
Abstract
Introduzione
Gli animali si affidano ai sistemi di regolazione genomica per dirigere l’espressione genica dinamica spazio-temporale e specifica del tipo di cellula che è essenziale per lo sviluppo e il mantenimento di uno stile di vita multicellulare. Tuttavia, non è ancora chiaro come tale sistema abbia avuto origine e si sia evoluto negli animali. Come l’ultimo antenato comune degli animali moderni possedeva già un vasto repertorio di geni regolatori, tra cui la maggior parte dei fattori di trascrizione e dei percorsi di segnalazione utilizzati nello sviluppo bilateriano(Srivastava et al., 2010; Larroux et al., 2008; Degnan et al., 2009; Larroux et al., 2006; Richards e Degnan, 2009; Ryan et al., 2013; Moroz et al., 2014; King et al., 2008; Sebé-Pedrós et al., 2011; de Mendoza et al., 2013; King et al., 2003; Richter e King, 2013), l’evoluzione della multicellularità animale richiedeva probabilmente più dell’origine dei nuovi geni. Altre caratteristiche normative, come il DNA cis-regolatorioe i modelli combinatoriali di modificazioni post-traslazionali (PTM) covalenti istoniche(Davidson e Peter, 2015), sarebbero state strumentali per dirigere l’espressione genica differenziale nei primi animali multicellulari. Per esempio, una recente analisi del genoma di Capsaspora, uno dei parenti unicellulari più vicini degli animali, rivela una mancanza di segni repressivi della cromatina, di tipi di promotori dello sviluppo e di elementi regolatori cis distali (potenziatori) tipicamente presenti negli animali complessi(cioè, eumetazoani)(Sebé-Pedrós et al., 2016).
Lo sviluppo di saggi di cromatina ad alto rendimento come l’immunoprecipitazione della cromatina accoppiato con il sequenziamento massivamente parallelo (ChIP-seq)(Robertson et al., 2007) ha permesso la dissezione di informazioni codificate con cromatina al di là della sequenza primaria del DNA, in particolare l’esame sistematico degli istoni PTM e il loro ruolo (s) nella regolazione trascrizionale(Zhou et al., 2011; Thurman et al., 2012; Kundaje et al., 2015; ENCODE Project Consortium, 2012). Sebbene i modelli combinatoriali di acetilazione e metilazione degli istoni siano componenti chiave dei meccanismi di regolazione genica alla base della formazione e del mantenimento degli eumetazoi(Schwaiger et al., 2014), non si sa se questo sistema sia limitato a questi animali o sia più antico.
I porifera (spugne) sono considerati uno dei più antichi lignaggi di animali filiformi sopravvissuti, divergenti dagli altri metazoi circa 700 Mya(Erwin et al., 2011). Nonostante siano uno degli animali morfologicamente più semplici, privi di intestino, nervi e muscoli, le spugne possiedono un vasto repertorio genetico per la regolazione trascrizionale richiesta nello sviluppo eumetazoico e nel modellamento del corpo(Srivastava et al., 2010; Larroux et al., 2008, 2006; Adamska et al., 2007; Gaiti et al., 2015; Nakanishi et al., 2014; Conaco et al., 2012; Riesgo et al., 2014; Grimson et al., 2008; Richards et al., 2008; Leininger et al., 2014; Fortunato et al., 2015, 2014; Bråte et al., 18212015). Qui, in seguito ai nostri recenti studi trascrittomici che hanno rivelato che la spugna Amphimedon queenslandica (qui Amphimedon) ha un’espressione genica dinamica dello sviluppo simile a quella degli eumetazoi(Gaiti et al., 2015; Fernandez-Valverde et al., 2015; Levin et al., 2016), ci siamo prefissati di determinare se questa complessità trascrizionale sia parallela alla complessità regolatoria codificata dai modelli di PTM dell’istone combinatorio. Analizzando un ampio compendio ChIP-seq di PTM dell’istone H3 in questa spugna, dimostriamo che un complesso panorama di regolazione genica composto da modifiche dell’istone combinatorio era già in atto all’alba degli animali. Inoltre, forniamo la prova dell’evoluzione e dell’espansione delle capacità genomiche regolatorie cis-distaliall’origine del regno animale.
Risultati
I principali stati della cromatina regolatoria di Anfimedone sono condivisi con gli eumetazoi
Abbiamo effettuato l’immunoprecipitazione della cromatina (ChIP) su adulti e larve di Amphimedon che si riproducono sessualmente utilizzando anticorpi contro specifici istoni H3 PTM che sono stati utilizzati per definire gli stati della cromatina nei modelli bilateriani(Zhou et al., 2011; Ho et al., 2014)(Figura 1A). Queste analisi sono state effettuate su miscele separate di tipi di cellule somatiche adulte e larvali e, quindi, una diversità di stati trascrizionali del gene. È importante notare che gli adulti e le larve di Anfimedone sono composti da diversi tipi di cellule con profili trascrizionali e stati normativi molto diversi(Gaiti et al., 2015; Conaco et al., 2012; Fernandez-Valverde et al., 2015; Degnan et al ., 2015). Mentre la nostra strategia di campionamento aumenta la complessità biologica degli stati della cromatina in toto, può diluire i segnali specifici del tipo di cellula. Questo contrasta con le analisi ChIP-seq eseguite su linee cellulari, embrioni con pochi tipi di cellule, o campioni di tessuto distinti, che incapsulano popolazioni cellulari e ambienti più omogenei(Sebé-Pedrós et al., 2016; Kundaje et al., 2015; Schwaiger et al., 2014; Gerstein et al., 2010; Pérez-Lluch et al., 2015a). Dato che l’attuale genoma di Amphimedon è una bozza di sequenza, le nostre analisi possono anche essere incomplete in regioni che hanno annotazioni incomplete e lacune nell’assemblaggio (13% dell’assemblaggio totale del genoma) (Srivastavaet al., 2010).10.7554/eLife.22194.003Figure 1.Chromatin states in Amphimedon.(A) Rappresentazione schematica del ciclo di vita di Amphimedon. Larve (di forma ovale, lunghe 300-500 µm) emergono dalle camere di covata materna e poi nuotano nella colonna d’acqua prima di sviluppare la competenza a stabilirsi e avviare la metamorfosi in un giovane. Il piano corporeo giovanile, che mostra i segni distintivi del piano corporeo adulto, compreso un sistema acquifero con canali, camere di choanocytes e oscula, è il risultato della drastica riorganizzazione della larva radialmente simmetrica, bi- o trilaterale. Questo giovane crescerà e maturerà poi in un adulto bentonico (che va da 10-30 cm3)(Degnanet al., 2015; Edgar et al., 2002).(B) Definizione e arricchimenti per un Modello Markov Nascosto a 9 stati basato su cinque istoni PTM (H3K4me3, H3K27ac, H3K4me1, H3K36me3 e H3K27me3) in Anfimedone adulto. Da sinistra a destra: definizioni dello stato della cromatina, abbreviazioni, probabilità di PTM dell’istone, copertura genomica, arricchimenti dell’annotazione funzionale del gene proteico codificante, espressi (Expr.) e repressi (Repr.) arricchimenti del gene proteico codificante. L’ombreggiatura blu indica l’intensità, scalata per colonna.(C) Adulti annotazioni di stato cromatina stato adulto sul ricco di gene altamente trascritto (attivo) scaffold (contig13500) che mostra la predominanza di ‘TssA’, ‘TxFlnk’, e ‘TxEnhA’ stati. Per la definizione degli stati della cromatina vedere il pannello(A). Sono mostrati i geni codificanti (viola) e gli RNA lunghi non codificanti (blu), insieme alle tracce di copertura del segnale che mostrano l’espressione CEL-seq nell’adulto. Una scala di grigio indica il livello di espressione CEL-seq: bianco (nessuna espressione); nero (espressione più alta).(D) Annotazioni sullo stato della cromatina per adulti su un’impalcatura prevalentemente silenziata (contig13522 da 500.000 a 1.500.000 bp) che mostrano la prevalenza degli stati ‘ReprPC’ e ‘ReprPCWk’. Per la definizione degli stati della cromatina si veda il pannello(A). I geni codificanti (viola) e gli RNA lunghi non codificanti (blu) sono mostrati, insieme alle tracce di copertura del segnale che mostrano l’espressione CEL-seq nell’adulto. Una scala di grigio indica il livello di espressione CEL-seq: bianco (nessuna espressione); nero (espressione più alta).DOI:http://dx.doi.org/10.7554/eLife.22194.00310.7554/eLife.22194.004Figure1-source data 1.Histone H3 modifiche covalenti post-traslazione e RNA polimerasi II indagati in questo studio e la loro tipica localizzazione genomica relativa ai geni codificanti e regioni di regolamentazione in organismi modello bilateriano.DOI:http://dx.doi.org/10.7554/eLife.22194.00410.7554/eLife.22194.005Figure1-source data 2.Histone H3 sequenze utilizzate per generare Figura 1-figure supplement 1.DOI:http://dx.doi.org/10.7554/eLife.22194.00510.7554/eLife.22194.006Figure1-source data 3.BLASTp risultato della ricerca delle metiltransferasi istoniche rilevanti e acetiltrasferasi contro leproteine di Amphimedon queenslandica(NCBI nr database; E-value <1e-09).DOI:http://dx.doi.org/10.7554/eLife.22194.00610.7554/eLife.22194.007Figure1-source data 4.Statistiche di sintesi e metriche di qualità dei set di dati ChIP-seq utilizzati in questo studio.Vedi anche Materiali e metodi per il pretrattamento della procedura dei set di dati ChIP-seq.DOI:http://dx.doi.org/10.7554/eLife.22194.00710.7554/eLife.22194.008Figure1-source data 5.Convalida dei risultati di ChIP-seq da parte di ChIP-quantitative PCRs (ChIP-qPCRs).DOI:http://dx.doi.org/10.7554/eLife.22194.00810.7554/eLife.22194.009FigureSupplemento a 1 cifra 1.Allineamento di sequenza multipla di varie proteine dell’istone eucariota H3 (1-136 amminoacidi), prodotte utilizzando ClustalO (RRID:SCR_001591) (Sieverset al., 2011).Si noti che l’intera sequenza di amminoacidi dell’istone H3 è altamente conservata attraverso gli eucarioti. La sequenza di spugna è evidenziata. Le sequenze di aminoacidi utilizzati per generare l’allineamento sono anche forniti in Figura 1-source data 2.DOI:http://dx.doi.org/10.7554/eLife.22194.00910.7554/eLife.22194.010Figure1-figure supplement 2.Assessment of reproducibility for biological replicates between histone modifications andRNA Polymerase II.(A) Pearson correlation coefficients between histone modifications and RNA Polymerase II (RNAPII). Esperimenti per adulti (repliche biologiche combinate) sono mostrati. I colori sottostanti indicano la somiglianza tra i diversi dataset. Si noti che H3K36me3 è stato segnalato per il segnale basso a rumore, spiegando potenzialmente la correlazione un po ‘alta con H3K27me3 (vedi Figura 1-dati sorgente 4). Tuttavia, questo non influisce in alcun modo sulle conclusioni del documento.(B) Annotazioni sullo stato della cromatina adulta su una regione prevalentemente silenziata. Per la definizione degli stati della cromatina vedi Figura 1A. I geni di codifica (viola) sono mostrati, insieme con l’input DNA-normalizzato copertura di ogni replicato biologico (R1 e R2) di diverse modificazioni dell’istone e l’espressione RNA-seq.(C) Come(B) per le regioni altamente trascritte. A parte il replicato RNAPII 1, che non ha superato la soglia di qualità richiesta per cui è stato escluso da tutte le ulteriori analisi (vedi Figura 1 – dati fonte 4), abbiamo ottenuto serie di dati altamente riproducibili.DOI:http://dx.doi.org/10.7554/eLife.22194.01010.7554/eLife.22194.011Figure1-figure supplement 3.Neighborhood positional enrichment plots of adult chromatin states around transcription start site (TSS) and transcription end site (TES) of protein-coding genes, prodotto da ChromHMM (Ernstand Kellis, 2012).per la definizione degli stati adulti della cromatina si veda la Figura 1A.(A) Arricchimenti posizionali in contenitori genomici da 100 bp intorno al TSS e TES (±1 kb) di geni a codifica proteica espressa in Amphimedon adulto .(B) Lo stesso che(A) per i geni codificanti proteine represse in Amphimedon adulto. L’ombreggiatura blu indica l’intensità.DOI:http://dx.doi.org/10.7554/eLife.22194.01110.7554/eLife.22194.012Figure1-figure supplemento 4.Chromatin stati 4.Chromatin stati in Amphimedonlarva . (A) Definizione e gli arricchimenti per un 9-stato Hidden Markov modello basato su quattro istoni PTMs (H3K4me3, H3K27ac, H3K27me1, H3K4me1, H3K27me3) in Amphimedon larva. Da sinistra a destra: definizioni dello stato della cromatina, abbreviazioni, probabilità di PTM dell’istone, copertura genomica, arricchimenti dell’annotazione funzionale del gene codificante delle proteine, arricchimenti del gene codificante delle proteine espressi (Expr.) e repressi (Repr.). L’ombreggiatura blu indica l’intensità, scalata per colonna.(B) Le annotazioni di stato cromatina su un ricco gene altamente trascritto (attivo) scaffold (contig13500) come in Figura 1. Per la definizione degli stati della cromatina vedere il pannello(A). I geni codificanti (viola) e RNA lunghi non codificanti (blu) sono mostrati, insieme a tracce di copertura del segnale che mostrano l’espressione CEL-seq in larva. Una scala di grigio indica il livello di espressione CEL-seq: bianco (nessuna espressione); nero (espressione più alta).(C) Annotazioni dello stato di cromatina su un’impalcatura prevalentemente silenziata (contig13522 da 500.000 a 1.500.000 bp) come in Figura 1. Per la definizione degli stati di cromatina vedi pannello(A). Geni codificanti (viola) e lunghi RNA non codificanti (blu) sono mostrati, insieme a tracce di copertura del segnale che mostrano l’espressione CEL-seq nell’adulto. Una scala di grigio indica il livello di espressione CEL-seq: bianco (nessuna espressione); nero (espressione più alta).(D) Arricchimenti posizionali di vicinato in bidoni genomici da 100 bp intorno al TSS e TES (±1 kb) di geni codificanti le proteine espresse nelle larve.(E) Lo stesso che(D) per i geni codificanti le proteine represse nelle larve. L’ombreggiatura blu indica l’intensità.DOI:http://dx.doi.org/10.7554/eLife.22194.012
Gli anticorpi utilizzati hanno come bersaglio il seguente istone H3 PTM: (i) lisina 4 monometilata (H3K4me1), associata ad elementi regolatori del cis distale come gli stimolatori; (ii) lisina 4 trimetilata (H3K4me3), arricchita in promotori attivi; (iii) lisina 36 trimetilata (H3K36me3), trovata con regioni attivamente trascritte; (iv) lisina 27 trimetilata (H3K27me3), arricchita nelle regioni silenziate da Policomb; e (v) lisina 27 acetilata (H3K27ac), che si trova intorno alle regioni regolatorie attivate. Abbiamo anche usato un anticorpo contro l’istone totale H3(Figura 1 – dati fonte 1). È stato incluso anche un anticorpo contro i residui non fosforilati di Ser2 non fosforilati della RNA polimerasi II (RNAPII 8WG16) nel dominio C-terminale(Brookes e Pombo, 2009)(Figura 1 – dati fonte 1). Poiché l’intera sequenza aminoacidica dell’istone H3 è perfettamente conservata in Amphimedon, insieme alle relative metiltransferasi istoniche e acetiltrasferasi, si prevede che questi anticorpi riconosceranno gli epitopi corretti(Figura 1-figure supplement 1; Figura 1-source data 2; Figura 1-source data 3). Questi anticorpi riconoscono gli epitopi corretti in organismi ancora più distanti tra loro(ad esempio, eucarioti non metazoici)(ad esempio,[Sebé-Pedrós et al., 2016; Ercan et al. , 2009; Barraza et al., 2015; Harmeyer et al., 2015; Liu et al., 2007; Eckalbar et al., 2016]).
Le letture di ChIP-seq generate da DNA immunoprecipitato e in ingresso (estratto di cellule intere) sono state allineate al genoma di Amphimedon(Srivastava et al., 2010), dando come risultato set di dati altamente riproducibili(Figura 1-figure supplement 2; Figura 1-source data 4; Figura 1-source data 5). Le letture mappate in modo univoco sono state successivamente utilizzate per identificare un insieme di stati distinti della cromatina basati sui cinque diversi istoni H3 PTM H3 che abbiamo analizzato. In particolare, gli stati di cromatina sono stati previsti in tutto l’addestramento del genoma un modello multivariato Hidden Markov con diversi stati a priori definiti (da 5 a 15) (Materiali e metodi). Abbiamo scelto di utilizzare un modello a 9 stati per tutte le analisi successive, in quanto riguardava tutti i principali componenti di codifica genica e di regolazione (promotore, potenziatore, corpo del gene) che ci aspettavamo di risolvere con questa selezione di histone H3 PTMs. Nonostante l’intrinseca eterogeneità cellulare del nostro materiale di partenza, siamo stati in grado di risolvere le specificità verso le componenti geniche tra questi nove stati della cromatina. Essi rientrano in due grandi categorie: una correlata con geni attivamente trascritti che includono promotori attivi (‘TssA’) ed esaltatori (‘TxEnhA’, ‘EnhWk’), e i confini 5′ e 3′ dei geni trascritti (‘TxFlnk’); e un’altra categoria con geni con trascrizione nulla o poco rilevabile; questi includono stati bivalenti o regolatori in bilico (‘BivTx’, ‘EnhP’), Policomb repressa (‘ReprPC’, ‘ReprPCWk’), e quiescenti (‘Quies’) (Figura 1B-D). I nove stati della cromatina differenzialmente associati a specifiche caratteristiche genomiche di Amphimedon. Ad esempio, lo stato ‘TssA’ (definito dalla presenza di H3K4me3) è stato arricchito intorno a siti di inizio trascrizione (TSS) di geni attivi. TxEnhA’ stato ‘TxEnhA’ (definito da H3K4me1, H3K27ac, e H3K36me3 arricchimento) associato con la codifica di esoni e introni che corrispondono a potenziali cis-elementi regolatori e brevi regioni intergeniche, che sono comuni nel genoma Amphimedon (Kundajeet al., 2015; Fernandez-Valverde et al., 2015; Kowalczyk et al., 2012; Ritter et al., 2012; Singer et al., 2015; Birnbaum et al., 2012; Zentner e Scacheri, 2012; Zentner et al., 2011; Fernandez-Valverde e Degnan, 2016). Al contrario, gli stati ‘ReprPC’ (definiti dall’arricchimento di H3K27me3) sono stati diffusi attraverso i corpi genici dei geni repressi, in linea con il ruolo noto di H3K27me3 nel silenziamento trascrizionale (Zhou et al., 2011; Ho et al., 2014) (Figura 1BD; Figura 1-figure supplement 2; Figura 1-figure supplement 3).
Nonostante sia composto da diversi tipi di cellule e abbia un profilo di espressione genica distinto dall’adulto, il genoma larvale possiede un insieme notevolmente simile di stati di cromatina(Figura 1-figure supplement 4). L’ottenimento di stati di cromatina consistenti sulla base dei dati ChIP-seq dell’istone PTMs ChIP-seq da due stadi marcatamente diversi del ciclo di vita di Amphimedon fornisce la prova corroborante che questa spugna possiede gli stessi stati regolatori presenti negli eumetazoi.

Figura 1-figura supplemento 4.Gli stati di cromatina in Amphimedon.Histone H3 covalente modifiche post-traslazione e RNA polimerasi II indagato in questo studio e la loro tipica localizzazione genomica rispetto ai geni di codifica e le regioni di regolamentazione in organismi modello bilateri.sequenze di Histone H3 utilizzati per generare Figura 1-figure supplemento 1.Risultati della ricerca BLASTp delle metiltransferasi istoniche e acetiltrasferasi rilevanti contro le proteine di Amphimedonqueenslandica (database NCBI nr; E-value <1e-09).statistiche di sintesi e metriche di qualità dei set di dati ChIP-seq utilizzati in questo studio.validazione dei risultati di ChIP-seq da parte di ChIP-quantitative PCRs (ChIP-qPCRs).Histone H3 modifiche covalenti post-traslazione e RNA polimerasi II indagato in questo studio e la loro tipica localizzazione genomica rispetto ai geni di codifica e le regioni di regolamentazione in organismi modello bilateri. sequenze di Histone H3 utilizzati per generare Figura 1-figure supplemento 1.BLASTp risultato della ricerca 1.BLASTp delle metiltransferasi histone rilevanti e acetiltrasferasi contro leproteine Amphimedonqueenslandica (NCBI nr database; E-valore <1e-09).Statistiche di sintesi e metriche di qualità dei set di dati ChIP-seq utilizzati in questo studio.Convalida dei risultati di ChIP-seq da parte di ChIP-quantitative PCRs (ChIP-qPCRs).allineamento di sequenze multiple di varie proteine H3 dell’istone eucariotico (1-136 aminoacidi), prodotte utilizzando ClustalO (RRID:SCR_001591) (Sievers et al., Valutazione della riproducibilità per i replicati biologici tra le modificazioni degli istoni e la polimerasi di RNA II.Tracce di arricchimento posizionale del vicinato di stati adulti di cromatina intorno al sito di inizio trascrizione (TSS) e al sito di fine trascrizione (TES) di geni che codificano le proteine, prodotte da ChromHMM(Ernst e Kellis, 2012).gli stati di cromatina in Amphimedonlarva.(A) Rappresentazione schematica del ciclo di vita di Amphimedon. Le larve (di forma ovale, lunghe 300-500 µm) emergono dalle camere di covata materna e poi nuotano nella colonna d’acqua prima di sviluppare la competenza a stabilirsi e avviare la metamorfosi in un giovane. Il piano corporeo giovanile, che mostra i segni distintivi del piano corporeo adulto, compreso un sistema acquifero con canali, camere di choanocytes e oscula, è il risultato della drastica riorganizzazione della larva radialmente simmetrica, bi- o trilaterale. Questo giovane crescerà e maturerà poi in un adulto bentonico (che va da 10-30 cm3)(Degnanet al., 2015; Edgar et al., 2002).(B) Definizione e arricchimenti per un Modello Markov Nascosto a 9 stati basato su cinque istoni PTM (H3K4me3, H3K27ac, H3K4me1, H3K36me3 e H3K27me3) in Anfimedone adulto. Da sinistra a destra: definizioni dello stato della cromatina, abbreviazioni, probabilità di PTM dell’istone, copertura genomica, arricchimenti dell’annotazione funzionale del gene proteico codificante, espressi (Expr.) e repressi (Repr.) arricchimenti del gene proteico codificante. L’ombreggiatura blu indica l’intensità, scalata per colonna.(C) Adulti annotazioni di stato cromatina stato adulto sul ricco di gene altamente trascritto (attivo) scaffold (contig13500) che mostra la predominanza di ‘TssA’, ‘TxFlnk’, e ‘TxEnhA’ stati. Per la definizione degli stati della cromatina vedere il pannello(A). Sono mostrati i geni codificanti (viola) e gli RNA lunghi non codificanti (blu), insieme alle tracce di copertura del segnale che mostrano l’espressione CEL-seq nell’adulto. Una scala di grigio indica il livello di espressione CEL-seq: bianco (nessuna espressione); nero (espressione più alta).(D) Annotazioni sullo stato della cromatina per adulti su un’impalcatura prevalentemente silenziata (contig13522 da 500.000 a 1.500.000 bp) che mostrano la prevalenza degli stati ‘ReprPC’ e ‘ReprPCWk’. Per la definizione degli stati della cromatina si veda il pannello(A). I geni codificanti (viola) e gli RNA lunghi non codificanti (blu) sono mostrati, insieme alle tracce di copertura del segnale che mostrano l’espressione CEL-seq nell’adulto. Una scala di grigio indica il livello di espressione CEL-seq: bianco (nessuna espressione); nero (espressione più alta).DOI:
