
Introduzione
Nel corso degli ultimi 30 anni, abbiamo imparato molto sulla genetica del morbo di Alzheimer (AD), ma i meccanismi patologici che causano l’insorgenza della malattia e che definiscono la sua progressione culminante nella neurodegenerazione massiva e nella demenza debilitante rimangono poco conosciuti. Sappiamo che l’amiloide-β (Aβ), un prodotto di trasformazione proteolitico incline all’aggregazione che consiste in sequenze di juxtamembrane e circa 2/3 del segmento transmembrana della proteina precursore dell’amiloide più grande (APP), gioca un ruolo centrale nelle fasi iniziali, mentre nelle fasi successive della malattia – attraverso meccanismi che sono completamente oscuri – la proteina microtubule-associata τ inizia a formare aggregati intraneuronali, cioè i grovigli neurofibrillari, che possono diffondersi transsinapticamente (Selkoe eHardy, 2016). È questa τ-aggregazione, non le placche amiloidi, che si verificano all’inizio del processo di malattia, che si pensa sia la principale responsabile della morte delle cellule neuronali e della perdita di massa cerebrale e che più si avvicina alla progressione della demenza. Prima che diventino visibili come le placche rivelatrici e i grovigli che cementano la diagnosi di AD per il patologo, Aβ e τ sono presenti come aggregati oligomerici più piccoli che possono compromettere direttamente e profondamente le funzioni neuronali, perturbando l’omeostasi sinaptica Ca2+ (Kuchibhotlaet al., 2008). La disfunzione sinaptica risultante è ampiamente considerata come il primo stadio del processo patogenetico dell’AD e come una delle principali cause della sua manifestazione clinica come lieve deterioramento cognitivo (MCI)(Palop e Mucke, 2010; Shankar e Walsh, 2009).
Sebbene l’AD comporti quasi certamente un accumulo precoce di Aβ, solo un numero sempre più esiguo di individui che soffrono della forma viziosa e dominante di AD ad esordio precoce porta mutazioni in APP (Campion et al., 1999). Queste mutazioni missense aumentano invariabilmente la produzione di Aβ e portano alla deposizione precoce della placca, mentre un numero altrettanto piccolo di persone ha una variazione genetica in APP che abbassa la produzione di Aβ e quindi protegge dalla forma tardiva molto più frequente di AD (LOAD) (Jonssonet al., 2012). Numerosi altri determinanti genetici contribuiscono alla maggior parte del LOAD, che si sviluppa tipicamente dopo il 6° decennio. Il più importante di questi è il genotipo Apolipoproteina E ε4 (ApoE4) (Corderet al., 1993; Strittmatter et al., 1993).
L’ApoE è una proteina che trasporta lipidi e colesterolo che viene prodotta principalmente dal fegato ed è responsabile dell’omeostasi dei lipidi plasmatici(Mahley, 1988). Si verifica in tre isoforme principali nell’uomo conosciute come ApoE2, ApoE3 e ApoE4, con ApoE3 che è l’allele più frequente (~77% omozigosi) seguito da ApoE4 (~15 – 20% di frequenza dell’allele) che è presente in >50% del CARICO (Liu et al., 2013). L’effetto di ApoE4 sull’accumulazione di Aβ attraverso il turnover Aβ alterato, l’aumento dell’aggregazione e quindi la formazione di placche è dipendente dal dosaggio dell’allele e questo può spiegare in parte il suo effetto sull’età di insorgenza precoce della malattia (Corderet al., 1993). Tuttavia, le ApoE4 possono compromettere in modo indipendente la funzione delle sinapsi e l’omeostasi Ca2+ perturbando il trasporto endocitico e il riciclaggio dei recettori ApoE sinaptici e dei recettori eccitatori AMPA e NMDA di tipo glutammato che sono regolati da questi recettori ApoE e che sono di conseguenza intrappolati con essi nelle stesse vescicole(Chen et al., 2010).
La maggior parte dei recettori ApoE, che sono tutti membri della famiglia dei recettori delle lipoproteine a bassa densità (LDL), sono espressi nel cervello e diversi sono componenti intrinseci delle sinapsi eccitatorie dove sono presenti nei compartimenti presinaptico e postsinaptico (recensione in Lane-Donovanet al., 2014; Pohlkamp et al., 2017). Di questi, il recettore 2 ApoE (Apoer2, alias LRP8) è il meglio caratterizzato. È presente sia pre- che postsinapticamente dove funziona principalmente come recettore per Reelin(Bal et al., 2013; Beffert et al., 2005; Lane-Donovan e Herz, 2017). La bobina è una proteina secreta di grandi dimensioni che è essenziale per la formazione degli strati corticali durante lo sviluppo embrionale del cervello, dove serve come molecola guida nella regolazione della migrazione neuronale(D’Arcangelo et al., 1995; Del Río et al., 1997). Mentre il cervello continua a svilupparsi e a maturare postnatalmente, il suo modello di espressione cambia e la bobina è ora prodotta da un sottoinsieme di interneuroni GABAergici che sono sparsi nella neocorteccia e nell’ippocampo(Alcántara et al., 1998; Pesold et al., 1998; Pohlkamp et al., 2014). Nel cervello adulto, questo Reelin secreto ora funziona come un neuromodulatore segnalando attraverso Apoer2 e il suo membro della famiglia Vldlr strettamente correlati per attivare Src-famiglia tirosina chinasi direttamente nella sinapsi, che si traduce in un aumento del flusso di Ca2 + attraverso i recettori NMDA e quindi l’elevazione robusta e il mantenimento del potenziamento sinaptico(Chen et al., 2005; Hiesberger et al., 1999; Wasser e Herz, 2017). Questo è l’evento chiave nel mantenimento dell’omeostasi sinaptica che è compromessa da ApoE4 e che si verifica indipendentemente dall’accumulo di Aβ (Chenet al., 2005). Infatti, alterazioni specifiche di ApoE4 nella struttura cerebrale sono state riscontrate in bambini di età inferiore ai 2 anni (Dean etal., 2014; Shaw et al., 2007).
La base molecolare con cui ApoE4 causa l’alterazione del normale trasporto e del riciclaggio delle vescicole endosomiche è molto probabilmente il risultato della sua propensione a dispiegarsi e ad assumere una conformazione ‘fuso-globule’ all’ingresso in un ambiente acido (Morrowet al., 2002). ApoE4 differisce da ApoE3 per un singolo aminoacido, che altera il suo punto isoelettrico per far coincidere il suo punto isoelettrico con il pH di ~6,5 che è presente nell’endosoma precoce (Caseyet al., 2010; Ordovas et al., 1987). Abbiamo ipotizzato che questa neutralizzazione della carica isoelettrica renderebbe ApoE4 incline all’aggregazione, che potrebbe essere la base molecolare del difetto di riciclaggio indotto da ApoE4 e dipendente dal dosaggio del gene.
Il pH nell’endosoma iniziale è mantenuto dalle funzioni opposte della pompa protonica, che diminuisce il pH vescicolare, e dallo scambiatore Na+/H+ NHE6, che lo aumenta(Fuster e Alexander, 2014). Qui, abbiamo studiato il ruolo dell’inibizione di NHE6 come mezzo per abbassare il pH endosomico, lontano dal punto isoelettrico di ApoE4. Abbiamo scoperto che questo semplice intervento farmacologico rilascia il blocco endosomico di ApoE4, ripristina il normale traffico di recettori ApoE e glutammato nei neuroni e corregge i difetti funzionali in vitro e in vivo. Questi risultati suggeriscono l’inibizione di NHE6 come un nuovo approccio terapeutico razionale per invertire il rischio di AD imposto da ApoE4.
Risultati
ApoE interagisce e colocalizza con Apoer2 nei neuroni
L’ApoE interagisce genericamente con le ripetizioni di tipo legante di tipo ligando ricco di cisteina che sono onnipresenti in tutti i membri della famiglia dei recettori LDL(Blacklow, 2007). Abbiamo prima studiato l’interazione tra ApoE e il suo legante-recettore Apoer2 utilizzando un test di interazione in fase solida. A questo scopo, abbiamo usato particelle ApoE naturalmente secrete contenenti le tre isoforme ApoE comuni negli esseri umani (ApoE2, ApoE3 e ApoE4). Abbiamo scoperto che ApoE3 e ApoE4 interagiscono fortemente con Apoer2, mentre il legame ApoE2 era molto più debole(Figura 1A e B). Questi risultati sono coerenti con il legame stabilito in modo simile ad alta affinità di ApoE3 e ApoE4 al recettore LDL e l’affinità 100 volte ridotta di ApoE2(Rall e Mahley, 1992; Weisgraber, 1994). Così, Apoer2 è un recettore ad alta affinità ApoE e l’analisi di immunofluorescenza dei neuroni primari trattati con GFP-tagged ApoE3 di conseguenza ha mostrato la co-localizzazione di Apoer2 e ApoE3 in endosomi(Figura 1C, Video 1 e 2).
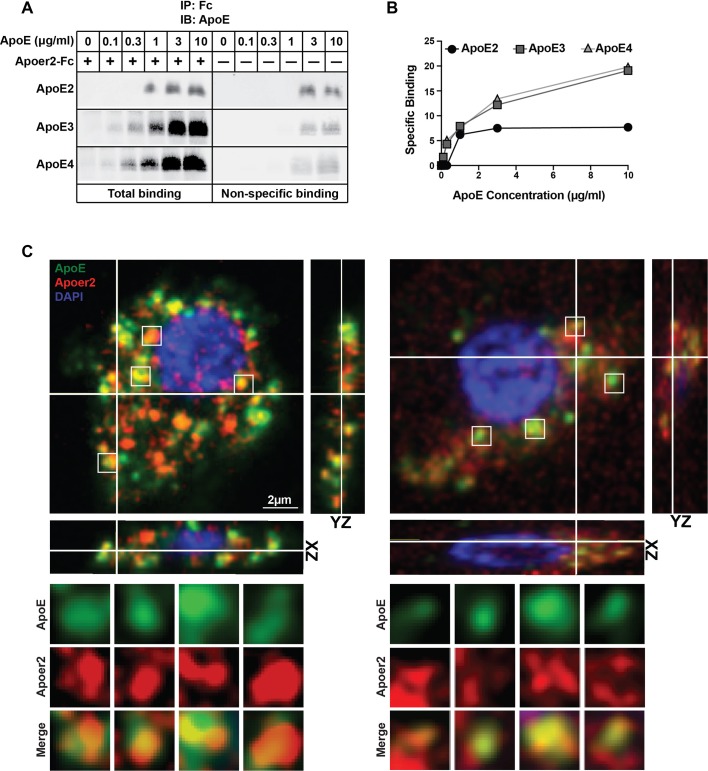
Figura 1 – Dati sorgente 1.2. Legame delle isoforme ApoE ad Apoer2.Legame delle isoforme ApoE ad Apoer2.Legame delle isoforme ApoE ad Apoer2.Legame delle isoforme ApoE ad Apoer2.Legame delle isoforme ApoE ad Apoer2.(A e B) Le isoforme ApoE interagiscono con il recettore 2 (Apoer2) come testato dalla co-immunoprecipitazione. ApoE3 e ApoE4 legano Apoer2 con affinità simile, mentre ApoE2 legano ad Apoer2 è scarsa. ApoE-condizionati media (0, 0,1, 0,3, 1, 3 e 10 µg/ml ApoE) sono stati incubati con Apoer2-Fc (secreto ectodominio Apoer2 ectodomain fuso a Fc) legato a proteina-G perline e tirato giù per eseguire immunoblotting per ApoE. Immagini rappresentative immunoblot(A) e la quantificazione(B) sono mostrati.(C) Apoer2 co-localizza con ApoE nei neuroni primari. I neuroni corticali primari sono stati infettati con mCherry-Apoer2 lentivirale (rosso) e successivamente trattati con ApoE3-GFP-condizionata media (verde). Un singolo piano di uno stack z è mostrato con le viste ortogonali xz- e yz-views come indicato. Le linee bianche indicano i tagli verticali e orizzontali. Le vescicole in scatola sono mostrate ingrandite nei pannelli sottostanti etichettati ApoE, Apoer2 e Merge. Inoltre, i filmati 3D delle celle sono forniti online(Video 1 e 2).10.7554/eLife.40048.004Figure 1-source data 1.Binding of ApoE Isoforms to Apoer2.

Video 1.2. Materiale di supporto per la Figura 1C. 2. Vista 3D di Apoer2 co-localizza con Apoe nei neuroni primari.N-terminale mCherry-labeled Apoer2 (rosso) e C-terminale GFP-labeled ApoE3 (verde) co-localizza intracellulare nei neuroni primari. Ratto neuroni corticali primari sono stati infettati con mCherry-Apoer2 lentivirale e successivamente esposti a ApoE3-GFP-condizionata media. Microscopia confocale è stata eseguita come descritto nella sezione Materiali e metodi.
Video 2.Materiale di supporto per laFigura 1C.Vista 3D di Apoer2 co-localizza con Apoe nei neuroni primari. N-terminale mCherry-labeled Apoer2 (rosso) e C-terminale GFP-labeled ApoE3 (verde) co-localizza intracellulare nei neuroni primari. Ratto neuroni corticali primari sono stati infettati con mCherry-Apoer2 lentivirale e successivamente esposti a ApoE3-GFP-condizionata media. Microscopia confocale è stata eseguita come descritto nella sezione Materiali e metodi.
Figura 1 – Dati fonte 1.2. Legame delle isoforme ApoE ad Apoer2.Legame delle isoforme ApoE ad Apoer2.Legame delle isoforme ApoE ad Apoer2.Legame delle isoforme ApoE ad Apoer2.Legame delle isoforme ApoE ad Apoer2.(A e B) Le isoforme ApoE interagiscono con il recettore 2 (Apoer2) come testato dalla co-immunoprecipitazione. ApoE3 e ApoE4 legano Apoer2 con affinità simile, mentre ApoE2 legano ad Apoer2 è scarsa. ApoE-condizionati media (0, 0,1, 0,3, 1, 3 e 10 µg/ml ApoE) sono stati incubati con Apoer2-Fc (secreto ectodominio Apoer2 ectodomain fuso a Fc) legato a proteina-G perline e tirato giù per eseguire immunoblotting per ApoE. Immagini rappresentative immunoblot(A) e la quantificazione(B) sono mostrati.(C) Apoer2 co-localizza con ApoE nei neuroni primari. I neuroni corticali primari sono stati infettati con mCherry-Apoer2 lentivirale (rosso) e successivamente trattati con ApoE3-GFP-condizionata media (verde). Un singolo piano di uno stack z è mostrato con le viste ortogonali xz- e yz-views come indicato. Le linee bianche indicano i tagli verticali e orizzontali. Le vescicole in scatola sono mostrate ingrandite nei pannelli sottostanti etichettati ApoE, Apoer2 e Merge. Inoltre, i filmati 3D delle celle sono forniti online(Video 1 e 2).10.7554/eLife.40048.004Figure 1-source data 1.Binding of ApoE Isoforms to Apoer2.
Video 1.2. Materiale di supporto per la Figura 1C. 2. Vista 3D di Apoer2 co-localizza con Apoe nei neuroni primari.N-terminale mCherry-labeled Apoer2 (rosso) e C-terminale GFP-labeled ApoE3 (verde) co-localizza intracellulare nei neuroni primari. Ratto neuroni corticali primari sono stati infettati con mCherry-Apoer2 lentivirale e successivamente esposti a ApoE3-GFP-condizionata media. Microscopia confocale è stata eseguita come descritto nella sezione Materiali e metodi.
Video 2.Materiale di supporto per laFigura 1C.Vista 3D di Apoer2 co-localizza con Apoe nei neuroni primari. N-terminale mCherry-labeled Apoer2 (rosso) e C-terminale GFP-labeled ApoE3 (verde) co-localizza intracellulare nei neuroni primari. Ratto neuroni corticali primari sono stati infettati con mCherry-Apoer2 lentivirale e successivamente esposti a ApoE3-GFP-condizionata media. Microscopia confocale è stata eseguita come descritto nella sezione Materiali e metodi.
ApoE4 riduce selettivamente l’espressione della superficie cellulare di Apoer2 neuronale
Abbiamo già segnalato in precedenza che la presenza di particelle di ApoE4 recettore che legano le particelle competenti ApoE4 a concentrazioni fisiologiche compromette il riciclaggio e di conseguenza l’espressione superficiale di Apoer2 neuronale(Chen et al., 2010). Per verificare se anche altri recettori non correlati che non interagiscono con ApoE possono essere influenzati nelle loro proprietà di espressione della membrana plasmatica e di riciclaggio, abbiamo eseguito gli esperimenti di biotinilazione della superficie cellulare descritti nella Figura 2A. In breve, i neuroni corticali primari di ratto sono stati incubati in assenza(Figura 2B, corsia 1) o la presenza di cellule di derivazione cellulare, naturalmente secreto ricombinante ApoE3 (corsia 2) o ApoE4 (corsia 3) particelle per 1 ora a 37 ˚ C, Reelin purificato è stato poi aggiunto per indurre l’endocitosi rapida di Apoer2, e dopo 30 minuti le cellule sono state trasferite a 4 ˚ C e lavate con PBS freddo come il ghiaccio. La biotinilazione della superficie cellulare è stata eseguita, le proteine biotinilate sono state isolate e rilevate tramite immunoblotting. Mentre Apoer2 rapidamente riciclato in presenza di ApoE3, la sua ricomparsa sulla superficie cellulare è stata notevolmente ritardata in presenza di ApoE4(Figura 2B,C). Al contrario, altri recettori della superficie cellulare endocitica che non legano ApoE, come il recettore dell’insulina (IR) o il recettore della transferrina (TfR), o che non interagiscono con la bobina e quindi non subiscono l’endocitosi indotta da ligandi (proteina 1 a bassa densità del recettore delle lipoproteine (Lrp1) e il recettore delle lipoproteine a bassa densità (Ldlr)), non sono stati significativamente influenzati dalla presenza di ApoE (Figura 2B,C).
