
Abstract
Background
Gli studi clinici forniscono prove sulla sicurezza e l’efficacia degli interventi (Tabella 1). Essi sono alla base della politica sanitaria e della regolamentazione, e informano il paziente e il fornitore di assistenza sanitaria sulle decisioni da prendere. Poiché molti studi non sono pubblicati[1- 6], e poiché molti rapporti pubblicati non includono tutte le informazioni necessarie per comprendere i metodi di sperimentazione [7- 10] e i risultati [11-17], ledecisioni basate sulle sole evidenze pubblicate potrebbero non portare al miglior equilibrio tra benefici e danni per i pazienti [18-21].Tabella 1Glossario dei terminiTermDefinitionApplication programming interface (API)Un insieme di metodi per facilitare la comunicazione tra i componenti software, come descritto nella Sezione 10 della Guida per l’Utente di PRS (https://prsinfo.clinicaltrials.gov/prs-users-guide.html#section10)Cancer centerUn’organizzazione specializzata nella diagnosi e nel trattamento del cancro, comprese le organizzazioni designate dal National Cancer Institute (vedi “National Cancer Institute cancer cancer center”)Studio clinico (“trial”)Uno studio in cui i partecipanti umani sono assegnati in prospettiva a ricevere uno o più interventi (ad es, diagnostici, terapeutici o di altro tipo) per valutare gli effetti dell’intervento (o degli interventi) sugli esiti sanitari. Per esempio, si veda[34, 36]Clinical and Translational Science Awards (CTSA)Awards finanziati dal National Center for Advancing Translational Sciences (NCATS), una parte del National Institutes of Health (NIH), per sostenere un consorzio di 64 organizzazioni di ricerca medica(https://ncats.nih.gov/ctsa)Food and Drug Administration Amendments Act of 2007 (FDAAA)US Public Law 110-85, che ha stabilito i requisiti per la registrazione e la segnalazione degli studi clinici (sezione 801)[31].Institutional review board (IRB)Un gruppo di persone con la responsabilità di garantire la protezione dei soggetti umani coinvolti nella ricerca. Per esempio, vedi[58- 60]InvestigatorA ricercatore coinvolto in uno studio clinico [34,36] .National Cancer Institute cancer cancer center (NCI cancer center)Una delle 69 organizzazioni designate dal National Cancer Institute (NCI) che sono specializzate nella diagnosi e nel trattamento del cancro(https://www.cancer.gov/research/nci-role/cancer-centers)Protocol Registration and Results System (PRS)Un sistema di inserimento dati basato sul web utilizzato per registrare gli studi su ClinicalTrials.gov e per presentare i risultati degli studi registrati Account dell’organizzazione PRS (“account”)Un account assegnato a un’organizzazione e utilizzato per inserire informazioni sugli studi clinici nel Protocol Registration and Results System. Un account dell’organizzazione può essere gestito da uno o più amministratori e può includere studi condotti da più sperimentatoriAmministratore PRS (“amministratore”)Una persona che gestisce un account dell’organizzazione nel Sistema di registrazione del protocollo e dei risultati. Gli amministratori sono in grado di creare account per i singoli sperimentatori, rivedere le informazioni sullo studio, modificare i permessi per la modifica delle informazioni sullo studio e controllare la presenza di problemiRegistrazione dello studio (registrazione)Il processo di inserimento di un set minimo di dati su uno studio clinico nel registro che è accessibile al pubblico (ad es, ClinicalTrials.gov)[34, 36].Parte responsabileLa persona o l’entità responsabile dell’invio di informazioni su uno studio clinico a ClinicalTrials.gov e dell’aggiornamento di tali informazioni[34, 36].RisultatiInformazioni sintetiche sugli effetti dell’intervento, inclusi il flusso dei partecipanti, le misure di outcome e gli eventi avversi[34, 36].SponsorLa persona o l’organizzazione che supervisiona uno studio clinico ed è responsabile dei dati dello studio[34, 36].The Final Rule (42 CFR 11)Un regolamento federale che implementa la Sezione 801 del Food and Drug Administration Amendments Act del 2007 (FDAAAA) ed espande i requisiti per la registrazione dello studio e la comunicazione dei risultati. La data di entrata in vigore è il 18 gennaio 2017 e la data di conformità è il 18 aprile 2017[34].Università/organizzazioneUn “tipo di organizzazione” utilizzato per classificare i conti delle organizzazioni PRS da www.ClinicalTrials.gov.
Per aiutare i partecipanti a iscriversi alle sperimentazioni, migliorare l’accesso alle informazioni e ridurre i pregiudizi, gli autori hanno da tempo proposto di registrare tutte le sperimentazioni in prospettiva[22-27]. Il Food and Drug Administration Modernization Act del 1997 ha portato alla creazione di ClinicalTrials.gov, un database pubblicamente accessibile gestito dalla National Library of Medicine (NLM), lanciato nel 2000[28]. Nel 2004, l’International Committee of Medical Journal Editors (ICMJE) ha annunciato che gli studi avviati a partire dal 2005 dovranno essere registrati per essere presi in considerazione per la pubblicazione[29, 30]. Il titolo VIII del Food and Drug Administration Amendments Act del 2007 (FDAAAA)[31] richiedeva la registrazione di alcuni studi clinici su farmaci, biologici e dispositivi medici e la pubblicazione dei risultati degli studi clinici dei prodotti approvati su ClinicalTrials.gov. L’FDAAAA ha anche autorizzato la Food and Drug Administration (FDA) ad emettere multe per non conformità, attualmente fino a 11.569 dollari al giorno per ogni sperimentazione[32]. “La regola finale”, entrata in vigore il 18 gennaio 2017, ha chiarito e ampliato i requisiti per la registrazione e la segnalazione (Box 1) [33, 34]; si prevedeva che le organizzazioni fossero in regola entro il 18 aprile 2017. In una politica complementare, i National Institutes of Health (NIH) hanno emanato requisiti più ampi che si applicano a tutte le sperimentazioni finanziate dal NIH, comprese le prime sperimentazioni e le sperimentazioni di interventi comportamentali[35, 36].
Ci sono poche prove su come le organizzazioni accademiche supportino la registrazione e la segnalazione degli studi, ma alcune prove suggeriscono che non sono pronte a soddisfare questi requisiti. Per esempio, le organizzazioni accademiche hanno ottenuto risultati peggiori rispetto all’industria nella registrazione prospettica degli studi[37, 38] e nel reporting dei risultati[39-46].
Metodi
Tra il 21 novembre 2016 e il 1° marzo 2017 abbiamo condotto un sondaggio online sulle organizzazioni accademiche negli Stati Uniti. Abbiamo intervistato gli amministratori che sono responsabili della gestione degli account delle organizzazioni su ClinicalTrials.gov. Per ogni account ClinicalTrials.gov idoneo, abbiamo chiesto a un amministratore di descrivere le politiche e le procedure e le risorse disponibili per supportare la registrazione e la rendicontazione dei trial presso la loro organizzazione (Box 2).
Identificazione dei conti PRS idonei
Il sistema online utilizzato per inserire le informazioni nel database di ClinicalTrials.gov si chiama Protocol Registration and Results System (PRS). Ogni studio registrato su ClinicalTrials.gov è associato ad un “record” di quello studio, e ogni record è assegnato ad un account dell’organizzazione PRS. Un record può includere o meno i risultati dello studio. Una singola organizzazione, come un’università o un sistema sanitario, può registrare gli studi utilizzando uno o più account. Ad esempio, “Yale University” è un unico account; in confronto, “Harvard Medical School” e “Harvard School of Dental Medicine” sono due account separati.