http://dx.doi.org/10.7554/eLife.22194.00310.7554/eLife.22194.004Figure 1-source data 1.Histone H3 modifiche covalenti post-traslazione e RNA polimerasi II indagati in questo studio e la loro tipica localizzazione genomica relativa ai geni codificanti e regioni di regolamentazione in organismi modello bilateri.DOI:http://dx.doi.org/10.7554/eLife.22194.004DOI:
http://dx.doi.org/10.7554/eLife.22194.00410.7554/eLife.22194.005Figure 1-source data 2.Histone Sequenze H3 utilizzate per generare Figura 1-figure supplement 1.DOI:http://dx.doi.org/10.7554/eLife.22194.005DOI:
http://dx.doi.org/10.7554/eLife.22194.00510.7554/eLife.22194.006Figure 1-source data 3.BLASTp risultato della ricerca degli istoni metiltransferasi e acetiltransferasi rilevanti contro leproteine di Amphimedonqueenslandica (NCBI nr database; E-value <1e-09).DOI:http://dx.doi.org/10.7554/eLife.22194.006DOI:
http://dx.doi.org/10.7554/eLife.22194.00610.7554/eLife.22194.007Figure 1-source data 4.Summary statistics and quality metrics of the ChIP-seq datasets used in this study.See also Materials and methods for preprocessing of ChIP-seq datasets procedure.DOI:http://dx.doi.org/10.7554/eLife.22194.007Vedere anche Materiali e metodi per la preelaborazione della procedura dei set di dati ChIP-seq.DOI:
http://dx.doi.org/10.7554/eLife.22194.00710.7554/eLife.22194.008Figure 1-source data 5.Validazione dei risultati di ChIP-seq da parte di ChIP-quantitative PCR (ChIP-qPCR).DOI:http://dx.doi.org/10.7554/eLife.22194.008DOI:
http://dx.doi.org/10.7554/eLife.22194.008DOI:
http://dx.doi.org/10.7554/eLife.22194.004DOI:
http://dx.doi.org/10.7554/eLife.22194.005DOI:
http://dx.doi.org/10.7554/eLife.22194.006Vedere anche Materiali e metodi per la preelaborazione della procedura dei set di dati ChIP-seq.DOI:
http://dx.doi.org/10.7554/eLife.22194.007DOI:
http://dx.doi.org/10.7554/eLife.22194.008Si noti che l’intera sequenza aminoacidica dell’istone H3 è altamente conservata attraverso gli eucarioti. La sequenza della spugna è evidenziata. Le sequenze di amminoacidi utilizzate per generare l’allineamento sono anche fornite nella Figura 1 – dati fonte 2.DOI:
http://dx.doi.org/10.7554/eLife.22194.009(A) Coefficienti di correlazione Pearson tra le modifiche dell’istone e la RNA polimerasi II (RNAPII). Esperimenti per adulti (repliche biologiche combinate) sono mostrati. I colori sottostanti indicano la somiglianza tra i diversi dataset. Si noti che H3K36me3 è stato segnalato per il segnale basso a rumore, spiegando potenzialmente la correlazione un po ‘alta con H3K27me3 (vedi Figura 1-dati sorgente 4). Tuttavia, questo non influisce in alcun modo sulle conclusioni del documento.(B) Annotazioni sullo stato della cromatina adulta su una regione prevalentemente silenziata. Per la definizione degli stati della cromatina vedi Figura 1A. I geni di codifica (viola) sono mostrati, insieme con l’input DNA-normalizzato copertura di ogni replicato biologico (R1 e R2) di diverse modifiche degli istoni e RNA-seq espressione.(C) Come(B) per le regioni altamente trascritte. Oltre al replicato RNAPII 1, che non ha superato la soglia di qualità richiesta per cui è stato escluso da tutte le ulteriori analisi (vedi Figura 1 – dati fonte 4), abbiamo ottenuto serie di dati altamente riproducibili.DOI:
http://dx.doi.org/10.7554/eLife.22194.010Per la definizione degli stati della cromatina adulta si veda la Figura 1A.(A) Arricchimenti posizionali in contenitori genomici da 100 bp intorno al TSS e TES (±1 kb) di geni codificanti le proteine espresse in Amphimedon adulto .(B) Lo stesso che(A) per i geni codificanti proteine represse in Amphimedon adulto. L’ombreggiatura blu indica l’intensità.DOI:
http://dx.doi.org/10.7554/eLife.22194.011(A) Definizione e arricchimenti per un Modello Markov Nascosto a 9 stati basato su quattro istoni PTM (H3K4me3, H3K27ac, H3K4me1, H3K27me3) in larve di Anfimedone. Da sinistra a destra: definizioni dello stato della cromatina, abbreviazioni, probabilità di PTM dell’istone, copertura genomica, arricchimenti dell’annotazione funzionale del gene codificante delle proteine, arricchimenti del gene codificante delle proteine espressi (Expr.) e repressi (Repr.). L’ombreggiatura blu indica l’intensità, scalata per colonna.(B) Le annotazioni di stato cromatina su un ricco gene altamente trascritto (attivo) scaffold (contig13500) come in Figura 1. Per la definizione degli stati della cromatina vedere il pannello(A). I geni codificanti (viola) e RNA lunghi non codificanti (blu) sono mostrati, insieme a tracce di copertura del segnale che mostrano l’espressione CEL-seq in larva. Una scala di grigio indica il livello di espressione CEL-seq: bianco (nessuna espressione); nero (espressione più alta).(C) Annotazioni dello stato di cromatina su un’impalcatura prevalentemente silenziata (contig13522 da 500.000 a 1.500.000 bp) come in Figura 1. Per la definizione degli stati di cromatina vedi pannello(A). Geni codificanti (viola) e lunghi RNA non codificanti (blu) sono mostrati, insieme a tracce di copertura del segnale che mostrano l’espressione CEL-seq nell’adulto. Una scala di grigio indica il livello di espressione CEL-seq: bianco (nessuna espressione); nero (espressione più alta).(D) Arricchimenti posizionali di vicinato in bidoni genomici da 100 bp intorno al TSS e TES (±1 kb) di geni codificanti le proteine espresse nelle larve.(E) Lo stesso che(D) per i geni codificanti le proteine represse nelle larve. L’ombreggiatura blu indica l’intensità.DOI:
http://dx.doi.org/10.7554/eLife.22194.012

Figura 1-figure supplement 1.Allineamento in sequenza multipla di varie proteine eucariotiche dell’istone H3 (1-136 aminoacidi), prodotte utilizzando ClustalO (RRID:SCR_001591) (Sieverset al., 2011).Si noti che l’intera sequenza di aminoacidi dell’istone H3 è altamente conservata attraverso gli eucarioti. La sequenza della spugna è evidenziata. Le sequenze di amminoacidi utilizzate per generare l’allineamento sono anche fornite nella Figura 1 – dati fonte 2.DOI:
http://dx.doi.org/10.7554/eLife.22194.009
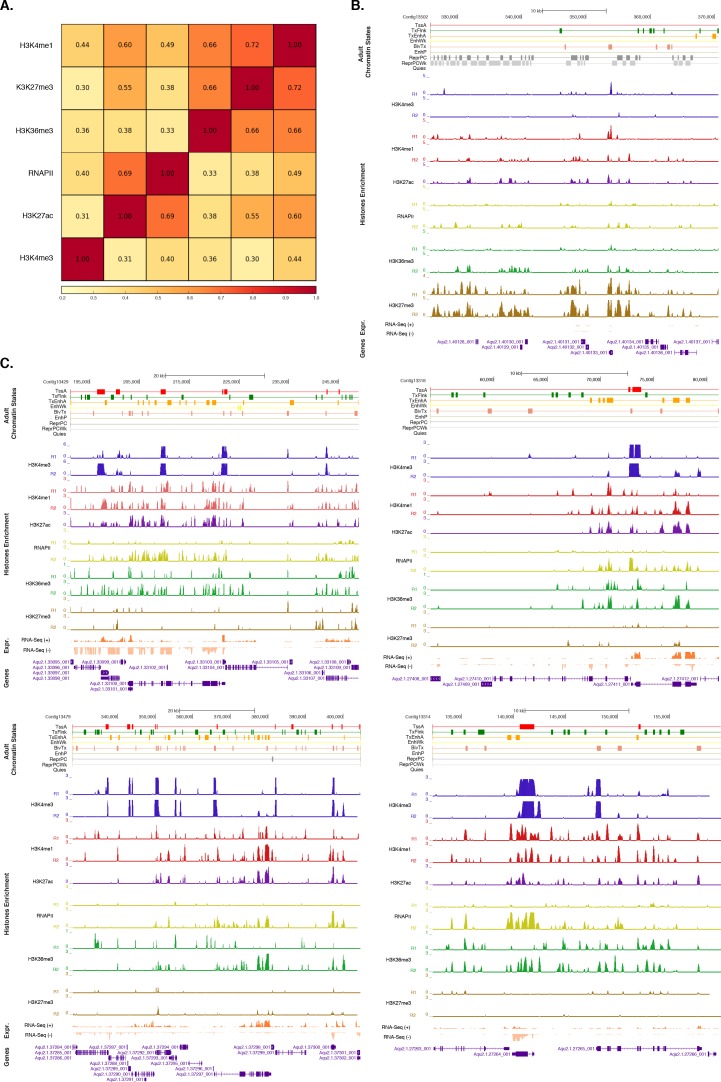
Figura 1-figure supplement 2.Valutazione della riproducibilità per i replicati biologici tra le modifiche dell’istone e la polimerasi di RNA II.(A) Coefficienti di correlazione Pearson tra le modificazioni dell’istone e la RNA polimerasi II (RNAPII). Esperimenti per adulti (repliche biologiche combinate) sono mostrati. I colori sottostanti indicano la somiglianza tra le diverse serie di dati. Si noti che H3K36me3 è stato segnalato per il segnale basso a rumore, spiegando potenzialmente la correlazione un po ‘alta con H3K27me3 (vedi Figura 1-dati sorgente 4). Tuttavia, questo non influisce in alcun modo sulle conclusioni del documento.(B) Annotazioni sullo stato della cromatina adulta su una regione prevalentemente silenziata. Per la definizione degli stati della cromatina vedi Figura 1A. I geni di codifica (viola) sono mostrati, insieme con l’input DNA-normalizzato copertura di ogni replicato biologico (R1 e R2) di diverse modifiche degli istoni e RNA-seq espressione.(C) Come(B) per le regioni altamente trascritte. Oltre al replicato RNAPII 1, che non ha superato la soglia di qualità richiesta per cui è stato escluso da tutte le ulteriori analisi (vedi Figura 1 – dati fonte 4), abbiamo ottenuto serie di dati altamente riproducibili.DOI:
http://dx.doi.org/10.7554/eLife.22194.010
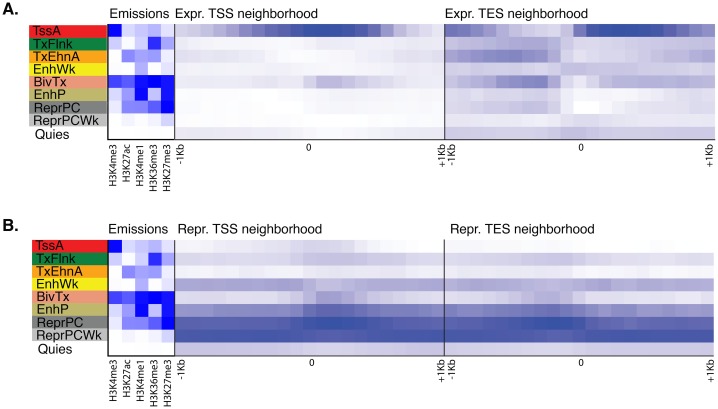
Figura 1-figure supplement 3.Figura 1—supplemento alla figura 3. Trame di arricchimento posizionale del vicinato di stati adulti di cromatina intorno al sito di inizio trascrizione (TSS) e al sito di fine trascrizione (TES) dei geni che codificano le proteine, prodotte da ChromHMM(Ernst e Kellis, 2012).Per la definizione degli stati della cromatina adulta si veda la Figura 1A.(A) Arricchimenti posizionali in contenitori genomici a 100 bp intorno al TSS e TES (± 1 kb) di geni a codifica proteica espressa in Amphimedon adulto .(B) Lo stesso che(A) per i geni codificanti proteine represse in Amphimedon adulto. L’ombreggiatura blu indica l’intensità.DOI:
http://dx.doi.org/10.7554/eLife.22194.011
Figura 1-figure supplement 4.Gli stati di cromatina in Amphimedonlarva.(A) Definizione e arricchimenti per un modello Markov nascosto a 9 stati basato su quattro istoni PTM (H3K4me3, H3K27ac, H3K4me1, H3K27me3) in Amphimedon larva. Da sinistra a destra: definizioni dello stato della cromatina, abbreviazioni, probabilità di PTM dell’istone, copertura genomica, arricchimenti dell’annotazione funzionale del gene codificante delle proteine, arricchimenti del gene codificante delle proteine espressi (Expr.) e repressi (Repr.). L’ombreggiatura blu indica l’intensità, scalata per colonna.(B) Le annotazioni di stato cromatina su un ricco gene altamente trascritto (attivo) scaffold (contig13500) come in Figura 1. Per la definizione degli stati della cromatina vedere il pannello(A). I geni codificanti (viola) e RNA lunghi non codificanti (blu) sono mostrati, insieme a tracce di copertura del segnale che mostrano l’espressione CEL-seq in larva. Una scala di grigi indica il livello di espressione CEL-seq: bianco (nessuna espressione); nero (espressione più alta).(C) Annotazioni dello stato di cromatina su un’impalcatura prevalentemente silenziata (contig13522 da 500.000 a 1.500.000 bp) come in Figura 1. Per la definizione degli stati di cromatina vedi pannello(A). Geni codificanti (viola) e lunghi RNA non codificanti (blu) sono mostrati, insieme a tracce di copertura del segnale che mostrano l’espressione CEL-seq nell’adulto. Una scala di grigio indica il livello di espressione CEL-seq: bianco (nessuna espressione); nero (espressione più alta).(D) Arricchimenti posizionali di vicinato in bidoni genomici da 100 bp intorno al TSS e TES (±1 kb) di geni codificanti le proteine espresse nelle larve.(E) Lo stesso che(D) per i geni codificanti le proteine represse nelle larve. L’ombreggiatura blu indica l’intensità.DOI:
http://dx.doi.org/10.7554/eLife.22194.012
Histone PTM e la messa a punto dell’espressione genica in Amphimedon
Per studiare la distribuzione degli istoni H3 PTM nei geni di Amphimedon, abbiamo calcolato l’arricchimento medio degli istoni H3 PTM e RNAPII rispetto ai TSS dei geni che codificano le proteine. Ingresso-normalizzato ChIP-seq lettura copertura ha rivelato un forte unimodale H3K4me3 picco unimodale posizionato immediatamente dopo il TSS di geni espressi che co-localizza con H3K27ac e RNAPII(Figura 2A; Figura 2-figure supplemento 1; Figura 2-figure supplemento 2A). Inoltre, H3K4me3 ha segnato (i) geni con orientamento testa a testa che possono essere sotto il controllo di un promotore bidirezionale (una caratteristica comune nel genoma di Amphimedon[Fernandez-Valverde e Degnan, 2016]), e (ii) TSSs alternativi(Figura 2-figure supplement 3). Ciò è coerente con il fatto che H3K4me3 è promoter-proximale e posizionato sul nucleosoma +1 (Zhouet al., 2011; Ho et al., 2014; Lenhard et al., 2012). È stata osservata una regione prominente di nucleosoma impoverito a monte del TSS dei geni espressi (probabilmente corrispondente al promotore prossimale) seguita da un nucleosoma strettamente localizzato (il nucleosoma +1) (vedi sotto Figura 2-figure supplement 4D), suggerendo che l’interazione tra il posizionamento del nucleosoma e la trascrizione è conservata nei promotori di spugna (Sebé-Pedróset al.,2016; Schwaiger et al., 2014; Roy et al., 2010; Bai e Morozov, 2010; Jiang e Pugh, 2009). Nel complesso, la distribuzione dell’istone H3 PTM in Amphimedon è correlata allo stato di espressione dei suoi geni, come negli eumetazoi(Schwaiger et al., 2014; Roy et al., 2010) (test esatto di Fisher, valore p corretto FDR<0,05) (Figura2B e C; Figura 2-figure supplement 2B-D).10.7554/eLife.22194.013Figure 2.Histone PTMs sono correlati con le variazioni di espressione genica durante lo sviluppo.(A) TSS centrato sulla media del DNA di ingresso TSS normalizzato lettura del diagramma di copertura di H3K4me3 attraverso i geni della proteina Amphimedon-coding. L’asse x si estende per ± 3 kb intorno a TSSs e rappresenta la posizione all’interno del gene rispetto a TSS. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per l’H3K4me3 ChIP-seq letto in Amphimedon adulto. Linea rosa: Geni non espressi. Linea blu: Geni a bassa espressione. Linea arancione: Geni espressi mediamente. Linea blu chiaro: Geni ad alta espressione. L’area grigia ombreggiata rappresenta la dimensione media delle sequenze di codifica di Amphimedon.(B) Esempio di geni codificanti contrassegnati da picchi H3K4me3. La finestra genomica mostra la copertura di H3K4me3 normalizzata del DNA in ingresso e l’espressione di RNA-seq sia nella larva che nell’adulto.(C) Viene mostrata l’associazione delle regioni di arricchimento di cinque istoni H3 PTMs (H3K4me3, H3K27ac, H3K4me1, H3K36me3 e H3K27me3) e RNAPII con liste di vari gruppi di espressione genica in adulti. La chiave di colore rappresenta il log2(odds ratio) e i significativi valori P aggiustati (il test esatto di Fisher) sono sovrapposti alle griglie. Un valore P di zero significa che la sovrapposizione è altamente significativa. N.S.: non significativo. Il rapporto delle quote rappresenta la forza dell’associazione.(D) TSS-centrato TSS media input DNA normalizzato normalizzato letto trame di copertura H3K4me3 e RNAPII attraverso ‘ad alta varianza’ e ‘a bassa varianza‘ proteina codifica geni. L’asse x si estende per ± 3 kb intorno a TSSs e rappresenta la posizione all’interno del gene rispetto a TSS. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per l’arricchimento di ChIP-seq letto in Amphimedon adulto. Azzurro: geni codificanti ad alta varianza. Linea arancione: geni codificanti a bassa varianza. L’area grigia ombreggiata rappresenta la dimensione media delle sequenze di codifica di Amphimedon.(E) I cinque termini più significativamente arricchiti di Ontologia Gene (GO) per i geni codificanti le proteine ad alta e bassa varianza ( valori P aggiustati tra parentesi, test ipergeometrico). La tabella completa GO è mostrata in Figura 2 dati fonte 1.DOI:http://dx.doi.org/10.7554/eLife.22194.01310.7554/eLife.22194.014Figure2 dati fonte 1.GO risultato arricchimento biologico processo termine arricchimento per i set di geni ad alta varianza e bassa varianza (test ipergeometrico, FDR<0.01).DOI:http://dx.doi.org/10.7554/eLife.22194.01410.7554/eLife.22194.015Figure2—dati fonte 2.KEGG percorsi 2.KEGG arricchiti in modo significativo nei geni a bassa e alta varianza.DOI:http://dx.doi.org/10.7554/eLife.22194.01510.7554/eLife.22194.016Figure2-figure supplement 1.TSS centrato 1.TSS media input DNA normalizzato lettura lettura media trame di copertura e mappe termiche di RNAPII, H3K27ac, H3K36me3, H3K4me1 e H3K27me3 attraverso la proteina Amphimedon geni codificanti. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per ChIP-seq letto in adulto. L’area grigia ombreggiata rappresenta la dimensione media delle sequenze di codifica di Amphimedon.DOI:http://dx.doi.org/10.7554/eLife.22194.01610.7554/eLife.22194.017Figure2-figure supplement 2.Histone PTMs e variazioni di espressione genica durante lo sviluppo. (A) TSS centrato su TSS media del DNA di ingresso normalizzata lettura normalizzata trama di copertura di H3K4me3 attraverso la proteina Amphimedon codifica geni Amphimedon. L’asse x si estende per ± 3 kb intorno a TSSs e rappresenta la posizione all’interno del gene rispetto a TSS. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per l’H3K4me3 ChIP-seq letto nelle larve. Linea blu: Geni non espressi. Linea arancione: secondo 500 geni espressi. Linea blu chiaro: primi 500 geni espressi. L’area grigia ombreggiata rappresenta la dimensione media delle sequenze di codifica di Amphimedon.(B) Viene mostrata l’associazione delle regioni di arricchimento di quattro istoni H3 PTMs (H3K4me3, H3K27ac, H3K4me1, e H3K27me3) e RNAPII con liste di vari gruppi di espressione genica nelle larve. La chiave di colore rappresenta il log2(odds ratio) e i significativi valori P regolati (test esatto di Fisher) sono sovrapposti alle griglie. Un valore P di zero significa che la sovrapposizione è altamente significativa. N.S.: non significativo.(C) Gene di codifica con espressione larvale specifica contrassegnata da H3K4me3. La finestra genomica mostra l’input-DNA normalizzato H3K4me3 copertura H3K4me3 e RNA-seq espressione sia in larva e adulto.(D) Come(C) per i geni codificanti con espressione adulto-specifica.DOI:http://dx.doi.org/10.7554/eLife.22194.01710.7554/eLife.22194.018FigureSupplemento a 2 cifre 3.H3K4me3 per l’arricchimento a 2 cifre dei geni con orientamento testa a testa e TSS alternativi.(A) Esempio di geni codificanti con TSS alternativi (Aqu2.1.39785_001 e Aqu2.1.396786_001) contrassegnati da successivi picchi di H3K4me3. La finestra genomica mostra la copertura di H3K4me3 normalizzata con DNA in ingresso e l’espressione RNA-seq sia nella larva che nell’adulto.(B) Uguale alla(A) per i geni strettamente localizzati testa a testa (Aqu2.1.30305_001 e Aqu2.1.30306_001). Sono mostrati i geni codificanti (viola) e le isoforme dei geni codificanti (azzurro).DOI:http://dx.doi.org/10.7554/eLife.22194.01810.7554/eLife.22194.019Figuresupplemento a 2 cifre 4.ChIP-seq profili di H3K4me3 e dell’istone totale H3 attraverso i geni ad alta e bassa varianza.(A) Profilo di espressione dello sviluppo, dalla scissione precoce all’adulto, dei geni ad alta varianza altamente espressi (n = 1066). I livelli di espressione sono stati misurati con CEL-seq e ridimensionati per riga. Il rosso indica un livello di espressione alto, l’azzurro un’espressione bassa. PS, post-settlement postlarva.(B) TSS-centrato TSS media di ingresso del DNA normalizzata lettura del diagramma di copertura H3K4me3 ad alta varianza di H3K4me3 attraverso la proteina ad alta varianza di codifica dei geni. L’asse x si estende per ± 3 kb intorno a TSSs e rappresenta la posizione all’interno del gene rispetto a TSS. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per H3K4me3 ChIP-seq in adulti. Linea viola: Geni a bassa espressione. Linea arancione: Geni espressi mediamente. Linea azzurra: Geni ad alta espressione.(C) Come(B) ma per i geni a bassa variazione.(D) TSS centrato sul DNA medio in ingresso normalizzato in lettura del DNA normalizzato copertura del diagramma di copertura totale dell’istone H3 attraverso i geni ad alta varianza e a bassa varianza della proteina di codifica. L’asse x si estende per ± 3 kb intorno ai TSS e rappresenta la posizione all’interno del gene rispetto ai TSS. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per ChIP-seq letto in adulto. Azzurro: Geni codificanti ad alta varianza. Linea arancione: Geni codificanti a bassa varianza. L’area grigia ombreggiata rappresenta la dimensione media delle sequenze di codifica di Amphimedon.DOI:http://dx.doi.org/10.7554/eLife.22194.019
Per indagare la dinamica degli istoni PTM nei geni regolati durante lo sviluppo di Amphimedon, abbiamo analizzato i dati CEL-seq(Levin et al., 2016; Hashimshony et al., 2012; Anavy et al., 2014), che comprendono 82 campioni di sviluppo di Amphimedon, dalla scissione precoce all’adulto compresso in 17 stadi, nel contesto dei profili ChIP-seq dell’istone totale H3, H3K4me3, e RNAPII. Abbiamo selezionato i geni con la più alta deviazione mediana assoluta per l’espressione genica attraverso questi 17 stadi di sviluppo di Amphimedon (misurando efficacemente l’ampiezza della variazione dei livelli di espressione per un dato gene), con il risultato di un insieme di 3.200 ‘ad alta varianza’ geni espressi (Figura 2-figuresupplement 4A). I restanti geni espressi sono stati definiti come geni ‘a bassa varianza‘(3.999) (vedi Materiali e metodi per l’elenco completo dei criteri di selezione). È degno di nota il fatto che i geni ad alta varianza sono stati espressi, in media, anche a livelli più alti rispetto ai geni a bassa varianza (espressione media degli adulti di 51 vs 7 CEL-seq conteggi normalizzati, rispettivamente). I TSS dei geni ad alta varianza erano fortemente marcati da H3K4me3 e occupati da RNAPII(Figura 2D; Figura 2-figure supplement 4B). Inoltre, hanno mostrato l’esaurimento del nucleosoma a monte dei TSS (visto come mancanza di segnale totale dell’istone H3), coerente con l’idea che H3K4me3 vicino ai TSS destabilizza l’interazione tra gli istoni e il DNA per dirigere RNAPII per facilitare il legame degli elementi regolatori del promotore e avviare la trascrizione(Jiang e Pugh, 2009; Ha et al., 2011; Boeger et al., 2003)(Figura 2-figure supplement 4D). Al contrario, livelli più bassi di H3K4me3 o RNAPII (test di Mann-Whitney U, p-valore=0,05287 e p-valore<2,2e-16, rispettivamente; Figura 2D; Figura 2-figure supplement 4C) ma una maggiore occupazione del nucleosoma caratterizza i geni a bassa varianza (vista come mancanza di esaurimento del nucleosoma a monte dei TSS; Figura 2-figure supplement 4D). Questi risultati sono coerenti con il fatto che H3K4me3 è predittivo dei livelli di espressione genica(Ha et al., 2011; Karlić et al., 2010).
I paesaggi distintivi degli istoni PTM in geni ad alta e bassa varianza sono anche correlati a distinti gruppi di geni funzionali correlati, come indicato dall’Ontologia Genetica (GO) e dalle analisi del percorso KEGG. I geni ad alta varianza, che includono anche un numero significativamente più elevato di famiglie di geni con fattore di trascrizione(ad esempio, JUN e ATF6 Jindriche Degnan [2016]) rispetto ai geni a bassa varianza (test esatto di Fisher, p-valore=3.872e-08), sono stati prevalentemente arricchiti nei percorsi di segnalazione (test ipergeometrico, FDR regolato p-valore p<0,01; Figura 2E; vedi Figura 2 dati 1 e Figura 2 dati 2 per l’ elenco completo). Al contrario, i geni a bassa varianza sono stati arricchiti per i termini metabolici GO(Figura 2E; vedi Figura 2—dati fonte 1 e Figura 2—dati fonte 2 per l’ elenco completo). Questo risultato è coerente con H3K4me3 essendo importante per la messa a punto dell’espressione genica dei geni dello sviluppo dinamicamente espressi, ad esempio, fattore di trascrizione e geni di segnalazione. Tuttavia, non è chiaro se l’H3K4me3 sia necessario per elevati livelli di espressione genica o se sia necessario o associato a frequenti passaggi di stato trascrizionale.