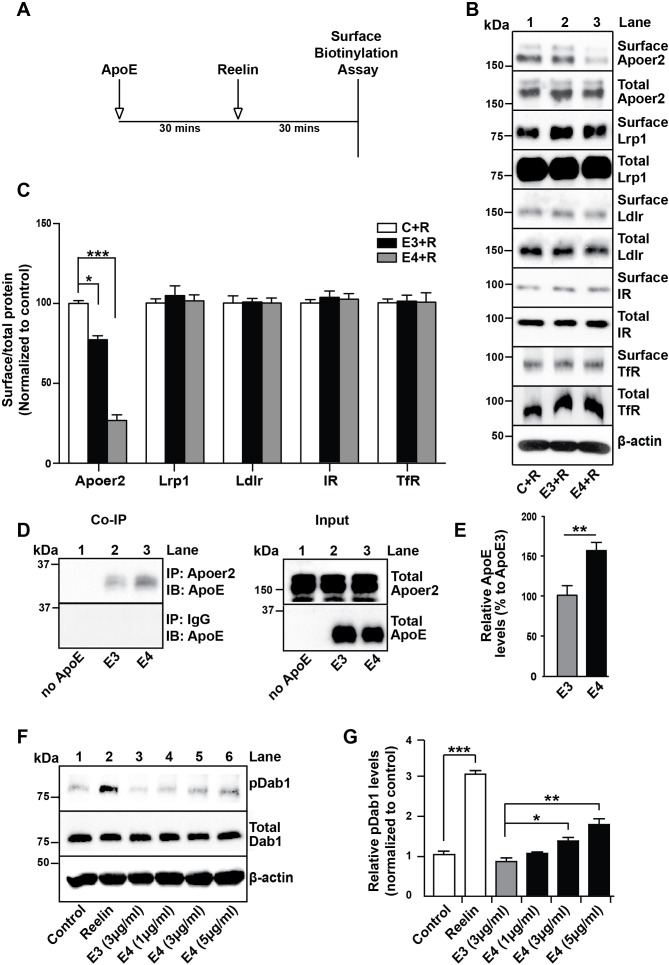
Figura 2 dati fonte 1.ApoE4 Impedisce il riciclaggio del recettore della bobina Apoer2.ApoE4 Impedisce il riciclaggio del recettore della bobina Apoer2.ApoE4 Impedisce il riciclaggio del recettore della bobina Apoer2.ApoE4 Impedisce il riciclaggio del recettore della bobina Apoer2.(A) Timeline per l’esperimento mostrato in B e C.(B e C) Le isoforme Apolipoproteine E (ApoE) riducono l’espressione superficiale di Apoer2. Il trattamento dei media condizionati con ApoE riduce l’espressione superficiale di Apoer2 in presenza di Reelin nei neuroni primari. I livelli di superficie di Apoer2 mostrano una riduzione maggiore con ApoE4 rispetto ad ApoE3. Altri recettori ApoE, come la proteina 1 (Lrp1) e il recettore delle lipoproteine a bassa densità (Ldlr), così come il recettore endocitico per la transferrina (TfR) e il recettore dell’insulina (IR) mostrano livelli superficiali comparabili in presenza di ApoE3 o ApoE4. I livelli di proteine di superficie e proteine totali sono stati analizzati mediante immunoblotting con anticorpi sollevati contro Apoer2, Lrp1, Ldlr, IR e TfR. L’analisi quantitativa del rapporto tra i livelli dei recettori superficiali e totali è mostrato(C).(D ed E) Le proteine dei neuroni primari incubati con mezzi condizionati con ApoE sono state immunoprecipitate con anti-Apoer2 o coniglio di controllo IgG e immunoblotate con anticorpo anti-ApoE. L’input è mostrato nel pannello di destra di(D) e la quantificazione in(E).(F e G) ApoE4, ma non ApoE3, induce la fosforilazione di Dab1 indipendentemente dalla bobina. I neuroni primari sono stati incubati con mezzi condizionati con ApoE o Reelin e testati per phospho-Dab1 e Dab1 totale. L’analisi quantitativa è mostrata(G). Tutti i dati sono espressi come media ± SEM da tre esperimenti indipendenti. *p<0.05, **p<0.01, ***p<0.001. L’analisi statistica è stata effettuata utilizzando il test a senso unico ANOVA e il test post-hoc di Dunnett (Ce G) o il t-test dello studente (E).10.7554/eLife.40048.008Cifra dati a 2 fonti 1ApoE4 Impedisce il riciclaggio del recettore della bobina Apoer2.
Figura 2 dati fonte 1.ApoE4 Impedisce il riciclaggio del recettore della bobina Apoer2.ApoE4 Impedisce il riciclaggio del recettore della bobina Apoer2.ApoE4 Impedisce il riciclaggio del recettore della bobina Apoer2.ApoE4 Impedisce il riciclaggio del recettore della bobina Apoer2.(A) Timeline per l’esperimento mostrato in B e C.(B e C) Le isoforme Apolipoproteine E (ApoE) riducono l’espressione superficiale di Apoer2. Il trattamento dei media condizionati con ApoE riduce l’espressione superficiale di Apoer2 in presenza di Reelin nei neuroni primari. I livelli di superficie di Apoer2 mostrano una riduzione maggiore con ApoE4 rispetto ad ApoE3. Altri recettori ApoE, come la proteina 1 (Lrp1) e il recettore delle lipoproteine a bassa densità (Ldlr), così come il recettore endocitico per la transferrina (TfR) e il recettore dell’insulina (IR) mostrano livelli superficiali comparabili in presenza di ApoE3 o ApoE4. I livelli di proteine di superficie e proteine totali sono stati analizzati mediante immunoblotting con anticorpi sollevati contro Apoer2, Lrp1, Ldlr, IR e TfR. L’analisi quantitativa del rapporto tra i livelli dei recettori superficiali e totali è mostrato(C).(D ed E) Le proteine dei neuroni primari incubati con mezzi condizionati con ApoE sono state immunoprecipitate con anti-Apoer2 o coniglio di controllo IgG e immunoblotate con anticorpo anti-ApoE. L’input è mostrato nel pannello di destra di(D) e la quantificazione in(E).(F e G) ApoE4, ma non ApoE3, induce la fosforilazione di Dab1 indipendentemente dalla bobina. I neuroni primari sono stati incubati con mezzi condizionati con ApoE o Reelin e testati per phospho-Dab1 e Dab1 totale. L’analisi quantitativa è mostrata(G). Tutti i dati sono espressi come media ± SEM da tre esperimenti indipendenti. *p<0.05, **p<0.01, ***p<0.001. L’analisi statistica è stata effettuata utilizzando il test a senso unico ANOVA e il test post-hoc di Dunnett (Ce G) o il t-test dello studente (E).10.7554/eLife.40048.008Cifra dati a 2 fonti 1ApoE4 Impedisce il riciclaggio del recettore della bobina Apoer2.
Ritenzione e attivazione prolungata di Apoer2 da parte di ApoE4
Il riciclaggio ritardato di Apoer2 è stato anche evidente dalla ritenzione prolungata di ApoE4 di derivazione cellulare rispetto a ApoE3 di derivazione cellulare in compartimenti intracellulari contenenti Apoer2 come mostrato dalla co-immunoprecipitazione dei lisati neuronali(Figura 2D,E). Che questa ritenzione prolungata può essere causata dal parziale dispiegamento di ApoE4 è ulteriormente supportato da un esperimento in cui sono state aggiunte quantità crescenti di particelle di ApoE4 naturalmente secreti, recettore vincolante competente ApoE4 sono stati aggiunti ai neuroni corticali primari e fosforilazione tirosina di Dab1 è stata misurata. Dab1 si lega al motivo NPxY nel dominio citoplasmatico di Apoer2 e quando i recettori sono raggruppati, ad esempio interagendo con Reelin, Dab1 subisce la transfosforilazione su residui di tirosina (Hiesbergeret al., 1999; Howell et al., 1997). Abbiamo ipotizzato che ApoE4 nel suo stato fuso-globulare, cioè in endosomi acidi, potrebbe analogamente indurre il raggruppamento dei recettori in modo dose-dipendente, mentre ApoE3 non lo farebbe. Quando abbiamo trattato i neuroni primari con ApoE4, Dab1 fosforilazione è stato effettivamente aumentato come previsto(Figura 2F,G).
Farmacologico NHE inibizione NHE e acidificazione del pH vescicolare ripristina il traffico Apoer2 in presenza di ApoE4
Il lavoro pionieristico di Weisgraber e colleghi ha rivelato la propensione di ApoE4 a diventare strutturalmente labile e a trasformarsi in uno stato fuso-globulare in un ambiente a basso pH, mentre ApoE2 e ApoE3 erano molto più resistenti allo sviluppo a basso pH indotto(Morrow et al., 2002). Inoltre, abbiamo notato che il punto isoelettrico (IEP) di ApoE4 si trova vicino al pH nel primo endosoma(Figura 3A). Molte proteine sono noti per perdere idrofilia vicino al loro IEP. Infatti, la prima purificazione dell’insulina dipendeva da questo fenomeno biofisico(Wintersteiner e Abramson, 1933). Abbiamo quindi ipotizzato che la labilità strutturale di ApoE4, combinata con una ridotta solubilità nell’ambiente endosomico acido, potrebbe essere un fattore trainante per il conseguente blocco di riciclaggio. La regolazione del pH nell’endosoma è ottenuta attraverso una combinazione di due meccanismi primari: l’attività della pompa protonica, cioè v-ATPase e lo scambio di protoni per Na+ o K+ attraverso l’attività degli scambiatori Na+/H+ (NHE) (Figura 3B). L’interruzione funzionale dell’isoforma endosomica NHE6 riduce così il pH endosomico(Brett et al., 2002) e si prevede che ripristini la solubilità di ApoE4 e quindi il traffico di vescicole(Figura 3C).
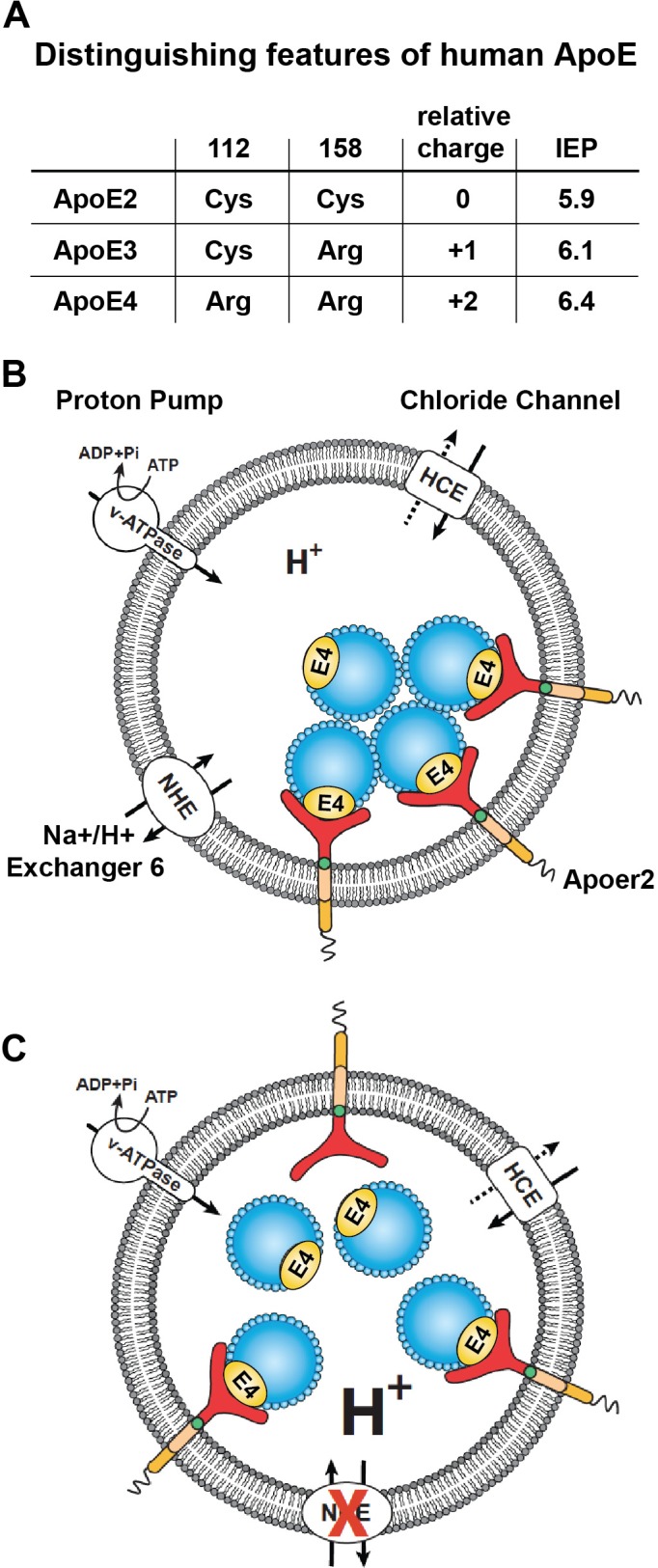
Figura 3.Modello di lavoro che illustra il meccanismo eptesico del difetto di traffico vescicolare causato dal traffico di Apoe4 umano.(A) Le cisteine/arginine ai residui 112 e 158 rappresentano la differenza tra la carica relativa e il punto isoelettrico (IEP) delle isoforme ApoE umane.(B) Gli aggregati endosomiali ApoE4/ApoE2 si formano dopo l’acidificazione. Il pH endosomico è regolato dal vacuolare di tipo H+-ATPase (vATPase, pompa protonica) e dagli scambiatori organellari Na+/H+ (NHE, perdita protonica). Dopo il legame con ApoE4, Apoer2 subisce l’endocitosi, viene sequestrato negli endosomi e il riciclaggio viene ritardato.(C) Gli endosomi ApoE4/ApoE4/Apoer2 si risolvono quando il pH viene ulteriormente abbassato. Acidificazione accelerata attraverso l’attività di inibizione NHE6 promuove la dissociazione di ApoE4 e Apoer2, con conseguente riciclaggio efficiente di Apoer2 torna alla membrana del plasma cellulare.
Gli inibitori specifici di NHE6 non esistono attualmente, tuttavia, esiste un gran numero di inibitori per l’abbondante isoforma NHE1, che si esprime sulla membrana plasmatica della maggior parte dei tipi di cellule e che regola il pH citosolico(Masereel et al., 2003). Gli inibitori di NHE1, come l’epiteliale sodio-antagonista e il derivato amiloride del guanidinio, sono stati utilizzati nella pratica clinica per mezzo secolo come diuretici, e numerosi analoghi sono stati sviluppati nel corso degli anni. Abbiamo ragionato sul fatto che alcuni di questi analoghi potrebbero inibire l’NHE6 in modo incrociato e quindi abbiamo deciso di testarli nel nostro saggio di riciclaggio Apoer2. Abbiamo trovato diversi analoghi dell’amiloride efficaci nel ripristinare il riciclaggio di Apoer2 e abbiamo scelto l’EMD87580 per un’analisi dettagliata(Figura 4A). Concentrazioni crescenti hanno progressivamente ripristinato il normale riciclaggio superficiale di Apoer2 in presenza di ApoE4(Figura 4B,C). Come mostrato nella Figura 4D ed E, anche le concentrazioni fisiologiche di ApoE3 (5 µg/ml) hanno compromesso il riciclo di Apoer2 in misura piccola ma significativa. Questo è stato anche impedito da EMD87580 alla stessa concentrazione (3 µM) alla quale l’effetto di ApoE4 sul riciclo di Apoer2 è stato completamente neutralizzato (Figura 4D,E), suggerendo che anche ApoE3, anche se in misura minore, può perdere la solubilità all’ingresso dell’endosoma precoce, e che questo è anche impedito da un abbassamento del pH. Per confermare che l’effetto dell’inibitore NHE è stato causato dall’alterazione del pH e non da una modalità d’azione non correlata abbiamo fatto l’esperimento inverso. Abbiamo incubato i neuroni con ApoE4 in presenza di 3 µM EMD87580 e di concentrazioni crescenti di Bafilomicina, un inibitore della pompa protonica. Figura 4F e G mostrano che l’aumento risultante del pH endosomico inverte la correzione del deficit di riciclaggio di ApoE4 da EMD87580 e si traduce in un rinnovato deterioramento del traffico di Apoer2. Ciò è stato ulteriormente confermato da un ulteriore esperimento in cui abbiamo incubato i neuroni in presenza di una concentrazione fissa di ApoE4 in presenza o meno di 50 nM Bafilomicina. In presenza di questa parziale inibizione dell’attività della pompa protonica, sono state richieste concentrazioni più elevate di EMD87580 per ripristinare il normale riciclaggio di Apoer2(Figura 4H) come risulta evidente dallo spostamento a destra della curva dose-risposta(Figura 4I). Presi insieme, questi dati mostrano che il pH vescicolare è il fattore trainante primario che determina in che misura ApoE4 altera il traffico endosomico.
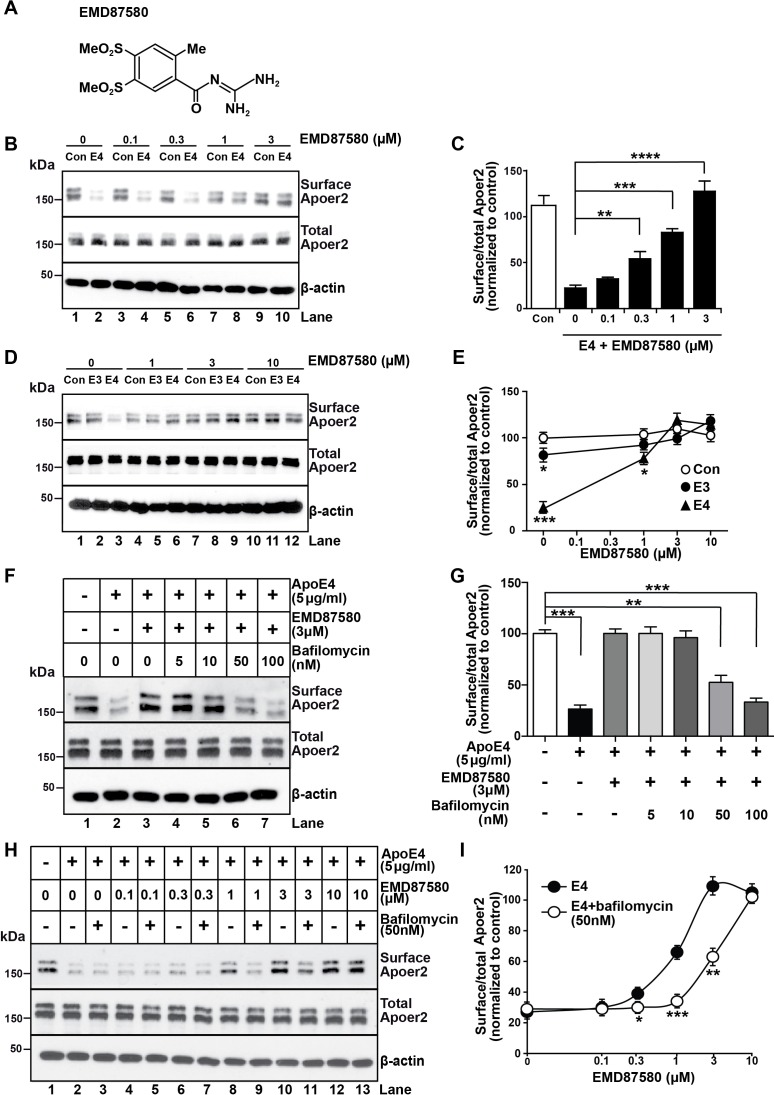
Figura 4 dati fonte 1.L’inibitore NHE EMD87580 impedisce l’intrappolamento intracellulare di ApoE4 e del suo recettore Apoer2.L’inibitore NHE EMD87580 impedisce l’intrappolamento intracellulare di ApoE4 e del suo recettore Apoer2.L’inibitore NHE EMD87580 impedisce l’intrappolamento intracellulare di ApoE4 e del suo recettore Apoer2.(A) Struttura chimica dell’inibitore NHE EMD87580.(B e C) EMD87580 aumenta l’espressione superficiale indotta dalla bobina di Apoer2 nei neuroni trattati con ApoE4 in modo dose-dipendente. I neuroni primari sono stati pretrattati con EMD87580 alle concentrazioni indicate e poi incubati con la bobina con o senza ApoE4 di derivazione cellulare. Livelli di superficie e totale Apoer2 sono stati analizzati mediante immunoblotting.(D ed E) L’effetto di EMD87580 sul traffico di Apoer2 indotto dalla bobina in presenza di ApoE3 o ApoE4. Le cellule neuronali primarie sono state trattate con EMD87580, Reelin e sia ApoE3- o ApoE4-media condizionata ApoE4.(F e G) Bafilomicina, un inibitore della pompa protonica, contrasta l’effetto di EMD87580 sul riciclaggio Apoer2 in modo dose-dipendente. I neuroni primari sono stati pretrattati con o senza bafilomicina in presenza o meno di EMD87580 e successivamente incubati con ApoE4 e Reelin. Livelli di superficie e totale Apoer2 sono stati analizzati mediante immunoblotting.(H e I) La bafilomicina sposta la curva di risposta della dose EMD87580 dell’espressione superficiale di Apoer2. Tutti i valori sono espressi come media ±SEM da tre esperimenti indipendenti. *p<0.05, **p<0.01, ***p<0.001. L’analisi statistica è stata effettuata utilizzando il test a senso unico ANOVA e il test post-hoc di Dunnett (C, Ee G) o il t-test dello studente (I).10.7554/eLife.40048.011Figure 4-source data 1.The NHE inibitore EMD87580 impedisce l’intrappolamento intracellulare di ApoE4 e il suo recettore Apoer2.