Abbiamo usato il conto PRS come unità di analisi perché i conti relativi alla stessa organizzazione spesso rappresentano scuole o dipartimenti che hanno politiche e procedure separate relative alla registrazione e al reporting di prova. Inoltre, non siamo a conoscenza di un metodo affidabile per associare i singoli conti all’organizzazione. Ad esempio, il conto “Johns Hopkins University” comprende per lo più registrazioni della Johns Hopkins University School of Medicine. Gli investigatori della Johns Hopkins University registrano anche le prove utilizzando i conti “Johns Hopkins Bloomberg School of Public Health”, “Johns Hopkins Children’s Hospital” e “Sidney Kimmel Comprehensive Cancer Center”. Le scuole e gli ospedali legati alla Johns Hopkins University hanno politiche, facoltà, personale amministrativo e comitati di revisione istituzionale (IRB) distinti.
Abbiamo incluso tutti i conti “attivi” classificati da ClinicalTrials.gov come “Università/Organizzazione” negli USA. Abbiamo ricevuto un foglio di calcolo dal NLM con il numero di registrazioni in ogni conto idoneo il 4 agosto 2016, e abbiamo ricevuto informazioni di contatto dell’amministratore PRS dal NLM il 28 settembre 2016 e il 12 dicembre 2016.
Progettazione del sondaggio
Abbiamo sviluppato una bozza di indagine basata sulla conoscenza dei contenuti degli investigatori e sulle prove di studi che ci erano noti all’epoca. Abbiamo organizzato le domande in tre settori: (1) caratteristiche dell’organizzazione, (2) politiche e pratiche di registrazione e risultati e (3) personale e risorse. Abbiamo anche invitato i partecipanti a descrivere gli eventuali sforzi di conformità che le nostre domande non hanno coperto. Abbiamo poi pilotato il sondaggio tra 14 membri della National Clinical Trials Registration and Results Reporting Taskforce. Il sondaggio finale ha utilizzato la logica del salto, in modo che i partecipanti vedessero solo le domande pertinenti in base alle loro precedenti risposte. Le risposte sono state salvate automaticamente e i partecipanti hanno potuto tornare al sondaggio in qualsiasi momento; ciò ha permesso ai partecipanti di discutere le loro risposte con i colleghi dell’organizzazione prima di presentare la domanda. Abbiamo condotto il sondaggio utilizzando il software Qualtrics(www.qualtrics.com/); una copia è disponibile come documento Word (file aggiuntivo 1) e sul sito web di Qualtrics (http://bit.ly/2tCSqyl).
Reclutamento dei partecipanti
Una o più persone, chiamate “amministratori PRS” da ClinicalTrials.gov, possono aggiungere o modificare i record in ogni account. Alcuni amministratori PRS sono impiegati specificamente per lavorare su ClinicalTrials.gov, ma molti amministratori PRS hanno poco o nessun tempo a disposizione delle loro organizzazioni per lavorare su ClinicalTrials.gov.
Per ogni account idoneo, abbiamo creato un indirizzo internet unico (URL) che abbiamo inviato per e-mail in una lettera d’invito a un amministratore. Per gli account con più di un amministratore, abbiamo prima contattato tutti gli amministratori e chiesto loro di selezionare l’amministratore appropriato per completare questo sondaggio; poi, abbiamo inviato il sondaggio all’amministratore nominato. Se un amministratore non ha completato il sondaggio, EMW ha inviato almeno due promemoria dal suo account e-mail dell’università dopo circa 2 settimane e 4 settimane. Per gli account con più amministratori, abbiamo inviato un’e-mail a tutti gli amministratori se l’amministratore designato non ha risposto dopo due solleciti. Abbiamo incaricato gli amministratori associati a più account di completare un sondaggio separato per ogni account.
I partecipanti hanno indicato il loro consenso continuando oltre la pagina di apertura e completando il sondaggio.
Analisi
Per analizzare i risultati, per prima cosa abbiamo escluso i conti che non hanno completato tre domande necessarie per sapere se avevano: (1) una politica di registrazione, (2) una politica di reporting dei risultati e (3) un software per gestire i loro registri. Abbiamo poi calcolato le statistiche descrittive utilizzando SPSS 24. Per i dati categorici, abbiamo calcolato il numero e la proporzione dei conti che hanno selezionato ogni risposta. Per i dati continui, abbiamo calcolato la mediana e l’intervallo interquartile (IQR) a seconda della distribuzione delle risposte.
Abbiamo condotto analisi di sottogruppo per determinare se le caratteristiche dell’organizzazione possono essere correlate alle politiche e alle risorse. Abbiamo confrontato: Conti affiliati con un Clinical and Translational Science Award (CTSA) rispetto ad altri contiConti affiliati con un centro oncologico rispetto ad altri contiConti con < 20 record, 20-99 record, e ≥ 100 record
Abbiamo condotto un’analisi di sensibilità per determinare se i risultati potrebbero essere sensibili alla mancata risposta, confrontando i conti che hanno risposto prima della data di entrata in vigore della Regola Finale (18 gennaio 2017) con i conti che hanno risposto il 18 gennaio 2017 o dopo l’entrata in vigore della Regola Finale.
Risultati
Caratteristiche dei conti ammissibili
Abbiamo identificato 783 conti idonei (file aggiuntivo 2), che ad agosto 2016 avevano 47.701 registrazioni. Il numero mediano di record per conto era di 7 (IQR = 3-36), che vanno da 1 (due conti) a 1563 (media = 61, deviazione standard (SD) = 155). Una minoranza dei conti è responsabile della maggior parte dei record; i conti 113/783 (14%) avevano un numero di record ≥ 100 entro agosto 2016, e questi conti erano responsabili di 38.311/47.701 (80%) record.
Il numero mediano di amministratori per conto era di 1 (IQR=1-3), e un’organizzazione aveva 182 amministratori registrati.
Partecipazione al sondaggio
Su 783 conti idonei, non abbiamo trovato i dati di contatto per 16 (2%) e abbiamo tentato di contattare 767 (98%). In quattro casi (<1%), non siamo stati in grado di identificare un indirizzo e-mail utilizzabile. Tra i conti idonei, il 10/783 (1%) ci ha inviato un’e-mail di rifiuto, il 306/783 (39%) non ha partecipato al sondaggio e l’81/783 (10%) non ha fornito informazioni sufficienti per essere incluso nell’analisi (Fig. 1). Due conti hanno riferito di avere più polizze relative allo stesso conto; abbiamo chiesto loro di completare le domande sulle caratteristiche del loro conto, ma non di completare le domande sulle loro specifiche politiche e risorse.Fig. 1Flowchart: Conti PRS inclusi nel sondaggio
I conti inclusi erano responsabili di 40.351/47.701 (85%) registrazioni registrate da conti idonei. Abbiamo ricevuto un’indagine parziale (43) o completa (323) per 366/783 (47%) conti idonei (file aggiuntivo 3).
Il primo conto ha completato il sondaggio il 21 novembre 2016 e l’ultimo conto ha completato il sondaggio il 21 marzo 2017; 31/366 (9%) conti hanno completato il sondaggio dopo il 17 gennaio 2017. A causa della logica del salto e del fatto che alcuni conti non hanno risposto a tutte le possibili domande, i conti hanno risposto tra le 6 e le 42 domande (mediana 19, IQR 17-29).
