
Introduzione
La nostra attuale domanda globale di energia è soddisfatta in gran parte utilizzando risorse finite, ovvero combustibili fossili. Tuttavia, entro il 2030, il consumo di energia aumenterà probabilmente di quasi il 20% fino a 23 TW. 1, 2 Se continuiamo a dipendere principalmente dai combustibili fossili per la produzione di energia, le grandi quantità di gas serra che saranno prodotte potrebbero portare a un inquinamento ambientale catastrofico e a un cambiamento climatico globale. Per evitare questa incombente crisi energetica e ambientale, c’è un sostegno sostanziale e crescente a livello mondiale per l’uso di fonti di energia più sostenibili e rinnovabili. In questo contesto, sforzi significativi sono stati dedicati alla creazione di sistemi fotosintetici artificiali non convenzionali che sfruttano l’energia solare per produrre sostanze chimiche e combustibili. 3 AP offre il vantaggio di immagazzinare direttamente l’energia della luce solare in forme più fungibili come i combustibili liquidi che possono essere introdotti direttamente nelle infrastrutture esistenti. Molti processi che hanno dominato l’attenzione della comunità AP sono semplici e scalabili, come la scissione dell’acqua, e spesso comprendono fasi elementari radicali. Anche se tali sistemi producono H2 prezioso come combustibile nella semi-reazione riduttiva, le attuali reazioni di ossidazione dei sistemi AP di solito generano prodotti di basso valore economico (ad esempio, O2 dall’ossidazione dell’acqua, figura1A), che ha ostacolato la commercializzazione di queste tecnologie.
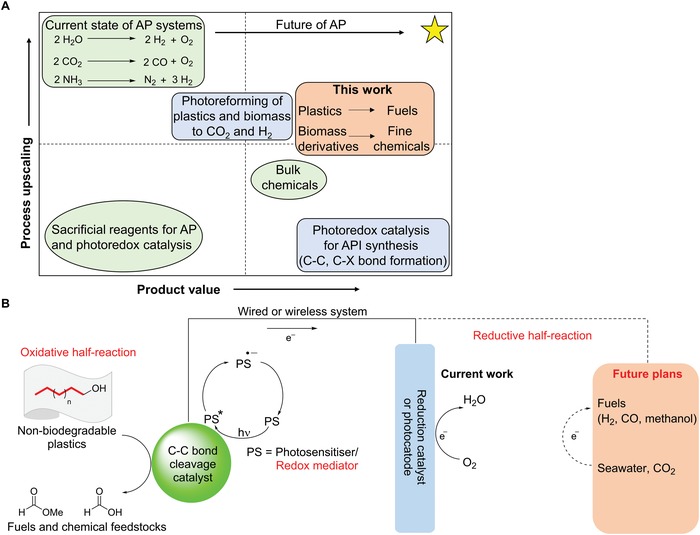
Figura 1.A) Confronti tra la catalisi AP e la catalisi del fotoredox nel contesto della scalabilità rispetto al valore del prodotto. Questo lavoro è un progresso verso il quadrante superiore destro, dove speriamo di ottenere processi sia su larga scala che economicamente attraenti, come indicato dalla stella gialla. Questo dato è stato adattato dalla referenza 24. B) Il nostro sistema AP proposto in cui l’ossidazione dell’acqua è sostituita da una scissione ossidativa del legame C-C per riciclare e valorizzare le plastiche non biodegradabili, mentre la mezza reazione riduttiva è attualmente la riduzione dell’O2, ma può potenzialmente essere altri processi fotocatalitici o fotoelettrocatalitici per produrre altri combustibili solari.
Recentemente, le reazioni basate sui radicali, sotto forma di catalisi fotoredox,4, 5, 6, 7, 8, 9, 10 sono emerse come interessanti, nuove vie per la sintesi organica e la produzione di prodotti chimici fini, che sono all’estremità opposta della catena del valore chimico ai sistemi AP ora (Figura 1A). I light harvester impiegati spesso includono costosi complessi di iridio o rutenio polipiridilici e loro derivati, e le reazioni sono di solito condotte su scale relativamente più piccole. I prodotti naturali o i principi attivi farmaceutici (API) di queste reazioni fotoredox sono spesso di pochi ordini di grandezza più costosi dei precursori, il che ha giustificato l’uso di costosi fotocatalizzatori metallici del gruppo del platino. Ad esempio, l’etamivan stimolante respiratorio costa 650 g-1 dollari USA e può essere sintetizzato a partire da 3,4-dimetossibenzaldeide (8 g-1 dollari USA), che mostriamo in questo articolo nelle sezioni seguenti. In particolare, a parte il lavoro seminale di MacMillan e dei collaboratori11 sulla decarbossilazione seguita da reazioni tandem C-C12 o C-X (X = O, N e S)13 di formazione del legame, la catalisi fotoredox è stata raramente utilizzata nel campo della catalisi selettiva C-La scissione del legame C nonostante il suo potenziale nella fase tardiva della decostruzione funzionale di molecole organiche complesse o la valorizzazione e il riciclaggio della lignina14, 15 e della plastica, per esempio. Wang e collaboratori16, 17 hanno riportato l’idrogenolisi fotocatalitica dei composti del modello della lignina, mentre Sarpong e collaboratori,18 Dong e collaboratori,18 Dong e collaboratori,19 Knowles e collaboratori,20 e Zuo e collaboratori21 hanno recentemente presentato l’attivazione C-C di ammine cicliche, chetoni o alcoli ciclici, seguita da derivatizzazione in condizioni fotoredox o termiche. Alcuni rapporti hanno descritto l’attivazione C-C accidentale in intermedi di reazione e substrati. 19, 22, 23 Tuttavia, l’applicazione intenzionale ad una vasta gamma di piccoli, aciclici, alcoli alifatici, e la catalisi a cascata di fotoredox catalisi da attivazione C-C e la funzionalizzazione di macromolecole rimane sconosciuta. Più criticamente, la catalisi del fotoredox rimane nel dominio dei processi di nicchia su piccola scala, come la produzione di API (Figura 1A).