Figura 3.Figura 3. Modello di lavoro che illustra il meccanismo eptesico del difetto di traffico vescicolare causato da Apoe4 umano.(A) Cisteine/arginine ai residui 112 e 158 rappresentano la differenza di carica relativa e punto isoelettrico (IEP) delle isoforme ApoE umane.(B) Gli aggregati endosomiali ApoE4/ApoE2 si formano dopo l’acidificazione. Il pH endosomico è regolato dal vacuolare di tipo H+-ATPase (vATPase, pompa protonica) e dagli scambiatori organellari Na+/H+ (NHE, perdita protonica). Dopo il legame con ApoE4, Apoer2 subisce l’endocitosi, viene sequestrato negli endosomi e il riciclaggio viene ritardato.(C) Gli endosomi ApoE4/ApoE4/Apoer2 si risolvono quando il pH viene ulteriormente abbassato. Acidificazione accelerata attraverso l’attività di inibizione NHE6 promuove la dissociazione di ApoE4 e Apoer2, con conseguente riciclaggio efficiente di Apoer2 torna alla membrana del plasma cellulare.
Figura 4 dati fonte 1.L’inibitore NHE EMD87580 impedisce l’intrappolamento intracellulare di ApoE4 e il suo recettore Apoer2.L’inibitore NHE EMD87580 impedisce l’intrappolamento intracellulare di ApoE4 e il suo recettore Apoer2.L’inibitore NHE EMD87580 impedisce l’intrappolamento intracellulare di ApoE4 e il suo recettore Apoer2.(A) Struttura chimica dell’inibitore NHE EMD87580.(B e C) EMD87580 aumenta l’espressione superficiale indotta dalla bobina di Apoer2 nei neuroni trattati con ApoE4 in modo dose-dipendente. I neuroni primari sono stati pretrattati con EMD87580 alle concentrazioni indicate e poi incubati con la bobina con o senza ApoE4 di derivazione cellulare. Livelli di superficie e totale Apoer2 sono stati analizzati mediante immunoblotting.(D ed E) L’effetto di EMD87580 sul traffico di Apoer2 indotto dalla bobina in presenza di ApoE3 o ApoE4. Le cellule neuronali primarie sono state trattate con EMD87580, Reelin e sia ApoE3- o ApoE4-media condizionata ApoE4.(F e G) Bafilomicina, un inibitore della pompa protonica, contrasta l’effetto di EMD87580 sul riciclaggio Apoer2 in modo dose-dipendente. I neuroni primari sono stati pretrattati con o senza bafilomicina in presenza o meno di EMD87580 e successivamente incubati con ApoE4 e Reelin. Livelli di superficie e totale Apoer2 sono stati analizzati mediante immunoblotting.(H e I) La bafilomicina sposta la curva di risposta della dose EMD87580 dell’espressione superficiale di Apoer2. Tutti i valori sono espressi come media ±SEM da tre esperimenti indipendenti. *p<0.05, **p<0.01, ***p<0.001. L’analisi statistica è stata effettuata utilizzando il test a senso unico ANOVA e il test post-hoc di Dunnett (C, Ee G) o il t-test dello studente (I).10.7554/eLife.40048.011Figure 4-source data 1.The NHE inibitore EMD87580 impedisce l’intrappolamento intracellulare di ApoE4 e il suo recettore Apoer2.
shRNA knockdown di NHE6 è sufficiente per ripristinare il normale traffico Apoer2 normale
Nei mammiferi esistono più di 10 diversi NHE(Fuster e Alexander, 2014), che funzionano nella regolazione del pH intracellulare, organellare ed extracellulare in tutte le cellule del corpo e in una varietà di organi. EMD87580, come la maggior parte degli altri inibitori NHE disponibili in commercio, è stato sviluppato con l’obiettivo di inibire l’NHE1(Chen et al., 2004) e la sua capacità di inibire in modo incrociato altre forme di NHE è sconosciuta. Poiché ApoE4 blocca Apoer2 e il riciclo dei recettori del glutammato, e l’endosoma precoce è il primo organulo acido che ApoE4 incontra durante l’endocitosi, abbiamo sospettato che l’effetto di EMD87580 per ripristinare il normale traffico dei recettori sia dovuto all’inibizione di NHE6. Tuttavia, non abbiamo potuto escludere che altri NHE vescicolari, in particolare l’NHE9, che risiede nel Golgi e l’endosoma tardivo, partecipino al rilascio del blocco di riciclaggio. Per determinare se l’inibizione dell’NHE6 è sufficiente a ripristinare il normale riciclaggio di Apoer2, abbiamo utilizzato shRNA progettati contro tutti gli NHE vescicolari presenti nei compartimenti intracellulari che ApoE e Apoer2 potrebbero incontrare durante il loro passaggio attraverso il percorso di riciclaggio. La figura 5A-D mostra che di tutti i NHE (da 1 e da 5 a 9) che sono stati presi di mira dall’inibizione degli shRNA, solo gli shRNA specifici per NHE6 sono stati in grado di ripristinare completamente l’espressione di Apoer2 alla membrana plasmatica in presenza di ApoE4 (figura 5A, corsia 6, 5C, corsie 8, 10, 12, e quantificati nei pannelli B e D). Tre diversi shRNA diretti contro NHE6 sono stati utilizzati in Figura 5C e D. Nessuno degli shRNA diretti contro NHE1, 5, 7, 8 o 9 ha avuto alcun effetto sulla superficie Apoer2 espressione, né in assenza di ApoE4 o in sua presenza.
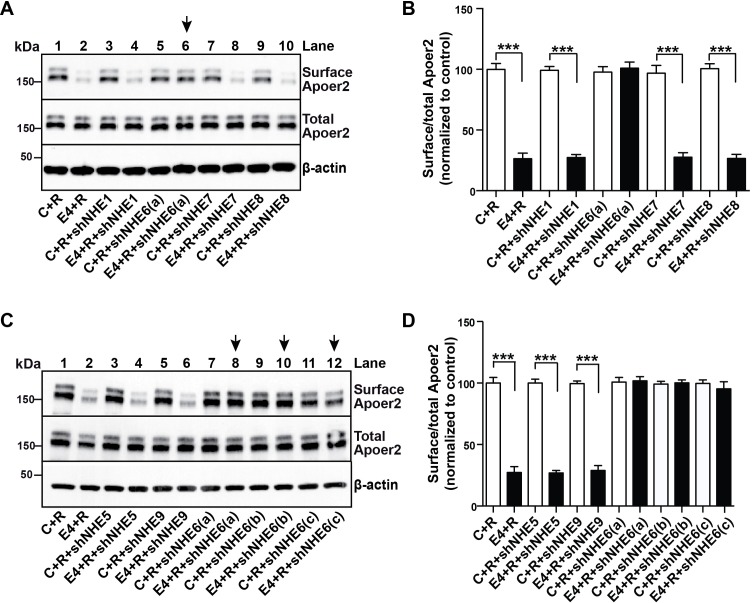
Figura 5 – Dati fonte 1.Figura 5—dati fonte 1. Un ruolo specifico per NHE6 nel traffico di Apoer2.Un ruolo specifico per NHE6 nel traffico di Apoer2.(A e C) shRNA abbattimento di NHE6, ma non altri NHE (NHE1, 5, 7, 8, 9) ripristina il riciclaggio di ApoE4-impaiati Apoer2. Gli shRNA mediati dal lentivirus che colpiscono NHE1, 5, 6, 7, 8 o 9 sono stati applicati ai neuroni primari. Le cellule sono state poi trattate con ApoE4-condizionati media e Reelin, e la superficie delle cellule e Apoer2 totale sono stati determinati da immunoblotting. Le frecce indicano le condizioni con livelli di superficie Apoer2 ripristinati. Tre diversi costrutti shRNA contro NHE6 hanno mostrato una significativa attenuazione dei livelli di superficie delle cellule Apoer2 (shNHE6 a, b, c).(B e D) Analisi quantitativa di(A) e(C). Tutti i dati sono espressi come media ±SEM di tre esperimenti indipendenti. ***p<0.001. L’analisi statistica è stata eseguita utilizzando il t-test dello studente (Be D).10.7554/eLife.40048.013Cifre dati a 5 fonti 1.A Ruolo specifico per NHE6 nel traffico Apoer2.
Figura 5 – dati fonte 1.Figura 5—dati fonte 1. Un ruolo specifico per NHE6 nel traffico Apoer2.(A e C) shRNA abbattimento di NHE6, ma non altri NHE (NHE1, 5, 7, 8, 9) ripristina il riciclaggio di ApoE4-impaiati Apoer2. Gli shRNA mediati dal lentivirus che colpiscono NHE1, 5, 6, 7, 8 o 9 sono stati applicati ai neuroni primari. Le cellule sono state poi trattate con ApoE4-condizionati media e Reelin, e la superficie delle cellule e Apoer2 totale sono stati determinati da immunoblotting. Le frecce indicano le condizioni con livelli di superficie Apoer2 ripristinati. Tre diversi costrutti shRNA contro NHE6 hanno mostrato una significativa attenuazione dei livelli di superficie delle cellule Apoer2 (shNHE6 a, b, c).(B e D) Analisi quantitativa di(A) e(C). Tutti i dati sono espressi come media ±SEM di tre esperimenti indipendenti. ***p<0.001. L’analisi statistica è stata eseguita utilizzando il t-test dello studente (Be D).10.7554/eLife.40048.013Cifre dati a 5 fonti 1.A Ruolo specifico per NHE6 nel traffico Apoer2.
Il knockdown shRNA di NHE6 ripristina il normale traffico di Apoer2, AMPA e recettori NMDA in presenza di ApoE4
Successivamente, abbiamo determinato se l’inibizione shRNA-mediata della funzione NHE6 avrebbe anche ripristinato il normale riciclaggio dei recettori del glutammato di tipo AMPA e NMDA nei neuroni primari trattati con ApoE4 e Reelin. Tre diversi shRNA che hanno ridotto in modo efficiente l’espressione della proteina NHE6 di almeno il 90%(Figura 6A) e un shRNA di controllo strapazzato sono stati utilizzati nel saggio di riciclaggio della superficie del recettore(Figura 6B, corsie 4 – 8). L’inibizione farmacologica della funzione NHE con EMD87580 è stata utilizzata come controllo (corsia 3). EMD87580 e il NHE6-shRNA specifici shRNA completamente restaurato Apoer2 e l’espressione superficiale del recettore del glutammato, mentre il shRNA strapazzato non ha avuto alcun effetto (pannello B e quantificato in pannelli C-F). Questi risultati hanno suggerito che l’inibizione di NHE6 potrebbe essere efficace nel ripristinare una normale risposta sinaptica in topi di sostituzione ApoE4 mirati, che abbiamo precedentemente dimostrato sono completamente resistenti al potenziamento a lungo termine (LTP) potenziamento da Reelin(Chen et al., 2010; Lane-Donovan et al., 2014).
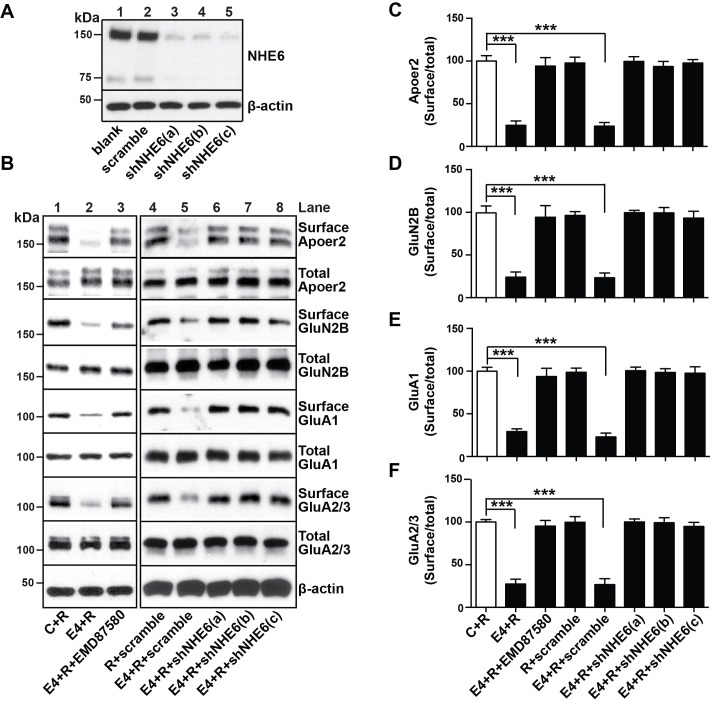
Figura 6 – dati fonte 1.Figura 6—dati fonte 1. Il knockdown NHE6 allevia i deficit di traffico di superficie indotti da ApoE4.NHE6 knockdown allevia i deficit di traffico di superficie indotti da ApoE4.NHE6 knockdown allevia i deficit di traffico di superficie indotti da ApoE4.NHE6 knockdown.(A) L’efficienza di knockdown shRNA lentivirale shRNA di espressione della proteina NHE6 nei neuroni corticali primari di ratto.(B) ShRNA lentivirale diretto contro NHE6 ripristina il deficit di traffico indotto da ApoE4 ApoE4 deficit di traffico di recettori di superficie. I neuroni corticali primari sono stati infettati con tre diversi shRNA lentivirali diretti contro NHE6 (shNHE6 a, b e c; corsie 6-7) o il controllo shRNA strapazzato (corsie 4, 5). Le colture infette sono state trattate senza (corsie 1 e 4) o con (corsie 2, 3, 5-8) ApoE4 e Reelin di derivazione cellulare (tutte le corsie) e il saggio di biotinilazione della superficie cellulare è stato eseguito per Apoer2, GluN2B, GluA1 e GluA2/3.(D-F) Analisi quantitativa del segnale immunoblot da (B). Tutti i dati sono espressi come media ±SEM da tre esperimenti indipendenti. *p<0.05, **p<0.01, ***p<0.001. L’analisi statistica è stata effettuata utilizzando l’ANOVA unidirezionale e il test post-hoc di Dunnett (C-F).10.7554/eLife.40048.015Figure 6-source data 1.NHE6 knockdown allevia i deficit di traffico in superficie indotti da ApoE4.
Figura 6 – dati fonte 1.Il knockdown di NHE6 allevia il deficit di traffico in superficie indotto da ApoE4.NHE6 riduce il deficit di traffico in superficie indotto da ApoE4.NHE6 riduce il deficit di traffico in superficie indotto da ApoE4.NHE6.(A) L’efficienza di knockdown shRNA lentivirale shRNA di espressione della proteina NHE6 nei neuroni corticali primari di ratto.(B) ShRNA lentivirale diretto contro NHE6 ripristina il deficit di traffico indotto da ApoE4 ApoE4 deficit di traffico di recettori di superficie. I neuroni corticali primari sono stati infettati con tre diversi shRNA lentivirali diretti contro NHE6 (shNHE6 a, b e c; corsie 6-7) o il controllo shRNA strapazzato (corsie 4, 5). Le colture infette sono state trattate senza (corsie 1 e 4) o con (corsie 2, 3, 5-8) ApoE4 e Reelin di derivazione cellulare (tutte le corsie) e il saggio di biotinilazione della superficie cellulare è stato eseguito per Apoer2, GluN2B, GluA1 e GluA2/3.(D-F) Analisi quantitativa del segnale immunoblot da (B). Tutti i dati sono espressi come media ±SEM da tre esperimenti indipendenti. *p<0.05, **p<0.01, ***p<0.001. L’analisi statistica è stata effettuata utilizzando l’ANOVA unidirezionale e il test post-hoc di Dunnett (C-F).10.7554/eLife.40048.015Figure 6-source data 1.NHE6 knockdown allevia i deficit di traffico in superficie indotti da ApoE4.