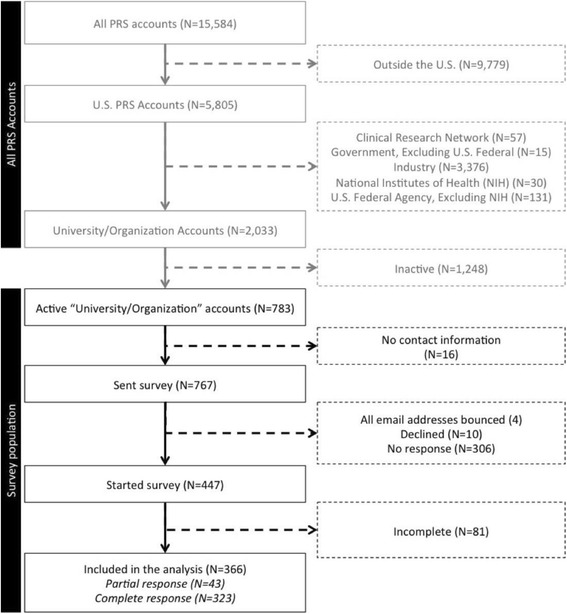
Fig. 1.Diagramma di flusso: Conti PRS inclusi nel sondaggio
Politiche e pratiche
Su 366 conti, 156 (43%) hanno dichiarato di avere una politica di registrazione e 129 (35%) hanno una politica di reporting dei risultati (Tabella 2). Le politiche sono entrate in vigore tra il 2000 e il 2016 (mediana=2013, IQR 2010-2015; modalità=2016).Tabella 2Politiche di registrazione di prova clinica e di segnalazione dei risultatiQUESTION (numero di partecipanti che hanno visto la domanda)N.PercentualePolitiche di registrazione di prova L’organizzazione ha una politica di registrazione?(N=366)aSi15643% No17347% Non so3710% La polizza copre gli investigatori che si iscrivono all’organizzazione?(N=156)b Sì5737% No7649% Non lo so2315% Respinto (non ha risposto)00% La polizza copre gli investigatori che lasciano l’organizzazione? (N=156)b Sì3824% No8756% Non lo so3120% Saltato (non ha risposto)00% Secondo la polizza, quando devono essere registrate le prove?(N= 139)b,c Prima dell’approvazione IRB1511% Prima dell’inizio dell’iscrizione4935%Con 21 giorni dall’inizio dell’iscrizione3122% I requisiti differiscono tra le prove2115%.Questo non viene affrontato nella polizza1813% Non lo so43% Non lo so43% Respinto (non ha risposto)11% Secondo la polizza, chi è responsabile di determinare se una prova deve essere registrata?(N= 129)b,c Indagatore principale7256% Consiglio di revisione istituzionale2016% Amministratore PRS3527% Altro1411% Questa responsabilità non è assegnata nella polizza129%.Non lo so00% Non lo so00% Non ha risposto (non ha risposto)00% Secondo la politica, gli investigatori possono essere penalizzati dall’organizzazione per non aver registrato un processo?(N=139)b Sì2723% No9191% Non lo so2121% Saltato (non ha risposto)00% Data di entrata in vigore della politica di registrazione del processo (N=139)=139)b,d,dMinimo (anno)2000Massimo (anno)2016Mediano (anno)2013 Politiche di reporting dei risultati L’organizzazione ha una politica di reporting dei risultati? (N=366)aSi12935%No19353% Non lo so4412% Secondo la politica, chi è responsabile del monitoraggio se i risultati sono riportati in tempo?(N=115)b,c Investigatore principale5447% Consiglio di revisione istituzionale54% Amministratore PRS6859% Altro1210% Questa responsabilità non è assegnata nella politica1816%.Non lo so11% Non ha risposto (non ha risposto)00% Secondo la politica, gli investigatori possono essere penalizzati dall’organizzazione per non aver segnalato un processo (N=114)eSì2118%No7566%Non lo so1816%Ha saltato (non ha risposto)00%aUna risposta a questa domanda era necessaria perché un account fosse incluso nell’analisi; i conti che non hanno visto o saltato questa domanda sono stati esclusi da tutte le analisibIl numero di risposte possibili (i.e., il denominatore) include i conti con una politica rilevante che ha visto questa domanda. Il numero di account che hanno visto ogni domanda è inferiore al numero totale di account nello studio perché (1) i partecipanti non hanno visto tutte le domande a causa della logica del salto, e (2) alcuni partecipanti hanno interrotto il sondaggio prima di vedere tutte le domandescPerché i partecipanti potevano “controllare tutto ciò che si applica”, la somma di tutte le categorie supera il numero di partecipanti che hanno risposto (cioè, alcuni partecipanti hanno selezionato risposte multiple)dBperché 50 (36%) hanno selezionato “Non so”, 89 account sono inclusi nell’analisiseCon 111 account che hanno visualizzato una domanda sulle penalità per (1) non essersi registrati o (2) non aver riportato una prova, 17 (15%) hanno risposto “Sì” ad entrambe le domande, e 31 (28%) hanno risposto “Sì” ad una o ad entrambe le domande
Tra i conti con polizze, la maggior parte delle polizze richiede la registrazione delle sperimentazioni applicabili ai sensi della FDAAA (118/140, 84%) e finanziate dal NIH (72/140, 51%) (file aggiuntivo 4). Le politiche prevedono diversi requisiti per il momento della registrazione (Tabella 3); la maggior parte richiede che gli studi siano registrati prima che venga concessa l’approvazione IRB (15/156; 11%), prima dell’inizio dell’arruolamento (49/156; 35%), o entro 21 giorni dall’inizio dell’arruolamento (31/156; 22%). Una minoranza di politiche si occupa della gestione degli studi associati all’adesione degli sperimentatori (57/156; 37%) e all’uscita dalle organizzazioni (38/156; 24%).Tabella 3Risorse a supporto della registrazione degli studi clinici e della comunicazione dei risultatiQUESTION (numero di partecipanti che hanno visualizzato la domanda)N.Percentuale L’organizzazione dispone di un sistema elettronico per la gestione della registrazione degli studi o della comunicazione dei risultati?(N=366)aSib6819% No27274% Non so267% Quali funzioni svolgono il personale che supporta la registrazione e la comunicazione dei risultati (N = 342)c Formazione di gruppo (es, Stile aula)6118% Formazione individuale15144%Inserire i dati per i principali investigatori (PI)17451% Mantenere un sito web educativo5717% Informare i PI su problemi o sanzioni24170%.Assistenza con l’analisi5817% Rispondere alle domande24170% Riesaminare i dati relativi ai problemi26277%Altri288% Non lo so226% Non lo so226% Ha saltato (non ha risposto)00% Qual è la qualifica più alta di un membro del personale?(N= 315)d Scuola media superiore113% Bachelors6822% Master12339% Grado più alto10935% Saltato (non ha risposto)41% L’organizzazione controlla la conformità ai requisiti di reporting dei risultati?(N= 116) Sì9985% No109% Non lo so76% Saltato (non ha risposto)00% Chi controlla la conformità ai requisiti di reporting dei risultati?(N=99)c,f Amministratore PRS8586% Consiglio di revisione istituzionale (IRB)g1111% Altro2020% Non sa00% Saltato (non ha risposto)00%Numero di personale equivalente a tempo pieno (FTE) (N =287)hMediano=0.08IQR= da 0,02 a 0.25aUna risposta a questa domanda era necessaria per includere un account nell’analisi; gli account che non hanno visto o saltato questa domanda sono stati esclusi da tutte le analisibDei 68 account che utilizzano un sistema di gestione elettronica (“software per computer”), 2 (3%) utilizzano un’interfaccia di programmazione di applicazioni (API) per comunicare con ClinicalTrials.govcPerché i partecipanti potevano “controllare tutto ciò che si applica,” la somma di tutte le categorie supera il numero di partecipanti che hanno risposto (cioè, alcuni partecipanti hanno selezionato più risposte)dIlnumero di risposte possibili (cioè il denominatore) comprende i conti con una politica pertinente che ha visto questa domanda. Il numero di account che hanno visto ogni domanda è inferiore al numero totale di account nello studio perché (1) i partecipanti non hanno visto tutte le domande a causa della logica di salto, e (2) alcuni partecipanti hanno interrotto il sondaggio prima di vedere tutte le domandeLe lauree più alte includono JD (N= 21, 7%), PhD (N = 69,22%), e MD (N = 32, 10%); 13 conti hanno selezionato 2 gradi superiori (8 sia PhD che JD, 5 sia PhD che MD)fIlnumero di risposte possibili è stato limitato ai conti che hanno riferito di aver monitorato la conformità con i loro risultati riportando policygOdegli 11 conti che hanno riferito che gli IRB monitorano la registrazione delle prove, 4 hanno indicato che l’IRB richiede la registrazione per l’approvazione per alcuni(N==3) o per tutte le prove (N==1) Irisultati sono lamedianae l’intervallo interquartile. Abbiamo anche calcolato la media = 0,3, la deviazione standard = 0,6
La responsabilità per la registrazione delle sperimentazioni è più spesso assegnata agli investigatori principali (72/129; 56%). La responsabilità di monitorare se i risultati vengono riportati in tempo è più spesso assegnata agli sperimentatori principali (54/115, 47%) e agli amministratori (68/115, 59%).
