
Introduzione
I tumori che nascono dallo stesso tessuto possono presentare grandi differenze nel comportamento clinico. Ad esempio, tra gli individui a cui è stato diagnosticato un cancro colorettale in stadio precoce, circa il 60% dei pazienti sarà curato con la sola chirurgia, mentre il restante 40% sperimenterà una recidiva che è spesso fatale(Mäkelä et al., 1995). Diversi biomarcatori patologici e molecolari sono tipicamente analizzati per valutare il rischio del paziente e aiutare il processo decisionale clinico. In generale, questi biomarcatori sono suddivisi in due classi: predittivi e prognostici(Nalejska et al., 2014). I biomarcatori predittivi identificano i pazienti che probabilmente rispondono a terapie specifiche, come le mutazioni dell ‘EGFR che sensibilizzano i tumori polmonari all’inibizione dell’EGFR(Paez et al., 2004). Al contrario, i biomarcatori prognostici forniscono informazioni sull’aggressività del cancro e sulla probabilità di morte del paziente. La de-differenziazione del tumore e l’infiltrazione dei linfonodi servono come biomarcatori prognostici prototipici a causa della loro forte associazione con gli esiti negativi(Connolly et al., 2003). Tuttavia, questi biomarcatori basati sulla patologia possono soffrire di bassi livelli di concordanza tra gli osservatori(Allsbrook et al., 2001; Coons et al., 1997; Elmore et al., 2015; Gilks et al., 2013), e anche una valutazione patologica perfetta fornisce informazioni incomplete sul decorso clinico più probabile di un paziente (Bijker et al.,2013; Nofech-Mozes et al. , 2005; Young, 2003; Zaniboni et al., 2004). Nuovi metodi per identificare i tumori aggressivi potrebbero portare a miglioramenti nella stratificazione del rischio del paziente, a una migliore gestione clinica e a una diminuzione di trattamenti eccessivi pericolosi e non necessari(Esserman et al., 2013).
I progressi nelle tecnologie ad alto rendimento hanno dato una visione senza precedenti della vasta gamma di cambiamenti genomici che si trovano all’interno di ogni cellula tumorale. Progetti come The Cancer Genome Atlas (TCGA) e l’International Cancer Genome Genome Consortium (ICGC) hanno caratterizzato la metilazione, la mutazione, il numero di copie e i modelli di espressione genica in tutti i tipi di cancro. Come risultato di questi studi, molte delle differenze genomiche tra cellule normali e trasformate sono state identificate e caratterizzate. Tuttavia, ci manca una comprensione simile delle differenze genomiche tra tumori indolenti e tumori maligni aggressivi. Poiché il costo del sequenziamento del DNA continua a diminuire, è diventato sempre più fattibile per gli ospedali implementare analisi di routine mirate e/o analisi genomiche dei tumori dei pazienti(Gagan e Van Allen, 2015; Sholl et al., 2016; Zehir et al., 2017). Ma, mentre sono stati scoperti diversi biomarcatori predittivi basati sul DNA e specifici per la terapia, le informazioni prognostiche contenute nei genomi dei tumori sono molto meno chiare.
I precedenti sforzi a livello genomico per scoprire nuovi biomarcatori prognostici si sono concentrati in gran parte sui cambiamenti di espressione genica associati alla mortalità dei pazienti(Anaya, 2016; Anaya et al., 2015; Gentles et al., 2015; Uhlen et al., 2017). Questi studi hanno identificato una serie di trascrizioni che codificano le proteine coinvolte nella progressione del ciclo cellulare che sono correlate con la ricorrenza e la morte in diversi tipi di cancro(Cuzick et al., 2011; Dancik e Theodorescu, 2015; Gentles et al., 2015; Mosley e Keri, 2008; Venet et al., 2011; Wang et al., 2012; Wistuba et al., 2013). Si sa relativamente meno di quanto i cambiamenti a livello di DNA influiscano sulla sopravvivenza dei pazienti. Le analisi delle mutazioni genetiche associate ai risultati sono state condotte prevalentemente su un numero limitato di oncogeni noti di singoli tipi di cancro e sono giunte a conclusioni divergenti. I rapporti della letteratura suggeriscono comunemente che le mutazioni negli oncogeni pilota sono associate a scarsi risultati, tra cui, ad esempio, le mutazioni KRAS nel cancro del polmone(Guan et al., 2013; Marabese et al., 2015; Sun et al., 2013), le mutazioni PIK3CA nel cancro al seno(Li et al., 2006; Oshiro et al., 2015) e le mutazioni BRAF nel cancro del colon-retto(Richman et al., 2009; Roth et al., 2010; Tol et al., 2009). Altri studi degli stessi geni negli stessi tipi di cancro non hanno osservato alcuna associazione significativa con l’esito(Bozhanov et al., 2010; Gonzalez-Angulo et al., 2009; Hutchins et al., 2011; Pang et al., 2014; Scoccianti et al., 2012). In generale, gli studi sui biomarcatori basati sulle mutazioni possono essere confusi da piccole dimensioni dei campioni, da test di ipotesi post-hoc, da endpoint clinici imprecisi e dal cosiddetto problema del “cassetto delle cartelle”, in cui è meno probabile che vengano pubblicati risultati negativi (Aronson,2005; Ensor, 2014; Goossens et al., 2015; Rosenthal, 1979; Scargle, 1999). Le informazioni prognostiche catturate dal sequenziamento degli oncogeni pilota rimangono sconosciute, e non è stata condotta un’analisi pan-cancro a livello di exomero delle mutazioni associate ai risultati.
Precedenti indagini sull’importanza prognostica delle alterazioni del numero di copie di DNA (CNA) hanno indicato che i tumori altamente anuploidi tendono ad avere esiti peggiori dei tumori diploidi(Friedlander et al., 1984; Kallioniemi et al., 1987; Kokal et al., 1986; Merkel e McGuire, 1990; Zimmerman et al., 1987). Tuttavia, queste analisi si sono concentrate in gran parte sui cambiamenti di lunghezza del braccio(Davoli et al., 2017; Roy et al., 2016) o sulle alterazioni che riguardano singoli oncogeni o soppressori tumorali(Deming et al., 2000; Shi et al., 2012; Srividya et al., 2011). L’importanza funzionale delle alterazioni del numero di copie nella maggior parte dei geni a livello di singolo gene è sconosciuta, e non è stata condotta un’analisi pan-cancro, gene per gene, delle alterazioni prognostiche del numero di copie. Al fine di ottenere una comprensione globale delle caratteristiche genomiche di un tumore primitivo che influenzano la prognosi del cancro, abbiamo raccolto e analizzato i profili molecolari da una serie di ‘scoperta’ di 9442 pazienti e da una serie di ‘validazione’ di 8618 pazienti con tumori solidi. La nostra analisi completa e centrata sui geni fa luce sui cambiamenti genomici che guidano la malattia aggressiva e fornirà una risorsa utile per lo sviluppo di strategie per migliorare la valutazione del rischio clinico. Inoltre, mettiamo a disposizione un portale web per facilitare l’accesso della comunità a questo ricco set di dati sui biomarcatori all’indirizzo http://survival.cshl.edu.
Un’analisi multipiattaforma e pan-cancro dei dati sulla sopravvivenza al cancro
Per determinare le differenze tra tumori benigni e tumori mortali, abbiamo prima analizzato più classi di dati genomici di 9442 pazienti con 16 tipi di tumore del TCGA (descritti nella Figura 1-figure supplement 1A; le abbreviazioni sono definite nella Figura 1-figure supplement 1B). Per ogni tipo di tumore e per ogni set di dati, abbiamo generato modelli di rischio proporzionali univariata Cox che collegano la presenza o l’espressione di una particolare caratteristica con l’esito clinico (descritto nel testo integrativo 1). Per ogni modello riportiamo il punteggio Z, che codifica sia la direzionalità che il significato di una particolare associazione. Se la presenza di una mutazione o di un’amplificazione del numero di copie è associata in modo significativo alla morte del paziente, allora un punteggio Z >1,96 corrisponde a un valore P < 0,05 (Figura 1-figuresupplement 2A-C). Al contrario, un punteggio Z inferiore a -1,96 indica che la presenza di una mutazione è associata alla sopravvivenza o che la delezione di un gene è significativamente associata alla morte del paziente.
Abbiamo estratto la mutazione, il numero di copie, l’espressione genica e le informazioni cliniche da 16 coorti TCGA (riassunti nel file supplementare 1 e discussi in dettaglio nel testo supplementare 2). Per valutare la validità della nostra pipeline di analisi dei dati, così come l’accuratezza delle annotazioni dei pazienti segnalati, abbiamo prima esaminato le curve di sopravvivenza complessive per i 16 tipi di tumore che abbiamo profilato. Come previsto, abbiamo osservato differenze significative nell’esito clinico in base al tessuto di origine del tumore (Figura 1-figure supplement 2D). Il tumore della prostata ha avuto il decorso clinico meno aggressivo, con un tempo di sopravvivenza mediano che non è stato raggiunto in questo dataset (>4600 giorni), mentre il tumore del pancreas ha conferito la prognosi peggiore (tempo di sopravvivenza mediano: 444 giorni). Complessivamente, le frequenze di sopravvivenza a 5 anni dei pazienti nel TCGA sono state molto simili alle medie nazionali riportate da NCI-SEER (R = 0,83, p < 0,0001), suggerendo che i pazienti inclusi in questa analisi sono ampiamente rappresentativi della popolazione generale (Figura 1-figuresupplement 2E). Successivamente, abbiamo dedotto il sesso del paziente sulla base di modelli di espressione genica cromosomica specifica(Gentles et al., 2015; van den Berge e Sijen, 2017). La nostra analisi ha mostrato una concordanza >99% con il sesso dichiarato del paziente, verificando ulteriormente l’accuratezza complessiva delle annotazioni cliniche e la nostra pipeline di elaborazione dei dati (Figura 1-figuresupplement 2F).
Le mutazioni tumorali trasmettono informazioni prognostiche limitate
Per prima cosa abbiamo cercato di scoprire se le mutazioni di codifica dei genomi tumorali erano associate all’esito del paziente. Abbiamo estratto le mutazioni non silenti in ogni tumore, e poi abbiamo identificato tutti i geni che sono stati mutati in ≥2% dei pazienti in ciascuna delle 16 coorti (discusso nel testo supplementare 1). Abbiamo poi eseguito l’analisi dei pericoli proporzionali di Cox per confrontare i tempi di sopravvivenza dei pazienti che ospitano copie mutanti o di tipo selvaggio di ciascun gene. Questa analisi ha scoperto pochissime mutazioni che sono state associate in modo significativo al risultato del paziente(figura 1 e file supplementare 2A-B). Ci siamo concentrati in primo luogo sugli oncogeni e sui soppressori tumorali conosciuti e abbiamo scoperto che tra i 30 geni guida del cancro più frequentemente mutati, solo due(EGFR e TP53) sono stati associati alla prognosi in più di due tipi di tumore(Figura 1C). Le mutazioni del TP53 erano legate all’esito in cinque dei 16 tipi di tumore, anche se le differenze nella sopravvivenza dei pazienti erano generalmente piccole(Figura 1-figure supplement 3A-B). Al contrario, molti altri geni guida del cancro non erano associati al tempo di sopravvivenza in nessun tipo di tumore. Mentre mutazioni in KRAS, PIK3CA, CDKN2A, BRAF, KMT2D, ATM, SMAD4, e molti altri geni sono stati osservati frequentemente, non sono mai stati significativamente legati all’esito del paziente(Figura 1C).
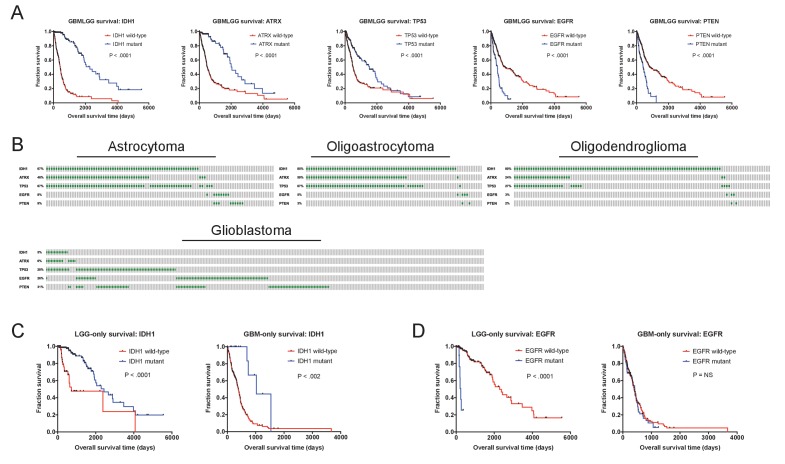
Figura 1-figure supplemento 7.Le mutazioni di una singola coppia di basi trasmettono informazioni prognostiche limitate.uno schema della pipeline di analisi della sopravvivenza del pan-cancro e dei set di dati utilizzati.l’analisi proporzionale dei rischi di sopravvivenza del Cox e l’accuratezza delle annotazioni cliniche TCGA.lo stato di mutazione del TP53 è associato all’esito in diversi tipi di cancro.Le mutazioni dei punti caldi e le mutazioni nei geni pilota di tumori multipli non sono generalmente associate alla prognosi clinica.Escludendo i pazienti con tumori ipermutati o quelli che sono stati trattati con terapie mirate, non sono in grado di rivelare mutazioni associate in modo significativo all’esito.le mutazioni con frequenze alleliche ad alta variante non sono più prognostiche delle mutazioni con frequenze alleliche a bassa variante.le mutazioni prognostiche nel glioma.(A) Schema delle mutazioni del TP53 e della sopravvivenza del paziente nella coorte di pazienti BLCA. I punti rossi indicano mutazioni missenso, i punti blu indicano mutazioni frameshift e i punti viola indicano mutazioni nonsenso.(B) Schema delle mutazioni RB1 e della sopravvivenza del paziente nella coorte dei pazienti BLCA. I punti rossi indicano mutazioni missenso, i punti blu indicano mutazioni frameshift, i punti viola indicano mutazioni nonsense e i punti verdi indicano mutazioni del sito di giunzione. Si noti che mentre 17 pazienti ospitano mutazioni RB1, 19 mutazioni sono visualizzate sulla trama del lecca lecca, mentre due pazienti ospitano due mutazioni nel gene RB1.(C) Una mappa termica delle associazioni di sopravvivenza significative tra i 30 geni driver del cancro più frequentemente mutilati in 16 tipi di tumore del TCGA sono visualizzati. I punteggi Z sono stati calcolati facendo regredire i tempi di sopravvivenza tra i pazienti che ospitano copie wild-type e le copie mutanti di un gene se un gene è stato mutato in ≥2% dei campioni per tipo di tumore. Ai fini della visualizzazione, vengono visualizzati solo i punteggi Z significativi. L’elenco completo dei punteggi Z è presentato nel file supplementare 2A.(D) Il numero di geni mutati in ≥2% dei campioni per tipo di tumore viene visualizzato.(E) Il numero di geni significativamente associati all’esito del paziente ad una soglia di falsa scoperta del 5% in ogni tipo di tumore sono visualizzati.(A) Viene presentato uno schema dell’elaborazione dei dati e delle analisi effettuate in questo rapporto. 16 tipi di tumore da TCGA sono stati utilizzati come coorte di scoperta. I dati di mutazione da ICGC e i dati sul numero di copie da coorti di pazienti curate da cBioportal sono stati utilizzati come coorti di convalida. Per identificare i biomarcatori di alta fiducia, un ulteriore set di pazienti sottoposti a sequenziamento mirato (MSKCC_2017) sono stati inclusi nell’analisi cross-cohort. Un elenco completo delle coorti di pazienti e dei tipi di dati analizzati è incluso nel file supplementare 1.(B e C) I tipi di cancro e le abbreviazioni di studio utilizzate nei set di dati TCGA e ICGC. Ulteriori informazioni su ogni coorte sono incluse nel file supplementare 1.(A) Viene visualizzata la relazione tra il punteggio Z e il valore P per -5 ≤ Z ≤ 5. 2.(B) I dati del campione per i valori di espressione genica per tre ipotetici geni (A, B e C) che illustrano diversi punteggi Z.(C) Grafici quantile-quantilitici che confrontano i punteggi Z e i valori del campione normalmente distribuiti per le CNA e per le mutazioni che utilizzano soglie diverse. Sulla base di questa analisi, solo i geni che ospitano mutazioni nel 2% o più dei campioni di una coorte sono stati inclusi per il calcolo della sopravvivenza (discusso nel testo integrativo 1).(D) I grafici Kaplan-Meier che mostrano il tempo di sopravvivenza post-diagnosi per gli otto tumori con gli esiti più favorevoli (in alto) e gli otto tumori con gli esiti più lugubri (in basso) nel dataset TCGA.(E) Viene visualizzato un confronto tra i tempi di sopravvivenza globale a 5 anni del TCGA e i tempi di sopravvivenza relativa a 5 anni per tipo di cancro del programma di sorveglianza, epidemiologia e risultati finali (SEER).(F) Vengono visualizzati i diagrammi di dispersione che mostrano l’espressione della trascrizione XIST codificata con il cromosoma X e la trascrizione RPS4Y1 codificata con il cromosoma Y nel cancro ovarico (OV), nel cancro alla prostata (PRAD) e in tutti i 16 tipi di tumore. Poiché lo XIST è espresso specificamente in cellule che contengono due o più copie del cromosoma X, questa combinazione di due geni si è dimostrata efficace nel discriminare il sesso cromosomico di una paziente sulla base dell’espressione genica (Gentleset al., 2015; van den Berge e Sijen, 2017). Di conseguenza, nella nostra analisi, quasi tutte le pazienti di sesso femminile hanno un’alta espressione XIST e una bassa espressione RPS4Y1, mentre quasi tutti i pazienti di sesso maschile hanno un’alta espressione RPS4Y1 e una bassa espressione XIST.(A) Trame Kaplan-Meier di tumori TP53-mutanti e TP53-WT in ogni tipo di tumore dal dataset TCGA. TP53 è associato al risultato in cinque dei 16 tipi di cancro (BRCA, GBMLGGG, HNSC, LUAD e PRAD). (B) Trame forestali di mutazioni TP53 in ogni coorte TCGA. I cerchi indicano il rapporto di pericolo, mentre le barre indicano il 95% di confidenza interna. (C) Trame Kaplan-Meier di tumori TP53-mutanti e TP53-WT in ogni tipo di tumore del set di dati ICGC. TP53 è associato all’esito in sette dei 16 tipi di cancro (EOPC-DE, LICA-FR, LINC-JP, LIRI-JP, PACA-AU, PACA-CA e PRAD-UK). (D) Trame forestali di mutazioni TP53 in ogni coorte ICGC. I cerchi indicano il rapporto di pericolo, mentre le barre indicano il 95% di confidenza interna. Si noti che i valori p non sono identici tra A e B e tra C e D, in quanto i valori p di Kaplan-Meier sono calcolati a partire da un test di log-rank, mentre i valori p delle aree forestali sono calcolati a partire da una regressione Cox.(A) Trame di lecca-lecca di mutazioni PIK3CA nella coorte BRCA. L’area di sinistra mostra tutte le mutazioni non silenti in PIK3CA, mentre l’area di destra mostra tutte le mutazioni in codoni “hotspot”, ad esempio i codoni che sono mutati in cinque o più pazienti nel set di dati TCGA. I punti rossi indicano mutazioni missenso, i punti blu indicano mutazioni frameshift, e i punti viola indicano mutazioni nonsenso.(B) Curve Kaplan-Meier che confrontano i pazienti BRCA con qualsiasi mutazione in PIK3CA rispetto a quelli senza mutazioni in PIK3CA (a sinistra), e pazienti COADREAD con qualsiasi mutazione in TP53 rispetto a quelli senza mutazioni in TP53 (a destra).(C) Curve Kaplan-Meier che confrontano i pazienti BRCA con varie mutazioni ‘hotspot’ in PIK3CA rispetto a quelli senza mutazioni ‘hotspot’ in PIK3CA (sinistra), e pazienti COADREAD con mutazioni ‘hotspot’ in TP53 rispetto a quelli senza mutazioni ‘ hotspot’ in TP53 (destra).(D) Viene visualizzata una heatmap delle associazioni di sopravvivenza significative tra i 30 codoni più frequentemente mutilati tra i 16 tipi di tumore del TCGA. I punteggi Z sono stati calcolati facendo regredire i tempi di sopravvivenza tra i pazienti che ospitano una mutazione in uno specifico codone e quelli che non ospitano una mutazione in quel particolare codone, se un codone è stato mutato in ≥2% dei campioni per tipo di tumore. Ai fini della visualizzazione, vengono visualizzati solo i punteggi Z significativi.(E) Tutti i codoni mutati in pazienti ≥5 (mutazioni “hotspot”) sono stati raggruppati insieme, e sono stati determinati i dieci geni che ospitano le mutazioni più “hotspot”. I punteggi Z sono stati calcolati confrontando i pazienti che ospitano qualsiasi mutazione ‘hotspot’ in un gene specificato e i pazienti che non hanno mutazioni ‘hotspot’ in quel gene. Una mappa termica mostra associazioni di sopravvivenza significative per i dieci geni con la maggior parte delle mutazioni “hotspot”.(F) I codici che sono stati mutati in ≥4% dei pazienti in ogni tipo di cancro sono stati identificati, e poi sono stati calcolati i punteggi Z confrontando i pazienti che ospitavano queste mutazioni ricorrenti rispetto a quelli che non lo facevano.(A) Escludendo i pazienti trattati con terapie mirate non riesce ad alterare significativamente i punteggi della mutazione Z. Le terapie mirate più comuni nella coorte TCGA erano gli inibitori BRAF (usati per trattare i melanomi BRAF-mutanti) e gli inibitori EGFR (usati per trattare i tumori polmonari EGFR-mutanti o i tumori colorettali di tipo KRAS-wild-type). Tuttavia, a causa dei tempi di raccolta dei campioni, che hanno consentito un follow-up del paziente superiore a 10 anni, solo un piccolo sottoinsieme di pazienti viene registrato come paziente che riceve queste terapie. Escludendo tutti i pazienti che hanno ricevuto queste terapie, non ha influito in modo significativo sulle curve di sopravvivenza risultanti.(B) Una heatmap delle associazioni significative tra mutazioni ed esito del paziente nel TCGA quando i tumori iper-mutati(Bailey et al., 2018) sono esclusi. Ai fini della visualizzazione, vengono visualizzati solo i punteggi Z significativi. L’elenco completo dei punteggi Z è presentato nel file supplementare 2C.(C) Le Heatmaps delle combinazioni di doppia mutazione in COADREAD e UCEC sono tracciate. Su 30 geni driver del cancro frequentemente mutilati, 26 sono mutati in ≥2% dei pazienti COADREAD e non sono individualmente correlati con la prognosi, mentre 17 sono mutati in ≥2% dei pazienti UCEC e non sono individualmente correlati con la prognosi. I punteggi Z sono stati calcolati confrontando i tempi di sopravvivenza dei pazienti che ospitano una mutazione non silente in due diversi geni guida del cancro rispetto ai pazienti che non ospitano una mutazione non silente in questi due geni. Le barre di colore blu e giallo indicano i punteggi Z.(A) Le frequenze alleliche della variante sono state calcolate per tutti i geni in 10 coorti TCGA. Box trame dei VAF di quattro driver di cancro comune (TP53, PIK3CA, ARID1A, e NF1) sono visualizzati. I box rappresentano il secondo e il terzo quartile VAF, mentre le barre di errore indicano il10° e il 90° percentile.(B) Curve Kaplan-Meier degli esiti dei pazienti rispetto alla mutazione VAF. Le mutazioni TP53 sono associate all’esito in HNSC, e questa relazione è vera sia per le mutazioni ad alta prevalenza (VAF ≥0.4) che per quelle a bassa prevalenza (VAF <0.4) (grafico in alto a sinistra). Allo stesso modo, nessuna informazione prognostica significativa è conferita dalle mutazioni TP53 in LUSC, dalle mutazioni KRAS in LUAD, o dalle mutazioni NF1 in SKCM, indipendentemente dalla prevalenza della mutazione.(C) Viene visualizzata una mappa termica delle associazioni di sopravvivenza significative tra i 30 geni guida del cancro più frequentemente mutilati in 16 tipi di tumore della TCGA. I punteggi Z sono stati calcolati facendo regredire i tempi di sopravvivenza tra i pazienti che ospitano copie wild-type e le copie mutanti di un gene se un gene è stato mutato in ≥2% dei campioni per tipo di tumore. Tuttavia, un gene è stato considerato “mutato” per questa analisi solo se il suo VAF era ≥0,4. Ai fini della visualizzazione, vengono visualizzati solo i punteggi Z significativi.(D) Una mappa termica di associazioni significative di sopravvivenza tra i 30 geni driver del cancro più frequentemente-mutate in 16 tipi di tumore dal TCGA sono visualizzati. I punteggi Z sono stati calcolati facendo regredire i tempi di sopravvivenza tra i pazienti che ospitano copie wild-type e le copie mutanti di un gene se un gene è stato mutato in ≥2% dei campioni per tipo di tumore. Per questa analisi del punteggio Z sono state incluse solo mutazioni con VAF maggiore o uguale al VAF mediano per un particolare gene in un particolare tipo di tumore. Ai fini della visualizzazione, vengono visualizzati solo i punteggi Z significativi.(E) Abbiamo confrontato i punteggi Z che risultano dalla regressione solo dei VAF sopra la media e i punteggi Z che risultano dalla regressione solo dei VAF sotto la media, per i 30 geni guida del cancro. In 62 casi, i punteggi Z più alti sono stati ottenuti considerando solo i VAF sopra la media, mentre in 68 casi, i punteggi Z più alti sono stati ottenuti considerando solo i VAF sotto la media.(A) Sono visualizzate le curve Kaplan-Meier dei cinque geni con le più forti associazioni di sopravvivenza in GBMLGG(ATRX, EGFR, IDH1, TP53 e PTEN).(B) Vengono visualizzati i modelli di mutazione secondo il sottotipo di glioma. Le caselle grigie corrispondono ai pazienti con un particolare sottotipo di glioma, mentre i trattini verdi indicano la presenza di una copia mutante del gene specificato.(C) Curve Kaplan-Meier delle mutazioni IDH1 in coorti LGG o GBM, analizzate separatamente. Le mutazioni IDH1 sono osservate in entrambe le coorti tumorali e sono associate alla sopravvivenza in entrambe le coorti tumorali.(D) Curve Kaplan-Meier delle mutazioni EGFR in coorti LGG o GBM, analizzate separatamente. Le mutazioni EGFR sono osservate in entrambe le coorti tumorali, ma sono prognostiche solo nei gliomi a basso grado. L’elenco completo dei punteggi Z è presentato nel file supplementare 2D.
