
Abstract
Introduzione
I vertebrati hanno sviluppato una vasta gamma di colori e modelli di colore epidermici, spesso in risposta alla selezione naturale, sessuale e artificiale. Numerosi studi hanno identificato geni chiave che determinano la variazione dei tipi di pigmenti prodotti dai melanociti (ad esempio, Hubbard et al., 2010; Manceau et al., 2010; Roulin e Ducrest, 2013; Domyan et al., 2014; Rosenblum et al., 2014). Al contrario, si conoscono molto meno i meccanismi genetici che determinano il pattern dei pigmenti in tutta l’epidermide e all’interno delle singole appendici epidermiche (ad esempio, piume, squame e peli)(Kelsh, 2004; Protas e Patel, 2008; Kelsh et al., 2009; Lin et al., 2009; Kaelin et al., 2012; Lin et al., 2013; Eom et al., 2015; Poelstra et al., 2015; Mallarino et al., 2016). Negli uccelli, i modelli di colore sono sorprendentemente diversi tra le diverse popolazioni e specie, e questi tratti hanno un profondo impatto sulla scelta del compagno, sulla cripsi e sulla comunicazione(Hill e McGraw, 2006).
Il piccione di roccia domestico(Columba livia) mostra un’enorme diversità fenotipica tra oltre 350 razze, compresa un’ampia varietà di modelli di pigmentazione del piumaggio che variano anche all’interno delle razze(Shapiro e Domyan, 2013; Domyan e Shapiro, 2017). Alcuni di questi fenotipi di pattern si trovano anche nelle popolazioni selvatiche e selvatiche(Johnston e Janiga, 1995). Un gran numero di loci genetici contribuiscono alla variazione del modello nei piccioni viaggiatori, compresi i geni che contribuiscono in modo additivo e altri che mascherano epistaticamente gli effetti di altri loci(Van Hoosen Jones, 1922; Hollander, 1937; Sell, 2012; Domyan et al., 2014). Nonostante la complessità genetica dell’intero spettro della diversità dei pattern di piumaggio dei piccioni, gli esperimenti genetici classici dimostrano che i principali fenotipi di pigmentazione dello scudo alare sono determinati da una serie allelica in un singolo locus(C, per il pattern ‘checker’) che produce quattro fenotipi: T-check(CT allele, detto anche T-pattern), checker(C), bar(+), e barless(c), in ordine decrescente di dominanza e melanismo(Figura 1A)(Bonhote e Smalley, 1911; Hollander, 1938a, 1983b; Levi, 1986; Sell, 2012). Il bar è il fenotipo ancestrale(Darwin, 1859; Darwin, 1868), ma il checker e il T-check possono verificarsi a frequenze più alte rispetto al bar nelle popolazioni selvatiche urbane, suggerendo un vantaggio in termini di fitness nelle aree a densità abitativa umana(Goodwin, 1952; Obukhova e Kreslavskii, 1984; Johnston e Janiga, 1995; Čanády e Mošanský, 2013).
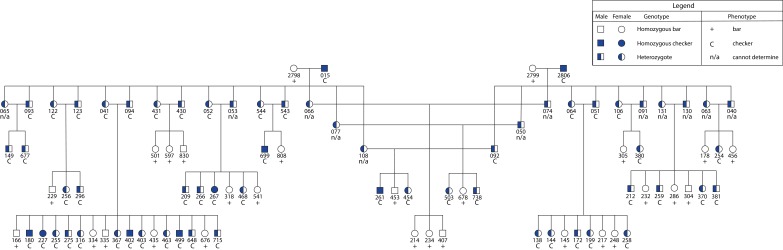
Figura 1-figure supplement 4.Una singola regione genomica è associata al pattern di pigmentazione delle ali dei piccioni viaggiatori(C. livia).Variazione del pattern di colore dello scudo alare tra i piccioni con alleli a scacchiera nella regione candidata di Scaffold 68.Confronto del genoma intero pFst per identificare una regione genomica candidata differenziata tra uccelli con diversi fenotipi di pattern alare.sequenze di aminoacidi EFHC2 di piccioni e altri amnioti (residui 525-604). I genotipi del locusC si separano con il fenotipo in un incrocio F2.(A) Quattro fenotipi classici di pigmentazione alare, mostrati in ordine decrescente di dominanza genetica e melanismo (da sinistra a destra): T-check, checker, bar e barless. Foto per gentile concessione del Genetics Science Learning Center(http://learn.genetics.utah.edu/content/pigeons).(B) Confronto tra i genomi dei piccioni di bar (n = 17) e di checker (n = 24). La linea rossa tratteggiata segna la soglia di significato a livello genomico (9,72e-10).(C) Il dettaglio del picco pFst mostra la regione di alta differenziazione su Scaffold 68. Cinque geni all’interno della regione sono mostrati in rosso. L’ombreggiatura blu segna la posizione del più piccolo aplotipo condiviso comune a tutti gli uccelli di controllo e di controllo T. L’omozigosi dell’aplotipo nella regione candidata si estende ulteriormente per gli uccelli checker e T-check (traccia blu) che per gli uccelli bar (grigio), una firma di selezione positiva per gli alleli derivati. L’omozigosi aplotipica estesa (EHHH) è stata misurata dalla posizione focale 1.751.072 secondo il metodo di Sabeti et al. (2007).(A-F) Gli uccelli rappresentativi sono visualizzati in un gradiente da meno (A)a più (F) pigmentati. Fila superiore: uccelli con le ali piegate ai loro lati in vista laterale sinistra. Fila centrale: l’ala destra è estesa per una visione più completa della pigmentazione del piumaggio. Fila inferiore: piume individuali da scudi alare. Le immagini sono state regolate per l’esposizione, il bilanciamento del bianco e la chiarezza utilizzando il plugin Adobe Camera Raw per Adobe Photoshop CC 2017 (Adobe Systems, San Jose, CA). Le barre di scala nella fila inferiore sono state inserite digitalmente.(A) Genoma intero pFst che confronta 32 bar e 27 checker e T-check uccelli.(B) Genoma intero pFst a confronto di 32 bar e nove uccelli senza barra.I residui di amminoacidi variabili sono marcati in magenta (residui simili) e verde (residui diversi). I checker C. livia, C. rupestris e C. guinea condividono 572C mentre la barra C. livia è fissa per 572Y (punta della freccia sinistra). Checker C . livia e C. guinea sono polimorfici per 584 hr/Y (punta di freccia a destra).Il pedigree raffigura un incrocio con quattro fondatori che separa la barra e la pedina nella generazione F2. I quadrati (maschio) e i cerchi (femmina) sono codificati per colore per il genotipo (vedi leggenda). Il fenotipo è riportato sotto il simbolo per ogni individuo (vedi leggenda). I numeri corrispondono agli identificatori individuali per ogni uccello.
La variazione del modello di colore è associata a diversi importanti tratti della storia della vita nelle popolazioni di piccioni selvatici. Ad esempio, gli uccelli checker e T-checker hanno una maggiore frequenza di fughe dal nido, stagioni riproduttive più lunghe (fino a tutto l’anno) e possono sequestrare metalli pesanti più tossici nei pigmenti del piumaggio attraverso la chelazione(Petersen e Williamson, 1949; Lofts et al., 1966; Murton et al., 1973; Janiga, 1991; Chatelain et al., 2014; 2016). Rispetto agli uccelli da bar, da dama e da T-check hanno anche una ridotta conservazione dei grassi e, forse di conseguenza, tassi di sopravvivenza degli adulti che svernano più bassi in ambienti rurali difficili(Petersen e Williamson, 1949a; Jacquin et al., 2012). Le femmine di piccioni preferiscono i compagni di dama ai bar, quindi la selezione sessuale probabilmente influenza anche le frequenze dei pattern di pigmentazione delle ali nelle popolazioni selvatiche(Burley, 1977; 1981; Johnston e Johnson, 1989). Al contrario, senza barra, il fenotipo recessivo e meno melanico, è raramente osservato nei piccioni selvatici(Johnston e Janiga, 1995). Nelle popolazioni domestiche, gli uccelli senza barra hanno una maggiore frequenza di difetti visivi, talvolta definiti “nebbiosi” (Hollandere Miller, 1981; Hollander , 1983b; Mangile, 1987), che potrebbero avere un impatto negativo sul fitness in natura.
In questo studio, indaghiamo la base molecolare e la storia evolutiva che sta alla base della diversità del modello alare nei piccioni. Scopriamo sia la codifica e la variazione regolatoria in un singolo gene candidato, sia un polimorfismo legato alla variazione del pattern all’interno e tra le specie che probabilmente è il risultato dell’ibridazione tra specie.
Figura 1-figure supplement 4.Figura 1—supplemento alla figura 4. Una singola regione genomica è associata al pattern di pigmentazione delle ali del piccione di roccia(C. livia).Variazione del pattern di colore dello scudo alare tra i piccioni con alleli a scacchiera nella regione candidata di Scaffold 68.Confronti di pFst del genoma intero del genoma per identificare una regione genomica candidata differenziata tra uccelli con diversi fenotipi di pattern alare.sequenze amminoacidiche EFHC2 di piccioni e altri amnioti (residui 525-604). I genotipi del locusC si separano con il fenotipo in un incrocio F2.(A) Quattro fenotipi classici di pigmentazione alare, mostrati in ordine decrescente di dominanza genetica e melanismo (da sinistra a destra): T-check, checker, bar e barless. Foto per gentile concessione del Genetics Science Learning Center(http://learn.genetics.utah.edu/content/pigeons).(B) Confronto tra i genomi dei piccioni di bar (n = 17) e di checker (n = 24). La linea rossa tratteggiata segna la soglia di significato a livello genomico (9,72e-10).(C) Il dettaglio del picco pFst mostra la regione di alta differenziazione su Scaffold 68. Cinque geni all’interno della regione sono mostrati in rosso. L’ombreggiatura blu segna la posizione del più piccolo aplotipo condiviso comune a tutti gli uccelli di controllo e di controllo T. L’omozigosi dell’aplotipo nella regione candidata si estende ulteriormente per gli uccelli checker e T-check (traccia blu) che per gli uccelli bar (grigio), una firma di selezione positiva per gli alleli derivati. L’omozigosi aplotipica estesa (EHHH) è stata misurata dalla posizione focale 1.751.072 secondo il metodo di Sabeti et al. (2007).(A-F) Gli uccelli rappresentativi sono visualizzati in un gradiente da meno (A)a più (F) pigmentati. Fila superiore: uccelli con le ali piegate ai loro lati in vista laterale sinistra. Fila centrale: l’ala destra è estesa per una visione più completa della pigmentazione del piumaggio. Fila inferiore: piume individuali da scudi alare. Le immagini sono state regolate per l’esposizione, il bilanciamento del bianco e la chiarezza utilizzando il plugin Adobe Camera Raw per Adobe Photoshop CC 2017 (Adobe Systems, San Jose, CA). Le barre di scala nella fila inferiore sono state inserite digitalmente.(A) Genoma intero pFst che confronta 32 bar e 27 checker e T-check uccelli.(B) Genoma intero pFst a confronto di 32 bar e nove uccelli senza barra.I residui di amminoacidi variabili sono marcati in magenta (residui simili) e verde (residui diversi). I checker C. livia, C. rupestris e C. guinea condividono 572C mentre la barra C. livia è fissa per 572Y (punta della freccia sinistra). Checker C . livia e C. guinea sono polimorfici per 584 hr/Y (punta di freccia a destra).Il pedigree raffigura un incrocio con quattro fondatori che separa la barra e la pedina nella generazione F2. I quadrati (maschio) e i cerchi (femmina) sono codificati per colore per il genotipo (vedi leggenda). Il fenotipo è riportato sotto il simbolo per ogni individuo (vedi leggenda). I numeri corrispondono agli identificatori individuali per ogni uccello.

Figura 1-figure supplement 1.Variazione del modello di colore dello scudo alare tra i piccioni con alleli a scacchiera nella regione candidata di Scaffold 68.(A-F) Gli uccelli rappresentativi sono visualizzati in un gradiente da meno (A)a più (F) pigmentato. Fila superiore: uccelli con le ali piegate ai loro lati in vista laterale sinistra. Fila centrale: l’ala destra è estesa per una visione più completa della pigmentazione del piumaggio. Fila inferiore: piume individuali da scudi alare. Le immagini sono state regolate per l’esposizione, il bilanciamento del bianco e la chiarezza utilizzando il plugin Adobe Camera Raw per Adobe Photoshop CC 2017 (Adobe Systems, San Jose, CA). Le barre di scala nella fila inferiore sono state inserite digitalmente.
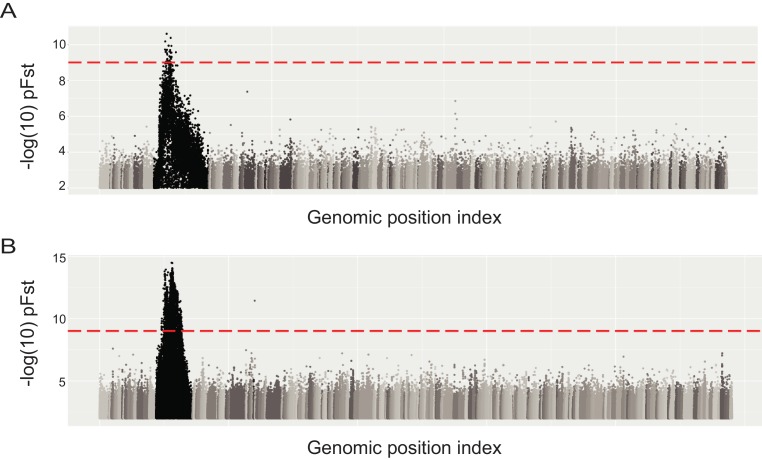
Figura 1-figure supplement 2.Figura 1—supplemento di figura 2. Confronti pFst del genoma intero per identificare una regione genomica candidata differenziata tra uccelli con diversi fenotipi di modello alare.(A) Genoma intero pFst genoma confrontando 32 bar e 27 uccelli di controllo e T-checker e T-check uccelli.(B) Genoma intero pFst a confronto di 32 bar e nove uccelli senza bar.

Figura 1-figure supplement 3.EFHC2 sequenze di aminoacidi di piccioni e altri amnioti (residui 525-604).I residui di amminoacidi variabili sono marcati in magenta (residui simili) e verde (residui diversi). Il checker C. livia, C. rupestris e C. guinea condividono 572C mentre la barra C. livia è fissa per 572Y (punta della freccia sinistra). Checker C . livia e C. gu inea sono polimorfici per 584 hr/Y (punta di freccia a destra).
Figura 1-figure supplement 4.I genotipi del locusC si separano con il fenotipo in un incrocio F2.Il pedigree rappresenta un incrocio con quattro fondatori che separa bar e checker nella generazione F2. I quadrati (maschio) e i cerchi (femmina) sono codificati per colore per il genotipo (vedi leggenda). Il fenotipo è riportato sotto il simbolo per ogni individuo (vedi leggenda). I numeri corrispondono agli identificatori individuali per ogni uccello.
Risultati e discussione
Una regione genomica su Scaffold 68 è associata al fenotipo del modello alare
Per identificare la regione genomica contenente il locus del pattern di pigmentazione alare principale, abbiamo utilizzato una misura probabilistica di differenziazione della frequenza allelica (pFst; Domyan et al., 2016) per confrontare i genomi risquenziati dei piccioni da barra con i genomi dei piccioni con pattern a scacchiera o a T (Figura 1A). Gli uccelli a scacchiera e quelli a T sono stati raggruppati insieme perché questi due modelli sono a volte difficili da distinguere, anche per gli hobbisti esperti. Gli uccelli di controllo sono tipicamente meno pigmentati degli uccelli di controllo a T, ma i modificatori genetici dei fenotipi dei modelli possono minimizzare questa differenza (vedi Figura 1-figure supplement 1 per esempi di variazione). Una scansione in due fasi del genoma intero (vedi Materiali e metodi; Figura 1B e C, Figura 1-figure supplement 2) ha identificato una singola regione di ~103 kb significativamente differenziata su Scaffold 68 che è stata condivisa da tutti i checker e T-check birds (posizione 1,702,691-1,805,600 del Cliv_1.0 montaggio del genoma del piccione, Shapiro et al. (2013); p=1,11e-16, soglia di significatività a livello di genoma = 9,72e-10). La regione minima condivisa è stata definita da breakpoint aplotipi in un checker omozigote e in un uccello da bar omozigote, ed è altamente differenziata dalla stessa regione in bar (63,28% di somiglianza di sequenza media nei siti informativi). Questa regione è qui di seguito indicata come l’aplotipo di dama minimo.
Come previsto per la serie allelica ben caratterizzata al locus C, abbiamo anche trovato che una regione ampiamente sovrapposta di Scaffold 68 era altamente differenziata tra i genomi degli uccelli bar e barless (p=3,11e-15, soglia di significatività a livello genomico = 9,71e-10; Figura 1-figure supplement 2). Insieme, questi confronti genomico-intero hanno identificato una singola regione genomica corrispondente al locus C a forma di ala.
Una variante del numero di copia è associata alla variazione del modello alare melanico
Per identificare le varianti genetiche associate al checker derivato e ai fenotipi T-check, abbiamo confrontato per la prima volta i geni di codifica delle proteine annotati in tutto il genoma. Abbiamo trovato un singolo, previsto, cambiamento fisso in EFHC2 (Y572C, Figura 1-figure supplement 3) in uccelli checker e T-check rispetto agli uccelli bar (VAAST; Yandell et al., 2011). Tuttavia, questa stessa sostituzione di aminoacidi si trova anche in Columba rupestris, una specie strettamente correlata a C. livia che ha un modello alare bar . Quindi, la sostituzione Y572C non è probabile che sia causativa per il checker o il modello di controllo T, né è probabile che abbia un forte impatto sulla funzione proteica (MutPred2 punteggio 0,468, nessun dominio riconosciuto interessato; PolyPhen-2 punteggio 0,036, benigno; Adzhubei et al., 2010; Pejaver et al., 2017).
