
Abstract
Introduzione
La replicazione del DNA metazoico richiede l’iniziazione a migliaia di origini di replicazione del DNA durante la fase S di ogni ciclo cellulare. Le origini sono loci genomiche in cui le elicasi di DNA prima di srotolare il DNA e la sintesi del DNA inizia. Le origini sono resi competenti per la replicazione durante la fase G1 di ogni ciclo cellulare con il caricamento di minicromosomi complessi di manutenzione (MCM) sul DNA. MCM è il componente principale dell’elicasi replicativa, e il processo di carico MCM è chiamato licenza di origine. I livelli totali di MCM rimangono costanti per tutto il ciclo cellulare, ma i livelli di MCM caricato sul DNA cambiano con il progredire delle cellule attraverso il ciclo cellulare. Le cellule possono iniziare il caricamento MCM già nel telopasi e il caricamento continua in tutto il G1 fino alla transizione G1/S, il punto di massimo MCM carico di DNA(Kimura et al., 1994; Todorov et al., 1995). Durante la fase S, i singoli complessi MCM sono attivati per lo srotolamento del DNA come origine ‘fuoco’. I complessi MCM viaggiano con le forchette di replicazione e vengono progressivamente scaricati man mano che le forchette di replicazione terminano(Figura 1a)(Deegan e Diffley, 2016; Remus e Diffley, 2009; Siddiqui et al., 2013).
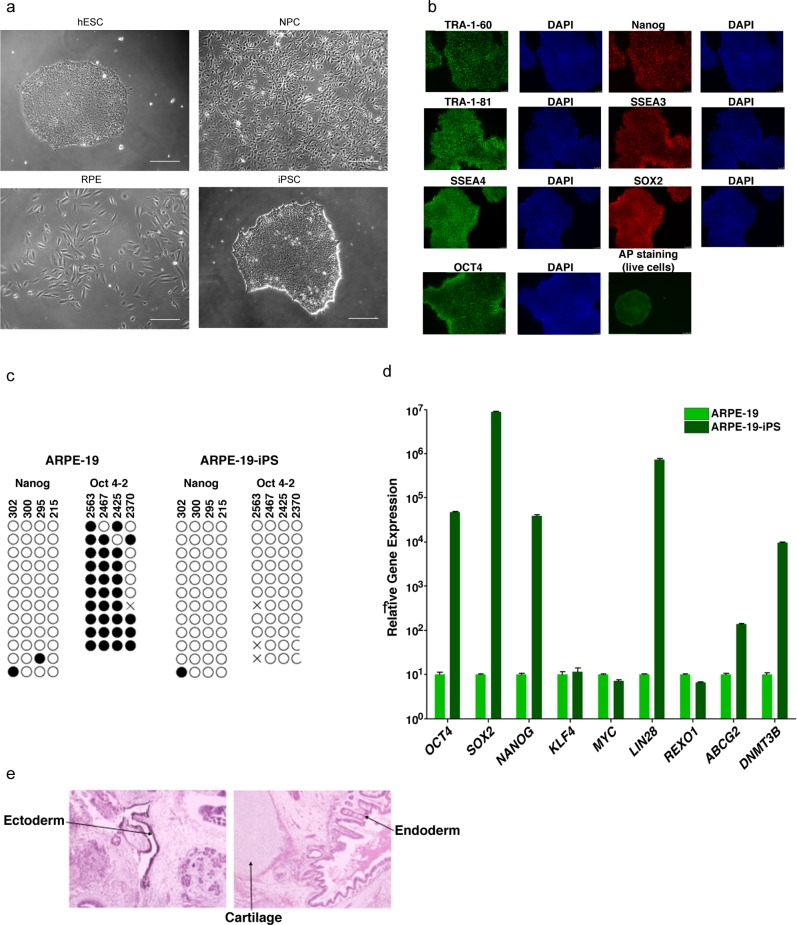
Figura 1-figure supplement 3.Figura 1—supplemento figura 3. Le cellule staminali pluripotenti caricano le MCM più velocemente delle cellule differenziate.citometria a flusso gating.validazione della citometria a flusso di cromatina.caratterizzazione delle cellule pluripotenti e differenziate.(a) I livelli di MCM caricati con DNA aumentano in G1 e diminuiscono in fase S, mentre i livelli totali di proteine MCM sono costanti per tutto il ciclo cellulare.(b) L’analisi citometrica a flusso di DNA caricato e MCM totale in cellule RPE1-hTERT asincrone proliferanti. Le fasi del ciclo cellulare sono definite dal contenuto di DNA e dalla sintesi del DNA. A sinistra: Le cellule sono state etichettate con EdU, estratto con detergente non ionico per rimuovere MCM non legato, fisso, e colorato con anti-MCM2 (un marcatore per il complesso MCM2-7), DAPI (DNA totale), e per l’incorporazione EdU (sintesi attiva del DNA). Le cellule arancioni sono S-fase con MCM carico di DNA, le cellule blu sono G1-fase con MCM carico di DNA, e le cellule grigie sono G1/G2/M cellule fase G1/G2/M senza MCM carico di DNA. Giusto: Le cellule sono state trattate come a sinistra, tranne che sono stati fissati prima dell’estrazione per rilevare MCM2 totale.(c) Le cellule T98G sono stati sincronizzati in G0 da inibizione di contatto e privazione di siero, poi rilasciato in G1 per 10 o 12 ore, o risincronizzato nei primi S con idrossiurea (HU), e rilasciato in S per 6 o 8 ore. MCM3 in frazioni arricchite con cromatina (Caricato) o lisati cellulari interi (Totale) è stato rilevato mediante immunoblotting.(d) Cromatografia a flusso di cromatina delle linee cellulari asincrone indicate che misurano il contenuto di DNA (DAPI), sintesi del DNA (incorporazione EdU), e MCM caricato (anti-MCM2). Le cellule blu sono G1-MCMDNA-positivo e EdU-negativo, arancione sono S fase MCMDNA-positivo; grigio sono G1/G2/M-MCMDNA-negativo.e) Barra grafica impilata della distribuzione di fase del ciclo cellulare dalle celle in(d); media con barre di errore ± SD (n = 3 repliche biologiche). La percentuale di cellule G1 in ogni popolazione è riportata nei settori verdi. I tempi di raddoppio sono stati calcolati sperimentalmente utilizzando l’analisi di regressione in GraphPad Prism.(a) Esempio di citometria a flusso gating con cromatina estratta ARPE-19 (RPE) cellule estratte dalla Figura 1d, misurando MCM caricato (anti-MCM2), sintesi del DNA (EdU incorporazione) e il contenuto di DNA (DAPI). Gating per discriminare le cellule da detriti era su FS-area vs SS-area, singlets (singole cellule) da ciuffi di cellule / doppie era su DAPI altezza vs DAPI area, fasi del ciclo cellulare sono stati determinati su DAPI vs EdU Non specifica colorazione di fondo da parte dell’anticorpo secondario è stata misurata utilizzando un campione di controllo negativo colorato senza anticorpo primario, solo l’anticorpo secondario. Questo cancello soglia di fondo è stato applicato a campioni sperimentali; G1-MCMDNA-positivo cellule sono state identificate prima dal contenuto di DNA e lo stato negativo EdU(G1) e poi dal segnale MCM2.(b) Esempio di citometria a flusso di colore gating con cromatina estratto ARPE-19 cellule dalla Figura 1d. A sinistra: controllo negativo per definire la colorazione di fondo con né la rilevazione EdU né l’anticorpo primario MCM2. Le cellule sono state colorate con DAPI, sottoposte a chimica di rilevazione EdU, e colorate con anticorpo secondario. Soglie di sfondo sono stati impostati utilizzando questi controlli e applicato a campioni sperimentali. Giusto: Campioni sperimentali in figura 1d principale che mostra il gating per identificare la sintesi del DNA (EdU), e MCM caricato (anti-MCM2). Gates definire schemi di colore per le trame di punti di colore.(a) Citometria a flusso di cromatina di cellule RPE colorate per MCM2 caricato (anti-MCM2), MCM3 caricato (anti-MCM3), e il contenuto di DNA (DAPI). Tutti e tre i grafici sono lo stesso campione. Le cellule colorate con anticorpo monoclonale MCM2 per MCM2 caricato co-stain altrettanto bene per un anticorpo policlonale MCM3 per MCM3 caricato.b) Citometria a flusso di cromatina di cellule RPE trattate con 100 nM siControl o un pool di 100 nM di siMCM3 per 72 ore, misura del contenuto di DNA (DAPI), sintesi del DNA (EdU incorporation), e MCM3 carico (anti-MCM3). I livelli di MCM3 caricato diminuiscono con il trattamento siMCM3, dimostrando la specificità degli anticorpi.(c) Immunoblot di cellule da(b).(d) Cellule RPE sincronizzate in G0 per inibizione da contatto. A sinistra: citometria a flusso proteico totale delle cellule G0 che misura il totale MCM (anti-MCM2), sintesi del DNA (incorporazione EdU) e contenuto di DNA (DAPI). A destra: Citometria a flusso di cromatina di cellule G0 che misura il MCM carico (anti-MCM2), la sintesi del DNA (incorporazione EdU) e il contenuto di DNA (DAPI). Le cellule G0 mancano di MCM caricato, dimostrando la specificità degli anticorpi.(e) Immunoblot di cellule per(d) dimostrare la sincronizzazione G0.(f) Immunoblot di cellule frazionate in proteine solubili e pellet (proteina cromatina associata) utilizzando un tampone CSK sale normale (100 mM NaCl) o ad alto contenuto di sale CSK (300 mM NaCl). NaCl ad alto contenuto di sale 300 mM NaCl elimina le proteine debolmente associate alla cromatina, compreso il Cdc6.g) Citometria a flusso di cromatina di cellule lisate con 100 mM di NaCl CSK normale o 300 mM di NaCl CSK ad alto contenuto salino.h) Istogrammi delle sole cellule G1-MCMDNA-positive di(g). Il MCM caricato in G1 rimane costante con alto sale, dimostrando la citometria a flusso di cromatina misura il DNA caricato MCM. Si noti la dura 300 mM sale mM altera la morfologia nucleare e la dispersione per citometria a flusso, per questo motivo, abbiamo usato l’estrazione di NaCl 100 mM per tutti gli altri esperimenti di citometria a flusso.(a) Immagini rappresentative di contrasto di fase in scala di grigi delle linee cellulari indicate dalla Figura 1d. Barra di scala è di 50 μm.(b) Analisi di immunofluorescenza delle cellule iPS visualizzando TRA-1-60, TRA-1-81, SSEA o OCT4 (in verde) e Nanog, SSEA3 o SOX2 (in rosso). La colorazione DAPI per il DNA indica i nuclei delle singole cellule in ogni colonia. La colorazione con fosfatasi alcalina viva (AP) è stata eseguita anche come indicatore positivo delle cellule staminali.(c) L’analisi del sequenziamento del bisolfito delle regioni promotrici NANOG e OCT4-2 nelle cellule ARPE-19 e ARPE-19-iPS è indicata. Le CpG metilate sono indicate con un cerchio chiuso, mentre le CpG non metilate sono indicate con un cerchio aperto. Una ‘X’ indica un disallineamento o uno spazio vuoto nella sequenza dei bisolfiti. La posizione CpG relativa al sito di partenza della trascrizione a valle è indicata sopra ogni riga.(d) Quantitativa RT-PCR analisi RT-PCR che mostra l’espressione genica relativa di OCT4, SOX2, NANOG, KLF4, MYC, LIN28, REXO1, ABCG2 e DNMT3B in ARPE-19 e ARPE-19-iPS cellule (normalizzato a GAPDH). Le barre di errore indicano un errore standard.e) La colorazione di ematossilina ed eosina di sezioni di teratoma da topi immunodeficienti iniettati con le cellule ARPE-19-iPS mostra cartilagine, così come l’endoderma e il teratoma ectoderma formato da ARPE-19-iPS.
Il controllo della licenza di origine è fondamentale per la stabilità del genoma. Le origini non devono essere ri-licenziate dopo l’inizio della fase S, perché tale ri-licenziamento può causare un fenomeno genotossico noto come ri-replicazione che può causare rotture del doppio filamento, amplificazione del gene, aneuploidia e instabilità generale del genoma(Arias e Walter ,2007; Truong e Wu, 2011). Per evitare la replicazione, il caricamento del MCM è strettamente limitato alla fase G1 da meccanismi multipli di sovrapposizione che distruggono o inattivano le proteine del caricamento del MCM per impedire qualsiasi licenza di nuova origine dopo l’inizio della fase S(Remus e Diffley, 2009; Arias e Walter, 2007; Truong e Wu, 2011). D’altra parte, le cellule tipicamente caricano da 5 a 10 volte più complessi MCM in G1 di quanto strettamente necessario in circostanze ideali, ed il carico addizionale MCM assicura la duplicazione tempestiva e completa del genoma anche se si incontrano ostacoli alla replicazione in fase S (Ibarra etal., 2008; Woodward et al. , 2006; Ge et al., 2007). E ‘possibile per le cellule di proliferare con meno di carico ottimale MCM, ma tali cellule sono ipersensibili ai danni del DNA e lo stress di replicazione(Blow et al., 2011; McIntosh e Blow, 2012).
Il carico di MCM alle origini della licenza è limitato a G1, ma la lunghezza G1 varia ampiamente tra i diversi tipi di cellule. Ad esempio, i cicli cellulari specializzati per lo sviluppo e le cellule immunitarie hanno lunghezze G1 minime di pochi minuti(O’Farrell et al., 2004; Kinjyo et al., 2015; Kermi et al., 2017). Nelle cellule staminali embrionali umane in coltura, G1 è solo 2-3 ore, e questo breve G1 è sia un segno distintivo di ed è stato implicato nel mantenimento della pluripotenza (Soufie Dalton, 2016; Kareta et al ., 2015a). G1 si allunga all’inizio della differenziazione, e nelle cellule somatiche coltivate è spesso maggiore di 12 ore(Calder et al., 2013). Così, diverse cellule proliferanti hanno quantità drasticamente diverse di tempo a disposizione per completare il carico MCM prima di fare il G1-to-S transizione di fase. Inoltre, le cellule staminali pluripotenti rispondono agli stimoli di differenziazione in particolare nella fase G1, suggerendo che l’equilibrio tra le fasi del ciclo cellulare influenza il potenziale di differenziazione(Gonzales et al., 2015; Pauklin e Vallier, 2013).
Dato che il carico MCM è limitato a G1 e l’ampia variazione delle lunghezze G1, abbiamo postulato che la quantità assoluta di MCM caricato in fase S è un prodotto sia del tempo trascorso in G1 che del tasso di carico MCM. La combinazione di questi due parametri ha implicazioni per la stabilità del genoma perché il carico più o meno MCM in G1 influenza la lunghezza della fase S e quanto efficacemente le cellule della fase S possono ospitare sia fonti endogene che esogene di stress da replicazione(Shima et al., 2007; Pruitt et al., 2007). Queste implicazioni sono rilevanti sia quando le distribuzioni del ciclo cellulare cambiano durante lo sviluppo che durante l’oncogenesi, poiché molte linee cellulari tumorali hanno anche brevi fasi G1. Il tasso effettivo di carico MCM nelle cellule umane non è stato ancora quantificato, tuttavia, e poco si sa su come la quantità, il tasso o la tempistica del carico MCM varia tra le cellule con diverse lunghezze G1. Qui, abbiamo utilizzato la citometria a flusso monocellulare per misurare i tassi di carico MCM in popolazioni asincrone di cellule pluripotenti e differenziate. Abbiamo scoperto che il carico rapido MCM è intrinseco alla pluripotenza, rallenta universalmente durante la differenziazione, e la licenza di replicazione rapida di replicazione sopprime la differenziazione. Questi risultati dimostrano che il tasso di carico MCM è soggetto a una regolamentazione dello sviluppo, e suggeriamo che la concessione di licenze di origine rapida è una nuova caratteristica della pluripotenza.
Figura 1-figure supplement 3.Figura 1—supplemento figura 3. Le cellule staminali pluripotenti caricano gli MCM più velocemente delle cellule differenziate.citometria a flusso gating.validazione della citometria a flusso della cromatina.caratterizzazione delle cellule pluripotenti e differenziate.(a) I livelli di MCM caricati con DNA aumentano in G1 e diminuiscono in fase S, mentre i livelli totali di proteine MCM sono costanti per tutto il ciclo cellulare.(b) L’analisi citometrica a flusso di DNA caricato e MCM totale in cellule RPE1-hTERT asincrone proliferanti. Le fasi del ciclo cellulare sono definite dal contenuto di DNA e dalla sintesi del DNA. A sinistra: Le cellule sono state etichettate con EdU, estratto con detergente non ionico per rimuovere MCM non legato, fisso, e colorato con anti-MCM2 (un marcatore per il complesso MCM2-7), DAPI (DNA totale), e per l’incorporazione EdU (sintesi attiva del DNA). Le cellule arancioni sono S-fase con MCM carico di DNA, le cellule blu sono G1-fase con MCM carico di DNA, e le cellule grigie sono G1/G2/M cellule fase G1/G2/M senza MCM carico di DNA. Giusto: Le cellule sono state trattate come a sinistra, tranne che sono stati fissati prima dell’estrazione per rilevare MCM2 totale.(c) Le cellule T98G sono stati sincronizzati in G0 da inibizione di contatto e privazione di siero, poi rilasciato in G1 per 10 o 12 ore, o risincronizzato nei primi S con idrossiurea (HU), e rilasciato in S per 6 o 8 ore. MCM3 in frazioni arricchite con cromatina (Caricato) o lisati cellulari interi (Totale) è stato rilevato mediante immunoblotting.(d) Cromatografia a flusso di cromatina delle linee cellulari asincrone indicate che misurano il contenuto di DNA (DAPI), sintesi del DNA (incorporazione EdU), e MCM caricato (anti-MCM2). Le cellule blu sono G1-MCMDNA-positivo e EdU-negativo, arancione sono S fase MCMDNA-positivo; grigio sono G1/G2/M-MCMDNA-negativo.e) Barra grafica impilata della distribuzione di fase del ciclo cellulare dalle celle in(d); media con barre di errore ± SD (n = 3 repliche biologiche). La percentuale di cellule G1 in ogni popolazione è riportata nei settori verdi. I tempi di raddoppio sono stati calcolati sperimentalmente utilizzando l’analisi di regressione in GraphPad Prism.(a) Esempio di citometria a flusso gating con cromatina estratta ARPE-19 (RPE) cellule estratte dalla Figura 1d, misurando MCM caricato (anti-MCM2), sintesi del DNA (EdU incorporazione) e il contenuto di DNA (DAPI). Gating per discriminare le cellule da detriti era su FS-area vs SS-area, singlets (singole cellule) da ciuffi di cellule / doppie era su DAPI altezza vs DAPI area, fasi del ciclo cellulare sono stati determinati su DAPI vs EdU Non specifica colorazione di fondo da parte dell’anticorpo secondario è stata misurata utilizzando un campione di controllo negativo colorato senza anticorpo primario, solo l’anticorpo secondario. Questo cancello soglia di fondo è stato applicato a campioni sperimentali; G1-MCMDNA-positivo cellule sono state identificate prima dal contenuto di DNA e lo stato negativo EdU(G1) e poi dal segnale MCM2.(b) Esempio di citometria a flusso di colore gating con cromatina estratto ARPE-19 cellule dalla Figura 1d. A sinistra: controllo negativo per definire la colorazione di fondo con né la rilevazione EdU né l’anticorpo primario MCM2. Le cellule sono state colorate con DAPI, sottoposte a chimica di rilevazione EdU, e colorate con anticorpo secondario. Soglie di sfondo sono stati impostati utilizzando questi controlli e applicato a campioni sperimentali. Giusto: Campioni sperimentali in figura 1d principale che mostra il gating per identificare la sintesi del DNA (EdU), e MCM caricato (anti-MCM2). Gates definire schemi di colore per le trame di punti di colore.(a) Citometria a flusso di cromatina di cellule RPE colorate per MCM2 caricato (anti-MCM2), MCM3 caricato (anti-MCM3), e il contenuto di DNA (DAPI). Tutti e tre i grafici sono lo stesso campione. Le cellule colorate con anticorpo monoclonale MCM2 per MCM2 caricato co-stain altrettanto bene per un anticorpo policlonale MCM3 per MCM3 caricato.b) Citometria a flusso di cromatina di cellule RPE trattate con 100 nM siControl o un pool di 100 nM di siMCM3 per 72 ore, misura del contenuto di DNA (DAPI), sintesi del DNA (EdU incorporation), e MCM3 carico (anti-MCM3). I livelli di MCM3 caricato diminuiscono con il trattamento siMCM3, dimostrando la specificità degli anticorpi.(c) Immunoblot di cellule da(b).(d) Cellule RPE sincronizzate in G0 per inibizione da contatto. A sinistra: citometria a flusso proteico totale delle cellule G0 che misura il totale MCM (anti-MCM2), sintesi del DNA (incorporazione EdU) e contenuto di DNA (DAPI). A destra: Citometria a flusso di cromatina di cellule G0 che misura il MCM carico (anti-MCM2), la sintesi del DNA (incorporazione EdU) e il contenuto di DNA (DAPI). Le cellule G0 mancano di MCM caricato, dimostrando la specificità degli anticorpi.(e) Immunoblot di cellule per(d) dimostrare la sincronizzazione G0.(f) Immunoblot di cellule frazionate in proteine solubili e pellet (proteina cromatina associata) utilizzando un tampone CSK sale normale (100 mM NaCl) o ad alto contenuto di sale CSK (300 mM NaCl). NaCl ad alto contenuto di sale 300 mM NaCl elimina le proteine debolmente associate alla cromatina, compreso il Cdc6.g) Citometria a flusso di cromatina di cellule lisate con 100 mM di NaCl CSK normale o 300 mM di NaCl CSK ad alto contenuto salino.h) Istogrammi delle sole cellule G1-MCMDNA-positive di(g). Il MCM caricato in G1 rimane costante con alto sale, dimostrando la citometria a flusso di cromatina misura il DNA caricato MCM. Si noti la dura 300 mM sale mM altera la morfologia nucleare e la dispersione per citometria a flusso, per questo motivo, abbiamo usato l’estrazione di NaCl 100 mM per tutti gli altri esperimenti di citometria a flusso.(a) Immagini rappresentative di contrasto di fase in scala di grigi delle linee cellulari indicate dalla Figura 1d. Barra di scala è di 50 μm.(b) Analisi di immunofluorescenza delle cellule iPS visualizzando TRA-1-60, TRA-1-81, SSEA o OCT4 (in verde) e Nanog, SSEA3 o SOX2 (in rosso). La colorazione DAPI per il DNA indica i nuclei delle singole cellule in ogni colonia. La colorazione con fosfatasi alcalina viva (AP) è stata eseguita anche come indicatore positivo delle cellule staminali.(c) L’analisi del sequenziamento del bisolfito delle regioni promotrici NANOG e OCT4-2 nelle cellule ARPE-19 e ARPE-19-iPS è indicata. Le CpG metilate sono indicate con un cerchio chiuso, mentre le CpG non metilate sono indicate con un cerchio aperto. Una ‘X’ indica un disallineamento o uno spazio vuoto nella sequenza dei bisolfiti. La posizione CpG relativa al sito di partenza della trascrizione a valle è indicata sopra ogni riga.(d) Quantitativa RT-PCR analisi RT-PCR che mostra l’espressione genica relativa di OCT4, SOX2, NANOG, KLF4, MYC, LIN28, REXO1, ABCG2 e DNMT3B in ARPE-19 e ARPE-19-iPS cellule (normalizzato a GAPDH). Le barre di errore indicano un errore standard.e) La colorazione di ematossilina ed eosina di sezioni di teratoma da topi immunodeficienti iniettati con le cellule ARPE-19-iPS mostra cartilagine, così come l’endoderma e il teratoma ectoderma formato da ARPE-19-iPS.