Ultimamente, Reisner24 ha ravvivato il concetto di avvicinare l’AP e la catalisi radicale per aumentare la scalabilità del processo, generando al contempo più prodotti a valore aggiunto (Figura 1A). Questo concetto è stato sperimentato da Meyer e dai suoi collaboratori, che hanno dimostrato il suo potenziale per l’ossidazione del cloruro25 e dell’alcol benzilico con l’evoluzione simultanea dell’H2. 26 Altri, tra cui Sun e collaboratori,27, 28, 29, 30 Meyer e collaboratori,31, 32, 33, 34, 35 Berlinguette e collaboratori,36, 37, 38 Reisner e collaboratori,14, 39, 40, 41 e Lei e co-lavoratori,42, 43, 44 hanno successivamente dimostrato che il divario tra AP e catalisi radicale può essere potenzialmente colmato attraverso la valorizzazione della biomassa, la funzionalizzazione delle ammine, l’ossidazione di altri alogenuri, la riduzione foto-elettrochimica della CO2 e persino la fotoringiovanimento delle materie plastiche.
Le materie plastiche sono diventate essenziali nella nostra vita quotidiana grazie al loro basso prezzo, alla loro versatilità e alla loro facilità di produzione. Tuttavia, molte di esse non sono biodegradabili e stanno causando problemi ambientali disastrosi sulla terraferma e anche negli oceani e nei corsi d’acqua. 45 Studi hanno dimostrato che le fibre microplastiche sono state trovate anche in organismi d’alto mare46 , riaffermando le conseguenze che le plastiche non biodegradabili possono avere anche su alcuni degli ecosistemi più remoti. Mentre il riciclaggio della plastica ha ottenuto un enorme sostegno in tutto il mondo negli ultimi anni, una grande porzione di polimeri non biodegradabili non viene recuperata e riciclata in modo eco-compatibile. Invece, i materiali plastici sono spesso degradati dalla combustione o pirolizzati per produrre H2 e piccole quantità di olefine a catena corta,47, 48, 49 con la generazione di significative quantità di CO2. In alternativa, i polimeri biodegradabili sono spesso più costosi da produrre e sono adatti solo per applicazioni specifiche. 50, 51 Queste problematiche evidenziano l’importanza di sviluppare strategie più sostenibili per non solo degradare, ma anche riutilizzare i rifiuti di plastica persistente senza generare nuovi inquinanti.
Sebbene la fotoringiovanimento della plastica per produrre carburante H2 come combustibile pulito presenti un approccio attraente per superare i problemi ambientali e contemporaneamente aggiungere valore all’AP (Figura 1A), tuttavia, gli studi precedenti hanno impiegato punti quantici tossici a base di Cd come fotocatalizzatori, e la plastica è stata convertita in miscele organiche intrattabili41 , entrambe indesiderabili. Inoltre, hanno ottenuto conversioni di circa il 40% per materie plastiche specifiche come il poliuretano e il polietilene tereftalato, e il loro processo richiedeva un pretrattamento alcalino per idrolizzare parzialmente il carbammato e i legami di estere. 41 Sebbene il lavoro più recente di Reisner e dei suoi collaboratori sulla fotoringiovanimento delle materie plastiche con CNxI catalizzatori /Ni2P presentano un progresso innovativo che ha superato la tossicità dei loro fotocatalizzatori originali contenenti Cd,52 il loro processo richiedeva ancora un pretrattamento alcalino, mentre le conversioni sono rimaste al di sotto del 50% e sono stati osservati prodotti multipli con una modesta selettività.
In questo contesto, il nostro gruppo ha perseguito attivamente i processi catalitici fotoredox che possono generare precursori organici a valore aggiunto53, 54 o degradare gli inquinanti,55 e possono eventualmente essere integrati in sistemi AP (Figura 1B). In particolare, abbiamo sviluppato fotocatalizzatori al vanadio (V) (2a-2f, Figura S1, Informazioni di supporto) in grado di scindere selettivamente il legame C-C attivato adiacente all’alcool benzilico nei substrati del modello di lignina (Figura 1B).2). 56, 57 Abbiamo usato l’etichettatura isotopica, l’analisi del prodotto, la spettroscopia, e la teoria funzionale della densità (DFT) calcoli per dimostrare che 2a- 2f operato come alcune delle prime istanze molecolari di fotocatalizzatori che assorbono la luce attraverso il trasferimento di carica ligand-to-metallo (LMCT) chimica a causa della noninnocenza redox del ligandi idrazone-immidare (Figura 2). 56 Il legame C-C di un alcol coordinato si fenderebbe per produrre un gruppo carbonilico come uno dei prodotti primari, mentre l’altro frammento radicale reagirebbe con l’O2 disciolto in aria per dare eventualmente un altro composto ossigenato. Questi prodotti iniziali potrebbero essere ulteriormente ossidati o idrolizzati in condizioni di reazione aerobica per diventare altri derivati ossigenati, tra cui alcoli, aldeidi, esteri e acidi carbossilici, che sono tutti precursori versatili e preziosi che possono subire trasformazioni successive attraverso reazioni stabilite. Ad esempio, uno dei nostri prodotti di reazione è il formiato fenilico, che costa circa 21 milioni di dollari USA-1, e può potenzialmente essere estratto dalla lignina da biomassa non alimentare che viene tipicamente scartata come rifiuto o bruciata come combustibile a basso costo.
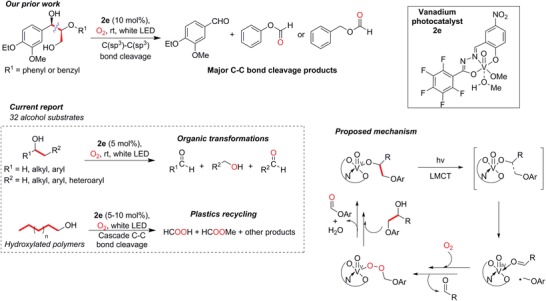
Figura 2.Il nostro gruppo ha riportato in precedenza studi sulla scissione del legame C-C nei modelli di lignina insieme al meccanismo proposto, la struttura del nostro fotocatalizzatore ottimale 2e, e le applicazioni di questo documento in diversi campi, compresa una dimostrazione nel riciclaggio di plastica non biodegradabile, come il polietilene, attraverso la scissione del legame C-C a cascata. I legami che subiscono la scissione sono evidenziati in grassetto e in rosso. I dettagli per le sintesi dei nostri fotocatalizzatori al vanadio sono rappresentati nella Figura S1 (Informazioni di supporto).