Successivamente, abbiamo esaminato la copertura delle sequenze attraverso l’aplotipo del checker e abbiamo scoperto una regione a numero variabile di copie (CNV) (breakpoint approssimativi alle posizioni 1.790.000 e 1.805.600 di Scaffold 68). Basandoci su letture normalizzate di uccelli risququenziati, abbiamo determinato che la regione CNV ha una, due o quattro copie per cromosoma. Gli uccelli bar (n = 12) nel nostro pannello di resequenziamento hanno sempre avuto un totale di due copie nella regione del CNV (una per ogni cromosoma), ma la maggior parte dei genomi checker (n = 5 di 7) e T-check (n = 2 di 2) esaminati avevano copie aggiuntive del CNV (Figura 2A). Utilizzando un test PCR per amplificare attraverso i breakpoint negli uccelli con più di una copia per cromosoma, abbiamo determinato che copie aggiuntive risultano da ripetizioni tandem. Non abbiamo trovato alcuna prova che l’aplotipo del checker contenga un’inversione basata sulla mappatura delle letture del paired-end ai breakpoint del CNV (WHAM; Kronenberg et al., 2015). Inoltre, siamo stati in grado di amplificare prodotti di PCR unici che coprono i breakpoint esterni del CNV (dati non mostrati), suggerendo che non ci sono inversioni all’interno della regione CNV.
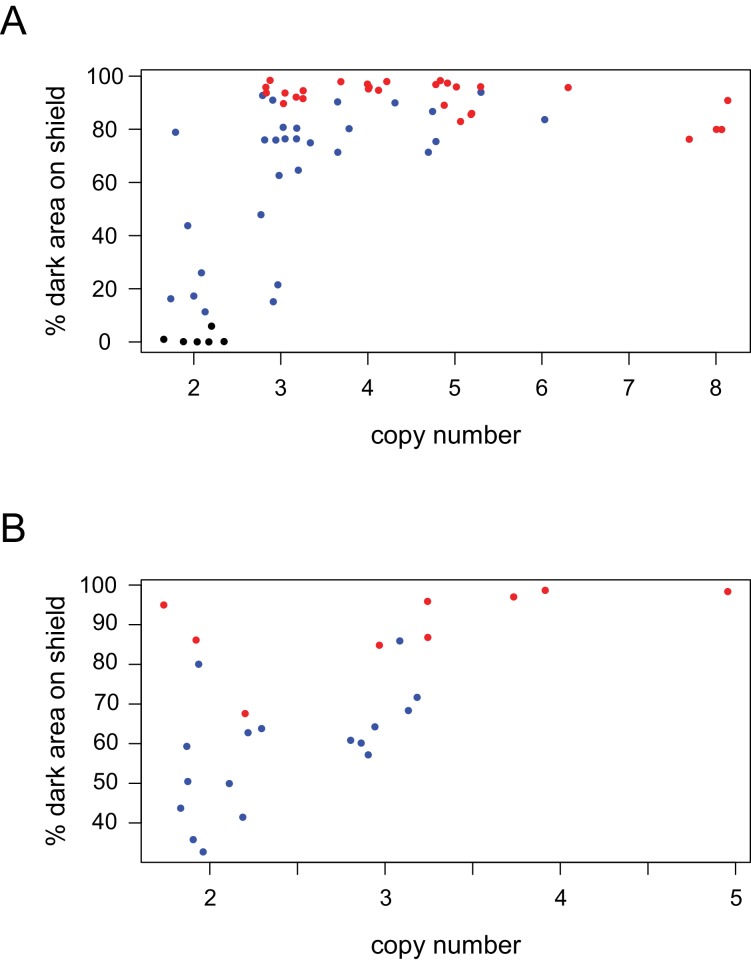
Figura 2-figure supplement 1.Una variante del numero di copia (CNV) nella regione candidata è associata ai risultati del test del numero di copia T-check e checker fenotipi.I risultati del test del numero di copia Taqman rappresentati nella Figura 2B.Taqman e i risultati della quantificazione del fenotipo rappresentati nella Figura 2C.Taqman sono associati alla pigmentazione dello scudo alare più scuro.(A) Le profondità di lettura normalizzate di uccelli resquenziati sono tracciate nella regione candidata tra EFHC2 e NDP su Scaffold 68. Le porzioni ispessite dei modelli genici rappresentano esoni e le porzioni sottili sono introni. Tracce rappresentative della profondità di lettura individuale sono mostrate per le seguenti: nero per la barra C. livia, grigio per gli individui di livia a scacchiera C. senza copie aggiuntive del CNV, blu per gli individui di livia a scacchiera C. con copie aggiuntive della regione CNV, rosso per la livia a T. livia.B) quantificazione CNV per 94 volatili (20 bar, 56 pedine e 18 pedine a T). I fenotipi del checker e del T-check (come riportato dagli allevatori) sono stati associati a un aumento del numero di copie (p=2,1e-05).(C) Quantificazione CNV e fenotipo per altri 84 uccelli, compresi 26 piccioni selvatici. L’aumento del numero di copie è stato associato ad un aumento dell’area scura sullo scudo alare (r2 = 0,46, regressione lineare). I punti sono colorati in base al fenotipo e all’origine segnalati: barra, nero; dama, blu; T-check, rosso; razze domestiche, punti di cerchio pieni; animali selvatici, punti di incrocio.10.7554/eLife.34803.010Cifra dati a 2 fonti 1.Taqman risultati del test del numero di copia rappresentati nella Figura 2B.10.7554/eLife.34803. 011Cifra i dati a 2 fonti 2.Taqman copy number test e i risultati di quantificazione del fenotipo rappresentati nella Figura 2C.CNV e quantificazione del fenotipo per(A) razze domestiche (n = 58) e (B) selvaggi catturati in natura (n = 26), analizzati dai dati della Figura 2C. I punti sono colorati in base al fenotipo e all’origine segnalati: barra, nero; dama, blu; T-check, rosso; razze domestiche, punti solidi; felini, punti di incrocio.
Coerentemente con il modello ereditario dominante del fenotipo, tutti gli uccelli a scacchiera e a T hanno almeno una copia dell’aplotipo a scacchiera. Tuttavia, il fatto che alcuni uccelli di razza a scacchiera avevano solo una copia della regione CNV su ogni cromosoma dimostra che un aumento del numero di copie non è necessario per produrre fenotipi melanici. L’analisi pedigree di una croce di laboratorio ha inoltre confermato la perfetta co-segregazione dell’aplotipo e del fenotipo del checker(Figura 1-figura supplemento 4, file supplementare 1). Pertanto, un aplotipo checker su almeno un cromosoma sembra essere necessario per i fenotipi melanici dominanti, ma non lo sono copie aggiuntive della regione CNV.
In un campione più ampio di piccioni, abbiamo trovato un’associazione significativa tra numero di copie e fenotipo (test TaqMan; test Wilcoxon a coppie, p=2.1e-05). I pattern Checker (n = 40 di 55) e T-check (n = 15 di 18) sono solitamente associati all’espansione del CNV, ma i piccioni con il pattern a barre (n = 20) non hanno mai avuto più di due copie in totale (una copia su ogni cromosoma; Figura 2B). Anche se copie aggiuntive del CNV si sono verificate solo nei checker e nei T-check birds, non abbiamo osservato un numero consistente di copie associate ad entrambi i fenotipi. Ciò potrebbe essere dovuto a una varietà di fattori, tra cui i modificatori che oscurano gli uccelli genotipici di controllo per assomigliare strettamente al T-checker(Van Hoosen Jones, 1922; Sell, 2012) e fattori ambientali come l’oscuramento dello scudo alare in funzione della temperatura durante lo sviluppo delle piume(Podhradsky, 1968).
A causa della potenziale ambiguità nella fenotipizzazione categorica, abbiamo poi misurato la percentuale di area pigmentata sullo scudo alare e abbiamo testato le associazioni tra il numero di copie e la percentuale di area pigmentata dello scudo alare. Abbiamo fenotipizzato e genotipizzato altri 63 uccelli di diverse razze domestiche e 26 uccelli selvatici, e abbiamo trovato che il numero stimato di copie nella regione variabile era correlato con la quantità di pigmento scuro sullo scudo alare (regressione non lineare dei minimi quadrati, seguita dal calcolo r2; r2 = 0,46) (Figura 2C). Questa correlazione si adattava meglio alla regressione quando i ferali sono stati esclusi (r2 = 0,68, Figura 2-figure supplement 1), forse perché numerosi modificatori della pigmentazione (ad esempio fuliggine e sporcizia ) si stanno separando nelle popolazioni di ferali (Hollander, 1938a; Johnston e Janiga, 1995). Insieme, le nostre analisi mostrano che l’aplotipo checker minimo è associato ad un aumento della pigmentazione sul piumaggio dello scudo alare, con conseguente variazione qualitativa tra fenotipi bar e checker (compreso il T-check). Inoltre, la variazione del numero di copie si trova solo negli aplotipi checker, e un numero maggiore di copie è associato ad un aumento quantitativo della pigmentazione nei soli uccelli checker e T-check.
Figura 2-figure supplemento 1.Una variante del numero di copie (CNV) nella regione candidata è associata ai risultati del test del numero di copie T-checker e dei fenotipi checker.I risultati del test del numero di copie Taqman rappresentati nella Figura 2B.Taqman e i risultati della quantificazione del fenotipo rappresentati nella Figura 2C.Taqman sono associati alla pigmentazione dello scudo alare più scuro.(A) Le profondità di lettura normalizzate di uccelli resquenziati sono tracciate nella regione candidata tra EFHC2 e NDP su Scaffold 68. Le porzioni ispessite dei modelli genici rappresentano esoni e le porzioni sottili sono introni. Tracce rappresentative della profondità di lettura individuale sono mostrate per le seguenti: nero per la barra C. livia, grigio per gli individui di livia a scacchiera C. senza copie aggiuntive del CNV, blu per gli individui di livia a scacchiera C. con copie aggiuntive della regione CNV, rosso per la livia a T. livia.B) quantificazione CNV per 94 volatili (20 bar, 56 pedine e 18 pedine a T). I fenotipi del checker e del T-check (come riportato dagli allevatori) sono stati associati a un aumento del numero di copie (p=2,1e-05).(C) Quantificazione CNV e fenotipo per altri 84 uccelli, compresi 26 piccioni selvatici. L’aumento del numero di copie è stato associato ad un aumento dell’area scura sullo scudo alare (r2 = 0,46, regressione lineare). I punti sono colorati in base al fenotipo e all’origine segnalati: barra, nero; dama, blu; T-check, rosso; razze domestiche, punti di cerchio pieni; animali selvatici, punti di incrocio.10.7554/eLife.34803.010Cifra dati a 2 fonti 1.Taqman risultati del test del numero di copia rappresentati nella Figura 2B.10.7554/eLife.34803. 011Cifra i dati a 2 fonti 2.Taqman copy number test e i risultati di quantificazione del fenotipo rappresentati nella Figura 2C.CNV e quantificazione del fenotipo per(A) razze domestiche (n = 58) e (B) selvaggi catturati in natura (n = 26), analizzati dai dati della Figura 2C. I punti sono colorati in base al fenotipo e all’origine segnalati: barra, nero; dama, blu; T-check, rosso; razze domestiche, punti solidi; felini, punti di incrocio.
Figura 2-figure supplement 1.Il CNV è associato alla pigmentazione dello scudo alare più scuro.2. CNV e quantificazione del fenotipo per(A) razze domestiche (n = 58) e (B) selvaggi catturati in natura (n = 26), analizzati dai dati della Figura 2C. I punti sono colorati in base al fenotipo e all’origine segnalati: barra, nero; dama, blu; T-check, rosso; razze domestiche, punti solidi; felini, punti di incrocio.
NDP è espresso in modo differenziato in gemme di piume di diversi fenotipi a forma di ala
Il CNV che è associato alla variazione del profilo alare risiede tra due geni, EFHC2 e NDP. EFHC2 è un componente delle ciglia mobili, e i mutanti dei topi hanno l’epilessia mioclonica giovanile(Linck et al., 2014). Negli esseri umani, la variazione allelica in EFHC2 è anche associata a risposte differenziali di paura e cognizione sociale(Weiss et al., 2007; Blaya et al., 2009; Startin et al., 2015; ma si veda Zinn et al., 2008). Tuttavia, l’EFHC2 non è stato implicato nei fenotipi di pigmentazione in nessun organismo. NDP codifica un ligando secreto che attiva la segnalazione WNT legandosi al suo unico recettore conosciuto FZD4 e al suo co-recettore LRP5(Smallwood et al., 2007; Hendrickx e Leyns, 2008; Deng et al., 2013; Ke et al., 2013). In particolare, il NDP è uno dei molti geni espressi in modo differenziato nelle piume di sottospecie di corvo strettamente imparentate che si differenziano, in parte, per l’intensità della pigmentazione del piumaggio(Poelstra et al., 2015). Inoltre, FZD4 è un noto marcatore di cellule staminali melanocitarie(Yamada et al., 2010). Così, sulla base della variazione di espressione nei diversi fenotipi di piumaggio del corvo e dell’espressione del suo recettore nei precursori delle cellule pigmentarie, il NDP è un forte candidato per la variazione dei pigmenti nei piccioni. NDP è un segnale a corto raggio(Niehrs, 2004), quindi sospettiamo che questo ligando sia secreto dai melanociti stessi o da cellule vicine ad essi.
Il CNV nello spazio intergenico tra EFHC2 e NDP nella regione candidata, insieme alla mancanza di varianti di codifica del candidato tra aplotipi a barre e aplotipi a scacchiera, ci ha portato a ipotizzare che la regione del CNV possa contenere variazioni normative che potrebbero alterare l’espressione di uno o di entrambi i geni vicini. Per testare questa possibilità, abbiamo eseguito la qRT-PCR sull’RNA raccolto dalle piume rigeneranti dello scudo alare di uccelli bar, checker e T-check. EFHC2 non è stato espresso in modo differenziato tra le piume di bar e le piume con disegno a scacchiera o a T (p=0,19, test di Wilcoxon a coppie, metodo di regolazione del valore p: fdr), anche se i livelli di espressione differivano leggermente tra le piume con disegno a scacchiera e quelle con disegno a T (p=0,046, Figura 3A). Anche i livelli di espressione di altri geni adiacenti alla regione minima dell’aplotipo a scacchiera non variavano in base al fenotipo(Figura 3-figure supplement 1).
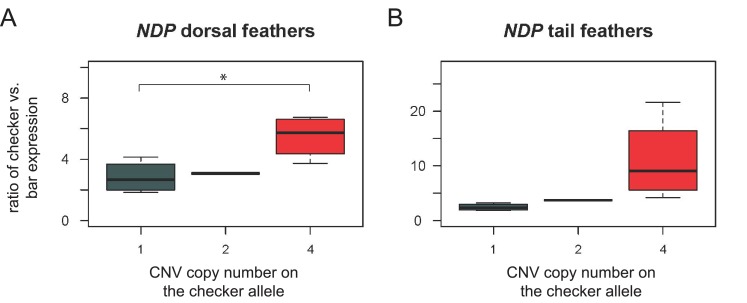
Figura 3-figure supplement 3.Le differenze di espressione in NDP, ma non EFHC2, indicano le differenze cis-regolatorie associate ai fenotipi di pigmentazione.qRT-PCR dati sorgente rappresentati nella Figura 3A, e Figura 3-figure supplement 1.Allele-specific expression assays source data rappresentata nella Figura 3B e Figura 3-figure supplement 3.qRT-PCR dati sorgente rappresentati nella Figura 3A, e Figura 3-figure supplement 1.Allele-specific expression assays dati sorgente dei saggi di espressione rappresentati nella Figura 3B e Figura 3-figure supplement 3.Expression of genes involved in pigmentation and genes in the candidate region. L’espressione diNDP varia in base al numero di copie e al fenotipo.il test di espressione allele-specifico per NDP nella rigenerazione delle piume del corpo dorsale e della coda.(A) I saggi qRT-PCR dimostrano una maggiore espressione di NDP nella rigenerazione delle piume degli uccelli di controllo e di controllo T rispetto agli uccelli da bar. I livelli di espressione di EFHC2 sono indistinguibili tra fenotipi bar e fenotipi melanici (p=0,19), sebbene checker e T-check differiscano l’uno dall’altro (p=0,046).(B) Saggio di espressione allele-specifico nella rigenerazione delle piume da uccelli eterozigoti bar/checker per NDP e EFHC2. Le copie della regione CNV sul cromosoma del checker sono state quantificate utilizzando un saggio Taqman personalizzato. Le caselle si estendono dal primo al terzo quartile, le barre si estendono fino ai valori minimi e massimi osservati, la linea nera indica la mediana. L’espressione degli alleli EFHC2 non erano significativamente diversi, e gli alleli di controllo del NDP hanno mostrato un’espressione più alta rispetto all’allele a barre; p=0,0028 per il test t a due campioni tra 1 e 4 copie, p=1,84e-06 per la regressione glm.10,7554/eLife.34803.016Figure 3-source data 1.qRT-PCR dati sorgente rappresentati nella Figura 3A, e Figura 3-figure supplemento 1.10.7554/eLife.34803.017Figure 3-source data 2.Allele-specific expression assays source data rappresentata nella Figura 3B e Figura 3-figure supplement 3.I livelli di espressione di Mitf(A), Sox10(B), MaoA(D), MaoB (E) e Fundc1(F) sono indistinguibili tra i fenotipi.(C) La tirosinasi mostra un aumento dell’espressione in T-check birds rispetto a bar (p=2.4e-04) e checker birds (p=3.8e-05). Le caselle si estendono dal primo al terzo quartile, le barre si estendono fino ai valori minimi e massimi osservati, la linea nera indica la mediana. I valori di espressione sono analizzati con il test di Wilcoxon Pairwise (metodo di regolazione del valore p: fdr).Il test di espressione qRT-PCR per NDP(Figura 3A) viene analizzato con il numero di copia nella regione CNV. Tutti gli uccelli checker (blu) e T-check (rosso), ad eccezione del singolo individuo con quattro copie totali (blu scuro), sono eterozigoti per bar. L’aumento dell’espressione NDP è correlato all’aumento del numero di copie nella regione CNV. Le caselle si estendono dal primo al terzo quartile, le barre si estendono fino ai valori minimi e massimi osservati, la linea nera indica la mediana. Lettere diverse indicano differenze significative a livello di coppia. Il test Wilcoxon a coppie (metodo di regolazione del valore di p: fdr) risulta per numero di copie: 2-3 copie, p=0,03788; 2-4 copie, p=0,04938; 2-5 copie, p=0,00015; due copie-barra, p=0.00432; 3-4 copie, p=0,03788; 3-5 copie, p=0,00122; tre copie-barra, p=1,9e-06; 4-5 copie, p=0,48485; quattro copie-barra, p=0,02179; cinque copie-barra, p=1,9e-06.Le copie della regione CNV sul cromosoma checker sono state quantificate come nella Figura 3B.(A) Nelle piume del corpo dorsale, gli alleli di controllo del NDP hanno mostrato un’espressione più alta rispetto all’allele a barre (p=0,025 per il test t a due campioni che confronta il rapporto 1-copia checker:bar con il rapporto 4-copia checker:bar, p=0,013 per la regressione glm, compresi i rapporti 1-, 2-, e 4-copia checker:bar). I rapporti di espressione degli alleli 1-copy checker:bar e 4-copy checker:bar erano significativamente diversi dal valore 1:1 previsto per l’espressione di uguale checker e bar; p<0,05 per ogni confronto.(B) Il rapporto di espressione nelle piume della coda non era significativamente diverso tra gli alleli di controllo con 1 o quattro copie (p=0,11, t-test; p=0,044 per la regressione glm compresi i rapporti 1-, 2-, e 4-copy checker:bar). I rapporti di espressione di 1-copy checker:bar erano significativamente diversi dal valore 1:1 previsto per il checker uguale e l’espressione a barre; p<0,05. I rapporti di espressione degli alleli 4-copy checker:bar non si discostano in modo significativo dal valore 1:1 previsto per equal checker e bar expression; p=0,07. Le caselle si estendono dal primo al terzo quartile, le barre si estendono fino ai valori minimi e massimi osservati, la linea nera indica la mediana.