L’inibizione delle NHE farmacologiche ripristina l’LTP potenziato con la bobina in presenza di ApoE4
Per verificare se il traffico di recettori del glutammato ripristinato da inibizione NHE6 migliora la funzione delle sinapsi, abbiamo misurato LTP ippocampale in fette acute di topi di ricambio ApoE umani mirati: ApoE3-knockin (ApoE3-KI) e ApoE4-knockin (ApoE4-KI). I topi sono stati trattati con o senza EMD87580 da applicazione simultanea intraperitoneale e intranasale, i cervelli sono stati successivamente raccolti e le registrazioni sul campo elettrofisiologico sono stati eseguiti. Abbiamo scelto di integrare le iniezioni intraperitoneali con la somministrazione intranasale a partire da EMD87580, in quanto tutti i NHE esistenti a base di guanidina, hanno una scarsa penetrazione della barriera emato-encefalica e la somministrazione intranasale di piccole molecole e peptidi, tra cui l’insulina, ha dimostrato di aumentare il loro effetto biologico nel cervello(Grassin-Delyle et al., 2012). Coerentemente con i risultati precedenti(Rönicke et al., 2009), abbiamo trovato che EMD87580 ha aumentato i rapporti input-output (I/O) nei topi ApoE3-KI (Figura 7A). Nei topi ApoE4-KI, i rapporti di base di I/O erano più alti e non rispondevano all’EMD87580(Figura 7B). Come mostrato in precedenza(Durakoglugil et al., 2009), LTP è stato aumentato nelle fette di ApoE3-KI trattate con Reelin(Figura 7C). Anche le fette di ApoE3-KI trattate con EMD87580 hanno mostrato un aumento dell’LTP(Figura 7C). È interessante notare che la bobina e l’EMD87580 non hanno alcun effetto sinergico aggiuntivo e di fatto aumentano l’LTP in misura minore rispetto all’EMD o alla sola bobina. Come mostrato in precedenza(Durakoglugil et al., 2009), la bobina non ha avuto alcun effetto sulla LTP nelle fette ApoE4-KI(Figura 7D). A differenza di ApoE3-KI, le fette di ApoE4-KI trattate con EMD87580 hanno mostrato un LTP ridotto. È importante notare che le fette di ApoE4-KI con EMD87580 hanno risposto prontamente alla bobina e l’LTP è stato aumentato. Pertanto, su sfondo ApoE4 knockin, EMD87580 ripristina i parametri elettrofisiologici paragonabili a quelli di ApoE3 e sfondo wild-type.
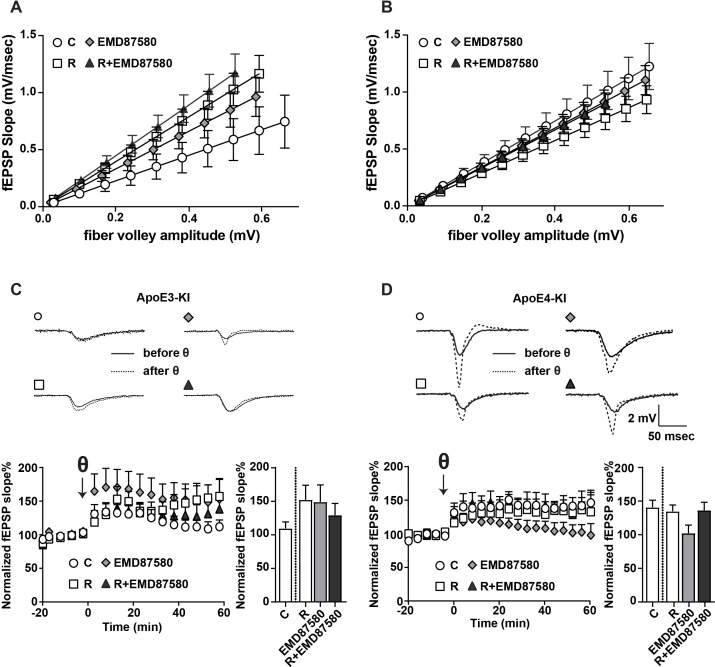
Figura 7 – dati fonte 1.Figura 7—dati sorgente 1. Il trattamento EMD87580 altera in modo differenziale la plasticità sinaptica nei topi ApoE3-KI e ApoE4-KI.Il trattamento EMD87580 altera in modo differenziale la plasticità sinaptica nei topi ApoE3-KI e ApoE4-KI.Il trattamento EMD87580 altera in modo differenziale la plasticità sinaptica nei topi ApoE3-KI e ApoE4-KI.I topi sono stati pretrattati con EMD87580 in vivo e le fette ippocampali acute sono state successivamente analizzate registrando i potenziali del campo extracellulare(A e B) Le curve di ingresso-uscita sono mostrate per ApoE3-KI (A) e ApoE4-KI(B).(A) Le fette ApoE3-KI trattate con Reelin o trattate con EMD87580 e Reelin hanno mostrato una maggiore pendenza di I/O rispetto al controllo (Ctrl: 1,127 ± 0.18; EMD87580: 1,653 ± 0,15; Reelin: 1,97 ± 0,14; Reelin ed EMD87580: 2,23 ± 0,16; F = 9,567, p<0,05).(B) Le curve di I/O sono state aumentate nelle fette ApoE4-KI alla linea di base (1,86 ± 0,16) rispetto al controllo ApoE3-KI (1,127 ± 0,18). Né l’EMD87580 né la bobina hanno influenzato in modo significativo le pendenze di I/O nelle fette ApoE4-KI (EMD: 1,705 ± 0,10; bobina: 1,43 ± 0,09; bobina e EMD: 1,67 ± 0,09; F = 1,8, p=0,14).(C e D) Risultati delle registrazioni LTP in ApoE3-KI(C) e ApoE4-KI (D). Tracce rappresentative prima (linea solida) e 40 minuti dopo (linea tratteggiata) la stimolazione theta-burst (TBS) per ogni paradigma di trattamento sono mostrati nei pannelli superiori. Pannelli in basso raffigurano le registrazioni LTP e la quantificazione delle risposte medie LTP tra 40 e 60 minuti dopo TBS (grafici a barre).(C) ApoE3-KI fette trattate con Reelin (152,4% ± 21,69, n = 5) e EMD87580 (149,30 ± 25).29, n = 7) aveva aumentato il LTP rispetto alle fette di controllo (109,7% ± 9,7, n = 6). Il trattamento combinato della bobina con EMD87580 ha aumentato l’LTP (129,4 ± 17,6, n = 7) rispetto al controllo.(D) Le fette ApoE4-KI non trattate hanno mostrato un aumento della LTP (140,80% ± 10,5, n = 12) rispetto alle fette ApoE3-KI non trattate (109,7% ± 9,7, n = 6). Le fette ApoE4-KI trattate con Reelin non hanno ulteriormente potenziato l’LTP (134,70% ± 9,63, n = 14), mentre le fette ApoE4-KI trattate con EMD87580 hanno mostrato un LTP ridotto (102 ± 11,9, n = 14). Le fette ApoE4-KI con EMD87580 mostrano un LTP maggiore quando trattate con Reelin (136,30 ± 11,87, n = 15) rispetto al solo trattamento EMD87580. Cerchi aperti: nessuna aggiunta; quadrati aperti: Rocchetto da solo; Diamanti grigi: EMD87580 da solo; Triangoli pieni: Bobina e trattamento EMD87580.10.7554/eLife.40048.017Figure 7-source data 1.EMD87580 trattamento altera in modo differenziato la plasticità sinaptica nei topi ApoE3-KI e ApoE4-KI.
Figura 7 – dati sorgente 1.Figura 7—dati sorgente 1. Il trattamento EMD87580 altera la plasticità sinaptica nei topi ApoE3-KI e ApoE4-KI.Il trattamento EMD87580 altera la plasticità sinaptica nei topi ApoE3-KI e ApoE4-KI.Il trattamento EMD87580 altera la plasticità sinaptica nei topi ApoE3-KI e ApoE4-KI.I topi sono stati pretrattati con EMD87580 in vivo e le fette ippocampali acute sono state successivamente analizzate registrando i potenziali del campo extracellulare(A e B) Le curve di ingresso-uscita sono mostrate per ApoE3-KI (A) e ApoE4-KI(B).(A) Le fette ApoE3-KI trattate con Reelin o trattate con EMD87580 e Reelin hanno mostrato una maggiore pendenza di I/O rispetto al controllo (Ctrl: 1,127 ± 0.18; EMD87580: 1,653 ± 0,15; Reelin: 1,97 ± 0,14; Reelin ed EMD87580: 2,23 ± 0,16; F = 9,567, p<0,05).(B) Le curve di I/O sono state aumentate nelle fette ApoE4-KI alla linea di base (1,86 ± 0,16) rispetto al controllo ApoE3-KI (1,127 ± 0,18). Né l’EMD87580 né la bobina hanno influenzato in modo significativo le pendenze di I/O nelle fette ApoE4-KI (EMD: 1,705 ± 0,10; bobina: 1,43 ± 0,09; bobina e EMD: 1,67 ± 0,09; F = 1,8, p=0,14).(C e D) Risultati delle registrazioni LTP in ApoE3-KI(C) e ApoE4-KI (D). Tracce rappresentative prima (linea solida) e 40 minuti dopo (linea tratteggiata) la stimolazione theta-burst (TBS) per ogni paradigma di trattamento sono mostrati nei pannelli superiori. Pannelli in basso raffigurano le registrazioni LTP e la quantificazione delle risposte medie LTP tra 40 e 60 minuti dopo TBS (grafici a barre).(C) ApoE3-KI fette trattate con Reelin (152,4% ± 21,69, n = 5) e EMD87580 (149,30 ± 25).29, n = 7) aveva aumentato il LTP rispetto alle fette di controllo (109,7% ± 9,7, n = 6). Il trattamento combinato della bobina con EMD87580 ha aumentato l’LTP (129,4 ± 17,6, n = 7) rispetto al controllo.(D) Le fette ApoE4-KI non trattate hanno mostrato un aumento della LTP (140,80% ± 10,5, n = 12) rispetto alle fette ApoE3-KI non trattate (109,7% ± 9,7, n = 6). Le fette ApoE4-KI trattate con Reelin non hanno ulteriormente potenziato l’LTP (134,70% ± 9,63, n = 14), mentre le fette ApoE4-KI trattate con EMD87580 hanno mostrato un LTP ridotto (102 ± 11,9, n = 14). Le fette ApoE4-KI con EMD87580 mostrano un LTP maggiore quando trattate con Reelin (136,30 ± 11,87, n = 15) rispetto al solo trattamento EMD87580. Cerchi aperti: nessuna aggiunta; quadrati aperti: Rocchetto da solo; Diamanti grigi: EMD87580 da solo; Triangoli pieni: Bobina e trattamento EMD87580.10.7554/eLife.40048.017Figure 7-source data 1.EMD87580 trattamento altera in modo differenziato la plasticità sinaptica nei topi ApoE3-KI e ApoE4-KI.
L’inibizione di NHE contrasta la soppressione LTP indotta da Aβ nei topi ApoE4-KI
Questi dati suggeriscono che l’inibizione endosomica di NHE può neutralizzare l’effetto di ApoE4 sul traffico di vescicole e sulla concomitante disfunzione sinaptica. Può anche invertire la soppressione sinaptica persistente causata dalla β-amiloide oligomerica in presenza di ApoE4?Gli oligomeri Aβ 42 sopprimono potentemente il potenziamento sinaptico (Townsendet al., 2006), ma questo può essere evitato con la preincubazione delle fette ippocampali con la bobina, che può da sola potenziare la sinapsi e quindi contrastare la soppressione indotta da Aβ (Durakoglugilet al., 2009). In Figura 8, abbiamo ripetuto questi esperimenti di nuovo in assenza così come in presenza di EMD87580. Come avevamo trovato in precedenza, estratti cerebrali di pazienti affetti da AD contenenti oligomeri Aβ, ma non gli estratti cerebrali di controllo, LTP potentemente soppresso in fette ippocampali da ApoE3-KI e nei topi ApoE4-KI. La bobina ha impedito questa soppressione nelle fette ApoE3, mentre le fette di topi ApoE4 erano quasi completamente resistenti alla bobina e LTP sono rimasti soppressi(Figura 8, triangoli solidi nei pannelli A e B). Al contrario, questa soppressione dell’LTP in presenza di Aβ e bobina nelle fette di ApoE4 è stata completamente abolita quando le fette sono state perfuse con EMD87580 per 4 ore prima dell’induzione dell’LTP. Nei dati qui non mostrati abbiamo osservato a 30 minuti di preperfusione con EMD87580 una tendenza ad alleviare la resistenza della bobina mediata da ApoE4 che, tuttavia, non era ancora significativa. Questo può suggerire che il sollievo del blocco di riciclaggio endosomico ApoE4 richiede un certo tempo, forse per “stanare” le vescicole che sono già bloccate e ostruire il percorso di riciclaggio.
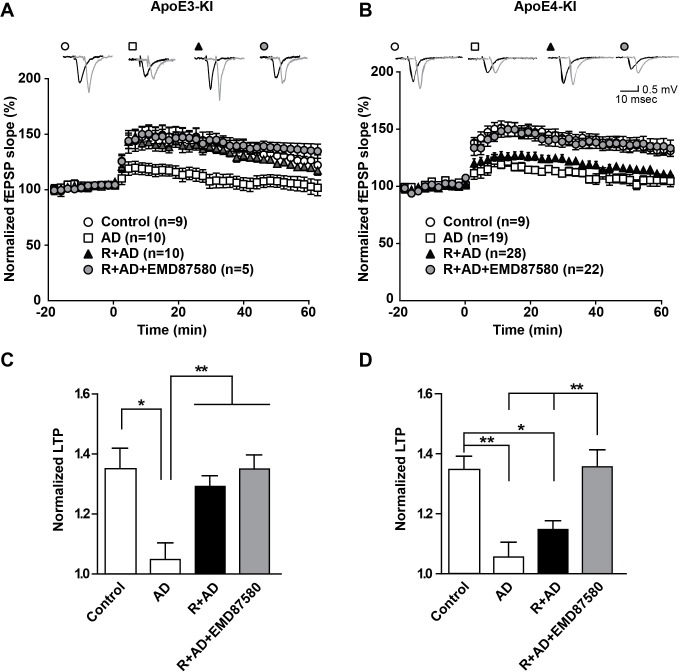
Figura 8 – dati fonte 1.Figura 8—dati fonte 1. L’inibizione di NHE contrasta la soppressione di LTP indotta da Aβ nei topi ApoE4-KI.L’inibizione di NHE contrasta la soppressione di LTP indotta da Aβ nei topi ApoE4-KI.L’inibizione di NHE contrasta la soppressione di LTP indotta da Aβ nei topi ApoE4-KI.(A-D) Il trattamento di fette di ippocampo con estratti cerebrali AD compromette il potenziamento a lungo termine (LTP) nei topi ApoE3-KI e ApoE4-KI. La bobina può attenuare i deficit di LTP indotti dagli estratti di AD in ApoE3-KI, ma non nei topi ApoE4-KI. L’inibizione di NHE contrasta i deficit di LTP indotti dall’estratto di AD nei topi ApoE4-KI. Fette di ippocampo sono stati preparati da 2 a 3 mesi ApoE3-KI e topi ApoE4-KI. Registrazioni di campo extracellulare sono state eseguite in fette trattate con estratto di cervello AD, Reelin e / o EMD87580. Fette di controllo sono stati trattati con estratto di cervello di controllo. Theta stimolazione scoppio (TBS) è stata eseguita 20 minuti dopo la linea di base stabile è stato raggiunto. Tracce rappresentative sono mostrati in ogni pannello, prima di induzione TBS (nero) e 40 minuti dopo TBS (grigio).(C, D) Analisi quantitativa delle pendenze fEPSP normalizzate a 40-60 min dopo TBS per (A), rispettivamente (B). Tutti i dati sono espressi come media ±SEM. *p<0,05, **p<0,01. L’analisi statistica è stata effettuata utilizzando ANOVA a senso unico seguito dal test post-hoc di Tukey (C, D).10.7554/eLife.40048.019Figure 8-source data 1.NHE inibizione 1.NHE contrasta la soppressione di LTP indotta da Aβ nei topi ApoE4-KI.
Figura 8 – dati sorgente 1.Figura 8—dati sorgente 1. L’inibizione NHE contrasta la soppressione LTP indotta da Aβ nei topi ApoE4-KI.L’inibizione NHE contrasta la soppressione LTP indotta da ApoE4-KI nei topi ApoE4-KI.L’inibizione NHE contrasta la soppressione LTP indotta da Aβ nei topi ApoE4-KI.(A-D) Il trattamento di fette di ippocampo con estratti cerebrali AD compromette il potenziamento a lungo termine (LTP) nei topi ApoE3-KI e ApoE4-KI. La bobina può attenuare i deficit di LTP indotti dagli estratti di AD in ApoE3-KI, ma non nei topi ApoE4-KI. L’inibizione di NHE contrasta i deficit di LTP indotti dall’estratto di AD nei topi ApoE4-KI. Fette di ippocampo sono stati preparati da 2 a 3 mesi ApoE3-KI e topi ApoE4-KI. Registrazioni di campo extracellulare sono state eseguite in fette trattate con estratto di cervello AD, Reelin e / o EMD87580. Fette di controllo sono stati trattati con estratto di cervello di controllo. Theta stimolazione scoppio (TBS) è stata eseguita 20 minuti dopo la linea di base stabile è stato raggiunto. Tracce rappresentative sono mostrati in ogni pannello, prima di induzione TBS (nero) e 40 minuti dopo TBS (grigio).(C, D) Analisi quantitativa delle pendenze fEPSP normalizzate a 40-60 min dopo TBS per (A), rispettivamente (B). Tutti i dati sono espressi come media ±SEM. *p<0,05, **p<0,01. L’analisi statistica è stata effettuata utilizzando ANOVA a senso unico seguito dal test post-hoc di Tukey (C, D).10.7554/eLife.40048.019Figure 8-source data 1.NHE inibizione 1.NHE contrasta la soppressione di LTP indotta da Aβ nei topi ApoE4-KI.
Discussione
Abbiamo utilizzato intuizioni sulla struttura molecolare e le proprietà biofisiche delle isoforme ApoE per sviluppare un nuovo approccio razionale di identificazione del farmaco per invertire l’aumento del rischio di AD inerente all’allele ApoE4. In studi precedenti, abbiamo scoperto che l’ApoE4 danneggia il riciclaggio delle vescicole endosomiche(Chen et al., 2010). Mentre indagavamo sulle basi molecolari di questo ritardo nel traffico, abbiamo riconosciuto che il punto isoelettrico previsto di ApoE4 corrisponde strettamente al pH prevalente nell’endosoma iniziale. Abbiamo ipotizzato che ApoE4, che è noto per assumere uno stato fuso-globulare in condizioni di pH basso, potrebbe perdere la solubilità quando entra nell’ambiente a pH più basso dell’endosoma precoce. Questo a sua volta potrebbe compromettere la propagazione delle vescicole attraverso il percorso di riciclaggio endosomico e causare il sequestro osservato dei recettori ApoE e dei recettori neurotrasmettitori eccitatori associati nei neuroni corticali(Chen et al., 2010). Abbiamo previsto che cambiando il pH endosomico lontano dal punto isoelettrico di ApoE4 dovrebbe impedire questa precipitazione isoelettrica e risolvere il blocco di riciclaggio. Dal momento che l’aumento del pH endosomico utilizzando agenti alcalinizzanti, come il cloruro di ammonio o clorochina, è noto per arrestare il traffico endosomico impedendo il rilascio di lipoproteine dai recettori lipoproteici(Goldstein et al., 1985), abbiamo studiato approcci per abbassare il pH endosomico invece. Due possibili meccanismi per questo sono i) l’attivazione della pompa protonica, o ii) la prevenzione dell’efflusso protonico dall’endosoma inibendo lo scambiatore endosomico sodio/idrogeno potassio NHE6. I nostri risultati mostrano ora che l’inibizione farmacologica e genetica dell’attività NHE nell’endosoma precoce è sufficiente per risolvere completamente il blocco di riciclaggio endosomico indotto da ApoE4 e ripristinare il normale tasso di riciclaggio della superficie cellulare del recettore sinaptico ApoE Apoer2 e dei recettori eccitatori del glutammato di tipo AMPA e NMDA che sono regolati da Apoer2 e che il traffico insieme ad Apoer2 attraverso i compartimenti di riciclaggio endosomici (illustrato dal modello mostrato in Figura 9).