Abbiamo poi considerato la possibilità che le mutazioni in specifici codoni possano avere un significato prognostico non catturato quando tutte le mutazioni in un gene sono messe insieme. Per testare questo, abbiamo identificato le 30 posizioni amminoacidiche più frequentemente mutate nelle coorti TCGA, e poi abbiamo chiesto se i pazienti che ospitano queste mutazioni hanno avuto esiti diversi rispetto a quelli che non le hanno avute. IDH1c132 mutazioni sono stati significativamente associati con una prognosi favorevole nel glioma, ma altri codoni ricorrenti-mutate (KRASc12, PIK3CAc1047, TP53c273, ecc) sono stati in gran parte disinformativi(Figura 1-figure supplemento 4A-D). Poi, abbiamo identificato i residui ‘hotspot’ che sono stati mutati in almeno cinque diversi pazienti in tutte le coorti. Considerando solo queste mutazioni ‘hotspot’ in ogni gene, anche queste mutazioni ‘hotspot’ non sono riuscite a scoprire robuste associazioni di sopravvivenza (Figura 1-figuresupplement 4E). Infine, abbiamo identificato mutazioni ricorrenti specifiche per il tipo di cancro, ma queste alterazioni (FGFR3c249 in BLCA, CTNNB1c37 in UCEC, ecc.) erano altrettanto disinformative(Figura 1-figure supplement 4F).
Successivamente, abbiamo cercato di verificare se l’uso di terapie mirate avesse attenuato gli effetti deleteri di alcune mutazioni dei driver (ad esempio, in BRAF o EGFR). Tuttavia, a causa dei tempi di raccolta dei campioni, pochissimi pazienti sono stati trattati con inibitori del BRAF o dell’EGFR, e la rimozione di quei pazienti che avevano ricevuto queste terapie non ha influito in modo significativo sui punteggi Z(Figura 1-figure supplement 5A). L’ipermutazione all’interno di un sottoinsieme di tumori potrebbe aumentare il “rumore” mutazionale e diminuire la nostra capacità di identificare le firme prognostiche, ma l’esclusione dei pazienti con tumori iper-mutati ha avuto un effetto minimo sul significato prognostico delle mutazioni del gene guida (Figura 1-figuresupplement 5B e Supplementary file 2C). Ci siamo quindi chiesti se la presenza di mutazioni nei geni driver di tumori multipli potesse cooperare per conferire un esito clinico peggiore. Abbiamo scoperto che, in generale, i pazienti che ospitano mutazioni in due geni pilota del cancro che non sono prognostici da soli avevano lo stesso rischio di morte dei pazienti con copie di uno o entrambi i geni(Figura 1-figure supplement 5C). Infine, abbiamo considerato la possibilità che la clonalità di una mutazione potesse influenzare il suo significato prognostico. Abbiamo calcolato la frequenza dell’allele variante (VAF) per ogni mutazione tumorale e abbiamo testato se le mutazioni presenti a livello clonale in singoli tumori erano più probabilmente associate all’esito. Abbiamo scoperto che limitando la nostra analisi alle mutazioni con VAF elevati non siamo riusciti a identificare più geni prognostici, indicando che la stratificazione del paziente è improbabile che venga migliorata valutando solo le mutazioni clonali(Figura 1-figure supplement 6).
Queste analisi hanno suggerito che, in generale, le mutazioni del gene guida del cancro mancano di un significativo potere di stratificazione del paziente. Questo ci ha portato ad indagare se le mutazioni in geni diversi dagli oncogeni a mutazioni ricorrenti e dai soppressori tumorali potessero influenzare la prognosi. Abbiamo quindi ampliato la nostra analisi per includere tutti i geni mutati in ≥2% dei pazienti con un particolare tipo di tumore. Per tenere conto di una notevole espansione del numero di geni testati, abbiamo applicato una correzione di Benjamini-Hochberg con un tasso di falsificazione del 5% ai punteggi individuali Z che abbiamo ottenuto. Abbiamo scoperto diversi geni che erano legati alla prognosi nel glioma, ma abbiamo trovato pochissimi geni significativamente associati alla morte o alla sopravvivenza negli altri 15 tipi di tumore(Figura 1D e file supplementare 2A). Ad esempio, nel carcinoma mammario e nell’adenocarcinoma polmonare, 128 e 3996 geni sono stati mutati rispettivamente in ≥2% delle pazienti, ma nessuna di queste mutazioni è stata correlata in modo significativo con l’esito del paziente ad un FDR del 5%. In totale, questi risultati indicano che la maggior parte delle mutazioni nei genomi tumorali manca di un significativo potere prognostico.
Figura 1-figure supplemento 7.Le mutazioni a coppia di base singola trasmettono informazioni prognostiche limitate.uno schema della pipeline di analisi della sopravvivenza del pan-cancro e dei set di dati utilizzati.l’analisi proporzionale dei rischi di sopravvivenza del Cox e l’accuratezza delle annotazioni cliniche del TCGA.lo stato della mutazione del TP53 è associato all’esito in diversi tipi di cancro.Le mutazioni dei punti caldi e le mutazioni nei geni pilota di tumori multipli non sono generalmente associate alla prognosi clinica.Escludendo i pazienti con tumori ipermutati o quelli che sono stati trattati con terapie mirate, non sono in grado di rivelare mutazioni associate in modo significativo all’esito.le mutazioni con frequenze alleliche ad alta variante non sono più prognostiche delle mutazioni con frequenze alleliche a bassa variante.le mutazioni prognostiche nel glioma.(A) Schema delle mutazioni del TP53 e della sopravvivenza del paziente nella coorte di pazienti BLCA. I punti rossi indicano mutazioni missenso, i punti blu indicano mutazioni frameshift e i punti viola indicano mutazioni nonsenso.(B) Schema delle mutazioni RB1 e della sopravvivenza del paziente nella coorte dei pazienti BLCA. I punti rossi indicano mutazioni missenso, i punti blu indicano mutazioni frameshift, i punti viola indicano mutazioni nonsense e i punti verdi indicano mutazioni del sito di giunzione. Si noti che mentre 17 pazienti ospitano mutazioni RB1, 19 mutazioni sono visualizzate sulla trama del lecca lecca, mentre due pazienti ospitano due mutazioni nel gene RB1.(C) Una mappa termica delle associazioni di sopravvivenza significative tra i 30 geni driver del cancro più frequentemente mutilati in 16 tipi di tumore del TCGA sono visualizzati. I punteggi Z sono stati calcolati facendo regredire i tempi di sopravvivenza tra i pazienti che ospitano copie wild-type e le copie mutanti di un gene se un gene è stato mutato in ≥2% dei campioni per tipo di tumore. Ai fini della visualizzazione, vengono visualizzati solo i punteggi Z significativi. L’elenco completo dei punteggi Z è presentato nel file supplementare 2A.(D) Il numero di geni mutati in ≥2% dei campioni per tipo di tumore viene visualizzato.(E) Il numero di geni significativamente associati all’esito del paziente ad una soglia di falsa scoperta del 5% in ogni tipo di tumore sono visualizzati.(A) Viene presentato uno schema dell’elaborazione dei dati e delle analisi effettuate in questo rapporto. 16 tipi di tumore da TCGA sono stati utilizzati come coorte di scoperta. I dati di mutazione da ICGC e i dati sul numero di copie da coorti di pazienti curate da cBioportal sono stati utilizzati come coorti di convalida. Per identificare i biomarcatori di alta fiducia, un ulteriore set di pazienti sottoposti a sequenziamento mirato (MSKCC_2017) sono stati inclusi nell’analisi cross-cohort. Un elenco completo delle coorti di pazienti e dei tipi di dati analizzati è incluso nel file supplementare 1.(B e C) I tipi di cancro e le abbreviazioni di studio utilizzate nei set di dati TCGA e ICGC. Ulteriori informazioni su ogni coorte sono incluse nel file supplementare 1.(A) Viene visualizzata la relazione tra il punteggio Z e il valore P per -5 ≤ Z ≤ 5. 2.(B) I dati del campione per i valori di espressione genica per tre ipotetici geni (A, B e C) che illustrano diversi punteggi Z.(C) Grafici quantile-quantilitici che confrontano i punteggi Z e i valori del campione normalmente distribuiti per le CNA e per le mutazioni che utilizzano soglie diverse. Sulla base di questa analisi, solo i geni che ospitano mutazioni nel 2% o più dei campioni di una coorte sono stati inclusi per il calcolo della sopravvivenza (discusso nel testo integrativo 1).(D) I grafici Kaplan-Meier che mostrano il tempo di sopravvivenza post-diagnosi per gli otto tumori con gli esiti più favorevoli (in alto) e gli otto tumori con gli esiti più lugubri (in basso) nel dataset TCGA.(E) Viene visualizzato un confronto tra i tempi di sopravvivenza globale a 5 anni del TCGA e i tempi di sopravvivenza relativa a 5 anni per tipo di cancro del programma di sorveglianza, epidemiologia e risultati finali (SEER).(F) Vengono visualizzati i diagrammi di dispersione che mostrano l’espressione della trascrizione XIST codificata con il cromosoma X e la trascrizione RPS4Y1 codificata con il cromosoma Y nel cancro ovarico (OV), nel cancro alla prostata (PRAD) e in tutti i 16 tipi di tumore. Poiché lo XIST è espresso specificamente in cellule che contengono due o più copie del cromosoma X, questa combinazione di due geni si è dimostrata efficace nel discriminare il sesso cromosomico di una paziente sulla base dell’espressione genica (Gentleset al., 2015; van den Berge e Sijen, 2017). Di conseguenza, nella nostra analisi, quasi tutte le pazienti di sesso femminile hanno un’alta espressione XIST e una bassa espressione RPS4Y1, mentre quasi tutti i pazienti di sesso maschile hanno un’alta espressione RPS4Y1 e una bassa espressione XIST.(A) Trame Kaplan-Meier di tumori TP53-mutanti e TP53-WT in ogni tipo di tumore dal dataset TCGA. TP53 è associato al risultato in cinque dei 16 tipi di cancro (BRCA, GBMLGGG, HNSC, LUAD e PRAD). (B) Trame forestali di mutazioni TP53 in ogni coorte TCGA. I cerchi indicano il rapporto di pericolo, mentre le barre indicano il 95% di confidenza interna. (C) Trame Kaplan-Meier di tumori TP53-mutanti e TP53-WT in ogni tipo di tumore del set di dati ICGC. TP53 è associato all’esito in sette dei 16 tipi di cancro (EOPC-DE, LICA-FR, LINC-JP, LIRI-JP, PACA-AU, PACA-CA e PRAD-UK). (D) Trame forestali di mutazioni TP53 in ogni coorte ICGC. I cerchi indicano il rapporto di pericolo, mentre le barre indicano il 95% di confidenza interna. Si noti che i valori p non sono identici tra A e B e tra C e D, in quanto i valori p di Kaplan-Meier sono calcolati a partire da un test di log-rank, mentre i valori p delle aree forestali sono calcolati a partire da una regressione Cox.(A) Trame di lecca-lecca di mutazioni PIK3CA nella coorte BRCA. L’area di sinistra mostra tutte le mutazioni non silenti in PIK3CA, mentre l’area di destra mostra tutte le mutazioni in codoni “hotspot”, ad esempio i codoni che sono mutati in cinque o più pazienti nel set di dati TCGA. I punti rossi indicano mutazioni missenso, i punti blu indicano mutazioni frameshift, e i punti viola indicano mutazioni nonsenso.(B) Curve Kaplan-Meier che confrontano i pazienti BRCA con qualsiasi mutazione in PIK3CA rispetto a quelli senza mutazioni in PIK3CA (a sinistra), e pazienti COADREAD con qualsiasi mutazione in TP53 rispetto a quelli senza mutazioni in TP53 (a destra).(C) Curve Kaplan-Meier che confrontano i pazienti BRCA con varie mutazioni ‘hotspot’ in PIK3CA rispetto a quelli senza mutazioni ‘hotspot’ in PIK3CA (sinistra), e pazienti COADREAD con mutazioni ‘hotspot’ in TP53 rispetto a quelli senza mutazioni ‘ hotspot’ in TP53 (destra).(D) Viene visualizzata una heatmap delle associazioni di sopravvivenza significative tra i 30 codoni più frequentemente mutilati tra i 16 tipi di tumore del TCGA. I punteggi Z sono stati calcolati facendo regredire i tempi di sopravvivenza tra i pazienti che ospitano una mutazione in uno specifico codone e quelli che non ospitano una mutazione in quel particolare codone, se un codone è stato mutato in ≥2% dei campioni per tipo di tumore. Ai fini della visualizzazione, vengono visualizzati solo i punteggi Z significativi.(E) Tutti i codoni mutati in pazienti ≥5 (mutazioni “hotspot”) sono stati raggruppati insieme, e sono stati determinati i dieci geni che ospitano le mutazioni più “hotspot”. I punteggi Z sono stati calcolati confrontando i pazienti che ospitano qualsiasi mutazione ‘hotspot’ in un gene specificato e i pazienti che non hanno mutazioni ‘hotspot’ in quel gene. Una mappa termica mostra associazioni di sopravvivenza significative per i dieci geni con la maggior parte delle mutazioni “hotspot”.(F) I codici che sono stati mutati in ≥4% dei pazienti in ogni tipo di cancro sono stati identificati, e poi sono stati calcolati i punteggi Z confrontando i pazienti che ospitavano queste mutazioni ricorrenti rispetto a quelli che non lo facevano.(A) Escludendo i pazienti trattati con terapie mirate non riesce ad alterare significativamente i punteggi della mutazione Z. Le terapie mirate più comuni nella coorte TCGA erano gli inibitori BRAF (usati per trattare i melanomi BRAF-mutanti) e gli inibitori EGFR (usati per trattare i tumori polmonari EGFR-mutanti o i tumori colorettali di tipo KRAS-wild-type). Tuttavia, a causa dei tempi di raccolta dei campioni, che hanno consentito un follow-up del paziente superiore a 10 anni, solo un piccolo sottoinsieme di pazienti viene registrato come paziente che riceve queste terapie. Escludendo tutti i pazienti che hanno ricevuto queste terapie, non ha influito in modo significativo sulle curve di sopravvivenza risultanti.(B) Una heatmap delle associazioni significative tra mutazioni ed esito del paziente nel TCGA quando i tumori iper-mutati(Bailey et al., 2018) sono esclusi. Ai fini della visualizzazione, vengono visualizzati solo i punteggi Z significativi. L’elenco completo dei punteggi Z è presentato nel file supplementare 2C.(C) Le Heatmaps delle combinazioni di doppia mutazione in COADREAD e UCEC sono tracciate. Su 30 geni driver del cancro frequentemente mutilati, 26 sono mutati in ≥2% dei pazienti COADREAD e non sono individualmente correlati con la prognosi, mentre 17 sono mutati in ≥2% dei pazienti UCEC e non sono individualmente correlati con la prognosi. I punteggi Z sono stati calcolati confrontando i tempi di sopravvivenza dei pazienti che ospitano una mutazione non silente in due diversi geni guida del cancro rispetto ai pazienti che non ospitano una mutazione non silente in questi due geni. Le barre di colore blu e giallo indicano i punteggi Z.(A) Le frequenze alleliche della variante sono state calcolate per tutti i geni in 10 coorti TCGA. Box trame dei VAF di quattro driver di cancro comune (TP53, PIK3CA, ARID1A, e NF1) sono visualizzati. I box rappresentano il secondo e il terzo quartile VAF, mentre le barre di errore indicano il10° e il 90° percentile.(B) Curve Kaplan-Meier degli esiti dei pazienti rispetto alla mutazione VAF. Le mutazioni TP53 sono associate all’esito in HNSC, e questa relazione è vera sia per le mutazioni ad alta prevalenza (VAF ≥0.4) che per quelle a bassa prevalenza (VAF <0.4) (grafico in alto a sinistra). Allo stesso modo, nessuna informazione prognostica significativa è conferita dalle mutazioni TP53 in LUSC, dalle mutazioni KRAS in LUAD, o dalle mutazioni NF1 in SKCM, indipendentemente dalla prevalenza della mutazione.(C) Viene visualizzata una mappa termica delle associazioni di sopravvivenza significative tra i 30 geni guida del cancro più frequentemente mutilati in 16 tipi di tumore della TCGA. I punteggi Z sono stati calcolati facendo regredire i tempi di sopravvivenza tra i pazienti che ospitano copie wild-type e le copie mutanti di un gene se un gene è stato mutato in ≥2% dei campioni per tipo di tumore. Tuttavia, un gene è stato considerato “mutato” per questa analisi solo se il suo VAF era ≥0,4. Ai fini della visualizzazione, vengono visualizzati solo i punteggi Z significativi.(D) Una mappa termica di associazioni significative di sopravvivenza tra i 30 geni driver del cancro più frequentemente-mutate in 16 tipi di tumore dal TCGA sono visualizzati. I punteggi Z sono stati calcolati facendo regredire i tempi di sopravvivenza tra i pazienti che ospitano copie wild-type e le copie mutanti di un gene se un gene è stato mutato in ≥2% dei campioni per tipo di tumore. Per questa analisi del punteggio Z sono state incluse solo mutazioni con VAF maggiore o uguale al VAF mediano per un particolare gene in un particolare tipo di tumore. Ai fini della visualizzazione, vengono visualizzati solo i punteggi Z significativi.(E) Abbiamo confrontato i punteggi Z che risultano dalla regressione solo dei VAF sopra la media e i punteggi Z che risultano dalla regressione solo dei VAF sotto la media, per i 30 geni guida del cancro. In 62 casi, i punteggi Z più alti sono stati ottenuti considerando solo i VAF sopra la media, mentre in 68 casi, i punteggi Z più alti sono stati ottenuti considerando solo i VAF sotto la media.(A) Sono visualizzate le curve Kaplan-Meier dei cinque geni con le più forti associazioni di sopravvivenza in GBMLGG(ATRX, EGFR, IDH1, TP53 e PTEN).(B) Vengono visualizzati i modelli di mutazione secondo il sottotipo di glioma. Le caselle grigie corrispondono ai pazienti con un particolare sottotipo di glioma, mentre i trattini verdi indicano la presenza di una copia mutante del gene specificato.(C) Curve Kaplan-Meier delle mutazioni IDH1 in coorti LGG o GBM, analizzate separatamente. Le mutazioni IDH1 sono osservate in entrambe le coorti tumorali e sono associate alla sopravvivenza in entrambe le coorti tumorali.(D) Curve Kaplan-Meier delle mutazioni EGFR in coorti LGG o GBM, analizzate separatamente. Le mutazioni EGFR sono osservate in entrambe le coorti tumorali, ma sono prognostiche solo nei gliomi a basso grado. L’elenco completo dei punteggi Z è presentato nel file supplementare 2D.
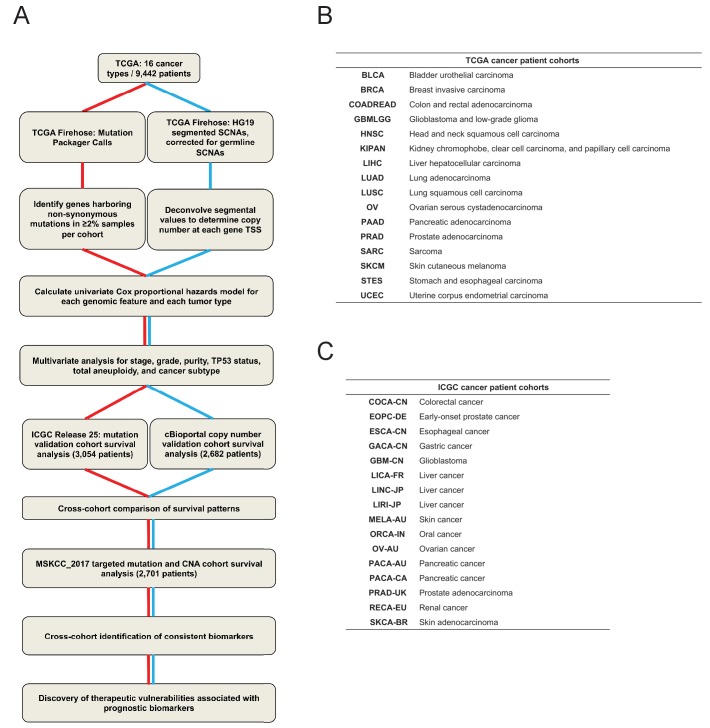
Figura 1-figura supplemento 1.Uno schema della pipeline di analisi della sopravvivenza pan-cancro e dei set di dati utilizzati.2.(A) Viene presentato uno schema dell’elaborazione dei dati e delle analisi effettuate in questo rapporto. 16 tipi di tumore da TCGA sono stati utilizzati come coorte di scoperta. Come coorti di validazione sono stati utilizzati i dati di mutazione da ICGC e i dati sul numero di copie di coorti di pazienti curate da cBioportal. Per identificare i biomarcatori di alta fiducia, un ulteriore set di pazienti sottoposti a sequenziamento mirato (MSKCC_2017) sono stati inclusi nell’analisi cross-cohort. Un elenco completo delle coorti di pazienti e dei tipi di dati analizzati è incluso nel file supplementare 1.(B e C) I tipi di cancro e le abbreviazioni di studio utilizzate nei set di dati TCGA e ICGC. Ulteriori informazioni su ogni coorte sono incluse nel file supplementare 1.
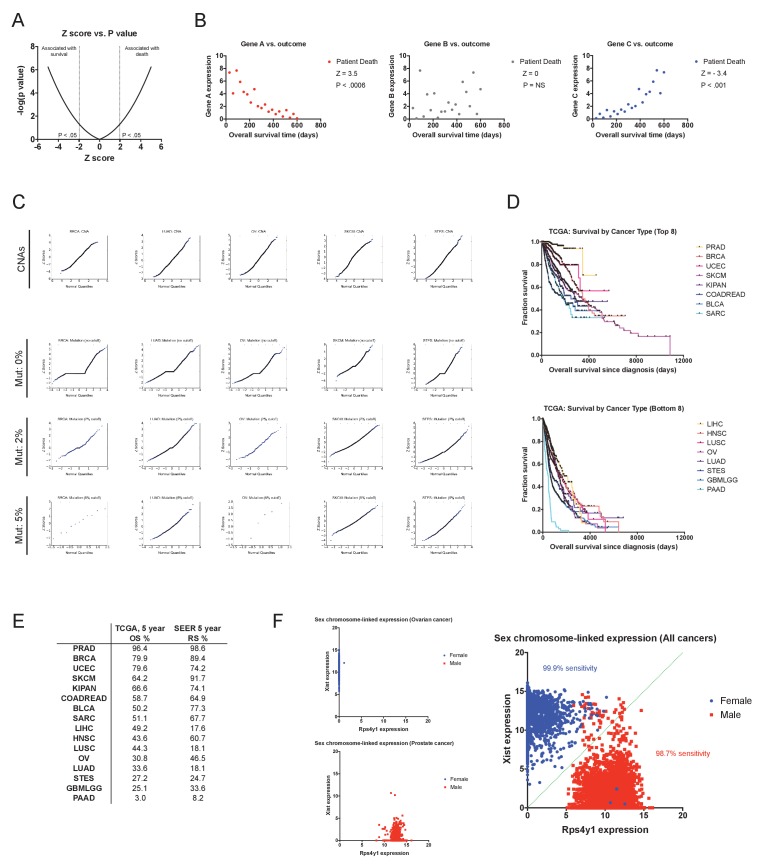
Figura 1-figura supplemento 2.2. Analisi dei rischi di sopravvivenza proporzionale Cox e l’accuratezza delle annotazioni cliniche TCGA.(A) Viene visualizzata la relazione tra il punteggio Z e il valore P per -5 ≤ Z ≤ 5.(B) I dati del campione per i valori di espressione genica per tre geni ipotetici (A, B e C) che illustrano diversi punteggi Z.(C) Grafici quantile-quantilitici che confrontano i punteggi Z e i valori del campione normalmente distribuiti per le CNA e per le mutazioni che utilizzano soglie diverse. Sulla base di questa analisi, solo i geni che ospitano mutazioni nel 2% o più dei campioni di una coorte sono stati inclusi per il calcolo della sopravvivenza (discusso nel testo integrativo 1).(D) I grafici Kaplan-Meier che mostrano il tempo di sopravvivenza post-diagnosi per gli otto tumori con gli esiti più favorevoli (in alto) e gli otto tumori con gli esiti più lugubri (in basso) nel dataset TCGA.(E) Viene visualizzato un confronto tra i tempi di sopravvivenza globale a 5 anni del TCGA e i tempi di sopravvivenza relativa a 5 anni per tipo di cancro del programma di sorveglianza, epidemiologia e risultati finali (SEER).(F) Vengono visualizzati i diagrammi di dispersione che mostrano l’espressione della trascrizione XIST codificata con il cromosoma X e la trascrizione RPS4Y1 codificata con il cromosoma Y nel cancro ovarico (OV), nel cancro alla prostata (PRAD) e in tutti i 16 tipi di tumore. Poiché lo XIST è espresso specificamente in cellule che contengono due o più copie del cromosoma X, questa combinazione di due geni si è dimostrata efficace nel discriminare il sesso cromosomico di una paziente sulla base dell’espressione genica (Gentleset al., 2015; van den Berge e Sijen, 2017). Di conseguenza, nella nostra analisi, quasi tutte le pazienti di sesso femminile hanno un’alta espressione XIST e una bassa espressione RPS4Y1, mentre quasi tutti i pazienti di sesso maschile hanno un’alta espressione RPS4Y1 e una bassa espressione XIST.