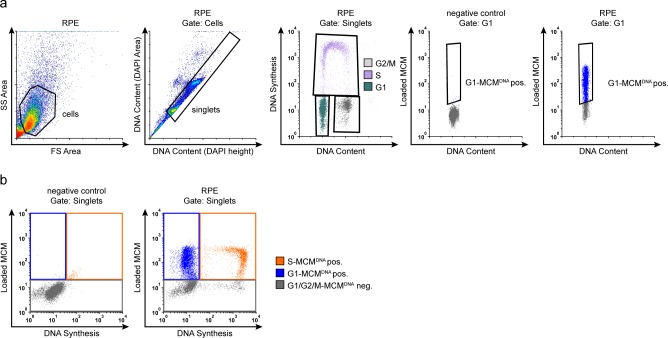
Figura 1-figure supplemento 1.Figura 1. Citometria a flusso gating.(a) Esempio di citometria a flusso gating con cromatina estratta ARPE-19 (RPE) cellule estratte dalla Figura 1d, misurando MCM caricato (anti-MCM2), sintesi del DNA (EdU incorporazione) e il contenuto di DNA (DAPI). Gating per discriminare le cellule da detriti era su FS-area vs SS-area, singlets (singole cellule) da ciuffi di cellule / doppie era su DAPI altezza vs DAPI area, fasi del ciclo cellulare sono stati determinati su DAPI vs EdU Non specifica colorazione di fondo da parte dell’anticorpo secondario è stata misurata utilizzando un campione di controllo negativo colorato senza anticorpo primario, solo l’anticorpo secondario. Questo cancello soglia di fondo è stato applicato a campioni sperimentali; G1-MCMDNA-positivo cellule sono state identificate prima dal contenuto di DNA e lo stato negativo EdU(G1) e poi dal segnale MCM2.(b) Esempio di citometria a flusso di colore gating con cromatina estratto ARPE-19 cellule dalla Figura 1d. A sinistra: controllo negativo per definire la colorazione di fondo con né la rilevazione EdU né l’anticorpo primario MCM2. Le cellule sono state colorate con DAPI, sottoposte a chimica di rilevazione EdU, e colorate con anticorpo secondario. Soglie di sfondo sono stati impostati utilizzando questi controlli e applicato a campioni sperimentali. Giusto: Campioni sperimentali in figura 1d principale che mostra il gating per identificare la sintesi del DNA (EdU), e MCM caricato (anti-MCM2). Gates definire schemi di colore per le trame di punti di colore.
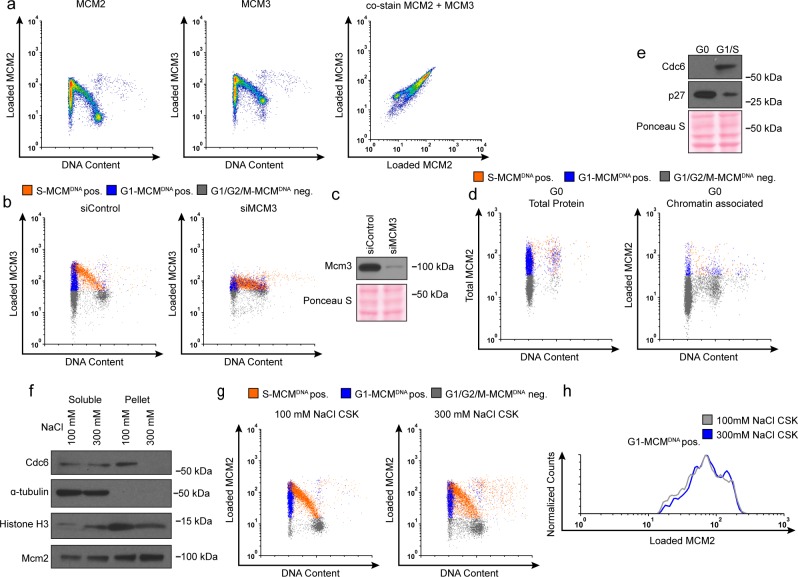
Figura 1-figure supplemento 2.Convalida della citometria a flusso di cromatina.(a) Citometria a flusso di cromatina di cellule RPE colorate per MCM2 caricato (anti-MCM2), MCM3 caricato (anti-MCM3), e il contenuto di DNA (DAPI). Tutti e tre i grafici sono lo stesso campione. Le cellule colorate con anticorpo monoclonale MCM2 per MCM2 caricato co-stain altrettanto bene per un anticorpo policlonale MCM3 per MCM3 caricato.b) Citometria a flusso di cromatina di cellule RPE trattate con 100 nM siControl o un pool di 100 nM di siMCM3 per 72 ore, misura del contenuto di DNA (DAPI), sintesi del DNA (EdU incorporation), e MCM3 carico (anti-MCM3). I livelli di MCM3 caricato diminuiscono con il trattamento siMCM3, dimostrando la specificità degli anticorpi.(c) Immunoblot di cellule da(b).(d) Cellule RPE sincronizzate in G0 per inibizione da contatto. A sinistra: citometria a flusso proteico totale delle cellule G0 che misura il totale MCM (anti-MCM2), sintesi del DNA (incorporazione EdU) e contenuto di DNA (DAPI). A destra: Citometria a flusso di cromatina di cellule G0 che misura il MCM carico (anti-MCM2), la sintesi del DNA (incorporazione EdU) e il contenuto di DNA (DAPI). Le cellule G0 mancano di MCM caricato, dimostrando la specificità degli anticorpi.(e) Immunoblot di cellule per(d) dimostrare la sincronizzazione G0.(f) Immunoblot di cellule frazionate in proteine solubili e pellet (proteina cromatina associata) utilizzando un tampone CSK sale normale (100 mM NaCl) o ad alto contenuto di sale CSK (300 mM NaCl). NaCl ad alto contenuto di sale 300 mM NaCl elimina le proteine debolmente associate alla cromatina, compreso il Cdc6.g) Citometria a flusso di cromatina di cellule lisate con 100 mM di NaCl CSK normale o 300 mM di NaCl CSK ad alto contenuto salino.h) Istogrammi delle sole cellule G1-MCMDNA-positive di(g). Il MCM caricato in G1 rimane costante con alto sale, dimostrando la citometria a flusso di cromatina misura il DNA caricato MCM. Si noti la dura 300 mM sale mM altera la morfologia nucleare e la dispersione per citometria a flusso, per questo motivo, abbiamo usato l’estrazione di NaCl 100 mM per tutti gli altri esperimenti di citometria a flusso.
Figura 1-figure supplement 3.Caratterizzazione di cellule pluripotenti e differenziate.(a) rappresentativo immagini di contrasto di fase in scala di grigi delle linee cellulari indicate dalla Figura 1d. Barra di scala è di 50 μm.(b) Analisi di immunofluorescenza delle cellule iPS visualizzando TRA-1-60, TRA-1-81, SSEA o OCT4 (in verde) e Nanog, SSEA3 o SOX2 (in rosso). La colorazione DAPI per il DNA indica i nuclei delle singole cellule in ogni colonia. La colorazione con fosfatasi alcalina viva (AP) è stata eseguita anche come indicatore positivo delle cellule staminali.(c) L’analisi del sequenziamento del bisolfito delle regioni promotrici NANOG e OCT4-2 nelle cellule ARPE-19 e ARPE-19-iPS è indicata. Le CpG metilate sono indicate con un cerchio chiuso, mentre le CpG non metilate sono indicate con un cerchio aperto. Una ‘X’ indica un disallineamento o uno spazio vuoto nella sequenza dei bisolfiti. La posizione CpG relativa al sito di partenza della trascrizione a valle è indicata sopra ogni riga.(d) Quantitativa RT-PCR analisi RT-PCR che mostra l’espressione genica relativa di OCT4, SOX2, NANOG, KLF4, MYC, LIN28, REXO1, ABCG2 e DNMT3B in ARPE-19 e ARPE-19-iPS cellule (normalizzato a GAPDH). Le barre di errore indicano un errore standard.e) La colorazione di ematossilina ed eosina di sezioni di teratoma da topi immunodeficienti iniettati con le cellule ARPE-19-iPS mostra cartilagine, così come l’endoderma e il teratoma ectoderma formato da ARPE-19-iPS.
Risultati
Le celle pluripotenti caricano MCM significativamente più velocemente delle celle differenziate
Abbiamo considerato due possibilità per come le celle con diverse lunghezze G1 caricano MCM sul DNA. Una possibilità è che le celle con brevi fasi G1 carichino MCM alla stessa velocità delle celle con lunghe fasi G1, con conseguente minor carico totale di MCM. In alternativa, le celle con brevi fasi G1 potrebbero caricare MCM più velocemente delle celle con lunghe fasi G1 e raggiungere livelli simili di MCM caricato. Per distinguere tra questi scenari, abbiamo sviluppato un saggio per misurare il MCM carico di DNA in singole cellule di popolazioni asincrone proliferanti adattando un metodo di citometria a flusso precedentemente segnalato (Hålandet al., 2015; Moreno et al., 2016). Abbiamo estratto cellule epiteliali immortalate (RPE1-hTERT) con detergente non ionico per rimuovere MCM solubile. Abbiamo poi fissato le restanti proteine legate alla cromatina per l’immunofluorescenza con anticorpo anti-MCM2 come marcatore del complesso MCM2-7, e per il contenuto di DNA (DAPI) e la sintesi del DNA (EdU) per misurare le fasi del ciclo cellulare(Figura 1b, a sinistra). Abbiamo usato un campione senza anticorpo primario come controllo (gli schemi di gating della citometria a flusso rilevanti sono mostrati in Figura 1-figure supplement 1). Per la citometria a flusso di cromatina, il segnale MCM al di sotto della soglia anticorpale è di colore grigio (‘G1/G2/M-MCMDNAneg’ ), mentre il segnale MCM rilevabile è di colore blu nelle cellule G1 (‘G1-MCMDNA pos’)o arancione nelle cellule di fase S (‘S-MCMDNA pos’). Come previsto, i livelli proteici totali MCM non cambiano sostanzialmente durante il ciclo cellulare(Figura 1b, a destra)(Todorov et al., 1995). Per confronto con i metodi di frazionamento cellulare comunemente utilizzati per valutare la dinamica MCM, abbiamo anche sondato immunoblot di frazioni arricchite di cromatina, e ha notato una simile espressione MCM, G1 carico, e la fase S modello di scarico(Figura 1c)(Cook et al., 2002; Méndez e Stillman, 2000). È interessante notare che le singole cellule G1 (striscia blu) hanno una gamma molto ampia di livelli di MCM caricato a DNA con una differenza di oltre 100 volte tra minimo e massimo(Figura 1b, a sinistra). MCM sono scaricati durante la fase S, che termina in G2/M con MCM non rilevabile sul DNA(Figura 1b, a sinistra). Inoltre, MCM caricato è resistente all’estrazione in tampone ad alto contenuto di sale che rimuove le proteine della cromatina legate in modo periferico (Figura 1-figure supplement 2f-h), simile ai complessi di lieviti MCM caricati in vitro (Bowers et al., 2004; Randell et al. , 2006). Abbiamo convalidato la specificità anticorpale MCM2 utilizzando cellule sincronizzate G0 quiescenti (MCM scaricato), e abbiamo anche osservato la stessa distribuzione del segnale G1 stesso ampio segnale utilizzando l’anticorpo MCM3(Figura 1-figure supplement 2a-d).
I complessi MCM caricati sono estremamente stabili sul DNA, sia in vivo che in vitro(Cocker et al., 1996; Evrin et al., 2009; Remus et al., 2009; Bowers et al., 2004). Nelle cellule umane, i MCM possono persistere sul DNA per più di 24 ore durante un arresto del ciclo cellulare e sono tipicamente scaricati solo durante la fase S(Kuipers et al., 2011). Queste proprietà si traducono in un carico di MCM che si verifica in modo unidirezionale durante la fase G1(Symeonidou et al., 2013). La natura unidirezionale del carico MCM significa che le cellule G1 con bassi livelli di MCM sono in G1 precoce, e le cellule G1 con alti livelli di MCM sono in G1 tardivo. Poiché abbiamo osservato un’ampia distribuzione del carico MCM in tutto il G1, comprese molte celle con bassi livelli di MCM caricato, concludiamo che le celle RPE1-hTERT caricano MCM relativamente lentamente durante le loro ~ 9 ore G1.
Abbiamo quindi utilizzato questo metodo per analizzare il carico MCM in celle asincrone con diverse lunghezze G1. H9 cellule staminali embrionali umane (hESC) hanno una breve fase G1 e trascorrere la maggior parte del ciclo cellulare in fase S. In contrasto con le cellule epiteliali differenziate, la maggior parte (~ 80%) di G1 hESCs aveva alti livelli di MCM caricato; pochissime cellule G1 aveva bassi livelli di MCM caricato (cellule blu, Figura 1d). Questa differenza suggerisce che gli hESCs caricano MCM rapidamente per ottenere abbondanti MCM caricato di DNA in breve tempo. Per verificare se il carico di MCM varia in cellule differenziate, abbiamo differenziato i CSE in cellule progenitrici neurali (PNG) per generare una coppia isogenica di cellule pluripotenti e differenziate. In contrasto con hESC, PNG differenziati aveva un tempo più lungo raddoppio e una vasta distribuzione di MCM carico di DNA in G1 (cellule blu, Figura 1d), ma anche trascorrere circa cinque volte più a lungo in G1(Figura 1e, ad esempio il 15% di un ciclo di 17 ore hESC cella hESC è di 2,5 ore in G1 vs 45% di un ciclo di 30 ore NPC cella NPC è 13,6 ore in G1). Dal momento che i PNG avevano molte celle con bassi livelli di DNA caricato MCM, si conclude che queste celle differenziate caricano MCM più lentamente rispetto alle hESC.
Per generare un’altra coppia isogenica di cellule pluripotenti e differenziate, abbiamo riprogrammato ARPE-19 cellule epiteliali pigmentate della retina primaria (RPE) in cellule staminali pluripotenti indotte (iPSC). Le iPSCs avevano caratteristiche distintive di pluripotenza come misurato al microscopio, il sequenziamento dei bisolfiti, l’espressione genica, e la formazione del teratoma (Figura 1-figure supplement 3), e le loro fasi G1 erano in genere sette volte più brevi rispetto ai loro genitori differenziati (Figura 1e). Come hESC, le iPSC pluripotenti avevano livelli prevalentemente elevati di MCM caricato di DNA in G1(Figura 1d). È importante notare che entrambe le linee cellulari pluripotenti hanno raggiunto livelli approssimativamente uguali di MCM caricato a DNA all’inizio della fase S, come le loro controparti differenziate, ma in meno tempo (le intensità assolute di carico MCM sono paragonabili quando i campioni vengono elaborati e analizzati con impostazioni identiche dello strumento). Presi insieme, questi dati dimostrano che le cellule pluripotenti caricano gli MCM rapidamente in G1, ma le cellule differenziate caricano gli MCM lentamente.
Abbiamo poi quantificato le relative velocità di carico MCM nelle celle pluripotenti e differenziate usando l’analisi della velocità ergodica, un metodo matematico che può derivare le velocità da popolazioni fisse eallo stato stazionario(Kafri et al., 2013). L’analisi ergodica può misurare qualsiasi parametro di tasso unidirezionale da una distribuzione a stato stazionario e non è limitata al ciclo cellulare (ad esempio, gli ingorghi delle auto)(Gray e Griffeath, 2001). L’analisi ergodica applicata al ciclo cellulare significa che all’interno di una popolazione allo stato stazionario con un tempo costante di raddoppio e distribuzione del ciclo cellulare, il numero di cellule in qualsiasi punto del ciclo cellulare è inversamente proporzionale alla velocità con cui si muovono attraverso quel punto. Per qualsiasi parametro misurato, la densità delle cellule indica la velocità: una bassa densità cellulare su un grafico di citometria a flusso indica una velocità veloce che passa attraverso quello stato del ciclo cellulare, mentre un’alta densità cellulare indica una velocità lenta. Questo fenomeno è analogo ad un’alta densità di auto che si muovono lentamente osservata in un determinato punto di una strada in un ingorgo rispetto ad una bassa densità di auto che si muovono velocemente su una strada aperta. Abbiamo visualizzato il carico MCM come istogrammi delle intensità MCMDNA solo nelle cellule G1 per l’analisi del tasso ergodico (G1-MCMDNA, Figura 2a,b e Figura 2-figure supplemento 1).
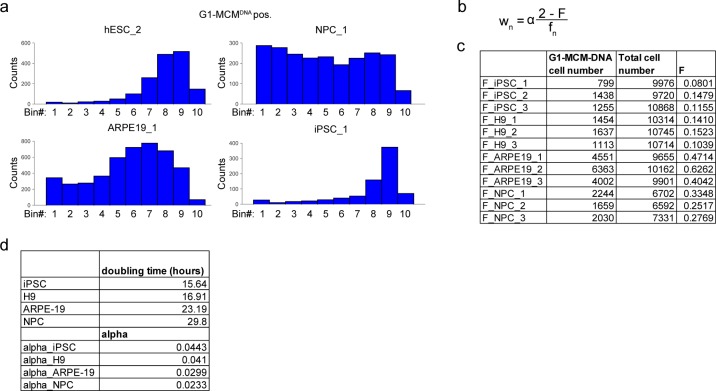
Figura 2-figure supplemento 1.Quantificazione del tasso di carico MCM mediante analisi del tasso ergodico.valori ERA grezzi.valori ERA grezzi.valori ERA grezzi.binning analisi del tasso ergodico.a) Schema di gating per la citometria a flusso di cromatina di iPSC che misura il contenuto di DNA (DAPI), sintesi del DNA (EdU incorporazione), e MCM caricato (anti-MCM2); questo campione è dalla Figura 1d.(b) Istogrammi di solo le cellule G1-MCMDNA-positive dei quattro campioni di citometria a flusso di cromatina in Figura 1d.(c) Calcolato tasso medio di carico MCM medio all’ora con l’analisi della velocità ergodica; media con barre di errore ± SD. (n = 3 repliche biologiche), non accoppiato due t-test a coda. **p=0.0049. ***p=0.001. Vedere Materiali e metodi per i dettagli. Vedere anche la figura 2 dati fonte 1.10.7554/eLife.30473.009Cifra 2 dati sorgente 1.Valori ERA grezzi.a) Istogrammi di campioni G1-MCMDNA positivi al G1-MCMDNA nella Figura 2B, raggruppati in 10 regioni per l’analisi della velocità ergodica (vedere Materiali e metodi).(b) Equazione del tasso ergodico basata su Kafri et al. (2013). wn è il tasso in ogni bin n, α è una contabilità costante per il raddoppio del tempo, F è la frazione di cellule in G1-MCMDNA positivo su tutte le cellule, fn è la frazione di cellule in ogni bin n su G1-MCMDNA positivo (vedi Materiali e metodi).(c) Valore di F per tre repliche biologiche per ogni linea cellulare, utilizzato per calcolare la Figura 2c.(d) Tempo di raddoppio e valori alfa utilizzati per calcolare i tassi nella Figura 2c.
Per calcolare la velocità di carico MCM per ora, abbiamo determinato sperimentalmente le distribuzioni del ciclo cellulare e raddoppiare i tempi di ogni popolazione di cellule(Figura 2-figure supplement 1). Le cellule pluripotenti hanno raggiunto livelli quasi uguali di MCM caricato alla transizione G1/S in meno tempo rispetto alle cellule differenziate. Per quantificare la differenza effettiva MCM tasso di carico MCM differenza, abbiamo suddiviso la popolazione G1-MCMDNA in 10 bidoni di uguali dimensioni, calcolato il tasso di carico MCM per ogni bidone, quindi il tasso di carico medio complessivo MCM per ogni popolazione G1. Questi calcoli hanno rivelato che gli hESC pluripotenti hanno caricato MCM 6,5 volte più velocemente all’ora rispetto ai PNG differenziati e gli iPSC pluripotenti hanno caricato MCM 3,9 volte più velocemente all’ora rispetto agli RPE differenziati(Figura 2c). Così, le celle pluripotenti con brevi fasi G1 caricano MCM significativamente più velocemente delle celle differenziate con lunghe fasi G1.
Figura 2-figure supplement 1.Quantificazione della velocità di carico delle MCM mediante l’analisi della velocità ergodica.Valori ERA grezzi.Valori ERA grezzi.Analisi della velocità ergodica binning.(a) Schema di gating per la citometria a flusso di cromatina di iPSCs che misura il contenuto di DNA (DAPI), sintesi del DNA (EdU incorporazione), e MCM caricato (anti-MCM2); questo campione è dalla Figura 1d.(b) Istogrammi di solo le cellule G1-MCMDNA-positive dei quattro campioni di citometria a flusso di cromatina in Figura 1d.(c) Calcolato tasso medio di carico MCM medio all’ora con l’analisi della velocità ergodica; media con barre di errore ± SD. (n = 3 repliche biologiche), non accoppiato due t-test a coda. **p=0.0049. ***p=0.001. Vedere Materiali e metodi per i dettagli. Vedere anche la figura 2 dati fonte 1.10.7554/eLife.30473.009Cifra 2 dati sorgente 1.Valori ERA grezzi.a) Istogrammi di campioni G1-MCMDNA positivi al G1-MCMDNA nella Figura 2B, raggruppati in 10 regioni per l’analisi della velocità ergodica (vedere Materiali e metodi).(b) Equazione del tasso ergodico basata su Kafri et al. (2013). wn è il tasso in ogni bin n, α è una contabilità costante per il raddoppio del tempo, F è la frazione di cellule in G1-MCMDNA positivo su tutte le cellule, fn è la frazione di cellule in ogni bin n su G1-MCMDNA positivo (vedi Materiali e metodi).(c) Valore di F per tre repliche biologiche per ogni linea cellulare, utilizzato per calcolare la Figura 2c.(d) Tempo di raddoppio e valori alfa utilizzati per calcolare i tassi nella Figura 2c.
Figura 2-figure supplement 1.1. Bombolatura ergonomica dell’analisi dei tassi.(a) Istogrammi da campioni G1-MCMDNA-positivi nella Figura 2B, binning in 10 regioni per l’analisi della velocità ergodica (vedi Materiali e metodi).(b) Equazione del tasso ergodico basata su Kafri et al. (2013). wn è il tasso in ogni bin n, α è una contabilità costante per il raddoppio del tempo, F è la frazione di cellule in G1-MCMDNA positivo su tutte le cellule, fn è la frazione di cellule in ogni bin n su G1-MCMDNA positivo (vedi Materiali e metodi).(c) Valore di F per tre repliche biologiche per ogni linea cellulare, utilizzato per calcolare la Figura 2c.(d) Tempo di raddoppio e valori alfa utilizzati per calcolare i tassi nella Figura 2c.
La differenziazione, la lunghezza G1 e la velocità di carico MCM sono accoppiate
Abbiamo ipotizzato che il carico MCM sia fondamentalmente legato alla pluripotenza perché il tasso di carico MCM è diminuito durante la differenziazione ed è aumentato durante la riprogrammazione. Questa idea prevede che il carico MCM rallentato è un fenomeno comune alla differenziazione verso tutti gli strati germinali. Per testare questa ipotesi, abbiamo iniziato la differenziazione in hESCs verso i tre strati germinali embrionali (neuroectoderma, mesoderma ed endoderma), raccogliendo le cellule a 24 ore e 48 ore dopo aver indotto la differenziazione(Figura 3-figure supplemento 1). Abbiamo confermato i progressi verso ogni lignaggio dai cambiamenti di espressione genica previsti, in particolare l’induzione di marcatori specifici del lignaggio e la modesta riduzione dei marcatori di pluripotenza – anche in questi punti di tempo molto presto (Figura 3c). Abbiamo valutato i tassi di carico MCM durante la differenziazione mediante citometria a flusso come prima. Il tasso di carico MCM chiaramente diminuito per tutti gli strati di germe rapidamente entro le prime 48 ore di iniziare la differenziazione(Figura 3a, confrontare gli istogrammi grigi per le cellule G1 indifferenziate con gli istogrammi verde e blu). La diminuzione del tasso di carico MCM ha coinciso anche con l’aumento della proporzione di cellule G1 per ogni lignaggio. Ad esempio, entro 24 ore di differenziazione neuroectoderma, G1 aveva già allungato e carico MCM aveva rallentato, ma durante mesoderma (BMP4) differenziazione sia G1 allungamento e rallentato carico MCM ha preso 48 ore(Figura 3a e b). I cambiamenti strettamente coordinati che abbiamo osservato universalmente durante la differenziazione suggeriscono che il tasso di carico MCM è accoppiato alla lunghezza G1. È importante sottolineare che questi risultati dimostrano che il tasso di origine delle licenze per il carico MCM è regolamentato dallo sviluppo.

Figura 3-figure supplemento 1.Differenziazione diminuisce universalmente il tasso di carico MCM.differenziazione delle cellule staminali.(a) Citometria a flusso di cromatina di hESC indotta a differenziare verso mesoderma (BMP4), neuroectoderma, mesoderma (GSK3βi), o endoderma per 24 o 48 ore. Gli istogrammi mostrano solo le cellule G1-MCMDNA positive come in Figura 2b. Vedi metodi per i protocolli di differenziazione. I conteggi delle cellule per 24 ore e 48 ore sono stati normalizzati rispetto ai campioni HESC corrispondenti.(b) Grafici a barre impilati della distribuzione del ciclo cellulare per le cellule in(a).(c) Analisi dell’espressione genica dei marcatori di differenziazione mediante PCR quantitativa dei campioni in(a); l’espressione log2 è relativa alle cellule indifferenziate. I dati sono la media ±SD di due repliche biologiche.(a) Immunoblots di differenziazione neuroectoderm, come in Figura 3a.(b) Immagini rappresentative di contrasto di fase durante i protocolli di differenziazione indicati dalla Figura 3a. La barra di scala è di 50 μm.