In precedenza, noi e altri hanno dimostrato che i topi ApoE4-KI mostrano LTP avanzata mentre la bobina neuromodulatore Reelin non ha alcun effetto potenziante sull’espressione LTP in questo genotipo(Durakoglugil et al., 2009), suggerendo che ApoE4 compromette la risposta fisiologica alla bobina. Qui, si dimostra che l’inibizione NHE con EMD87580 nei topi ApoE4-KI inverte la resistenza della bobina indotta da ApoE4(Figura 7) e ripristina la capacità della bobina di proteggere contro la tossicità Aβ (Figura 8). ApoE3-KI fette trattate con EMD87580 mostrano un aumento di LTP coerente con i precedenti risultati che hanno dimostrato che l’inibitore NHE etilisopropilamiloruro (EIPA) può migliorare l’LTP indotta da theta-burst (Rönicke et al., 2009). L’attività NHE è stata proposta come un meccanismo di feedback negativo che può regolare l’eccitabilità neuronale e la plasticità. Nei topi ApoE4-KI, EMD87580 ha ridotto l’LTP a livelli di controllo di base dei topi ApoE3-KI e soprattutto l’LTP è diventato ora reattivo alla Reelin-facilitazione. Questa osservazione suggerisce che è il ripristino del normale rilascio di ApoE4 da Apoer2 in compartimenti endosomici acidificati e la successiva normalizzazione del traffico endosomico che ristabilisce l’omeostasi sinaptica ottimale in presenza di ApoE4.
Sebbene ad oggi siano stati scoperti più di 20 loci genetici che modificano il rischio di AD ad esordio tardivo(Karch et al., 2014), il genotipo ApoE4 è di gran lunga il principale fattore di rischio genetico per l’AD ad esordio tardivo oltre all’invecchiamento, interessando quasi 1/5 della popolazione umana, e quindi è clinicamente il più importante. I meccanismi molecolari con cui l’ApoE4 impone questo rischio sono ancora oggetto di dibattito. I primi lavori successivi alla scoperta di questa sorprendente associazione genetica da parte del gruppo Roses(Corder et al., 1993) si sono concentrati sulla capacità differenziale delle isoforme ApoE di interagire con l’Aβe di influenzare la formazione di fibrille. Gli sforzi nel nostro laboratorio si sono basati sulla logica che i recettori ApoE, cioè la famiglia di geni del recettore LDL, sono molto probabilmente coinvolti nel processo della malattia. Questa ipotesi è stata il driver iniziale che ha definito una pletora di funzioni sorprendenti di proteine correlate ai recettori LDL (LRPs) nel sistema nervoso centrale e periferico(Bal et al., 2013; Beffert et al., 2005; Bell et al., 2012; Choi et al., 2013; Kim et al., 2008; Lane-Donovan e Herz, 2017; Liu et al., 2013; Liu et al., 2007; Liu et al., 2011; May et al., 2004; Nakajima et al., 2013; Pohlkamp et al., 2015; Pohlkamp et al., 2017; Trommsdorff et al., 1999; Wasser e Herz, 2017; Wasser et al., 2014; Weeber et al., 2002; Zhang et al., 2008; Zhao et al., 2017). Essi includono ruoli senza precedenti come recettori a trasduttore diretto di segnale(Hiesberger et al., 1999; Trommsdorff et al., 1999) e regolatori di trasmissione sinaptica centrale e periferica(Beffert et al., 2005; Choi et al., 2013; Weeber et al., 2002) e hanno stabilito una forte base razionale e meccanicistica per cui le isoforme ApoE e i recettori ApoE possono influenzare direttamente l’omeostasi sinaptica, la sopravvivenza neuronale e quindi il deterioramento cognitivo e la neurodegenerazione progressiva che sono alla base di LOAD.
Il riciclaggio delle vescicole presinaptiche e postsinaptiche è un elemento centrale della trasmissione sinaptica(Harris et al., 2012; Kawasaki et al., 2000; Robinson et al., 1993; Sontag et al., 1994; Sudhof, 2004). È curioso notare che anche i compartimenti endosomici allargati e le funzioni endolisosomiche compromesse sono una caratteristica importante dell’espressione, dell’elaborazione e dell’inizio dell’AD di APP(Cataldo et al., 2000; Decourt et al., 2013; Ishigaki et al., 2000; Nixon et al., 2001; Salehi et al., 2006). La nostra scoperta originale che le isoforme ApoE possono alterare in modo differenziato le funzioni sinaptiche intrappolando le vescicole di riciclaggio postsinaptiche (ed eventualmente anche presinaptiche) che contengono recettori ApoE è stata ispirata dalle osservazioni originali di Heeren, Beisiegel e colleghi che per primi hanno descritto una ritenzione intracellulare prolungata di ApoE4 in una linea cellulare di epatoma(Heeren et al., 2004). In una serie di esperimenti, abbiamo dimostrato che ApoE4 compromette l’attivazione del recettore NMDA e la conduttanza neuronale Ca2 + dal neuromodulatore e il ligando del recettore ApoE Reelin. Ciò è stato causato dal drammatico ritardo del riciclaggio del recettore della bobina e del regolatore del recettore del glutammato Apoer2 in presenza di ApoE4 e, in particolare, anche in misura minore da ApoE3 e meno da ApoE2(Chen et al., 2010). La cinetica di riciclaggio drammaticamente alterata in presenza di ApoE4 porta a uno stato alterato di omeostasi sinaptica, che abbiamo definito “resistenza alla bobina” (Lane-Donovanet al., 2014) e che impedisce alla sinapsi di adattarsi adeguatamente ai crescenti livelli di oligomeri Aβ soppressivi delle sinapsi che si accumulano nel cervello che invecchia. L’aumento compensativo dell’attività sinaptica e di rete(Palop e Mucke, 2010) guiderebbe ulteriormente la produzione di Aβ (Cirritoet al., 2008), accelerando così un ciclo auto-rinforzante che si prevede contribuisca all’accumulo di amiloide nei portatori di ApoE4. Inoltre, un secondo meccanismo attraverso il quale si prevede che il riciclo delle vescicole contenenti ApoE4 compromessa contribuirebbe ad accelerare l’accumulo di amiloide e la deposizione della placca(Fagan et al., 2002) comporta la riduzione del tasso di turnover di Aβ nel cervello dei topi sostitutivi di ApoE4 (Castellano etal., 2011) e degli esseri umani (Wildsmith et al.,2012).
Recentemente, il laboratorio Bu ha riportato una interazione diretta di ApoE con i recettori dell’insulina nel cervello, che ha anche portato alla loro ritenzione intracellulare e alla loro segnalazione di insulina alterata(Zhao et al., 2017). Anche se nei nostri esperimenti non abbiamo rilevato il traffico di recettori dell’insulina compromessa(Figura 2B e C), e non vi è anche alcuna prova che i portatori di ApoE4 siano predisposti alla prevista resistenza all’insulina che ci si aspetterebbe derivi dalla trappola intracellulare dei recettori dell’insulina, questi dati aggiungono tuttavia un ulteriore supporto al nostro modello che postula il traffico di vescicole endosomiali neuronali compromesse come la causa principale dell’aumento del rischio di AD imposto da ApoE4(Chen et al., 2005; Chen et al., 2010; Lane-Donovan e Herz, 2017; Lane-Donovan et al., 2014).
I nostri dati indicano che ApoE4 induce deficit endosomici di traffico. Coerentemente, i farmaci alcalinizzanti, come il cloruro di ammonio o la clorochina, che aumentano il pH vescicolare nella cellula, portano ad un arresto del traffico endosomico impedendo il rilascio di lipoproteine dai loro recettori(Goldstein et al., 1985). Abbiamo quindi scelto di esplorare l’effetto dell’acidificazione degli endosomi come mezzo per superare i problemi di traffico causati da ApoE4. Al contrario, un recente studio sugli astrociti ha proposto che la presenza di ApoE4 porta all’acidificazione degli endosomi causata dalla riduzione di NHE6, che porta alla compromissione dell’Aβ-clearance (Prasade Rao, 2018). Questi autori hanno inoltre riportato che l’aumento dell’espressione di NHE6, che eleverebbe il pH endosomico, indotta dall’inibizione dell’HDAC, ha alleviato la compromissione della liberazione di Aβ.
Qui mostriamo che la compromissione sinaptica indotta da Aβ potrebbe essere abolita dall’inibitore NHE EMD87580 (Figura 8). Entrambi gli studi mostrano che la manipolazione del pH endosomico può influenzare il traffico endosomico di ApoE. Mostriamo che nei neuroni questo altera i livelli di espressione della superficie cellulare Apoer2 e quindi la plasticità sinaptica. Prasad e Rao (2018) ha mostrato che negli astrociti un meccanismo simile può mediare la clearance Aβ che coinvolge il recettore ApoE Lrp1. Il punto isoelettrico di ApoE4, ma non ApoE2 e ApoE3, corrisponde al pH fisiologico presente nei primi endosomi e la ridotta solubilità a o vicino al loro punto isoelettrico è una proprietà generale di molte proteine tra cui l’insulina(Wintersteiner e Abramson, 1933). A differenza di ApoE2 e ApoE3, la conformazione di ApoE4 nell’endosoma precoce potrebbe essere particolarmente vulnerabile, perché da sola ha dimostrato di essere incline a dispiegarsi in condizioni di pH basso con conseguente stato fuso-globulare(Morrow et al., 2002).
Sulla stessa linea, ApoE4 ha aumentato il numero e la dimensione degli endosomi precoci nei pazienti con AD(Cataldo et al., 2000) e nei neuroni derivati da cellule staminali pluripotenti indotte(Lin et al., 2018). Questi dati suggeriscono che la manipolazione del pH endosomico rappresenta un promettente obiettivo terapeutico per migliorare i deficit di traffico endosomico indotti da ApoE4.
Al fine di affrontare pienamente le funzioni fisiologiche di NHE6 e comprendere il suo ruolo nei processi neurodegenerativi indotti da ApoE4, sarà necessario sviluppare inibitori specifici NHE6 in grado di modulare farmacologicamente NHE6 – al contrario di accenderlo o spegnerlo in modo binario – per raggiungere il pH ottimale per il traffico di ApoE4 senza indebite interferenze con i processi essenziali di trasporto cellulare o la funzione di altri NHE. Tali inibitori devono anche essere in grado di attraversare la barriera emato-encefalica, cosa che le classi di inibitori non specifici attualmente disponibili non sono in grado di fare.
In sintesi, abbiamo dimostrato che il cambiamento conformazionale di ApoE4 in uno stato di globule fuso(Morrow et al., 2002) in un ambiente a basso pH, che è correlato con il riciclaggio delle vescicole endosomiche compromesse in presenza di ApoE4(Chen et al., 2010; Heeren et al., 2004; Lane-Donovan e Herz, 2017; Lane-Donovan et al., 2014; Rellin et al., 2008), può essere invertito cambiando il pH endosomico(Herz et al., 2010). Come base meccanicistica proponiamo la propensione di molte proteine a perdere idrofilia al loro punto isoelettrico o in prossimità di esso, che le rende inclini all’aggregazione e alla precipitazione, una scoperta seminale che ha reso possibile la purificazione dell’insulina su scala industriale(Wintersteiner e Abramson, 1933). Alterando il pH endosomico, ApoE4 mantiene la sua solubilità e si evita il blocco di riciclaggio. Questa semplice proprietà biofisica può spiegare in modo semplice molte osservazioni con cui le isoforme ApoE influenzano in modo differenziato le funzioni neuronali e i meccanismi rilevanti per l’AD. Le nostre scoperte puntano anche verso gli inibitori specifici di NHE6 come base razionale per un nuovo approccio per cancellare il rischio di AD imposto da ApoE4.
Materiali e metodi
| Tipo di reagente (specie) o risorsa | Designazione | Fonte o riferimento | Identificatori | Ulteriori informazioni |
|---|---|---|---|---|
| Linea cellulare(Homo sapiens) | HEK293 | Thermo Fisher | R70507, RRID: CVCL_0045 | Testato mycoplasma libero ogni anno, ultimo test 16 gennaio 2018 |
| Linea cellulare(Homo sapiens) | HEK293T | ATCC | CRL-3216, RRID: CVCL_0063 | Testato mycoplasma libero ogni anno, ultimo test 16 gennaio 2018 |
| Ceppo, fondo di ceppo(Mus muscolo muscoloso) | Mouse/ApoE3ki (B6.129P2- Apoetm2 (APOE*3)Mae N8) | (Knouff et al., 1999; Sullivan et al., 1997) | Originariamente fornito dal Dr. Nobuyo Maeda | |
| Ceppo, fondo di ceppo(Mus muscolo muscoloso) | Mouse/ApoE4ki (B6.129P2- Apoetm2 (APOE*4)Mae N8) | (Knouff et al., 1999; Sullivan et al., 1997) | Originariamente fornito dal Dr. Nobuyo Maeda | |
| Ceppo, fondo di ceppo(Rattus norvegicus) | Topo SD | Fiume Carlo | SC:400 | |
| Anticorpo | capra anti- ApoE, pAb | EMD Millipore | 178479, RRID: AB_10682965 | 1:1000 (WB) |
| Anticorpo | coniglio anti-Apore2 | Herz Lab, #2561 | 1:1000 (WB) | |
| Anticorpo | coniglio anti-Dab1 | Herz Lab, #5091 | 1:1000 (WB) | |
| Anticorpo | mouse anti-FLAG M2 | Sigma-Aldrich | F3165, RRID: AB_259529 | 1:1000 (WB) |
| Anticorpo | coniglio anti-GluA1 | Abcam | ab31232, RRID: AB_2113447 | 1:1000 (WB) |
| Anticorpo | coniglio anti-GluA2/3 | EMD Millipore | 07-598, RRID: AB_310741 | 1:1000 (WB) |
| Anticorpo | coniglio anti-GluN2B | Tecnologia di segnalazione cellulare | 4207S, RRID: AB_1264223 | 1:1000 (WB) |
| Anticorpo | coniglio anti-insulina Recettore B (4B8), mAb | Tecnologia di segnalazione cellulare | 3025, RRID: AB_2280448 | 1:1000 (WB) |
| Anticorpo | coniglio anti-Lrp1 | Laboratorio Herz | 1:5000 (WB) | |
| Anticorpo | coniglio anti-Ldlr | Laboratorio Herz | 1:1000 (WB) | |
| Anticorpo | coniglio anti -NHE6 (C-termino) | Laboratorio Herz | 1:1000 (WB) | |
| Anticorpo | antifosfotirosina del mouse (4G10) mAb | EMD Millipore | 05-321, RRID: AB_309678 | 1:1000 (WB) |
| Anticorpo | coniglio recettore anti-Transferrina | Abcam | Ab61134, RRID: AB_943620 | 1:1000 (WB) |
| Anticorpo | coniglio anti-B-Actin | Abcam | Ab8227, RRID: AB_2305186 | 1:3000 (WB) |
| Peptide, proteina ricombinante | 6-ciano-7- nitrochinossalina-2,3-dione, CNQX | Sigma-Aldrich | C127 | |
| Peptide, proteina ricombinante | ApoE3, umano | Sigma-Aldrich | SRP4696 | |
| Composto chimico, farmaco | B-27 Supplemento (50X), senza siero | Thermo Fisher | 17504044 | |
| Composto chimico, farmaco | Bafilomicina A1 | Cayman Chemical | CAS88899-55-2 | |
| Composto chimico, farmaco | DMEM | Sigma-Aldrich | D6046 | |
| Composto chimico, farmaco | FuGENE | Promega | E2311 | |
| Composto chimico, farmaco | HBSS (1X) | Gibco | 14175 | |
| Composto chimico, farmaco | Acido L-glutammico (glutammato) | Sigma-Aldrich | G1251 | |
| Composto chimico, farmaco | Neurobasale Medio (1X) Liquido senza fenolo rosso | Thermo Fisher | 12348017 | |
| Composto chimico, farmaco | NeutrAvidin Agarosio | Thermo Fisher | 29201 | |
| Composto chimico, farmaco | Nimodipina | Sigma-Aldrich | N3764 | |
| Composto chimico, farmaco | NP-40 Alternativa | EMD Millipore | 492016 | |
| Composto chimico, farmaco | 32% Paraformaldeide soluzione AQ | Fisher Scientific | 15714S | |
| Composto chimico, farmaco | PBS (1X) | Sigma-Aldrich | D8537 | |
| Composto chimico, farmaco | Penicillina – soluzione di streptomicina, 100X | Corning | 30-002 CI | |
| Composto chimico, farmaco | Cocktail di Inibitori della fosfatasi | Thermo Fisher | 78420 | |
| Composto chimico, farmaco | Poly-D -Soluzione di Lisina | Sigma-Aldrich | A-003-M | |
| Composto chimico, farmaco | Proteina A- Sepharosio 4B | Thermo Fisher | 101042 | |
| Composto chimico, farmaco | Proteina G- Sepharosio 4B | Thermo Fisher | 101142 | |
| Composto chimico, farmaco | Cocktail di Inibitori delle Proteinasi | Sigma-Aldrich | P8340 | |