Al contrario, l’espressione di NDP è stata significativamente aumentata nelle piume a scacchiera – e ancora più alta nelle piume a T – rispetto alle piume a barre (Figura 3A) (confronto tra le barre a scacchiera, p=1,9e-05; barra-T a scacchiera, p=1,0e-08; barra-T a scacchiera, p=0,0071; test di Wilcoxon a coppie, tutti i confronti sono stati significativi con un tasso di falsificazione di 0,05). Inoltre, quando i dati di espressione qRT-PCR per le piume checker e T-checker sono stati raggruppati per numero di copie invece che per fenotipo categorico, il numero di copie CNV è stato associato positivamente al livello di espressione NDP(Figura 3-figure supplement 2). Pertanto, l’espressione di NDP è stata associata positivamente sia con l’aumento del melanismo (fenotipo a pigmento categorico) che con il genotipo CNV.
L’aumento dell’espressione di NDP potrebbe essere il risultato di almeno due meccanismi molecolari. In primo luogo, uno o più elementi regolatori nella regione del CNV (o altrove sullo stesso filamento di DNA) potrebbero aumentare l’espressione di NDP in cis. Tali cambiamenti influenzerebbero l’espressione dell’allele solo sullo stesso cromosoma(Wittkopp et al., 2004). In secondo luogo, i fattori trans-attori trans-attori codificati all’interno dell’aplotipo checker minimo (ad esempio, EFHC2 o una caratteristica non preannotata) potrebbero aumentare l’espressione del NDP, con conseguente upregulation degli alleli NDP su entrambi i cromosomi.
Per distinguere queste possibilità, abbiamo effettuato dei test di espressione allele-specifici(Domyan et al., 2014; 2016) sulle piume rigeneranti dello scudo alare degli uccelli che erano eterozigoti per gli alleli bar e checker nella regione candidata (alleli checker con una, due o quattro copie del CNV). Nel comune ambiente cellulare ad azione transociale degli uccelli eterozigoti, gli alleli di controllo del NDP erano più altamente espressi rispetto agli alleli bar, e queste differenze sono state ulteriormente amplificate negli alleli di controllo con più copie del CNV(Figura 3B) (p=0.0028 per il test t a due campioni tra 1 e 4 copie, p=1,84e-06 per la regressione generalizzata del modello lineare; i rapporti di espressione checker:bar per gli alleli checker a 1 e 4 copie erano significativamente diversi da 1:1, p≤0,002 per ogni confronto). In confronto, le trascrizioni di EFHC2 dagli alleli checker e bar non sono state espresse in modo differenziato nello sfondo eterozigote(Figura 3B) (p=0,55 per il test t-test a due campioni tra 1 e 4 copie, p=0,47 per la regressione lineare; i rapporti di espressione checker:bar per gli alleli checker a 1 e 4 copie non sono stati significativamente diversi da 1:1, p>0,3 per ogni confronto). Gli alleli checker di NDP erano anche più altamente espressi in piume provenienti da altre regioni del corpo (coda e dorso, Figura 3-figure supplement 3), anche se il pattern di pigmento su queste regioni è generalmente simile in uccelli bar e checker (ad esempio, entrambi i fenotipi hanno una banda scura sulla coda). Insieme, i nostri studi sull’espressione indicano che un cambiamento normativo ad azione cis guida una maggiore espressione di NDP nei piccioni con pattern di piumaggio più melanico, ma non altera l’espressione di EFHC2 o di altri geni vicini. Inoltre, poiché l’espressione del NDP aumenta con l’aggiunta di copie aggiuntive del CNV, l’elemento regolatore probabilmente risiede all’interno del CNV stesso.
Per cercare gli stimolatori conosciuti nella regione del CNV, abbiamo mappato gli elementi del VISTA(Visel et al., 2007) e del REPTILE(He et al., 2017) al genoma del piccione. Non abbiamo trovato alcun riscontro all’interno dell’aplotipo minimo dal set di dati VISTA e 12 riscontri dal set di dati REPTILE(file supplementare 2). Di questi 12, un riscontro è avvenuto nella regione CNV (Scaffold 68: 1.795.453-1.795.511). Tuttavia, questo potenziatore per topi solitari (ENSMUSR0000000084784, http://uswest.ensembl.org/Mus_musculus/) non è noto per regolare l’EFHC2 o l’NDP nei topi, e si trova su un cromosoma di topo che non è ortologico per il piccione Scaffold 68. Sarà necessario un ulteriore lavoro funzionale per valutare se questa o altre sequenze nella regione del CNV agiscono come elementi normativi in C. livia.
Figura 3-figure supplement 3.Le differenze di espressione in NDP, ma non EFHC2, indicano differenze di regolamentazione cis associate ai fenotipi di pigmentazione.qRT-PCR dati sorgente rappresentati nella Figura 3A, e Figura 3-figure supplement 1.Allele-specific expression assays source data rappresentata nella Figura 3B e Figura 3-figure supplement 3.qRT-PCR dati sorgente rappresentati nella Figura 3A, e Figura 3-figure supplement 1.Allele-specific expression assays dati sorgente dei saggi di espressione rappresentati nella Figura 3B e Figura 3-figure supplement 3.Expression of genes involved in pigmentation and genes in the candidate region. L’espressione diNDP varia in base al numero di copie e al fenotipo.il test di espressione allele-specifico per NDP nella rigenerazione delle piume del corpo dorsale e della coda.(A) I saggi qRT-PCR dimostrano una maggiore espressione di NDP nella rigenerazione delle piume degli uccelli di controllo e di controllo T rispetto agli uccelli da bar. I livelli di espressione di EFHC2 sono indistinguibili tra fenotipi bar e fenotipi melanici (p=0,19), sebbene checker e T-check differiscano l’uno dall’altro (p=0,046).(B) Saggio di espressione allele-specifico nella rigenerazione delle piume da uccelli eterozigoti bar/checker per NDP e EFHC2. Le copie della regione CNV sul cromosoma del checker sono state quantificate utilizzando un saggio Taqman personalizzato. Le caselle si estendono dal primo al terzo quartile, le barre si estendono fino ai valori minimi e massimi osservati, la linea nera indica la mediana. L’espressione degli alleli EFHC2 non erano significativamente diversi, e gli alleli di controllo del NDP hanno mostrato un’espressione più alta rispetto all’allele a barre; p=0,0028 per il test t a due campioni tra 1 e 4 copie, p=1,84e-06 per la regressione glm.10,7554/eLife.34803.016Figure 3-source data 1.qRT-PCR dati sorgente rappresentati nella Figura 3A, e Figura 3-figure supplemento 1.10.7554/eLife.34803.017Figure 3-source data 2.Allele-specific expression assays source data rappresentata nella Figura 3B e Figura 3-figure supplement 3.I livelli di espressione di Mitf(A), Sox10(B), MaoA(D), MaoB (E) e Fundc1(F) sono indistinguibili tra i fenotipi.(C) La tirosinasi mostra un aumento dell’espressione in T-check birds rispetto a bar (p=2.4e-04) e checker birds (p=3.8e-05). Le caselle si estendono dal primo al terzo quartile, le barre si estendono fino ai valori minimi e massimi osservati, la linea nera indica la mediana. I valori di espressione sono analizzati con il test di Wilcoxon Pairwise (metodo di regolazione del valore p: fdr).Il test di espressione qRT-PCR per NDP(Figura 3A) viene analizzato con il numero di copia nella regione CNV. Tutti gli uccelli checker (blu) e T-check (rosso), ad eccezione del singolo individuo con quattro copie totali (blu scuro), sono eterozigoti per bar. L’aumento dell’espressione NDP è correlato all’aumento del numero di copie nella regione CNV. Le caselle si estendono dal primo al terzo quartile, le barre si estendono fino ai valori minimi e massimi osservati, la linea nera indica la mediana. Lettere diverse indicano differenze significative a livello di coppia. Il test Wilcoxon a coppie (metodo di regolazione del valore di p: fdr) risulta per numero di copie: 2-3 copie, p=0,03788; 2-4 copie, p=0,04938; 2-5 copie, p=0,00015; due copie-barra, p=0.00432; 3-4 copie, p=0,03788; 3-5 copie, p=0,00122; tre copie-barra, p=1,9e-06; 4-5 copie, p=0,48485; quattro copie-barra, p=0,02179; cinque copie-barra, p=1,9e-06.Le copie della regione CNV sul cromosoma checker sono state quantificate come nella Figura 3B.(A) Nelle piume del corpo dorsale, gli alleli di controllo del NDP hanno mostrato un’espressione più alta rispetto all’allele a barre (p=0,025 per il test t a due campioni che confronta il rapporto 1-copia checker:bar con il rapporto 4-copia checker:bar, p=0,013 per la regressione glm, compresi i rapporti 1-, 2-, e 4-copia checker:bar). I rapporti di espressione degli alleli 1-copy checker:bar e 4-copy checker:bar erano significativamente diversi dal valore 1:1 previsto per l’espressione di uguale checker e bar; p<0,05 per ogni confronto.(B) Il rapporto di espressione nelle piume della coda non era significativamente diverso tra gli alleli di controllo con 1 o quattro copie (p=0,11, t-test; p=0,044 per la regressione glm compresi i rapporti 1-, 2-, e 4-copy checker:bar). I rapporti di espressione di 1-copy checker:bar erano significativamente diversi dal valore 1:1 previsto per il checker uguale e l’espressione a barre; p<0,05. I rapporti di espressione degli alleli 4-copy checker:bar non si discostano in modo significativo dal valore 1:1 previsto per equal checker e bar expression; p=0,07. Le caselle si estendono dal primo al terzo quartile, le barre si estendono fino ai valori minimi e massimi osservati, la linea nera indica la mediana.
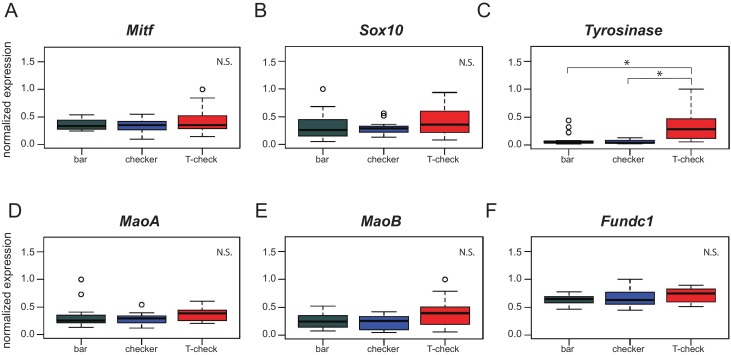
Figura 3-figure supplement 1.Espressione dei geni coinvolti nella pigmentazione e dei geni nella regione candidata.I livelli di espressione di Mitf(A), Sox10(B), MaoA(D), MaoB (E) e Fundc1(F) sono indistinguibili tra i fenotipi.(C) La tirosinasi mostra un aumento dell’espressione in T-check birds rispetto a bar (p=2.4e-04) e checker birds (p=3.8e-05). Le caselle si estendono dal primo al terzo quartile, le barre si estendono fino ai valori minimi e massimi osservati, la linea nera indica la mediana. I valori di espressione sono analizzati con il test di Wilcoxon Pairwise (metodo di regolazione del valore p: fdr).
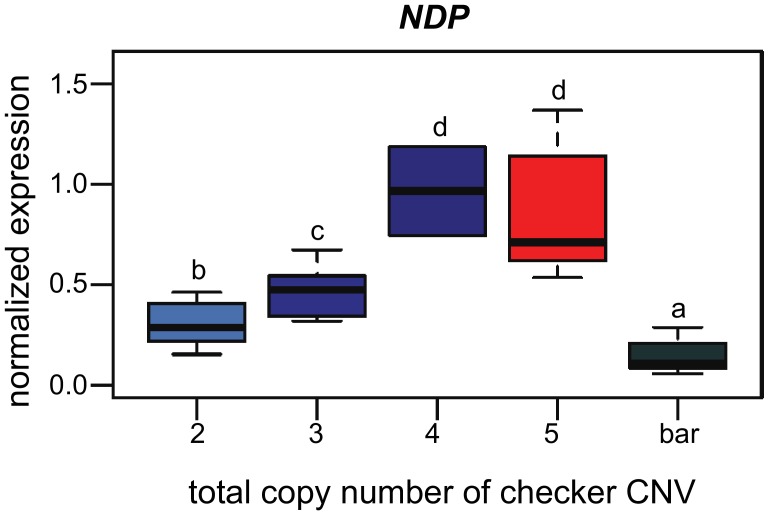
Figura 3-figure supplement 2.L’espressioneNDP varia in base al numero di copie e al fenotipo.Il test di espressione qRT-PCR per NDP(Figura 3A) viene analizzato in base al numero di copie nella regione CNV. Tutti gli uccelli di controllo (blu) e T-check (rosso), ad eccezione del singolo individuo con quattro copie totali (blu scuro), sono eterozigoti per bar. L’aumento dell’espressione NDP è correlato all’aumento del numero di copie nella regione CNV. Le caselle si estendono dal primo al terzo quartile, le barre si estendono fino ai valori minimi e massimi osservati, la linea nera indica la mediana. Lettere diverse indicano differenze significative a livello di coppia. Il test Wilcoxon a coppie (metodo di regolazione del valore di p: fdr) risulta per numero di copie: 2-3 copie, p=0,03788; 2-4 copie, p=0,04938; 2-5 copie, p=0,00015; due copie-barra, p=0.00432; 3-4 copie, p=0,03788; 3-5 copie, p=0,00122; tre copie-barra, p=1,9e-06; 4-5 copie, p=0,48485; quattro copie-barra, p=0,02179; cinque copie-barra, p=1,9e-06.
Figura 3-figure supplemento 3.Figura 3—supplemento di figura 3. Saggio di espressione allele-specifico per NDP nella rigenerazione delle piume del corpo dorsale e della coda.Le copie della regione CNV sul cromosoma checker sono state quantificate come nella Figura 3B.(A) Nelle piume del corpo dorsale, gli alleli di controllo del NDP hanno mostrato un’espressione più alta rispetto all’allele a barre (p=0,025 per il test t-test a due campioni che confronta il rapporto 1-copia checker:bar con il rapporto 4-copia checker:bar, p=0,013 per la regressione glm che include i rapporti 1-, 2-, e 4-copia checker:bar). I rapporti di espressione degli alleli 1-copy checker:bar e 4-copy checker:bar erano significativamente diversi dal valore 1:1 previsto per l’espressione di uguale checker e bar; p<0,05 per ogni confronto.(B) Il rapporto di espressione nelle piume della coda non era significativamente diverso tra gli alleli di controllo con 1 o quattro copie (p=0,11, t-test; p=0,044 per la regressione glm compresi i rapporti 1-, 2-, e 4-copy checker:bar). I rapporti di espressione di 1-copy checker:bar erano significativamente diversi dal valore 1:1 previsto per il checker uguale e l’espressione a barre; p<0,05. I rapporti di espressione degli alleli 4-copy checker:bar non si discostano in modo significativo dal valore 1:1 previsto per equal checker e bar expression; p=0,07. Le caselle si estendono dal primo al terzo quartile, le barre si estendono fino ai valori minimi e massimi osservati, la linea nera indica la mediana.
Una mutazione missenso al codone iniziale di NDP è associata a barless
Negli esseri umani, mutazioni in NDP può provocare la malattia di Norrie, un disturbo ereditario recessivo caratterizzato da una serie di sintomi tra cui carenze della vista, disturbi intellettuali e motori, e carenze uditive(Norrie, 1927; Warburg, 1961; Holmes, 1971; Chen et al., 1992; Sims et al., 1992). Le mutazioni che codificano le proteine nel PND, comprese le mutazioni identiche che si separano all’interno di pedigree monofamiliari, danno luogo a risultati fenotipici variabili, compresa la penetrazione incompleta(Meindl et al., 1995; Berger, 1998; Allen et al., 2006). Intrigante, piccioni senza barra hanno anche una maggiore incidenza di carenze di visione e, come negli esseri umani con alcuni alleli mutanti di NDP, questo fenotipo non è completamente penetrante(Hollander, 1983b). Così, sulla base del noto allelismo al locus C, la nomina di modifiche normative al NDP come candidati per il C e CT alleli, e i sintomi correlati alla visione della malattia di Norrie, NDP è anche un forte candidato per il fenotipo senza barra( allelec ).
Per testare questa previsione, abbiamo usato VAAST per analizzare i genomi sequenziati di 9 piccioni senza barra e abbiamo trovato che tutti erano omozigoti per un cambiamento nonsinonimo di codifica della proteina al codone di partenza del NDP che era perfettamente associato con il fenotipo del modello di ala senza barra(Figura 4, Figura 1-figure supplement 2). Non abbiamo rilevato nessun altro gene con cambiamenti di codifica fissi o regioni di differenziazione significativa della frequenza dell’allele (pFst) altrove nel genoma. Abbiamo genotipizzato altri 14 uccelli senza barra e abbiamo trovato che tutti erano omozigoti per la stessa mutazione del codice di partenza. La mutazione senza barra è prevista per troncare il capolinea amminoacido della proteina NDP da 11 amminoacidi, interrompendo così la sequenza di segnali peptidici a 24 amminoacidi(www.uniprot.org, Q00604 NDP_Human). NDP è ancora trascritto e rilevabile dalla RT-PCR nella rigenerazione delle piume senza barra; pertanto, si ipotizza che la mutazione del codice di partenza potrebbe alterare la normale secrezione della proteina nella matrice extracellulare(Gierasch, 1989).