Abbiamo poi chiesto se la lunghezza G1 e il tasso di carico MCM sono obbligatoriamente accoppiati, o se il collegamento può essere cortocircuitato facendo avanzare artificialmente la transizione G1/S. Per distinguere queste possibilità, abbiamo costruito un derivato RPE1-hTERT con un cDNA CICLINO E1 cDNA sotto controllo doxiciciclina. La sovrapproduzione di CYCLIN E1 ha ridotto in modo riproducibile la lunghezza di G1, in linea con gli studi precedenti(Figura 4a,b)(Resnitzky et al., 1994; Ekholm-Reed et al., 2004). Sorprendentemente, le cellule sovrapproduzione Cyclin E1 (designato come ‘↑Cyclin E1’) non solo ha trascorso meno tempo in G1, ma anche iniziato fase S con quantità molto più bassa di MCM caricato rispetto al controllo, questa nuova sottopopolazione apparso nella regione centrale triangolare delle trame che è tipicamente chiaro di cellule fase S (Figura 4c,darancione S-MCMDNApos ). Ciclina E1 sovrapproduzione E1 drammaticamente aumentato la proporzione di questi MCM-basso prime cellule fase S di sei volte dal 9,9% delle cellule di controllo fase S al 63,6% di ↑Cyclin E1, cellule fase S (Figura 4c-e). Concludiamo anche che il tasso di carico MCM non è aumentato per accogliere il G1 più breve perché il modello di carico MCM in G1 è rimasto costante e le cellule ↑Cyclin E1 aveva in media almeno due volte meno MCM carico di DNA nella fase S iniziale delle cellule di controllo (Figura 4f-h). Inoltre, il ↑Cyclin E, MCM-cellule basse MCM incorporato significativamente meno EdU per unità di tempo rispetto alle cellule MCM-alto ha fatto (1,6 volte più bassa media, 1,8 volte più bassa mediana), indicando che i bassi livelli di MCM caricato sono insufficienti per la progressione normale fase S (Figura 4i). Le prime cellule della fase S con il minimo MCM caricato aveva anche la sintesi del DNA meno da intensità EdU (dati non mostrati). Così, l’accorciamento della lunghezza G1 senza aumentare il tasso di carico MCM fa sì che le cellule G1 entrino prematuramente nella fase S senza il complemento completo di MCM caricato con DNA.
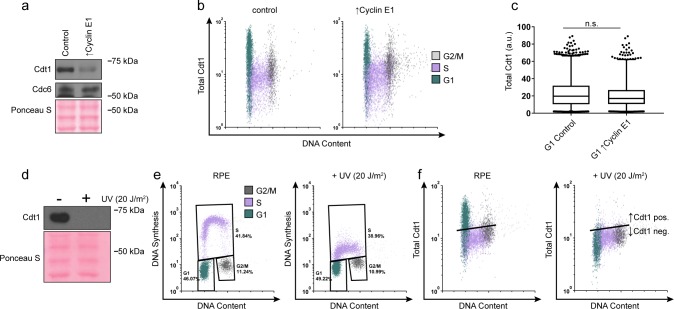
Figura 4-figure supplemento 1.Ciclina E sovrapproduzione disaccoppia il carico MCM e G1 length.G1 Cdt1 livelli di Cdt1 non sono interessati dalla sovrapproduzione Cyclin E.(a) Immunoblots di un derivato stabile di RPE1-hTert cellule RPE1-hTert con un integrato ciclina doxiciclina inducibile ciclina E costruire trattato con 100 ng / ml doxiciclina per 72 ore per sovrapprodurre Cyclin E1 (↑Cyclin E1) o con il controllo del veicolo.b) Barre grafiche impilate della distribuzione del ciclo cellulare misurata mediante citometria a flusso per le cellule indicate ina); media con barre di errore ± SD (n = 3 repliche biologiche).(c) Cromatografia a flusso di cromatina di cellule di controllo o ciclina E-overproducendo cellule che misurano il contenuto di DNA (DAPI), sintesi del DNA (incorporazione EdU), e MCM caricato (anti-MCM2).(d) S fase-MMMDNA-positivo cellule S da campioni in(c) diviso in popolazioni che hanno iniziato la fase S con MCMDNA alto o basso. Le prime cellule S sono cellule di fase S con contenuto di DNA G1.(e) La percentuale di MCMDNA positivo, ma-bassa intensità del segnale MCM cellule fase S di tutte le cellule S-MCMDNA-positive da tre repliche biologiche; media con barre di errore ± SD, non accoppiato due coda t-test. ***p=0.002.(f) Media MCM caricato nella fase iniziale S, (S-MCMDNA positivo, G1 contenuto di DNA) da tre repliche biologiche; media con barre di errore ± SD, non accoppiato due coda t-test. ***p=0.0004.g) Istogramma delle cellule G1-MCMDNA-positive di G1-MCMDNA da campioni mostrati in(c). I conteggi per ↑Cyclin E1 sono normalizzati al controllo.(h) Istogramma delle cellule S iniziali da campioni mostrati in(d). I conteggi per ↑Cyclin E1 sono normalizzati al controllo. (Questi dati sono uno dei replicati quantificati in(f).). (i) intensità EdU da ↑Cyclin E1, MCM-cellule alte o MCM-basse da (d) come casella e baffi trame. La linea centrale è mediana, i bordi della scatola esterna sono il 25° e il 75° percentile, i bordi dei baffi sono il1° e il 99° percentile, i singoli punti di dati sono il più basso e il più alto 1%, rispettivamente. Mediana EdU incorporazione EdU di MCM-alto ↑Cyclin cellule E1 è 1,8 volte maggiore di MCM-basso, medio EdU incorporazione è 1,6 volte maggiore in MCM-alto di MCM-basso, media di tre repliche biologiche. I campioni confrontati da non accoppiato, due coda t-test, **p = 0,0027, **p = 0,0033, rispettivamente.(a) Immunoblot di lisati di cellule intere di cellule asincrone trattate come in(Figura 4a).(b) Cdt1 totale rilevato mediante citometria a flusso di cellule trattate come nella Figura 4a che misura il contenuto di DNA (DAPI), sintesi del DNA (incorporazione EdU), e Cdt1 (anti-Cdt1). Verde sono le cellule G1, Viola sono le cellule di fase S (EdU positivo), Grigio sono il contenuto di DNA G2/M.(c) Box-and-whiskers trame di G1 Cdt1 concentrazione per cella da(b). La linea centrale è mediana, i bordi esterni della scatola sono il 25° e il 75° percentile, i bordi dei baffi sono il1° e il 99° percentile, i punti di dati individuali sono il più basso e il più alto 1%, rispettivamente. G1 L’intensità Cdt1 nei controlli è 1,1 volte maggiore della media e 1,2 volte maggiore della mediana rispetto a G1 ↑Cyclin E1. I campioni non sono stati significativamente diversi, rispetto a due code, non accoppiato t test, p = 0,1907, p = 0,3525, rispettivamente; media di tre repliche biologiche.d) Immunoblot di lisato cellulare intero da cellule RPE1-htert trattate con o senza 20 J/m2 di irradiazione UV, raccolto 1 ora dopo l’irradiazione. L’irradiazione UV induce il carico di PCNA accoppiato per la riparazione del DNA e successivamente il Cdt1 per la degradazione(Arias e Walter, 2005).e) Citometria a flusso delle cellule di controllo e delle cellule irradiate UV come in(a), misurazione del contenuto di DNA (DAPI), sintesi del DNA (incorporazione EdU), e Cdt1 (anti-Cdt1). I grafici dimostrano una diminuzione della sintesi del DNA dopo l’irradiazione UV.(f) Gli stessi campioni come in(e). G1 Cdt1 è degradato dopo la riparazione del DNA indotta da UV, dimostrando la specificità degli anticorpi Cdt1 per la citometria a flusso di immunofluorescenza. La linea nera indica lo sfondo definito dalla colorazione dei controlli con il solo anticorpo secondario. Le cellule al di sotto della linea (come le cellule della fase S) sono Cdt1 negative. Le cellule sopra la linea sono Cdt1 positive.
Studi precedenti hanno dimostrato che i CDK possono inibire il caricamento del MCM inibendo direttamente i fattori di caricamento del MCM, come ad esempio stimolando la degradazione del Cdt1 (Cdc10-dipendente trascrizione 1), una proteina essenziale per il caricamento del MCM(Ekholm-Reed et al., 2004; Sugimoto et al., 2004; Tanaka e Diffley, 2002). I livelli di Cdt1 nei lisati di cellule asincrone sono effettivamente diminuiti in seguito alla sovrapproduzione di Cyclin E1(Figura 4-figure supplemento 1a). D’altra parte, dal momento che Cdt1 è stabile in fase G1 e degradato in fase S, il segnale Cdt1 inferiore potrebbe avere riflesso meno Cdt1 nel lisato a causa della maggiore proporzione di cellule di fase S, questo effetto indiretto potrebbe applicarsi a qualsiasi proteina ciclo cellulare regolata in popolazioni cellulari con diverse distribuzioni del ciclo cellulare. Per testare questa idea, abbiamo misurato i livelli totali di proteina Cdt1 totale specificamente in G1 per citometria a flusso(Figura 4-figure supplemento 1b). La sovrapproduzione di Cyclin E non ha ridotto in modo significativo i livelli di Cdt1 G1 rispetto al controllo (1,1 volte più alta media, 1,2 mediana più alta, Figura 4-figure supplement 1c). Abbiamo convalidato l’anticorpo Cdt1 per la citometria a flusso in immunofluorescenza(Figura 4-figure supplement 1d-f). Concludiamo che Cyclin E/Cdk2 inibisce il carico MCM indirettamente, almeno in parte, accorciando G1 e diminuendo il tempo disponibile per il carico MCM.
Figura 3-figure supplement 1.La differenziazione diminuisce universalmente il tasso di carico MCM.(a) Citometria a flusso di cromatina di hESC indotta a differenziare verso mesoderma (BMP4), neuroectoderma, mesoderma (GSK3βi), o endoderma per 24 o 48 ore. Gli istogrammi mostrano solo le cellule G1-MCMDNA positive come in Figura 2b. Vedi metodi per i protocolli di differenziazione. I conteggi delle cellule per 24 ore e 48 ore sono stati normalizzati rispetto ai campioni HESC corrispondenti.(b) Grafici a barre impilati della distribuzione del ciclo cellulare per le cellule in(a).(c) Analisi dell’espressione genica dei marcatori di differenziazione mediante PCR quantitativa dei campioni in(a); l’espressione log2 è relativa alle cellule indifferenziate. I dati sono la media ±SD di due repliche biologiche.(a) Immunoblots di differenziazione neuroectoderm, come in Figura 3a.(b) Immagini rappresentative di contrasto di fase durante i protocolli di differenziazione indicati dalla Figura 3a. La barra di scala è di 50 μm.
Figura 3-figure supplemento 1.1. Differenziazione delle cellule staminali.(a) Immunoblot di differenziazione neuroectoderm, come nella Figura 3a.(b) Immagini rappresentative di contrasto di fase durante i protocolli di differenziazione indicati dalla Figura 3a. La barra di scala è di 50 μm.
Figura 4-figure supplemento 1.Cyclin E sovrapproduzione disaccoppia il carico MCM e G1 length.G1 Cdt1 livelli non sono interessati dalla sovrapproduzione Cyclin E.(a) Immunoblots di un derivato stabile di cellule RPE1-hTert con un integrato doxiciciclina induttivo ciclina E costruito trattato con 100 ng / ml doxiciclina per 72 ore per sovrapprodurre Cyclin E1 (↑Cyclin E1) o con il controllo del veicolo.b) Barre grafiche impilate della distribuzione del ciclo cellulare misurata mediante citometria a flusso per le cellule indicate ina); media con barre di errore ± SD (n = 3 repliche biologiche).(c) Cromatografia a flusso di cromatina di cellule di controllo o ciclina E-overproducendo cellule che misurano il contenuto di DNA (DAPI), sintesi del DNA (incorporazione EdU), e MCM caricato (anti-MCM2).(d) S fase-MMMDNA-positivo cellule S da campioni in(c) diviso in popolazioni che hanno iniziato la fase S con MCMDNA alto o basso. Le prime cellule S sono cellule di fase S con contenuto di DNA G1.(e) La percentuale di MCMDNA positivo, ma-bassa intensità del segnale MCM cellule fase S di tutte le cellule S-MCMDNA-positive da tre repliche biologiche; media con barre di errore ± SD, non accoppiato due coda t-test. ***p=0.002.(f) Media MCM caricato nella fase iniziale S, (S-MCMDNA positivo, G1 contenuto di DNA) da tre repliche biologiche; media con barre di errore ± SD, non accoppiato due coda t-test. ***p=0.0004.g) Istogramma delle cellule G1-MCMDNA-positive di G1-MCMDNA da campioni mostrati in(c). I conteggi per ↑Cyclin E1 sono normalizzati al controllo.(h) Istogramma delle cellule S iniziali da campioni mostrati in(d). I conteggi per ↑Cyclin E1 sono normalizzati al controllo. (Questi dati sono uno dei replicati quantificati in(f).). (i) intensità EdU da ↑Cyclin E1, MCM-cellule alte o MCM-basse da (d) come casella e baffi trame. La linea centrale è mediana, i bordi della scatola esterna sono il 25° e il 75° percentile, i bordi dei baffi sono il1° e il 99° percentile, i singoli punti di dati sono il più basso e il più alto 1%, rispettivamente. Mediana EdU incorporazione EdU di MCM-alto ↑Cyclin cellule E1 è 1,8 volte maggiore di MCM-basso, medio EdU incorporazione è 1,6 volte maggiore in MCM-alto di MCM-basso, media di tre repliche biologiche. I campioni confrontati da non accoppiato, due coda t-test, **p = 0,0027, **p = 0,0033, rispettivamente.(a) Immunoblot di lisati di cellule intere di cellule asincrone trattate come in(Figura 4a).(b) Cdt1 totale rilevato mediante citometria a flusso di cellule trattate come nella Figura 4a che misura il contenuto di DNA (DAPI), sintesi del DNA (incorporazione EdU), e Cdt1 (anti-Cdt1). Verde sono le cellule G1, Viola sono le cellule di fase S (EdU positivo), Grigio sono il contenuto di DNA G2/M.(c) Box-and-whiskers trame di G1 Cdt1 concentrazione per cella da(b). La linea centrale è mediana, i bordi esterni della scatola sono il 25° e il 75° percentile, i bordi dei baffi sono il1° e il 99° percentile, i punti di dati individuali sono il più basso e il più alto 1%, rispettivamente. G1 L’intensità Cdt1 nei controlli è 1,1 volte maggiore della media e 1,2 volte maggiore della mediana rispetto a G1 ↑Cyclin E1. I campioni non sono stati significativamente diversi, rispetto a due code, non accoppiato t test, p = 0,1907, p = 0,3525, rispettivamente; media di tre repliche biologiche.d) Immunoblot di lisato cellulare intero da cellule RPE1-htert trattate con o senza 20 J/m2 di irradiazione UV, raccolto 1 ora dopo l’irradiazione. L’irradiazione UV induce il carico di PCNA accoppiato per la riparazione del DNA e successivamente il Cdt1 per la degradazione(Arias e Walter, 2005).e) Citometria a flusso delle cellule di controllo e delle cellule irradiate UV come in(a), misurazione del contenuto di DNA (DAPI), sintesi del DNA (incorporazione EdU), e Cdt1 (anti-Cdt1). I grafici dimostrano una diminuzione della sintesi del DNA dopo l’irradiazione UV.(f) Gli stessi campioni come in(e). G1 Cdt1 è degradato dopo la riparazione del DNA indotta da UV, dimostrando la specificità degli anticorpi Cdt1 per la citometria a flusso di immunofluorescenza. La linea nera indica lo sfondo definito dalla colorazione dei controlli con il solo anticorpo secondario. Le cellule al di sotto della linea (come le cellule della fase S) sono Cdt1 negative. Le cellule sopra la linea sono Cdt1 positive.
Figura 4-figure supplemento 1.G1 I livelli di Cdt1 non sono influenzati dalla sovrapproduzione di Cyclin E.a) Immunoblot di lisati di cellule intere di cellule asincrone trattate come in(Figura 4a).(b) Cdt1 totale rilevato dalla citometria a flusso delle cellule trattate come nella Figura 4a che misura il contenuto di DNA (DAPI), sintesi del DNA (incorporazione EdU), e Cdt1 (anti-Cdt1). Verde sono le cellule G1, Viola sono le cellule di fase S (EdU positivo), Grigio sono il contenuto di DNA G2/M.(c) Box-and-whiskers trame di G1 Cdt1 concentrazione per cella da(b). La linea centrale è mediana, i bordi esterni della scatola sono il 25° e il 75° percentile, i bordi dei baffi sono il1° e il 99° percentile, i punti di dati individuali sono il più basso e il più alto 1%, rispettivamente. G1 L’intensità Cdt1 nei controlli è 1,1 volte maggiore della media e 1,2 volte maggiore della mediana rispetto a G1 ↑Cyclin E1. I campioni non sono stati significativamente diversi, rispetto a due code, non accoppiato t test, p = 0,1907, p = 0,3525, rispettivamente; media di tre repliche biologiche.d) Immunoblot di lisato cellulare intero da cellule RPE1-htert trattate con o senza 20 J/m2 di irradiazione UV, raccolto 1 ora dopo l’irradiazione. L’irradiazione UV induce il carico di PCNA accoppiato per la riparazione del DNA e successivamente il Cdt1 per la degradazione(Arias e Walter, 2005).e) Citometria a flusso delle cellule di controllo e delle cellule irradiate UV come in(a), misurazione del contenuto di DNA (DAPI), sintesi del DNA (incorporazione EdU), e Cdt1 (anti-Cdt1). I grafici dimostrano una diminuzione della sintesi del DNA dopo l’irradiazione UV.(f) Gli stessi campioni come in(e). G1 Cdt1 è degradato dopo la riparazione del DNA indotta da UV, dimostrando la specificità degli anticorpi Cdt1 per la citometria a flusso di immunofluorescenza. La linea nera indica lo sfondo definito dalla colorazione dei controlli con il solo anticorpo secondario. Le cellule al di sotto della linea (come le cellule della fase S) sono Cdt1 negative. Le cellule sopra la linea sono Cdt1 positive.
Gli HESC a caricamento rapido hanno più Cdt1 in G1
Il caricamento di complessi MCM sul DNA richiede le sei sottounità Origin Recognition Complex (ORC), Cdc6, e Cdt1. Abbiamo ipotizzato che il caricamento veloce di MCM nelle cellule staminali pluripotenti si ottiene con l’aumento dei livelli delle proteine di caricamento. Per testare questa idea, abbiamo sondato i lisati proteici delle cellule asincrone per confrontare la quantità di proteine di carico MCM tra le linee cellulari isogeniche. I livelli totali di proteine Mcm2 e ORC sono rimasti costanti(Figura 5a). Gli altri fattori di carico MCM normalmente cambiano nella loro abbondanza durante il ciclo cellulare a causa della proteolisi regolata. I livelli di proteina Cdc6 sono bassi in G1 e alti in fase S (Figura 5b). Al contrario, i livelli di proteina Cdt1 sono alti in G1 e bassi in fase S (Figura 5b)(Mailand e Diffley, 2005; Pozo e Cook, 2016). Poiché una popolazione asincrona di cellule pluripotenti passa molto più tempo in fase S rispetto alle cellule differenziate, ci aspettavamo che i livelli di Cdc6 fossero più alti nelle cellule pluripotenti asincrone rispetto alle loro controparti isogeniche. Il Cdc6 era infatti più alto nelle cellule pluripotenti, così come il Geminin, una proteina regolata in modo simile al Cdc6(Figura 5a)(McGarry e Kirschner, 1998). Con nostra sorpresa, anche se la maggior parte delle cellule pluripotenti asincrone erano in fase S, un tempo in cui il Cdt1 è degradato, i livelli di Cdt1 erano più alti nelle cellule pluripotenti asincrone che nelle cellule differenziate isogeniche(Figura 5a). Un’osservazione simile è stata riportata per le cellule staminali embrionali di topo(Ballabeni et al., 2011). Questi dati implicano che i livelli di Cdt1 sono più alti nella fase G1 delle cellule pluripotenti che in quella G1 delle cellule differenziate, fornendo una potenziale spiegazione per il rapido caricamento di MCM nelle cellule pluripotenti.
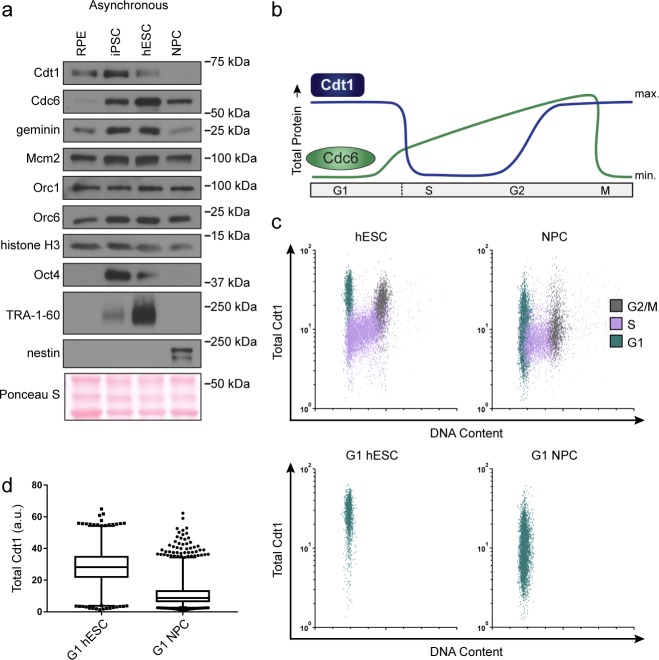
Figura 5.hESC hanno livelli elevati di Cdt1 in G1.(a) Immunoblot di lisati di cellule intere dalle linee cellulari asincrone indicate.(b) Cambiamenti previsti nei livelli di proteina totale di Cdt1 e Cdc6 durante il ciclo cellulare umano.(c) Cdt1 totale rilevato nelle cellule asincrone mediante citometria a flusso che misura il contenuto di DNA (DAPI), sintesi del DNA (incorporazione EdU) e Cdt1 (anti-Cdt1). Le cellule verdi sono G1, le cellule viola sono fase S (EdU positivo), le cellule grigie sono G2/M.(d) Box-and-whiskers trame di G1 Cdt1 concentrazione per cella da(C). La linea centrale è mediana, i bordi esterni della scatola sono il 25° e il 75° percentile, i bordi dei baffi sono il1° e il 99° percentile, i singoli punti di dati sono il più basso e il più alto 1%, rispettivamente. La mediana G1 Cdt1 nei PNG è 2,9 volte maggiore, la media è 2,2 volte maggiore di G1 Cdt1 nei PNG, la media p=0,0504 mediana p=0,0243, media di tre repliche biologiche. I diagrammi di flusso sono solo cellule G1 (verde) da(c).
Per misurare direttamente i livelli di Cdt1 specificamente in G1, abbiamo raccolto hESC asincroni e PNG asincroni poi fissati e colorati per Cdt1, EdU e DAPI. La degradazione del Cdt1 in fase S in hESC è simile alle cellule differenziate con livelli molto bassi in fase S (viola, Figura 5c). Al contrario, i PNG avevano una vasta popolazione di cellule con alti livelli di Cdt1 in G1 (cellule verdi) e quantità significative di Cdt1 in fase G2/M (cellule grigie), mentre i PNG avevano una distribuzione ampia e complessivamente inferiore di Cdt1 in G1 e molto poco di Cdt1 in G2/M(Figura 5d). Il G1 hESCs costantemente ospitato Cdt1 significativamente più Cdt1 rispetto al G1 NPCs ha fatto (2,9 volte più alta mediana, 2,2 volte più alta media, tre repliche [Figura 5d]). Notiamo che il mRNA CDT1 mRNA è modestamente ma costantemente più alto negli hESC asincroni rispetto ai derivati differenziati, e che i livelli di proteina Cdt1 diminuiscono durante la differenziazione precoce in coincidenza con il rallentamento del tasso di licenza, ma prima della perdita di Oct4(Figura 3c e Figura 3-figure supplement 1). Noi ipotizziamo che la maggiore quantità di questa proteina essenziale di carico MCM specificamente in G1 contribuisce al rapido tasso di carico MCM negli hESC.
