
Abstract
Introduzione
Uno dei principali obiettivi della ricerca sulla genetica umana è quello di comprendere i contributi genetici a malattie complesse, in particolare i meccanismi molecolari attraverso i quali le varianti comuni del DNA hanno un impatto sull’eziologia della malattia. La maggior parte degli studi di associazione a livello genomico (GWAS) implica varianti non codificanti che sono lontane dai geni, complicando l’interpretazione della loro modalità d’azione e la corretta identificazione del gene target(Maurano et al., 2012). Le prove di montaggio suggeriscono che le varianti di malattia disturbano la funzione degli elementi regolatori che agiscono sul cis, come gli stimolatori, che a loro volta influenzano l’espressione del gene o dei geni specifici che sono bersagli funzionali di questi elementi(Wright et al., 2010; Musunuru et al., 2010; Cowper-Sal lari et al., 2012; Smemo et al., 2014; Claussnitzer et al., 2015). Tuttavia, poiché gli elementi regolatori ad azione cispossono essere localizzati a distanza di kilobase (kb) dai loro geni target, l’identificazione dei veri obiettivi funzionali degli elementi regolatori rimane una sfida(Smemo et al., 2014).
Le tecniche di cattura della conformazione del cromosoma come Hi-C(Lieberman-Aiden et al., 2009) consentono la mappatura a livello genomico dei contatti a lungo raggio della cromatina e rappresentano quindi una strategia promettente per identificare i bersagli genici distali di varianti genetiche associate alla malattia. Recentemente, le mappe Hi-C sono state generate in numerosi tipi di cellule umane, tra cui le cellule staminali embrionali e i primi lignaggi embrionali(Dixon et al., 2012, 2015), le cellule immunitarie (Rao et al., 2014), i fibroblasti(Jin et al., 2013) e altri tipi di tessuto primario(Schmitt et al., 2016). Tuttavia, nonostante la crescente abbondanza di mappe Hi-C, la maggior parte dei set di dati sono di risoluzione limitata (>40 kb) e non identificano con precisione le regioni genomiche a contatto con i promotori genici.
Più recentemente, è stata sviluppata la cattura dei promotori Hi-C (PCHi-C) che aumenta notevolmente il potere di rilevare le interazioni che coinvolgono le sequenze dei promotori(Schoenfelder et al., 2015; Mifsud et al., 2015). Il PCHi-C in diversi tipi di cellule ha identificato migliaia di contatti tra i promotori e ha rivelato ampie differenze nell’architettura del promotore tra i vari tipi di cellule e nella differenziazione(Schoenfelder et al., 2015; Mifsud et al., 2015; Javierre et al., 2016; Freire-Pritchett et al., 2017; Rubin et al., 2017; Siersbæk et al., 2017). Questi studi hanno dimostrato collettivamente che l’architettura del genoma riflette l’identità delle cellule, suggerendo che i tipi di cellule rilevanti per la malattia sono fondamentali per il successo dell’interrogazione dei meccanismi di regolazione genica dei loci della malattia.
A sostegno di questa idea, diversi studi recenti hanno utilizzato mappe di interazione ad alta risoluzione del promotore per identificare i geni bersaglio specifici dei tessuti delle associazioni GWAS. Javierre et al. hanno generato dati di cattura del promotore Hi-C in 17 tipi di cellule primarie del sangue umano e hanno identificato 2604 geni potenzialmente causali per i disturbi del sistema immunitario e del sangue, compresi molti geni con ruoli non previsti in queste malattie(Javierre et al., 2016). Allo stesso modo, Mumbach et al. hanno interrogato i GWAS SNPs associati a malattie autoimmuni utilizzando HiChIP dove hanno identificato ~10.000 interazioni promotore-alimentatore che collegavano diverse centinaia di SNPs ai geni bersaglio, la maggior parte dei quali non erano il gene più vicino (Mumbachet al., 2017). È importante notare che entrambi gli studi hanno riportato la specificità del tipo di cellula delle interazioni tra geni target SNP e SNP.
Le malattie cardiovascolari, tra cui l’aritmia cardiaca, l’insufficienza cardiaca e l’infarto del miocardio, continuano ad essere la principale causa di morte in tutto il mondo. Oltre 50 GWAS sono stati condotti solo per questi specifici fenotipi cardiovascolari, con più di 500 loci implicati nel rischio di malattie cardiovascolari (catalogo NHGRI GWAS, https://www.ebi.ac.uk/gwas/), la maggior parte dei quali mappano le regioni genomiche non codificanti. Per iniziare a sezionare i meccanismi molecolari con cui le varianti genetiche contribuiscono al rischio di CVD, è necessaria una mappa completa di regolazione genica delle cellule cardiache umane. Qui, vi presentiamo le mappe ad alta risoluzione di interazione del promotore ad alta risoluzione di iPSC umani e cardiomiociti derivati da iPSC (CMs). Utilizzando PCHi-C, abbiamo identificato centinaia di migliaia di interazioni del promotore in ogni tipo di cellula. Dimostriamo la rilevanza fisiologica di questi set di dati interrogando funzionalmente la relazione tra l’espressione genica e le interazioni a lungo raggio del promotore, e dimostriamo l’utilità dei dati di interazione a lungo raggio della cromatina per risolvere i target funzionali dei loci associati alla malattia.
Risultati
I cardiomiociti derivati dall’iPSC forniscono un modello efficace per studiare l’architettura della genetica CVD
Abbiamo usato le CMs derivate da iPSC(Burridge et al., 2014) come modello per studiare la regolazione genica cardiovascolare e la genetica delle malattie. I CMs generati in questo studio erano puri all’86-94% sulla base dell’espressione della proteina T della troponina cardiaca ed esibivano un battito spontaneo e uniforme (Figura 1-figure supplement 1A, Video 1) . Per dimostrare che iPSC e CMs ricapitolare i profili trascrizionali ed epigenetici di cellule primarie abbinate, abbiamo condotto RNA-seq e ChIP-seq per il marchio attivo enhancer H3K27ac in entrambi i tipi di cellule e confrontato questi dati con i tipi di cellule simili dal progetto Epigenome Roadmap Project(Kundaje et al., 2015). RNA-seq profili di iPSCs clusterizzati strettamente con cellule staminali embrionali H1, mentre CMs clusterizzati con entrambi i ventricolo sinistro (LV) e il cuore fetale (FH) profili(Figura 1-figure supplement 1B). Inoltre, abbiamo osservato che i tipi di cellule abbinate hanno mostrato una sovrapposizione tre volte maggiore nel numero di promotori-distale H3K27ac ChIP-seq picchi rispetto ai tipi di cellule non abbinate(Figura 1-figure supplement 1C,D), indicando che sia iPSC e CMs ricapitolare gli stati epigenetici specifici dei tessuti delle cellule staminali umane e cardiomiociti primari, rispettivamente.
Per convalidare ulteriormente il nostro sistema, abbiamo analizzato i geni espressi in modo differenziato tra iPSC e CMs. Tra il top 10% dei geni sovra-espressi nelle CMs erano geni direttamente correlati alla funzione cardiaca, compresi i fattori essenziali di trascrizione cardiaca(GATA4, MEIS1, TBX5 e TBX20) e prodotti di differenziazione(TNNT2, MYH7B, MYL7, ACTN2, NPPA, HCN4 e RYR2) (fold-change >1.5,Padj<0,05, Figura 1-figure supplement 2A-C). Gene Ontologia (GO) l’analisi di arricchimento per i geni sovra-espressi in CMs rispetto a iPSCs ulteriormente confermato i fenotipi cardiaci specifici di queste cellule con termini top relativi allo sviluppo del sistema di conduzione cardiaca e la contrazione delle cellule del muscolo cardiaco(Figura 1-figure supplement 2D).
Promoter-capture Hi-C identifica gli elementi regolatori distali in iPSC e CMs
Per mappare in modo completo gli elementi regolatori a lungo raggio in iPSC e CM, abbiamo eseguito Hi-C in situ(Rao et al., 2014) in triplice differenziazione iPSC-CM; soprattutto, abbiamo usato l’enzima di restrizione MboI a quattro taglienti che genera frammenti di legatura con una dimensione media di 422 bp, consentendo una risoluzione a livello di potenziatore dei contatti del promotore. Abbiamo arricchito le librerie iPSC e CM in situ Hi-C per le interazioni del promotore attraverso l’ibridazione con un set di 77.476 sonde di RNA biotinilato (“esche”) che mirano a 22.600 promotori umani RefSeq protein-coding (vedi Materiali e metodi) e sequenziato ogni libreria ad una profondità media di ~ 413 milioni (M) paired-end leggi. Dopo aver rimosso i duplicati e le coppie di lettura che non hanno mappato su un’esca, abbiamo ottenuto una media di 31M e 41M coppie di lettura per ogni replicato per iPSC e CM, rispettivamente. Abbiamo usato CHiCAGO(Cairns et al., 2016), una pipeline di calcolo che tiene conto dei bias dalla cattura della sequenza, per identificare le interazioni significative e ulteriormente filtrate per quelle significative in almeno due dei tre replicati (vedi Materiali e metodi). Infine, ci siamo concentrati esclusivamente sulle interazioni che sono state separate da una distanza di almeno 10 kb. Questo criterio affronta l’alta frequenza di evet di legatura di prossimità nei dati Hi-C, che sono difficili da distinguere come contatti browniani casuali o interazioni funzionali della cromatina(Cairns et al., 2016). In totale, abbiamo identificato 350.062 interazioni del promotore in iPSC e 401.098 in CM. Una grande proporzione (~ 55%) delle interazioni sono state condivise tra i due tipi di cellule, indicando che anche ad alta risoluzione molte interazioni a lungo raggio sono stabili tra i tipi di cellule(Figura 1A). Circa il 20% di tutte le interazioni sono state tra due promotori, dimostrando l’elevata connettività tra i geni e sostenendo il ruolo recentemente suggerito dei promotori che agiscono come input di regolamentazione per i geni distali(Dao et al., 2017; Diao et al., 2017)(Figura 1B). La maggior parte delle interazioni sono state promotore-distale, con una mediana di ~170 kb tra il promotore e la regione distale che interagisce (Figura 1C).
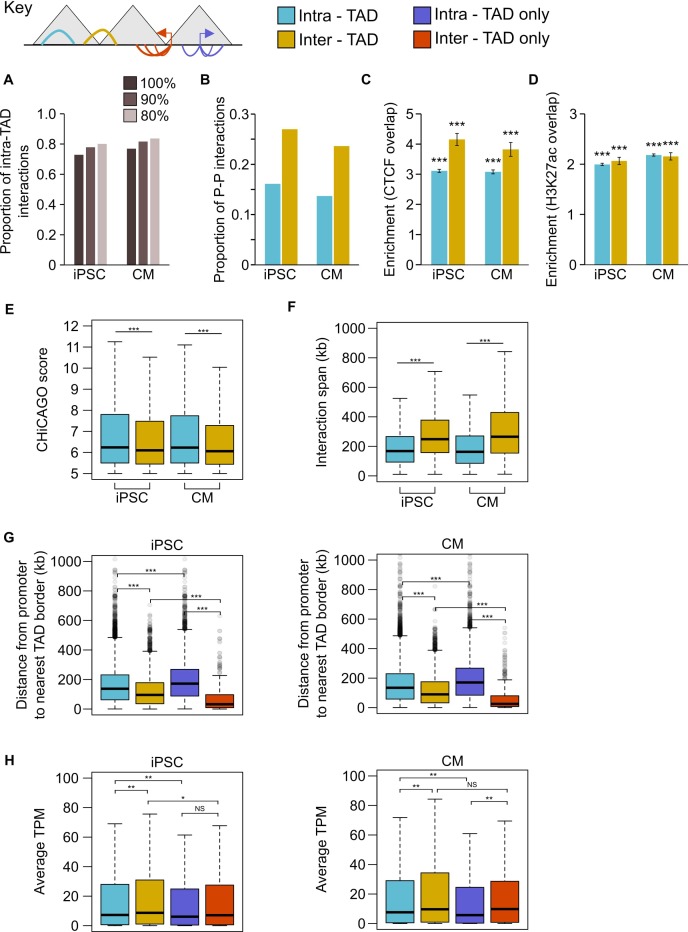
Figura 1-figure supplement 3.Caratteristiche generali delle interazioni del promotore.Controllo di qualità di iPSC-CMs.Analisi dell’RNA-seq in iPSC e iPSC-CMs.Analisi delle interazioni PCHi-C nel contesto dei TAD.(A) Diagramma di Venn che mostra il numero di interazioni del promotore specifiche per tipo di cella e condivise in ogni tipo di cella.(B) Proporzione delle interazioni in ogni categoria di distanza: promotore (P)-promoter (entrambe le estremità che interagiscono si sovrappongono ad un sito di inizio trascrizione (TSS)); P-prossimale (l’estremità non promotrice si sovrappone alla regione catturata ma non al TSS); P-distale (l’estremità non promotrice è al di fuori della regione catturata). Si noti che tutte le interazioni del promotore sono separate da almeno 10 kb.(C) Distribuzione delle distanze che coprono ogni interazione in iPSC e CM. La linea rossa rappresenta la mediana (170 kb in iPSCs, 164 kb in CMs); la linea nera rappresenta la media (208 kb in iPSCs, 206 kb in CMs).(D) Una regione di ~ 2 Mb del cromosoma 8 che comprende il gene GATA4 è mostrata insieme alla pre-captazione (genoma intero) Hi-C mappe di interazione Hi-C a 40 kb di risoluzione per iPSC (in alto) e CMs (in basso). TAD chiamati con TopDom sono mostrati come barre colorate (dimensione mediana TAD = 640 kb in entrambi i tipi di cellule, dimensione media TAD = 742 kb in iPSCs e 743 kb in CMs) e significative interazioni PCHi-C come archi colorati.(E) Vista ingrandita del locus GATA4 (promotore evidenziato in giallo) in iPSCs (in alto) e CMs (in basso) insieme ai corrispondenti dati RNA-seq generati come parte di questo studio, e dati ChIP-seq per H3K27ac, H3K4me1, H3K27me3 e CTCF dal progetto Epigenome Roadmap Project/ENCODE (H1 e ventricolo sinistro per iPSC e CM, rispettivamente). I conteggi di lettura GATA4 filtrati utilizzati da CHiCAGO sono visualizzati in blu con le corrispondenti interazioni significative mostrate sotto forma di archi. Per chiarezza, vengono mostrate solo le interazioni GATA4. Le regioni evidenziate in grigio mostrano le interazioni che si sovrappongono agli stimolatori cardiaci convalidati in vivo (scatole rosa), con embrioni rappresentativi E11,5 per ogni elemento stimolatore(Visel et al., 2007). La punta della freccia rossa indica il cuore.(A) Citometria a flusso di cardiomiociti derivati da iPSC. Immagine rappresentativa dei dati di flusso per i cardiomiociti (a sinistra) e percentuale di troponina cardiaca T (cTnT) positiva per ogni differenziazione (a destra). Le cellule sono state prima gated sul vivo / morto e poi sulla colorazione cTnT.(B) Analisi dei componenti principali dei dati RNA-seq in iPSC e CMs insieme a cellule staminali embrionali H1, cellule ventricolari sinistre (LV), cellule cardiache fetali (FH), e cellule linfoblastoidi linea cellulare (LCL). LCLs cluster indipendentemente da iPSC e CM, indicando che iPSC sono stati riprogrammati fedelmente.(C) Percentuale di Epigenoma Roadmap H3K27ac ChIP-seq picchi sovrapposti iPSC e CM H3K27ac. Sono mostrati i picchi sovrapposti per tutti i picchi e solo i picchi non promoter. LV, ventricolo sinistro; H1, H1 linea di cellule staminali embrionali.(D) Tre scatti del browser del genoma che mostrano il paesaggio epigenetico in CMs rispetto al ventricolo sinistro, atri destro, fegato adulto e ippocampo cerebrale della tabella di marcia dell’epigenoma.(A) Cluster analysis of RNA-seq data from each triplicate of iPSC and CM.(B) Numero di geni espressi in modo differenziato in ogni tipo di cellula.(C) Geni selezionati sovraesposti in CMs rispetto agli iPSC. (D) Gene Ontologia analisi di arricchimento dei processi biologici associati ai 4802 geni sovraespressi in cardiomiociti.In questa analisi, le interazioni sono state classificate come intra-TAD (entrambe le estremità dell’interazione completamente all’interno di un singolo TAD) o inter-TAD (ogni estremità dell’interazione è in un TAD diverso). Sono state omesse le interazioni che rientrano parzialmente o totalmente nei “confini” o “vuoti” della TAD come definiti dal TopDom (vedi Materiali e metodi).(A) Percentuale di interazioni che sono intra-TAD a diversi cut-off. Tutte le analisi hanno utilizzato interazioni che erano al 100% all’interno di un TAD.(B) Percentuale delle interazioni promotore-promotori nell’insieme delle interazioni intra-TAD e inter-TAD.(C,D) Arricchimento delle interazioni intra-TAD e interTAD per sovrapporre i picchi CTCF(C) o H3K27ac (D). Sono stati analizzati solo i picchi ChIP-seq del promotore-distale. ***p<2,2 × 10-16, test Z.(E) Punteggio CHiCAGO e(F) intervallo di interazione delle interazioni intra- e inter-TAD. ***p<2.2 × 10-16, Wilcoxon rank-sum test.(G,H) Considerare i promotori con un’interazione intra-TAD, un’interazione inter-TAD, o esclusivamente interazioni intra-TAD o inter-TAD:(G) distanza dal promotore TSS al più vicino confine TAD e(H) valore medio TPM del promotore. ***p<2,2 × 10-16, **p<0,01, *p<0,05, NS= non significativo, test del grado Wilcoxon.

Video 1.Video di cardiomiociti di derivazione iPSC che mostrano un battito spontaneo al giorno 20 della differenziazione (giorno di raccolta delle cellule).
Per confrontare le mappe PCHi-C con le caratteristiche note dell’organizzazione del genoma, abbiamo sequenziato le nostre librerie Hi-C di pre-cattura ad una profondità media di 665M di lettura per tipo di cellula e abbiamo identificato i domini topologicamente associati (TAD) con TopDom (vedi Materiali e metodi). I TAD sono unità organizzative di cromosomi definiti da blocchi genomici <1 megabase (Mb) che mostrano alte frequenze autointerattive con una frequenza di interazione molto bassa attraverso i confini del TAD (Dixon etal., 2012; Nora et al., 2012). In particolare, si ritiene che questa organizzazione vincoli l’attività degli elementi di regolazione cis per indirizzare i geni all’interno dello stesso TAD, poiché è stato dimostrato che l’interruzione dei confini del TAD porta all’attivazione aberrante dei geni nei TAD vicini(Nora et al., 2012; Lupiáñez et al., 2015; Franke et al., 2016; Symmons et al., 2016; Tsujimura et al., 2015 ). Abbiamo trovato che la maggior parte delle interazioni PCHi-C si è verificata all’interno di TAD (73 e 77% in iPSC e CM, rispettivamente; Figura 1D e Figura 1-figure supplement 3A). Le interazioni TAD-crossing (“inter-TAD”) contenevano proporzionalmente più interazioni promotore-promotori rispetto alle interazioni intra-TAD, ed erano più suscettibili di sovrapporsi ai siti CTCF distali promotore-distali; tuttavia, erano analogamente arricchite per il looping ai siti distali H3K27ac, un marchio di cromatina attiva (Figura 1-figuresupplement 3B-D). Le interazioni inter-TAD avevano punteggi CHiCAGO leggermente più bassi, che riflettono un numero inferiore di letture a supporto di queste interazioni, e si estendevano a distanze genomiche maggiori rispetto alle interazioni intra-TAD(Figura 1-figure supplement 3E,F). Inoltre, i promotori con interazioni inter-TAD erano preferibilmente localizzati vicino ai confini del TAD(Figura 1-figure supplement 3G) e avevano livelli di espressione più elevati rispetto ai promotori con interazioni intra-TAD, in particolare nei CM (Figura 1-figure supplement 3H). Queste osservazioni sono coerenti con studi precedenti che hanno dimostrato che i geni altamente espressi, in particolare i geni domestici, sono arricchiti ai confini del TAD(Dixon et al., 2012).