Figura 1-figure supplement 3.Lo stato di mutazione del TP53 è associato all’esito in diversi tipi di cancro.(A) Kaplan-Meier trame di tumori TP53-mutanti e TP53-WT in ogni tipo di cancro dal dataset TCGA. TP53 è associato all’esito in cinque dei 16 tipi di cancro (BRCA, GBMLGGG, HNSC, LUAD e PRAD). (B) Trame forestali di mutazioni TP53 in ogni coorte TCGA. I cerchi indicano il rapporto di pericolo, mentre le barre indicano il 95% di confidenza interna. (C) Trame Kaplan-Meier di tumori TP53-mutanti e TP53-WT in ogni tipo di tumore del set di dati ICGC. TP53 è associato all’esito in sette dei 16 tipi di cancro (EOPC-DE, LICA-FR, LINC-JP, LIRI-JP, PACA-AU, PACA-CA e PRAD-UK). (D) Trame forestali di mutazioni TP53 in ogni coorte ICGC. I cerchi indicano il rapporto di pericolo, mentre le barre indicano il 95% di confidenza interna. Si noti che i valori p non sono identici tra A e B e tra C e D, in quanto i valori p di Kaplan-Meier sono calcolati a partire da un test di log-rank, mentre i valori p delle aree forestali sono calcolati a partire da una regressione Cox.
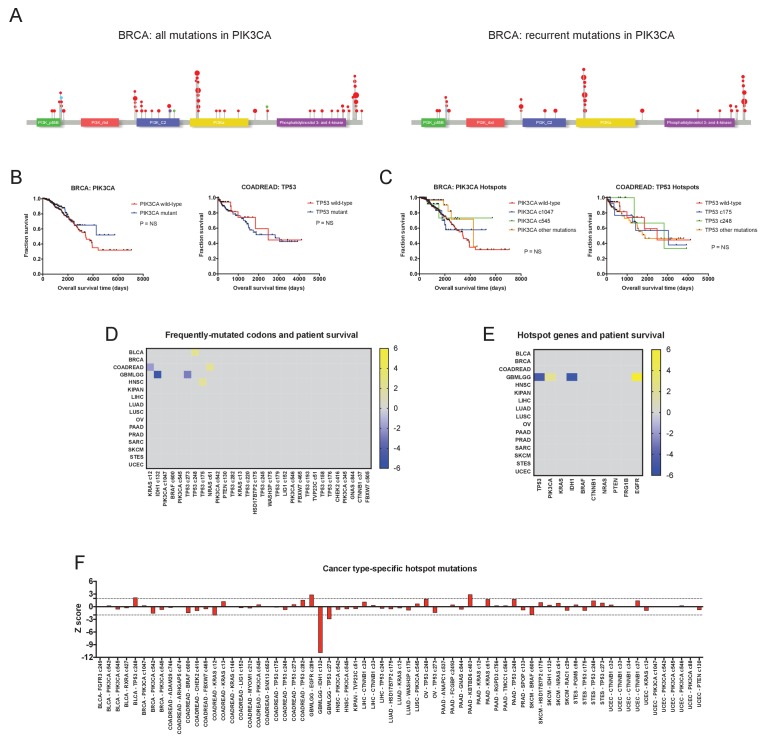
Figura 1-figure supplement 4.Le mutazioni dei punti caldi e le mutazioni nei geni guida del cancro multiplo non sono generalmente associate alla prognosi clinica.(A) Lollipop trame di mutazioni PIK3CA nella coorte BRCA. Il grafico di sinistra mostra tutte le mutazioni non silenti in PIK3CA, mentre il grafico di destra mostra tutte le mutazioni nei codoni ‘hotspot’, ad esempio, i codoni che sono mutati in cinque o più pazienti nel set di dati TCGA. I punti rossi indicano mutazioni missenso, i punti blu indicano mutazioni frameshift, e i punti viola indicano mutazioni nonsenso.(B) Curve Kaplan-Meier che confrontano i pazienti BRCA con qualsiasi mutazione in PIK3CA rispetto a quelli senza mutazioni in PIK3CA (a sinistra), e pazienti COADREAD con qualsiasi mutazione in TP53 rispetto a quelli senza mutazioni in TP53 (a destra).(C) Curve Kaplan-Meier che confrontano i pazienti BRCA con varie mutazioni ‘hotspot’ in PIK3CA rispetto a quelli senza mutazioni ‘hotspot’ in PIK3CA (sinistra), e pazienti COADREAD con mutazioni ‘hotspot’ in TP53 rispetto a quelli senza mutazioni ‘ hotspot’ in TP53 (destra).(D) Viene visualizzata una heatmap delle associazioni di sopravvivenza significative tra i 30 codoni più frequentemente mutilati tra i 16 tipi di tumore del TCGA. I punteggi Z sono stati calcolati facendo regredire i tempi di sopravvivenza tra i pazienti che ospitano una mutazione in uno specifico codone e quelli che non ospitano una mutazione in quel particolare codone, se un codone è stato mutato in ≥2% dei campioni per tipo di tumore. Ai fini della visualizzazione, vengono visualizzati solo i punteggi Z significativi.(E) Tutti i codoni mutati in pazienti ≥5 (mutazioni “hotspot”) sono stati raggruppati insieme, e sono stati determinati i dieci geni che ospitano le mutazioni più “hotspot”. I punteggi Z sono stati calcolati confrontando i pazienti che ospitano qualsiasi mutazione ‘hotspot’ in un gene specificato e i pazienti che non hanno mutazioni ‘hotspot’ in quel gene. Una mappa termica mostra associazioni di sopravvivenza significative per i dieci geni con la maggior parte delle mutazioni “hotspot”.(F) I codici che sono stati mutati in ≥4% dei pazienti in ogni tipo di cancro sono stati identificati, e poi sono stati calcolati i punteggi Z confrontando i pazienti che ospitavano queste mutazioni ricorrenti rispetto a quelli che non lo facevano.
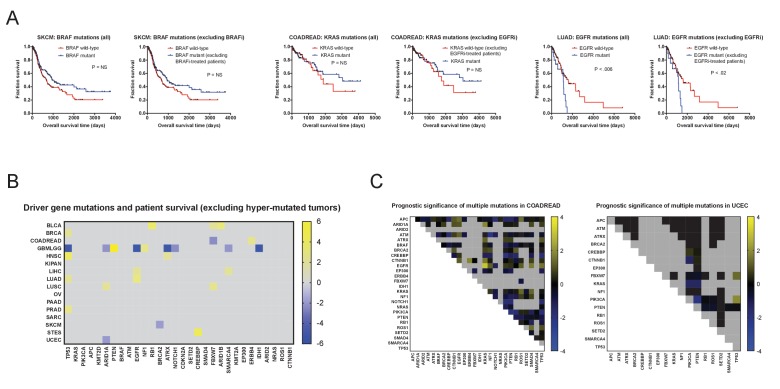
Figura 1-figure supplemento 5.Escludendo i pazienti con tumori ipermutati o quelli che sono stati trattati con terapie mirate non riesce a rivelare mutazioni significativamente associate all’esito.(A) Escludendo i pazienti trattati con terapie mirate non riesce ad alterare significativamente i punteggi della mutazione Z. Le terapie mirate più comuni nella coorte TCGA erano gli inibitori BRAF (usati per trattare i melanomi BRAF-mutanti) e gli inibitori EGFR (usati per trattare i tumori polmonari EGFR-mutanti o i tumori colorettali di tipo KRAS-wild-type). Tuttavia, a causa dei tempi di raccolta dei campioni, che hanno consentito un follow-up del paziente superiore a 10 anni, solo un piccolo sottoinsieme di pazienti viene registrato come paziente che riceve queste terapie. Escludendo tutti i pazienti che hanno ricevuto queste terapie, non ha influito in modo significativo sulle curve di sopravvivenza risultanti.(B) Una heatmap delle associazioni significative tra mutazioni ed esito del paziente nel TCGA quando i tumori iper-mutati(Bailey et al., 2018) sono esclusi. Ai fini della visualizzazione, vengono visualizzati solo i punteggi Z significativi. L’elenco completo dei punteggi Z è presentato nel file supplementare 2C.(C) Le Heatmaps delle combinazioni di doppia mutazione in COADREAD e UCEC sono tracciate. Su 30 geni driver del cancro frequentemente mutilati, 26 sono mutati in ≥2% dei pazienti COADREAD e non sono individualmente correlati con la prognosi, mentre 17 sono mutati in ≥2% dei pazienti UCEC e non sono individualmente correlati con la prognosi. I punteggi Z sono stati calcolati confrontando i tempi di sopravvivenza dei pazienti che ospitano una mutazione non silente in due diversi geni guida del cancro rispetto ai pazienti che non ospitano una mutazione non silente in questi due geni. Le barre di colore blu e giallo indicano i punteggi Z.
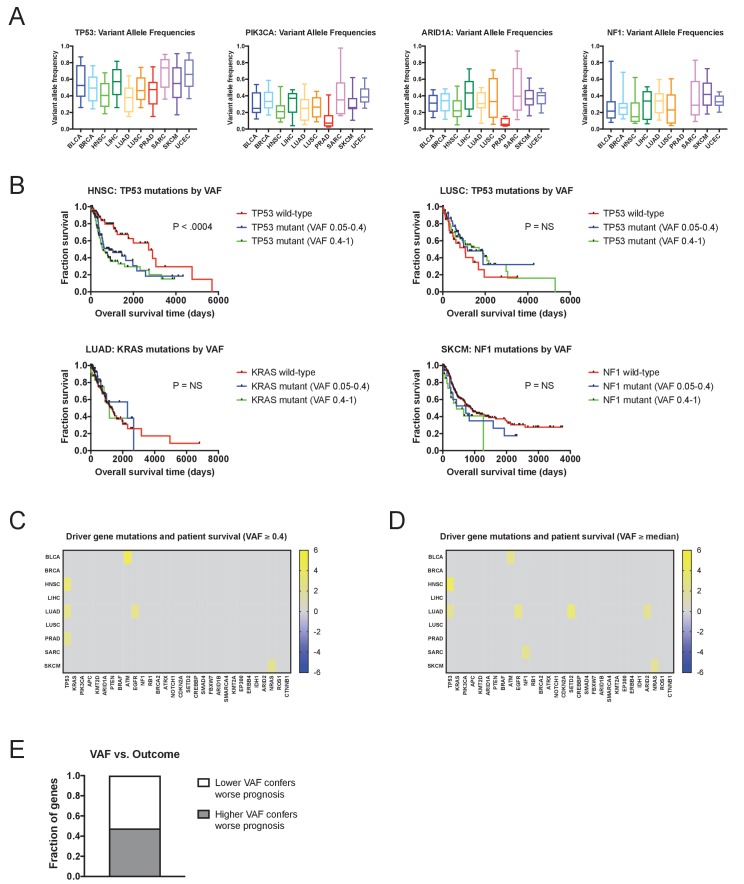
Figura 1-figure supplemento 6.Le mutazioni con frequenze alleliche ad alta variante non sono più prognostiche delle mutazioni con frequenze alleliche a bassa variante.(A) Le frequenze alleliche varianti sono state calcolate per tutti i geni in 10 coorti TCGA. Sono visualizzati i diagrammi dei VAF di quattro comuni driver per il cancro (TP53, PIK3CA, ARID1A e NF1). I box rappresentano il secondo e il terzo quartile VAF, mentre le barre di errore indicano il10° e il 90° percentile.(B) Curve Kaplan-Meier degli esiti dei pazienti rispetto alla mutazione VAF. Le mutazioni TP53 sono associate all’esito in HNSC, e questa relazione è vera sia per le mutazioni ad alta prevalenza (VAF ≥0.4) che per quelle a bassa prevalenza (VAF <0.4) (grafico in alto a sinistra). Allo stesso modo, nessuna informazione prognostica significativa è conferita dalle mutazioni TP53 in LUSC, dalle mutazioni KRAS in LUAD, o dalle mutazioni NF1 in SKCM, indipendentemente dalla prevalenza della mutazione.(C) Viene visualizzata una mappa termica delle associazioni di sopravvivenza significative tra i 30 geni guida del cancro più frequentemente mutilati in 16 tipi di tumore della TCGA. I punteggi Z sono stati calcolati facendo regredire i tempi di sopravvivenza tra i pazienti che ospitano copie wild-type e le copie mutanti di un gene se un gene è stato mutato in ≥2% dei campioni per tipo di tumore. Tuttavia, un gene è stato considerato “mutato” per questa analisi solo se il suo VAF era ≥0,4. Ai fini della visualizzazione, vengono visualizzati solo i punteggi Z significativi.(D) Una mappa termica di associazioni significative di sopravvivenza tra i 30 geni driver del cancro più frequentemente-mutate in 16 tipi di tumore dal TCGA sono visualizzati. I punteggi Z sono stati calcolati facendo regredire i tempi di sopravvivenza tra i pazienti che ospitano copie wild-type e le copie mutanti di un gene se un gene è stato mutato in ≥2% dei campioni per tipo di tumore. Per questa analisi del punteggio Z sono state incluse solo mutazioni con VAF maggiore o uguale al VAF mediano per un particolare gene in un particolare tipo di tumore. Ai fini della visualizzazione, vengono visualizzati solo i punteggi Z significativi.(E) Abbiamo confrontato i punteggi Z che risultano dalla regressione solo dei VAF sopra la media e i punteggi Z che risultano dalla regressione solo dei VAF sotto la media, per i 30 geni guida del cancro. In 62 casi, i punteggi Z più alti sono stati ottenuti considerando solo i VAF sopra la media, mentre in 68 casi, i punteggi Z più alti sono stati ottenuti considerando solo i VAF sotto la media.
Figura 1-figure supplemento 7.Mutazioni prognostiche nel glioma.(A) Sono visualizzate le curve Kaplan-Meier dei cinque geni con le più forti associazioni di sopravvivenza in GBMLGG(ATRX, EGFR, IDH1, TP53 e PTEN).(B) Vengono visualizzati i modelli di mutazione secondo il sottotipo di glioma. Le caselle grigie corrispondono ai pazienti con un particolare sottotipo di glioma, mentre i trattini verdi indicano la presenza di una copia mutante del gene specificato.(C) Curve Kaplan-Meier delle mutazioni IDH1 in coorti LGG o GBM, analizzate separatamente. Le mutazioni IDH1 sono osservate in entrambe le coorti tumorali e sono associate alla sopravvivenza in entrambe le coorti tumorali.(D) Curve Kaplan-Meier delle mutazioni EGFR in coorti LGG o GBM, analizzate separatamente. Le mutazioni EGFR sono osservate in entrambe le coorti tumorali, ma sono prognostiche solo nei gliomi a basso grado. L’elenco completo dei punteggi Z è presentato nel file supplementare 2D.
Mutazioni prognostiche sottotipo-indipendenti e sottotipo-indipendenti nei gliomi
Nella nostra analisi di cui sopra, abbiamo notato che i cinque geni con le associazioni di sopravvivenza più forti sono stati tutti osservati nella coorte GBMLGG (pan-glioma). Poiché il glioma sembrava essere un’eccezione alla nostra constatazione generale che le mutazioni sono raramente prognostiche, abbiamo indagato ulteriormente questa coorte. Tra i geni con il punteggio più alto, abbiamo trovato che le mutazioni di PTEN ed EGFR conferiscono una prognosi sfavorevole, mentre le mutazioni in IDH1, TP53 e ATRX sono associate a una prognosi favorevole(Figura 1-figure supplement 7A). Le mutazioni in questi geni sono state precedentemente collegate a sottotipi di gliomi distinti(Ceccarelli et al., 2016; Kannan et al., 2012; Suzuki et al., 2015), e abbiamo verificato che le mutazioni in IDH1, TP53, e ATRX sono state osservate più frequentemente nei glioblastomi di basso grado, mentre le mutazioni in PTEN e EGFR sono state osservate più frequentemente nei glioblastomi di alto grado(Figura 1-figure supplement 7B). Tuttavia, quando abbiamo analizzato separatamente i glioblastomi e i glioblastomi di basso grado, molte di queste alterazioni sono rimaste prognostiche(file supplementare 2D). Per esempio, mentre le mutazioni dell’IDH1 erano più comuni nei gliomi a basso grado, sono state occasionalmente osservate anche nei tumori di alto grado, e sono state associate indipendentemente alla sopravvivenza prolungata in entrambe le coorti(Figura 1-figure supplement 7C). Al contrario, quando le mutazioni EGFR sono state osservate nei glioblastomi di basso grado, sono state associate a scarsi risultati, ma le mutazioni EGFR non erano prognostiche nei glioblastomi di alto grado(Figura 1-figure supplement 7D). Quindi, nei glioblastomi, le mutazioni contengono sia informazioni prognostiche sottotipo-dipendenti che sottotipo-indipendenti. Tuttavia, al di fuori di questo tipo di cancro e del soppressore tumorale TP53, le mutazioni nella maggior parte dei geni guida del cancro non sono prognostiche.
I CNA dei geni guida sono comunemente associati alla mortalità dei pazienti affetti da cancro
Poiché le mutazioni erano in gran parte disinformative, ci siamo poi impegnati a determinare se il numero di copie geniche trasmetteva informazioni prognostiche. Abbiamo determinato il numero di copie di ogni gene nel suo sito di inizio trascrizionale e abbiamo regredito questo valore rispetto all’esito del paziente in ogni coorte tumorale. Abbiamo poi esaminato l’impatto clinico dei CNA che interessano gli stessi 30 geni guida del cancro che abbiamo studiato in precedenza. Sorprendentemente, abbiamo scoperto che il numero di copie di questi oncogeni e soppressori tumorali era spesso collegato all’esito del paziente(Figura 2 e file supplementare 3A-B). L’amplificazione di EGFR, PIK3CA e BRAF e la cancellazione di CDKN2A, RB1 ed EP300 erano fortemente associati a tempi di sopravvivenza del paziente più brevi in quattro o più tipi di cancro ciascuno. Il numero di copie era prognostico anche per i geni in cui le mutazioni non erano legate all’esito: ad esempio, mentre le mutazioni nel PIK3CA non erano mai informative, il numero di copie del PIK3CA era associato all’esito nei tumori della mammella, del colorettale, del glioma, del polmone squamoso, del pancreas e della prostata(Figura 2B e D). Nel complesso, tra i 30 geni guida del cancro più frequentemente mutilati, abbiamo rilevato 108 associazioni significative tra numero di copie geniche ed esito, rispetto alle 23 associazioni tra mutazione ed esito. Per 28 dei 30 geni guida, il numero di copie di DNA era prognostico in più tipi di cancro rispetto allo stato di mutazione. Concludiamo che determinare il numero di copie di oncogeni e soppressori tumorali in un tumore primitivo può stratificare meglio il rischio del paziente rispetto alla valutazione delle mutazioni di una singola coppia di basi.
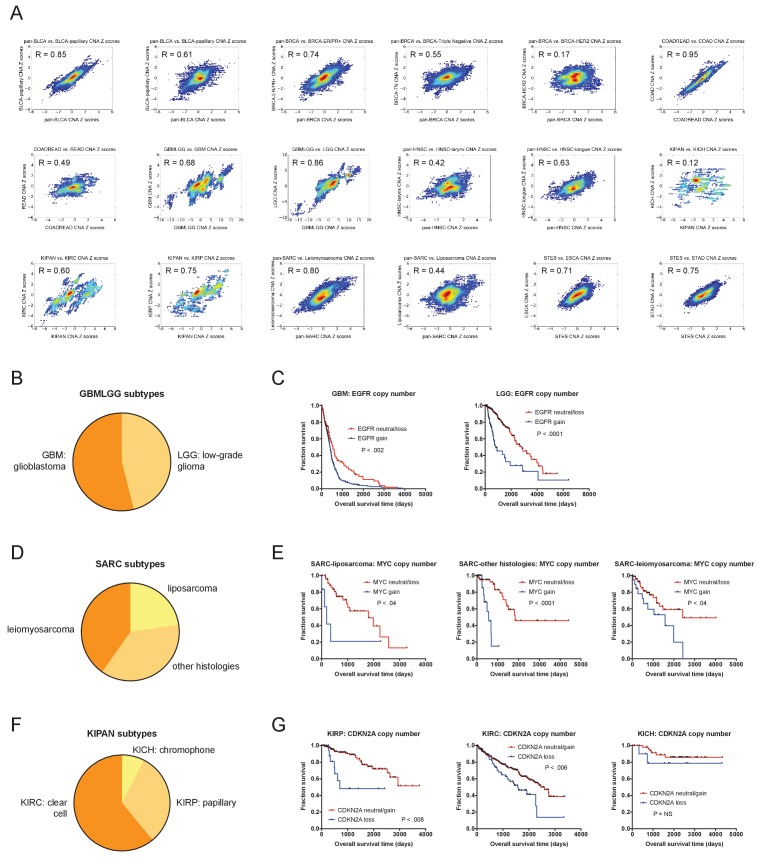
Figura 2-figure supplemento 4.I valori discreti del numero di copie mantengono ancora un significativo potere prognostico. Il valore prognostico degli oncogeni e dei soppressori tumorali è indipendente dalla purezza del campione tumorale.(A) Esempi di CNA del gene guida associati all’esito del paziente. Il numero di copie di CDKN2A, EGFR e BRCA2 nelle coorti di pazienti indicate sono visualizzati, così come le curve Kaplan-Meier di sopravvivenza del paziente secondo il numero di copie del gene. Le amplificazioni e le delezioni corrispondono a CNA > |0,3|, mentre le delezioni profonde e i guadagni ad alta copia corrispondono a CNA > |1|.(B) Viene visualizzata una mappa termica delle associazioni di sopravvivenza significative tra i 30 geni driver del cancro più frequentemente mutilati in 16 tipi di tumore del TCGA. I punteggi Z sono stati calcolati facendo regredire il numero di copie di geni rispetto all’esito del paziente all’interno di ciascun tipo di tumore. L’elenco completo dei punteggi Z è presentato nel file supplementare 3A.(C) I punteggi Z di 16 tipi di tumore del TCGA sono stati combinati usando il metodo di Stouffer, e poi i risultanti punteggi meta-Z sono stati tracciati contro la posizione cromosomica. I geni sono stati raggruppati con il punteggio medio Z in gruppi di 5 per la visualizzazione. I nomi dei geni indicano i geni candidati alla guida che si trovano all’interno di picchi e valli associati alla sopravvivenza.(D) Le curve Kaplan-Meier sono state tracciate per due oncogeni, PIK3CA (a sinistra) e KRAS (a destra), confrontando la rilevanza prognostica delle mutazioni in quei geni rispetto alle alterazioni del numero di copie in questi geni. Le amplificazioni corrispondono a CNA > 0,3, mentre i guadagni ad alta copia corrispondono a CNA > 1.(A) Una mappa termica delle associazioni di sopravvivenza significative tra i 30 geni driver del cancro più frequentemente mutilati in 16 tipi di tumore dal TCGA sono visualizzati. I punteggi di Z sono stati calcolati facendo regredire il numero di copie del gene, trifotomizzati in ‘delezioni’ (<-0.3), ‘amplificazioni’ (>0.3), e ‘copia-neutro’ (≥-0.3 e ≤0.3), rispetto all’esito del paziente all’interno di ciascun tipo di tumore.(B) Le curve Kaplan-Meier sono tracciate per due soppressori tumorali, SMARCA4 (in alto) e CDKN2A. (in basso), confrontando la rilevanza prognostica delle mutazioni in questi geni rispetto alle alterazioni del numero di copie di questi geni. Le delezioni corrispondono a CNA < -0,3.(A) Un grafico a barre che mostra i punteggi Z ottenuti regredendo la purezza del campione, misurata da IHC, rispetto alla sopravvivenza del paziente. Le linee tratteggiate indicano punteggi Z di 1,96 e -1,96, corrispondenti a un valore P < 0,05.(B) Rappresentativo Kaplan-Meier trame rappresentative della sopravvivenza del paziente in BRCA e UCEC, diviso in base alla purezza del campione analizzato.(C) punteggi Z da modelli multivariati tra cui il numero di copie e la purezza del tumore dai 16 tipi di cancro dal TCGA sono stati combinati utilizzando il metodo di Stouffer, e poi i punteggi risultanti meta-Z sono stati tracciate contro la posizione cromosomica. I geni sono stati raggruppati con il punteggio medio Z in gruppi di 5 per la visualizzazione.(D) I CNA rimangono prognostici anche in campioni di tumore puro. La sopravvivenza dei pazienti OV secondo il numero di copie CCNE1 è stata tracciata, utilizzando i dati di tutti i pazienti (a sinistra) o solo dei pazienti con campioni tumorali con purezza ≥85%. L’elenco completo dei punteggi Z è presentato nel file supplementare 3C-E.(A) I punteggi Z sono stati calcolati per i modelli di pericolo proporzionali multivariati Cox che includono sia il numero di copie geniche che lo stadio o il grado del tumore. I diagrammi di densità mostrano la correlazione tra i punteggi Z generati da modelli univariati e questi modelli bivariati. I punteggi Z completi sono elencati nel file supplementare 4.(B) La frazione di caratteristiche identificate nei modelli univariati che rimangono significative nei modelli di stadi e gradi corretti viene mostrata. Si noti che le informazioni sui voti erano disponibili solo per otto coorti TCGA.(C) Trame Kaplan-Meier che dimostrano l’impatto prognostico indipendente dallo stadio delle delezioni ACTL9 in HNSC.(D) Trame Kaplan-Meier che dimostrano l’impatto prognostico indipendente dallo stadio delle amplificazioni ZNF546 in OV. Le amplificazioni e le delezioni corrispondono a CNA > |0,3|.(E) Trame Kaplan-Meier che dimostrano l’impatto prognostico stadio-dipendente delle mutazioni TP53 in HNSC e PRAD. Le mutazioni TP53 sono associate all’esito solo in HNSC in stadio avanzato e PRAD in fase iniziale.(A) Il valore prognostico dei CNA a livello genico all’interno di 18 diversi sottotipi di cancro presenti all’interno del TCGA sono stati analizzati. I grafici di densità mostrano le correlazioni tra i punteggi Z ottenuti dall’analisi univariata che analizza la coorte tumorale parentale e l’analisi univariata del sottotipo tumorale indicato. I risultati completi sono elencati nel file supplementare 4G.(B) Le percentuali di tumori GBM e LGG all’interno della coorte GBMLGG vengono visualizzate.(C) Sono visualizzate le curve Kaplan-Meier delle amplificazioni EGFR nelle coorti GBM e LGG.(D) Sono visualizzate le percentuali di liposarcomi, leiomiosarcomi e sarcomi indifferenziati con altre istologie all’interno della coorte SARC.(E) Le curve Kaplan-Meier delle amplificazioni di MYC nel liposarcoma, leiomiosarcoma e sarcomi con altre coorti istologiche sono visualizzati.(F) Le percentuali di carcinomi a cellule chiare, carcinomi papillari e carcinomi cromofobici all’interno della coorte KIPAN sono visualizzati.(G) Le curve Kaplan-Meier delle delezioni di CDKN2A nelle coorti cellulari, papillari e cromofobiche chiare sono visualizzate. Le amplificazioni e le delezioni corrispondono a CNA > |0,3|.
