
Abstract
Cosa c’è di nuovo?
- Questa revisione basata sull’evidenza dei geni che causano la Sindrome di Brugada (BrS) e test clinici di routine nei pazienti indica che 20 dei 21 geni non hanno prove genetiche sufficienti a supportare la loro causalità per la BrS.
- Solo il gene SCN5A è classificato come avente prove definitive come causa della BrS.
Quali sono le implicazioni cliniche?
- La valutazione genetica di routine di geni diversi da SCN5A non è attualmente garantita nella cura clinica dei pazienti con BrS.
- La valutazione genetica dei geni senza prove sufficienti a sostegno della causalità per la BrS può portare ad una errata interpretazione di varianti rare di questi geni e a conclusioni diagnostiche o interventi inappropriati per i pazienti e i membri della famiglia.
Editoriale, vedi pag . 1206
Il Progetto Genoma Umano ha permeato la scienza e la medicina con il progetto per la salute e le malattie umane.1 Negli ultimi 25 anni, l’evoluzione di questo storico risultato è stata straordinaria, consentendo di chiarire il fondamento genetico e molecolare di migliaia di malattie umane. L’impatto sull’erogazione della sanità è stato ampio e ha definito una nuova era della medicina di precisione. Ora i pazienti possono avere la loro diagnosi clinica geneticamente confermata, in alcuni casi gestita in modo specifico in base al genotipo, e possono scegliere di condividere le loro informazioni genetiche con i membri della famiglia a rischio per consentire test genetici presintomatici per il rischio di malattia. La fornitura di servizi di test genetici per le cure cliniche è ora ampiamente disponibile, e attualmente 54-057 test genetici offerti da 503 laboratori sono registrati dal Registro dei test genetici del Centro Nazionale per l’Informazione sulle Biotecnologie(https://www.ncbi.nlm.nih.gov/gtr/ interrogato il 12 ottobre 2017), esaminando 16-236 geni per 10-889 condizioni.
Il successo dell’implementazione della medicina di precisione, alla sua base, richiede che le associazioni di malattie geniche segnalate siano basate su prove affidabili per garantire l’applicazione appropriata delle informazioni genetiche nell’ottimizzazione delle cure, prevenendo al contempo conclusioni imprecise che possono causare danni. Rimane una notevole variabilità nel livello delle prove genetiche e sperimentali delle associazioni di malattie geniche segnalate, che sollevano dubbi sulla validità clinica di alcuni geni e potenziali preoccupazioni sulla loro inclusione nei test genetici clinici. Sono disponibili database di malattie geniche come Online Mendelian Inheritance in Man,2 anche se prezioso, manca il rigoroso approccio critico per esaminare la validità clinica delle associazioni proposte. La necessità di un metodo sistematico e basato sull’evidenza per la cura delle associazioni gene-malattie ha stimolato lo sviluppo del ClinGen (Clinical Genome Resource),3 un consorzio internazionale di genetisti, scienziati genomici ed esperti in ambito clinico, finanziato dall’Istituto Nazionale della Salute, con l’obiettivo comune di definire quadri di riferimento standardizzati e basati sull’evidenza per la valutazione delle associazioni genetiche segnalate per l’uso nella medicina di precisione.
Qui, riportiamo la prima applicazione del quadro di riferimento per la cura genica basata sull’evidenza ClinGen per le malattie del ritmo cardiaco genetico e la morte cardiaca improvvisa. La sindrome di Brugada (BrS) ha una prevalenza stimata di 1:2000.4,5 Quando è familiare, segue una modalità di ereditarietà autosomica dominante.6 Ad oggi, sono stati segnalati più di 20 geni associati a BrS6 e vengono regolarmente testate come cause monogene per questa condizione su una varietà di test genetici clinici in tutto il mondo. Poiché la penetrazione della malattia per la BrS è incompleta e legata all’età, i test genetici possono essere utilizzati per scopi diagnostici e per lo screening dei membri della famiglia a rischio. In questo contesto, le informazioni genetiche possono portare all’etichettatura della malattia nei singoli individui, influenzare il processo decisionale dei medici nel guidare i trattamenti preventivi per mezzo di un defibrillatore cardioverter impiantabile, o portare allo screening a cascata dei membri della famiglia. In considerazione dell’impatto significativo che una diagnosi di BrS (o di qualsiasi malattia genetica) può avere su un individuo e sulla sua famiglia, è fondamentale che solo i geni con una solida validità clinica siano valutati nella cura dei pazienti per minimizzare il rischio di un’errata interpretazione delle informazioni genetiche che possono in ultima analisi causare danni indebiti o ansia.
Metodi
Al fine di replicare i risultati o il processo di questo studio, i metodi analitici e il materiale di studio per questo studio sono descritti e referenziati di conseguenza, e tutti i dati sono disponibili per altri ricercatori nel supplemento di dati solo on-line(punteggi).
Selezione dei geni per la cura
I geni sono stati selezionati per la cura se soddisfano tutti i seguenti criteri: (1) ≥2 pubblicazioni nella letteratura medica peer-reviewed che suggeriscono la causalità di un singolo gene per il BrS, (2) letteratura riferita che presenta sia dati genetici che sperimentali, e (3) presenti su ≥3 gruppi di test genetici clinici BrS provenienti da laboratori diagnostici accreditati. Va notato che un certo numero di geni valutati per BrS sono stati implicati anche in altre malattie; tuttavia, questo sforzo non ha valutato la validità di alcun gene per disturbi diversi da BrS.
Quadro di cura dei geni
Abbiamo formato 3 team di cura genica per curare in modo indipendente ogni gene. Ogni team di cura era guidato da un medico genetista certificato dal consiglio e comprendeva altri 2 membri. Tutti i membri del team di cura dei geni dovevano essere laureati in genetica umana (3 master, 3 scienziati MD/PhD e 3 genetisti clinici MD). I team di cura genica hanno lavorato alla cieca rispetto ad altri team di cura e hanno utilizzato la struttura di cura genica ClinGen recentemente proposta.7 I membri del team di cura dovevano rivedere una procedura operativa standard per la cura dei geni utilizzando questo framework(https://www.clinicalgenome.org/curation-activities/gene-disease-validity/educational-and-training-materials/standard-operating-procedures/) e hanno ricevuto una presentazione webinar che illustrava l’applicazione del processo analitico.
Recentemente è stata pubblicata una descrizione dettagliata del quadro di classificazione della validità delle malattie geniche ClinGen.7 Questo quadro fornisce un approccio sistematico e basato sull’evidenza per valutare le associazioni di malattie geniche segnalate. Utilizzando un sistema di punteggio semiquantitativo, ogni relazione gene-malattia è classificata in 4 livelli di classificazione della validità clinica in base alla somma delle prove che la accompagnano: definitiva (12-18 punti e letteratura replicata), forte (12-18 punti), moderata (7-11 punti) e limitata (1-6 punti).