Alcune politiche consentono alle organizzazioni di penalizzare gli sperimentatori che non registrano gli studi (27/115; 18%) o che non riportano i risultati (21/114; 18%). Un account (<1%) ha riferito che la loro organizzazione ha penalizzato uno sperimentatore per non conformità.
Risorse
Pochi conti utilizzano software informatici per la gestione dei loro registri (68/366; 19%). Tra quelli che usano software per computer, due usano l’interfaccia di programmazione delle applicazioni (API) per connettersi con ClinicalTrials.gov (Tabella 3).
Tra i conti 287/366 (78%) che assegnano il personale per soddisfare i requisiti di registrazione e segnalazione di ClinicalTrials.gov, il numero mediano del personale equivalente a tempo pieno (FTE) è 0,08 (IQR=0,02-0,25). Tra il personale che supporta i requisiti di registrazione e reporting di ClinicalTrials.gov, il personale con il più alto livello di istruzione ha un diploma di laurea (232/411; 75%) più spesso di un diploma di laurea (68/411; 22%) o un diploma di scuola superiore (11/411; 3%). Al momento di questa indagine, 34/338 (10%) prevedeva di assumere più personale, mentre 217/338 (64%) e 87/338 (26%) non prevedevano rispettivamente di assumere più personale o non lo sapevano. Tra i conti affiliati a una CTSA, 24/109 (22%) ricevono supporto per la conformità di ClinicalTrials.gov dalla CTSA.
Il personale svolge vari ruoli, tra cui quello di educare gli investigatori individualmente (151/342; 44%) e in gruppo (61/42; 18%), inserire i dati per gli investigatori principali (174/342; 51%), mantenere i siti web educativi (57/342; 17%), notificare i problemi agli investigatori (241/342; 70%), assistere gli investigatori nell’analisi (58/342; 17%), rispondere alle domande (241/342; 70%) e rivedere le registrazioni dei problemi (262/342; 77%).
Analisi dei sottogruppi
Le politiche di registrazione e di rendicontazione sono più comuni tra i seguenti conti: (1) quelli con molte registrazioni, (2) quelli affiliati ai CTSA e (3) quelli affiliati ai centri oncologici (Tabella 4). Ad esempio, la maggior parte dei centri oncologici ha una politica di registrazione (61/97; 63%) e una politica di rendicontazione (52/97; 54%); una minoranza degli altri conti ha una politica di registrazione (94/267; 35%) o una politica di rendicontazione (77/267; 28%).Tabella 4Analisi dei sottogruppiCTSA affiliationCancer center affiliationaAccount sizeTotalQUESTION (numero di partecipanti che hanno visto la domanda)CTSA (N= 109)Non CTSA (N = 257)Cancer center (N = 97)Non cancer center (N = 97)Non cancer center (N = 97)Non cancer center (N =)267)≥ 100 record (N = 98)20-99 record (N = 77)< 20 record (N = 191)(N = 366)Numero di record29,07611,27524,97014,94035,2693756132640,351 L’organizzazione ha una politica di registrazione?(N=366)SÌ5853%9838%6163%9435%6364%3343%6031%15643%No4037%13352%3031%14253%2930%3951%10555%17347%Non so1110%2610%2610%66%3112%66%66%56%2614%2614%3710% L’organizzazione ha una politica di reporting dei risultati?(N=366)□Sì5248%7730%5254%7728%5556%2634%4825%12935%No4642%14757%3738%15458%36458%3637%4457%11359%19353%Dont know1110%3313%88%3614%77%77%79%3016%4412% L’organizzazione dispone di un sistema elettronico per la gestione della registrazione di prova o la comunicazione dei risultati?(N=366)□Sì2725%4116%2627%4115%2627%79%3518%6819%No7367%19977%6870%20376%6869%6686%6686%13872%272727274%Dont know98%177%33%239%239%181818%45%42%42%267%Numero di personale equivalente a tempo pieno (FTE) (N= 288)bN = 94Mean (SD) =0.59 (0,83)N= 193Mezzo (SD) =0. 17 (0,39)N = 84Mezzo (SD) =0,56 (0,81)N =201Mezzo (SD) =0.25 (0,72)N= 88Mean (SD) =0,69 (0,83)N = 61Mean (SD) =0,13 (0,24)N = 138Mean (SD) =0.14 (0,41)N==287Mean (SD) =0,31 (0,60)Mediana (IQR) =0,25 (0,05 a 0,95)Mediana (IQR) =0,05 (0,02 a 0.15)Mediana (IQR) =0,25 (0,05 a 0,85)Mediana (IQR) =0,05 (0,02 a 0,15)Mediana (IQR) =0,42 (0,15 a 1,00)Mediana (IQR) =0,06 (0,02 a 0.15)Mediana (IQR) =0,05 (0,01 a 0,10)Mediana (IQR) =0,08 (0,02 a 0,25)Range0 a 5,6Range0 a 4Range0 a 5,6Range0 a 4Range0 a 5,6Range0 a 5,6Range0 a 1,58Range0 a 4Range0 a 4Range0 a 5,6CTSA Conti affiliati a un Clinical and Translational Science Award (CTSA). Il numero di conti affiliati CTSA supera il numero di CTSA perché a volte più conti sono stati affiliati alla stessa CTSA. Non conti CTSA non affiliati a una CTSA. Conti di un centro oncologico affiliato a un centro oncologico del National Cancer Institute (NCI) o a un altro centro oncologico. Non centro oncologico Conti non affiliati a un NCI o a un altro centro oncologico. ≥ 100 registrazioni Conti con 100 o più studi registrati negli USA per i quali l’organizzazione è stata indicata come “sponsor principale”. 20-99 registrazioni Conti con un numero di studi registrati compreso tra 20 e 99. < 20 registrazioni Conti con meno di 20 studi registratiesaDue conti non hanno riportato se sono affiliati a un centro oncologico; non sono inclusi in questo sottogruppo analysisb I risultati sono per i conti che hanno risposto a questa domanda. Nella nostra analisi iniziale abbiamo trovato dati potenzialmente non validi; ad esempio, alcuni partecipanti hanno inserito “0,5” anziché “50%”. Questo si è verificato perché un bug del software ci ha impedito di applicare una regola di convalida dei dati nel sondaggio. Per verificare questi risultati, abbiamo inviato un’e-mail agli amministratori che hanno indicato che il personale ha speso ≤1% del suo tempo per la registrazione e la reportistica di prova. Post hoc, abbiamo escluso due outlier perché sembravano riportare il numero totale del personale impiegato presso l’organizzazione piuttosto che il numero del personale che supporta la registrazione di prova e la segnalazione dei risultati.