Nella nostra analisi finora, abbiamo trattato le mutazioni come variabile binaria (“mutante” vs. “non mutante”), mentre le alterazioni del numero di copie sono trattate come valori continui. Così, il maggiore significato prognostico dei CNA tumorali potrebbe riflettere il fatto che le misurazioni dei singoli CNA contengono intrinsecamente più informazioni. Per testare questa possibilità, abbiamo trifotomizzato i valori del CNA in ‘delezioni’ (<-0.3), ‘amplificazioni’ (>0.3), e ‘copia-neutro’ (≥-0.3 e≤0.3). Abbiamo quindi calcolato le regressioni Cox agli stessi 30 loci utilizzando i valori discreti del numero di copie. Questa analisi ha portato a 94 associazioni di sopravvivenza significative, più di quattro volte il numero di caratteristiche significative come quando le mutazioni sono state analizzate, e paragonabile al numero di caratteristiche significative che sono risultate utilizzando i valori continui del CNA(Figura 2-figure supplement 1). Questa analisi suggerisce che la maggiore significatività prognostica dei CNA non è semplicemente una conseguenza della natura continua dei dati del numero di copie.
Abbiamo poi indagato se questi CNA oncogeni e soppressori tumorali erano suscettibili di guidare la mortalità dei pazienti, o se erano geni passeggeri che cambiavano nel numero di copie insieme ad altri driver sconosciuti. Per valutare questa domanda, abbiamo combinato i punteggi Z di diversi tipi di cancro utilizzando il metodo di Stouffer (Stouffer, 1949), e poi abbiamo tracciato i punteggi meta-Z del pan-cancro lungo ogni cromosoma (Figura 2C). Questa analisi ha rivelato molteplici picchi e valli taglienti nei dati che si sono sovrapposti a mutazioni note dei driver. Le più significative variazioni del numero di copie di sopravvivenza associate alla sopravvivenza, a livello genomico, sono state trovate sul cromosoma 9p in una valle che includeva precisamente il soppressore tumorale CDKN2A. I picchi di punteggio Z sono stati trovati in loci che includono oncogeni PIK3CA, EGFR, MYC, CCNE1, e altri. Questa sovrapposizione suggerisce che, in molti casi, il numero di copie di questi oncogeni e soppressori tumorali influenza direttamente il rischio di morte dei pazienti affetti da cancro.
Figura 2-figure supplemento 4.I valori discreti del numero di copie mantengono ancora un significativo potere prognostico. Il valore prognostico degli oncogeni e dei soppressori tumorali è indipendente dalla purezza del campione tumorale.(A) Esempi di CNA del gene guida associati all’esito del paziente. Il numero di copie di CDKN2A, EGFR e BRCA2 nelle coorti di pazienti indicate sono visualizzati, così come le curve Kaplan-Meier di sopravvivenza del paziente secondo il numero di copie del gene. Le amplificazioni e le delezioni corrispondono a CNA > |0,3|, mentre le delezioni profonde e i guadagni ad alta copia corrispondono a CNA > |1|.(B) Viene visualizzata una mappa termica delle associazioni di sopravvivenza significative tra i 30 geni driver del cancro più frequentemente mutilati in 16 tipi di tumore del TCGA. I punteggi Z sono stati calcolati facendo regredire il numero di copie di geni rispetto all’esito del paziente all’interno di ciascun tipo di tumore. L’elenco completo dei punteggi Z è presentato nel file supplementare 3A.(C) I punteggi Z di 16 tipi di tumore del TCGA sono stati combinati usando il metodo di Stouffer, e poi i risultanti punteggi meta-Z sono stati tracciati contro la posizione cromosomica. I geni sono stati raggruppati con il punteggio medio Z in gruppi di 5 per la visualizzazione. I nomi dei geni indicano i geni candidati alla guida che si trovano all’interno di picchi e valli associati alla sopravvivenza.(D) Le curve Kaplan-Meier sono state tracciate per due oncogeni, PIK3CA (a sinistra) e KRAS (a destra), confrontando la rilevanza prognostica delle mutazioni in quei geni rispetto alle alterazioni del numero di copie in questi geni. Le amplificazioni corrispondono a CNA > 0,3, mentre i guadagni ad alta copia corrispondono a CNA > 1.(A) Una mappa termica delle associazioni di sopravvivenza significative tra i 30 geni driver del cancro più frequentemente mutilati in 16 tipi di tumore dal TCGA sono visualizzati. I punteggi di Z sono stati calcolati facendo regredire il numero di copie del gene, trifotomizzati in ‘delezioni’ (<-0.3), ‘amplificazioni’ (>0.3), e ‘copia-neutro’ (≥-0.3 e ≤0.3), rispetto all’esito del paziente all’interno di ciascun tipo di tumore.(B) Le curve Kaplan-Meier sono tracciate per due soppressori tumorali, SMARCA4 (in alto) e CDKN2A. (in basso), confrontando la rilevanza prognostica delle mutazioni in questi geni rispetto alle alterazioni del numero di copie di questi geni. Le delezioni corrispondono a CNA < -0,3.(A) Un grafico a barre che mostra i punteggi Z ottenuti regredendo la purezza del campione, misurata da IHC, rispetto alla sopravvivenza del paziente. Le linee tratteggiate indicano punteggi Z di 1,96 e -1,96, corrispondenti a un valore P < 0,05.(B) Rappresentativo Kaplan-Meier trame rappresentative della sopravvivenza del paziente in BRCA e UCEC, diviso in base alla purezza del campione analizzato.(C) punteggi Z da modelli multivariati tra cui il numero di copie e la purezza del tumore dai 16 tipi di cancro dal TCGA sono stati combinati utilizzando il metodo di Stouffer, e poi i punteggi risultanti meta-Z sono stati tracciate contro la posizione cromosomica. I geni sono stati raggruppati con il punteggio medio Z in gruppi di 5 per la visualizzazione.(D) I CNA rimangono prognostici anche in campioni di tumore puro. La sopravvivenza dei pazienti OV secondo il numero di copie CCNE1 è stata tracciata, utilizzando i dati di tutti i pazienti (a sinistra) o solo dei pazienti con campioni tumorali con purezza ≥85%. L’elenco completo dei punteggi Z è presentato nel file supplementare 3C-E.(A) I punteggi Z sono stati calcolati per i modelli di pericolo proporzionali multivariati Cox che includono sia il numero di copie geniche che lo stadio o il grado del tumore. I diagrammi di densità mostrano la correlazione tra i punteggi Z generati da modelli univariati e questi modelli bivariati. I punteggi Z completi sono elencati nel file supplementare 4.(B) La frazione di caratteristiche identificate nei modelli univariati che rimangono significative nei modelli di stadi e gradi corretti viene mostrata. Si noti che le informazioni sui voti erano disponibili solo per otto coorti TCGA.(C) Trame Kaplan-Meier che dimostrano l’impatto prognostico indipendente dallo stadio delle delezioni ACTL9 in HNSC.(D) Trame Kaplan-Meier che dimostrano l’impatto prognostico indipendente dallo stadio delle amplificazioni ZNF546 in OV. Le amplificazioni e le delezioni corrispondono a CNA > |0,3|.(E) Trame Kaplan-Meier che dimostrano l’impatto prognostico stadio-dipendente delle mutazioni TP53 in HNSC e PRAD. Le mutazioni TP53 sono associate all’esito solo in HNSC in stadio avanzato e PRAD in fase iniziale.(A) Il valore prognostico dei CNA a livello genico all’interno di 18 diversi sottotipi di cancro presenti all’interno del TCGA sono stati analizzati. I grafici di densità mostrano le correlazioni tra i punteggi Z ottenuti dall’analisi univariata che analizza la coorte tumorale parentale e l’analisi univariata del sottotipo tumorale indicato. I risultati completi sono elencati nel file supplementare 4G.(B) Le percentuali di tumori GBM e LGG all’interno della coorte GBMLGG vengono visualizzate.(C) Sono visualizzate le curve Kaplan-Meier delle amplificazioni EGFR nelle coorti GBM e LGG.(D) Sono visualizzate le percentuali di liposarcomi, leiomiosarcomi e sarcomi indifferenziati con altre istologie all’interno della coorte SARC.(E) Le curve Kaplan-Meier delle amplificazioni di MYC nel liposarcoma, leiomiosarcoma e sarcomi con altre coorti istologie sono visualizzati.(F) Le percentuali di carcinomi a cellule chiare, carcinomi papillari e carcinomi cromofobici all’interno della coorte KIPAN sono visualizzati.(G) Le curve Kaplan-Meier delle delezioni di CDKN2A nelle coorti cellulari, papillari e cromofobiche chiare sono visualizzate. Le amplificazioni e le delezioni corrispondono a CNA > |0,3|.
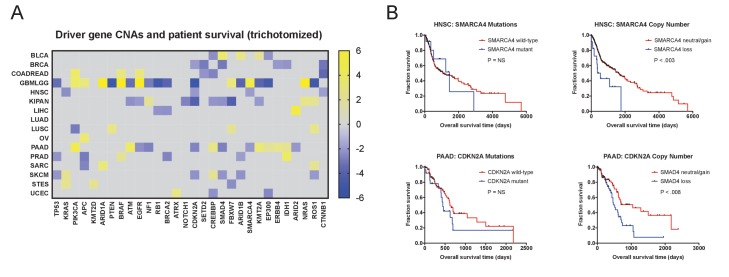
Figura 2-figure supplemento 1.I valori discreti del numero di copie mantengono ancora un significativo potere prognostico.(A) Una mappa termica di associazioni significative di sopravvivenza tra i 30 geni driver del cancro più frequentemente mutilati in 16 tipi di tumore dal TCGA sono visualizzati. I punteggi di Z sono stati calcolati facendo regredire il numero di copie del gene, tricotomizzati in ‘delezioni’ (<-0.3), ‘amplificazioni’ (>0.3), e ‘copia-neutro’ (≥-0.3 e ≤0.3), rispetto all’esito del paziente all’interno di ciascun tipo di tumore.(B) Le curve Kaplan-Meier sono tracciate per due soppressori tumorali, SMARCA4 (in alto) e CDKN2A. (in basso), confrontando la rilevanza prognostica delle mutazioni in questi geni rispetto alle alterazioni del numero di copie di questi geni. Le delezioni corrispondono a CNA < -0,3.
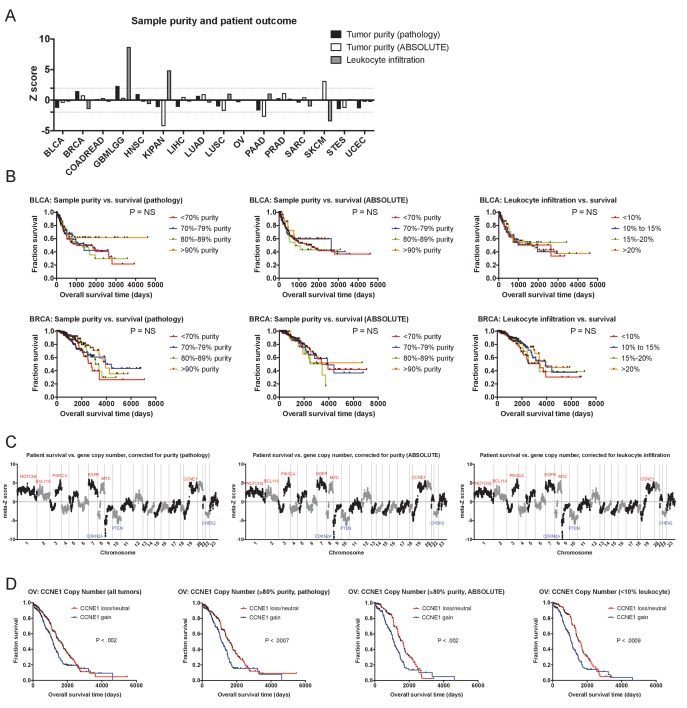
Figura 2-figure supplemento 2.Il valore prognostico dei CNA del cancro è indipendente dalla purezza del campione tumorale.(A) Un grafico a barre che mostra i punteggi Z ottenuti regredendo la purezza del campione, misurata da IHC, rispetto alla sopravvivenza del paziente. Le linee tratteggiate indicano punteggi Z di 1,96 e -1,96, corrispondenti a un valore P < 0,05.(B) Rappresentativo Kaplan-Meier trame rappresentative della sopravvivenza del paziente in BRCA e UCEC, diviso in base alla purezza del campione analizzato.(C) punteggi Z da modelli multivariati tra cui il numero di copie e la purezza del tumore dai 16 tipi di cancro dal TCGA sono stati combinati utilizzando il metodo di Stouffer, e poi i punteggi risultanti meta-Z sono stati tracciate contro la posizione cromosomica. I geni sono stati raggruppati con il punteggio medio Z in gruppi di 5 per la visualizzazione.(D) I CNA rimangono prognostici anche in campioni di tumore puro. La sopravvivenza dei pazienti OV secondo il numero di copie CCNE1 è stata tracciata, utilizzando i dati di tutti i pazienti (a sinistra) o solo dei pazienti con campioni tumorali con purezza ≥85%. L’elenco completo dei punteggi Z è presentato nel file supplementare 3C-E.
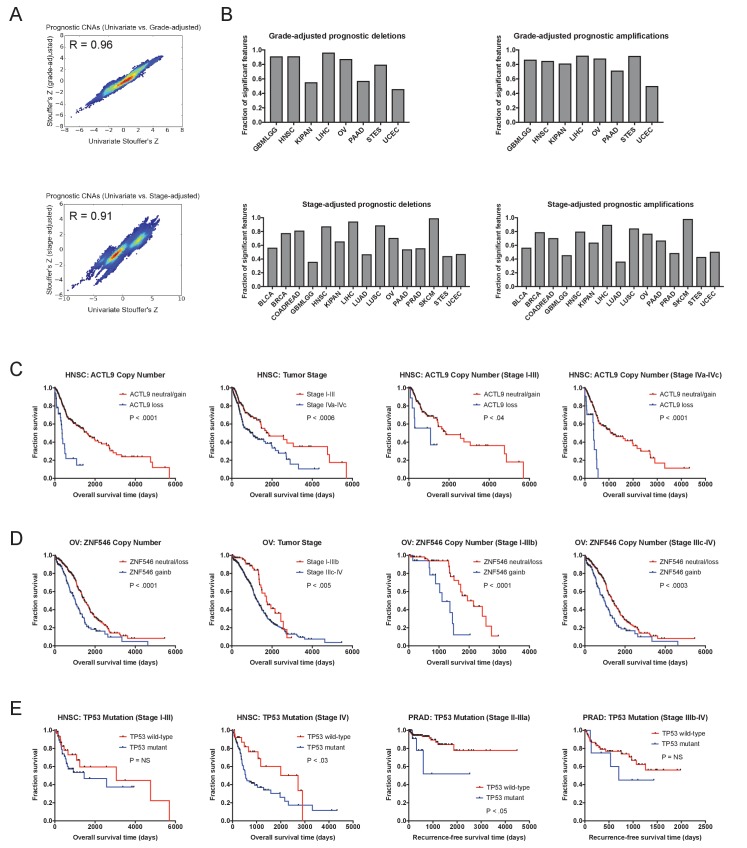
Figura 2-figure supplementary 3.I CNA rimangono prognostici dopo la correzione dello stadio e del grado del tumore.(A) I punteggi Z sono stati calcolati per i modelli di pericolo proporzionali multivariati Cox che includono sia il numero di copie geniche che lo stadio o il grado del tumore. I diagrammi di densità mostrano la correlazione tra i punteggi Z generati dai modelli univariati e questi modelli bivariati. I punteggi Z completi sono elencati nel file supplementare 4.(B) La frazione di caratteristiche identificate nei modelli univariati che rimangono significative nei modelli di stadi e gradi corretti viene mostrata. Si noti che le informazioni sui voti erano disponibili solo per otto coorti TCGA.(C) Trame Kaplan-Meier che dimostrano l’impatto prognostico indipendente dallo stadio delle delezioni ACTL9 in HNSC.(D) Trame Kaplan-Meier che dimostrano l’impatto prognostico indipendente dallo stadio delle amplificazioni ZNF546 in OV. Le amplificazioni e le delezioni corrispondono a CNA > |0,3|.(E) Trame Kaplan-Meier che dimostrano l’impatto prognostico stadio-dipendente delle mutazioni TP53 in HNSC e PRAD. Le mutazioni TP53 sono associate all’esito solo in HNSC in stadio avanzato e PRAD in fase iniziale.
Figura 2-figure supplemento 4.I CNA rimangono prognostici dopo la correzione del sottotipo tumorale.(A) Il valore prognostico dei CNA a livello genico all’interno di 18 diversi sottotipi di cancro presenti all’interno del TCGA sono stati analizzati. I grafici di densità mostrano le correlazioni tra i punteggi Z ottenuti dall’analisi univariata che analizza la coorte tumorale parentale e l’analisi univariata del sottotipo tumorale indicato. I risultati completi sono elencati nel file supplementare 4G.(B) Le percentuali di tumori GBM e LGG all’interno della coorte GBMLGG vengono visualizzate.(C) Sono visualizzate le curve Kaplan-Meier delle amplificazioni EGFR nelle coorti GBM e LGG.(D) Sono visualizzate le percentuali di liposarcomi, leiomiosarcomi e sarcomi indifferenziati con altre istologie all’interno della coorte SARC.(E) Le curve Kaplan-Meier delle amplificazioni di MYC nel liposarcoma, leiomiosarcoma e sarcomi con altre coorti istologie sono visualizzati.(F) Le percentuali di carcinomi a cellule chiare, carcinomi papillari e carcinomi cromofobici all’interno della coorte KIPAN sono visualizzati.(G) Le curve Kaplan-Meier delle delezioni di CDKN2A nelle coorti cellulari, papillari e cromofobiche chiare sono visualizzate. Le amplificazioni e le delezioni corrispondono a CNA > |0,3|.
Il significato prognostico dei CNA è indipendente dalla purezza del campione tumorale e dall’infiltrazione immunitaria
L’aneuploidia del cromosoma intero è stata precedentemente collegata ad una diminuzione dell’infiltrazione delle cellule immunitarie(Davoli et al., 2017; Taylor et al., 2018). Abbiamo quindi considerato la possibilità che i CNA siano prognostici attraverso un meccanismo indiretto, cioè che si trovano nei tumori che mancano di una robusta infiltrazione immunitaria, e questa risposta immunitaria carente era di per sé la causa della mortalità dei pazienti. Tuttavia, molteplici linee di prova sostengono contro questa interpretazione. In primo luogo, abbiamo valutato l’associazione tra la sopravvivenza del paziente e tre diverse misure della purezza del campione tumorale: la frazione cellulare tumorale valutata dal patologo, la purezza del campione come giudicata da ABSOLUTE(Carter et al., 2012; Taylor et al., 2018), e l’infiltrazione dei leucociti, come giudicata dall’analisi della metilazione(Taylor et al., 2018). Abbiamo scoperto che la purezza del campione era associata in modo incoerente con l’esito del paziente(Figura 2-figure supplement 2). Ad esempio, una maggiore purezza del tumore determinata dall’analisi patologica o ASSOLUTA è stata associata ad un risultato peggiore in una sola delle 16 coorti, ciascuna(Figura 2-figure supplement 2A-B). La mancanza di una forte correlazione tra popolazioni cellulari infiltrate e prognosi clinica suggerisce che la purezza dell’analita è insufficiente a spiegare la relazione tra CNA e sopravvivenza del paziente. In secondo luogo, abbiamo generato modelli Cox multivariati che includevano il numero di copie geniche e queste tre misurazioni della purezza del tumore, e abbiamo trovato che i CNA dei geni guida rimangono ampiamente prognostici in questi modelli bivariati(Figura 2-figure supplement 2C e Supplementary file 3C-E). Per esempio, abbiamo scoperto che l’amplificazione del Ciclino E1 è associata ad una prognosi sfavorevole nel cancro ovarico, e questo è rimasto vero anche quando la nostra analisi è stata limitata a campioni di tumore ad alta purezza e a campioni che mancavano di una significativa presenza di leucociti(Figura 2-figure supplement 2D). Quindi, mentre l’interrelazione tra aneuploidia e tolleranza immunologica gioca probabilmente un ruolo importante nello sviluppo del tumore, questa analisi suggerisce che non è il driver primario della mortalità della paziente associata al CNA.
L’analisi del numero di copie migliora la stratificazione del paziente conferita dai parametri clinici comuni
La valutazione patologica dello stadio e del grado del tumore è una fonte importante di informazioni prognostiche, anche se le valutazioni in cieco rivelano una significativa discordanza tra gli osservatori(Allsbrook et al., 2001; Coons et al., 1997; Elmore et al., 2015; Gilks et al., 2013). Abbiamo quindi testato se i biomarcatori CNA che abbiamo scoperto potrebbero influenzare la stratificazione conferita da questi parametri. Abbiamo trovato che i punteggi Z generati da modelli univariati o multivariati che includevano lo stadio o il grado erano altamente correlati (R = 0,91 e R = 0,96 rispettivamente; Figura 2-figure supplement 3A e Supplementary file 4). Nel complesso, il 71% dei CNA prognostici nei singoli tipi di cancro è rimasto prognostico in questi modelli multivariati(Figura 2-figure supplement 3B). Quindi, includere la valutazione del numero di copie a livello genico può migliorare significativamente la stratificazione del rischio del paziente oltre i parametri clinici standard(Figura 2-figure supplement 3C-D). Alcune mutazioni sono state analogamente in grado di fornire informazioni prognostiche in modo indipendente dallo stadio e dal grado. Tuttavia, a causa della minore significatività complessiva della maggior parte delle mutazioni che abbiamo identificato, i miglioramenti nella stratificazione dei pazienti sono stati generalmente più modesti(Figura 2-figure supplement 3E e Supplementary file 4E).
Anche i valori del numero di copie a livello genico sono rimasti prognostici quando si è proceduto alla separazione delle coorti TCGA per sottotipo di cancro(Figura 2-figure supplement 4 e Supplementary file 4F-G). Per esempio, i valori dei punteggi CNA Z erano altamente correlati tra la coorte GBMLGG di massa e i singoli sottotipi GBM e LGG (R = 0,68 e R = 0,86, rispettivamente; Figura 2-figure supplement 4A). Mentre si analizzava la coorte GBM separatamente abolito il significato prognostico delle mutazioni EGFR(Figura 2-figure supplement 4D), amplificazioni EGFR è rimasto associato con il risultato in entrambe le coorti LGG e GBM (Figura 2-figure supplement 4B-C). Le amplificazioni in MYC e PIK3CA sono state prognostiche in modo simile in più sottotipi tumorali(Figura 2-figure supplement 4D-E e Supplementary file 4G). In altri loci, il basso numero di pazienti di alcuni sottotipi può oscurare il rilevamento di biomarcatori specifici. Ad esempio, all’interno della coorte KIPAN, il 67% dei tumori sono carcinomi a cellule chiare, il 23% dei tumori sono carcinomi a cellule papillari e il 10% dei tumori sono carcinomi cromofobici. La delezione di CDKN2A è un forte indicatore di una prognosi sfavorevole nella coorte dei reni, nei carcinomi a cellule chiare e nei carcinomi a cellule papillari, ma non ha raggiunto una significatività statistica quando i carcinomi cromofobici renali sono stati analizzati in modo indipendente(Figura 2-figure supplement 4F-G). In totale, questi risultati sottolineano la capacità dei CNA del gene guida di migliorare la stratificazione del paziente quando si controlla l’identità del tumore, anche se un numero maggiore di coorti può essere necessario per identificare i biomarcatori più forti nei sottotipi di cancro rari.
