
Abstract
Introduzione
Il virus dell’epatite B (HBV) è uno degli agenti patogeni umani più diffusi, con oltre 250 milioni di persone infettate in tutto il mondo e un bilancio annuo di circa 1 milione di morti in tutto il mondo(OMS, 2017). L’infezione delle cellule epatiche da HBV porta all’epatite B acuta, che si autolimita in circa il 90-95% dei casi. In circa il 5-10% degli individui infetti la clearance del virus non riesce e i pazienti sviluppano un’infezione cronica da epatite B, che li mette a rischio per tutta la vita per la cirrosi epatica e il cancro al fegato (carcinoma epatocellulare). L’HBV si trasmette di solito per contatto con sangue infetto, in paesi altamente endemici spesso durante il parto(OMS, 2017).
L’HBV ha un genoma del DNA circolare, parzialmente a doppio filamento, di circa 3,2 kbp, che codifica quattro cornici di lettura aperte sovrapposte (P, pre-S/S/S, pre-C/C e X). Sulla base della diversità della sequenza genomica, gli HBV sono attualmente classificati in otto genotipi (A-H) e numerosi sottogenotipi che mostrano distribuzioni geografiche distinte(Castelhano et al., 2017). Si ipotizza che tutti i genotipi siano principalmente il risultato di eventi di ricombinazione(Littlejohn et al., 2016; Simmonds e Midgley, 2005). In misura minore, l’evoluzione dell’HBV è anche guidata dall’accumulo di mutazioni puntiformi(Schaefer, 2007; Araujo, 2015).
Nonostante sia diffusa e ben studiata, l’origine e la storia evolutiva dell’HBV sono ancora poco chiare e controverse(Littlejohn et al., 2016; Souza et al., 2014). Gli HBV nei primati non umani (NHP), per esempio negli scimpanzé e nei gorilla, sono filogeneticamente strettamente correlati, eppure distinti dagli isolati umani di HBV, a sostegno della nozione di origine africana del virus(Souza et al., 2014). Le analisi basate sull’orologio molecolare che datano l’origine dell’HBV hanno portato a stime contrastanti con alcune recenti quanto circa 400 anni fa(Zhou e Holmes, 2007; Souza et al., 2014). Queste osservazioni hanno sollevato dubbi sull’idoneità degli approcci di datazione molecolare per ricostruire l’evoluzione dell’HBV(Bouckaert et al., 2013; Souza et al., 2014). Inoltre, le ricerche sul DNA antico (aDNA) sulle mummie infette da HBV delXVI secolo d.C. hanno rivelato una relazione molto stretta tra i genomi dell’HBV antico e moderno(Kahila Bar-Gal et al., 2012; Patterson Ross et al., 2018), indicando una sorprendente mancanza di cambiamenti genetici temporali del virus negli ultimi 500 anni(Patterson Ross et al., 2018). Sono quindi necessari studi diacronici sull’aDNA HBV, in cui vengano indagati sia i cambiamenti del genoma virale nel tempo, sia la provenienza e l’età dei campioni archeologici, per comprendere meglio l’origine e la storia evolutiva del virus.
Qui riportiamo l’analisi di tre genomi completi di HBV recuperati da resti scheletrici umani del Neolitico preistorico e del Medioevo in Europa centrale. I nostri risultati mostrano che l’HBV circolava già nella popolazione europea più di 7000 anni fa. Sebbene le forme antiche mostrino un rapporto con gli isolati moderni, esse sembrano rappresentare lignaggi distinti che non hanno parenti moderni stretti e che oggi sono probabilmente estinti.
Risultati e discussione
Abbiamo rilevato prove della presenza di HBV antico in tre campioni di denti umani come parte di uno screening metagenomico per gli agenti patogeni virali che è stato eseguito su dati di sequenziamento del fucile da caccia da 53 scheletri utilizzando il software di allineamento metagenomico MALT(Vågene et al., 2018). I resti degli individui sono stati scavati dai siti neolitici di Karsdorf (Linearbandkeramik [LBK], 5056-4959 cal BC) e Sorsum (gruppo Tiefstichkeramik della cultura Funnel Beaker, 3335-3107 cal BC), nonché dal cimitero medievale di Petersberg/Kleiner Madron (1020-1116 cal AD), tutti situati in Germania (Figura 1,Figura 1-figure supplementi 1- 3). Dopo che i tre estratti di aDNA erano apparsi positivi all’HBV nello screening iniziale del virus, sono stati sottoposti a un sequenziamento profondo senza alcun arricchimento preliminare che ha portato a 367-419 milioni di letture per campione(Tabella 1). Un’analisi dei componenti principali (PCA) del DNA umano recuperato da Karsdorf (copertura genomica triplice) ha rivelato che il campione è strettamente legato ad altri individui contemporanei del primo Neolitico dal LBK(Figura 1-figure supplement 4). La composizione genetica dei primi agricoltori LBK è stata precedentemente trovata abbastanza distinta dai precedenti cacciatori-raccoglitori occidentali d’Europa. Lo spostamento genetico tra le due popolazioni è stato interpretato come il risultato della migrazione dei primi agricoltori dall’Anatolia occidentale all’Europa centrale, introducendo l’agricoltura (Lazaridiset al., 2014; Haak et al., 2015). L’individuo Sorsum più giovane di quasi 2000 anni (copertura genomica 1,2 volte superiore) si concentra nell’APC più strettamente con individui della cultura contemporanea Funnel Beaker che ha abitato la Germania settentrionale alla fine del quarto millennio a.C.(Figura 1-figure supplement 4). Questa popolazione si è dimostrata in precedenza piuttosto confusa, come risultato di una sovrapposizione spaziale e temporale di agricoltori del primo Neolitico e di cacciatori-raccoglitori occidentali rimasti per quasi 2000 anni(Bollongino et al., 2013; Haak et al., 2015). L’individuo di Petersberg (copertura genomica 2,9 volte superiore), tuttavia, ha mostrato affinità genetiche nell’APC con le moderne popolazioni dell’Europa centrale. Tutti e tre gli antichi individui umani sono quindi in accordo con le prove archeologiche e le date radiocarboniche per la loro rispettiva epoca di origine. Insieme ai tipici modelli di danno da aDNA(Figura 1-figure supplements 5- 6), l’indagine genetica della popolazione umana supporta l’origine antica dei dataset ottenuti.
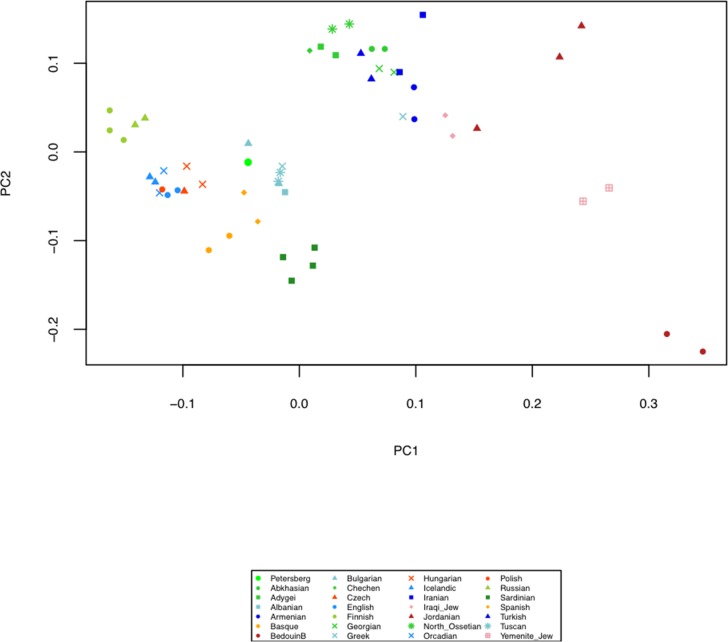
Figura 1-figura supplemento 9.Il cranio dell’individuo Karsdorf 537 indagato proviene da un maschio con un’età alla morte di circa 25-30 anni.il frammento di mandibola dell’individuo Sorsum XLVII 11 analizzato in questo studio proviene da un maschio.il cranio dell’individuo Petersberg analizzato dalla tomba 820 proviene da un maschio con un’età alla morte di circa 65-70 anni.Principal Component Analysis (PCA) dei campioni umani Karsdorf e Sorsum insieme a popolazioni antiche precedentemente pubblicate proiettate su 27 popolazioni moderne dell’Eurasia occidentale (non mostrate) sulla base di un insieme di 1,23 milioni di SNPs(Mathieson et al., 2015).trame di danno che mostrano modelli di deaminazione di letture specifiche per Hg19 per le biblioteche trattate con HalfUDG di(a) Karsdorf,(b) Sorsum,(c) Petersberg.trame di danno che mostrano modelli di deaminazione di letture specifiche per HBV per le biblioteche trattate con HalfUDG di(a) Karsdorf,(b) Sorsum, (c) Petersberg.MS / MS spettro del proteotypic HBV-peptide DLLDTASALYR dal HBV-proteina antigene del nucleo esterno (residui 58-68).analisi dei componenti principali (PCA) dei campioni umani Karsdorf e Sorsum insieme con le popolazioni antiche precedentemente pubblicato proiettato su 27 moderni giorni popolazioni dell’Eurasia occidentale (mostrato in grigio) sulla base di una serie di 1,23 milioni di SNPs (Mathiesonet al., Analisi dei componenti principali (PCA) del campione umano di Petersberg proiettato su 27 popolazioni moderne dell’Eurasia occidentale sulla base di un set di 1,23 milioni di SNP (Mathiesonet al.,2015).Posizione geografica dei campioni da cui sono stati isolati i genomi dell’HBV antico. Le date radiocarboniche dei campioni sono indicate in due serie di sigma. Le icone indicano il materiale del campione (dente o mummia). I genomi HBV ottenuti in questo studio sono indicati da un riquadro nero.Per effettuare l’allineamento sono stati utilizzati i riferimenti indicati nel file supplementare 1.([M + 2 hr]2+): m/z = 1237.6429 Da. Precisione di massa del peptide precursore = 0,56 ppm.
| *Merged legge | Lunghezza della sequenza di consenso HBV | Copertura media HBV | Lacune nella sequenza di consenso in posizione nt | *Mappato legge HBV | *Mappato legge umano | Copertura umana media | Genomi umani/HBVVgenomi | |
|---|---|---|---|---|---|---|---|---|
| Karsdorf | 386,780,892 | 3183 | 104X | 2157–2175; 3107–3128; 3133–3183 | 10,718 | 122,568,310 | 2.96X | 1: 35.1 |
| Sorsum | 367,574,767 | 3182 | 47X | – | 3249 | 9,856,001 | 1.17X | 1: 40.2 |
| Petersberg | 419,413,082 | 3161 | 46X | 880–1000; 1232–1329; 1331–1415; 1420–1581; 1585–1598 | 2125 | 105,476,677 | 2.88X | 1: 16 |
Per una ricostruzione riuscita del genoma dell’HBV, abbiamo mappato tutte le sequenze metagenomiche su 16 genomi di riferimento dell’HBV (otto genotipi umani (A-H) e 8 NHP provenienti dall’Africa e dall’Asia) che sono rappresentativi dell’attuale diversità dei ceppi HBV(file supplementare 6). Le letture mappate sono state utilizzate per un assemblaggio de novo, con il risultato di contigui da cui è stata costruita una sequenza di consenso HBV antica per campione. I genomi di consenso sono 3161 (copertura 46 volte), 3182 (copertura 47 volte), e 3183 (copertura 104 volte) nucleotidi in lunghezza, che rientra nella gamma di lunghezza dei moderni genomi HBV e suggerisce che abbiamo ricostruito con successo l’intero antico genoma HBV (Tabella 1, Figura 2-figure supplementi 1- 3). Inoltre, quando abbiamo condotto la cromatografia liquida – spettrometria di massa (LC-MS) basata sulla proteomica bottom-up su materiale dentale dei tre individui, abbiamo identificato nei campioni di Karsdorf e Petersberg un peptide che è parte della proteina del nucleo HBV molto stabile, che supporta la presenza e la replicazione attiva dell’HBV nel sangue degli individui (Figura 1-figure supplement 7).
