si è verificata la mortalità, 13 individui non osservati con segni clinici durante il periodo dell’epidemia che sono scomparsi sono stati codificati come morti e come positivi ai segni respiratori. Poiché nessuno di questi individui era una femmina adolescente (l’unica categoria di scimpanzé di sesso maschile che può migrare da una comunità all’altra), la loro improvvisa scomparsa (e il fatto che da allora non sono mai stati più visti) conferma che in realtà erano morti.
Diagnostica molecolare
Abbiamo raccolto campioni fecali da singoli scimpanzé di entrambe le comunità durante il periodo dell’epidemia. Due millilitri di feci sono stati immessi immediatamente nel tampone RNAlater (Thermo Fisher Scientific, Waltham, MA, USA) con un rapporto 1:1, omogeneizzati e conservati a -20°C fino all’esportazione negli Stati Uniti. I campioni fecali accoppiati erano disponibili da 25 scimpanzé clinicamente malati (11 da Ngogo e 14 da Kanyawara) durante e prima (quarto trimestre del 2016) del periodo dell’epidemia, e li abbiamo testati per una serie di agenti patogeni respiratori utilizzando il pannello patogeno respiratorio NxTAG (Luminex Corporation, Austin, TX, USA) come precedentemente descritto[10], che testa il virus dell’influenza A (sottotipi multipli), virus sinciziali respiratori umani A e B, coronavirus (sottotipi multipli), metapneumovirus umani, rinovirus/enterovirus, adenovirus, virus parainfluenzali 1-4, bocavirus, e gli agenti patogeni batterici Chlamydophila pneumoniae e Mycoplasma pneumoniae.
Le necroscopie sono state eseguite su due scimpanzé recuperati subito dopo la morte (le uniche due carcasse recuperate, nonostante gli sforzi profusi) da veterinari addestrati che indossavano adeguati dispositivi di protezione individuale. Uno scimpanzé di Ngogo è morto il 16 gennaio 2017 (“Stella”, una femmina di 20 anni) e l’altro scimpanzé di Kanyawara è morto il 13 gennaio 2017 (“Ruanda”, una femmina di 22 anni). In quest’ultimo caso, l’individuo aveva tossito 10 giorni prima ed era stato attaccato ripetutamente da conspecifici mentre era ancora debole; il trauma risultante sembrava essere la causa immediata della morte. I campioni (tamponi sterili con fusto in plastica e punte di dacron) sono stati raccolti dal naso, dalla laringe, dalla trachea, dai bronchi e dal parenchima polmonare di entrambi gli individui e conservati in un tampone RNAlater 0.25 ml a -20°C. Tamponi sono stati omogeneizzati e RNA totale è stato estratto e convertito in librerie cDNA sul campo, poi preparato per il sequenziamento su uno strumento Illumina MiSeq (Illumina, San Diego, CA, USA) utilizzando 300 bp paired-end leggere chimica come descritto in precedenza [10].
Sequenziamento del genoma del virus e analisi filogenetiche
La reazione a catena della polimerasi (PCR) e il sequenziamento Sanger sono stati utilizzati per completare i genomi virali e per caratterizzare i virus rilevati dal saggio Luminex nei campioni fecali (Tabella S1). Per dedurre le relazioni filogenetiche tra i due virus degli scimpanzé e le sequenze virali pubblicate, abbiamo analizzato gli allineamenti genomici completi di tutti i virus disponibili in GenBank (lunghezze di allineamento 13.234 e 15.330 posizioni, rispettivamente) utilizzando PhyML 3.0[52] con 1000 repliche di bootstrap dei dati per valutare la fiducia statistica nei clades, e abbiamo visualizzato gli alberi risultanti utilizzando FigTree 1.4.3[53,54].
Disponibilità dei dati: I dati sono disponibili in GenBank con i numeri di adesione MH428626-MH428633.