I CNA del gene pilota contengono informazioni prognostiche non catturate dallo stato di mutazione del TP53 o dall’aneuploidia totale
I tumori altamente aneuploidi tendono ad ospitare mutazioni nella TP53, e sia le mutazioni della TP53 che l’aneuploidia al braccio sono state precedentemente associate a scarsi risultati clinici(Davoli et al., 2017; Petitjean et al., 2007). Utilizzando un ‘punteggio di aneuploidia’ per ciascun tumore basato sul numero totale di alterazioni di lunghezza del braccio (Tayloret al., 2018), abbiamo verificato che i tumori TP53-mutanti presentano più aneuploidia rispetto ai tumori di tipo TP53-paura (Figura 3-figuresupplement 1A), e che l’aneuploidia totale è un fattore di prognosi sfavorevole in diversi tipi di cancro (Figura 3-figuresupplement 1C). Per indagare la relazione tra i CNA prognostici a livello genico, lo stato del TP53 e l’aneuploidia del braccio, abbiamo selezionato una serie di 40 amplificazioni prognostiche e delezioni per ulteriori analisi(Figura 3-figure supplement 2A). Nei modelli multivariati che includevano lo stato di mutazione del TP53, 33 di 40 (83%) CNA a livello genico sono rimasti prognostici, dimostrando che questi CNA non sono legati alla morte a causa di un’associazione indiretta con lo stato del TP53(Figura 3-figure supplement 2A-B). Allo stesso modo, nei modelli multivariati che includevano l’aneuploidia tumorale totale, l’80% di questi CNA erano ancora associati all’esito(Figura 3-figure supplement 2C-D). Infine, come proxy del carico di alterazione strutturale totale, abbiamo sommato il numero di breakpoint (come indicato dai valori del numero di copie discrete lungo un cromosoma) in ogni tumore(Figura 3-figure supplement 1B). Questa metrica è stata associata all’esito in più tipi di tumore(Figura 3-figure supplement 1C), ma il 75% dei CNA del gene pilota è rimasto prognostico in modelli multivariati che includevano questo punteggio (Figura 3-figure supplement 2E-F). Questi risultati indicano che la valutazione dei CNA a livello genico dei tumori può fornire maggiori informazioni prognostiche rispetto al semplice screening per le mutazioni del TP53 o alla misurazione dei livelli di massa dell’aneuploidia tumorale(file supplementare 5).
I CNA focali in genere lasciano presagire una prognosi peggiore di quella di un ampio CNA
Ci siamo quindi posti l’obiettivo di determinare se le alterazioni del numero di copie focali e le alterazioni generali del numero di copie possano avere effetti distinti sull’esito del paziente. Per indagare questa possibilità, abbiamo confrontato il potere prognostico delle CNA focali (definite come un’alterazione ≤3 Mb di lunghezza; Krijgsman et al., 2014) e delle CNA ampie (definite come tutte le alterazioni >3 Mb di lunghezza). Tra i loci in cui sono state osservate sia le alterazioni ampie che quelle focali, abbiamo spesso riscontrato che le CNA ampie erano associate ad esiti moderatamente peggiori, mentre le CNA focali erano associate a bruschi cali di sopravvivenza(Figura 3A-B). In alcuni loci, le CNA ampie hanno avuto esiti indistinguibili dai tumori copy-neutrali, mentre solo le CNA focali sono state associate alla morte(Figura 3C). Raramente abbiamo rilevato casi in cui le CNA ampie indicavano una prognosi peggiore di un’alterazione focale(Figura 3A). Interpretiamo questi risultati come un riflesso delle sanzioni di fitness indotte dall’aneuploidia(Sheltzer et al., 2017; Sheltzer e Amon, 2011): le grandi alterazioni del numero di copie cambiano il dosaggio di più geni contemporaneamente e possono compromettere la crescita del tumore, mentre le alterazioni mirate che colpiscono specificamente il numero di copie del gene guida massimizzano il potenziale maligno.
Figura 3-figure supplemento 3.Effetti della dimensione dell’amplicone e dello stato di mutazione genica sui CNA prognostici. CNA a livello genico, stato del TP53, aneuploidia totale del tumore e carico di alterazione totale.(A) 20 amplificazioni prognostiche e 20 delezioni prognostiche sono state selezionate per ulteriori analisi (vedi anche Figura 3-figure supplemento 2). Di questi 40, 14 avevano almeno cinque pazienti che avevano CNA focali (≤3 Mb) e almeno cinque pazienti che avevano CNA ampi (>3 Mb). Sono stati costruiti modelli di pericolo proporzionali univariata Cox confrontando la presenza o l’assenza di qualsiasi CNA nel luogo indicato, o confrontando la presenza o l’assenza di una CNA di una particolare dimensione.(B e C) Le curve Kaplan-Meier sono state tracciate a quattro loci prognostici confrontando tumori con CNA focali (≤3 Mb), tumori con CNA ampi (>3 Mb) e tumori che mancano di CNA in quel locus. Le amplificazioni e le delezioni corrispondono alle CNA > |0.3|.(D) Sono stati costruiti modelli di pericolo proporzionali Multivariati Cox che includono sia il numero di copie del gene indicato che lo stato mutazionale di quel gene. Vengono visualizzati i punteggi Z per i modelli univariati (solo CNA) o multivariati (CNA + stato di mutazione).(E) Curve Kaplan-Meier che confrontano lo stato della mutazione genica e il numero di copie del gene per le alterazioni EGFR e MLH1 nel cancro colorettale. L’amplificazione dell’EGFR e la delezione dell’MLH1 sono associate ad una prognosi sfavorevole, indipendentemente dal fatto che il tumore contenga una mutazione EGFR o MLH1. Nel grafico in basso, si noti che nessun tumore ospita sia delezioni che mutazioni dell’MLH1.(A) Il numero totale di aneuploidie di lunghezza del braccio per campione è tracciato per i tumori TP53-tipo selvatico e TP53-mutanti di ogni coorte TCGA. Le caselle rappresentano il secondo e il terzo quartile, mentre le barre di errore indicano il10° e il 90° percentile. *, p < 0,05; **, p < 0,005; ***, p < 0,0005 (test Mann-Whitney U).(B) L’onere totale di alterazione strutturale in ogni campione è tracciato per i tumori TP53-tipo selvatico e TP53-mutanti di ogni coorte TCGA. Le caselle rappresentano il secondo e il terzo quartile, mentre le barre di errore indicano il10° e il 90° percentile. *, p < 0,05; **, p < 0,005; ***, p < 0,0005 (test Mann-Whitney U).(C) I punteggi Z dai modelli di pericolo proporzionali univariata Cox sono tracciati per l’aneuploidia totale del tumore, il carico di alterazione totale e lo stato di mutazione TP53.(A) Sono state selezionate 20 delezioni prognostiche e 20 amplificazioni prognostiche per ulteriori studi. Sono stati costruiti modelli di pericolo proporzionali multivariati Cox che includono sia il numero di copie del gene indicato sia lo stato di mutazione della TP53. Sono stati visualizzati i punteggi Z sia per i modelli univariati (CNAs da soli) che per i modelli multivariati (CNAs + stato di TP53).(B) Curve Kaplan-Meier che confrontano il numero di copie e lo stato del TP53 dei geni indicati. Le amplificazioni nei diagrammi Kaplan-Meier corrispondono alle CNA > 0,3.(C) Sono stati costruiti modelli di pericolo proporzionali Multivariati Cox che includono sia il numero di copie del gene indicato che l’aneuploidia tumorale totale. Vengono visualizzati i punteggi Z sia per i modelli univariati (CNAs da soli) che per i modelli multivariati (CNAs + aneuploidia totale).(D) Curve Kaplan-Meier che confrontano il numero di copie geniche e l’aneuploidia tumorale totale. I campioni di aneuploidia “bassa” e “alta” sono stati determinati in base al livello medio di aneuploidia in ogni coorte. Le amplificazioni e le delezioni corrispondono alle CNA > |0.3|.(E) Sono stati costruiti modelli di pericolo proporzionali Multivariati Cox che includono sia il numero di copie del gene indicato che il carico totale di alterazione strutturale. Z punteggi per i modelli univariati (CNAs solo) o i modelli multivariati (CNAs + carico di alterazione totale) sono visualizzati.(F) Curve Kaplan-Meier che confrontano il numero di copie geniche e il carico di alterazione. Il carico di alterazione “basso” e “alto” è stato determinato in base al numero medio di alterazioni strutturali in ogni coorte. Amplificazioni e delezioni corrispondono alle CNA > |0.3|.(A) I coefficienti di correlazione Pearson tra numero di copia del gene e l’espressione del gene a 40 loci prognostici sono visualizzati.(B) Un diagramma di dispersione che mostra la corrispondenza tra il numero di copia CCND1 e l’espressione nella coorte HNSC.(C) Un diagramma di dispersione che mostra la corrispondenza tra il numero di copie RB1 e l’espressione nella coorte BRCA.
Figura 3-figure supplement 3.Figura 3— supplemento alla figura 3. Effetti della dimensione dell’amplicone e dello stato di mutazione genica sui CNA prognostici. CNA a livello genico, stato del TP53, aneuploidia totale del tumore e carico di alterazione totale.(A) 20 amplificazioni prognostiche e 20 delezioni prognostiche sono state selezionate per ulteriori analisi (vedi anche Figura 3-figure supplemento 2). Di questi 40, 14 avevano almeno cinque pazienti che avevano CNA focali (≤3 Mb) e almeno cinque pazienti che avevano CNA ampi (>3 Mb). Sono stati costruiti modelli di pericolo proporzionali univariata Cox confrontando la presenza o l’assenza di qualsiasi CNA nel luogo indicato, o confrontando la presenza o l’assenza di una CNA di una particolare dimensione.(B e C) Le curve Kaplan-Meier sono state tracciate a quattro loci prognostici confrontando tumori con CNA focali (≤3 Mb), tumori con CNA ampi (>3 Mb) e tumori che mancano di CNA in quel locus. Le amplificazioni e le delezioni corrispondono alle CNA > |0.3|.(D) Sono stati costruiti modelli di pericolo proporzionali Multivariati Cox che includono sia il numero di copie del gene indicato che lo stato mutazionale di quel gene. Vengono visualizzati i punteggi Z per i modelli univariati (solo CNA) o multivariati (CNA + stato di mutazione).(E) Curve Kaplan-Meier che confrontano lo stato della mutazione genica e il numero di copie del gene per le alterazioni EGFR e MLH1 nel cancro colorettale. L’amplificazione dell’EGFR e la delezione dell’MLH1 sono associate ad una prognosi sfavorevole, indipendentemente dal fatto che il tumore contenga una mutazione EGFR o MLH1. Nel grafico in basso, si noti che nessun tumore ospita sia delezioni che mutazioni dell’MLH1.(A) Il numero totale di aneuploidie di lunghezza del braccio per campione è tracciato per i tumori TP53 di tipo selvatico e TP53-mutanti di ogni coorte TCGA. Le caselle rappresentano il secondo e il terzo quartile, mentre le barre di errore indicano il10° e il 90° percentile. *, p < 0,05; **, p < 0,005; ***, p < 0,0005 (test Mann-Whitney U).(B) L’onere totale di alterazione strutturale in ogni campione è tracciato per i tumori TP53-tipo selvatico e TP53-mutanti di ogni coorte TCGA. Le caselle rappresentano il secondo e il terzo quartile, mentre le barre di errore indicano il10° e il 90° percentile. *, p < 0,05; **, p < 0,005; ***, p < 0,0005 (test Mann-Whitney U).(C) I punteggi Z dai modelli di pericolo proporzionali univariata Cox sono tracciati per l’aneuploidia totale del tumore, il carico di alterazione totale e lo stato di mutazione TP53.(A) Sono state selezionate 20 delezioni prognostiche e 20 amplificazioni prognostiche per ulteriori studi. Sono stati costruiti modelli di pericolo proporzionali multivariati Cox che includono sia il numero di copie del gene indicato sia lo stato di mutazione della TP53. Sono stati visualizzati i punteggi Z sia per i modelli univariati (CNAs da soli) che per i modelli multivariati (CNAs + stato di TP53).(B) Curve Kaplan-Meier che confrontano il numero di copie e lo stato del TP53 dei geni indicati. Le amplificazioni nei diagrammi Kaplan-Meier corrispondono alle CNA > 0,3.(C) Sono stati costruiti modelli di pericolo proporzionali Multivariati Cox che includono sia il numero di copie del gene indicato che l’aneuploidia tumorale totale. Vengono visualizzati i punteggi Z sia per i modelli univariati (CNAs da soli) che per i modelli multivariati (CNAs + aneuploidia totale).(D) Curve Kaplan-Meier che confrontano il numero di copie geniche e l’aneuploidia tumorale totale. I campioni di aneuploidia “bassa” e “alta” sono stati determinati in base al livello medio di aneuploidia in ogni coorte. Le amplificazioni e le delezioni corrispondono alle CNA > |0.3|.(E) Sono stati costruiti modelli di pericolo proporzionali Multivariati Cox che includono sia il numero di copie del gene indicato che il carico totale di alterazione strutturale. Z punteggi per i modelli univariati (CNAs solo) o i modelli multivariati (CNAs + carico di alterazione totale) sono visualizzati.(F) Curve Kaplan-Meier che confrontano il numero di copie geniche e il carico di alterazione. Il carico di alterazione “basso” e “alto” è stato determinato in base al numero medio di alterazioni strutturali in ogni coorte. Amplificazioni e delezioni corrispondono alle CNA > |0.3|.(A) I coefficienti di correlazione Pearson tra numero di copia del gene e l’espressione del gene a 40 loci prognostici sono visualizzati.(B) Un diagramma di dispersione che mostra la corrispondenza tra il numero di copia CCND1 e l’espressione nella coorte HNSC.(C) Un diagramma di dispersione che mostra la corrispondenza tra il numero di copie RB1 e l’espressione nella coorte BRCA.
Figura 3-figure supplement 1.CNA a livello genico, stato TP53, aneuploidia tumorale totale e carico di alterazione totale.(A) Il numero totale di aneuploidie di lunghezza del braccio per campione è tracciato per i tumori TP53-tipo selvatico e TP53-mutanti da ogni coorte TCGA. Le caselle rappresentano il secondo e il terzo quartile, mentre le barre di errore indicano il10° e il 90° percentile. *, p < 0,05; **, p < 0,005; ***, p < 0,0005 (test Mann-Whitney U).(B) L’onere totale di alterazione strutturale in ogni campione è tracciato per i tumori TP53-tipo selvatico e TP53-mutanti di ogni coorte TCGA. Le caselle rappresentano il secondo e il terzo quartile, mentre le barre di errore indicano il10° e il 90° percentile. *, p < 0,05; **, p < 0,005; ***, p < 0,0005 (test Mann-Whitney U).(C) I punteggi Z dai modelli di pericolo proporzionali univariata Cox sono tracciati per l’aneuploidia totale del tumore, il carico di alterazione totale e lo stato di mutazione TP53.
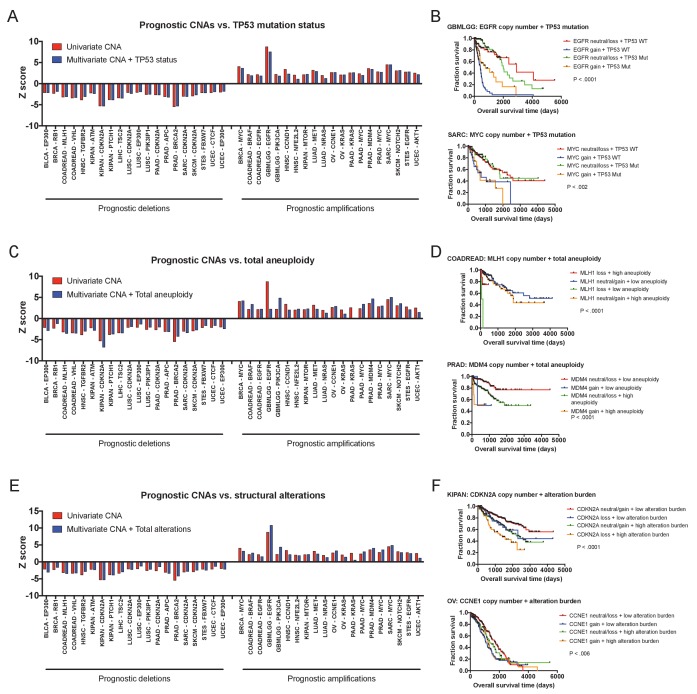
Figura 3-figure supplemento 2.Analisi multivariata dei CNA prognostici.(A) 20 delezioni prognostiche e 20 amplificazioni prognostiche sono state selezionate per ulteriori studi. Sono stati costruiti modelli di pericolo proporzionali multivariati Cox che includono sia il numero di copie del gene indicato sia lo stato di mutazione del TP53. Sono stati visualizzati i punteggi Z sia per i modelli univariati (CNAs da soli) che per i modelli multivariati (CNAs + stato di TP53).(B) Curve Kaplan-Meier che confrontano il numero di copie e lo stato del TP53 dei geni indicati. Le amplificazioni nei diagrammi Kaplan-Meier corrispondono alle CNA > 0,3.(C) Sono stati costruiti modelli di pericolo proporzionali Multivariati Cox che includono sia il numero di copie del gene indicato che l’aneuploidia tumorale totale. Vengono visualizzati i punteggi Z sia per i modelli univariati (CNAs da soli) che per i modelli multivariati (CNAs + aneuploidia totale).(D) Curve Kaplan-Meier che confrontano il numero di copie geniche e l’aneuploidia tumorale totale. I campioni di aneuploidia “bassa” e “alta” sono stati determinati in base al livello medio di aneuploidia in ogni coorte. Le amplificazioni e le delezioni corrispondono alle CNA > |0.3|.(E) Sono stati costruiti modelli di pericolo proporzionali Multivariati Cox che includono sia il numero di copie del gene indicato che il carico totale di alterazione strutturale. Z punteggi per i modelli univariati (CNAs solo) o i modelli multivariati (CNAs + carico di alterazione totale) sono visualizzati.(F) Curve Kaplan-Meier che confrontano il numero di copie geniche e il carico di alterazione. Il carico di alterazione “basso” e “alto” è stato determinato in base al numero medio di alterazioni strutturali in ogni coorte. Amplificazioni e delezioni corrispondono alle CNA > |0.3|.
Figura 3-figure supplemento 3.CNA prognostici alterano l’espressione del gene che comprendono.(A) I coefficienti di correlazione Pearson tra il numero di copia del gene e l’espressione del gene a 40 loci prognostici sono visualizzati.(B) Un diagramma di dispersione che mostra la corrispondenza tra il numero di copia CCND1 e l’espressione nella coorte HNSC.(C) Un diagramma di dispersione che mostra la corrispondenza tra il numero di copie RB1 e l’espressione nella coorte BRCA.
Le CNA focali influenzano l’esito del paziente modificando i livelli di espressione dei geni wild-type
Le alterazioni del numero di copie genetiche si traducono tipicamente in una variazione proporzionale dell’espressione dei loci interessati(Pollack et al., 2002; Stingele et al., 2012; Williams et al., 2008), sebbene siano stati segnalati casi di compensazione del dosaggio(Gonçalves et al., 2017). Per testare gli effetti dei CNA prognostici sull’espressione genica, abbiamo confrontato i livelli di trascrizione e le variazioni del numero di copie geniche a 40 loci prognostici e abbiamo trovato una correlazione significativa tra i due al 98% dei geni analizzati(Figura 3-figure supplement 3). Successivamente, abbiamo cercato di scoprire se queste alterazioni del numero di copie erano mortali perché aumentavano o diminuivano l’espressione dei prodotti genici mutanti. In altre parole, abbiamo potuto osservare che l’amplificazione di un gene pilota è prognostica solo nei tumori in cui anche quel gene pilota è mutato. È interessante notare che non è così: al 95% dei nostri loci di prova, il numero di copie geniche è rimasto prognostico in modelli multivariati che includevano anche lo stato di mutazione genica(Figura 3D). Per esempio, nel cancro colorettale, l’amplificazione dell’EGFR era associata alla morte anche nei tumori che non presentavano mutazioni EGFR(Figura 3E). In totale, questi risultati indicano che anche in loci con mutazioni ricorrenti, i cambiamenti nell’espressione del gene wild-type possono avere un profondo effetto sul comportamento delle cellule tumorali. Insieme alla nostra osservazione che i cambiamenti focali tendono a conferire una prognosi peggiore rispetto ai cambiamenti generali, questi risultati supportano il modello recentemente proposto di “isola del gene del cancro” dell’evoluzione del genoma tumorale (discusso in dettaglio più avanti; Solimini et al., 2012).
Coorti di pazienti indipendenti verificano il significato prognostico dei CNA del gene guida
Per determinare la generalità delle nostre scoperte, abbiamo raccolto coorti di pazienti indipendenti che ospitano mutazioni o copiano dati numerici legati all’esito della sopravvivenza(file supplementare 1). Abbiamo quindi eseguito un’analisi dei pericoli proporzionali Cox univariata su queste coorti di “validazione” e abbiamo confrontato i risultati con i punteggi Z ottenuti dal nostro set di dati TCGA “discovery”. In primo luogo, abbiamo identificato le mutazioni prognostiche all’interno di un insieme di 16 coorti di pazienti dell’International Cancer Genome Consortium (ICGC), che comprende 3054 pazienti analizzati con il sequenziamento del genoma intero o dell’intero esoma. Nel complesso, le frequenze di mutazione e i punteggi Z delle mutazioni ricorrenti erano molto simili tra le coorti ICGC e TCGA (R = 0,67, p < 0,0001, e R = 0,56, p < 0,0001, rispettivamente; Figura 4A-B). Coerentemente con la nostra analisi TCGA, le mutazioni nel TP53 sono state associate al risultato in più coorti di pazienti rispetto a qualsiasi altro gene(Figura 4C e Figura 1-figure supplement 3C-D). Altre mutazioni, anche nei geni guida del cancro noti, sono state raramente associate all’esito in singoli tipi di cancro e hanno avuto un significato minimo di pan-cancro(Figura 4D-F e file supplementare 6). Mutazioni in KRAS, PIK3CA, BRAF, APC, PTEN, CDKN2A, e molte altre sono state osservate frequentemente, ma non sono mai state correlate con l’esito(Figura 4D). Abbiamo poi analizzato 2431 ulteriori pazienti con dati di CNA curata da cBioportal, e trovato numerose amplificazioni e delezioni associate alla mortalità del paziente(file supplementare 6C). Nel cancro al seno, abbiamo trovato amplificazioni prognostiche che erano incentrate sugli oncogeni, tra cui ERBB2, MYC e MDM2, mentre le delezioni prognostiche comprendevano i soppressori tumorali CDKN2A, PTEN e TP53 (Figura 4G). Nel complesso, abbiamo osservato una correlazione altamente significativa tra i punteggi meta-Z ottenuti dai set di dati TCGA e cBioportal (R = 0,42; Figura 4H) . Infine, nelle coorti di pazienti sottoposti sia all’analisi delle mutazioni che all’analisi del numero di copie, abbiamo verificato che le CNA nei geni pilota hanno comunemente un significato prognostico maggiore rispetto alle mutazioni in quegli stessi geni(Figura 4I). Ad esempio, nel carcinoma mammario, tra i 25 geni frequentemente mutilati, le mutazioni in soli due geni(TP53 e GATA3) hanno mostrato un significato prognostico, mentre i CNA in 12 di quegli stessi geni sono stati associati all’esito del paziente(Figura 4J). In totale, queste analisi suggeriscono che i modelli di sopravvivenza scoperti nel set di dati TCGA sono conservati in coorti indipendenti di pazienti affetti da cancro. In particolare, mentre le mutazioni nella maggior parte dei geni guida del cancro sono non prognostiche, le alterazioni del numero di copie in questi stessi geni sono strettamente legate all’esito del paziente.
Figura 4.Il numero di copie del gene pilota, ma non le mutazioni del gene pilota, sono associati alla sopravvivenza in coorti di pazienti indipendenti.(A) I geni mutati in ≥10% dei pazienti in ogni tipo di tumore nel TCGA sono stati identificati e poi confrontati con la frequenza di mutazione di questi geni nella coorte o coorti ICGC corrispondente. L’elenco completo dei punteggi Z è presentato nel file supplementare 6A.(B) I punteggi Z dei 10 geni più frequentemente mutilati per tipo di tumore nell’ICGC sono stati identificati e poi confrontati con i punteggi Z dello stesso gene della coorte o delle coorti TCGA corrispondenti.(C) Sono stati contati i punteggi Z significativi (>1,96 o<-1,96) per gene, e poi è stato tracciato il numero di coorti significative del TCGA e dell’ICGC. Mentre la stragrande maggioranza dei geni frequentemente mutilati sono significativi in zero o in un tipo di cancro, lo stato di mutazione del TP53 è associato alla prognosi in 12 delle 32 coorti totali di pazienti.(D) Viene mostrata una mappa termica delle associazioni di sopravvivenza significative tra i 30 geni guida del cancro più frequentemente mutilati in 16 coorti di pazienti dell’ICGC. I punteggi Z sono stati calcolati facendo regredire i tempi di sopravvivenza tra i pazienti che ospitano copie wild-type e le copie mutanti di un gene se un gene è stato mutato in ≥2% dei campioni per tipo di tumore. Ai fini della visualizzazione, vengono visualizzati solo i punteggi Z significativi. L’elenco completo dei punteggi Z è presentato nel file supplementare 6A.(E) Il numero di geni mutati in ≥2% dei campioni per tipo di tumore viene visualizzato.(F) Il numero di geni significativamente associati all’esito del paziente ad una soglia di falsa scoperta del 5% in ogni tipo di tumore sono visualizzati.(G) I punteggi Z per il numero di copie di ciascun gene della coorte TCGA BRCA e della coorte cBioportale METABRIC sono tracciati l’uno contro l’altro. L’elenco completo dei punteggi Z è presentato nel file supplementare 6C.(H) I punteggi meta-Z da set di dati curati da cBioportal sono tracciati contro i punteggi meta-Z dei corrispondenti quattro tipi di cancro del TCGA (BLCA, BRCA, LIHC e LUAD). L’elenco completo dei punteggi Z è presentato nel file supplementare 6C.(I) Curve Kaplan-Meier che confrontano le mutazioni e le CNA in ERBB3 e PTEN nella coorte cBioportale METABRIC.(J) Un grafico a barre dei punteggi Z per le mutazioni e le CNA in 25 geni pilota nella coorte cBioportale METABRICO. Mentre le mutazioni in soli due geni sono associate alla prognosi, le CNA in 12 di questi stessi geni sono associate alla prognosi.