L’analisi della rete filogenetica è stata effettuata con un set di dati composto da 493 ceppi di HBV moderni che rappresentano la piena diversità genetica. Sorprendentemente, i genomi neolitici HBV non si sono raggruppati con alcun ceppo umano nella filogenesi. Invece, essi si ramificavano in due lignaggi ed erano più strettamente correlati ai genomi africani NHP (Figura 2, 93% di somiglianza). Anche se i due ceppi neolitici sono stati recuperati da esseri umani che avevano vissuto a circa 2000 anni di distanza l’uno dall’altro, hanno mostrato una maggiore somiglianza genomica tra loro rispetto a qualsiasi altro genotipo umano o NHP. Tuttavia, i loro genomi differivano del 6% l’uno dall’altro e possono quindi essere considerati rappresentanti di due lignaggi separati. Tuttavia, essi differivano meno dell’8% dai ceppi NHP africani e non dovrebbero quindi essere chiamati genotipi separati(Figura 2-figure supplement 4). Il genoma dell’individuo di Petersberg, di 1000 anni, si compone dei moderni genotipi D4.
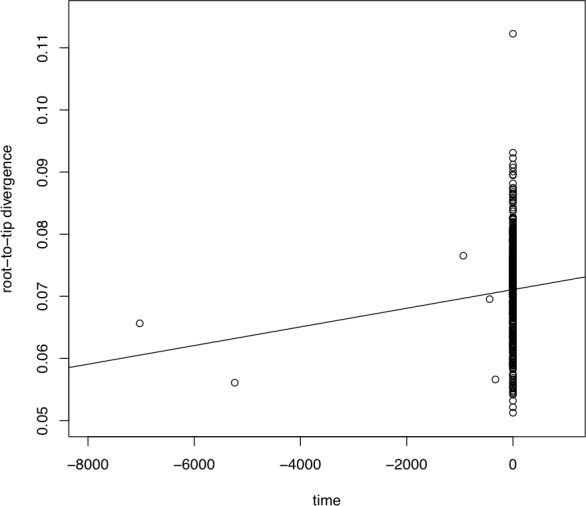
Figura 2-figure supplemento 9.Figura 2—supplemento alla figura 9. Rete.risultati dell’analisi di ricombinazione con i metodi RDP, GENECOV, Chimera, MaxChi, BootScan, SiScan, 3Seq all’interno del pacchetto software RDP v4 con tutti i moderni genomi di riferimento completi (n = 493) e cinque genomi antichi.allineamento di sequenza multipla del rappresentante 493 e cinque antichi genomi HBV.albero di massima probabilità basato sull’allineamento di sequenza multipla del rappresentante 493 e cinque antichi genomi HBV con 2000 repliche.Neighbour-Joining tree based on the multiple sequence alignment of the 493 representative modern and five ancient HBV genomes with 10000 replicates.Results of the recombination analysis using the methods RDP, GENECOV, Chimera, MaxChi, BootScan, SiScan, 3Seq within the RDP v4 software package with all modern full reference genomes (n = 493) and five ancient genomes.Allineamento sequenziale multiplo del rappresentante 493 e di cinque genomi HBV antichi.albero di massima probabilità basato sull’allineamento sequenziale multiplo del rappresentante 493 e di cinque genomi HBV antichi con 2000 repliche.albero di collegamento tra vicini basato sull’allineamento sequenziale multiplo del rappresentante 493 moderno e di cinque genomi HBV antichi con 10000 repliche.sequenza di consenso del genoma HBV di Karsdorf.sequenza di consenso del genoma HBV di Sorsum.Sequenza di consenso del genoma Petersberg HBV. Distanza genetica (hamming) dei nostri tre genomi HBV antichi rispetto a tutti i 493 genomi di riferimento.analisi BootScan della sequenza Karsdorf.analisi BootScan della sequenza Sorsum.analisi BootScan della sequenza Petersberg.SimPlot di (a)Karsdorf, (b) Sorsum e (c) Petersberg.Plot della distanza filogenetica dalla radice alla punta rispetto al tempo di campionamento (TempEst).Rete di 493 moderni, due genomi antichi pubblicati (scatola gialla chiara), e tre antichi virus dell’epatite B (HBV) ottenuti in questo studio (scatola grigia). I colori indicano gli otto genotipi umani del virus HBV(A-H), due genotipi di scimmie (Scimmie I, scimmie africane e scimmie II, scimmie asiatiche) e genomi antichi (rosso).10.7554/eLife.3666666.023Figure 2-source data 1.Results of the recombination analysis using the methods RDP, GENECOV, Chimera, MaxChi, BootScan, SiScan, 3Seq within the RDP v4 software package with all modern full reference genomes (n = 493) and five ancient genomes.10.7554/eLife.3666666.024Figure 2.Multiple sequence alignment of the 493 representative and five ancient HBV genomes.The multiple sequence alignment was stripped of any sites that had gaps in more than 95%.L’allineamento a sequenza multipla è stato eliminato da tutti i siti che avevano lacune in più del 95%.10.7554/eLife.3666666.025Figure 2-source data 3.Maximum-likelihood tree based on the multiple sequence alignment of the 493 representative and five ancient HBV genomes with 2000 replicates.10.7554/eLife.3666666.026Figure 2—dati fonte 4.Neighbour-Joining tree basato sull’allineamento di sequenze multiple dei 493 rappresentativi moderni e di cinque antichi genomi HBV con 10000 replicati.L’allineamento a sequenza multipla è stato eliminato da tutti i siti che avevano lacune in più del 95%.Sono indicate l’organizzazione di cornici di lettura aperte sovrapposte e la posizione approssimativa della porzione a singolo filamento del filamento più. Gli spazi vuoti nella sequenza sono contrassegnati in rosso. La trama verde rappresenta la copertura della mappatura delle letture grezze contro il consenso. Le trame circolari sono state generate utilizzando circos-0.69-6 e le informazioni di copertura della rimappatura.Sono indicate l’organizzazione dei fotogrammi di lettura aperti sovrapposti e la posizione approssimativa della porzione di filo singolo di filo più. Le lacune nella sequenza sono contrassegnate in rosso. La trama verde rappresenta la copertura della mappatura delle letture grezze contro il consenso. Le trame circolari sono state generate utilizzando circos-0.69-6 e le informazioni di copertura della rimappatura.Sono indicate l’organizzazione dei fotogrammi di lettura aperti sovrapposti e la posizione approssimativa della porzione di filo singolo di filo più. Le lacune nella sequenza sono contrassegnate in rosso. La trama verde rappresenta la copertura della mappatura delle letture grezze contro il consenso. Le trame circolari sono state generate utilizzando circos-0.69-6 e le informazioni di copertura della rimappatura.Le lacune o i siti non chiamati (“N”) sono stati ignorati.In ogni caso, frammenti di sequenza di 200 basi incrementando di 20 basi, 100 repliche bootstrap, sono stati confrontati con gruppi di sequenza di(a) gli otto genotipi umani, due genotipi di primati, e quattro genomi antichi e(b) gli otto genotipi umani, due genotipi di primati (codificati a colori come descritto nella leggenda).In ogni caso, frammenti di sequenza di 200 basi incrementando di 20 basi, 100 repliche bootstrap, sono stati confrontati con gruppi di sequenza o sequenze di consenso del 50% di(a) gli otto genotipi umani, due genotipi di primati, e quattro genomi antichi e(b) gli otto genotipi umani, due genotipi di primati (codificati a colori come descritto nella leggenda).In ogni caso, frammenti di sequenza di 200 basi incrementando di 20 basi, 100 repliche bootstrap, sono stati confrontati con gruppi di sequenza o sequenze di consenso del 50% di(a) gli otto genotipi umani, due genotipi di primati, e quattro genomi antichi e(b) gli otto genotipi umani, due genotipi di primati (codificati a colori come descritto nella leggenda).In ogni caso, frammenti di sequenza di 200 basi incrementando di 20 basi, 100 repliche bootstrap, sono stati confrontati con gruppi di sequenza degli otto genotipi umani, quattro genotipi di primati e quattro genotipi antichi (codificati a colori come descritto nella leggenda).Ogni punto rappresenta un campione.
A causa della ricombinazione continua nel tempo, diversi segmenti o moduli genici dei genomi ancestrali possono apparire in varie generazioni di virus successive. Tali precursori sono stati postulati(Simmonds e Midgley, 2005) e la loro esistenza è supportata dai risultati della nostra analisi di ricombinazione(Figura 2-figure supplements 5- 8,Figura 2-figure data 1). Alcuni frammenti delle sequenze di Karsdorf sono apparsi molto simili ai genotipi umani moderni (G, E) e ai genotipi africani NHP, e il genoma di Sorsum ha mostrato in parte un’elevata somiglianza con i genotipi umani G, E e B.(Figura 2-figure supplements 5- 8,Figura 2 dati fonte 1). Data la stretta relazione tra i due genomi del virus neolitico, è anche ipotizzabile che il vecchio HBV di Karsdorf potesse essere una fonte lontana per il virus Sorsum più giovane(Figura 2-figure supplements 5- 8,Figura 2–dati fonte 1). La relazione più stretta tra il Neolitico e i ceppi NHP rispetto ad altri ceppi umani è degna di nota e può aver comportato una trasmissione reciproca tra specie in una o forse più volte in passato(Simmonds e Midgley, 2005; Souza et al., 2014; Rasche et al ., 2016).
Nel complesso, i nostri risultati dimostrano che l’HBV esisteva già in Europa 7000 anni fa e che la sua struttura genomica assomigliava molto a quella dei moderni virus dell’epatite B. Entrambi i virus neolitici si collocano tra l’attuale moderno umano e la nota diversità dell’NHP. Pertanto, si può ipotizzare che, sebbene i due ceppi HBV del Neolitico non siano più osservati oggi e quindi possano riflettere due distinti ceppi che si sono estinti, essi potrebbero essere ancora strettamente correlati ai remoti antenati dei genotipi attuali, il che è supportato da segni di antichi eventi di ricombinazione. I precursori più antichi, i ceppi intermedi e moderni di entrambi gli esseri umani e le PSN devono essere sequenziati per districarsi nella complessa evoluzione dell’HBV. Poiché questa evoluzione è caratterizzata da ricombinazione e mutazioni puntiformi e può essere ulteriormente complicata dall’attraversamento della barriera dell’ospite scimmia umana(Simmonds e Midgley, 2005; Souza et al., 2014; Rasche et al., 2016), la datazione genetica non dovrebbe dare risultati significativi. Questo è inoltre supportato da un’analisi TempEst(Rambaut et al., 2016) che mostra un segnale temporale molto limitato(Figura 2-figure supplement 9). Va tuttavia notato che il genoma più antico (Karsdorf) è stato trovato in un individuo che apparteneva ad una popolazione di primi agricoltori che erano emigrati nel corso delle precedenti centinaia di anni dal Vicino Oriente all’Europa centrale. Si potrebbe ipotizzare che la stretta vicinanza agli animali recentemente addomesticati, i cambiamenti nella strategia di sussistenza e lo stile di vita sedentario adottato possano aver contribuito alla diffusione dell’HBV tra le popolazioni umane del Neolitico.
In base alle nostre analisi, l’HBV DNA può essere rilevato in modo affidabile in campioni di denti che hanno fino a 7000 anni. L’HBV antico è stato finora identificato solo nei tessuti molli di due mummie delXVI secolo(Kahila Bar-Gal et al., 2012; Patterson Ross et al., 2018). L’analisi aDNA dell’HBV da scheletri preistorici, che facilita gli studi evolutivi su una vasta scala temporale, non è stata finora descritta. Una spiegazione della difficoltà di una diagnosi molecolare di HBV nelle ossa è che l’infezione da virus non lascia lesioni sui resti scheletrici che permetterebbero ai ricercatori di selezionare a priori gli individui colpiti, come nel caso, ad esempio, della lebbra(Schuenemann et al., 2013). La diagnosi di un’infezione da HBV nelle popolazioni scheletriche è puramente casuale ed è quindi più probabile in uno screening su larga scala.