Per illustrare l’utilità delle mappe di interazione PCHi-C ad alta risoluzione, si evidenzia il locus GATA4 nella Figura 1D e E. GATA4 è un regolatore principale dello sviluppo del cuore(Watt et al., 2004; Pikkarainen et al., 2004) e il gene GATA4 si trova in una struttura TAD che è relativamente stabile tra iPSC e CM(Figura 1D). Tuttavia, PCHi-C ha identificato un aumento delle frequenze di interazione tra il promotore GATA4 e diverse regioni contrassegnate da H3K27ac, tra cui quattro stimolatori cardiaci in vivo convalidati dal browser Vista enhancer(Visel et al., 2007), in particolare in CMs e in coincidenza con una forte up-regolazione di GATA4(Figura 1-figure supplement 2C). Anche se le analisi basate su TAD aiutano a definire il panorama normativo cis di un gene, i dati ad alta risoluzione dell’interazione del promotore forniscono la risoluzione necessaria per mappare con precisione le interazioni tra il promotore e l’evidenziatore nel contesto della differenziazione cellulare.
Per convalidare la mappa di interazione CM come risorsa per la genetica delle malattie cardiovascolari abbiamo poi ampiamente caratterizzato diversi aspetti importanti dell’architettura genetica in CMs. Abbiamo confrontato le CM con le iPSC in ogni analisi come misura della specificità del tipo di cellula. Queste analisi servono come parametri di riferimento che si basano su caratteristiche consolidate dell’organizzazione del genoma e aiutano le interpretazioni dei ruoli che le interazioni a lungo raggio svolgono nella regolazione dei geni.
Figura 1-figure supplement 3.Caratteristiche generali delle interazioni del promotore.Controllo di qualità delle iPSC-CMs.Analisi dell’RNA-seq nelle iPSC e nelle iPSC-CMs.Analisi delle interazioni PCHi-C nel contesto delle TAD.(A) Diagramma di Venn che mostra il numero di interazioni del promotore specifiche per tipo di cella e condivise in ogni tipo di cella.(B) Proporzione delle interazioni in ogni categoria di distanza: promotore (P)-promoter (entrambe le estremità che interagiscono si sovrappongono ad un sito di inizio trascrizione (TSS)); P-prossimale (l’estremità non promotrice si sovrappone alla regione catturata ma non al TSS); P-distale (l’estremità non promotrice è al di fuori della regione catturata). Si noti che tutte le interazioni del promotore sono separate da almeno 10 kb.(C) Distribuzione delle distanze che coprono ogni interazione in iPSC e CM. La linea rossa rappresenta la mediana (170 kb in iPSCs, 164 kb in CMs); la linea nera rappresenta la media (208 kb in iPSCs, 206 kb in CMs).(D) Una regione di ~ 2 Mb del cromosoma 8 che comprende il gene GATA4 è mostrata insieme alla pre-captazione (genoma intero) Hi-C mappe di interazione Hi-C a 40 kb di risoluzione per iPSC (in alto) e CMs (in basso). TAD chiamati con TopDom sono mostrati come barre colorate (dimensione mediana TAD = 640 kb in entrambi i tipi di cellule, dimensione media TAD = 742 kb in iPSCs e 743 kb in CMs) e significative interazioni PCHi-C come archi colorati.(E) Vista ingrandita del locus GATA4 (promotore evidenziato in giallo) in iPSCs (in alto) e CMs (in basso) insieme ai corrispondenti dati RNA-seq generati come parte di questo studio, e dati ChIP-seq per H3K27ac, H3K4me1, H3K27me3 e CTCF dal progetto Epigenome Roadmap Project/ENCODE (H1 e ventricolo sinistro per iPSC e CM, rispettivamente). I conteggi di lettura GATA4 filtrati utilizzati da CHiCAGO sono visualizzati in blu con le corrispondenti interazioni significative mostrate sotto forma di archi. Per chiarezza, vengono mostrate solo le interazioni GATA4. Le regioni evidenziate in grigio mostrano le interazioni che si sovrappongono agli stimolatori cardiaci convalidati in vivo (scatole rosa), con embrioni rappresentativi E11,5 per ogni elemento stimolatore(Visel et al., 2007). La punta della freccia rossa indica il cuore.(A) Citometria a flusso di cardiomiociti derivati da iPSC. Immagine rappresentativa dei dati di flusso per i cardiomiociti (a sinistra) e percentuale di troponina cardiaca T (cTnT) positiva per ogni differenziazione (a destra). Le cellule sono state prima gated sul vivo / morto e poi sulla colorazione cTnT.(B) Analisi dei componenti principali dei dati RNA-seq in iPSC e CMs insieme a cellule staminali embrionali H1, cellule ventricolari sinistre (LV), cellule cardiache fetali (FH), e cellule linfoblastoidi linea cellulare (LCL). LCLs cluster indipendentemente da iPSC e CM, indicando che iPSC sono stati riprogrammati fedelmente.(C) Percentuale di Epigenoma Roadmap H3K27ac ChIP-seq picchi sovrapposti iPSC e CM H3K27ac. Sono mostrati i picchi sovrapposti per tutti i picchi e solo i picchi non promoter. LV, ventricolo sinistro; H1, H1 linea di cellule staminali embrionali.(D) Tre scatti del browser del genoma che mostrano il paesaggio epigenetico in CMs rispetto al ventricolo sinistro, atri destro, fegato adulto e ippocampo cerebrale della tabella di marcia dell’epigenoma.(A) Cluster analysis of RNA-seq data from each triplicate of iPSC and CM.(B) Numero di geni espressi in modo differenziato in ogni tipo di cellula.(C) Geni selezionati sovraesposti in CMs rispetto agli iPSC. (D) Gene Ontologia analisi di arricchimento dei processi biologici associati ai 4802 geni sovraespressi in cardiomiociti.In questa analisi, le interazioni sono state classificate come intra-TAD (entrambe le estremità dell’interazione completamente all’interno di un singolo TAD) o inter-TAD (ogni estremità dell’interazione è in un TAD diverso). Sono state omesse le interazioni che rientrano parzialmente o totalmente nei “confini” o “vuoti” della TAD come definiti dal TopDom (vedi Materiali e metodi).(A) Percentuale di interazioni che sono intra-TAD a diversi cut-off. Tutte le analisi hanno utilizzato interazioni che erano al 100% all’interno di un TAD.(B) Percentuale delle interazioni promotore-promotori nell’insieme delle interazioni intra-TAD e inter-TAD.(C,D) Arricchimento delle interazioni intra-TAD e interTAD per sovrapporre i picchi CTCF(C) o H3K27ac (D). Sono stati analizzati solo i picchi ChIP-seq del promotore-distale. ***p<2,2 × 10-16, test Z.(E) Punteggio CHiCAGO e(F) intervallo di interazione delle interazioni intra- e inter-TAD. ***p<2.2 × 10-16, Wilcoxon rank-sum test.(G,H) Considerare i promotori con un’interazione intra-TAD, un’interazione inter-TAD, o esclusivamente interazioni intra-TAD o inter-TAD:(G) distanza dal promotore TSS al più vicino confine TAD e(H) valore medio TPM del promotore. ***p<2,2 × 10-16, **p<0,01, *p<0,05, NS= non significativo, test del grado Wilcoxon.
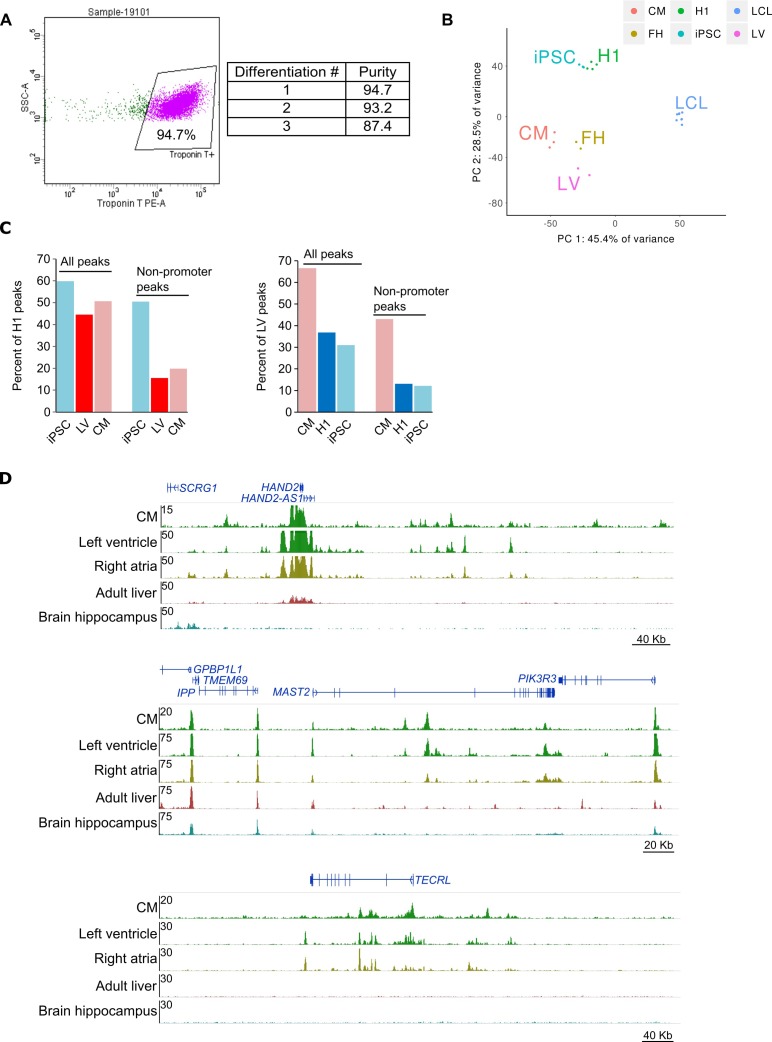
Figura 1-figure supplement 1.Controllo di qualità delle iPSC-CM.(A) Citometria a flusso di cardiomiociti derivati da iPSC. Immagine rappresentativa dei dati di flusso per i cardiomiociti (a sinistra) e la percentuale di troponina cardiaca T (cTnT) positivo per ogni differenziazione (a destra). Le cellule sono state prima gated sul vivo / morto e poi sulla colorazione cTnT.(B) Analisi dei componenti principali dei dati RNA-seq in iPSC e CMs insieme a cellule staminali embrionali H1, cellule ventricolari sinistre (LV), cellule cardiache fetali (FH), e cellule linfoblastoidi linea cellulare (LCL). LCLs cluster indipendentemente da iPSC e CM, indicando che iPSC sono stati riprogrammati fedelmente.(C) Percentuale di Epigenoma Roadmap H3K27ac ChIP-seq picchi sovrapposti iPSC e CM H3K27ac. Sono mostrati i picchi sovrapposti per tutti i picchi e solo i picchi non promoter. LV, ventricolo sinistro; H1, H1 linea di cellule staminali embrionali.(D) Tre scatti del browser del genoma che mostrano il paesaggio epigenetico in CMs rispetto al ventricolo sinistro, atri destro, fegato adulto e ippocampo cerebrale della tabella di marcia dell’epigenoma.
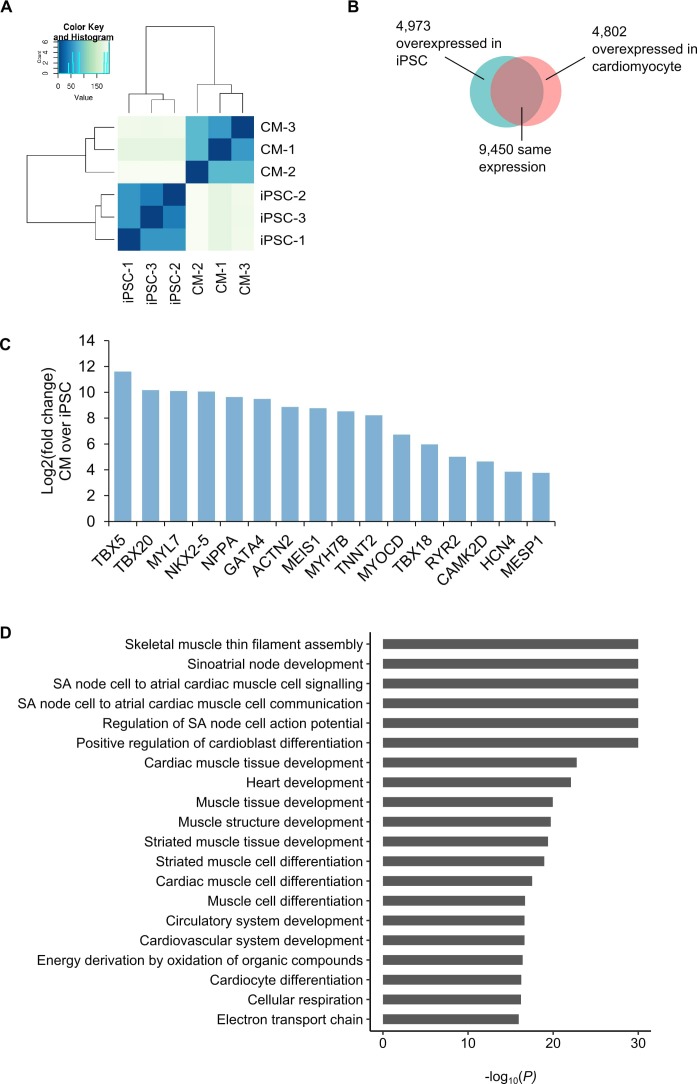
Figura 1-figure supplemento 2.Analisi di RNA-seq in iPSC e iPSC-CMs.(A) Cluster analisi dei dati di RNA-seq da ogni triplice copia di iPSC e CM.(B) Numero di geni espressi in modo differenziato in ciascun tipo di cellula.(C) Geni selezionati sovraesposti in CMs rispetto alle iPSC. (D) Gene Ontologia analisi di arricchimento dei processi biologici associati ai 4802 geni sovraespressi in cardiomiociti.
Figura 1-figure supplemento 3.Analisi delle interazioni PCHi-C nel contesto di TAD.In questa analisi, le interazioni sono state classificate come intra-TAD (entrambe le estremità dell’interazione completamente all’interno di un singolo TAD) o inter-TAD (ogni estremità dell’interazione è in un TAD diverso). Sono state omesse le interazioni che rientrano parzialmente o totalmente nei “confini” o nelle “lacune” delle DAT definite da TopDom (vedi Materiali e metodi).(A) Percentuale di interazioni che sono intra-TAD a diversi cut-off. Tutte le analisi hanno utilizzato interazioni che erano al 100% all’interno di un TAD.(B) Percentuale delle interazioni promotore-promotori nell’insieme delle interazioni intra-TAD e inter-TAD.(C,D) Arricchimento delle interazioni intra-TAD e interTAD per sovrapporre i picchi CTCF(C) o H3K27ac (D). Sono stati analizzati solo i picchi ChIP-seq del promotore-distale. ***p<2,2 × 10-16, test Z.(E) Punteggio CHiCAGO e(F) intervallo di interazione delle interazioni intra- e inter-TAD. ***p<2.2 × 10-16, Wilcoxon rank-sum test.(G,H) Considerare i promotori con un’interazione intra-TAD, un’interazione inter-TAD, o esclusivamente interazioni intra-TAD o inter-TAD:(G) distanza dal promotore TSS al più vicino confine TAD e(H) valore medio TPM del promotore. ***p<2,2 × 10-16, **p<0,01, *p<0,05, NS= non significativo, test del grado Wilcoxon.
Video 1.Video di cardiomiociti di derivazione iPSC che mostrano un battito spontaneo al giorno 20 della differenziazione (giorno di raccolta delle cellule).
Le interazioni del promotore sono arricchite per motivi di fattori di trascrizione specifici dei tessuti
Gli stimolatori distali attivano i geni target attraverso il DNA looping, un meccanismo che permette ai fattori di trascrizione legati a distanza di entrare in contatto con il macchinario di trascrizione dei promotori target(Pennacchio et al., 2013; Miele e Dekker, 2008; Deng et al., 2012). Per valutare se questa caratteristica della regolazione genica si è riflessa nelle interazioni iPSC e CM, abbiamo condotto un’analisi dei motivi utilizzando HOMER(Heinz et al., 2010) sull’insieme delle sequenze di interazione promotore-distale in ogni tipo di cellula. Inizialmente ci siamo concentrati sulle interazioni per i geni espressi in modo differenziato tra iPSC e CM (fold-change >1.5, Padj <0 .05). Abbiamo identificato CTCF come il motivo più arricchito in ogni caso(Figura 2A,B), coerente con il ruolo noto di questo fattore nel mediare le interazioni genomiche a lungo raggio a lungo raggio(Phillips and Corces, 2009; Phillips-Cremins et al., 2013; Nora et al ., 2017). Tra gli altri motivi top, abbiamo identificato i motivi del fattore di pluripotenza OCT4-SOX2-TCF-NANOG (OSN) e SOX2 come preferenzialmente arricchiti in sequenze distali in looping ai geni sovraespressi in iPSC(Figura 2A,C), mentre i motivi top in sequenze distali in looping ai geni sovraespressi in CMs incluso TBX20, ESRRB e MEIS1(Figura 2B,C). I fattori di trascrizione TBX20 e MEIS1 sono importanti regolatori dello sviluppo e della funzione del cuore(Cai et al., 2005; Sakabe et al., 2012; Mahmoud et al., 2013) e l’ESRRB è stato precedentemente identificato come un potenziale partner vincolante del TBX20 nei cardiomiociti dei topi adulti(Shen et al., 2011). Abbiamo anche osservato che le interazioni distali uniche per iPSC o CM sono state arricchite in modo simile per motivi di fattori di trascrizione specifici dei tessuti(Figura 2D). In linea con un recente rapporto che AP-1 contribuisce alla formazione di loop dinamici durante lo sviluppo dei macrofagi(Phanstiel et al., 2017), sia le interazioni iPSC e CM-specifiche sono stati arricchiti per motivi AP-1(Figura 2D), suggerendo che i fattori di trascrizione AP-1 può rappresentare un complesso organizzatore del genoma precedentemente non riconosciuto.
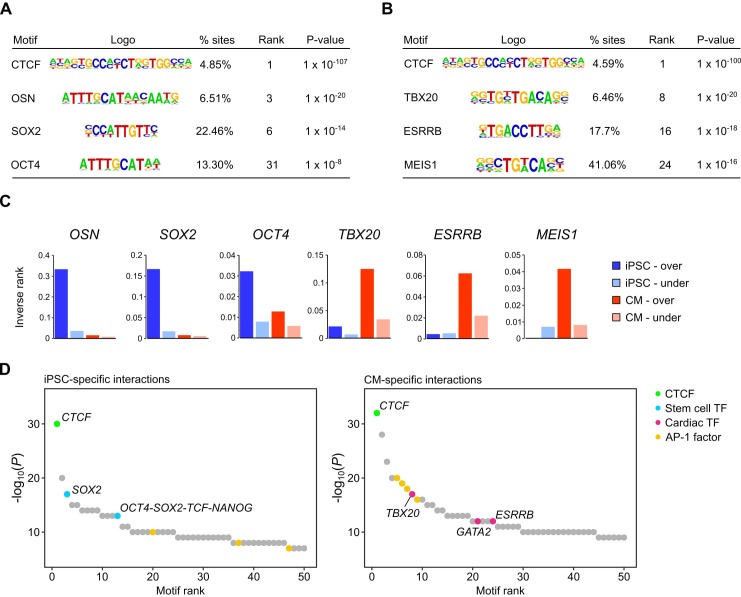
Figura 2.Figura 2. Arricchimento del motivo del fattore di trascrizione nelle regioni distali che interagiscono.(A,B) Selezionato fattore di trascrizione (TF) motivi identificati utilizzando HOMER nelle sequenze di interazione promotore-distale per tutti i geni sovraespressi in(A) iPSCs e(B) CMs (fold change > 1,5, Padj < 0,05).‘% siti’ si riferisce alla percentuale di interazioni distali che si sovrappongono al motivo; rango si basa sulla p-valore significato.(C) Per confrontare i ranghi del motivo tra i set di geni, l’inverso del rango è tracciato per motivi selezionati identificati in interazioni distali da geni sovra o sotto espressi sia in iPSCs che in CMs.(D) I primi 50 motivi identificati nelle interazioni specifiche del tipo di cellula. OSN, motivo OCT4-SOX2-TCF-NANOG.
Figura 2.Figura 2. Arricchimento del motivo del fattore di trascrizione nelle regioni distali che interagiscono.(A,B) Selezionato fattore di trascrizione (TF) motivi identificati utilizzando HOMER nelle sequenze di interazione promotore-distale per tutti i geni sovraespressi in(A) iPSCs e(B) CMs (fold change > 1,5, Padj < 0,05).‘% siti’ si riferisce alla percentuale di interazioni distali che si sovrappongono al motivo; rango è basato sul p-valore significato.(C) Per confrontare i ranghi del motivo tra i set di geni, l’inverso del rango è tracciato per motivi selezionati identificati in interazioni distali da geni sovra o sotto espressi sia in iPSCs che in CMs.(D) I primi 50 motivi identificati nelle interazioni specifiche del tipo di cellula. OSN, motivo OCT4-SOX2-TCF-NANOG.