Figura 4.Il numero di copia del gene pilota, ma non le mutazioni del gene pilota, sono associati alla sopravvivenza in coorti di pazienti indipendenti.(A) I geni mutati in ≥10% dei pazienti in ogni tipo di tumore nel TCGA sono stati identificati, e poi confrontati con la frequenza di mutazione di questi geni nella coorte o coorti ICGC corrispondente. L’elenco completo dei punteggi Z è presentato nel file supplementare 6A.(B) I punteggi Z dei 10 geni più frequentemente mutilati per tipo di tumore nell’ICGC sono stati identificati e poi confrontati con i punteggi Z dello stesso gene della coorte o delle coorti TCGA corrispondenti.(C) Sono stati contati i punteggi Z significativi (>1,96 o<-1,96) per gene, e poi è stato tracciato il numero di coorti significative del TCGA e dell’ICGC. Mentre la stragrande maggioranza dei geni frequentemente mutilati sono significativi in zero o in un tipo di cancro, lo stato di mutazione del TP53 è associato alla prognosi in 12 delle 32 coorti totali di pazienti.(D) Viene mostrata una mappa termica delle associazioni di sopravvivenza significative tra i 30 geni guida del cancro più frequentemente mutilati in 16 coorti di pazienti dell’ICGC. I punteggi Z sono stati calcolati facendo regredire i tempi di sopravvivenza tra i pazienti che ospitano copie wild-type e le copie mutanti di un gene se un gene è stato mutato in ≥2% dei campioni per tipo di tumore. Ai fini della visualizzazione, vengono visualizzati solo i punteggi Z significativi. L’elenco completo dei punteggi Z è presentato nel file supplementare 6A.(E) Il numero di geni mutati in ≥2% dei campioni per tipo di tumore viene visualizzato.(F) Il numero di geni significativamente associati all’esito del paziente ad una soglia di falsa scoperta del 5% in ogni tipo di tumore sono visualizzati.(G) I punteggi Z per il numero di copie di ciascun gene della coorte TCGA BRCA e della coorte cBioportale METABRIC sono tracciati l’uno contro l’altro. L’elenco completo dei punteggi Z è presentato nel file supplementare 6C.(H) I punteggi meta-Z da set di dati curati da cBioportal sono tracciati contro i punteggi meta-Z dei corrispondenti quattro tipi di cancro del TCGA (BLCA, BRCA, LIHC e LUAD). L’elenco completo dei punteggi Z è presentato nel file supplementare 6C.(I) Curve Kaplan-Meier che confrontano le mutazioni e le CNA in ERBB3 e PTEN nella coorte cBioportale METABRIC.(J) Un grafico a barre dei punteggi Z per le mutazioni e le CNA in 25 geni pilota nella coorte cBioportale METABRICO. Mentre le mutazioni in soli due geni sono associate alla prognosi, le CNA in 12 di questi stessi geni sono associate alla prognosi.
Identificazione incrociata di biomarcatori prognostici ad alta fiducia
Al fine di scoprire i biomarcatori con la maggiore rilevanza clinica potenziale, abbiamo poi identificato le mutazioni individuali e i CNA che sono stati costantemente associati al risultato attraverso coorti di pazienti indipendenti. Per aumentare la nostra capacità di rilevare queste alterazioni genetiche, abbiamo eseguito un’analisi di sopravvivenza su un ulteriore set di 2701 tumori primari sottoposti a sequenziamento mirato e analisi del numero di copie (MSKCC_2017; file supplementare 7)(Zehir et al., 2017), su 2431 pazienti di coorti cBioportali i cui tumori erano stati sequenziati(file supplementare 6D), e su 628 pazienti di coorti ICGC sottoposti ad analisi del numero di copie(file supplementare 6B). Il nostro set di dati combinati dei pazienti comprendeva quindi da due a sei coorti indipendenti da ciascuno dei 13 tipi di cancro comuni, per un totale di 16.580 pazienti. Queste coorti sono state raccolte in diversi luoghi, in diverse popolazioni di pazienti, utilizzando diversi disegni di studio, e i campioni sono stati analizzati utilizzando diverse tecnologie genomiche. Abbiamo ragionato che le alterazioni che sono state associate in modo coerente con l’esito, nonostante queste differenze significative, rappresenterebbero biomarcatori altamente penetranti della prognosi dei pazienti. Per identificare tali alterazioni, abbiamo esaminato i biomarcatori che erano associati all’esito (|Z| > 1,96) in ≥2 coorti indipendenti, e che erano altamente significativi (|meta-Z| > 3,3) in tutte le coorti disponibili. Questo approccio ha rivelato molteplici biomarcatori genetici di alta fiducia del risultato della paziente che, a nostra conoscenza, erano nuovi, tra cui amplificazioni MDM4 nel cancro alla prostata, amplificazioni NOTCH2 nel melanoma, e 2q32 delezioni nel cancro ovarico(file supplementare 8). Questi robusti biomarcatori hanno permesso una notevole stratificazione del rischio per la paziente, e i CNA con punteggio massimo sono rimasti prognostici in modelli multivariati che includevano criteri prognostici comunemente misurati (punteggio di Gleason nel cancro della prostata, sierologia dell’epatite nel cancro del fegato, ecc; Figura 5-figure supplement 1). Coerentemente con le nostre analisi a coorte singola, i CNA prognostici a coorte incrociata erano significativamente più comuni delle mutazioni prognostiche, e il TP53 era l’unico gene il cui stato di mutazione era associato all’esito in più di un tipo di cancro(Figura 5-figure supplement 2A).
Alcuni biomarcatori prognostici sono anche associati a vulnerabilità terapeutiche uniche
Abbiamo ipotizzato che alcune alterazioni genetiche sufficienti ad influenzare la sopravvivenza complessiva del paziente potrebbero avere un impatto anche su altri aspetti del comportamento oncologico, tra cui, potenzialmente, la sensibilità ai farmaci. In altre parole, i biomarcatori che contengono informazioni prognostiche significative potrebbero potenzialmente contenere anche informazioni predittive. Abbiamo quindi cercato di scoprire se le alterazioni genetiche che hanno guidato la malattia aggressiva potessero anche sensibilizzare i tumori dei pazienti a specifici regimi terapeutici. Analizzando una coorte di 1000 xenotrapianti derivati da pazienti (PDX), abbiamo identificato diversi casi in cui i biomarcatori ad alta fiducia erano associati alla vulnerabilità a particolari agenti anti-cancro(Gao et al., 2015a). Per esempio, abbiamo identificato le delezioni di Chr9 che comprendevano il CDKN2A come un biomarcatore robusto per una prognosi sfavorevole nel cancro al seno(file supplementare 8). Abbiamo scoperto che i PDX che ospitano delezioni di CDKN2A erano profondamente sensibili alla terapia combinata con un inibitore CDK4/6 e un inibitore mTOR(Figura 5-figure supplement 2B), coerente con il fatto che una proteina codificata da CDKN2A , p16, funziona come un inibitore naturale di CDK4/6 (Serranoet al., 1993), p. 4). Al contrario, altri biomarcatori associati ad una prognosi sfavorevole nel cancro al seno non sono riusciti a prevedere la sensibilità a questa combinazione di trattamento, ma sono invece correlati alla sensibilità ad altri agenti(fascicolo supplementare 8). A causa del numero limitato di farmaci testati nei PDX, abbiamo ampliato la nostra ricerca di target per includere un profilo farmacogenomico recentemente descritto delle linee cellulari tumorali e abbiamo scoperto diverse vulnerabilità aggiuntive dei biomarcatori(Figura 5A-B). Per esempio, abbiamo identificato le mutazioni in STAG2 come un biomarcatore di alta fiducia di una prognosi sfavorevole nel glioma, e abbiamo scoperto che i gliomi STAG2-mutanti sono squisitamente sensibili al trattamento con l’inibitore PARP olaparib (Figura 5A). In totale, abbiamo identificato vulnerabilità terapeutiche altamente significative per il 49% dei biomarcatori prognostici scoperti dalla nostra analisi integrata, fornendo potenziali strategie per trattare un sottoinsieme di pazienti che hanno i tumori più aggressivi.
Figura 5-figure supplement 2.Robusti biomarcatori prognostici associati alla sensibilità ai farmaci nelle linee cellulari tumorali.analisi multivariata di biomarcatori ad alta fiducia con criteri clinici standard.robusti biomarcatori prognostici associati alla sensibilità ai farmaci nelle linee cellulari tumorali.(A) Mutazioni e CNA associati all’esito del paziente in più coorti di gliomo/glioblastoma. Le mutazioni in STAG2 sono associate alla sensibilità all’inibitore PARP olaparib, mentre le delezioni CDKN2A sono associate alla sensibilità al palbociclib dell’inibitore CDK4/6 nelle linee cellulari del glioma(Iorio et al., 2016).(B) Le mutazioni e i CNA associati all’esito del paziente in coorti multiple di tumore della vescica sono visualizzati. Le mutazioni in RB1 sono associate alla sensibilità all’inibitore SYK BAY-61-3606 nelle linee cellulari del tumore della vescica (Iorioet al., 2016). L’elenco completo dei biomarcatori di alta fiducia e delle potenziali vulnerabilità è elencato nel file supplementare 8.(A-D) Trame forestali dei CNA indicati in diverse coorti in modelli Cox multivariati che includono variabili cliniche comunemente misurate. I cerchi indicano il rapporto di rischio, mentre le barre indicano il 95% di confidenza interna.(A) Viene visualizzato il numero di biomarcatori genetici di alta fiducia identificati in ciascuno dei tipi di tumore indicati. L’elenco completo dei biomarcatori è presentato nel file supplementare 8.(B) Le mutazioni e i CNA associati all’esito clinico in coorti di pazienti con tumore al seno multiplo sono visualizzati. I grafici a cascata mostrano la variazione percentuale del volume di PDX tra i PDX che ospitano le delezioni di CDKN2A, le delezioni di 17p12 o le amplificazioni di Chr20q13 trattate con gli agenti indicati(Gao et al., 2015a). LEE011 è un inibitore CDK4/CDK6, everolimus è un inibitore mTOR, BKM120 è un inibitore pan-PI3K, LJC049 è un inibitore TNKS, BYL719 è un inibitore PI3KCA, cetuximab è un inibitore EGFR, ed encorafenib è un inibitore BRAF. L’elenco completo dei biomarcatori è presentato nel file supplementare 8.
Figura 5-figure supplement 2.Robusti biomarcatori prognostici associati alla sensibilità ai farmaci nelle linee cellulari tumorali.analisi multivariata di biomarcatori ad alta fiducia con criteri clinici standard.robusti biomarcatori prognostici associati alla sensibilità ai farmaci nelle linee cellulari tumorali.(A) Mutazioni e CNA associati all’esito del paziente in più coorti di gliomo/glioblastoma. Le mutazioni in STAG2 sono associate alla sensibilità all’inibitore PARP olaparib, mentre le delezioni CDKN2A sono associate alla sensibilità al palbociclib dell’inibitore CDK4/6 nelle linee cellulari del glioma(Iorio et al., 2016).(B) Le mutazioni e i CNA associati all’esito del paziente in coorti multiple di tumore della vescica sono visualizzati. Le mutazioni in RB1 sono associate alla sensibilità all’inibitore SYK BAY-61-3606 nelle linee cellulari del tumore della vescica (Iorioet al., 2016). L’elenco completo dei biomarcatori di alta fiducia e delle potenziali vulnerabilità è elencato nel file supplementare 8.(A-D) Trame forestali dei CNA indicati in diverse coorti in modelli Cox multivariati che includono variabili cliniche comunemente misurate. I cerchi indicano il rapporto di rischio, mentre le barre indicano il 95% di confidenza interna.(A) Viene visualizzato il numero di biomarcatori genetici di alta fiducia identificati in ciascuno dei tipi di tumore indicati. L’elenco completo dei biomarcatori è presentato nel file supplementare 8.(B) Le mutazioni e i CNA associati all’esito clinico in coorti di pazienti con tumore al seno multiplo sono visualizzati. I grafici a cascata mostrano la variazione percentuale del volume di PDX tra i PDX che ospitano le delezioni di CDKN2A, le delezioni di 17p12 o le amplificazioni di Chr20q13 trattate con gli agenti indicati(Gao et al., 2015a). LEE011 è un inibitore CDK4/CDK6, everolimus è un inibitore mTOR, BKM120 è un inibitore pan-PI3K, LJC049 è un inibitore TNKS, BYL719 è un inibitore PI3KCA, cetuximab è un inibitore EGFR, ed encorafenib è un inibitore BRAF. L’elenco completo dei biomarcatori è presentato nel file supplementare 8.
Figura 5-figure supplement 1.Analisi multivariata di biomarcatori ad alta fiducia con criteri clinici standard.2.(A-D) Trame forestali dei CNA indicati in diverse coorti in modelli Cox multivariati che includono variabili cliniche comunemente misurate. I cerchi indicano il rapporto di rischio, mentre le barre indicano il 95% di confidenza interna.
Figura 5-figure supplement 2.Robusti biomarcatori prognostici associati alla sensibilità al farmaco nelle linee cellulari tumorali.(A) Viene visualizzato il numero di biomarcatori genetici ad alta fiducia identificati in ciascuno dei tipi di tumore indicati. L’elenco completo dei biomarcatori è presentato nel file supplementare 8.(B) Le mutazioni e i CNA associati all’esito clinico in coorti di pazienti con tumore al seno multiplo sono visualizzati. I grafici a cascata mostrano la variazione percentuale del volume di PDX tra i PDX che ospitano le delezioni di CDKN2A, le delezioni di 17p12 o le amplificazioni di Chr20q13 trattate con gli agenti indicati(Gao et al., 2015a). LEE011 è un inibitore CDK4/CDK6, everolimus è un inibitore mTOR, BKM120 è un inibitore pan-PI3K, LJC049 è un inibitore TNKS, BYL719 è un inibitore PI3KCA, cetuximab è un inibitore EGFR, ed encorafenib è un inibitore BRAF. L’elenco completo dei biomarcatori è presentato nel file supplementare 8.
Discussione
La medicina moderna ha notevolmente prolungato la sopravvivenza di individui con diagnosi di cancro(Johnson et al., 2017). Tuttavia, l’evidenza crescente suggerisce che ampi sottoinsiemi di pazienti ricevono cure non ottimali e sono sottoposti a un trattamento eccessivo o insufficiente rispetto al loro livello di rischio(Bhatt e Klotz, 2016; Esserman et al., 2013; Swaminathan e Swaminathan, 2015). Ad oggi, molte delle alterazioni genetiche che differenziano i tumori mortali e quelli benigni sono rimaste oscure. La nostra analisi dei biomarcatori prognostici di 17.879 pazienti fa luce su queste differenze genetiche, identifica un sottoinsieme di pazienti che possono beneficiare maggiormente di un intervento aggressivo e suggerisce strategie terapeutiche per i tumori che ospitano alcune alterazioni associate a una prognosi sfavorevole. Un portale web per facilitare l’accesso a questi risultati è disponibile all’indirizzo http://survival.cshl.edu/.
Poiché i tumori si manifestano a causa dell’accumulo di mutazioni negli oncogeni promotori della crescita e nei soppressori tumorali inibitori della crescita, ci si può aspettare che la presenza e la diversità di queste mutazioni detengano il decorso clinico di un tumore. Tuttavia, i nostri dati suggeriscono che in molti casi non è così. In letteratura esistono sostanziali disaccordi sul valore dei biomarcatori prognostici basati sulle mutazioni, in quanto gli stessi oncogeni driver sono stati segnalati in modo indipendente come caratteristiche prognostiche avverse o non significative(Guan et al., 2013; Marabese et al., 2015; Scoccianti et al., 2012; Sun et al., 2013). In questo manoscritto, abbiamo eseguito un’analisi imparziale a livello genomico di set di dati pubblici con dimensioni del campione prestabilite. Questo approccio può quindi aggirare alcuni problemi, tra cui il test di ipotesi post-hoc, il bias di selezione dei pazienti e il “problema del cassetto dei file”, che può confondere gli studi mirati sui biomarcatori (Aronson,2005; Ensor, 2014; Goossens et al., 2015; Rosenthal, 1979; Scargle, 1999). Riteniamo possibile che, con campioni di dimensioni maggiori o sottotipi tumorali più specifici, si possano identificare ulteriori mutazioni prognostiche. È importante notare che nella maggior parte delle coorti di pazienti che abbiamo raccolto, i tumori sono stati analizzati su piattaforme genomiche multiple, e i CNA erano comunemente prognostici nelle stesse coorti in cui le mutazioni geniche non lo erano. Questi risultati sottolineano la nostra capacità di individuare con successo i biomarcatori in coorti di queste dimensioni e suggeriscono che, in un confronto testa a testa, le alterazioni del numero di copie forniscono informazioni prognostiche più utili rispetto alle mutazioni di un singolo gene.
Mentre abbiamo identificato pochissime mutazioni associate all’esito del paziente, diverse linee di evidenza sottolineano i potenziali benefici di un continuo sforzo di sequenziamento clinico. In primo luogo, la nostra analisi ha rivelato un sottoinsieme di mutazioni con potere prognostico specifico dei tessuti, tra cui mutazioni TP53 nel cancro al seno, mutazioni RB1 nel cancro della vescica e mutazioni FBXW7 nel cancro colorettale. In secondo luogo, la maggior parte dei pazienti delle coorti TCGA sono stati trattati con farmaci citotossici standard. Poiché le terapie mirate e le immunoterapie sono sempre più adottate nella clinica, le mutazioni oncogenetiche non prognostiche nei set di dati qui analizzati possono essere in grado di prevedere la sensibilità a specifici agenti terapeutici(Gagan e Van Allen, 2015). In terzo luogo, i tumori stessi sono composti da popolazioni subclonali che ospitano serie distinte di mutazioni, e recenti evidenze suggeriscono che l’eterogeneità del cancro può influenzare il decorso clinico(Jamal-Hanjani et al., 2017). Quindi, interrogando lo spettro delle mutazioni a livello subclonale si possono identificare mutazioni prognostiche non distinte nelle analisi di massa.
Sebbene i cambiamenti su larga scala nella ploidia tumorale siano stati precedentemente riconosciuti come un indicatore di scarso esito(Friedlander et al., 1984; Kallioniemi et al., 1987; Kokal et al., 1986; Merkel e McGuire, 1990; Zimmerman et al., 1987), i contributi delle alterazioni del numero di copie nella maggior parte dei singoli geni sono rimasti inesplorati. Nonostante il limitato valore di stratificazione delle mutazioni nei geni guida del cancro, abbiamo scoperto che le alterazioni del numero di copie di molti di questi stessi geni sono ampiamente prognostiche. I CNA focali tendevano a conferire una prognosi peggiore di quella di un ampio CNA, in linea con un modello in cui gli squilibri di dosaggio dei geni su larga scala innescano uno stress proteotossico e impongono una penalità di fitness alle cellule tumorali(Santaguida e Amon, 2015; Sheltzer e Amon, 2011). Inoltre, mentre i CNA prognostici comunemente causavano cambiamenti proporzionali nell’espressione del gene target, la maggior parte dei CNA restavano prognostici indipendentemente dal fatto che influissero o meno sull’espressione di un gene mutato. Questi risultati supportano un modello di tumorigenesi “isola genica del cancro” o “aneuploidia cumulativa”, in cui i tumori accumulano una serie di cambiamenti limitati del numero di copie che interessano regioni aplo-sensibili e triplo-sensibili (Davoli et al.,2013; Solimini et al., 2012). L’identificazione delle conseguenze funzionali di questi CNA prognostici sulla fisiologia tumorale è un obiettivo chiave per il futuro.
I pazienti i cui tumori ospitano alterazioni genetiche che causano la mortalità hanno urgente bisogno di migliorare le opzioni di trattamento. Abbiamo scoperto molti casi in cui i biomarcatori ad alta fiducia della malattia aggressiva hanno sensibilizzato i tumori a specifiche terapie antitumorali. Approfittando di queste vulnerabilità, un approccio di precisione-medicina potrebbe essere applicato sia per stratificare il rischio del paziente sia per identificare le combinazioni di farmaci più probabili per fornire un beneficio clinico. Diverse sensibilità previste dal nostro lavoro hanno un supporto clinico o meccanicistico, tra cui l’uso di inibitori CDK4/6 per il trattamento dei tumori CDKN2A, l’uso di inibitori PARP per il trattamento dei tumori STAG2-mutanti, e l’uso di inibitori SYK per il trattamento dei tumori RB1-mutanti(Bailey et al., 2014; Gao et al., 2015b; Zhang et al., 2012). Il trattamento con agenti mirati altera significativamente il paesaggio epigenetico e genetico cellulare, spesso culminando nello sviluppo di resistenze alle terapie applicate(Holohan et al., 2013). Si ipotizza che le alterazioni secondarie che i tumori evolvono per tollerare questi farmaci potrebbero anche alterare o smussare il fenotipo aggressivo causato dall’alterazione del driver originale. In questo modo, prendere di mira un biomarcatore che conferisce una prognosi sfavorevole potrebbe sia direttamente portare a un miglioramento degli esiti dei pazienti, innescando una robusta risposta clinica, sia indirettamente aiutare i pazienti, costringendo l’evoluzione del tumore ad abbandonare la dipendenza da un driver di malattia aggressivo.
Materiali e metodi
Fonti di dati
Le coorti di pazienti analizzate in questo studio sono elencate nel file supplementare 1. Per l’analisi TCGA sono stati utilizzati i file pre-processati dal Broad Institute TCGA Firehose(https://gdac.broadinstitute.org/). Per l’analisi del numero di copie genomiche TCGA, abbiamo usato gli SCNA segmentati HG19, corretti per gli SCNA germline. Il tempo di sopravvivenza globale è stato utilizzato come endpoint clinico per tutti i tipi di cancro ad eccezione del PRAD. La sopravvivenza globale è stata scelta perché riflette un evento obiettivo e non ambiguo, è lo standard d’oro per gli studi clinici oncologici ed è ampiamente disponibile in diversi studi(Driscoll e Rixe, 2009). Tuttavia, poiché meno del 2% dei pazienti della coorte PRAD è deceduto durante il periodo di follow-up, come endpoint surrogato è stato utilizzato il termine “giorni alla ricorrenza biochimica”. Per tutti i tumori, la sopravvivenza o il tempo di follow-up dalla diagnosi sono stati corretti per i giorni al prelievo del campione. I tumori primari (indicati con un ’01’ nel codice a barre del paziente) sono stati utilizzati per ogni tipo di tumore ad eccezione della SKCM; per questo tumore, erano disponibili pochi campioni primari, per cui sono stati inclusi campioni metastatici (indicati con un ’06’ nel codice a barre) per i pazienti in cui non era disponibile un tumore primario. Per ulteriori discussioni sui campioni TCGA, vedere il testo supplementare 2. La frazione di cellule tumorali valutata in base alla patologia è stata ottenuta dai file clinici del TCGA sotto “Percent_tumor_cells”. Lo stadio e il grado del tumore sono stati ottenuti in modo simile dai file clinici TCGA appropriati.
La mutazione, il numero di copia e i dati clinici della Release 25 dell’International Genome Consortium sono stati scaricati dal Portale Dati ICGC(Zhang et al., 2011). La sopravvivenza globale è stata utilizzata come endpoint clinico per tutte le coorti ad eccezione dell’EOPC-DE; a causa dei pochi decessi in questa coorte, la sopravvivenza senza recidiva è stata utilizzata come endpoint. Le coorti sono state scelte in base alla disponibilità di dati WGS o WES, e sono state incluse se provengono da un tipo di cancro paragonabile ai tipi studiati nella nostra analisi TCGA.
Il numero di copie, la mutazione e i dati clinici di cBioportal sono stati scaricati come file pre-processati da www.cbioportal.org(Gao et al., 2013). Per i pazienti descritti in Zehir et al. (2017) (le coorti cBioportal/MSKCC_2017), sono stati inclusi solo i tumori primari per tutti i tipi di cancro ad eccezione del melanoma.
Strategia di analisi globale
Tutte le elaborazioni e le analisi sono state eseguite utilizzando Python. L’analisi del pericolo proporzionale Cox ha utilizzato il pacchetto di sopravvivenza R(https://cran.r-project.org/web/packages/survival/index.html) per calcolare i punteggi Z e i valori p. La giustificazione e ulteriori spiegazioni per l’uso della modellazione dei pericoli proporzionali Cox si trovano nel testo supplementare 1. Il progetto rpy2 è stato utilizzato per controllare R dal pitone, consentendo una perfetta integrazione dei calcoli dei punteggi Z con l’elaborazione dei dati e l’analisi del pan-cancro. I DataFrame Pandas sono stati utilizzati come struttura primaria per la memorizzazione e la manipolazione dei dati. Inoltre, sono stati utilizzati occasionalmente metodi e array nativi numpy per l’archiviazione efficiente di dati strettamente numerici, ad esempio, come input per i modelli di pericolo proporzionali Cox. Il pacchetto statsmodels(www.statsmodels.org) è stato utilizzato per la correzione delle false scoperte con la procedura Benjamini-Hochberg. Microsoft Excel è stato usato occasionalmente per l’elaborazione e l’esame finale dei dati, per cui è stato aggiunto un singolo apostrofo prima dei nomi dei geni nelle fasi intermedie di elaborazione dei dati per proteggere i geni dall’autoformattazione(Zeeberg et al., 2004).
Il codice è stato strutturato in modo da consentire la facilità di riutilizzo interno e la riproducibilità dei risultati. I metodi di analisi di Cox univariate proportional hazards, Cox univariate proportional hazards, Cox multivariate proportional hazards, Kaplan-Meier e Stouffers sono stati inseriti in una libreria di analisi, prendendo come input i dati necessari per eseguire il calcolo come array numpy o panda DataFrames.
Analisi TCGA
Oltre al codice per le analisi statistiche, il codice per l’elaborazione dei file clinici TCGA è stato inserito in una biblioteca comune. Questo approccio ha permesso di eseguire lo stesso codice per l’elaborazione dei file clinici TCGA attraverso una varietà di analisi di piattaforma, garantendo un comportamento identico per ogni piattaforma. Il codice di elaborazione clinica TCGA ha selezionato gli endpoint clinici rilevanti e i dati di approvvigionamento del campione. L’elaborazione ha tradotto i dati clinici disponibili nel formato richiesto per i modelli di pericolo proporzionali Cox: un valore di endpoint/tempo di sopravvivenza e un valore di censura per ogni paziente. In questa libreria è stato incluso anche il codice per selezionare i campioni tumorali in base al tipo di tumore.