Nel complesso, le biomolecole dell’HBV sembrano essere ben conservate nei denti: Evitando i pregiudizi derivanti dalla cattura del DNA e dalla mappatura basata sul riferimento, abbiamo potuto ricostruire tre genomi dell’HBV con un assemblaggio de novo a partire dai dati del fucile da caccia e persino osservando i peptidi specifici dell’HBV. Il rapporto tra i genomi dell’HBV e il genoma umano nei nostri campioni era piuttosto alto e simile in tutti e tre i campioni (Karsdorf 35:1, Sorsum 40.2:1 e Petersberg 16:1). Poiché non vi sono prove che il DNA HBV sia più resistente alla degradazione post-mortem rispetto al DNA umano, l’alto tasso di HBV rispetto al DNA umano può riflettere lo stato di malattia negli individui infetti al momento della morte. Un elevato numero di copie di DNA virale nel sangue degli individui infetti è associato a un’infezione acuta da HBV o alla riattivazione dell’HBV cronico. Pertanto, sembra probabile che la morte degli individui antichi sia correlata all’infezione da HBV, ma potrebbe non essere la causa diretta della morte in quanto l’insufficienza epatica fulminante è piuttosto rara nei pazienti moderni. L’infezione da HBV potrebbe invece aver contribuito ad altre forme di insufficienza epatica letale come la cirrosi epatica o il cancro al fegato.
In considerazione dell’inaspettata complessità delle nostre scoperte, prevediamo futuri studi diacronici sull’HBV che vanno oltre l’ambito temporale e geografico del nostro lavoro attuale.
Figura 1-figura supplemento 9.Il cranio dell’individuo di Karsdorf 537 indagato proviene da un maschio con un’età alla morte di circa 25-30 anni.il frammento di mandibola dell’individuo di Sorsum XLVII 11 analizzato in questo studio proviene da un maschio.il cranio dell’individuo di Petersberg analizzato dalla tomba 820 proviene da un maschio con un’età alla morte di circa 65-70 anni.Principal Component Analysis (PCA) dei campioni umani Karsdorf e Sorsum insieme a popolazioni antiche precedentemente pubblicate proiettate su 27 popolazioni moderne dell’Eurasia occidentale (non mostrate) sulla base di un insieme di 1,23 milioni di SNPs(Mathieson et al., 2015).trame di danno che mostrano modelli di deaminazione di letture specifiche per Hg19 per le biblioteche trattate con HalfUDG di(a) Karsdorf,(b) Sorsum,(c) Petersberg.trame di danno che mostrano modelli di deaminazione di letture specifiche per HBV per le biblioteche trattate con HalfUDG di(a) Karsdorf,(b) Sorsum, (c) Petersberg.MS / MS spettro del proteotypic HBV-peptide DLLDTASALYR dal HBV-proteina antigene del nucleo esterno (residui 58-68).analisi dei componenti principali (PCA) dei campioni umani Karsdorf e Sorsum insieme con le popolazioni antiche precedentemente pubblicato proiettato su 27 moderni giorni popolazioni dell’Eurasia occidentale (mostrato in grigio) sulla base di una serie di 1,23 milioni di SNPs (Mathiesonet al., Analisi dei componenti principali (PCA) del campione umano di Petersberg proiettato su 27 popolazioni moderne dell’Eurasia occidentale sulla base di un set di 1,23 milioni di SNP (Mathiesonet al.,2015).Posizione geografica dei campioni da cui sono stati isolati i genomi dell’HBV antico. Le date radiocarboniche dei campioni sono indicate in due serie di sigma. Le icone indicano il materiale del campione (dente o mummia). I genomi HBV ottenuti in questo studio sono indicati da un riquadro nero.Per effettuare l’allineamento sono stati utilizzati i riferimenti indicati nel file supplementare 1.([M + 2 hr]2+): m/z = 1237.6429 Da. Precisione di massa del peptide precursore = 0,56 ppm.
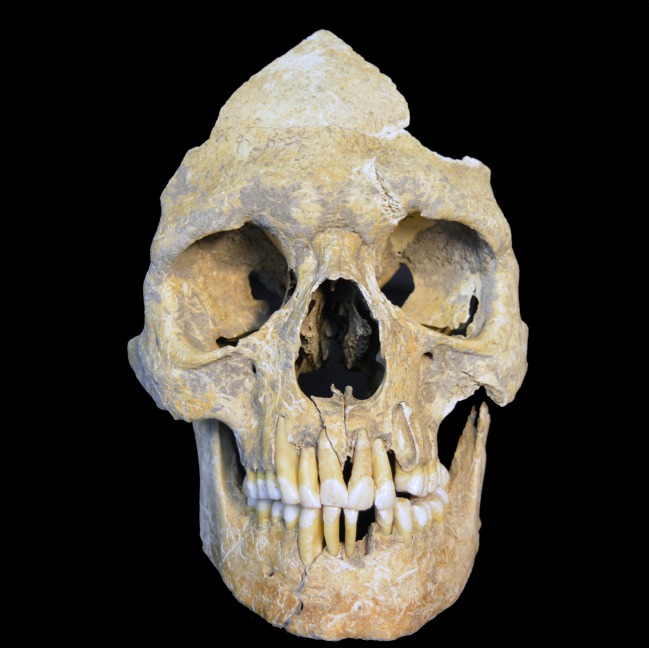
Figura 1-figure supplement 1.Il cranio dell’individuo indagato Karsdorf 537 è di un maschio con un’età alla morte di circa 25-30 anni.
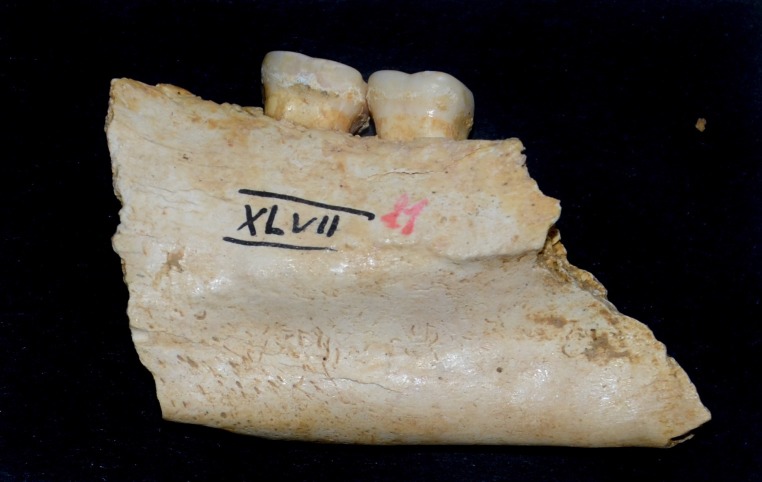
Figura 1-figura supplemento 2.Il frammento di mandibola del Sorsum individuale XLVII 11 analizzato in questo studio è di un maschio.
Figura 1-figura supplemento 3.Il cranio dell’individuo Petersberg analizzato dalla tomba 820 è di un maschio con un’età alla morte di circa 65-70 anni.
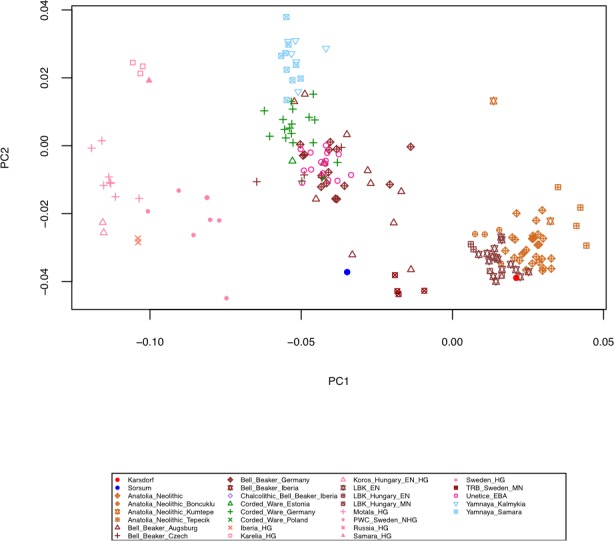
Figura 1-figure supplemento 4.Figura 1—supplemento di figura 4. Analisi dei componenti principali (PCA) dei campioni umani di Karsdorf e Sorsum insieme a popolazioni antiche precedentemente pubblicate proiettate su 27 popolazioni moderne dell’Eurasia occidentale (non mostrate) sulla base di un insieme di 1,23 milioni di SNP(Mathieson et al., 2015).
Figura 1-figure supplemento 5.Figura 1—supplemento di figura 5. Trame di danno che mostrano i modelli di deaminazione di hg19-specifiche letture per le biblioteche trattate con HalfUDG di(a) Karsdorf,(b) Sorsum,(c) Petersberg.
Figura 1-figure supplement 6.Figura 1—supplemento alla figura 6. Trame di danno che mostrano modelli di deaminazione di letture HBV-specifiche per le biblioteche trattate con HalfUDG di(a) Karsdorf, (b) Sorsum, (c) Petersberg.Per effettuare l’allineamento sono stati usati i riferimenti mostrati nel file supplementare 1.
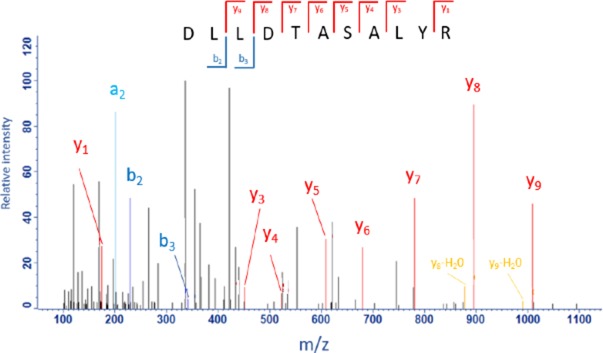
Figura 1-figura supplemento 7.MS / MS spettro del proteotipico HBV-peptide DLLDTASALYR dal HBV-proteina antigene del nucleo esterno (residui 58-68).([M + 2 hr]2+): m/z = 1237.6429 Da. Precisione di massa del peptide precursore = 0,56 ppm.
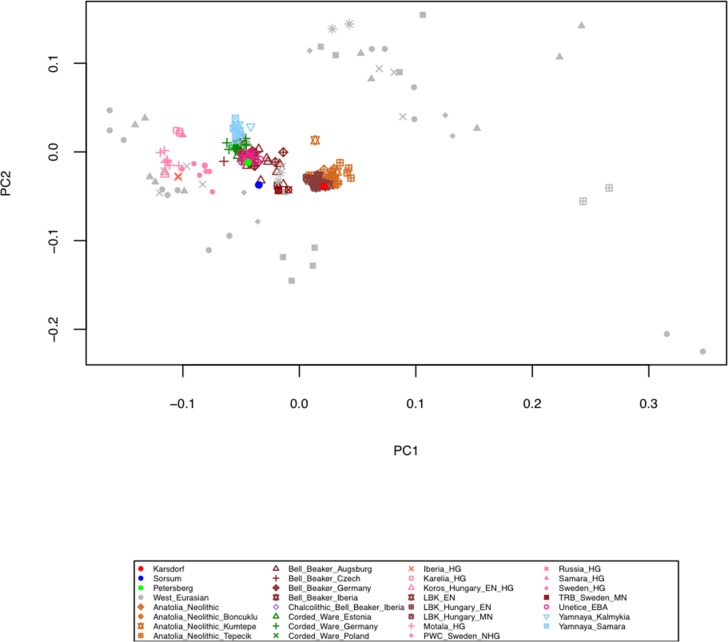
Figura 1-figure supplement 8.Analisi dei componenti principali (PCA) dei campioni umani di Karsdorf e Sorsum insieme a popolazioni antiche precedentemente pubblicate proiettate su 27 popolazioni dell’Eurasia occidentale moderna (mostrate in grigio) sulla base di un set di 1,23 milioni di SNPs(Mathieson et al., 2015).
Figura 1-figure supplemento 9.Figura 1—supplemento alla figura 9. Principal Component Analysis (PCA) del campione umano di Petersberg proiettato su 27 popolazioni moderne dell’Eurasia occidentale sulla base di un set di 1,23 milioni di SNP(Mathieson et al., 2015).