Le interazioni a lungo raggio del promotore sono arricchite per gli elementi cis-regolatori attivi e corrispondono alle dinamiche di espressione genica
Gli elementi regolatori cis funzionalmente attivi sono caratterizzati dalla presenza di specifiche modificazioni dell’istone; gli esaltatori attivi sono generalmente associati a H3K4me1 e H3K27ac(Creyghton et al., 2010; Heintzman et al., 2009), mentre gli elementi inattivi (ad es. in bilico o silenziati) sono spesso associati a H3K27me3(Rada-Iglesias et al., 2011; Erceg et al., 2017). A sostegno della funzione di regolazione genica delle interazioni a lungo raggio, abbiamo trovato che i frammenti MboI promotori-distali coinvolti nelle interazioni significative del promotore sono stati arricchiti per queste tre modifiche degli istoni sia in iPSCs che in CMs(Figura 3A-C). Quando i promotori sono stati raggruppati per livello di espressione, abbiamo osservato che questo arricchimento è aumentato con l’aumento di espressione per H3K27ac e H3K4me1, e diminuito con l’aumento di espressione per H3K27me3, coerente con una natura additiva di enhancer-promoter interazioni(Schoenfelder et al., 2015; Javierre et al., 2016), e convalidando che PCHi-C arricchisce per i probabili contatti funzionali a lungo raggio cromatina.
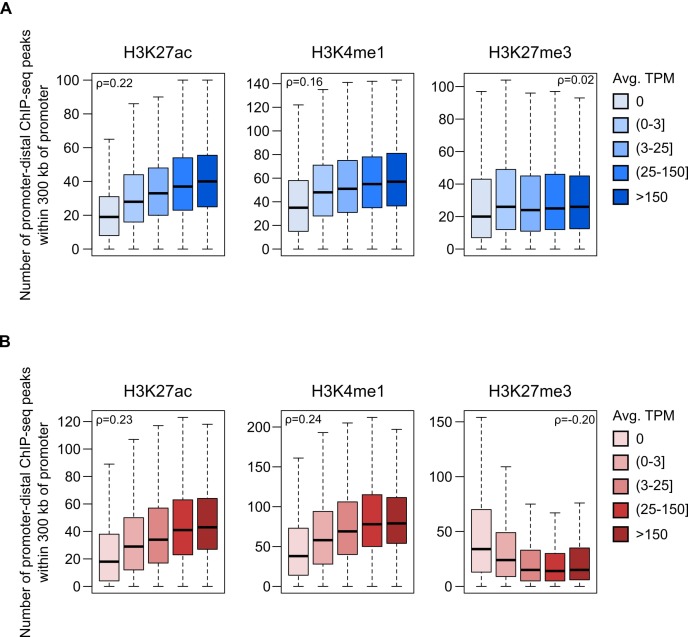
Figura 3-figure supplemento 1.Correlazione tra il numero di picchi di ChIP-seq dell’istone entro 300 kb di promotori e il livello di espressione genica.(A,B) Percentuale delle interazioni promotore-distale che si sovrappongono ad un picco dell’istone ChIP-seq rispetto ai frammenti MboI di controllo casuale (vedi Materiali e metodi). Le interazioni iPSC sono state sovrapposte ai dati H1 ESC ChIP-seq; le interazioni CM sono state sovrapposte ai dati ChIP-seq del ventricolo sinistro del progetto Epigenome Roadmap Project(file supplementare 10).(C) Arricchimento dei dati presentati in(A) e(B).(D) Arricchimento delle interazioni promotore-distale in base al livello di espressione del promotore. I promotori sono stati raggruppati in cinque cassonetti in base ai loro valori medi di TPM. La linea tratteggiata indica l’assenza di arricchimento.(E) Arricchimento delle interazioni cella-tipo specifico e condiviso (colonne) in base alle caratteristiche della cromatina specifica del tessuto e condivisa (righe).(F) Esempio del gene NPPA in iPSC (in alto) e CM (in basso). Gray box evidenzia le interazioni CM-specifiche CM alle caratteristiche cromato-specifiche della cromatina e un potenziatore cardiaco in vivo(Visel et al., 2007). Per chiarezza, sono mostrate solo le interazioni per NPPA. *p<0.00001, #p=0.0017, Z-test.Numero di picchi ChIP-seq dell’istone promotore-distale dell’istone ChIP-seq entro 300 kb di promotori in iPSC(A) e CM(B). Il rho di Spearman (ρ) è stato calcolato sull’insieme completo dei valori di espressione del promotore/contatori di picco per tutti i promotori con almeno una interazione significativa nel rispettivo tipo di cellula (12.926 geni per iPSC e 13.555 geni per CM; vedere Materiali e metodi). I dati sono raggruppati per categoria di espressione per sottolineare la tendenza. Le barre orizzontali indicano la mediana per ogni categoria di espressione. Tutte le stime di correlazione sono significative a p<2,2 × 10-16 ad eccezione di H3K27me3 in iPSC (p=0,06).
Una forte correlazione (coefficiente di correlazione di Pearson r> 0,7) tra il grado di modificazione degli istoni e l’espressione genica è stata segnalata per la prima volta quasi 10 anni fa (Karlić et al., 2010); tuttavia, tale analisi ha considerato solo le modificazioni degli istoni entro 2 kb di promotori. Per capire se questa relazione si estende oltre le regioni prossimali del promotore, abbiamo correlato il numero di picchi di ChIP-seq dell’istone entro 300 kb di promotori con il livello di espressione del promotore (Figura 3-figure supplement 1A,B). H3K27ac e H3K4me1 entrambi correlati positivamente con il livello di espressione (Spearman ρ = 0,22 e 0,16, rispettivamente in iPSC e ρ = 0,23 e 0 ,24, rispettivamente in CMs, p<2.2-16); al contrario, H3K27me3 è correlato negativamente con il livello di espressione in CMs (Spearman’s ρ = -0.20, p<2.2-16); tuttavia, questa relazione non era presente in iPSCs (Spearman’s ρ = 0.02, p=0.06). Sebbene moderate, queste correlazioni potrebbero in parte spiegare perché i geni espressi più alti mostrano un arricchimento più forte per le interazioni del promotore che si sovrappongono ai picchi degli istoni quando si usa un modello di sfondo a livello genomico (vedi Materiali e metodi), e danno supporto all’idea che i geni attivi si trovano in ambienti genomici generalmente attivi(Stevens et al., 2017; Gilbert et al., 2004).
Abbiamo poi studiato la relazione tra le interazioni specifiche del tipo di cellula e l’arricchimento per i marchi CTCF specifici per i tessuti, H3K27ac, e H3K27me3, ipotizzando che le interazioni uniche per iPSC o CM sarebbero più arricchite per le caratteristiche della cromatina specifiche per i tessuti. Infatti, abbiamo osservato che le interazioni specifiche per tipo di cellula preferibilmente coinvolti picchi H3K27ac dal tipo di cellula corrispondente, e sono stati o non arricchito (iPSC) o impoverito (CM) per i marchi H3K27ac che erano specifici per il tipo di cellula non corrispondente(Figura 3E, pannello centrale). Tuttavia, l’arricchimento più forte è stato per le interazioni specifiche del tipo di cellula per sovrapporre le caratteristiche della cromatina che erano presenti in entrambi i tipi di cellule(Figura 3E). Inoltre, le interazioni che sono stati condivisi tra iPSC e CMs sono stati più arricchiti per le caratteristiche cromatina condivisa. Questi risultati suggeriscono che tutte le interazioni, sia condivise o uniche per un tipo di cellula, preferibilmente contattare le regioni regolatorie che sono attive in entrambi i tipi di cellule, mentre le interazioni specifiche del tipo di cellula non sono suscettibili di verificarsi in regioni specificamente contrassegnate nel tipo di cellula non corrispondente.
Un esempio di un gene che comprende queste osservazioni è il gene peptidico natriuretico atriale NPPA(Figura 3F) che è espresso specificamente nelle cellule dell’atrio cardiaco ed è upregolato in CMs(Figura 1-figure supplement 2C). NPPA fa numerose cellule di tipo specifico interazioni con una regione distale che è solo contrassegnato con cromatina attiva (H3K27ac e H3K4me1) in CMs, inoltre, la caratterizzazione funzionale ha dimostrato che questa regione corrisponde ad un enhancer in vivo che ricapitola l’espressione endogena di NPPA nel cuore in via di sviluppo (Viselet al., 2007). Presi nel loro insieme, questi risultati illuminano la complessa relazione tra le interazioni a lungo raggio del promotore e la regolazione del gene e forniscono la prova che l’architettura del promotore riflette l’espressione genica specifica del tipo di cellula.
Figura 3-figure supplement 1.Correlazione tra il numero di picchi di ChIP-seq dell’istone entro 300 kb di promotori e il livello di espressione genica.(A,B) Percentuale delle interazioni promotore-distale che si sovrappongono ad un picco dell’istone ChIP-seq rispetto ai frammenti MboI di controllo casuale (vedi Materiali e metodi). Le interazioni iPSC sono state sovrapposte ai dati H1 ESC ChIP-seq; le interazioni CM sono state sovrapposte ai dati ChIP-seq del ventricolo sinistro del progetto Epigenome Roadmap Project(file supplementare 10).(C) Arricchimento dei dati presentati in(A) e(B).(D) Arricchimento delle interazioni promotore-distale in base al livello di espressione del promotore. I promotori sono stati raggruppati in cinque cassonetti in base ai loro valori medi di TPM. La linea tratteggiata indica l’assenza di arricchimento.(E) Arricchimento di interazioni specifiche per le cellule e condivise (colonne) con caratteristiche cromatografiche specifiche per i tessuti e condivise (righe).(F) Esempio del gene NPPA in iPSC (in alto) e CM (in basso). Gray box evidenzia le interazioni CM-specifiche CM alle caratteristiche cromato-specifiche della cromatina e un potenziatore cardiaco in vivo(Visel et al., 2007). Per chiarezza, sono mostrate solo le interazioni per NPPA. *p<0.00001, #p=0.0017, Z-test.Numero di picchi ChIP-seq dell’istone promotore-distale dell’istone ChIP-seq entro 300 kb di promotori in iPSC(A) e CM(B). Il rho di Spearman (ρ) è stato calcolato sull’insieme completo dei valori di espressione del promotore/contatori di picco per tutti i promotori con almeno una interazione significativa nel rispettivo tipo di cellula (12.926 geni per iPSC e 13.555 geni per CM; vedere Materiali e metodi). I dati sono raggruppati per categoria di espressione per sottolineare la tendenza. Le barre orizzontali indicano la mediana per ogni categoria di espressione. Tutte le stime di correlazione sono significative a p<2,2 × 10-16 ad eccezione di H3K27me3 in iPSC (p=0,06).
Figura 3-figure supplement 1.Correlazione tra il numero di picchi di ChIP-seq dell’istone entro 300 kb di promotori e il livello di espressione genica.2. Numero di picchi ChIP-seq dell’istone promotore-distale dell’istone ChIP-seq entro 300 kb di promotori in iPSC(A) e CM(B). Il rho di Spearman (ρ) è stato calcolato sull’insieme completo dei valori di espressione del promotore/contatori di picco per tutti i promotori con almeno una interazione significativa nel rispettivo tipo di cellula (12.926 geni per iPSC e 13.555 geni per CM; vedere Materiali e metodi). I dati sono raggruppati per categoria di espressione per sottolineare la tendenza. Le barre orizzontali indicano la mediana per ogni categoria di espressione. Tutte le stime di correlazione sono significative a p<2,2 × 10-16 ad eccezione di H3K27me3 in iPSC (p=0,06).
I cambiamenti dinamici nella compartimentazione genomica coinvolgono un sottoinsieme di geni specifici del cuore
Come benchmark finale dei nostri set di dati, abbiamo analizzato le differenze su larga scala nell’organizzazione del genoma tra iPSC e CM. I primi studi Hi-C hanno rivelato che il genoma è organizzato in due grandi compartimenti, A e B, che corrispondono rispettivamente a regioni aperte e chiuse dei cromosomi(Lieberman-Aiden et al., 2009; Rao et al., 2014). Anche se la maggior parte dei compartimenti sono stabili tra i diversi tipi di cellule, alcuni compartimenti cambiano stato in modo specifico per il tipo di cellula che può riflettere importanti cambiamenti nella regolazione dei geni(Dixon et al., 2015). Per valutare se l’acquisizione di dati Hi-C, che è più conveniente in termini di costi per l’acquisizione di interazioni incentrate sul promotore, è in grado di identificare i compartimenti A/B, abbiamo confrontato i nostri dati di acquisizione Hi-C con le biblioteche Hi-C di pre-acquisizione a livello genomico. I compartimenti A/B identificati utilizzando HOMER(Heinz et al., 2010) erano notevolmente simili nei set di dati del genoma intero e PCHi-C (corrispondenza del 97%, Figura 4A, pannello superiore, e Figura 4-figure supplements 1 e 2), dimostrando che i dati PCHi-C contengono informazioni sufficienti per identificare le regioni del genoma ampiamente attive e inattive. Come esempio, si evidenzia una regione di 10 Mb sul cromosoma 4 contenente il locus del gene CAMK2D(Figura 4A). I compartimenti erano relativamente stabili in questa regione in iPSC e CMs; tuttavia, il gene CAMK2D stesso si trovava in un compartimento dinamico che passava da inattivo in iPSCs ad attivo in CMs. Corrispondentemente, questo gene è stato altamente upregulated durante la differenziazione a CMs(Figura 4A, inserto).
Figura 4-figure supplemento 3.Figura 4—supplemento figura 3. A / B commutazione comparazione dei compartimenti A / B corrisponde all’attivazione di geni specifici dei tessuti.Comparazione di A / B compartimenti in Hi-C e PCHi-C.Esempio di A / B compartimenti.GO analisi sui geni di commutazione da attivi compartimenti A in iPSC a compartimenti B inattivi in CMs.(A) Pannello superiore: Regione di 10 Mb sul cromosoma quattro che mostra i compartimenti A (verde) e B (blu) in base alla prima analisi dei componenti principali calcolata da HOMER(Heinz et al., 2010) dell’intero genoma Hi-C e cattura i dati di interazione Hi-C. Pannello in basso: ingrandito il locus CAMK2D; sono mostrati solo i compartimenti Hi-C A/B catturati. Inset: livello di espressione di CAMK2D in iPSC e CM attraverso i tre replicati.(B) Livello di espressione (TPM) dei geni situati nel comparto A (verde) o B (blu) in ogni replicato di iPSC (sinistra) o CM (destra).(C) Differenza nel livello di espressione (log2 fold change relativo agli iPSC) dei geni che passano da iPSC a CM o che rimangono in compartimenti stabili.(D) Analisi ontologica dei processi biologici associati ai geni che passano da compartimenti B ad A durante la differenziazione iPSC-CM. ***p<2.2 × 10-16, test di Wilcoxon rank-sum.Correlazione tra il punteggio del compartimento A/B (analisi dei componenti principali dei dati di interazione, PC-1) nell’intero genoma Hi-C (asse y) e l’acquisizione del promotore Hi-C (asse x) negli iPSC (in alto) e CM (in basso). Spearman ρ > 0,98, p<2,2 × 10-16 in tutti i casi.Istantanea del browser del genoma di una regione di ~53 Mb sul cromosoma quattro che mostra i compartimenti A/B in tutti e tre i replicati di iPSC e CM utilizzando sia i dati del genoma intero (WG) che quelli dell’acquisizione Hi-C del promotore.
Abbiamo osservato questo effetto a livello globale, in quanto i geni situati nei compartimenti A sono stati espressi a livelli significativamente più elevati rispetto ai geni situati nei compartimenti B sia negli iPSC che nelle CM(Figura 4B). Inoltre, i geni che hanno scambiato i compartimenti A/B tra i tipi di cellule sono stati corrispondentemente regolati in alto o in basso(Figura 4C). L’analisi GO dei 1008 geni che sono passati dai compartimenti B a quelli A durante la differenziazione iPSC-CM ha rivelato un arricchimento per termini come “sviluppo del sistema cardiovascolare” e “contrazione cardiaca” (Figura 4D, file supplementare 5). È importante notare che questi geni sono stati identificati esclusivamente in base alla loro posizione in un compartimento genomico dinamico e non dai dati di espressione genica. Analisi GO per i geni che sono passati da compartimenti A a compartimenti B durante la differenziazione iPSC-CM relativi a processi non cardiaci, come lo sviluppo della pelle, la differenziazione delle cellule epiteliali e la determinazione del sesso(Figura 4-figure supplement 3, file supplementare 5 e 6). Questi dati mostrano che la PCHi-C ha catturato accuratamente le interazioni tissutali specifiche e indicano che la compartimentazione dei geni nelle regioni spazialmente regolate del nucleo può essere un meccanismo per garantire l’espressione genica tissutale specifica(Dixon et al., 2015). In sintesi, le nostre analisi hanno dimostrato che le interazioni del promotore CM ricapitolano le caratteristiche chiave della regolazione e della funzione dei geni cardiaci, convalidando la mappa CM come strumento importante per indagare la genetica CVD.
Figura 4-figure supplement 3.Figura 4—supplemento alla figura 3. La commutazione dei compartimenti A/B corrisponde all’attivazione dei geni specifici dei tessuti.Confronto dei compartimenti A/B in Hi-C e PCHi-C.Esempio di compartimenti A/B.Analisi di GO sui geni che passano da compartimenti A attivi in iPSC a compartimenti B inattivi in CM.(A) Pannello superiore: Regione di 10 Mb sul cromosoma quattro che mostra i compartimenti A (verde) e B (blu) in base alla prima analisi dei componenti principali calcolata da HOMER(Heinz et al., 2010) dell’intero genoma Hi-C e cattura i dati di interazione Hi-C. Pannello in basso: ingrandito il locus CAMK2D; sono mostrati solo i compartimenti Hi-C A/B catturati. Inset: livello di espressione di CAMK2D in iPSC e CM attraverso i tre replicati.(B) Livello di espressione (TPM) dei geni situati nel comparto A (verde) o B (blu) in ogni replicato di iPSC (sinistra) o CM (destra).(C) Differenza nel livello di espressione (log2 fold change relativo agli iPSC) dei geni che passano da iPSC a CM o che rimangono in compartimenti stabili.(D) Analisi ontologica dei processi biologici associati ai geni che passano da compartimenti B ad A durante la differenziazione iPSC-CM. ***p<2.2 × 10-16, test di Wilcoxon rank-sum.Correlazione tra il punteggio del compartimento A/B (analisi dei componenti principali dei dati di interazione, PC-1) nell’intero genoma Hi-C (asse y) e l’acquisizione del promotore Hi-C (asse x) negli iPSC (in alto) e CM (in basso). Spearman ρ > 0,98, p<2,2 × 10-16 in tutti i casi.Istantanea del browser del genoma di una regione di ~53 Mb sul cromosoma quattro che mostra i compartimenti A/B in tutti e tre i replicati di iPSC e CM utilizzando sia i dati del genoma intero (WG) che quelli dell’acquisizione Hi-C del promotore.
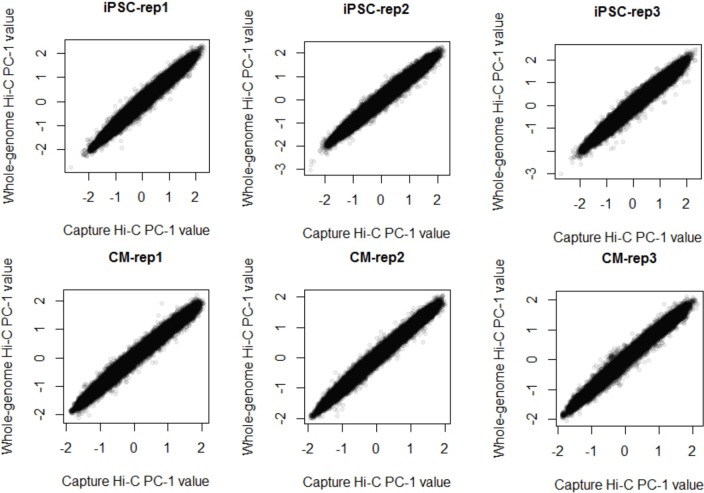
Figura 4-figure supplemento 1.Confronto dei compartimenti A/B in Hi-C e PCHi-C.Correlazione tra il punteggio del compartimento A/B (analisi dei componenti principali dei dati di interazione, PC-1) nel genoma intero Hi-C (asse y) e l’acquisizione del promotore Hi-C (asse x) negli iPSC (in alto) e CM (in basso). Spearman ρ > 0,98, p<2,2 × 10-16 in tutti i casi.