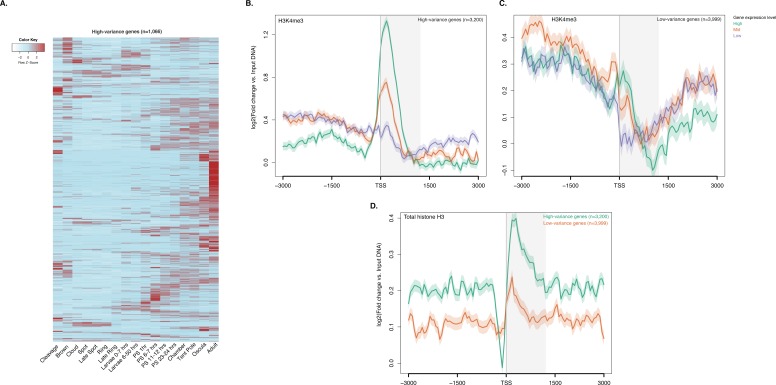
Figura 2-figure supplement 4.Figura 2—supplemento alla figura 4. I PTM Histone sono correlati con le variazioni dell’espressione genica durante lo sviluppo.GO risultato dell’arricchimento con il termine di processo biologico per i set di geni ad altae bassa varianza (test ipergeometrico, FDR<0.01).percorsi KEGG significativamente arricchiti in geni a bassa e alta varianza.GO risultato dell’arricchimento con il termine di processo biologico per i set di geni ad alta e bassa varianza (test ipergeometrico, FDR<0.01).KEGG percorsi significativamente arricchito in bassa varianza e ad alta varianza geni.TSS centrato DNA media di ingresso del DNA normalizzata lettura normalizzata trame di copertura e mappe termiche di RNAPII, H3K27ac, H3K36me3, H3K4me1 e H3K27me3 attraverso la proteina Amphimedon codifica geni.Histone PTMs e variazioni di espressione genica durante lo sviluppo.H3K4me3 arricchimento a geni con orientamento testa a testa e TSSs alternativi.ChIP-seq profili di H3K4me3 e histone H3 totale attraverso geni ad alta e bassa varianza.(A) TSS centrato su TSS media di ingresso DNA normalizzato in ingresso lettura copertura di copertura normalizzata di H3K4me3 attraverso i geni di codifica della proteina Amphimedon. L’asse x si estende per ± 3 kb intorno a TSSs e rappresenta la posizione all’interno del gene rispetto a TSS. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per l’H3K4me3 ChIP-seq letto in Amphimedon adulto. Linea rosa: Geni non espressi. Linea blu: Geni a bassa espressione. Linea arancione: Geni espressi mediamente. Linea blu chiaro: Geni ad alta espressione. L’area grigia ombreggiata rappresenta la dimensione media delle sequenze di codifica di Amphimedon.(B) Esempio di geni codificanti contrassegnati da picchi H3K4me3. La finestra genomica mostra la copertura di H3K4me3 normalizzata del DNA in ingresso e l’espressione di RNA-seq sia nella larva che nell’adulto.(C) Viene mostrata l’associazione delle regioni di arricchimento di cinque istoni H3 PTMs (H3K4me3, H3K27ac, H3K4me1, H3K36me3 e H3K27me3) e RNAPII con liste di vari gruppi di espressione genica in adulti. La chiave di colore rappresenta il log2(odds ratio) e i significativi valori P aggiustati (il test esatto di Fisher) sono sovrapposti alle griglie. Un valore P di zero significa che la sovrapposizione è altamente significativa. N.S.: non significativo. Il rapporto delle quote rappresenta la forza dell’associazione.(D) TSS-centrato TSS media input DNA normalizzato normalizzato letto trame di copertura H3K4me3 e RNAPII attraverso ‘ad alta varianza’ e ‘a bassa varianza‘ proteina codifica geni. L’asse x si estende per ± 3 kb intorno a TSSs e rappresenta la posizione all’interno del gene rispetto a TSS. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per l’arricchimento di ChIP-seq letto in Amphimedon adulto. Azzurro: geni codificanti ad alta varianza. Linea arancione: geni codificanti a bassa varianza. L’area grigia ombreggiata rappresenta la dimensione media delle sequenze di codifica di Amphimedon.(E) I cinque termini più significativamente arricchiti di Ontologia Gene (GO) per i geni codificanti le proteine ad alta e bassa varianza ( valori P aggiustati tra parentesi, test ipergeometrico). La tabella GO completa è mostrata in Figura 2 dati fonte 1.1. DOI:
http://dx.doi.org/10.7554/eLife.22194.01310.7554/eLife.22194.014Cifra 2 dati a 2 fonti 1.GO risultato dell’arricchimento con il termine di processo biologico per i set di geni ad alta e bassa varianza (test ipergeometrico, FDR<0.01).DOI:http://dx.doi.org/10.7554/eLife.22194.014DOI:
http://dx.doi.org/10.7554/eLife.22194.01410.7554/eLife.22194.015Cifra dati a 2 fonti 2.KEGG percorsi arricchiti in modo significativo nei geni a bassa e alta varianza.DOI:http://dx.doi.org/10.7554/eLife.22194.015DOI:
http://dx.doi.org/10.7554/eLife.22194.015DOI:
http://dx.doi.org/10.7554/eLife.22194.014DOI:
http://dx.doi.org/10.7554/eLife.22194.015L’asse x si estende per ± 3 kb intorno ai TSS e rappresenta la posizione all’interno del gene rispetto ai TSS. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per ChIP-seq letto in adulto. L’area grigia ombreggiata rappresenta la dimensione media delle sequenze di codifica di Amphimedon.DOI:
http://dx.doi.org/10.7554/eLife.22194.016(A) TSS-centrato TSS media di ingresso DNA normalizzato letto trama di copertura di H3K4me3 attraverso i geni di codifica della proteina Amphimedon. L’asse x si estende per ± 3 kb intorno a TSSs e rappresenta la posizione all’interno del gene rispetto a TSS. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per l’H3K4me3 ChIP-seq letto nelle larve. Linea blu: Geni non espressi. Linea arancione: secondo 500 geni espressi. Linea blu chiaro: primi 500 geni espressi. L’area grigia ombreggiata rappresenta la dimensione media delle sequenze di codifica di Amphimedon.(B) Viene mostrata l’associazione delle regioni di arricchimento di quattro istoni H3 PTMs (H3K4me3, H3K27ac, H3K4me1, e H3K27me3) e RNAPII con liste di vari gruppi di espressione genica nelle larve. La chiave di colore rappresenta il log2(odds ratio) e i significativi valori P regolati (test esatto di Fisher) sono sovrapposti alle griglie. Un valore P di zero significa che la sovrapposizione è altamente significativa. N.S.: non significativo.(C) Gene di codifica con espressione larvale specifica contrassegnata da H3K4me3. La finestra genomica mostra l’input-DNA normalizzato H3K4me3 copertura H3K4me3 e RNA-seq espressione sia in larva e adulto.(D) Come(C) per i geni codificanti con espressione adulto-specifica.DOI:
http://dx.doi.org/10.7554/eLife.22194.017(A) Esempio di geni codificanti con TSS alternativi (Aqu2.1.39785_001 e Aqu2.1.396786_001) contrassegnati da successivi picchi H3K4me3. La finestra genomica mostra la copertura di H3K4me3 normalizzata con DNA in ingresso e l’espressione RNA-seq sia nella larva che nell’adulto.(B) Uguale alla(A) per i geni strettamente localizzati testa a testa (Aqu2.1.30305_001 e Aqu2.1.30306_001). Sono mostrati i geni codificanti (viola) e le isoforme dei geni codificanti (azzurro).DOI:
http://dx.doi.org/10.7554/eLife.22194.018(A) Profilo di espressione dello sviluppo, dalla scissione precoce all’adulto, dei geni ad alta varianza altamente espressi (n = 1066). I livelli di espressione sono stati misurati con CEL-seq e ridimensionati per riga. Il rosso indica un livello di espressione alto, l’azzurro un’espressione bassa. PS, post-settlement postlarva.(B) TSS-centrato TSS media di ingresso del DNA normalizzata lettura del diagramma di copertura H3K4me3 ad alta varianza di H3K4me3 attraverso la proteina ad alta varianza di codifica dei geni. L’asse x si estende per ± 3 kb intorno a TSSs e rappresenta la posizione all’interno del gene rispetto a TSS. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per H3K4me3 ChIP-seq in adulti. Linea viola: Geni a bassa espressione. Linea arancione: Geni espressi mediamente. Linea azzurra: Geni ad alta espressione.(C) Come(B) ma per i geni a bassa variazione.(D) TSS centrato sul DNA medio in ingresso normalizzato in lettura del DNA normalizzato copertura del diagramma di copertura totale dell’istone H3 attraverso i geni ad alta varianza e a bassa varianza della proteina di codifica. L’asse x si estende per ± 3 kb intorno ai TSS e rappresenta la posizione all’interno del gene rispetto ai TSS. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per ChIP-seq letto in adulto. Azzurro: Geni codificanti ad alta varianza. Linea arancione: Geni codificanti a bassa varianza. L’area grigia ombreggiata rappresenta la dimensione media delle sequenze di codifica di Amphimedon.DOI:
http://dx.doi.org/10.7554/eLife.22194.019
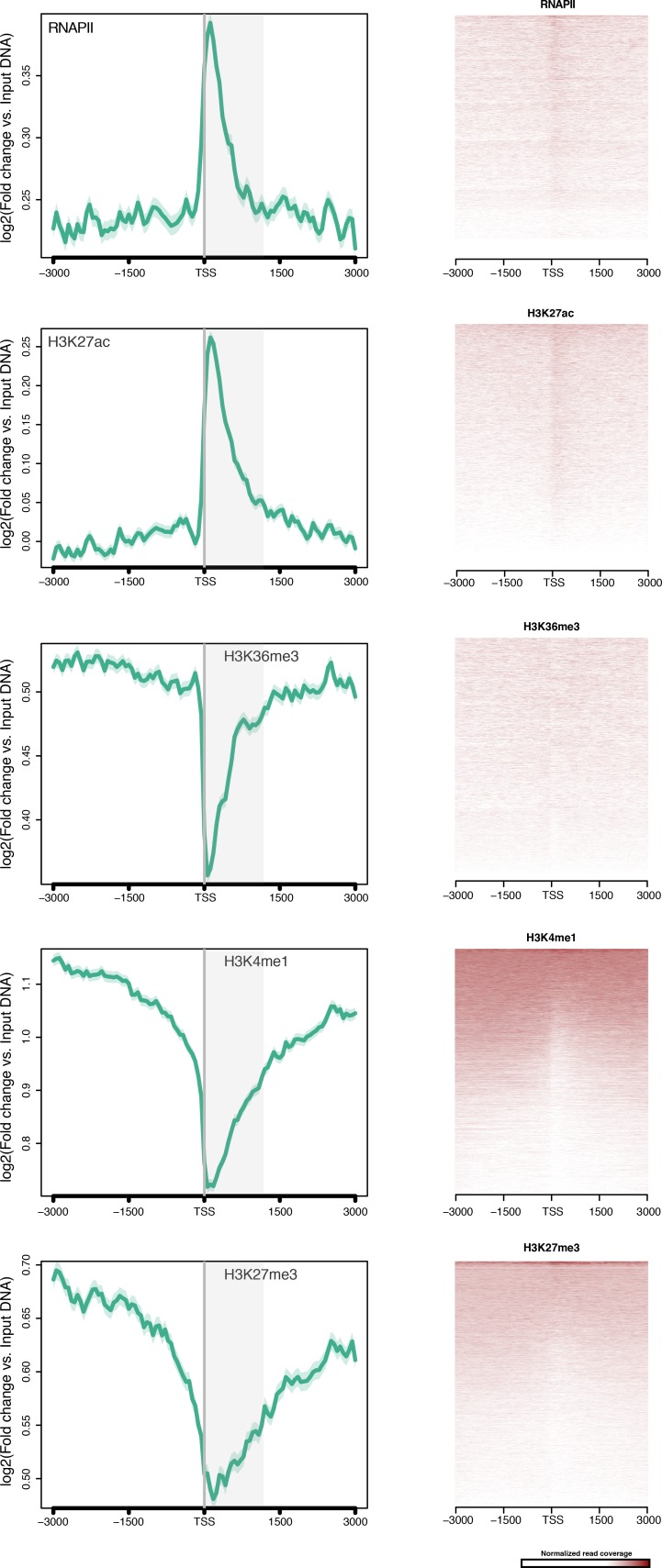
Figura 2-figure supplement 1.TSS centrata sul DNA medio in ingresso normalizzato in ingresso trame di copertura lettura normalizzata e mappe termiche di RNAPII, H3K27ac, H3K36me3, H3K4me1 e H3K27me3 attraverso i geni di codifica della proteina Amphimedon.L’asse x si estende per ± 3 kb intorno ai TSS e rappresenta la posizione all’interno del gene rispetto ai TSS. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per ChIP-seq letto in adulto. L’area grigia ombreggiata rappresenta la dimensione media delle sequenze di codifica di Amphimedon.DOI:
http://dx.doi.org/10.7554/eLife.22194.016

Figura 2-figure supplement 2.Histone PTM e variazioni di espressione genica durante lo sviluppo.(A) TSS-centrato TSS media di ingresso DNA normalizzato letto trama di copertura di H3K4me3 attraverso i geni di codifica della proteina Amphimedon. L’asse x si estende per ± 3 kb intorno a TSSs e rappresenta la posizione all’interno del gene rispetto a TSS. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per l’H3K4me3 ChIP-seq letto nelle larve. Linea blu: Geni non espressi. Linea arancione: secondo 500 geni espressi. Linea blu chiaro: primi 500 geni espressi. L’area grigia ombreggiata rappresenta la dimensione media delle sequenze di codifica di Amphimedon.(B) Viene mostrata l’associazione delle regioni di arricchimento di quattro istoni H3 PTMs (H3K4me3, H3K27ac, H3K4me1, e H3K27me3) e RNAPII con liste di vari gruppi di espressione genica nelle larve. La chiave di colore rappresenta il log2(odds ratio) e i significativi valori P regolati (test esatto di Fisher) sono sovrapposti alle griglie. Un valore P di zero significa che la sovrapposizione è altamente significativa. N.S.: non significativo.(C) Gene di codifica con espressione larvale specifica contrassegnata da H3K4me3. La finestra genomica mostra l’input-DNA normalizzato H3K4me3 copertura H3K4me3 e RNA-seq espressione sia in larva e adulto.(D) Come(C) per i geni codificanti con espressione adulto-specifica.DOI:
http://dx.doi.org/10.7554/eLife.22194.017

Figura 2-figure supplement 3.H3K4me3 arricchimento ai geni con orientamento testa a testa e TSS alternativi.(A) Esempio di geni codificanti con TSS alternativi (Aqu2.1.39785_001 e Aqu2.1.396786_001) contrassegnati da successivi picchi di H3K4me3. La finestra genomica mostra la copertura di H3K4me3 normalizzata con DNA in ingresso e l’espressione RNA-seq sia nella larva che nell’adulto.(B) Uguale alla(A) per i geni strettamente localizzati testa a testa (Aqu2.1.30305_001 e Aqu2.1.30306_001). Sono mostrati i geni codificanti (viola) e le isoforme dei geni codificanti (azzurro).DOI:
http://dx.doi.org/10.7554/eLife.22194.018
Figura 2-figure supplement 4.ChIP-seq profili di H3K4me3 e dell’istone totale H3 attraverso i geni ad alta e bassa varianza.(A) Profilo di espressione dello sviluppo, dalla scissione precoce all’adulto, dei geni ad alta varianza altamente espressi (n = 1066). I livelli di espressione sono stati misurati con CEL-seq e ridimensionati per riga. Il rosso indica un livello di espressione alto, l’azzurro un’espressione bassa. PS, post-settlement postlarva.(B) TSS-centrato TSS media di ingresso del DNA normalizzata lettura del diagramma di copertura H3K4me3 ad alta varianza di H3K4me3 attraverso la proteina ad alta varianza di codifica dei geni. L’asse x si estende per ± 3 kb intorno a TSSs e rappresenta la posizione all’interno del gene rispetto a TSS. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per H3K4me3 ChIP-seq in adulti. Linea viola: Geni a bassa espressione. Linea arancione: Geni espressi mediamente. Linea azzurra: Geni ad alta espressione.(C) Come(B) ma per i geni a bassa variazione.(D) TSS centrato sul DNA medio in ingresso normalizzato in lettura del DNA normalizzato copertura del diagramma di copertura totale dell’istone H3 attraverso i geni ad alta varianza e a bassa varianza della proteina di codifica. L’asse x si estende per ± 3 kb intorno ai TSS e rappresenta la posizione all’interno del gene rispetto ai TSS. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per ChIP-seq letto in adulto. Azzurro: Geni codificanti ad alta varianza. Linea arancione: Geni codificanti a bassa varianza. L’area grigia ombreggiata rappresenta la dimensione media delle sequenze di codifica di Amphimedon.DOI:
http://dx.doi.org/10.7554/eLife.22194.019
L’assenza di H3K4me3 nei geni fortemente regolati in termini di sviluppo sembra essere una caratteristica conservata dai metazoiti
La recente scoperta che la trascrizione di una sottopopolazione di geni espressi in modo estremamente dinamico – tipicamente espressa in un solo stadio di sviluppo – in Drosophila e C. elegans avviene in assenza di H3K4me3 ha messo in discussione il ruolo canonico degli istoni PTM nella modulazione dell’espressione genica (Pérez-Lluchet al., 2015a). Per verificare se questa caratteristica appena scoperta è conservata nei non-bilateriani, abbiamo interrogato i suddetti dati CEL-seq(Levin et al., 2016; Hashimshony et al., 2012; Anavy et al., 2014), che comprendono 82 campioni di sviluppo di Anfimedone, dalla scissione precoce all’adulto compresso in 17 stadi, e selezionati arbitrariamente, in modo simile a Pérez-Lluch et al. (2015a), i 1.000 geni con i più bassi coefficienti di variazione (“stabili”geni) espressi con piccole variazioni durante lo sviluppo. Al contrario, i 1.000 geni con i coefficienti di variazione più alti sono stati definiti come geni “regolati”. In particolare, i geni ‘regolati’ consistevano in una piccola popolazione di geni che differivano dai geni ‘ad alta varianza‘ descritti in precedenza per avere modelli di espressione molto più ristretti, principalmente espressi nella fase tardo-giovanile e/o adulta (Figura 3-figuresupplement 1). Anche se i geni stabili e regolati avevano livelli simili di RNAPII e l’istone totale H3(Figura 3-figure supplement 1B e C ), i geni stabili erano fortemente marcati da H3K4me3 e i geni regolati avevano livelli significativamente più bassi di H3K4me3 (Mann-Whitney U test, p-value=7.431e-05; Figura 3A), suggerendo che la riduzione dei livelli di H3K4me3 non influisce sull’espressione dei geni regolati (Pérez-Lluchet al., 2015a).10.7554/eLife.22194.020Figure 3.Expression senza H3K4me3 in geni fortemente regolati per lo sviluppo.(A) TSS centrato sul DNA di ingresso media di ingresso normalizzato letto trama di copertura di H3K4me3 attraverso i geni di codifica delle proteine ‘regolati’ e ‘ stabile’ durante lo sviluppo di Amphimedon. L’asse x si estende ± 3 kb intorno a TSSs e rappresenta la posizione all’interno del gene rispetto a TSS. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per l’H3K4me3 ChIP-seq letto in Amphimedon adulto. Linea azzurra: primi 500 geni regolati. Linea arancione: secondi 500 geni regolati. Linea viola: primi 500 geni stabili. Linea rosa: secondi 500 geni stabili. L’area grigia ombreggiata rappresenta la dimensione media delle sequenze di codifica di Amphimedon.(B) Ingresso DNA-normalizzato H3K4me3 copertura H3K4me3 e RNA-seq espressione in adulto per Aqu2.1.40735_001, un gene espresso in modo stabile durante lo sviluppo di Amphimedon, Aqu2.1.39666_001, un gene regolato con espressione adulto-specifica, e Aqu2.1.34366_001, un gene regolato con espressione larvale specifica.(C) TSS-centrato TSS media di ingresso del DNA normalizzata lettura trama di copertura di H3K4me3 attraverso i geni di codifica della proteina ‘regolata’ e ‘ stabile’ durante lo sviluppo di Nematostella vectensis. L’asse x si estende ± 3 kb intorno a TSSs e rappresenta la posizione all’interno del gene rispetto a TSS. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per l’H3K4me3 ChIP-seq, che si legge nei polipi femminili adulti di Nematostella. L’area grigia ombreggiata rappresenta la dimensione media delle sequenze di codifica di Nematostella.DOI:http://dx.doi.org/10.7554/eLife.22194.02010.7554/eLife.22194.021Figuresupplemento a 3 cifre 1.ChIP-seq profili di RNAPII, istone totale H3, H3K36me3 e H3K27me3 attraverso i geni regolati e stabili. (A) Profilo di espressione dello sviluppo, dalla scissione precoce all’adulto, dei geni di codifica delle proteine regolati e stabili (vedi testo principale e Materiali e metodi per i dettagli). I livelli di espressione sono stati misurati da CEL-seq e ridimensionato per riga. Il rosso indica un livello di espressione alto, l’azzurro un’espressione bassa. Si noti che i geni ‘regolati’ mostrano modelli di espressione molto più limitata, essendo in genere espresso solo in uno o due stadi di sviluppo (oscula e / o adulti), che sia‘stabile’e ‘ad alta varianza’ geni(vedi Figura 2-figure supplemento 4A per un confronto). PS, post-sedimentazione postlarva.(B) TSS centrato sul DNA di ingresso media di ingresso normalizzata normalizzata lettura trama copertura RNAPII,(C) totale H3,(D) H3K36me3 e(E) H3K27me3 attraverso ‘regolamentato‘ e ‘stabile’proteina codifica dei geni durante lo sviluppo di Amphimedon. L’asse x si estende per ± 3 kb intorno a TSSs e rappresenta la posizione all’interno del gene rispetto a TSS. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per ChIP-seq letto in adulto. Linea azzurra: primi 1.000 geni regolati. Linea arancione: primi 1.000 geni stabili. L’area grigia ombreggiata rappresenta la dimensione media delle sequenze di codifica di Amphimedon.DOI:http://dx.doi.org/10.7554/eLife.22194.021
Abbiamo confrontato il modello di H3K4me3 tra uno dei tre principali geni espressi in modo stabile durante lo sviluppo della spugna (Aqu2.1.40735_001, un E3 ubiquitin-proteina ligasi), e il gene con il più alto coefficiente di variazione (Aqu2.1.39666_001, un gene specifico della spugna putativo espresso specificamente nell’adulto)(Figura 3B). Il primo ha mostrato un forte arricchimento H3K4me3 al TSS, mentre il secondo mancava di qualsiasi marcatura, anche se la sua espressione nell’adulto era ~ 70 volte superiore al gene stabile (33 vs 2361 CEL-seq conteggi normalizzati nell’adulto, rispettivamente). Questa mancanza di H3K4me3 al TSS di geni regolati è stata osservata in modo simile nella larva, esemplificata qui da un gene regolato specifico per la larva (Aqu2.1.34366_001) espresso 3,5 volte superiore al gene stabile di cui sopra (Aqu2.1.40735_001) (147 vs 43 CEL-seq conteggi normalizzati in larva, rispettivamente)(Figura 3B). Inoltre, come mostrato in Drosophila(Pérez-Lluch et al., 2015a), i geni regolati hanno mostrato livelli più elevati di H3K27me3 (Mann-Whitney U test, p-valore<6.517e-06) e livelli più bassi di H3K36me3 (Mann-Whitney U test, p-valore<9.235e-08) rispetto ai geni stabili (Figura 3-figure supplemento 1D ed E). Analizzando l’espressione genica basata sull’RNA-seq attraverso lo sviluppo della Nematostella vectensis cnidariana ( Helmet al., 2013) e i set di dati ChIP-seq precedentemente pubblicati nei polipi femminili adulti di Nematostella (Schwaigeret al., 2014), abbiamo ottenuto lo stesso modello (test Mann-Whitney U, p-value<2.2e-16; Figura 3C).
Questi risultati suggeriscono che l’H3K4me3 potrebbe non essere strumentale per l’espressione estremamente dinamica dello sviluppo e rafforzano la nostra interpretazione che è necessaria per la sintonizzazione dei livelli di espressione genica, un pattern che sembra essere una caratteristica metazoica conservata (Pérez-Lluchet al., 2015a).
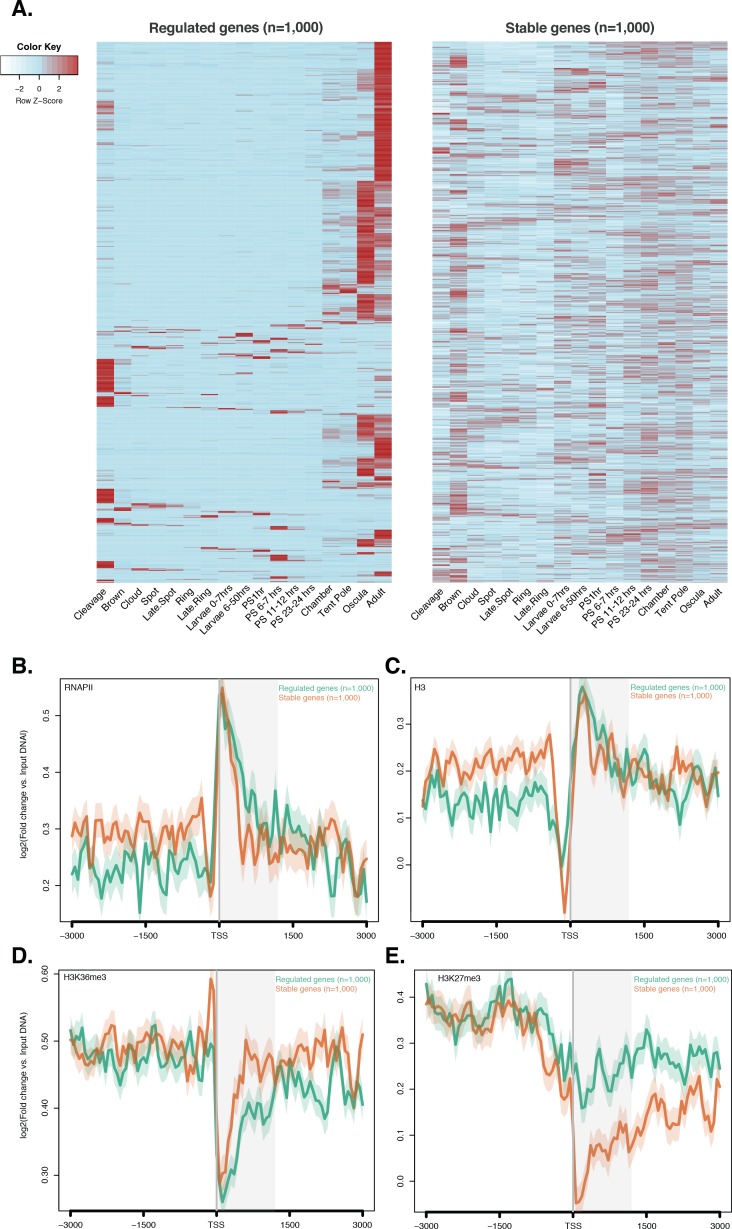
Figura 3-figure supplement 1.Espressione senza H3K4me3 in geni fortemente regolati in sviluppo.ChIP-seq profili di RNAPII, istone totale H3, H3K36me3 e H3K27me3 attraverso geni regolati e stabili.(A) TSS-centrato TSS media di ingresso DNA normalizzato letto trama di copertura media di H3K4me3 attraverso i geni di codifica della proteina ‘regolata’ e ‘ stabile’ durante lo sviluppo di Amphimedon. L’asse x abbraccia ± 3 kb intorno a TSSs e rappresenta la posizione all’interno del gene rispetto a TSS. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per l’H3K4me3 ChIP-seq letto in Amphimedon adulto. Linea azzurra: primi 500 geni regolati. Linea arancione: secondi 500 geni regolati. Linea viola: primi 500 geni stabili. Linea rosa: secondi 500 geni stabili. L’area grigia ombreggiata rappresenta la dimensione media delle sequenze di codifica di Amphimedon.(B) Ingresso DNA-normalizzato H3K4me3 copertura H3K4me3 e RNA-seq espressione in adulto per Aqu2.1.40735_001, un gene espresso in modo stabile durante lo sviluppo di Amphimedon, Aqu2.1.39666_001, un gene regolato con espressione adulto-specifica, e Aqu2.1.34366_001, un gene regolato con espressione larvale specifica.(C) TSS-centrato TSS media di ingresso del DNA normalizzata lettura trama di copertura di H3K4me3 attraverso i geni di codifica della proteina ‘regolata’ e ‘ stabile’ durante lo sviluppo di Nematostella vectensis. L’asse x si estende ± 3 kb intorno a TSSs e rappresenta la posizione all’interno del gene rispetto a TSS. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per l’H3K4me3 ChIP-seq, che si legge nei polipi femminili adulti di Nematostella. L’area grigia ombreggiata rappresenta la dimensione media delle sequenze di codifica di Nematostella.DOI:
http://dx.doi.org/10.7554/eLife.22194.020(A) Profilo di espressione dello sviluppo, dalla scissione precoce all’adulto, dei geni di codifica delle proteine regolati e stabili (vedi testo principale e Materiali e metodi per i dettagli). I livelli di espressione sono stati misurati da CEL-seq e ridimensionati per riga. Il rosso indica un livello di espressione alto, l’azzurro un’espressione bassa. Si noti che i geni ‘regolati’ mostrano modelli di espressione molto più limitata, essendo in genere espresso solo in uno o due stadi di sviluppo (oscula e / o adulti), che sia‘stabile’e ‘ad alta varianza’ geni(vedi Figura 2-figure supplemento 4A per un confronto). PS, post-sedimentazione postlarva.(B) TSS centrato sul DNA di ingresso media di ingresso normalizzato normalizzato letto trama copertura RNAPII,(C) totale H3,(D) H3K36me3 e(E) H3K27me3 attraverso ‘regolamentato‘ e ‘stabile’proteina codifica dei geni durante lo sviluppo di Amphimedon. L’asse x si estende per ± 3 kb intorno a TSSs e rappresenta la posizione all’interno del gene rispetto a TSS. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per ChIP-seq letto in adulto. Linea azzurra: primi 1.000 geni regolati. Linea arancione: primi 1.000 geni stabili. L’area grigia ombreggiata rappresenta la dimensione media delle sequenze di codifica di Amphimedon.DOI:
http://dx.doi.org/10.7554/eLife.22194.021
Figura 3-figure supplement 1.ChIP-seq profili di RNAPII, istone totale H3, H3K36me3 e H3K27me3 attraverso geni regolati e stabili.(A) Profilo di espressione dello sviluppo, dalla scissione precoce all’adulto, dei geni di codifica delle proteine regolati e stabili (vedi testo principale e Materiali e metodi per i dettagli). I livelli di espressione sono stati misurati da CEL-seq e ridimensionati per riga. Il rosso indica un livello di espressione alto, l’azzurro un’espressione bassa. Si noti che i geni ‘regolati’ mostrano modelli di espressione molto più limitata, essendo in genere espresso solo in uno o due stadi di sviluppo (oscula e / o adulti), che sia‘stabile’e ‘ad alta varianza’ geni(vedi Figura 2-figure supplemento 4A per un confronto). PS, post-sedimentazione postlarva.(B) TSS centrato sul DNA di ingresso media di ingresso normalizzato normalizzato letto trama copertura RNAPII,(C) totale H3,(D) H3K36me3 e(E) H3K27me3 attraverso ‘regolamentato‘ e ‘stabile’proteina codifica dei geni durante lo sviluppo di Amphimedon. L’asse x si estende per ± 3 kb intorno a TSSs e rappresenta la posizione all’interno del gene rispetto a TSS. L’asse y rappresenta l’arricchimento normalizzato del DNA in ingresso per ChIP-seq letto in adulto. Linea azzurra: primi 1.000 geni regolati. Linea arancione: primi 1.000 geni stabili. L’area grigia ombreggiata rappresenta la dimensione media delle sequenze di codifica di Amphimedon.DOI:
http://dx.doi.org/10.7554/eLife.22194.021
Il complesso repressivo Polycomb 2 (PRC2) è conservato ad Amphimedon e i suoi siti di rilegatura contengono presunti motivi di rilegatura a fattore GAGA
PRC2 è responsabile della trimetilazione della lisina 27 dell’istone H3 (H3K27me3), uno degli istoni repressivi H3 PTM meglio caratterizzati(Margueron e Reinberg, 2011). Come passo per indagare un meccanismo putativo di silenziamento mediato dalla PRC2 in Amphimedon, abbiamo identificato gli omologhi di spugna dei componenti della Drosophila PRC2 e abbiamo scoperto che il genoma di Amphimedon contiene quattro copie di omologhi E(z), due copie di omologhi ESC e una copia per ciascuno dei restanti componenti, SU(z)12 e Nurf55(Figura 4A; Figura 4 dati fonte 1).10.7554/eLife.22194.022Figure 4.DNA motivi 4.DNA sovrarappresentati in H3K27me3 regioni trascrizionalmente silenziate.(A) Diagramma che rappresenta la composizione del complesso Drosophila PRC2 e dei suoi quattro componenti principali: la sottounità catalitica del complesso E(z), la proteina di zinco dito SU(z)12, la proteina WD-ripetizione ESC e la proteina legante lo storico Nurf55. E(z) è responsabile della principale attività enzimatica della PRC2, che è quella di trimetilare l’istone H3 alla lisina 27, producendo H3K27me3. Adattato da(Vissers et al., 2012). Viene mostrata la presenza (verde) o l’assenza (arancione) di PRC2 e dei suoi componenti principali nelle diverse specie di opistoconte rappresentate nell’albero filogenetico (a sinistra). L’anfimedone è evidenziato in verde.(B) Loghi di sequenza di un sottoinsieme dei motivi del DNA determinati dall’analisi MEME-ChIP per essere significativamente arricchito nelle regioni trascrizionalmente silenziate contrassegnate da H3K27me3 in Amphimedon adulto. Per ogni motivo, il miglior TOMTOM corrisponde a un motivo nei database dei mouse JASPAR CORE e UniPROBE, rispettivamente il valore E e il numero di siti che contribuiscono alla costruzione del motivo. Il motivo abbinato è mostrato in alto e il motivo di interrogazione è mostrato in basso.DOI:http://dx.doi.org/10.7554/eLife.22194.02210.7554/eLife.22194.023Figure4 dati fonte 1.Putative orthologs of Drosophila PcG components and associated factors in yeast, Capsaspora , sponge, nematode, and human genome.Table of PcG protein is adapted from(http://www.igh.cnrs.fr/equip/cavalli/link.PolycombTeaching.html).DOI:http://dx.doi.org/10.7554/eLife.22194.02310.7554/eLife.22194.024Figure4-figure supplement 1.Matching loghi sequenza dei motivi del DNA determinati dall’analisi MEME-ChIP per essere significativamente arricchito nelle regioni trascrizionalmente silenziate contrassegnate da H3K27me3 sia in adulti che in larve.il valore E (log likelihood ratio di ogni motivo) e il numero di siti che contribuiscono alla costruzione del motivo sono mostrati, rispettivamente, il valore E (log likelihood ratio di ogni motivo) e il numero di siti che contribuiscono alla costruzione del motivo.DOI:http://dx.doi.org/10.7554/eLife.22194.024
Il reclutamento di PRC2 è stato caratterizzato al meglio a Drosophila, dove le proteine PRC2 reprimono i loro geni target attraverso il reclutamento in Polycomb Response Elements (PRE), che contengono siti di legame per le proteine di legame del DNA specifiche della sequenza, compreso il fattore GAGA e i membri della famiglia dei fattori simili a Krüppel (Müllere Kassis, 2006; Brown et al ., 2005; Strutt et al., 1997; Simon e Kingston, 2009; Kassis e Brown, 2013). Per verificare se i complessi Amphimedon PRC2 potrebbero essere reclutati attraverso un meccanismo simile, abbiamo usato le regioni trascrizionalmente silenziate contrassegnate da H3K27me3 in un’analisi del motivo de novo (Materiali e metodi). Abbiamo cercato motivi brevi (6-15 bp) sulla base del fatto che i siti di interazione noti delle proteine PREbinding in Drosophila sono approssimativamente di questa lunghezza (~8 bp). Motivi vincolanti conservati simili ai fattori GAGA e Krüppel-like, oltre a motivi vincolanti simili ai regolatori dello sviluppo contenenti omeodominio (ad esempio, i membri della famigliaIrx ), sono stati significativamente arricchiti (E-valore<0,05) nel DNA associato alle regioni silenziate H3K27me3 sia negli adulti che nelle larve (Figura4B; Figura 4-figure supplement 1). Come negli eumetazoi, questo risultato suggerisce che i complessi di Amphimedon PRC2 sono probabilmente reclutati attraverso sequenze simili a PRE e possono mirare a regolatori dello sviluppo per la deposizione di H3K27me3 e il silenziamento trascrizionale(Margueron e Reinberg ,2011; Di Croce e Helin, 2013; Boyer et al., 2006).