In breve, la struttura basata sull’evidenza valuta separatamente i dati genetici e quelli sperimentali e fornisce una metrica di punteggio basata sul livello di evidenza fornito nella letteratura pubblicata per il gene. I punteggi delle prove genetiche sono stati ponderati in base al disegno e alla qualità dello studio genetico. Per esempio, i geni implicati in studi con dati familiari, la segregazione delle varianti e il logaritmo significativo dei punteggi delle probabilità ricevono un punteggio assegnato maggiore rispetto ai geni implicati attraverso approcci genici candidati con coorti di piccole dimensioni.
I punteggi delle prove sperimentali si sono basati sull’interpretazione e sulla rilevanza fenotipica dei saggi in vitro che valutano le alterazioni funzionali delle varianti geniche complicate dalla malattia, così come sugli studi di organismi modello o di salvataggio, come proposto da MacArthur et al.8
I dettagli di questa matrice di punteggio e di un foglio di calcolo modello sono accessibili online7 https://www.clinicalgenome.org/working-groups/gene-curation/projects-initiatives/gene-disease-clinical-validity-scoring-matrix/.
Un gruppo di esperti di dominio clinico, composto da altri 9 individui con decine di anni di esperienza nell’assistenza clinica o nella ricerca nel campo della BrS, è stato incaricato di eseguire una valutazione finale e una classificazione gene-by-gene. Questi membri del panel avevano la possibilità di modificare la classificazione di ciascun gene (upgrade, no change, e downgrade) in base alla loro esperienza collettiva e alla valutazione indipendente della letteratura medica e delle prove scientifiche dopo la revisione dei riassunti del team di curatori. Ogni individuo del panel ha esaminato in modo indipendente i dati di ciascun gene e insieme il panel ha discusso le classificazioni di ciascun gene in teleconferenza e in incontri faccia a faccia(Figura I nel supplemento di dati solo online). Alla fine, il presidente del panel ha chiesto che ogni individuo fornisse un voto riservato su una classificazione finale per ogni gene.
Risultati
Identificare i geni di Brugada per la cura
Utilizzando una ricerca PubMed (termini di ricerca: gene [da scaffale] per PubMed [ricerca gene della sindrome di Brugada]), abbiamo identificato 23 geni che si riferiscono ad essere associati a BrS (Tabella 1). A partire da settembre 2017, abbiamo identificato 30 laboratori accreditati che offrono specifici panel multigene BrS, 15 negli Stati Uniti, 15 a livello internazionale (NCBI Genetic Testing Registry: https://www.ncbi.nlm.nih.gov/gtr/ interrogato il 29 giugno 2017; sito web GeneTests, https://www.genetests.org/disorders/?disid=33991, visitato il 9 settembre 2017). Questi pannelli variano nel numero di geni testati (range 3-23 geni, mediana 11 geni per pannello). Solo 4 geni erano presenti su tutti i pannelli di prova(SCN5A, GPD1L, CACNA1C e CACNB2). Dei 23 geni, 21 sono stati riportati in letteratura in pubblicazioni ≥2 peer-reviewed ed erano presenti su pannelli ≥3 BrS test genetici clinici (range 3-29 pannelli, mediana 15 pannelli per gene) (Figura 1). Questi 21 geni sono stati selezionati per la cura genica.
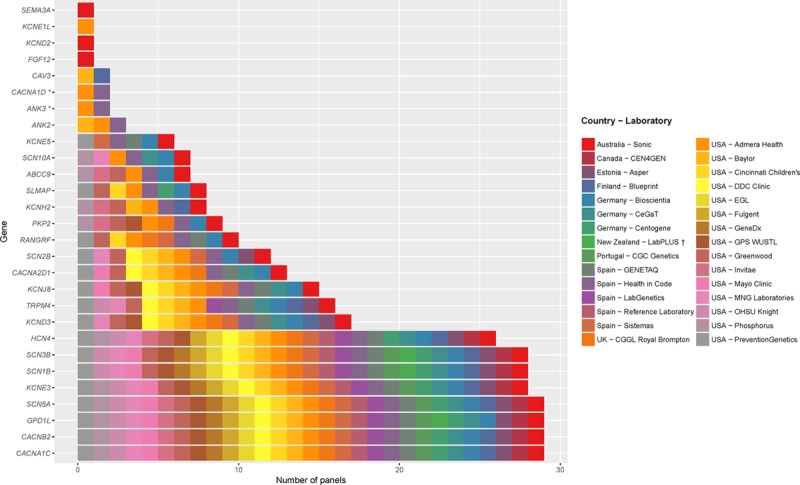
Figura 1.Geni inclusi nei pannelli di test genetici clinici per la sindrome di Brugada. Sono stati analizzati trenta pannelli multigene offerti in esclusiva per la sindrome di Brugada. L’asse x mostra il numero di pannelli che includono ogni gene. Le barre sono codificate a colori in base ai laboratori che offrono il test. L’elenco dei laboratori è riportato nella legenda a destra del grafico. *I 2 pannelli per il test della sindrome di Brugada per ANK3 e CACNA1D li hanno contrassegnati come “geni candidati senza prove, ma probabilmente correlati al fenotipo”. LabPLUS offre un pannello per i BrS di tipo 2, 5, 7, che non include SCN5A, CACNA1C e CACNB2.
Figura 1.Figura 1.Geni inclusi nei pannelli di test genetici clinici per la sindrome di Brugada. Sono stati analizzati trenta pannelli multigene offerti in esclusiva per la sindrome di Brugada. L’asse x mostra il numero di pannelli che includono ogni gene. Le barre sono codificate a colori in base ai laboratori che offrono il test. L’elenco dei laboratori è riportato nella legenda a destra del grafico. *I 2 pannelli per il test della sindrome di Brugada per ANK3 e CACNA1D li hanno contrassegnati come “geni candidati senza prove, ma probabilmente correlati al fenotipo”. LabPLUS offre un pannello per i BrS di tipo 2, 5, 7, che non include SCN5A, CACNA1C e CACNB2.
Cura dei geni
Nell’arco di 6 mesi, 3 team di cura genica hanno esaminato in modo indipendente la letteratura pubblicata per ogni gene e applicato il quadro analitico, spendendo 318-5 ore (14-5±5-9 ore per gene, media±SD). Un totale di 130 pubblicazioni sono state esaminate e registrate dai team di cura (in media 7 pubblicazioni per gene, range 2-19). Un elenco completo delle pubblicazioni esaminate per ogni gene può essere trovato nel supplemento dati online.