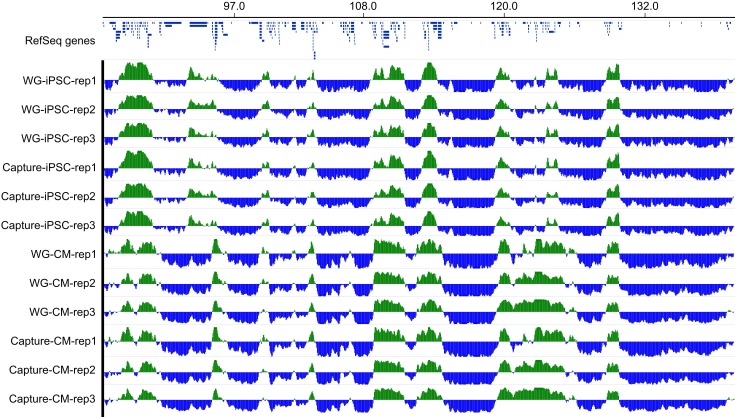
Figura 4-figure supplement 2.Esempio di scomparti A/B.Istantanea del browser del genoma di una regione di ~53 Mb sul cromosoma quattro che mostra i compartimenti A/B in tutti e tre i replicati di iPSC e CM che utilizzano sia i dati del genoma intero (WG) che quelli dell’acquisizione Hi-C del promotore.
Figura 4-figure supplemento 3.Analisi GO sui geni che passano da compartimenti A attivi in iPSC a compartimenti B inattivi in CM.
Le interazioni del promotore CM collegano i GWAS SNPs ai geni target
Un’applicazione particolarmente rilevante delle mappe di interazione del promotore ad alta risoluzione è quella di guidare gli studi post-GWAS identificando i geni target delle varianti associate alla malattia. Abbiamo utilizzato questo approccio per collegare i GWAS SNPs per diverse importanti malattie cardiovascolari al loro gene target (o ai loro geni target) utilizzando la mappa di interazione CM. Abbiamo compilato 524 SNPs di piombo dal database NHGRI(https://www.ebi.ac.uk/gwas/) per tre importanti classi di CVD: aritmie cardiache, insufficienza cardiaca e infarto del miocardio(Tabella 1, file supplementari 7 e 8). A causa dei modelli di disequilibrio di collegamento (LD), il vero SNP causale potrebbe essere qualsiasi SNP in LD elevato con la variante di piombo. Pertanto, abbiamo ampliato questo insieme di SNPs per includere tutte le varianti in alta LD (r2 >0,9, entro 50 kb di SNP di piombo), aumentando il numero di varianti putatilmente causali a 10.475 (di seguito chiamati SNPs di LD). Abbiamo scoperto che nel 1999 (19%) degli SNP LD si trovavano in frammenti MboI promotori-distali che interagivano con i promotori di 347 geni in CMs(file supplementare 8), di seguito denominati geni bersaglio. La maggior parte (89%) delle coppie di geni target LD SNP-target sono stati localizzati all’interno dello stesso TAD, con una distanza mediana di 185 kb tra ogni coppia di geni target SNP(Figura 5A). È importante notare che il 90,4% delle interazioni geniche SNP-target ha saltato almeno un promotore genico e il 42% degli SNP ha interagito con almeno due diversi promotori(Figura 5B).
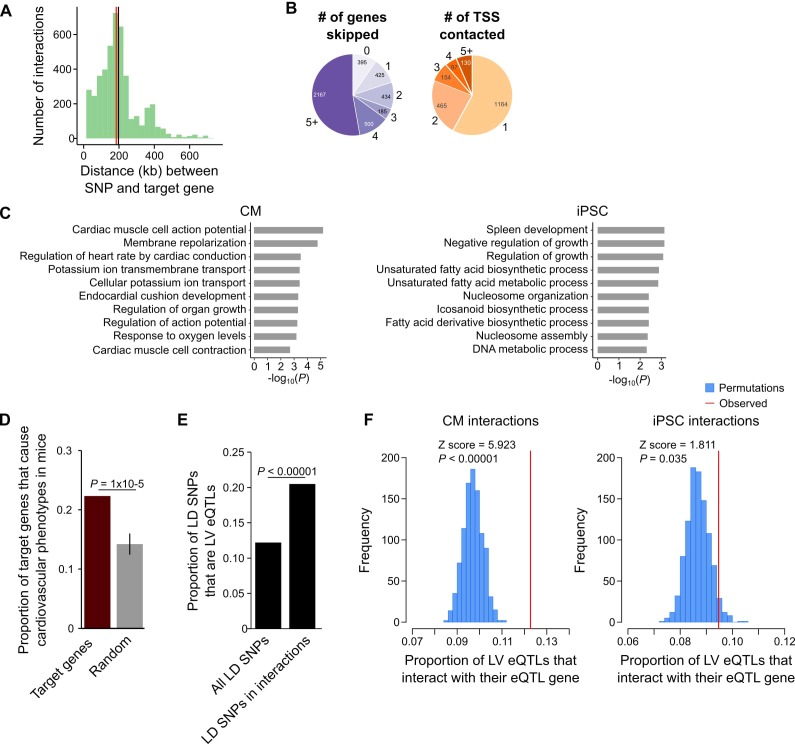
Figura 5.Le interazioni del promotore CM collegano i GWAS SNP di CVD GWAS ai geni target.(A) Distribuzione delle distanze genomiche che separano le interazioni geniche SNP-target (linea rossa, mediana = 185 kb; linea nera, media = 197 kb).(B) Grafico a torta che mostra il numero di TSS saltati per ogni interazione genica SNP-target (a sinistra) e il numero di geni contattati da ogni SNP (a destra).(C) GO analisi di arricchimento per l’arricchimento dei geni per il looping dei geni agli SNP LD utilizzando i dati di interazione del promotore CM (pannello di sinistra) o i dati di interazione del promotore iPSC (pannello di destra).(D) Percentuale dei geni target che risultano in un fenotipo cardiovascolare quando vengono eliminati nel mouse (database MGI[Blake et al., 2017]), rispetto a un set di controllo casuale. p-Valore calcolato con un test Z.(E) Percentuale di GWAS LD SNPs che sono eQTLs nel ventricolo sinistro (LV) se si considera l’insieme completo di LD SNPs, o il sottoinsieme che si sovrappongono alle interazioni del promotore CM. p-Valore calcolato con il test esatto di Fisher.(F) Percentuale di LV eQTL (a livello genomico) che mappano all’interno di un’interazione del promotore per il gene associato al gene eQTL (indicato dalla linea rossa). Le permutazioni casuali sono state ottenute riassegnando l’insieme di interazioni di ogni promotore ad un nuovo promotore e calcolando la proporzione di eQTLs in interazioni casuali che interagiscono con il loro gene eQTL-associato. Le proporzioni considerano solo gli eQTL che si sovrappongono ad un’interazione promotore-distale. I valori P calcolati con un test Z.
| Aritmia | Infarto del miocardio | Insufficienza cardiaca | Combinato | |
|---|---|---|---|---|
| Numero di studi | 30 | 11 | 11 | 50 |
| Tag SNPs | 358 | 86 | 80 | 524 |
| SNPs in LD | 6555 | 1822 | 2098 | 10,475 |
| SNPs in looping ai geni | 1152 | 357 | 490 | 1999 |
| Geni bersaglio | 237 | 72 | 53 | 347 |
Per confermare che le interazioni CM PCHi-C collegavano gli SNP ai geni target rilevanti per il CVD, abbiamo eseguito un’analisi GO e abbiamo scoperto che i geni target erano altamente e specificamente arricchiti per i processi biologici legati alla funzione cardiaca, come la ripolarizzazione della membrana e la conduzione cardiaca(Figura 5C, pannello sinistro e file supplementare 5 e 6). Come controllo, abbiamo usato le interazioni iPSC per collegare gli stessi SNPs ai geni target e abbiamo osservato un insieme completamente diverso di processi biologici non correlati per questi geni(Figura 5C, pannello di destra). Per caratterizzare ulteriormente la rilevanza biologica dei geni target, abbiamo estratto i dati di knock-out del mouse dal database Mouse Genome Informatics (MGI)(Blake et al., 2017), che ha rivelato che un numero statisticamente significativo di geni target ha portato ad un fenotipo cardiovascolare quando è stato messo knock-out nel mouse (78 geni (22,4%), p=1 × 10-5, Figura 5D) . Infine, abbiamo esaminato i dati quantitativi di espressione dei tratti loci (eQTL) del tessuto del ventricolo sinistro umano (LV) ottenuti nell’ambito del progetto Genotype-Tissue Expresion (GTEx)(Carithers et al., 2015) e abbiamo trovato che, del 1999 LD SNPs nelle interazioni, 410 (20,5%) corrispondevano a LV eQTLs; in confronto, solo il 12,2% del set completo di LD SNPs corrispondeva a LV eQTLs (p<0,00001, Figura 5E). Abbiamo poi valutato se gli eQTLs si collegano al loro gene associato. Per questa analisi, abbiamo considerato l’intero set di LV eQTLs, in quanto i 410 LD SNP eQTLs rappresentano una percentuale troppo piccola di una parte dell’intero set (<0,1% di tutti i LV eQTLs) per accertare pienamente la significatività. A livello genomico, i LV eQTL nelle interazioni promotore-distale erano significativamente più probabili di loop al loro gene associato rispetto a quanto previsto per caso (p<0,00001, Figura 5F, pannello sinistro). È importante notare che questo significato è diminuito quando i LV eQTLs sono stati analizzati con interazioni promoter iPSC (p=0,035, Figura 5F, pannello di destra). Presi nel loro insieme, questi risultati indicano che le interazioni del promotore CM identificano un sottoinsieme di SNPs rilevanti per la malattia più probabilmente funzionali e supportano l’uso della mappa CM per assegnare SNPs distali associati a CVD ai geni target putativi.
Figura 5.Figura 5. Le interazioni del promotore CM collegano i CVD GWAS SNPs CVD ai geni bersaglio.(A) Distribuzione delle distanze genomiche che separano le interazioni geniche SNP-target (linea rossa, mediana = 185 kb; linea nera, media = 197 kb).(B) Grafico a torta che mostra il numero di TSS saltati per ogni interazione genica SNP-target (a sinistra) e il numero di geni contattati da ogni SNP (a destra).(C) GO analisi di arricchimento per l’arricchimento dei geni per il looping dei geni agli SNP LD utilizzando i dati di interazione del promotore CM (pannello di sinistra) o i dati di interazione del promotore iPSC (pannello di destra).(D) Percentuale dei geni target che risultano in un fenotipo cardiovascolare quando vengono eliminati nel mouse (database MGI[Blake et al., 2017]), rispetto a un set di controllo casuale. p-Valore calcolato con un test Z.(E) Percentuale di GWAS LD SNPs che sono eQTLs nel ventricolo sinistro (LV) se si considera l’insieme completo di LD SNPs, o il sottoinsieme che si sovrappongono alle interazioni del promotore CM. p-Valore calcolato con il test esatto di Fisher.(F) Percentuale di LV eQTL (a livello genomico) che mappano all’interno di un’interazione del promotore per il gene associato al gene eQTL (indicato dalla linea rossa). Le permutazioni casuali sono state ottenute riassegnando l’insieme di interazioni di ogni promotore ad un nuovo promotore e calcolando la proporzione di eQTLs in interazioni casuali che interagiscono con il loro gene eQTL-associato. Le proporzioni considerano solo gli eQTL che si sovrappongono ad un’interazione promotore-distale. I valori P calcolati con un test Z.
Uso dell’espressione genica come metrica per l’interpretazione della rilevanza della malattia dei geni bersaglio appena identificati
Sulla base di un arricchimento dei geni target con funzione cardiaca nota, abbiamo poi valutato se il livello di espressione è una metrica informativa per dare ulteriore priorità agli studi di follow-up funzionale. Abbiamo esaminato il livello di espressione dei 347 geni target e abbiamo trovato che erano moderatamente sovra-espressi nelle CM rispetto alle iPSC (mediana log2 fold change = 1,08, media log2 fold change = 1,44, valori medi TPM erano 40,6 nelle iPSC e 60,1 nelle CM, p = 0,12, Figura 6A e B). Anche se non significativo, questo risultato riflette l’arricchimento di geni noti legati al cuore che interagiscono con i loci CVD. Tuttavia, poiché un sottoinsieme di geni target è stato sovraespresso nelle iPSC rispetto alle CMs(Figura 6C), abbiamo previsto che il livello di espressione genica da solo potrebbe essere una metrica insufficiente per valutare la rilevanza dei geni target per la biologia CVD. Infatti, abbiamo scoperto che 21 dei 78 geni target (27%) che causano fenotipi cardiovascolari quando sono stati messi fuori uso nei topi erano sovraespressi nelle iPSC rispetto alle CMs(file supplementare 8). Questo risultato indica che i geni putativi causali possono non apparire come candidati ovvi sulla base dei soli dati di espressione genica.
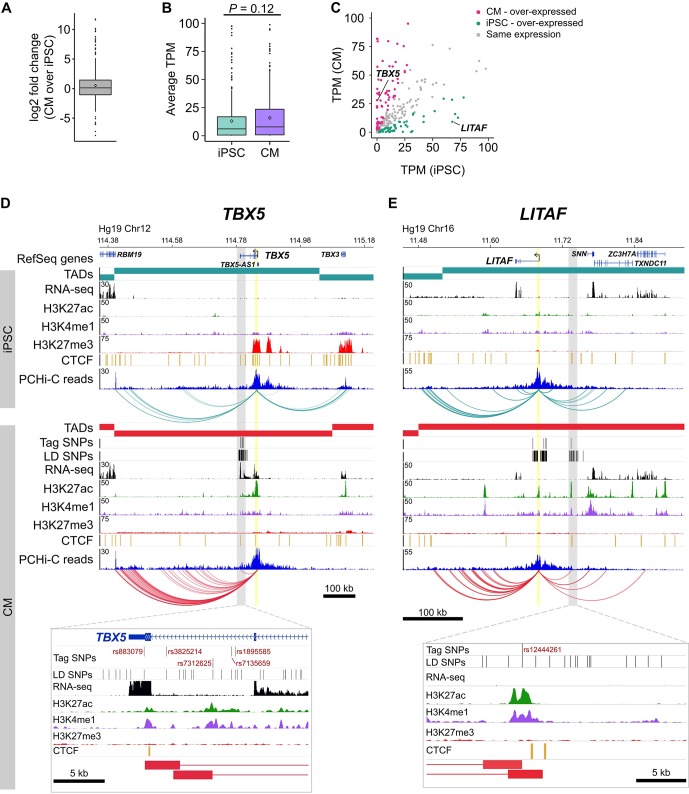
Figura 6.Figura 6. Caratterizzazione dei geni target in base al livello di espressione.(A) Log2 fold change del livello di espressione dei geni target nelle CMs rispetto alle iPSC (la barra orizzontale indica la mediana, 1,08; il diamante indica la media, 1,44).(B) Valori medi di TPM dei geni target in iPSC e CMs (p=0,12, test di Wilcoxon rank-sum). I diamanti indicano il valore medio (40,6 per iPSC, 60,1 per CM).(C) Confronto dei valori medi di TPM per i geni target nelle CM e nelle iPSC. Vedere il file supplementare 8 per l’elenco completo dei geni e dei valori TPM.(D,E) Esempi di looping dei geni per l’aritmia cardiaca GWAS SNPs in CMs.(D) Il gene TBX5 interagisce con un locus di aritmia validato funzionalmente(Smemo et al., 2012).(E) Il gene LITAF interagisce con un locus identificato in(Arking et al., 2014). La regione evidenziata in giallo indica il promotore; il riquadro grigio e il pannello dello zoom mostrano le regioni che interagiscono con il promotore (riquadri rosa) che si sovrappongono alle aritmie SNPs. Per chiarezza, vengono mostrate solo le interazioni per il promotore indicato.
Per illustrare questo punto, si evidenziano due geni: TBX5, un gene direttamente collegato all’aritmia cardiaca(Figura 6D)(Smemo et al., 2012; Arnolds et al., 2012), e LITAF, un gene che, fino a poco tempo fa, non aveva un ruolo evidente nella biologia cardiaca(Moshal et al., 2017)(Figura 6E). Entrambi i geni formavano interazioni a lungo raggio a LD SNPs identificati in aritmia GWAS, rendendo entrambi i geni candidati bersagli funzionali delle associazioni GWAS. TBX5, che è sovra-espresso in CMs(Figura 6C), è il gene target più probabile dei SNPs LD nelle vicinanze sulla base dei dati di interazione, ma anche a causa del suo ruolo noto nel dirigere il corretto sviluppo del sistema di conduzione cardiaca. LITAF, d’altra parte, è stato sovraespresso in iPSCs rispetto alle CMs(Figura 6C) e non era noto per contribuire alla funzione cardiaca fino a quando un recente studio ha identificato questo gene come regolatore dell’eccitazione cardiaca nei cuori dei pesci zebra(Moshal et al., 2017).
Figura 6.Caratterizzazione dei geni target in base al livello di espressione.(A) Log2 fold change del livello di espressione dei geni target nelle CMs rispetto alle iPSC (la barra orizzontale indica la mediana, 1,08; il diamante indica la media, 1,44).(B) Valori medi di TPM dei geni target in iPSC e CMs (p=0,12, test di Wilcoxon rank-sum). I diamanti indicano il valore medio (40,6 per iPSC, 60,1 per CM).(C) Confronto dei valori medi di TPM per i geni target nelle CM e nelle iPSC. Vedere il file supplementare 8 per l’elenco completo dei geni e dei valori TPM.(D,E) Esempi di looping dei geni per l’aritmia cardiaca GWAS SNPs in CMs.(D) Il gene TBX5 interagisce con un locus di aritmia validato funzionalmente(Smemo et al., 2012).(E) Il gene LITAF interagisce con un locus identificato in(Arking et al., 2014). La regione evidenziata in giallo indica il promotore; il riquadro grigio e il pannello dello zoom mostrano le regioni che interagiscono con il promotore (riquadri rosa) che si sovrappongono alle aritmie SNPs. Per chiarezza, vengono mostrate solo le interazioni per il promotore indicato.
Le interazioni del promotore CM sono informative per le associazioni cardiovascolari che non coinvolgono direttamente i cardiomiociti
Poiché le tre classi di malattia che abbiamo analizzato rappresentano diverse patologie, abbiamo previsto che i geni bersaglio identificati per ogni classe individualmente possono riferirsi a diversi processi biologici. In particolare, abbiamo considerato che le aritmie cardiache – che derivano direttamente da difetti nei cardiomiociti specializzati per la conduzione elettrica – possono scoprire i geni target più rilevanti per il cuore rispetto all’insufficienza cardiaca e all’infarto del miocardio, due CVD che coinvolgono anche sistemi non cardiaci. Se suddiviso nelle rispettive classi di malattia, abbiamo confermato che la maggior parte dell’arricchimento del GO per i termini cardiaci era guidato dall’aritmia cardiaca SNPs(Figura 7A), con termini direttamente correlati al sistema di conduzione cardiaca. Le analisi dell’infarto miocardico(Figura 7B) e dell’insufficienza cardiaca(Figura 7C) hanno evidenziato un insieme di geni che sono stati leggermente arricchiti per la regolazione della crescita e della morfogenesi, rispettivamente.