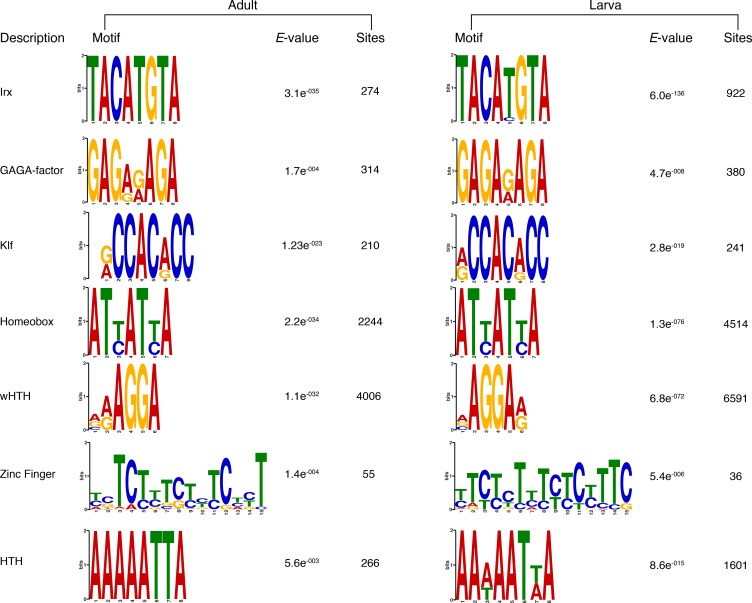
Figura 4-figure supplement 1.Motivi del DNA sovrarappresentati in H3K27me3 regioni trascrizionalmente silenziati.ortologi putativo di componenti Drosophila PcG Drosophila e fattori associati in lievito, Capsaspora, spugna, nematodo, e il genoma umano.Ortologie putativo di componenti di Drosophila PcG e fattori associati in lievito, Capsaspora, spugna, nematode e genoma umano.loghi di sequenza di corrispondenza dei motivi del DNA determinati dall’analisi MEME-ChIP per essere significativamente arricchito nelle regioni trascrizionalmente silenziate contrassegnate da H3K27me3 sia in adulti che in larve.(A) Diagramma che rappresenta la composizione del complesso di Drosophila PRC2 e dei suoi quattro componenti principali: la sottounità catalitica del complesso E (z), la proteina di zinco dito SU (z) 12, la proteina WD-ripetizione ESC e la proteina istone-legante Nurf55. E(z) è responsabile della principale attività enzimatica della PRC2, che è quella di trimetilare l’istone H3 alla lisina 27, producendo H3K27me3. Adattato da(Vissers et al., 2012). Viene mostrata la presenza (verde) o l’assenza (arancione) di PRC2 e dei suoi componenti principali nelle diverse specie di opistoconte rappresentate nell’albero filogenetico (a sinistra). L’anfimedone è evidenziato in verde.(B) Loghi di sequenza di un sottoinsieme dei motivi del DNA determinati dall’analisi MEME-ChIP per essere significativamente arricchito nelle regioni trascrizionalmente silenziate contrassegnate da H3K27me3 in Amphimedon adulto. Per ogni motivo, il miglior TOMTOM corrisponde a un motivo nei database dei mouse JASPAR CORE e UniPROBE, rispettivamente il valore E e il numero di siti che contribuiscono alla costruzione del motivo. Il motivo abbinato è mostrato in alto e il motivo di interrogazione è mostrato in basso.DOI:
http://dx.doi.org/10.7554/eLife.22194.02210.7554/eLife.22194.023Figure 4-source data 1.Putative orthologs of Drosophila PcG components and associated factors in yeast, Capsaspora , sponge, nematode, and human genome.Table of PcG protein is adapted from(http://www.igh.cnrs.fr/equip/cavalli/link.PolycombTeaching.html).DOI:http://dx.doi.org/10.7554/eLife.22194.023La tabella delle proteine PcG è adattata da(http://www.igh.cnrs.fr/equip/cavalli/link.PolycombTeaching.html).DOI:
http://dx.doi.org/10.7554/eLife.22194.023La tabella delle proteine PcG è adattata da(http://www.igh.cnrs.fr/equip/cavalli/link.PolycombTeaching.html).DOI:
http://dx.doi.org/10.7554/eLife.22194.023Sono indicati rispettivamente il valore E(log likelihood ratio di ogni motivo) e il numero di siti che contribuiscono alla costruzione del motivo.DOI:
http://dx.doi.org/10.7554/eLife.22194.024
Figura 4-figure supplement 1.Loghi di sequenza di corrispondenza dei motivi del DNA determinati dall’analisi MEME-ChIP per essere significativamente arricchito nelle regioni trascrizionalmente silenziate segnate da H3K27me3 sia nell’adulto che nella larva.Sono indicati rispettivamente il valore E(log likelihood ratio di ciascun motivo) e il numero di siti che contribuiscono alla costruzione del motivo.DOI:
http://dx.doi.org/10.7554/eLife.22194.024
Il sottoinsieme dei lincRNA di Amphimedon è associato ad uno stato di cromatina simile ad un potenziatore
Un ulteriore livello di complessità normativa nello sviluppo dell’eumetazoano è fornito da lunghi RNA intergenici non codificanti (lincRNA)(Ulitsky, 2016; Hezroni et al., 2015; Quinn e Chang, 2016), di cui è stato recentemente dimostrato lo sviluppo espresso in spugne(Gaiti et al., 2015; Bråte et al., 2015). Qui, abbiamo esteso queste analisi e analizzato gli stati della cromatina degli ncRNA intergenici di Amphimedon long intergenic ncRNAs (lincRNAs)(Gaiti et al., 2015), evitando gli lncRNAs in introni di sequenza di codifica delle proteine o antisenso ai geni codificanti, che possono produrre segnali ambigui.
Studi precedenti hanno dimostrato che il rapporto di H3K4me1-to-H3K4me3 segna intorno TSSs può separare lincRNA in lincRNA di tipo potenziatore (elincRNA; alto rapporto H3K4me1-to-H3K4me3) e lincRNA di tipo promotore canonico (plincRNA; basso rapporto H3K4me1-to-H3K4me3)(Sebé-Pedrós et al.,2016; Marques et al., 2013; IIott et al., 2014). Così, per esplorare se i lincRNA di spugna possono provenire da regioni di potenziamento, abbiamo interrogato i nostri set di dati ChIP-seq e calcolato il rapporto relativo di H3K4me1-to-H3K4me3 in una finestra di 4 kb centrata su lincRNA TSSs. In questa analisi sono stati utilizzati solo lincRNA in impalcature più grandi di 10 kb che si sovrappongono alle regioni di arricchimento di H3K4me1, H3K4me3 e RNAPII (n = 217). Analogamente a IIott et al. (2014), abbiamo arbitrariamente adottato un rapporto H3K4me1-to-H3K4me3 di >1,2 e <0,8 per definire rispettivamente gli elincRNA e i plincRNA. Sulla base di questi criteri, abbiamo trovato 153 elincRNA putativi (70%) significativamente arricchiti per H3K4me1 rispetto a H3K4me3 (test Mann-Whitney U, p-valore=2,272e-05) e 21 (10%) plincRNA putativi con firma del promotore canonico, cioè, maggiore arricchimento di H3K4me3 rispetto a H3K4me1 (test Mann-Whitney U, p-valore=1,925e-07). 43 (20%) lincRNAs non potevano essere assegnati ad entrambi i gruppi, cioè, 0,8 < H3K4me1-to-H3K4me3 < 1,2 (Figura 5A-D; Figura5-dati fonte1; Figura 5-figure supplement 1).10. 7554/eLife.22194.025Figure 5.Amphimedon popolazioni di lincRNA definite dalle firme dell’istone PTM.(A) Heatmap che mostra la copertura media letta normalizzata di H3K4me1, H3K4me3 e il loro rapporto in Amphimedon adulti attraverso un intervallo di 4 kb centrato su TSS di lincRNA. Ogni linea delle mappe termiche rappresenta un singolo lincRNA (asse y). I profili sono ordinati in base alle differenze di arricchimento tra H3K4me1 e DNA in ingresso, e H3K4me3 e DNA in ingresso, rispettivamente. Viene fornito anche il log2(rapporto) H3K4me1:H3K4me3 intorno ai TSS.(B) Arricchimento di H3K4me1 (sinistra) e H3K4me3 (destra) (ChIP contro input) a plincRNA e elincRNA. I valori P sono indicati per il test Mann-Whitney U.(C) Esempio di lincRNA con firma di cromatina simile a quella del promotore (plincRNA). Per la definizione degli stati della cromatina adulta si veda la Figura 1A. Promoter-like lincRNAs (blu) sono mostrati, insieme con ingresso DNA-normalizzato copertura di diverse modifiche degli istoni e RNA-seq espressione nell’adulto.(D) Come(C), ma per i lincRNA con la firma della cromatina simile a quella del promotore (elincRNA).DOI:http://dx.doi.org/10.7554/eLife.22194.02510.7554/eLife.22194.026Figuredati a 5 fonti 1.Annotazione di elincRNA e plincRNA presunti.DOI:http://dx.doi.org/10.7554/eLife.22194.02610.7554/eLife.22194.027Figuresupplemento a 5 cifre 1.Ulteriori esempi di plincRNA ed elincRNA.(A) Esempio di un lincRNA con firma di cromatina simile a quella del promotore (plincRNA). Per la definizione di stati adulti cromatina vedi Figura 1A. Promoter-like lincRNA (blu) è mostrato, insieme con ingresso DNA-normalizzato copertura di diverse modifiche degli istoni e RNA-seq espressione nell’adulto.(B) Lo stesso di(A), ma per un lincRNA con enhancer-come firma cromatina (elincRNA).(C) Come(B) ma per lincRNA non espressi con firma di cromatina simile a quella dell’esaltatore (elincRNA). Si noti la prevalenza dello stato di cromatina “ReprPC”, “ReprPCWk” e “EnhP” in queste regioni.DOI:http://dx.doi.org/10.7554/eLife.22194.027
Questi risultati indicano che i lincRNA di spugna possono essere separati in due popolazioni distinte di poli(A)+ trascrizioni basate sullo stato della cromatina ai loro TSS. Sebbene queste due popolazioni assomiglino a quelle presenti nei lincRNA umani, nei topi e nei lincRNA di Capsaspora(Sebé-Pedrós et al., 2016; Marques et al. , 2013; IIott et al., 2014), il loro significato funzionale è ancora da determinare.
Figura 5-figure supplement 1.Figura 5—supplemento alla figura 1.Amphimedon lincRNA popolazioni di lincRNA definite dall’istone PTM signatures.annotazione di elincRNA putativi e plincRNA.annotazione di elincRNA putativi e plincRNA.ulteriori esempi di plincRNA e di elincRNA.(A) Heatmap che mostra la copertura media letta normalizzata di H3K4me1, H3K4me3 e il loro rapporto in Amphimedon adulti attraverso un intervallo di 4 kb centrato su TSSs di lincRNA. Ogni linea delle mappe termiche rappresenta un singolo lincRNA (asse y). I profili sono ordinati in base alle differenze di arricchimento tra H3K4me1 e DNA in ingresso, e H3K4me3 e DNA in ingresso, rispettivamente. Viene fornito anche il log2(rapporto) H3K4me1:H3K4me3 intorno ai TSS.(B) Arricchimento di H3K4me1 (sinistra) e H3K4me3 (destra) (ChIP contro input) a plincRNA e elincRNA. I valori P sono indicati per il test Mann-Whitney U.(C) Esempio di lincRNA con firma di cromatina simile a quella del promotore (plincRNA). Per la definizione degli stati della cromatina adulta si veda la Figura 1A. Promoter-like lincRNAs (blu) sono mostrati, insieme con ingresso DNA-normalizzato copertura di diverse modifiche degli istoni e RNA-seq espressione nell’adulto.(D) Come(C), ma per i lincRNA con la firma della cromatina simile a quella del promotore (elincRNA).DOI:
http://dx.doi.org/10.7554/eLife.22194.02510.7554/eLife.22194.026Figure 5-source data 1.Annotazione di elincRNA e plincRNA putativi.DOI:http://dx.doi.org/10.7554/eLife.22194.026DOI:
http://dx.doi.org/10.7554/eLife.22194.026DOI:
http://dx.doi.org/10.7554/eLife.22194.026(A) Esempio di un lincRNA con firma in cromatina simile a quella del promotore (plincRNA). Per la definizione degli stati della cromatina adulta si veda la Figura 1A. Promoter-like lincRNA (blu) è mostrato, insieme con ingresso DNA-normalizzato copertura di diverse modifiche degli istoni e RNA-seq espressione nell’adulto.(B) Lo stesso di(A), ma per un lincRNA con enhancer-come firma cromatina (elincRNA).(C) Come(B) ma per lincRNA non espressi con firma di cromatina simile a quella dell’esaltatore (elincRNA). Si noti la prevalenza dello stato di cromatina “ReprPC”, “ReprPCWk” e “EnhP” in queste regioni.DOI:
http://dx.doi.org/10.7554/eLife.22194.027
Figura 5-figure supplement 1.Ulteriori esempi di plincRNA e di elincRNA.(A) Esempio di un lincRNA con firma di cromatina simile a quella del promotore (plincRNA). Per la definizione di stati adulti cromatina vedi Figura 1A. Promoter-like lincRNA (blu) è mostrato, insieme con ingresso DNA-normalizzato copertura di diverse modifiche degli istoni e RNA-seq espressione nell’adulto.(B) Lo stesso di(A), ma per un lincRNA con enhancer-come firma cromatina (elincRNA).(C) Come(B) ma per lincRNA non espressi con firma di cromatina simile a quella dell’esaltatore (elincRNA). Si noti la prevalenza dello stato di cromatina “ReprPC”, “ReprPCWk” e “EnhP” in queste regioni.DOI:
http://dx.doi.org/10.7554/eLife.22194.027
Identificazione degli elementi potenziatori in Amphimedon
Per identificare gli elementi potenziatori putativi di Amphimedon in silico, abbiamo selezionato regioni distali di arricchimento H3K4me1 di H3K4me1 (regioni ad alta confidenza, che rappresentano eventi riproducibili attraverso veri replicati biologici) che non si sono sovrapposte a TSS (±200 bp) di geni che codificano le proteine e lncRNA, ma si sono sovrapposte a regioni designate come in uno stato di cromatina potenziatrice in base all’analisi ChromHMM (“.TxEnhA’ o ‘EnhWk’ o ‘EnhP’ in stato adulto; ‘TxEnhA1’ o ‘TxEnhA2’ o ‘.EnhWk’ o ‘EnhP’ stato in larve, che consistono in tipici modelli di eumetazoani enhancer histone H3 PTM) (Figura 6A). Un sottoinsieme di queste regioni è stato segnato anche da H3K27ac, e quindi è probabile che sia trascrizionalmente attivo(Figura 6A e B; Figura 6-dati fonte 1). Queste regioni attivate-simili agli stimolatori hanno mostrato un arricchimento significativo di H3K4me1 e H3K27ac rispetto all’H3K4me3 (test di Mann-Whitney U, valore p<2.2e-16; Figura 6C; Figura 6-figure supplement 1), una firma biochimica tipica degli stimolatori di eumetazoani (Schwaigeret al., 2014). È interessante notare che RNAPII ha occupato alcuni di questi Anfimedone predetto elementi simili agli stimolatori attivati (35% e 41% in adulti e larve, rispettivamente), suggerendo che gli RNA poli(A)+ stimolatori potrebbero essere trascritti da queste regioni(Natoli e Andrau, 2012; Li et al., 2016; Kim et al., 2010)(Figura 6A-D; Figura 6-figure supplement 2). In alternativa, ma non esclusivamente, questo potrebbe rappresentare il risultato del looping della cromatina e la simultanea riduzione sia degli stimolatori che dei promotori con l’anticorpo RNAPII(Shlyueva et al., 2014).10.7554/eLife.22194.028Figure 6.Distal enhancer regulation at the dawn of animals.(A) Panoramica della pipeline di filtraggio computazionale adottata per prevedere gli elementi simil-elementi stimolatori attivati dall’anfimedone presunto. Vedere il testo principale e Materiali e metodi per i dettagli.(B) Heatmap che mostra l’arricchimento di diverse modifiche dell’istone in corrispondenza di elementi simili agli stimolatori attivati previsti (±2 kb delle regioni adiacenti).(C) Boxplot che mostra l’arricchimento di diverse modifiche dell’istone (ChIP rispetto all’input) in corrispondenza di elementi simili agli stimolatori attivati previsti, mostrando che gli elementi simili agli stimolatori attivati hanno livelli di H3K4me1 superiori a quelli di H3K4me3, una caratteristica tipica degli stimolatori di eumetazoani. Quattro asterischi (****) indicano i valori p<2,2e-16 per il test di Mann-Whitney U tra H3K4me3 e H3K27ac, tra H3K4me3 e H3K4me1, e tra H3K4me3 e RNAPII, rispettivamente.(D) Esempio di elementi simili a potenziatori attivati previsti. I geni di codifica delle proteine (viola) sono mostrati, insieme con l’input DNA-normalizzato copertura di DNA-normalizzato di diverse modifiche degli istoni e RNA-seq espressione nell’adulto. Le regioni di arricchimento (picchi di confidenza elevati, che rappresentano eventi riproducibili attraverso veri replicati biologici) corrispondenti agli elementi simili agli stimolatori attivati previsti sono evidenziati in grigio.(E) Loghi di sequenza dei motivi del DNA determinati dall’analisi MEME-ChIP arricchiti nelle sequenze di potenziatori attivati previsti nell’adulto. Per ogni motivo, vengono mostrati, rispettivamente, la migliore corrispondenza con un motivo nei database dei mouse JASPAR CORE e UniPROBE, il valore E e il numero di siti che contribuiscono alla costruzione del motivo. Il motivo abbinato è mostrato in alto e il motivo di interrogazione è mostrato in basso.(F) Sono mostrate le attività di arricchimento dell’ontologia genica (GO) dei geni vicini che codificano le proteine dei geni dell’adulto che ha previsto l’attivazione di elementi simili agli stimolatori. La lunghezza della barra indica il significato dell’arricchimento (test ipergeometrico; -log10[valore P- regolato]). Solo i primi dieci termini del processo biologico GO sono mostrati. Vedere la Figura 6-dati sorgente 2 per l ‘elenco completo.(G) Boxplot che mostra la dimensione degli introni che ospitano gli elementi di tipo potenziatore attivati da adulti rispetto a tutti gli introni del genoma. L’asse y indica la dimensione degli introni (bp) in scala log. Il valore P è indicato per il test di Mann-Whitney U.DOI:http://dx.doi.org/10.7554/eLife.22194.02810.7554/eLife.22194.029Figure6-source data 1.Posizione genomica di tutti gli elementi simili al potenziatore attivato previsto e la loro distanza dal TSS più vicino.DOI:http://dx.doi.org/10.7554/eLife.22194.02910.7554/eLife.22194.030Figure6 dati fonte 2.Annotazione funzionale dei geni vicini più vicini dei geni dell’adulto che ha previsto l’attivazione di elementi simili agli stimolatori.DOI:http://dx.doi.org/10.7554/eLife.22194.03010.7554/eLife.22194.031Figure6—dati fonte 3.Annotazione funzionale dei geni vicini più vicini dei geni della larva ha previsto elementi simili agli stimolatori attivati.DOI:http://dx.doi.org/10.7554/eLife.22194.03110.7554/eLife.22194.032Figure6—dati fonte 4.GO esito dell’arricchimento con il termine arricchimento per i geni vicini più vicini dei geni dell’adulto ha previsto elementi simili agli stimolatori attivati (test ipergeometrico, FDR<0.01).DOI:http://dx.doi.org/10.7554/eLife.22194.03210.7554/eLife.22194.033Figure6-source data 5.GO termine risultato dell’arricchimento per i più vicini geni vicini dei geni della larva predetti elementi simili a potenziatori attivati (test ipergeometrico, FDR<0.01).DOI:http://dx.doi.org/10.7554/eLife.22194.03310.7554/eLife.22194.034FigureSupplemento a 6 cifre 1.Gli elementi simili agli stimolatori attivati hanno livelli di H3K4me1 più alti rispetto ai livelli di H3K4me3.(A)Arricchimento di H3K4me1 (rosso) e H3K4me3 (blu) (ChIP rispetto all’input) agli elementi simili agli stimolatori attivati previsti (sinistra) e TSS (destra), mostrando che gli elementi simili agli stimolatori attivati hanno livelli di H3K4me1 più alti rispetto ai livelli di H3K4me3, una caratteristica tipica degli stimolatori di eumetazoani. I valori Psono indicati per il test Mann-Whitney U.(B) Distribuzione della distanza relativa osservata tra H3K4me1 (rosso) e H3K4me3 (blu) regioni di arricchimento (picchi) e TSS. Se non esiste una correlazione spaziale tra i due insiemi, ci si aspetterebbe che le distanze relative siano distribuite uniformemente tra le distanze relative che vanno da 0 a 0,5, come osservato per i picchi di H3K4me1 e i TSS. Se, tuttavia, gli intervalli tendono ad essere molto più vicini di quanto ci si aspettasse per caso, la distribuzione delle distanze relative osservate verrebbe spostata verso bassi valori di distanza relativa, come osservato per i picchi H3K4me3 e i TSS.(C) La distribuzione della distanza relativa osservata tra i picchi di H3K4me1 (rosso) e H3K4me3 (blu) e gli elementi tipo potenziatori attivati previsti, indica la correlazione spaziale tra i picchi di H3K4me1 e gli elementi tipo potenziatori attivati previsti.DOI:http://dx.doi.org/10.7554/eLife.22194.03410.7554/eLife.22194.035FigureSupplemento a 6 cifre 2.Esempi di espressione CEL-seq o RNA-seq rilevata in siti putativo attivato enhancer-come, suggerendo che gli eRNA 1D, che sono generalmente poliadenilati (Natolie Andrau, 2012; Li et al., 2016), potrebbero essere trascritti da queste regioni.geni di codifica delle proteine (viola) sono mostrati, insieme a tracce di copertura del DNA di ingresso-normalizzato tracce di diverse modifiche degli istoni. Le regioni di arricchimento (picchi di confidenza elevati, che rappresentano eventi riproducibili attraverso veri replicati biologici) corrispondenti a presunti siti simili a potenziatori attivati sono evidenziati in grigio. Se non diversamente specificato, vengono mostrati i dati degli adulti.DOI:http://dx.doi.org/10.7554/eLife.22194.03510.7554/eLife.22194.036FigureSupplemento a 6 cifre 3.Loghi di sequenze aggiuntive dei motivi del DNA determinati dall’analisi MEME-ChIP per essere arricchiti in modo significativo nelle sequenze simil-animatori attivati previsti per gli adulti. Il motivo abbinato è mostrato in alto e il motivo della query è mostrato in basso.DOI:http://dx.doi.org/10.7554/eLife.22194.03610.7554/eLife.22194.037FigureSupplemento a 6 cifre 4.Loghi delle sequenze corrispondenti dei motivi del DNA determinati dall’analisi MEME-ChIP per essere significativamente arricchiti nelle previste sequenze attivate simili a potenziatori sia nell’adulto che nella larva.il valore E (log likelihood ratio di ogni motivo) e il numero di siti che contribuiscono alla costruzione del motivo sono mostrati, rispettivamente.DOI:http://dx.doi.org/10.7554/eLife.22194.03710.7554/eLife.22194.038Figuresupplemento a 6 cifre 5.Esempi di elementi simili al potenziatore previsto in prossimità di ben noti geni dello sviluppo e del fattore di trascrizione.sono mostrati i geni che codificano le proteine (viola), insieme alla copertura del DNA in ingresso normalizzato di diverse modifiche dell’istone e l’espressione dell’RNA-seq. Le regioni di arricchimento (picchi di confidenza elevati, che rappresentano eventi riproducibili attraverso i veri replicati biologici) corrispondenti a siti simil-investitori adulti putativi sono evidenziati in grigio.DOI:http://dx.doi.org/10.7554/eLife.22194.038
Negli eumetazoi, i geni che codificano i regolatori trascrizionali sono a loro volta regolati da elementi multipli potenziatori(Schwaiger et al., 2014; Shlyueva et al., 2014; Nègre et al., 2011; Bogdanovic et al., 2012; Woolfe et al., 2005; Heintzman et al., 2009). Abbiamo quindi eseguito l’analisi dei motivi de novo e, nonostante il limitato potere di rilevazione dei motivi a causa dell’intrinseca eterogeneità cellulare del nostro materiale di partenza, siamo stati in grado di dimostrare che i motivi di legame di consenso delle principali famiglie di fattori di trascrizione dello sviluppo erano sovrarappresentati nelle sequenze di intensificatori attivati previsti per gli adulti, tra cui i motivi di legame di Zinco, Irx, SOX e POU (Figura 6E; Figura 6-figure supplement 3). È degno di nota il fatto che le dita di zinco possono anche essere coinvolte in ruoli che potrebbero essere estranei alla regolazione diretta dell’espressione genica in sé, ad esempio il rimodellamento della cromatina(Wysocka et al., 2006). Motivi di legame simili sono stati ottenuti analizzando le larve previste sequenze attivate simili a quelle degli stimolatori(Figura 6-figure supplement 4). Successivamente, abbiamo esaminato se la spugna ha predetto elementi simili agli stimolatori attivati erano preferibilmente localizzati accanto ai geni che codificano le proteine coinvolti nello sviluppo e/o nella regolazione trascrizionale. Cercando i TSS più vicini a ciascuno degli elementi simili agli stimolatori attivati previsti ad Amphimedon, abbiamo nominato i geni target presunti che codificano le proteine. Analogamente agli eumetazoi, questi geni vicini più vicini sono stati significativamente arricchiti per l’ontologia genica (GO) termini associati con l’attività del fattore di trascrizione e processi di sviluppo (test ipergeometrico, FDR regolato p-valore<0,01) (Figura 6F), e comprendeva diversi fattori di trascrizione, tra cui SOX2, FOS e NF-kB (Figura6D; Figura 6-figure supplement 5; Figura 6-source data 2- 5).
I vertebrati presentano ampie regioni intergeniche in cui si trova la maggior parte degli stimolatori previsti(ENCODE Project Consortium, 2012; Djebali et al., 2012). Al contrario, ad Amphimedon, che ha un genoma altamente compatto con regioni intergeniche minime(Fernandez-Valverde e Degnan, 2016), gli elementi simili agli stimolatori attivati previsti erano prevalentemente intragenici, con solo una minoranza presente nelle regioni intergeniche (9% e 20% rispettivamente in adulti e larve)(Fernandez-Valverde e Degnan, 2016). Questo, insieme al forte arricchimento degli stati di cromatina tipicamente associati agli stimolatori di eumetazoani – ‘TxEnhA’ e ‘EnhWk’ – negli introni (Figura 1A; Figura 1-figure supplement 4), suggerisce una simile distribuzione genomica complessiva tra gli elementi stimolatori di Amphimedon, Nematostella e Drosophila (Schwaiger et al.,2014; Nègre et al., 2011; Arnold et al., 2013).
Una maggiore lunghezza dell’introne si associa spesso alla presenza di elementi non codificanti altamente conservati(Irimia et al., 2011). Abbiamo, quindi, estratto gli introni che porto previsto elementi simili a potenziatori attivati e confrontato la loro distribuzione dimensionale con le dimensioni di tutte le regioni introniche presenti nel genoma. I primi erano significativamente più lunghi della dimensione genomica media degli introni, con una media di 332 bp e 256 bp, e una mediana di 99 bp e 71 bp, rispettivamente (test Ansari-Bradley, valore p=0,06151; test Mann-Whitney U, valore p=1,927e-06).(Figura 6G), suggerendo che un’espansione cis-regolatoria sembra essersi verificata principalmente nelle regioni introniche piuttosto che intergeniche di Amphimedon.
