
Contesto
Il naltrexone è un antagonista dell’oppioide puro con attività a più recettori umani oppioidi e non oppioidi. I suoi usi su licenza sono come un aiuto per prevenire le ricadute nei disturbi da consumo di alcol (AUDs) e la dipendenza da oppioidi dopo l’astinenza, e nella combinazione compressa naltrexone-bupropione per l’obesità [1]. Queste condizioni sono tutti i principali problemi di salute a livello globale, con tassi crescenti di disabilità e di decesso che si verificano in molti paesi [2, 3]. Nonostante la preoccupazione per l’impatto di queste malattie e la necessità di cure, il naltrexone è attualmente sottoutilizzato nella maggior parte dei paesi, in particolare per i deficit di AUD [4, 6].
A dosi normali o superiori (≥50 mg), il naltrexone viene utilizzato anche off-label per diverse dipendenze e disturbi del controllo degli impulsi che attualmente non hanno trattamenti farmacologici autorizzati, come la dipendenza da anfetamine e cocaina [7, 8], i disturbi del controllo degli impulsi [9-11], i disturbi alimentari [12] e i disturbi dello spettro autistico [13].
In seguito ai risultati sperimentali che basse dosi di naltrexone provocano la soppressione della crescita tumorale [14] e la modulazione immunitaria [15], viene sempre più utilizzato a dosi di circa 4,5 mg. Questo è noto come naltrexone a basse dosi (LDN). Studi clinici su piccola scala di LDN sono stati condotti, per esempio, nella malattia di Crohn, nella sclerosi multipla, nella fibromialgia e nell’infezione da HIV, dove le prove hanno dimostrato efficacia e/o bassa tossicità [16- 19]. Altre condizioni per le quali viene utilizzato l’LDN, come la sindrome da fatica cronica (nota anche come encefalomielite mialgica), la sindrome del dolore regionale complesso e i disturbi autoimmuni, sono ancora in attesa di studi clinici randomizzati (RCT) [20- 22]. L’LDN è ora autorizzato come coadiuvante nell’infezione da HIV per le vendite da banco in Kenya e in Nigeria [23]. In Norvegia, è stato associato a una riduzione delle prescrizioni per i trattamenti più convenzionali in alcune condizioni [24, 25]. Nel Regno Unito, ogni anno vengono emesse circa 1.400 prescrizioni NHS per l’LDN ([26]; comunicazione personale di D. Steinke, ottobre 2016: Uso di LDN nel CPRD), mentre oltre 12.000 persone hanno ricevuto una prescrizione privata negli ultimi 10 anni (comunicazione personale di S. Dickson, ottobre 2017: Prescrizioni private di LDN dispensate da Dicksons Chemist Glasgow negli ultimi 10 anni).
Problemi di sicurezza noti per il naltrexone
Il naltrexone è controindicato in coloro che attualmente utilizzano gli oppiacei a causa della possibilità di gravi eventi avversi (SAE) di astinenza da oppiacei troppo rapida o di overdose di oppiacei, che possono essere pericolosi per la vita [1, 27]. Questi eventi avversi gravi sono di natura diversa da quelli che si verificano negli utilizzatori di oppiacei non drogati.
Le preoccupazioni sulla tossicità epatica causata dal naltrexone hanno avuto origine da diversi studi ad alte dosi (fino a 300 mg) negli anni ’80 [28]. A causa di questi risultati, la Food and Drug Administration (FDA) statunitense ha inizialmente richiesto un “avviso in scatola nera” sull’epatotossicità nel foglietto illustrativo del naltrexone; la FDA specifica tali avvertimenti per richiamare l’attenzione su rischi gravi o pericolosi per la vita. Tuttavia, poiché non sono noti casi di insufficienza epatica dovuta al naltrexone [29, 30], l’avvertenza è stata infine rimossa nel 2013 [27]. Il British National Formulary mette in guardia contro i casi di epatite acuta, insufficienza epatica o grave insufficienza renale e nei casi di grave insufficienza renale. Gli effetti collaterali noti includono nausea, vomito, dolori addominali, diminuzione dell’appetito, vertigini, letargia, mal di testa e disturbi del sonno [1, 29].
Sicurezza dei farmaci negli studi clinici
La qualità della registrazione e della segnalazione dei danni negli studi clinici è stata storicamente meno rigorosa di quella dell’efficacia [31, 32]. Il progresso è stato favorito dall’introduzione di definizioni standard per gli eventi avversi (Box 1); l’obbligo di tenere registrazioni dettagliate degli eventi avversi (AE) negli studi clinici, introdotto nel 2001; l’International Committee of Medical Journal Editors’. l’approvazione degli standard di segnalazione suggeriti nell’estensione degli standard consolidati di segnalazione degli eventi avversi (CONSORT) per i danni pubblicati nel 2004 [33], e la lista di controllo degli eventi avversi per le revisioni sistematiche e le meta-analisi preferite (PRISMA) pubblicata nel 2016 [34]; e l’obbligo di registrare gli esiti, compresi gli EA e i SAE, per gli RCT registrati nei registri degli studi clinici dal 2014 nell’Unione Europea (UE) [35] e dal 2017 negli USA [36]. Una sintesi delle prove di danno (SAE e AE) contribuirebbe a fornire un profilo di sicurezza più accurato del naltrexone.
Perché è importante fare questa recensione?
Ci sono state diverse revisioni descrittive e non sistematiche della sicurezza del naltrexone recentemente [37, 38], ma nessuna si è finora concentrata su AE e SAE negli studi clinici sul naltrexone. Includendo studi provenienti da un’ampia gamma di condizioni e concentrandosi solo su uno specifico risultato avverso che ha una definizione normativa, dovrebbe consentire di raccogliere una grande quantità di dati nocivi di alta qualità. Le persone con dipendenze sono riluttanti ad assumere, e i terapeuti a prescrivere, un farmaco per superare la dipendenza da un altro farmaco, incluso l’alcol [39, 40], e i medici rimangono preoccupati per il rischio di tossicità epatica con il naltrexone [41- 43]. Pertanto, sono necessarie prove sulla sicurezza del naltrexone. I pazienti che assumono naltrexone e LDN possono farlo per periodi di tempo prolungati; pertanto, stabilire la sicurezza a lungo termine del naltrexone è particolarmente importante. Può essere possibile scoprire tassi aumentati o diminuiti di alcuni SAE, in particolare eventi cardiovascolari o cerebrovascolari o tumori nelle meta-analisi, a causa dei loro tassi di fondo generalmente bassi [44].
Lo scopo principale di questa revisione era quello di esaminare i SAE che si verificano negli studi clinici sul naltrexone orale, dato per qualsiasi condizione, a parte l’uso di oppioidi o ex oppioidi, rispetto al placebo. La nostra attenzione per i SAE è in linea con la recente enfasi posta sulla comprensione e la prevenzione di danni permanenti o permanenti al paziente (piuttosto che sull’esame di ogni AE), come evidenziato, ad esempio, nella revisione Dalton del dovere di candore [45]. Ulteriori obiettivi sono stati quelli di indagare i possibili fattori di rischio di SAE per il naltrexone attraverso l’analisi di sottogruppi di gruppo di malattie, il dosaggio e la durata dello studio; di esaminare SAE specifici (decessi, eventi cardiovascolari o cerebrovascolari e tumori); e di esaminare i ritiri e i ritiri dovuti a SAE negli stessi studi clinici. Un obiettivo secondario era quello di esaminare gli AA per il naltrexone rispetto al placebo.
Metodi
La revisione ha seguito il Cochrane Handbook for guidance per tutta la durata [46] e il PRISMA danneggia l’estensione [34]. Il protocollo è stato registrato sul sito web di PROSPERO nel gennaio 2017, numero di registrazione CRD42017054421. È possibile accedervi all’indirizzo https://www.crd.york.ac.uk/PROSPERO/display_record.asp?ID=CRD42017054421.