Figura 4.Figura 4. Piccioni senza barra hanno una mutazione senza senso al sito di inizio di traduzione altamente conservato di NDP.I piccioni viaggiatori senza barre sono omozigoti per una mutazione senza senso che tronca il capolinea degli aminoacidi di NDP di 13 aminoacidi; la stessa posizione del codice di partenza è influenzata da una mutazione in due famiglie umane con cecità ereditaria (rosso, fondo degli allineamenti).
Negli esseri umani, le mutazioni di codifica in NDP sono spesso associate a una serie di deficit neurologici. Nei piccioni, tuttavia, solo l’esaurimento dei pigmenti delle ali e difetti della vista sono segnalati in omozigoti senza barra. Sorprendentemente, due famiglie umane che separano la malattia di Norrie hanno solo difetti della vista, e come i piccioni senza barra, questi individui hanno mutazioni del codice di inizio in NDP(Figura 4)(Isashiki et al., 1995). Pertanto, le mutazioni peptidiche del segnale potrebbero interessare uno specifico sottoinsieme dei processi di sviluppo regolati dal NDP, lasciando intatte altre funzioni (in gran parte neurologiche). L’NDP è fondamentale per la formazione vascolare della retina(Xu et al., 2004) e per la proliferazione dei progenitori retinici dipendenti dai porcospini(McNeill et al., 2013) nei mammiferi, e si ipotizza che uno o entrambi questi processi siano influenzati dalle mutazioni dei codoni di partenza anche nei piccioni. In sintesi, i fenotipi del modello alare nei piccioni sono associati all’evoluzione di entrambi i cambiamenti normativi (checker, T-check) e di codifica (senza barra) nello stesso gene, e i piccioni senza barra condividono un deficit visivo parzialmente penetrante con i pazienti umani che hanno sostituzioni del codice di partenza.
Il lavoro futuro verificherà se le mutazioni del codice di partenza senza barra (e umane) influenzano la secrezione extracellulare di NDP e come l’espressione di NDP regola direttamente o indirettamente l’attività dei melanociti. I confini netti definiscono le aree fortemente pigmentate delle piume di dama(Figura 1, Figura 1-figure supplement 1), simili ai modelli intra-piuma in altre specie che sono mediate sia dall’attività dei melanociti che dalla distribuzione topologica dei loro progenitori (Linet al., 2013; Chen et al., 2015). Si sa molto di più sul controllo molecolare della struttura del piumaggio e del colore rispetto al modello di pigmentazione, basato in parte su esperimenti per manipolare l’espressione genica in vivo attraverso l’infezione virale e negli espianti attraverso la cattiva espressione delle proteine(Harris et al., 2002; Yu et al., 2002; Harris et al., 2005; Chen et al., 2015; Boer et al., 2017). Ci aspettiamo che l’identificazione di NDP come gene di patterning apra nuove strade di esperimenti funzionali simili per capire come la distribuzione dei pigmenti sia mediata.
Figura 4.Figura 4. I piccioni senza sbarre hanno una mutazione senza senso nel sito di inizio della traduzione altamente conservato di NDP.I piccioni viaggiatori senza barre sono omozigoti per una mutazione senza senso che tronca il capolinea degli amminoacidi di NDP di 13 amminoacidi; la stessa posizione del codice di partenza è influenzata da una mutazione in due famiglie umane con cecità ereditaria (rosso, fondo degli allineamenti).
Firme di introgressione dell’aplotipo a scacchiera
Gli appassionati di piccioni hanno a lungo ipotizzato che il modello a dama nel piccione di roccia(Columba livia) sia il risultato di un evento di ibridazione tra specie incrociate con il piccione maculato(C. guinea,Figura 5D), una specie con un modello ad ala a dama (G. Hochlan, G. Young, comunicazione personale)(Hollander, 1983b). Stimiamo che C. liv ia e C. guinea divergevano 4-5 milioni di anni fa (MYA): le specie di colombari (piccioni e colombe) divergono l’una dall’altra in sequenza mitocondriale citocromo b nucleotide a 1,96% per MY (Weire Schluter, 2008), e C. livia e C. guinea differiscono a questo gene del 8,0%. Le stime delle date di divergenza per queste due specie basate sulle sequenze del genoma nucleare variano tra 3,2 e 6,7 MYA (K.P.J., risultati non pubblicati). Nonostante questo tempo di divergenza di diversi MY, gli incroci interspecie tra C. livia e C. guinea possono produrre ibridi fertili(Whitman, 1919; Irwin et al., 1936; Taibel, 1949; Miller, 1953). Inoltre, l’ibridoF1 e la progenie di backcross tra C. gu inea e la barra C. livia hanno le ali a scacchiera, proprio come C. livia con l’allele C(Whitman, 1919; Taibel, 1949). Taibel (1949) ha mostrato che, sebbene le femmine ibrideF1 fossero sterili, altre due generazioni di maschi ibridi backcrossing a C. liv ia potevano produrre figli a scacchi di entrambi i sessi che erano completamente fertili. In breve, Taibel introdusse il tratto della dama da C. guinea a C. livia in sole tre generazioni.
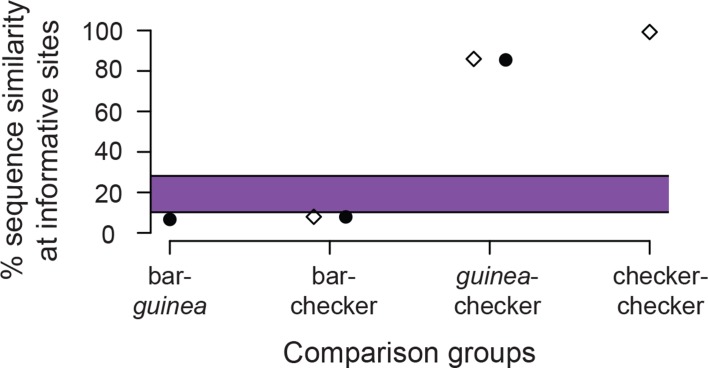
Figura 5-figure supplemento 1.Firme di introgressione dell’aplotipo a scacchiera da C. guinea a C. livia. 2. Numeri di SNPs tra le diverse combinazioni a coppie di barra omozigote, checker, e C. gu inea rappresentata in Figura 5C.Numeri di SNPs tra le diverse combinazioni a coppie di barra omozigote, checker, e C. guinea rappresentata in Figura 5C.Previsto (barra viola) e la proporzione osservata di siti di segregazione condivisi su 1.458 SNPs nella regione aplotipo minimo per diversi confronti pairwise tra gli assemblaggi del genoma de novo da breve lettura dati di resequencing per la barra, checker, e C. guinea.(A) ABBA-BABA test con C. liv ia (bar), C. livia (checker), C . guinea, e C. palumbus mostra elevata D-statistica nella regione candidato Scaffold 68. Sono mostrati tre test ABBA-BABA rappresentativi e la linea rossa tratteggiata segna la media D-statistica a livello genomico per 10 × 10 diverse combinazioni di bar e checker birds (ARC-STA, MAP-ORR, IRT-STA sono mostrati, dove ARC, MAP, e IRT sono campioni checker e STA e ORR sono campioni bar; vedere Metodi).(B) HybridCheck mostra una somiglianza di sequenza a coppie tra i siti informativi di una terzina di sequenza. Viene mostrata una terzina rappresentativa di bar (Fer_VA), checker (ARC), e C. guinea confronto. La traccia blu mostra la somiglianza di sequenza tra la barra e la pedina, la traccia viola mostra la somiglianza tra la barra e C. guinea, e la traccia gialla mostra la somiglianza di sequenza tra la pedina e C. guinea.(C) Aspettativa (barra viola) e proporzione osservata di siti di segregatura condivisi su 4261 SNP totali nella regione aplotipo minimo per diversi confronti a coppie tra e tra 16 barre, 11 zigrinatura e 1 C. guinea.(D) Piccione maculato(Columba guinea). Foto per gentile concessione di Kjeuring (licenza CC BY 3.0, https://creativecommons.org/licenses/by/3.0/legalcode). Foto ritagliata da ‘Speckled pigeon Columba guinea Table Mountain Table Mountain Cape Town’, https://en.wikipedia.org/wiki/Speckled_pigeon#/media/File:Speckledpigeon.JPG. Immagine con inserto in piuma degli autori.10.7554/eLife.34803.021Figure 5-source data 1.Numeri di SNPs tra le diverse combinazioni a coppie di barra omozigote, checker, e C. guinea rappresentata in Figura 5C .I cerchi riempiti denotano la somiglianza della sequenza nei confronti a coppie di assemblaggi de novo, e i diamanti denotano i confronti a coppie tra gli assemblaggi de novo e l’assemblaggio di riferimento, che ha l’aplotipo checker. La percentuale di somiglianza percentuale è molto più bassa in questo grafico rispetto alla Figura 5C perché il numero di polimorfismi tra i confronti a coppie è simile, ma il numero totale di siti di separazione è molto più basso in questo piccolo dataset rispetto al confronto tra molti genomi sequenziati. Nel dataset più grande rappresentato nella Figura 5C, il numero più alto di SNP è stato determinato in gran parte dalla diversità di sequenza tra i 16 bar di uccelli. Abbiamo identificato 105 SNPs negli assemblaggi de novo che non abbiamo chiamato nei dati di resequenziamento. Di questi, 31 differiscono nel confronto a coppie tra il checker e C. guinea assemblies, 84 differiscono tra checker e bar, e 98 differiscono tra bar e C. guinea. Totale dei siti polimorfici tra gli assemblaggi de novo nell’intersezione di 92.199 bp tra tutti e tre gli assemblaggi de novo: bar-checker, 1343; checker-C. guinea, 212; bar-C.guinea, 1361. Totale dei siti polimorfici tra gli assemblaggi de novo e il genoma di riferimento (checker): bar-reference, 1342; checker-reference, 11; C. guinea-reference, 205.
Per valutare la possibilità di un evento di introgressione antico, abbiamo sequenziato un genoma individuale C. guinea a 33X di copertura e mappato le letture al gruppo di riferimento C. livia. Abbiamo calcolato quattro tassonomi D-statistici(‘ABBA-BABA’ test; Durand et al., 2011) per testare le deviazioni dalla somiglianza di sequenza prevista tra C. guinea e C. livia, utilizzando ungenoma di colombaccio (C. palumbus) come outgroup (file supplementare3). In questo caso, l’aspettativa nulla è che la regione candidata C sia più simile tra la barra conspecifica e il checker C . livia di quanto lo sia la stessa regione in C. guinea. Cioè, la filogenesi della regione candidata dovrebbe essere congruente con la filogenesi della specie. Tuttavia, abbiamo trovato che il D-statistico si avvicina ad uno nella regione candidata (n = 10 ciascuno per la barra e la zigrinatura C. livia), indicando che la zigrinatura C. livia sono più simili a C. guinea che agli uccelli della barra conspecifica in questa regione (Figura 5A). La statistica D media del genoma a livello di genoma era vicina a zero (0,021), indicando che le sequenze di barre e pedine sono più simili l’una all’altra in tutto il genoma rispetto a C. guinea.
Questa somiglianza tra C. gu inea e checker C. livia nella regione candidata al modello è stata ulteriormente supportata dall’analisi della sequenza utilizzando HybridCheck(Ward e van Oosterhout, 2016). Al di fuori della regione candidata, gli uccelli checker hanno una elevata somiglianza di sequenza con gli uccelli bar conspecifici e una bassa somiglianza con C. guinea(Figura 5B). All’interno della regione candidata, tuttavia, questa relazione mostra un’impressionante inversione di tendenza, e le sequenze di zigolo e di C. guinea sono più simili tra loro. Inoltre, anche se la statistica D-statistica a livello genomico era relativamente bassa, l’intervallo di confidenza del 95% (CI) era maggiore di zero (da 0,021 a 0,022), fornendo ulteriori prove per uno o più eventi di introgressione da C. guinea in genomi checker e T-check. A differenza di molti checker e T-check C. livia, non abbiamo trovato ulteriori copie della regione CNV in C. guinea. Questo potrebbe indicare che il CNV si è espanso in C. livia, o che il CNV è presente in un sottoinsieme di C. guinea ma non è stato ancora campionato. Presi nel loro insieme, questi modelli di somiglianza di sequenza e di divergenza supportano l’ipotesi che l’aplotipo del candidato checker nei piccioni viaggiatori abbia avuto origine dall’introgressione di C . guinea.
Mentre l’introgressione post-divergenza è un’ipotesi allettante per spiegare la somiglianza di sequenza tra la scacchiera C. livia e C. guinea, un’altra possibilità formale è che la somiglianza di sequenza tra questi gruppi sia dovuta ad un ordinamento incompleto del lignaggio. In un esempio analogo, alleli chiari e scuri della pigmentazione dell’abbronzatura sono stati probabilmente segregati nell’antenato della Drosophila americana e della D. novamexicana, e l’allele chiaro è stato successivamente fissato in quest’ultima specie(Wittkopp et al., 2009). Tuttavia, gli alleli chiari e scuri continuano a segregarsi nella D. americana, e l’allele chiaro in questa specie ha la stessa origine ancestrale di quello fissato nella D. novamexicana. Allo stesso modo, abbiamo voluto verificare se l’aplotipo a scacchiera minima poteva essere presente nell’ultimo antenato comune di C. guinea e C. livia, ma ora si segrega solo in C. livia.
Abbiamo misurato le differenze nucleotidiche tra i diversi alleli dell’aplotipo minimo e abbiamo confrontato questi conteggi con i tassi di polimorfismo che ci si aspetta si accumulino nel tempo di 4-5 MY di divergenza tra C. livia e C. guinea (Figura 5C, barraviola, vedi Materiali e metodi). guinea si è avvicinato al numero previsto di accumuli in questa regione in 4-5 MY (59,90% di somiglianza di sequenza a siti di separazione, SD = 2,6%, 1708 ± 109 SNPs media, Figura 5C), ma anche confronti intraspecifici tra la barra e il checker C. livia (63,28%, SD = 2,3%, 1564 ± 99). Al contrario, le sequenze di C. guinea e C. livia checker hanno avuto differenze significativamente inferiori a quelle che ci si aspetterebbe si accumulino tra le due specie (90,96%, SD = 0,13%, 384 ± 6, p<2,2e-16, t-test). Questi risultati supportano un evento di introgressione da C. guinea a C. livia, piuttosto che un allele condiviso ereditato da un antenato comune prima della divergenza. Tra le 11 sequenze di aplotipo checker, abbiamo trovato una somiglianza di sequenza notevolmente elevata (99,39%, SD = 0,18%, 26 ± 8 differenze medie), corrispondente ad un tempo di divergenza aplotipo di 89 ± 27 mila anni (KY), basato sul tasso di mutazione.
Il genoma di riferimento del piccione di roccia contiene l’aplotipo checker, che potrebbe influenzare la scoperta di SNP nei nostri genomi sequenziati. Abbiamo quindi eseguito degli assemblaggi de novo utilizzando le letture di Illumina shotgun da C. guinea e da individui ad alta copertura di barra e checker, quindi abbiamo confrontato le sequenze nucleotidiche nelle regioni dell’aplotipo minimo in cui tutti e tre gli assemblaggi si sono sovrapposti (92.199 su 102.909 bp, ovvero 89,6%). Abbiamo trovato modelli simili di divergenza tra gli assemblaggi de novo e i genomi sequenziati che sono stati mappati al riferimento, indicando che la scoperta di SNP non era fortemente influenzata dal nostro approccio di mappatura a lettura breve (Figura 5-figuresupplement 1). Sulla base dei polimorfismi a coppie tra il riferimento checker e l’assemblaggio de novo checker (11 differenze), il tempo di divergenza dell’aplotipo è di 42 KY. Questa cifra è più recente della nostra stima basata su più individui, ma i risultati chiave sono che entrambe le stime sono circa 2 ordini di grandezza più recenti del tempo di divergenza tra le specie, e la somiglianza tra le sequenze di dama e C. guinea è caratteristica delle variazioni all’interno della specie piuttosto che tra specie .
Infine, fino ad oggi l’evento o gli eventi presunti di introgressione, abbiamo stimato l’età dell’aplotipo minimo di zigrino sulla base del modello di decadimento del disequilibrio di collegamento(Voight et al., 2006). Utilizzando un tasso di ricombinazione calcolato per i piccioni viaggiatori(Holt et al., 2018), l’aplotipo a scacchiera ha avuto origine in C. liv ia tra 429 e 857 anni fa, assumendo da una a due generazioni all’anno. I corrispondenti intervalli di confidenza del 95% sono da 267 a 716 anni fa, assumendo una generazione all’anno e da 534 a 1.432 anni fa, assumendo due generazioni all’anno.
Insieme, queste molteplici linee di evidenza supportano l’ipotesi che l’aplotipo a scacchiera sia stato introdotto da C. guinea in C. livia dopo l’ addomesticamento del piccione viaggiatore (~5000 anni fa). I valori statistici D-statistici a quattro tassonomi si avvicinano a quello del locus NDP(Figura 5A), indicando che l’aplotipo a scacchiera C . liv ia è molto più strettamente legato a C . gu inea che alla barra C. livia in questo locus. Inoltre, le differenze a coppie tra C. gu inea e aplotipi di controllo sono incompatibili con l’ordinamento incompleto del lignaggio(Figura 5C), supponendo un tempo di divergenza di 4-5 MY specie e nessun flusso genico successivo.La mancanza di diversità nucleotidica singola tra gli aplotipi checker, con solo 26 ± 8 differenze medie e una divergenza genica stimata di 89 KY, è insolitamente bassa per la diversità tipicamente osservata in grandi popolazioni di piccioni in libertà (Shapiro et al.,2013). Le differenze tra le stime basate sulla mutazione (89 KY) e quelle basate sulla LD (da 0,4 a 0,9 KY) dell’età dell’aplotipo checker sono una conseguenza prevista dell’incrocio e della selezione artificiale, dato che la prima è una stima dell’età dell’antenato comune più recente nella popolazione di origine, mentre la seconda è una stima con limite inferiore per la data di introgressione. Incongruenze di questa entità sono inaspettate in assenza di introgressione. Inoltre, la D-statistica del genoma a livello di genoma che confronta C. gu inea e barra con C. guinea e checker è bassa ma significativamente maggiore di 0, indicando che il flusso genico da C. guinea a checker è stato superiore a quello da C. guinea a barra in tutto il genoma. In particolare, il risultato non zero D-statistico tiene quando il locus NDP è escluso da questo calcolo. Questi risultati sono attesi se gli aplotipi di checker sono stati recentemente introdotti in C. livia da allevatori di piccioni, e l’incrocio tra popolazioni di checker e bar non è stato completamente casuale. Coerentemente con questa aspettativa, l’accoppiamento non casuale è osservato nelle popolazioni selvatiche, e gli allevatori di piccioni spesso impongono la selezione del modello di colore ai loro uccelli(Darwin, 1868; Burley, 1977; 1981; Johnston e Johnson, 1989; National_Pigeon_Association, 2010).