Figura 6-figure supplemento 5.Figura 6— supplemento alla figura 5. Regolazione distale del potenziatore all’alba degli animali.posizione genomica di tutti gli elementi simili al potenziatore attivato previsto e la loro distanza dalla più vicina TSS.annotazione funzionale dei geni vicini più vicini dell’adulto ha previsto elementi simili al potenziatore attivato.annotazione funzionale dei geni vicini più vicini della larva ha previsto elementi simili al potenziatore attivato.GO termine risultato dell’arricchimento per i geni vicini più vicini dei geni dell’adulto ha previsto elementi simili agli stimolatori attivati (test ipergeometrico, FDR<0.01).GO termine risultato dell’arricchimento per i geni vicini più vicini dei geni della larva ha previsto elementi simili agli stimolatori attivati (test ipergeometrico, FDR<0.01).posizione genomica di tutti gli elementi simili agli stimolatori attivati previsti e la loro distanza dal TSS più vicino.Annotazione funzionale dei geni vicini più vicini dell’adulto ha previsto elementi simili agli stimolatori attivati.annotazione funzionale dei geni vicini più vicini della larva ha previsto elementi simili agli stimolatori attivati.risultato dell’arricchimento con il termine GO per i geni vicini più vicini dell’adulto ha previsto elementi simili agli stimolatori attivati (test ipergeometrico, FDR<0.01).GO termine risultato dell’arricchimento per i geni vicini più vicini dei geni della larva predetto elementi simili a stimolatori attivati (test ipergeometrico, FDR<0.01).gli elementi simili a stimolatori attivati hanno livelli più alti di H3K4me1 rispetto ai livelli di H3K4me3.Esempi di espressione CEL-seq o RNA-seq rilevata in siti simili a stimolatori presunti attivati, suggerendo che gli eRNA 1D, che sono generalmente poliadenilati (Natolie Andrau, 2012; Li et al., Loghi di sequenze aggiuntive dei motivi del DNA determinati dall’analisi MEME-ChIP per essere significativamente arricchiti nelle sequenze di stimolatori attivati previsti per gli adulti.Loghi di sequenze corrispondenti dei motivi del DNA determinati dall’analisi MEME-ChIP per essere significativamente arricchiti nelle sequenze di stimolatori attivati previsti sia per gli adulti che per le larve.Esempi di elementi simili agli stimolatori previsti in prossimità di ben noti geni del fattore di sviluppo e di trascrizione.(A) Panoramica della pipeline di filtraggio computazionale adottata per predire il putativo Amphimedon attivato elementi simili agli stimolatori. Vedere il testo principale e Materiali e metodi per i dettagli.(B) Heatmap che mostra l’arricchimento di diverse modifiche dell’istone in corrispondenza di elementi simili agli stimolatori attivati previsti (±2 kb delle regioni adiacenti).(C) Boxplot che mostra l’arricchimento di diverse modifiche dell’istone (ChIP rispetto all’input) in corrispondenza di elementi simili agli stimolatori attivati previsti, mostrando che gli elementi simili agli stimolatori attivati hanno livelli di H3K4me1 superiori a quelli di H3K4me3, una caratteristica tipica degli stimolatori di eumetazoani. Quattro asterischi (****) indicano i valori p<2,2e-16 per il test di Mann-Whitney U tra H3K4me3 e H3K27ac, tra H3K4me3 e H3K4me1, e tra H3K4me3 e RNAPII, rispettivamente.(D) Esempio di elementi simili a potenziatori attivati previsti. I geni di codifica delle proteine (viola) sono mostrati, insieme con l’input DNA-normalizzato copertura di DNA-normalizzato di diverse modifiche degli istoni e RNA-seq espressione nell’adulto. Le regioni di arricchimento (picchi di confidenza elevati, che rappresentano eventi riproducibili attraverso veri replicati biologici) corrispondenti agli elementi simili agli stimolatori attivati previsti sono evidenziati in grigio.(E) Loghi di sequenza dei motivi del DNA determinati dall’analisi MEME-ChIP arricchiti nelle sequenze di potenziatori attivati previsti nell’adulto. Per ogni motivo, vengono mostrati, rispettivamente, la migliore corrispondenza con un motivo nei database dei mouse JASPAR CORE e UniPROBE, il valore E e il numero di siti che contribuiscono alla costruzione del motivo. Il motivo abbinato è mostrato in alto e il motivo di interrogazione è mostrato in basso.(F) Sono mostrate le attività di arricchimento dell’ontologia genica (GO) dei geni vicini che codificano le proteine dei geni dell’adulto che ha previsto l’attivazione di elementi simili agli stimolatori. La lunghezza della barra indica il significato dell’arricchimento (test ipergeometrico; -log10[valore P- regolato]). Solo i primi dieci termini del processo biologico GO sono mostrati. Vedere la Figura 6-dati sorgente 2 per l ‘elenco completo.(G) Boxplot che mostra la dimensione degli introni che ospitano gli elementi di tipo potenziatore attivati da adulti rispetto a tutti gli introni del genoma. L’asse y indica la dimensione degli introni (bp) in scala log. Il valore P è indicato per il test di Mann-Whitney U.DOI:
http://dx.doi.org/10.7554/eLife.22194.02810.7554/eLife.22194.029Figure 6-source data 1.Genomic location of all the predicted activated enhancer-like elements and their distance to the closest TSS.DOI:http://dx.doi.org/10.7554/eLife.22194.029DOI:
http://dx.doi.org/10.7554/eLife.22194.02910.7554/eLife.22194.030Figure 6-source data 2.Functional annotation of nearest neighbors genes of the adult predicted activated enhancer-like elements.DOI:http://dx.doi.org/10.7554/eLife.22194.030DOI:
http://dx.doi.org/10.7554/eLife.22194.03010.7554/eLife.22194.031Figure 6-source data 3.Functional annotation of nearest neighbors genes of the larva predicted activated enhancer-like elements.DOI:http://dx.doi.org/10.7554/eLife.22194.031DOI:
http://dx.doi.org/10.7554/eLife.22194.03110.7554/eLife.22194.032Figure 6-source data 4.GO risultato dell’arricchimento a 6 fonti per i geni vicini più vicini dell’adulto ha previsto l’attivazione di elementi simili al potenziatore (test ipergeometrico, FDR<0.01).DOI:http://dx.doi.org/10.7554/eLife.22194.032DOI:
http://dx.doi.org/10.7554/eLife.22194.03210.7554/eLife.22194.033Figure 6-source data 6-source data 5.GO esito dell’arricchimento a termine per i più vicini geni vicini della larva previsto attivato elementi simili a potenziatori (test ipergeometrico, FDR<0.01).DOI:http://dx.doi.org/10.7554/eLife.22194.033DOI:
http://dx.doi.org/10.7554/eLife.22194.033DOI:
http://dx.doi.org/10.7554/eLife.22194.029DOI:
http://dx.doi.org/10.7554/eLife.22194.030DOI:
http://dx.doi.org/10.7554/eLife.22194.031DOI:
http://dx.doi.org/10.7554/eLife.22194.032DOI:
http://dx.doi.org/10.7554/eLife.22194.033(A) Arricchimento dell’H3K4me1 (rosso) e dell’H3K4me3 (blu) (ChIP rispetto all’input) a elementi simili agli stimolatori attivati previsti (sinistra) e TSS (destra), mostrando che gli elementi simili agli stimolatori attivati hanno livelli di H3K4me1 superiori a quelli dell’H3K4me3, una caratteristica tipica degli stimolatori di eumetazoani. I valori Psono indicati per il test Mann-Whitney U.(B) Distribuzione della distanza relativa osservata tra H3K4me1 (rosso) e H3K4me3 (blu) regioni di arricchimento (picchi) e TSS. Se non esiste una correlazione spaziale tra i due insiemi, ci si aspetterebbe che le distanze relative siano distribuite uniformemente tra le distanze relative che vanno da 0 a 0,5, come osservato per i picchi di H3K4me1 e i TSS. Se, tuttavia, gli intervalli tendono ad essere molto più vicini di quanto ci si aspettasse per caso, la distribuzione delle distanze relative osservate verrebbe spostata verso bassi valori di distanza relativa, come osservato per i picchi H3K4me3 e i TSS.(C) La distribuzione della distanza relativa osservata tra i picchi di H3K4me1 (rosso) e H3K4me3 (blu) e gli elementi tipo potenziatori attivati previsti, indica la correlazione spaziale tra i picchi di H3K4me1 e gli elementi tipo potenziatori attivati previsti.DOI:
http://dx.doi.org/10.7554/eLife.22194.034Sono mostrati i geni della codifica delle proteine (viola), insieme alle tracce di copertura del DNA normalizzato in ingresso di diverse modifiche dell’istone. Le regioni di arricchimento (picchi di confidenza elevati, che rappresentano eventi riproducibili attraverso veri replicati biologici) corrispondenti a presunti siti simili a potenziatori attivati sono evidenziati in grigio. Se non diversamente specificato, vengono mostrati i dati degli adulti.DOI:
http://dx.doi.org/10.7554/eLife.22194.035Sono indicati rispettivamente il valore E(log likelihood ratio di ogni motivo) e il numero di siti che contribuiscono alla costruzione del motivo. Il motivo abbinato è mostrato in alto e il motivo di interrogazione è mostrato in basso.DOI:
http://dx.doi.org/10.7554/eLife.22194.036Sono indicati rispettivamente il valore E(log likelihood ratio di ogni motivo) e il numero di siti che contribuiscono alla costruzione del motivo.DOI:
http://dx.doi.org/10.7554/eLife.22194.037Sono mostrati i geni della codifica delle proteine (viola), insieme alla copertura del DNA in ingresso, normalizzata per il DNA, delle diverse modifiche degli istoni e dell’espressione dell’RNA-seq. Le regioni di arricchimento (picchi di confidenza elevati, che rappresentano eventi riproducibili attraverso i veri replicati biologici) corrispondenti a siti di putativi adulti simili a potenziatori sono evidenziati in grigio.DOI:
http://dx.doi.org/10.7554/eLife.22194.038
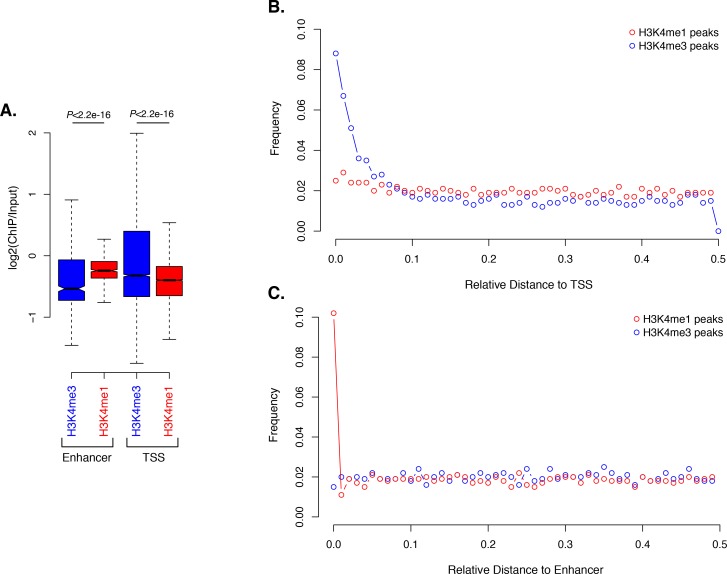
Figura 6-figure supplement 1.Gli elementi attivi simili a potenziatori hanno livelli di H3K4me1 superiori a quelli dell’H3K4me3.(A) Arricchimento di H3K4me1 (rosso) e H3K4me3 (blu) (ChIP rispetto all’input) in corrispondenza di elementi simili agli stimolatori attivati previsti (sinistra) e TSS (destra), mostrando che gli elementi simili agli stimolatori attivati hanno livelli di H3K4me1 più alti rispetto ai livelli di H3K4me3, una caratteristica tipica degli stimolatori di eumetazoani. I valori Psono indicati per il test Mann-Whitney U.(B) Distribuzione della distanza relativa osservata tra H3K4me1 (rosso) e H3K4me3 (blu) regioni di arricchimento (picchi) e TSS. Se non esiste una correlazione spaziale tra i due insiemi, ci si aspetterebbe che le distanze relative siano distribuite uniformemente tra le distanze relative che vanno da 0 a 0,5, come osservato per i picchi di H3K4me1 e i TSS. Se, tuttavia, gli intervalli tendono ad essere molto più vicini di quanto ci si aspettasse per caso, la distribuzione delle distanze relative osservate verrebbe spostata verso bassi valori di distanza relativa, come osservato per i picchi H3K4me3 e i TSS.(C) La distribuzione della distanza relativa osservata tra i picchi di H3K4me1 (rosso) e H3K4me3 (blu) e gli elementi tipo potenziatori attivati previsti, indica la correlazione spaziale tra i picchi di H3K4me1 e gli elementi tipo potenziatori attivati previsti.DOI:
http://dx.doi.org/10.7554/eLife.22194.034

Figura 6-figure supplement 2.Esempi di espressione CEL-seq o RNA-seq rilevata in siti simili a potenziatori presunti attivati, suggerendo che gli eRNA 1D, che sono generalmente poliadenilati(Natoli e Andrau, 2012; Li et al., 2016), potrebbero essere trascritti da queste regioni.I geni che codificano le proteine (viola) sono mostrati, insieme alle tracce di copertura del DNA normalizzato del DNA in ingresso di diverse modifiche degli istoni. Le regioni di arricchimento (picchi di confidenza elevati, che rappresentano eventi riproducibili attraverso veri replicati biologici) corrispondenti a presunti siti simili a potenziatori attivati sono evidenziati in grigio. Se non diversamente specificato, vengono mostrati i dati degli adulti.DOI:
http://dx.doi.org/10.7554/eLife.22194.035
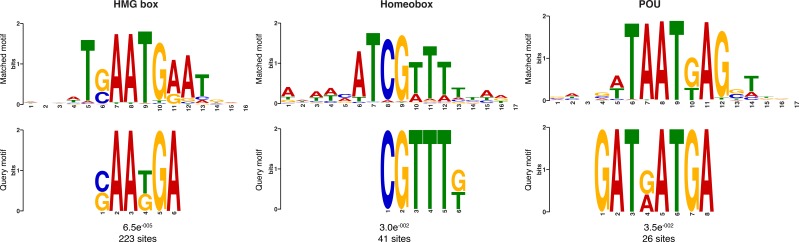
Figura 6-figure supplement 3.Loghi di sequenza aggiuntivi dei motivi del DNA determinati dall’analisi MEME-ChIP per essere significativamente arricchiti nelle sequenze di potenziatori attivati previsti per gli adulti.Il valore E(rapporto di probabilità di log di ogni motivo) e il numero di siti che contribuiscono alla costruzione del motivo sono mostrati, rispettivamente, il valore E(rapporto di probabilità di log di ogni motivo) e il numero di siti che contribuiscono alla costruzione del motivo. Il motivo abbinato è mostrato in alto e il motivo di interrogazione è mostrato in basso.DOI:
http://dx.doi.org/10.7554/eLife.22194.036
Figura 6-figure supplement 4.Loghi di sequenza di corrispondenza dei motivi del DNA determinati dall’analisi MEME-ChIP per essere significativamente arricchito nelle previste sequenze attivate come stimolatori, sia nell’adulto che nella larva.Il valore E(rapporto di probabilità di log di ogni motivo) e il numero di siti che contribuiscono alla costruzione del motivo sono mostrati, rispettivamente, il valore E(rapporto di probabilità di log di ogni motivo) e il numero di siti che contribuiscono alla costruzione del motivo.DOI:
http://dx.doi.org/10.7554/eLife.22194.037
Figura 6-figure supplement 5.Esempi di elementi simili a potenziatori previsti in prossimità di ben noti geni del fattore di sviluppo e di trascrizione.I geni che codificano le proteine (viola) sono mostrati, insieme alla copertura di input DNA-normalizzato di diverse modifiche degli istoni e l’espressione di RNA-seq. Regioni di arricchimenti (picchi di confidenza elevati, che rappresentano eventi riproducibili attraverso i veri replicati biologici) corrispondenti a siti putativi adulti simili a potenziatori sono evidenziati in grigio.DOI:
http://dx.doi.org/10.7554/eLife.22194.038
La regolamentazione Cis limita le architetture del genoma oltre 700 Myr di evoluzione
Gli elementi normativi non codificanti altamente conservati sono spesso associati non solo alla maggiore lunghezza degli introni, ma anche ai geni che codificano i regolatori dello sviluppo(Woolfe et al., 2005; Vavouri et al., 2007; Sandelin et al., 2004). Particolarmente interessanti sono le coppie microsinteniche ancestrali conservate (qui presenti unità microsinteniche) che consistono di (i) due geni vicini che condividono elementi cis-regolatori comuni, oppure (ii) un regolatore dello sviluppo e geni vicini funzionalmente non correlati, i cui introni ospitano elementi cis-regolatori conservati(Kikuta et al., 2007; Irimia et al., 2013; Engström et al., 2007; Irimia et al., 2012; Naville et al., 2015). Sono state fornite prove sperimentali dell’esistenza di questo tipo di regolamentazione cis nei vertebrati(Irimia et al., 2012; Naville et al., 2015).
Per verificare se si tratta di un antico meccanismo di regolazione cis mantenuto attraverso l’evoluzione animale, abbiamo valutato la relazione spaziale tra i geni di ciascuna delle 80 unità microsinteniche precedentemente segnalate come presenti nel genoma di Amphimedon(Irimia et al., 2012) e ne abbiamo chiarito l’ortologia, confermando la presenza di 60 unità microsinteniche non ambigue. Sorprendentemente, 43 di queste 60 microsintensie metazoiche evolutive conservate contenevano firme simili a potenziatori presunti negli adulti di Amphimedon(Figura 7A; Figura 7 – dati fonte 1; Figura 7 – dati fonte 1; Figura 7 – supplemento 1). Si trattava di una frazione molto più elevata rispetto ad un set di controllo costituito da 60 coppie di due geni vicini non sintenici selezionati in modo casuale (1.000 iterazioni; valore p<0,00001). Questo modello è stato comprovato dal ritrovamento di firme simili a quelle degli stimolatori di larve in 16 delle 60 unità microsinteniche, sette delle quali contenevano sia larve che elementi simili agli stimolatori previsti per gli adulti(Figura 7A; Figura 7 – dati fonte 1; Figura 7 – dati fonte 1 ; Figura 7 – supplemento 1).10.7554/eLife.22194.039Figure 7.Amphimedon elementi simili agli stimolatorianfimedoni sono arricchiti in unità microsinteniche metazoano-specifiche.(A) Segnature presunte di adulti e larve simili agli stimolatori identificati nelle 60 coppie microsinteniche metazoano-specifiche studiate.(B) Il cladogramma rappresenta la distribuzione filogenetica nota della coppia di geni microsintenici Isl2-Scaper attraverso opistoconti. L’orientamento della freccia corrisponde all’orientamento del gene. Isl2-Scaper non è conservato nel lievito, Capsaspora, Nematostella e C. elegans.(C) Elementi miglioratori nel locus della coppia di geni microsintenici Isl-Scaper in Amphimedon. Scaper e Isl geni (viola) sono mostrati, insieme con ingresso DNA-normalizzato copertura di H3K4me3 e H3K4me1 e RNA-seq espressione sia in adulti e larve. Regioni di arricchimenti (picchi di confidenza elevati, che rappresentano eventi riproducibili attraverso i veri replicati biologici) corrispondenti agli elementi simili a potenziatori previsti che si trovano all’interno degli introni di Scaper sono evidenziati in grigio.DOI:http://dx.doi.org/10.7554/eLife.22194.03910.7554/eLife.22194.040Figure7-source data 1.60 unità microsinteniche che rappresentano i collegamenti genici funzionali e la presenza-assenza di stati di cromatina contenenti i tipici modelli PTM dell’istone dell’enhancer eumetazoano (‘EnhP’,’ EnhWk’,’ TxEnhA’) (solo per adulti) e/o in elementi simili all’enhancer predetto in silico (sia larve che adulti).DOI:http://dx.doi.org/10.7554/eLife.22194.04010.7554/eLife.22194.041FigureSupplemento a 7 cifre 1.Ulteriori esempi di elementi simili al potenziatore predetto in unità microsinteniche conservate. Sono mostrati i geni che codificano le proteine (viola), insieme alla copertura di input DNA-normalizzato di diverse modifiche dell’istone e all’espressione RNA-seq/CEL-seq. Le regioni di arricchimento (picchi di confidenza elevati, che rappresentano eventi riproducibili attraverso veri replicati biologici) corrispondenti a siti simili a potenziatori presunti situati all’interno delle unità microsinteniche sono evidenziati in grigio. Se non diversamente specificato, vengono mostrati i dati degli adulti.DOI:http://dx.doi.org/10.7554/eLife.22194.041
Un caso eclatante di legame genico conservato riguarda il gene omeobox Islet LIM(Isl), che svolge ruoli conservati nello sviluppo animale(Thor e Thomas, 1997; Liang et al., 2011), e Scaper (proteina associata alla ciclina S-fase A nella ER)(Figura 7B). L’Amphimedon Scaper contiene 25 introni, alcuni dei quali sono notevolmente più lunghi (> 1 kb) rispetto alla dimensione media degli introni (Fernandez-Valverde et al., 2015), e prevede elementi simili a potenziatori situati all’interno del suo introne 10, 17 e 21 (Figura 7C). Allo stesso modo, la microsintenia di Tfap4 (fattore di trascrizione AP-4)(Simionato et al., 2007) e Glis2 (famiglia GLIS zinc finger 2) è profondamente conservata. Simile ad un’osservazione nei vertebrati(Abbasi et al., 2007), la spugna Glis2 contiene due introni, di cui il secondo ospita diversi adulti predetti elementi stimolanti attivati come enhancer(Figura 7-source dati 1; Figura 7-figure supplement 1). Insieme, questi risultati suggeriscono che la posizione genomica di alcuni elementi cis-regolatori pone probabilmente dei vincoli all’evoluzione dei geni vicini, portando alla comparsa di blocchi genici microsintenici conservati in tutto il regno animale.

Figura 7-figure supplemento 1.Anfimedone elementi simili a potenziatori sono arricchiti in unità microsinteniche metazoano-specifiche.60 unità microsinteniche che rappresentano i legami genici funzionali e la presenza-assenza di stati di cromatina contenenti i tipici schemi PTM dell’istone dell’enhancer eumetazoano (‘EnhP’,’ EnhWk’,’ TxEnhA’) (solo per adulti) e/o in elementi simili all’enhancer predetto in silico (sia larve che adulti).60 unità microsinteniche che rappresentano i legami genici funzionali e la presenza-assenza di stati di cromatina contenenti i tipici schemi PTM dell’istone dell’enhancer eumetazoano (‘EnhP’,’ EnhWk’,’ TxEnhA’) (solo per adulti) e/o in elementi simil-stimolatori predetti in silico (sia larve che adulti).ulteriori esempi di elementi simil-stimolatori predetti in unità microsinteniche conservate.(A) Firme presunte di elementi simili agli stimolatori di larve e adulti identificate nelle 60 coppie microsinteniche metazoano-specifiche studiate.(B) Il cladogramma rappresenta la distribuzione filogenetica nota della coppia di geni microsintenici Isl2-Scaper attraverso opistoconti. L’orientamento della freccia corrisponde all’orientamento del gene. Isl2-Scaper non è conservato nel lievito, Capsaspora, Nematostella e C. elegans.(C) Elementi miglioratori nel locus della coppia di geni microsintenici Isl-Scaper in Amphimedon. Scaper e Isl geni (viola) sono mostrati, insieme con ingresso DNA-normalizzato copertura di H3K4me3 e H3K4me1 e RNA-seq espressione sia in adulti e larve. Regioni di arricchimenti (picchi di confidenza elevati, che rappresentano eventi riproducibili attraverso i veri replicati biologici) corrispondenti agli elementi simili a potenziatori previsti che si trovano all’interno degli introni di Scaper sono evidenziati in grigio.DOI:
http://dx.doi.org/10.7554/eLife.22194.03910.7554/eLife.22194.040Figure 7-source data 1.60 unità microsinteniche che rappresentano i legami genici funzionali e l’assenza di presenza di stati cromatici contenenti i tipici pattern PTM dell’istone dell’enhancer eumetazoano (‘EnhP’,’ EnhWk’,’ TxEnhA’) (solo per adulti) e/o in elementi simili all’enhancer predetto in silico (sia larve che adulti).DOI:http://dx.doi.org/10.7554/eLife.22194.040DOI:
http://dx.doi.org/10.7554/eLife.22194.040DOI:
http://dx.doi.org/10.7554/eLife.22194.040Sono mostrati i geni della codifica delle proteine (viola), insieme alla copertura normalizzata del DNA in ingresso delle diverse modifiche degli istoni e all’espressione RNA-seq/CEL-seq. Le regioni di arricchimento (picchi di confidenza elevati, che rappresentano eventi riproducibili attraverso i veri replicati biologici) corrispondenti a siti simili a potenziatori presunti situati all’interno delle unità microsinteniche sono evidenziate in grigio. Se non diversamente specificato, vengono mostrati i dati degli adulti.DOI:
http://dx.doi.org/10.7554/eLife.22194.041
Figura 7-figure supplement 1.1. Ulteriori esempi di elementi simili a potenziatori previsti in unità microsinteniche conservate.I geni di codifica delle proteine (viola) sono mostrati, insieme con l’input DNA-normalizzato copertura del DNA di ingresso di diverse modifiche degli istoni e RNA-seq/CEL-seq espressione. Le regioni di arricchimento (picchi di confidenza elevati, che rappresentano eventi riproducibili attraverso veri replicati biologici) corrispondenti a siti simili a potenziatori presunti situati all’interno delle unità microsinteniche sono evidenziati in grigio. Se non diversamente specificato, vengono mostrati i dati degli adulti.DOI:
http://dx.doi.org/10.7554/eLife.22194.041
Discussione
Dal loro punto di divergenza oltre 700 Mya, le spugne e gli eumetazoi hanno avuto storie evolutive radicalmente diverse, con l’antenato eumetazoiano che ha dato origine a una serie di piani corporei morfologicamente complessi, e l’antenato spugna che ha dato origine a un piano corporeo morfologicamente semplice di base. Poiché entrambi questi lignaggi condividono un repertorio notevolmente simile di famiglie geniche di sviluppo(Srivastava et al., 2010; Larroux et al., 2008; Richards et al., 2008), queste diverse traiettorie evolutive devono ancora essere riconciliate in termini di contenuto e organizzazione del genoma. Recentemente è stato dimostrato che, pur avendo un genoma altamente compatto con regioni intergeniche minime(Fernandez-Valverde e Degnan, 2016), Amphimedon mostra un’espressione genica dinamica dello sviluppo simile a quella degli eumetazoi(Gaiti et al., 2015; Levin et al., 2016). Generando le prime, a nostra conoscenza, mappe complete a livello genomico dell’istone H3 PTM e degli elementi potenziatori putativi in un animale non eumetazoico, determiniamo che questa complessità trascrizionale è parallela alla complessità normativa codificata dai modelli dell’istone combinatorio H3 PTM in questa spugna.
L’istone H3 PTM ha conservato il ruolo (o i ruoli) nella modulazione dell’espressione genica attraverso i metazoi
Nonostante la semplicità morfologica di Amphimedon, troviamo in questa spugna una forte evidenza dell’esistenza di una serie di stati normativi che sono alla base dello sviluppo eumetazoico. Per esempio, l’analisi del promotore genomico dell’H3K4me3 – l’istone eucariotico canonico e diffuso H3 PTM di trascrizione attiva – rivela una complessa correlazione tra l’occupazione del nucleosoma contenente H3K4me3 e l’espressione genica negli adulti e nelle larve di Amphimedon, coerente con un ruolo attivo e finemente sintonizzato per l’H3K4me3 nella modulazione dell’attività trascrizionale e della variabilità di espressione dei geni dello sviluppo. Inaspettatamente, identifichiamo una piccola sottopopolazione di geni altamente e specificamente espressi che sfidano questa premessa e sono trascritti in assenza di H3K4me3 in Amphimedon e Nematostella. Questa sottopopolazione di geni si differenzia dalla maggior parte degli altri geni espressi in fase di sviluppo che possiedono il marchio H3K4me3, per avere profili di espressione molto più limitati in uno stadio; in questa analisi, la maggior parte sono espressi solo in uno stadio di sviluppo. Anche se si potrebbe sostenere che questa apparente assenza di H3K4me3 è la conseguenza dell’espressione di geni regolati che sono spazialmente limitati a specifiche popolazioni cellulari, limitando così potenzialmente la nostra sensibilità di rilevazione con la nostra miscela cellulare ChIP-seq, questi risultati sono paralleli alla recente scoperta di Pérez-Lluch et al. (2015a) che Drosophila e C. elegans mostrano lo stesso modello, suggerendo che questa caratteristica appena scoperta è conservata in tutto il regno animale. Poiché l’espressione dei geni regolati dallo sviluppo è richiesta solo per un periodo limitato, l’assenza del marchio H3K4me3 consentirebbe la loro rapida attivazione e disattivazione. Meccanismi alternativi, come il legame transitorio dei fattori di trascrizione, sembrano svolgere un ruolo importante nella regolazione dell’espressione di questi geni(Pérez-Lluch et al., 2015a, 2015b) .
Modello di un meccanismo evolutivo conservato del silenziamento del gene mediato da PRC2
Il Polycomb Repressive Complex 2 (PRC2) è stato conservato durante tutta l’evoluzione degli opistoconti, con le sue sottounità fondamentali (E(z), SU(z)12, ESC e Nurf55) presenti negli animali, nei choanoflagellati e nei funghi multicellulari, ma assenti nella Capsaspora, e nei lieviti in boccio e di fissione(Sebé-Pedrós et al.., 2016; Margueron e Reinberg, 2011; Shaver et al., 2010; Jamieson et al., 2013; Connolly et al., 2013; Ikeuchi et al., 2015; Whitcomb et al., 2007)(Figura 4A). Ciò è coerente con il fatto che il complesso PRC2 si perde in diversi lignaggi unicellulari. Uno dei ruoli ancestrali della PCR2 negli opistoconti può essere stato nella risposta di difesa contro virus ed elementi trasponibili, o l’inserimento di nuovi geni(Jamieson et al., 2013), prima di essere cooptato per la regolazione dello sviluppo specifico di tipo cellulare negli animali, dove H3K27me3 e PRC2 sono necessari per trasmettere la memoria della repressione attraverso le generazioni e durante lo sviluppo(Margueron e Reinberg, 2011; Shaver et al., 2010; Gaydos et al., 2014; Barski et al., 2007). Infatti, PRC2 spesso regola la deposizione dei marchi H3K27me3 presso i regolatori di sviluppo dei loci codificati(Ha et al., 2011; Margueron e Reinberg, 2011; Barski et al., 2007). Il ritrovamento di brevi depositi di fattori di trascrizione dello sviluppo conservati in breve tempo nelle regioni silenziate di Amphimedon H3K27me3 è coerente con questo scenario evolutivo. Analogamente alle recenti scoperte nelle piante(Deng et al., 2013; Hecker et al., 2015), l’identificazione di un motivo arricchito nelle regioni silenziate di H3K27me3 simile al sito di legame del fattore GAGA, una componente degli elementi di risposta del gruppo Drosophila Polycomb, suggerisce un ruolo per i siti di legame del fattore GAGA nel rafforzamento del reclutamento di PRC2 per i geni target(Müller e Kassis, 2006; Simon e Kingston, 2009; Kassis e Brown, 2013). È degno di nota che una spugna omologa del fattore GAGA di Drosophila non è stata identificata nell’attuale assemblaggio del genoma di Amphimedon(Figura 4 dati fonte 1), suggerendo la cooptazione convergente di altre proteine leganti del DNA con ruolo(i) analogo(i) nel reclutamento di PRC2.