| Composto chimico, farmaco | Inibitore dello scambiatore sodio-idrogeno | Merck KGaA | EMD87580 | |
| Composto chimico, farmaco | Sulfo-NHS- SS-biotina | Pierce | 21331 | |
| Composto chimico, farmaco | Tetrodotossina | Sigma-Aldrich | T8024 | |
| Composto chimico, farmaco | Tritone X-100 | Sigma-Aldrich | CAS9002-93-1 | |
| Composto chimico, farmaco | Vectashield con DAPI | Laboratori Vettoriali | H-1200 | |
| Reagente DNA ricombinante | pcDNA3.1-Zeo | Invitrogen | V79020 | |
| Reagente DNA ricombinante | psPAX2 | Addgene | 12260 | |
| Reagente DNA ricombinante | pMD2.G | Addgene | 12259 | |
| Reagente DNA ricombinante | pLKO.1 | Addgene | 10878 | |
| Reagente DNA ricombinante | pLVXCMV100 | (Dean et al., 2017) | N/A | |
| Costruttore trasformato(Mus muscolo muscoloso) | pCrl, vettore di espressione della bobina | (D’Arcangelo et al., 1997) | N/A | |
| Costruttore trasformato(Mus muscolo muscoloso) | pcDNA3.1-Apoer2-Fc | (Hiesberger et al., 1999) | N/A | |
| Costruttore trasformato(Homo sapiens) | pcDNA3.1-ApoE2 | (Chen et al., 2010) | N/A | progenitore pcDNA3.1-Zeo |
| Costruttore trasformato(Homo sapiens) | pcDNA3.1-ApoE3 | (Chen et al., 2010) | N/A | progenitore pcDNA3.1-Zeo |
| Costruttore trasformato(Homo sapiens) | pcDNA3.1-ApoE4 | (Chen et al., 2010) | N/A | progenitore pcDNA3.1-Zeo |
| Costrutto trasformato(costrutto shRNA) | pLKO.1-shRNA scramble | questo documento | N/A | progenitore pLKO.1 |
| Costruttore trasformato(costrutto shRNA) | pLKO.1-shNHE1 | Biosistema aperto | TRCN000000044651 | progenitore pLKO.1 |
| Costruttore trasformato(costrutto shRNA) | pLKO.1-shNHE5 | questo documento | N/A | progenitore pLKO.1 |
| Costruttore trasformato(costrutto shRNA) | pLKO.1-shNHE6 a | Biosistema aperto | TRC N000 0068828 | progenitore pLKO.1 |
| Costruttore trasformato(costrutto shRNA) | pLKO.1-shNHE6 b | Biosistema aperto | TRCN000000068830 | progenitore pLKO.1 |
| Costruttore trasformato(costrutto shRNA) | pLKO.1-shNHE6 c | Biosistema aperto | TRCN000000068832 | progenitore pLKO.1 |
| Costruttore trasformato(costrutto shRNA) | pLKO.1-shNHE7 | Biosistema aperto | TRCN000000068812 | progenitore pLKO.1 |
| Costruttore trasformato(costrutto shRNA) | pLKO.1-shNHE8 | questo documento | N/A | progenitore pLKO.1 |
| Costruttore trasformato(costrutto shRNA) | pLKO.1-shNHE9 | Biosistema aperto | TRCN000000068856 | progenitore pLKO.1 |
| Costruttore trasformato(Mus muscolo muscoloso) | pLVX-mCherry- Apoer2 | questo documento | N/A | progenitore pLVXCMV100 |
| Costruttore trasformato(Homo sapiens) | pcDNA3.1- ApoE3-GFP | questo documento | N/A | progenitore pcDNA3.1-Zeo |
| Reagente a base di sequenza (oligo) | Scramble shRNA in avanti | IDT Tecnologie del DNA inegrate | N/A | 5′-CCGGCCTAAGGTTAAGTCGCCCCCCCCC-3′. |
| Reagente a base di sequenza (oligo) | Scramble shRNA inverso | IDT Tecnologie del DNA inegrate | N/A | 5′-GAGCCGGAGGGGGCGGACTTAACCTTAGG TTTTTTTG-3′. |
| Reagente a base di sequenza (oligo) | shRNA anti NHE1 (SLC9A1) in avanti | Biosistema aperto | TRCN000000044651 | |
| Reagente a base di sequenza (oligo) | shRNA anti NHE1 (SLC9A1) inverso | Biosistema aperto | TRCN000000044651 | 5′-AGTAAGGAAGGAAGGGAAGGGATTTTTTOO-3”. |
| Reagente a base di sequenza (oligo) | shRNA anti NHE5 (SLC9A5) in avanti | IDT Tecnologie del DNA inegrate | N/A | 5′-CCGGAAGGAAGGACCACTCATTCATTTTAG TCTCG-3′. |
| Reagente a base di sequenza (oligo) | shRNA anti NHE5 (SLC9A5) inverso | IDT Tecnologie del DNA inegrate | N/A | 5′-AGACTAAGATGATGAGTGTGTGGTTTTCCTTT TTTTG-3′. |
| Reagente a base di sequenza (oligo) | shRNA anti NHE6 (SLC9A6) -a avanti | Biosistema aperto | TRCN000000068828 | 5′-CCGGGGCCGTTTATATATATATATGGCATAGGAACTC-3”. |
| Reagente a base di sequenza (oligo) | shRNA anti NHE6 (SLC9A6)- un inverso | Biosistema aperto | TRCN000000068828 | 5′-GAGTTTTTATGCCTATGCCATATATAAACGGTTTTTTTTTG-3”. |
| Reagente a base di sequenza (oligo) | shRNA anti NHE6 (SLC9A6)-b avanti | Biosistema aperto | TRCN000000068830 | 5′-CCGGCCCTTTTTTTTTTTAATTTTAATTTTOATTTTOATTTTOATTTTOATT-3”. |
| Reagente a base di sequenza (oligo) | shRNA anti NHE6 (SLC9A6)-b reverse | Biosistema aperto | TRCN000000068830 | 5′-AGATTAAGTAAGAGAGAGAGAGACAAGGGTTTTTTTOO-3”. |
| Reagente a base di sequenza (oligo) | shRNA anti NHE6 (SLC9A6)-c avanti | Biosistema aperto | TRCN000000068832 | 5′-CCCCCCCCTTTTTTTTATTTATTTTAGCATACTCG-3”. |
| Reagente a base di sequenza (oligo) | shRNA anti NHE6 (SLC9A6)-c inverso | Biosistema aperto | TRCN000000068832 | 5′-AGTATGCTAAGATAGATAGACCCAAGGGTTTTTOO-3”. |
| Reagente a base di sequenza (oligo) | shRNA anti NHE7 (SLC9A7) in avanti | Biosistema aperto | TRCN000000068812 | 5′-CCGGCCATTGATTGTACT ATCCTCGTCTACTCG-3′. |
| Reagente a base di sequenza (oligo) | shRNA anti NHE7 (SLC9A7) inverso | Biosistema aperto | TRCN000000068812 | 5′-AGTAGACGAGGATAGGGATAGGGGGTTTTTTOO-3”. |
| Reagente a base di sequenza (oligo) | shRNA anti NHE8 (SLC9A8) in avanti | IDT Tecnologie del DNA inegrate | N/A | 5′-CCGGAAGGCTTTTTTTTTTTOOCCCCC-3”. |
| Reagente a base di sequenza (oligo) | shRNA anti NHE8 (SLC9A8) inverso | IDT Tecnologie del DNA inegrate | N/A | 5′-GAGCATCCAACCACAT GAAGCCCCTTTTTTTTTTTTTOO-3”. |
| Reagente a base di sequenza (oligo) | shRNA anti NHE9 (SLC9A9) in avanti | Biosistema aperto | TRCN000000068856 | 5′-CCGGCTGGGCAGAAA GCAGAAGATTATTC-3”. |
| Reagente a base di sequenza (oligo) | shRNA anti NHE9 (SLC9A9) inverso | Biosistema aperto | TRCN000000068856 | 5′-GAGAATTTTTTOO DDII CTGCCCTTTTTTTOO-3”. |
| Reagente a base di sequenza (oligo) | Apoer2 NT sito di clonazione (sdm) in avanti | Tecnologie del DNA inegrate | N/A | 5′-TACAAATCTAGATCTAGATCCATCCG CTGCCGGGGCGGCCAAG-3”. |
| Reagente a base di sequenza (oligo) | Apoer2 NT sito di clonazione (sdm) reverese | Tecnologie del DNA inegrate | N/A | 5′-ACTCATGTCGACCGATG CGGAGAGATG CTGAAGATG CTGAAGATG-3”. |
| Reagente a base di sequenza (oligo) | mCherry (per mCherry-Apoer2) in avanti | Tecnologie del DNA inegrate | N/A | 5′-AAATTCGTCGTCGACATGGGACATTG AGCAAGGGCGA GGAGGATAAC-3”. |
| Reagente a base di sequenza (oligo) | mCherry (per mCherry-Apoer2) reverse | Tecnologie del DNA inegrate | N/A | 5′-GGGAACGGTCTAGTAGAG GACTTGTACAGCTC GTCCATG-3′. |
| Reagente a base di sequenza (oligo) | Apoer2 in avanti | Tecnologie del DNA inegrate | N/A | 5′-TGGAGCGCGCGCTAGCGGGC CACCATGGGGGCCCCCCC AGAACTGG-3”. |
| Reagente a base di sequenza (oligo) | Apoer2 inverso | Tecnologie del DNA inegrate | N/A | 5′-AACCCCCGGAATTCTCA GGGCAGTCCAT CATCTTCAAGAC-3”. |
| Reagente a base di sequenza (oligo) | Rimozione del sito NheI (sdm) in avanti | Tecnologie del DNA inegrate | N/A | 5′-GTTTTTACCGTCGA CCTCTAGCTAGCTAG-3′. |
| Reagente a base di sequenza (oligo) | Rimozione del sito NheI (sdm) inverso | Tecnologie del DNA inegrate | N/A | 5′-AATGTCAAGGTTTTTTTOOCCCCCCCTTTOO-3”. |
| Reagente a base di sequenza (oligo) | CMVfull in avanti | Tecnologie del DNA inegrate | N/A | 5′-CAGTTTATTTATCGATATCGGGG GCCAGATATATACGG TTGACATTG-3′. |
| Reagente a base di sequenza (oligo) | CMVfull inverso | Tecnologie del DNA inegrate | N/A | 5′-TTTTTCCGGGCTAGCGGATCC CCCTATAGGGATTTTTTTTTTTTOO DDDII CCCTATAGGGATTTTOO DDDII CCCTATAGGGATTTTOO 3”. |
| Reagente a base di sequenza (oligo) | ApoE3 (ApoE3-GFP) in avanti( | Tecnologie del DNA inegrate | N/A | 5′-ATCAGGGAATTCAAC CATGAAGGTTTTTOO TGGGGTTGGCCCG-3”. |
| Reagente a base di sequenza (oligo) | Inversione delle PFL (ApoE3-PFL) | Tecnologie del DNA inegrate | N/A | 5′-ATTGGTGGATGGATCCGCCGT GATTGTCGGCTG GGCACAG-3”. |
| Software, algoritmo | Adobe Creative Cloud | Adobe | RRID: SCR_010279 | |
| Software, algoritmo | GraphPad Prisma 7.0 | Software GraphPad | RRID: SCR_002798 | |
| Software, algoritmo | Fiji/ImageJ | NIH | RRID: SCR_002285 | |
| Software, algoritmo | LabView7.0 | Strumenti nazionali | RRID: SCR_014325 | |
| Software, algoritmo | Odyssey Imaging System | LI-COR | RRID: SCR_014579 | |
| Software, algoritmo | Clustal Omega | EMBL-EBI | RRID: SCR_001591 | |
| Leica TCS SPE | Leica | RRID: SCR_002140 |
Contatto per la condivisione di reagenti e risorse
La richiesta di reagenti deve essere indirizzata a Joachim Herz (Joachim.Herz@utsouthwestern.edu).
Animali
I topi ApoE3-KI o ApoE4-KI retroincrociati a C57BL/6 sono stati generosamente forniti da Nobuyo Maeda e Patrick Sullivan(Knouff et al., 1999; Sullivan et al., 1997). Gli animali sono stati ospitati in gruppo su un ciclo standard di 12 ore luce/buio e alimentati ad libitum chow standard per topi (Diet 7001; Harlan Teklad, Madison, WI, USA). Dichiarazione etica: Tutte le procedure sperimentali sono state eseguite secondo le linee guida approvate per la cura degli animali Institutional Animal Care and Use Committee (IACUC) presso l’Università del Texas Southwestern Medical Center di Dallas (Numero di approvazione: A3472-01; 2015-101088).
Estratti di cervello corticale umano
Estratti di cervello corticale umano deidentificato (IRB esenti) da un non-AD, soggetto normale (Controllo) e un caso di malattia di Alzheimer clinicamente e istopatologicamente confermato (AD) sono stati preparati come descritto in precedenza (Durakoglugilet al., 2009). In breve, l’estratto cerebrale di controllo conteneva Aβ monomerico, ma non oligomeri rilevabili e solo una traccia di aggregati di ordine superiore; al contrario, l’estratto cerebrale di AD conteneva Aβ monomerico in aggiunta a dimeri Aβ e aggregati di ordine superiore. Un grammo di tessuto cerebrale è stato omogeneizzato in 4 ml di tampone Tris salino e centrifugato a 175.000 x g come descritto da Shankar et al. (2008). Il supernatante è stato designato “estratto”. Il genotipo del tessuto cerebrale era ApoE3/3 per il controllo e ApoE3/4 per il tessuto AD.
Neuroni primari
Ratto primario (Sprague-Dawley) neuroni corticali (E18) sono stati preparati come descritto in precedenza(Chen et al., 2005) e coltivati in piastre a sei pozzetti (1 milione di neuroni/9 cm2) o su coprivetrini rivestiti in Poly-D-Lisina (30.000 neuroni/1,1 cm2) in mezzo Neurobasal/B27 a 37°C e 5% di CO2. A 9-14 giorni in vitro (DIV) neuroni primari sono stati utilizzati per esperimenti.
Costruzioni di plasmide
Tutti i primer utilizzati per la clonazione sono elencati nella tabella delle risorse chiave.
pcDNA3.1-Apoer2-Fc: Il costrutto del mouse ApoER2-Fc (ectodominio secreto di Apoer2), etichettato con l’epitopo V5 e Fc, è stato descritto in precedenza(Hiesberger et al., 1999). Per produrre Apoer2-Fc ricombinante, sono state trasfettate 293 cellule HEK e il mezzo è stato raccolto come descritto(Chen et al., 2010).
pLKO.1-shRNA costruisce: pLKO.1-costruzioni contenenti shRNA targeting diversi sottotipi NHE sono stati acquistati da Sigma o creati inserendo oligo allineati (per la sequenza shRNA fare riferimento alla tabella delle risorse chiave) nel pLKO.1-TRC, come descritto altrove(Moffat et al., 2006). pLVX-mCherry-Apoer2: I siti di clonazione SalI e XbaI sono stati inseriti in un plasmide contenente il cDNA a lunghezza piena Apoer2 immediatamente a valle del peptide del segnale Apoer2 (N-terminus, NT) mediante mutagenesi diretta al sito. In una seconda fase, mCherry amplificato in PCR è stato inserito nei siti SalI e XbaI di nuova creazione. NT-mCherry-Apoer2 è stato poi amplificato mediante PCR e clonato nei siti NheI ed EcoRI del vettore lentivirale che è stato generato modificando pLVXCMV100 rimuovendo un sito NheI attraverso la mutagenesi site-directed e sostituendo il CMV100 troncato con un promotore CMV a tutta lunghezza (inserito nei siti ClaI e NheI). pcDNA3.1-ApoE3-GFP: A poE3 è stato amplificato tramite PCR e inserito in pEGFP-N1 utilizzando i siti di clonazione EcoR1 e BamHI.
Generazione di proteine ricombinanti
Bobina
HEK 293 cellule che esprimono stabilmente la bobina (pCrl) sono state coltivate in medium DMEM(D’Arcangelo et al., 1995; Förster et al., 1998). Il terreno contenente Reelin è stato raccolto e purificato come descritto in precedenza(Weeber et al., 2002). Autenticato dalla loro capacità di secernere la bobina. Stato del micoplasma accertato annualmente, ultimo risultato negativo dopo la presentazione di questo manoscritto.
L’ApoE utilizzato in questo studio era contenuto in particelle di lipoproteine naturalmente secrete da cellule trasfettate in uno stato minimamente lipidico e prodotte come segue: Le cellule HEK 293 sono state trasfettate (FuGENE) con il vettore pcDNA3.1 contenente ApoE umano a lunghezza piena (ApoE2, ApoE3, ApoE4, ApoE4P, ApoE3 mutante o ApoE4 mutante) cDNA o pcDNA3.1 vuoto (controllo). Il mezzo DMEM post-trasformazione 24 ore su 24 è stato sostituito con un mezzo Neurobasale. Dopo 3 giorni, il mezzo contenente le diverse isoforme ApoE è stato raccolto e analizzato su SDS-PAGE per calcolare le concentrazioni di ApoE utilizzando ApoE3 umano commerciale come standard. Le proteine sono state immunoblotate per ApoE e rilevate con anticorpo secondario IRDye 800CW (Li-Cor). I livelli di ApoE sono stati quantificati Odyssey imaging a infrarossi.
Macchie occidentali
Dopo il trattamento, i neuroni sono stati lavati tre volte con PBS a freddo, e lisati in tampone RIPA (50 mM Tris-HCl, pH 8.0; 150 mM NaCl; 1% Nonidet P-40; inibitori della fosfatasi e della proteasi) per 20 minuti in ghiaccio. I detriti cellulari sono stati rimossi per centrifugazione per 10 minuti a 14.000 rpm e 4°C in una centrifuga Eppendorff. Le concentrazioni di proteine sono state misurate utilizzando il Bradford Protein Assay (Bio-Rad). Dopo aver aggiunto 4x tampone di carico SDS (0,1 M Tris-HCl, pH 6,8, 2% SDS, 5% β-mercaptoetanolo, 10% glicerolo, e 0,05% blu di bromphenol) i campioni sono stati bolliti a 95 ° C per 10 min. Per l’immunoblotting di NHE6, i campioni sono stati incubati per 30 minuti a temperatura ambiente invece di ebollizione. 4x SDS tampone di carico è stato aggiunto al media per rilevare le proteine secrete. Dopo l’ebollizione (95°C per 10 min) i campioni sono stati caricati su SDS-PAGE. Le proteine sono state trasferite su una membrana di nitrocellulosa per il Western Blotting con gli anticorpi indicati.
Microscopia confocale
Per l’imaging DIV9 neuroni DIV9 su coprioggetti da 12 mm sono stati infettati con DNA lentivirale codificante NT-mCherry-Apoer2. Il mezzo contenente il lentivirus è stato rimosso 14 ore dopo l’infezione. Su DIV12 ApoE3-GFP contenente supernatante di 293 cellule trasfettate con pcDNA3.1-ApoE3-GFP (FuGENE) è stato aggiunto. Su DIV13 neuroni sono stati lavati 2x con PBS e fissato con il 4% PFA. I coprioggetti sono stati montati utilizzando Vectashield Antifade Mounting Medium con DAPI. Le immagini Z-stack sono state ottenute utilizzando un microscopio Confocal Zeiss LSM880 Airyscan e un obiettivo 63x e un passo di 1 µm. Le proiezioni 3D e le viste ortogonali sono state generate utilizzando il software NIH Fiji/ImageJ.