Infine, il limite superiore della stima dell’età basata sulle LD dell’aplotipo a scacchiera di 1.432 anni fa indica che l’aplotipo a scacchiera è stato introdotto in C. liv ia ben dopo l’addomesticamento dei piccioni viaggiatori. Poiché gli intervalli di C. liv ia e C. guinea si sovrappongono in Africa settentrionale(del Hoyo et al., 2017), è possibile che si siano verificati eventi di introgressione nelle popolazioni in libertà. Tuttavia, la spiegazione più probabile è che gli aplotipi di C . gu inea sono stati introdotti in C. livia dagli allevatori di piccioni. Una volta generati gli ibridi maschili, ciò può essere realizzato in poche generazioni(Taibel, 1949). Così, gli esseri umani potrebbero aver intenzionalmente selezionato questo fenotipo, che è legato a tratti della storia della vita che sono vantaggiosi negli ambienti urbani, e poi costruito habitat urbani ideali per farli prosperare(Jerolmack, 2008).
Figura 5-figure supplement 1.Firme di introgressione dell’aplotipo a scacchiera da C. guinea a C. livia. 2. Numeri di SNPs tra le diverse combinazioni a coppie di barra omozigote, checker, e C. gu inea rappresentata in Figura 5C.Numeri di SNPs tra le diverse combinazioni a coppie di barra omozigote, checker, e C. guinea rappresentata in Figura 5C.Previsto (barra viola) e la proporzione osservata di siti di segregazione condivisi su 1.458 SNPs nella regione aplotipo minimo per diversi confronti pairwise tra gli assemblaggi del genoma de novo da breve lettura dati di resequencing per la barra, checker, e C. guinea.(A) ABBA-BABA test con C. liv ia (bar), C. livia (checker), C . guinea, e C. palumbus mostra elevata D-statistica nella regione candidato Scaffold 68. Sono mostrati tre test ABBA-BABA rappresentativi e la linea rossa tratteggiata segna la media D-statistica a livello genomico per 10 × 10 diverse combinazioni di bar e checker birds (ARC-STA, MAP-ORR, IRT-STA sono mostrati, dove ARC, MAP, e IRT sono campioni checker e STA e ORR sono campioni bar; vedere Metodi).(B) HybridCheck mostra una somiglianza di sequenza a coppie tra i siti informativi di una terzina di sequenza. Viene mostrata una terzina rappresentativa di bar (Fer_VA), checker (ARC), e C. guinea confronto. La traccia blu mostra la somiglianza di sequenza tra la barra e la pedina, la traccia viola mostra la somiglianza tra la barra e C. guinea, e la traccia gialla mostra la somiglianza di sequenza tra la pedina e C. guinea.(C) Aspettativa (barra viola) e proporzione osservata di siti di segregatura condivisi su 4261 SNP totali nella regione aplotipo minimo per diversi confronti a coppie tra e tra 16 barre, 11 zigrinatura e 1 C. guinea.(D) Piccione maculato(Columba guinea). Foto per gentile concessione di Kjeuring (licenza CC BY 3.0, https://creativecommons.org/licenses/by/3.0/legalcode). Foto ritagliata da ‘Speckled pigeon Columba guinea Table Mountain Table Mountain Cape Town’, https://en.wikipedia.org/wiki/Speckled_pigeon#/media/File:Speckledpigeon.JPG. Immagine con inserto in piuma degli autori.10.7554/eLife.34803.021Figure 5-source data 1.Numeri di SNPs tra le diverse combinazioni a coppie di barra omozigote, checker, e C. guinea rappresentata in Figura 5C .I cerchi riempiti denotano la somiglianza della sequenza nei confronti a coppie di assemblaggi de novo, e i diamanti denotano i confronti a coppie tra gli assemblaggi de novo e l’assemblaggio di riferimento, che ha l’aplotipo checker. La percentuale di somiglianza percentuale è molto più bassa in questo grafico rispetto alla Figura 5C perché il numero di polimorfismi tra i confronti a coppie è simile, ma il numero totale di siti di separazione è molto più basso in questo piccolo dataset rispetto al confronto tra molti genomi sequenziati. Nel dataset più grande rappresentato nella Figura 5C, il numero più alto di SNP è stato determinato in gran parte dalla diversità di sequenza tra i 16 bar di uccelli. Abbiamo identificato 105 SNPs negli assemblaggi de novo che non abbiamo chiamato nei dati di resequenziamento. Di questi, 31 differiscono nel confronto a coppie tra il checker e C. guinea assemblies, 84 differiscono tra checker e bar, e 98 differiscono tra bar e C. guinea. Totale dei siti polimorfici tra gli assemblaggi de novo nell’intersezione di 92.199 bp tra tutti e tre gli assemblaggi de novo: bar-checker, 1343; checker-C. guinea, 212; bar-C.guinea, 1361. Totale dei siti polimorfici tra gli assemblaggi de novo e il genoma di riferimento (checker): bar-reference, 1342; checker-reference, 11; C. guinea-reference, 205.
Figura 5-figure supplement 1.Figura 5—supplemento alla figura 1. Prevista (barra viola) e percentuale osservata di siti di segregazione condivisi su 1.458 SNPs nella regione aplotipo minimo per diversi confronti a coppie tra gli assemblaggi del genoma de novo a partire da dati di ricerca di breve lettura per barra, checker e C. guinea.I cerchi riempiti indicano la somiglianza della sequenza nei confronti a coppie degli assemblaggi de novo, e i diamanti indicano i confronti a coppie tra gli assemblaggi de novo e l’assemblaggio di riferimento, che ha l’aplotipo checker. La percentuale di somiglianza percentuale è molto più bassa in questo grafico rispetto alla Figura 5C perché il numero di polimorfismi tra i confronti a coppie è simile, ma il numero totale di siti di separazione è molto più basso in questo piccolo dataset rispetto al confronto tra molti genomi sequenziati. Nel dataset più grande rappresentato nella Figura 5C, il numero più alto di SNP è stato determinato in gran parte dalla diversità di sequenza tra i 16 bar di uccelli. Abbiamo identificato 105 SNPs negli assemblaggi de novo che non abbiamo chiamato nei dati di resequenziamento. Di questi, 31 differiscono nel confronto a coppie tra il checker e C. guinea assemblies, 84 differiscono tra checker e bar, e 98 differiscono tra bar e C. guinea. Totale dei siti polimorfici tra gli assemblaggi de novo nell’intersezione di 92.199 bp tra tutti e tre gli assemblaggi de novo: bar-checker, 1343; checker-C. guinea, 212; bar-C.guinea, 1361. Totale dei siti polimorfici tra gli assemblaggi de novo e il genoma di riferimento (checker): bar-reference, 1342; checker-reference, 11; C. guinea-reference, 205.
Introgressione e pleiotropia
I tratti adattativi possono sorgere attraverso nuove mutazioni o variazioni permanenti all’interno di una specie, e un numero crescente di studi indica introgressioni adattive tra vertebrati e altri organismi(Hedrick, 2013; Martin e Orgogozo, 2013; Harrison e Larson, 2014; Zhang et al., 2016). In alcuni casi, i loci introgressi sono associati a tratti adattativi nelle specie riceventi, tra cui la tolleranza ad alta quota nelle popolazioni umane tibetane di Denisovans(Huerta-Sánchez et al., 2014), la resistenza ai pesticidi anticoagulanti nel topo d’appartamento del topo algerino (Song et al.,2011; Liu et al., 2015), e la morfologia del becco tra le diverse specie di fringuelli di Darwin (Lamichhaney et al., 2015). Tra gli uccelli domestici, le introgressioni sono responsabili dei tratti di colore della pelle e del piumaggio nei polli e nei canarini, rispettivamente(Eriksson et al., 2008; Lopes et al., 2016). Gli alleli sottoposti a selezione artificiale in una specie addomesticata possono essere vantaggiosi anche in natura, così come nell’introgressione del colore scuro del mantello dai cani domestici ai lupi(Anderson et al., 2009) (tuttavia, il colore potrebbe in realtà essere un marker visivo per un tratto fisiologico vantaggioso conferito dallo stesso allele; Coulson et al., 2011).
In questo studio, abbiamo identificato una presunta introgressione in C. livia di C. guinea che è vantaggiosa sia in ambienti urbani artificiali (selezione da parte degli allevatori) sia in ambienti urbani a vita libera (selezione sessuale e naturale). Un cambiamento nel pattern di colore del piumaggio è una conseguenza fenotipica immediatamente evidente dell’allele a scacchiera, ma altri tratti sono collegati a questo pattern di pigmentazione. Ad esempio, i piccioni di razza checker e T-check hanno stagioni riproduttive più lunghe, fino a tutto l’anno in alcune località(Lofts et al., 1966; Murton et al., 1973), e C. guinea si riproduce tutto l’anno anche nella maggior parte della sua gamma nativa(del Hoyo et al., 2017). Forse non a caso, l’NDP è espresso nei tessuti delle gonadi di C. livia adulta(MacManes et al., 2017) e nel tratto riproduttivo di altre amnioti(Paxton et al., 2010). L’abrogazione dell’espressione o della funzione di NDP o del suo recettore FZD4 è associata a infertilità e difetti delle gonadi(Luhmann et al., 2005; Kaloglu et al., 2011; Ohlmann et al., 2012; Ohlmann e Tamm, 2012). Inoltre, gli uccelli di controllo e quelli di controllo a T depositano meno grasso durante i mesi invernali normalmente quiescenti per la riproduzione. Nell’uomo, i livelli di espressione di FZD4 e del co-recettore LRP5 nel tessuto adiposo rispondono a vari livelli di insulina(Karczewska-Kupczewska et al., 2016), e LRP5 regola la quantità e la posizione del deposito di tessuto adiposo(Loh et al., 2015; Karczewska-Kupczewska et al., 2016). Pertanto, sulla base dei suoi ruoli riproduttivi e metabolici nei piccioni e in altri amnioti, l’NDP è un valido candidato non solo per la variazione del pattern di colore, ma anche per la serie di altri tratti osservati nei piccioni checker (selvatici e selvatici) e nei piccioni T-check. Infatti, i potenziali effetti pleiotropici del NDP aumentano la possibilità che la produzione riproduttiva e altri vantaggi fisiologici siano obiettivi secondari o addirittura primari di selezione, e i fenotipi melanici sono segnali genetici onesti di un insieme di tratti adattativi controllati da un singolo locus.
Il cambiamento adattivo cis-regolatorio è anche un tema importante nell’evoluzione dei vertebrati e di altri animali(Shapiro et al., 2004; Miller et al., 2007; Wray, 2007; Carroll, 2008; Chan et al., 2010; Wittkopp e Kalay, 2011; O’Brown et al., 2015; Signor e Nuzhdin, 2018). Questo tema è particolarmente importante negli studi sulla variazione del colore nella Drosophila, in cui la variazione normativa ha un impatto sia sul tipo che sul modello dei pigmenti sul corpo e sulle ali(Gompel et al., 2005; Prud’homme et al., 2006; Rebeiz et al., 2009). In alcuni casi, l’evoluzione di più elementi regolatori dello stesso gene può mettere a punto fenotipi, come il colore del mantello del topo e la distribuzione dei tricomi nei moscerini della frutta(McGregor et al., 2007; Linnen et al., 2013). Nei casi di geni che hanno ruoli di sviluppo multipli, l’introgressione può portare al trasferimento simultaneo di molteplici tratti vantaggiosi(Rieseberg, 2011). Il ruolo potenziale del NDP sia nel piumaggio che nella variazione fisiologica dei piccioni potrebbe rappresentare un esempio lampante di effetti regolatori pleiotropici.
Modelli di pigmentazione alare che assomigliano a checker sono presenti in molte specie selvatiche all’interno e all’esterno di Columbidae tra cui Patagioenas maculosa (Piccione alato Spot), Spilopelia chinensis (Colomba maculata), Geopelia cuneata (Colomba diamante), Gyps rueppelli (Avvoltoio di Rüppell), e Pygiptila stellaris ( Picchio alato Spot). Sulla base dei nostri risultati nei piccioni, il NDP e i suoi bersagli a valle possono servire come geni candidati iniziali per chiedere se meccanismi molecolari simili generano modelli convergenti in altre specie.
Materiali e metodi
Dichiarazione etica
L’allevamento degli animali e le procedure sperimentali sono state eseguite in conformità con i protocolli approvati dal Comitato istituzionale per la cura e l’utilizzo degli animali dell’Università dello Utah (protocolli 10-05007, 13-04012 e 16-03010).
Raccolta ed estrazione del campione di DNA
Campioni di sangue sono stati raccolti nello Utah in occasione di mostre locali di piccioni, nelle case degli allevatori di piccioni locali, dai piccioni nel laboratorio di Shapiro e da animali selvatici catturati a Salt Lake City, Utah. Le foto di ogni uccello sono state scattate al momento della raccolta dei campioni per la nostra documentazione e per la verifica del fenotipo. Campioni di tessuto di C. rupestris, C. guinea e C. palumbus sono stati forniti rispettivamente dal Museo Burke dell’Università di Washington, dal Louisiana State University Museum of Natural Science e dalla voliera Tracy. Gli allevatori al di fuori dello Utah sono stati contattati via e-mail o per telefono per ottenere campioni di piume. Agli allevatori sono stati inviati pacchetti di raccolta di piume e istruzioni e i campioni di piume sono stati rispediti all’Università dello Utah insieme a informazioni fenotipiche dettagliate. Gli allevatori sono stati istruiti a inviare solo campioni che non erano imparentati con i nonni. Il DNA è stato poi estratto da sangue, tessuti e piume come descritto in precedenza(Stringham et al., 2012).
Determinazione del colore e del fenotipo del modello di uccelli adulti
Fenotipi di piume e colori degli uccelli sono stati designati dai rispettivi allevatori. Agli uccelli che sono stati allevati nella nostra struttura presso l’Università dello Utah o raccolti da popolazioni selvatiche è stato assegnato un fenotipo utilizzando riferimenti standard(Levi, 1986; Sell, 2012).
Analisi genomiche
I file BAM di un pannello di uccelli precedentemente sequenziati sono stati combinati con i file BAM di altri otto uccelli senza bar, 23 bar e 23 uccelli a dama(22 selvatici, 24 domestici), un singolo C. guinea, e un singolo C. palumbus. SNV e piccoli indel sono stati chiamati usando il Genome Analysis Toolkit (Unified Genotyper and LeftAlignAnd TrimVariants functions, default settings; McKenna et al., 2010). Le varianti sono state filtrate come descritto in precedenza(Domyan et al., 2016) e il successivo file VCF (Variant call format) è stato utilizzato per le analisi pFst e ABBA-BABA come parte della libreria software VCFLIB(https://github.com/vcflib) e VAAST(Yandell et al., 2011) come descritto in precedenza(Shapiro et al., 2013).
pFst è stata eseguita per la prima volta su genomi interi di 32 bar e 27 checker birds. Alcuni degli uccelli a scacchiera e dei bar sono stati sequenziati a bassa copertura (~1X), quindi non siamo stati in grado di definire con sicurezza i confini dell’aplotipo condiviso. Per ovviare a questo problema, abbiamo utilizzato il nucleo dell’aplotipo per identificare ulteriori uccelli a barre e zigrini da un insieme di uccelli che erano già stati sequenziati a una copertura più alta(Shapiro et al., 2013). Questi uccelli aggiuntivi non sono stati inclusi nella scansione iniziale perché i loro fenotipi del modello alare erano nascosti da altri tratti di colore e modello che sono epistatiche per barrare e controllare i fenotipi. Ad esempio, i loci rossi recessivi (e) e diffusi(S) producono un pigmento uniforme su tutto il corpo, oscurando così qualsiasi barra o dama(Van Hoosen Jones, 1922; Hollander, 1938a; Sell, 2012; Domyan et al., 2014). Sebbene il disegno delle ali principali non sia visibile in questi uccelli, la presenza o l’assenza dell’aplotipo della pedina di base ci ha permesso di caratterizzarli come barre o pedine. Abbiamo quindi ri-ran pFst utilizzando 17 bar e 24 uccelli checker/T-checker con almeno 8X copertura media di profondità di lettura(Figura 1B) e abbiamo trovato un aplotipo checker minimo condiviso di ~100 kb (Scaffold 68 posizione 1.702.691-1.805.600), come definito dai punti di rottura dell’aplotipo in un checker omozigote e un uccello bar omozigote (NCBI BioSamples SAMN01057561 e SAMN01057543, rispettivamente; BioProject PRJNA167554). pFst è stato anche utilizzato per confrontare i genomi di 32 bar e nove uccelli senza bar. I nuovi dati di sequenza per C. livia sono depositati nel database SRA del NCBI sotto BioProject PRJNA428271 con i numeri di adesione BioSample SAMN08286792- SAMN08286844. I nuovi dati di sequenza per C. guinea e C. palumbus sono depositati nella banca dati dell’NCBI SRA con i numeri di adesione SRS1416880 e SRS1416881, rispettivamente.
Pedigree di un controllore e di una barra di separazione intercrossa F2
Abbiamo genotipizzato e fenotipizzato un incrocio di laboratorio che separa i modelli a barre e a quadretti nella generazione F2. Abbiamo generato un pedigree da questa famiglia per gli individui F2 di cui abbiamo potuto identificare i fenotipi come bar o checker (n = 62). Non siamo riusciti a determinare i fenotipi a barra o a scacchiera per tutti gli individui perché anche altri pattern di pigmento che mascherano epistaticamente barra e scacchiera – mandorla (Stlocus ), spread (S), e rosso recessivo (E)– si stanno separando nella croce. Gli individui F2 sono stati esclusi dall’analisi se avevano uno di questi fenotipi di mascheramento, ma i genitoriF1 sono stati mantenuti se hanno prodotto la prole F2 con fenotipi a scacchiera o a barra. Abbiamo usato i primer che amplificano all’interno dell’aplotipo minimo (primer AV17, vedi file supplementare 1) per genotipo tutti gli individui F2, i loro genitoriF1 (n = 26) e i fondatori (n = 4) mediante il sequenziamento Sanger per l’aplotipo checker per valutare se l’aplotipo checker era segregato con fenotipo ad ala.