La Figura 2 riassume le classificazioni di validità clinica e i punteggi semiquantitativi per le prove genetiche e sperimentali dei team di cura per ogni gene. C’è stata una completa concordanza tra i 3 team di cura nelle classificazioni di validità clinica di tutti i 21 geni. SCN5A è stato l’unico gene che ha raggiunto il livello di evidenza definitiva per BrS. Tutti gli altri geni (20 su 21) sono stati classificati come prove limitate dai team di curatori.
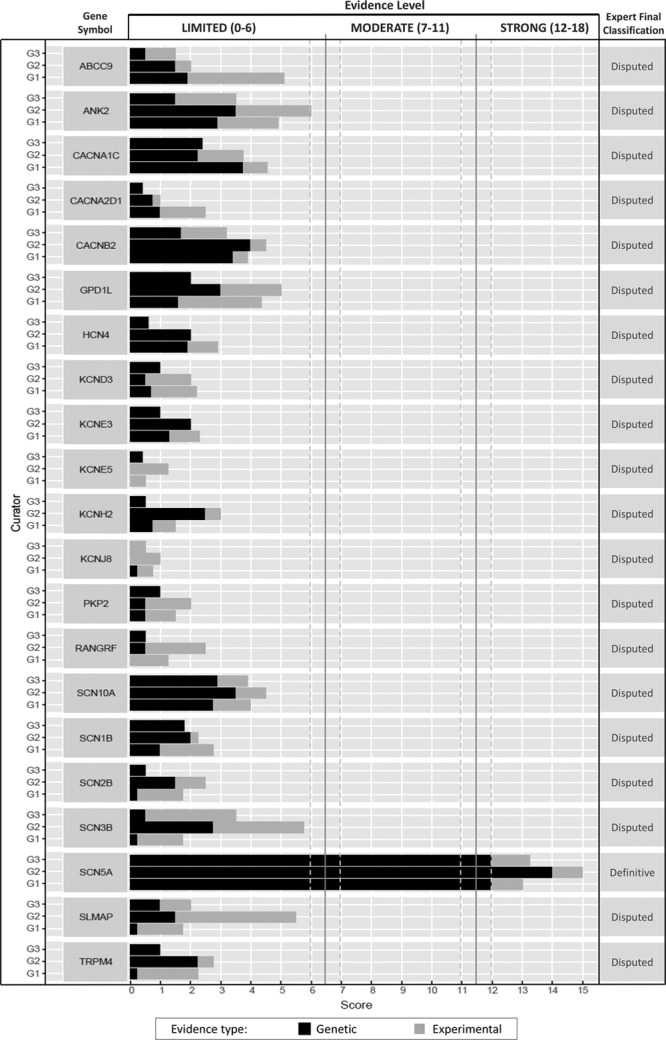
Figura 2.Classificazioni di validità clinica e punteggi della matrice per le associazioni geniche della sindrome di Brugada. Dei 21 geni della sindrome di Brugada (asse y) curati, solo SCN5A ha raggiunto la classificazione definitiva; tutti gli altri geni sono stati classificati come limitati. Ogni barra rappresenta i punteggi di un singolo gruppo di curatori (G) (G1, G2, G3).
Il panel di esperti di dominio clinico, pur concordando con l’applicazione del modello di punteggio da parte dei team di curatori, ha riclassificato tutti i 20 geni di prova limitata come controversi, concludendo che la letteratura attualmente pubblicata non è sufficiente per affermare la causalità per BrS per uno qualsiasi di questi 20 geni. Il consenso del gruppo di esperti è stato unanime (votato 9-0) per la riclassificazione basata su questioni specifiche relative alla metodologia degli studi genetici, la mancanza di prove statistiche di supporto, l’assenza di modelli animali geneticamente modificati che ricapitolano la malattia e l’interpretazione incerta dei dati sperimentali in vitro come correlati al fenotipo della malattia. Questi concetti sono discussi in dettaglio nella sezione Discussione. Tabelle riassuntive su una base gene per gene sono disponibili nel supplemento di dati online (punteggi).
Figura 2.Classificazioni di validità clinica e punteggi delle matrici per le associazioni geniche della sindrome di Brugada. Dei 21 geni della sindrome di Brugada (asse y) curati, solo SCN5A ha raggiunto la classificazione definitiva; tutti gli altri geni sono stati classificati come limitati. Ogni barra rappresenta i punteggi di un singolo gruppo di curatori (G) (G1, G2, G3).
Richiesta di presentazione di ClinVar
Per valutare l’impatto potenziale dei panel di test disponibili sulle interpretazioni delle varianti geniche, abbiamo analizzato il numero di presentazioni di ClinVar per BrS da parte dei laboratori di test clinici. La nostra query (sindrome di Brugada come [malattia/fenotipo] filtri: test clinici [queried 1 novembre 2017], cioè, esclusi solo i contributi di ricerca e letteratura) ha restituito 1223 variazioni per BrS in 21 diversi geni, il 33% in SCN5A (Figura 3A). Di varianti classificate come patogene e probabili patogene, il 6% era nei 20 geni di prova controversi, mentre il resto era in SCN5A. In totale, i geni di prova controversi più comunemente avevano presentato varianti classificate come incertezze di significato o con interpretazioni contrastanti (56% o 420/747 delle varianti presentate) (Figura 3B).