Biotinilazione di superficie/riciclopoer2
I neuroni primari sono stati pretrattati per 30 minuti con terreno condizionato con ApoE (5 μg/ml, se non diversamente specificato), quindi incubati con Reelin (2 μg/ml) per altri 30 minuti (vedere la linea temporale in Figura 2A). Dopo il trattamento le cellule sono state lavate con PBS a freddo e incubate in PBS contenente sulfo-NHS-SS-biotina (1,0 mg/ml) per 30 minuti a 4°C. Reagente in eccesso è stato estinto risciacquando i neuroni con PBS freddo contenente 100 mM di glicina. I neuroni sono stati lisati in 160 µl/9 cm2 tampone di lisi (PBS con 0,1% SDS, 1% Triton X-100, e inibitori della proteasi) a 4 ° C per 20 min. I detriti cellulari sono stati rimossi per centrifugazione per 10 minuti a 14.000 rpm a 4°C in una centrifuga Eppendorff. La concentrazione di proteine è stata misurata utilizzando il Bradford Protein Assay (Bio-Rad) e 100 µg di proteine totali sono stati incubati con 50 µl di NeutrAvidina agarosio a 4°C per 1 ora. Pellet di agarosio sono stati lavati tre volte con tampone di lavaggio (500 mM NaCl; 15 mM Tris-HCl, pH 8,0; 0,5% Triton X-100), le proteine di superficie biotinilate sono stati eluiti da perle di agarosio da ebollizione in 2x SDS tampone di carico del campione SDS e caricato su SDS-PAGE per l’analisi western blot. Per i trattamenti farmacologici, le cellule sono state preincubate con EMD87580 e/o Bafilomicina per 1 ora prima dell’aggiunta di ApoE e Reelin. 3 μM EMD87580 (3 mM di stock in PBS) è stato utilizzato se non diversamente specificato.
Co-immunoprecipitazione
Interazione Apoer2-Fc/ApoE: Proteina G sefarosio (50 µl) è stato aggiunto a 1 ml di supernatante di coltura contenente ApoER2-Fc da cellule HEK 293 a 4 ° C durante la notte. Perline sono stati poi sedimentati da centrifugazione breve, e poi 500 µl supernatante coltura contenente ApoE e CaCl2 (concentrazione finale 1 mM) è stato aggiunto alle perline. La miscela è stata incubata per altre 4 ore a 4°C. Le perle sono stati lavati tre volte con tampone di lavaggio (500 mM NaCl, 15 mM Tris.HCl, 0,5% Triton X-100 (pH 8,0)), e le proteine legate sono stati separati su 4 – 15% SDS-PAGE e immunoblotted per ApoE.
Apoer2-ApoE interazione: I neuroni trattati con ApoE (5 µg/ml per 3 ore a 37°C) sono stati lavati con PBS ghiacciato e lisati in buffer RIPA. Per l’immunoprecipitazione 600 µg di lisato sono stati co-incubati con siero di coniglio anti-Apoer2 o siero di coniglio di controllo e proteine A-Sefarosio perline a 4 ° C durante la notte. Perle precipitati sono stati lavati 3x in buffer RIPA, risospesi in 2x SDS campione buffer e bollito a 95 ° C per 10 min. Le proteine eluite sono state sondate per Apoer2 e ApoE.
Fosforilazione di Dab1
I neuroni primari sono stati trattati con Reelin (2 µg/ml per 30 min) o ApoE3 o ApoE4 a 37°C per 3 ore. Dopo il trattamento, i neuroni sono stati lisati e preparati per il Western Blotting come descritto in precedenza e sondati con anticorpi sollevati contro la Dab1 totale e la fosfotirosina (4G10) per identificare la fosfo-Dab1(Masereel et al., 2003).
Preparazione di particelle lentivirali
HEK 293 cellule T sono stati co-trasferiti con psPAX2, pMD2.G, ed i costrutti di trasferimento individuale (pLKO.1-shRNA o pLVX-mCherry-Apoer2). I media sono stati sostituiti dopo 12 – 15 ore. I supporti contenenti particelle virali sono stati raccolti e i detriti delle cellule sono stati filati. Il virus è stato concentrato da ultra-centrifugazione e risospensione in DMEM (1/10 di volume). Per trasfettare i neuroni 100 µl di virus concentrato è stato aggiunto per ml di terreno nella piastra di coltura. I mezzi di trasduzione sono stati sostituiti 12-15 ore dopo l’infezione e neuroni sono stati incubati per 3 giorni prima che gli esperimenti sono stati condotti.
Trattamento in vivo EMD87580
Per il trattamento in vivo, i topi sono stati iniettati per via intraperitoneale con EMD87580 (1 mg/ml in PBS) alla dose di 5 mg/kg. Inoltre, i topi hanno ricevuto un’applicazione intranasale di 10 µl di 1 mg/ml di EMD87580. Gli animali sono stati trattati due volte al giorno per 2 giorni consecutivi (mattina e sera, intervallo di 12 ore). Il terzo giorno, i topi sono stati trattati con EMD87580 al mattino e sacrificati 2 ore dopo per le registrazioni extracellulare campo extracellulare di fette di ippocampo.
Registrazioni di campo extracellulare
Le fette di ippocampo sono state preparate da topi ApoE3-KI o ApoE4-KI di 2 o 3 mesi. I cervelli sono stati rapidamente rimossi e messi in soluzione fredda di taglio ad alto contenuto di saccarosio (110 mM saccarosio, 60 mM NaCl, 3 mM KCl, 1,25 mM NaH2PO4, 28 mM NaHCO3, 0,5 mM CaCl2, 5 mM glucosio, 0,6 mM acido ascorbico, 7 mM MgSO4). Le sezioni trasversali di 400 µm sono state tagliate con un vibratore. Le fette sono stati poi trasferiti in una camera di incubazione contenente il 50% di aCSF (124 mM NaCl, 3 mM KCl, 1,25 mM NaH2PO4, 26 mM NaHCO3, 10 mM D-glucosio, 2 mM CaCl2, 1 mM MgSO4) e il 50% di soluzione di taglio del saccarosio. Per le registrazioni elettrofisiologiche è stata utilizzata una concentrazione finale di 3 µM EMD87580. Per il trattamento combinato con Reelin, estratto cerebrale AD e EMD87580(Figura 8), fette sono stati pretrattati con EMD87580 per 3 ore e che la concentrazione è stata mantenuta durante la registrazione in presenza o assenza di Reelin e / o estratto AD. Le fette sono state trasferite nella camera di registrazione dell’interfaccia dove sono state tenute in aCSF a 31°C e ad una portata di 2 – 3 ml/min. Nella camera di registrazione, diverse combinazioni di trattamento con estratto di cervello AD, Reelin, e EMD87580 sono stati utilizzati. Le fette sono state perfuse con i diversi componenti finali per un ulteriore ~ 30 minuti fino a quando non si sono stabilizzati nella camera di registrazione prima dell’applicazione della stimolazione theta burst e un’ora dopo per tutto il periodo di registrazione. Per la stimolazione sono stati utilizzati elettrodi bipolari concentrici (FHC, Catalogo no CBBRC75, 1201 Main St Bowdoin, ME 04287, USA) e posto nella falda radiatum. L’intensità dello stimolo è stato impostato a 40 – 60% di risposta massima e consegnato attraverso uno stimolatore di impulsi isolato (A-M Systems, modello 2100. Un programma scritto personalizzato in Labview 7.0 è stato utilizzato per la registrazione e l’analisi di esperimenti LTP (per gentile concessione del dottor Jay Gibson). Un burst theta (TBS; treno di 4 impulsi a 100 Hz ripetuto 10 volte con intervalli di 200 ms e di nuovo ripetuto 5 volte a intervalli di 10 s) è stato utilizzato come stimolo di condizionamento. Per l’analisi di ingresso/uscita i dati sono stati binnati e montati in modo lineare. Le pendenze sono state calcolate utilizzando l’analisi di regressione.
Quantificazione e analisi statistica
I dati sono stati espressi come media ± SEM e valutati utilizzando il test t Student’s t a due code per due gruppi con una variabile testata e varianze uguali, o l’analisi a senso unico della varianza (ANOVA) con il post-hoc di Dunnett per gruppi multipli con la sola variabile testata. Le differenze sono state considerate significative a p<0,05 (*p<0,05, **p<0,01, ***p<0,001).
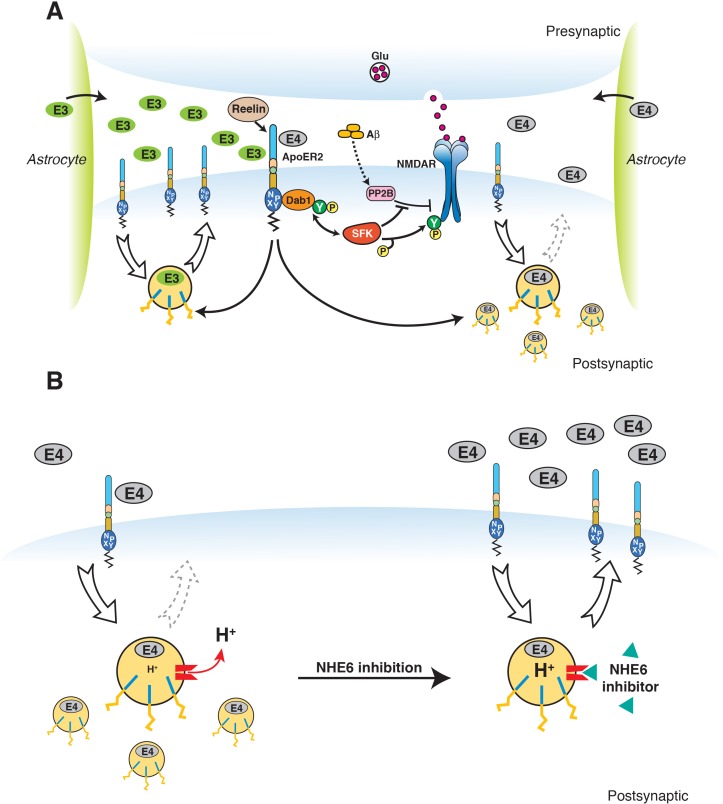
Figura 9.Ripristino del traffico vescicolare e dell’omeostasi sinaptica da inibizione NHE6 in presenza di ApoE4.(A) Effetto delle isoforme ApoE sulla segnalazione del recettore ApoE alla sinapsi. Apoer2 induce NMDAR tirosina fosforilazione tirosina attivando SFKs in risposta alla bobina nel neurone postsinaptico. Astrociti derivati ApoE3 (ovali verdi) o ApoE4 (ovali grigi) si legano ad Apoer2 e sono costitutivamente ma lentamente interiorizzati. Apoer2 subisce un’endocitosi accelerata in risposta alla segnalazione della bobina. ApoE4 sequesters Apoer2 in compartimenti intracellulari riducendo così la capacità del neurone postsinaptico di fosforilato (attivare) recettori NMDA in risposta alla Reelin (mostrato a destra), mentre ApoE2 o ApoE3 riciclare in modo efficiente alla superficie cellulare e quindi esaurire i livelli di superficie Apoer2 in misura minore (illustrato a sinistra per ApoE3). Gli oligomeri Aβ interferiscono con la fosforilazione della tirosina NMDAR attivando le fosfatasi tirosiniche. Questo pannello è stato riprodotto da Chen et al. (2010).(B) Quando NHE6 (rosso) funziona normalmente, il pH endosomico iniziale viene mantenuto a ~6,5 dall’azione della pompa protonica e dalla perdita di protoni attraverso NHE6. Questo livello di pH è vicino al punto isoelettrico (pI) di ApoE4, che è particolarmente sensibile al dispiegamento strutturale in uno stato globoso fuso all’ingresso nei compartimenti acidi(Morrow et al., 2002). La conseguente ridotta solubilità può ostacolare il rilascio efficiente di ApoE4 dai suoi recettori (come Apoer2 nei neuroni). Su inibizione NHE6, il pH endosomico è ulteriormente ridotto, con conseguente rilascio di ApoE4 dai suoi recettori attraverso una migliore solubilità e le forze di repulsione delle proteine si esercitano l’una sull’altra quando il pH è inferiore al loro pI. Così, il traffico e il riciclaggio di Apoer2 e dei recettori dei neurotrasmettitori a cui è associato viene ripristinato. Va notato che questa proprietà di ApoE4 di subire il traffico di ApoE4 attraverso i compartimenti endosomici è universale e non è limitata ai neuroni. Pertanto, l’inibizione dell’NHE6 può anche aiutare la clearance dell’amiloideβ nel cervello e la rimozione delle LDL nel fegato.
Figura 9.Figura 9. Ripristino del traffico vescicolare e omeostasi sinaptica da inibizione NHE6 in presenza di ApoE4.(A) Effetto delle isoforme ApoE sulla segnalazione del recettore ApoE alla sinapsi. Apoer2 induce NMDAR tirosina fosforilazione tirosina attivando SFKs in risposta alla bobina nel neurone postsinaptico. Astrociti derivati ApoE3 (ovali verdi) o ApoE4 (ovali grigi) si legano ad Apoer2 e sono costitutivamente ma lentamente interiorizzati. Apoer2 subisce un’endocitosi accelerata in risposta alla segnalazione della bobina. ApoE4 sequesters Apoer2 in compartimenti intracellulari riducendo così la capacità del neurone postsinaptico di fosforilato (attivare) recettori NMDA in risposta alla Reelin (mostrato a destra), mentre ApoE2 o ApoE3 riciclare in modo efficiente alla superficie cellulare e quindi esaurire i livelli di superficie Apoer2 in misura minore (illustrato a sinistra per ApoE3). Gli oligomeri Aβ interferiscono con la fosforilazione della tirosina NMDAR attivando le fosfatasi tirosiniche. Questo pannello è stato riprodotto da Chen et al. (2010).(B) Quando NHE6 (rosso) funziona normalmente, il pH endosomico iniziale viene mantenuto a ~6,5 dall’azione della pompa protonica e dalla perdita di protoni attraverso NHE6. Questo livello di pH è vicino al punto isoelettrico (pI) di ApoE4, che è particolarmente sensibile al dispiegamento strutturale in uno stato globoso fuso all’ingresso nei compartimenti acidi(Morrow et al., 2002). La conseguente ridotta solubilità può ostacolare il rilascio efficiente di ApoE4 dai suoi recettori (come Apoer2 nei neuroni). Su inibizione NHE6, il pH endosomico è ulteriormente ridotto, con conseguente rilascio di ApoE4 dai suoi recettori attraverso una migliore solubilità e le forze di repulsione delle proteine si esercitano l’una sull’altra quando il pH è inferiore al loro pI. Così, il traffico e il riciclaggio di Apoer2 e dei recettori dei neurotrasmettitori a cui è associato viene ripristinato. Va notato che questa proprietà di ApoE4 di subire il traffico di ApoE4 attraverso i compartimenti endosomici è universale e non è limitata ai neuroni. Pertanto, l’inibizione dell’NHE6 può anche aiutare la clearance dell’amiloideβ nel cervello e la rimozione delle LDL nel fegato.