Figura 2-figure supplemento 9.Figura 2—supplemento alla figura 9. Rete.risultati dell’analisi di ricombinazione utilizzando i metodi RDP, GENECOV, Chimera, MaxChi, BootScan, SiScan, 3Seq all’interno del pacchetto software RDP v4 con tutti i moderni genomi di riferimento completo (n = 493) e cinque genomi antichi.allineamento di sequenza multipla del rappresentante 493 e cinque antichi genomi HBV.albero di massima probabilità basato sull’allineamento di sequenza multipla del rappresentante 493 e cinque antichi genomi HBV con 2000 repliche.Neighbour-Joining tree based on the multiple sequence alignment of the 493 representative modern and five ancient HBV genomes with 10000 replicates.Results of the recombination analysis using the methods RDP, GENECOV, Chimera, MaxChi, BootScan, SiScan, 3Seq within the RDP v4 software package with all modern full reference genomes (n = 493) and five ancient genomes.Allineamento sequenziale multiplo del rappresentante 493 e di cinque genomi HBV antichi.albero di massima probabilità basato sull’allineamento sequenziale multiplo del rappresentante 493 e di cinque genomi HBV antichi con 2000 repliche.albero di collegamento tra vicini basato sull’allineamento sequenziale multiplo del rappresentante 493 moderno e di cinque genomi HBV antichi con 10000 repliche.sequenza di consenso del genoma HBV di Karsdorf.sequenza di consenso del genoma HBV di Sorsum.Sequenza di consenso del genoma Petersberg HBV. Distanza genetica (hamming) dei nostri tre genomi HBV antichi rispetto a tutti i 493 genomi di riferimento.analisi BootScan della sequenza Karsdorf.analisi BootScan della sequenza Sorsum.analisi BootScan della sequenza Petersberg.SimPlot di (a)Karsdorf, (b) Sorsum e (c) Petersberg.Plot della distanza filogenetica dalla radice alla punta rispetto al tempo di campionamento (TempEst).Rete di 493 moderni, due genomi antichi pubblicati (scatola gialla chiara), e tre antichi virus dell’epatite B (HBV) ottenuti in questo studio (scatola grigia). I colori indicano gli otto genotipi umani del virus HBV(A-H), due genotipi di scimmie (Scimmie I, scimmie africane e scimmie II, scimmie asiatiche) e genomi antichi (rosso).10.7554/eLife.3666666.023Figure 2-source data 1.Results of the recombination analysis using the methods RDP, GENECOV, Chimera, MaxChi, BootScan, SiScan, 3Seq within the RDP v4 software package with all modern full reference genomes (n = 493) and five ancient genomes.10.7554/eLife.3666666.024Figure 2.Multiple sequence alignment of the 493 representative and five ancient HBV genomes.The multiple sequence alignment was stripped of any sites that had gaps in more than 95%.L’allineamento a sequenza multipla è stato eliminato da tutti i siti che avevano lacune in più del 95%.10.7554/eLife.3666666.025Figure 2-source data 3.Maximum-likelihood tree based on the multiple sequence alignment of the 493 representative and five ancient HBV genomes with 2000 replicates.10.7554/eLife.3666666.026Figure 2—dati fonte 4.Neighbour-Joining tree basato sull’allineamento di sequenze multiple dei 493 rappresentativi moderni e di cinque antichi genomi HBV con 10000 replicati.L’allineamento a sequenza multipla è stato eliminato da tutti i siti che avevano lacune in più del 95%.Sono indicate l’organizzazione di cornici di lettura aperte sovrapposte e la posizione approssimativa della porzione a singolo filamento del filamento più. Gli spazi vuoti nella sequenza sono contrassegnati in rosso. La trama verde rappresenta la copertura della mappatura delle letture grezze contro il consenso. Le trame circolari sono state generate utilizzando circos-0.69-6 e le informazioni di copertura della rimappatura.Sono indicate l’organizzazione dei fotogrammi di lettura aperti sovrapposti e la posizione approssimativa della porzione di filo singolo di filo più. Le lacune nella sequenza sono contrassegnate in rosso. La trama verde rappresenta la copertura della mappatura delle letture grezze contro il consenso. Le trame circolari sono state generate utilizzando circos-0.69-6 e le informazioni di copertura della rimappatura.Sono indicate l’organizzazione dei fotogrammi di lettura aperti sovrapposti e la posizione approssimativa della porzione di filo singolo di filo più. Le lacune nella sequenza sono contrassegnate in rosso. La trama verde rappresenta la copertura della mappatura delle letture grezze contro il consenso. Le trame circolari sono state generate utilizzando circos-0.69-6 e le informazioni di copertura dalla rimappatura.Le lacune o i siti non chiamati (“N”) sono stati ignorati.In ogni caso, frammenti di sequenza di 200 basi incrementando di 20 basi, 100 repliche bootstrap, sono stati confrontati con gruppi di sequenza di(a) gli otto genotipi umani, due genotipi di primati, e quattro genomi antichi e(b) gli otto genotipi umani, due genotipi di primati (codificati a colori come descritto nella leggenda).In ogni caso, frammenti di sequenza di 200 basi incrementando di 20 basi, 100 repliche bootstrap, sono stati confrontati con gruppi di sequenza o sequenze di consenso del 50% di(a) gli otto genotipi umani, due genotipi di primati, e quattro genomi antichi e(b) gli otto genotipi umani, due genotipi di primati (codificati a colori come descritto nella leggenda).In ogni caso, frammenti di sequenza di 200 basi incrementando di 20 basi, 100 repliche bootstrap, sono stati confrontati con gruppi di sequenza o sequenze di consenso del 50% di(a) gli otto genotipi umani, due genotipi di primati, e quattro genomi antichi e(b) gli otto genotipi umani, due genotipi di primati (codificati a colori come descritto nella leggenda).In ogni caso, frammenti di sequenza di 200 basi incrementando di 20 basi, 100 repliche bootstrap, sono stati confrontati con gruppi di sequenza degli otto genotipi umani, quattro genotipi di primati e quattro genotipi antichi (codificati a colori come descritto nella leggenda).Ogni punto rappresenta un campione.
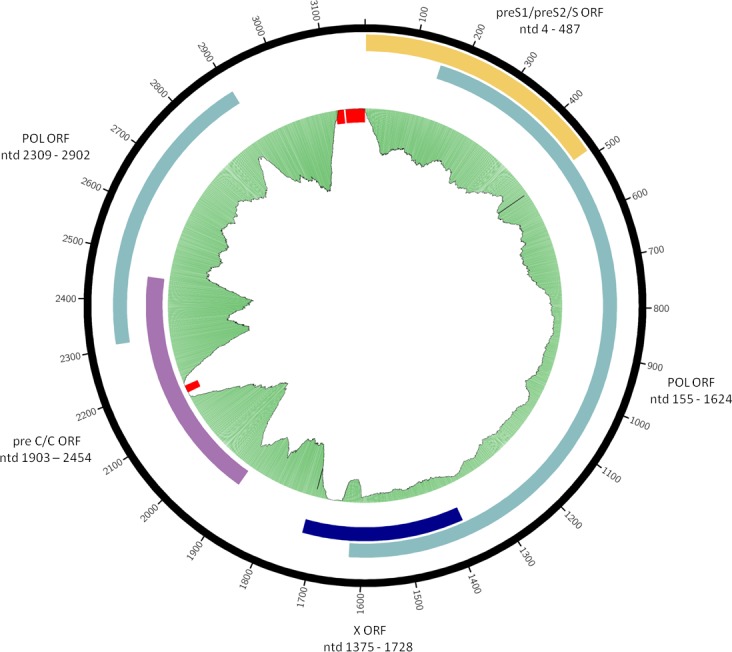
Figura 2-figure supplemento 1.Sequenza di consenso del genoma HBV di Karsdorf.Sono indicate l’organizzazione dei fotogrammi di lettura aperti sovrapposti e la posizione approssimativa della porzione a singolo filamento del filamento più. Le lacune nella sequenza sono contrassegnate in rosso. La trama verde rappresenta la copertura della mappatura delle letture grezze contro il consenso. Le trame circolari sono state generate utilizzando circos-0.69-6 e le informazioni di copertura dalla rimappatura.
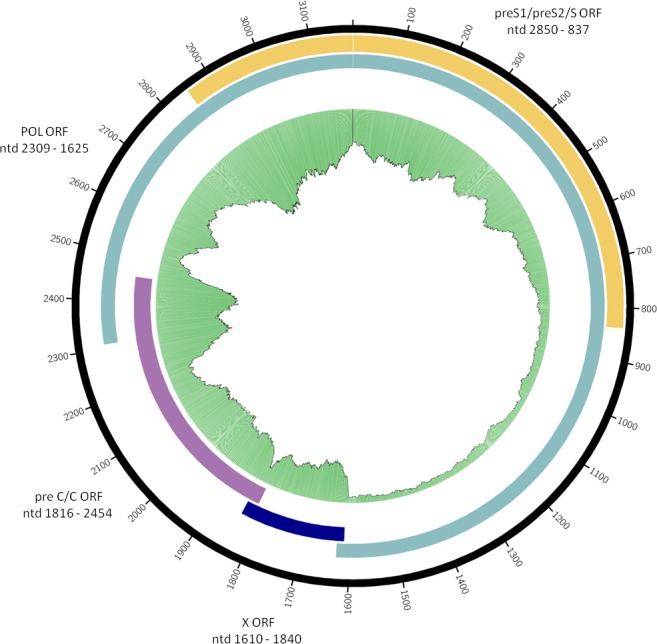
Figura 2-figure supplemento 2.Sequenza di consenso del genoma del Sorsum HBV.Sono indicate l’organizzazione dei fotogrammi di lettura aperti sovrapposti e la posizione approssimativa della porzione a singolo filamento del filamento più. Le lacune nella sequenza sono contrassegnate in rosso. La trama verde rappresenta la copertura della mappatura delle letture grezze contro il consenso. Le trame circolari sono state generate utilizzando circos-0.69-6 e le informazioni di copertura dalla rimappatura.
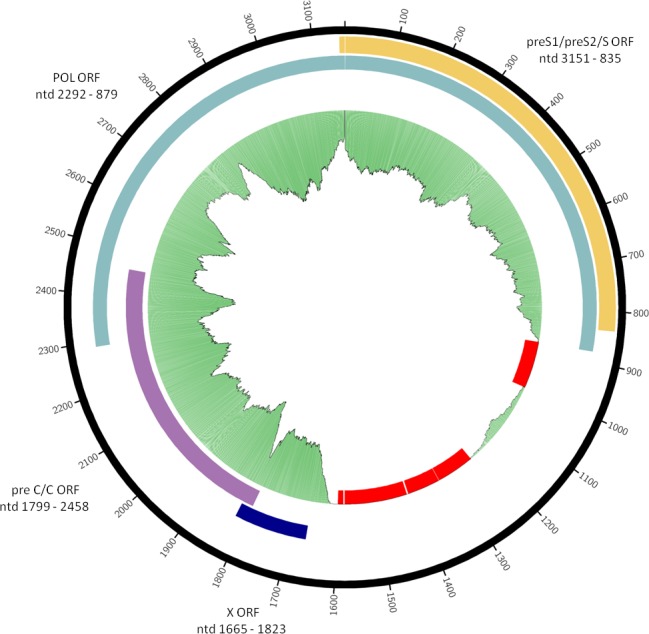
Figura 2-figure supplemento 3.Sequenza di consenso del genoma di Petersberg HBV.4. Organizzazione dei fotogrammi di lettura aperti sovrapposti e posizione approssimativa della porzione di filo singolo di filo più sono indicati. Le lacune nella sequenza sono contrassegnate in rosso. La trama verde rappresenta la copertura della mappatura delle letture grezze contro il consenso. Le trame circolari sono state generate utilizzando circos-0.69-6 e le informazioni di copertura dalla rimappatura.