Criteri di selezione
Sono stati inclusi tutti gli RCT di progettazione parallela di durata superiore a 4 settimane, in partecipanti di qualsiasi età e per qualsiasi condizione, in cui il naltrexone orale è stato confrontato con il placebo. Gli studi in cui l’uso di oppioidi o ex oppioidi è stato specificato nel protocollo sono stati esclusi a causa della possibilità di interazioni oppioidi / antagonisti oppioidi che si verificano. Sono stati inclusi solo gli studi pubblicati dopo il 1° gennaio 2001, a causa della diffusa introduzione di norme che richiedono la registrazione degli AA e la segnalazione degli AA negli RCT di quell’anno [47].
Risultati
La principale misura di risultato è stata il numero di partecipanti con un SAE registrato nel braccio naltrexone rispetto al braccio placebo. Il giudizio dell’investigatore sul fatto che si sia verificato un SAE e che sia stata seguita una causalità, come suggerito dalla Conferenza Internazionale sull’Armonizzazione (ICH) (Box 1). Laddove non è stata data alcuna definizione, per sostenere il nostro giudizio è stata utilizzata la definizione o le definizioni della guida dettagliata CT-3 per l’UE [48] e dalla FDA per gli USA [49], come riassunto nel riquadro 1. Il risultato secondario è stato il tipo di AA riportato in entrambi i rami del trattamento.
Metodi di ricerca per l’identificazione degli studi
Sono state ricercate le seguenti banche dati elettroniche: Cochrane Central Register of Controlled Trials (CENTRAL), PubMed MEDLINE, EMBASE (via OVID), Web of Science Core Collection, PsycINFO (via OVID) e International Pharmaceutical Abstracts via OVID (file aggiuntivo 1). Non ci sono state restrizioni linguistiche. Non sono stati inclusi termini per l’AE o per gli effetti collaterali per evitare una selezione troppo restrittiva degli studi con il potenziale rischio di distorsione della segnalazione degli esiti [51- 53]. La data finale delle ricerche era maggio 2018.
Ulteriori fonti sono state le rilevanti revisioni sistematiche contenenti studi clinici sul naltrexone e gli articoli di riviste che sono stati valutati per l’inclusione in questa revisione. Il Registro Internazionale degli Studi Clinici dell’Organizzazione Mondiale della Sanità, il Registro degli Studi Clinici degli Stati Uniti, clinicaltrials.gov e il Registro degli Studi Clinici dell’Unione Europea EudraCT sono stati ricercati utilizzando la parola “naltrexone”. Queste sono buone fonti di studi clinici non pubblicati ma completati [36]. Quando uno studio è apparso inedito, lo sperimentatore principale è stato contattato per confermarlo. Sono stati registrati gli studi in corso, per consentire il futuro aggiornamento di questa revisione sistematica. La letteratura grigia è stata inclusa nella revisione dai registri degli studi clinici, gli abstract delle conferenze elencate in CENTRAL, i documenti normativi presentati alla FDA statunitense per le licenze dei farmaci e gli studi non pubblicati provenienti da precedenti revisioni sistematiche.
Raccolta e gestione dei dati
Tutti gli screening e l’estrazione dei dati sono stati effettuati da due ricercatori indipendenti (MB e SB per lo screening e MB e AM per l’estrazione dei dati), e i risultati sono stati confrontati per stilare una lista finale. Eventuali differenze sono state risolte mediante discussione, con l’occasionale contributo di un terzo revisore (HvM, MP, SR o LR). Lo screening iniziale ha eliminato gli studi utilizzando il titolo e l’abstract, con documenti completi esaminati per selezionare gli studi finali inclusi. Tutte le ricerche sono state scaricate nel software di riferimento Endnote, dove sono stati rimossi i duplicati dei documenti, e sono stati identificati più documenti collegati allo stesso studio. Sono stati registrati i numeri trovati in ogni fase e le motivazioni delle decisioni prese.
I dati sono stati registrati su moduli di estrazione dati. I dati quantitativi per gli esiti primari e secondari, i numeri di iscrizione e i ritiri (numeri e motivi), i SAE (sia il numero di partecipanti con un SAE che il numero totale di SAE e le descrizioni) e gli AE (numeri totali per il termine preferito del Medical Dictionary for Regulatory Activities (MedDRA)) sono stati estratti su un foglio di calcolo Excel. Le appendici dei siti web, gli studi sussidiari e gli eventuali protocolli pubblicati sono stati esaminati per ottenere informazioni rilevanti. I risultati su clinicaltrials.gov e su EudraCT sono stati sottoposti a un controllo incrociato con i dati disponibili nel rapporto dello studio.
Valutazione della qualità
Lo strumento Cochrane risk of-bias [54] è stato adattato per le misure di esito di questa revisione, evidenziando otto aree di conduzione degli studi e di segnalazione. L’estensione CONSORT per i danni [33] è stata utilizzata per informare la scelta dei criteri. Le aree scelte sono state: generazione di sequenze casuali (bias di selezione)occultamento delle assegnazioni (bias di selezione)Accecamento dei partecipanti e del personale alla randomizzazione (bias di performance)Accecamento della valutazione dell’esito (bias di rilevazione)Adeguata segnalazione dei dati di esito (bias di attrito)Adeguata raccolta di EA e SAE (bias di attrito)Adeguata segnalazione di SAE (bias di segnalazione)Altre bias (per esempio sponsorizzazione commerciale, periodi di rodaggio con placebo)
È stata redatta una tabella del rischio di parzialità che comprendeva i commenti tratti direttamente dai documenti, seguiti da un giudizio per ogni studio. I giudizi sono stati espressi da MB e tutte le decisioni sono state esaminate da SR, con discussioni occasionali con un terzo revisore al fine di raggiungere un consenso. I risultati sono stati utilizzati per identificare gli studi a basso rischio di distorsione in tutte le otto categorie (studi a basso rischio), mentre gli studi rimanenti con almeno una categoria giudicata non a basso rischio.
Misure dell’effetto del trattamento
Una corrispondente meta-analisi è stata eseguita utilizzando dati estratti da pubblicazioni su riviste e altre fonti (clinicaltrials.gov e dati forniti dagli autori), laddove rilevanti. Il pooled risk ratio (RR) è stato confrontato tra gli studi che riportano i SAE nel braccio naltrexone rispetto al braccio placebo per gli eventi registrati durante il trattamento attivo (naltrexone o placebo). Poiché i partecipanti possono avere più SAE identici in uno studio clinico, o più SAE che potrebbero essere correlati, l’RR è stato analizzato per partecipante piuttosto che per evento. È stata eseguita un’analisi di sensibilità della differenza di rischio (RD) perché utilizza i dati di tutti gli studi, compresi quelli senza eventi in entrambi i bracci. Sono state inoltre eseguite meta-analisi per la RR dei singoli EA a termine preferenziale di MedDRA, ritiri, ritiri dovuti a EA e decessi. Gli intervalli di confidenza associati sono stati registrati al 95%.
Sintesi dei dati e valutazione dell’eterogeneità
Sebbene ci fosse un’eterogeneità clinica tra gli studi, la meta-analisi era appropriata perché il comparatore e la misura dell’esito era la stessa per tutti gli studi e la direzione dell’effetto era probabilmente simile [55]. Il programma R è stato utilizzato per tutte le meta-analisi (R Foundation for Statistical Computing, Vienna, Austria). Gli studi con eventi in un solo braccio sono stati inclusi applicando la correzione di continuità di aggiungere 0,5 a tutte le celle di una tabella di risultati 2 × 2 per ogni studio [56, 57]. Gli studi con doppio zero (cioè gli studi che riportano eventi zero in ogni braccio di trattamento) sono stati esclusi dall’analisi, come raccomandato nel Cochrane Handbook. I dati sono stati analizzati in base all’intenzione di trattamento. L’eterogeneità clinica è stata riconosciuta utilizzando modelli di effetti casuali in tutte le analisi. L’eterogeneità statistica è stata esaminata con la statistica I2. Valori inferiori al 25% rappresentano una bassa eterogeneità e superiori al 75% rappresentano un’elevata eterogeneità [55]. È stata utilizzata una meta-regolazione univariata e multivariata per esplorare ulteriori cause di eterogeneità che coinvolgono le covariate, tra cui l’età, il sesso, l’anno di pubblicazione, la durata dello studio e la qualità dello studio (cioè basso o alto rischio di distorsioni).