Identificazione del breakpoint CNV e analisi della profondità di lettura
I breakpoint approssimativi della regione CNV sono stati identificati alle posizioni 1.790.000 e 1.805.600 di Scaffold 68 utilizzando WHAM in genomi sequenziati di uccelli omozigoti da bar o da dama con una copertura superiore a 8x(Kronenberg et al., 2015). Per 12 bar, sette checker, e 2 T-check genomi risquenziati, sono stati generati 68 g di file di profondità Scaffold utilizzando VCFtools(Danecek et al., 2011). Sono stati specificati due sottoinsiemi: il primo conteneva il CNV e il secondo era esterno al CNV ed è stato utilizzato per la normalizzazione (posizioni 1.500.000-2.000.000.000 e 800.000-1.400.000, rispettivamente). La profondità di lettura nel CNV è stata normalizzata dividendo la profondità di lettura in questa regione per la profondità media di lettura dalla seconda regione (non-CNV), quindi moltiplicata per due per normalizzare per la diploidia.
Saggio Taqman per la variazione del numero di copie
La variazione del numero di copie è stata stimata utilizzando un test personalizzato Taqman Copy Number Assay (ID del test: cnvtaq1_CC1RVED; Applied Biosystems, Foster City, CA) per 93 uccelli fenotipizzati per categoria di pattern di pigmento delle ali e 89 uccelli la cui pigmentazione è stata quantificata dall’analisi delle immagini. Dopo l’estrazione del DNA, i campioni sono stati diluiti a 5 ng/μL. I campioni sono stati eseguiti in quadruplice secondo il protocollo del produttore.
Quantificazione del fenotipo del modello di pigmento
Al momento della raccolta del campione di sangue è stato fotografato lo scudo dell’ala destra (immagini in formato RAW di una Nikon D70 o di una fotocamera digitale Sony a6000). Utilizzando il software Photoshop (Adobe Systems, San Jose, CA), lo scudo dell’ala, compresa la barra (sulle piume secondarie nascoste) è stato isolato dal file RAW originale. Le immagini sono state regolate per rimuovere le ombre e il contrasto è stato impostato al 100%. L’immagine isolata dello scudo alare regolato è stata poi importata in ImageJ (imagej.nih.gov/) in formato JPEG. La profondità dell’immagine è stata impostata a 8 bit e abbiamo poi applicato il comando di soglia. La soglia è stata ulteriormente regolata a mano per catturare la zigrinatura e le particelle sono state analizzate utilizzando una dimensione minima di 50 pixel. Questa procedura ha calcolato l’area di pigmentazione del piumaggio scuro sullo scudo dell’ala. L’area totale dello scudo è stata calcolata utilizzando l’impostazione della soglia Huang e analizzando le particelle come prima (dimensione minima dei pixel di 50). Le particelle dell’area scura sono state divise per le particelle dell’area totale dell’ala, e poi moltiplicate per 100 per ottenere la percentuale di area scura sullo scudo dell’ala. Le misurazioni sono state effettuate in triplice copia per ogni uccello, e le percentuali medie di area scura per ogni uccello sono state usate per testare le associazioni tra numero di copie e fenotipo usando una regressione non lineare dei minimi quadrati.
qRT-PCR analisi dell’espressione genica
Due piume secondarie nascoste delle ali, ciascuna dagli scudi alare di 8 bar, sette checker, e 8 T-check uccelli sono stati spennati per stimolare la rigenerazione delle piume per gli esperimenti qRT-PCR. Nove giorni dopo la spennatura, rigenerante gemme di piume sono stati rimossi, il prossimale 5 mm è stato tagliato longitudinalmente, e gli esemplari sono stati conservati in RNAlater (Qiagen, Valencia, CA) a 4 ° C per un massimo di tre giorni. Successivamente, le cellule del collare sono stati rimossi, l’RNA è stato isolato, e mRNA è stato trascritto al contrario di cDNA come descritto in precedenza(Domyan et al., 2014). I primer Intron-spanning (vedi file supplementare 1) sono stati utilizzati per amplificare ogni target utilizzando uno strumento CFX96 qPCR e iTaq Universal Syber Green Supermix (Bio-Rad, Hercules, CA). I campioni sono stati eseguiti in duplicato e normalizzati a β-actina. Il valore medio è stato determinato e i risultati sono presentati come media ± S.E. per ogni fenotipo. I risultati sono stati confrontati utilizzando un test Wilcoxon Rank Sum e le differenze di espressione sono state considerate statisticamente significative se p<0,05.
Test di espressione allele-specifico
SNPs in NDP e EFHC2 sono stati identificati come diagnostici del bar o checker/T-check haplotypes da uccelli sequenziati. Gli uccelli eterozigoti sono stati identificati con il sequenziamento Sanger nella regione dell’aplotipo checker minimo (primer AV17, vedi file supplementare 1). Dodici uccelli eterozigoti checker e T-check sono stati poi verificati da ulteriori reazioni di Sanger (AV54 per NDP e AV97 per EFHC2, vedi file supplementare 1) per essere eterozigoti per gli SNP diagnostici in NDP e EFHC2. I saggi PyroMark Custom (Qiagen, Valencia, CA) sono stati progettati per ogni SNP utilizzando il software del produttore (file supplementare1). Il Pyrosequencing è stato eseguito su gDNA derivato da sangue e cDNA derivato da cellule del collare da 9 giorni di rigenerazione delle piume dello scudo alare utilizzando uno strumento PyroMark Q24 (Qiagen, Valencia, CA). Ulteriori pirosequenziamento è stato eseguito per 9 dei 12 degli uccelli originali da 9 giorni di rigenerazione delle piume dorsali e della coda seguendo lo stesso protocollo. Rapporti di intensità del segnale dai campioni di cDNA sono stati normalizzati ai rapporti dai campioni di gDNA corrispondenti per il controllo per il bias nell’amplificazione allelica. Rapporti normalizzati sono stati analizzati da Wilcoxon Rank Sum test. Abbiamo confrontato i rapporti di espressione di 1-copia checker:bar a 4-copia checker:bar per determinare se copie aggiuntive del CNV sono state associate ad una maggiore espressione dell’allele checker:bar. Abbiamo anche confrontato i rapporti di espressione 1-copy checker:bar e quattro rapporti di espressione copyer:bar con un rapporto 1:1 (espressione uguale di entrambi gli alleli) utilizzando il test Wilcoxon Rank Sum per determinare se i rapporti checker:bar misurati erano significativamente diversi dall’ipotesi nulla di espressione uguale di alleli bar e checker. Il rapporto 2-copy checker:bar non è stato confrontato in queste analisi perché c’era un solo campione. I rapporti di espressione degli alleli sono stati analizzati insieme per le copie 1, 2 e 4 utilizzando una regressione glm per determinare se il numero di copie CNV era associato ad un aumento dell’espressione dell’allele di controllo. I risultati sono stati considerati significativi se p<0,05.
Ricerca di sequenza migliorata
VISTA(https://enhancer.lbl.gov/)(Visel et al., 2007) e REPTILE(He et al., 2017) sono stati mappati con il genoma di riferimento del piccione utilizzando il bwa-mem(Li e Durbin, 2009). I file di output BAM sono stati filtrati per le regioni ortogonali di alta qualità e ulteriormente filtrati per gli allineamenti all’interno dell’aplotipo checker minimo su Scaffold 68(file supplementare 2).
Genotipizzazione e allineamenti NDP
Gli esoniNDP sono stati sequenziati utilizzando i primer nel file supplementare 1. Le coppie di primer sono state progettate utilizzando il genoma di riferimento dei piccioni viaggiatori (Cliv_1.0)(Shapiro et al., 2013). 2. I prodotti PCR sono stati purificati utilizzando un kit di purificazione QIAquick PCR (Qiagen, Valencia, CA) e Sanger sequenziato. Le sequenze di ogni esone sono state poi modificate per la qualità con Sequencher v.5.1 (GeneCodes, Ann Arbor, MI). Le sequenze sono state tradotte e allineate con SIXFRAME e CLUSTALW in SDSC Biology Workbench(http://workbench.sdsc.edu). Le sequenze di aminoacidi al di fuori di Columbidae sono state scaricate da Ensembl(www.ensembl.org).
Calcoli statistici D
L’intero genoma ABBA-BABA(https://github.com/vcflib) è stato eseguito su 10 × 10 combinazioni di bar e dama (file supplementare3) uccelli nella disposizione: bar, dama, C. guinea, C. palumbus. VCFLIB(https://github.com/vcflib) è stato utilizzato per lisciare i risultati grezzi ABBA-BABA in finestre da 1000 kb o 100 kb per le analisi del genoma intero o Scaffold 68 rispettivamente. Per ogni combinazione 10 × 10. Abbiamo calcolato la media Dstatisticaattraverso il genoma. Questi sono stati poi mediati per generare una media del genoma intero di D= 0,0212, contrassegnata come linea tratteggiata nella Figura 5A. Gli intervalli di fiducia sono stati generati tramite blocchi mobili bootstrap(Kunsch, 1989). Le dimensioni dei blocchi sono uguali alle finestre sopra riportate, con valori D-statistici ricampionati con sostituzione un numero di volte pari al numero di finestre in un campione. Nella Figura 5A, sono mostrati tre test rappresentativi ABBA-BABA per diverse combinazioni di bar e checker birds. I checker e i bar birds usati in ogni confronto rappresentativo sono: ARC-STA, SRS346901 e SRS346887; MAP-ORR, SRS346893 e SRS346881; IRT-STA, SRS346892 e SRS346887 rispettivamente. ARC, MAP e IRT sono omozigoti per l’aplotipo checker. STA e ORR sono omozigoti per l’aplotipo bar.
Fase dell’aplotipo e analisi HybridCheck
I file VCF contenenti i genotipi di Scaffold 68 per 16 bar, 11 checker omozigoti, e 1 C. guinea sono stati scaglionati utilizzando la versione 3.3 di Beagle(Browning e Browning, 2007). I VCF sono stati poi convertiti in formato fasta usando vcf2fasta in vcf-lib(https://github.com/vcflib). HybridCheck(Ward and van Oosterhout, 2016)(https://github.com/Ward9250/HybridCheck) è stato eseguito per visualizzare le somiglianze di sequenza a coppie tra i trii di bar, checker e le sequenze di C. gu inea attraverso Scaffold 68 utilizzando le impostazioni predefinite.
Confronti SNP a coppie
I file VCF a fasi per 16 bar omozigoti, 11 checker omozigoti e 1 C. guinea sono stati sottoinsimati alla regione aplotipo minimo checker (posizioni 1.702.691-1.805.600) con tabix (Li,2011). Il modulo software vcf-compare software (VCFtools,(Danecek et al., 2011) è stato utilizzato per eseguire confronti a coppie tra bar, checker e C. guinea (176 bar-checker, 16 bar-guinea e 11 checker-guinea) così come tra bar e checker (120 bar-bar e 55 checker-checker). Il numero totale di differenze per ogni gruppo è stato confrontato con il numero di differenze che si prevede si accumuleranno durante un tempo di divergenza di 4-5 MY in una regione di 102.909 bp (la dimensione dell’aplotipo minimo di zigrino) con il tasso di mutazione μ = 1,42e-9 (Shapiro et al.,2013) utilizzando l’equazione acoalescenza: Tempo= #SNPs/(2xμx lunghezza della regione). Le differenze osservate a coppie e il numero di differenze attese sono state valutate con test t a due campioni e tutti i gruppi sono stati considerati statisticamente diversi dall’aspettativa di 4-5 MY (1169.05-1461.31). Vi erano 4261 siti totali di segregazione nella regione a aplotipo minimo tra tutti gli uccelli utilizzati per il confronto a coppie. Mezzi e deviazioni standard per ogni gruppo sono stati calcolati in R(R_Development_Core_Team, 2008).
Confronti SNP in assemblaggi de novo di genomi di bar, checker e C. guinea
Per garantire che la chiamata SNP non fosse influenzata dall’uso di un riferimento che ha l’aplotipo del checker, abbiamo eseguito assemblaggi de novo di una barra (SRS346895), un checker (SRS346878), e un individuo C. guinea (SRS1416880) utilizzando CLC Genomics Workbench (Qiagen, Valencia, CA). Questi individui C. liv ia sono stati scelti perché avevano la più alta copertura di profondità media di lettura a livello genomico per ogni fenotipo a 14X (bar) e 15X (checker; il campione di C. guinea C. è stato sequenziato a 33X). Gli assemblaggi genomo-intero sono stati mappati sul genoma di riferimento e le varianti (varianti a singolo nucleotide, varianti strutturali, indel) sono state chiamate da SMARTIE-SV(https://github.com/zeeev/smartie-sv), che utilizza l’allineatore BLASR(Chaisson e Tesler, 2012), utilizzando parametri predefiniti. Abbiamo identificato le regioni in cui tutti e tre i nuovi assiemi si sono intersecati con l’assieme di riferimento. Abbiamo quindi contato gli SNP attraverso l’aplotipo minimo in cui tutti e tre gli assiemi si sono intersecati (92.199 di 102.909 bp; 12 contigui che si intersecano in lunghezza da 678 a 21565 bp, mediana = 5047,5).
Le varianti identificate negli assemblaggi de novo per gli individui di checker, bar o C. guinea sono state filtrate manualmente per rimuovere le varianti in cui l’allele alternativo era ‘N’ o una serie di coppie di base ‘N’. Le varianti che si estendono su più coppie di base in ogni singolo file sono state identificate e divise manualmente in più polimorfismi nucleotidici singoli. Le chiamate di varianti filtrate e suddivise in schede delimitate tra ogni assemblaggio de novo e il genoma di riferimento sono state lette in R v.3.3.2(R_Development_Core_Team, 2008). Per ogni file di chiamata di variante è stata estratta la posizione di partenza. I confronti a coppie di posizioni per gli assiemi checker, bar e C. guinea de novo sono stati fatti usando il comando ‘setdiff’ per generare liste di varianti che sono state osservate solo in un individuo su una data coppia (checker vs. bar, checker vs. C. guinea , bar vs. C. guinea ). Queste liste di posizioni sono state poi utilizzate per sottoinsimare i file di chiamata della variante originale e per assemblare liste di differenze di coppia. Per esempio, gli SNP che differiscono tra checker e bar includerebbero varianti che differiscono dal riferimento in checker, ma non bar, più varianti che differiscono dal riferimento in bar, ma non checker.
Inoltre, il comando ‘intersect’ è stato utilizzato per identificare le varianti in più assiemi de novo. Per le varianti che apparivano in più di un assemblaggio de novo, sono stati confrontati alleli alternativi per ogni assemblaggio. Nella maggior parte dei casi, entrambi gli assemblaggi de novo mostravano lo stesso allele alternativo, e quindi non differivano l’uno dall’altro.
Abbiamo trovato 1458 posizioni totali SNP basate sul confronto dei tre assemblaggi de novo. Nel confronto descritto sopra e mostrato nelle Figure 5C, sono stati identificati 362 SNP nella stessa regione. Questo numero più elevato di SNP è stato determinato dalla dimensione del campione molto più grande e dalla diversità di aplotipi tra i 16 uccelli bar.
Trascrizione dell’amplificazione dell’allele senza barra di NDP
Al fine di determinare se l’allele senza barra di NDP è trascritto e persiste nelle cellule del collare o è degradato (ad esempio, da decadimento non mediato dal senso), abbiamo progettato un test PCR per amplificare le trascrizioni degli mRNA di NDP. Le piume di quattro uccelli senza barra, 2 bar, due checker e 2 T-check sono state pizzicate per stimolare la rigenerazione. Abbiamo poi raccolto le piume rigenerate dopo 9 giorni, estratto l’RNA dalle cellule del collare, e sintetizzato il cDNA come descritto sopra. Abbiamo poi generato ampliconi da ogni campione utilizzando primer intron-spanning (AV200 primer, vedi file supplementare 1). I primer sono stati ancorati nell’esone contenente la mutazione del codice di partenza senza barre e l’esone 3′ ad esso, così questo saggio testato sia per la presenza di trascrizioni e di splicing coerente tra alleli e fenotipi.
Allineamenti EFHC2
EFHC2 sequenze esoniche da barra omozigote risquenziata (n = 16), controllo omozigote o T (n = 11), e senza barra (n = 9) Columba livia; C. rupestris (n = 1); C. guinea (n = 1); e C. palumbus (n = 1) sono stati estratti utilizzando il browser IGV (Thorvaldsdóttir et al.,2013). Le sequenze di esoni per ogni gruppo sono state tradotte utilizzando SIXFRAME in SDSC Biology Workbench(http://workbench.sdsc.edu). Le sequenze di peptidi sono state poi allineate alle sequenze di aminoacidi EFHC2 di altre specie scaricate da ensembl(http://www.ensembl.org) utilizzando CLUSTALW(Thompson et al., 1994) in SDSC Biology Workbench. Le sequenze di esoni da ulteriori C. livia (n = 17 checker o T-checker, e n = 14 bar) e C. guinea (n = 5) uccelli sono stati determinati da Sanger sequenziamento.
Stima del tasso di ricombinazione
Le stime della frequenza di ricombinazione sono state generate da una mappa genetica basata su un incrocio F2 di due razze di C. livia divergenti, un Puter Pomerania e uno Scandaroon(Domyan et al., 2016). In breve, per la costruzione della mappa genetica, i dati di genotipizzazione mediante sequenziamento (GBS) sono stati generati, tagliati e filtrati come descritto(Domyan et al., 2016), quindi mappati all’assemblaggio del genoma del piccione(Holt et al., 2018) utilizzando Bowtie2(Langmead e Salzberg, 2012). I genotipi sono stati chiamati utilizzando stack(Catchen et al., 2011), e la costruzione della mappa genetica è stata eseguita utilizzando R/qtl(www.rqtl.org)(Broman et al., 2003). Sono state calcolate frequenze di ricombinazione a coppie per tutti i marcatori basati sui genotipi GBS. All’interno delle singole impalcature, i marcatori sono stati filtrati per rimuovere i loci che mostravano una distorsione di segregazione (Chi-quadrato, p<0,01) o un probabile errore di genotipizzazione. In particolare, i marcatori sono stati rimossi se la caduta del marcatore ha portato ad un aumento del punteggio LOD, o se la rimozione di un marcatore non terminale ha portato ad una diminuzione della lunghezza di >10 cM che non era supportata dalla distanza fisica.Sono stati rimossianche i singoli genotipi con punteggi LOD di errore > 5 (Lincoln e Lander, 1992). Per stimare l’età dell’evento di introgressione tra C. guinea e C. livia (Scaffold 68, posizioni dei marcatori 1.017.014 e 1 . 971.666; file supplementare 4) sono state utilizzate le frequenze di ricombinazione a coppie per i marcatori che affiancano la regione candidata e che sono stati mantenuti nella mappa di collegamento finale.