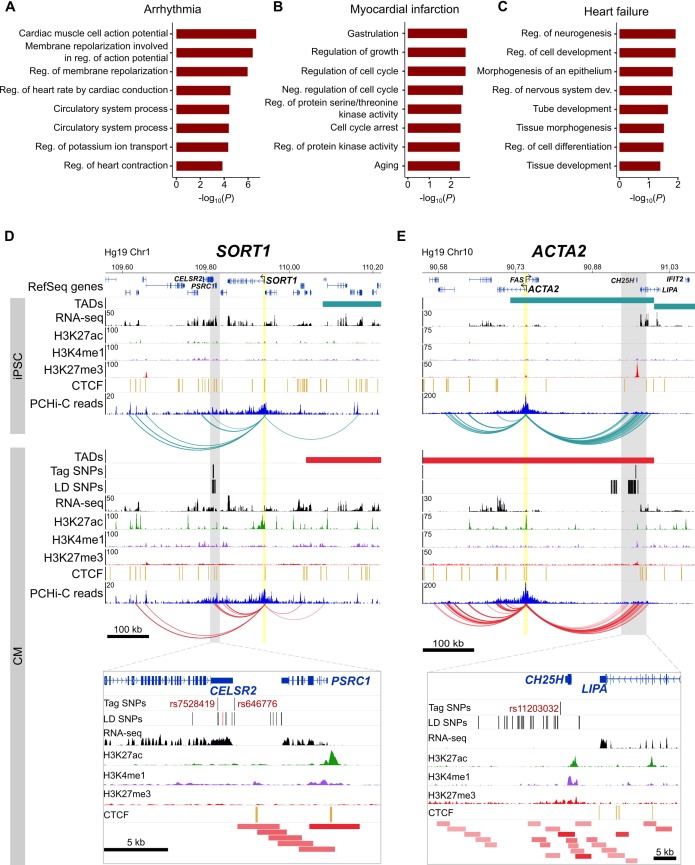
Figura 7.Rilevanza delle interazioni del promotore CM per l’aritmia cardiaca, l’infarto del miocardio e l’insufficienza cardiaca.(A-C) Gene Ontologia analisi per i geni target looping per (A) aritmia cardiaca SNPs, (B) SNPs infarto delmiocardio, e (C) insufficienza cardiaca SNPs.(D) Il promotore SORT1 loops a un locus infarto miocardico distale(Musunuru et al., 2010). Il rs12740374 SNP mostrato per interrompere un sito di legame C/EBP in(Musunuru et al., 2010) è colorato di rosso.(E) Il promotore ACTA2 si collega al locus 10q21 per lo scompenso cardiaco(Smith et al., 2010). Le trame dello zoom rappresentano l’intera regione interagente che si sovrappone ai GWAS LD SNPs. Per chiarezza, sono mostrate solo le interazioni per il gene indicato.
Nonostante questi processi apparentemente non specifici, ogni set di geni target conteneva importanti candidati rilevanti per la malattia. Ad esempio, una delle associazioni più forti per l’infarto del miocardio si trova tra i geni CELSR2 e PSRC1 sul cromosoma 1p13, ma uno schermo attento di geni la cui espressione è stata influenzata dall’allele di rischio implicava il gene SORT1 più distale(Musunuru et al., 2010). SORT1 codifica un recettore di ordinamento che si esprime in molti tessuti e che ha dimostrato di agire nel fegato per regolare i livelli di colesterolo(Petersen et al., 1997; Musunuru et al., 2010). Nonostante il funzionamento nel fegato, abbiamo identificato interazioni multiple del promotore tra SORT1 e il locus GWAS dell’infarto miocardico nelle CMs(Figura 7D), implicando direttamente SORT1 come gene target e dando ulteriore supporto alla validazione sperimentale di questo locus come potenziatore SORT1(Musunuru et al., 2010). Inoltre, il gene ACTA2 si trova a 220 kb di distanza dal locus GWAS per l’insufficienza cardiaca prossimale ai geni CH25H e LIPA sul cromosoma 10q21(Smith et al., 2010)(Figura 7E). ACTA2 codifica la proteina dell’actina specifica delle cellule muscolari lisce e le mutazioni di questo gene hanno dimostrato di causare, tra le altre malattie vascolari, una malattia coronarica(Guo et al., 2009). Nonostante la sua posizione ad una notevole distanza dall’associazione GWAS, le interazioni della cromatina forniscono un importante livello di evidenza che ACTA2 è un gene causale putativo nello sviluppo dello scompenso cardiaco. Pertanto, la mappa di interazione CM non è solo utile per interrogare malattie direttamente correlate ai cardiomiociti, come nel caso delle aritmie cardiache, ma aiuta anche l’interpretazione dei geni target che possono agire nei tessuti non cardiaci.
Figura 7.Rilevanza delle interazioni del promotore CM per l’aritmia cardiaca, l’infarto del miocardio e l’insufficienza cardiaca.(A-C) Gene Ontologia analisi per i geni target looping per (A) aritmia cardiaca SNPs, (B) SNPs infarto delmiocardio, e (C) insufficienza cardiaca SNPs.(D) Il promotore SORT1 loops a un locus infarto miocardico distale(Musunuru et al., 2010). Il rs12740374 SNP mostrato per interrompere un sito di legame C/EBP in(Musunuru et al., 2010) è colorato di rosso.(E) Il promotore ACTA2 si collega al locus 10q21 per lo scompenso cardiaco(Smith et al., 2010). Le trame dello zoom rappresentano l’intera regione interagente che si sovrappone ai GWAS LD SNPs. Per chiarezza, sono mostrate solo le interazioni per il gene indicato.
Discussione
La comprensione incompleta della regolazione dei geni a lungo raggio è un ostacolo importante nella traduzione dei loci associati al GWAS in biologia della malattia. Le sfide principali in questo processo includono l’identificazione di una presunta mappatura delle varianti causali all’interno degli elementi regolatori e il collegamento funzionale di questi elementi regolatori ai loro geni bersaglio. Per delineare le interazioni gene-regolamentazione tra SNPs associati a CVD e geni causali presunti, abbiamo generato mappe ad alta risoluzione delle interazioni del promotore nelle iPSC umane e nelle CM derivate da iPSC. Abbiamo dimostrato che i promotori interagiscono con un insieme diversificato di elementi del DNA distale in entrambi i tipi di cellule, comprese le sequenze di promotori noti, che riflettono l’identità delle cellule e corrispondono all’espressione genica specifica del tessuto. Per dimostrare l’utilità della mappa CM, abbiamo collegato 1.999 SNPs associati CVD a geni bersaglio causali presunti che hanno identificato sia i geni convalidati e potenzialmente nuovi importanti per la biologia delle malattie cardiovascolari. Per convalidare la rilevanza biologica delle nostre mappe, abbiamo affrontato diverse caratteristiche importanti delle interazioni a lungo raggio cromatina nelle analisi comparative.
I promotori contattano le regioni distali arricchite per motivi di fattori di trascrizione specifici dei tessuti
La regolazione dei geni mediante elementi regolatori a distanza comporta il collegamento di sequenze di DNA separate linearmente, ad esempio tra un promotore e i suoi stimolatori distali, attraverso meccanismi di looping della cromatina(Spitz e Furlong, 2012). A sostegno di questo modello, riportiamo un arricchimento dei motivi dei fattori di trascrizione che definiscono i tessuti nelle sequenze che interagiscono distalmente di promotori espressi in modo differenziato sia per le CM e le iPSC, fornendo un importante livello di evidenza per convalidare la rilevanza funzionale delle interazioni iPSC e CM. Una spiegazione di questo arricchimento è che le nostre mappe di interazione sono ad alta risoluzione. Abbiamo generato librerie Hi-C con il cutter MboI da 4 bp, che genera frammenti con una dimensione media di 422 bp; questa maggiore specificità della regione catturata porta probabilmente ad una migliore risoluzione della sottostante sequenza di enhancer e, di conseguenza, ad una maggiore potenza per rilevare brevi motivi di legame del fattore di trascrizione.
Influenza delle interazioni attive e repressive del promotore a livello di espressione genica
La maggior parte degli studi di cattura Hi-C finora hanno riportato che il livello di espressione genica è correlato con l’arricchimento di vari segni istonici. Abbiamo osservato la stessa tendenza nei nostri dati, con geni altamente espressi che mostrano un forte arricchimento per il looping nelle regioni distali marcate H3K4me1 e H3K27ac, e geni poco espressi che mostrano un forte arricchimento per il looping nelle regioni marcate H3K27me3. Questi dati sono coerenti con un modello in cui il numero di interazioni a lungo raggio con gli stimolatori o i repressori contribuisce ulteriormente al livello di espressione genica(Schoenfelder et al., 2015; Javierre et al., 2016). Le forze che guidano una maggiore associazione tra promotori ed elementi regolatori cis distali non sono completamente comprese e sono state oggetto di indagine nel campo dell’organizzazione del genoma e della biologia della cromatina per diversi anni(Dekker e Mirny, 2016; Calo e Wysocka, 2013). Una possibilità è che questo arricchimento crescente sia guidato dalla compartimentazione genomica della cromatina attiva e inattiva. Abbiamo dimostrato che il livello di espressione di un gene è correlato al numero di picchi di ChIP-seq dell’istone all’interno di una grande finestra (300 kb) che circonda ogni promotore. Così, i geni altamente espressi sono più propensi a contattare le regioni di cromatina attiva rispetto ai geni poco espressi, che corrispondono all’arricchimento crescente osservato dei contatti e dell’espressione che noi e altri abbiamo riportato. Questo aumento locale della cromatina attiva o repressiva può essere una delle forze trainanti alla base dell’aumento del livello di espressione dipendente dall’associazione tra i promotori e gli elementi regolatori cis, simile ad un modello di separazione di fase mediato da un modello di interazioni tra promotori e promotori(Hnisz et al., 2017).
Una mappa di interazione del promotore per la genetica delle malattie cardiovascolari
Abbiamo dimostrato diversi modi in cui i dati di interazione del promotore possono essere utilizzati per comprendere meglio la genetica delle malattie, affrontando in particolare il requisito principale per una mappa ad alta risoluzione della rete di regolazione genica nei cardiomiociti umani. Sebbene le CMs derivate da iPSC siano note per essere relativamente immature e non riflettano pienamente i diversi aspetti strutturali e funzionali delle cellule cardiache adulte(Gherghiceanu et al., 2011; Karakikes et al., 2015), la difficoltà di ottenere sottopopolazioni pure di cardiomiociti primari ad alta integrità richiede l’uso di un sistema in vitro. Abbiamo dimostrato che le CM usate in questo studio erano altamente pure e ricapitolano le proprietà di regolazione genica note dei cardiomiociti primari. Grazie a questa purezza, siamo stati in grado di integrare SNPs associati a CVD con interazioni del promotore CM con alta fiducia, assegnando quasi il 20% delle varianti in alta LD con queste associazioni a 347 geni target.
Sostenendo la rilevanza fisiologica delle CMs al sistema di conduzione cardiaca, abbiamo trovato che i geni target erano più rilevanti per i loci GWAS associati alle aritmie cardiache, in linea con le precedenti scoperte nelle cellule immunitarie che molte interazioni dei geni target erano uniche per i sottotipi di cellule immunitarie rilevanti(Javierre et al., 2016; Mumbach et al., 2017). I nostri dati hanno anche rivelato che anche per le malattie la cui eziologia coinvolge tipi di cellule diverse dai cardiomiociti, come l’infarto del miocardio e l’insufficienza cardiaca, abbiamo identificato interazioni che coinvolgono loci associati a queste malattie che ricapitolano le interazioni stimolatore-promotori in tipi di cellule non cardiache. Come esempio, abbiamo dimostrato che un locus convalidato di infarto miocardico interagisce con il promotore distale SORT1 nelle CM anche se questo locus è stato ampiamente caratterizzato nel contesto del metabolismo del colesterolo negli epatociti. Pertanto, le interazioni del promotore che osserviamo collegando il locus di malattia al SORT1 possono rappresentare un’architettura genomica invariante dei tessuti, che riflette probabilmente l’organizzazione del genoma in generale è relativamente stabile(Dixon et al., 2015; Jin et al., 2013; Ghavi-Helm et al., 2014). Mentre sosteniamo l’uso della mappa CM per lo studio dei meccanismi di regolazione genica delle malattie legate alla biologia dei cardiomiociti, sottolineiamo anche che, laddove identificata, qualsiasi interazione tra un promotore e una regione genomica associata a una malattia putativa serve come un importante livello di evidenza per dare priorità a quel gene per i futuri studi di follow-up.
Limitazioni delle mappe PCHi-C
La tecnica PCHi-C promette di identificare ad alta risoluzione e di produrre tutti gli elementi di regolazione genica in qualsiasi tessuto o stadio di sviluppo di interesse. Tuttavia, a causa di limitazioni tecniche e biologiche, ci sono importanti avvertimenti alla PCHi-C che dovrebbero essere presi in considerazione quando si interpretano i dati di interazione iPSC o CM. L’avvertenza più importante è che ci sono probabilmente molti falsi negativi, o interazioni “mancanti”. Anche se la fase di cattura arricchisce notevolmente per il promotore contenente frammenti di legatura in una biblioteca Hi-C, il panorama totale dei contatti del promotore in una popolazione di cellule è ancora sottocampionata, anche con una profondità di sequenziamento di ~ 400M legge per ogni replicato condotto per questo studio. Ciò è dovuto a diversi fattori, tra cui l’efficienza di ibridazione di ogni esca, la capacità di progettare un numero sufficiente di esche per promotore e la natura transitoria di molte interazioni normative. Quest’ultimo aspetto è confuso dall’effetto dipendente dalla distanza sulla frequenza di legatura: all’aumentare della distanza tra due frammenti, aumenta anche la profondità di lettura necessaria per identificare in modo robusto tale interazione. La fattibilità di sequenziamenti più profondi e di modifiche alle condutture computazionali continuerà a migliorare la copertura e la risoluzione dei dati Hi-C.
Inoltre, poiché il programma CHiCAGO non incorpora i confini del TAD nel suo modello di sfondo, potrebbe sottovalutare leggermente il numero previsto di letture corrispondenti alle interazioni intra-TAD che potrebbero portare a potenziali falsi positivi. Tuttavia, notiamo che esiste una forte corrispondenza tra le TAD chiamate su dati Hi-C pre-catturati e le interazioni PCHi-C identificate con CHiCAGO(Figura 1-figure supplement 3A); questo suggerisce che la contabilizzazione dei confini TAD può migliorare solo marginalmente la nostra capacità di identificare le interazioni significative.
Una considerazione finale è l’interpretazione delle interazioni che coinvolgono i geni inattivi. Anche se la maggior parte degli elementi normativi sono considerati come attivanti, è possibile che le interazioni a lungo raggio possano anche contribuire al silenziamento genico; ciò è supportato dall’osservazione che i geni silenziosi sono arricchiti per le interazioni a lungo raggio fino alle regioni marcate H3K27me3(Figura 3D). In alternativa, i geni silenziosi possono entrare in contatto con elementi regolatori che non sono attivi nel tipo di cellula analizzata o nella fase di sviluppo; questi possono rappresentare loop ‘preformati’ tra i geni e i loro elementi regolatori, come descritto in Ghavi-Helm et al. (2014).
Nonostante queste limitazioni, i set di dati che forniamo qui rappresentano un insieme altamente arricchito di ~350.000 e ~400.000 interazioni del promotore in iPSC e CMs, rispettivamente; anche se ci sono probabilmente interazioni mancanti, le interazioni che abbiamo identificato devono essere considerate come molto alta fiducia, in quanto sono stati identificati indipendentemente in almeno due repliche biologiche e mostrano un forte segnale di arricchimento per le caratteristiche note dell’architettura del genoma e la regolazione del gene. In conclusione, le mappe di interazione del promotore che abbiamo generato in questo studio rappresentano risorse importanti per qualsiasi indagine sui meccanismi di regolazione genica alla base dei tratti delle malattie cardiovascolari. L’elenco delle varianti di regolazione dei candidati e dei loro geni bersaglio può servire come punto di ingresso per diverse ipotesi relative al GWAS CVD, e può essere prontamente testato in contesti sperimentali. Per fornire sia le mappe iPSC che le mappe CM come risorsa accessibile, abbiamo ospitato l’insieme completo dei dati presentati in questo studio come un hub di pista pubblico presso il WashU EpiGenome Browser(Zhou et al., 2015), accessibile al seguente link: http://epigenomegateway.wustl.edu/browser/?genome=hg19&publichub=Lindsey. Inoltre, forniamo i significativi file di interazione PCHi-C utilizzati in tutte le analisi del materiale supplementare(file supplementari 1 e 2); questi possono essere applicati alle future analisi multiomiche della regolazione genica e della genetica delle malattie.
Materiali e metodi
| Tipo di reagente (specie) o risorsa | Designazione | Fonte o riferimento | Identificatori | Ulteriori informazioni |
|---|---|---|---|---|
| Linea cellulare(H. sapiens, maschio) | H19101 iPSC | 10.1101/gr.224436.117 | ||
| Anticorpo | Anti-acetilico Histone H3 (Lys27) (monoclonale del mouse) | Wako Chemicals (USA) | 306–34849 | H3K27ac ChIP-seq |
| Anticorpo | Troponina T anticardiaca (monoclonale del topo) | BD Bioscienze | 564767 | Citometria a flusso CM |
| Composto chimico, farmaco | ROCK Y-27632 diidrocloruro | Abcam | ab120129, 10 mg | Cultura tissutale iPSC |
| Composto chimico, farmaco | CHIR-99021 triidrocloruro | Tocris | 4953 | Differenziazione CM |
| Composto chimico, farmaco | Wnt-C59 | Tocris | 5148 | Differenziazione CM |
| Saggio commerciale o kit | TruSeq RNA libarary prep kit V2 | Illumina | RS-122-2001 | RNA-seq |
| Saggio commerciale o kit | NEBNext Multiplex Oligos per Illumina | NEB | E7335S | Ciao-C |
| Saggio commerciale o kit | Kit di trascrizione MEGAshortscript T7 | Thermo Fisher | AM135 | Generazione di sonde |
| Reagente a base di sequenza | Primer A | IDT | 5′-CTGGGAATCGGAATCGCACCCCCCCGTGTGT-3′ | Generazione di sonde |
| Reagente a base di sequenza | Primer B | IDT | 5′-CGTATGAGGATGAGCCGGCCAGTG-3′ | Generazione di sonde |
| Reagente a base di sequenza | Primer A T7 | IDT | 5′-GGATTCTAATACATACGACTCACT ATAGGGATCGATCGCACCCACCCCCCGTGTGT-3′. | Generazione di sonde |
| Reagente a base di sequenza | primer di bloccaggio P5 | IDT | 1016184 | Cattura Hi-C |
| Reagente a base di sequenza | primer di bloccaggio P7 | IDT | 1016186 | Cattura Hi-C |
Cultura tissutale degli iPSC
Abbiamo utilizzato la linea iPSC Yoruban iPSC 19101, gentilmente fornita dal laboratorio di Yoav Gilad. Questa linea iPSC è stato riprogrammato da cellule linfoblastoidi come parte di un precedente studio, dove è stato dimostrato di differenziare in tutti e tre gli strati di germe, ha mostrato un cariotipo normale, ed espresso marcatori caratteristici di pluripotenza(Banovich et al., 2018). iPSCs sono stati coltivati in Essential 8 (E8) Medium (Thermo Fisher #A1517001) integrato con 1X Penicillin-Streptomicina (Pen/Strep, Gibco) su Matrigel rivestiti piatti di coltura dei tessuti (Corning #354277). Le cellule sono state passate quando erano ~ 80% confluenti con soluzione di dissociazione senza enzimi (30 mM NaCl, 0,5 mM EDTA, 1X PBS meno magnesio e calcio) e mantenuto in E8 Medium con 10 μM Y-27632 diidrocloruro (Abcam #ab120129) per 24 ore. Medium è stato sostituito ogni giorno. colture iPSC regolarmente testato negativo per la contaminazione da micoplasma utilizzando il kit universale di rilevamento del micoplasma (ATCC # 30-1012K).
Differenziazione dei cardiomiociti
Le differenziazioni dei cardiomiociti erano basate sul protocollo di Burridge et al. (2014) con le modifiche descritte in Banovich et al .(Banovich et al., 2018). Gli iPSC sono stati espansi in piatti da 60 mm in media E8 fino a raggiungere una confluenza del 60-70%, al momento in cui la differenziazione è stata avviata (giorno 0). Il giorno 0, E8 media è stato sostituito con 10 mL di media cardiaci di base/12 μM GSK-3 inibitore CHIR-99021 triidrocloruro (Tocris #4953)/Matrigel overlay [media cardiaci di base: RPMI 1640 meno L-glutammina (HyClone #SH30096.01) con 1X GlutaMax (Life Technologies #11879020) integrato con 1X B27 meno insulina (Thermo Fisher #A1895601) e 1X Pen/Strep; la sovrapposizione Matrigel è stata realizzata sciogliendo Matrigel in 50 mL di media cardiaca di base ad una concentrazione di 0,5X secondo il fattore di diluizione specifico del lotto]. Dopo 24 ore (giorno 1), l’inibitore GSK-3 è stato rimosso sostituendo la media con 10 mL di media cardiaca di base. Il giorno 3, la media è stata sostituita con 10 mL di media cardiaca di base integrata con 2 μM Wnt-C59 (Tocris #5148). Il 5° giorno (48 ore dopo), i media sono stati sostituiti con 10 ml di media cardiaci di base. Il giorno 7, le cellule sono state lavate una volta con 1X PBS e poi sono stati aggiunti 15 mL di media cardiaca di base. I supporti sono stati sostituiti ogni due giorni in questo modo fino al 15° giorno in cui sono stati selezionati i cardiomiociti sostituendo i supporti cardiaci di base con 10 mL di lattato (RPMI 1640 meno D-glucosio, più L-glutammina (Life Technologies #11879020), integrata con 0.5 mg/mL di albumina umana ricombinante (Sigma 70024-90-7), 5 mM di sodio DL-lattato (Sigma 72-17-3), 213 μg/mL di acido L-ascorbico 2-fosfato (Sigma 70024-90-7) e 1X Pen/Strep). Lattato media è stato sostituito ogni due giorni fino al giorno 20 a quel punto sono stati raccolti cardiomiociti. Le cellule provenienti da differenziazioni di successo hanno mostrato un battito spontaneo intorno ai giorni 7-10.