Studi con gruppi di trattamento multipli
Sono stati inclusi studi per la sperimentazione di più farmaci o terapie (ad esempio, farmaco più naltrexone rispetto al farmaco, o un disegno fattoriale a quattro bracci) quando c’era un braccio placebo adatto per il confronto con il naltrexone. Non sono stati inclusi gli studi con una combinazione fissa di naltrexone e un altro farmaco in cui il comparatore era un singolo placebo. Questo escludeva la compressa di combinazione di naltrexone-bupropione a rilascio lento. Negli studi con più bracci di naltrexone e un solo braccio placebo (ad esempio, se sono stati sperimentati diversi dosaggi di naltrexone), i dati del braccio placebo sono stati divisi in base alle proporzioni dei partecipanti reclutati per ogni braccio di naltrexone. Questo ha evitato un doppio conteggio del braccio placebo. Nelle prove con più interventi psicoterapeutici in diversi bracci, i risultati di questi potevano essere combinati, a patto che gli stessi interventi fossero nei bracci placebo.
Dati mancanti
Se i dati erano mancanti o ambigui (ad esempio, se non era chiaro da un documento se si erano verificati dei SAE o se c’erano discrepanze nei dati tra il documento e il sito web clinicaltrials.gov), gli autori principali sono stati contattati per ulteriori informazioni. Sono state registrate tutte le corrispondenze di questo tipo, anche quando ciò ha comportato modifiche ai dati. Negli studi privi di commenti specifici sui SAE, talvolta era necessario un giudizio per determinare la presenza o l’assenza di SAE, a seconda dell’entità delle informazioni fornite sugli EA. I motivi delle decisioni sono stati registrati, citando il testo o la tabella pertinente dello studio. Tutti gli studi che prevedevano giudizi sui dati sono stati giudicati poco chiari per il rischio di una distorsione della comunicazione.
Sottogruppo e analisi di sensibilità
Le analisi dei sottogruppi di malattia o condizione e dose sono state definite a priori con una motivazione per cui tali differenze nei tassi di SAE possono esistere [55]. Inoltre, la durata dello studio è stata aggiunta come analisi post hoc a causa del suo potenziale effetto di modifica dei tassi di EAS. Le analisi di sensibilità, che comprendono solo studi a basso rischio di bias in tutte le categorie, hanno esplorato la robustezza dei risultati rispetto ai rischi di bias [55]. Altre analisi di sensibilità sono state esplorate in seguito ai risultati dell’analisi dei dati per testare la robustezza dei risultati.
Valutazione degli errori di segnalazione
Questa recensione ha cercato di ridurre i pregiudizi di pubblicazione utilizzando strategie di ricerca ad ampio raggio, includendo le pubblicazioni che non erano in inglese, e cercando gli studi clinici non dichiarati nei registri degli studi clinici. Il bias di segnalazione è stato valutato visivamente per ogni meta-analisi utilizzando trame ad imbuto e le relative analisi statistiche.
Risultati
Flusso di prova: diagramma di flusso e numeri
Le ricerche elettroniche hanno identificato 7873 citazioni, e altre 995 registrazioni individuate da siti web di studi clinici (821), revisioni sistematiche (157) e riferimenti in altri documenti (17). L’eliminazione di doppi riferimenti ha ridotto il numero a 4738 record, di cui 4390 sono stati esclusi in base all’esame degli abstract. Sono stati ottenuti articoli a testo integrale per 348 citazioni. Da questi, 96 citazioni sono state escluse e 163 erano documenti sussidiari. Sono stati così individuati 89 studi primari (file aggiuntivo 2). I numeri identificati in ogni fase, dalla ricerca iniziale alle analisi quantitative, e le ragioni per l’esclusione degli studi, sono riportati in un diagramma di flusso PRISMA 2009 (Fig. 1) [58].Fig 1PRISMA 2009 diagramma di flusso. Organizzazione Mondiale della Sanità dell’OMS; EudraCT European Clinical Trials Database
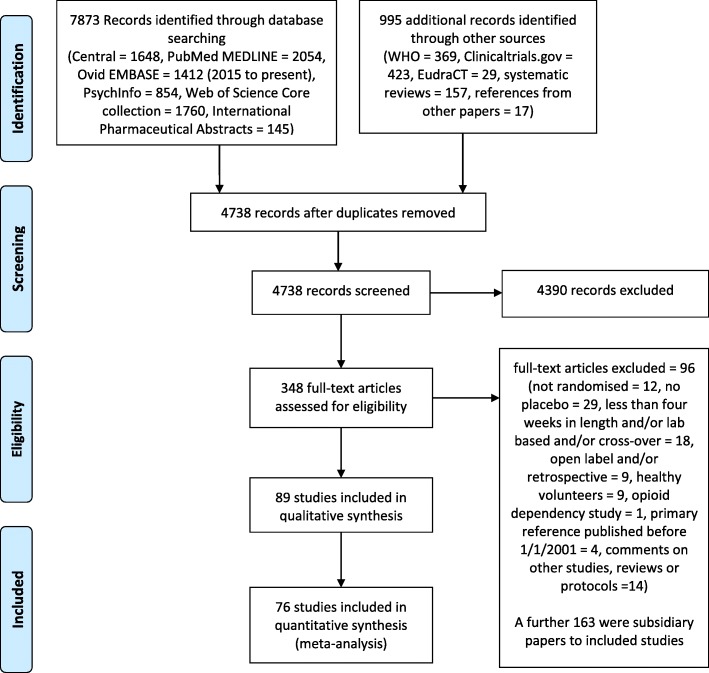
Fig. 1.Diagramma di flusso PRISMA 2009. Organizzazione Mondiale della Sanità dell’OMS; EudraCT European Clinical Trials Database
Caratteristiche degli studi inclusi
Sono stati trovati ottantanove studi (11.194 partecipanti) che hanno soddisfatto i criteri di revisione, compresa la pubblicazione dopo il 1° gennaio 2001. Tre studi sono stati esclusi perché hanno fornito solo il numero totale dei partecipanti, lasciando 86 studi (10.957 partecipanti) dai quali è stato possibile estrarre i dati per l’analisi. La tabella 1 riassume le caratteristiche degli studi inclusi per categorie generali, mentre il file aggiuntivo 3: la tabella S1 fornisce i dettagli di ogni studio. La dose target di naltrexone variava da 3 mg a 250 mg. Le condizioni più frequenti sono state le AUD (36 studi). In altri 21 studi, tra cui studi sull’infezione da HIV, disturbi psichiatrici, dipendenze e fumo, i partecipanti hanno avuto una doppia diagnosi che includeva le AUD. Altri studi hanno riguardato vari disturbi psichiatrici, disturbi del controllo degli impulsi, altre dipendenze, obesità, malattia di Crohn, fibromialgia e tumori. I gruppi di pazienti in molti degli studi tendevano ad avere problemi complessi, multi-morbilità, dipendenze o malattie multiple presenti o passate e ad assumere più farmaci, così come i farmaci di prova.Tabella 1Riepilogo delle caratteristiche degli studi inclusiCategoriaCaratteristiche dei partecipanti allo studioNumero di studiMalattia o condizioneAUD38Tossicodipendenza da farmaci o fumo ± AUD18Disturbi psichiatrici ± AUD13Disturbi del controllo degli impulsi9Obesità o disturbi alimentari6Disturbi infiammatori3malignità2Dose target di naltrexonea (mg)≤4.5516-497506110012>1008Età media dove givenb (anni)10 a <20220 a <30230 a <30230 a <401140 a <506250 a <603≥602Lunghezza dello studioc (settimane)4-758-111712-154216-251826-526AUD studi sull’uso di alcol disordera3 sono stati studi di dosaggio multibracciob82 studic88 studic88
Risultati della valutazione della qualità
I risultati della valutazione del rischio Cochrane di distorsioni per tutti gli studi sono riassunti nel file aggiuntivo 4: Tabella S2. Dodici studi sono stati giudicati a basso rischio di distorsione in tutte le otto categorie. Questi studi hanno iscritto un totale di 2.540 partecipanti (28%). Diciotto studi (20%) erano a basso rischio per sei o sette delle categorie, e 14 studi (16%) erano a basso rischio in due o meno categorie.