Figura 5.hESCs hanno alti livelli di Cdt1 in G1.(a) Immunoblot di lisati di cellule intere dalle linee cellulari asincrone indicate.b) Cambiamenti previsti nei livelli di proteina totale di Cdt1 e Cdc6 durante il ciclo cellulare umano.(c) Cdt1 totale rilevato nelle cellule asincrone mediante citometria a flusso che misura il contenuto di DNA (DAPI), sintesi del DNA (incorporazione EdU) e Cdt1 (anti-Cdt1). Le cellule verdi sono G1, le cellule viola sono fase S (EdU positivo), le cellule grigie sono G2/M.(d) Box-and-whiskers trame di G1 Cdt1 concentrazione per cella da(C). La linea centrale è mediana, i bordi esterni della scatola sono il 25° e il 75° percentile, i bordi dei baffi sono il1° e il 99° percentile, i singoli punti di dati sono il più basso e il più alto 1%, rispettivamente. La mediana G1 Cdt1 nei PNG è 2,9 volte maggiore, la media è 2,2 volte maggiore di G1 Cdt1 nei PNG, la media p=0,0504 mediana p=0,0243, media di tre repliche biologiche. I diagrammi di flusso sono solo cellule G1 (verde) da(c).
Manipolare i fattori di carico MCM altera i tassi di carico MCM
Il Cdt1 è essenziale per il caricamento MCM(Pozo e Cook, 2016); pertanto, la riduzione dei livelli di Cdt1 dovrebbe rallentare il caricamento MCM. Se la velocità di caricamento MCM è legata alla lunghezza G1, allora rallentare il caricamento MCM riducendo i livelli di Cdt1 potrebbe anche allungare il G1. Per testare questa previsione, abbiamo usato il siRNA per ridurre il Cdt1 negli hESC e abbiamo misurato le variazioni sia del tasso di carico MCM che della lunghezza G1(Figura 6a,b). Come previsto, l’esaurimento del Cdt1 ha ridotto il tasso di carico MCM negli hESC(Figura 6c). Incredibilmente, la lunghezza G1 è aumentata in coincidenza con la diminuzione del tasso di carico MCM(Figura 6d). Questi dati corroborano lo stretto legame tra il tasso di carico MCM e la lunghezza G1 nei PMS.
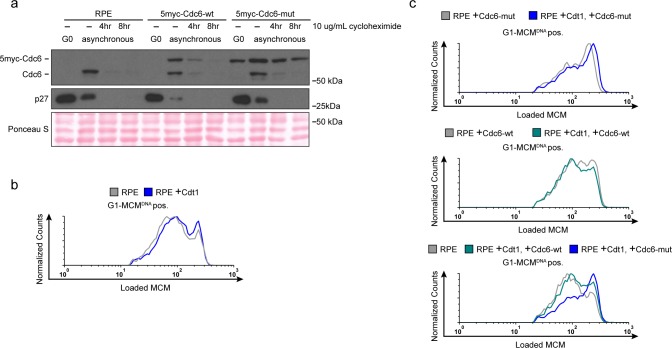
Figura 6-figure supplement 1.La manipolazione dei fattori di carico del MCM altera i tassi di carico del MCM.la manipolazione dei fattori di carico del MCM altera i tassi di carico del MCM.a) Citometria a flusso di cromatina per i CSE trattati con 25 nM siCdt1 o 100 nM siControl per 24 ore ed etichettati con EdU per 30 minuti prima del raccolto.b) Immunoblot di proteine totali dalle cellule di cuialla lettera a).c) Barra grafica impilata delle distribuzioni del ciclo cellulare per i campioni in(a); rappresentativa di due repliche biologiche. La percentuale di cellule G1 in ogni popolazione è riportata nei settori verdi.(d) Istogrammi di MCM caricato in cellule G1-MCMDNA. I conteggi per siCdt1 sono normalizzati al corrispondente campione siControl.(e) Immunoblot di Cdt1 e Cdc6 in cellule RPE con combinazioni delle seguenti: produzione costitutiva di 5Myc-Cdc6-wt o 5myc-Cdc6-mut (non mirato per la degradazione da APCCDH1: R56A, L59A, K81A, K81A, E82A, N83A) e un integrato doxiciciclina inducibile Cdt1-HA costruire trattato con 100 ng / ml doxiciciclina per 48 ore per sovraprodurre Cdt1-HA.(f) Grafici a barre impilati della distribuzione del ciclo cellulare misurata mediante citometria a flusso per le cellule indicate in(e); media con barre di errore ± SD (n = 3 repliche biologiche). La percentuale di cellule G1 in ogni popolazione è riportata nei settori verdi.(g) Istogramma di MCM caricato in cellule G1-MCMDNA-positive da(e). I conteggi di Cdc6-wt e Cdc6-mut sono normalizzati ai controlli RPE genitore.(a) Convalida del 5myc-Cdc6-mut. Immunoblot di lisati cellulari totali di RPE, RPE +5myc-Cdc6-wt o RPE +5myc-Cdc6-mut. Le cellule sono state sincronizzate in G0 per inibizione del contatto. Le cellule asincrone sono state trattate con 10 ug / ml di cicloesossimide per 4 o 8 ore. Cdc6-mut è più stabile in cicloesmide ed è resistente alla degradazione indotta da APCCDH1 indotta nelle cellule G0.b) Istogramma di MCM caricato in G1-MCMDNA cellule positive in cellule RPE che esprimono Cdt1 ectopica rispetto al controllo. C’è poco aumento del tasso di carico MCM. Conteggi di Cdt1-cellule che esprimono Cdt1 (blu,+Cdt1) sono normalizzati ai controlli RPE genitore (grigio).(c) Istogramma di MCM caricato in G1-MCMDNA-positivo cellule in cellule RPE da(Figura 6e) con le combinazioni indicate di Cdc6 e Cdt1 espressione Cdt1. I conteggi nei campioni sperimentali (verde e blu) sono normalizzati ai rispettivi controlli (grigio).
Come complemento al rallentamento del carico MCM in hESC con un breve G1, abbiamo anche cercato di accelerare la licenza di origine nelle cellule con un lungo G1 sovrapproduzione di proteine essenziali di licenza. Abbiamo prima costruito un derivato RPE_hTERT con ~due volte la sovrapproduzione di Cdt1 inducibile, ma questa manipolazione era insufficiente per accelerare il caricamento MCM (Figura 6-figure supplemento 1b). Abbiamo anche testato ectopica myc-tagged Cdc6 espresso costituzionalmente in cellule RPE(Figura 6e), questa aggiunta ha avuto solo effetti minimi sul tasso di carico MCM(Figura 6g, confrontare gli istogrammi grigio e verde). Abbiamo considerato, tuttavia, che l’umana Cdc6 è instabile in gran parte della fase G1 perché è destinato alla degradazione da APCCdh1 Mailande Diffley, 2005; Petersen et al., Abbiamo quindi espresso un mutante Cdc6 precedentemente descritto che è resistente alla distruzione mediata da APCCDH1 sia da solo che in combinazione con Cdt1 inducibile (Figura 6e e Figura 6-figure supplement 1a) (Mailand e Diffley, 2005; Petersen et al., 2000). In precedenza abbiamo dimostrato che i tagged Cdc6 e Cdt1 sono funzionali(Cook et al., 2002; Coleman et al., 2015; Chandrasekaran et al., 2011). L’espressione della stabile Cdc6-mut era sufficiente per aumentare i tassi di carico MCM(Figura 6g, confrontare l’istogramma blu con gli istogrammi grigio e verde); sovrapproduzione di Cdt1 ha avuto poco effetto additivo sul tasso di carico MCM nelle cellule RPE(Figura 6-figure supplemento 1b). È interessante notare che l’accelerazione del carico MCM con questo metodo non ha ridotto il G1 in RPE(Figura 6f), dimostrando ulteriormente che la lunghezza della fase G1 e il tasso di carico MCM alle origini della licenza può essere disaccoppiato. Un caricamento MCM lento può ritardare l’ingresso della fase S attraverso il punto di controllo della licenza(Teer et al., 2006; Shreeram et al., 2002; Nevis et al., 2009; Ge e Blow, 2009), ma un caricamento MCM rapido non è sufficiente a far scattare l’ingresso della fase S.
Figura 6-figure supplement 1.La manipolazione dei fattori di carico del MCM altera i tassi di carico del MCM.la manipolazione dei fattori di carico del MCM altera i tassi di carico del MCM.a) Citometria a flusso di cromatina per hESC trattati con 25 nM siCdt1 o 100 nM siControl per 24 ore ed etichettati con EdU per 30 minuti prima del raccolto.b) Immunoblot di proteine totali dalle cellule di cuialla lettera a).c) Barra grafica impilata delle distribuzioni del ciclo cellulare per i campioni in(a); rappresentativa di due repliche biologiche. La percentuale di cellule G1 in ogni popolazione è riportata nei settori verdi.(d) Istogrammi di MCM caricato in cellule G1-MCMDNA. I conteggi per siCdt1 sono normalizzati al corrispondente campione siControl.(e) Immunoblot di Cdt1 e Cdc6 in cellule RPE con combinazioni delle seguenti: produzione costitutiva di 5Myc-Cdc6-wt o 5myc-Cdc6-mut (non mirato per la degradazione da APCCDH1: R56A, L59A, K81A, K81A, E82A, N83A) e un integrato doxiciciclina inducibile Cdt1-HA costruire trattato con 100 ng / ml doxiciciclina per 48 ore per sovraprodurre Cdt1-HA.(f) Grafici a barre impilati della distribuzione del ciclo cellulare misurata mediante citometria a flusso per le cellule indicate in(e); media con barre di errore ± SD (n = 3 repliche biologiche). La percentuale di cellule G1 in ogni popolazione è riportata nei settori verdi.(g) Istogramma di MCM caricato in cellule G1-MCMDNA-positive da(e). I conteggi di Cdc6-wt e Cdc6-mut sono normalizzati ai controlli RPE genitore.(a) Convalida del 5myc-Cdc6-mut. Immunoblot di lisati cellulari totali di RPE, RPE +5myc-Cdc6-wt o RPE +5myc-Cdc6-mut. Le cellule sono state sincronizzate in G0 per inibizione del contatto. Le cellule asincrone sono state trattate con 10 ug / ml di cicloesossimide per 4 o 8 ore. Cdc6-mut è più stabile in cicloesmide ed è resistente alla degradazione indotta da APCCDH1 indotta nelle cellule G0.b) Istogramma di MCM caricato in G1-MCMDNA cellule positive in cellule RPE che esprimono Cdt1 ectopica rispetto al controllo. C’è poco aumento del tasso di carico MCM. Conteggi di Cdt1-cellule che esprimono Cdt1 (blu,+Cdt1) sono normalizzati ai controlli RPE genitore (grigio).(c) Istogramma di MCM caricato in G1-MCMDNA-positivo cellule in cellule RPE da(Figura 6e) con le combinazioni indicate di Cdc6 e Cdt1 espressione Cdt1. I conteggi nei campioni sperimentali (verde e blu) sono normalizzati ai rispettivi controlli (grigio).
Figura 6-figure supplemento 1.La manipolazione dei fattori di carico MCM altera i tassi di carico MCM.a) Convalida del 5myc-Cdc6-mut. Immunoblots di lisati cellulari totali di RPE, RPE +5myc-Cdc6-wt o RPE +5myc-Cdc6-mut. Le cellule sono state sincronizzate in G0 per inibizione del contatto. Le cellule asincrone sono state trattate con 10 ug / ml di cicloesossimide per 4 o 8 ore. Cdc6-mut è più stabile in cicloesmide ed è resistente alla degradazione indotta da APCCDH1 indotta nelle cellule G0.b) Istogramma di MCM caricato in G1-MCMDNA cellule positive in cellule RPE che esprimono Cdt1 ectopica rispetto al controllo. C’è poco aumento del tasso di carico MCM. Conteggi di Cdt1-cellule che esprimono Cdt1 (blu,+Cdt1) sono normalizzati ai controlli RPE genitore (grigio).(c) Istogramma di MCM caricato in G1-MCMDNA-positivo cellule in cellule RPE da(Figura 6e) con le combinazioni indicate di Cdc6 e Cdt1 espressione Cdt1. I conteggi nei campioni sperimentali (verde e blu) sono normalizzati ai rispettivi controlli (grigio).
Caricamento rapido MCM protegge hESC pluripotenza
La nostra dimostrazione che un caricamento MCM più lento si verifica universalmente durante la differenziazione precoce ha suggerito un collegamento funzionale tra il tasso di caricamento MCM e la manutenzione pluripotenza. Abbiamo considerato che rallentare il carico MCM potrebbe promuovere la differenziazione. Per esplorare questa idea, abbiamo rallentato prematuramente il carico MCM nei Cdt1 prima di indurne la differenziazione(Figura 7e). Dopo l’esaurimento del Cdt1, abbiamo stimolato la differenziazione verso i mesodermi con BMP4(Bernardo et al., 2011). Dopo 48 ore, abbiamo quantificato Oct4 e Cdx2 mediante immunocolorazione(Figura 7a, Figura 7-figure supplement 1). Il fattore di trascrizione pluripotenza Oct4 e il fattore di trascrizione omeobox Cdx2 reprimono reciprocamente l’espressione dell’altro, creando una chiara distinzione tra cellule pluripotenti Oct4-positive Cdx2-negative e cellule differenzianti Oct4-negative Cdx2-positive (Niwa et al.,2005). Abbiamo quantificato l’intensità media di fluorescenza sia di Oct4 che di Cdx2 in >18.000 cellule per condizione con una pipeline CellProfiler personalizzata e automatizzata, tracciando le intensità di segnale per ogni cellula in un diagramma di dispersione della densità (Figura 7b,c). Stimolando gli hESC di controllo con 10 ng/mL di BMP4 si è leggermente spostata la popolazione verso la differenziazione, ma la maggior parte delle cellule è rimasta pluripotente con alti livelli di Oct4 in questo punto di tempo. Sorprendentemente, hESC pretrattati con Cdt1 siRNA al caricamento MCM prematuramente lento ha guadagnato una popolazione sostanziale di Oct4 negativo-Cdx2 cellule differenzianti positive rispetto ai controlli che sono stati trattati in modo simile. Per quantificare il grado di differenziazione, abbiamo diviso l’intensità del Cdx2 di ogni cellula per la sua intensità di Oct4, creando un unico punteggio di differenziazione(Figura 7d). Dopo 10 ng / ml di trattamento BMP4, Cdt1-impoverito hESC aveva punteggi significativamente più alti, indicando che prematuramente rallentando il carico MCM promosso differenziazione (p<0,0001, a due code Mann-Whitney test a due code). Sia le cellule di controllo e le cellule Cdt1-impoverito Cdt1 differenziato più pienamente ad una maggiore concentrazione di 50 ng / ml BMP4, ma le cellule Cdt1-impoverito ancora differenziato ulteriormente rispetto ai controlli (p<0,0001, a due code Mann-Whitney test, Figura 7b e Figura 7-figure supplemento 2a). Altre combinazioni di concentrazioni BMP4 o tempi di trattamento hanno anche portato ad un consistente, significativo aumento della differenziazione nelle cellule pretrattate per rallentare il carico MCM (p<0,0001, test Mann-Whitney a due code, dati non mostrati). È importante notare che il fenotipo è stato conservato attraverso i lignaggi di differenziazione multipla, come prematuramente rallentare il carico MCM prima della differenziazione endodermica anche aumentato il numero di cellule positive per il fattore di trascrizione endodermico Sox17 rispetto ai controlli allo stesso punto di tempo(Figura 7-figure supplemento 1b). Per verificare se il mantenimento della pluripotenza era dovuto al ruolo del Cdt1 nella concessione di licenze di origine e non alle sue funzioni mitotiche o di altro tipo (Varmaet al., 2012), abbiamo rallentato la concessione di licenze esaurendo la proteina di carico ortogonale MCM, Cdc6 (Figura 7-figure supplement 3a-d). Un più modesto knockdown di Cdc6 è correlato con un effetto più debole, ma rilevabile sul carico MCM. È interessante notare che questo grado di inibizione della licenza non ha avuto alcun effetto sulla lunghezza G1. Nonostante la breve lunghezza G1, rallentando il carico MCM con l’esaurimento del Cdc6 si è favorita in modo significativo la differenziazione(Figura 7-figure supplement 2b-f, p<0.0001, test Mann-Whitney a due code. Così, si conclude che il caricamento lento del MCM promuoveva generalmente la differenziazione e per estensione, che il caricamento rapido del MCM preserva la pluripotenza.
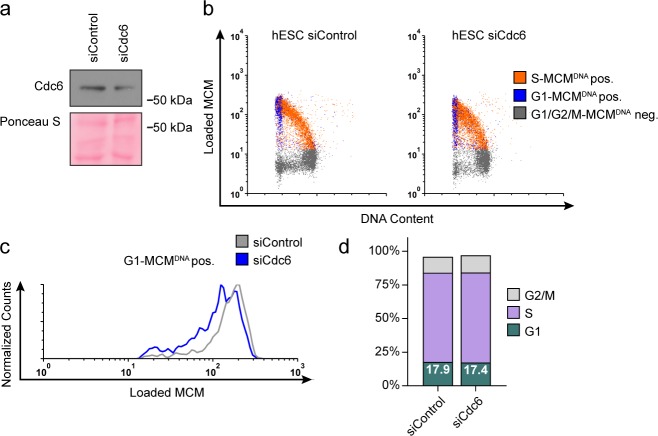
Figura 7-figure supplement 3.Figura 7—supplemento alla figura 3. Il caricamento lento del MCM promuove la differenziazione.Valori dell’immagine grezza.Valori dell’immagine grezza.Valori dell’immagine grezza.Completa serie di dati di microscopia e differenziazione dell’endoderma.Il caricamento lento del MCM promuove la differenziazione.Ridurre il tasso di caricamento del MCM con un siRNA alternativo che punta al Cdc6 invece che al Cdt1.(a) Microscopia a immunofluorescenza di hESC trattati con 100 nM di siControl o 100 nM di siCdt1 per 20 ore e poi trattati con BMP4 come indicato. Le cellule sono state fissate e colorate con DAPI (blu), anticorpo Cdx2 (verde) e anticorpo Oct4 (rosso). Le immagini sono una regione di 18 campi di vista per condizione; la barra della scala è di 100 μm (vedi Materiali e metodi).(b) Densità scatterplot di intensità di fluorescenza media (unità arbitrarie) di colorazione Oct4 e Cdx2 per ogni cellula in ogni condizione, > 18.000 cellule sono state quantificate per condizione. Si veda anche la Figura 7 – dati fonte 1.(c) Diagramma della relazione tra Oct4 e Cdx2 in cellule pluripotenti e differenziate come riportato in (b); barra di colore per gli scatterplotter in(b).d) Istogramma del rapporto di intensità media di fluorescenza Cdx2/Oct4 per tutte le cellule in siControl e siCdt1 trattate con 10 ng/mL BMP4 per 48 ore. Il contenitore dell’istogramma più a destra contiene tutti i valori superiori a 3,5. L’inserto è un diagramma a scatola e baffi degli stessi dati, la linea centrale è mediana, i bordi esterni della scatola sono il 25° e il 75° percentile, i bordi dei baffi sono il1° e il 99° percentile, i singoli punti dei dati sono rispettivamente il più basso e il più alto 1%. Le mediane sono 0,3722, 0,9319, e le medie sono 0,4285, 1,194 per siControl e siCdt1, rispettivamente. Campioni confrontati da due test di Mann-Whitney a coda, ****p<0.0001. Si veda anche la Figura 7-dati sorgente 1.e) Immunoblot per Cdt1 in lisati cellulari interi a 20 ore di trattamento con siRNA, prima del trattamento BMP4.f) Illustrazione del rapporto tra differenziazione e variazioni del tasso di carico del MCM.10.7554/eLife.30473.021Figure 7-source data 1.Raw image values.a) Dati completi di microscopia ad immunofluorescenza per la figura 7. Le caselle bianche segnano le aree mostrate in Figura 7b; le barre della scala sono 100 μM.b) Immunoblot per Cdt1 in lisati cellulari interi di HESC trattati con 100 nM siControl o siCdt1 per 24 ore prima di iniziare la differenziazione verso l’endoderma, due repliche biologiche. Il grafico a barre indica la percentuale di cellule Sox17-positive, n > 2.300 cellule per condizione. Si veda anche la Figura 7-dati sorgente 1.a) Istogramma del rapporto medio di intensità di fluorescenza Cdx2/Oct4 per tutte le cellule in siControl e siCdt1 trattate con 50 ng/mL BMP4 per 36 o 48 ore. Il contenitore dell’istogramma più a destra contiene tutti i valori superiori a 3,0 o 4,0, rispettivamente. L’inserto è un diagramma a scatola e baffi degli stessi dati, la linea centrale è mediana, i bordi esterni della scatola sono il 25° e il 75° percentile, i bordi dei baffi sono il1° e il 99° percentile, i singoli punti dei dati sono rispettivamente il più basso e il più alto 1%. Le mediane per 50 ng/mL 36 ore sono 0. 0,5418, 0,9465, e le medie sono 0. 0,5993, 1,05 per siControl e siCdt1, rispettivamente. Le mediane per 50 ng/mL 48 ore sono 0,916, 1,368, e i mezzi sono 1,055, 1,387 per siControl e siCdt1, rispettivamente. Campioni confrontati da due test di Mann-Whitney a coda, ****p<0.0001. Si veda anche la Figura 7-dati sorgente 1.b) Microscopia a immunofluorescenza di hESC trattati con 100 nM di siControl o 100 nM di siCdc6 per 32 ore e poi trattati con BMP4 come indicato. Le cellule sono state fissate e colorate con DAPI (blu), anticorpo Cdx2 (verde) e anticorpo Oct4 (rosso). Le immagini sono una regione di 27 campi di vista per condizione; la barra della scala è di 100 μm. (vedere Materiali e metodi).(c) Densità scatterplot di intensità di fluorescenza media (unità arbitrarie) di colorazione Oct4 e Cdx2 per ogni cellula in ogni condizione,>7.900 cellule sono state quantificate per condizione. Si veda anche la Figura 7 – dati fonte 1.d) Diagramma della relazione tra Oct4 e Cdx2 in cellule pluripotenti e differenziate come riportato in(b); barra di colore per gli scatterplotter in(b).e) Istogramma del rapporto medio di intensità di fluorescenza Cdx2/Oct4 per tutte le cellule in siControl e siCdc6 trattate con 100 ng/mL BMP4 per 36 ore. Il contenitore dell’istogramma più a destra contiene tutti i valori superiori a 5,0. L’inserto è un diagramma a scatola e baffi degli stessi dati, la linea centrale è mediana, i bordi esterni della scatola sono il 25° e il 75° percentile, i bordi dei baffi sono il1° e il 99° percentile, i singoli punti dei dati sono rispettivamente il più basso e il più alto 1%. Le mediane sono 1,296, 1,58, e le medie sono 1,433, 1,728 per siControl e siCdc6, rispettivamente. Campioni confrontati da due test di Mann-Whitney a coda, ****p<0.0001. Si veda anche la Figura 7-dati sorgente 1.e) Immunoblot per Cdc6 in lisati cellulari interi a 32 ore di trattamento con siRNA, prima del trattamento BMP4.2.a) Immunoblot di proteine totali per hESC trattati con 100 nM siControl per 32 ore o 100 nM siCdc6 pool per 32 ore e pulse-labeled con EdU per 30 minuti prima del raccolto.b) Citometria a flusso di cromatina di cellule provenienti daa) colorate con DAPI e anti-MCM2 e sottoposte a rilevamento EdU.c) Istogramma MCMDNA intensità positiva in cellule G1. I conteggi per siCdc6 sono normalizzati al corrispondente campione siControl.d) Barra grafica impilata delle distribuzioni del ciclo cellulare per i campioni in(a).