Stima minima dell’età dell’aplotipo
L’età minima di aplotipo è stata stimata dopo Voight et al. (2006). Si assume una filogenesi a forma di stella, in cui tutti i campioni con l’aplotipo minimo sono identici all’evento di ricombinazione più vicino e si differenziano subito dopo di esso. Scegliendo una variante al centro dell’aplotipo minimo, abbiamo calcolato l’EHH, e stimato l’età utilizzando l’aplotipo più grande con una probabilità di omozigosi appena inferiore a 0,25. Si noti chePr[homoz]= e-2rgwhere r è la distanza della mappa genetica, e g è il numero di generazioni dall’introgressione/inizio della selezione. Thereforeg=-100log(Pr[homoz])2r
L’intervallo di confidenza intorno a g è stato stimato assumingN∼Binom(n=22, p=0,204)
Qui, N è una variabile casuale distribuita binomialmente per il numero di campioni che non sono ricombinati ad una distanza della mappa pari a 2 r. Poi, Pr[homoz]=N/22. La probabilità che un campione non abbia un evento di ricombinazione entro 2 r della SNP focale è p = (Pr[homoz | sinistra]+Pr[homoz | destra])/2 è derivata dai dati. Sia a sinistra che a destra dell’SNP focale abbiamo scelto la fine dell’aplotipo al primo SNP che ha portato Pr[homoz]<0,25.
References
- Adzhubei IA, Schmidt S, Peshkin L, Ramensky VE, Gerasimova A, Bork P, Kondrashov AS, Sunyaev SR. A method and server for predicting damaging missense mutations. Nature Methods. 2010; 7:248-249. DOI | PubMed
- Allen RC, Russell SR, Streb LM, Alsheikheh A, Stone EM. Phenotypic heterogeneity associated with a novel mutation (Gly112Glu) in the norrie disease protein. Eye. 2006; 20:234-241. DOI | PubMed
- Anderson TM, vonHoldt BM, Candille SI, Musiani M, Greco C, Stahler DR, Smith DW, Padhukasahasram B, Randi E, Leonard JA, Bustamante CD, Ostrander EA, Tang H, Wayne RK, Barsh GS. Molecular and evolutionary history of melanism in North American gray wolves. Science. 2009; 323:1339-1343. DOI | PubMed
- Berger W. Molecular dissection of norrie disease. Cells Tissues Organs. 1998; 162:95-100. DOI
- Blaya C, Moorjani P, Salum GA, Gonçalves L, Weiss LA, Leistner-Segal S, Manfro GG, Smoller JW. Preliminary evidence of association between EFHC2, a gene implicated in fear recognition, and harm avoidance. Neuroscience Letters. 2009; 452:84-86. DOI | PubMed
- Boer EF, Van Hollebeke HF, Shapiro MD. Genomic determinants of epidermal appendage patterning and structure in domestic birds. Developmental Biology. 2017; 429:409-419. DOI | PubMed
- Bonhote JL, Smalley FW. On colour and colour-pattern inheritance in pigeons. Proceedings of the Zoological Society of London. 1911; 81:601-626. DOI
- Broman KW, Wu H, Sen S, Churchill GA. R/qtl: qtl mapping in experimental crosses. Bioinformatics. 2003; 19:889-890. DOI | PubMed
- Browning SR, Browning BL. Rapid and accurate haplotype phasing and missing-data inference for whole-genome association studies by use of localized haplotype clustering. The American Journal of Human Genetics. 2007; 81:1084-1097. DOI | PubMed
- Burley N. Parental investment, mate choice, and mate quality. PNAS. 1977; 74:3476-3479. DOI | PubMed
- Burley N. Mate choice by multiple criteria in a monogamous species. The American Naturalist. 1981; 117:515-528. DOI
- Carroll SB. Evo-devo and an expanding evolutionary synthesis: a genetic theory of morphological evolution. Cell. 2008; 134:25-36. DOI | PubMed
- Catchen JM, Amores A, Hohenlohe P, Cresko W, Postlethwait JH. Stacks : building and genotyping loci de novo from Short-Read sequences. G3: Genes|Genomes|Genetics. 2011; 1:171-182. DOI | PubMed
- Chaisson MJ, Tesler G. Mapping single molecule sequencing reads using basic local alignment with successive refinement (BLASR): application and theory. BMC Bioinformatics. 2012; 13DOI | PubMed
- Chan YF, Marks ME, Jones FC, Villarreal G, Shapiro MD, Brady SD, Southwick AM, Absher DM, Grimwood J, Schmutz J, Myers RM, Petrov D, Jónsson B, Schluter D, Bell MA, Kingsley DM. Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science. 2010; 327:302-305. DOI | PubMed
- Chatelain M, Gasparini J, Frantz A. Do trace metals select for darker birds in urban areas? An experimental exposure to lead and zinc. Global Change Biology. 2016; 22:2380-2391. DOI | PubMed
- Chatelain M, Gasparini J, Jacquin L, Frantz A. The adaptive function of melanin-based plumage coloration to trace metals. Biology Letters. 2014; 10DOI | PubMed
- Chen CF, Foley J, Tang PC, Li A, Jiang TX, Wu P, Widelitz RB, Chuong CM. Development, regeneration, and evolution of feathers. Annual Review of Animal Biosciences. 2015; 3:169-195. DOI | PubMed
- Chen ZY, Hendriks RW, Jobling MA, Powell JF, Breakefield XO, Sims KB, Craig IW. Isolation and characterization of a candidate gene for norrie disease. Nature Genetics. 1992; 1:204-208. DOI | PubMed
- Coulson T, MacNulty DR, Stahler DR, vonHoldt B, Wayne RK, Smith DW. Modeling effects of environmental change on wolf population dynamics, trait evolution, and life history. Science. 2011; 334:1275-1278. DOI | PubMed
- Čanády A, Mošanský L. Population size and plumage polymorphism of feral pigeon (Columba livia forma urbana) from urban environment of Košice city (Slovakia). Zoology and Ecology. 2013; 23:104-110. DOI
- Danecek P, Auton A, Abecasis G, Albers CA, Banks E, DePristo MA, Handsaker RE, Lunter G, Marth GT, Sherry ST, McVean G, Durbin R, 1000 Genomes Project Analysis Group. The variant call format and VCFtools. Bioinformatics. 2011; 27:2156-2158. DOI | PubMed
- Darwin C. On the Origin of Species by Means of Natural Selection. John Murray: London; 1859.
- Darwin CR. The Variation of Animals and Plants Under Domestication. John Murray: London; 1868.
- Handbook of the Birds of the World Alive. Lynx Edicions: Barcelona; 2017.
- Deng C, Reddy P, Cheng Y, Luo CW, Hsiao CL, Hsueh AJ. Multi-functional norrin is a ligand for the LGR4 receptor. Journal of Cell Science. 2013; 126:2060-2068. DOI | PubMed
- Domyan ET, Guernsey MW, Kronenberg Z, Krishnan S, Boissy RE, Vickrey AI, Rodgers C, Cassidy P, Leachman SA, Fondon JW, Yandell M, Shapiro MD. Epistatic and combinatorial effects of pigmentary gene mutations in the domestic pigeon. Current Biology. 2014; 24:459-464. DOI | PubMed
- Domyan ET, Kronenberg Z, Infante CR, Vickrey AI, Stringham SA, Bruders R, Guernsey MW, Park S, Payne J, Beckstead RB, Kardon G, Menke DB, Yandell M, Shapiro MD. Molecular shifts in limb identity underlie development of feathered feet in two domestic avian species. eLife. 2016; 5DOI | PubMed
- Domyan ET, Shapiro MD. Pigeonetics takes flight: evolution, development, and genetics of intraspecific variation. Developmental Biology. 2017; 427:241-250. DOI | PubMed
- Durand EY, Patterson N, Reich D, Slatkin M. Testing for ancient admixture between closely related populations. Molecular Biology and Evolution. 2011; 28:2239-2252. DOI | PubMed
- Eom DS, Bain EJ, Patterson LB, Grout ME, Parichy DM. Long-distance communication by specialized cellular projections during pigment pattern development and evolution. eLife. 2015; 4DOI | PubMed
- Eriksson J, Larson G, Gunnarsson U, Bed’hom B, Tixier-Boichard M, Strömstedt L, Wright D, Jungerius A, Vereijken A, Randi E, Jensen P, Andersson L. Identification of the yellow skin gene reveals a hybrid origin of the domestic chicken. PLoS Genetics. 2008; 4DOI | PubMed
- Gierasch LM. Signal sequences. Biochemistry. 1989; 28:923-930. DOI | PubMed
- Gompel N, Prud’homme B, Wittkopp PJ, Kassner VA, Carroll SB. Chance caught on the wing: cis-regulatory evolution and the origin of pigment patterns in Drosophila. Nature. 2005; 433:481-487. DOI | PubMed
- Goodwin D. The colour-varieties of feral pigeons. London Bird Report. 1952; 16:35-36.
- Harris MP, Fallon JF, Prum RO. Shh-Bmp2 signaling module and the evolutionary origin and diversification of feathers. Journal of Experimental Zoology. 2002; 294:160-176. DOI | PubMed
- Harris MP, Williamson S, Fallon JF, Meinhardt H, Prum RO. Molecular evidence for an activator-inhibitor mechanism in development of embryonic feather branching. PNAS. 2005; 102:11734-11739. DOI | PubMed
- Harrison RG, Larson EL. Hybridization, introgression, and the nature of species boundaries. Journal of Heredity. 2014; 105:795-809. DOI | PubMed
- He Y, Gorkin DU, Dickel DE, Nery JR, Castanon RG, Lee AY, Shen Y, Visel A, Pennacchio LA, Ren B, Ecker JR. Improved regulatory element prediction based on tissue-specific local epigenomic signatures. PNAS. 2017; 114:E1633-E1640. DOI | PubMed
- Hedrick PW. Adaptive introgression in animals: examples and comparison to new mutation and standing variation as sources of adaptive variation. Molecular Ecology. 2013; 22:4606-4618. DOI | PubMed
- Hendrickx M, Leyns L. Non-conventional frizzled ligands and wnt receptors. Development, Growth & Differentiation. 2008; 50:229-243. DOI | PubMed
- Bird Coloration, Volume 2: Function and Evolution. Harvard University Press; 2006.
- Hollander WF, Miller WJ. Hereditary variants of behavior and vision in the pigeon. Iowa State Journal of Research. 1981; 55:323-331.
- Hollander WF. Hereditary Interrelationships of Certain Factors in Pigeons. Department of Genetics. University of Wisconsin; 1937.
- Hollander WF. Inheritance of certain "blue-black" patterns and "bleached" colorations in the domestic pigeon. Genetics. 1938a; 23:12-23. PubMed
- Hollander WF. Origins and Excursions in Pigeon Genetics. The Ink Spot, Burton: Kansas; 1983b.
- Holmes LB. Norrie’s disease: an X-linked syndrome of retinal malformation, mental retardation, and deafness. The Journal of Pediatrics. 1971; 79:89-92. DOI | PubMed
- Holt C, Campbell M, Keays DA, Edelman N, Kapusta A, Maclary E, T Domyan E, Suh A, Warren WC, Yandell M, Gilbert MTP, Shapiro MD. Improved genome assembly and annotation for the rock pigeon (Columba livia). G3: Genes|Genomes|Genetics. 2018; 8:1391-1398. DOI | PubMed
- Hubbard JK, Uy JA, Hauber ME, Hoekstra HE, Safran RJ. Vertebrate pigmentation: from underlying genes to adaptive function. Trends in Genetics. 2010; 26:231-239. DOI | PubMed
- Huerta-Sánchez E, Jin X, AsanBianba Z, Peter BM, Vinckenbosch N, Liang Y, Yi X, He M, Somel M, Ni P, Wang B, Ou X, HuasangLuosang J, Cuo ZX, Li K, Gao G, Yin Y, Wang W, Zhang X, Xu X, Yang H, Li Y, Wang J, Wang J, Nielsen R. Altitude adaptation in tibetans caused by introgression of Denisovan-like DNA. Nature. 2014; 512:194-197. DOI | PubMed
- Irwin MR, Cole LJ, Gordon CD. Immunogenetic studies of species and of species hybrids in pigeons, and the separation of species-specific characters in backcross generations. Journal of Experimental Zoology. 1936; 73:285-308. DOI
- Isashiki Y, Ohba N, Yanagita T, Hokita N, Doi N, Nakagawa M, Ozawa M, Kuroda N. Novel mutation at the initiation codon in the norrie disease gene in two Japanese families. Human Genetics. 1995; 95:105-108. DOI | PubMed
- Jacquin L, Récapet C, Bouche P, Leboucher G, Gasparini J. Melanin-based coloration reflects alternative strategies to cope with food limitation in pigeons. Behavioral Ecology. 2012; 23:907-915. DOI
- Janiga M. Nesting Mortality of Granivorous Birds Due to Microorganisms and Toxic Substances. Polish Ecological Publ: Warsaw; 1991.
- Jerolmack C. How pigeons became rats: the Cultural-Spatial logic of problem animals. Social Problems. 2008; 55:72-94. DOI
- Johnson KP. Deletion Bias in avian introns over evolutionary timescales. Molecular Biology and Evolution. 2004; 21:599-602. DOI | PubMed
- Johnston RF, Janiga M. Feral Pigeons. Oxford University Press; 1995.
- Johnston RF, Johnson SG. Nonrandom mating in feral pigeons. The Condor. 1989; 91:23-29. DOI
- Kaelin CB, Xu X, Hong LZ, David VA, McGowan KA, Schmidt-Küntzel A, Roelke ME, Pino J, Pontius J, Cooper GM, Manuel H, Swanson WF, Marker L, Harper CK, van Dyk A, Yue B, Mullikin JC, Warren WC, Eizirik E, Kos L, O’Brien SJ, Barsh GS, Menotti-Raymond M. Specifying and sustaining pigmentation patterns in domestic and wild cats. Science. 2012; 337:1536-1541. DOI | PubMed
- Kaloglu C, Cesur I, Bulut HE. Norrin immunolocalization and its possible functions in rat endometrium during the estrus cycle and early pregnancy. Development, Growth & Differentiation. 2011; 53:887-896. DOI | PubMed
- Karczewska-Kupczewska M, Stefanowicz M, Matulewicz N, Nikołajuk A, Strączkowski M. Wnt signaling genes in adipose tissue and skeletal muscle of humans with different degrees of insulin sensitivity. The Journal of Clinical Endocrinology & Metabolism. 2016; 101:3079-3087. DOI | PubMed
- Ke J, Harikumar KG, Erice C, Chen C, Gu X, Wang L, Parker N, Cheng Z, Xu W, Williams BO, Melcher K, Miller LJ, Xu HE. Structure and function of norrin in assembly and activation of a frizzled 4-Lrp5/6 complex. Genes & Development. 2013; 27:2305-2319. DOI | PubMed
- Kelsh RN, Harris ML, Colanesi S, Erickson CA. Stripes and belly-spots — a review of pigment cell morphogenesis in vertebrates. Seminars in Cell & Developmental Biology. 2009; 20:90-104. DOI | PubMed
- Kelsh RN. Genetics and evolution of pigment patterns in fish. Pigment Cell Research. 2004; 17:326-336. DOI | PubMed
- Kronenberg ZN, Osborne EJ, Cone KR, Kennedy BJ, Domyan ET, Shapiro MD, Elde NC, Yandell M. Wham: identifying structural variants of biological consequence. PLOS Computational Biology. 2015; 11DOI | PubMed
- Kunsch HR. The jackknife and the bootstrap for general stationary observations. The Annals of Statistics. 1989; 17:1217-1241. DOI
- Lamichhaney S, Berglund J, Almén MS, Maqbool K, Grabherr M, Martinez-Barrio A, Promerová M, Rubin CJ, Wang C, Zamani N, Grant BR, Grant PR, Webster MT, Andersson L. Evolution of Darwin’s finches and their beaks revealed by genome sequencing. Nature. 2015; 518:371-375. DOI | PubMed
- Langmead B, Salzberg SL. Fast gapped-read alignment with bowtie 2. Nature Methods. 2012; 9:357-359. DOI | PubMed
- Levi WM. The Pigeon. Levi Publishing Co., Inc: Sumter, SC; 1986.