Prevalenza e natura di eventi avversi gravi
In eventi attribuiti ad un particolare braccio di studio, naltrexone o placebo, sono stati registrati un totale di 315 SAE su 260 partecipanti. Il numero di partecipanti con almeno un SAE è stato di 119 nel braccio naltrexone e di 141 nel braccio placebo. Tra i 315 SAE, sono stati registrati nove decessi, tre nelle braccia naltrexone e sei nelle braccia placebo. Sebbene l’esame della natura e della causalità delle SAE fosse al di là dell’ambito di questo studio, ovunque tali dati siano stati forniti, sono stati estratti. Il nostro esame descrittivo di questi dati limitati ha suggerito che non ci sono differenze tra i due bracci di trattamento in termini di natura delle SAE. Tra gli studi inclusi, gli EA sono stati riportati attraverso 20 confronti indipendenti. Sono stati identificati un totale di 7.017 AA (che comprendono 188 eventi a termine preferenziali di MedDRA): 3.938 nel braccio naltrexone e 3.079 nel braccio placebo (file aggiuntivo 5: tabella S3). Tutti gli EA sono stati segnalati come di natura mite-moderata.
Test statistici e risultati
Eventi avversi gravi
Non c’erano prove di alcuna differenza tra naltrexone e placebo nella meta-analisi della RR delle SAE. In totale sono stati analizzati 31 confronti tra i 26 studi che registrano il numero di EAS per braccio di studio. Il RR messo in comune per il numero di partecipanti che hanno sperimentato almeno un SAE per il naltrexone rispetto al placebo non era statisticamente significativo (RR 0,84, 95% CI 0,66-1,06). I test per l’eterogeneità hanno mostrato una bassa eterogeneità statistica (I2 = 0%). La parcella forestale per questo risultato è mostrata in Fig. 2. Il RD messo in comune per il numero di partecipanti che hanno sperimentato almeno un SAE per il naltrexone rispetto al placebo non è stato significativo (RD -0,01, 95% CI -0,02-0,00). L’eterogeneità è stata bassa (I2 = 7%). La trama forestale per RR di morte non ha mostrato un aumento del rischio di morte per naltrexone rispetto al placebo (RR 0,79, 95% CI 0,33-1,91). Anche se specificato nel protocollo, nessuna meta-analisi dei SAE specifici a causa di eventi cardiovascolari o cerebrovascolari o tumori è stata intrapresa a causa del basso numero di eventi registrati. L’analisi di meta-regressione univariata e multivariata non ha rivelato alcun significato per nessuna delle covariate.Fig. 2Il diagramma di riferimento del rapporto di rischio (RR) di eventi avversi gravi in RCT di naltrexone vs placebo. I dati tra parentesi mostrano la media o la gamma di età dei partecipanti e la percentuale di partecipanti maschi o femmine. Sono stati esclusi dalla meta-analisi gli studi a doppio zero (cioè quelli che hanno riportato eventi zero in ogni gruppo di trattamento). Questo vale anche per tutte le analisi dei sottogruppi (per dose, malattia e tempo)
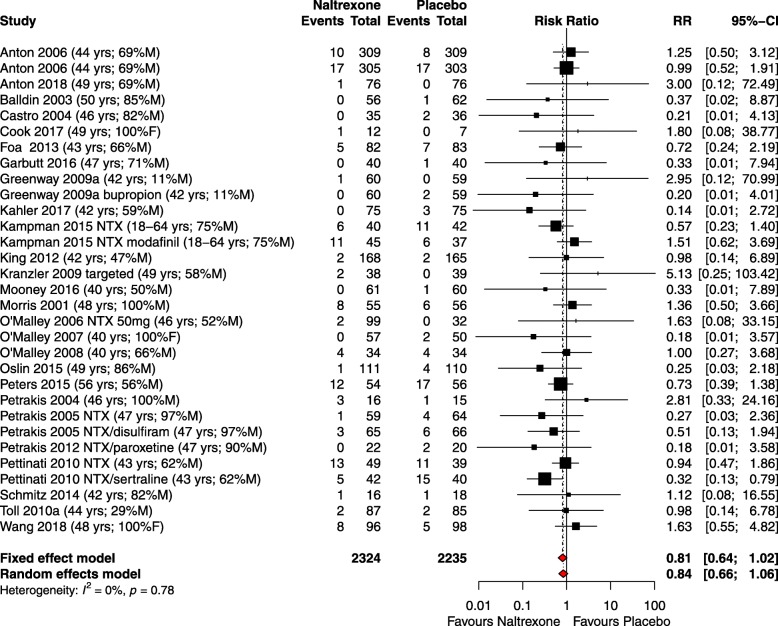
Fig 2.Trama forestale del rapporto di rischio (RR) di eventi avversi gravi in RCT di naltrexone vs placebo. I dati tra parentesi mostrano la media o la gamma di età dei partecipanti e la percentuale di partecipanti maschi o femmine. Sono stati esclusi dalla meta-analisi gli studi a doppio zero (cioè quelli che hanno riportato eventi zero in ogni gruppo di trattamento). Questo vale anche per tutte le analisi dei sottogruppi (per dose, malattia e tempo)
Eventi avversi
Un’analisi secondaria di 188 AA (file aggiuntivo 5: tabella S3) ha rivelato solo sei AA a termine preferenziale MedDRA statisticamente significativi. Si tratta di diminuzione dell’appetito (RR 1,44, 95% IC 1,09-1,91), vertigini (RR 1,45, 95% CI 1,15-1,83), nausea (RR 1,59, 95% CI 1,37-1).84), sonnolenza (RR 1,45, 95% CI 1,07-1,97), sudorazione (RR 1,89, 95% CI 1,25-2,87) e vomito (RR 1,91, 95% CI 1,51-2,42). Tuttavia, l’analisi di sensibilità ha rivelato che questi sono solo di natura lieve e comuni a tutti i pazienti.
Prelievi e prelievi dovuti agli AA
Non c’erano prove di una differenza tra naltrexone e placebo nella meta-analisi di RR dei prelievi (RR 0,99, 95% CI 0,93-1,05, I2 = 8%), mentre c’era un aumento del rischio di prelievo a causa di AE (RR 1,33, 95% CI 1,06-1,67, I2 = 0%).