I dati di input grezzi per l’analisi della mutazione necessitavano di un ulteriore pretrattamento prima di poter costruire i modelli di pericolo proporzionali di Cox. Questo pretrattamento comprendeva la rimozione delle intestazioni per paziente in tutto il dato e la trasposizione di alcuni dati. Per tutte le analisi che utilizzano i dati delle mutazioni TCGA, sono state escluse le mutazioni annotate come mute. I geni sono stati inclusi nelle analisi a valle solo se sono stati mutati nel 2% o più dei pazienti in una coorte di tipo tumorale.
Anche i dati di input grezzi per l’analisi del numero di copie hanno richiesto una sostanziale pre-elaborazione. I dati di input del numero di copia sono costituiti da mappe di localizzazione per paziente e per cromosoma dei numeri di copia (hg19 scaricato dal Genoma Browser UCSC; Tyner et al., 2017). Queste mappe sono state convertite in un singolo valore di numero di copia per ogni gene. Abbiamo creato un albero di intervallo (usando il pacchetto intervaltree python, https://pypi.python.org/pypi/intervaltree) delle mappe di localizzazione per ogni cromosoma e abbiamo usato l’HGNC appropriato per convertire le posizioni dei cromosomi in geni per ogni paziente. Abbiamo usato la posizione del sito di partenza trascrizionale del gene per cercare nell’albero degli intervalli il valore del numero di copia di un gene. Questa analisi ha prodotto un file intermedio di forma simile alle altre piattaforme TGCA, che ha permesso una semplice analisi Cox. Si noti che i modelli di pericolo proporzionali Cox sono un metodo indipendente dalla soglia per eseguire l’analisi di sopravvivenza, e quindi non è stata specificata alcuna soglia minima o massima per l’alterazione del numero di copie.
Un tumore è stato definito come avente un’amplificazione o una delezione focale se il suo numero di copie era superiore a 0,3 o inferiore a -0,3, e l’intervallo cromosomico con un numero di copie superiore all’80% del numero di copie al gene di interesse era inferiore o uguale a 3 Mb (Krijgsmanet al., 2014).
Per calcolare il numero di alterazioni strutturali per tumore, è stato sommato per ogni paziente il numero di valori distinti del numero di copie per cromosoma nel file di segmentazione del DNA.
Analisi TCGA pan-cancro
Per ogni piattaforma e tipo di analisi, abbiamo eseguito un’analisi pan-cancro. Questa analisi ha creato un singolo punteggio Z per ogni gene combinando i punteggi Z per ogni tipo di cancro utilizzando il metodo Stouffer. Per eseguire il metodo Stouffer, abbiamo preso la somma dei punteggi Z per un singolo gene e abbiamo diviso tale somma per la radice quadrata del numero di tipi di cancro con i punteggi Z per il gene (Stouffer, 1949). Questo punteggio meta-Z è stato poi confrontato con i punteggi meta-Z ottenuti in modo simile da altre analisi della piattaforma.
Ulteriori analisi della mutazione TCGA
Abbiamo eseguito diverse analisi aggiuntive sui dati delle mutazioni, tra cui i punteggi della combinazione di doppia mutazione Z, i punteggi del codice hotspot Z e i punteggi Z corretti per i VAF. Per i punteggi Z della doppia mutazione, abbiamo preso i 30 geni più comuni dei driver per il cancro ed eseguito combinazioni a coppie. Abbiamo poi calcolato i rischi proporzionali di Cox per ogni coppia di geni, in cui si considerava che un paziente avesse una mutazione a coppie se e solo se entrambi i geni erano stati mutati in modo non silenzioso per quel paziente. I punteggi Z sono stati calcolati per una coppia solo se (1) nessuno dei due geni della coppia era statisticamente significativo da solo nell’analisi univariata e (2) se entrambi i geni sono stati mutati insieme in almeno 10 pazienti.
I punteggi Z per codice sono stati calcolati per un insieme selezionato di codici hotspot. La maggior parte dei tipi di cancro erano disponibili in HG37, quindi le posizioni di mutazione HG37 sono stati utilizzati per individuare i codoni. Le mutazioni per OV e COADREAD erano disponibili solo in HG36, quindi le posizioni dei geni sono state convertite in HG37 prima dell’elaborazione del codone. I punteggi per il codone Z sono stati calcolati identificando prima i pazienti con mutazioni nel gene rilevante, quindi selezionando da quell’insieme di pazienti quelli le cui mutazioni erano nel codone di interesse. Se il 2% o più dei pazienti aveva mutazioni nel codone selezionato, è stato calcolato un punteggio Z.
I VAF sono stati calcolati per 10 dei tipi di cancro TCGA. Abbiamo analizzato i dati VAF in due modi. In primo luogo, abbiamo calcolato i punteggi Z, contando solo un gene come mutato se il suo VAF era maggiore o uguale a 0,4. In secondo luogo, abbiamo identificato il punteggio VAF mediano per ogni gene, e abbiamo calcolato i punteggi Z contando solo un gene come mutato se il suo VAF era uguale o superiore al VAF mediano per quel gene.
Analisi CBioPortale
CBioPortal è stato strutturato in modo simile alle analisi TCGA, anche se l’elaborazione dei dati non è stata considerata in una libreria indipendente, poiché ciascuno di questi set di dati è stato utilizzato in una sola analisi. I dati dei numeri di copia da un tipo di cancro del CBioPortal, blca_mskcc, richiedevano una pre-elaborazione iniziale nel modo descritto sopra per i numeri di copia TCGA. Le mutazioni sono state incluse se sono state annotate come uno di questi tipi: In_Frame_Ins, Nonstop_Mutation, Translation_Start_Site, In_Frame_Del, Splice_Region, Frame_Shift_Ins, Frame_Shift_Del, Splice_Site, Nonsense_Mutation, o Missense_Mutation.
Analisi ICGC
L’analisi ICGC è stata strutturata in modo simile all’analisi CBioPortal. Le mutazioni sono state incluse nelle analisi a valle solo se sono state annotate come uno di questi tipi: cancellazione dirompente dell’inframe, inserimento dirompente dell’inframe, variante frameshift, cancellazione dell’inframe, variante missense, variante accettatore di giunzioni, variante donatore di giunzioni, stop guadagnato, o stop perso. I punteggi Z sono stati calcolati se un gene è stato mutato nel 2% o più dei pazienti di una particolare coorte.
Identificazione di biomarcatori di alta fiducia associati a sensibilità ai farmaci
Attraverso set di dati indipendenti, sono state identificate coorti di pazienti con tipi di cancro correlati. Sono state determinate mutazioni o CNA significativamente associate alla prognosi del paziente (Z > 1,96 o Z < -1,96) in due o più coorti indipendenti da ciascun tipo di cancro. Poi, il sottoinsieme di queste alterazioni che sono rimaste altamente significative (Z > 3,3 o Z < -3,3) in tutte le coorti dello stesso tipo di cancro sono stati classificati come biomarcatori di alta fiducia. In alcuni casi, le amplificazioni che si estendevano a regioni cromosomiche continue sono risultate correlate alla prognosi del paziente. Questi segmenti sono stati identificati manualmente. Per la determinazione della sensibilità terapeutica descritta di seguito, il gene con il punteggio minimo di meta-Z (per le delezioni) o massimo di meta-Z (per le amplificazioni) all’interno di un segmento è stato scelto per rappresentare il segmento nel suo insieme.
I dati di sensibilità terapeutica per i PDX sono stati acquisiti da(Gao et al., 2015a). Per identificare le mutazioni correlate alla sensibilità terapeutica, per ogni farmaco o combinazione di farmaci, è stato effettuato un confronto se cinque o più PDX avevano una mutazione in un gene di interesse e se cinque o più PDX erano di tipo selvaggio per un gene di interesse. Per i geni e le terapie che rispondevano a questi criteri, abbiamo poi identificato i casi in cui la terapia ha portato a una risposta clinica nella popolazione mutante, definita come una crescita media della “migliore risposta media”<15% di crescita tumorale tra i PDX con una mutazione nel gene di interesse. Infine, per i geni e le terapie che soddisfano questi criteri, abbiamo eseguito un t-test per la ‘Miglior risposta media’ tra PDX con copie mutanti e wild-type di un gene di interesse. Abbiamo riportato terapie in cui questi criteri sono stati soddisfatti e i tumori con mutazione erano più sensibili alla terapia rispetto ai tumori con copie wild-type del gene di interesse (p < 0,01).
Per identificare i CNA correlati alla sensibilità della terapia nella coorte PDX, sono state chiamate le amplificazioni e le delezioni (CNA >|.3|), che sono state poi considerate separatamente. Come sopra, le CNA sono state incluse se cinque o più PDX mostravano un’alterazione, e se cinque o più PDX non mostravano tale alterazione. Per i geni e le terapie che si adattavano a questi criteri, abbiamo poi identificato i casi in cui la terapia risultava in una risposta clinica nella popolazione alterata, definita come una media di ‘Migliore risposta media’<15% di crescita tumorale tra i PDX con un’amplificazione o delezione nel gene di interesse. Infine, per i geni e le terapie che soddisfano questi criteri, abbiamo eseguito un t-test per la ‘Miglior risposta media’ tra PDX con copie mutanti e wild-type di un gene di interesse. Abbiamo riportato terapie in cui questi criteri sono stati soddisfatti e i tumori con una mutazione erano più sensibili alla terapia rispetto ai tumori con copie wild-type del gene di interesse (p < 0,01).
I dati sulla sensibilità terapeutica delle linee cellulari tumorali sono stati acquisiti da(Iorio et al., 2016). Per questi dati sono stati utilizzati due diversi confronti. In primo luogo, i calcoli descritti di seguito sono stati eseguiti per le linee cellulari dal tipo di cancro specifico che il biomarcatore di alta fiducia è stato identificato in. Se questa analisi non ha prodotto vulnerabilità significative, i calcoli sono stati ripetuti per tutti i tipi di cancro (pan-cancro).
Le mutazioni di alta fiducia sono state valutate se cinque o più linee cellulari nell’insieme di interesse avevano una mutazione non sinonima in quel gene, e se cinque o più linee cellulari avevano copie wild-type di quel gene. Le CNA sono state valutate se cinque o più linee cellulari avevano un’alterazione (delezione o amplificazione) di quel gene, e se cinque o più linee cellulari non avevano tale alterazione. Per ogni confronto, i test T sono stati eseguiti tra il valore log(IC50) di ogni composto testato. Per le analisi di tipo monocancro, è stata utilizzata una soglia di p < 0,01 per identificare la significatività, mentre per le analisi di tipo pan-cancro, è stata utilizzata una soglia di p < 0,0001 per identificare la significatività.
Codice
Il codice è disponibile su GitHub all’indirizzo https://github.com/joan-smith/genomic-features-survival ( Smith, 2018; copia archiviata all’indirizzo https://github.com/elifesciences-publications/genomic-features-survival).
Analisi Kaplan-Meier
Le trame di Kaplan-Meier sono state generate utilizzando il prisma del Graphpad. Le cancellazioni e le amplificazioni nei grafici Kaplan-Meier corrispondono a CNA > |0.3|; le cancellazioni profonde e i guadagni ad alta copia corrispondono a CNA > |1|. I valori P riportati nei grafici KM sono stati generati dal test log-rank in Prisma. Si noti che i grafici Kaplan-Meier sono visualizzati in questo manoscritto principalmente per la facilità di visualizzazione dei risultati dei pazienti. I punteggi Z sono stati sempre generati con la modellazione dei pericoli proporzionali Cox, che non richiede la selezione di cut-off artificiali o soglie per i dati continui.
Ulteriori fonti di dati e strumenti
I 30 geni guida del cancro frequentemente mutilati sono stati acquisiti da(Zehir et al., 2017). Le statistiche NCI-SEER sono state scaricate da https://seer.cancer.gov. I punteggi totali di aneuploidia tumorale, i valori di purezza determinati in modo ASSOLUTO e l’infiltrazione dei leucociti sono stati ottenuti da(Taylor et al., 2018). Campioni iper-mutate sono stati ottenuti da(Bailey et al., 2018). Le trame dei lecca-lecca sono state generate utilizzando il software Lollipops(Jay e Brouwer, 2016). I diagrammi di densità sono stati generati con script Python utilizzando matplotlib(https://matplotlib.org/). Le mutazioni di una singola coppia di basi sono state mappate su codici utilizzando PolyPhen-2(Adzhubei et al., 2010).
Testo supplementare 1. Modellazione dei pericoli proporzionali Cox
Sono state sviluppate diverse tecniche statistiche per eseguire analisi di sopravvivenza o di “time-to-failure” (riviste in Kleinbaum e Klein, 2012). Queste includono l’analisi Kaplan-Meier, la regressione proporzionale dei pericoli Cox, la modellazione accelerata del tempo di guasto e molte altre. In questo articolo, abbiamo scelto di applicare la regressione proporzionale dei pericoli Cox per analizzare i dati sulla sopravvivenza al cancro. Il modello Cox è rappresentato dalla seguente funzione:ht,X=h0(t)e∑i=1nβiXi
Dove t è il tempo di sopravvivenza, h(t, X) è la funzione di pericolo, h0(t) è il pericolo di base,Xi è una potenziale variabile prognostica, e β i indica la forza dell’associazione tra una variabile prognostica e la sopravvivenza. In questo modello, i pazienti hanno un rischio di morte di base, dipendente dal tempo [h0(t)], modificato da caratteristiche prognostiche indipendenti dal tempo che aumentano (β i>0) o diminuiscono (β i<0) il rischiodi morte. In questo lavoro, riportiamo i punteggi Z, che sono calcolati dividendo il coefficiente di regressione (β i)per il suo errore standard.
La modellazione dei pericoli proporzionali Cox è stata scelta per diversi motivi. In primo luogo, a differenza dell’analisi Kaplan-Meier, i modelli Cox non richiedono la selezione di una soglia o di un cut-off, quindi i dati continui come i valori di espressione genica non devono essere dicotomizzati. (Si noti che in questo manoscritto, le trame di Kaplan-Meier sono fornite a scopo di visualizzazione, ma i punteggi Z riportati sono sempre da modelli Cox). In secondo luogo, i modelli Cox possono accettare sia dati di input continui che discreti, consentendo di utilizzare questo approccio per analizzare sia le caratteristiche genomiche binarie (ad esempio, mutante vs. non mutante) che quelle continue (ad esempio, numero di copia del gene). In terzo luogo, i modelli Cox sono suscettibili sia di analisi univariate (i = 1) che multivariate (i > 1). In quarto luogo, la regressione Cox ci permette di calcolare i punteggi Z e un valore p per ogni associazione, poiché i punteggi Z rappresentano il numero di deviazioni standard dalla media di una distribuzione normale. Quinto, i punteggi Z codificano la direzionalità di un’associazione: i fattori prognostici poveri mostreranno valori di β i superiori a 0, mentre i fattori prognostici favorevoli mostreranno valori di β i inferiori a 0. Questo permette di confrontare direttamente le caratteristiche di sopravvivenza “positive” e “negative”. In sesto luogo, i punteggi Z sono utili per le meta-analisi, in quanto possono essere combinati utilizzando il Metodo Stouffer (Stouffer, 1949):Z= ∑i=1nZik
Settimo, la modellazione dei pericoli proporzionali di Cox è comunemente utilizzata sia nelle precedenti analisi di sopravvivenza a livello genomico che in numerosi studi clinici sui biomarcatori(Dhanasekaran et al., 2001; Fukuoka et al., 2011; Gentles et al., 2015; Parker et al., 2009; Wang et al., 2005), facilitando il confronto con altri sforzi di scoperta di biomarcatori.
Per verificare la normalità sottostante dei punteggi Z, abbiamo generato dei grafici qq per i valori del numero di copie geniche(Figura 1-figure supplement 2C). Le distribuzioni risultanti per le CNA erano generalmente lineari, come previsto, con spalle occasionali con punteggi Z bassi e alti. Abbiamo calcolato in modo simile i punteggi Z per tutti i geni che ospitano mutazioni di codifica in sequenza; tuttavia, abbiamo scoperto che questo ha portato a plateau intorno all’origine in più tipi di cancro. Queste aberrazioni sono state causate dal verificarsi di mutazioni rare e casuali in geni multipli che non avevano alcun potere prognostico. Per eliminare questi altipiani, abbiamo sperimentato diverse soglie per l’analisi delle mutazioni. Considerando solo le mutazioni che si sono verificate in una certa percentuale di pazienti affetti da cancro, la comparsa degli altipiani è diminuita, ma soglie elevate hanno anche eliminato dalla considerazione le mutazioni in un certo numero di fattori cancerogeni noti. Abbiamo selezionato una soglia del 2% per bilanciare il mantenimento della normalità della distribuzione del punteggio Z mantenendo anche mutazioni poco frequenti ma significative nei geni dei driver.
Si noti che in molti documenti di analisi della sopravvivenza è inclusa una fase di “selezione delle caratteristiche” per identificare un numero minimo di caratteristiche in grado di identificare accuratamente i pazienti a rischio. Abbiamo eseguito un’analisi imparziale e completa del genoma senza selezione delle caratteristiche, per generare un punteggio Z per ogni gene e per ogni tipo di caratteristica del genoma. In questo lavoro non viene applicata nessuna fase di selezione delle caratteristiche.
Testo supplementare 2. 2. Analisi di sopravvivenza nelle coorti TCGA
Le coorti di pazienti che sono state assemblate per il TCGA sono state raccolte per consentire un’analisi molecolare dei principali sottotipi di cancro riscontrati negli Stati Uniti. Sebbene siano state raccolte informazioni cliniche per quasi tutti i pazienti, queste coorti non sono state scelte specificamente per condurre studi di sopravvivenza. Riteniamo che la nostra analisi di sopravvivenza sia appropriata per diversi motivi. In primo luogo, abbiamo verificato che i tempi di sopravvivenza complessiva dei pazienti all’interno del TCGA sono altamente coerenti con i dati epidemiologici nazionali raccolti dal NCI(Figura 1-figure supplement 2D-E). In secondo luogo, abbiamo scoperto che molti biomarcatori ben consolidati hanno un’importanza prognostica nelle coorti TCGA, comprese le mutazioni IDH1 nel glioma(Figura 1-figure supplement 7), le mutazioniTP53 nel cancro al seno (Figura1-figure supplement 3), lo stadio e il grado deltumore in diversi tipi di cancro (Figura 2-figure supplement 3),e altro ancora. In terzo luogo, abbiamo convalidato i modelli di sopravvivenza che descriviamo nel TCGA in diverse coorti di pazienti indipendenti, indicando che questi non sono fenomeni specifici del TCGA(Figura 4). In quarto luogo, in un’analisi indipendente della qualità delle annotazioni cliniche nel TCGA(Liu et al., In quinto luogo, i nostri sforzi si basano su un solido lavoro che ha anche eseguito analisi di sopravvivenza su coorti TCGA, e, in alcuni casi, risultati simili convalidati dal TCGA in popolazioni di pazienti indipendenti (Andor et al.,2016; Davoli et al., 2017; Gentles et al., 2015; Guinney et al., 2015; Uhlen et al., 2017) . Infine, notiamo che il TCGA presenta diversi vantaggi rispetto agli studi di sopravvivenza standard avviati dagli sperimentatori. I campioni dei pazienti sono stati raccolti e analizzati in modo imparziale, precludendo la possibilità del “problema del cassetto di archiviazione” (mancata pubblicazione di risultati negativi) o dell’adeguamento post-hoc delle dimensioni del campione (terminando l’arruolamento del paziente quando si trova un risultato significativo). Sono disponibili un numero significativamente maggiore di dati molecolari sui tumori TCGA rispetto a qualsiasi altro set di dati di dimensioni comparabili, il che consente di effettuare analisi multivariate e correlazionali di diverse sfaccettature dei genomi tumorali. Tutti i dati del TCGA e tutto il codice di questo manoscritto sono disponibili al pubblico, consentendo una facile replicazione ed estensione su questa analisi.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature Methods. 2010; 7:248-249. DOI | PubMed
- Allsbrook WC, Mangold KA, Johnson MH, Lane RB, Lane CG, Epstein JI. Interobserver reproducibility of Gleason grading of prostatic carcinoma: general pathologist. Human Pathology. 2001; 32:81-88. DOI | PubMed
- Anaya J, Reon B, Chen WM, Bekiranov S, Dutta A. A pan-cancer analysis of prognostic genes. PeerJ. 2015; 3DOI | PubMed
- Anaya J. OncoRank: A pan-cancer method of combining survival correlations and its application to mRNAs, miRNAs, and lncRNAs. PeerJ Preprints. 2016; 4DOI
- Andor N, Graham TA, Jansen M, Xia LC, Aktipis CA, Petritsch C, Ji HP, Maley CC. Pan-cancer analysis of the extent and consequences of intratumor heterogeneity. Nature Medicine. 2016; 22:105-113. DOI | PubMed
- Aronson JK. Biomarkers and surrogate endpoints. British Journal of Clinical Pharmacology. 2005; 59:491-494. DOI | PubMed
- Bailey ML, O’Neil NJ, van Pel DM, Solomon DA, Waldman T, Hieter P. Glioblastoma cells containing mutations in the cohesin component STAG2 are sensitive to PARP inhibition. Molecular Cancer Therapeutics. 2014; 13:724-732. DOI | PubMed
- Bailey MH, Tokheim C, Porta-Pardo E, Sengupta S, Bertrand D, Weerasinghe A, Colaprico A, Wendl MC, Kim J, Reardon B, Ng PK, Jeong KJ, Cao S, Wang Z, Gao J, Gao Q, Wang F, Liu EM, Mularoni L, Rubio-Perez C, Nagarajan N, Cortés-Ciriano I, Zhou DC, Liang WW, Hess JM, Yellapantula VD, Tamborero D, Gonzalez-Perez A, Suphavilai C, Ko JY, Khurana E, Park PJ, Van Allen EM, Liang H, Lawrence MS, Godzik A, Lopez-Bigas N, Stuart J, Wheeler D, Getz G, Chen K, Lazar AJ, Mills GB, Karchin R, Ding L, MC3 Working Group, Cancer genome atlas research network. Comprehensive characterization of cancer driver genes and mutations. Cell. 2018; 173:371-385. DOI | PubMed
- Bhatt JR, Klotz L. Overtreatment in cancer – is it a problem?. Expert Opinion on Pharmacotherapy. 2016; 17:1-5. DOI | PubMed
- Bijker N, Donker M, Wesseling J, den Heeten GJ, Rutgers EJ. Is DCIS breast cancer, and how do I treat it?. Current Treatment Options in Oncology. 2013; 14:75-87. DOI | PubMed
- Bozhanov SS, Angelova SG, Krasteva ME, Markov TL, Christova SL, Gavrilov IG, Georgieva EI. Alterations in p53, BRCA1, ATM, PIK3CA, and HER2 genes and their effect in modifying clinicopathological characteristics and overall survival of Bulgarian patients with breast cancer. Journal of Cancer Research and Clinical Oncology. 2010; 136:1657-1669. DOI | PubMed
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, Beroukhim R, Pellman D, Levine DA, Lander ES, Meyerson M, Getz G. Absolute quantification of somatic DNA alterations in human cancer. Nature Biotechnology. 2012; 30:413-421. DOI | PubMed
- Ceccarelli M, Barthel FP, Malta TM, Sabedot TS, Salama SR, Murray BA, Morozova O, Newton Y, Radenbaugh A, Pagnotta SM, Anjum S, Wang J, Manyam G, Zoppoli P, Ling S, Rao AA, Grifford M, Cherniack AD, Zhang H, Poisson L, Carlotti CG, Tirapelli DP, Rao A, Mikkelsen T, Lau CC, Yung WK, Rabadan R, Huse J, Brat DJ, Lehman NL, Barnholtz-Sloan JS, Zheng S, Hess K, Rao G, Meyerson M, Beroukhim R, Cooper L, Akbani R, Wrensch M, Haussler D, Aldape KD, Laird PW, Gutmann DH, Noushmehr H, Iavarone A, Verhaak RG, TCGA Research Network. Molecular profiling reveals biologically discrete subsets and pathways of progression in Diffuse Glioma. Cell. 2016; 164:550-563. DOI | PubMed
- Connolly JL, Schnitt SJ, Wang HH, Longtine JA, Dvorak A, Dvorak HF. Holland-Frei Cancer Medicine. Decker Inc; 2003.