Mancata risposta
Abbiamo trovato prove dirette e indirette di un pregiudizio di non risposta, il che suggerisce che i nostri risultati potrebbero sovrastimare la quantità di supporto disponibile presso le organizzazioni accademiche. Per esempio, un amministratore che ha rifiutato di partecipare ha risposto che la sua organizzazione “non ha uno staff centrale che gestisce clinicaltrials.gov e non utilizza un account istituzionale”.
La dimensione del conto era legata alla partecipazione al sondaggio e molti conti partecipanti erano grandi (Tabella 5). Di questi conti che abbiamo invitato a completare il sondaggio che comprendeva < 20 record, 171/532 (32%) hanno partecipato. A titolo di confronto, hanno partecipato 98/113 (87%) conti con ≥ 100 record.Tabella 5Caratteristiche dei partecipantiQUESTION (numero di partecipanti che hanno visto la domanda)No.PercentualeConti ammissibili(N= 783) a<20 recordb53268%20-99 record13818%≥100 record11314%Partecipanti (N=366)<20 record19152%20-99 record7721%≥100 record9827% Il conto PRS è affiliato ad una CTSA?(N=366) Sì (selezionato un CTSA)10930% No (selezionato “Non applicabile”)21158% Respinto (non ha risposto)4613% Il conto PRS è affiliato a uno o più dei seguenti conti?(N=366)un centro oncologico NCI6217% Altro centro oncologico (non designato NCI)c3710% Scuola medica10027% Ospedale d’insegnamento13838% Altra scuolasd10729% Altro17247%Selezionato”.Non lo so”195% Saltato (non ha risposto)2<1%aPerché i partecipanti potevano “controllare tutto ciò che si applica,” la somma di tutte le categorie supera il numero di conti che hanno partecipato (i.e., alcuni partecipanti hanno selezionato risposte multiple)b I recordincludono studi per i quali l’organizzazione era elencata come “sponsor principale” e lo studio è stato condotto negli USA; abbiamo escluso i record per i quali lo sperimentatore principale (PI) era lo “sponsor principale”, e abbiamo escluso gli studi fatti al di fuori degli USAcTwo accounts ha selezionato sia un “NCI cancer center” che un “Altro centro oncologico”; così, 97 accounts sono stati affiliati con un centro oncologico “Altre scuole“ includono: scuola di sanità pubblica(N= 59, 16%), scuola di assistenza sociale (N = 41,11%), scuola di arti e scienze (N = 56, 15%), scuola di infermieristica (N = 72, 20%), scuola diodontoiatria(N = 40, 11%)
La partecipazione potrebbe essere stata correlata alle risorse dell’organizzazione. Quasi tutti i CTSA (62/64; 97%) e la maggior parte dei centri oncologici del National Cancer Institute (NCI) (55/69; 80%) hanno partecipato all’indagine (Tabella 5), compresi 48 conti affiliati sia a un centro oncologico che a un CTSA. Inoltre, alcuni dei conti inclusi erano collegati; ad esempio, 107 conti erano affiliati a uno dei 62 CTSA.
In un’analisi di sensibilità (file aggiuntivo 5), non abbiamo trovato chiare differenze tra le politiche e il software per computer nel confronto tra chi ha risposto in anticipo e chi ha risposto in ritardo. La maggior parte dei partecipanti ha completato il sondaggio prima della data effettiva, quindi i ritardatari hanno incluso solo 31/366 (8%) conti.
Discussione
Sintesi dei risultati
A nostra conoscenza, questa è la più grande e completa indagine delle organizzazioni che registrano e segnalano gli studi clinici su ClinicalTrials.gov. Abbiamo avuto un alto tasso di partecipazione, e i conti che hanno completato il sondaggio conducono la stragrande maggioranza dei trial clinici registrati dalle organizzazioni accademiche negli Stati Uniti. Abbiamo scoperto che alcune organizzazioni erano pronte a soddisfare i requisiti per la registrazione e la segnalazione degli studi clinici prima che la Regola Finale entrasse in vigore, ma c’è un’ampia variazione nella pratica. La maggior parte delle organizzazioni non ha politiche per la registrazione e la rendicontazione degli studi clinici. La maggior parte delle politiche esistenti sono coerenti con la FDAAA; tuttavia, la maggior parte non è coerente con la politica di registrazione ICMJE. Quasi la metà delle politiche esistenti non richiede la registrazione di tutte le sperimentazioni finanziate dall’NIH, anche se le organizzazioni potrebbero adattare le loro politiche in risposta ai nuovi requisiti dell’NIH. Poche politiche includono sanzioni per gli investigatori che non registrano o non segnalano le loro sperimentazioni. Sebbene alcune organizzazioni utilizzino software informatici per monitorare la registrazione e la reportistica degli studi, solo due hanno sistemi che si collegano direttamente con ClinicalTrials.gov (ad esempio, utilizzando le API). La maggior parte del personale che supporta la registrazione e la reportistica della sperimentazione ha altre responsabilità, e la maggior parte delle organizzazioni non prevede di assumere altro personale per supportare la registrazione e la reportistica della sperimentazione.
Implicazioni
I nostri risultati suggeriscono che la maggior parte delle organizzazioni assegnano la responsabilità della registrazione e del reporting delle prove ai singoli investigatori e forniscono poca supervisione. Studi precedenti indicano che gli investigatori senior spesso delegano questa responsabilità ai loro colleghi più giovani[47].
Per quanto ne sappiamo, la FDA non ha mai valutato una sanzione pecuniaria civile per la mancata registrazione o segnalazione di uno studio, e il NIH non ha mai penalizzato un’organizzazione per non aver soddisfatto i loro requisiti. La politica dell’ICMJE non è applicata in modo uniforme[48], e molti studi pubblicati non sono ancora registrati in modo prospettico e completo[37, 49- 52]. Le organizzazioni possono essere più propense a conformarsi a questi requisiti se sono ritenute responsabili di ciò dalle riviste, dalla FDA e dai finanziatori (vedi, ad esempio, http://www.who.int/ictrp/results/jointstatement/en).