Sottogruppo e analisi di sensibilità
Nelle analisi di sottogruppi predefiniti di RR di SAE, non c’è stata alcuna differenza nei risultati per diverse dosi (Fig. 3) di naltrexone o per diversi gruppi di malattie/condizioni. A causa del numero limitato di studi con dosaggi <26 mg rispetto agli altri gruppi di dosaggio, si specifica che questi risultati devono essere interpretati con cautela. La valutazione dei SAE per gruppo di malattie è riportata nel file aggiuntivo 6: Figura S1. Questa analisi non ha mostrato alcun significato statistico. Un’analisi post hoc per durata dello studio non ha mostrato alcuna differenza di rischio tra studi di durata ≤15 settimane (RR 0,74, 95% CI 0,53-1,02, I2 = 0%) rispetto a studi di durata >15 settimane (RR 0,96, 95% CI 0,69-1,34, I2 = 0%). L’analisi di sensibilità del basso rischio di studi di bias (RR 0.97, 95% CI 0.61-1.54, I2 = 0%) non ha mostrato alcuna differenza di rischio rispetto agli studi a più alto rischio di bias (RR 0.80, 95% CI 0.61-1.05, I2 = 0%) (File addizionale 7: Figura S2).Fig 3Trama del sottogruppo analisi per dose del rapporto di rischio (RR) di eventi avversi gravi in RCT di naltrexone vs placebo. I dati tra parentesi mostrano la media o la gamma di età dei partecipanti e la percentuale di partecipanti maschi o femmine. Sono stati esclusi dalla meta-analisi gli studi a doppio zero (cioè quelli che hanno riportato eventi pari a zero in ogni gruppo di trattamento)
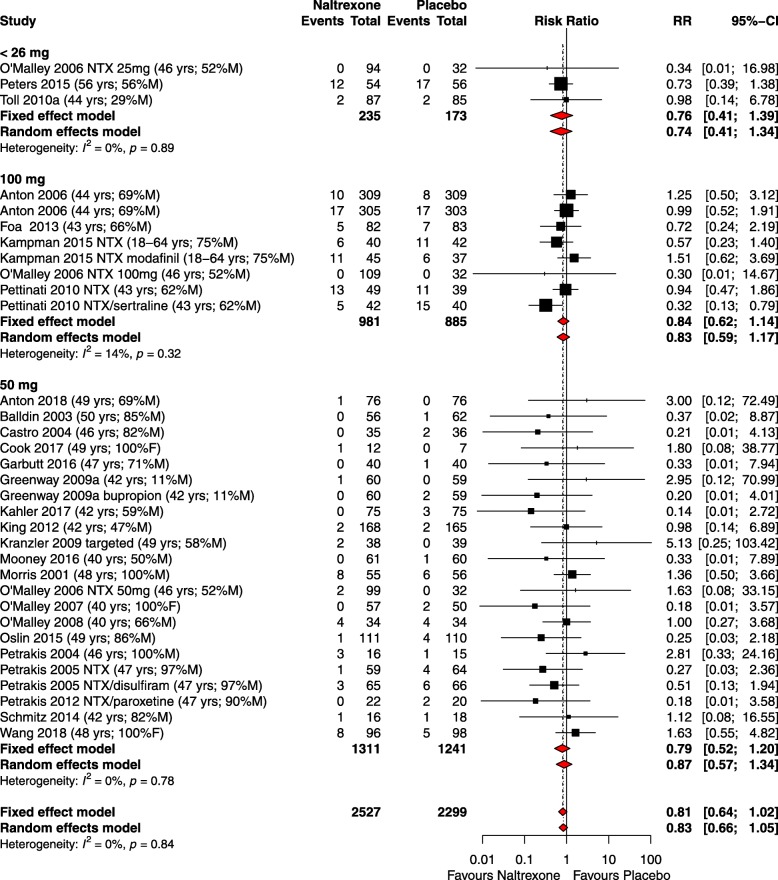
Fig. 3.Trama forestale del sottogruppo analisi per dose del rapporto di rischio (RR) di eventi avversi gravi in RCT di naltrexone vs placebo. I dati tra parentesi mostrano la media o la gamma di età dei partecipanti e la percentuale di partecipanti maschi o femmine. Sono stati esclusi dalla meta-analisi gli studi a doppio zero (cioè quelli che hanno riportato eventi pari a zero in ogni gruppo di trattamento)
Valutazione per il pregiudizio di pubblicazione
Non vi erano prove di asimmetria del diagramma ad imbuto per indicare un’asimmetria di pubblicazione per il RR degli EA o per il RR dei ritiri o dei ritiri dovuti agli EA. Il grafico ad imbuto per l’analisi principale è incluso come Fig. 4.Fig 4Il grafico ad imbuto del rapporto di rischio ponderato (RR) di eventi avversi gravi in RCT di naltrexone vs placebo vs errore standard
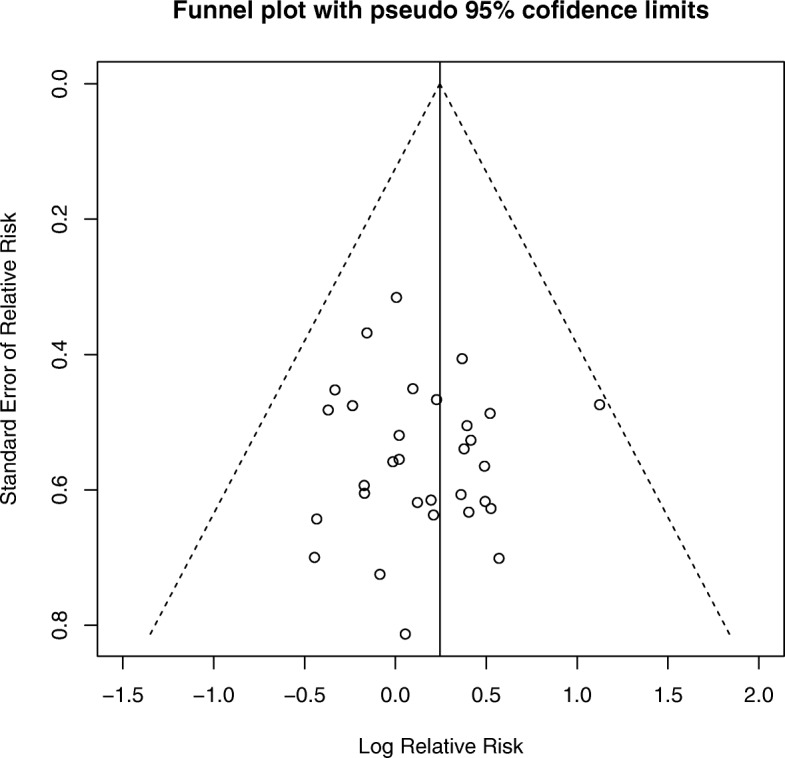
Fig. 4.Fig. 4. Trama ad imbuto del rapporto di rischio ponderato (RR) di eventi avversi gravi in RCT di naltrexone vs placebo vs errore standard
Discussione
Sintesi dei principali risultati
Questa meta-analisi di 89 RCT basata su 11.194 partecipanti non ha mostrato alcuna evidenza di un aumento del rischio di SAE per il naltrexone rispetto al placebo. Questi risultati sono stati coerenti tra gli studi con durata, dosaggi e condizioni di indice variabili, suggerendo che il naltrexone è sicuro da usare in un’ampia varietà di indicazioni autorizzate e non autorizzate. Abbiamo scoperto che gli EA come vertigini, nausea e vomito sono potenzialmente più comuni per il naltrexone rispetto al placebo. Tuttavia, questa constatazione deve essere interpretata con cautela perché i dati relativi agli EA sono stati scarsi (meno di 21 studi hanno contribuito alle analisi degli EA).
Punti di forza e limitazioni
Ci sono stati diversi punti di forza di questa recensione. Uno era la dimensione, che era sufficientemente grande sia nel numero di partecipanti che nel numero di studi che avrebbe permesso di individuare danni specifici dovuti a un farmaco. Papanikolaou e Ioannidis [59] hanno calcolato la dimensione del campione di una revisione sistematica necessaria per rilevare un evento raro (0,25%) che si verifica in circa l’1% dei soggetti come 4000 soggetti (80% di potenza e α = 0,05), e questa revisione sistematica conteneva oltre 10.000 soggetti di 89 studi. Inoltre, questa revisione comprendeva un’ampia gamma di studi provenienti da diversi paesi, contesti e gruppi di malattie, compresi pazienti con patologie multiple o dipendenze. Questi ultimi complessi scenari riflettono più fedelmente la pratica clinica rispetto ai consueti criteri di ingresso restrittivi degli studi clinici. Di conseguenza, la dimensione relativa dell’effetto riscontrato è probabilmente generalizzabile [60]. La nostra metodologia per l’esame delle misure di outcome, che non erano le misure di outcome primarie in nessuno degli studi clinici, ma che ora fanno parte della reportistica standard degli studi clinici, ha ridotto il rischio di distorsione della reportistica e della pubblicazione [61], così come l’uso dei registri degli studi clinici.