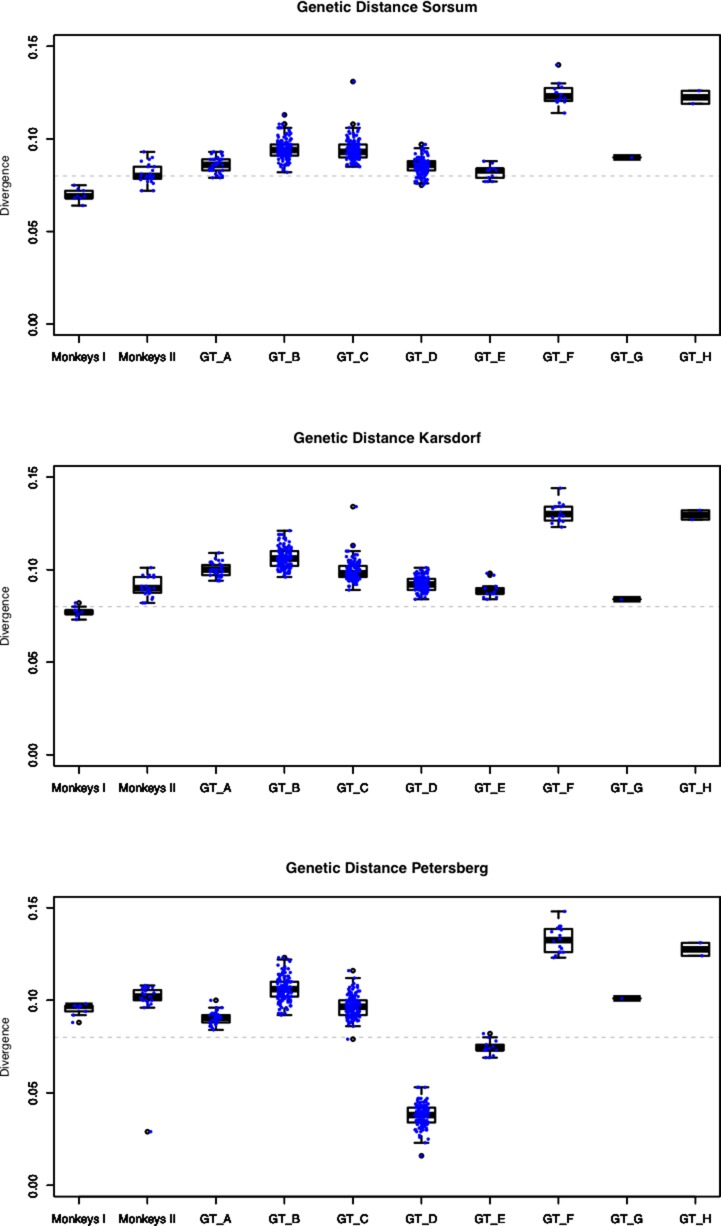
Figura 2-figure supplemento 4.Distanza genetica (hamming) dei nostri tre antichi genomi HBV rispetto a tutti i 493 genomi di riferimento.Le lacune o i siti non chiamati (‘N’) sono stati ignorati.
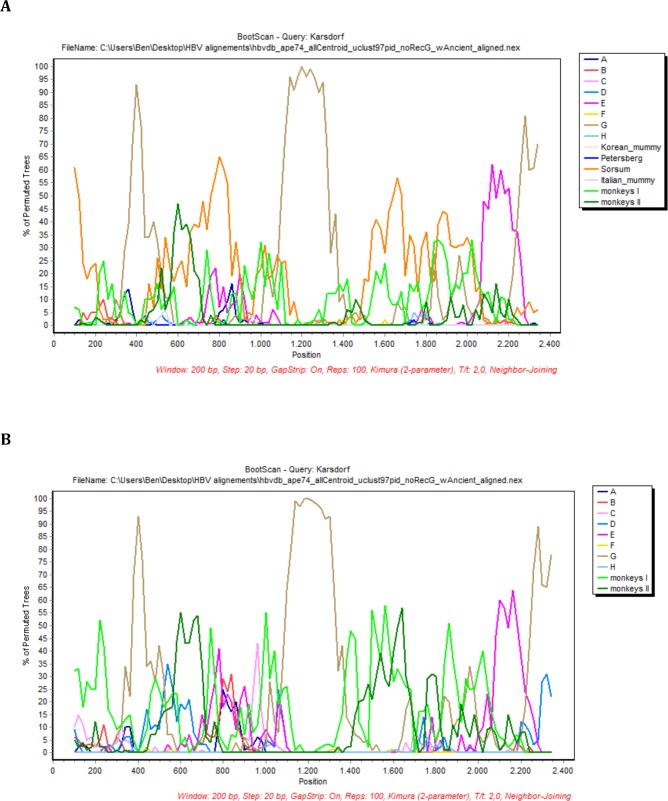
Figura 2-figure supplement 5.Analisi BootScan della sequenza Karsdorf.In ogni caso, frammenti di sequenza di 200 basi incrementando di 20 basi, 100 repliche bootstrap, sono stati confrontati con gruppi di sequenza di(a) gli otto genotipi umani, due genotipi di primati, e quattro genomi antichi e(b) gli otto genotipi umani, due genotipi di primati (codificati a colori come descritto nella leggenda).
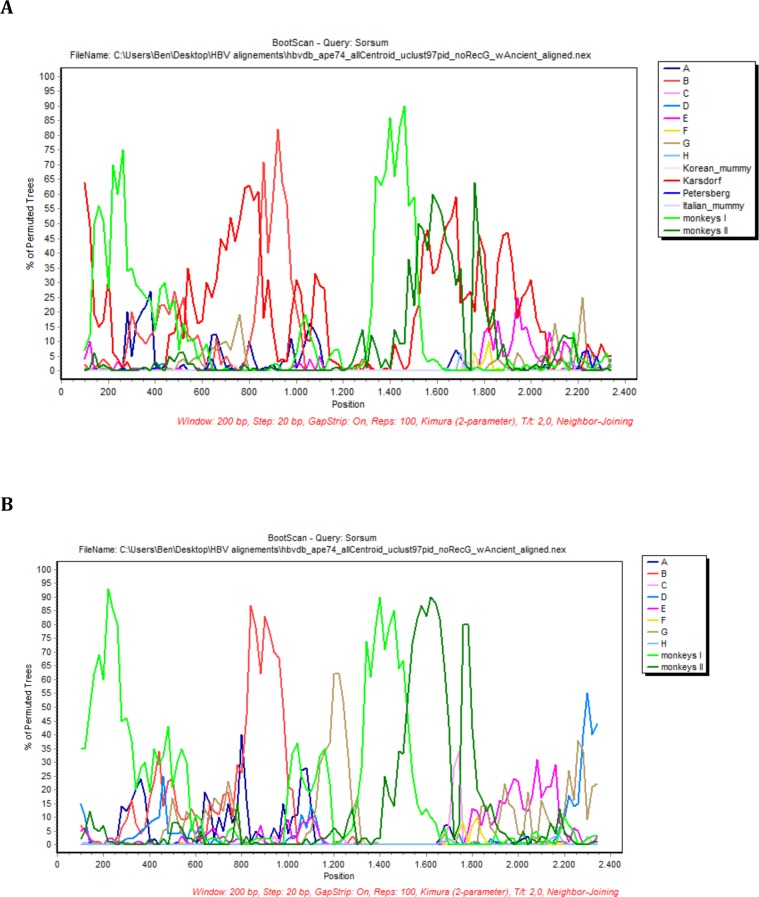
Figura 2-figure supplemento 6.Analisi BootScan della sequenza Sorsum.In ogni caso, frammenti di sequenza di 200 basi incrementando di 20 basi, 100 repliche bootstrap, sono stati confrontati con gruppi di sequenza o il 50% di sequenze di consenso di(a) gli otto genotipi umani, due genotipi di primati, e quattro genomi antichi e(b) gli otto genotipi umani, due genotipi di primati (codificati a colori come descritto nella leggenda).
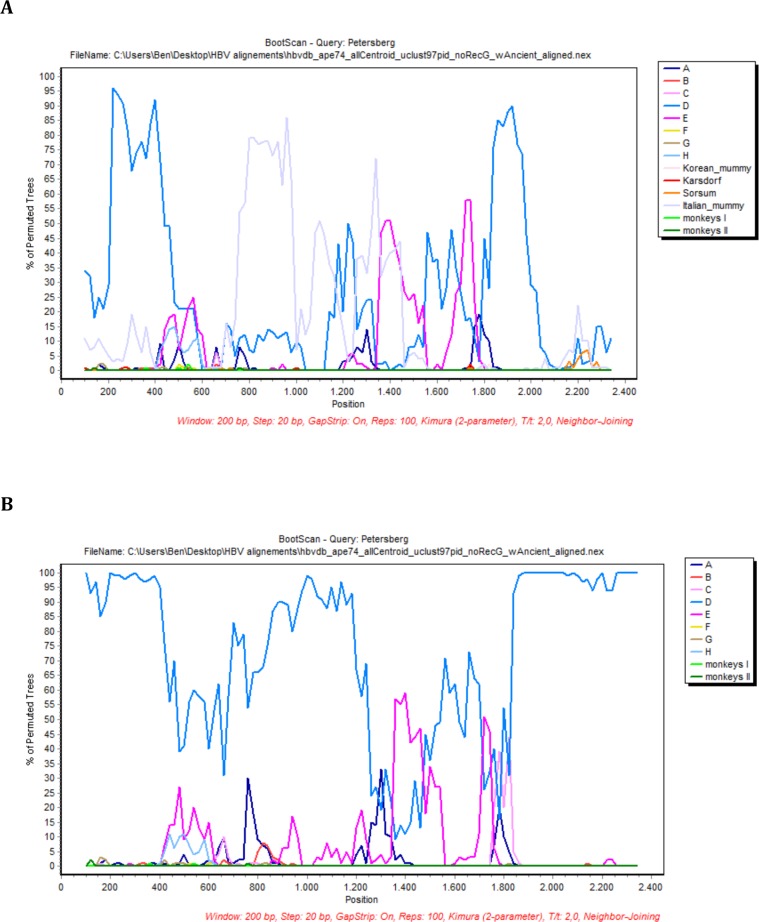
Figura 2-figure supplemento 7.BootScan analisi della sequenza Petersberg.In ogni caso, frammenti di sequenza di 200 basi incrementando di 20 basi, 100 repliche bootstrap, sono stati confrontati con gruppi di sequenza o il 50% di sequenze di consenso di(a) gli otto genotipi umani, due genotipi di primati, e quattro genomi antichi e(b) gli otto genotipi umani, due genotipi di primati (codificati a colori come descritto nella leggenda).
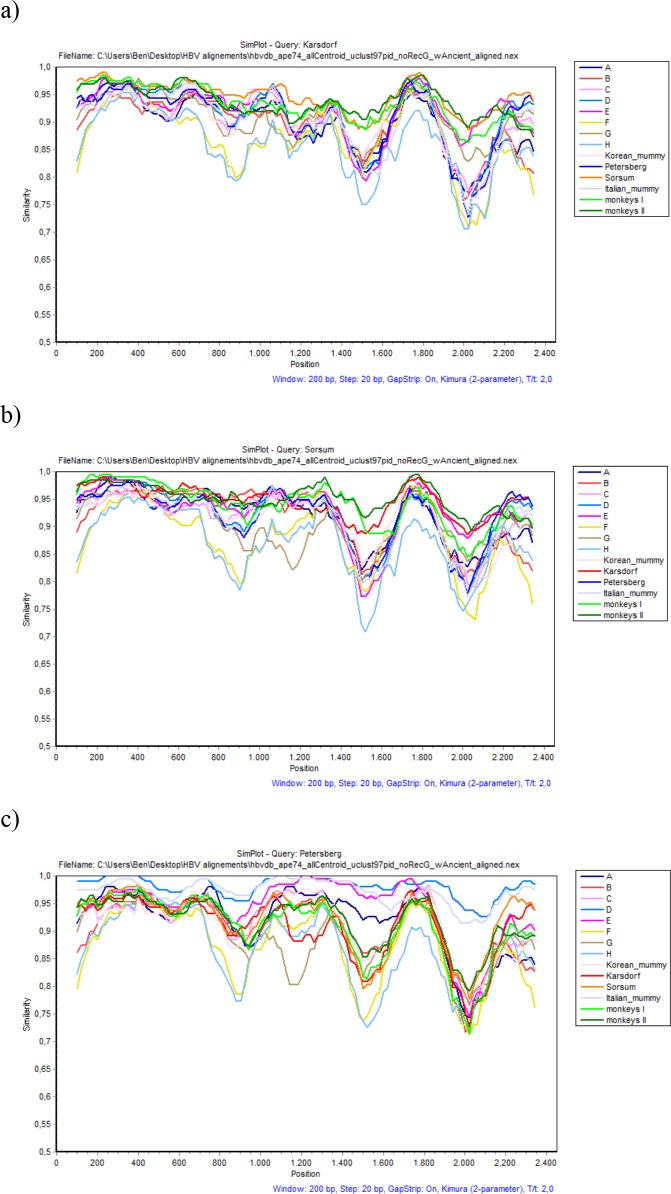
Figura 2-figure supplemento 8.Analisi SimPlot di(a) Karsdorf,(b) Sorsum e(c) Petersberg.In ogni caso, frammenti di sequenza di 200 basi incrementando di 20 basi, 100 repliche bootstrap, sono stati confrontati con gruppi di sequenza degli otto genotipi umani, quattro genotipi di primati e quattro genomi antichi (codificati a colori come descritto nella leggenda).