- Coons SW, Johnson PC, Scheithauer BW, Yates AJ, Pearl DK. Improving diagnostic accuracy and interobserver concordance in the classification and grading of primary gliomas. Cancer. 1997; 79:1381-1393. DOI | PubMed
- Cuzick J, Swanson GP, Fisher G, Brothman AR, Berney DM, Reid JE, Mesher D, Speights VO, Stankiewicz E, Foster CS, Møller H, Scardino P, Warren JD, Park J, Younus A, Flake DD, Wagner S, Gutin A, Lanchbury JS, Stone S, Transatlantic Prostate Group. Prognostic value of an RNA expression signature derived from cell cycle proliferation genes in patients with prostate cancer: a retrospective study. The Lancet Oncology. 2011; 12:245-255. DOI | PubMed
- Dancik GM, Theodorescu D. The prognostic value of cell cycle gene expression signatures in muscle invasive, high-grade bladder cancer. Bladder Cancer. 2015; 1:45-63. DOI
- Davoli T, Xu AW, Mengwasser KE, Sack LM, Yoon JC, Park PJ, Elledge SJ. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell. 2013; 155:948-962. DOI | PubMed
- Davoli T, Uno H, Wooten EC, Elledge SJ. Tumor aneuploidy correlates with markers of immune evasion and with reduced response to immunotherapy. Science. 2017; 355DOI | PubMed
- Deming SL, Nass SJ, Dickson RB, Trock BJ. C-myc amplification in breast cancer: a meta-analysis of its occurrence and prognostic relevance. British Journal of Cancer. 2000; 83:1688-1695. DOI | PubMed
- Dhanasekaran SM, Barrette TR, Ghosh D, Shah R, Varambally S, Kurachi K, Pienta KJ, Rubin MA, Chinnaiyan AM. Delineation of prognostic biomarkers in prostate cancer. Nature. 2001; 412:822-826. DOI | PubMed
- Driscoll JJ, Rixe O. Overall survival: still the gold standard: why overall survival remains the definitive end point in cancer clinical trials. Cancer journal. 2009; 15:401-405. DOI | PubMed
- Elmore JG, Longton GM, Carney PA, Geller BM, Onega T, Tosteson AN, Nelson HD, Pepe MS, Allison KH, Schnitt SJ, O’Malley FP, Weaver DL. Diagnostic concordance among pathologists interpreting breast biopsy specimens. JAMA. 2015; 313:1122-1132. DOI | PubMed
- Ensor JE. Biomarker validation: common data analysis concerns. The Oncologist. 2014; 19:886-891. DOI | PubMed
- Esserman LJ, Thompson IM, Reid B. Overdiagnosis and overtreatment in cancer: an opportunity for improvement. JAMA. 2013; 310:797-798. DOI | PubMed
- Friedlander ML, Hedley DW, Taylor IW, Russell P, Coates AS, Tattersall MH. Influence of cellular DNA content on survival in advanced ovarian cancer. Cancer Research. 1984; 44:397-400. PubMed
- Fukuoka M, Wu YL, Thongprasert S, Sunpaweravong P, Leong SS, Sriuranpong V, Chao TY, Nakagawa K, Chu DT, Saijo N, Duffield EL, Rukazenkov Y, Speake G, Jiang H, Armour AA, To KF, Yang JC, Mok TS, Y-l W, K-f T, Tsk M. Biomarker analyses and final overall survival results from a phase III, randomized, open-label, first-line study of gefitinib versus carboplatin/paclitaxel in clinically selected patients with advanced non-small-cell lung cancer in Asia (IPASS). Journal of Clinical Oncology. 2011; 29:2866-2874. DOI | PubMed
- Gagan J, Van Allen EM. Next-generation sequencing to guide cancer therapy. Genome Medicine. 2015; 7DOI | PubMed
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, Cerami E, Sander C, Schultz N. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Science Signaling. 2013; 6DOI | PubMed
- Gao H, Korn JM, Ferretti S, Monahan JE, Wang Y, Singh M, Zhang C, Schnell C, Yang G, Zhang Y, Balbin OA, Barbe S, Cai H, Casey F, Chatterjee S, Chiang DY, Chuai S, Cogan SM, Collins SD, Dammassa E, Ebel N, Embry M, Green J, Kauffmann A, Kowal C, Leary RJ, Lehar J, Liang Y, Loo A, Lorenzana E, Robert McDonald E, McLaughlin ME, Merkin J, Meyer R, Naylor TL, Patawaran M, Reddy A, Röelli C, Ruddy DA, Salangsang F, Santacroce F, Singh AP, Tang Y, Tinetto W, Tobler S, Velazquez R, Venkatesan K, Von Arx F, Wang HQ, Wang Z, Wiesmann M, Wyss D, Xu F, Bitter H, Atadja P, Lees E, Hofmann F, Li E, Keen N, Cozens R, Jensen MR, Pryer NK, Williams JA, Sellers WR. High-throughput screening using patient-derived tumor xenografts to predict clinical trial drug response. Nature Medicine. 2015a; 21:1318-1325. DOI | PubMed
- Gao J, Adams RP, Swain SM. Does CDKN2A loss predict palbociclib benefit?. Current Oncology. 2015b; 22:498-e501. DOI | PubMed
- Gentles AJ, Newman AM, Liu CL, Bratman SV, Feng W, Kim D, Nair VS, Xu Y, Khuong A, Hoang CD, Diehn M, West RB, Plevritis SK, Alizadeh AA. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nature Medicine. 2015; 21:938-945. DOI | PubMed
- Gilks CB, Oliva E, Soslow RA. Poor interobserver reproducibility in the diagnosis of high-grade endometrial carcinoma. The American Journal of Surgical Pathology. 2013; 37:874-881. DOI | PubMed
- Gonçalves E, Fragoulis A, Garcia-Alonso L, Cramer T, Saez-Rodriguez J, Beltrao P. Widespread post-transcriptional attenuation of genomic copy-number variation in cancer. Cell Systems. 2017; 5:386-398. DOI | PubMed
- Gonzalez-Angulo AM, Stemke-Hale K, Palla SL, Carey M, Agarwal R, Meric-Berstam F, Traina TA, Hudis C, Hortobagyi GN, Gerald WL, Mills GB, Hennessy BT. Androgen receptor levels and association with PIK3CA mutations and prognosis in breast cancer. Clinical Cancer Research. 2009; 15:2472-2478. DOI | PubMed
- Goossens N, Nakagawa S, Sun X, Hoshida Y. Cancer biomarker discovery and validation. Translational Cancer Research. 2015; 4:256-269. DOI | PubMed
- Guan JL, Zhong WZ, An SJ, Yang JJ, Su J, Chen ZH, Yan HH, Chen ZY, Huang ZM, Zhang XC, Nie Q, Wu YL. KRAS mutation in patients with lung cancer: a predictor for poor prognosis but not for EGFR-TKIs or chemotherapy. Annals of Surgical Oncology. 2013; 20:1381-1388. DOI | PubMed
- Guinney J, Dienstmann R, Wang X, de Reyniès A, Schlicker A, Soneson C, Marisa L, Roepman P, Nyamundanda G, Angelino P, Bot BM, Morris JS, Simon IM, Gerster S, Fessler E, De Sousa E Melo F, Missiaglia E, Ramay H, Barras D, Homicsko K, Maru D, Manyam GC, Broom B, Boige V, Perez-Villamil B, Laderas T, Salazar R, Gray JW, Hanahan D, Tabernero J, Bernards R, Friend SH, Laurent-Puig P, Medema JP, Sadanandam A, Wessels L, Delorenzi M, Kopetz S, Vermeulen L, Tejpar S. The consensus molecular subtypes of colorectal cancer. Nature Medicine. 2015; 21:1350-1356. DOI | PubMed
- Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nature Reviews Cancer. 2013; 13:714-726. DOI | PubMed
- Hutchins G, Southward K, Handley K, Magill L, Beaumont C, Stahlschmidt J, Richman S, Chambers P, Seymour M, Kerr D, Gray R, Quirke P. Value of mismatch repair, KRAS, and BRAF mutations in predicting recurrence and benefits from chemotherapy in colorectal cancer. Journal of Clinical Oncology. 2011; 29:1261-1270. DOI | PubMed
- Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Gonçalves E, Barthorpe S, Lightfoot H, Cokelaer T, Greninger P, van Dyk E, Chang H, de Silva H, Heyn H, Deng X, Egan RK, Liu Q, Mironenko T, Mitropoulos X, Richardson L, Wang J, Zhang T, Moran S, Sayols S, Soleimani M, Tamborero D, Lopez-Bigas N, Ross-Macdonald P, Esteller M, Gray NS, Haber DA, Stratton MR, Benes CH, Wessels LFA, Saez-Rodriguez J, McDermott U, Garnett MJ. A landscape of pharmacogenomic interactions in cancer. Cell. 2016; 166:740-754. DOI | PubMed
- Jamal-Hanjani M, Wilson GA, McGranahan N, Birkbak NJ, Watkins TBK, Veeriah S, Shafi S, Johnson DH, Mitter R, Rosenthal R, Salm M, Horswell S, Escudero M, Matthews N, Rowan A, Chambers T, Moore DA, Turajlic S, Xu H, Lee SM, Forster MD, Ahmad T, Hiley CT, Abbosh C, Falzon M, Borg E, Marafioti T, Lawrence D, Hayward M, Kolvekar S, Panagiotopoulos N, Janes SM, Thakrar R, Ahmed A, Blackhall F, Summers Y, Shah R, Joseph L, Quinn AM, Crosbie PA, Naidu B, Middleton G, Langman G, Trotter S, Nicolson M, Remmen H, Kerr K, Chetty M, Gomersall L, Fennell DA, Nakas A, Rathinam S, Anand G, Khan S, Russell P, Ezhil V, Ismail B, Irvin-Sellers M, Prakash V, Lester JF, Kornaszewska M, Attanoos R, Adams H, Davies H, Dentro S, Taniere P, O’Sullivan B, Lowe HL, Hartley JA, Iles N, Bell H, Ngai Y, Shaw JA, Herrero J, Szallasi Z, Schwarz RF, Stewart A, Quezada SA, Le Quesne J, Van Loo P, Dive C, Hackshaw A, Swanton C, TRACERx Consortium. Tracking the evolution of non-small-cell lung cancer. New England Journal of Medicine. 2017; 376:2109-2121. DOI | PubMed
- Jay JJ, Brouwer C. Lollipops in the clinic: information dense mutation plots for precision medicine. PLoS One. 2016; 11DOI | PubMed
- Johnson SB, Gross CP, Park HS, Yu JB. Use of alternative medicine for cancer and its impact on survival. Journal of Clinical Oncology. 2017; 35DOI
- Kallioniemi OP, Hietanen T, Mattila J, Lehtinen M, Lauslahti K, Koivula T. Aneuploid DNA content and high S-phase fraction of tumour cells are related to poor prognosis in patients with primary breast cancer. European Journal of Cancer and Clinical Oncology. 1987; 23:277-282. DOI | PubMed
- Kannan K, Inagaki A, Silber J, Gorovets D, Zhang J, Kastenhuber ER, Heguy A, Petrini JH, Chan TA, Huse JT. Whole-exome sequencing identifies ATRX mutation as a key molecular determinant in lower-grade glioma. Oncotarget. 2012; 3:1194-1203. DOI | PubMed
- Kleinbaum DG, Klein M. Statistics for Biology and Health. Springer-Verlag: New York; 2012.
- Kokal W, Sheibani K, Terz J, Harada JR. Tumor DNA content in the prognosis of colorectal carcinoma. JAMA: The Journal of the American Medical Association. 1986; 255:3123-3127. DOI | PubMed
- Krijgsman O, Carvalho B, Meijer GA, Steenbergen RDM, Ylstra B. Focal chromosomal copy number aberrations in cancer—Needles in a genome haystack. Biochimica et Biophysica Acta (BBA) – Molecular Cell Research. 2014; 1843:2698-2704. DOI | PubMed
- Li SY, Rong M, Grieu F, Iacopetta B. PIK3CA mutations in breast cancer are associated with poor outcome. Breast Cancer Research and Treatment. 2006; 96:91-95. DOI | PubMed
- Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV, Omberg L, Wolf DM, Shriver CD, Thorsson V, Hu H, Jianfang L, Caesar-Johnson SJ, Demchok JA, Felau I, Kasapi M, Ferguson ML, Hutter CM, Sofia HJ, Tarnuzzer R, Wang Z, Yang L, Zenklusen JC, Zhang J, Cancer Genome Atlas Research Network. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018; 173:400-416. DOI | PubMed
- Mäkelä JT, Laitinen SO, Kairaluoma MI. Five-year follow-up after radical surgery for colorectal cancer. Results of a prospective randomized trial. Archives of Surgery. 1995; 130:1062-1067. PubMed
- Marabese M, Ganzinelli M, Garassino MC, Shepherd FA, Piva S, Caiola E, Macerelli M, Bettini A, Lauricella C, Floriani I, Farina G, Longo F, Bonomi L, Fabbri MA, Veronese S, Marsoni S, Broggini M, Rulli E. KRAS mutations affect prognosis of non-small-cell lung cancer patients treated with first-line platinum containing chemotherapy. Oncotarget. 2015; 6:34014-34022. DOI | PubMed
- Merkel DE, McGuire WL. Ploidy, proliferative activity and prognosis. DNA flow cytometry of solid tumors. Cancer. 1990; 65:1194-1205. DOI | PubMed
- Mosley JD, Keri RA. Cell cycle correlated genes dictate the prognostic power of breast cancer gene lists. BMC Medical Genomics. 2008; 1DOI | PubMed
- Nalejska E, Mączyńska E, Lewandowska MA. Prognostic and predictive biomarkers: tools in personalized oncology. Molecular Diagnosis & Therapy. 2014; 18:273-284. DOI | PubMed
- Nofech-Mozes S, Spayne J, Rakovitch E, Hanna W. Prognostic and predictive molecular markers in DCIS: a review. Advances in Anatomic Pathology. 2005; 12:256-264. DOI | PubMed
- Oshiro C, Kagara N, Naoi Y, Shimoda M, Shimomura A, Maruyama N, Shimazu K, Kim SJ, Noguchi S. PIK3CA mutations in serum DNA are predictive of recurrence in primary breast cancer patients. Breast Cancer Research and Treatment. 2015; 150:299-307. DOI | PubMed
- Paez JG, Jänne PA, Lee JC, Tracy S, Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, Naoki K, Sasaki H, Fujii Y, Eck MJ, Sellers WR, Johnson BE, Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science. 2004; 304:1497-1500. DOI | PubMed
- Pang B, Cheng S, Sun SP, An C, Liu ZY, Feng X, Liu GJ. Prognostic role of PIK3CA mutations and their association with hormone receptor expression in breast cancer: a meta-analysis. Scientific Reports. 2014; 4DOI | PubMed
- Parker JS, Mullins M, Cheang MC, Leung S, Voduc D, Vickery T, Davies S, Fauron C, He X, Hu Z, Quackenbush JF, Stijleman IJ, Palazzo J, Marron JS, Nobel AB, Mardis E, Nielsen TO, Ellis MJ, Perou CM, Bernard PS. Supervised risk predictor of breast cancer based on intrinsic subtypes. Journal of Clinical Oncology. 2009; 27:1160-1167. DOI | PubMed
- Petitjean A, Achatz MI, Borresen-Dale AL, Hainaut P, Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007; 26:2157-2165. DOI | PubMed
- Pollack JR, Sørlie T, Perou CM, Rees CA, Jeffrey SS, Lonning PE, Tibshirani R, Botstein D, Børresen-Dale AL, Brown PO. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. PNAS. 2002; 99:12963-12968. DOI | PubMed
- Richman SD, Seymour MT, Chambers P, Elliott F, Daly CL, Meade AM, Taylor G, Barrett JH, Quirke P. KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: results from the MRC FOCUS trial. Journal of Clinical Oncology. 2009; 27:5931-5937. DOI | PubMed
- Rosenthal R. The file drawer problem and tolerance for null results. Psychological Bulletin. 1979; 86:638-641. DOI
- Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D, Dietrich D, Biesmans B, Bodoky G, Barone C, Aranda E, Nordlinger B, Cisar L, Labianca R, Cunningham D, Van Cutsem E, Bosman F. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. Journal of Clinical Oncology. 2010; 28:466-474. DOI | PubMed
- Roy DM, Walsh LA, Desrichard A, Huse JT, Wu W, Gao J, Bose P, Lee W, Chan TA. Integrated genomics for pinpointing survival loci within arm-level somatic copy number alterations. Cancer Cell. 2016; 29:737-750. DOI | PubMed
- Santaguida S, Amon A. Short- and long-term effects of chromosome mis-segregation and aneuploidy. Nature Reviews Molecular Cell Biology. 2015; 16:473-485. DOI | PubMed
- Scargle JD. Publication Bias (The “File-Drawer Problem”) in Scientific Inference. arXiv. 1999. Publisher Full Text
- Scoccianti C, Vesin A, Martel G, Olivier M, Brambilla E, Timsit JF, Tavecchio L, Brambilla C, Field JK, Hainaut P, European Early Lung Cancer Consortium. Prognostic value of TP53, KRAS and EGFR mutations in nonsmall cell lung cancer: the EUELC cohort. European Respiratory Journal. 2012; 40:177-184. DOI | PubMed
- Serrano M, Hannon GJ, Beach D. A new regulatory motif in cell-cycle control causing specific inhibition of cyclin D/CDK4. Nature. 1993; 366:704-707. DOI | PubMed
- Sheltzer JM, Amon A. The aneuploidy paradox: costs and benefits of an incorrect karyotype. Trends in Genetics. 2011; 27:446-453. DOI | PubMed
- Sheltzer JM, Ko JH, Replogle JM, Habibe Burgos NC, Chung ES, Meehl CM, Sayles NM, Passerini V, Storchova Z, Amon A, Jh K. Single-chromosome gains commonly function as tumor suppressors. Cancer Cell. 2017; 31:240-255. DOI | PubMed
- Shi J, Yao D, Liu W, Wang N, Lv H, Zhang G, Ji M, Xu L, He N, Shi B, Hou P. Highly frequent PIK3CA amplification is associated with poor prognosis in gastric cancer. BMC Cancer. 2012; 12DOI | PubMed
- Sholl LM, Do K, Shivdasani P, Cerami E, Dubuc AM, Kuo FC, Garcia EP, Jia Y, Davineni P, Abo RP, Pugh TJ, van Hummelen P, Thorner AR, Ducar M, Berger AH, Nishino M, Janeway KA, Church A, Harris M, Ritterhouse LL, Campbell JD, Rojas-Rudilla V, Ligon AH, Ramkissoon S, Cleary JM, Matulonis U, Oxnard GR, Chao R, Tassell V, Christensen J, Hahn WC, Kantoff PW, Kwiatkowski DJ, Johnson BE, Meyerson M, Garraway LA, Shapiro GI, Rollins BJ, Lindeman NI, MacConaill LE. Institutional implementation of clinical tumor profiling on an unselected cancer population. JCI Insight. 2016; 1:e87062. DOI | PubMed
- Smith J. GitHub. 2018. Publisher Full Text
- Solimini NL, Xu Q, Mermel CH, Liang AC, Schlabach MR, Luo J, Burrows AE, Anselmo AN, Bredemeyer AL, Li MZ, Beroukhim R, Meyerson M, Elledge SJ. Recurrent hemizygous deletions in cancers may optimize proliferative potential. Science. 2012; 337:104-109. DOI | PubMed
- Srividya MR, Thota B, Shailaja BC, Arivazhagan A, Thennarasu K, Chandramouli BA, Hegde AS, Santosh V. Homozygous 10q23/PTEN deletion and its impact on outcome in glioblastoma: a prospective translational study on a uniformly treated cohort of adult patients. Neuropathology. 2011; 31:376-383. DOI | PubMed
- Stingele S, Stoehr G, Peplowska K, Cox J, Mann M, Storchova Z. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Molecular Systems Biology. 2012; 8DOI | PubMed
- Stouffer SA. The American Soldier. Princeton University Press; 1949.
- Sun JM, Hwang DW, Ahn JS, Ahn MJ, Park K. Prognostic and predictive value of KRAS mutations in advanced non-small cell lung cancer. PLoS One. 2013; 8DOI | PubMed
- Suzuki H, Aoki K, Chiba K, Sato Y, Shiozawa Y, Shiraishi Y, Shimamura T, Niida A, Motomura K, Ohka F, Yamamoto T, Tanahashi K, Ranjit M, Wakabayashi T, Yoshizato T, Kataoka K, Yoshida K, Nagata Y, Sato-Otsubo A, Tanaka H, Sanada M, Kondo Y, Nakamura H, Mizoguchi M, Abe T, Muragaki Y, Watanabe R, Ito I, Miyano S, Natsume A, Ogawa S. Mutational landscape and clonal architecture in grade II and III gliomas. Nature Genetics. 2015; 47:458-468. DOI | PubMed
- Swaminathan D, Swaminathan V. Geriatric oncology: problems with under-treatment within this population. Cancer Biology & Medicine. 2015; 12:275-283. DOI | PubMed
- Taylor AM, Shih J, Ha G, Gao GF, Zhang X, Berger AC, Schumacher SE, Wang C, Hu H, Liu J, Lazar AJ, Cherniack AD, Beroukhim R, Meyerson M. Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell. 2018; 33:676-689. DOI | PubMed
- Tol J, Nagtegaal ID, Punt CJ. BRAF mutation in metastatic colorectal cancer. New England Journal of Medicine. 2009; 361:98-99. DOI | PubMed
- Tyner C, Barber GP, Casper J, Clawson H, Diekhans M, Eisenhart C, Fischer CM, Gibson D, Gonzalez JN, Guruvadoo L, Haeussler M, Heitner S, Hinrichs AS, Karolchik D, Lee BT, Lee CM, Nejad P, Raney BJ, Rosenbloom KR, Speir ML, Villarreal C, Vivian J, Zweig AS, Haussler D, Kuhn RM, Kent WJ. The UCSC genome browser database: 2017 update. Nucleic acids research. 2017; 45:D626-D634. DOI | PubMed
- Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, Sanli K, von Feilitzen K, Oksvold P, Lundberg E, Hober S, Nilsson P, Mattsson J, Schwenk JM, Brunnström H, Glimelius B, Sjöblom T, Edqvist PH, Djureinovic D, Micke P, Lindskog C, Mardinoglu A, Ponten F. A pathology atlas of the human cancer transcriptome. Science. 2017; 357DOI | PubMed
- van den Berge M, Sijen T. A male and female RNA marker to infer sex in forensic analysis. Forensic Science International: Genetics. 2017; 26:70-76. DOI | PubMed
- Venet D, Dumont JE, Detours V. Most random gene expression signatures are significantly associated with breast cancer outcome. PLoS Computational Biology. 2011; 7DOI | PubMed
- Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu J, Jatkoe T, Berns EM, Atkins D, Foekens JA. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. The Lancet. 2005; 365:671-679. DOI | PubMed
- Wang L, Hurley DG, Watkins W, Araki H, Tamada Y, Muthukaruppan A, Ranjard L, Derkac E, Imoto S, Miyano S, Crampin EJ, Print CG. Cell cycle gene networks are associated with melanoma prognosis. PLoS One. 2012; 7DOI | PubMed
- Williams BR, Prabhu VR, Hunter KE, Glazier CM, Whittaker CA, Housman DE, Amon A. Aneuploidy affects proliferation and spontaneous immortalization in mammalian cells. Science. 2008; 322:703-709. DOI | PubMed
- Wistuba II, Behrens C, Lombardi F, Wagner S, Fujimoto J, Raso MG, Spaggiari L, Galetta D, Riley R, Hughes E, Reid J, Sangale Z, Swisher SG, Kalhor N, Moran CA, Gutin A, Lanchbury JS, Barberis M, Kim ES. Validation of a proliferation-based expression signature as prognostic marker in early stage lung adenocarcinoma. Clinical Cancer Research. 2013; 19:6261-6271. DOI | PubMed
- Young RC. Early-stage ovarian cancer: to treat or not to treat. JNCI Journal of the National Cancer Institute. 2003; 95:94-95. DOI | PubMed
- Zaniboni A, Labianca R, Gruppo Italiano per lo Studio e la Cura dei Tumori del Digerente. Adjuvant therapy for stage II colon cancer: an elephant in the living room?. Annals of Oncology. 2004; 15:1310-1318. DOI | PubMed
- Zeeberg BR, Riss J, Kane DW, Bussey KJ, Uchio E, Linehan WM, Barrett JC, Weinstein JN. Mistaken identifiers: gene name errors can be introduced inadvertently when using Excel in bioinformatics. BMC Bioinformatics. 2004; 5DOI | PubMed
- Zehir A, Benayed R, Shah RH, Syed A, Middha S, Kim HR, Srinivasan P, Gao J, Chakravarty D, Devlin SM, Hellmann MD, Barron DA, Schram AM, Hameed M, Dogan S, Ross DS, Hechtman JF, DeLair DF, Yao J, Mandelker DL, Cheng DT, Chandramohan R, Mohanty AS, Ptashkin RN, Jayakumaran G, Prasad M, Syed MH, Rema AB, Liu ZY, Nafa K, Borsu L, Sadowska J, Casanova J, Bacares R, Kiecka IJ, Razumova A, Son JB, Stewart L, Baldi T, Mullaney KA, Al-Ahmadie H, Vakiani E, Abeshouse AA, Penson AV, Jonsson P, Camacho N, Chang MT, Won HH, Gross BE, Kundra R, Heins ZJ, Chen HW, Phillips S, Zhang H, Wang J, Ochoa A, Wills J, Eubank M, Thomas SB, Gardos SM, Reales DN, Galle J, Durany R, Cambria R, Abida W, Cercek A, Feldman DR, Gounder MM, Hakimi AA, Harding JJ, Iyer G, Janjigian YY, Jordan EJ, Kelly CM, Lowery MA, Morris LGT, Omuro AM, Raj N, Razavi P, Shoushtari AN, Shukla N, Soumerai TE, Varghese AM, Yaeger R, Coleman J, Bochner B, Riely GJ, Saltz LB, Scher HI, Sabbatini PJ, Robson ME, Klimstra DS, Taylor BS, Baselga J, Schultz N, Hyman DM, Arcila ME, Solit DB, Ladanyi M, Berger MF. Mutational landscape of metastatic cancer revealed from prospective clinical sequencing of 10,000 patients. Nature Medicine. 2017; 23:703-713. DOI | PubMed
- Zhang J, Baran J, Cros A, Guberman JM, Haider S, Hsu J, Liang Y, Rivkin E, Wang J, Whitty B, Wong-Erasmus M, Yao L, Kasprzyk A. International cancer genome consortium data portal–a one-stop shop for cancer genomics data. Database. 2011; 2011:bar026. DOI | PubMed
- Zhang J, Benavente CA, McEvoy J, Flores-Otero J, Ding L, Chen X, Ulyanov A, Wu G, Wilson M, Wang J, Brennan R, Rusch M, Manning AL, Ma J, Easton J, Shurtleff S, Mullighan C, Pounds S, Mukatira S, Gupta P, Neale G, Zhao D, Lu C, Fulton RS, Fulton LL, Hong X, Dooling DJ, Ochoa K, Naeve C, Dyson NJ, Mardis ER, Bahrami A, Ellison D, Wilson RK, Downing JR, Dyer MA. A novel retinoblastoma therapy from genomic and epigenetic analyses. Nature. 2012; 481:329-334. DOI | PubMed
- Zimmerman PV, Hawson GA, Bint MH, Parsons PG. Ploidy as a prognostic determinant in surgically treated lung cancer. The Lancet. 1987; 2:530-533. DOI | PubMed
Fonte
Smith JC, Sheltzer JM, Settleman J, Settleman J () Systematic identification of mutations and copy number alterations associated with cancer patient prognosis. eLife 7e39217. https://doi.org/10.7554/eLife.39217