È probabile che alcuni studi non abbiano riportato e/o registrato in modo adeguato gli AA. Pertanto abbiamo controllato e registrato eventuali casi di discrepanze nei dati simili ai rapporti precedenti [62]. Riteniamo improbabile che i SAE mancanti o registrati in modo errato avrebbero modificato le conclusioni della meta-analisi, perché non vi erano differenze sistematiche tra questi studi che riportassero i SAE in modo adeguato e inadeguato e perché le analisi di sensibilità, in particolare quella che includeva solo gli studi con un rischio di distorsione complessivamente basso, supportavano la conclusione principale. Se il follow-up fosse stato scarso, si sarebbe potuta verificare una sottovalutazione dei SAE in studi con alti tassi di logoramento. Inoltre, poiché l’adesione all’estensione CONSORT per le raccomandazioni sui danni [33] è stata scarsa in molti studi, in particolare nell’uso di definizioni standardizzate e nella descrizione degli eventi, non siamo stati in grado di effettuare alcuna analisi qualitativa dei risultati.
Questa revisione è stata limitata agli studi sul naltrexone per via orale, esclusi gli studi che riguardano la dipendenza o l’uso attuale o precedente di oppioidi. La nostra valutazione dei SAE per gruppo di malattie dovrebbe essere considerata solo come esplorativa, perché la classificazione delle popolazioni in gruppi di malattie specifiche non era chiara a causa della predominanza di AUD anche in studi di altre malattie.
Mentre lo scopo principale di questo studio era quello di esaminare i dati SAE provenienti dagli RCT, abbiamo esaminato gli EA in un’analisi secondaria, ma questa analisi si è basata su dati limitati identificati nella pubblicazione della rivista e nel rapporto del registro. Le prove precedenti hanno anche dimostrato che la valutazione e la segnalazione delle AA è spesso incoerente e incompleta in tutti gli studi. Ad esempio, un’ampia analisi della sicurezza di 44 studi [63] sul naltrexone per le AUD ha rilevato che spesso le AA non venivano raccolte utilizzando misure standardizzate, che i metodi per l’acquisizione sistematica delle AA spesso non venivano segnalati e che la segnalazione delle AA era altamente selettiva.
La registrazione di AA può essere ostacolata dalla presenza di effetti nocebo (dannosi) (cioè il peggioramento dei sintomi durante il trattamento con placebo), che possono variare da malattia a malattia. In particolare nella dipendenza da alcol e droghe, i meccanismi placebo e nocebo possono avere un impatto sugli esiti terapeutici e sugli effetti collaterali dei trattamenti [64]. Anche se meno probabile nella registrazione dei SAE a causa della loro gravità [64], questo potrebbe aver avuto un impatto anche sui nostri risultati.
Infine, sono stati necessari alcuni perfezionamenti del protocollo, ma questi sono avvenuti come raccomandato prima di qualsiasi raccolta di dati [65]. Il cambiamento principale è stato l’esclusione dalla revisione degli studi di laboratorio, degli studi di durata inferiore a 4 settimane e degli studi incrociati. L’esercizio di scoping iniziale non aveva rivelato il gran numero di tali studi e tentare un’analisi di tutti questi avrebbe superato le risorse disponibili.
Confronto con la letteratura esistente
A nostra conoscenza, questa è la prima grande revisione sistematica di SAE in persone che assumono naltrexone, escludendo solo le persone che assumono oppioidi. Due grandi revisioni sistematiche precedenti del naltrexone in AUD sono state condotte da Rösner et al. [66] per la Cochrane Collaboration e Jonas et al. [63] per l’Agenzia per la ricerca e la qualità sanitaria. Entrambi hanno esaminato le AA, ma in un minor numero di studi. Rösner et al. [66] hanno analizzato nove studi, tra cui due che utilizzavano naltrexone iniettabile, e hanno calcolato la RD di sperimentare le SAE come -0,02 (95% CI -0,05-0,00). Utilizzando una gamma più ampia di studi e criteri di inclusione e limitando le date di pubblicazione dopo il 1° gennaio 2001, questa revisione è stata in grado di fornire una valutazione più accurata del rischio di EAS rispetto a qualsiasi revisione precedente.
Implicazioni per i ricercatori, i clinici e i responsabili politici
I risultati di questa revisione sono a sostegno di un uso più ampio del naltrexone e hanno un potenziale realistico di impatto sulle linee guida cliniche. I responsabili politici (ad esempio la Task Force Preventiva degli Stati Uniti e il National Institute of Clinical Excellence) sono incoraggiati ad utilizzare i risultati di questa revisione in combinazione con altri studi incentrati sui benefici e sul rapporto costo-efficacia del naltrexone per elaborare/revisionare raccomandazioni basate sull’evidenza per quanto riguarda l’uso autorizzato del naltrexone in una più ampia gamma di condizioni. Il trattamento degli AUD, per i quali il naltrexone è attualmente sottoutilizzato, è un’area chiave di considerazione. Le stime suggeriscono che circa il 58% delle persone che dipendono dall’alcol in Inghilterra vogliono ridurre il loro consumo di alcol [67]. L’aumento dell’uso della farmacoterapia per le AUD si è dimostrato efficace in termini di costi e potrebbe ridurre i decessi [68-70].
Questa revisione mostra i vantaggi di esaminare sia i profili di beneficio che di rischio per i farmaci e la necessità di una registrazione coerente e adeguata degli EA e dei SAE nei rapporti degli RCT. Gli studi recenti inclusi in questa revisione non hanno ancora riportato in modo coerente i danni allo standard suggerito nell’estensione CONSORT per i danni [33, 71], e le differenze di giudizio su ciò che costituisce un SAE erano evidenti tra gli studi. La ricerca sull’efficacia del naltrexone per la maggior parte delle malattie, a parte gli AUD e l’abuso di oppioidi, è attualmente carente; il naltrexone sembrerebbe un ottimo candidato per il riuso, dato che è sicuro ed economico, essendo da tempo fuori brevetto. È anche possibile che il naltrexone possa essere associato a cambiamenti nei tassi di tumori e ad eventi cardiovascolari o cerebrovascolari, date le complesse interazioni degli oppioidi nel corpo [72, 73]. Pertanto, sono necessari sia studi clinici pragmatici su larga scala di indicazioni potenzialmente nuove per il naltrexone, sia valutazioni sistematiche attraverso studi farmaco-epidemiologici che utilizzano dati di sicurezza a lungo termine (ad esempio il Clinical Practice Research Datalink del Regno Unito (https://www.cprd.com/home/).
Conclusioni
Questa revisione sistematica e meta-analisi non ha trovato alcuna evidenza di una differenza di rischio di SAE per il naltrexone orale rispetto al placebo. Questa evidenza supporta l’uso del naltrexone nella sua forma attualmente autorizzata e fornisce un solido supporto agli sforzi contemporanei per lo studio del naltrexone dove attualmente non è autorizzato.
File aggiuntivi
File aggiuntivo 1:Strategie di ricerca specifiche per le banche dati (DOCX 23 kb)File aggiuntivo 2:Studi ammissibili per la revisione (DOCX 32 kb)File aggiuntivo 3:Tabella S1. Caratteristiche degli studi inclusi (DOCX 43 kb)File aggiuntivo 4:Tabella S2. Riepilogo del rischio di parzializzazione (PDF 420 kb)File aggiuntivo 5:Tabella S3. Meta-analisi dei dati degli eventi avversi negli studi (DOCX 24 kb)File aggiuntivo 6:Figura S1. Analisi di sottogruppo dei SAE per tipo di malattia (PDF 13 kb)File aggiuntivo 7:Figura S2. Trama forestale dei SAE per rischio di sbieco (PDF 13 kb)
References
- British National Formulary (BNF) 75 (March 2018). BMJ Publishing Group Ltd and Royal Pharmaceutical Society: London; 2018.