Figura 2-figure supplemento 9.Trama della distanza filogenetica dalla radice alla punta rispetto al tempo di campionamento (TempEst).Ogni punto rappresenta un campione.
Materiali e metodi
Resti umani
L’insediamento LBK di Karsdorf, Sassonia-Anhalt, Germania, si trova nella valle del fiume Unstrut. Tra il 1996 e il 2010 sono stati condotti a Karsdorf scavi sistematici che hanno portato alla scoperta di insediamenti e tombe dal Neolitico all’età del ferro (Behnke, 2007; 2011; 2012) . Il LBK è rappresentato da 24 case lunghe in orientamento nord-ovest a sud-est che sono state associate a sepolture di insediamenti(Veit, 1996). L’individuo indagato 537 è un maschio con un’età alla morte di circa 25-30 anni (Figura 1-figure supplement 1), datato a 5056-4959 cal BC (KIA 40357-6116 ± 32 BP) (Brandt et al., 2014; Nicklisch,2017).
La tomba della galleria di Sorsum, Bassa Sassonia, Germania, è tipologicamente datata al Tiefstichkeramik (gruppo della cultura Funnelbeaker). Il Sorsum è eccezionale in quanto è stato costruito nella roccia. Durante gli scavi (1956-1960) della camera mortuaria sono stati recuperati circa 105 individui (Claus, 1983; Czarnetzki , 1966). L’individuo XLVII 11 analizzato in questo studio è un maschio(Figura 1-figure supplement 2) e risale a 3335-3107 cal BC (MAMS 33641-4501 ± 19 BP).
Il cimitero medievale di Petersberg/Kleiner Madron, in Baviera, Germania, si trova sulla cima di una collina a 850 metri sul livello del mare e 400 metri sopra il pavimento della valle dell’Inn. Nella parte orientale del cimitero sono stati sepolti i membri di un priorato che molto probabilmente fu fondato alla fine delX secolo. Fonti scritte ne documentano l’esistenza dal 1132 in poi(Meier, 1998). Durante gli scavi sistematici (1997-2004) nella parte sud-orientale del cimitero sono stati scoperti i resti di individui sepolti in 99 tombe. L’individuo esaminato nella tomba 820 è un maschio con un’età alla morte di circa 65-70 anni (Lösch, 2009 – Figura 1-figure supplement 3) risalente al 1020-1116 cal AD (MAMS 33642-982 ± 17 BP).
Estrazione del DNA e sequenziamento
Le estrazioni di DNA e le fasi di pre-PCR sono state effettuate in camere bianche dedicate alla ricerca sull’aDNA. Per le analisi sono stati utilizzati i denti. I campioni di Petersberg e Sorsum sono stati elaborati nel Laboratorio di DNA Antico dell’Università di Kiel e il campione di Karsdorf nel Laboratorio di DNA Antico del Max Planck Institute for the Science of Human History (MPI SHH) di Jena. Tutte le procedure hanno seguito le linee guida sul controllo della contaminazione negli studi sull’aDNA(Warinner et al., 2017; Key et al., 2017). I denti sono stati puliti in soluzione di candeggina pura per rimuovere potenziali contaminazioni prima della polverizzazione. Cinquanta milligrammi di polvere sono stati utilizzati per l’estrazione seguendo un protocollo a base di silice(Dabney et al., 2013). I controlli negativi sono stati inclusi in tutte le fasi.
Da ogni campione, le librerie di sequenziamento del DNA a doppio filamento (UDGhalf) sono state preparate secondo un protocollo stabilito per il sequenziamento multiplex ad alta velocità(Meyer e Kircher, 2010). Gli indici specifici del campione sono stati aggiunti ad entrambi gli adattatori della biblioteca tramite amplificazione con due primer di indice. L’estrazione e gli spazi vuoti della biblioteca sono stati trattati allo stesso modo. Per lo screening iniziale, la biblioteca dell’individuo di Karsdorf è stata sequenziata su 1/50 di una corsia sull’HiSeq 3000 (2 × 75 bp) presso l’MPI SHH di Jena e le biblioteche di Petersberg e Sorsum sono state sequenziate sulla piattaforma Illumina HiSeq 4000 (2 × 75 bp) presso l’Istituto di Biologia Molecolare Clinica dell’Università di Kiel, utilizzando la chimica HiSeq v4 e il protocollo del produttore per il sequenziamento multiplex. Il deep-sequencing per ognuno dei tre campioni è stato effettuato su due corsie sulla piattaforma Illumina HiSeq 4000 presso l’Istituto di Biologia Molecolare Clinica dell’Università di Kiel.
Clip e fusione
I set di dati prodotti per tutti i campioni antichi contenevano paired-end di lettura con un numero variabile di nucleotidi sovrapposti e sequenze di adattatori artificiali. Abbiamo utilizzato la versione 1.7.3 di ClipAndMerge, un modulo della pipeline EAGER(Peltzer et al., 2016), per clippare le sequenze di adattatori, unire le letture di paired-end corrispondenti in regioni sovrapposte e tagliare le letture risultanti. Abbiamo utilizzato le opzioni predefinite con il seguente comando:
java -jar ClipAndMerge.jar -in1 $FASTQ1 -in2 $FASTQ2 \
-f AGATCGGGAAGAGAGCACACACGACTCTGAACTCCAGTCAC \\x22
-r AGATCGGGAAGAGCGGTCGGTGTGTAGGGAAAGAGGGAAAGTGTA \\\x22
-l 25 -qt -q 20 -o $output_file
dove $FASTQ1 e $FASTQ2 sono i due file di input FASTQ con gzip
Taglio dell’adattatore
ClipAndMerge utilizza un allineamento di sovrapposizione del rispettivo adattatore in avanti o indietro con l’estremità di 3′ di ogni lettura per rimuovere le sequenze dell’adattatore di sequenziamento. Le regioni alla fine dei 3′ di ogni lettura che erano contenute nell’allineamento sono state ritagliate. Le letture che erano più corte di 25 nucleotidi dopo il clipping dell’adattatore o che contenevano solo sequenze di adattatori (dimeri dell’adattatore) sono state rimosse. Tutte le letture rimanenti sono state poi utilizzate nella fase di fusione.
Fusione di letture accoppiate sovrapposte
La fusione è stata eseguita per tutte le rimanenti letture accoppiate con una sovrapposizione minima di 10 nucleotidi e al massimo il 5% di disallineamenti nella regione di sovrapposizione. L’algoritmo ha selezionato la sovrapposizione massima che soddisfa questi criteri. La sequenza di consenso è stata generata utilizzando i nucleotidi nelle regioni di sovrapposizione dalla lettura con il punteggio di qualità PHRED più alto, massimizzando la qualità della lettura risultante.
Rifilatura della qualità
In una fase finale, ClipAndMerge ha eseguito il taglio di qualità delle letture e tutti i nucleotidi con punteggi PHRED inferiori a 20 sono stati tagliati dalla fine di 3′ di ogni lettura. Infine, tutte le letture con meno di 25 nucleotidi dopo il trimming di qualità sono state rimosse. Le letture di alta qualità risultanti sono state usate per l’allineamento.
Screening dei virus
Lo screening dei set di dati è stato effettuato con il software MALT utilizzando come riferimento il database ncbi-viral. È stata impostata una soglia di identità della sequenza dell’85% e la modalità di allineamento è stata cambiata in SemiGlobal. L’analisi è stata effettuata con il seguente comando:
malt-run –mode BlastN -e 0.001 -id 85 –alignmentType SemiGlobal –index $index –inFile $FASTQCM –output $OUT
dove $index è il file indice, $FASTQCM è il file ritagliato e unito e $OUT è l’output del file.
Gli allineamenti risultanti sono stati ispezionati visivamente usando MEGAN 6. Le letture di mappatura del riferimento dell’epatite B nel database (NC_003977.2) sono state estratte e verificate usando un megablast discontiguo contro il taxa del virus (taxid: 10239) con parametri predefiniti.
Allineamento HBV
Per l’identificazione dei genotipi, i campioni sono stati allineati rispetto a un riferimento per ciascuno degli otto genotipi dell’epatite B disponibili nel progetto di genotipizzazione dell’epatite B della NCBI (https://www.ncbi.nlm.nih.gov/projects/genotyping/view.cgi?db=2)(File supplementare 1). Inoltre, sono stati utilizzati otto ceppi NHP. Tutti i riferimenti sono stati combinati in un unico file FASTA ed è stata eseguita una mappatura competitiva utilizzando il BWA. La mappatura è stata effettuata utilizzando il seguente comando:
bwa aln -n 0.01 l 300 $INDEX $FASTQCM $OUT
dove $INDEX è il riferimento, $FASTQCM è il file di ingresso e $OUT è il file di uscita.
La qualità minima della mappatura è stata impostata a 0.
Rimozione duplicati
Abbiamo utilizzato DeDup versione 0.11.3, parte della pipeline EAGER,Peltzer et al., 2016 per identificare e rimuovere tutte le letture duplicate nei file BAM specifici del campione(file supplementare 2) con le opzioni predefinite e il seguente comando:
java -jar DeDup.jar -i $IN -o $OUT
dove $IN è il file BAM in ingresso e $OUT è il file BAM in uscita.
L’estrazione mappata legge
Dopo la rimozione dei duplicati, i file BAM risultanti sono stati convertiti in file SAM utilizzando SAMtools versione 0.1.19-96b5f2294a con parametri di default e il seguente comando:
vista samtools -h -o $OUT $IN
dove $OUT è il file di uscita SAM e $IN è il file di ingresso BAM. Legge dal file SAM dove viene convertito in FASTQ utilizzando il seguente script awk:
awk ‘/^[FMR]/{print ‘@”$1’\n’$10’\n’$10’\n+\n’$11}’ $IN > $OUT dove $IN è il file SAM e $OUT è il file FASTQ risultante contenente tutte le letture mappate.
Assemblaggio De novo
L’assemblaggio de novo è stato eseguito utilizzando l’assemblatore del genoma SPAdes versione v3.9.0(Bankevich et al., 2012) con le seguenti impostazioni:
picche.py -t 20 m 500 k
11,13,15,17,19,21,23,25,27,29,31,33,35,37,39,41,43,45,47,49,51,53,55,57,59,61,63,65,67,69,71,73,75,77,79,81,83,85,87,89,91,93,95,97,99,101,103,105,107,109,111,113,115,117,119,121,123,125,127 s $IN -o $OUT
dove $IN è un file FASTQ contenente le letture mappate e $OUT è la cartella di output per SPAdes.
I contig risultanti per ogni valore K sono stati controllati e quello che ha generato il contig più lungo è stato selezionato per un’ulteriore elaborazione(file supplementare 3).
Mappatura dei contig
I vincoli sono stati mappati rispetto al file multi FASTA contenente tutti e 16 i riferimenti. È stato utilizzato il seguente comando:
bwa mem $INDEX $IN $OUT
dove $INDEX è il riferimento, $IN il file contenente i conti/conti e $OUT il file BAM risultante.
Generazione del consenso
Per la ricostruzione genomica degli antichi ceppi HBV, i risultati degli allineamenti sono stati esaminati visivamente con IGV versione 2.3.92(Thorvaldsdóttir et al., 2013). Per la costruzione di una sequenza di consenso sono state utilizzate informazioni sull’ordine di contig e sulla direzione. Le basi che erano state tagliate in modo morbido nell’allineamento sono state tagliate utilizzando il software SeqKit versione 0.7.0 e riallineate ai 16 riferimenti come descritto sopra. Questo è stato fatto a causa della struttura circolare del genoma di HBV. I grandi contigui dovevano essere divisi per preservare l’ordine genomico rispetto alle sequenze di riferimento(file supplementare 4).