Figura 7-figure supplemento 3.Figura 7—supplemento di figura 3. Il caricamento lento di MCM promuove la differenziazione.Raw image values.Raw image values.Raw image values.Complete microscopy dataset e differenziazione endoderm.Slow MCM loading promuove la differenziazione.Reducing MCM loading rate by an alternative siRNA targeting Cdc6 invece di Cdt1.(a) Microscopia a immunofluorescenza di hESC trattati con 100 nM di siControl o 100 nM di siCdt1 per 20 ore e poi trattati con BMP4 come indicato. Le cellule sono state fissate e colorate con DAPI (blu), anticorpo Cdx2 (verde) e anticorpo Oct4 (rosso). Le immagini sono una regione di 18 campi di vista per condizione; la barra della scala è di 100 μm (vedi Materiali e metodi).(b) Densità scatterplot di intensità di fluorescenza media (unità arbitrarie) di colorazione Oct4 e Cdx2 per ogni cellula in ogni condizione, > 18.000 cellule sono state quantificate per condizione. Si veda anche la Figura 7 – dati fonte 1.(c) Diagramma della relazione tra Oct4 e Cdx2 in cellule pluripotenti e differenziate come riportato in (b); barra di colore per gli scatterplotter in(b).d) Istogramma del rapporto di intensità media di fluorescenza Cdx2/Oct4 per tutte le cellule in siControl e siCdt1 trattate con 10 ng/mL BMP4 per 48 ore. Il contenitore dell’istogramma più a destra contiene tutti i valori superiori a 3,5. L’inserto è un diagramma a scatola e baffi degli stessi dati, la linea centrale è mediana, i bordi esterni della scatola sono il 25° e il 75° percentile, i bordi dei baffi sono il1° e il 99° percentile, i singoli punti dei dati sono rispettivamente il più basso e il più alto 1%. Le mediane sono 0,3722, 0,9319, e le medie sono 0,4285, 1,194 per siControl e siCdt1, rispettivamente. Campioni confrontati da due test di Mann-Whitney a coda, ****p<0.0001. Si veda anche la Figura 7-dati sorgente 1.e) Immunoblot per Cdt1 in lisati cellulari interi a 20 ore di trattamento con siRNA, prima del trattamento BMP4.f) Illustrazione del rapporto tra differenziazione e variazioni del tasso di carico del MCM.10.7554/eLife.30473.021Figure 7-source data 1.Raw image values.a) Dati completi di microscopia ad immunofluorescenza per la figura 7. Le caselle bianche segnano le aree mostrate in Figura 7b; le barre della scala sono 100 μM.b) Immunoblot per Cdt1 in lisati cellulari interi di HESC trattati con 100 nM siControl o siCdt1 per 24 ore prima di iniziare la differenziazione verso l’endoderma, due repliche biologiche. Il grafico a barre indica la percentuale di cellule Sox17-positive, n > 2.300 cellule per condizione. Si veda anche la Figura 7-dati sorgente 1.a) Istogramma del rapporto medio di intensità di fluorescenza Cdx2/Oct4 per tutte le cellule in siControl e siCdt1 trattate con 50 ng/mL BMP4 per 36 o 48 ore. Il contenitore dell’istogramma più a destra contiene tutti i valori superiori a 3,0 o 4,0, rispettivamente. L’inserto è un diagramma a scatola e baffi degli stessi dati, la linea centrale è mediana, i bordi esterni della scatola sono il 25° e il 75° percentile, i bordi dei baffi sono il1° e il 99° percentile, i singoli punti dei dati sono rispettivamente il più basso e il più alto 1%. Le mediane per 50 ng/mL 36 ore sono 0. 0,5418, 0,9465, e le medie sono 0. 0,5993, 1,05 per siControl e siCdt1, rispettivamente. Le mediane per 50 ng/mL 48 ore sono 0,916, 1,368, e i mezzi sono 1,055, 1,387 per siControl e siCdt1, rispettivamente. Campioni confrontati da due test di Mann-Whitney a coda, ****p<0.0001. Si veda anche la Figura 7-dati sorgente 1.b) Microscopia a immunofluorescenza di hESC trattati con 100 nM di siControl o 100 nM di siCdc6 per 32 ore e poi trattati con BMP4 come indicato. Le cellule sono state fissate e colorate con DAPI (blu), anticorpo Cdx2 (verde) e anticorpo Oct4 (rosso). Le immagini sono una regione di 27 campi di vista per condizione; la barra della scala è di 100 μm. (vedere Materiali e metodi).(c) Densità scatterplot di intensità di fluorescenza media (unità arbitrarie) di colorazione Oct4 e Cdx2 per ogni cellula in ogni condizione,>7.900 cellule sono state quantificate per condizione. Si veda anche la Figura 7 – dati fonte 1.d) Diagramma della relazione tra Oct4 e Cdx2 in cellule pluripotenti e differenziate come riportato in(b); barra di colore per gli scatterplotter in(b).e) Istogramma del rapporto medio di intensità di fluorescenza Cdx2/Oct4 per tutte le cellule in siControl e siCdc6 trattate con 100 ng/mL BMP4 per 36 ore. Il contenitore dell’istogramma più a destra contiene tutti i valori superiori a 5,0. L’inserto è un diagramma a scatola e baffi degli stessi dati, la linea centrale è mediana, i bordi esterni della scatola sono il 25° e il 75° percentile, i bordi dei baffi sono il1° e il 99° percentile, i singoli punti dei dati sono rispettivamente il più basso e il più alto 1%. Le mediane sono 1,296, 1,58, e le medie sono 1,433, 1,728 per siControl e siCdc6, rispettivamente. Campioni confrontati da due test di Mann-Whitney a coda, ****p<0.0001. Si veda anche la Figura 7-dati sorgente 1.e) Immunoblot per Cdc6 in lisati cellulari interi a 32 ore di trattamento con siRNA, prima del trattamento BMP4.2.a) Immunoblot di proteine totali per hESC trattati con 100 nM siControl per 32 ore o 100 nM siCdc6 pool per 32 ore e pulse-labeled con EdU per 30 minuti prima del raccolto.b) Citometria a flusso di cromatina di cellule provenienti daa) colorate con DAPI e anti-MCM2 e sottoposte a rilevamento EdU.c) Istogramma MCMDNA intensità positiva in cellule G1. I conteggi per siCdc6 sono normalizzati al corrispondente campione siControl.d) Barra grafica impilata delle distribuzioni del ciclo cellulare per i campioni in(a).
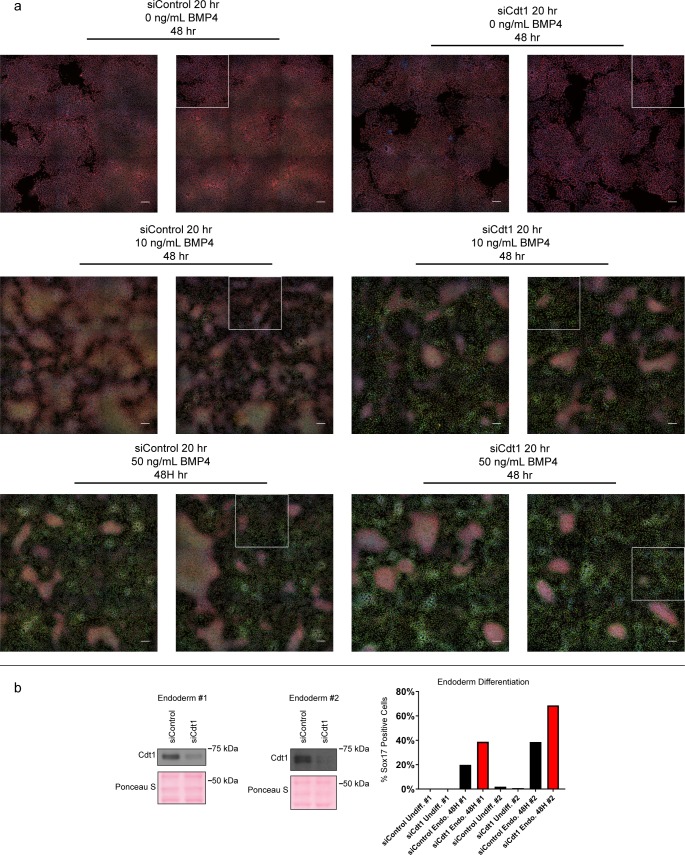
Figura 7-figure supplemento 1.1. Set completo di dati di microscopia e differenziazione endodermica.a) Dati completi di microscopia a immunofluorescenza per la figura 7. Le caselle bianche contrassegnano le aree mostrate nella Figura 7b; le barre della scala sono di 100 μM.b) Immunoblot per Cdt1 in lisati cellulari interi di HESC trattati con 100 nM siControl o siCdt1 per 24 ore prima di iniziare la differenziazione verso l’endoderma, due repliche biologiche. Il grafico a barre indica la percentuale di cellule Sox17-positive, n > 2.300 cellule per condizione. Si veda anche la Figura 7-dati sorgente 1.
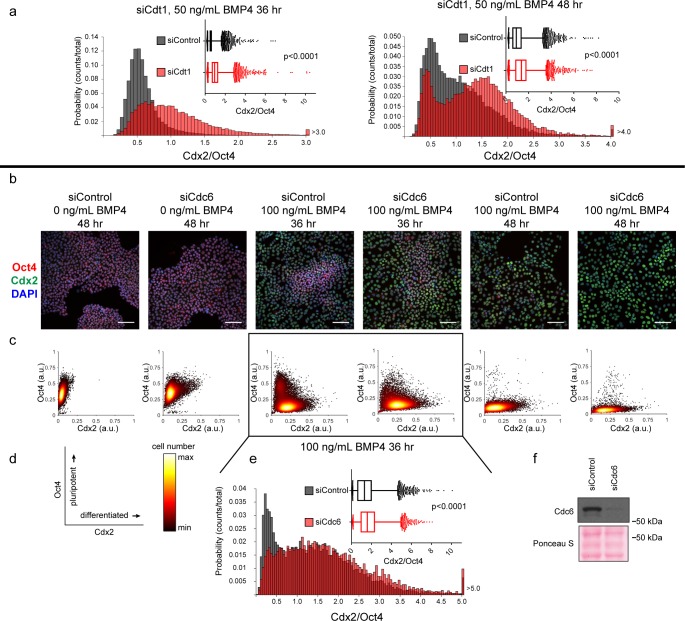
Figura 7-figure supplement 2.Il caricamento lento di MCM promuove la differenziazione.a) Istogramma del rapporto medio di intensità di fluorescenza Cdx2/Oct4 per tutte le cellule in siControl e siCdt1 trattate con 50 ng/mL BMP4 per 36 o 48 ore. Il contenitore dell’istogramma più a destra contiene tutti i valori superiori a 3,0 o 4,0, rispettivamente. L’inserto è un diagramma a scatola e baffi degli stessi dati, la linea centrale è mediana, i bordi esterni della scatola sono il 25° e il 75° percentile, i bordi dei baffi sono il1° e il 99° percentile, i singoli punti dei dati sono rispettivamente il più basso e il più alto 1%. Le mediane per 50 ng/mL 36 ore sono 0. 0,5418, 0,9465, e le medie sono 0. 0,5993, 1,05 per siControl e siCdt1, rispettivamente. Le mediane per 50 ng/mL 48 ore sono 0,916, 1,368, e i mezzi sono 1,055, 1,387 per siControl e siCdt1, rispettivamente. Campioni confrontati da due test di Mann-Whitney a coda, ****p<0.0001. Si veda anche la Figura 7-dati sorgente 1.b) Microscopia a immunofluorescenza di hESC trattati con 100 nM di siControl o 100 nM di siCdc6 per 32 ore e poi trattati con BMP4 come indicato. Le cellule sono state fissate e colorate con DAPI (blu), anticorpo Cdx2 (verde) e anticorpo Oct4 (rosso). Le immagini sono una regione di 27 campi di vista per condizione; la barra della scala è di 100 μm. (vedere Materiali e metodi).(c) Densità scatterplot di intensità di fluorescenza media (unità arbitrarie) di colorazione Oct4 e Cdx2 per ogni cellula in ogni condizione,>7.900 cellule sono state quantificate per condizione. Si veda anche la Figura 7 – dati fonte 1.d) Diagramma della relazione tra Oct4 e Cdx2 in cellule pluripotenti e differenziate come riportato in(b); barra di colore per gli scatterplotter in(b).e) Istogramma del rapporto medio di intensità di fluorescenza Cdx2/Oct4 per tutte le cellule in siControl e siCdc6 trattate con 100 ng/mL BMP4 per 36 ore. Il contenitore dell’istogramma più a destra contiene tutti i valori superiori a 5,0. L’inserto è un diagramma a scatola e baffi degli stessi dati, la linea centrale è mediana, i bordi esterni della scatola sono il 25° e il 75° percentile, i bordi dei baffi sono il1° e il 99° percentile, i singoli punti dei dati sono rispettivamente il più basso e il più alto 1%. Le mediane sono 1,296, 1,58, e le medie sono 1,433, 1,728 per siControl e siCdc6, rispettivamente. Campioni confrontati da due test di Mann-Whitney a coda, ****p<0.0001. Si veda anche la Figura 7-dati sorgente 1.e) Immunoblot per Cdc6 in lisati cellulari interi a 32 ore di trattamento con siRNA, prima del trattamento BMP4.
Figura 7-figure supplement 3.Figura 7—figura 3. Riduzione del tasso di carico MCM con un siRNA alternativo che punta al Cdc6 invece che al Cdt1.a) Immunoblot di proteine totali per hESC trattati con 100 nM siControl per 32 ore o 100 nM siCdc6 pool per 32 ore e pulse-labeled con EdU per 30 minuti prima del raccolto.b) Citometria a flusso di cromatina di cellule provenienti daa) colorate con DAPI e anti-MCM2 e sottoposte a rilevamento EdU.c) Istogramma MCMDNA intensità positiva in cellule G1. I conteggi per siCdc6 sono normalizzati al corrispondente campione siControl.d) Barra grafica impilata delle distribuzioni del ciclo cellulare per i campioni in(a).
Discussione
In questo studio, dimostriamo che il caricamento rapido di MCM per autorizzare le origini di replicazione è una proprietà intrinseca delle cellule pluripotenti. Le cellule staminali embrionali umane hanno un tasso di caricamento MCM notevolmente veloce, e la riprogrammazione per creare cellule staminali pluripotenti indotte aumenta il tasso di caricamento MCM. Inoltre, il caricamento MCM rallenta in concomitanza con l’allungamento G1 e rimodellamento del ciclo cellulare esteso che accompagnano le prime fasi di differenziazione(Figura 7f). A nostra conoscenza, questa è la prima dimostrazione che il tasso di carico MCM è regolato in modo evolutivo. La regolazione dello sviluppo del tasso di carico MCM è coerente con il lavoro precedente che mostrava livelli più elevati di Cdt1 totale in Cdt1 nei CSE asincroni del mouse rispetto alle cellule differenziate(Ballabeni et al., 2011). La diminuzione regolata del tasso di carico MCM è fondamentale durante la differenziazione, in quanto il carico MCM rapido protegge la pluripotenza, e rallentare prematuramente il carico MCM promuove la differenziazione.
Le cellule staminali Pluripotenti caricano rapidamente i complessi MCM per raggiungere simili quantità totali di MCM caricato alla transizione G1/S in meno tempo rispetto alle loro controparti differenziate isogeniche. Anche se non abbiamo rilevato un sostanziale carico di MCM in telopasi, come suggerito in precedenza(Dimitrova et al., 2002), è chiaro che il carico di telopasi è un’opzione in alcune cellule e un requisito in cellule senza G1 rilevabile come S. pombe e nelle prime divisioni nucleari in D. melanogaster(Nishitani et al., 2000; Farrell e O’Farrell, 2014). Le cellule staminali raggiungono un caricamento MCM più veloce, almeno in parte, da livelli di Cdt1 particolarmente elevati in G1. Questi livelli elevati sono raggiunti non solo da una modesta differenza in mRNA CDT1 mRNA(Figura 3c), ma anche post-trascrizionale da specifica riaccumulo di Cdt1 proteina Cdt1 nella precedente fase G2(Figura 5c). La stabilità del Cdt1 nella fase G2 è stata attribuita alla protezione mediata dalla geminina dalla SCFSkp2 E3 ubiquitina ligasi in cellule non staminali(Tsunematsu et al., 2013; Ballabeni et al., 2004; Clijsters et al., 2013). Non è chiaro, tuttavia, che i livelli di geminina siano particolarmente elevati nelle cellule staminali rispetto alle cellule differenziate (a parte le differenze previste dai cambiamenti nella distribuzione del ciclo cellulare(Figura 5a)(Ballabeni et al., 2011)), quindi sembra improbabile che la geminina porti a livelli più elevati di Cdt1 nelle cellule staminali. Anche i livelli di Skp2 non cambiano nelle prime fasi della differenziazione delle cellule staminali(Egozi et al., 2007). Il Cdt1 è protetto nella tarda fase S e G2 dall’attività della ciclina A/Cdk1(Rizzardi et al., 2015), e quindi riteniamo probabile che l’elevata attività CDK documentata nelle cellule staminali contribuisca alla stabilizzazione del Cdt1 in G2(Sela et al., 2012). L’accumulo anticipato di Cdt1 per promuovere il carico di MCM in G1 è stato originariamente proposto da esperimenti su linee cellulari derivate dal cancro(Ballabeni et al., 2004; Clijsters et al., 2013). Le nostre osservazioni nelle cellule staminali suggeriscono che questa strategia è impiegata da cellule non trasformate durante le fasi di sviluppo che richiedono G1 a breve termine.
Altri fattori oltre all’accumulo di Cdt1 possono anche accelerare il carico di MCM. Gli hESC che abbiamo saggiato hanno 2-3 volte maggiori livelli di proteina Cdt1 in G1 rispetto ai PNG, ma caricano MCM 6,5 volte più velocemente all’ora rispetto ai PNG. Una molecola di Cdt1 può (in vitro) caricare più complessi MCM dal momento che il Cdt1 viene rilasciato nella fase solubile immediatamente dopo aver completato una reazione di carico(Ticau et al., 2015). Le cellule staminali possono sperimentare una minore degradazione di Cdc6 nei primi anni di G1 a causa di attività ciclica quasi costitutiva E / Cdk2 e / o attività APCCdh1 attenuata, corroborata dalla nostra osservazione che un mutante Cdc6 che non è preso di mira da APCCdh1 aumenta la velocità di carico MCM nelle cellule con lunghe fasi G1 (Figura 6g)(Ballabeni et al., 2011; Neganova et al., 2009; Filipczyk et al., 2007). Inoltre, le cellule staminali sono arricchite per l’euchromatina, un ambiente che può essere particolarmente permissivo per un rapido caricamento MCM(Chen e Dent, 2014).
Il carico rapido MCM può contribuire a meccanismi che mantengono brevi fasi G1 nelle cellule pluripotenti. Il punto di controllo delle licenze di origine collega la quantità di MCM caricato alla lunghezza G1 controllando l’attività Cdk2. A questo proposito, la sovrapproduzione di Cyclin E ha “cortocircuitato” il checkpoint di licenza in celle differenziate a caricamento lento. Questo punto di controllo è stato finora dimostrato solo in p53-profitta di cellule differenziate di mammiferi(Ge e Blow, 2009), ma l’allungamento G1 di hESC dopo l’esaurimento del Cdt1 suggerisce che le cellule staminali pluripotenti hanno anche un punto di controllo di licenza di funzionamento. Le cellule con un veloce caricamento MCM potrebbero soddisfare questo checkpoint rapidamente, attivare Cyclin E/Cdk2, e quindi passare meno tempo in G1. Meccanismi che supportano la breve lunghezza del G1 preservano la pluripotenza nei CSE (e promuovono la riprogrammazione in iPSC), poiché le cellule sono più sensibili ai segnali di differenziazione in G1; a questo proposito, l’estensione della fase G1 nei CSE può aumentare la propensione alla differenziazione(Soufi e Dalton, 2016; Pauklin e Vallier, 2013; Filipczyk et al., 2007; Coronado et al., 2013). Recenti lavori con topi knockout quintuplo privi di tutti i ciclini di tipo D ed E hanno anche riportato che il Cyclin E/Cdk2 contribuisce ulteriormente a mantenere la pluripotenza stabilizzando i fattori di trascrizione di Oct4, Sox2 e Nanog(Liu et al., 2017). Il caricamento veloce MCM può essersi evoluto come una proprietà intrinseca delle cellule pluripotenti per mantenere alta l’attività Cdk2 e mantenere la fase G1 breve.
Ciclina / attività Cdkk non è l’unica connessione tra il ciclo cellulare e pluripotenza. Non-CDK proteine associate al ciclo cellulare regolano l’espressione dei geni chiave della pluripotenza tra cui SOX2 e NANOG(Gonzales et al., 2015; Pauklin et al., 2016; Li et al., 2012). I fattori di trascrizione della pluripotenza regolano l’espressione dei geni del ciclo cellulare, compresi quelli che codificano le cicline, gli inibitori CDK e E2F3a(Lee et al., 2010; Kanai et al., 2015; Choi et al., 2012). D’altra parte, la pluripotenza e le funzioni del ciclo cellulare possono essere disaccoppiate geneticamente negli esperimenti in cui la manipolazione del ciclo cellulare non ha alterato la pluripotenza e viceversa (Scognamiglio et al.,2016; Kareta et al., 2015b). Osserviamo che l’inibizione della licenza può accelerare la differenziazione anche senza allungare notevolmente G1(Figura 7-figure supplements 2,3) che può indicare un ulteriore collegamento diretto e indipendente dal ciclo cellulare tra il tasso di carico MCM e la differenziazione.
Notiamo che la differenziazione precoce non è l’unica impostazione in cui può essere rilevante un rapido caricamento MCM durante un breve G1. Come gli hESC, le cellule T attivate hanno cicli cellulari molto veloci con brevi fasi G1(Kinjyo et al., 2015). La trasformazione oncogena è anche frequentemente associata all’accorciamento del G1. Può essere che i percorsi che collegano la differenziazione alla velocità di carico MCM sono anche cooptato in alcuni tumori per indurre la licenza rapida. D’altra parte, un sottoinsieme di tumori può proliferare in uno stato di perpetua sottolicenza che contribuisce all’instabilità del genoma caratteristica delle cellule trasformate. Le indagini future chiariranno le relazioni molecolari tra i percorsi di segnalazione dello sviluppo, il tasso di carico MCM e il rimodellamento del ciclo cellulare.