- GBD 2015 Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016; 388:1545-1602. DOI | PubMed
- GBD 2015 Mortality and Causes of Death Collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980–2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016; 388:1459-1544. DOI | PubMed
- Bramness JG, Mann K, Wurst FM. Marketing status and perceived efficacy of drugs for supporting abstinence and reducing alcohol intake in alcohol use disorders: a survey among European Federation of Addiction Societies in Europe. Eur Addict Res.. 2016; 22:318-321. DOI | PubMed
- Thompson A, Ashcroft DM, Owens L, van Staa TP, Pirmohamed M. Drug therapy for alcohol dependence in primary care in the UK: A Clinical Practice Research Datalink study. PLoS One.. 2017; 12:e0173272. DOI | PubMed
- Witkiewitz K, Vowles KE. Alcohol and opioid use, co-use, and chronic pain in the context of the opioid epidemic: a critical review. Alcohol Clin Exp Res.. 2018; 42:478-488. DOI | PubMed
- Jayaram-Lindström N, Hammarberg A, Beck O, Franck J. Naltrexone for the treatment of amphetamine dependence: a randomized, placebo-controlled trial. Am J Psychiatry.. 2008; 165:1442-1448. DOI | PubMed
- Kampman K, Pettinati H, Lynch K, Plebani J, Lachewitz J, Feeney K. Modafinil and naltrexone for the treatment of comorbid cocaine and alcohol dependence. Drug Alcohol Depend.. 2015; 146:e152. DOI
- Grant JE, Kim SW. Hartman BK. A double-blind, placebo-controlled study of the opiate antagonist naltrexone in the treatment of pathological gambling urges. J Clin Psychiatry.. 2008; 69:783-789. DOI | PubMed
- Grant JE, Kim SW. Odlaug BL. A double-blind, placebo-controlled study of the opiate antagonist, naltrexone, in the treatment of kleptomania. Biol Psychiatry.. 2009; 65:600-606. DOI | PubMed
- Papay K, Xie SX, Stern M, Hurtig H, Siderowf A, Duda JE. Naltrexone for impulse control disorders in Parkinson disease: a placebo-controlled study. Neurology.. 2014; 83:826-833. DOI | PubMed
- O’Malley SS, Sinha R, Grilo CM, Capone C, Farren CK, McKee SA. Naltrexone and cognitive behavioral coping skills therapy for the treatment of alcohol drinking and eating disorder features in alcohol-dependent women: a randomized controlled trial. Alcohol Clin Exp Res.. 2007; 31:625-634. PubMed
- Roy A, Roy M, Deb S, Unwin G, Roy A. Are opioid antagonists effective in attenuating the core symptoms of autism spectrum conditions in children: a systematic review. J Intellect Disabil Res.. 2015; 59:293-306. DOI | PubMed
- Zagon IS, McLaughlin PJ. Duration of opiate receptor blockade determines tumorigenic response in mice with neuroblastoma: a role for endogenous opioid systems in cancer. Life Sci.. 1984; 35:409-416. DOI | PubMed
- Yi Z, Guo S, Hu X, Wang X, Zhang X, Griffin N. Functional modulation on macrophage by low dose naltrexone (LDN). Int Immunopharmacol.. 2016; 39:397-402. DOI | PubMed
- Cree BAC, Kornyeyeva E, Goodin DS. Pilot trial of low-dose naltrexone and quality of life in multiple sclerosis. Ann Neurol.. 2010; 68:145-150. PubMed
- Traore AK, Thiero O, Dao S, Kounde FFC, Cisse M, Mccandless JB. Impact of low dose naltrexone (LDN) on antiretroviral therapy (ART) treated HIV + adults in Mali: a single blind randomized clinical trial. J AIDS HIV Res.. 2011; 3:189-198.
- Smith JP, Bingaman SI, Ruggiero F, Mauger DT, Mukherjee A, McGovern CO. Therapy with the opioid antagonist naltrexone promotes mucosal healing in active Crohn’s disease: a randomized placebo-controlled trial. Dig Dis Sci.. 2011; 56:2088-2097. DOI | PubMed
- Younger J, Noor N, McCue R, MacKey S. Low-dose naltrexone for the treatment of fibromyalgia: findings of a small, randomized, double-blind, placebo-controlled, counterbalanced, crossover trial assessing daily pain levels. Arthritis Rheum.. 2013; 65:529-538. DOI | PubMed
- Sturn KM, Collin M. Low-dose naltrexone: a new therapy option for complex regional pain syndrome type I patients. Int J Pharm Compd.. 2016; 20:197-201. PubMed
- Weinstock LB, Myers TL, Shetty A. Low-dose naltrexone for the treatment of sarcoidosis. Sarcoidosis Vasc Diffuse Lung Dis.. 2017; 34:184-187.
- Cao S, Lilly E, Chen ST. Variable response to naltrexone in patients with Hailey-Hailey disease. JAMA Dermatology.. 2018; 154:362-363. DOI | PubMed
- Immune Therapeutics Inc. Immune Therapeutics, Inc. announces exclusive agreement for sale of lodonal in Kenya valued at over $31 million. 2017.
- Raknes G, Småbrekke L. A sudden and unprecedented increase in low dose naltrexone (LDN) prescribing in Norway. Patient and prescriber characteristics, and dispense patterns. A drug utilization cohort study. Pharmacoepidemiol Drug Saf.. 2017; 26:136-142. DOI | PubMed
- Raknes G, Simonsen P, Småbrekke L. The effect of low dose naltrexone on medication in inflammatory bowel disease: a quasi experimental before-and-after prescription database study. J Crohns Colitis.. 2018; 12:677-686. DOI | PubMed
- . Accessed 12 Dec 2018Publisher Full Text
- . Accessed 7 Oct 2017Publisher Full Text
- Pfohl DN, Allen JI, Atkinson RLRL, Knopman DS, Malcolm RJ, Mitchell JE. Naltrexone hydrochloride (Trexan): a review of serum transaminase elevations at high dosage. NIDA Res Monogr.. 1986; 67:66-72. PubMed
- Croop RS, Faulkner EB, Labriola DF. for the Naltrexone Usage Study Group. The safety profile of naltrexone in the treatment of alcoholism. Results from a multicentre usage study. The Naltrexone Usage Study Group. Arch Gen Psychiatry.. 1997; 54:1130-1135. DOI | PubMed
- Brewer C, Wong VS. Naltrexone: report of lack of hepatotoxicity in acute viral hepatitis, with a review of the literature. Addict Biol.. 2004; 9:81-87. DOI | PubMed
- Ioannidis JP, Lau J. Completeness of safety reporting in randomized trials: an evaluation of 7 medical areas. JAMA.. 2001; 285:437-443. DOI | PubMed
- Loke YK, Derry S. Reporting of adverse drug reactions in randomised controlled trials – a systematic survey. BMC Clin Pharmacol.. 2001; 1:3. DOI | PubMed
- Ioannidis JPA, Evans SJW, Gøtzsche PC, O’Neill RT, Altman DG, Schulz K. Better reporting of harms in randomized trials: an extension of the CONSORT statement. Ann Intern Med.. 2004; 141:781-788. DOI | PubMed
- Zorzela L, Loke YK, Ioannidis JP, Golder S, Santaguida P, Altman DG. PRISMA harms checklist: improving harms reporting in systematic reviews. BMJ.. 2016; i157:352.
- European Commission. Commission Guideline – Guidance on posting and publication of result-related information on clinical trials in relation to the implementation of Article 57(2) of Regulation (EC) No 726/2004 and Article 41(2) of Regulation (EC) No 1901/2006. J Eur Union. 2012; C302:7-10.