References
- Köndgen S, Kühl H, N’Goran PK. Pandemic human viruses cause decline of endangered great apes. Curr Biol. 2008; 18:260-264. DOI | PubMed
- Dunay E, Apakupakul K, Leard S. Pathogen transmission from humans to great apes is a growing threat to primate conservation. Ecohealth. 2018; 15:148-162. DOI | PubMed
- Williams JM, Lonsdorf EV, Wilson ML. Causes of death in the Kasekela chimpanzees of Gombe National Park, Tanzania. Am J Primatol. 2008; 70:766-777. DOI | PubMed
- Lonsdorf EV, Gillespie TR, Wolf TM. Socioecological correlates of clinical signs in two communities of wild chimpanzees (Pan troglodytes) at Gombe National Park, Tanzania. Am J Primatol. 2018; 80:e22562. DOI
- Emery Thompson M, Machanda ZP, Scully EJ. Risk factors for respiratory illness in a community of wild chimpanzees (Pan troglodytes schweinfurthii). R Soc Open Sci. 2018; 5:180840. DOI | PubMed
- Kondgen S, Schenk S, Pauli G. Noninvasive monitoring of respiratory viruses in wild chimpanzees. Ecohealth. 2010; 7:332-341. DOI | PubMed
- Kaur T, Singh J, Tong S. Descriptive epidemiology of fatal respiratory outbreaks and detection of a human-related metapneumovirus in wild chimpanzees (Pan troglodytes) at Mahale Mountains National Park, western Tanzania. Am J Primatol. 2008; 70:755-765. DOI | PubMed
- Grutzmacher KS, Köndgen S, Keil V. Codetection of respiratory syncytial virus in habituated wild western lowland gorillas and humans during a respiratory disease outbreak. Ecohealth. 2016; 13:499-510. DOI | PubMed
- Grutzmacher KS, Keil V, Metzger S. Human respiratory syncytial virus and Streptococcus pneumoniae infection in wild bonobos. Ecohealth. 2018; 15:462-466. DOI | PubMed
- Scully EJ, Basnet S, Wrangham RW. Lethal respiratory disease associated with human rhinovirus C in wild chimpanzees, Uganda, 2013. Emerg Infect Dis. 2018; 24:267-274. DOI | PubMed
- Patrono LV, Samuni L, Corman VM. Human coronavirus OC43 outbreak in wild chimpanzees, Cote d Ivoire, 2016. Emerg Microbes Infect. 2018; 7:118. DOI | PubMed
- Davies TJ, Pedersen AB.. Phylogeny and geography predict pathogen community similarity in wild primates and humans. Proc Biol Sci. 2008; 275:1695-1701. DOI | PubMed
- Gilardi KV. Best practice Guidelines for health monitoring and disease control in great Ape populations. IUCN SSC Primate Specialist Group: Gland; 2015.
- Muehlenbein MP, Martinez LA, Lemke AA. Unhealthy travelers present challenges to sustainable primate ecotourism. Travel Med Infect Dis. 2010; 8:169-175. DOI | PubMed
- Ryan SJ, Walsh PD.. Consequences of non-intervention for infectious disease in African great apes. PLoS One. 2011; 6:e29030. DOI | PubMed
- Leendertz FH, Ellerbrok H, Boesch C. Anthrax kills wild chimpanzees in a tropical rainforest. Nature. 2004; 430:451-452. DOI | PubMed
- Calvignac-Spencer S, Leendertz SA, Gillespie TR. Wild great apes as sentinels and sources of infectious disease. Clin Microbiol Infect. 2012; 18:521-527. DOI | PubMed
- Chapman CA, Struhsaker TT, Lambert JE.. Thirty years of research in Kibale National Park, Uganda, Reveals a Complex Picture for Conservation. Int J Primatol. 2005; 26:539-555. DOI
- Muller MN, Wrangham RW.. Mortality rates among Kanyawara chimpanzees. J Hum Evol. 2014; 66:107-114. DOI | PubMed
- Wood BM, Watts DP, Mitani JC. Favorable ecological circumstances promote life expectancy in chimpanzees similar to that of human hunter-gatherers. J Hum Evol. 2017; 105:41-56. DOI | PubMed
- Palacios G, Lowenstine LJ, Cranfield MR. Human metapneumovirus infection in wild mountain gorillas, Rwanda. Emerg Infect Dis. 2011; 17:711-713. DOI | PubMed
- Kondgen S, Calvignac-Spencer S, Grützmacher K. Evidence for human Streptococcus pneumoniae in wild and captive chimpanzees: a potential threat to wild populations. Sci Rep. 2017; 7:14581. DOI | PubMed
- Papenburg J, Carbonneau J, Isabel S. Genetic diversity and molecular evolution of the major human metapneumovirus surface glycoproteins over a decade. J Clin Virol. 2013; 58:541-547. DOI | PubMed
- Sasaki M, Ishii A, Orba Y. Human parainfluenza virus type 3 in wild nonhuman primates, Zambia. Emerg Infect Dis. 2013; 19:121404. DOI
- Kumar S, Collins PL, Samal SK.. Identification of simian agent 10 as human parainfluenza virus type 3 suggests transmission of a human virus to an African monkey. J Virol. 2010; 84:13068-13070. DOI | PubMed
- Kahn JS.. Epidemiology of human metapneumovirus. Clin Microbiol Rev. 2006; 19:546-557. DOI | PubMed
- Haas LE, Thijsen SF, van Elden L. Human metapneumovirus in adults. Viruses. 2013; 5:87-110. DOI | PubMed
- Nkogue CN, Horie M, Fujita S. Molecular epidemiological study of adenovirus infecting western lowland gorillas and humans in and around Moukalaba-Doudou National Park (Gabon). Virus Genes. 2016; 52:671-678. DOI | PubMed
- Seimon TA, Olson SH, Lee KJ. Adenovirus and herpesvirus diversity in free-ranging great apes in the Sangha region of the Republic of Congo. PLoS One. 2015; 10:e0118543. DOI | PubMed
- Wevers D, Metzger S, Babweteera F. Novel adenoviruses in wild primates: a high level of genetic diversity and evidence of zoonotic transmissions. J Virol. 2011; 85:10774-10784. DOI | PubMed
- Hoppe E, Pauly M, Gillespie TR. Multiple cross-species transmission events of human adenoviruses (HAdV) during hominine evolution. Mol Biol Evol. 2015; 32:2072-2084. DOI | PubMed
- Tong S, Yip CCY, Tong S. Identification of adenoviruses in fecal specimens from wild chimpanzees (Pan trogylodytes schweinfurthii) in western Tanzania. Am J Trop Med Hyg. 2010; 82:967-970. DOI | PubMed
- Goodall J.. The chimpanzees of Gombe: patterns of Behavior. Harvard University Press: Cambridge (MA; 1986.