Materiali e metodi
| Tipo di reagente (specie) o risorsa | Designazione | Fonte o riferimento | Identificatori | Ulteriori informazioni |
|---|---|---|---|---|
| Ceppo, ceppo di fondo(Escherichia coli) | E. coli: DH5α | Invitrogen | Cat#11319019 | |
| Reagente genetico(Homo sapiens) | Rc/CMV ciclino E | Hinds et al. (1992) PMID: 1388095 | Addgene 8963 | |
| Reagente genetico(Homo sapiens) | pInducer20 | Meerbrey et al., 2011PMID: 21307310 | Addgene 44012 | |
| Reagente genetico(Homo sapiens) | ΔNRF | Dr. J. Bear | N/A | |
| Reagente genetico(Homo sapiens) | VSVG | Dr. J. Bear | N/A | |
| Reagente genetico(Homo sapiens) | pInducer20-Cyclin E1 | Questo documento | N/A | vedi Materiali e metodi |
| Reagente genetico(Homo sapiens) | pDONR221 | Invitrogen | Cat#12536017 | |
| Reagente genetico(Homo sapiens) | pENTR221-Ciclo E1 | Questo documento | N/A | |
| Reagente genetico(Homo sapiens) | PCR4-TOPO | Invitrogen | Cat# 450030 | |
| Reagente genetico(Homo sapiens) | pInducer20-blast | Questo documento | N/A | vedi Materiali e metodi |
| Reagente genetico(Homo sapiens) | pInducer20-blast-Cdt1-HA | Questo documento | N/A | vedi Materiali e metodi |
| Reagente genetico(Homo sapiens) | CLXSN-5myc-Cdc6-wt | Questo documento | N/A | vedi Materiali e metodi |
| Reagente genetico(Homo sapiens) | CLXSN-5myc-Cdc6-mut | Questo documento | N/A | vedi Materiali e metodi |
| Linea cellulare(Homo sapiens) maschio | T98G | ATCC | Cat#CRL-1690 | |
| Linea cellulare(Homo sapiens) femmina | RPE1-hTERT | ATCC | Cat#CRL-4000 | |
| Linea cellulare(Homo sapiens) maschio | ARPE-19 | ATCC | Cat#CRL-2302 | |
| Linea cellulare(Homo sapiens) femmina | H9 hESC (WA09) | WiCell | hPSCReg ID: WAe009-A | |
| Linea cellulare(Homo sapiens) maschio | NPC | Questo documento | N/A | vedi Materiali e metodi |
| Linea cellulare(Homo sapiens) femmina | HEK293T | ATCC | Cat# CRL-3216 | |
| Linea cellulare(Homo sapiens) maschio | ARPE-iPSC | Questo documento | N/A | vedi Materiali e metodi |
| Anticorpo | Anti-Mcm2, mouse monoclonale (BM28) | BD Bioscienze | Cat#610700;RRID: AB_2141952 | 1:10.000 (IB) 1:200 (FC) |
| Anticorpo | Anti-Mcm3, coniglio policlonale | Laboratori Bethyl | Cat#A300-192A; RRID: AB_162726 | 1:10.000 (IB) 1:200 (FC) |
| Anticorpo | Anti-Cdt1, coniglio monoclonale (D10F11) (immunoblot) | Tecnologie di segnalazione cellulare | Cat#8064S; RRID: AB_10896851 | 1:10.000 (IB) |
| Anticorpo | Anti-Cdt1, monoclonale di coniglio (EPR17891) (citometria a flusso) | Abcam | Cat#ab20202067; RRID:AB_2651122 | 1:100 (FC) |
| Anticorpo | Anti-Cdc6, mouse monoclonale (180,2) | Santa Cruz Biotecnologia | Cat#sc-9964; RRID: AB_627236 | 1:2000 (IB) |
| Anticorpo | Anti-Oct4, coniglio policlonale (immunoblot) | Abcam | Cat#ab19857; RRID: AB_445175 | 1:4000 (IB) |
| Anticorpo | Anti-Oct4, mouse monoclonale (9B7) (microscopia) | Millipore | Cat#:MABD76; RRID: AB_10919170 | 1:1000 (IF) |
| Anticorpo | Anti-Cdx2, coniglio monoclonale (EPR2764Y) | Abcam | Cat#ab76541; RRID: AB_1523334 | 1:1000 (IF) |
| Anticorpo | Anti-Sox17, capra policlonale | Sistemi R e D | Cat#AF1924; RRID: AB_355060 | 1:500 (IF) |
| Anticorpo | Anticiclina E1, coniglio policlonale | Santa Cruz Biotecnologia | Cat#sc-198; RRID: AB_631346 | 1:2000 (IB) |
| Anticorpo | Anti-Orc1, coniglio policlonale | Laboratori Bethyl | Cat#A301-892A; AB_1524103 | 1:1000 (IB) |
| Anticorpo | Anti-Orc6, ratto monoclonale (3A4) | Santa Cruz Biotecnologia | Cat#sc-32735; RRID: AB_670295 | 1:5000 (IB) |
| Anticorpo | Anti-geminino, coniglio policlonale | Santa Cruz Biotecnologia | Cat#sc-13015; RRID: AB_2263394 | 1:3000 (IB) |
| Anticorpo | Anti-Histone H3, coniglio monoclonale (D1H2) | Tecnologie di segnalazione cellulare | Cat#4499S; RRID: AB_10544537 | 1:10.000 (IB) |
| Anticorpo | Anti-TRA-1-60, mouse monoclonale (cl.A) | Invitrogeno | Cat#41-1000; RRID: AB_605376 | 1:5000 (IB) |
| Anticorpo | Anti-nestina, mouse monoclonale (10 C2) | Abcam | Cat#ab22035; RRID: AB_446723 | 1:10000 (IB) |
| Anticorpo | Topo anti-TRA-1-60 (immunofluorescenza) | Millipore/Chemicon | Cat# MAB4360; RRID: AB_2119183 | 1:400 (IF) |
| Anticorpo | Topo anti-TRA-81 (immunofluorescenza) | Millipore/Chemicon | Cat# MAB4381; RRID:AB_177638 | 1:400 (IF) |
| Anticorpo | Mouse anti-SSEA-4 (MC-813-70) (immunofluorescenza) | Millipore/Chemicon | Cat# MAB4304; RRID:AB_177629 | 1:200 (IF) |
| anticorpo | Coniglio anti-SSEA3 (MC-631) (immunofluorescenza) | Millipore/Chemicon | Cat# MAB4303; RRID:AB_177628 | 1:200 (IF) |
| Anticorpo | Anti-Oct3/4 capra policlonale (immunofluorescenza) | Abcam | Cat# ab27985; RRID:AB_776898 | 1:200 (IF) |
| Anticorpo | Policlonale di capra anti-NANOG (immunofluorescenza) | Everest Biotech | Cat# EB068601; RRID:AB_2150379 | 1:200 (IF) |
| Anticorpo | Anti-p27 coniglio policlonale | Santa Cruz Biotecnologia | Cat#sc-528; RRID:AB_632129 | 1:2000 (IB) |
| Anticorpo | Anti-α-tubulina | Sigma Aldrich | Cat#9026 | 1:50000 (IB) |
| Anticorpo | Capra anti-Mouse-HRP | Jackson ImmunoRicerca | Cat#115-035-146; RRID: AB_2307392 | 1:10000 (IB) |
| Anticorpo | Asino anti-coniglio-HRP | Jackson ImmunoRicerca | Cat#711-035-152; RRID: AB_10015282 | 1:10000 (IB) |
| Anticorpo | Bovino anti-Goat-HRP | Jackson ImmunoRicerca | Cat#805-035-180; RRID: AB_2340874 | 1:10000 (IB) |
| Anticorpo | Asino anti-Rat-HRP | Jackson ImmunoRicerca | Cat#712-035-153; RRID: AB_2340639 | 1:10000 (IB) |
| Anticorpo | Donkey anti-Goat-Alexa 594 | Jackson ImmunoRicerca | Cat#705-585-147; RRID: AB_2340433 | 1:1000 (IF) |
| Anticorpo | Asino anti-coniglio-Alexa 488 | Tecnologie per la vita | Cat#A21206; RRID: AB_2535792 | 1:1000 (SE) (FC) |
| Anticorpo | Capra anti-Topo-Alexa 594 | Tecnologie per la vita | Cat#A11032; RRID: AB_2535792 | 1:1000 (IF) |
| Anticorpo | Asino anti-coniglio-Alexa 647 | Jackson ImmunoRicerca | Cat#711-605-152; RRID: AB_2492288 | 1:1000 (FC) |
| Anticorpo | Donkey anti-Mouse-Alexa 488 | Jackson ImmunoRicerca | Cat#715-545-150; RRID: AB_2340845 | 1:1000 (FC) |
| Reagente a base di sequenza | siCdt1- CCUACGUCAAGGACGGACAATT | Nevis et al. (2009) PMCID: PMC2972510 | N/A | |
| Reagente a base di sequenza | siCdc6-2534- CACCAUGCUCAGCCCCAUUAAGGUUAUAU | Nevis et al. (2009) PMCID: PMC2972510 | N/A | |
| Reagente a base di sequenza | siCdc6-2144- UCUAGCCAAUGUGUGUGUUGUAUGUA | Nevis et al. (2009) PMCID: PMC2972510 | N/A | |
| Reagente a base di sequenza | siControllo (Luciferasi)- CUUUACGCUGAGUACUUCGA | Coleman et al. (2015) PMID: 26272819 | N/A | |
| Reagente a base di sequenza | siMCM3-2859 5′- augacuauugcaucuucuucauug | Questo documento | sintetizzato da invitrogeno | |
| Reagente a base di sequenza | siMCM3-2936 5′- aacauaugacuucugugaguacu | Questo documento | sintetizzato da invitrogeno | |
| Reagente a base di sequenza | POU5F1-F: 5′-CCTGAAGCAGAAGAGGATCACC, | Eton Bioscience | ||
| Reagente a base di sequenza | POU5F1-R 5′-AAAGCCGGCAGATGGTCGTTTTTGG, | Eton Bioscience | ||
| Reagente a base di sequenza | CDX2-F 5′-ACAGTCGCCTACATCACCATCCATCCATCCG, | Eton Bioscience | ||
| Reagente a base di sequenza | CDX2-R 5′-CCTCTTTTTTTTTTTTGTTTTGCGGTTTTC, | Eton Bioscience | ||
| Reagente a base di sequenza | T-F 5′-CTTCCAGAAGTCAAAGTCAAGTTCAAGTTTOO, | Eton Bioscience | ||
| Reagente a base di sequenza | T-R 5′-TGAACTGGGTGTCAGGGAAGCA, | Eton Bioscience | ||
| Reagente a base di sequenza | SOX17-F 5′-ACGCTTTTTTTTTTTGGGGCTAAG, | Eton Bioscience | ||
| Reagente a base di sequenza | SOX17-R 5′-GTCAGCCGCCCCCCCCCCACTTTTTOO, | Eton Bioscience | ||
| Reagente a base di sequenza | CDT1-F 5′-GGAGGTCAGATTACCCCAGCTCAC, | Eton Bioscience | ||
| Reagente a base di sequenza | CDT1-R, 5′-TTGACGTGTGGGCCACCCCCCCAGTTTOO, | Eton Bioscience | ||
| Reagente a base di sequenza | SOX2-F 5′-CTACAGCATGATGATGCAGGACCA, | Eton Bioscience | ||
| Reagente a base di sequenza | SOX2-R 5′-TCTGCGCGGAGCTGGTCATGGAGT, | Eton Bioscience | ||
| Reagente a base di sequenza | PAX6-F 5′-AATCAGAGAAGACAGGCCA, | Eton Bioscience | ||
| Reagente a base di sequenza | PAX6-R 5′-GTGGTAGGTATCATAACTC, | Eton Bioscience | ||
| Reagente a base di sequenza | ACTB-F 5′-CACCATTGGCAATGAGCGGTTTTC, | Eton Bioscience | ||
| Reagente a base di sequenza | ACTB-R 5′-AGGTCTTTTTGCGGATGTCCACGT | Eton Bioscience | ||
| Reagente a base di sequenza | CDC6-KEN-F: 5- ctccaccaccaccaaagggccaaggcaaggcggggccccccttttttcacatacacatacac | Eurofins | ||
| Reagente a base di sequenza | CDC6-KEN-R: 5- GTGTATGTGGGGGGGGGGACCCCCCCCCCCCCCCCTTTTGTTTTTTTTTTTTTGGGGTGGGGGGGGAG | Eurofins | ||
| Reagente a base di sequenza | CDC6-DBOX-F: 5- aagccctgccttccccccccccccccgccaaacgtggggggatgatgacaacctatgcaa | Eurofins | ||
| Reagente a base di sequenza | CDC6-DBOX-R: 5- TTGCATAGGTTGGCCCCCCCATTTTTTTTGGGGGGGGGGGGGGGGGGGGCAGAGAGGGCAGGGTTTTT | Eurofins | ||
| Reagente a base di sequenza | AgeI-rta3-F: 5- gctcggatctccaccccccccgggattttttttttgcagtcgaattcac | Eurofins | ||
| Reagente a base di sequenza | AgeI-IRES-blast-R: 5-ACAAAAGGGTGGCCATGGCCATGGTTTTTTTTTTTTTTTTTCA | Eurofins | ||
| Reagente a base di sequenza | Blast-F:5- tgaAaaaaacacacgatgataagggatgataagtttaaaccaccatggccaagcccctttttttgt | Eurofins | ||
| Reagente a base di sequenza | Blast-AgeI-Ind-R: 5-GTTCAATCATGGGTGGGACCGG CTATTAGCCCTCCCACACACATAACCA | Eurofins | ||
| Reagente a base di sequenza | BP-cycE-F 5’GGGGGACAAGTTGTGTACAAAAAAAAGCAGGGCTACCATGAAGGAGGAGGAGGACGGGCGGCGCGC | Eurofins | ||
| Reagente a base di sequenza | BP-cycE-R 5’GGGGGACCACCACTTTGTACAAGAAGGGGGGTTCACGCCATTTCCGGGCCCCCCCGCT | Eurofins | ||
| Software, algoritmo | MATLAB | MathWorks | https://www.mathworks.com/ | |
| Software, algoritmo | GraphPad Prisma 7 | Software GraphPad | https://www.graphpad.com/scientific-software/prism/ | |
| Software, algoritmo | Software di ricerca avanzata NIS-Elements | Nikon | https://www.nikoninstruments.com/Products/Software/NIS-Elements-Advanced-Research | |
| Software, algoritmo | CellProfiler | Carpenter et al., 2006 PMC1794559 | http://cellprofiler.org/ | |
| Software, algoritmo | FCS Express 6 | De Novo Software | https://www.denovosoftware.com/ | |
| Software, algoritmo | FCSExtract Utility | Conte F Glynn | http://research.stowers.org/mcm/efg/ScientificSoftware/Utility/FCSExtract/index.htm | |
| Software, algoritmo | QUMA | RIKEN | http://quma.cdb.riken.jp | |
| Software, algoritmo | Adobe Photoshop CS6 | Adobe | http://www.adobe.com/products/photoshop.html | |
| Saggio commerciale o kit | Kit di riprogrammazione CytoTune-iPS 2.0 Sendai | Invitrogen | Cat#A16517 | |
| Saggio commerciale o kit | DNeasy Kit sangue e tessuti | Qiagen | Cat#69504 | |
| Saggio commerciale o kit | RNeasy Mini kit | Qiagen | Cat#74104 | |
| Saggio commerciale o kit | Kit Epitect Bisulfite | Qiagen | Cat#59104 | |
| Saggio commerciale o kit | Kit di purificazione dell’RNA totale di Norgen Biotek | Norgen Biotek | Cat#37500 | |
| Saggio commerciale o kit | RNA ad alta capacità applicata di Biosystem da RNA a CDNA | Biosistema applicato | Cat#4387406 | |
| Saggio commerciale o kit | Kit per il rilevamento della fosfatasi alcalina | Millipore | Cat# SCR004 | |
| Saggio commerciale o kit | Kit di estrazione del gel QIAquick | Qiagen | Cat# 28704 | |
| Composto chimico, farmaco | DAPI | Tecnologie per la vita | Cat#D1306 | |
| Composto chimico, farmaco | EdU | Santa Cruz Biotecnologia | Cat#sc-284628 | |
| Composto chimico, farmaco | Ponceau S | Sigma Aldrich | Cat#P7170-1L | |
| Peptide, proteina ricombinante | Proteina BMP4 | Sistemi R e D | Cat#314 BP-010 | |
| Peptide, proteina ricombinante | Attivare una proteina | Sistemi R e D | Cat#338-AC-010 | |
| Composto chimico, farmaco | Y-27632 2HCl | Selleck Chemicals | Cat#S1049 | |
| Composto chimico, farmaco | CHIR-99021 | Selleck Chemicals | Cat#S2924 | |
| Composto chimico, farmaco | mTESR1 | Tecnologie delle cellule staminali | Cat#05850 | |
| Composto chimico, farmaco | STEMdiff Neural Induction Medium | Tecnologie delle cellule staminali | Cat#05835 | |
| Composto chimico, farmaco | STEMdiff Progenitore Neurale Medio | Tecnologie delle cellule staminali | Cat#05833 | |
| Composto chimico, farmaco | Essenziale 8 Medio | Tecnologie per la vita | Cat#A1517001 | |
| Composto chimico, farmaco | Doxiciclina | CalBiochem | Cat#324385 | |
| Composto chimico, farmaco | Alexa 647-azide | Tecnologie per la vita | Cat#A10277 | |
| Composto chimico, farmaco | Alexa 488-azide | Tecnologie per la vita | Cat#A10266 | |
| Composto chimico, farmaco | Hydroxyurea | Alfa Aesar | Cat#A10831 | |
| Composto chimico, farmaco | Matrice a membrana in GFR Matrigel Corning Matrigel | Corning | Cat#CB-40230 | |
| Composto chimico, farmaco | Poli-L-Ornitina | Sigma Aldrich | Cat#P4957-50ML | |
| Composto chimico, farmaco | Lamininin | Sigma Aldrich | Cat#L2020-1MG | |
| Composto chimico, farmaco | ReLesR | Tecnologie delle cellule staminali | Cat#05872 |
Cultura cellulare
Le linee cellulari sono state autenticate mediante profilatura STR (ATCC, Manassas, VA) e confermate come negative al micoplasma. T98G, HEK293T, e RPE1-hTERT sono stati coltivati in Dulbecco Modified Eagle Medium (DMEM) integrato con 2 mM L-glutammina e 10% di siero bovino fetale (FBS) e incubato in 5% di CO2 a 37 ° C. ARPE-19 (maschio) sono stati coltivati in 1:1 DMEM:F12 integrato con 2 mM L-glutammina e 10% di siero bovino fetale e incubati in 5% di CO2 a 37 ° C. T98G, HEK293T, HEK293T, RPE1-hTERT, e ARPE-19 cellule erano dal ATCC e sono stati passati con tripsina e non ha permesso di raggiungere la confluenza. WA09 (H9 hESCs) sono stati coltivati in mTeSR1 (StemCell Technologies) con media cambia ogni 24 ore su Matrigel (Corning, New York, NY) piatti rivestiti e incubati in 5% di CO2 a 37 ° C. Gli H9 avevano un cariotipo diploide normale al passaggio 32 e sono stati utilizzati dal passaggio 32-42. Le ARPE-iPSC sono state coltivate in Essential 8 (Life Technologies, Grand Island, NY) con cambi di media ogni 24 ore su piatti rivestiti Matrigel (Corning) e incubate in 5% di CO2 a 37°C. Le iPSC sono state usate dal passaggio 20-25 e avevano cariotipo normale. Sia hESCs e iPSCs sono stati regolarmente passati ogni 4 giorni come aggregati utilizzando ReLeSR, secondo le istruzioni del produttore (StemCell Technologies, Canada). Gli hESC e iPSC sono stati solo passati come cellule singole in 10 μM Y-27632 2HCl (Selleck Chemicals, Houston, TX) per esperimenti, come descritto in precedenza (Watanabe etal., 2007). NPC sono stati coltivati in Neural Progenitor Medium (StemCell Technologies) con cambi di media ogni 24 ore su poli-L-ornitina /aminina (Sigma Aldrich, St. Louis, MO) piatti rivestiti e incubati in 5% di CO2 a 37 ° C. I PNG sono stati passati con StemPro Accutase (Gibco, Waltham, MA) settimanalmente.
Totale lisato e il frazionamento della cromatina
Le cellule sono state raccolte tramite tripsinizzazione. Per i lisati proteici totali, le cellule sono state lisate su ghiaccio per 20 minuti in tampone CSK (300 mM saccarosio, 100 mM NaCl, 3 mM MgCl2, 10 mM PIPES pH 7,0) con 0,5% di tritone x-100 e proteasi e inibitori della fosfatasi (0.1 mM AEBSF, 1 µg/ mL pepstatina A, 1 µg/ mL leupeptina, 1 µg/ mL aprotinina, 10 µg/ ml fosvitina, 1 mM β-fosfato di glicerolo, 1 mM Na-ortovanadato). Le cellule sono state centrifugate a 13.000 xg a 4°C per 5 minuti, poi i supernatanti sono stati trasferiti in una nuova provetta per un test di Bradford (Biorad, Hercules, CA) utilizzando una curva standard BSA. Il frazionamento della cromatina per l’immunoblotting è stato eseguito come descritto in precedenza(Cook et al., 2002; Méndez e Stillman, 2000), utilizzando il buffer CSK con 1 mM ATP, 5 mM CaCl2, 0,5% tritone x-100 e inibitori della proteasi e della fosfatasi per isolare le proteine insolubili e la nucleasi S7 (Roche) per rilasciare le proteine legate al DNA. Un test di Bradford (Biorad) è stato eseguito a parità di carico. Per 100 mM o 300 mM NaCl solubile / frazionamento del pellet, le cellule sono state lisate in CSK standard (100 mM NaCl) o CSK ad alto contenuto di sale (300 mM NaCl) con 0,5% di tritone X-100 con proteasi e inibitori della fosfatasi per 5 minuti su ghiaccio. Poi le cellule sono state centrifugate a 2000 xg per 3 minuti, supernatanti trasferiti in un nuovo tubo come la frazione solubile. Il pellet rimanente è stato sospeso in 2x SDS tampone di carico SDS (2% SDS, 5% 2-mercaptoetanolo, 0,1% blu di bromofenolo, 50 mM Tris pH 6,8, 10% glicerolo) come la frazione di pellet. Il test di Bradford è stato eseguito sulla frazione solubile a parità di carico.
Immunoblotting
I campioni sono stati diluiti con buffer di caricamento SDS (finale: 1% SDS, 2,5% 2-mercaptoetanolo, 0,1% blu di bromofenolo, 50 mM Tris pH 6,8, 10% glicerolo) e bollito. I campioni sono stati eseguiti su gel SDS-PAGE, poi le proteine trasferite su membrane di difluoruro di polivinilidene (Thermo Fisher, Waltham, MA) o nitrocellulosa (GE Healthcare, Chicago, IL). Le membrane sono state bloccate a temperatura ambiente per 1 ora in latte al 5% o al 5% di BSA in Tris-Buffered-Saline-0,1%-tween-20 (TBST). Dopo il blocco, le membrane sono stati incubati in anticorpo primario durante la notte a 4 ° C a 4 ° C sia nel latte 1,25% o 5% BSA in TBST con 0,01% sodio azide. I blot sono stati lavati con TBST poi incubati in anticorpo secondario coniugato con HRP in 2,5% latte o 5% BSA in TBST per 1 ora, lavati con TBST, e poi le membrane sono state incubate con ECL Prime (Amersham, Regno Unito) ed esposte a film autoadiografico (Denville, Holliston, MA). Carico proteico proteico uguale è stato verificato dalla colorazione Ponceau S (Sigma Aldrich). Gli anticorpi utilizzati per l’immunocolorazione erano: Mcm2, (BD Bioscienze, San Jose, CA, Cat#610700), Mcm3, (Bethyl Laboratories, Montgomery, TX, Cat#A300-192A), Cdt1, (Cell Signaling Technologies, Beverly,MA, Cat#8064S), Cdc6, (Santa Cruz Biotechnology, Santa Cruz, CA, Cat#sc-9964), Oct4, (Abcam, Cambridge, MA, Cat#ab19857), Cyclin E1, (Santa Cruz Biotechnology, Cat#sc-198), Orc1, (Bethyl Laboratories, Cat#A301-892A), Orc6, (Santa Cruz Biotechnology, Cat#sc-32735), geminin, (Santa Cruz Biotechnology, Cat#sc-13015), Histone H3, (Cell Signaling Technologies, Cat#4499S), TRA-1-60, (Invitrogen, Cat#41-1000), nestin, (Abcam, Cat#ab22035), p27 (Santa Cruz Biotechnology, Cat#sc-528), α-tubulin (Sigma Aldrich, Cat#9026).
Citometria a flusso
Per i campioni etichettati con il marchio EdU, le cellule sono state incubate con 10 uM EdU (Santa Cruz Biotechnology) per 30 minuti prima del prelievo. Per la citometria a flusso proteico totale, le cellule sono state raccolte con tripsina e risospese come cellule singole, lavate con PBS, e fissate con il 4% di paraformaldeide (Electron Microscopy Sciences, Hatfield, PA) in PBS per 15 minuti a temperatura ambiente, poi l’1% BSA-PBS è stato aggiunto, miscelato e le cellule sono state centrifugate a 1000 xg per 7 minuti (e per tutte le successive fasi di centrifugazione), poi lavate con 1% BSA-PBS e centrifugato. Le cellule fisse sono state permeabalizzate con 0,5% di tritone x-100 in 1% di BSA-PBS a temperatura ambiente per 15 minuti, centrifugate, poi lavate una volta con 1% di BSA, PBS e centrifugate di nuovo prima dell’etichettatura. Per la citometria a flusso di cromatina, le cellule sono state raccolte con tripsina e risospese come cellule singole, lavate con PBS, e poi lisate su ghiaccio per 5 minuti in tampone CSK con 0,5% di tritone x-100 con proteasi e inibitori della fosfatasi. Successivamente, l’1% di BSA-PBS è stato aggiunto e miscelato, poi le cellule sono state centrifugate per 3 minuti a 1000 xg, poi fissate in paraformaldeide al 4% in PBS per 15 minuti a temperatura ambiente. Per 100 mM NaCl vs 300 mM NaCl CSK, le cellule sono state elaborate come nella citometria a flusso di cromatina di cui sopra, tranne CSK contenente 300 mM NaCl è stato utilizzato al posto del normale 100 mM NaCl. Successivamente, l’1% BSA-PBS è stato aggiunto, mescolato e le cellule sono state centrifugate e poi lavate di nuovo prima di etichettare. I metodi di etichettatura per i campioni di proteine totali e campioni di cromatina erano identici. Per la sintesi del DNA (EdU), i campioni sono stati centrifugati e incubati in PBS con 1 mM CuSO4, 1 μM fluoroforo azide, e 100 mM acido ascorbico (fresco) per 30 minuti a temperatura ambiente al buio. 1% BSA-PBS +0,1% NP-40 è stato aggiunto, miscelato e centrifugato. I campioni sono stati risospesi in anticorpo primario in 1% BSA-PBS +0,1% NP-40 e incubati a 37 ° C per 1 ora al buio. Successivamente, l’1% BSA-PBS +0,1% NP-40 è stato aggiunto, miscelato e centrifugato. I campioni sono stati risospesi in anticorpo secondario in 1% BSA-PBS +0,1% NP-40 e incubati a 37 ° C per 1 ora al buio. Successivamente, l’1% BSA-PBS +0,1% NP-40 è stato aggiunto, miscelato e centrifugato. Infine, le cellule sono state risospese in 1% BSA-PBS +0,1% NP-40 con 1 μg/mL DAPI (Life Technologies) e 100 μg/mL RNAse A (Sigma Aldrich) e incubati durante la notte a 4 ° C al buio. I campioni sono stati eseguiti su un citometro a flusso CyAn ADP (Beckman Coulter, Brea, CA) e analizzati con il software FCS Express sei (De Novo, Glendale, CA). Sono state utilizzate le seguenti combinazioni anticorpo/fluoroforo: (1): Alexa 647-azide (Life Technologies), primario: Mcm2 (BD Biosciences, Cat#610700), secondario: Donkey anti-mouse-Alexa 488 (Jackson ImmunoResearch), DAPI. (2): Alexa 488-azide (Life Technologies), primario: Cdt1 (Abcam, Cat#610700), secondario: Donkey anti-rabbit-Alexa 647 (Jackson ImmunoResearch), DAPI. (3): Alexa 647-azide (Life Technologies), primario: Mcm3 (Bethyl Cat#A300-192A), secondario: Donkey anti-rabbit-Alexa 488 (Jackson ImmunoResearch), DAPI. (4): primario: Mcm3 (Bethyl Cat#A300-192A), Mcm2 (BD Biosciences, Cat#610700), secondario: Donkey anti-mouse-Alexa 488, Donkey anti-rabbit-Alexa 647 (Jackson ImmunoResearch, West Grove, PA), DAPI. Le cellule sono state gated sulla zona FS contro la zona SS. Le canottiere sono state cancellate sull’area DAPI contro l’altezza del DAPI. I cancelli positivi / negativi per EdU e MCM sono stati gated su un campione di controllo negativo, che è stato trattato con EdU né anticorpo primario, ma incubato con 647-azide e l’anticorpo secondario Donkey antimouse-Alex 488 e DAPI per tener conto della colorazione di fondo(Figura 1-figure supplemento 1).