- Zarin DA, Tse T, Williams RJ, Carr S. Special report. Trial reporting in ClinicalTrials.gov – the final rule. N Engl J Med.. 2016; 375:1998-2004. DOI | PubMed
- Sinclair JMA, Chambers SE, Shiles CJ, Baldwin DS. Safety and tolerability of pharmacological treatment of alcohol dependence: comprehensive review of evidence. Drug Saf.. 2016; 39:627-645. DOI | PubMed
- Patten DK, Schultz BG, Berlau DJ. The safety and efficacy of low-dose naltrexone in the management of chronic pain and inflammation in multiple sclerosis, fibromyalgia, Crohn’s disease, and other chronic pain disorders. Pharmacotherapy. 2018; 38:382-389. DOI | PubMed
- Johnson RA, Lukens JM, Kole JW, Sisti DA. Views about responsibility for alcohol addiction and negative evaluations of naltrexone. Subst Abuse Treat Prev Policy.. 2015; 10:10. DOI | PubMed
- Brown S-E, Vagenas P, Konda KA, Clark JL, Lama JR, Gonzales P. Men who have sex with men in Peru: acceptability of medication-assisted therapy for treating alcohol use disorders. Am J Mens Health.. 2017; 11:1269-1278. DOI | PubMed
- Yen MH, Ko HC, Tang FI, Lu RB, Hong JS. Study of hepatotoxicity of naltrexone in the treatment of alcoholism. Alcohol.. 2006; 38:117-120. DOI | PubMed
- McDonough M. Naltrexone and liver disease. Aust Prescr.. 2015; 38:151. PubMed
- Crowley P. Author’s response. Aust Prescr.. 2015; 38:151. DOI | PubMed
- McPherson K, Hemminki E. Synthesising licensing data to assess drug safety. BMJ.. 2004; 328:518-520. DOI | PubMed
- Dalton D, Williams N. Building a culture of candour: a review of the threshold for the duty of candour and of the incentives for care organisations to be candid. Royal College of Surgeons of England: London; 2014.
- Higgins JP, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions version 5.1.0 (updated March 2011). Chichester: Wiley; 2011.Publisher Full Text
- European Commission. EudraLex volume 10: Clinical trials. EudraLex. Publications Office of the European Union: Luxembourg; 2006.
- European Commission. Detailed guidance on the collection, verification and presentation of adverse event/reaction reports arising from clinical trials on medicinal products for human use (“CT-3”). J Eur Union. 2011; C172:01-13.
- Center for Drug Evaluation and Research, Center for Biologics Evaluation and Research. Guidance for industry and investigators. Safety reporting requirements for INDs and BA/BE studies. FDA: Silver Springs; 2012.
- Harmonised Tripartite Guideline ICH. Clinical safety data management: definitions and standards for expedited reporting E2A. ICH: Geneva; 1994.
- Loke YK, Price D, Herxheimer A, the Cochrane Adverse Effects Methods Group. Systematic reviews of adverse effects: framework for a structured approach. BMC Med Res Methodol.. 2007; 7:32. DOI | PubMed
- .Publisher Full Text
- Derry S, Loke YK, Aronson JK. Incomplete evidence: the inadequacy of databases in tracing published adverse drug reactions in clinical trials. BMC Med Res Methodol.. 2001; 1:7. DOI | PubMed
- Higgins J, Altman D, Sterne JAC, editors. Chapter 8: Assessing risk of bias in included studies. In: Higgins J, Green S, editors. Cochrane Handbook for Systematic Reviews of Interventions. Version 5. London: The Cochrane Collaboration; 2011.Publisher Full Text
- .Publisher Full Text
- Sweeting MJ, Sutton AJ, Lambert PC. What to add to nothing? Use and avoidance of continuity corrections in meta-analysis of sparse data. Stat Med.. 2004; 23:1351-1375. DOI | PubMed
- Diamond GA, Bax L, Kaul S. Uncertain effects of rosiglitazone on the risk for myocardial infarction and cardiovascular death. Ann Intern Med.. 2007; 147:578-581. DOI | PubMed
- Moher D, Liberati A, Tetzlaff J, Altman DG. the PRISMA group. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. Ann Intern Med.. 2009; 151:264-269. DOI | PubMed
- Papanikolaou PN, Ioannidis JPA. Availability of large-scale evidence on specific harms from systematic reviews of randomized trials. Am J Med.. 2004; 117:582-589. DOI | PubMed
- Rothwell PM. External validity of randomised controlled trials: “to whom do the results of this trial apply?”. Lancet.. 2005; 365:82-93. DOI | PubMed
- Kirkham JJ, Dwan KM, Altman DG, Gamble C, Dodd S, Smyth R. The impact of outcome reporting bias in randomised controlled trials on a cohort of systematic reviews. BMJ.. 2010; 340:c365. DOI | PubMed
- Becker JE, Krumholz HM, Ben-Josef G, Ross JS. Reporting of results in clinicaltrials.gov and high-impact journals. JAMA.. 2014; 311:1063-1065. DOI | PubMed
- Jonas DE, Amick HR, Feltner C, Bobashev G, Thomas K, Wines R. Pharmacotherapy for adults with alcohol use disorders in outpatient settings. JAMA.. 2014; 311:1889-1900. DOI | PubMed
- Spagnolo PA, Colloca L, Heilig M. The role of expectation in the therapeutic outcomes of alcohol and drug addiction treatments. Alcohol Alcohol.. 2015; 50:282-285. DOI | PubMed
- Page MJ, McKenzie JE, Kirkham J, Dwan K, Kramer S, Green S. Bias due to selective inclusion and reporting of outcomes and analyses in systematic reviews of randomised trials of healthcare interventions. Cochrane Database Syst Rev.. 2014; 10:MR000035.
- Rösner S, Hackl-Herrwerth A, Leucht S, Vecchi S, Srisurapanont M, Soyka M. Opioid antagonists for alcohol dependence. Cochrane Database Syst Rev.. 2010; 12:CD001867.
- Pryce R, Buykx P, Gray L, Stone T, Drummond C, Brennan A. Estimates of alcohol dependence in England based on APMS 2014, including estimates of children living in a household with an adult with alcohol dependence. Prevalence, trends and amenability to treatment. Public Health England: London; 2017.
- Walters D, Connor JP, Feeney GFX, Young RM. The cost effectiveness of naltrexone added to cognitive-behavioral therapy in the treatment of alcohol dependence. J Addict Dis.. 2009; 28:137-144. DOI | PubMed
- Dunlap LJ, Zarkin GA, Bray JW, Mills M, Kivlahan DR, McKay JR. Revisiting the cost-effectiveness of the COMBINE study for alcohol dependent patients: the patient perspective. Med Care.. 2010; 48:306-313. DOI | PubMed
- Rehm J, Shield KD, Gmel G, Rehm MX, Frick U. Modeling the impact of alcohol dependence on mortality burden and the effect of available treatment interventions in the European Union. Eur Neuropsychopharmacol.. 2013; 23:89-97. DOI | PubMed
- Hodkinson A, Kirkham JJ, Tudur-Smith C, Gamble C. Reporting of harms data in RCTs: a systematic review of empirical assessments against the CONSORT harms extension. BMJ Open.. 2013; 3:e003436. DOI
- Afsharimani B, Doornebal CW, Cabot PJ, Hollmann MW, Parat MO. Comparison and analysis of the animal models used to study the effect of morphine on tumour growth and metastasis. Br J Pharmacol.. 2015; 172:251-259. DOI | PubMed
- Budzyński J, Rybakowski J, Swiatkowski M, Torliński L, Kłopocka M, Kosmowski W. Naltrexone exerts a favourable effect on plasma lipids in abstinent patients with alcohol dependence. Alcohol Alcohol.. 2000; 35:91-97. DOI | PubMed
Fonte
Bolton M, Hodkinson A, Boda S, Mould A, Panagioti M, et al. (2019) Serious adverse events reported in placebo randomised controlled trials of oral naltrexone: a systematic review and meta-analysis. BMC Medicine 1710. https://doi.org/10.1186/s12916-018-1242-0