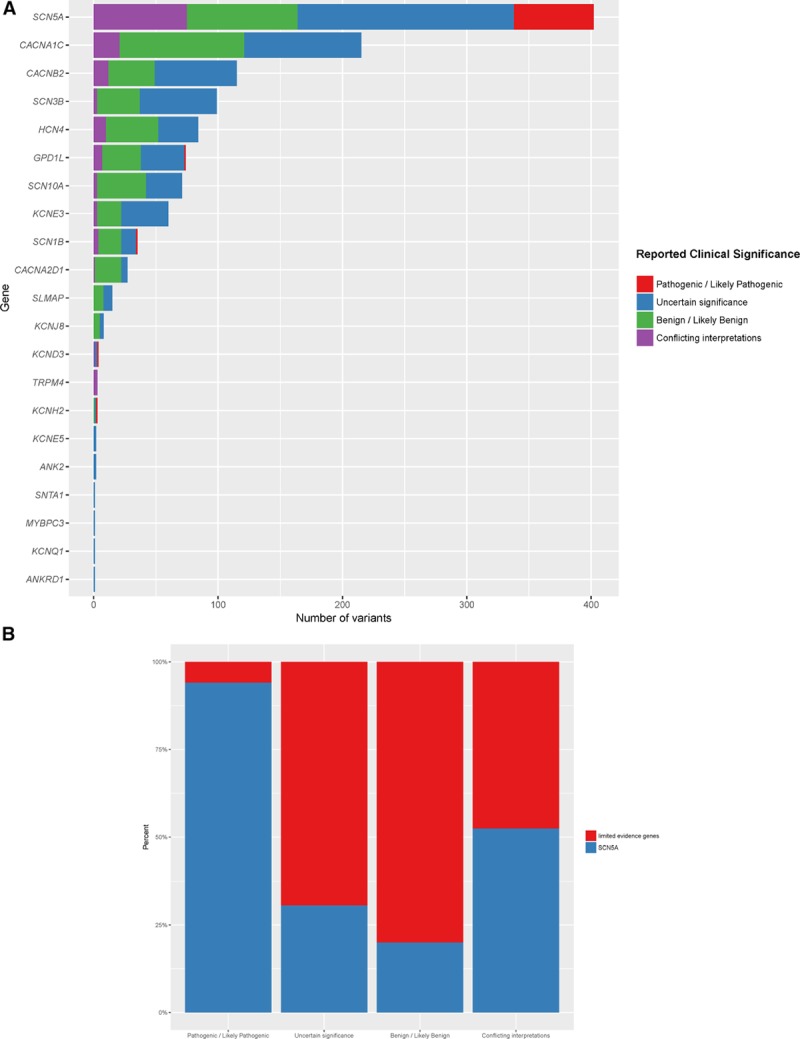
Figura 3.Varianti ClinVar per la sindrome di Brugada per gene e interpretazione clinica. A, Tutte le varianti presentate a ClinVar(http://clinvar.com/) dai laboratori clinici per la sindrome di Brugada sono state tracciate (N=1223, escludendo 182 varianti che erano solo letteratura o ricerca). I geni sono elencati lungo l’asse y, mentre l’asse x mostra il conteggio delle varianti per ogni gene. Le barre sono codificate a colori in base alla classificazione clinica delle varianti (vedi legenda). B, La proporzione relativa delle classificazioni di interpretazione delle varianti presentate per i geni SCN5A e Limited evidence.
Figura 3.Varianti ClinVar per la sindrome di Brugada per gene e interpretazione clinica. A, Tutte le varianti presentate a ClinVar(http://clinvar.com/) dai laboratori clinici per la sindrome di Brugada sono state tracciate (N=1223, escludendo 182 varianti che erano solo letteratura o ricerca). I geni sono elencati lungo l’asse y, mentre l’asse x mostra il conteggio delle varianti per ogni gene. Le barre sono codificate a colori in base alla classificazione clinica delle varianti (vedi legenda). B, La proporzione relativa delle classificazioni di interpretazione delle varianti presentate per i geni SCN5A e Limited evidence.
Discussione
In questo studio, abbiamo eseguito una cura basata sull’evidenza di 21 geni segnalati come cause a singolo gene per il BrS che vengono regolarmente testati in pazienti da laboratori accreditati per questa condizione di morte aritmica improvvisa. Evidentemente, solo 1(SCN5A) di 21 geni è stato classificato come gene di prova definitivo per il BrS. Tutti gli altri geni (20 su 21) hanno ricevuto una classificazione finale di contestazione per quanto riguarda le affermazioni sulla causalità per BrS da parte di un panel di esperti di dominio clinico.
I risultati di questo studio contestano l’inclusione di 20 geni, su 21, attualmente offerti per test genetici clinici per BrS. Tutti e 3 i team di curatori e il panel di esperti di dominio clinico hanno concordato che solo SCN5A ha prove sufficienti per giustificare l’inclusione di BrS nei panel di test genetici clinici e che le prove attuali non supportano la causalità o i test clinici dei 20 geni aggiuntivi curati. Il gruppo di esperti ha citato i seguenti fatti che da soli o in combinazione tra loro hanno portato alla conclusione della classificazione dei geni controversi: (1) i dati familiari o di segregazione dei casi affetti da varianti rare erano insufficienti a supportare la causalità del gene nella maggior parte degli studi; quando erano presenti dati sufficienti per il legame familiare con una regione genomica, non sono stati forniti dati di sequenziamento completi di tutte le varianti rare condivise tra gli individui affetti all’interno della regione genomica condivisa; (2) i geni riportati erano implicati di avere una causalità per BrS sulla base delle varianti di geni rari identificati nelle coorti BrS, un’osservazione che è insufficiente per rivendicare la causalità alla luce dell’osservazione ormai nota di frequenti e benigne variazioni genetiche rare in popolazioni umane sane comuni alla maggior parte dei geni; (3) la frequenza delle varianti geniche rare segnalate nei casi di BrS che portano a rivendicazioni di causalità è stata successivamente determinata essere grossolanamente sottovalutata con frequenze osservate nella popolazione generale che sono incompatibili con la causalità della malattia basata sulla prevalenza della malattia; (4) i dati funzionali di supporto sono stati limitati agli esperimenti in vitro, suggerendo un meccanismo molecolare plausibile ma senza ricapitolazione fenotipica e in assenza di saggi convalidati che dimostrino di distinguere le varianti genetiche rare che causano la malattia dalle varianti genetiche rare benigne; e (5) i dati esistenti non dimostrano l’evidenza statistica di un eccesso di varianti genetiche rare per il gene nei casi BrS rispetto ai controlli sani.