L’origine della regolazione del potenziatore distale animale
L’analisi del DNA cis-regolatore e degli istoni PTM hanno rivelato che alcuni meccanismi cis-regolatori, come quelli associati ai promotori prossimali, sono presenti negli olozoi non animali, mentre altri sembrano essersi evoluti successivamente sul gambo che porta ai metazoi della corona, in particolare gli stimolatori distali(Sebé-Pedrós et al., 2016; Schwaiger et al., 2014). Quest’ultimo è stato posto come uno dei fattori chiave che contribuiscono alla coordinazione spaziale e temporale della differenziazione cellulare che definisce lo sviluppo animale(Levine, 2010; Levine et al., 2014; Levine e Tjian, 2003; Peter e Davidson, 2011). La nostra previsione in silico degli elementi potenziatori di Anfimedone basata sui modelli di co-localizzazione dell’istone H3 PTM è coerente con questi elementi che si evolvono lungo il fusto metazoanico al passaggio alla multicellularità(Sebé-Pedrós et al., 2016). È interessante notare che gli elementi di regolazione del DNA del promotore per consentire il contesto e l’espressione del gene specifico del tipo di cellula sembravano evolvere anche nei metazoi del fusto (Fernandez-Valverdee Degnan, 2016), suggerendo che questi sono anche una componente critica del paesaggio animale cis-regolatorio. Anfimedone predetto enhancer-come elementi sono caratterizzati dalla stessa combinazione di istone H3 PTMs come in eumetazoani, che sembrano essere carenti di parenti olozoici unicellulari degli animali(Sebé-Pedrós et al., 2016; Bulger e Groudine, 2011). La loro associazione preferenziale con i regolatori dello sviluppo e delle trascrizioni suggerisce che gli elementi potenziatori dell’anfimedone sono in grado di regolare i geni dello sviluppo in modo simile agli eumetazoi(Schwaiger et al., 2014; Shlyueva et al., 2014; Nègre et al., 2011; Bogdanovic et al., 2012; Woolfe et al., 2005; Heintzman et al., 2009). Gli elementi potenziatori sono noti per essere associati alla trascrizione di RNA poli(A)- e poli(A)+ potenziatori sia brevi che lunghi (rispettivamente 2D e 1D eRNA)(Natoli e Andrau, 2012; Li et al., 2016; Kim et al., 2010). La presenza di RNAPII e la rilevazione dell’espressione in un sottoinsieme degli elementi simili agli stimolatori attivati da Amphimedon è coerente con questa nozione(Figura 6-figure supplement 2). Anche se la trascrizione non codificante in questi stimolatori dovrà essere studiata in dettaglio, questa co-occupazione di elementi stimolatori e RNAPII è stata osservata anche in Nematostella e nei bilateriani(Schwaiger et al., 2014; Li et al., 2016; Kim et al., 2010; De Santa et al., 2010; Chen et al., 2013), dove questi elementi potrebbero interagire fisicamente con il complesso di iniziazione della trascrizione al TSS del loro gene(i) target (Schwaiger et al., 2014).
A differenza dei bilateriani, dove il fattore di legame trascrizionale del repressore CCCTC (CTCF) si localizza con il genoma della coesina a livello del genoma ed è coinvolto nelle interazioni a lungo raggio tra potenziatore e promotore e nella struttura di cromatina di ordine superiore(Lee e Iyer, 2012; Seitan et al., 2013; Merkenschlager e Odom, 2013), Amphimedon manca di CTCF(Heger et al., 2012). Questo probabilmente limita le interazioni del potenziatore di Amphimedon con i macchinari trascrizionali del promotore prossimale a brevi distanze. Il looping della cromatina degli stimolatori ai loro promotori bersaglio in questa spugna potrebbe quindi avvenire attraverso un meccanismo di legame della coesina indipendente dal CTCF, come proposto negli cnidari, che mancano anche del CTCF(Schwaiger et al., 2014). In alternativa, ma non esclusivamente, il RNAPII e i suoi macchinari trascrizionali associati possono tracciare attraverso il DNA che interviene tra gli stimolatori e i promotori(Li et al., 2016), e potrebbe essere il meccanismo preferito delle interazioni stimolatore-promotori in questa spugna. La co-occupazione di elementi simili agli stimolatori di Anfimedone e RNAPII supporta questo meccanismo di attivazione trascrizionale. Studi futuri dell’architettura del genoma 3D saranno cruciali per chiarire il meccanismo di interazione potenziatore-promotori in questa spugna e altri animali non-bilateriani precoci di ramificazione privi di questa proteina architettonica(Gaiti et al., 2016).
Infine, troviamo una forte evidenza per gli elementi cis-regolatori che sono importanti per il mantenimento dei blocchi di geni microsintenici metazoano-specifici microsintenici oltre 700 Myr dell’evoluzione. L’emergere di una regolazione distale del potenziatore prima della cladogenesi metazoica potrebbe spiegare la pervasività dei blocchi di regolazione sintensiva conservati nei genomi animali e l’assenza di questi blocchi nei loro parenti unicellulari(Srivastava et al., 2010; Sebé-Pedrós et al., 2016; Irimia et al., 2013, 2012; Bulger e Groudine, 2011; Putnam et al., 2007; Duan et al., 2010). La forte evidenza che gli elementi potenziatori siano arricchiti in unità microsinteniche specifiche per metazoo profondamente conservate suggerisce che la loro posizione genomica potrebbe limitare l’architettura del genoma, portando alla comparsa di microsinteni conservati in tutto il regno animale(Irimia et al., 2013, 2012).
In conclusione, un paesaggio di regolazione genica conservato simile a quello degli eumetazoi morfologicamente complessi sembra essere già presente all’alba degli animali, e quindi probabilmente ha dato origine ad almeno 700 Mya. In particolare, sembrano esserci stati cambiamenti fondamentali nell’architettura cis-regolatoria del genoma lungo il fusto metazoico, in concomitanza con l’evoluzione della multicellularità animale, compresa l’apparente origine degli stimolatori distali e dei tipi promotori per la specificità del tipo di cellula e la regolazione dello sviluppo. Con questo in mente, proponiamo uno scenario evolutivo in cui le differenze quantitative piuttosto che qualitative nei meccanismi di regolazione probabilmente guidano l’evoluzione e la diversificazione dei piani corporei eumetazoici(Figura 8).10.7554/eLife.22194.042Figure 8.Origin of animal cis-regulatory complexity.The phylogenetic relationship of representative animal lineages and unicellular holozoans is shown here. In evidenza sono le principali innovazioni genomiche che si correlano con l’emergere e la precoce diversificazione degli animali. Alcuni componenti del panorama regolatorio dei metazoi possono essere anteriori alla scissione dei lignaggi metazoi e olozoi, tra cui le interazioni regolatorie del nucleo TF-TF e i lunghi RNA intergenici non codificanti, che sono stati recentemente identificati in parenti unicellulari di animali(Sebé-Pedrós et al., 2016; de Mendoza et al., 2015), ma la cui origine evolutiva non è ancora chiara. Con un complesso panorama di regolamentazione genica già in atto all’alba degli animali, l’espansione delle famiglie geniche evolutive (fattori di trascrizione codificanti e componenti dei percorsi di segnalazione), DNA cis-regolatorio e RNA non codificanti, insieme all’emergere della proteina architettonica CTCF per consentire interazioni più complesse tra potenziatore e promotore, sembrano essere alla base della diversificazione evolutiva dei piani corporei eumetazoici.DOI:http://dx.doi.org/10.7554/eLife.22194.042
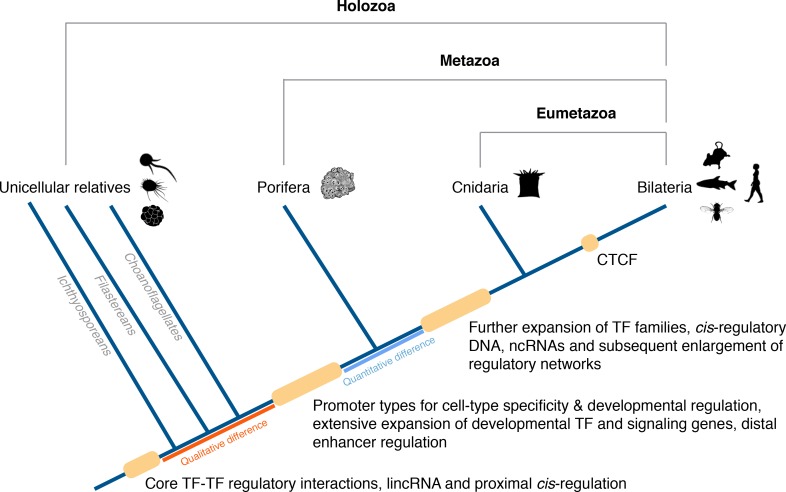
Figura 8.Origine della complessità normativa cis-animale.Il rapporto filogenetico di lignaggi animali rappresentativi e olozoi unicellulari è mostrato qui. In evidenza sono le principali innovazioni genomiche che sono correlate con l’emergere e la diversificazione precoce degli animali. Alcuni componenti del panorama regolatorio dei metazoi possono precedere la scissione dei lignaggi metazoi e olozoi, tra cui le interazioni regolatorie del nucleo TF-TF e i lunghi RNA intergenici non codificanti, che sono stati recentemente identificati in parenti unicellulari di animali(Sebé-Pedrós et al., 2016; de Mendoza et al., 2015), ma la cui origine evolutiva non è ancora chiara. Con un complesso panorama di regolamentazione genica già in atto all’alba degli animali, l’espansione delle famiglie geniche evolutive (fattori di trascrizione codificanti e componenti dei percorsi di segnalazione), DNA cis-regolatorio e RNA non codificanti, insieme all’emergere della proteina architettonica CTCF per consentire interazioni più complesse tra potenziatore e promotore, sembrano essere alla base della diversificazione evolutiva dei piani corporei eumetazoici.DOI:
http://dx.doi.org/10.7554/eLife.22194.042
Materiali e metodi
Raccolta di animali
Gli adulti e le larve diAnfimedon queenslandica sono stati raccolti da Heron Island Reef, Great Barrier Reef, Queensland, Australia, eallevati come descritto in precedenza(Leys et al., 2008).
Anticorpi
Abbiamo usato un anticorpo monoclonale di topo contro la ripetizione non fosforilata del C-terminale di RNA polimerasi II (RRID:AB_492629) (clone 8WG16, #05-952, Merck Millipore, Billerica, MA), un anticorpo policlonale di coniglio contro H3K4me3 (RRID:AB_1977252) (#07-473, Merck Millipore), un anticorpo policlonale di coniglio contro H3K27me3 (RRID:AB_310624) (#07-449, Merck Millipore), un anticorpo monoclonale di topo contro H3K4me1 (RRID:AB_10806625) (#17-676, Merck Millipore), un anticorpo policlonale di coniglio contro H3K27ac (RRID:AB_310550) (#07-360, Merck Millipore), un anticorpo monoclonale di coniglio contro H3K36me3 (RRID:AB_10615601) (#17-10032, Merck Millipore), e un anticorpo policlonale di coniglio contro l’istone H3 (RRID:AB_417398) (#07-690, Merck Millipore) (Figura 1 –dati fonte1). L’intera sequenza amminoacidica dell’istone H3 è perfettamente conservata tra Amphimedon e altri eucarioti dove questi anticorpi sono stati usati con successo(Sebé-Pedrós et al., 2016; Ercan et al., 2009; Barraza et al., 2015; Harmeyer et al., 2015; Liu et al., 2007; Eckalbar et al., 2016).(Figura 1-figure supplement 1).
Saggi di immunoprecipitazione della cromatina (ChIP)
Circa un cm3 di tessuto di spugna adulto è stato spremuto attraverso un panno fine e le cellule (~ 107) sono stati reticolati in formaldeide al 2% per 5 minuti a temperatura ambiente (RT). Larve (~ 350) sono state messe in comune, omogeneizzate e reticolate come sopra. Una procedura simile è stata poi adottata per entrambe le fasi di sviluppo. In particolare, la reticolazione è stata estinta con 125 mM di glicina per 5 minuti a temperatura ambiente (RT). Le cellule sono state lavate due volte in 0,22 µm di acqua di mare filtrata e centrifugate a 500 g per 5 min. Cellule in pellet sono stati lisati in tampone di lisi SDS (10 mM EDTA, 50 mM Tris-HCl a pH 8.0, 1% SDS, più proteasi e inibitori della fosfatasi), incubati per almeno 10 min su ghiaccio, e sonicati per 12 min (12 cicli, ciascuno 30 s ‘ON’, 30 s ‘OFF’) in un Bioruptor Sonicator (Diagenode, Seraing, Belgio) per generare 200-300 bp frammenti. Le condizioni ottimali di sonicazione sono state precedentemente determinate testando una gamma di cicli di sonicazione (da 5 a 30); 12 cicli sono stati considerati ottimali. Il materiale non solubile è stato rimosso dal lisato mediante centrifugazione a 12.000 g per 10 minuti a 4°C. Un’aliquota del materiale solubile è stata rimossa per il DNA in ingresso e conservata a -20°C. Per ridurre la concentrazione di SDS allo 0,1%, il restante materiale solubile è stato diluito 10 volte in tampone di diluizione ChIP (1,2 mM EDTA, 16,7 mM Tris-HCl a pH 8,0, 167 mM NaCl, 1,1% Triton X-100, 0,01% SDS, più PhosSTOP inibitore della fosfatasi e cocktail di inibitori della proteasi cOmpleta [Roche, Basilico, Svizzera]). Per ridurre il background non specifico, il materiale solubile diluito è stato pre-applicato con le perle della proteina G Dynabeads (#10003D, ThermoFisher, Waltham, MA), e, allo stesso tempo, gli anticorpi sono stati collegati alle perle della proteina G Dynabeads (#10003D, ThermoFisher) ruotando per un’ora a 4°C. A questo punto, il materiale solubile diluito e diluito è stato incubato con le miscele di perle anticorpali, ruotando a 4°C durante la notte. Il materiale immunoprecipitato è stato lavato tre volte con tampone di lavaggio a basso contenuto di sale (2 mM EDTA, 20 mM Tris-HCl a pH 8,0, 150 mM NaCl, 1% Triton X-100, 0,1% SDS), tre volte con tampone di lavaggio ad alto contenuto di sale (2 mM EDTA, 20 mM Tris-HCl a pH 8.0, 500 mM NaCl, 1% Triton X-100, 0,1% SDS), tre volte con tampone di lavaggio LiCl (1 mM EDTA, 1 mM Tris-HCl a pH 8,0, 1% DOC, 1% NP-40, 250 mM LiCl), e tre volte con tampone TE (10 mM Tris-Cl, pH 8,0; 1 mM EDTA). Complessi di DNA sono stati eluiti 30 minuti a 65 ° C con TE-SDS (10 mM Tris-Cl, pH 8,0; 1 mM EDTA; 1% SDS) e decrosslinked durante la notte a 65 ° C, insieme con DNA in ingresso, con l’aggiunta di 125 mM NaCl. Complessi di DNA decrosslinked e DNA di ingresso sono stati trattati con RNaseA, e successivamente con proteinasi K. Infine, immunoprecipitato e DNA di ingresso sono stati purificati con fenolo: cloroformio: estrazione isoamilica (25:24:1), recuperato per precipitazione con etanolo in presenza di 300 mM NaOAc pH 5,2 e 2 µl di vettore glicogeno (10 mg/ml), e risospeso in UltraPure DNase/RNase-Free Distilled Water (ThermoFisher) per un uso successivo. Le biblioteche di DNA immunoprecipitato e DNA in ingresso sono state preparate utilizzando il NEBNext ChIP-seq Library Prep Master Mix Set per Illumina (#E6240, New England Biolabs, Ipswich, MA) secondo il protocollo del produttore. La qualità e il profilo delle biblioteche è stato analizzato utilizzando Agilent High Sensitivity DNA Kit (#5067-4626, Agilent, Santa Clara, CA) e quantificato utilizzando KAPA Library Quantification Kit (#KK4824, Kapa Biosystems, Wilmington, MA). Il sequenziamento profondo (100 bp paired-end) delle biblioteche per adulti – due repliche biologiche per H3K4me3, H3K4me1, H3K36me3, H3K27me3, RNAPII, DNA in ingresso e nessuna replica biologica per H3K27ac e istone totale H3 – è stato eseguito dall’unità NGS Macrogen Oceania NGS sullo strumento Illumina HiSeq 2000 (Illumina, San Diego, CA, Stati Uniti). Il sequenziamento profondo (40 bp paired-end) delle biblioteche di larve – nessun replicato biologico per H3K4me3, H3K4me1, H3K27me3, H3K27ac, RNAPII, DNA in ingresso – è stato eseguito dalla Central Analytical Research facility (CARF), Brisbane, Queensland, Australia, sullo strumento Illumina NextSeq 500 (Illumina, San Diego, CA, Stati Uniti).
Analisi dei dati ChIP-seq
Le letture delle sequenze di Illumina grezze per adulti sono state controllate utilizzando FastQC v0.52(http://www.bioinformatics.babraham.ac.uk/projects/fastqc/) e la qualità è stata filtrata utilizzando Trimmomatic v1.0.0.0 (SLIDINGWINDOW: 4:15, LEADING: 3, TRAILING: 3, HEADCROP: 5, MINLEN: 50) (RRID:SCR_011848)(Bolger et al., 2014). Le letture di qualità del sequenziamento di Illumina paired-end filtrate sono state poi allineate al genoma di Amphimedon(Srivastava et al., 2010) utilizzando Bowtie v1.1.2 (RRID:SCR_005476)(Langmead et al., 2009) con parametri -m 1, -n 2, -X 500, –best (letture mappate in modo univoco e un massimo di due disallineamenti all’interno del seme). Le letture non allineate sono state rimosse utilizzando SAMtools v0.1.19 (RRID:SCR_002105)(Li et al., 2009). Per tutti i set di dati ChIP-seq, sono state utilizzate misure di correlazione incrociata dei fili per stimare i rapporti segnale-rumore utilizzando SPP v1.11.0 (RRID:SCR_001790). I set di dati ChIP-seq per ciascun marchio sono stati segnalati se i punteggi erano al di sotto di una soglia normalizzata del coefficiente di correlazione incrociata dei trefoli (NSC) di 1,05, come descritto nelle linee guida modENCODE e ENCODE(ENCODE Project Consortium, 2012; Landt et al., 2012; Kellis et al., 2014; Kharchenko et al., 2008). Queste analisi sono state eseguite sul server Galaxy-qld(http://galaxy-qld.genome.edu.au/galaxy) sviluppato nell’ambito del progetto GVL(Afgan et al., 2015, 2016) e mantenuto dal Research Computing Centre, University of Queensland, Australia.
I coefficienti di correlazione di Pearson (Pearson r) dei segnali di arricchimento delle pieghe del genoma (FE) (vedi sotto) sono stati calcolati per le repliche biologiche ed è stata richiesta una soglia minima di 0,5, come da Ho et al . (2014). Inoltre, per garantire la coerenza tra i replicati biologici, è stato inoltre richiesto un Irreproducible Discovery Rate (IDR) di almeno 0,5 (vedi sotto), come descritto nelle linee guida modENCODE e ENCODE(ENCODE Project Consortium, 2012; Landt et al., 2012; Kellis et al., 2014; Kharchenko et al., 2008). I set di dati ChIP-seq che soddisfacevano questi criteri sono stati poi fusi in repliche biologiche (vedi ,).
Le regioni di arricchimento Histone PTM rispetto ai corrispondenti controlli sequenziali del DNA in ingresso sono stati determinati utilizzando MACS2 v2.1.0 (RRID:SCR_013291)(Zhang et al., 2008) secondo le linee guida dei consorzi modENCODE, ENCODE e Roadmap Epigenomics consortiums(Kundaje et al., 2015; ENCODEProject Consortium, 2012; Landt et al.,2012; Kellis et al.,2014; Kharchenko et al.,2008). In particolare, MACS2 è stato utilizzato in modalità broadpeak con una soglia di valore P broadpeak di 0,1 e una soglia narrowpeak di 0,01 (-p 0,01, –broad, –nomodello, –extsize 146, -g 1,45e8). Le regioni arricchite sono state valutate su singoli replicati (R1 e R2), sui dati raggruppati (letture raggruppate su repliche biologiche) (P) e su pseudoreplicati sottocampionati (ottenuti raggruppando le letture da repliche biologiche e sottocampionamento casuale, senza sostituzione, due pseudoreplicati con la metà del numero totale di letture raggruppate). (PR1 e PR2). Per ogni istone PTM, abbiamo definito “R” come l’insieme dei picchi in P che si sovrappongono in R1 e R2, e “PR” come l’insieme dei picchi in P che si sovrappongono in PR1 e PR2. Successivamente, abbiamo definito ‘M’ come l’insieme dei picchi che corrispondono esattamente in R e PR, e ‘T’ come l’insieme dei picchi che corrispondono esattamente in R e PR e quelli che sono unici in R o unici in PR. Per una dichiarazione sulla riproducibilità abbiamo richiesto che il rapporto M-to-T sia almeno 0,5(Figura 1 – dati sorgente 4). Per ottenere regioni di arricchimento affidabili, abbiamo limitato tutte le ulteriori analisi alle regioni arricchite identificate utilizzando dati raggruppati che sono stati anche identificati in modo indipendente sia nei replicati che negli pseudoplicati (il set ‘M’). Queste regioni di arricchimento possono essere interpretate come regioni ad alta confidenza, che rappresentano eventi riproducibili attraverso veri replicati biologici. Per l’H3K27ac, per il quale non era disponibile alcuna replica, abbiamo usato la colonna del valore P per classificare i picchi e abbiamo mantenuto solo i picchi con un valore p<0,001. Abbiamo usato la rappresentazione gappedPeak per gli istoni PTM con modelli di arricchimento relativamente compatti, tra cui H3K4me3, H3K27ac e H3K4me1. I picchi gapped sono ampi domini (che passano il valore P 0,1) che contengono almeno un picco stretto che passa un valore P di 0,01. Per l’istone diffuso PTM – H3K36me3 e H3K27me3 – abbiamo usato la rappresentazione broadPeak. I picchi RNAPII sono stati rilevati utilizzando il software peakzilla (RRID:SCR_007471)(Bardet et al., 2013), utilizzando il DNA in ingresso come controllo (-c 1.5, -s 3). È stata calcolata anche la frazione di letture che rientrano nelle regioni di picco (FRiP) (vedi Figura 1-dati sorgente 4). In linea con le linee guida ENCODE(ENCODE Project Consortium, 2012; Landt et al., 2012; Kellis et al., 2014; Kharchenko et al., 2008), tutti i nostri set di dati hanno un arricchimento del FRiP pari o superiore all’1%.
Per ogni coppia di set di dati ChIP allineati e di DNA in ingresso, abbiamo anche usato MACS2 (Zhang et al., 2008) per generare tracce di copertura del segnale a livello genomico per ogni posizione nel genoma di Amphimedon(Srivastava et al., 2010). Il DNA in ingresso è stato usato come controllo per la normalizzazione del segnale per la copertura dell’istone ChIP-seq. I tre tipi di statistiche di punteggio del segnale calcolati per base sono i seguenti: (i) rapporto di arricchimento delle pieghe dei conteggi di ChIP-seq rispetto ai conteggi di fondo attesi locali (FE); (ii) log10 negativo del log10 del valore P di Poisson dei conteggi di ChIP-seq rispetto ai conteggi di fondo attesi locali (ppois); e (iii) sottrazione del rumore dal campione di trattamento (sottrazione).
I set di dati di Larva ChIP-seq sono stati analizzati come descritto sopra, con le seguenti piccole modifiche. La contaminazione dell’adattatore prima del filtraggio della qualità di lettura è stata rimossa utilizzando Cutadapt (RRID:SCR_011841)(Martin, 2011). Le letture sono state poi filtrate in qualità utilizzando Trimmomatic v1.0.0 (SLIDINGWINDOW: 4:15, LEADING: 3, TRAILING: 3, HEADCROP: 3, MINLEN: 20) (RRID:SCR_011848)(Bolger et al., 2014). Le regioni di arricchimento Histone PTM e RNAPII relative ai controlli sequenziali del DNA in ingresso sono state determinate utilizzando MACS2 v2.1.0 (RRID:SCR_013291)(Zhang et al., 2008) in modalità broadpeak con una soglia del valore q broadpeak di 0,1 e una soglia narrowpeak di 0,05 (-q 0,05, –broad, –nomodello, –extsize 146, -g 1.45e8).
In entrambi gli stadi, gli stati della cromatina in tutto il genoma sono stati definiti utilizzando ChromHMM v1.10(Ernst e Kellis, 2012), che si basa su un modello multivariato Hidden Markov Model, utilizzando parametri predefiniti. Per ogni set di dati ChIP-seq, i conteggi delle letture sono stati calcolati in bins non sovrapposti di 200 bp attraverso il genoma di Amphimedon(Srivastava et al., 2010). Ogni bin è stato discrezionalizzato in due livelli, uno che indica l’arricchimento e 0 che indica l’assenza di arricchimento. La binarizzazione è stata eseguita confrontando i conteggi delle letture di ChIP-seq con i corrispondenti conteggi delle letture di controllo del DNA in ingresso all’interno di ciascun bin e utilizzando una soglia del valore P di Poisson di 1e-4 (la soglia di discretizzazione predefinita in ChromHMM). Abbiamo addestrato diversi modelli in modalità parallela con il numero di stati che vanno da 5 a 15 stati e abbiamo scelto un modello a 9 stati come miglior modello che cattura tutte le interazioni chiave tra i segni della cromatina e copre tutte le possibili posizioni genomiche (promotore, potenziatore, corpo del gene) che ci aspettavamo di risolvere data la selezione degli istoni PTM che abbiamo usato (H3K4me3, H3K27ac, H3K36me3, H3K4me1, H3K27me3 nell’adulto; e H3K4me3, H3K27ac, H3K27ac, H3K4me1, H3K27me3 in larva). Per assegnare biologicamente significativo mnemonica ai nove stati, ChromHMM è stato utilizzato per calcolare la sovrapposizione e gli arricchimenti di quartiere di ogni stato rispetto a vari tipi di annotazioni funzionali(Figura 1B; Figura 1-figure supplement 2; Figura 1-figure supplement 3; Figura 1-figure supplement 4). L’arricchimento degli stati in diverse caratteristiche genomiche è stato calcolato dividendo la percentuale di nucleotidi occupati da uno stato in una particolare caratteristica genomica per la percentuale di nucleotidi che questa caratteristica genomica rappresenta nell’intero genoma. Per le trame di arricchimento di sovrapposizione nelle figure, gli arricchimenti per ogni caratteristica genomica (colonna) in tutti gli stati sono normalizzati sottraendo il valore minimo dalla colonna e dividendo poi per il massimo della colonna. Quindi, i valori vanno sempre da 0 (bianco) a 1 (blu scuro)(cioè una scala relativa di colonna). Per i grafici di arricchimento posizionale del quartiere, la normalizzazione viene effettuata su tutte le colonne(cioè, il valore minimo su tutta la matrice viene sottratto da ogni valore e diviso per il massimo su tutta la matrice). Le annotazioni funzionali utilizzate sono state le seguenti: (1) Isole CpG ottenute utilizzando i modelli Hidden Markov come descritto in Wu et al. (2010). (2) Esoni, geni, introni, siti di inizio trascrizione (TSS) e siti di fine trascrizione (TES), finestre di 200 bp intorno ai TSS e finestre di 200 bp intorno ai TES basate sulle annotazioni del modello genico Aqu2.1 (Fernandez-Valverde et al., 2015). (3) Geni espressi e repressi, i loro TSS e TES. I geni sono stati classificati nella classe espressi (conteggi CEL-seq normalizzati > 0,5) e repressi (conteggi CEL-seq normalizzati < 0,5) in base ai loro livelli di espressione CEL-seq nello stadio rilevante (larva o adulto) (Levin et al., 2016; Hashimshony et al., 2012; Anavy et al., 2014).
Regioni di arricchimento dei vari istoni H3 PTM e RNAPII sono stati sovrapposti con geni codificanti le proteine e il pacchetto Bioconductor R GeneOverlap v1.14.0(https://www.bioconductor.org/packages/release/bioc/html/GeneOverlap.html) è stato utilizzato per testare e visualizzare la loro associazione con le liste di vari gruppi di espressione genica(R Core Team, 2014)(Figura 2B; Figura 2-figure supplement 2B). I geni che codificano le proteine sono stati classificati in “alto”, “medio”, “basso” e “non espresso” in base ai loro livelli di espressione CEL-seq nello stadio rilevante (larva o adulto) (Levin et al., 2016; Hashimshonyet al., 2012; Anavy et al., 2014 ). I geni espressi sono stati definiti liberamente come geni che avevano un numero di CEL-seq letti > 0 nello stadio rilevante. In particolare, per definire i geni espressi “alti”, “medi”, “bassi”, tutti i geni che codificano le proteine espressi nella fase rilevante sono stati ordinati in base ai valori dei dati CEL-seq e separati in tre contenitori di un numero uguale di geni, in modo simile alle analisi precedenti (Schwaiger et al., 2014).
Gli elementi potenziatori sono stati previsti come regioni di arricchimento H3K4me1 affidabili, che non si sovrappongono ai TSS (nessuna intersezione con 200 bp a monte o 200 bp a valle dei TSS di geni che codificano le proteine e gli lncRNA), ma si sovrappongono alle regioni designate come regioni in stato di cromatina potenziatrice (”Enhancer chromatin state”).TxEnhA’ o ‘EnhWk’ o ‘EnhP’ in stato adulto; ‘TxEnhA1’ o ‘TxEnhA2’ o ‘EnhWk’ o ‘EnhP’ o ‘EnhP’ in stato larvale) in base all’analisi ChromHMM. Gli elementi potenziatori attivati sono stati predetti intersecando gli elementi potenziatori con picchi significativi di H3K27ac, richiedendo una frazione di sovrapposizione minima del 50%. BEDTools v2.23.0 (RRID:SCR_006646)(Quinlan e Hall, 2010) è stato utilizzato per calcolare le sovrapposizioni tra le regioni di arricchimento e gli stati della cromatina con le diverse caratteristiche genomiche, nonché per identificare il TSS più vicino per ciascuno degli elementi attivatori attivi.