Tempo di raddoppio
Il tempo di raddoppio è stato calcolato placcando lo stesso numero di celle come descritto sopra e contando il numero di celle nel tempo utilizzando un contatore automatico di celle Luna II (Logos Biosystems, Corea del Sud) a 24, 48 e 72 ore dopo la placcatura. Tre o quattro pozzi sono stati contati come repliche tecniche in ogni punto temporale. L’analisi di regressione di GraphPad Prism è stata utilizzata per calcolare il tempo di raddoppio, e più repliche biologiche sono state mediate per un tempo di raddoppio medio finale. Gli ARPE-19 sono stati contati quattro volte, gli HESC e gli NPC tre volte, e gli iPSC due volte.
Sincronizzazione cellulare e trattamenti
Per sincronizzare le cellule in G1, le cellule T98G sono state coltivate al 100% di confluenza, lavate con PBS, e incubate per 72 ore in 0,1% FBS, DMEM, L-glutammina. Dopo la fame di siero, le cellule sono state ri-stimolate passando 1:3 con tripsina a nuovi piatti in 20% FBS, DMEM, L-glutammina, raccogliendo le cellule 10 ore e 12 ore post-stimolazione. Per sincronizzare le cellule T98G nella fase iniziale S, le cellule sono state trattate come per G1, tranne 1 mM Hydroxyurea (Alfa Aesar, Haverhill, MA) è stato aggiunto ai media su ri-stimolazione e le cellule sono state raccolte 18 ore post-stimolazione. Per sincronizzare le cellule a metà tardi S, le cellule sono state trattate come nei primi anni S, poi a 18 ore post-stimolazione cellule sono state lavate con PBS e rilasciato in 10% FBS, DMEM, L-Glutammina, raccogliendo 6 ore, 8 ore dopo il rilascio.
Per sincronizzare le cellule RPE1-hTERT in G0, le cellule sono state coltivate al 100% di confluenza, poi incubate per 48 ore in 10% FBS, DMEM, L-glutammina. Per RPE1 in G1/S, le cellule G0 sono state tripsinizzate e passate 1:6 con tripsina a nuovi piatti in 10% FBS, DMEM, L-glutammina, e raccolti con tripsina 22 ore dopo. Per il trattamento di cicloesossimide (Sigma), le cellule RPE asincrone sono state trattate con 10 ug / ml per 4 ore o 8 ore come indicato. Per l’irradiazione UV, le cellule RPE asincrone sono state trattate con 20 J/m2 di UV con uno Stratalinker (Stratagene, San Diego, CA) e raccolte 1 ora dopo.
Clonazione
Il plasmide pInducer20-Cyclin E è stato costruito utilizzando il metodo di clonazione Gateway (Invitrogen). I siti attBsono stati aggiunti al Cyclin E1 cDNA tramite PCR utilizzando il plasmide Rc/CMV cyclin E come template e i primer BP-cycE-F (5′ GGGGGACAAGTTTGTACAAAAAAAGCAGGCTACCATGAGGGACGGACGGGCCGGCGCGC) e BP-cycE-R (5′ GGGGACCACCACTTTGTGTACAAGAAGCTGGGTTCACGCCATCCGGCCCCCGGCT)(Hinds et al..,1992). Il prodotto PCR è stato ricombinato con il plasmide pDONR221 utilizzando la clonasi BP (Invitrogen) secondo le istruzioni del produttore e trasformato in DH5α per creare pENTR221-Cyclin E1. Poi la reazione LR è stata eseguita tra pInducer20 e pENTR221-Cyclin E1 utilizzando LR Clonase (Invitrogen, Carlsbad, CA) secondo le istruzioni del produttore e trasformata in DH5α per creare pInducer20-Cyclin E1. Il plasmide pInducer20 è stato convertito in resistenza alla blasticidina (pInducer20-blast2) da Gibson Assembly (New England Biolabs, Ipswitch, MA) seguendo il protocollo del produttore. pInducer20 è stato tagliato con AgeI e assemblato con prodotti PCR con i seguenti primer:
AgeI-rta3-F: 5- gctcggatctccaccccccggattttttgcagtcgaattcac
AgeI-IRES-blast-R: 5- ACAAAGGCTTGGCCATGGATGGATGGTTTTTTTTTTTTTTCA
Blast-F:5- tgaAaaaaacacacgatgataagggatgataagtttaaaccatggccaagccccttttttgt
Blast-AgeI-Ind-R: 5- GTTCAATCATGGGTGGGACCGG CTATTAGCCCTCCCACACACATAACCA
Il plasmide pLenti CMV blast plasmid era un modello per il gene della resistenza alla blasticidina. Un Cdt1-HA marcato con il tag Cdt1-HA è stato clonato in pInducer20-blast utilizzando la clonazione Gateway come descritto sopra.
Il mutante Cdc6 incapace di legare APCCDH1 (5myc-Cdc6-mut) è stato descritto in precedenza: R56A, L59A, K81A, E82A, N83A(Petersen et al., 2000). pCLXSN-5myc-Cdc6-wt è stato clonato in 5myc-Cdc6-mut da due gruppi sequenziali Gibson (NEB) secondo le istruzioni del produttore. Primer utilizzati:
CDC6-KEN-F: 5- ctccaccaccaccaaagggccaaggcaaggcggggccccttttttcacatacatacac
CDC6-KEN-R: 5- GTGTATGTGGGGGGGGGGACCCCCCCCCCCCCCCCTTTTGTTTTTTTTTTTTTGGGGTGGGGGGGGAG
CDC6-DBOX-F: 5- aagccctgccttccccccccccccccgccaaacgtggggggatgatgacaacctatgcaa
CDC6-DBOX-R: 5- TTGCATAGGTTGGCCCCCCCATTTTTTTTGGGGGGGGGGGGGGGGGGGGCAGAGAGGGCAGGGTTTTT
Costruzione di linee cellulari e produzione di proteine inducibili
Per confezionare il retrovirus, pCLXSN 5myc-Cdc6 wt o mut sono stati co-trasformati con i plasmidi pCI-GPZ e VSVG in HEK293T utilizzando 50 μg/mL Polyethylenimine-Max (Aldrich Chemistry).Per confezionare lentivirus, pInducer20-Cyclin E1 o pInducer20-blast2-Cdt1-HA sono stati co-trasferiti con ΔNRF e plasmidi VSVG in HEK293T utilizzando 50 μg/mL Polyethylenimine-Max (Aldrich Chemistry). Supernatante virale è stato trasdotto con 8 ug / ml di polibrene (Millipore, Burlington, MA) su cellule RPE1-hTERT durante la notte. Le cellule sono state selezionate con 500 ug/mL di neomicina (Gibco) o 5 μg/mL di blasticidina (Research Products International, Mount Prospect, IL) per 1 settimana. Per sovrapprodurre la ciclina E1, le cellule sono state trattate con 100 ng/mL doxiciciclina (CalBiochem, San Diego, CA) per 72 ore in 10% FBS, DMEM, L-glutammina. Le cellule di controllo sono stati i Inducer20-Ciclina E1 senza doxiciclina. Per sovrapprodurre Cdt1, le cellule sono state trattate con 100 ng / ml doxiciciclina per 48 ore in 10% FBS, DMEM, L-glutammina, le cellule di controllo erano senza doxiciciclina.
trasfezioni di siRNA
Per il trattamento con siRNA, Dharmafect 1 (Dharmacon, Lafayette, CO) è stato miscelato in mTeSR1 con il siRNA appropriato secondo le istruzioni del produttore, poi diluito con mTeSR1 e aggiunto alle cellule dopo l’aspirazione dei vecchi media. Le concentrazioni finali di siRNA erano: 100 nM siControllo (Luciferasi), 25 o 100 nM siCdt1, o una miscela di due siCdc6 (2144 e 2534 a 50 nM ciascuno). La miscela di siRNA Cdt1 è stata incubata su cellule per 20 o 24 ore, poi cambiata in mTeSR1 nuovo senza siRNA. La miscela di siRNA Cdc6 siRNA è stato incubato su cellule per 24 ore, poi cambiato a nuovo mTeSR1 senza siRNA per 8 ore (32 ore totali). Il Cdt1, Cdc6 e Luciferase siRNA sono stati descritti in precedenza(Coleman et al., 2015; Nevis et al., 2009). Per il trattamento di siRNA delle cellule RPE, Dharmafect 1 (Dharmacon) è stato miscelato in Optimem (Gibco) con il siRNA appropriato secondo le istruzioni del produttore, poi diluito con DMEM, 10% FBS, L-glutammina e aggiunto alle cellule dopo l’aspirazione di vecchi media. Il giorno successivo, la miscela di siRNA è stata aspirata e sostituita con DMEM fresco, 10% FBS, L-glutammina, raccogliendo campioni 72 ore dopo l’inizio del trattamento con siRNA. I siRNA erano siControl (Luciferasi) a 100 nM o una miscela di due siRNA MCM3 (2859 e 2936 a 50 nM ciascuno).
siMCM3-2859 5′- augacuauugcaucuucuucauugdTdT
siMCM3-2936 5′- aacauaugacuucuucaguacudTdT
Differenziazione
Mesoderma (BMP4): hESC sono stati passati come cellule singole a 7 × 103/cm2 inmTeSR1 con 10 μM Y-27632 2HCl su piastre rivestite di Matrigel. 24 ore dopo, il supporto è stato cambiato per iniziare la differenziazione con mTeSR1 fresco con 100 ng/mL BMP4 (Sistemi R e D, Minneapolis, MN), e 24 ore dopo il supporto è stato cambiato in mTeSR1 fresco con 100 ng/mL BMP4 per 48 ore totali di differenziazione.
Neuroectoderm: hESCs sono stati differenziati utilizzando un protocollo monostrato basato su un protocollo a induzione neurale medio: hESCs sono stati passati come singole cellule a 5,2 × 104/cm2 in STEMdiff Neural Induction Medium (StemCell Technologies) con 10 micron Y-27632 2HCl su piastre rivestite Matrigel, e placcatura iniziato la differenziazione. 24 ore dopo, il supporto è stato cambiato in un nuovo Neural Induction Medium per altre 24 ore per una differenziazione totale di 48 ore. Per derivare i PNG, gli hESC sono stati differenziati in mezzo di induzione neurale (StemCell Technologies) utilizzando il protocollo di induzione neurale del corpo embrionale secondo le istruzioni del produttore, in modo simile ai rapporti precedenti (Robinsonet al., 2016). Una volta generati, gli NPC sono stati mantenuti in Neural Progenitor Medium (StemCell Technologies).
Mesoderma (GSK3βi): hESC sono stati passati come cellule singole a 3 × 104/cm2 in mTeSR1 con 10 μM Y-27632 2HCl su piastre rivestite di Matrigel. 24 ore dopo, i supporti sono stati modificati per iniziare la differenziazione. Le cellule sono stati lavati con Advanced RPMI 1640 (Gibco), poi incubati in Advanced RPMI 1640 con B27 meno insulina (Gibco), 2 mM L-glutammina, e 8 micron CHIR-99021 (Selleck Chemicals). A 24 ore dopo aver cambiato i media, le cellule sono state lavate con Advanced RPMI 1640 e poi incubate in Advanced RPMI 1640 con B27 meno insulina (Gibco), 2 mM L-glutammina, senza CHIR-99021 per 24 ore per un totale di 48 ore di differenziazione.
Endoderm: hESC sono stati passati come cellule singole a 4 × 103/cm2 inmTeSR1 con 10 μM Y-27632 2HCl su piastre rivestite in Matrigel. Il giorno dopo, il supporto è stato cambiato in mTeSR1 fresco senza Y-27632 2HCl. 24 ore dopo, i supporti sono stati cambiati per iniziare la differenziazione. Le cellule sono state lavate con Advanced RPMI 1640 (Gibco), poi incubate in Advanced RPMI 1640 con 0,2% FBS, 2 mM L-glutammina, 100 ng/mL Activin A (R e D Systems) e 2,5 μM CHIR-99021. A 24 ore dopo aver cambiato il supporto, le cellule sono state lavate con Advanced RPMI 1640 poi incubate in Advanced RPMI 1640 con 0,2% FBS, 2 mM L-glutammina, 100 ng/mL di Activin A, senza CHIR-99021 per 24 ore per un totale di 48 ore di differenziazione.
Microscopia a contrasto di fase
Le immagini a contrasto di fase sono state acquisite con un microscopio invertito Axiovert 40 CFL, obiettivo 20x (Zeiss, Germania).
Microscopia a immunofluorescenza
Per la microscopia a immunofluorescenza, gli hESC sono stati placcati come cellule singole in mTeSR1 con 10 μM Y-27632 2HCl in Matrigel rivestito, 24 pozzetti, # 1,5 piastre di fondo in vetro (Cellvis) a 7 × 103/cm2 persiCdt1, Mesoderm (BMP4), a 5 × 103 per siCdc6, Mesoderm (BMP4), e a 4 × 103/cm2 per siCdt1, Endoderm. Le cellule sono state incubate con siCdt1 per 20 ore (Mesoderm (BMP4)), 24 ore (Endoderm) o siCdc6 per 32 ore (Mesoderm [BMP4]), tutte in parallelo con siControl come descritto sopra (trasfezioni siRNA). Dopo il trattamento con siRNA, le cellule sono state differenziate come descritto sopra (Differenziazione) con le seguenti modifiche: Per Mesoderm (BMP4), sono state utilizzate più concentrazioni di BMP4 e tempi di trattamento come indicato(Figura 7, Figura 7-figure supplement 1). Per il trattamento meno di 48 ore, le cellule sono state incubate in mTeSR1 dopo il trattamento con siRNA fino all’inizio della differenziazione. (Esempio: 12 ore di mTeSR1 poi 36 ore di BMP4, per un totale di 48 ore). Per l’endoderma, il primo RPMI/Activin/CHIR-99021 è stato subito dopo il siRNA, senza un giorno di incubazione in mTeSR1. Dopo la differenziazione, le cellule sono state fissate in paraformaldeide al 4% in PBS per 15 minuti a temperatura ambiente, lavate con PBS, e permeabalizzate con 5% BSA, PBS, 0,3% di tritone x-100 a 4°C durante la notte. Successivamente, le cellule sono state incubate in anticorpo primario in 5% BSA, PBS, 0,3% di tritone x-100 a 4 ° C durante la notte. Le cellule sono state lavate con PBS a temperatura ambiente, poi incubate in anticorpo secondario in 5% BSA, PBS, 0,3% di tritone x-100 a temperatura ambiente per 1 ora. Le cellule sono state lavate con PBS, poi incubate in 1 μg/mL DAPI in PBS per 10 minuti a temperatura ambiente, poi lavate con PBS. Per Mesoderm (BMP4) gli anticorpi primari erano Oct4 (Millipore, Cat#MABD76) e Cdx2 coniglio (Abcam, Cat#ab76541), gli anticorpi secondari erano capra anti-mouse-Alexa 594, asino anti-rabbit-Alexa 488. Per l’endoderma l’anticorpo primario era Sox17 (Sistemi R e D, Cat#AF1924), l’anticorpo secondario era asino anti-capra-Alexa 594 (Jackson ImmunoResearch). Le cellule sono state riprese in PBS su un microscopio invertito Nikon Ti Eclipse con un rivelatore Andor Zyla 4.2 sCMOS. Le immagini sono state scattate come scansione 3 × 3 di 20x campi con un obiettivo 0,75 NA, cucite con il 15% di sovrapposizione tra i campi utilizzando il software di ricerca avanzata NIS-Elements (Nikon, Giappone). La correzione dell’ombreggiatura è stata applicata all’interno del software NIS-Elements prima dell’acquisizione delle immagini. Le immagini grezze sono state quantificate utilizzando una pipeline CellProfiler personalizzata.
qPCR (Figura 3)
I lisati di RNA sono stati preparati utilizzando il kit di purificazione dell’RNA totale di Norgen Biotek (Cat. 37500). I lisati sono stati prima trattati con Promega RQ1 RNase-Free DNase (Promega, Madison, WI), e poi convertiti in cDNA utilizzando il kit ad alta capacità RNA-to-cDNA di Applied Biosystem (Cat. 4387406). La PCR quantitativa in tempo reale (qPCR) con SYBR Green (Bio-Rad; SsoAdvanced Universal SYBR Green Supermix, Cat. 1725271) è stata effettuata per valutare l’espressione genica. Tutti i risultati sono stati normalizzati in ACTB. I primer per qPCR sono stati ordinati a Eton Bioscience, San Diego, CA. Primer:
POU5F1-F: 5′-CCTGAAGCAGAAGAGGATCACC,
POU5F1-R 5′-AAAGCCGGCAGATGGTCGTTTTTGG,
CDX2-F 5′-ACAGTCGCCTACATCACCATCCATCCATCCG,
CDX2-R 5′-CCTCTTTTTTTTTTTTGTTTTGCGGTTTTC,
T-F 5′-CTTCCAGAAGTCAAAGTCAAGTTCAAGTTTOO,
T-R 5′-TGAACTGTGTGTCAGGGAAGCA,
SOX17-F 5′-ACGCTTTTTTTTTTTGGGGCTAAG,
SOX17-R 5′-GTCAGCCGCCCCCCCCCCACTTTTTOO,
CDT1-F 5′-GGAGGTCAGATTACCCCAGCTCAC,
CDT1-R, 5′-TTGACGTGTGGGCCACCCCCCCAGTTTOO,
SOX2-F 5′-CTACAGCATGATGATGCAGGACCA,
SOX2 -R 5′-TCTGCGCGGAGCTGGTCATGGAGT,
PAX6-F 5′-AATCAGAGAAGACAGGCCA,
PAX6-R 5′-GTGGTAGGTATCATAACTC,
ACTB-F 5′-CACCATTGGCAATGAGCGGTTTTC,
ACTB-R 5′-AGGTCTTTTTGCGGATGTCCACGT.
Generazione di celle ARPE-19-iPS
Le cellule ARPE-19-iPS sono state derivate dalla linea cellulare epiteliale del pigmento retinico umano ARPE-19 riprogrammando con il kit di riprogrammazione CytoTune-iPS 2.0 Sendai (Invitrogen) seguendo le istruzioni del produttore.
In breve, due giorni prima della trasduzione del virus Sendai, 100.000 cellule ARPE-19 sono state placcate in un pozzetto di una piastra a 6 pozzetti con ATCC-formulato DMEM:F12 medio e sono stati trasdotti con il CytoTune 2.0 Sendai vettori riprogrammazione Sendai al MOI raccomandato dal produttore 48 ore dopo (d0). Il mezzo è stato sostituito con un mezzo fresco ogni due giorni a partire da un giorno dopo la trasduzione (d1). Al giorno 7, le cellule trasdotte sono state replicate su piastre a sei pozzetti rivestite di Matrigel. Le cellule sono state alimentate con mezzo Essential otto ogni giorno. Le colonie hanno iniziato a formarsi in 2-3 settimane ed erano pronte per il trasferimento dopo un’ulteriore settimana. Le colonie indifferenziate venivano raccolte manualmente e trasferite su piastre a sei pozzetti rivestite di Matrigel per l’espansione. Dopo due cicli di subclonazione ed espansione (dopo il passaggio 10), RT-PCR è stato utilizzato per verificare se le cellule iPS erano vettoriali con le sequenze di primer pubblicate nel manuale del produttore.
Dopo che le cellule iPS sono diventate libere da virus, sono state sottoposte al laboratorio citogenomico dell’Università del Minnesota per l’analisi del cariotipo. Questa analisi ha indicato che le cellule ARPE-19-iPS hanno cariotipi normali.
Caratterizzazione di immunofluorescenza delle cellule ARPE-19-iPS
Per esaminare i marcatori di pluripotenza, le cellule iPS sono state fissate con il 4% di paraformaldeide per 20 minuti. Se era necessaria una permeazione nucleare, le cellule sono state trattate con lo 0,2% di triton-x-100 in soluzione salina tamponata con fosfato (PBS) per 30 minuti, bloccate in albumina di siero bovino al 3% in PBS per 2 ore, e incubate con l’anticorpo primario durante la notte a 4˚C. Sono stati utilizzati anticorpi che hanno come bersaglio i seguenti antigeni: TRA1-60 (MAB4360, 1:400), TRA1-81 (MAB4381, 1:400), antigene embrionale stadio-specifico-4 (MAB4304, 1:200) e antigene embrionale specifico stadio-3 (MAB-4303, 1:200), tutti da Millipore/Chemicon (Billerica, MA), OCT3/4 (AB27985, 1:200) da Abcam (Cambridge, MA), e NANOG (EB068601:100) da Everest (Upper Heyford, Oxfordshire, UK). Le cellule sono state incubate con anticorpi secondari Alexa Fluor Series (tutti 1:500, Invitrogen) per 1 ora a temperatura ambiente e poi con DAPI per 10 min. Le immagini sono state esaminate con un microscopio confocale rovesciato Olympus FluoView 1000 m IX81 e analizzate con Adobe Photoshop CS6. L’attività della fosfatasi alcalina diretta (AP) è stata analizzata secondo le raccomandazioni del produttore (Millipore).
Bisolfito sequenziamento e analisi di metilazione
Il DNA genomico è stato isolato utilizzando il kit DNeasy Blood and Tissue (Qiagen, Germania) secondo le raccomandazioni del produttore per l’isolamento dalle cellule dei mammiferi. La conversione di Bisolfito è stata eseguita utilizzando il kit di Bisolfito Epitect (Qiagen) secondo il protocollo del produttore per basse quantità di DNA. Single-step PCR amplificazione delle regioni promotore NANOG e OCT4 sono stati condotti utilizzando Accuprime Supermix II (Invitrogen). I prodotti di amplificazione sono stati visualizzati mediante elettroforesi su gel e le bande sono state escisse e purificate utilizzando il kit QIAquick Gel Extraction (Qiagen). I prodotti PCR purificati sono stati inseriti nel vettore PCR4-TOPO (Invitrogen) e i singoli cloni sono stati sequenziati. L’allineamento e l’analisi di metilazione sono stati eseguiti utilizzando il programma online QUMA(http://quma.cdb.riken.jp/). I cloni sequenziati con almeno il 90% di conversione della citosina non CpG e almeno il 90% di omologia della sequenza sono stati mantenuti per l’analisi.
PCR quantitativa della trascrittasi inversa (Figura 1-figure supplement 2)
L’RNA è stato isolato utilizzando il kit RNeasy Mini (Qiagen) e trattato con TURBO DNA-free (Ambion, Austin, TX). Il cDNA del primo filamento è stato sintetizzato utilizzando un SuperMix di sintesi del primo filamento SuperMix Superscript III (Invitrogen). La trascrittasi inversa-PCR è stata eseguita utilizzando TaqMan Gene Expression Assays e TaqMan Universal PCR Master Mix, No AmpErase UNG (Applied Biosystems, Carslbad, CA) come da protocollo del produttore.
I test di espressione genica TaqMan utilizzati sono stati OCT4 (Hs04260367-gH), SOX2 (Hs01053049-sl), NANOG (Hs04399610-g1), KLF4 (Hs00358836-m1), MYC (Hs00153408-m1), LIN28 (Hs00702808-s1), REXO1 (Hs00810654-m1), ABCG2 (Hs1053790-m1), DNMT3 (Hs00171876-m1), con GAPDH (Hs9999999905-m1) utilizzato come controllo endogeno. I livelli di espressione sono stati misurati in duplicato. Per i geni con espressione al di sotto della soglia di fluorescenza, la soglia del ciclo (Ct) è stata impostata a 40 per calcolare l’espressione relativa. L’analisi è stata effettuata utilizzando un sistema di rilevamento della sequenza ABI PRISM 7500 (Applied Biosystems).