Dei 20 geni classificati come contestati, 19 su 20 sono stati originariamente riportati in studi preconcetti sui geni candidati sulla base della plausibilità biologica, in contrasto con un legame familiare a priori o altra metodologia imparziale a livello genomico, approcci raccomandati per la scoperta del gene.8 Manoscritti seminali per 13 dei 19 geni hanno riportato varianti rare solo in casi sporadici senza dati di segregazione o evidenza di eccesso di varianti rare rispetto ai soggetti di controllo(ABCC9, ANK2, CACNA2D1, HCN4, KCND3, KCNH2, KCNJ8, RANGRF, SCN1B, SCN2B, SCN3B, SLMAP, TRPM4). Sono stati riportati tre geni con una segregazione limitata in soli 2 individui in 2 generazioni(CACNA1C, KCNE5, e SCN10A). Solo 3 geni(CACNB2, KCNE3, PKP2) sono stati segnalati per avere >2 segregazioni all’interno di una famiglia.9–11 Anche se la descrizione originale di una famiglia che ospita una variante CACNB2 identificata attraverso un approccio genico candidato indicava 6 segregazioni,9 i nostri curatori hanno notato una persona colpita che non portava la variante segnalata nel CACNB2. Ulteriori rapporti indipendenti non sono stati identificati con prove genetiche o statistiche sufficienti a giustificare la classificazione al di là del livello di prova contestato per questo gene. Allo stesso modo, 4 e 3 segregazioni sono state riportate nei manoscritti originali usando approcci preconcetti al gene candidato che implicano rispettivamente KCNE3 e PKP2, ma senza sufficienti dati statistici, famiglie aggiuntive o prove di supporto in letteratura.10,11
Il gene SCN5A è stato l’unico gene, tra i 21 curati, ad essere classificato come definitivo da tutti e 3 i team di cura. È interessante notare che il manoscritto originale che riportava l’associazione di questo gene con BrS era anche uno studio genico candidato in piccoli pedigree.12 Tuttavia, i documenti successivi riportavano pedigree più grandi con segregazione e sufficienti prove statistiche a sostegno della causalità genica.13,14 Inoltre, i curatori e il gruppo di esperti hanno citato la densa letteratura che riportava varianti proteico-trunzanti in questo gene che segregano con il fenotipo, e una variante rara pubblicata (minore frequenza allelica <0,001) analisi del carico di geni segnalati per essere associati a BrS, che ha identificato un significativo eccesso di varianti SCN5A nei casi BrS rispetto ai controlli sani (20-4% contro il 2-4%, P<1-4×10-7) .15 Nella stessa analisi, gli autori hanno valutato 18 dei 20 geni di prova controversi e non hanno trovato alcun arricchimento significativo di varianti rare nei casi BrS.15
In particolare, il gene GPD1L ha ricevuto una classificazione limitata delle prove da parte di tutti e 3 i team di curatori e una classificazione finale dei controversi, nonostante la sua identificazione attraverso l’uso del legame genetico. Sebbene i geni identificati con una significativa segregazione e un apparente collegamento al fenotipo possano ricevere un punteggio più alto nell’applicazione del modello basato sull’evidenza, il punteggio assegnato dipende dal fatto che il sequenziamento completo di tutti i geni nella regione genomica collegata sia stato eseguito rispetto allo screening selezionato di soli geni specifici. Nel caso del GPD1L, i curatori e il gruppo di esperti hanno notato l’ampia dimensione della regione genomica collegata, il sequenziamento selezionato di solo un numero limitato di geni all’interno di questa grande regione (≈ 24 milioni di coppie di base) con la mancanza di un sequenziamento completo della regione da valutare per varianti geniche alternative, l’osservazione che la variante segnalata in questo gene è ora riconosciuta come presente in 1/5000 individui in database pubblici(http://gnomad.broadinstitute.org/gene/ENSG00000152642), e l’assenza di qualsiasi successiva descrizione familiare dopo la pubblicazione seminale nel 2002.16,17
Solo recentemente è stato riconosciuto che molte varianti benigne possono essere estremamente rare in una popolazione, e quindi la scoperta di una rara variazione di un gene in pazienti con una condizione genetica è ben lungi dall’essere una prova sufficiente per affermare la causalità. Tredici dei 20 geni di prova controversi sono stati pubblicati prima del 2013 prima della disponibilità di grandi database pubblici che indicano le frequenze delle varianti geniche in migliaia di individui, dove non c’è motivo di prevedere una sovrarappresentazione di questo particolare disturbo con una prevalenza di 1 nel 2000. In assenza di grandi database per confrontare le frequenze delle varianti riscontrate nei casi di malattia, le segnalazioni di malattie geniche precoci hanno tipicamente valutato una piccola coorte di controlli (100-1000 campioni) per decidere la rarità della variante. Due geni curati in questo studio sono stati originariamente implicati sulla base di varianti suggerite come rare negli studi originali, solo per essere successivamente notato di avere frequenza eterozigosi nei database pubblici maggiore o uguale alla prevalenza di BrS(KCNJ8 p.Ser422Leu, 1/250; SCN3B p.Leu10Pro, p.Val110Ile, entrambi 1/2500). Questo numero è ulteriormente evidenziato in una pubblicazione di Risgaard et al.18 valutare la prevalenza di mutazioni causate da malattie precedentemente concluse da pubblicazioni originali che implicano 12 geni BrS. Hanno trovato che su ≈4000 esomi individuali resi disponibili al pubblico nel 2012 dal National Heart, Lung, and Blood Institute GO Exome Sequencing Project, 1 su 23 individui era portatore di una mutazione BrS originariamente segnalata. Nonostante la classificazione come gene di prova definitiva, si stima che tra il 10% e il 20% delle mutazioni putativi segnalate in SCN5A possa essere stato erroneamente classificato, evidenziando le difficoltà di interpretazione delle varianti anche per i geni definitivi in assenza di grandi database.19
Infine, i geni BrS riportati, curati in questo studio, hanno tipicamente fornito dati funzionali in vitro per suggerire un meccanismo molecolare plausibile per la malattia causata da varianti rare. Tuttavia, una funzione alterata in vitro dimostrata in linee cellulari non cardiache, immortalate in assenza di ricapitolazione fenotipica in un animale intatto non è sinonimo di prova della causalità della malattia. Inoltre, nessuna delle metodologie sperimentali in vitro utilizzate per questi 20 geni controversi è stata convalidata per distinguere la funzione di varianti rare benigne note per esistere in popolazioni sane dalle varianti rare segnalate che causano la malattia. Per illustrare questo problema, la variante KCNJ8 segnalata p.Ser422Leu, la variante KCNJ8 originariamente implicata come causa di malattia per il BrS, è stata segnalata per avere un guadagno di 2 volte della funzione della corrente ionica in vitro rispetto al tipo selvatico, nonostante la sua presenza in 1/250 individui nella popolazione generale, una frequenza troppo comune per essere una causa di BrS.20,21 Analogamente, sono state segnalate differenze funzionali significative per le varianti segnalate in SCN3B come causa di BrS, nonostante la frequente presenza di queste stesse varianti in popolazioni presumibilmente sane.22,23 Nel contesto del ritmo cardiaco e delle caratteristiche elettrocardiografiche nell’uomo, ci si aspetterebbe che entrambe le varianti di geni rari e comuni conferiscano differenze funzionali che spieghino l’ampia gamma di variazione normale della frequenza cardiaca, dei parametri QRS e QT. Infatti, è riconosciuto che le variazioni geniche comuni (frequenza allelica minore >0,5% al 20%) in geni che causano malattie che causano canalopatie come KCNH2 e SCN5A possono provocare alterazioni funzionali in vitro simili a mutazioni putativi in quegli stessi geni e non portare ad un fenotipo associato.24,25
Dati i risultati di questo sforzo di cura genica, è importante considerare attentamente il motivo per cui i laboratori accreditati includono i geni nei panel di test BrS che non hanno prove sufficienti per la causalità della malattia. In primo luogo, gli organismi di accreditamento non richiedono ai laboratori di giustificare l’inclusione di geni nei panel per i test clinici. È anche possibile che il mercato sempre più concorrenziale della genetica di laboratorio abbia motivato un approccio più basato sui geni, che ha portato a una rapida espansione dei panel di geni. Le implicazioni pratiche dell’inclusione di questi geni di prova controversi sui panel per i test clinici sono potenzialmente dannose. I medici e i consulenti genetici possono confidare nel fatto che l’inclusione dei geni nei panel di malattie da parte di laboratori accreditati implica che essi abbiano valide associazioni con le malattie. Tuttavia, il test di geni con prove insufficienti di causalità crea inutili sfide per l’interpretazione delle varianti, in particolare per le varianti di perdita di funzionalità previste che possono essere erroneamente ritenute patogene e aumenta la possibilità di interpretazioni falso-positive, uno scenario che può portare a una previsione di rischio inappropriata nei membri della famiglia, a test clinici non necessari, a terapie profilattiche e a un significativo disagio all’interno di una famiglia. Ciò riguarda in particolare le malattie genetiche legate all’età con una penetranza incompleta, come il BrS, dove le osservazioni genetiche possono essere eccessivamente persuasive verso le conclusioni diagnostiche. Queste preoccupazioni non sono solo ipotetiche. La nostra interrogazione di ClinVar ha indicato che tra le varianti presentate classificate come patogene o probabili patogene per BrS, il 6% rappresentava varianti nei geni controversi. Inoltre, >50% delle varianti presentate in questi geni controversi erano classificate come di significato incerto o avevano interpretazioni contrastanti. Le ramificazioni di queste interpretazioni per le varianti in geni con prove insufficienti a sostenere la causalità per questa condizione di morte improvvisa sono preoccupanti e possono aver portato a cure inappropriate in alcune famiglie.