Il miglioramento della trasparenza della ricerca a lungo termine richiederà cambiamenti nelle norme e nella cultura. Le organizzazioni potrebbero adottare quattro misure immediate per migliorare la registrazione e la rendicontazione delle sperimentazioni. In primo luogo, le organizzazioni potrebbero offrire formazione per aiutare gli sperimentatori a comprendere questi requisiti. In secondo luogo, le organizzazioni potrebbero implementare politiche e procedure a sostegno della registrazione e della rendicontazione della sperimentazione. Per esempio, le organizzazioni potrebbero richiedere che gli sperimentatori rispondano a domande sulle richieste di IRB per identificare gli studi clinici che richiedono la registrazione. Le organizzazioni potrebbero anche richiedere che gli sperimentatori forniscano i numeri di registrazione degli studi prima di permettere l’inizio degli studi. In terzo luogo, le organizzazioni potrebbero identificare gli studi che non soddisfano i requisiti di registrazione e di reporting e aiutare i singoli sperimentatori a renderli conformi. In particolare, il software potrebbe fornire promemoria automatici quando le informazioni sullo studio devono essere aggiornate[53] o quando i risultati saranno dovuti, e il software potrebbe aiutare le organizzazioni ad identificare i problemi che richiedono attenzione da parte dei leader. I potenziali promemoria consentirebbero agli amministratori e agli sperimentatori di aggiornare le informazioni prima che diventino non conformi ai requisiti di segnalazione. Infine, le organizzazioni potrebbero assicurare che ci siano conseguenze per gli investigatori che non soddisfano i requisiti di registrazione e di segnalazione delle prove. Per esempio, le organizzazioni potrebbero interrompere l’iscrizione alle sperimentazioni in corso o impedire agli sperimentatori di ottenere nuove sovvenzioni[54].
Limitazioni
Anche se abbiamo inviato diversi solleciti e abbiamo dato ai partecipanti mesi di tempo per rispondere, i nostri risultati potrebbero essere influenzati dalla mancata risposta e dalla desiderabilità sociale. Tuttavia, tali pregiudizi ci porterebbero a sopravvalutare il supporto per la registrazione e la segnalazione delle prove di ricerca. Gli account partecipanti conducono più prove rispetto agli account non partecipanti e sembrano avere più probabilità di avere politiche e risorse a sostegno della trasparenza.
Poiché abbiamo analizzato i risultati per account, i nostri risultati non sono direttamente comparabili con gli studi che raggruppano gli studi utilizzando i campi dati “finanziatore” [39, 40,43], “sponsor” [41, 44], “collaboratore” [41], o “affiliazione“[42]. Abbiamo analizzato i risultati per account perché (1) l’account dovrebbe di solito rappresentare la “parte responsabile”, che è la persona o l’organizzazione legalmente responsabile dell’adempimento dei requisiti di registrazione e di segnalazione degli studi, e (2) perché non eravamo a conoscenza di un altro metodo per identificare tutti gli studi, o anche tutti gli account, associati ad ogni organizzazione.
Non sempre siamo stati in grado di determinare quali studi sono stati associati a specifiche organizzazioni, e le organizzazioni potrebbero non sapere quali account utilizzano i loro investigatori. Le organizzazioni potrebbero lavorare con ClincalTrials.gov per identificare gli indirizzi e-mail non funzionanti, aggiornare le informazioni di contatto degli amministratori, assegnare e identificare un amministratore responsabile della supervisione di ogni account e creare una relazione uno a uno tra ogni account e l’organizzazione. Ad esempio, ClinicalTrials.gov potrebbe identificare più account gestiti da amministratori della stessa organizzazione e aiutare le organizzazioni a spostare le informazioni in un unico account. Le organizzazioni dovrebbero prepararsi prima di centralizzare le loro registrazioni; l’amministrazione centralizzata potrebbe ridurre la registrazione e la rendicontazione delle prove se gli amministratori non hanno il tempo, la formazione e le risorse per gestire questi compiti in modo efficace.
Abbiamo richiesto informazioni a un amministratore per ogni organizzazione e gli amministratori potrebbero non essere a conoscenza di politiche e pratiche che riguardano altre parti delle loro organizzazioni (ad esempio, IRB, gestione delle sovvenzioni). Infine, alcune organizzazioni sono state erroneamente classificate su ClinicalTrials.gov (ad esempio, organizzazioni non statunitensi); non sappiamo quante organizzazioni sono state inavvertitamente incluse o escluse a causa di un’errata classificazione.
Ricerca futura
Sono necessarie ulteriori ricerche per determinare come sostenere la registrazione e la rendicontazione delle prove presso diversi tipi di organizzazioni. Alcune grandi organizzazioni registrano diverse prove ogni settimana, mentre altre ne registrano alcune ogni anno. Per le piccole organizzazioni, l’assunzione di personale per sostenere la registrazione e la rendicontazione delle prove potrebbe essere proibitiva. Ulteriori ricerche qualitative potrebbero esplorare come i diversi tipi di organizzazioni rispondono a questi requisiti.
I futuri sondaggi potrebbero esaminare i predittori di conformità con i requisiti di registrazione e segnalazione delle prove. Anche se ci sono importanti variazioni nella politica e nella pratica, ulteriori analisi quantitative avrebbero poco valore immediato perché la maggior parte delle organizzazioni ha una bassa conformità[37-45]. Invece, studi di casi dettagliati potrebbero essere più utili per identificare le migliori pratiche. Ad esempio, la Duke Medicine ha sviluppato un approccio centralizzato[55], e il Dipartimento degli Affari dei Veterani degli Stati Uniti (VA) ha descritto molteplici sforzi per sostenere la trasparenza, compreso un “sistema di portale interno basato sul web” [54]. La National Clinical Trials Registration and Results Reporting Taskforce è una rete di amministratori che si incontrano mensilmente in teleconferenza, condividono le risorse (ad esempio, materiale didattico) e forniscono una formazione informale tra pari. Poiché l’industria sembra fare meglio del mondo accademico[37, 39- 44], potrebbe essere utile per le organizzazioni accademiche comprendere i metodi utilizzati dall’industria per monitorare e segnalare la conformità (vedi, ad esempio, [56]).
Abbiamo intervistato le organizzazioni dopo la pubblicazione della Regola Finale, e la maggior parte degli account ha completato l’indagine prima che la Regola Finale entrasse in vigore, diversi mesi prima della data di conformità[34]. I nostri risultati dovrebbero essere considerati una “linea di base” per studi futuri che indagano se le organizzazioni adottano nuove politiche e procedure e se assegnano nuove risorse per soddisfare i requisiti di registrazione e di reporting. Il governo federale stima che i costi di conformità per le organizzazioni saranno di 70.287.277 dollari all’anno[34]. Questo sondaggio, e i futuri aggiornamenti, potrebbero essere utilizzati per migliorare le stime dei costi di conformità.
Conclusioni
Per sostenere la registrazione degli studi clinici e la comunicazione dei risultati, le organizzazioni dovrebbero prendere in seria considerazione l’adozione di politiche appropriate, l’assegnazione di risorse per l’implementazione di tali politiche e la garanzia che vi siano conseguenze per gli sperimentatori che non si registrano e non comunicano i risultati della loro ricerca.
Box 1: Requisiti per la registrazione e la comunicazione dei risultati degli studi clinici
Comitato internazionale dei redattori di riviste mediche (ICMJE)
Per essere presi in considerazione per la pubblicazione, gli studi clinici devono essere registrati nel registro pubblico prima di iscrivere i partecipanti [29, 30].
I rapporti degli studi clinici devono includere una dichiarazione di condivisione dei dati [57].
Food and Drug Administration Amendments Act del 2007 (FDAAA) e The Final Rule[31, 34].