Tra questi composti ossigenati, gli alcoli, molti dei quali possono essere ottenuti commercialmente da biomasse e prodotti naturali come materie prime a prezzi accessibili, sono interessanti precursori di altri derivati a valle a valore aggiunto. Uno dei metodi più comuni per funzionalizzare gli alcoli è l’ossidazione ad aldeidi e acidi carbossilici, che comporta principalmente reazioni di attivazione del legame C-H. Al contrario, l’ossidazione selettiva, catalitica degli alcoli tramite scissione a legame singolo C-C è rara ed è stato un problema formidabile nella chimica sintetica, ma può essere potenzialmente un potente strumento nella fase tardiva di funzionalizzazione o di riciclaggio degli alcoli in molecole complesse e macromolecole. Diverse sfide includono la scarsa selettività dell’attivazione del legame C-C58, 59, 60 rispetto alle reazioni in altri gruppi funzionali, la mancanza di processi di formazione del legame che compensano energeticamente e l’inerzia cinetica del legame C-C non polare. Di conseguenza, gli esempi precedenti si sono spesso basati sul rilascio di ceppi ad anello in piccole molecole cicliche come il ciclopropano, il ciclolobutano, e persino il ciclopentano,61 o l’aromatizzazione di substrati prearomatici. 62 Inoltre, le condizioni termiche sono tipicamente utilizzate su substrati specializzati con gruppi funzionali appositamente costruiti per prevenire la decomposizione indesiderata. 58
Qui, applichiamo 2e nel fotoriduttore, scissione controllata del legame C-C e ossigenazione degli alcoli alifatici non attivati, in condizioni atmosferiche ambientali con l’aria come ossidante terminale. I prodotti includono idrocarburi ossigenati, che sono precursori auspicabili per la sintesi organica. Dimostriamo inoltre che questa strategia può essere utilizzata in più campi, non solo per le sintesi organiche, ma anche per lo smontaggio del legame C-C a cascata in polimeri biodegradabili e non biodegradabili terminati con alcol, che rappresentano un significativo progresso nella catalisi del fotoredox e nella bonifica ambientale.
Figura 1.A) Confronti tra AP e catalisi del fotoredox nel contesto della scalabilità rispetto al valore del prodotto. Questo lavoro è un progresso verso il quadrante superiore destro, dove speriamo di ottenere processi sia su larga scala che economicamente attraenti, come indicato dalla stella gialla. Questo dato è stato adattato dalla referenza 24. B) Il nostro sistema AP proposto in cui l’ossidazione dell’acqua è sostituita da una scissione ossidativa del legame C-C per riciclare e valorizzare le plastiche non biodegradabili, mentre la mezza reazione riduttiva è attualmente la riduzione dell’O2, ma può potenzialmente essere altri processi fotocatalitici o fotoelettrocatalitici per produrre altri combustibili solari.
Figura 2.Il nostro gruppo ha precedentemente riportato studi sulla scissione del legame C-C nei modelli di lignina insieme al meccanismo proposto, la struttura del nostro fotocatalizzatore ottimale 2e, e le applicazioni di questa carta in diversi campi, compresa una dimostrazione nel riciclaggio di plastiche non biodegradabili, come il polietilene, attraverso la scissione del legame C-C a cascata. I legami che subiscono la scissione sono evidenziati in grassetto e in rosso. I dettagli per le sintesi dei nostri fotocatalizzatori al vanadio sono rappresentati nella Figura S1 (Informazioni di supporto).
Risultati e discussione
Ottimizzazione delle condizioni e valutazione della tolleranza di gruppo funzionale
A causa del gran numero di impurità e dei diversi gruppi funzionali nelle macromolecole e nelle materie plastiche, abbiamo cercato di valutare la tolleranza dei gruppi funzionali del nostro fotocatalizzatore per determinare i limiti del nostro sistema. La nostra ottimizzazione delle condizioni è iniziata con 1-fenilpropan-2-olo (3) (Figura S2, Informazioni di supporto), un ingrediente intermedio e aromatico farmaceutico disponibile in commercio. Gratificantemente, il substrato è stato prontamente sottoposto a scissione del legame C-C raggiungendo l’89% di conversione entro 6 h in presenza di 5 mol% di 2e. I prodotti primari della reazione dovrebbero essere l’alcool benzilico (4) da ossigenazione con aria e acetaldeide (5), che sono stati ottenuti in rese moderate del 32% e 47%, rispettivamente (Figura S2 (voce 1), Informazioni di supporto). Abbiamo osservato che l’ossidazione in situ di 4 ha portato alla benzaldeide (6) in quantità significative (34%).