La conclusione del Progetto Genoma Umano nel 2003 è stata accolta con grande entusiasmo in tutti i campi della medicina e ha portato alla rapida segnalazione di migliaia di geni nell’ultimo decennio come causa di malattia. Il nostro studio mette in evidenza la crescente soglia ora necessaria per concludere la causalità delle malattie geniche alla luce dell’evoluzione delle conoscenze sulle variazioni genetiche naturali nella popolazione e la necessità di un’interpretazione prudente dei saggi funzionali che non ricapitolano il fenotipo della malattia e non sono convalidati per distinguere le varianti rare benigne da quelle rare che causano la malattia. Anche se la maggior parte delle associazioni di malattie geniche segnalate nel nostro studio non ha attualmente prove sufficienti per giustificare la loro inclusione nei test genetici clinici per la cura diretta dei pazienti e delle famiglie con BrS, molti di questi geni dovrebbero rimanere nel regno della ricerca, e alcuni di questi geni potrebbero in ultima analisi guadagnare validità clinica nei futuri sforzi di cura genica. Tuttavia, ulteriori ricerche volte a promuovere i geni controversi o a invocare nuove cause monogeniche per la malattia dovrebbero fornire prove genetiche utilizzando un’analisi imparziale dei dati genomici con prove statistiche di supporto. Questi possono includere studi su famiglie numerose o su centinaia di persone, che purtroppo non sono comuni in BrS. In alternativa, grandi coorti di controllo dei casi che dimostrano un eccesso statisticamente significativo di varianti rare in un gene di interesse tra i casi potrebbero soddisfare una forte evidenza genetica. Alla luce dell’osservazione comune di casi sporadici e non familiari di BrS, l’eredità oligogenica può giocare un ruolo significativo e creare un paesaggio genetico più impegnativo da studiare.
Un’implementazione di successo della medicina di precisione richiede che l’inclusione dei geni nei panel di test diagnostici rifletta accuratamente le associazioni di malattie geniche clinicamente valide. In assenza di validità clinica dei geni testati, test o interventi clinici non necessari, costosi e potenzialmente dannosi possono essere ordinati e utilizzati nella cura e nel processo decisionale clinico per un paziente o una famiglia. Se la medicina di precisione deve ottimizzare la cura dei pazienti e delle famiglie, è essenziale che i medici, i consulenti e i laboratori diagnostici si riuniscano per garantire l’inclusione più appropriata dei geni per i test diagnostici e la successiva interpretazione. Le nostre scoperte giustificano un approccio sistematico e basato sull’evidenza per valutare la validità delle associazioni di malattie geniche segnalate prima dell’uso nella cura del paziente.
Riconoscimenti
M.H.G. ha progettato lo studio. S.U. e M.H.G. hanno condotto una prima ricerca in letteratura e hanno determinato i geni da curare. S.M.H., G.C., R.J., E.L., S.M.J., M.S., C.F.M., S.B. e R.H.K. hanno eseguito la biocurazione genica basata sulle prove. S.M.H. ha riassunto i dati, ha condotto le analisi statistiche e ha generato i dati. J.G., M.C., A.C.S.S., V.N., J.S.W., R.E.H.R., M.J.A., A.A.M.W. e M.H.G. facevano parte del gruppo di esperti di eomi clinici e hanno esaminato i dati e le analisi della biocurazione. M.H.G. ha scritto l’articolo. Tutti gli autori hanno rivisto e curato l’articolo. Gli autori ringraziano Jennifer Goldstein, PhD, C. Lisa Kurtz, PhD, e Roozbeh Manshaei, PhD, per la loro assistenza.
Fonti di finanziamento
L’Istituto nazionale americano di ricerca sul genoma umano ha parzialmente finanziato questo studio. I membri dell’agenzia di finanziamento non hanno avuto alcun ruolo nella progettazione dello studio o nella raccolta, analisi e interpretazione dei dati o nella scrittura del manoscritto. L’autore corrispondente aveva pieno accesso a tutti i dati dello studio e aveva la responsabilità finale della decisione di presentare il manoscritto per la pubblicazione. Il Clinical Genome Resource Consortium è finanziato dal National Human Genome Research Institute (U41 HG006834, U01 HG007436). Il Dr Ware è sostenuto dal Wellcome Trust (107469/Z15/Z) e dalla British Heart Foundation. Il dottor Wilde è sostenuto dalla Netherlands Cardiovascular Research Initiative (Predict). Il dottor Gollob è sostenuto dal Mid-Career Scientist Award della Heart and Stroke Foundation (MC7449) e dalla Peter Munk Research Chair in Cardiovascular Molecular Medicine (Toronto General Hospital, Università di Toronto).
Informativa
Nessuna.