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009; 25:1754-1760. DOI | PubMed
- Li H. Tabix: fast retrieval of sequence features from generic TAB-delimited files. Bioinformatics. 2011; 27:718-719. DOI | PubMed
- Lin CM, Jiang TX, Baker RE, Maini PK, Widelitz RB, Chuong CM. Spots and stripes: pleomorphic patterning of stem cells via p-ERK-dependent cell chemotaxis shown by feather morphogenesis and mathematical simulation. Developmental Biology. 2009; 334:369-382. DOI | PubMed
- Lin SJ, Foley J, Jiang TX, Yeh CY, Wu P, Foley A, Yen CM, Huang YC, Cheng HC, Chen CF, Reeder B, Jee SH, Widelitz RB, Chuong CM. Topology of feather melanocyte progenitor niche allows complex pigment patterns to emerge. Science. 2013; 340:1442-1445. DOI | PubMed
- Linck R, Fu X, Lin J, Ouch C, Schefter A, Steffen W, Warren P, Nicastro D. Insights into the structure and function of ciliary and flagellar doublet microtubules: tektins, Ca2+-binding proteins, and stable protofilaments. The Journal of Biological Chemistry. 2014; 289:17427-17444. DOI | PubMed
- Lincoln SE, Lander ES. Systematic detection of errors in genetic linkage data. Genomics. 1992; 14:604-610. DOI | PubMed
- Linnen CR, Poh YP, Peterson BK, Barrett RD, Larson JG, Jensen JD, Hoekstra HE. Adaptive evolution of multiple traits through multiple mutations at a single gene. Science. 2013; 339:1312-1316. DOI | PubMed
- Liu KJ, Steinberg E, Yozzo A, Song Y, Kohn MH, Nakhleh L. Interspecific introgressive origin of genomic diversity in the house mouse. PNAS. 2015; 112:196-201. DOI | PubMed
- Lofts B, Murton RK, Westwood NJ. Gonadal cycles and the evolution of breeding seasons in British Columbidae. Journal of Zoology. 1966; 150:249-272. DOI
- Loh NY, Neville MJ, Marinou K, Hardcastle SA, Fielding BA, Duncan EL, McCarthy MI, Tobias JH, Gregson CL, Karpe F, Christodoulides C. LRP5 regulates human body fat distribution by modulating adipose progenitor biology in a dose- and depot-specific fashion. Cell Metabolism. 2015; 21:262-273. DOI | PubMed
- Lopes RJ, Johnson JD, Toomey MB, Ferreira MS, Araujo PM, Melo-Ferreira J, Andersson L, Hill GE, Corbo JC, Carneiro M. Genetic basis for red coloration in birds. Current Biology. 2016; 26:1427-1434. DOI | PubMed
- Luhmann UF, Meunier D, Shi W, Lüttges A, Pfarrer C, Fundele R, Berger W. Fetal loss in homozygous mutant norrie disease mice: a new role of norrin in reproduction. Genesis. 2005; 42:253-262. DOI | PubMed
- MacManes MD, Austin SH, Lang AS, Booth A, Farrar V, Calisi RM. Widespread patterns of sexually dimorphic gene expression in an avian hypothalamic-pituitary-gonadal (HPG) axis. Scientific Reports. 2017; 7DOI | PubMed
- Mallarino R, Henegar C, Mirasierra M, Manceau M, Schradin C, Vallejo M, Beronja S, Barsh GS, Hoekstra HE. Developmental mechanisms of stripe patterns in rodents. Nature. 2016; 539:518-523. DOI | PubMed
- Manceau M, Domingues VS, Linnen CR, Rosenblum EB, Hoekstra HE. Convergence in pigmentation at multiple levels: mutations, genes and function. Philosophical Transactions of the Royal Society B: Biological Sciences. 2010; 365:2439-2450. DOI | PubMed
- Mangile RJ. What is "foggy" vision?. American Pigeon Journal. 1987;27-28.
- Martin A, Orgogozo V. The loci of repeated evolution: a catalog of genetic hotspots of phenotypic variation. Evolution. 2013; 22:1235-1250. DOI
- McGregor AP, Orgogozo V, Delon I, Zanet J, Srinivasan DG, Payre F, Stern DL. Morphological evolution through multiple cis-regulatory mutations at a single gene. Nature. 2007; 448:587-590. DOI | PubMed
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. The genome analysis toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Research. 2010; 20:1297-1303. DOI | PubMed
- McNeill B, Mazerolle C, Bassett EA, Mears AJ, Ringuette R, Lagali P, Picketts DJ, Paes K, Rice D, Wallace VA. Hedgehog regulates norrie disease protein to drive neural progenitor self-renewal. Human Molecular Genetics. 2013; 22:1005-1016. DOI | PubMed
- Meindl A, Lorenz B, Achatz H, Hellebrand H, Schmitz-Valckenberg P, Meitinger T. Missense mutations in the NDP gene in patients with a less severe course of norrie disease. Human Molecular Genetics. 1995; 4:489-490. DOI | PubMed
- Miller CT, Beleza S, Pollen AA, Schluter D, Kittles RA, Shriver MD, Kingsley DM. cis-Regulatory changes in kit ligand expression and parallel evolution of pigmentation in sticklebacks and humans. Cell. 2007; 131:1179-1189. DOI | PubMed
- Miller WJ. The time of appearance of Species-Specific antigens of Columba guinea in the Embryos of Backcross Hybrids. Physiological Zoology. 1953; 26:124-131. DOI
- Murton RK, Westwood NJ, Thearle RJ. Polymorphism and the evolution of a continuous breeding season in the pigeon, Columba livia. Journal of Reproduction and Fertility. Supplement. 1973; 19:563-577. PubMed
- National_Pigeon_Association. 2010 National Pigeon Association Book of Standards. Purebred Pigeon Publishing: Goodlettsville, TN; 2010.
- Niehrs C. Norrin and frizzled; a new vein for the eye. Developmental Cell. 2004; 6:453-454. PubMed
- Norrie G. Causes of blindness in children. Acta Ophthalmologica. 1927; 5:357-386. DOI
- O’Brown NM, Summers BR, Jones FC, Brady SD, Kingsley DM. A recurrent regulatory change underlying altered expression and wnt response of the stickleback armor plates gene EDA. eLife. 2015; 4DOI | PubMed
- Obukhova NY, Kreslavskii AG. Izmenchivost’ i nasledovanie okraski u sizykh golubei. Zoologicheskiĭ Zhurnal. 1984; 64:1685-1694.
- Ohlmann A, Merkl R, Tamm ER. Focus on molecules: norrin. Experimental Eye Research. 2012; 102:109-110. DOI | PubMed
- Ohlmann A, Tamm ER. Norrin: molecular and functional properties of an angiogenic and neuroprotective growth factor. Progress in Retinal and Eye Research. 2012; 31:243-257. DOI | PubMed
- Paxton CN, Bleyl SB, Chapman SC, Schoenwolf GC. Identification of differentially expressed genes in early inner ear development. Gene Expression Patterns. 2010; 10:31-43. DOI | PubMed
- Pejaver V, Urresti J, Lugo-Martinez J, Pagel KA, Lin GN, Nam H-J, Mort M, Cooper DN, Sebat J, Iakoucheva LM, Mooney SD, Radivojac P. MutPred2: inferring the molecular and phenotypic impact of amino acid variants. bioRxiv. 2017. DOI
- Petersen N, Williamson K. Polymorphism and breeding of the rock dove in the faroe islands. The Ibis. 1949a; 91:17-23.
- Podhradsky V. Influence of some exongenous factors on the pigmentation of some domesticated pigeon phenotypes. Biológia. 1968; 23:113-123.
- Poelstra JW, Vijay N, Hoeppner MP, Wolf JB. Transcriptomics of colour patterning and coloration shifts in crows. Molecular Ecology. 2015; 24:4617-4628. DOI | PubMed
- Protas ME, Patel NH. Evolution of coloration patterns. Annual Review of Cell and Developmental Biology. 2008; 24:425-446. DOI | PubMed
- Prud’homme B, Gompel N, Rokas A, Kassner VA, Williams TM, Yeh SD, True JR, Carroll SB. Repeated morphological evolution through cis-regulatory changes in a pleiotropic gene. Nature. 2006; 440:1050-1053. DOI | PubMed
- R_Development_Core_Team. R Foundation for Statistical Computing. Vienna, Austria; 2008.
- Rebeiz M, Pool JE, Kassner VA, Aquadro CF, Carroll SB. Stepwise modification of a modular enhancer underlies adaptation in a Drosophila population. Science. 2009; 326:1663-1667. DOI | PubMed
- Rieseberg L. Adaptive introgression: the seeds of resistance. Current Biology. 2011; 21:R581-R583. DOI | PubMed
- Rosenblum EB, Parent CE, Brandt EE. The molecular basis of phenotypic convergence. Annual Review of Ecology, Evolution, and Systematics. 2014; 45:203-226. DOI
- Roulin A, Ducrest AL. Genetics of colouration in birds. Seminars in Cell & Developmental Biology. 2013; 24:594-608. DOI | PubMed
- Sabeti PC, Varilly P, Fry B, Lohmueller J, Hostetter E, Cotsapas C, Xie X, Byrne EH, McCarroll SA, Gaudet R, Schaffner SF, Lander ES, Frazer KA, Ballinger DG, Cox DR, Hinds DA, Stuve LL, Gibbs RA, Belmont JW, Boudreau A, Hardenbol P, Leal SM, Pasternak S, Wheeler DA, Willis TD, Yu F, Yang H, Zeng C, Gao Y, Hu H, Hu W, Li C, Lin W, Liu S, Pan H, Tang X, Wang J, Wang W, Yu J, Zhang B, Zhang Q, Zhao H, Zhao H, Zhou J, Gabriel SB, Barry R, Blumenstiel B, Camargo A, Defelice M, Faggart M, Goyette M, Gupta S, Moore J, Nguyen H, Onofrio RC, Parkin M, Roy J, Stahl E, Winchester E, Ziaugra L, Altshuler D, Shen Y, Yao Z, Huang W, Chu X, He Y, Jin L, Liu Y, Shen Y, Sun W, Wang H, Wang Y, Wang Y, Xiong X, Xu L, Waye MM, Tsui SK, Xue H, Wong JT, Galver LM, Fan JB, Gunderson K, Murray SS, Oliphant AR, Chee MS, Montpetit A, Chagnon F, Ferretti V, Leboeuf M, Olivier JF, Phillips MS, Roumy S, Sallée C, Verner A, Hudson TJ, Kwok PY, Cai D, Koboldt DC, Miller RD, Pawlikowska L, Taillon-Miller P, Xiao M, Tsui LC, Mak W, Song YQ, Tam PK, Nakamura Y, Kawaguchi T, Kitamoto T, Morizono T, Nagashima A, Ohnishi Y, Sekine A, Tanaka T, Tsunoda T, Deloukas P, Bird CP, Delgado M, Dermitzakis ET, Gwilliam R, Hunt S, Morrison J, Powell D, Stranger BE, Whittaker P, Bentley DR, Daly MJ, de Bakker PI, Barrett J, Chretien YR, Maller J, McCarroll S, Patterson N, Pe’er I, Price A, Purcell S, Richter DJ, Sabeti P, Saxena R, Schaffner SF, Sham PC, Varilly P, Altshuler D, Stein LD, Krishnan L, Smith AV, Tello-Ruiz MK, Thorisson GA, Chakravarti A, Chen PE, Cutler DJ, Kashuk CS, Lin S, Abecasis GR, Guan W, Li Y, Munro HM, Qin ZS, Thomas DJ, McVean G, Auton A, Bottolo L, Cardin N, Eyheramendy S, Freeman C, Marchini J, Myers S, Spencer C, Stephens M, Donnelly P, Cardon LR, Clarke G, Evans DM, Morris AP, Weir BS, Tsunoda T, Johnson TA, Mullikin JC, Sherry ST, Feolo M, Skol A, Zhang H, Zeng C, Zhao H, Matsuda I, Fukushima Y, Macer DR, Suda E, Rotimi CN, Adebamowo CA, Ajayi I, Aniagwu T, Marshall PA, Nkwodimmah C, Royal CD, Leppert MF, Dixon M, Peiffer A, Qiu R, Kent A, Kato K, Niikawa N, Adewole IF, Knoppers BM, Foster MW, Clayton EW, Watkin J, Gibbs RA, Belmont JW, Muzny D, Nazareth L, Sodergren E, Weinstock GM, Wheeler DA, Yakub I, Gabriel SB, Onofrio RC, Richter DJ, Ziaugra L, Birren BW, Daly MJ, Altshuler D, Wilson RK, Fulton LL, Rogers J, Burton J, Carter NP, Clee CM, Griffiths M, Jones MC, McLay K, Plumb RW, Ross MT, Sims SK, Willey DL, Chen Z, Han H, Kang L, Godbout M, Wallenburg JC, L’Archevêque P, Bellemare G, Saeki K, Wang H, An D, Fu H, Li Q, Wang Z, Wang R, Holden AL, Brooks LD, McEwen JE, Guyer MS, Wang VO, Peterson JL, Shi M, Spiegel J, Sung LM, Zacharia LF, Collins FS, Kennedy K, Jamieson R, Stewart J, International HapMap Consortium. Genome-wide detection and characterization of positive selection in human populations. Nature. 2007; 449:913-918. DOI | PubMed
- Sell A. Pigeon Genetics: Applied Genetics in the Domestic Pigeon. Sell Publishing: Achim, Germany; 2012.
- Shapiro MD, Domyan ET. Domestic pigeons. Current Biology. 2013; 23:R302-R303. DOI | PubMed
- Shapiro MD, Kronenberg Z, Li C, Domyan ET, Pan H, Campbell M, Tan H, Huff CD, Hu H, Vickrey AI, Nielsen SC, Stringham SA, Hu H, Willerslev E, Gilbert MT, Yandell M, Zhang G, Wang J. Genomic diversity and evolution of the head crest in the rock pigeon. Science. 2013; 339:1063-1067. DOI | PubMed
- Shapiro MD, Marks ME, Peichel CL, Blackman BK, Nereng KS, Jónsson B, Schluter D, Kingsley DM. Genetic and developmental basis of evolutionary pelvic reduction in threespine sticklebacks. Nature. 2004; 428:717-723. DOI | PubMed
- Signor SA, Nuzhdin SV. The evolution of gene expression in Cis and trans. Trends in Genetics. 2018. DOI | PubMed
- Sims KB, Lebo RV, Benson G, Shalish C, Schuback D, Chen ZY, Bruns G, Craig IW, Golbus MS, Breakefield XO. The Norrie disease gene maps to a 150 kb region on chromosome Xp11.3. Human Molecular Genetics. 1992; 1:83-89. DOI | PubMed
- Smallwood PM, Williams J, Xu Q, Leahy DJ, Nathans J. Mutational analysis of Norrin-Frizzled4 recognition. Journal of Biological Chemistry. 2007; 282:4057-4068. DOI | PubMed
- Song Y, Endepols S, Klemann N, Richter D, Matuschka FR, Shih CH, Nachman MW, Kohn MH. Adaptive introgression of anticoagulant rodent poison resistance by hybridization between old world mice. Current Biology. 2011; 21:1296-1301. DOI | PubMed
- Startin CM, Fiorentini C, de Haan M, Skuse DH. Variation in the X-linked EFHC2 gene is associated with social cognitive abilities in males. PLoS One. 2015; 10DOI | PubMed
- Stringham SA, Mulroy EE, Xing J, Record D, Guernsey MW, Aldenhoven JT, Osborne EJ, Shapiro MD. Divergence, convergence, and the ancestry of feral populations in the domestic rock pigeon. Current Biology. 2012; 22:302-308. DOI | PubMed
- Taibel AM. Nuovi risultati d’incrocio diretto e reciproco fra Columba livia Domestica e Columba guinea. Archivio Zoologico Italiano. 1949; 34:431-476.
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Research. 1994; 22:4673-4680. DOI | PubMed
- Thorvaldsdóttir H, Robinson JT, Mesirov JP. Integrative genomics viewer (IGV): high-performance genomics data visualization and exploration. Briefings in Bioinformatics. 2013; 14:178-192. DOI | PubMed
- Van Hoosen Jones S. Studies on inheritance in pigeons. IV. Checks and bars and other modificaitons of black. Genetics. 1922; 7:466-507. PubMed
- Visel A, Minovitsky S, Dubchak I, Pennacchio LA. VISTA enhancer browser–a database of tissue-specific human enhancers. Nucleic Acids Research. 2007; 35:D88-D92. DOI | PubMed
- Voight BF, Kudaravalli S, Wen X, Pritchard JK. A map of recent positive selection in the human genome. PLoS Biology. 2006; 4DOI | PubMed
- Warburg M. Norrie’s Disease: A new hereditary bilateral pseudotumour of the retina. Acta Ophthalmologica. 1961; 399:757-772.
- Ward BJ, van Oosterhout C. HYBRIDCHECK: software for the rapid detection, visualization and dating of recombinant regions in genome sequence data. Molecular Ecology Resources. 2016; 16:534-539. DOI | PubMed
- Weir JT, Schluter D. Calibrating the avian molecular clock. Molecular Ecology. 2008; 17:2321-2328. DOI | PubMed
- Weiss LA, Purcell S, Waggoner S, Lawrence K, Spektor D, Daly MJ, Sklar P, Skuse D. Identification of EFHC2 as a quantitative trait locus for fear recognition in Turner syndrome. Human Molecular Genetics. 2007; 16:107-113. DOI | PubMed
- Whitman CO. Orthogenic Evolution of Pigeons. Posthumous Works of C.O. Whitman. Carnegie Inst: Washington, DC; 1919.
- Wittkopp PJ, Haerum BK, Clark AG. Evolutionary changes in cis and trans gene regulation. Nature. 2004; 430:85-88. DOI | PubMed
- Wittkopp PJ, Kalay G. Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nature Reviews Genetics. 2011; 13:59-69. DOI | PubMed
- Wittkopp PJ, Stewart EE, Arnold LL, Neidert AH, Haerum BK, Thompson EM, Akhras S, Smith-Winberry G, Shefner L. Intraspecific polymorphism to interspecific divergence: genetics of pigmentation in Drosophila. Science. 2009; 326:540-544. DOI | PubMed
- Wray GA. The evolutionary significance of cis-regulatory mutations. Nature Reviews Genetics. 2007; 8:206-216. DOI | PubMed
- Xu Q, Wang Y, Dabdoub A, Smallwood PM, Williams J, Woods C, Kelley MW, Jiang L, Tasman W, Zhang K, Nathans J. Vascular development in the retina and inner ear: control by norrin and Frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004; 116:883-895. PubMed
- Yamada T, Akamatsu H, Hasegawa S, Inoue Y, Date Y, Mizutani H, Yamamoto N, Matsunaga K, Nakata S. Melanocyte stem cells express receptors for canonical Wnt-signaling pathway on their surface. Biochemical and Biophysical Research Communications. 2010; 396:837-842. DOI | PubMed
- Yandell M, Huff C, Hu H, Singleton M, Moore B, Xing J, Jorde LB, Reese MG. A probabilistic disease-gene finder for personal genomes. Genome Research. 2011; 21:1529-1542. DOI | PubMed
- Yu M, Wu P, Widelitz RB, Chuong CM. The morphogenesis of feathers. Nature. 2002; 420:308-312. DOI | PubMed
- Zhang W, Dasmahapatra KK, Mallet J, Moreira GR, Kronforst MR. Genome-wide introgression among distantly related Heliconius butterfly species. Genome Biology. 2016; 17DOI | PubMed
- Zinn AR, Kushner H, Ross JL. EFHC2 SNP rs7055196 is not associated with fear recognition in 45,X turner syndrome. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics. 2008; 147B:507-509. DOI | PubMed
Fonte
Vickrey AI, Bruders R, Kronenberg Z, Mackey E, Bohlender RJ, et al. () Introgression of regulatory alleles and a missense coding mutation drive plumage pattern diversity in the rock pigeon. eLife 7e34803. https://doi.org/10.7554/eLife.34803





{kind=link}