Le analisi di arricchimento dei motiviDe novo sono state eseguite utilizzando MEME-ChIP contro JASPAR CORE e UniPROBE Mouse database (-meme-minw 6, -meme-maxw 15, meme-nmotifs 20, -dreme-e 0.05, -meme-mod zoops) (RRID:SCR_001783)(Machanick e Bailey, 2011). Ogni motivo è stato rinominato in base al motivo più simile presente nel database TOMTOM o nella letteratura, se presente.
Le analisi di arricchimento funzionale dell’ontologia genica (GO) sono state eseguite con il plugin Cytoscape BiNGO (RRID:SCR_005736)(Maere et al., 2005; Shannon et al., 2003) con annotazione personalizzata e un cut-off del valore P regolato FDR di 0,01. Tutti i peptidi previsti da Amphimedon(Fernandez-Valverde et al., 2015) sono stati annotati utilizzando BLASTp (RRID:SCR_001010)(Altschul et al., 1990)(valore E di 0,001) rispetto al database di proteine NCBI non ridondante (nr). Tutte le proteine sono state anche ricercate per motivi proteici e peptidi di segnale utilizzando InterProScan 5(Jones et al., 2014) con parametri predefiniti. Le annotazioni del percorso KEGG sono state ottenute sul server web BlastKOALA per il gruppo tassonomico ‘Animali’ contro il file di database ‘family_eukaryotes + genus_prokaryotes’, utilizzando le impostazioni predefinite. Le analisi dei percorsi sono state eseguite con i file di annotazione BlastKOALA utilizzando lo strumento KEGG Mapper – Reconstruct pathhway tool (Kanehisaet al., 2016).
I profili di copertura e le heatmaps sono stati calcolati utilizzando ngs.plot v2.61 (RRID:SCR_011795)(Shen et al., 2014) e deepTools v2.4.1(Ramírez et al., 2016). Come sopra, i geni che codificano le proteine sono stati classificati in ‘alto’, ‘medio’, ‘basso’ e ‘non espresso’ in base ai loro livelli di espressione CEL-seq nello stadio rilevante (larva o adulto) (Levin et al., 2016; Hashimshony et al., 2012; Anavy et al., 2014 ). I geni espressi sono stati definiti liberalmente come geni che hanno avuto un numero di CEL-seq letti >0 nello stadio rilevante. In particolare, per definire i geni espressi “alti”, “medi”, “bassi”, i geni che codificano le proteine espressi nella fase rilevante sono stati ordinati in base ai valori dei dati CEL-seq e separati in tre contenitori di un numero uguale di geni, in modo simile alle analisi precedenti (Schwaiger et al., 2014).
Per tutte le analisi sono stati utilizzati solo lincRNA trovati in impalcature più grandi di 10 kbe, dato il genoma compatto di Amphimedon(Fernandez-Valverde e Degnan, 2016), tutte le analisi TSS sono state limitate ai geni non sovrapponibili che codificano le proteine con una distanza intergenica >1 kb che sono stati trovati in impalcature più grandi di 10 kb.
Tutti i dati del browser del genoma sono stati generati utilizzando un’istanza locale del browser del genoma UCSC (RRID:SCR_005780)(Kuhn et al., 2013).
ChIP-quantitative PCRs (ChIP-qPCRs)
Le PCR ChIP-quantitative PCR (ChIP-qPCR) sono state eseguite utilizzando la piattaforma LightCycler 480 (Roche, Basil, Svizzera). Le librerie ChIP (H3K4me1, H3K27ac, H3K4me3, H3K27me3) e Input DNA sono state diluite in acqua, combinate con la master mix LightCycler 480 SYBR green I (Roche, Basil, Svizzera) e i primer 0,2 µM, poi ciclate con il seguente profilo: 95°C per 10 min, 40 cicli di 95°C per 10 s, 60°C per 10 s, 72°C per 20 s. Le sequenze di primer sono disponibili nel file supplementare 1.
I valori dei cicli di quantificazione (Cq) sono stati estrapolati dal software del produttore (versione 1.5.1.6.1 SP2) utilizzando le impostazioni di High Confidence. Sono stati inoltre eseguiti una curva di fusione e nessun controllo di template (ntc) per assicurare che i singoli ampliconi fossero responsabili del segnale fluorescente. Il valore numerico 3,32 (log210, che rappresenta il 10% della cromatina in ingresso) è stato sottratto dal valore Cq del campione in ingresso per generare l’ingresso regolato Cq. Come controlli negativi sono state utilizzate due diverse regioni intergeniche non vincolate dal nostro istone PTM di interesse. È stata calcolata l’analisi Cq a doppio delta (dd) (vedi Figura 1 – dati sorgente 5).
In particolare, sono state utilizzate le seguenti formule per calcolare l’aumento di piega del segnale su sfondo:
dCq_IP = Cq_IP – Cq_Intergenico
dCq_Input = Cq_Input – Cq_intergenic
ddCq = dCq_IP – dCq_Input
Cambio di piega = 2∧ (-ddCq)
Geni ad alta e bassa varianza in Amphimedon
Le letture grezze CEL-seq sono state elaborate e mappate sul genoma di Amphimedon utilizzando Bowtie (RRID:SCR_005476)(Langmead et al., 2009). Abbiamo poi compresso gli 82 campioni di sviluppo di Amphimedon, dalla scissione precoce all’adulto, in 17 stadi, facendo la media dei replicati biologici per ogni stadio di sviluppo attraverso di essi. Gli stadi larvali sono stati combinati in due gruppi diversi (Larve 0-7 ore e Larve 6-50 ore), in quanto questi punti di sviluppo hanno un solo replicato per punto temporale. Per ridurre il rumore, sono stati scartati i geni che codificano le proteine e gli RNA lunghi non codificanti con un’espressione complessiva di meno di 100 CEL-seq conteggi grezzi durante tutto il corso del tempo di sviluppo. I conteggi dei geni grezzi CEL-seq sono stati poi normalizzati utilizzando la trasformazione di stabilizzazione della varianza in DEseq2 1.6.3 (RRID:SCR_000154)(Love et al., 2014) e i 15.000 geni più variabili (14.698 geni codificanti le proteine + 301 lncRNA) sono stati estratti utilizzando la deviazione assoluta mediana. I 14.698 geni codificanti le proteine sono stati poi filtrati per trattenere solo i geni codificanti le proteine non sovrapposti con espressione rilevabile allo stadio adulto (conteggi CEL-seq normalizzati > 0) con una distanza intergenica > 1 kb che sono stati trovati in scaffali più grandi di 10 kb. Ciò ha portato ad un numero totale di 3.200 geni ad ‘alta varianza’. Il rimanente espresso (CEL-seq conteggi normalizzati > 0 nell’adulto) non sovrapponendo i geni di codifica delle proteine con una distanza intergenica > 1 kb che sono stati trovati in impalcature più grandi di 10 kb sono stati considerati ‘a bassa varianza’ geni (n= 3.999). Per definire basso, medio e alto, i 3.200 geni ad alta varianza e i 3.999 geni a bassa varianza sono stati ordinati in base ai valori dei dati CELseq e separati in tre contenitori di un numero uguale di geni.
Geni regolati e stabili in Amphimedon
Le letture grezze CEL-seq sono state elaborate e mappate sul genoma di Amphimedon utilizzando Bowtie (RRID:SCR_005476)(Langmead et al., 2009). I conteggi delle letture sono stati normalizzati dividendo per il numero totale delle letture contate e moltiplicando per106. Abbiamo poi compresso gli 82 campioni di sviluppo di Amphimedon, dalla scissione precoce all’adulto, in 17 stadi, facendo la media dei replicati biologici per ogni stadio di sviluppo attraverso di essi. Gli stadi larvali sono stati combinati in due gruppi diversi (Larve 0-7 ore e Larve 6-50 ore), poiché questi punti di sviluppo hanno un solo replicato per punto temporale. Per ridurre il rumore, sono stati mantenuti solo i geni codificanti le proteine con un’espressione di almeno quattro conteggi normalizzati CEL-seq in almeno due punti temporali di sviluppo. Per definire la stabilità trascrizionale dei geni codificanti le proteine, è stato calcolato il coefficiente di variazione dell’espressione genica per ogni gene codificante le proteine (n = 15.146), come riportato da Pérez-Lluch et al. (2015a). Per le trame di copertura TSS input DNA-normalizzato, questi 15.146 geni codificanti le proteine sono stati poi filtrati per mantenere solo espresso (CEL-seq conteggi normalizzati > 0 nel relativo stadio [larva o adulto]) non sovrapponendo i geni codificanti le proteine con una distanza intergenica > 1 kb che sono stati trovati in scaffali più grandi di 10 kb. Infine, dalla classifica completa di questi geni codificanti le proteine espresse, abbiamo definito i 1.000 geni inferiori con la più bassa variazione di espressione durante lo sviluppo come geni ‘stabili’ e i 1.000 geni superiori con la più alta variazione di espressione comegeni fortemente sviluppati ‘regolati’.
Geni regolati e stabili in Nematostella vectensis
Sono stati utilizzati i set di dati ChIP-seq disponibili sui polipi femminili adulti per H3K4me3 e i corrispondenti controlli del DNA in ingresso(Schwaiger et al., 2014). I set di dati di ChIP e i corrispondenti set di dati del DNA in ingresso e i geni stabili e regolati dal punto di vista dello sviluppo sono stati generati utilizzando le stesse procedure della spugna (vedi sopra). Per ottenere quantificazioni di geni e trascrizioni, abbiamo mappato i set di dati RNA-seq disponibili(Helm et al., 2013) su modelli di geni NveGenes2.0 (http://www.cnidariangenomes.org/) utilizzando kallisto(Bray et al., 2016).
Identificazione degli ortologi e filogenesi
Gli ortologi dei componenti della Drosophila PcG e dei fattori associati sono stati identificati utilizzando le ricerche BLASTp (RRID:SCR_001010)(Altschul et al., 1990) contro i proteomi previsti delle specie selezionate(Figura 4 dati fonte 1) con un valore soglia E di 0,001 e prendendo un massimo di 5 hit per specie. Tutti i risultati ottenuti sono stati allineati utilizzando MAFFT con la modalità L-INS-i (RRID:SCR_011811)(Katoh e Standley, 2013). Gli allineamenti sono stati automaticamente tagliati con trimAl v1.4 (151) in modalità -automatizzata1. Gli allineamenti risultanti tagliati sono stati poi utilizzati per l’inferenza filogenetica utilizzando FastTree2(Price et al., 2010) con parametri -wag -cat 8 -gamma. Gli alberi filogenetici sono stati ispezionati manualmente per discriminare quali colpi BLASTp hanno formato clades monofiletici con le sequenze di query Drosophila. La stessa metodologia è stata utilizzata per identificare le coppie microsinteniche ancestrali conservate prese da Irimia et al. (2012), ma utilizzando le sequenze di Homo sapiens come proteine di query. Le coppie ortologiche di Amphimedon convalidate con filogenesi sono state controllate manualmente per verificare la contiguità nel genoma e quelle trovate in diverse impalcature o con più di due geni intervenienti sono state rimosse dalle analisi successive.
Accesso ai dati
I set di datiAmphimedon ChIP-seq sono stati depositati presso la NCBI Gene Expression Omnibus (GEO)(RRID:SCR_007303)(Edgar et al., 2002) con il numero di adesione GSE79645. Nel corso dello studio è stato utilizzato l’ampQue1 per l’assemblaggio del genoma di Amphimedon. I set di dati CEL-seq possono essere ottenuti dal NCBI GEO (GSE54364)(Anavy et al., 2014). I set di dati Amphimedon RNA-seq possono essere scaricati dall’SRA del NCBI (RRID:SCR_004891) con l’adesione SRP044247(Fernandez-Valverde et al., 2015). I set di dati Nematostella vectensis RNA-seq possono essere scaricati dall’SRA del BCN con l’adesione SRP018739(Helm et al., 2013). I set di dati N. vectensis ChIP-seq possono essere ottenuti dal NCBI GEO (GSE46488)(Schwaiger et al., 2014). Per tutte le analisi abbiamo utilizzato i seguenti set di dati del modello genico. A. queenslandica: modelli Aqu2.1 (http://amphimedon.qcloud.qcif.edu.au/) (ultimo accesso il 25 febbraio 2017)(Fernandez-Valverde et al., 2015), lncRNAs(http://amphimedon.qcloud.qcif.edu.au/lncRNAs/) (ultimo accesso il 25 febbraio 2017) (Gaiti et al., 2015); N. vectensis: Modelli NveGenes2.0 (http://www.cnidariangenomes.org/) (ultimo accesso il 25 febbraio 2017).
References
- Abbasi AA, Paparidis Z, Malik S, Goode DK, Callaway H, Elgar G, Grzeschik KH. Human GLI3 intragenic conserved non-coding sequences are tissue-specific enhancers. PLoS One. 2007; 2DOI | PubMed
- Adamska M, Degnan SM, Green KM, Adamski M, Craigie A, Larroux C, Degnan BM. Wnt and TGF-beta expression in the sponge Amphimedon queenslandica and the origin of metazoan embryonic patterning. PLoS One. 2007; 2DOI | PubMed
- Afgan E, Baker D, van den Beek M, Blankenberg D, Bouvier D, Čech M, Chilton J, Clements D, Coraor N, Eberhard C, Grüning B, Guerler A, Hillman-Jackson J, Von Kuster G, Rasche E, Soranzo N, Turaga N, Taylor J, Nekrutenko A, Goecks J. The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2016 update. Nucleic Acids Research. 2016; 44:W3-W10. DOI | PubMed
- Afgan E, Sloggett C, Goonasekera N, Makunin I, Benson D, Crowe M, Gladman S, Kowsar Y, Pheasant M, Horst R, Lonie A. Genomics virtual laboratory: a practical bioinformatics workbench for the cloud. Plos One. 2015; 10DOI | PubMed
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. Journal of Molecular Biology. 1990; 215:403-410. DOI | PubMed
- Anavy L, Levin M, Khair S, Nakanishi N, Fernandez-Valverde SL, Degnan BM, Yanai I. BLIND ordering of large-scale transcriptomic developmental timecourses. Development. 2014; 141:1161-1166. DOI | PubMed
- Arnold CD, Gerlach D, Stelzer C, Boryń ŁM, Rath M, Stark A. Genome-wide quantitative enhancer activity maps identified by STARR-seq. Science. 2013; 339:1074-1077. DOI | PubMed
- Bai L, Morozov AV. Gene regulation by nucleosome positioning. Trends in Genetics. 2010; 26:476-483. DOI | PubMed
- Bardet AF, Steinmann J, Bafna S, Knoblich JA, Zeitlinger J, Stark A. Identification of transcription factor binding sites from ChIP-seq data at high resolution. Bioinformatics. 2013; 29:2705-2713. DOI | PubMed
- Barraza A, Cabrera-Ponce JL, Gamboa-Becerra R, Luna-Martínez F, Winkler R, Álvarez-Venegas R. The Phaseolus vulgaris PvTRX1h gene regulates plant hormone biosynthesis in embryogenic callus from common bean. Frontiers in Plant Science. 2015; 6DOI | PubMed
- Barski A, Cuddapah S, Cui K, Roh TY, Schones DE, Wang Z, Wei G, Chepelev I, Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007; 129:823-837. DOI | PubMed
- Birnbaum RY, Clowney EJ, Agamy O, Kim MJ, Zhao J, Yamanaka T, Pappalardo Z, Clarke SL, Wenger AM, Nguyen L, Gurrieri F, Everman DB, Schwartz CE, Birk OS, Bejerano G, Lomvardas S, Ahituv N. Coding exons function as tissue-specific enhancers of nearby genes. Genome Research. 2012; 22:1059-1068. DOI | PubMed
- Boeger H, Griesenbeck J, Strattan JS, Kornberg RD. Nucleosomes unfold completely at a transcriptionally active promoter. Molecular Cell. 2003; 11:1587-1598. DOI | PubMed
- Bogdanovic O, Fernandez-Miñán A, Tena JJ, de la Calle-Mustienes E, Hidalgo C, van Kruysbergen I, van Heeringen SJ, Veenstra GJ, Gómez-Skarmeta JL. Dynamics of enhancer chromatin signatures mark the transition from pluripotency to cell specification during embryogenesis. Genome Research. 2012; 22:2043-2053. DOI | PubMed
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014; 30:2114-2120. DOI | PubMed
- Boyer LA, Plath K, Zeitlinger J, Brambrink T, Medeiros LA, Lee TI, Levine SS, Wernig M, Tajonar A, Ray MK, Bell GW, Otte AP, Vidal M, Gifford DK, Young RA, Jaenisch R. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature. 2006; 441:349-353. DOI | PubMed
- Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nature Biotechnology. 2016; 34:525-527. DOI | PubMed
- Bråte J, Adamski M, Neumann RS, Shalchian-Tabrizi K, Adamska M. Regulatory RNA at the root of animals: dynamic expression of developmental lincRNAs in the calcisponge Sycon ciliatum. Proceedings. Biological Sciences. 2015; 282DOI | PubMed
- Brookes E, Pombo A. Modifications of RNA polymerase II are pivotal in regulating gene expression states. EMBO Reports. 2009; 10:1213-1219. DOI | PubMed
- Brown JL, Grau DJ, DeVido SK, Kassis JA. An Sp1/KLF binding site is important for the activity of a polycomb group response element from the Drosophila engrailed gene. Nucleic Acids Research. 2005; 33:5181-5189. DOI | PubMed
- Bulger M, Groudine M. Functional and mechanistic diversity of distal transcription enhancers. Cell. 2011; 144:327-339. DOI | PubMed
- Capella-Gutiérrez S, Silla-Martínez JM, Gabaldón T. trimAl: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics. 2009; 25:1972-1973. DOI | PubMed
- Chen RA, Down TA, Stempor P, Chen QB, Egelhofer TA, Hillier LW, Jeffers TE, Ahringer J. The landscape of RNA polymerase II transcription initiation in C. elegans reveals promoter and enhancer architectures. Genome Research. 2013; 23:1339-1347. DOI | PubMed
- Conaco C, Neveu P, Zhou H, Arcila ML, Degnan SM, Degnan BM, Kosik KS. Transcriptome profiling of the demosponge Amphimedon queenslandica reveals genome-wide events that accompany major life cycle transitions. BMC Genomics. 2012; 13DOI | PubMed
- Connolly LR, Smith KM, Freitag M. The Fusarium graminearum histone H3 K27 methyltransferase KMT6 regulates development and expression of secondary metabolite gene clusters. PLoS Genetics. 2013; 9DOI | PubMed
- Davidson EH, Peter IS. Genomic Control Process. Oxford: Academic Press; 2015.
- de Mendoza A, Sebé-Pedrós A, Šestak MS, Matejcic M, Torruella G, Domazet-Loso T, Ruiz-Trillo I. Transcription factor evolution in eukaryotes and the assembly of the regulatory toolkit in multicellular lineages. PNAS. 2013; 110:E4858-E4866. DOI | PubMed
- de Mendoza A, Suga H, Permanyer J, Irimia M, Ruiz-Trillo I. Complex transcriptional regulation and independent evolution of fungal-like traits in a relative of animals. eLife. 2015; 4DOI | PubMed
- De Santa F, Barozzi I, Mietton F, Ghisletti S, Polletti S, Tusi BK, Muller H, Ragoussis J, Wei CL, Natoli G. A large fraction of extragenic RNA pol II transcription sites overlap enhancers. PLoS Biology. 2010; 8DOI | PubMed
- Degnan BM, Adamska M, Richards GS, Larroux C, Leininger S, Bergum B. Evolutionary Developmental Biology of Invertebrates 1: Introduction, Non-Bilateria, Acoelomorpha, Xenoturbellida, Chaetognatha. Springer: Vienna, Austria; 2015.
- Degnan BM, Vervoort M, Larroux C, Richards GS. Early evolution of metazoan transcription factors. Current Opinion in Genetics & Development. 2009; 19:591-599. DOI | PubMed
- Deng W, Buzas DM, Ying H, Robertson M, Taylor J, Peacock WJ, Dennis ES, Helliwell C. Arabidopsis polycomb repressive complex 2 binding sites contain putative GAGA factor binding motifs within coding regions of genes. BMC Genomics. 2013; 14DOI | PubMed
- Di Croce L, Helin K. Transcriptional regulation by Polycomb group proteins. Nature Structural & Molecular Biology. 2013; 20:1147-1155. DOI | PubMed
- Djebali S, Davis CA, Merkel A, Dobin A, Lassmann T, Mortazavi A, Tanzer A, Lagarde J, Lin W, Schlesinger F, Xue C, Marinov GK, Khatun J, Williams BA, Zaleski C, Rozowsky J, Röder M, Kokocinski F, Abdelhamid RF, Alioto T, Antoshechkin I, Baer MT, Bar NS, Batut P, Bell K, Bell I, Chakrabortty S, Chen X, Chrast J, Curado J, Derrien T, Drenkow J, Dumais E, Dumais J, Duttagupta R, Falconnet E, Fastuca M, Fejes-Toth K, Ferreira P, Foissac S, Fullwood MJ, Gao H, Gonzalez D, Gordon A, Gunawardena H, Howald C, Jha S, Johnson R, Kapranov P, King B, Kingswood C, Luo OJ, Park E, Persaud K, Preall JB, Ribeca P, Risk B, Robyr D, Sammeth M, Schaffer L, See LH, Shahab A, Skancke J, Suzuki AM, Takahashi H, Tilgner H, Trout D, Walters N, Wang H, Wrobel J, Yu Y, Ruan X, Hayashizaki Y, Harrow J, Gerstein M, Hubbard T, Reymond A, Antonarakis SE, Hannon G, Giddings MC, Ruan Y, Wold B, Carninci P, Guigó R, Gingeras TR. Landscape of transcription in human cells. Nature. 2012; 489:101-108. DOI | PubMed
- Duan Z, Andronescu M, Schutz K, McIlwain S, Kim YJ, Lee C, Shendure J, Fields S, Blau CA, Noble WS. A three-dimensional model of the yeast genome. Nature. 2010; 465:363-367. DOI | PubMed
- Eckalbar WL, Schlebusch SA, Mason MK, Gill Z, Parker AV, Booker BM, Nishizaki S, Muswamba-Nday C, Terhune E, Nevonen KA, Makki N, Friedrich T, VanderMeer JE, Pollard KS, Carbone L, Wall JD, Illing N, Ahituv N. Transcriptomic and epigenomic characterization of the developing bat wing. Nature Genetics. 2016; 48:528-536. DOI | PubMed
- Edgar R, Domrachev M, Lash AE. Gene expression omnibus: ncbi gene expression and hybridization array data Repository. Nucleic Acids Research. 2002; 30:207-210. DOI | PubMed
- ENCODE Project Consortium. An integrated encyclopedia of DNA elements in the human genome. Nature. 2012; 489:57-74. DOI | PubMed
- Engström PG, Ho Sui SJ, Drivenes O, Becker TS, Lenhard B. Genomic regulatory blocks underlie extensive microsynteny conservation in insects. Genome Research. 2007; 17:1898-1908. DOI | PubMed
- Ercan S, Dick LL, Lieb JD. The C. elegans dosage compensation complex propagates dynamically and independently of X chromosome sequence. Current Biology. 2009; 19:1777-1787. DOI | PubMed
- Ernst J, Kellis M. ChromHMM: automating chromatin-state discovery and characterization. Nature Methods. 2012; 9:215-216. DOI | PubMed
- Erwin DH, Laflamme M, Tweedt SM, Sperling EA, Pisani D, Peterson KJ. The Cambrian conundrum: early divergence and later ecological success in the early history of animals. Science. 2011; 334:1091-1097. DOI | PubMed
- Fernandez-Valverde SL, Calcino AD, Degnan BM. Deep developmental transcriptome sequencing uncovers numerous new genes and enhances gene annotation in the sponge Amphimedon queenslandica. BMC Genomics. 2015; 16DOI | PubMed
- Fernandez-Valverde SL, Degnan BM. Bilaterian-like promoters in the highly compact Amphimedon queenslandica genome. Scientific Reports. 2016; 6DOI | PubMed
- Fortunato SA, Adamski M, Adamska M. Comparative analyses of developmental transcription factor repertoires in sponges reveal unexpected complexity of the earliest animals. Marine Genomics. 2015; 24:121-129. DOI | PubMed
- Fortunato SA, Adamski M, Ramos OM, Leininger S, Liu J, Ferrier DE, Adamska M. Calcisponges have a ParaHox gene and dynamic expression of dispersed NK homeobox genes. Nature. 2014; 514:620-623. DOI | PubMed
- Gaiti F, Calcino AD, Tanurdžić M, Degnan BM. Origin and evolution of the metazoan non-coding regulatory genome. Developmental Biology. 2016; pii: S0012- 1606:30573-30575. DOI | PubMed
- Gaiti F, Fernandez-Valverde SL, Nakanishi N, Calcino AD, Yanai I, Tanurdzic M, Degnan BM. Dynamic and widespread lncRNA expression in a sponge and the origin of animal complexity. Molecular Biology and Evolution. 2015; 32:2367-2382. DOI | PubMed
- Gaydos LJ, Wang W, Strome S. Gene repression. H3K27me and PRC2 transmit a memory of repression across generations and during development. Science. 2014; 345:1515-1518. DOI | PubMed
- Gerstein MB, Lu ZJ, Van Nostrand EL, Cheng C, Arshinoff BI, Liu T, Yip KY, Robilotto R, Rechtsteiner A, Ikegami K, Alves P, Chateigner A, Perry M, Morris M, Auerbach RK, Feng X, Leng J, Vielle A, Niu W, Rhrissorrakrai K, Agarwal A, Alexander RP, Barber G, Brdlik CM, Brennan J, Brouillet JJ, Carr A, Cheung MS, Clawson H, Contrino S, Dannenberg LO, Dernburg AF, Desai A, Dick L, Dosé AC, Du J, Egelhofer T, Ercan S, Euskirchen G, Ewing B, Feingold EA, Gassmann R, Good PJ, Green P, Gullier F, Gutwein M, Guyer MS, Habegger L, Han T, Henikoff JG, Henz SR, Hinrichs A, Holster H, Hyman T, Iniguez AL, Janette J, Jensen M, Kato M, Kent WJ, Kephart E, Khivansara V, Khurana E, Kim JK, Kolasinska-Zwierz P, Lai EC, Latorre I, Leahey A, Lewis S, Lloyd P, Lochovsky L, Lowdon RF, Lubling Y, Lyne R, MacCoss M, Mackowiak SD, Mangone M, McKay S, Mecenas D, Merrihew G, Miller DM, Muroyama A, Murray JI, Ooi SL, Pham H, Phippen T, Preston EA, Rajewsky N, Rätsch G, Rosenbaum H, Rozowsky J, Rutherford K, Ruzanov P, Sarov M, Sasidharan R, Sboner A, Scheid P, Segal E, Shin H, Shou C, Slack FJ, Slightam C, Smith R, Spencer WC, Stinson EO, Taing S, Takasaki T, Vafeados D, Voronina K, Wang G, Washington NL, Whittle CM, Wu B, Yan KK, Zeller G, Zha Z, Zhong M, Zhou X, Ahringer J, Strome S, Gunsalus KC, Micklem G, Liu XS, Reinke V, Kim SK, Hillier LW, Henikoff S, Piano F, Snyder M, Stein L, Lieb JD, Waterston RH, modENCODE Consortium. Integrative analysis of the Caenorhabditis elegans genome by the modENCODE project. Science. 2010; 330:1775-1787. DOI | PubMed
- Grimson A, Srivastava M, Fahey B, Woodcroft BJ, Chiang HR, King N, Degnan BM, Rokhsar DS, Bartel DP. Early origins and evolution of microRNAs and Piwi-interacting RNAs in animals. Nature. 2008; 455:1193-1197. DOI | PubMed
- Ha M, Ng DW, Li WH, Chen ZJ. Coordinated histone modifications are associated with gene expression variation within and between species. Genome Research. 2011; 21:590-598. DOI | PubMed
- Harmeyer KM, South PF, Bishop B, Ogas J, Briggs SD. Immediate chromatin immunoprecipitation and on-bead quantitative PCR analysis: a versatile and rapid ChIP procedure. Nucleic Acids Research. 2015; 43DOI | PubMed
- Hashimshony T, Wagner F, Sher N, Yanai I. CEL-Seq: single-cell RNA-Seq by multiplexed linear amplification. Cell Reports. 2012; 2:666-673. DOI | PubMed
- Hecker A, Brand L H, Peter S, Simoncello N, Kilian J, Harter K. The Arabidopsis GAGA-binding factor BPC6 recruits PRC1 component LHP1 to GAGA DNA-motifs. Plant Physiology. 2015; 168:1013-1024. DOI | PubMed
- Heger P, Marin B, Bartkuhn M, Schierenberg E, Wiehe T. The chromatin insulator CTCF and the emergence of metazoan diversity. PNAS. 2012; 109:17507-17512. DOI | PubMed
- Heintzman ND, Hon GC, Hawkins RD, Kheradpour P, Stark A, Harp LF, Ye Z, Lee LK, Stuart RK, Ching CW, Ching KA, Antosiewicz-Bourget JE, Liu H, Zhang X, Green RD, Lobanenkov VV, Stewart R, Thomson JA, Crawford GE, Kellis M, Ren B. Histone modifications at human enhancers reflect global cell-type-specific gene expression. Nature. 2009; 459:108-112. DOI | PubMed