I cardiomiociti sono stati raccolti lavando una volta con 1X PBS seguito da incubazione in 4 mL TrypLE (Life Technologies 12604-021) a 37°C per 5 min. Dopo l’incubazione, 4 mL di lattato è stato aggiunto al TrypLE e una pipetta da 1 mL è stata utilizzata per dislocare le cellule. Le cellule sono state filtrate una volta con un filtro da 100 μM e poi una volta con un filtro da 40 μM. Le cellule sono state pellettate a 500xg e poi risospese in PBS e contate. Per ogni lotto di differenziazione, 5 milioni di cellule sono state prese per la cattura del promotore Hi-C e 1 milione di cellule sono state prese per l’RNA-seq. Per valutare la purezza, 2 milioni di cellule sono state prese per l’analisi citometrica a flusso utilizzando un anticorpo per la troponina T cardiaca (BD Biosciences 564767). Tutte le cellule utilizzate negli esperimenti a valle erano almeno l’86% di Troponina T positiva(Figura 1-figure supplement 1A). Abbiamo effettuato tre differenziazioni indipendenti della stessa linea iPSC e generato promotore-cattura Hi-C e RNA-seq librerie in iPSC e CM da ogni triplice copia.
Acquisizione del promotore Hi-C
Reticolazione delle celle
Gli iPSC o cardiomiociti sono stati raccolti da piatti per la coltura dei tessuti e contati. Le cellule sono state risospese in 1X PBS ad una concentrazione di 1 milione di cellule/mL e il 37% di formaldeide è stato aggiunto ad una concentrazione finale dell’1%. La reticolazione è stata effettuata per 10 minuti a temperatura ambiente su una piattaforma oscillante. La glicina è stata aggiunta ad una concentrazione finale di 0,2 M per estinguere la reazione. Le cellule sono state pellettate, congelate a scatto in azoto liquido e conservate a -80°C fino a quando non sono state pronte per la lavorazione Hi-C.
in situ Hi-C
Abbiamo preparato tutte le librerie di cattura Hi-C del promotore in un unico lotto utilizzando tre pellet reticolati di 5 milioni di cellule sia per iPSC che per i cardiomiociti derivati da iPSC, che rappresentano tre differenziazioni indipendenti di cardiomiociti. La fase Hi-C in situ è stata eseguita come in Rao et al. (2014) con una singola modifica in cui sono stati utilizzati i reagenti NEBNext del kit NEBNext Multiplex Oligos for Illumina (NEB #E7335S) al posto degli adattatori Illumina, seguendo le istruzioni del produttore. Le librerie Hi-C sono state amplificate direttamente dalle perline T1 (Life Technologies #65602) utilizzando i primer NEBNext e sei cicli di PCR.
Cattura del promotore – progettazione e generazione della sonda
Le sonde di cattura Hi-C sono state progettate per mirare a quattro estremità di frammenti di restrizione MboI (120 bp) vicino al TSS dei geni RefSeq di codifica delle proteine(O’Leary et al., 2016) mappati a hg19 nel Genome Browser UCSC(Speir et al., 2016). Per selezionare i frammenti di restrizione, abbiamo mantenuto solo frammenti di restrizione MboI più lunghi di 200 bp e sovrapposti di 10 kb intorno a un RefSeq TSS. Per i TSS più vicini a 1 kb l’uno dall’altro, ne è stato mantenuto solo uno, in quanto le loro interazioni erano probabilmente catturate dagli altri TSS RefSeq. Le quattro estremità dei frammenti di restrizione MboI più vicine ad ogni RefSeq TSS sono state selezionate come sonde putativo. Le sequenze da 120 bp sono state sottoposte al software proprietario SureDesign di Agilent per la selezione delle sonde, che può spostare leggermente la posizione e rimuovere le sonde. In totale, abbiamo ordinato una libreria di 77.476 oligo-dna a filamento singolo da CustomArray, Inc.(www.customarrayinc.com). Ogni oligo consisteva della sequenza 5′-ATCGCGCACCAGGGTGTGTGTN120CACTGCGGGCTCCTCA-3′ (Gnirkeet al., 2009) dove N120 rappresenta i 120 nucleotidi adiacenti al sito di taglio MboI. L’elenco completo delle sonde oligo e il nome del gene corrispondente è fornito nel file supplementare 9.1.
Gli oligo sono arrivati come un pool contenente 1000 ng di materiale. Abbiamo usato 16 ng del pool di oligo in una reazione PCR per renderli a doppio filamento utilizzando primer 5′-CTGGGAATCGCCACCAGCGGTGTGT-3′ (Primer A), e 5′-CGTGGGATGAGGAGGAGGCCGCAGGTG-3′ (Primer B) come in (Gnirke et al.,2009). La reazione di PCR è stata pulita con perle AMPure XP (Agencourt #A6388) ed eluita con 20 μl di acqua. Per aggiungere il promotore T7 completo all’estremità 5′ degli oligo, è stata effettuata una seconda reazione di PCR utilizzando 10 ng del prodotto di PCR pulito di primo turno con il primer forward 5′-GGGATTCTAATACGACTCACTATAGGGGGATCGCACCAGGGTGTGT-3′ (Primer A T7). Abbiamo purificato il prodotto di PCR corrispondente a 176 bp utilizzando un kit di estrazione del gel Qiagen (#28704). Per generare esche di RNA biotinilato, abbiamo eseguito la trascrizione in vitro sulla libreria a doppio filamento utilizzando il MEGAshortscript T7 Transcription Kit (Thermo Fisher #AM135) con Biotin-16-dUTP (Sigma #11388908910). Dopo il trattamento DNase la reazione di trascrizione è stata pulita con il kit MEGAclear (Thermo Fisher #AM1908) ed eluita con 50 μl di tampone di eluizione. Abbiamo confermato la corretta dimensione dell’esca su un gel denaturante.
Cattura del promotore – ibridazione con la libreria Hi-C
Per isolare i frammenti contenenti frammenti del promotore dalla biblioteca Hi-C in situ dell’intero genoma, abbiamo ibridato la piscina di esche di RNA biotinilato con la biblioteca Hi-C come segue. Una miscela contenente 500 ng della biblioteca Hi-C, 2,5 microlitri di DNA Cot-1 umana (Invitrogen # 15279-011), 2,5 microlitri di DNA di sperma di salmone (Invitrogen # 15632-011), 0,5 microlitri di primer di blocco P5 (IDT #1016184), e 0,5 microlitri di primer di blocco P7 (IDT #1016186) è stato riscaldato per 5 min. a 95°, tenuto a 65° e miscelato con 13 μl di tampone di ibridazione preriscaldato (10X SSPE, 10X Denhardt’s, 10 mM EDTA e 0,2% SDS) e una miscela preriscaldata di 6 μl di 500 ng di esca RNA biotinilata e 20U SUPERase-In (Thermo Fisher #AM2694). La miscela di ibridazione è stata incubata per 24 ore a 65°C. Per isolare i frammenti catturati, abbiamo preparato 500 ng di perle magnetiche rivestite di streptavidina (Dynabeads MyOne Streptavidin T1, Thermo Fisher # 65601) in 200 μl di tampone legante (1M NaCl, 10 mM Tris-HCl pH 7,5, 1 mM EDTA). La miscela di ibridazione è stata aggiunta alle perle di Streptavidina e ruotata per 30 minuti a temperatura ambiente. Le perle contenenti i frammenti di Hi-C catturati sono stati lavati con 1X SSC, 0,1% SDS per 15 min a temperatura ambiente, seguito da tre lavaggi (10 min ciascuno) a 65 ° C con 0,1X SSC / 0,1% SDS. Dopo il lavaggio finale, le perle sono state risospese in 22 μl di acqua e si è proceduto alla post-captazione PCR. La reazione PCR è stata eseguita come prima, con 11 μl di ‘cattura Hi-C perline’ e 8 cicli di amplificazione. Una purificazione delle perle AMPure XP è stata utilizzata per pulire la reazione PCR e il DNA è stato quantificato utilizzando il sistema QuantiFluor dsDNA System (Promega #E2670) e un bioanalizzatore ad alta sensibilità. Le librerie Hi-C di acquisizione finale sono state sottoposte a 100 bp di sequenziamento paired-end su una macchina Illumina HiSeq 4000. I riepiloghi del conteggio letti sono forniti nel file supplementare 9.2.
9.2. Interazione che chiama
Abbiamo usato HiCUP v0.5.9(Wingett et al., 2015) per allineare e filtrare le letture Hi-C (i conteggi delle letture totali e filtrate sono presentati nel file supplementare 9.2). Le letture uniche sono state date a CHiCAGO versione 1.2.0(Cairns et al., 2016) e sono state chiamate interazioni significative con parametri predefiniti. In questo studio ci siamo concentrati esclusivamente sulle interazioni cisin quanto l’evidenza che le interazioni transcromosomiche contribuiscono alla regolazione dell’espressione genica è limitata. CHiCAGO riporta le interazioni per ogni frammento di restrizione catturato; per riassumere le interazioni per gene, abbiamo considerato l’intervallo che abbraccia tutti i frammenti catturati (cioè l’insieme di sonde che abbracciano ogni TSS) come la regione promotrice (“TSS fusa”). Ciò significa che le regioni promotrici create hanno lunghezze variabili. Nei casi in cui più geni sono stati annotati nella stessa regione promotrice, riportiamo l’interazione per ogni gene individualmente. Questa annotazione ci ha permesso di eseguire analisi a livello genico, ad esempio in base al livello di espressione. Abbiamo eliminato questa ridondanza se necessario, ad esempio nelle analisi di arricchimento del motivo dei frammenti che interagiscono con il promotore. Usando i file di interazione “merged TSS”, abbiamo filtrato le interazioni per mantenere quelle mappate entro 1 kb l’una dall’altra in almeno due repliche. In particolare, abbiamo esteso ogni frammento di promotore-interazione di 1 kb per ogni estremità e poi abbiamo usato la funzionalità pairToPair di BEDTools(Quinlan e Hall, 2010) per identificare le interazioni in cui entrambe le estremità corrispondevano tra i replicati. Per identificare le interazioni specifiche del tipo di cella, abbiamo richiesto che l’interazione (con l’estensione di 1 kb) non fosse presente in nessuno dei tre replicati dell’altro tipo di cella. Il numero di coppie di lettura per promotore e il numero corrispondente di interazioni significative identificate è presentato nel file supplementare 9.3. Le analisi TAD, l’arricchimento del motivo, l’arricchimento del picco ChIP-seq e le analisi eQTL (relative alle Figure 1, 2, 3 e 5) sono state condotte con interazioni a livello di frammentazione (nessuna estensione 1 kb). Le analisi GWAS SNP sono state condotte con interazioni con estensione 1kb, in quanto si è cercato di essere il più inclusivi possibile nel collegare gli SNP CVD ai geni target.
Le interazioni PCHi-C, TAD, RNA-seq, ChIP-seq disponibili al pubblico, e GWAS SNPs sono ospitate dal WashU EpiGenome Browser(Zhou et al., 2015) come hub pubblico. È possibile accedere a questo sito web all’indirizzo http://epigenomegateway.wustl.edu/browser/. L’hub pubblico (“A promoter interaction map for cardiovascular disease genetics”) si trova sotto il browser Human Hg19.
Trame in stile 4C
Per generare i conteggi di lettura dei geni visualizzati nelle figure del navigatore genome-browser, tutte le coppie di lettura mappate per catturare i frammenti MboI per un determinato promotore sono state sommate attraverso i replicati. In particolare, abbiamo sommato le letture per ogni frammento MboI in cui la lettura era parte di una coppia di lettura che mappata ad un’esca per il gene dato. Gli archi che sono visualizzati sotto la trama in stile 4C rappresentano interazioni significative che sono state identificate in almeno due repliche come descritto sopra in ‘Interaction calling’.
Analisi TAD
Per identificare i TAD, abbiamo raggruppato le letture attraverso i replicati per ogni tipo di cella utilizzando i dati Hi-C di pre-acquisizione (600M letture per iPSC e 733M letture per CM) e utilizzato HiCUP v0.5.9(Wingett et al., 2015) per allineare e filtrare le letture Hi-C. HOMER v4.8.3(Heinz et al., 2010) è stato utilizzato per generare matrici di interazione normalizzate ad una risoluzione di 40 kb e poi TopDom v0.0.2(Shin et al., 2016) è stato utilizzato con una dimensione di finestra w = 10 per identificare domini topologici, confini e lacune. Abbiamo considerato solo i domini per le analisi di questo documento. Abbiamo considerato un’interazione di cattura del promotore Hi-C come ‘intra-TAD’ se l’intero arco dell’interazione era completamente contenuto in un singolo dominio. Le interazioni ‘Inter-TAD’ sono definite come interazioni in cui ogni estremità mappa un dominio diverso.
Scomparti A/B
Il programma runHiCpca.pl del pacchetto HOMER(Heinz et al., 2010) v4.8.3 è stato utilizzato per chiamare i compartimenti A/B con -res 50000 sia per il genoma intero che per l’acquisizione di dati Hi-C.
RNA-seq
L’RNA totale è stato estratto da pellet congelati in flash di 1 milione di cellule utilizzando TRI Reagent (Sigma #T9424) e un omogeneizzatore seguito dall’isolamento dell’RNA e dalla pulizia con il Direct-zol RNA Kit (Zymo Research #11-331). Le librerie di RNA-seq sono state generate con il kit Illumina TruSeq V2 (Illumina, RS-122-2001) e 1 μg di RNA, seguendo le istruzioni del produttore. Le librerie sono state realizzate con RNA isolato da tre differenziazioni indipendenti iPSC-CM (triplicati di iPSC e di cardiomiociti). Le librerie sono state sequenziate su un Illumina HiSeq 4000.
I conteggi dei geni sono stati quantificati con Salmon 0.7.2(Patro et al., 2017) e importati con tximport 1.2.0(Soneson et al., 2015) in DESeq2 1.12.4(Love et al., 2014) per chiamare i geni espressi differenzialmente. Per selezionare i geni espressi differenzialmente per le analisi a valle è stata richiesta una differenza minima di 1,5 volte tra CM e triplicati iPSC e un valore p minimo regolato di 0,05 per selezionare i geni espressi differenzialmente per le analisi a valle. TPMs (trascrizioni per milione) sono stati anche stimati da Salmon. Poiché i campioni chiaramente raggruppati in base ai loro tessuti noti di origine(Figura 1-figure supplement 2A), non è stata eseguita alcuna correzione per gli effetti del lotto.
H3K27ac ChIP-seq per il confronto con i campioni della tabella di marcia epigenoma
Abbiamo eseguito ChIP-seq su 2,5 milioni di cellule ciascuna per iPSC e CMs utilizzando anticorpi H3K27ac (Wako #306-34849). In breve, le cellule sono state reticolate con l’1% di formaldeide per 10 minuti a temperatura ambiente, estinte con glicina 0,2M per 5 minuti, pellettate e congelate a scatto in azoto liquido. Le cellule sono state lisate in Lysis Buffer 1 (50 mM HEPES-KOH, pH 7,5, 140 mM NaCl, 1 mM EDTA, 10% glicerolo, 0,5% NP-40, 0,25% Triton X-100). Cromatina reticolata è stato tosato ad una dimensione media di 300 bp utilizzando un Bioruptor con 30 “on / 30” off ad alta impostazione e poi incubato durante la notte a 4 ° C con 1 microgrammo di anticorpo. Dynabeads M-280 Sheep Anti-Mouse IgG (ThermoFisher # 11201D) sono stati utilizzati per tirare giù la cromatina e ChIP DNA è stato eluito e preparato per il sequenziamento utilizzando il kit di preparazione NEBNext Ultra II DNA Library (NEB #E7645S). Le letture ChIP-seq sono state allineate con Bowtie 2-2.2.3 (Langmeade Salzberg, 2012) e i picchi sono stati chiamati con HOMER (Heinzet al., 2010) v4.8.3 su letture uniche con qualità di mappatura >10 utilizzando i parametri dell’istone -regione e -stile -. Picchi significativi sono stati sovrapposti con picchi di H3K27ac da campioni di Epigenome Roadmap che hanno dimostrato un’elevata concordanza tra i tipi di tessuto corrispondenti(Figura 1-figure supplement 2C,D). Poiché abbiamo eseguito un basso livello di sequenziamento, non abbiamo identificato tanti picchi quanti sono i campioni della Roadmap. Pertanto, abbiamo utilizzato i dati Roadmap ChIP-seq in tutte le nostre analisi.
Analisi dell’ontologia genica
Le associazioni di Gene Ontologia umana (GO) dei termini GO(Ashburner et al., 2000) ai geni e il database GO sono stati scaricati il 22 gennaio 2016 da http://geneontology.org/gene-associations. I termini GO sono stati associati ai geni RefSeq tramite simboli genici. Utilizzando il grafico di annotazione GO, tutti i termini dei genitori sono stati assegnati ai termini annotati a un gene. Un test ipergeometrico è stato utilizzato per calcolare la significatività statistica della differenza del numero di geni associati a un dato termine GO in un particolare set di geni e l’universo di tutti i geni RefSeq (p<0,05). p-Valori p sono stati corretti con la funzione p.adjust del pacchetto R utilizzando il metodo ‘fdr’.
Per due dei gruppi di malattie GWAS (insufficienza cardiaca e infarto del miocardio), l’elenco dei geni che fanno il looping dei geni LD SNPs comprendeva molti geni istonici. Questo perché c’è un tag SNP situato al centro di un gruppo di geni istonici (contenente >30 geni istonici situati vicini) in ogni caso. Dopo aver espanso il tag SNP a tutti gli SNP in LD, molti dei geni degli istoni in quel cluster si sono collegati in loop agli SNP LD, ottenendo un’alta rappresentazione di questi geni nella lista finale dei geni. L’analisi di arricchimento dell’ontologia genica risultante ha dato termini relativi all’organizzazione del nucleosoma e della cromatina a causa di questa sovrarappresentazione. Abbiamo quindi scelto di rimuovere questi geni dalle liste genetiche finali dei geni bersaglio dell’insufficienza cardiaca e dell’infarto del miocardio.
Analisi del motivo
Il programma findMotifsGenome.pl del pacchetto HOMER(Heinz et al., 2010) v4.8.3 è stato utilizzato con il parametro -size given parameter per identificare i motivi sovrarappresentati nelle sequenze di interazioni distali (non promoter) che interagiscono con i promotori. Come detto in precedenza, questa analisi è stata eseguita su sequenze frammentarie a livello di promotore-interazione.
Analisi dell’arricchimento di Histone ChIP-seq
Abbiamo ottenuto i dati di ChIP-seq disponibili al pubblico sotto forma di chiamate di picco elaborate per H3K27ac, H3K4me1 e H3K27me3 dal Roadmap Epigenomics Project(Kundaje et al., 2015), e per CTCF da ENCODE(ENCODE Project Consortium, 2012)(file supplementare 10). Abbiamo considerato solo i picchi mappati al di fuori della regione catturata dai promotori per garantire che i nostri risultati non fossero guidati dal forte segnale di picco sulla maggior parte dei promotori. Come proxy per le iPSC, abbiamo usato i dati della linea di cellule staminali embrionali H1 e per le CM abbiamo usato i dati del tessuto del ventricolo sinistro. Abbiamo raggruppato i geni in cinque categorie di espressione in base ai valori medi di TPM: gruppo 1 (0 TPM), gruppo 2 (TPM 0-3), gruppo 3 (TPM 3-25), gruppo 4 (TPM 25-150) e gruppo 5 (TPM >150) e per ogni gruppo di geni, abbiamo calcolato l’arricchimento per le interazioni del promotore per sovrapporre una data caratteristica. Per calcolare l’arricchimento delle interazioni che si sovrappongono ad una caratteristica epigenetica, abbiamo confrontato la proporzione osservata di frammenti MboI in interazioni significative che si sovrappongono ad una caratteristica con la proporzione di frammenti MboI casuali che si sovrappongono alla caratteristica. In particolare, abbiamo selezionato in modo casuale frammenti MboI da un insieme che escludeva la mappatura dei frammenti all’interno delle regioni catturate (promotori) o all’interno di regioni genomiche non mappabili (lacune). Il numero di frammenti selezionati casualmente corrispondeva al numero di frammenti interagenti considerati per l’analisi. Abbiamo eseguito 100 iterazioni di frammenti casuali sovrapposti con una caratteristica e abbiamo riportato l’arricchimento medio delle pieghe. Ci riferiamo a questo metodo di arricchimento come modello di sfondo ‘a livello genomico’, perché per ogni gruppo di espressione genica, la proporzione osservata di frammenti contenenti un picco viene confrontata con frammenti selezionati casualmente dall’intero genoma.