La riapplicazione del grezzo va contro la sequenza di consenso
Le letture grezze di ogni campione sono state mappate sulla corrispondente sequenza di consenso utilizzando il software CircularMapper versione 1.93.4 e la seguente riga di comando:
java -jar CircularGenerator.jar -e $E -i $IN -s ‘$N’.
dove $E è la lunghezza dell’allungamento, $IN è il file di input e $N è il nome della sequenza di destinazione.
bwa aln -t 8 $IN $R -n 0.01-l 300-f $OUT
dove $IN è la sequenza di consenso allungata, $R è il file contenente le letture ritagliate e unite e $OUT è il file di uscita.
bwa samse $RE $RE $IN $R -f $OUT
dove $RE è il riferimento allungato, $IN è l’uscita bwa aln, $ R è il file contenente le letture ritagliate e unite e $OUT è il file di uscita.
java -jar realign-1.93.4.jar -e $E -i $IN -r $OR
dove $E è la lunghezza dell’allungamento, $IN è l’output di bwa samse e $OR è il consenso non modificato.
Analisi filogenetica
I ceppi di riferimento dell’epatite B per le scimmie sono stati raccolti utilizzando edirect con il seguente comando:
esearch -db pubmed -query ‘epatite B e orangutan O epatite B e Gibbon O epatite B e gorilla O epatite B e scimpanzé O epatite B e orangutan’ | elink -target nuccore | efetch -format fasta > $OUT
dove $OUT è il file di output in formato fasta contenente tutte le sequenze dei documenti che contengono le chiavi di ricerca.
Per controllare le sequenze ricevute è stato effettuato un allineamento di sequenze multiple utilizzando l’algoritmo linsi contenuto nella versione 7.310 del MAFFT. È stato utilizzato il seguente comando:
linsi $IN > $OUT
dove $IN è il file di input contenente le sequenze recuperate e $OUT è l’allineamento delle sequenze multiple.
L’allineamento è stato ispezionato visivamente in AliView (v. 1.18.1) e le sequenze che differivano dalla maggioranza sono state rimosse. Questo passo è stato necessario a causa del comando di ricerca senza restrizioni che, per caso, poteva anche restituire sequenze non primarie. Dopo il filtraggio il set conteneva 74 scimmie che infettavano i ceppi HBV.
Utilizzando i 74 ceppi di scimmie e 5497 genomi non ricombinanti disponibili su hpvdb(https://hbvdb.ibcp.fr/HBVdb/HBVdbDataset?seqtype=0) è stato effettuato il clustering con UClust v 1.1.579 (Edgar, 2010). Il clustering con una soglia di identità del 97% ha prodotto 493 genomi rappresentativi dell’HBV. Combinandoli con i cinque ceppi antichi è stato effettuato un allineamento di sequenze multiple utilizzando la versione 10.1.2 di Geneious(Kearse et al., 2012) con una matrice di costo simile al 65%, una penalità di 12 e una penalità di 3. L’allineamento di sequenze multiple è stato eliminato da tutti i siti (colonne) che avevano lacune in più del 95% delle sequenze. L’allineamento completo che include tutti i genomi moderni e antichi come multi-fasta è disponibile in Figura 2. L’allineamento è stato utilizzato per costruire una rete con il software SplitsTree v4(Huson e Bryant, 2006), creando una NeighborNet(Bryant e Moulton, 2004) con distanze P non corrette.
Lo stesso allineamento di sequenze multiple è stato utilizzato per la generazione di alberi di Maximum-Likelihood (ML) e Neighbour-Joining (NJ). È stata utilizzata la versione MEGA7 7170509-x86_64 con la seguente riga di comando:
Megacc -a $MAO -d $IN -o $OUT
dove $MAO è il file di configurazione del megacc, $IN è l’allineamento multiplo e $OUT è la directory di uscita. Per entrambi gli alberi sono stati utilizzati 1408 siti informativi e il modello di sostituzione Jukes-Cantor. I replicati di Bootstrap sono 2000 per ML e 10000 per NJ. Gli alberi sono forniti in Figura 2 dati sorgente 3 e 4.
Analisi dell’orologio molecolare
L’evoluzione del virus dell’epatite B nel tempo non è chiara per quanto riguarda il suo tasso evolutivo e il ruolo della ricombinazione. Studi precedenti hanno tentato di rilevare una firma molecolare simile ad un orologio senza successo. Indaghiamo se i genomi antichi qui presentati permettono un’analisi dell’orologio molecolare utilizzando TempEst v1.5.1(Rambaut et al., 2016). Il set di dati mostra una correlazione poco positiva tra la divergenza genetica e il tempo di campionamento (coefficiente di correlazione 0,075) e c’è molto poco segnale temporale (TempEst R^2 = 0,006, vedi Figura 2-figure supplement 9). Pertanto, ci asteniamo da ulteriori analisi di datazione.
Analisi della ricombinazione
Abbiamo eseguito l’analisi di ricombinazione utilizzando tutti i moderni genomi di riferimento completi (n = 493) e cinque genomi antichi utilizzati per l’analisi della rete (vedi sopra in Analisi Filogenetica). I metodi RDP, GENECOV, GENECOV, Chimera, MaxChi, BootScan, SiScan, 3Seq all’interno di RDP v4(Martin et al., 2015) con una dimensione della finestra di 100 nt e il parametro impostato sul genoma circolare con e senza riferimento all’outgroup (i risultati sono forniti in Figura 2 dati fonte 1) e SimPlot v 3.5.1 (Loleet al., 1999, Figura 2-figure supplements 5- 8) sono stati applicati al set di dati.
Analisi LC-MS/MS e ricerche nel database
Le proteine sono state estratte da campioni di denti in polvere (50 mg) utilizzando un protocollo modificato di preparazione del campione filtrato (FASP) come descritto in precedenza(Warinner et al., 2014; Cappellini et al., 2014). I campioni digeriti con tripsiina in-filtro sono stati analizzati su un Dionex Ultimate 3000 nano-HPLC accoppiato ad uno spettrometro di massa Q Exactive (Thermo Scientific, Brema). I campioni sono stati lavati su una colonna trappola (Acclaim Pepmap 100 C18, 10 mm ×300 μm, 3 μm, 100 Å, Dionex) per 5 minuti con 3% acetonitrile (ACN)/0,1% TFA ad una portata di 30 μL/min prima della separazione dei peptidi su una colonna analitica Acclaim PepMap 100 C18 (15 cm ×75 μm, 3 μm, 100 Å, Dionex). Una portata di 300 nL/min utilizzando l’eluente A (0,05% di acido formico (FA)) e l’eluente B (80% ACN/0,04% FA) è stato utilizzato per la separazione a gradiente come segue: gradiente lineare 5 ± 50% B in 60 min, 50 ± 95% B in 5 min, 95% B per 10 min, 95 ± 5% B in 1 min, e l’equilibrio al 5% B per 12 min. La tensione di spruzzatura applicata su un emettitore PicoTip rivestito in metallo (dimensione della punta 30 μm, New Objective, Woburn, Massachusetts, US) era di 1,25 kV, con una temperatura della sorgente di 250°C. Gli spettri MS a scansione completa sono stati acquisiti da 5 a 145 min tra 300 e 2.000 m/z ad una risoluzione di 60.000 a m/z 400 (obiettivo di controllo automatico del guadagno [AGC] di 1E6; tempo massimo di iniezione ionica [IIT] di 500 ms). I cinque precursori più intensi con stati di carica 2 + utilizzati sono stati selezionati con una finestra di isolamento di 1,6 m/z e frammentati da HCD con energie di collisione normalizzate di 25. La tolleranza di massa del precursore è stata impostata a 10 ppm, e l’esclusione dinamica (30 s) è stata attivata.
Gli spettri acquisiti sono stati analizzati mediante ricerche nel database utilizzando Proteome Discoverer (PD) 2.2.0.388 con i motori di ricerca SequestHT (Thermo Scientific). Le ricerche sono state effettuate su un database combinato costruito dalla combinazione del database completo delle proteine svizzere (468.716 voci, scaricate da Uniprot,21 dicembre 2017), del database dell’epatite B (sette voci, scaricate da Uniprot,7 dicembre 2017) e dei comuni contaminanti di laboratorio (115 voci, scaricate da Uniprot,15 agosto 2014). Per la ricerca sono state utilizzate le seguenti impostazioni: specificità semi-triptica; due siti di scissione mancanti; tolleranze di massa di 10 ppm per i precursori e per le masse dei frammenti 0,02 Da (HCD) e 0,5 Da (CID); modifiche statiche: carbamidometilazione su Cys; modifiche dinamiche: ossidazione di Met, Lys e Pro. Un’ulteriore ricerca è stata effettuata utilizzando 12 file FASTA da sequenze di DNA tradotte in silico. Le sequenze di DNA sono state ottenute da precedenti sequenze di DNA dei campioni.
Una serie quasi completa di y-ion e due frammenti di b-ion permettono di assegnare la sequenza peptidica completa. Il peptide è stato identificato nel campione biologico di Petersberg con quattro corrispondenze spettrali peptidiche, dimostrando che il rilevamento di questo peptide non è un evento casuale. Inoltre, lo stesso peptide potrebbe essere identificato anche nel secondo campione biologico di Karsdorf (non mostrato); le corse in bianco tra le corse LC-MS/MS dei due campioni escludono potenziali artefatti dovuti al carryover del campione.
Si noti che il metodo MS/MS qui applicato non permette di distinguere i residui di leucina (L) o isoleucina (I). La permutazione manuale dei residui di leucina nella sequenza sopra indicata seguita da una ricerca BLAST (parametri di ricerca predefiniti) ha portato all’identificazione dell’antigene del nucleo esterno della proteina HBV in tutti i casi ad eccezione delle combinazioni DIIDTASALYR e DLLDTASAIYR, queste due varianti sono state segnalate dalla ricerca BLAST come le proteine ipotetiche proteina CR988_04570[Treponema sp.] e anticorpo anti-GFP [costrutto sintetico] con l’antigene del nucleo esterno della proteina HBV-proteina elencati al rango 3. Tuttavia, queste proteine non sono state trovate nei dati genomici. Quindi, nonostante l’incertezza dell’assegnazione I/L, i dati MS/MS supportano la scoperta genomica di un’infezione da HBV.
Analisi genetiche della popolazione umana
La mappatura dei file FASTQ tagliati con l’adattatore e uniti al genoma umano di riferimento hg19 è stata effettuata utilizzando BWA(Li e Durbin, 2010) con la seguente riga di comando:
bwa aln -n 0.01 l 300 $INDEX $FASTQCM $OUT
dove $INDEX è il riferimento, $FASTQCM è il file di ingresso e $OUT è il file di uscita. La rimozione dei duplicati dopo la mappatura è stata eseguita come descritto sopra.
I dati di sequenziamento mappati sono stati trasformati nel formato Eigenstrat(Price et al., 2006) e fusi con un set di dati di 1.233.013 SNPs(Haak et al., 2015; Mathieson et al., 2015). Utilizzando il software Smartpca(Patterson et al., 2006) i tre campioni e le popolazioni antiche precedentemente pubblicate sono stati proiettati su una mappa di base della variazione genetica calcolata su 32 popolazioni dell’Eurasia occidentale(Figura 1-figure supplements 4, 8 e 9).
Determinazione del sesso
Il sesso è stato valutato in base al rapporto tra le sequenze che si allineano ai cromosomi X e Y rispetto agli autosomi(Skoglund et al., 2013).
References
- Araujo NM. Hepatitis B virus intergenotypic recombinants worldwide: an overview. Infection, Genetics and Evolution. 2015; 36:500-510. DOI
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology. 2012; 19:455-477. DOI | PubMed
- Behnke HJ. Archäologie in Sachsen-Anhalt N.F. 2007; 4:63-86.
- Behnke HJ. Archäologie in Sachsen-Anhalt N.F. 2011; 5:184-199.
- Behnke HJ. Archäologie in Sachsen-Anhalt N. F. 2012; 6:35-70.