Materiale supplementare

References
- Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W, Funke R, Gage D, Harris K, Heaford A, Howland J, Kann L, Lehoczky J, LeVine R, McEwan P, McKernan K, Meldrim J, Mesirov JP, Miranda C, Morris W, Naylor J, Raymond C, Rosetti M, Santos R, Sheridan A, Sougnez C, Stange-Thomann Y, Stojanovic N, Subramanian A, Wyman D, Rogers J, Sulston J, Ainscough R, Beck S, Bentley D, Burton J, Clee C, Carter N, Coulson A, Deadman R, Deloukas P, Dunham A, Dunham I, Durbin R, French L, Grafham D, Gregory S, Hubbard T, Humphray S, Hunt A, Jones M, Lloyd C, McMurray A, Matthews L, Mercer S, Milne S, Mullikin JC, Mungall A, Plumb R, Ross M, Shownkeen R, Sims S, Waterston RH, Wilson RK, Hillier LW, McPherson JD, Marra MA, Mardis ER, Fulton LA, Chinwalla AT, Pepin KH, Gish WR, Chissoe SL, Wendl MC, Delehaunty KD, Miner TL, Delehaunty A, Kramer JB, Cook LL, Fulton RS, Johnson DL, Minx PJ, Clifton SW, Hawkins T, Branscomb E, Predki P, Richardson P, Wenning S, Slezak T, Doggett N, Cheng JF, Olsen A, Lucas S, Elkin C, Uberbacher E, Frazier M, Gibbs RA, Muzny DM, Scherer SE, Bouck JB, Sodergren EJ, Worley KC, Rives CM, Gorrell JH, Metzker ML, Naylor SL, Kucherlapati RS, Nelson DL, Weinstock GM, Sakaki Y, Fujiyama A, Hattori M, Yada T, Toyoda A, Itoh T, Kawagoe C, Watanabe H, Totoki Y, Taylor T, Weissenbach J, Heilig R, Saurin W, Artiguenave F, Brottier P, Bruls T, Pelletier E, Robert C, Wincker P, Smith DR, Doucette-Stamm L, Rubenfield M, Weinstock K, Lee HM, Dubois J, Rosenthal A, Platzer M, Nyakatura G, Taudien S, Rump A, Yang H, Yu J, Wang J, Huang G, Gu J, Hood L, Rowen L, Madan A, Qin S, Davis RW, Federspiel NA, Abola AP, Proctor MJ, Myers RM, Schmutz J, Dickson M, Grimwood J, Cox DR, Olson MV, Kaul R, Raymond C, Shimizu N, Kawasaki K, Minoshima S, Evans GA, Athanasiou M, Schultz R, Roe BA, Chen F, Pan H, Ramser J, Lehrach H, Reinhardt R, McCombie WR, de la Bastide M, Dedhia N, Blöcker H, Hornischer K, Nordsiek G, Agarwala R, Aravind L, Bailey JA, Bateman A, Batzoglou S, Birney E, Bork P, Brown DG, Burge CB, Cerutti L, Chen HC, Church D, Clamp M, Copley RR, Doerks T, Eddy SR, Eichler EE, Furey TS, Galagan J, Gilbert JG, Harmon C, Hayashizaki Y, Haussler D, Hermjakob H, Hokamp K, Jang W, Johnson LS, Jones TA, Kasif S, Kaspryzk A, Kennedy S, Kent WJ, Kitts P, Koonin EV, Korf I, Kulp D, Lancet D, Lowe TM, McLysaght A, Mikkelsen T, Moran JV, Mulder N, Pollara VJ, Ponting CP, Schuler G, Schultz J, Slater G, Smit AF, Stupka E, Szustakowki J, Thierry-Mieg D, Thierry-Mieg J, Wagner L, Wallis J, Wheeler R, Williams A, Wolf YI, Wolfe KH, Yang SP, Yeh RF, Collins F, Guyer MS, Peterson J, Felsenfeld A, Wetterstrand KA, Patrinos A, Morgan MJ, de Jong P, Catanese JJ, Osoegawa K, Shizuya H, Choi S, Chen YJ, Szustakowki J, International Human Genome Sequencing Consortium. Initial sequencing and analysis of the human genome.. Nature. 2001; 409:860-921. PubMed
- Amberger JS, Bocchini CA, Schiettecatte F, Scott AF, Hamosh A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders.. Nucleic Acids Res. 2015; 43(Database issue):D789-D798. PubMed
- Rehm HL, Berg JS, Brooks LD, Bustamante CD, Evans JP, Landrum MJ, Ledbetter DH, Maglott DR, Martin CL, Nussbaum RL, Plon SE, Ramos EM, Sherry ST, Watson MS, ClinGen. ClinGen: the Clinical Genome Resource.. N Engl J Med. 2015; 372:2235-2242. PubMed
- Wilde AA, Antzelevitch C, Borggrefe M, Brugada J, Brugada R, Brugada P, Corrado D, Hauer RN, Kass RS, Nademanee K, Priori SG, Towbin JA, Study Group on the Molecular Basis of Arrhythmias of the European Society of Cardiology. Proposed diagnostic criteria for the Brugada syndrome: consensus report.. Circulation. 2002; 106:2514-2519. PubMed
- Postema PG. About Brugada syndrome and its prevalence.. Europace. 2012; 14:925-928. PubMed
- Watanabe H, Minamino T. Genetics of Brugada syndrome.. J Hum Genet. 2016; 61:57-60. PubMed
- Strande NT, Riggs ER, Buchanan AH, Ceyhan-Birsoy O, DiStefano M, Dwight SS, Goldstein J, Ghosh R, Seifert BA, Sneddon TP, Wright MW, Milko LV, Cherry JM, Giovanni MA, Murray MF, O’Daniel JM, Ramos EM, Santani AB, Scott AF, Plon SE, Rehm HL, Martin CL, Berg JS. Evaluating the clinical validity of gene-disease associations: an evidence-based framework developed by the clinical genome resource.. Am J Hum Genet. 2017; 100:895-906. PubMed
- MacArthur DG, Manolio TA, Dimmock DP, Rehm HL, Shendure J, Abecasis GR, Adams DR, Altman RB, Antonarakis SE, Ashley EA, Barrett JC, Biesecker LG, Conrad DF, Cooper GM, Cox NJ, Daly MJ, Gerstein MB, Goldstein DB, Hirschhorn JN, Leal SM, Pennacchio LA, Stamatoyannopoulos JA, Sunyaev SR, Valle D, Voight BF, Winckler W, Gunter C. Guidelines for investigating causality of sequence variants in human disease.. Nature. 2014; 508:469-476. PubMed
- Antzelevitch C, Pollevick GD, Cordeiro JM, Casis O, Sanguinetti MC, Aizawa Y, Guerchicoff A, Pfeiffer R, Oliva A, Wollnik B, Gelber P, Bonaros EP, Burashnikov E, Wu Y, Sargent JD, Schickel S, Oberheiden R, Bhatia A, Hsu LF, Haïssaguerre M, Schimpf R, Borggrefe M, Wolpert C. Loss-of-function mutations in the cardiac calcium channel underlie a new clinical entity characterized by ST-segment elevation, short QT intervals, and sudden cardiac death.. Circulation. 2007; 115:442-449. PubMed