Per calcolare la correlazione tra l’espressione e l’istone ChIP-seq densità di picco, abbiamo calcolato la correlazione del rango di Spearman tra il valore di espressione per ogni gene (il valore medio TPM) e il numero di picchi mappatura entro 300 kb di ogni gene TSS. Abbiamo considerato solo i geni con almeno una interazione significativa nel rispettivo tipo di cellula per consentire generalizzazioni all’analisi di arricchimento presentata in Figura 3.
Analisi GWAS
Abbiamo compilato SNPs significativi a livello genomico associati al GWAS per l’aritmia cardiaca, l’insufficienza cardiaca e l’infarto del miocardio dal database NHGRI-EBI(http://www.ebi.ac.uk/gwas/); vedere il file supplementare 7 per l’elenco dei termini utilizzati per identificare specifici GWAS. Abbiamo esteso ogni set di SNP a tutti gli SNP in alta LD (r2 >0,9) utilizzando i dati della fase 3 del progetto 1000 genomi (Nikpayet al., 2015) (Filesupplementare3). Per ogni SNP di piombo dal GWAS che abbiamo analizzato, abbiamo selezionato un intervallo di 100 kb centrato sull’SNP (SNP ± 50 kb). Per ogni intervallo di 100 kb, è stato usato il Tabix(Li, 2011) per recuperare i genotipi. Abbiamo poi usato PLINK(Purcell et al., 2007) v1.90p sui dati della fase tre del progetto 1000 genomi(Nikpay et al., 2015) (ftp.1000genomes.ebi.ac.uk/vol1/ftp/release/20130502, v5a) per selezionare gli SNP in LD (r2 >0.9) con il tag SNP e una frequenza minima di allele di 0.01. Abbiamo incluso solo le popolazioni studiate principalmente nei GWAS: CEU (Europa centrale), ASW (Afroamericana) e JPT (Giapponese). Abbiamo assegnato tutti gli SNP nelle interazioni promotore-distale ai loro geni interagenti (“geni bersaglio”) utilizzando i dati di cattura Hi-C del promotore dei cardiomiociti. Non abbiamo richiesto l’SNP per la mappatura delle regioni associate a cromatina aperta o marchi di rinforzo, in quanto questi tipi di dati sono altamente specifici per il tipo di cellula e non abbiamo voluto escludere gli SNP in regioni che possono essere attive in tipi di cellule non saggiate.
Notiamo che uno dei principali GWAS per la cardiomiopatia dilatativa non è stato incluso nel database NHGRI-EBI(Meder et al., 2014), probabilmente perché c’è un errore nell’ottenere i metodi online del documento. Dopo un’attenta ispezione dello studio, abbiamo concluso che il GWAS soddisfaceva i criteri NHGRI-EBI e abbiamo incluso nella nostra analisi le associazioni di quello studio. Un elenco completo di tutti gli studi utilizzati in questa analisi si trova nel file supplementare 8.
Analisi MGI
Per calcolare l’arricchimento dei geni target per causare fenotipi cardiovascolari quando vengono eliminati nei topi (Mouse Genome Genome Informatics database), abbiamo selezionato a caso 347 geni dalla lista dei geni di partenza (cioè geni con almeno un’interazione promotore-distale nelle CMs, il che significa che potrebbe essere un gene target), e calcolato la proporzione che ha causato un fenotipo cardiovascolare nei topi. Abbiamo eseguito questa selezione randomizzata per 1000 iterazioni per generare i valori randomizzati (attesi). I geni casuali non dovevano essere espressi, in quanto l’insieme dei geni target contiene geni che non sono espressi. p-Valore è stato calcolato con un test Z.
analisi eQTL
Per gli eQTL utilizzati nei confronti delle varianti GWAS e delle interazioni Hi-C, abbiamo utilizzato l’insieme degli eQTL GTEx v7 identificati come significativi nel ventricolo sinistro del cuore(Carithers et al., 2015). Gli eQTL sono stati chiamati significativi se q < 0,05 dopo la correzione del tasso di falsa scoperta (Storey eTibshirani, 2003). Abbiamo considerato solo gli eQTL promotori-distali che erano almeno 10 kb dal loro gene associato per consentire a quell’eQTL di mappare un’interazione con il suo gene associato.
Per calcolare l’arricchimento per gli eQTL da collegare al loro gene associato, abbiamo usato un modello di sfondo in cui l’insieme delle interazioni di ogni promotore è stato mappato di nuovo su un promotore diverso, mantenendo la distanza e l’orientamento del filo coerenti. Abbiamo eseguito questa mappatura di tutte le interazioni del promotore 1000 volte e calcolato la proporzione di tutti gli eQTL che mappano le interazioni per il loro gene associato ad eQTL in ogni permutazione. Abbiamo usato le interazioni CM o le interazioni iPSC con lo stesso insieme di eQTL del ventricolo sinistro per confrontare la specificità del tipo di cellula dei dati di interazione del promotore.
Disponibilità dei dati
I dati di sequenziamento grezzi ed elaborati sono forniti su ArrayExpress attraverso i numeri di adesione E-MTAB-6014 (Hi-C) e E-MTAB-6013 (RNA-seq).
References
- Arking DE, Pulit SL, Crotti L, van der Harst P, Munroe PB, Koopmann TT, Sotoodehnia N, Rossin EJ, Morley M, Wang X, Johnson AD, Lundby A, Gudbjartsson DF, Noseworthy PA, Eijgelsheim M, Bradford Y, Tarasov KV, Dörr M, Müller-Nurasyid M, Lahtinen AM, Nolte IM, Smith AV, Bis JC, Isaacs A, Newhouse SJ, Evans DS, Post WS, Waggott D, Lyytikäinen LP, Hicks AA, Eisele L, Ellinghaus D, Hayward C, Navarro P, Ulivi S, Tanaka T, Tester DJ, Chatel S, Gustafsson S, Kumari M, Morris RW, Naluai ÅT, Padmanabhan S, Kluttig A, Strohmer B, Panayiotou AG, Torres M, Knoflach M, Hubacek JA, Slowikowski K, Raychaudhuri S, Kumar RD, Harris TB, Launer LJ, Shuldiner AR, Alonso A, Bader JS, Ehret G, Huang H, Kao WH, Strait JB, Macfarlane PW, Brown M, Caulfield MJ, Samani NJ, Kronenberg F, Willeit J, Smith JG, Greiser KH, Meyer Zu Schwabedissen H, Werdan K, Carella M, Zelante L, Heckbert SR, Psaty BM, Rotter JI, Kolcic I, Polašek O, Wright AF, Griffin M, Daly MJ, Arnar DO, Hólm H, Thorsteinsdottir U, Denny JC, Roden DM, Zuvich RL, Emilsson V, Plump AS, Larson MG, O’Donnell CJ, Yin X, Bobbo M, D’Adamo AP, Iorio A, Sinagra G, Carracedo A, Cummings SR, Nalls MA, Jula A, Kontula KK, Marjamaa A, Oikarinen L, Perola M, Porthan K, Erbel R, Hoffmann P, Jöckel KH, Kälsch H, Nöthen MM, den Hoed M, Loos RJ, Thelle DS, Gieger C, Meitinger T, Perz S, Peters A, Prucha H, Sinner MF, Waldenberger M, de Boer RA, Franke L, van der Vleuten PA, Beckmann BM, Martens E, Bardai A, Hofman N, Wilde AA, Behr ER, Dalageorgou C, Giudicessi JR, Medeiros-Domingo A, Barc J, Kyndt F, Probst V, Ghidoni A, Insolia R, Hamilton RM, Scherer SW, Brandimarto J, Margulies K, Moravec CE, del Greco M F, Fuchsberger C, O’Connell JR, Lee WK, Watt GC, Campbell H, Wild SH, El Mokhtari NE, Frey N, Asselbergs FW, Mateo Leach I, Navis G, van den Berg MP, van Veldhuisen DJ, Kellis M, Krijthe BP, Franco OH, Hofman A, Kors JA, Uitterlinden AG, Witteman JC, Kedenko L, Lamina C, Oostra BA, Abecasis GR, Lakatta EG, Mulas A, Orrú M, Schlessinger D, Uda M, Markus MR, Völker U, Snieder H, Spector TD, Ärnlöv J, Lind L, Sundström J, Syvänen AC, Kivimaki M, Kähönen M, Mononen N, Raitakari OT, Viikari JS, Adamkova V, Kiechl S, Brion M, Nicolaides AN, Paulweber B, Haerting J, Dominiczak AF, Nyberg F, Whincup PH, Hingorani AD, Schott JJ, Bezzina CR, Ingelsson E, Ferrucci L, Gasparini P, Wilson JF, Rudan I, Franke A, Mühleisen TW, Pramstaller PP, Lehtimäki TJ, Paterson AD, Parsa A, Liu Y, van Duijn CM, Siscovick DS, Gudnason V, Jamshidi Y, Salomaa V, Felix SB, Sanna S, Ritchie MD, Stricker BH, Stefansson K, Boyer LA, Cappola TP, Olsen JV, Lage K, Schwartz PJ, Kääb S, Chakravarti A, Ackerman MJ, Pfeufer A, de Bakker PI, Newton-Cheh C, CARe Consortium, COGENT Consortium, DCCT/EDIC, eMERGE Consortium, HRGEN Consortium. Genetic association study of QT interval highlights role for calcium signaling pathways in myocardial repolarization. Nature Genetics. 2014; 46:826-836. DOI | PubMed
- Arnolds DE, Liu F, Fahrenbach JP, Kim GH, Schillinger KJ, Smemo S, McNally EM, Nobrega MA, Patel VV, Moskowitz IP. TBX5 drives Scn5a expression to regulate cardiac conduction system function. Journal of Clinical Investigation. 2012; 122:2509-2518. DOI | PubMed
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G. Gene ontology: tool for the unification of biology. the gene ontology consortium. Nature Genetics. 2000; 25:25-29. DOI | PubMed
- Banovich NE, Li YI, Raj A, Ward MC, Greenside P, Calderon D, Tung PY, Burnett JE, Myrthil M, Thomas SM, Burrows CK, Romero IG, Pavlovic BJ, Kundaje A, Pritchard JK, Gilad Y. Impact of regulatory variation across human iPSCs and differentiated cells. Genome Research. 2018; 28:122-131. DOI | PubMed
- Blake JA, Eppig JT, Kadin JA, Richardson JE, Smith CL, Bult CJ, the Mouse Genome Database Group. Mouse Genome Database (MGD)-2017: community knowledge resource for the laboratory mouse. Nucleic Acids Research. 2017; 45:D723-D729. DOI | PubMed
- Burridge PW, Matsa E, Shukla P, Lin ZC, Churko JM, Ebert AD, Lan F, Diecke S, Huber B, Mordwinkin NM, Plews JR, Abilez OJ, Cui B, Gold JD, Wu JC, Jc W. Chemically defined generation of human cardiomyocytes. Nature Methods. 2014; 11:855-860. DOI | PubMed
- Cai CL, Zhou W, Yang L, Bu L, Qyang Y, Zhang X, Li X, Rosenfeld MG, Chen J, Evans S. T-box genes coordinate regional rates of proliferation and regional specification during cardiogenesis. Development. 2005; 132:2475-2487. DOI | PubMed
- Cairns J, Freire-Pritchett P, Wingett SW, Várnai C, Dimond A, Plagnol V, Zerbino D, Schoenfelder S, Javierre BM, Osborne C, Fraser P, Spivakov M. CHiCAGO: robust detection of DNA looping interactions in Capture Hi-C data. Genome Biology. 2016; 17DOI | PubMed
- Calo E, Wysocka J. Modification of enhancer chromatin: what, how, and why?. Molecular Cell. 2013; 49:825-837. DOI | PubMed
- Carithers LJ, Ardlie K, Barcus M, Branton PA, Britton A, Buia SA, Compton CC, DeLuca DS, Peter-Demchok J, Gelfand ET, Guan P, Korzeniewski GE, Lockhart NC, Rabiner CA, Rao AK, Robinson KL, Roche NV, Sawyer SJ, Segrè AV, Shive CE, Smith AM, Sobin LH, Undale AH, Valentino KM, Vaught J, Young TR, Moore HM, GTEx Consortium. A novel approach to high-quality postmortem tissue procurement: The GTEx project. Biopreservation and Biobanking. 2015; 13:311-319. DOI | PubMed
- Claussnitzer M, Dankel SN, Kim KH, Quon G, Meuleman W, Haugen C, Glunk V, Sousa IS, Beaudry JL, Puviindran V, Abdennur NA, Liu J, Svensson PA, Hsu YH, Drucker DJ, Mellgren G, Hui CC, Hauner H, Kellis M. FTO obesity variant circuitry and Adipocyte Browning in humans. New England Journal of Medicine. 2015; 373:895-907. DOI | PubMed
- Cowper-Sal lari R, Zhang X, Wright JB, Bailey SD, Cole MD, Eeckhoute J, Moore JH, Lupien M. Breast cancer risk-associated SNPs modulate the affinity of chromatin for FOXA1 and alter gene expression. Nature Genetics. 2012; 44:1191-1198. DOI | PubMed
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, Boyer LA, Young RA, Jaenisch R. Histone H3K27ac separates active from poised enhancers and predicts developmental state. PNAS. 2010; 107:21931-21936. DOI | PubMed
- Dao LTM, Galindo-Albarrán AO, Castro-Mondragon JA, Andrieu-Soler C, Medina-Rivera A, Souaid C, Charbonnier G, Griffon A, Vanhille L, Stephen T, Alomairi J, Martin D, Torres M, Fernandez N, Soler E, van Helden J, Puthier D, Spicuglia S. Genome-wide characterization of mammalian promoters with distal enhancer functions. Nature Genetics. 2017; 49:1073-1081. DOI | PubMed
- Dekker J, Mirny L. The 3D genome as moderator of chromosomal communication. Cell. 2016; 164:1110-1121. DOI | PubMed
- Deng W, Lee J, Wang H, Miller J, Reik A, Gregory PD, Dean A, Blobel GA, a BG. Controlling long-range genomic interactions at a native locus by targeted tethering of a looping factor. Cell. 2012; 149:1233-1244. DOI | PubMed
- Diao Y, Fang R, Li B, Meng Z, Yu J, Qiu Y, Lin KC, Huang H, Liu T, Marina RJ, Jung I, Shen Y, Guan KL, Ren B. A tiling-deletion-based genetic screen for cis-regulatory element identification in mammalian cells. Nature Methods. 2017; 14:629-635. DOI | PubMed
- Dixon JR, Jung I, Selvaraj S, Shen Y, Antosiewicz-Bourget JE, Lee AY, Ye Z, Kim A, Rajagopal N, Xie W, Diao Y, Liang J, Zhao H, Lobanenkov VV, Ecker JR, Thomson JA, Ren B. Chromatin architecture reorganization during stem cell differentiation. Nature. 2015; 518:331-336. DOI | PubMed
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature. 2012; 485:376-380. DOI | PubMed
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57-74. DOI | PubMed
- Erceg J, Pakozdi T, Marco-Ferreres R, Ghavi-Helm Y, Girardot C, Bracken AP, Furlong EE. Dual functionality of cis-regulatory elements as developmental enhancers and Polycomb response elements. Genes & Development. 2017; 31:590-602. DOI | PubMed
- Franke M, Ibrahim DM, Andrey G, Schwarzer W, Heinrich V, Schöpflin R, Kraft K, Kempfer R, Jerković I, Chan WL, Spielmann M, Timmermann B, Wittler L, Kurth I, Cambiaso P, Zuffardi O, Houge G, Lambie L, Brancati F, Pombo A, Vingron M, Spitz F, Mundlos S. Formation of new chromatin domains determines pathogenicity of genomic duplications. Nature. 2016; 538:265-269. DOI | PubMed
- Freire-Pritchett P, Schoenfelder S, Várnai C, Wingett SW, Cairns J, Collier AJ, García-Vílchez R, Furlan-Magaril M, Osborne CS, Fraser P, Rugg-Gunn PJ, Spivakov M. Global reorganisation of cis-regulatory units upon lineage commitment of human embryonic stem cells. eLife. 2017; 6DOI | PubMed
- Ghavi-Helm Y, Klein FA, Pakozdi T, Ciglar L, Noordermeer D, Huber W, Furlong EE. Enhancer loops appear stable during development and are associated with paused polymerase. Nature. 2014; 512:100. DOI | PubMed
- Gherghiceanu M, Barad L, Novak A, Reiter I, Itskovitz-Eldor J, Binah O, Popescu LM. Cardiomyocytes derived from human embryonic and induced pluripotent stem cells: comparative ultrastructure. Journal of Cellular and Molecular Medicine. 2011; 15:2539-2551. DOI | PubMed
- Gilbert N, Boyle S, Fiegler H, Woodfine K, Carter NP, Bickmore WA. Chromatin architecture of the human genome: gene-rich domains are enriched in open chromatin fibers. Cell. 2004; 118:555-566. DOI | PubMed
- Gnirke A, Melnikov A, Maguire J, Rogov P, LeProust EM, Brockman W, Fennell T, Giannoukos G, Fisher S, Russ C, Gabriel S, Jaffe DB, Lander ES, Nusbaum C. Solution hybrid selection with ultra-long oligonucleotides for massively parallel targeted sequencing. Nature Biotechnology. 2009; 27:182-189. DOI | PubMed
- Guo DC, Papke CL, Tran-Fadulu V, Regalado ES, Avidan N, Johnson RJ, Kim DH, Pannu H, Willing MC, Sparks E, Pyeritz RE, Singh MN, Dalman RL, Grotta JC, Marian AJ, Boerwinkle EA, Frazier LQ, LeMaire SA, Coselli JS, Estrera AL, Safi HJ, Veeraraghavan S, Muzny DM, Wheeler DA, Willerson JT, Yu RK, Shete SS, Scherer SE, Raman CS, Buja LM, Milewicz DM. Mutations in smooth muscle alpha-actin (ACTA2) cause coronary artery disease, stroke, and Moyamoya disease, along with thoracic aortic disease. The American Journal of Human Genetics. 2009; 84:617-627. DOI | PubMed
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, Ching KA, Antosiewicz-Bourget JE, Liu H, Zhang X, Green RD, Lobanenkov VV, Stewart R, Thomson JA, Crawford GE, Kellis M, Ren B. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009; 459:108-112. DOI | PubMed
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Molecular Cell. 2010; 38:576-589. DOI | PubMed
- Hnisz D, Shrinivas K, Young RA, Chakraborty AK, Sharp PA. A phase separation model for transcriptional control. Cell. 2017; 169:13-23. DOI | PubMed
- Javierre BM, Burren OS, Wilder SP, Kreuzhuber R, Hill SM, Sewitz S, Cairns J, Wingett SW, Várnai C, Thiecke MJ, Burden F, Farrow S, Cutler AJ, Rehnström K, Downes K, Grassi L, Kostadima M, Freire-Pritchett P, Wang F, Stunnenberg HG, Todd JA, Zerbino DR, Stegle O, Ouwehand WH, Frontini M, Wallace C, Spivakov M, Fraser P, BLUEPRINT Consortium. Lineage-Specific genome architecture links enhancers and Non-coding disease variants to target gene promoters. Cell. 2016; 167:1369-1384. DOI | PubMed
- Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY, Yen CA, Schmitt AD, Espinoza CA, Ren B. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature. 2013; 503:290-294. DOI | PubMed
- Karakikes I, Ameen M, Termglinchan V, Wu JC. Human induced pluripotent stem cell-derived cardiomyocytes: insights into molecular, cellular, and functional phenotypes. Circulation Research. 2015; 117:80-88. DOI | PubMed
- Karlić R, Chung HR, Lasserre J, Vlahovicek K, Vingron M. Histone modification levels are predictive for gene expression. PNAS. 2010; 107:2926-2931. DOI | PubMed
- Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu YC, Pfenning AR, Wang X, Claussnitzer M, Liu Y, Coarfa C, Harris RA, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, Hawkins RD, Lister R, Hong C, Gascard P, Mungall AJ, Moore R, Chuah E, Tam A, Canfield TK, Hansen RS, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh KH, Feizi S, Karlic R, Kim AR, Kulkarni A, Li D, Lowdon R, Elliott G, Mercer TR, Neph SJ, Onuchic V, Polak P, Rajagopal N, Ray P, Sallari RC, Siebenthall KT, Sinnott-Armstrong NA, Stevens M, Thurman RE, Wu J, Zhang B, Zhou X, Beaudet AE, Boyer LA, De Jager PL, Farnham PJ, Fisher SJ, Haussler D, Jones SJ, Li W, Marra MA, McManus MT, Sunyaev S, Thomson JA, Tlsty TD, Tsai LH, Wang W, Waterland RA, Zhang MQ, Chadwick LH, Bernstein BE, Costello JF, Ecker JR, Hirst M, Meissner A, Milosavljevic A, Ren B, Stamatoyannopoulos JA, Wang T, Kellis M, Roadmap Epigenomics Consortium. Integrative analysis of 111 reference human epigenomes. Nature. 2015; 518:317-330. DOI | PubMed
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nature Methods. 2012; 9:357-359. DOI | PubMed
- Li H. Tabix: fast retrieval of sequence features from generic TAB-delimited files. Bioinformatics. 2011; 27:718-719. DOI | PubMed
- Lieberman-Aiden E, van Berkum NL, Williams L, Imakaev M, Ragoczy T, Telling A, Amit I, Lajoie BR, Sabo PJ, Dorschner MO, Sandstrom R, Bernstein B, Bender MA, Groudine M, Gnirke A, Stamatoyannopoulos J, Mirny LA, Lander ES, Dekker J. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science. 2009; 326:289-293. DOI | PubMed
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology. 2014; 15DOI | PubMed
- Lupiáñez DG, Kraft K, Heinrich V, Krawitz P, Brancati F, Klopocki E, Horn D, Kayserili H, Opitz JM, Laxova R, Santos-Simarro F, Gilbert-Dussardier B, Wittler L, Borschiwer M, Haas SA, Osterwalder M, Franke M, Timmermann B, Hecht J, Spielmann M, Visel A, Mundlos S. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell. 2015; 161:1012-1025. DOI | PubMed
- Mahmoud AI, Kocabas F, Muralidhar SA, Kimura W, Koura AS, Thet S, Porrello ER, Sadek HA. Meis1 regulates postnatal cardiomyocyte cell cycle arrest. Nature. 2013; 497:249-253. DOI | PubMed
- Maurano MT, Humbert R, Rynes E, Thurman RE, Haugen E, Wang H, Reynolds AP, Sandstrom R, Qu H, Brody J, Shafer A, Neri F, Lee K, Kutyavin T, Stehling-Sun S, Johnson AK, Canfield TK, Giste E, Diegel M, Bates D, Hansen RS, Neph S, Sabo PJ, Heimfeld S, Raubitschek A, Ziegler S, Cotsapas C, Sotoodehnia N, Glass I, Sunyaev SR, Kaul R, Stamatoyannopoulos JA. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012; 337:1190-1195. DOI | PubMed
- Meder B, Rühle F, Weis T, Homuth G, Keller A, Franke J, Peil B, Lorenzo Bermejo J, Frese K, Huge A, Witten A, Vogel B, Haas J, Völker U, Ernst F, Teumer A, Ehlermann P, Zugck C, Friedrichs F, Kroemer H, Dörr M, Hoffmann W, Maisch B, Pankuweit S, Ruppert V, Scheffold T, Kühl U, Schultheiss HP, Kreutz R, Ertl G, Angermann C, Charron P, Villard E, Gary F, Isnard R, Komajda M, Lutz M, Meitinger T, Sinner MF, Wichmann HE, Krawczak M, Ivandic B, Weichenhan D, Gelbrich G, El-Mokhtari NE, Schreiber S, Felix SB, Hasenfuß G, Pfeufer A, Hübner N, Kääb S, Arbustini E, Rottbauer W, Frey N, Stoll M, Katus HA. A genome-wide association study identifies 6p21 as novel risk locus for dilated cardiomyopathy. European Heart Journal. 2014; 35:1069-1077. DOI | PubMed
- Miele A, Dekker J. Long-range chromosomal interactions and gene regulation. Molecular BioSystems. 2008; 4DOI | PubMed
- Mifsud B, Tavares-Cadete F, Young AN, Sugar R, Schoenfelder S, Ferreira L, Wingett SW, Andrews S, Grey W, Ewels PA, Herman B, Happe S, Higgs A, LeProust E, Follows GA, Fraser P, Luscombe NM, Osborne CS. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nature Genetics. 2015; 47:598-606. DOI | PubMed
- Moshal K, Roder K, Werdich AA, Dural NT, Kim TY, Cooper LL, Yc L, Choi B-R, Terentyev D, MacRae C, Koren G. LITAF, A novel regulator of cardiac excitation. FASEB Journal : Official Publication of the Federation of American Societies for Experimental Biology. 2017; 31
- Mumbach MR, Satpathy AT, Boyle EA, Dai C, Gowen BG, Cho SW, Nguyen ML, Rubin AJ, Granja JM, Kazane KR, Wei Y, Nguyen T, Greenside PG, Corces MR, Tycko J, Simeonov DR, Suliman N, Li R, Xu J, Flynn RA, Kundaje A, Khavari PA, Marson A, Corn JE, Quertermous T, Greenleaf WJ, Chang HY. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nature Genetics. 2017; 49:1602-1612. DOI | PubMed
- Musunuru K, Strong A, Frank-Kamenetsky M, Lee NE, Ahfeldt T, Sachs KV, Li X, Li H, Kuperwasser N, Ruda VM, Pirruccello JP, Muchmore B, Prokunina-Olsson L, Hall JL, Schadt EE, Morales CR, Lund-Katz S, Phillips MC, Wong J, Cantley W, Racie T, Ejebe KG, Orho-Melander M, Melander O, Koteliansky V, Fitzgerald K, Krauss RM, Cowan CA, Kathiresan S, Rader DJ. From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 2010; 466:714-719. DOI | PubMed
- Nikpay M, Goel A, Won HH, Hall LM, Willenborg C, Kanoni S, Saleheen D, Kyriakou T, Nelson CP, Hopewell JC, Webb TR, Zeng L, Dehghan A, Alver M, Armasu SM, Auro K, Bjonnes A, Chasman DI, Chen S, Ford I, Franceschini N, Gieger C, Grace C, Gustafsson S, Huang J, Hwang SJ, Kim YK, Kleber ME, Lau KW, Lu X, Lu Y, Lyytikäinen LP, Mihailov E, Morrison AC, Pervjakova N, Qu L, Rose LM, Salfati E, Saxena R, Scholz M, Smith AV, Tikkanen E, Uitterlinden A, Yang X, Zhang W, Zhao W, de Andrade M, de Vries PS, van Zuydam NR, Anand SS, Bertram L, Beutner F, Dedoussis G, Frossard P, Gauguier D, Goodall AH, Gottesman O, Haber M, Han BG, Huang J, Jalilzadeh S, Kessler T, König IR, Lannfelt L, Lieb W, Lind L, Lindgren CM, Lokki ML, Magnusson PK, Mallick NH, Mehra N, Meitinger T, Memon FU, Morris AP, Nieminen MS, Pedersen NL, Peters A, Rallidis LS, Rasheed A, Samuel M, Shah SH, Sinisalo J, Stirrups KE, Trompet S, Wang L, Zaman KS, Ardissino D, Boerwinkle E, Borecki IB, Bottinger EP, Buring JE, Chambers JC, Collins R, Cupples LA, Danesh J, Demuth I, Elosua R, Epstein SE, Esko T, Feitosa MF, Franco OH, Franzosi MG, Granger CB, Gu D, Gudnason V, Hall AS, Hamsten A, Harris TB, Hazen SL, Hengstenberg C, Hofman A, Ingelsson E, Iribarren C, Jukema JW, Karhunen PJ, Kim BJ, Kooner JS, Kullo IJ, Lehtimäki T, Loos RJF, Melander O, Metspalu A, März W, Palmer CN, Perola M, Quertermous T, Rader DJ, Ridker PM, Ripatti S, Roberts R, Salomaa V, Sanghera DK, Schwartz SM, Seedorf U, Stewart AF, Stott DJ, Thiery J, Zalloua PA, O’Donnell CJ, Reilly MP, Assimes TL, Thompson JR, Erdmann J, Clarke R, Watkins H, Kathiresan S, McPherson R, Deloukas P, Schunkert H, Samani NJ, Farrall M. A comprehensive 1,000 Genomes-based genome-wide association meta-analysis of coronary artery disease. Nature Genetics. 2015; 47:1121-1130. DOI | PubMed
- Nora EP, Goloborodko A, Valton AL, Gibcus JH, Uebersohn A, Abdennur N, Dekker J, Mirny LA, Bruneau BG. Targeted degradation of CTCF decouples local insulation of chromosome domains from genomic compartmentalization. Cell. 2017; 169:930-944. DOI | PubMed
- Nora EP, Lajoie BR, Schulz EG, Giorgetti L, Okamoto I, Servant N, Piolot T, van Berkum NL, Meisig J, Sedat J, Gribnau J, Barillot E, Blüthgen N, Dekker J, Heard E. Spatial partitioning of the regulatory landscape of the X-inactivation centre. Nature. 2012; 485:381-385. DOI | PubMed
- O’Leary NA, Wright MW, Brister JR, Ciufo S, Haddad D, McVeigh R, Rajput B, Robbertse B, Smith-White B, Ako-Adjei D, Astashyn A, Badretdin A, Bao Y, Blinkova O, Brover V, Chetvernin V, Choi J, Cox E, Ermolaeva O, Farrell CM, Goldfarb T, Gupta T, Haft D, Hatcher E, Hlavina W, Joardar VS, Kodali VK, Li W, Maglott D, Masterson P, McGarvey KM, Murphy MR, O’Neill K, Pujar S, Rangwala SH, Rausch D, Riddick LD, Schoch C, Shkeda A, Storz SS, Sun H, Thibaud-Nissen F, Tolstoy I, Tully RE, Vatsan AR, Wallin C, Webb D, Wu W, Landrum MJ, Kimchi A, Tatusova T, DiCuccio M, Kitts P, Murphy TD, Pruitt KD. Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Research. 2016; 44:D733-D745. DOI | PubMed
- Patro R, Duggal G, Love MI, Irizarry RA, Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nature Methods. 2017; 14:417-419. DOI | PubMed
- Pennacchio LA, Bickmore W, Dean A, Nobrega MA, Bejerano G. Enhancers: five essential questions. Nature Reviews Genetics. 2013; 14:288-295. DOI | PubMed
- Petersen CM, Nielsen MS, Nykjaer A, Jacobsen L, Tommerup N, Rasmussen HH, Roigaard H, Gliemann J, Madsen P, Moestrup SK. Molecular identification of a novel candidate sorting receptor purified from human brain by receptor-associated protein affinity chromatography. Journal of Biological Chemistry. 1997; 272:3599-3605. DOI | PubMed
- Phanstiel DH, Van Bortle K, Spacek D, Hess GT, Shamim MS, Machol I, Love MI, Aiden EL, Bassik MC, Snyder MP. Static and dynamic DNA loops form AP-1-Bound activation hubs during macrophage development. Molecular Cell. 2017; 67:1037-1048. DOI | PubMed
- Phillips JE, Corces VG. CTCF: master weaver of the genome. Cell. 2009; 137:1194-1211. DOI | PubMed
- Phillips-Cremins JE, Sauria ME, Sanyal A, Gerasimova TI, Lajoie BR, Bell JS, Ong CT, Hookway TA, Guo C, Sun Y, Bland MJ, Wagstaff W, Dalton S, McDevitt TC, Sen R, Dekker J, Taylor J, Corces VG. Architectural protein subclasses shape 3D organization of genomes during lineage commitment. Cell. 2013; 153:1281-1295. DOI | PubMed
- Pikkarainen S, Tokola H, Kerkelä R, Ruskoaho H. GATA transcription factors in the developing and adult heart. Cardiovascular Research. 2004; 63:196-207. DOI | PubMed
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MA, Bender D, Maller J, Sklar P, de Bakker PI, Daly MJ, Sham PC. PLINK: a tool set for whole-genome association and population-based linkage analyses. The American Journal of Human Genetics. 2007; 81:559-575. DOI | PubMed
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841-842. DOI | PubMed
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, Wysocka J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature. 2011; 470:279-283. DOI | PubMed
- Rao SS, Huntley MH, Durand NC, Stamenova EK, Bochkov ID, Robinson JT, Sanborn AL, Machol I, Omer AD, Lander ES, Aiden EL. A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell. 2014; 159:1665-1680. DOI | PubMed
- Rubin AJ, Barajas BC, Furlan-Magaril M, Lopez-Pajares V, Mumbach MR, Howard I, Kim DS, Boxer LD, Cairns J, Spivakov M, Wingett SW, Shi M, Zhao Z, Greenleaf WJ, Kundaje A, Snyder M, Chang HY, Fraser P, Khavari PA. Lineage-specific dynamic and pre-established enhancer-promoter contacts cooperate in terminal differentiation. Nature Genetics. 2017; 49:1522-1528. DOI | PubMed
- Sakabe NJ, Aneas I, Shen T, Shokri L, Park SY, Bulyk ML, Evans SM, Nobrega MA. Dual transcriptional activator and repressor roles of TBX20 regulate adult cardiac structure and function. Human Molecular Genetics. 2012; 21:2194-2204. DOI | PubMed
- Schmitt AD, Hu M, Jung I, Xu Z, Qiu Y, Tan CL, Li Y, Lin S, Lin Y, Barr CL, Ren B. A compendium of chromatin contact maps reveals spatially active regions in the human genome. Cell Reports. 2016; 17:2042-2059. DOI | PubMed
- Schoenfelder S, Furlan-Magaril M, Mifsud B, Tavares-Cadete F, Sugar R, Javierre BM, Nagano T, Katsman Y, Sakthidevi M, Wingett SW, Dimitrova E, Dimond A, Edelman LB, Elderkin S, Tabbada K, Darbo E, Andrews S, Herman B, Higgs A, LeProust E, Osborne CS, Mitchell JA, Luscombe NM, Fraser P. The pluripotent regulatory circuitry connecting promoters to their long-range interacting elements. Genome Research. 2015; 25:582-597. DOI | PubMed
- Shen T, Aneas I, Sakabe N, Dirschinger RJ, Wang G, Smemo S, Westlund JM, Cheng H, Dalton N, Gu Y, Boogerd CJ, Cai CL, Peterson K, Chen J, Nobrega MA, Evans SM. Tbx20 regulates a genetic program essential to adult mouse cardiomyocyte function. Journal of Clinical Investigation. 2011; 121:4640-4654. DOI | PubMed
- Shin H, Shi Y, Dai C, Tjong H, Gong K, Alber F, Zhou XJ. TopDom: an efficient and deterministic method for identifying topological domains in genomes. Nucleic Acids Research. 2016; 44DOI | PubMed
- Siersbæk R, Madsen JGS, Javierre BM, Nielsen R, Bagge EK, Cairns J, Wingett SW, Traynor S, Spivakov M, Fraser P, Mandrup S. Dynamic rewiring of Promoter-Anchored chromatin loops during adipocyte differentiation. Molecular Cell. 2017; 66:420-435. DOI | PubMed
- Smemo S, Campos LC, Moskowitz IP, Krieger JE, Pereira AC, Nobrega MA. Regulatory variation in a TBX5 enhancer leads to isolated congenital heart disease. Human Molecular Genetics. 2012; 21:3255-3263. DOI | PubMed
- Smemo S, Tena JJ, Kim KH, Gamazon ER, Sakabe NJ, Gómez-Marín C, Aneas I, Credidio FL, Sobreira DR, Wasserman NF, Lee JH, Puviindran V, Tam D, Shen M, Son JE, Vakili NA, Sung HK, Naranjo S, Acemel RD, Manzanares M, Nagy A, Cox NJ, Hui CC, Gomez-Skarmeta JL, Nóbrega MA. Obesity-associated variants within FTO form long-range functional connections with IRX3. Nature. 2014; 507:371-375. DOI | PubMed
- Smith NL, Felix JF, Morrison AC, Demissie S, Glazer NL, Loehr LR, Cupples LA, Dehghan A, Lumley T, Rosamond WD, Lieb W, Rivadeneira F, Bis JC, Folsom AR, Benjamin E, Aulchenko YS, Haritunians T, Couper D, Murabito J, Wang YA, Stricker BH, Gottdiener JS, Chang PP, Wang TJ, Rice KM, Hofman A, Heckbert SR, Fox ER, O’Donnell CJ, Uitterlinden AG, Rotter JI, Willerson JT, Levy D, van Duijn CM, Psaty BM, Witteman JC, Boerwinkle E, Vasan RS. Association of genome-wide variation with the risk of incident heart failure in adults of European and African ancestry: a prospective meta-analysis from the cohorts for heart and aging research in genomic epidemiology (CHARGE) consortium. Circulation: Cardiovascular Genetics. 2010; 3:256-266. DOI | PubMed
- Soneson C, Love MI, Robinson MD. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research. 2015; 4DOI | PubMed
- Speir ML, Zweig AS, Rosenbloom KR, Raney BJ, Paten B, Nejad P, Lee BT, Learned K, Karolchik D, Hinrichs AS, Heitner S, Harte RA, Haeussler M, Guruvadoo L, Fujita PA, Eisenhart C, Diekhans M, Clawson H, Casper J, Barber GP, Haussler D, Kuhn RM, Kent WJ. The UCSC genome browser database: 2016 update. Nucleic Acids Research. 2016; 44:D717-D725. DOI | PubMed
- Spitz F, Furlong EE. Transcription factors: from enhancer binding to developmental control. Nature Reviews Genetics. 2012; 13:613-626. DOI | PubMed
- Stevens TJ, Lando D, Basu S, Atkinson LP, Cao Y, Lee SF, Leeb M, Wohlfahrt KJ, Boucher W, O’Shaughnessy-Kirwan A, Cramard J, Faure AJ, Ralser M, Blanco E, Morey L, Sansó M, Palayret MGS, Lehner B, Di Croce L, Wutz A, Hendrich B, Klenerman D, Laue ED. 3D structures of individual mammalian genomes studied by single-cell Hi-C. Nature. 2017; 544:59-64. DOI | PubMed
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. PNAS. 2003; 100:9440-9445. DOI | PubMed
- Symmons O, Pan L, Remeseiro S, Aktas T, Klein F, Huber W, Spitz F. The shh topological domain facilitates the action of remote enhancers by reducing the effects of genomic distances. Developmental Cell. 2016; 39:529-543. DOI | PubMed
- Tsujimura T, Klein FA, Langenfeld K, Glaser J, Huber W, Spitz F. A discrete transition zone organizes the topological and regulatory autonomy of the adjacent tfap2c and bmp7 genes. PLoS Genetics. 2015; 11DOI | PubMed
- Visel A, Minovitsky S, Dubchak I, Pennacchio LA. VISTA Enhancer Browser–a database of tissue-specific human enhancers. Nucleic Acids Research. 2007; 35:D88-D92. DOI | PubMed
- Watt AJ, Battle MA, Li J, Duncan SA. GATA4 is essential for formation of the proepicardium and regulates cardiogenesis. PNAS. 2004; 101:12573-12578. DOI | PubMed
- Wingett S, Ewels P, Furlan-Magaril M, Nagano T, Schoenfelder S, Fraser P, Andrews S. HiCUP: pipeline for mapping and processing Hi-C data. F1000Research. 2015; 4DOI | PubMed
- Wright JB, Brown SJ, Cole MD. Upregulation of c-MYC in cis through a large chromatin loop linked to a cancer risk-associated single-nucleotide polymorphism in colorectal cancer cells. Molecular and Cellular Biology. 2010; 30:1411-1420. DOI | PubMed
- Zhou X, Li D, Zhang B, Lowdon RF, Rockweiler NB, Sears RL, Madden PA, Smirnov I, Costello JF, Wang T. Epigenomic annotation of genetic variants using the roadmap epigenome browser. Nature Biotechnology. 2015; 33:345-346. DOI | PubMed
Fonte
Montefiori LE, Sobreira DR, Sakabe NJ, Aneas I, Joslin AC, et al. () A promoter interaction map for cardiovascular disease genetics. eLife 7e35788. https://doi.org/10.7554/eLife.35788