References
- Alcántara S, Ruiz M, D’Arcangelo G, Ezan F, de Lecea L, Curran T, Sotelo C, Soriano E. Regional and cellular patterns of reelin mRNA expression in the forebrain of the developing and adult mouse. The Journal of Neuroscience. 1998; 18:7779-7799. DOI | PubMed
- Bal M, Leitz J, Reese AL, Ramirez DM, Durakoglugil M, Herz J, Monteggia LM, Kavalali ET. Reelin mobilizes a VAMP7-dependent synaptic vesicle pool and selectively augments spontaneous neurotransmission. Neuron. 2013; 80:934-946. DOI | PubMed
- Beffert U, Weeber EJ, Durudas A, Qiu S, Masiulis I, Sweatt JD, Li WP, Adelmann G, Frotscher M, Hammer RE, Herz J. Modulation of synaptic plasticity and memory by reelin involves differential splicing of the lipoprotein receptor Apoer2. Neuron. 2005; 47:567-579. DOI | PubMed
- Bell RD, Winkler EA, Singh I, Sagare AP, Deane R, Wu Z, Holtzman DM, Betsholtz C, Armulik A, Sallstrom J, Berk BC, Zlokovic BV. Apolipoprotein E controls cerebrovascular integrity via cyclophilin A. Nature. 2012; 485:512-516. DOI | PubMed
- Blacklow SC. Versatility in ligand recognition by LDL receptor family proteins: advances and frontiers. Current Opinion in Structural Biology. 2007; 17:419-426. DOI | PubMed
- Brett CL, Wei Y, Donowitz M, Rao R. Human Na(+)/H(+) exchanger isoform 6 is found in recycling endosomes of cells, not in mitochondria. American Journal of Physiology. Cell Physiology. 2002; 282:C1031-C1041. DOI | PubMed
- Campion D, Dumanchin C, Hannequin D, Dubois B, Belliard S, Puel M, Thomas-Anterion C, Michon A, Martin C, Charbonnier F, Raux G, Camuzat A, Penet C, Mesnage V, Martinez M, Clerget-Darpoux F, Brice A, Frebourg T. Early-onset autosomal dominant alzheimer disease: prevalence, genetic heterogeneity, and mutation spectrum. The American Journal of Human Genetics. 1999; 65:664-670. DOI | PubMed
- Casey JR, Grinstein S, Orlowski J. Sensors and regulators of intracellular pH. Nature Reviews Molecular Cell Biology. 2010; 11:50-61. DOI | PubMed
- Castellano JM, Kim J, Stewart FR, Jiang H, DeMattos RB, Patterson BW, Fagan AM, Morris JC, Mawuenyega KG, Cruchaga C, Goate AM, Bales KR, Paul SM, Bateman RJ, Holtzman DM. Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Science Translational Medicine. 2011; 3DOI | PubMed
- Cataldo AM, Peterhoff CM, Troncoso JC, Gomez-Isla T, Hyman BT, Nixon RA. Endocytic pathway abnormalities precede amyloid beta deposition in sporadic alzheimer’s disease and Down syndrome: differential effects of APOE genotype and presenilin mutations. The American Journal of Pathology. 2000; 157:277-286. PubMed
- Chen L, Chen CX, Gan XT, Beier N, Scholz W, Karmazyn M. Inhibition and reversal of myocardial infarction-induced hypertrophy and heart failure by NHE-1 inhibition. American Journal of Physiology-Heart and Circulatory Physiology. 2004; 286:H381-H387. DOI | PubMed
- Chen Y, Beffert U, Ertunc M, Tang TS, Kavalali ET, Bezprozvanny I, Herz J. Reelin modulates NMDA receptor activity in cortical neurons. Journal of Neuroscience. 2005; 25:8209-8216. DOI | PubMed
- Chen Y, Durakoglugil MS, Xian X, Herz J. ApoE4 reduces glutamate receptor function and synaptic plasticity by selectively impairing ApoE receptor recycling. PNAS. 2010; 107:12011-12016. DOI | PubMed
- Choi HY, Liu Y, Tennert C, Sugiura Y, Karakatsani A, Kröger S, Johnson EB, Hammer RE, Lin W, Herz J. APP interacts with LRP4 and agrin to coordinate the development of the neuromuscular junction in mice. eLife. 2013; 2DOI | PubMed
- Cirrito JR, Kang JE, Lee J, Stewart FR, Verges DK, Silverio LM, Bu G, Mennerick S, Holtzman DM. Endocytosis is required for synaptic activity-dependent release of amyloid-beta in vivo. Neuron. 2008; 58:42-51. DOI | PubMed
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of alzheimer’s disease in late onset families. Science. 1993; 261:921-923. DOI | PubMed
- D’Arcangelo G, Miao GG, Chen SC, Soares HD, Morgan JI, Curran T. A protein related to extracellular matrix proteins deleted in the mouse mutant reeler. Nature. 1995; 374:719-723. DOI | PubMed
- D’Arcangelo G, Nakajima K, Miyata T, Ogawa M, Mikoshiba K, Curran T. Reelin is a secreted glycoprotein recognized by the CR-50 monoclonal antibody. The Journal of Neuroscience. 1997; 17:23-31. DOI | PubMed
- Dean DC, Jerskey BA, Chen K, Protas H, Thiyyagura P, Roontiva A, O’Muircheartaigh J, Dirks H, Waskiewicz N, Lehman K, Siniard AL, Turk MN, Hua X, Madsen SK, Thompson PM, Fleisher AS, Huentelman MJ, Deoni SC, Reiman EM. Brain differences in infants at differential genetic risk for late-onset alzheimer disease: a cross-sectional imaging study. JAMA Neurology. 2014; 71:11-22. DOI | PubMed
- Dean KM, Roudot P, Welf ES, Pohlkamp T, Garrelts G, Herz J, Fiolka R. Imaging subcellular dynamics with fast and Light-Efficient volumetrically parallelized microscopy. Optica. 2017; 4:263-271. DOI | PubMed
- Decourt B, Mobley W, Reiman E, Shah RJ, Sabbagh MN. Recent perspectives on APP, secretases, endosomal pathways and how they influence Alzheimer’s Related Pathological Changes in Down Syndrome. Journal of Alzheimer’s Disease & Parkinsonism Suppl. 2013; 7
- Del Río JA, Heimrich B, Borrell V, Förster E, Drakew A, Alcántara S, Nakajima K, Miyata T, Ogawa M, Mikoshiba K, Derer P, Frotscher M, Soriano E. A role for Cajal-Retzius cells and reelin in the development of hippocampal connections. Nature. 1997; 385:70-74. DOI | PubMed
- Durakoglugil MS, Chen Y, White CL, Kavalali ET, Herz J. Reelin signaling antagonizes beta-amyloid at the synapse. PNAS. 2009; 106:15938-15943. DOI | PubMed
- Fagan AM, Watson M, Parsadanian M, Bales KR, Paul SM, Holtzman DM. Human and murine ApoE markedly alters A beta metabolism before and after plaque formation in a mouse model of alzheimer’s disease. Neurobiology of Disease. 2002; 9:305-318. DOI | PubMed
- Förster E, Kaltschmidt C, Deng J, Cremer H, Deller T, Frotscher M. Lamina-specific cell adhesion on living slices of Hippocampus. Development. 1998; 125:3399-3410. PubMed
- Fuster DG, Alexander RT. Traditional and emerging roles for the SLC9 na+/H+ exchangers. Pflügers Archiv – European Journal of Physiology. 2014; 466:61-76. DOI | PubMed
- Goldstein JL, Brown MS, Anderson RG, Russell DW, Schneider WJ. Receptor-mediated endocytosis: concepts emerging from the LDL receptor system. Annual Review of Cell Biology. 1985; 1:1-39. DOI | PubMed
- Grassin-Delyle S, Buenestado A, Naline E, Faisy C, Blouquit-Laye S, Couderc LJ, Le Guen M, Fischler M, Devillier P. Intranasal drug delivery: an efficient and non-invasive route for systemic administration: focus on opioids. Pharmacology & Therapeutics. 2012; 134:366-379. DOI | PubMed
- Harris JJ, Jolivet R, Attwell D. Synaptic Energy Use and Supply. Neuron. 2012; 75:762-777. DOI | PubMed
- Heeren J, Grewal T, Laatsch A, Becker N, Rinninger F, Rye KA, Beisiegel U. Impaired recycling of apolipoprotein E4 is associated with intracellular cholesterol accumulation. Journal of Biological Chemistry. 2004; 279:55483-55492. DOI | PubMed
- Herz j, Xian X, Yang Y. Promoting Cycling of ApoE4 Isoform. University of Texas, Board of Regents: USA; 2010.
- Hiesberger T, Trommsdorff M, Howell BW, Goffinet A, Mumby MC, Cooper JA, Herz J. Direct binding of reelin to VLDL receptor and ApoE receptor 2 induces tyrosine phosphorylation of disabled-1 and modulates tau phosphorylation. Neuron. 1999; 24:481-489. DOI | PubMed
- Howell BW, Gertler FB, Cooper JA. Mouse disabled (mDab1): a src binding protein implicated in neuronal development. The EMBO Journal. 1997; 16:121-132. DOI | PubMed
- Ishigaki Y, Oikawa S, Suzuki T, Usui S, Magoori K, Kim DH, Suzuki H, Sasaki J, Sasano H, Okazaki M, Toyota T, Saito T, Yamamoto TT. Virus-mediated transduction of apolipoprotein E (ApoE)-sendai develops lipoprotein glomerulopathy in ApoE-deficient mice. Journal of Biological Chemistry. 2000; 275:31269-31273. DOI | PubMed
- Jonsson T, Atwal JK, Steinberg S, Snaedal J, Jonsson PV, Bjornsson S, Stefansson H, Sulem P, Gudbjartsson D, Maloney J, Hoyte K, Gustafson A, Liu Y, Lu Y, Bhangale T, Graham RR, Huttenlocher J, Bjornsdottir G, Andreassen OA, Jönsson EG, Palotie A, Behrens TW, Magnusson OT, Kong A, Thorsteinsdottir U, Watts RJ, Stefansson K. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature. 2012; 488:96-99. DOI | PubMed
- Karch CM, Cruchaga C, Goate AM. Alzheimer’s disease genetics: from the bench to the clinic. Neuron. 2014; 83:11-26. DOI | PubMed
- Kawasaki F, Hazen M, Ordway RW. Fast synaptic fatigue in shibire mutants reveals a rapid requirement for dynamin in synaptic vesicle membrane trafficking. Nature Neuroscience. 2000; 3:859-860. DOI | PubMed
- Kim N, Stiegler AL, Cameron TO, Hallock PT, Gomez AM, Huang JH, Hubbard SR, Dustin ML, Burden SJ. Lrp4 is a receptor for agrin and forms a complex with MuSK. Cell. 2008; 135:334-342. DOI | PubMed
- Knouff C, Hinsdale ME, Mezdour H, Altenburg MK, Watanabe M, Quarfordt SH, Sullivan PM, Maeda N. Apo E structure determines VLDL clearance and atherosclerosis risk in mice. Journal of Clinical Investigation. 1999; 103:1579-1586. DOI | PubMed
- Kuchibhotla KV, Goldman ST, Lattarulo CR, Wu HY, Hyman BT, Bacskai BJ. Abeta plaques lead to aberrant regulation of calcium homeostasis in vivo resulting in structural and functional disruption of neuronal networks. Neuron. 2008; 59:214-225. DOI | PubMed
- Lane-Donovan C, Philips GT, Herz J. More than cholesterol transporters: lipoprotein receptors in CNS function and neurodegeneration. Neuron. 2014; 83:771-787. DOI | PubMed
- Lane-Donovan C, Herz J. ApoE, ApoE receptors, and the synapse in Alzheimer’s Disease. Trends in Endocrinology & Metabolism. 2017; 28:273-284. DOI | PubMed
- Lin YT, Seo J, Gao F, Feldman HM, Wen HL, Penney J, Cam HP, Gjoneska E, Raja WK, Cheng J, Rueda R, Kritskiy O, Abdurrob F, Peng Z, Milo B, Yu CJ, Elmsaouri S, Dey D, Ko T, Yankner BA, Tsai LH. APOE4 causes widespread molecular and cellular alterations associated with Alzheimer’s Disease Phenotypes in Human iPSC-Derived Brain Cell Types. Neuron. 2018; 98:1141-1154. DOI | PubMed
- Liu Q, Zerbinatti CV, Zhang J, Hoe HS, Wang B, Cole SL, Herz J, Muglia L, Bu G. Amyloid precursor protein regulates brain apolipoprotein E and cholesterol metabolism through lipoprotein receptor LRP1. Neuron. 2007; 56:66-78. DOI | PubMed
- Liu Q, Zhang J, Zerbinatti C, Zhan Y, Kolber BJ, Herz J, Muglia LJ, Bu G. Lipoprotein receptor LRP1 regulates leptin signaling and energy homeostasis in the adult central nervous system. PLoS Biology. 2011; 9DOI | PubMed
- Liu CC, Kanekiyo T, Xu H, Bu G. Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nature Reviews Neurology. 2013; 9:106-118. DOI | PubMed
- Mahley RW. Apolipoprotein E: cholesterol transport protein with expanding role in cell biology. Science. 1988; 240:622-630. DOI | PubMed
- Masereel B, Pochet L, Laeckmann D. An overview of inhibitors of na(+)/H(+) exchanger. European Journal of Medicinal Chemistry. 2003; 38:547-554. DOI | PubMed
- May P, Rohlmann A, Bock HH, Zurhove K, Marth JD, Schomburg ED, Noebels JL, Beffert U, Sweatt JD, Weeber EJ, Herz J. Neuronal LRP1 functionally associates with postsynaptic proteins and is required for normal motor function in mice. Molecular and Cellular Biology. 2004; 24:8872-8883. DOI | PubMed
- Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, Carpenter AE, Foo SY, Stewart SA, Stockwell BR, Hacohen N, Hahn WC, Lander ES, Sabatini DM, Root DE. A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell. 2006; 124:1283-1298. DOI | PubMed
- Morrow JA, Hatters DM, Lu B, Hochtl P, Oberg KA, Rupp B, Weisgraber KH. Apolipoprotein E4 forms a molten globule. A potential basis for its association with disease. The Journal of Biological Chemistry. 2002; 277:50380-50385. DOI | PubMed
- Nakajima C, Kulik A, Frotscher M, Herz J, Schäfer M, Bock HH, May P. Low density lipoprotein receptor-related protein 1 (LRP1) modulates N-methyl-D-aspartate (NMDA) receptor-dependent intracellular signaling and NMDA-induced regulation of postsynaptic protein complexes. Journal of Biological Chemistry. 2013; 288:21909-21923. DOI | PubMed
- Nixon RA, Mathews PM, Cataldo AM. The neuronal endosomal-lysosomal system in Alzheimer’s disease. Journal of Alzheimer’s Disease. 2001; 3:97-107. DOI | PubMed
- Ordovas JM, Litwack-Klein L, Wilson PW, Schaefer MM, Schaefer EJ. Apolipoprotein E isoform phenotyping methodology and population frequency with identification of apoE1 and apoE5 isoforms. Journal of Lipid Research. 1987; 28:371-380. PubMed
- Palop JJ, Mucke L. Amyloid-beta-induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nature Neuroscience. 2010; 13:812-818. DOI | PubMed
- Pesold C, Impagnatiello F, Pisu MG, Uzunov DP, Costa E, Guidotti A, Caruncho HJ. Reelin is preferentially expressed in neurons synthesizing gamma-aminobutyric acid in cortex and Hippocampus of adult rats. PNAS. 1998; 95:3221-3226. DOI | PubMed
- Pohlkamp T, Dávid C, Cauli B, Gallopin T, Bouché E, Karagiannis A, May P, Herz J, Frotscher M, Staiger JF, Bock HH. Characterization and distribution of Reelin-positive interneuron subtypes in the rat barrel cortex. Cerebral Cortex. 2014; 24:3046-3058. DOI | PubMed
- Pohlkamp T, Durakoglugil M, Lane-Donovan C, Xian X, Johnson EB, Hammer RE, Herz J. Lrp4 domains differentially regulate limb/brain development and synaptic plasticity. Plos One. 2015; 10DOI | PubMed
- Pohlkamp T, Wasser CR, Herz J. Functional roles of the interaction of APP and lipoprotein receptors. Frontiers in Molecular Neuroscience. 2017; 10DOI | PubMed
- Prasad H, Rao R. Amyloid clearance defect in ApoE4 astrocytes is reversed by epigenetic correction of endosomal pH. PNAS. 2018; 115:E6640-E6649. DOI | PubMed
- Rall SC, Mahley RW. The role of apolipoprotein E genetic variants in lipoprotein disorders. Journal of Internal Medicine. 1992; 231:653-659. DOI | PubMed
- Rellin L, Heeren J, Beisiegel U. Recycling of apolipoprotein E is not associated with cholesterol efflux in neuronal cells. Biochimica Et Biophysica Acta (BBA) – Molecular and Cell Biology of Lipids. 2008; 1781:232-238. DOI | PubMed
- Robinson PJ, Sontag J-M, Liu J-P, Fykse EM, Slaughter C, McMahontt H, Südhof TC. Dynamin GTPase regulated by protein kinase C phosphorylation in nerve terminals. Nature. 1993; 365:163-166. DOI | PubMed
- Rönicke R, Schröder UH, Böhm K, Reymann KG. The Na+/H+ exchanger modulates long-term potentiation in rat hippocampal slices. Naunyn-Schmiedeberg’s Archives of Pharmacology. 2009; 379:233-239. DOI
- Salehi A, Delcroix JD, Belichenko PV, Zhan K, Wu C, Valletta JS, Takimoto-Kimura R, Kleschevnikov AM, Sambamurti K, Chung PP, Xia W, Villar A, Campbell WA, Kulnane LS, Nixon RA, Lamb BT, Epstein CJ, Stokin GB, Goldstein LS, Mobley WC. Increased app expression in a mouse model of down’s syndrome disrupts NGF transport and causes cholinergic neuron degeneration. Neuron. 2006; 51:29-42. DOI | PubMed
- Selkoe DJ, Hardy J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Molecular Medicine. 2016; 8:595-608. DOI | PubMed
- Shankar GM, Li S, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nature Medicine. 2008; 14:837-842. DOI | PubMed
- Shankar GM, Walsh DM. Alzheimer’s disease: synaptic dysfunction and Abeta. Molecular Neurodegeneration. 2009; 4DOI | PubMed
- Shaw P, Lerch JP, Pruessner JC, Taylor KN, Rose AB, Greenstein D, Clasen L, Evans A, Rapoport JL, Giedd JN. Cortical morphology in children and adolescents with different apolipoprotein E gene polymorphisms: an observational study. The Lancet Neurology. 2007; 6:494-500. DOI | PubMed
- Sontag JM, Fykse EM, Ushkaryov Y, Liu JP, Robinson PJ, Südhof TC. Differential expression and regulation of multiple dynamins. The Journal of Biological Chemistry. 1994; 269:4547-4554. PubMed
- Strittmatter WJ, Weisgraber KH, Huang DY, Dong LM, Salvesen GS, Pericak-Vance M, Schmechel D, Saunders AM, Goldgaber D, Roses AD. Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset alzheimer disease. PNAS. 1993; 90:8098-8102. DOI | PubMed
- Sudhof TC. The synaptic vesicle cycle. Annual Review of Neuroscience. 2004; 27:509-547. DOI | PubMed
- Sullivan PM, Mezdour H, Aratani Y, Knouff C, Najib J, Reddick RL, Quarfordt SH, Maeda N. Targeted replacement of the mouse apolipoprotein E gene with the common human APOE3 allele enhances diet-induced hypercholesterolemia and atherosclerosis. Journal of Biological Chemistry. 1997; 272:17972-17980. DOI | PubMed
- Townsend M, Shankar GM, Mehta T, Walsh DM, Selkoe DJ. Effects of secreted oligomers of amyloid beta-protein on hippocampal synaptic plasticity: a potent role for trimers. The Journal of Physiology. 2006; 572:477-492. DOI | PubMed
- Trommsdorff M, Gotthardt M, Hiesberger T, Shelton J, Stockinger W, Nimpf J, Hammer RE, Richardson JA, Herz J. Reeler/Disabled-like disruption of neuronal migration in knockout mice lacking the VLDL receptor and ApoE receptor 2. Cell. 1999; 97:689-701. DOI | PubMed
- Wasser CR, Masiulis I, Durakoglugil MS, Lane-Donovan C, Xian X, Beffert U, Agarwala A, Hammer RE, Herz J. Differential splicing and glycosylation of Apoer2 alters synaptic plasticity and fear learning. Science Signaling. 2014; 7DOI | PubMed
- Wasser CR, Herz J. Reelin: neurodevelopmental architect and homeostatic regulator of excitatory synapses. Journal of Biological Chemistry. 2017; 292:1330-1338. DOI | PubMed
- Weeber EJ, Beffert U, Jones C, Christian JM, Forster E, Sweatt JD, Herz J. Reelin and ApoE receptors cooperate to enhance hippocampal synaptic plasticity and learning. Journal of Biological Chemistry. 2002; 277:39944-39952. DOI | PubMed
- Weisgraber KH. Apolipoprotein E: structure-function relationships. Advances in Protein Chemistry. 1994; 45:249-302. PubMed
- Wildsmith KR, Basak JM, Patterson BW, Pyatkivskyy Y, Kim J, Yarasheski KE, Wang JX, Mawuenyega KG, Jiang H, Parsadanian M, Yoon H, Kasten T, Sigurdson WC, Xiong C, Goate A, Holtzman DM, Bateman RJ. In vivo human apolipoprotein E isoform fractional turnover rates in the CNS. PLoS ONE. 2012; 7DOI | PubMed
- Wintersteiner O, Abramson HA. The isoelectric point of insulin: electrical properties of adsorbed and crystalline insulin. The Journal of Biological Chemistry. 1933; 99:741-753.
- Zhang B, Luo S, Wang Q, Suzuki T, Xiong WC, Mei L. LRP4 serves as a coreceptor of agrin. Neuron. 2008; 60:285-297. DOI | PubMed
- Zhao N, Liu CC, Van Ingelgom AJ, Martens YA, Linares C, Knight JA, Painter MM, Sullivan PM, Bu G. Apolipoprotein E4 impairs neuronal insulin signaling by trapping insulin receptor in the endosomes. Neuron. 2017; 96:115-129. DOI | PubMed
Fonte
Xian X, Pohlkamp T, Durakoglugil MS, Wong CH, Beck JK, et al. () Reversal of ApoE4-induced recycling block as a novel prevention approach for Alzheimer’s disease. eLife 7e40048. https://doi.org/10.7554/eLife.40048