Per prevenire l’overoxidation di 4 e preservare la selettività della reazione, agenti di trasferimento di atomi di idrogeno (HAT) come il trifenilmetano, 9,10-diidroantracene (9,10-DHA), e 1,4-cicloesadiene (1,4-CHD) sono stati esplorati come antiossidanti per inibire le specie reattive dell’ossigeno (ROS) derivanti dall’uso dell’aria. I loro effetti sulla selettività sono riassunti nella Figura S2 (voci 2-5) (Informazioni di supporto). Antiossidante 1,4-CHD ha fornito i migliori risultati nel ridurre al minimo la sovraossidazione di 4, pur mantenendo la conversione quantitativa e una maggiore selettività dei prodotti primari. Sebbene fosse necessaria una quantità moderata di 1,4-CHD, gli additivi o i reagenti sacrificali stechiometrici nella catalisi del fotoredox, come acidi, basi o reagenti redox terminali, sono comunemente impiegati per migliorare la chemioselettività. Per la chimica fine e la produzione di API da parte della chimica organica sintetica, le scale sono più piccole e il costo aggiuntivo di questi additivi può essere giustificato dal valore aggiunto dei prodotti. I parametri aggiuntivi che abbiamo vagliato comprendevano l’uso di O2 al posto dell’aria, il caricamento del catalizzatore e i catalizzatori nella nostra libreria esistente (Figura S2 (voci 6-13), Informazioni di supporto). Inoltre, abbiamo condotto una fotolisi su scala più ampia di 3 a dieci volte (136 mg di substrato) le condizioni standard ottimizzate e ancora osservato la conversione completa del substrato e le rese di prodotto rispettabili, anche se con il doppio del tempo di reazione (Figura S2 (Voce 14), Informazioni di supporto). Anche gli esperimenti di controllo condotti in assenza di catalizzatore, luce e aria confermano che ognuno di questi è vitale. Questi ed altri dettagli di ottimizzazione sono compilati nelle Tabelle S1-S4 (Informazioni di supporto).
Inoltre, abbiamo studiato se gli accettatori di elettroni diversi dall’O2 possano essere utilizzati per il nostro sistema fotocatalitico, per verificare la fattibilità di combinare questa singolare scissione ossidativa degli alcoli con una diversa reazione di riduzione per la produzione di combustibili solari in un futuro sistema integrato AP. Abbiamo scelto il perossido di idrogeno, l’idroperossido di terz-butile (TBHP) e il rame (II) triflato come ossidanti rappresentativi per esaminare questa possibilità (Tabella S5, Informazioni di supporto). Mentre i perossidi erano alternative adeguate all’O2 per la scissione fotocatalitica del legame C-C in 3, l’uso di rame(II) triflato ha portato ad una reattività totalmente diversa senza prodotti dalla scissione del legame C-C prevista di 3. In particolare, la conversione di 3 in presenza dei perossidi (Tabella S5 (Voci 1 e 2), Informazioni di supporto) è stata più lenta delle reazioni con l’O2. Pertanto, le nostre condizioni ottimali di reazione includono l’uso di acetonitrile come solvente (0,50 mL), 0,10 × 10-3m di substrato, temperature ambiente, 5 mol% di catalizzatore 2e, 0,40 × 10-3m di 1,4-CHD, O2 come ossidante terminale, e un diodo emettitore di luce bianca 48 W (LED) come sorgente luminosa. In tutti gli esperimenti con gli altri substrati, le reazioni fotocatalitiche sono state condotte almeno due volte e di solito tre volte, con deviazioni dalle condizioni ottimali indicate, se del caso.
Con le condizioni di reazione ottimizzate in mano, si è proceduto ad esplorare la tolleranza del gruppo funzionale e gli effetti di vari sostituti sulla reattività e sulla distribuzione del prodotto. Poiché molti substrati complessi e macromolecole contengono molteplici funzionalità, è imperativo esaminare le prestazioni del catalizzatore in presenza di diversi gruppi funzionali. A questo scopo, abbiamo impiegato un metodo di screening del substrato basato su un meccanismo, recentemente riportato da Glorius e collaboratori con additivi piuttosto che con substrati reali. 63 Invece di sintetizzare tutti i derivati di 3 che contengono diversi gruppi funzionali, il derivato del benzene corrispondente è stato aggiunto alla miscela di reazione in quantità stechiometriche. Le reazioni di scissione del legame C-C fotocatalizzato hanno proceduto con conversioni essenzialmente quantitative e rendimenti da moderati ad elevati dei prodotti aldeidici e alcolici in presenza di un’ampia gamma di gruppi funzionali sia elettronico-donanti che -relevanti (Figura S3, Informazioni di supporto). Sono stati tollerati gli elettroni che donano metile e metossi, così come gli esteri di estrazione degli elettroni, gli alogeni, il nitro e il nitrile. In particolare, la fattibilità dei composti aromatici alogenati offre l’opportunità di una successiva trasformazione con la tradizionale chimica di transizione di accoppiamento metallo-catalizzata. Anche se le aniline sono state trovate per inibire il catalizzatore, senza alcun miglioramento delle conversioni dopo tempi di reazione prolungati, l’ammina potrebbe essere facilmente protetta da un gruppo terz-butiloxicarbonile (Boc) e la reazione procede senza problemi (Figura S3 (voce 8), Informazioni di supporto). L’ampia tolleranza del gruppo funzionale del nostro sistema fotocatalitico ci ha incoraggiato ad indagare sulla sua applicabilità agli alcoli più impegnativi.
Guidati dalle intuizioni dello screening basato sul meccanismo, abbiamo poi esaminato la reattività dei derivati di 3. Abbiamo ipotizzato che i substrati che generano anche radicali benzilici stabilizzati sulla scissione del legame C-C dovrebbero subire facili reazioni fotocatalitiche. Per testare questa idea, abbiamo esaminato il substrato 7 (Figura3A, voce 1) e ha utilizzato i suoi derivati per espandere rapidamente la libreria di substrati introducendo varie funzionalità su uno dei suoi anelli aryl. Gratificamente, 7 è stato completamente convertito in 4 e 6 entro 14 h, e la resa di oltre il 100% di 6 probabilmente è nata dalla sovraossidazione in situ di 4. Inoltre, come anticipato dai risultati dello screening meccanicistico, i sostituenti di elettroni che ritirano gli elettroni ostacolavano la reattività e portavano a tempi di reazione più lunghi, ma la conversione quantitativa era ancora ottenibile (Figura 3A, substrati 8-10 e 14-16). Allo stesso modo, i substrati con gruppi metilici e metossici che donano elettroni al para, meta, e anche le posizioni ortopediche stericamente esigenti hanno reagito prontamente per fornire rendimenti da moderati ad elevati dei prodotti attesi in condizioni standard (Figura 3A, substrati 11-13).