Gli studi clinici applicabili devono essere registrati su ClinicalTrials.gov entro 21 giorni dall’iscrizione del primo partecipante
Le registrazioni di prova devono includere i risultati primari e secondari, comprese le misure specifiche e i punti di tempo che saranno utilizzati per valutare i risultati della prova
I risultati devono essere comunicati entro 12 mesi dalla raccolta finale dei dati a sostegno dell’esito primario. (La Regola Finale ha ampliato questo requisito per includere sia i prodotti approvati che quelli non approvati).
I risultati devono includere gli esiti primari e secondari, tutti gli eventi avversi gravi, la mortalità di tutte le cause e gli eventi avversi che si verificano nel 5% dei partecipanti.
I risultati devono includere informazioni di base sull’età e sul sesso e, se raccolti, per razza o gruppo etnico
Le registrazioni devono essere aggiornate come segue: entro 15 giorni dalle modifiche allo stato di approvazione o di autorizzazione; entro 30 giorni dal raggiungimento della data di completamento primaria; ed entro 30 giorni dalle modifiche allo stato di reclutamento dello studio, allo stato di comitato di revisione per la protezione dei soggetti umani o alla parte responsabile
Istituti Nazionali di Sanità (NIH)[36].
I requisiti per le sperimentazioni cliniche finanziate dall’NIH rispecchiano i requisiti per le sperimentazioni applicabili ai sensi della FDAAA
Tutti gli studi clinici finanziati da NIH (in tutto o in parte) devono essere registrati e i loro risultati devono essere riportati su ClinicalTrials.gov
Box 2: Argomenti del sondaggio
Politiche e procedure
L’organizzazione ha una politica che richiede agli investigatori di registrare le loro prove? Una politica di segnalazione dei risultati?
Quali processi sono coperti da queste politiche?
Quando sono entrate in vigore queste politiche?
Queste politiche descrivono i processi per gli investigatori che entrano e escono dall’organizzazione?
Ci sono sanzioni per gli investigatori che non registrano le loro sperimentazioni o non riportano i loro risultati?
Personale e supporto
Quali funzioni svolgono i membri del personale (ad esempio, inserire i risultati, controllare i registri, educare gli investigatori)?
Quanti membri dello staff sono assegnati a supporto della registrazione della sperimentazione e della comunicazione dei risultati? Quanto tempo dedicano a queste attività?
Ci sono piani per assumere altro personale in futuro?
Sistemi di monitoraggio
L’organizzazione dispone di un sistema di monitoraggio della registrazione delle prove e dei risultati? Per notificare agli investigatori quando i risultati sono dovuti?
L’IRB controlla se le sperimentazioni sono registrate e segnalate?
File aggiuntivi
File aggiuntivo 1:Strumento di rilevamento. (DOCX 500 kb)File aggiuntivo 2:Conti ammissibili. (DOCX 452 kb)File aggiuntivo 3:Conti partecipanti. (DOCX 476 kb)File aggiuntivo 4:Risultati supplementari del sondaggio. (DOCX 440 kb)File aggiuntivo 5:Analisi di sensibilità. (DOCX 436 kb)
References
- Ross JS, Mulvey GK, Hines EM, Nissen SE, Krumholz HM. Trial publication after registration in ClinicalTrials.Gov: a cross-sectional analysis. PLoS Med. 2009; 6(9):e1000144. DOI | PubMed
- Dwan K, Gamble C, Williamson PR, Kirkham JJ, Reporting Bias Group. Systematic review of the empirical evidence of study publication bias and outcome reporting bias – an updated review. PLoS One. 2013; 8(7):e66844. DOI | PubMed
- Song F, Parekh S, Hooper L, Loke YK, Ryder J, Sutton AJ. Dissemination and publication of research findings: an updated review of related biases. Health Technol Assess. 2010; 14(8):iii, ix-xi, 1–193. DOI | PubMed
- Cruz ML, Xu J, Kashoki M, Lurie P. Publication and Reporting of the Results of Postmarket Studies for Drugs Required by the US Food and Drug Administration, 2009 to 2013. JAMA Intern Med. 2017; 177(8):1207-1210. DOI | PubMed
- Jones CW, Handler L, Crowell KE, Keil LG, Weaver MA, Platts-Mills TF. Non-publication of large randomized clinical trials: cross sectional analysis. BMJ. 2013; 347:f6104. DOI | PubMed
- Pica N, Bourgeois F. Discontinuation and nonpublication of randomized clinical trials conducted in children. Pediatrics. 2016;138(3)
- Turner L, Shamseer L, Altman DG, Weeks L, Peters J, Kober T. Consolidated Standards of Reporting Trials (CONSORT) and the completeness of reporting of randomised controlled trials (RCTs) published in medical journals. Cochrane Database Syst Rev. 2012; 11:MR000030. PubMed
- Wieseler B, Kerekes MF, Vervoelgyi V, McGauran N, Kaiser T. Impact of document type on reporting quality of clinical drug trials: a comparison of registry reports, clinical study reports, and journal publications. BMJ. 2012; 344:d8141. DOI | PubMed
- Dechartres A, Trinquart L, Atal I, Moher D, Dickersin K, Boutron I. Evolution of poor reporting and inadequate methods over time in 20 920 randomised controlled trials included in Cochrane reviews: research on research study. BMJ. 2017; 357:j2490. DOI | PubMed
- Glasziou P, Meats E, Heneghan C, Shepperd S. What is missing from descriptions of treatment in trials and reviews?. BMJ. 2008; 336(7659):1472-1474. DOI | PubMed
- Dwan K, Altman DG, Cresswell L, Blundell M, Gamble CL, Williamson PR. Comparison of protocols and registry entries to published reports for randomised controlled trials. Cochrane Database Syst Rev. 2011; 1:MR000031.
- Hart B, Lundh A, Bero L. Effect of reporting bias on meta-analyses of drug trials: reanalysis of meta-analyses. BMJ. 2012; 344:d7202. DOI | PubMed
- Rising K, Bacchetti P, Bero L. Reporting bias in drug trials submitted to the Food and Drug Administration: review of publication and presentation. PLoS Med. 2008; 5(11):e217. DOI | PubMed
- Baudard M, Yavchitz A, Ravaud P, Perrodeau E, Boutron I. Impact of searching clinical trial registries in systematic reviews of pharmaceutical treatments: methodological systematic review and reanalysis of meta-analyses. BMJ. 2017; 356:j448. DOI | PubMed
- Jones CW, Keil LG, Holland WC, Caughey MC, Platts-Mills TF. Comparison of registered and published outcomes in randomized controlled trials: a systematic review. BMC Med. 2015; 13:282. DOI | PubMed
- Pranic S, Marusic A. Changes to registration elements and results in a cohort of ClinicalTrials.gov trials were not reflected in published articles. J Clin Epidemiol. 2016; 70:26-37. DOI | PubMed
- Tang E, Ravaud P, Riveros C, Perrodeau E, Dechartres A. Comparison of serious adverse events posted at ClinicalTrials.gov and published in corresponding journal articles. BMC Med. 2015; 13:189. DOI | PubMed
- Chalmers I. Underreporting research is scientific misconduct. JAMA. 1990; 263(10):1405-1408. DOI | PubMed
- Chan AW, Song F, Vickers A, Jefferson T, Dickersin K, Gotzsche PC. Increasing value and reducing waste: addressing inaccessible research. Lancet. 2014; 383(9913):257-266. DOI | PubMed
- Glasziou P, Altman DG, Bossuyt P, Boutron I, Clarke M, Julious S. Reducing waste from incomplete or unusable reports of biomedical research. Lancet. 2014; 383(9913):267-276. DOI | PubMed
- Maund E, Tendal B, Hrobjartsson A, Jorgensen KJ, Lundh A, Schroll J. Benefits and harms in clinical trials of duloxetine for treatment of major depressive disorder: comparison of clinical study reports, trial registries, and publications. BMJ. 2014; 348:g3510. DOI | PubMed
- Simes R. Publication bias: the case for an international registry of clinical trials. J Clin Oncol. 1986; 4(10):1529-1541. DOI | PubMed
- Meinert CL. Toward prospective registration of clinical trials. Control Clin Trials. 1988; 9(1):1-5. DOI | PubMed
- Chalmers TC. Randomize the first patient!. N Engl J Med. 1977; 296(2):107. PubMed
- Levine J, Guy W, Cleary PA. Therapeutic trials of psychopharmacologic agents: 1968–1972. In: McMahon FG, editor. Principles and techniques of human research and therapeutics. VIII. Futura Publishing Co: Mt. Kisco; 1974.