- Helm RR, Siebert S, Tulin S, Smith J, Dunn CW. Characterization of differential transcript abundance through time during Nematostella vectensis development. BMC Genomics. 2013; 14:266-10. DOI | PubMed
- Hezroni H, Koppstein D, Schwartz MG, Avrutin A, Bartel DP, Ulitsky I, Schwartz Matthew G, Bartel David P. Principles of long noncoding RNA evolution derived from direct comparison of transcriptomes in 17 species. Cell Reports. 2015; 11:1110-1122. DOI | PubMed
- Ho JW, Jung YL, Liu T, Alver BH, Lee S, Ikegami K, Sohn KA, Minoda A, Tolstorukov MY, Appert A, Parker SC, Gu T, Kundaje A, Riddle NC, Bishop E, Egelhofer TA, Hu SS, Alekseyenko AA, Rechtsteiner A, Asker D, Belsky JA, Bowman SK, Chen QB, Chen RA, Day DS, Dong Y, Dose AC, Duan X, Epstein CB, Ercan S, Feingold EA, Ferrari F, Garrigues JM, Gehlenborg N, Good PJ, Haseley P, He D, Herrmann M, Hoffman MM, Jeffers TE, Kharchenko PV, Kolasinska-Zwierz P, Kotwaliwale CV, Kumar N, Langley SA, Larschan EN, Latorre I, Libbrecht MW, Lin X, Park R, Pazin MJ, Pham HN, Plachetka A, Qin B, Schwartz YB, Shoresh N, Stempor P, Vielle A, Wang C, Whittle CM, Xue H, Kingston RE, Kim JH, Bernstein BE, Dernburg AF, Pirrotta V, Kuroda MI, Noble WS, Tullius TD, Kellis M, MacAlpine DM, Strome S, Elgin SC, Liu XS, Lieb JD, Ahringer J, Karpen GH, Park PJ. Comparative analysis of metazoan chromatin organization. Nature. 2014; 512:449-452. DOI | PubMed
- IIott NE, Heward JA, Roux B, Tsitsiou E, Fenwick PS, Lenzi L, Goodhead I, Hertz-Fowler C, Heger A, Hall N, Donnelly LE, Sims D, Lindsay MA. Long non-coding RNAs and enhancer RNAs regulate the lipopolysaccharide-induced inflammatory response in human monocytes. Nature Communications. 2014; 5DOI | PubMed
- Ikeuchi M, Iwase A, Rymen B, Harashima H, Shibata M, Ohnuma M, Breuer C, Morao AK, de Lucas M, De Veylder L, Goodrich J, Brady SM, Roudier F, Sugimoto K. PRC2 represses dedifferentiation of mature somatic cells in Arabidopsis. Nature Plants. 2015; 1DOI | PubMed
- Irimia M, Maeso I, Burguera D, Hidalgo-Sánchez M, Puelles L, Roy SW, Garcia-Fernàndez J, Ferran JL. Contrasting 5′ and 3′ evolutionary histories and frequent evolutionary convergence in Meis/hth gene structures. Genome Biology and Evolution. 2011; 3:551-564. DOI | PubMed
- Irimia M, Maeso I, Roy SW, Fraser HB. Ancient cis-regulatory constraints and the evolution of genome architecture. Trends in Genetics. 2013; 29:521-528. DOI | PubMed
- Irimia M, Tena JJ, Alexis MS, Fernandez-Miñan A, Maeso I, Bogdanovic O, de la Calle-Mustienes E, Roy SW, Gómez-Skarmeta JL, Fraser HB. Extensive conservation of ancient microsynteny across metazoans due to cis-regulatory constraints. Genome Research. 2012; 22:2356-2367. DOI | PubMed
- Jamieson K, Rountree MR, Lewis ZA, Stajich JE, Selker EU. Regional control of histone H3 lysine 27 methylation in Neurospora. PNAS. 2013; 110:6027-6032. DOI | PubMed
- Jiang C, Pugh BF. Nucleosome positioning and gene regulation: advances through genomics. Nature Reviews Genetics. 2009; 10:161-172. DOI | PubMed
- Jindrich K, Degnan BM. The diversification of the basic leucine zipper family in eukaryotes correlates with the evolution of multicellularity. BMC Evolutionary Biology. 2016; 16:1-12. DOI | PubMed
- Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, Pesseat S, Quinn AF, Sangrador-Vegas A, Scheremetjew M, Yong SY, Lopez R, Hunter S. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014; 30:1236-1240. DOI | PubMed
- Kanehisa M, Sato Y, Morishima K. BlastKOALA and GhostKOALA: kegg tools for functional characterization of genome and metagenome sequences. Journal of Molecular Biology. 2016; 428:726-731. DOI | PubMed
- Karlić R, Chung HR, Lasserre J, Vlahovicek K, Vingron M. Histone modification levels are predictive for gene expression. PNAS. 2010; 107:2926-2931. DOI | PubMed
- Kassis JA, Brown JL. Polycomb group response elements in Drosophila and vertebrates. Advances in Genetics. 2013; 81:83-118. DOI | PubMed
- Katoh K, Standley DM. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Molecular Biology and Evolution. 2013; 30:772-780. DOI | PubMed
- Kellis M, Wold B, Snyder MP, Bernstein BE, Kundaje A, Marinov GK, Ward LD, Birney E, Crawford GE, Dekker J, Dunham I, Elnitski LL, Farnham PJ, Feingold EA, Gerstein M, Giddings MC, Gilbert DM, Gingeras TR, Green ED, Guigo R, Hubbard T, Kent J, Lieb JD, Myers RM, Pazin MJ, Ren B, Stamatoyannopoulos JA, Weng Z, White KP, Hardison RC. Defining functional DNA elements in the human genome. PNAS. 2014; 111:6131-6138. DOI | PubMed
- Kharchenko PV, Tolstorukov MY, Park PJ. Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nature Biotechnology. 2008; 26:1351-1359. DOI | PubMed
- Kikuta H, Laplante M, Navratilova P, Komisarczuk AZ, Engström PG, Fredman D, Akalin A, Caccamo M, Sealy I, Howe K, Ghislain J, Pezeron G, Mourrain P, Ellingsen S, Oates AC, Thisse C, Thisse B, Foucher I, Adolf B, Geling A, Lenhard B, Becker TS. Genomic regulatory blocks encompass multiple neighboring genes and maintain conserved synteny in vertebrates. Genome Research. 2007; 17:545-555. DOI | PubMed
- Kim TK, Hemberg M, Gray JM, Costa AM, Bear DM, Wu J, Harmin DA, Laptewicz M, Barbara-Haley K, Kuersten S, Markenscoff-Papadimitriou E, Kuhl D, Bito H, Worley PF, Kreiman G, Greenberg ME. Widespread transcription at neuronal activity-regulated enhancers. Nature. 2010; 465:182-187. DOI | PubMed
- King N, Hittinger CT, Carroll SB. Evolution of key cell signaling and adhesion protein families predates animal origins. Science. 2003; 301:361-363. DOI | PubMed
- King N, Westbrook MJ, Young SL, Kuo A, Abedin M, Chapman J, Fairclough S, Hellsten U, Isogai Y, Letunic I, Marr M, Pincus D, Putnam N, Rokas A, Wright KJ, Zuzow R, Dirks W, Good M, Goodstein D, Lemons D, Li W, Lyons JB, Morris A, Nichols S, Richter DJ, Salamov A, Sequencing JG, Bork P, Lim WA, Manning G, Miller WT, McGinnis W, Shapiro H, Tjian R, Grigoriev IV, Rokhsar D. The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans. Nature. 2008; 451:783-788. DOI | PubMed
- Kowalczyk MS, Hughes JR, Garrick D, Lynch MD, Sharpe JA, Sloane-Stanley JA, McGowan SJ, De Gobbi M, Hosseini M, Vernimmen D, Brown JM, Gray NE, Collavin L, Gibbons RJ, Flint J, Taylor S, Buckle VJ, Milne TA, Wood WG, Higgs DR, Lynch Magnus D, Sharpe Jacqueline A, Sloane-Stanley Jacqueline A. Intragenic enhancers act as alternative promoters. Molecular Cell. 2012; 45:447-458. DOI | PubMed
- Kuhn RM, Haussler D, Kent WJ. The UCSC genome browser and associated tools. Briefings in Bioinformatics. 2013; 14:144-161. DOI | PubMed
- Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, Ziller MJ, Amin V, Whitaker JW, Schultz MD, Ward LD, Sarkar A, Quon G, Sandstrom RS, Eaton ML, Wu YC, Pfenning AR, Wang X, Claussnitzer M, Liu Y, Coarfa C, Harris RA, Shoresh N, Epstein CB, Gjoneska E, Leung D, Xie W, Hawkins RD, Lister R, Hong C, Gascard P, Mungall AJ, Moore R, Chuah E, Tam A, Canfield TK, Hansen RS, Kaul R, Sabo PJ, Bansal MS, Carles A, Dixon JR, Farh KH, Feizi S, Karlic R, Kim AR, Kulkarni A, Li D, Lowdon R, Elliott G, Mercer TR, Neph SJ, Onuchic V, Polak P, Rajagopal N, Ray P, Sallari RC, Siebenthall KT, Sinnott-Armstrong NA, Stevens M, Thurman RE, Wu J, Zhang B, Zhou X, Beaudet AE, Boyer LA, De Jager PL, Farnham PJ, Fisher SJ, Haussler D, Jones SJ, Li W, Marra MA, McManus MT, Sunyaev S, Thomson JA, Tlsty TD, Tsai LH, Wang W, Waterland RA, Zhang MQ, Chadwick LH, Bernstein BE, Costello JF, Ecker JR, Hirst M, Meissner A, Milosavljevic A, Ren B, Stamatoyannopoulos JA, Wang T, Kellis M, Roadmap Epigenomics Consortium. Integrative analysis of 111 reference human epigenomes. Nature. 2015; 518:317-330. DOI | PubMed
- Landt SG, Marinov GK, Kundaje A, Kheradpour P, Pauli F, Batzoglou S, Bernstein BE, Bickel P, Brown JB, Cayting P, Chen Y, DeSalvo G, Epstein C, Fisher-Aylor KI, Euskirchen G, Gerstein M, Gertz J, Hartemink AJ, Hoffman MM, Iyer VR, Jung YL, Karmakar S, Kellis M, Kharchenko PV, Li Q, Liu T, Liu XS, Ma L, Milosavljevic A, Myers RM, Park PJ, Pazin MJ, Perry MD, Raha D, Reddy TE, Rozowsky J, Shoresh N, Sidow A, Slattery M, Stamatoyannopoulos JA, Tolstorukov MY, White KP, Xi S, Farnham PJ, Lieb JD, Wold BJ, Snyder M. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Research. 2012; 22:1813-1831. DOI | PubMed
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biology. 2009; 10DOI | PubMed
- Larroux C, Fahey B, Liubicich D, Hinman VF, Gauthier M, Gongora M, Green K, Wörheide G, Leys SP, Degnan BM. Developmental expression of transcription factor genes in a demosponge: insights into the origin of metazoan multicellularity. Evolution &Development. 2006; 8:150-173. DOI | PubMed
- Larroux C, Luke GN, Koopman P, Rokhsar DS, Shimeld SM, Degnan BM. Genesis and expansion of metazoan transcription factor gene classes. Molecular Biology and Evolution. 2008; 25:980-996. DOI | PubMed
- Lee BK, Iyer VR. Genome-wide studies of CCCTC-binding factor (CTCF) and cohesin provide insight into chromatin structure and regulation. Journal of Biological Chemistry. 2012; 287:30906-30913. DOI | PubMed
- Leininger S, Adamski M, Bergum B, Guder C, Liu J, Laplante M, Bråte J, Hoffmann F, Fortunato S, Jordal S, Rapp HT, Adamska M. Developmental gene expression provides clues to relationships between sponge and eumetazoan body plans. Nature Communications. 2014; 5DOI | PubMed
- Lenhard B, Sandelin A, Carninci P. Metazoan promoters: emerging characteristics and insights into transcriptional regulation. Nature Reviews Genetics. 2012; 13:233-245. DOI | PubMed
- Levin M, Anavy L, Cole AG, Winter E, Mostov N, Khair S, Senderovich N, Kovalev E, Silver DH, Feder M, Fernandez-Valverde SL, Nakanishi N, Simmons D, Simakov O, Larsson T, Liu SY, Jerafi-Vider A, Yaniv K, Ryan JF, Martindale MQ, Rink JC, Arendt D, Degnan SM, Degnan BM, Hashimshony T, Yanai I. The mid-developmental transition and the evolution of animal body plans. Nature. 2016; 531:637-641. DOI | PubMed
- Levine M, Cattoglio C, Tjian R. Looping back to leap forward: transcription enters a new era. Cell. 2014; 157:13-25. DOI | PubMed
- Levine M, Tjian R. Transcription regulation and animal diversity. Nature. 2003; 424:147-151. DOI | PubMed
- Levine M. Transcriptional enhancers in animal development and evolution. Current Biology. 2010; 20:R754-R763. DOI | PubMed
- Leys SP, Degnan BM. Embryogenesis and metamorphosis in a haplosclerid demosponge: gastrulation and transdifferentiation of larval ciliated cells to choanocytes. Invertebrate Biology. 2002; 121:171-189. DOI
- Leys SP, Larroux C, Gauthier M, Adamska M, Fahey B, Richards GS, Degnan SM, Degnan BM. Isolation of Amphimedon developmental material. Cold Spring Harbor Protocols. 2008; 2008DOI | PubMed
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. The sequence alignment/Map format and SAMtools. Bioinformatics. 2009; 25:2078-2079. DOI | PubMed
- Li W, Notani D, Rosenfeld MG. Enhancers as non-coding RNA transcription units: recent insights and future perspectives. Nature Reviews Genetics. 2016; 17:207-223. DOI | PubMed
- Liang X, Song MR, Xu Z, Lanuza GM, Liu Y, Zhuang T, Chen Y, Pfaff SL, Evans SM, Sun Y. Isl1 is required for multiple aspects of motor neuron development. Molecular and Cellular Neuroscience. 2011; 47:215-222. DOI | PubMed
- Liu Y, Taverna SD, Muratore TL, Shabanowitz J, Hunt DF, Allis CD. RNAi-dependent H3K27 methylation is required for heterochromatin formation and DNA elimination in Tetrahymena. Genes & Development. 2007; 21:1530-1545. DOI | PubMed
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology. 2014; 15:1-21. DOI | PubMed
- Machanick P, Bailey TL. MEME-ChIP: motif analysis of large DNA datasets. Bioinformatics. 2011; 27:1696-1697. DOI | PubMed
- Maere S, Heymans K, Kuiper M. BiNGO: a cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics. 2005; 21:3448-3449. DOI | PubMed
- Margueron R, Reinberg D. The polycomb complex PRC2 and its mark in life. Nature. 2011; 469:343-349. DOI | PubMed
- Marques AC, Hughes J, Graham B, Kowalczyk MS, Higgs DR, Ponting CP. Chromatin signatures at transcriptional start sites separate two equally populated yet distinct classes of intergenic long noncoding RNAs. Genome Biology. 2013; 14DOI | PubMed
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011; 17:10-12. DOI
- Merkenschlager M, Odom DT. CTCF and cohesin: linking gene regulatory elements with their targets. Cell. 2013; 152:1285-1297. DOI | PubMed
- Moroz LL, Kocot KM, Citarella MR, Dosung S, Norekian TP, Povolotskaya IS, Grigorenko AP, Dailey C, Berezikov E, Buckley KM, Ptitsyn A, Reshetov D, Mukherjee K, Moroz TP, Bobkova Y, Yu F, Kapitonov VV, Jurka J, Bobkov YV, Swore JJ, Girardo DO, Fodor A, Gusev F, Sanford R, Bruders R, Kittler E, Mills CE, Rast JP, Derelle R, Solovyev VV, Kondrashov FA, Swalla BJ, Sweedler JV, Rogaev EI, Halanych KM, Kohn AB. The ctenophore genome and the evolutionary origins of neural systems. Nature. 2014; 510:109-114. DOI | PubMed
- Müller J, Kassis JA. Polycomb response elements and targeting of Polycomb group proteins in Drosophila. Current Opinion in Genetics & Development. 2006; 16:476-484. DOI | PubMed
- Nakanishi N, Sogabe S, Degnan BM. Evolutionary origin of gastrulation: insights from sponge development. BMC Biology. 2014; 12DOI | PubMed
- Natoli G, Andrau JC. Noncoding transcription at enhancers: general principles and functional models. Annual Review of Genetics. 2012; 46:1-19. DOI | PubMed
- Naville M, Ishibashi M, Ferg M, Bengani H, Rinkwitz S, Krecsmarik M, Hawkins TA, Wilson SW, Manning E, Chilamakuri CS, Wilson DI, Louis A, Lucy Raymond F, Rastegar S, Strähle U, Lenhard B, Bally-Cuif L, van Heyningen V, FitzPatrick DR, Becker TS, Roest Crollius H. Long-range evolutionary constraints reveal cis-regulatory interactions on the human X chromosome. Nature Communications. 2015; 6DOI | PubMed
- Nègre N, Brown CD, Ma L, Bristow CA, Miller SW, Wagner U, Kheradpour P, Eaton ML, Loriaux P, Sealfon R, Li Z, Ishii H, Spokony RF, Chen J, Hwang L, Cheng C, Auburn RP, Davis MB, Domanus M, Shah PK, Morrison CA, Zieba J, Suchy S, Senderowicz L, Victorsen A, Bild NA, Grundstad AJ, Hanley D, MacAlpine DM, Mannervik M, Venken K, Bellen H, White R, Gerstein M, Russell S, Grossman RL, Ren B, Posakony JW, Kellis M, White KP. A cis-regulatory map of the Drosophila genome. Nature. 2011; 471:527-531. DOI | PubMed
- Peter IS, Davidson EH. Evolution of gene regulatory networks controlling body plan development. Cell. 2011; 144:970-985. DOI | PubMed
- Pérez-Lluch S, Blanco E, Tilgner H, Curado J, Ruiz-Romero M, Corominas M, Guigó R. Absence of canonical marks of active chromatin in developmentally regulated genes. Nature Genetics. 2015a; 47:1158-1167. DOI | PubMed
- Pérez-Lluch S, Guigó R, Corominas M. Active transcription without histone modifications. Oncotarget. 2015b; 6DOI | PubMed
- Price MN, Dehal PS, Arkin AP. FastTree 2–approximately maximum-likelihood trees for large alignments. PLoS One. 2010; 5DOI | PubMed
- Putnam NH, Srivastava M, Hellsten U, Dirks B, Chapman J, Salamov A, Terry A, Shapiro H, Lindquist E, Kapitonov VV, Jurka J, Genikhovich G, Grigoriev IV, Lucas SM, Steele RE, Finnerty JR, Technau U, Martindale MQ, Rokhsar DS. Sea Anemone genome reveals ancestral eumetazoan gene repertoire and genomic organization. Science. 2007; 317:86-94. DOI | PubMed
- Quinlan AR, Hall IM. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010; 26:841-842. DOI | PubMed
- Quinn JJ, Chang HY. Unique features of long non-coding RNA biogenesis and function. Nature Reviews Genetics. 2016; 17:47-62. DOI | PubMed
- R Core Team. R Foundation for Statistical Computing: Vienna, Austria; 2014.
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, Manke T. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Research. 2016; 44:W160-W165. DOI | PubMed
- Richards GS, Degnan BM. The dawn of developmental signaling in the metazoa. Cold Spring Harbor Symposia on Quantitative Biology. 2009; 74:81-90. DOI | PubMed
- Richards GS, Simionato E, Perron M, Adamska M, Vervoort M, Degnan BM. Sponge genes provide new insight into the evolutionary origin of the neurogenic circuit. Current Biology. 2008; 18:1156-1161. DOI | PubMed
- Richter DJ, King N. The genomic and cellular foundations of animal origins. Annual Review of Genetics. 2013; 47:509-537. DOI | PubMed
- Riesgo A, Farrar N, Windsor PJ, Giribet G, Leys SP. The analysis of eight transcriptomes from all poriferan classes reveals surprising genetic complexity in sponges. Molecular Biology and Evolution. 2014; 31:1102-1120. DOI | PubMed
- Ritter DI, Dong Z, Guo S, Chuang JH. Transcriptional enhancers in protein-coding exons of vertebrate developmental genes. PLoS One. 2012; 7DOI | PubMed
- Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, Thiessen N, Griffith OL, He A, Marra M, Snyder M, Jones S. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nature Methods. 2007; 4:651-657. DOI | PubMed
- Roy S, Ernst J, Kharchenko PV, Kheradpour P, Negre N, Eaton ML, Landolin JM, Bristow CA, Ma L, Lin MF, Washietl S, Arshinoff BI, Ay F, Meyer PE, Robine N, Washington NL, Di Stefano L, Berezikov E, Brown CD, Candeias R, Carlson JW, Carr A, Jungreis I, Marbach D, Sealfon R, Tolstorukov MY, Will S, Alekseyenko AA, Artieri C, Booth BW, Brooks AN, Dai Q, Davis CA, Duff MO, Feng X, Gorchakov AA, Gu T, Henikoff JG, Kapranov P, Li R, MacAlpine HK, Malone J, Minoda A, Nordman J, Okamura K, Perry M, Powell SK, Riddle NC, Sakai A, Samsonova A, Sandler JE, Schwartz YB, Sher N, Spokony R, Sturgill D, van Baren M, Wan KH, Yang L, Yu C, Feingold E, Good P, Guyer M, Lowdon R, Ahmad K, Andrews J, Berger B, Brenner SE, Brent MR, Cherbas L, Elgin SC, Gingeras TR, Grossman R, Hoskins RA, Kaufman TC, Kent W, Kuroda MI, Orr-Weaver T, Perrimon N, Pirrotta V, Posakony JW, Ren B, Russell S, Cherbas P, Graveley BR, Lewis S, Micklem G, Oliver B, Park PJ, Celniker SE, Henikoff S, Karpen GH, Lai EC, MacAlpine DM, Stein LD, White KP, Kellis M, modENCODE Consortium. Identification of functional elements and regulatory circuits by Drosophila modENCODE. Science. 2010; 330:1787-1797. DOI | PubMed
- Ryan JF, Pang K, Schnitzler CE, Nguyen AD, Moreland RT, Simmons DK, Koch BJ, Francis WR, Havlak P, Smith SA, Putnam NH, Haddock SH, Dunn CW, Wolfsberg TG, Mullikin JC, Martindale MQ, Baxevanis AD, NISC Comparative Sequencing Program. The genome of the ctenophore Mnemiopsis leidyi and its implications for cell type evolution. Science. 2013; 342DOI | PubMed
- Sandelin A, Bailey P, Bruce S, Engström PG, Klos JM, Wasserman WW, Ericson J, Lenhard B. Arrays of ultraconserved non-coding regions span the loci of key developmental genes in vertebrate genomes. BMC Genomics. 2004; 5:99-9. DOI | PubMed
- Schwaiger M, Schönauer A, Rendeiro AF, Pribitzer C, Schauer A, Gilles AF, Schinko JB, Renfer E, Fredman D, Technau U. Evolutionary conservation of the eumetazoan gene regulatory landscape. Genome Research. 2014; 24:639-650. DOI | PubMed
- Sebé-Pedrós A, Ballaré C, Parra-Acero H, Chiva C, Tena JJ, Sabidó E, Gómez-Skarmeta JL, Di Croce L, Ruiz-Trillo I. The dynamic regulatory genome of Capsaspora and the origin of animal multicellularity. Cell. 2016; 165:1224-1237. DOI | PubMed
- Sebé-Pedrós A, de Mendoza A, Lang BF, Degnan BM, Ruiz-Trillo I. Unexpected repertoire of metazoan transcription factors in the unicellular holozoan Capsaspora owczarzaki. Molecular Biology and Evolution. 2011; 28:1241-1254. DOI | PubMed
- Seitan VC, Faure AJ, Zhan Y, McCord RP, Lajoie BR, Ing-Simmons E, Lenhard B, Giorgetti L, Heard E, Fisher AG, Flicek P, Dekker J, Merkenschlager M. Cohesin-based chromatin interactions enable regulated gene expression within preexisting architectural compartments. Genome Research. 2013; 23:2066-2077. DOI | PubMed
- Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Research. 2003; 13:2498-2504. DOI | PubMed
- Shaver S, Casas-Mollano JA, Cerny RL, Cerutti H. Origin of the polycomb repressive complex 2 and gene silencing by an E(z) homolog in the unicellular alga Chlamydomonas. Epigenetics. 2010; 5:301-312. DOI | PubMed
- Shen L, Shao N, Liu X, Nestler E. Ngs.plot: quick mining and visualization of next-generation sequencing data by integrating genomic databases. BMC Genomics. 2014; 15DOI | PubMed
- Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nature Reviews Genetics. 2014; 15:272-286. DOI | PubMed
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Söding J, Thompson JD, Higgins DG. Fast, scalable generation of high-quality protein multiple sequence alignments using clustal omega. Molecular Systems Biology. 2011; 7DOI | PubMed
- Simionato E, Ledent V, Richards G, Thomas-Chollier M, Kerner P, Coornaert D, Degnan BM, Vervoort M. Origin and diversification of the basic helix-loop-helix gene family in metazoans: insights from comparative genomics. BMC Evolutionary Biology. 2007; 7:33-18. DOI | PubMed
- Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nature Reviews Molecular Cell Biology. 2009; 10:697-708. DOI | PubMed
- Singer M, Kosti I, Pachter L, Mandel-Gutfreund Y. A diverse epigenetic landscape at human exons with implication for expression. Nucleic Acids Research. 2015; 43:3498-3508. DOI | PubMed
- Srivastava M, Simakov O, Chapman J, Fahey B, Gauthier ME, Mitros T, Richards GS, Conaco C, Dacre M, Hellsten U, Larroux C, Putnam NH, Stanke M, Adamska M, Darling A, Degnan SM, Oakley TH, Plachetzki DC, Zhai Y, Adamski M, Calcino A, Cummins SF, Goodstein DM, Harris C, Jackson DJ, Leys SP, Shu S, Woodcroft BJ, Vervoort M, Kosik KS, Manning G, Degnan BM, Rokhsar DS. The Amphimedon queenslandica genome and the evolution of animal complexity. Nature. 2010; 466:720-726. DOI | PubMed
- Strutt H, Cavalli G, Paro R. Co-localization of polycomb protein and GAGA factor on regulatory elements responsible for the maintenance of homeotic gene expression. The EMBO Journal. 1997; 16:3621-3632. DOI | PubMed
- Thor S, Thomas JB. The Drosophila islet gene governs axon pathfinding and neurotransmitter identity. Neuron. 1997; 18:397-409. DOI | PubMed
- Thurman RE, Rynes E, Humbert R, Vierstra J, Maurano MT, Haugen E, Sheffield NC, Stergachis AB, Wang H, Vernot B, Garg K, John S, Sandstrom R, Bates D, Boatman L, Canfield TK, Diegel M, Dunn D, Ebersol AK, Frum T, Giste E, Johnson AK, Johnson EM, Kutyavin T, Lajoie B, Lee BK, Lee K, London D, Lotakis D, Neph S, Neri F, Nguyen ED, Qu H, Reynolds AP, Roach V, Safi A, Sanchez ME, Sanyal A, Shafer A, Simon JM, Song L, Vong S, Weaver M, Yan Y, Zhang Z, Zhang Z, Lenhard B, Tewari M, Dorschner MO, Hansen RS, Navas PA, Stamatoyannopoulos G, Iyer VR, Lieb JD, Sunyaev SR, Akey JM, Sabo PJ, Kaul R, Furey TS, Dekker J, Crawford GE, Stamatoyannopoulos JA. The accessible chromatin landscape of the human genome. Nature. 2012; 489:75-82. DOI | PubMed
- Ulitsky I. Evolution to the rescue: using comparative genomics to understand long non-coding RNAs. Nature Reviews Genetics. 2016; 17:601-614. DOI | PubMed
- Vavouri T, Walter K, Gilks WR, Lehner B, Elgar G. Parallel evolution of conserved non-coding elements that target a common set of developmental regulatory genes from worms to humans. Genome Biology. 2007; 8:R15-14. DOI | PubMed
- Vissers JH, van Lohuizen M, Citterio E. The emerging role of Polycomb repressors in the response to DNA damage. Journal of Cell Science. 2012; 125:3939-3948. DOI | PubMed
- Whitcomb SJ, Basu A, Allis CD, Bernstein E. Polycomb Group proteins: an evolutionary perspective. Trends in Genetics. 2007; 23:494-502. DOI | PubMed
- Woolfe A, Goodson M, Goode DK, Snell P, McEwen GK, Vavouri T, Smith SF, North P, Callaway H, Kelly K, Walter K, Abnizova I, Gilks W, Edwards YJ, Cooke JE, Elgar G. Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biology. 2005; 3DOI | PubMed
- Wu H, Caffo B, Jaffee HA, Irizarry RA, Feinberg AP. Redefining CpG islands using Hidden Markov models. Biostatistics. 2010; 11:499-514. DOI | PubMed
- Wysocka J, Swigut T, Xiao H, Milne TA, Kwon SY, Landry J, Kauer M, Tackett AJ, Chait BT, Badenhorst P, Wu C, Allis CD. A PHD finger of NURF couples histone H3 lysine 4 trimethylation with chromatin remodelling. Nature. 2006; 442:86-90. DOI | PubMed
- Zentner GE, Scacheri PC. The chromatin fingerprint of gene enhancer elements. Journal of Biological Chemistry. 2012; 287:30888-30896. DOI | PubMed
- Zentner GE, Tesar PJ, Scacheri PC. Epigenetic signatures distinguish multiple classes of enhancers with distinct cellular functions. Genome Research. 2011; 21:1273-1283. DOI | PubMed
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS. Model-based analysis of ChIP-Seq (MACS). Genome Biology. 2008; 9DOI | PubMed
- Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nature Reviews Genetics. 2011; 12:7-18. DOI | PubMed
Fonte
Gaiti F, Jindrich K, Fernandez-Valverde SL, Roper KE, Degnan BM, et al. () Landscape of histone modifications in a sponge reveals the origin of animal . eLife 6e22194. https://doi.org/10.7554/eLife.22194