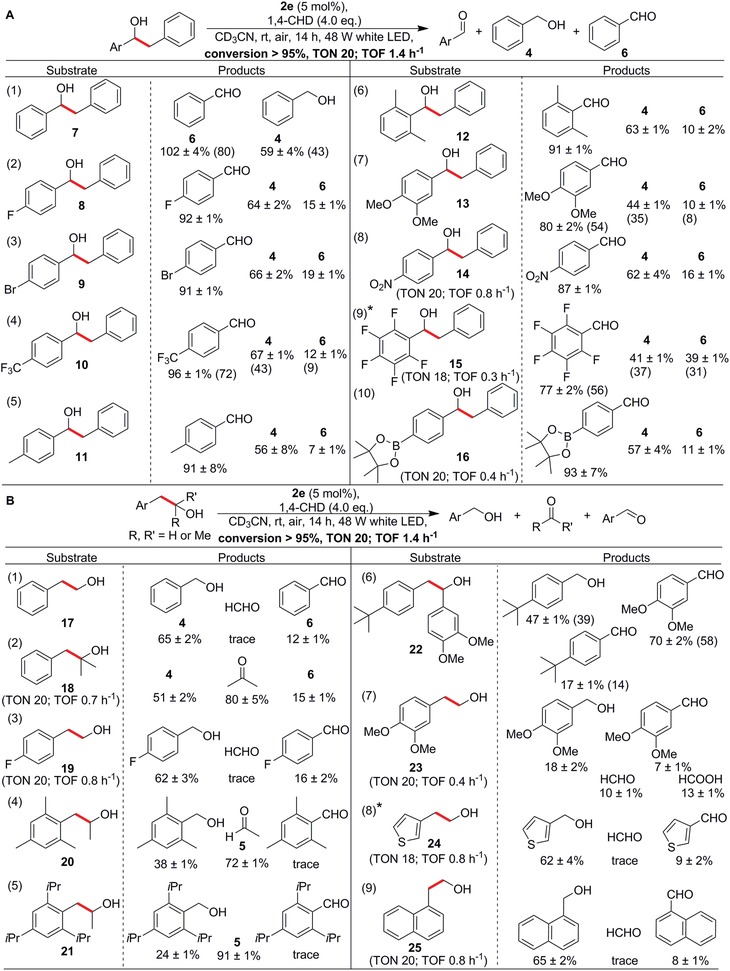
Figura 3.A) Effetti di diversi gruppi funzionali sul tasso e la distribuzione del prodotto della scissione fotocatalitica legame C-C in 7 e suoi derivati. B) Esempi di substrati stericamente esigenti 20 e 21, così come la presenza di un eteroatomea in 24 e anelli fusi in 25. I rendimenti del prodotto sono stati determinati dalla spettroscopia 1H NMR con 1,1,2,2,2-tetracloroetano come standard interno. I tempi di reazione sono stati di 24 ore per i substrati 14, 19 e 25; 68 ore per 15; 48 ore per 16; 30 ore per 18; e 46 ore per 23. I legami che subiscono la scissione sono evidenziati in grassetto e in rosso. I numeri di turnover (TON) e le frequenze di turnover (TOF) sono mostrati sotto la freccia di reazione o specificati quando diversi. Alcuni rendimenti rappresentativi isolati sono indicati tra parentesi. *Conversione 90%.
Inoltre, abbiamo scelto i substrati 17 e 18 (un alcool primario e un alcool terziario, rispettivamente), che genererebbero anche i radicali benzilici, e le reazioni hanno raggiunto elevate conversioni entro 14 e 30 h, rispettivamente (Figura 3B, voci 1 e 2), nonostante l’impedimento sterico previsto da quest’ultimo. La reazione con substrati contenenti mesitile, 2,4,6-triisopropilfenile, e 4-terz-butilfenile sostitutivi (substrati 20-22, rispettivamente) ha raggiunto la piena conversione e moderata resa del prodotto entro 14 h, indicando che la reazione procede anche con l’impedimento sterico. Substrati di alcool primario con fluoro carente di elettroni (19), metossi-donazione di elettroni (23), zolfo in 2-(tiofen-3-il)etan-1-olo (24), e anelli arilici fusi in 2-(tiofen-3-il)etan-1-olo (24), e anelli arilici fusi in 2-(naftalen-1-yl)ethan-1-ol (25) erano tutti adatti, con elevate conversioni e rese ragionevoli, a conferma che la ramificazione sul carbonio alcolico non era critica in questa reazione. Questi risultati hanno ulteriormente rafforzato la possibilità di applicare il nostro fotocatalizzatore per l’attivazione del legame C-C in diversi substrati, ricchi di elettroni, carenti di elettroni e persino stericamente impegnativi.
Infine, abbiamo esplorato la possibilità della sintesi API utilizzando alcuni dei prodotti di reazione come precursori. Ad esempio, si è scoperto che 13 forniscono selettivamente 3,4-dimetossibenzaldeide ad alto rendimento. Abbiamo poi utilizzato 3,4-dimetossibenzaldeide nella sintesi di etamivan (26), che è un API impiegato nel trattamento di malattie croniche del sistema respiratorio e di overdose di barbiturici. I dettagli riguardanti la sintesi di 26 sono riassunti nelle Informazioni di supporto.
Figura 3.A) Effetti di diversi gruppi funzionali sul tasso e sulla distribuzione del prodotto della scissione fotocatalitica del legame C-C fotocatalitico in 7 e suoi derivati. B) Esempi di substrati stericamente esigenti 20 e 21, così come la presenza di un eteroatomea in 24 e anelli fusi in 25. I rendimenti del prodotto sono stati determinati dalla spettroscopia 1H NMR con 1,1,2,2,2-tetracloroetano come standard interno. I tempi di reazione sono stati di 24 ore per i substrati 14, 19 e 25; 68 ore per 15; 48 ore per 16; 30 ore per 18; e 46 ore per 23. I legami che subiscono la scissione sono evidenziati in grassetto e in rosso. I numeri di turnover (TON) e le frequenze di turnover (TOF) sono mostrati sotto la freccia di reazione o specificati quando diversi. Alcuni rendimenti rappresentativi isolati sono indicati tra parentesi. *Conversione 90%.