- Delpón E, Cordeiro JM, Núñez L, Thomsen PE, Guerchicoff A, Pollevick GD, Wu Y, Kanters JK, Larsen CT, Hofman-Bang J, Burashnikov E, Christiansen M, Antzelevitch C. Functional effects of KCNE3 mutation and its role in the development of Brugada syndrome.. Circ Arrhythm Electrophysiol. 2008; 1:209-218. PubMed
- Cerrone M, Lin X, Zhang M, Agullo-Pascual E, Pfenniger A, Chkourko Gusky H, Novelli V, Kim C, Tirasawadichai T, Judge DP, Rothenberg E, Chen HS, Napolitano C, Priori SG, Delmar M. Missense mutations in plakophilin-2 cause sodium current deficit and associate with a Brugada syndrome phenotype.. Circulation. 2014; 129:1092-1103. PubMed
- Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, Ortiz-Lopez R, Wang Z, Antzelevitch C, O’Brien RE, Schulze-Bahr E, Keating MT, Towbin JA, Wang Q. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation.. Nature. 1998; 392:293-296. PubMed
- Bezzina C, Veldkamp MW, van Den Berg MP, Postma AV, Rook MB, Viersma JW, van Langen IM, Tan-Sindhunata G, Bink-Boelkens MT, van Der Hout AH, Mannens MM, Wilde AA. A single Na(+) channel mutation causing both long-QT and Brugada syndromes.. Circ Res. 1999; 85:1206-1213. PubMed
- Hong K, Guerchicoff A, Pollevick GD, Oliva A, Dumaine R, de Zutter M, Burashnikov E, Wu YS, Brugada J, Brugada P, Brugada R. Cryptic 5’ splice site activation in SCN5A associated with Brugada syndrome.. J Mol Cell Cardiol. 2005; 38:555-560. PubMed
- Le Scouarnec S, Karakachoff M, Gourraud JB, Lindenbaum P, Bonnaud S, Portero V, Duboscq-Bidot L, Daumy X, Simonet F, Teusan R, Baron E, Violleau J, Persyn E, Bellanger L, Barc J, Chatel S, Martins R, Mabo P, Sacher F, Haïssaguerre M, Kyndt F, Schmitt S, Bézieau S, Le Marec H, Dina C, Schott JJ, Probst V, Redon R. Testing the burden of rare variation in arrhythmia-susceptibility genes provides new insights into molecular diagnosis for Brugada syndrome.. Hum Mol Genet. 2015; 24:2757-2763. PubMed
- Weiss R, Barmada MM, Nguyen T, Seibel JS, Cavlovich D, Kornblit CA, Angelilli A, Villanueva F, McNamara DM, London B. Clinical and molecular heterogeneity in the Brugada syndrome: a novel gene locus on chromosome 3.. Circulation. 2002; 105:707-713. PubMed
- London B, Michalec M, Mehdi H, Zhu X, Kerchner L, Sanyal S, Viswanathan PC, Pfahnl AE, Shang LL, Madhusudanan M, Baty CJ, Lagana S, Aleong R, Gutmann R, Ackerman MJ, McNamara DM, Weiss R, Dudley SC. Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias.. Circulation. 2007; 116:2260-2268. PubMed
- Risgaard B, Jabbari R, Refsgaard L, Holst AG, Haunsø S, Sadjadieh A, Winkel BG, Olesen MS, Tfelt-Hansen J. High prevalence of genetic variants previously associated with Brugada syndrome in new exome data.. Clin Genet. 2013; 84:489-495. PubMed
- Kapplinger JD, Giudicessi JR, Ye D, Tester DJ, Callis TE, Valdivia CR, Makielski JC, Wilde AA, Ackerman MJ. Enhanced classification of Brugada syndrome-associated and long-QT syndrome-associated genetic variants in the SCN5A-encoded Na(v)1.5 cardiac sodium channel.. Circ Cardiovasc Genet. 2015; 8:582-595. PubMed
- Barajas-Martínez H, Hu D, Ferrer T, Onetti CG, Wu Y, Burashnikov E, Boyle M, Surman T, Urrutia J, Veltmann C, Schimpf R, Borggrefe M, Wolpert C, Ibrahim BB, Sánchez-Chapula JA, Winters S, Haïssaguerre M, Antzelevitch C. Molecular genetic and functional association of Brugada and early repolarization syndromes with S422L missense mutation in KCNJ8.. Heart Rhythm. 2012; 9:548-555. PubMed
- Medeiros-Domingo A, Tan BH, Crotti L, Tester DJ, Eckhardt L, Cuoretti A, Kroboth SL, Song C, Zhou Q, Kopp D, Schwartz PJ, Makielski JC, Ackerman MJ. Gain-of-function mutation S422L in the KCNJ8-encoded cardiac K(ATP) channel Kir6.1 as a pathogenic substrate for J-wave syndromes.. Heart Rhythm. 2010; 7:1466-1471. PubMed
- Hu D, Barajas-Martinez H, Burashnikov E, Springer M, Wu Y, Varro A, Pfeiffer R, Koopmann TT, Cordeiro JM, Guerchicoff A, Pollevick GD, Antzelevitch C. A mutation in the beta 3 subunit of the cardiac sodium channel associated with Brugada ECG phenotype.. Circ Cardiovasc Genet. 2009; 2:270-278. PubMed
- Ishikawa T, Takahashi N, Ohno S, Sakurada H, Nakamura K, On YK, Park JE, Makiyama T, Horie M, Arimura T, Makita N, Kimura A. Novel SCN3B mutation associated with brugada syndrome affects intracellular trafficking and function of Nav1.5.. Circ J. 2013; 77:959-967. PubMed
- Anson BD, Ackerman MJ, Tester DJ, Will ML, Delisle BP, Anderson CL, January CT. Molecular and functional characterization of common polymorphisms in HERG (KCNH2) potassium channels.. Am J Physiol Heart Circ Physiol. 2004; 286:H2434-H2441. PubMed
- Tan BH, Valdivia CR, Rok BA, Ye B, Ruwaldt KM, Tester DJ, Ackerman MJ, Makielski JC. Common human SCN5A polymorphisms have altered electrophysiology when expressed in Q1077 splice variants.. Heart Rhythm. 2005; 2:741-747. PubMed
Fonte
Hosseini SM, Kim R, Udupa S, Costain G, Jobling R, et al. (2018) Reappraisal of Reported Genes for Sudden Arrhythmic Death. Circulation 138(12): . https://doi.org/10.1161/CIRCULATIONAHA.118.035070