Analisi del teratoma
Le cellule ARPE-19-iPS contenute in una miscela di DMEM/F12, Matrigel e collagene sono state impiantate sul fianco posteriore dei topi NSG (n = 5) fino alla formazione di una massa palpabile. Il tessuto del teratoma è stato asportato per l’esame istologico dopo l’incorporazione e la colorazione con ematossilina ed eosina. Gli esperimenti sono stati condotti con l’approvazione del Comitato Istituzionale per la cura degli animali e la ricerca presso l’Università del Minnesota.
Analisi del tasso ergonomico
L’Analisi Ergodica del Tasso per i dati del ciclo cellulare si basava su lavori pubblicati in precedenza(Kafri et al., 2013). In primo luogo, i dati grezzi dei file di citometria a flusso sono stati estratti utilizzando FCSExtract Utility (Earl F Glynn) per i file con valore separato da virgole (.csv). I dati nei file .csv sono stati poi gated in FCS Express 6 (De Novo Software) e i dati per il solo G1-MCMDNAsono stati esportati. Le celle negative MCM sono state escluse in base al campione di controllo negativo (vedi Citometria a flusso). Il tasso medio di caricamento MCM è stato calcolato in MATLAB (MathWorks). Per calcolare il tasso medio di carico MCM, il G1-MCMDNA sono stati suddivisi in 10 contenitori di uguali dimensioni, con tasso calcolato per ogni contenitore, e tutti i 10 tassi sono stati mediati insieme per un tasso medio di carico MCM. Il calcolo del tasso è stato basato sulla formula di Kafri et al: wn=α2-Ffnwn = tasso dicarico MCM nel bin n
α = ln(2)/tempo di raddoppio (Figura 2-figuresupplement 1)
F = numero di G1-MCMDNAcellule/numero totale di cellule nel campione. F è stato calcolato da FCS Express ed è stato inserito manualmente in MATLAB(Figura 2-figure supplemento 1).
fn = numero di cellule nel bin n / numero di cellule G1-MCMDNA
I contenitori sono stati creati in MATLAB(Figura 2-figure supplement 1). Per controllare le piccole differenze giorno per giorno nei dati grezzi delle intensità di colorazione, i bordi dell’istogramma sono stati definiti con il primo bidone che inizia con il valore MCM più basso e l’ultimo bidone che termina con il valore MCM più alto, diviso in 10 bidoni di uguali dimensioni tra il valore MCM più basso e quello più alto. I 10 wn sono stati poi mediati per una media finale di w per campione. Codice MATLAB del campione:
alpha_iPSC = log(2)/15.64;F_iPSC_1 = 0.0801;%calcolare i valori MCM più bassi e più alti%maxMCM_iPSC_1 = max(iPSC_1(:,1));minMCM_iPSC_1 = min(iPSC_1(::,1));%creare l’istogramma con 10 bidoni e specificare i limiti di primo e ultimo bidone% h10_iPSC_1 = istogramma(iPSC_1(:,1), ‘NumBins’, 10, ‘BinLimits’, [minMCM_iPSC_1, maxMCM_iPSC_1]);%calcolare fn all’interno di ogni bin%totalf_iPSC_1 = dimensione(iPSC_1);fn10_iPSC_1=(h10_iPSC_1.Valori)/totalf_iPSC_1(1,1);%calcolare la media w% w10mean_iPSC_1 = media(alpha_iPSC_1. *(2> F_iPSC_1)./fn10_iPSC_1);
Il tasso medio di caricamento MCM è stato calcolato per tre repliche biologiche per ogni linea cellulare, e le repliche sono state mediate utilizzando GraphPad Prism per ulteriori analisi statistiche. Non possiamo utilizzare l’analisi del tasso ergodico su cellule che differenziano attivamente (ad esempio la figura 3) perché non sono allo stato stazionario.
Quantificazione e analisi statistica
L’analisi statistica è stata eseguita con GraphPad Prism seven utilizzando il test t non accoppiato a due code (visualizzato come media ±SD) o il test Mann-Whitney a due code come indicato nelle leggende delle figure. I livelli di significatività sono stati impostati a *p≤0,05, **p≤0,01, ***p≤0,001, ****p≤0,0001. Tutti gli esperimenti sono stati eseguiti almeno due volte e i dati rappresentativi sono mostrati in figure.
References
- Arias EE, Walter JC. Replication-dependent destruction of Cdt1 limits DNA replication to a single round per cell cycle in Xenopus egg extracts. Genes & Development. 2005; 19:114-126. DOI | PubMed
- Arias EE, Walter JC. Strength in numbers: preventing rereplication via multiple mechanisms in eukaryotic cells. Genes & Development. 2007; 21:497-518. DOI | PubMed
- Ballabeni A, Melixetian M, Zamponi R, Masiero L, Marinoni F, Helin K. Human geminin promotes pre-RC formation and DNA replication by stabilizing CDT1 in mitosis. The EMBO Journal. 2004; 23:3122-3132. DOI | PubMed
- Ballabeni A, Park IH, Zhao R, Wang W, Lerou PH, Daley GQ, Kirschner MW. Cell cycle adaptations of embryonic stem cells. PNAS. 2011; 108:19252-19257. DOI | PubMed
- Bernardo AS, Faial T, Gardner L, Niakan KK, Ortmann D, Senner CE, Callery EM, Trotter MW, Hemberger M, Smith JC, Bardwell L, Moffett A, Pedersen RA. BRACHYURY and CDX2 mediate BMP-induced differentiation of human and mouse pluripotent stem cells into embryonic and extraembryonic lineages. Cell Stem Cell. 2011; 9:144-155. DOI | PubMed
- Blow JJ, Ge XQ, Jackson DA. How dormant origins promote complete genome replication. Trends in Biochemical Sciences. 2011; 36:405-414. DOI | PubMed
- Bowers JL, Randell JC, Chen S, Bell SP. ATP hydrolysis by ORC catalyzes reiterative Mcm2-7 assembly at a defined origin of replication. Molecular Cell. 2004; 16:967-978. DOI | PubMed
- Calder A, Roth-Albin I, Bhatia S, Pilquil C, Lee JH, Bhatia M, Levadoux-Martin M, McNicol J, Russell J, Collins T, Draper JS. Lengthened G1 phase indicates differentiation status in human embryonic stem cells. Stem Cells and Development. 2013; 22:279-295. DOI | PubMed
- Carpenter AE, Jones TR, Lamprecht MR, Clarke C, Kang IH, Friman O, Guertin DA, Chang JH, Lindquist RA, Moffat J, Golland P, Sabatini DM. CellProfiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biology. 2006; 7:R100-R100.10. DOI | PubMed
- Chandrasekaran S, Tan TX, Hall JR, Cook JG. Stress-stimulated mitogen-activated protein kinases control the stability and activity of the Cdt1 DNA replication licensing factor. Molecular and Cellular Biology. 2011; 31:4405-4416. DOI | PubMed
- Chen T, Dent SY. Chromatin modifiers and remodellers: regulators of cellular differentiation. Nature Reviews Genetics. 2014; 15:93-106. DOI | PubMed
- Choi SC, Choi JH, Park CY, Ahn CM, Hong SJ, Lim DS. Nanog regulates molecules involved in stemness and cell cycle-signaling pathway for maintenance of pluripotency of P19 embryonal carcinoma stem cells. Journal of Cellular Physiology. 2012; 227:3678-3692. DOI | PubMed
- Clijsters L, Ogink J, Wolthuis R. The spindle checkpoint, APC/C(Cdc20), and APC/C(Cdh1) play distinct roles in connecting mitosis to S phase. The Journal of Cell Biology. 2013; 201:1013-1026. DOI | PubMed
- Cocker JH, Piatti S, Santocanale C, Nasmyth K, Diffley JF. An essential role for the Cdc6 protein in forming the pre-replicative complexes of budding yeast. Nature. 1996; 379:180-182. DOI | PubMed
- Coleman KE, Grant GD, Haggerty RA, Brantley K, Shibata E, Workman BD, Dutta A, Varma D, Purvis JE, Cook JG. Sequential replication-coupled destruction at G1/S ensures genome stability. Genes & Development. 2015; 29:1734-1746. DOI | PubMed
- Cook JG, Park CH, Burke TW, Leone G, DeGregori J, Engel A, Nevins JR. Analysis of Cdc6 function in the assembly of mammalian prereplication complexes. PNAS. 2002; 99:1347-1352. DOI | PubMed
- Coronado D, Godet M, Bourillot PY, Tapponnier Y, Bernat A, Petit M, Afanassieff M, Markossian S, Malashicheva A, Iacone R, Anastassiadis K, Savatier P. A short G1 phase is an intrinsic determinant of naïve embryonic stem cell pluripotency. Stem Cell Research. 2013; 10:118-131. DOI | PubMed
- Deegan TD, Diffley JF. MCM: one ring to rule them all. Current Opinion in Structural Biology. 2016; 37:145-151. DOI | PubMed
- Dimitrova DS, Prokhorova TA, Blow JJ, Todorov IT, Gilbert DM. Mammalian nuclei become licensed for DNA replication during late telophase. Journal of Cell Science. 2002; 115:51-59. PubMed
- Egozi D, Shapira M, Paor G, Ben-Izhak O, Skorecki K, Hershko DD. Regulation of the cell cycle inhibitor p27 and its ubiquitin ligase Skp2 in differentiation of human embryonic stem cells. The FASEB Journal. 2007; 21:2807-2817. DOI | PubMed
- Ekholm-Reed S, Méndez J, Tedesco D, Zetterberg A, Stillman B, Reed SI. Deregulation of cyclin E in human cells interferes with prereplication complex assembly. The Journal of Cell Biology. 2004; 165:789-800. DOI | PubMed
- Evrin C, Clarke P, Zech J, Lurz R, Sun J, Uhle S, Li H, Stillman B, Speck C. A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. PNAS. 2009; 106:20240-20245. DOI | PubMed
- Farrell JA, O’Farrell PH. From egg to gastrula: how the cell cycle is remodeled during the Drosophila mid-blastula transition. Annual Review of Genetics. 2014; 48:269-294. DOI | PubMed
- Filipczyk AA, Laslett AL, Mummery C, Pera MF. Differentiation is coupled to changes in the cell cycle regulatory apparatus of human embryonic stem cells. Stem Cell Research. 2007; 1:45-60. DOI | PubMed
- Ge XQ, Jackson DA, Blow JJ. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes & Development. 2007; 21:3331-3341. DOI | PubMed
- Ge XQ, Blow JJ. The licensing checkpoint opens up. Cell Cycle. 2009; 8:2320-2322. PubMed
- Gonzales KA, Liang H, Lim YS, Chan YS, Yeo JC, Tan CP, Gao B, Le B, Tan ZY, Low KY, Liou YC, Bard F, Ng HH. Deterministic Restriction on Pluripotent State Dissolution by Cell-Cycle Pathways. Cell. 2015; 162:564-579. DOI | PubMed
- Gray L, Griffeath D. The ergodic theory of traffic jams. Journal of Statistical Physics. 2001; 105:413-452. DOI
- Håland TW, Boye E, Stokke T, Grallert B, Syljuåsen RG. Simultaneous measurement of passage through the restriction point and MCM loading in single cells. Nucleic Acids Research. 2015; 43DOI | PubMed
- Hinds PW, Mittnacht S, Dulic V, Arnold A, Reed SI, Weinberg RA. Regulation of retinoblastoma protein functions by ectopic expression of human cyclins. Cell. 1992; 70:993-1006. DOI | PubMed
- Ibarra A, Schwob E, Méndez J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. PNAS. 2008; 105:8956-8961. DOI | PubMed
- Kafri R, Levy J, Ginzberg MB, Oh S, Lahav G, Kirschner MW. Dynamics extracted from fixed cells reveal feedback linking cell growth to cell cycle. Nature. 2013; 494:480-483. DOI | PubMed
- Kanai D, Ueda A, Akagi T, Yokota T, Koide H. Oct3/4 directly regulates expression of E2F3a in mouse embryonic stem cells. Biochemical and Biophysical Research Communications. 2015; 459:374-378. DOI | PubMed
- Kareta MS, Sage J, Wernig M. Crosstalk between stem cell and cell cycle machineries. Current Opinion in Cell Biology. 2015a; 37:68-74. DOI | PubMed
- Kareta MS, Gorges LL, Hafeez S, Benayoun BA, Marro S, Zmoos AF, Cecchini MJ, Spacek D, Batista LF, O’Brien M, Ng YH, Ang CE, Vaka D, Artandi SE, Dick FA, Brunet A, Sage J, Wernig M. Inhibition of pluripotency networks by the Rb tumor suppressor restricts reprogramming and tumorigenesis. Cell Stem Cell. 2015b; 16:39-50. DOI | PubMed
- Kermi C, Lo Furno E, Maiorano D. Regulation of DNA Replication in Early Embryonic Cleavages. Genes. 2017; 8DOI | PubMed
- Kimura H, Nozaki N, Sugimoto K. DNA polymerase alpha associated protein P1, a murine homolog of yeast MCM3, changes its intranuclear distribution during the DNA synthetic period. The EMBO journal. 1994; 13:4311-4320. PubMed
- Kinjyo I, Qin J, Tan SY, Wellard CJ, Mrass P, Ritchie W, Doi A, Cavanagh LL, Tomura M, Sakaue-Sawano A, Kanagawa O, Miyawaki A, Hodgkin PD, Weninger W. Real-time tracking of cell cycle progression during CD8+ effector and memory T-cell differentiation. Nature Communications. 2015; 6DOI | PubMed
- Kuipers MA, Stasevich TJ, Sasaki T, Wilson KA, Hazelwood KL, McNally JG, Davidson MW, Gilbert DM. Highly stable loading of Mcm proteins onto chromatin in living cells requires replication to unload. The Journal of Cell Biology. 2011; 192:29-41. DOI | PubMed
- Lee J, Go Y, Kang I, Han YM, Kim J. Oct-4 controls cell-cycle progression of embryonic stem cells. Biochemical Journal. 2010; 426:171-181. DOI | PubMed
- Li H, Collado M, Villasante A, Matheu A, Lynch CJ, Cañamero M, Rizzoti K, Carneiro C, Martínez G, Vidal A, Lovell-Badge R, Serrano M. p27(Kip1) directly represses Sox2 during embryonic stem cell differentiation. Cell Stem Cell. 2012; 11:845-852. DOI | PubMed
- Liu L, Michowski W, Inuzuka H, Shimizu K, Nihira NT, Chick JM, Li N, Geng Y, Meng AY, Ordureau A, Kołodziejczyk A, Ligon KL, Bronson RT, Polyak K, Harper JW, Gygi SP, Wei W, Sicinski P. G1 cyclins link proliferation, pluripotency and differentiation of embryonic stem cells. Nature Cell Biology. 2017; 19:177-188. DOI | PubMed
- Mailand N, Diffley JF. CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell. 2005; 122:915-926. DOI | PubMed
- McGarry TJ, Kirschner MW. Geminin, an inhibitor of DNA replication, is degraded during mitosis. Cell. 1998; 93:1043-1053. DOI | PubMed
- McIntosh D, Blow JJ. Dormant origins, the licensing checkpoint, and the response to replicative stresses. Cold Spring Harbor Perspectives in Biology. 2012; 4:a012955. DOI | PubMed
- Meerbrey KL, Hu G, Kessler JD, Roarty K, Li MZ, Fang JE, Herschkowitz JI, Burrows AE, Ciccia A, Sun T, Schmitt EM, Bernardi RJ, Fu X, Bland CS, Cooper TA, Schiff R, Rosen JM, Westbrook TF, Elledge SJ. The pINDUCER lentiviral toolkit for inducible RNA interference in vitro and in vivo. Proceedings of the National Academy of Sciences. 2011; 108:3665-3670. DOI | PubMed
- Méndez J, Stillman B. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Molecular and Cellular Biology. 2000; 20:8602-8612. DOI | PubMed
- Moreno A, Carrington JT, Albergante L, Al Mamun M, Haagensen EJ, Komseli ES, Gorgoulis VG, Newman TJ, Blow JJ. Unreplicated DNA remaining from unperturbed S phases passes through mitosis for resolution in daughter cells. PNAS. 2016; 113:E5757-E5764. DOI | PubMed
- Neganova I, Zhang X, Atkinson S, Lako M. Expression and functional analysis of G1 to S regulatory components reveals an important role for CDK2 in cell cycle regulation in human embryonic stem cells. Oncogene. 2009; 28:20-30. DOI | PubMed
- Nevis KR, Cordeiro-Stone M, Cook JG. Origin licensing and p53 status regulate Cdk2 activity during G(1). Cell Cycle. 2009; 8:1952-1963. DOI | PubMed
- Nishitani H, Lygerou Z, Nishimoto T, Nurse P. The Cdt1 protein is required to license DNA for replication in fission yeast. Nature. 2000; 404:625-628. DOI | PubMed
- Niwa H, Toyooka Y, Shimosato D, Strumpf D, Takahashi K, Yagi R, Rossant J. Interaction between Oct3/4 and Cdx2 determines trophectoderm differentiation. Cell. 2005; 123:917-929. DOI | PubMed
- O’Farrell PH, Stumpff J, Tin Su T, Su TT. Embryonic cleavage cycles: How is a mouse like a fly?. Current Biology. 2004; 14:R35-R45. DOI | PubMed
- Pauklin S, Madrigal P, Bertero A, Vallier L. Initiation of stem cell differentiation involves cell cycle-dependent regulation of developmental genes by Cyclin D. Genes & Development. 2016; 30:421-433. DOI | PubMed
- Pauklin S, Vallier L. The cell-cycle state of stem cells determines cell fate propensity. Cell. 2013; 155:135-147. DOI | PubMed
- Petersen BO, Wagener C, Marinoni F, Kramer ER, Melixetian M, Lazzerini Denchi E, Gieffers C, Matteucci C, Peters JM, Helin K. Cell cycle- and cell growth-regulated proteolysis of mammalian CDC6 is dependent on APC-CDH1. Genes & Development. 2000; 14:2330-2343. DOI | PubMed
- Pozo P, Cook J. Regulation and function of cdt1; a key factor in cell proliferation and genome stability. Genes. 2016; 8DOI
- Pruitt SC, Bailey KJ, Freeland A. Reduced Mcm2 expression results in severe stem/progenitor cell deficiency and cancer. Stem Cells. 2007; 25:3121-3132. DOI | PubMed
- Randell JC, Bowers JL, Rodríguez HK, Bell SP. Sequential ATP hydrolysis by Cdc6 and ORC directs loading of the Mcm2-7 helicase. Molecular Cell. 2006; 21:29-39. DOI | PubMed
- Remus D, Beuron F, Tolun G, Griffith JD, Morris EP, Diffley JF. Concerted loading of Mcm2-7 double hexamers around DNA during DNA replication origin licensing. Cell. 2009; 139:719-730. DOI | PubMed
- Remus D, Diffley JF. Eukaryotic DNA replication control: lock and load, then fire. Current Opinion in Cell Biology. 2009; 21:771-777. DOI | PubMed
- Resnitzky D, Gossen M, Bujard H, Reed SI. Acceleration of the G1/S phase transition by expression of cyclins D1 and E with an inducible system. Molecular and Cellular Biology. 1994; 14:1669-1679. DOI | PubMed
- Rizzardi LF, Coleman KE, Varma D, Matson JP, Oh S, Cook JG. CDK1-dependent inhibition of the E3 ubiquitin ligase CRL4CDT2 ensures robust transition from S Phase to Mitosis. Journal of Biological Chemistry. 2015; 290:556-567. DOI | PubMed
- Robinson M, Chapani P, Styan T, Vaidyanathan R, Willerth SM. Functionalizing ascl1 with novel intracellular protein delivery technology for promoting neuronal differentiation of human induced pluripotent stem cells. Stem Cell Reviews and Reports. 2016; 12:476-483. DOI | PubMed
- Scognamiglio R, Cabezas-Wallscheid N, Thier MC, Altamura S, Reyes A, Prendergast ÁM, Baumgärtner D, Carnevalli LS, Atzberger A, Haas S, von Paleske L, Boroviak T, Wörsdörfer P, Essers MA, Kloz U, Eisenman RN, Edenhofer F, Bertone P, Huber W, van der Hoeven F, Smith A, Trumpp A. Myc depletion induces a pluripotent dormant state mimicking diapause. Cell. 2016; 164:668-680. DOI | PubMed
- Sela Y, Molotski N, Golan S, Itskovitz-Eldor J, Soen Y. Human embryonic stem cells exhibit increased propensity to differentiate during the G1 phase prior to phosphorylation of retinoblastoma protein. Stem Cells. 2012; 30:1097-1108. DOI | PubMed
- Shima N, Alcaraz A, Liachko I, Buske TR, Andrews CA, Munroe RJ, Hartford SA, Tye BK, Schimenti JC. A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nature Genetics. 2007; 39:93-98. DOI | PubMed
- Shreeram S, Sparks A, Lane DP, Blow JJ. Cell type-specific responses of human cells to inhibition of replication licensing. Oncogene. 2002; 21:6624-6632. DOI | PubMed
- Siddiqui K, On KF, Diffley JF. Regulating DNA replication in eukarya. Cold Spring Harbor Perspectives in Biology. 2013; 5:a012930. DOI | PubMed
- Soufi A, Dalton S. Cycling through developmental decisions: how cell cycle dynamics control pluripotency, differentiation and reprogramming. Development. 2016; 143:4301-4311. DOI | PubMed
- Sugimoto N, Tatsumi Y, Tsurumi T, Matsukage A, Kiyono T, Nishitani H, Fujita M. Cdt1 phosphorylation by cyclin A-dependent kinases negatively regulates its function without affecting geminin binding. Journal of Biological Chemistry. 2004; 279:19691-19697. DOI | PubMed
- Symeonidou IE, Kotsantis P, Roukos V, Rapsomaniki MA, Grecco HE, Bastiaens P, Taraviras S, Lygerou Z. Multi-step loading of human minichromosome maintenance proteins in live human cells. Journal of Biological Chemistry. 2013; 288:35852-35867. DOI | PubMed
- Tanaka S, Diffley JF. Deregulated G1-cyclin expression induces genomic instability by preventing efficient pre-RC formation. Genes & Development. 2002; 16:2639-2649. DOI | PubMed
- Teer JK, Machida YJ, Labit H, Novac O, Hyrien O, Marheineke K, Zannis-Hadjopoulos M, Dutta A. Proliferating human cells hypomorphic for origin recognition complex 2 and pre-replicative complex formation have a defect in p53 activation and Cdk2 kinase activation. Journal of Biological Chemistry. 2006; 281:6253-6260. DOI | PubMed
- Ticau S, Friedman LJ, Ivica NA, Gelles J, Bell SP. Single-molecule studies of origin licensing reveal mechanisms ensuring bidirectional helicase loading. Cell. 2015; 161:513-525. DOI | PubMed
- Todorov IT, Attaran A, Kearsey SE. BM28, a human member of the MCM2-3-5 family, is displaced from chromatin during DNA replication. The Journal of Cell Biology. 1995; 129:1433-1445. DOI | PubMed
- Truong LN, Wu X. Prevention of DNA re-replication in eukaryotic cells. Journal of Molecular Cell Biology. 2011; 3:13-22. DOI | PubMed
- Tsunematsu T, Takihara Y, Ishimaru N, Pagano M, Takata T, Kudo Y. Aurora-A controls pre-replicative complex assembly and DNA replication by stabilizing geminin in mitosis. Nature Communications. 2013; 4:1-11. DOI
- Varma D, Chandrasekaran S, Sundin LJ, Reidy KT, Wan X, Chasse DA, Nevis KR, DeLuca JG, Salmon ED, Cook JG. Recruitment of the human Cdt1 replication licensing protein by the loop domain of Hec1 is required for stable kinetochore-microtubule attachment. Nature Cell Biology. 2012; 14:593-603. DOI | PubMed
- Watanabe K, Ueno M, Kamiya D, Nishiyama A, Matsumura M, Wataya T, Takahashi JB, Nishikawa S, Nishikawa S, Muguruma K, Sasai Y. A ROCK inhibitor permits survival of dissociated human embryonic stem cells. Nature Biotechnology. 2007; 25:681-686. DOI | PubMed
- Woodward AM, Göhler T, Luciani MG, Oehlmann M, Ge X, Gartner A, Jackson DA, Blow JJ. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. The Journal of Cell Biology. 2006; 173:673-683. DOI | PubMed
Fonte
Matson JP, Dumitru R, Coryell P, Baxley RM, Chen W, et al. () Rapid DNA replication origin licensing protects stem cell pluripotency. eLife 6e30473. https://doi.org/10.7554/eLife.30473