Scioglimento a cascata C-C per applicazioni di energia sostenibile e di bonifica ambientale
A parte i substrati che generano radicali benzilici stabilizzati sulla scissione del legame C-C, eravamo intenti ad espandere questa reazione unica ad alcoli prontamente disponibili, non attivati, aciclici e alifatici che avrebbero formato radicali terziari, secondari o anche primari più reattivi. Gratificamente, i substrati che hanno portato a questi radicali progressivamente meno stabili procedevano (Figura4, substrati 27-30), anche se i tempi di reazione sono gradualmente aumentati con una concomitante riduzione della conversione. Alcuni dei prodotti di questi alcoli a catena più corta, come formaldeide, metanolo, propionaldeide e acetone, sono volatili, e la formaldeide è notoriamente instabile. Sotto l’irradiazione prolungata con il nostro setup, questi composti potrebbero subire reazioni collaterali come l’oligomerizzazione o evaporare a temperatura ambiente, tenendo conto della diminuzione dei rendimenti del prodotto, nonostante le conversioni relativamente elevate.
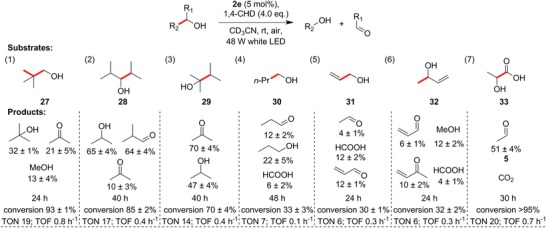
Figura 4.Reattività degli alcoli semplici di origine commerciale. I legami che subiscono la scissione sono evidenziati in grassetto e in rosso. I substrati 27 e 30 indicano una possibilità di scissione a cascata dei legami C-C, mentre 31 e 32 suggeriscono l’assenza di reazioni correlate a 1O2. Il substrato 33 evidenzia la possibilità di convertire i composti derivati dalla biomassa in prodotti chimici di base. I TON e TOF sono mostrati sotto ogni substrato.
Sorprendentemente, per 30, abbiamo osservato che in assenza dell’antiossidante 1,4-CHD, uno dei prodotti primari, 1-propanolo, sembrava subire una successiva scissione del legame C-C in situ per generare etanolo. Per escludere la possibilità dell’intermediazione dell’ossigeno singoletto (1O2) generato dal trasferimento di energia da 2e, abbiamo usato gli alcoli allilici, 31 e 32, come substrati (Figura 4, voci 5 e 6). Gli alcoli allilici sono noti per reagire con 1O2 per produrre idroperossidi o epossidi attraverso reazioni di tipo ene. Tuttavia, in presenza di 2e a basse conversioni, abbiamo rilevato principalmente prodotti di scissione del legame C-C, e piccole quantità di prodotti carbonilici, indicando che 1O2 era improbabile che fosse la specie attiva predominante. In tutti gli esempi fino ad ora, abbiamo osservato che ovunque il prodotto carbonilico primario sia stabile, i rendimenti sono molto elevati, mentre i rendimenti combinati dell’altro prodotto ossigenato rimangono rispettabili.
Oltre ai substrati che generano semplici radicali alchilici sulla scissione del legame C-C, abbiamo indagato se si possono formare altri tipi di radicali. Abbiamo scelto l’acido lattico racemico (33), che di solito si ottiene dalla fermentazione microbica degli zuccheri, ed è il monomero dell’acido polilattico plastico biodegradabile (PLA). Notevolmente, 33 ha subito l’attivazione del legame C-C quantitativamente in una reazione pulita (Figura 4, voce 7), producendo quantità moderate del volatile 5 e presumibilmente CO2, anche se quest’ultimo non è stato quantificato. Questo risultato ha identificato un altro tipo di substrato vitale per la nostra reazione, in cui si formano i radicali acilici. Inoltre, questo esempio evidenzia la possibilità di convertire l’acido lattico derivato dalla biomassa o di riciclare il PLA di nuovo a 5 come materia prima chimica.
La scissione del legame C-C a cascata in 1-butanolo osservata in assenza di 1,4-CHD ci ha ispirato a prendere di mira gli alcoli a catena più lunga e persino i substrati macromolecolari come i polimeri (figura5). Allo stesso modo, abbiamo condotto le reazioni utilizzando gli alcoli macromolecolari senza 1,4-CHD in modo che non avremmo avuto bisogno di additivi stechiometrici e non si sarebbero generati sottoprodotti inutili. Abbiamo iniziato con il polietilenglicole (PEG) poiché si scioglie facilmente a temperatura ambiente in acetonitrile, il nostro solvente di reazione. Di conseguenza, PEG 400 (34) è stato sottoposto a conversione completa entro 2,5 giorni nelle nostre condizioni ambientali ottimizzate, in assenza di 1,4-CHD per produrre quantità significative di acido formico, piccole quantità di formiato di metile, altri formiato alchilico, e prodotti oligomerici. Crediamo che la formaldeide inizialmente generata possa oligomerizzare alla paraformaldeide o sia stata ulteriormente ossidata ad acido formico durante la reazione, che potrebbe poi essere diventata esterificata.
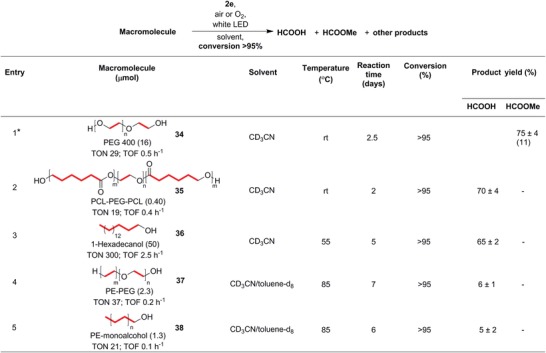
Figura 5.Scissione a cascata del legame C-C in polimeri macromolecolari idrossilati funzionalizzati biodegradabili (34-36) e non biodegradabili (37 e 38). In tutti i substrati polimerici, i siti di scissione del legame C-C sono evidenziati in rosso, con legami C-C di colore rosso multiplo che indicano l’accorciamento a cascata della catena polimerica. Sono indicati i rendimenti NMR dei prodotti identificati. I TON e TOF mostrati sono calcolati in base al numero di legami C-C nel substrato. *Condotto con 2 eq. di metanolo rispetto all’unità monomerica. Il rendimento isolato del formiato di metile dal trasferimento sotto vuoto è mostrato tra parentesi.