- CDC Center for Global Health. Uganda’s rapid response to avian influenza virus outbreak in birds. Global Health Protection and Security Summer 2017. 2017; 16Publisher Full Text
- Tang JW, Lam TT, Zaraket H. Global epidemiology of non-influenza RNA respiratory viruses: data gaps and a growing need for surveillance. Lancet Infect Dis. 2017; 17:e320-e326. DOI | PubMed
- van den Hoogen BG, van Doornum G , Fockens J. Prevalence and clinical symptoms of human metapneumovirus infection in hospitalized patients. J Infect Dis. 2003; 188:1571-1577. DOI | PubMed
- Huck B. Human metapneumovirus induces more severe disease and stronger innate immune response in BALB/c mice as compared with respiratory syncytial virus. Respir Res. 2007; 8(6)Publisher Full Text
- Skiadopoulos MH, Biacchesi S, Buchholz UJ. The two major human metapneumovirus genetic lineages are highly related antigenically, and the fusion (F) protein is a major contributor to this antigenic relatedness. J Virol. 2004; 78:6927-6937. DOI | PubMed
- Branche AR, Falsey AR.. Parainfluenza virus infection. Semin Respir Crit Care Med. 2016; 37:538-554. DOI | PubMed
- Ohsawa K, Yamada A, Takeuchi K. Genetic characterization of parainfluenza virus 3 derived from Guinea pigs. J Vet Med Sci. 1998; 60:919-922. DOI | PubMed
- Malherbe H, Harwin R.. The cytopathic effects of vervet monkey viruses. S Afr Med J. 1963; 37:407-411. PubMed
- Hawthorne JD, Lorenz D, Albrecht P.. Infection of marmosets with parainfluenza virus types 1 and 3. Infect Immun. 1982; 37:1037-1041. PubMed
- van Wyke Coelingh KL. Antibody responses of humans and nonhuman primates to individual antigenic sites of the hemagglutinin-neuraminidase and fusion glycoproteins after primary infection or reinfection with parainfluenza type 3 virus. J Virol. 1990; 64:3833-3843. PubMed
- Jones EE. Predisposition to invasive pneumococcal illness following parainfluenza type 3 virus infection in chimpanzees. J Am Vet Med Assoc. 1984; 185:1351-1353. PubMed
- Coverstone AM, Wilson B, Burgdorf D. Recurrent wheezing in children following human metapneumovirus infection. J Allergy Clin Immunol. 2018; 142:297-301. DOI | PubMed
- Sidhu RS, Sharma A, Paterson ID. Influenza H1N1 infection leading to cardiac tamponade in a previously healthy patient: a case report. Res Cardiovasc Med. 2016; 5:e31546. DOI | PubMed
- Goldberg TL, Paige SB, Chapman CA.. New Directions in conservation Medicine: Applied cases of Ecological health. Oxford University Press: New York; 2012.
- Lonsdorf EV, Murray CM, Lonsdorf EV. A retrospective analysis of factors correlated to chimpanzee (Pan troglodytes schweinfurthii) respiratory health at Gombe National Park, Tanzania. Ecohealth. 2011; 8:26-35. DOI | PubMed
- Althaus CL.. Estimating the reproduction number of Ebola virus (EBOV) during the 2014 outbreak in West Africa. PLoS Curr. 2014; 6Publisher Full Text
- R Core Team. R: A language and environment for statistical computing. R Foundation for Statistical Computing: Vienna; 2013.
- Burnham KP, Anderson DR, Huyvaert KP.. AIC model selection and multimodel inference in behavioral ecology: some background, observations, and comparisons. Behav Ecol Sociobiol. 2011; 49:8-17.
- Guindon S, Dufayard J-F, Lefort V. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010; 59:307-321. DOI | PubMed
- Rambaut A.. 2016. Publisher Full Text
- Toohey-Kurth K, Sibley SD, Goldberg TL.. Metagenomic assessment of adventitious viruses in commercial bovine sera. Biologicals. 2017; 47:64-68. DOI | PubMed
Fonte
Negrey JD, Reddy RB, Scully EJ, Phillips-Garcia S, Owens LA, et al. (2019) Simultaneous outbreaks of respiratory disease in wild chimpanzees caused by distinct viruses of human origin. Emerging Microbes & Infections 8(1): . https://doi.org/10.1080/22221751.2018.1563456