- Bollongino R, Nehlich O, Richards MP, Orschiedt J, Thomas MG, Sell C, Fajkosová Z, Powell A, Burger J. 2000 years of parallel societies in stone age central Europe. Science. 2013; 342:479-481. DOI | PubMed
- Bouckaert R, Alvarado-Mora MV, Pinho JR. Evolutionary rates and HBV: issues of rate estimation with Bayesian molecular methods. Antiviral Therapy. 2013; 18:497-503. DOI | PubMed
- Brandt G, Knipper C, Nicklisch N, Ganslmeier R, Klamm M, Alt KW. Early Farmers. The View from Archaeology and Science. Oxford University Press, British Academy; 2014.
- Bryant D, Moulton V. Neighbor-net: an agglomerative method for the construction of phylogenetic networks. Molecular Biology and Evolution. 2004; 21:255-265. DOI | PubMed
- Cappellini E, Gentry A, Palkopoulou E, Ishida Y, Cram D, Roos A-M, Watson M, Johansson US, Fernholm B, Agnelli P, Barbagli F, Littlewood DTJ, Kelstrup CD, Olsen JV, Lister AM, Roca AL, Dalén L, Gilbert MTP. Resolution of the type material of the Asian elephant, Elephas maximus Linnaeus, 1758 (Proboscidea, Elephantidae). Zoological Journal of the Linnean Society. 2014; 170:222-232. DOI
- Castelhano N, Araujo NM, Arenas M. Heterogeneous recombination among Hepatitis B virus genotypes. Infection, Genetics and Evolution. 2017; 54:486-490. DOI | PubMed
- Claus M. Das neolithische Felsenkammergrab auf dem Halsberg bei Sorsum, Stadt Hildesheim. Die Kunde N. F. . 1983;91-122.
- Czarnetzki A. Die menschlichen Skelettreste aus vier neolithischen Steinkisten Hessens und Niedersachsens. Ungedr. Diss. Univ. Tübingen: Tübingen; 1966.
- Dabney J, Knapp M, Glocke I, Gansauge MT, Weihmann A, Nickel B, Valdiosera C, García N, Pääbo S, Arsuaga JL, Meyer M. Complete mitochondrial genome sequence of a middle pleistocene cave bear reconstructed from ultrashort DNA fragments. PNAS. 2013; 110:15758-15763. DOI | PubMed
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010; 26:2460-2461. DOI | PubMed
- Haak W, Lazaridis I, Patterson N, Rohland N, Mallick S, Llamas B, Brandt G, Nordenfelt S, Harney E, Stewardson K, Fu Q, Mittnik A, Bánffy E, Economou C, Francken M, Friederich S, Pena RG, Hallgren F, Khartanovich V, Khokhlov A, Kunst M, Kuznetsov P, Meller H, Mochalov O, Moiseyev V, Nicklisch N, Pichler SL, Risch R, Rojo Guerra MA, Roth C, Szécsényi-Nagy A, Wahl J, Meyer M, Krause J, Brown D, Anthony D, Cooper A, Alt KW, Reich D. Massive migration from the steppe was a source for Indo-European languages in Europe. Nature. 2015; 522:207-211. DOI | PubMed
- Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Molecular Biology and Evolution. 2006; 23:254-267. DOI | PubMed
- Kahila Bar-Gal G, Kim MJ, Klein A, Shin DH, Oh CS, Kim JW, Kim TH, Kim SB, Grant PR, Pappo O, Spigelman M, Shouval D. Tracing hepatitis B virus to the 16th century in a Korean mummy. Hepatology. 2012; 56:1671-1680. DOI | PubMed
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, Thierer T, Ashton B, Meintjes P, Drummond A. Geneious basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics. 2012; 28:1647-1649. DOI | PubMed
- Key FM, Posth C, Krause J, Herbig A, Bos KI. Mining metagenomic data sets for ancient DNA: recommended protocols for authentication. Trends in Genetics. 2017; 33:508-520. DOI | PubMed
- Lazaridis I, Patterson N, Mittnik A, Renaud G, Mallick S, Kirsanow K, Sudmant PH, Schraiber JG, Castellano S, Lipson M, Berger B, Economou C, Bollongino R, Fu Q, Bos KI, Nordenfelt S, Li H, de Filippo C, Prüfer K, Sawyer S, Posth C, Haak W, Hallgren F, Fornander E, Rohland N, Delsate D, Francken M, Guinet JM, Wahl J, Ayodo G, Babiker HA, Bailliet G, Balanovska E, Balanovsky O, Barrantes R, Bedoya G, Ben-Ami H, Bene J, Berrada F, Bravi CM, Brisighelli F, Busby GB, Cali F, Churnosov M, Cole DE, Corach D, Damba L, van Driem G, Dryomov S, Dugoujon JM, Fedorova SA, Gallego Romero I, Gubina M, Hammer M, Henn BM, Hervig T, Hodoglugil U, Jha AR, Karachanak-Yankova S, Khusainova R, Khusnutdinova E, Kittles R, Kivisild T, Klitz W, Kučinskas V, Kushniarevich A, Laredj L, Litvinov S, Loukidis T, Mahley RW, Melegh B, Metspalu E, Molina J, Mountain J, Näkkäläjärvi K, Nesheva D, Nyambo T, Osipova L, Parik J, Platonov F, Posukh O, Romano V, Rothhammer F, Rudan I, Ruizbakiev R, Sahakyan H, Sajantila A, Salas A, Starikovskaya EB, Tarekegn A, Toncheva D, Turdikulova S, Uktveryte I, Utevska O, Vasquez R, Villena M, Voevoda M, Winkler CA, Yepiskoposyan L, Zalloua P, Zemunik T, Cooper A, Capelli C, Thomas MG, Ruiz-Linares A, Tishkoff SA, Singh L, Thangaraj K, Villems R, Comas D, Sukernik R, Metspalu M, Meyer M, Eichler EE, Burger J, Slatkin M, Pääbo S, Kelso J, Reich D, Krause J. Ancient human genomes suggest three ancestral populations for present-day Europeans. Nature. 2014; 513:409-413. DOI | PubMed
- Li H, Durbin R. Fast and accurate long-read alignment with burrows-wheeler transform. Bioinformatics. 2010; 26:589-595. DOI | PubMed
- Littlejohn M, Locarnini S, Yuen L. Origins and evolution of hepatitis B virus and hepatitis D virus. Cold Spring Harbor Perspectives in Medicine. 2016; 6DOI | PubMed
- Lole KS, Bollinger RC, Paranjape RS, Gadkari D, Kulkarni SS, Novak NG, Ingersoll R, Sheppard HW, Ray SC. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. Journal of Virology. 1999; 73:152-160. PubMed
- Lösch S. Medizinischen Fakultät der Universität München; 2009.
- Martin DP, Murrell B, Golden M, Khoosal A, Muhire B. RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evolution. 2015; 1DOI | PubMed
- Mathieson I, Lazaridis I, Rohland N, Mallick S, Patterson N, Roodenberg SA, Harney E, Stewardson K, Fernandes D, Novak M, Sirak K, Gamba C, Jones ER, Llamas B, Dryomov S, Pickrell J, Arsuaga JL, de Castro JM, Carbonell E, Gerritsen F, Khokhlov A, Kuznetsov P, Lozano M, Meller H, Mochalov O, Moiseyev V, Guerra MA, Roodenberg J, Vergès JM, Krause J, Cooper A, Alt KW, Brown D, Anthony D, Lalueza-Fox C, Haak W, Pinhasi R, Reich D. Genome-wide patterns of selection in 230 ancient Eurasians. Nature. 2015; 528:499-503. DOI | PubMed
- Meier T. Das Archäologische Jahr in Bayern. 1998.
- Meyer M, Kircher M. Illumina sequencing library preparation for highly multiplexed target capture and sequencing. Cold Spring Harbor Protocols. 2010; 2010DOI | PubMed
- Nicklisch N. Forschungsberichte des Landesmuseums für Vorgeschichte Halle. 2017; 11
- Patterson N, Price AL, Reich D. Population structure and eigenanalysis. PLoS Genetics. 2006; 2DOI | PubMed
- Patterson Ross Z, Klunk J, Fornaciari G, Giuffra V, Duchêne S, Duggan AT, Poinar D, Douglas MW, Eden JS, Holmes EC, Poinar HN. The paradox of HBV evolution as revealed from a 16th century mummy. PLoS Pathogens. 2018; 14DOI | PubMed
- Peltzer A, Jäger G, Herbig A, Seitz A, Kniep C, Krause J, Nieselt K. EAGER: efficient ancient genome reconstruction. Genome Biology. 2016; 17DOI | PubMed
- Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nature Genetics. 2006; 38:904-909. DOI | PubMed
- Rambaut A, Lam TT, Max Carvalho L, Pybus OG. Exploring the temporal structure of heterochronous sequences using TempEst (formerly Path-O-Gen). Virus Evolution. 2016; 2DOI | PubMed
- Rasche A, Souza B, Drexler JF. Bat hepadnaviruses and the origins of primate hepatitis B viruses. Current Opinion in Virology. 2016; 16:86-94. DOI | PubMed
- Schaefer S. Hepatitis B virus taxonomy and hepatitis B virus genotypes. World Journal of Gastroenterology. 2007; 13:14-21. DOI | PubMed
- Schuenemann VJ, Singh P, Mendum TA, Krause-Kyora B, Jäger G, Bos KI, Herbig A, Economou C, Benjak A, Busso P, Nebel A, Boldsen JL, Kjellström A, Wu H, Stewart GR, Taylor GM, Bauer P, Lee OY, Wu HH, Minnikin DE, Besra GS, Tucker K, Roffey S, Sow SO, Cole ST, Nieselt K, Krause J. Genome-wide comparison of medieval and modern Mycobacterium leprae. Science. 2013; 341:179-183. DOI | PubMed
- Simmonds P, Midgley S. Recombination in the genesis and evolution of hepatitis B virus genotypes. Journal of Virology. 2005; 79:15467-15476. DOI | PubMed
- Skoglund P, Storå J, Götherström A, Jakobsson M. Accurate sex identification of ancient human remains using DNA shotgun sequencing. Journal of Archaeological Science. 2013; 40:4477-4482. DOI
- Souza BF, Drexler JF, Lima RS, Rosário MO, Netto EM. Theories about evolutionary origins of human hepatitis B virus in primates and humans. The Brazilian Journal of Infectious Diseases. 2014; 18:535-543. PubMed
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Briefings in Bioinformatics. 2013; 14:178-192. DOI | PubMed
- Vågene ÅJ, Herbig A, Campana MG, Robles García NM, Warinner C, Sabin S, Spyrou MA, Andrades Valtueña A, Huson D, Tuross N, Bos KI, Krause J. Salmonella enterica genomes from victims of a major sixteenth-century epidemic in Mexico. Nature Ecology & Evolution. 2018; 2:520-528. DOI | PubMed
- Veit U. Studien zum Problem der Siedlungsbestattungen im europäischen Neolithikum, Münster. Waxmann: New York; 1996.
- Warinner C, Herbig A, Mann A, Fellows Yates JA, Weiß CL, Burbano HA, Orlando L, Krause J. A robust framework for microbial archaeology. Annual Review of Genomics and Human Genetics. 2017; 18:321-356. DOI | PubMed
- Warinner C, Rodrigues JF, Vyas R, Trachsel C, Shved N, Grossmann J, Radini A, Hancock Y, Tito RY, Fiddyment S, Speller C, Hendy J, Charlton S, Luder HU, Salazar-García DC, Eppler E, Seiler R, Hansen LH, Castruita JA, Barkow-Oesterreicher S, Teoh KY, Kelstrup CD, Olsen JV, Nanni P, Kawai T, Willerslev E, von Mering C, Lewis CM, Collins MJ, Gilbert MT, Rühli F, Cappellini E. Pathogens and host immunity in the ancient human oral cavity. Nature Genetics. 2014; 46:336-344. DOI | PubMed
- WHO. Global Hepatitis Report. 2017.
- Zhou Y, Holmes EC. Bayesian estimates of the evolutionary rate and age of hepatitis B virus. Journal of Molecular Evolution. 2007; 65:197-205. DOI | PubMed
Fonte
Krause-Kyora B, Susat J, Key FM, Kühnert D, Bosse E, et al. () Neolithic and medieval virus genomes reveal complex evolution of hepatitis B. eLife 7e36666. https://doi.org/10.7554/eLife.36666