In un esperimento successivo, abbiamo aggiunto due equivalenti (eq.) di metanolo rispetto all’unità monomerica per intrappolare la formaldeide come prodotti più stabili. In particolare, i dati 1H NMR hanno mostrato una reazione molto più selettiva, con quantità significative di formiato di metile come prodotto principale (39, figura6A; Figura S4, Informazioni di supporto). Per verificare se 39 ha avuto origine dalla condensazione tra metanolo e acido formico per scissione del legame C-C dal PEG 400, o era interamente da ossidazione parziale del metanolo esogeno, abbiamo ripetuto l’esperimento ma con due eq. di metanolo deuterato (MeOD-d4) rispetto all’unità monomerica invece. Di conseguenza, abbiamo osservato l’assenza dei protoni metilici dell’estere ma la ritenzione del protone formiato nello spettro 1H NMR (Figura 6B). Inoltre, la comparsa di un picco a 3,69 ppm nella 2H{1H} NMR (Figura 6C) e un multipletto tra 50 e 52 ppm ma prevalentemente un singoletto a 162,9 ppm nello spettro 13C{1H} (Figura 6D) suggeriscono che 40 era effettivamente HC(O)OCD3 formato sia da MeOD-d4 che da acido formico derivato dal PEG 400. Questo promettente risultato ci permette di riciclare il PEG in preziosi precursori come gli esteri e gli acidi carbossilici.
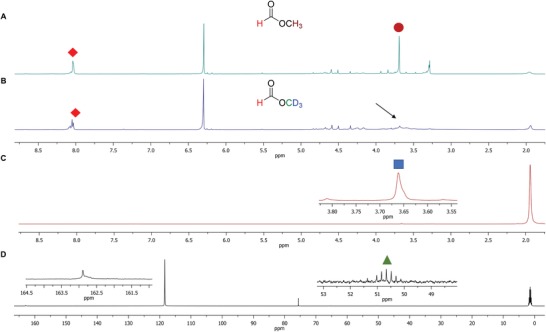
Figura 6.Figura 6. Conversione del PEG in formiato di metile in presenza di MeOH o MeOD-d4 in condizioni di reazione al fotoredox. A) Lo spettro 1H NMR della miscela di reazione con MeOH dopo 2,5 giorni di irraggiamento luminoso. B) 1H, C) 2H{1H}, e D) 13C{1H}. Spettri NMR della miscela di reazione con MeOD-d4 invece di MeOH dopo lo stesso tempo di irradiazione. La freccia nera indica l’assenza del picco CH3 quando si usa MeOD-d4 al posto di MeOH. Il quadrato rosso inclinato e il cerchio marrone rappresentano il formiato H e il CH3, mentre il quadrato blu e il triangolo verde corrispondono rispettivamente al CD3 e a un carbonio proveniente da MeOD-d4. I picchi a 6,30 ppm negli spettri 1H NMR hanno origine dallo standard interno di 1,1,2,2,2-tetracloroetilene, mentre i suoi carboni appaiono a 75,6 ppm nel 13C{1H}. Spettro NMR. Gli esperimenti NMR sono stati tutti condotti in CD3CN. Gli inserti mostrano sezioni dello spettro espanse verticalmente. L’osservazione del formiato di metile e di altri prodotti instabili alla conversione del substrato inferiore è mostrata nella Figura S4 (Informazioni di supporto).
In seguito al nostro successo con 34, abbiamo poi applicato questa scissione del legame C-C a cascata e l’ossigenazione al poli-ε-caprolattone.blocco-polietilenglicole-blocco-poli-ε-caprolattone (PCL-PEG-PCL) copolimero triblock (35). La reazione è avvenuta senza problemi con la trasformazione completa del polimero entro 2 giorni in condizioni ambientali, con l’acido formico come prodotto principale (Figura 5, voce 2). Analogamente, in un esperimento successivo, l’aggiunta di metanolo ha permesso di ottenere prevalentemente 39 come prodotto principale. Con questi due esempi di polimeri in mano, si è proceduto allo screening del PLA e dell’alcool polivinilico (PVA) per la reattività. Tuttavia, entrambi i polimeri hanno mostrato una limitata solubilità in acetonitrile. Inoltre, trattandosi di polimeri biodegradabili, abbiamo scelto di concentrare la nostra attenzione sui polimeri non biodegradabili, dannosi per l’ambiente e comunemente presenti negli inquinanti microplastici.
Incoraggiati dai rispettabili rendimenti del prodotto dal riciclo del biodegradabile 34 e del suo block-copolimero 35, abbiamo poi esaminato se anche gli analoghi non biodegradabili potessero essere substrati. Le materie plastiche basate sulla spina dorsale del polietilene, come il polipropilene e il polistirolo, purtroppo sono proliferate e sono diventate rifiuti recalcitranti che inquinano gli oceani. 64, 65, 66 Come substrato modello per il polietilene, abbiamo selezionato un alcool a catena lunga, 1-esadecanolo (36). A causa della sua scarsa solubilità in acetonitrile a temperatura ambiente, 36 ha dovuto essere riscaldato a 55 °C durante la reazione per consentire la completa dissoluzione. Gratificantemente, dopo 5 giorni di irradiazione con luce visibile e 2e, più del 95% dei 36 ha reagito per generare buoni rendimenti di acido formico come uno dei prodotti principali. Esperimenti di controllo condotti al buio o in assenza di 2e a 55 °C hanno dimostrato che 36 non hanno reagito affatto nello stesso periodo di tempo.