- Dickersin K, Rennie D. The evolution of trial registries and their use to assess the clinical trial enterprise. JAMA. 2012; 307(17):1861-1864. DOI | PubMed
- Dickersin K, Rennie D. Registering clinical trials. JAMA. 2003; 290(4):516-523. DOI | PubMed
- Zarin DA, Tse T, Williams RJ, Califf RM, Ide NC. The ClinicalTrials.gov results database—update and key issues. N Engl J Med. 2011; 364(9):852-860. DOI | PubMed
- De Angelis CD, Drazen JM, Frizelle FA, Haug C, Hoey J, Horton R. Is this clinical trial fully registered? A statement from the International Committee of Medical Journal Editors. JAMA. 2005; 293(23):2927-2929. DOI | PubMed
- DeAngelis CD, Drazen JM, Frizelle FA, Haug C, Hoey J, Horton R. Clinical trial registration: a statement from the International Committee of Medical Journal Editors. JAMA. 2004; 292(11):1363-1364. DOI | PubMed
- 121 Stat. 823. Food and Drug Administration Amendments Act (FDAAA) of 2007. Public Law 110–85.
- 45 CFR 102.3. Adjustment of civil monetary penalties for inflation. Federal Register.
- Zarin DA, Tse T, Williams RJ, Carr S. Trial reporting in ClinicalTrials.gov — The Final Rule. N Engl J Med. 2016; 375:1998. DOI | PubMed
- 42 CFR 11. Clinical trials registration and results information submission; Final rule. Federal Register.
- Hudson KL, Lauer MS, Collins FS. Toward a new era of trust and transparency in clinical trials. JAMA. 2016; 316(13):1353-1354. DOI | PubMed
- NOT-OD-16-149. National Institutes of Health. NIH policy on dissemination of NIH-funded clinical trial information. Federal Register.
- Zarin DA, Tse T, Williams RJ, Rajakannan T. Update on trial registration 11 years after the ICMJE policy was established. N Engl J Med. 2017; 376(4):383-391. DOI | PubMed
- Law MR, Kawasumi Y, Morgan SG. Despite law, fewer than one in eight completed studies of drugs and biologics are reported on time on ClinicalTrials.gov. Health Aff (Millwood). 2011; 30(12):2338-2345. DOI | PubMed
- Prayle AP, Hurley MN, Smyth AR. Compliance with mandatory reporting of clinical trial results on ClinicalTrials.gov: cross sectional study. BMJ. 2012; 344:d7373. DOI | PubMed
- Ross JS, Tse T, Zarin DA, Xu H, Zhou L, Krumholz HM. Publication of NIH funded trials registered in ClinicalTrials.gov: cross sectional analysis. BMJ. 2012; 344:d7292. DOI | PubMed
- . Accessed 24 Oct 2017.Publisher Full Text
- Chen R, Desai NR, Ross JS, Zhang W, Chau KH, Wayda B. Publication and reporting of clinical trial results: cross sectional analysis across academic medical centers. BMJ. 2016; 352:i637. DOI | PubMed
- Anderson ML, Chiswell K, Peterson ED, Tasneem A, Topping J, Califf RM. Compliance with results reporting at ClinicalTrials.gov. N Engl J Med. 2015; 372(11):1031-1039. DOI | PubMed
- Zarin DA, Tse T, Ross JS. Trial-results reporting and academic medical centers. N Engl J Med. 2015; 372(24):2371-2372. DOI | PubMed
- TranspariMED. Medical Research Ethics at Top UK Universities: Performance, Policies and Future Plans. 2017.
- . Accessed 13 Feb 2018.Publisher Full Text
- Weber WE, Merino JG, Loder E. Trial registration 10 years on. BMJ. 2015; 351:h3572. DOI | PubMed
- Dal-Re R, Ross JS, Marusic A. Compliance with prospective trial registration guidance remained low in high-impact journals and has implications for primary end point reporting. J Clin Epidemiol. 2016; 75:100-107. DOI | PubMed
- Scott A, Rucklidge JJ, Mulder RT. Is mandatory prospective trial registration working to prevent publication of unregistered trials and selective outcome reporting? An observational study of five psychiatry journals that mandate prospective clinical trial registration. PLoS One. 2015; 10(8):e0133718. DOI | PubMed
- Cybulski L, Mayo-Wilson E, Grant S. Improving transparency and reproducibility through registration: the status of intervention trials published in clinical psychology journals. J Consult Clin Psychol. 2016; 10.1037/ccp0000115.
- Viergever RF, Karam G, Reis A, Ghersi D. The quality of registration of clinical trials: still a problem. PLoS One. 2014; 9(1):e84727. DOI | PubMed
- Huic M, Marusic M, Marusic A. Completeness and changes in registered data and reporting bias of randomized controlled trials in ICMJE journals after trial registration policy. PLoS One. 2011; 6(9):e25258. DOI | PubMed
- Maruani A, Boutron I, Baron G, Ravaud P. Impact of sending email reminders of the legal requirement for posting results on ClinicalTrials.gov: cohort embedded pragmatic randomized controlled trial. BMJ. 2014; 349:g5579. DOI | PubMed
- Huang GD, Altemose JK, O’Leary TJ. Public access to clinical trials: lessons from an organizational implementation of policy. Contemp Clin Trials. 2017; 57:87-89. DOI | PubMed
- O’Reilly EK, Hassell NJ, Snyder DC, Natoli S, Liu I, Rimmler J. ClinicalTrials.gov reporting: strategies for success at an academic health center. Clin Transl Sci. 2015; 8(1):48-51. DOI | PubMed
- Evoniuk G, Mansi B, DeCastro B, Sykes J. Impact of study outcome on submission and acceptance metrics for peer reviewed medical journals: six year retrospective review of all completed GlaxoSmithKline human drug research studies. BMJ. 2017; 357:j1726. DOI | PubMed
- Taichman DB, Sahni P, Pinborg A, Peiperl L, Laine C, James A. Data sharing statements for clinical trials: a requirement of the International Committee of Medical Journal Editors. JAMA. 2017; 317:2491-2492. DOI | PubMed
- ICH. Guideline for good clinical practice. International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH). ICH Expert Working Group: Geneva; 1996.
- 46.102(g) C. Protection of Human Subjects. Code of Federal Regulations.
- 56.102 C. Institutional Review Boards. Code of Federal Regulations.
Fonte
Mayo-Wilson E, Heyward J, Keyes A, Reynolds J, White S, et al. (2018) Clinical trial registration and reporting: a survey of academic organizations in the United States. BMC Medicine 1660. https://doi.org/10.1186/s12916-018-1042-6