Dopo aver stabilito le condizioni di reazione per questo modello di polietilene, abbiamo preso di mira il copolimero polietilene-blocco-polietilene glicole (PE-PEG, 37). Anche se la reazione doveva essere condotta a 85 °C in una miscela di acetonitrile e toluene per dissolvere completamente il substrato, è stata osservata una conversione completa di entrambe le unità PE e PEG nello spettro 1H NMR della miscela di reazione dopo ≈7 giorni. I prodotti includevano acido formico e alchil formiato a catena più corta. Ci aspettiamo che gran parte dell’acido formico (punto di ebollizione di 101 °C) sia evaporato a 85 °C nelle condizioni di reazione, anche se si possono usare reattori sigillati ad alta pressione per trattenere i prodotti volatili. Allo stesso modo, gli esperimenti di controllo hanno confermato che 37 era stabile a 85 °C al buio o in assenza di 2e in condizioni di reazione altrimenti identiche.
Infine, abbiamo preso di mira il polietilene stesso, come rappresentante dei polimeri non biodegradabili derivati dalla spina dorsale del polietilene. Prevediamo che la scissione fotocatalitica C-C di altri derivati del polietilene (ad esempio, il polistirolo) dovrebbe essere ancora più facile, poiché i radicali intermedi si stabilizzerebbero nel punto di ramificazione e 2e può tollerare un’ampia gamma di sostituti. Abbiamo utilizzato il polietilene monoalcolico (38) in condizioni simili a quelle del 37. Notevolmente, dopo 6 giorni, è stata osservata una conversione completa del substrato, a differenza dell’esempio precedentemente riportato per la fotoringiovanimento delle materie plastiche, dove una conversione fino a solo il 50% è stata riportata dopo 18 giorni. 41, 52 Inoltre, il nostro sistema non richiede un pretrattamento alcalino per avviare l’idrolisi parziale dei polimeri per accelerare la reazione. 41, 52 Ci aspettiamo che la reazione proceda attraverso la scissione del legame C-C a cascata simile agli altri substrati macromolecolari che abbiamo testato, e un meccanismo di reazione plausibile basato sui prodotti osservati e sui nostri studi precedenti56, 57 è rappresentato nella Figura S5 (Informazioni di supporto).
Simili agli altri polimeri esaminati, l’acido formico e i formirati alchilici sono stati identificati come alcuni dei prodotti. Inoltre, la CO2 nello spazio di testa è stata rilevata anche dalla gascromatografia (quantità stimata del 62% della resa teorica), che potrebbe rappresentare la diminuzione della resa del prodotto nonostante le elevate conversioni. Al contrario, quando sono state condotte reazioni di 36 a temperature più basse di 55 °C, sono state ottenute rese moderate di acido formico, indicando che l’eccessiva ossidazione dell’acido formico a CO2 potrebbe essere il risultato delle elevate temperature di reazione di cui avevamo bisogno per solubilizzare i polimeri. D’altra parte, gli esperimenti di controllo con 38 condotti in assenza di luce o senza 2e alle stesse temperature elevate non hanno mostrato alcuna perdita osservabile di 38. Poiché l’acido formico può essere alimentato direttamente nelle celle a combustibile o può essere usato come deposito liquido per H2,67, 68, 69 mentre i formirati alchilici sono solventi, refrigeranti e prodotti chimici della piattaforma,70 il nostro approccio fotocatalitico per riadattare le plastiche non biodegradabili in utili combustibili e materie prime di piccole molecole può essere una semireazione ossidativa prospettica e scalabile in un sistema AP integrato (Figura 1B).
Figura 4.Reattività di semplici alcoli di origine commerciale. I legami che subiscono la scissione sono evidenziati in grassetto e in rosso. I substrati 27 e 30 indicano la possibilità di scissione a cascata dei legami C-C, mentre 31 e 32 suggeriscono l’assenza di reazioni correlate 1O2. Il substrato 33 evidenzia la possibilità di convertire i composti derivati dalla biomassa in prodotti chimici di base. I TON e TOF sono mostrati sotto ogni substrato.
Figura 5.Scissione a cascata del legame C-C in polimeri macromolecolari idrossilati funzionalizzati biodegradabili (34- 36) e non biodegradabili (37 e 38). In tutti i substrati polimerici, i siti di scissione del legame C-C sono evidenziati in rosso, con legami C-C di colore rosso multiplo che indicano l’accorciamento a cascata della catena polimerica. Sono indicati i rendimenti NMR dei prodotti identificati. I TON e TOF mostrati sono calcolati in base al numero di legami C-C nel substrato. *Condotto con 2 eq. di metanolo rispetto all’unità monomerica. Il rendimento isolato del formiato di metile dal trasferimento sotto vuoto è mostrato tra parentesi.
Figura 6.Figura 6. Conversione di PEG in formiato di metile prevalentemente in presenza di MeOH o MeOD-d4 nelle nostre condizioni di reazione del fotoredox. A) Lo spettro 1H NMR della miscela di reazione con MeOH dopo 2,5 giorni di irraggiamento luminoso. B) 1H, C) 2H{1H}, e D) 13C{1H}. Spettri NMR della miscela di reazione con MeOD-d4 invece di MeOH dopo lo stesso tempo di irradiazione. La freccia nera indica l’assenza del picco CH3 quando si usa MeOD-d4 al posto di MeOH. Il quadrato rosso inclinato e il cerchio marrone rappresentano il formiato H e il CH3, mentre il quadrato blu e il triangolo verde corrispondono rispettivamente al CD3 e a un carbonio proveniente da MeOD-d4. I picchi a 6,30 ppm negli spettri 1H NMR hanno origine dallo standard interno di 1,1,2,2,2-tetracloroetilene, mentre i suoi carboni appaiono a 75,6 ppm nel 13C{1H}. Spettro NMR. Gli esperimenti NMR sono stati tutti condotti in CD3CN. Gli inserti mostrano sezioni dello spettro espanse verticalmente. L’osservazione del formiato di metile e di altri prodotti instabili alla conversione del substrato inferiore è mostrata nella Figura S4 (Informazioni di supporto).
Conclusioni
In conclusione, abbiamo dimostrato la diversa applicabilità di una scissione selettiva alifatica C-C a legame singolo sotto l’irradiazione della luce visibile e condizioni di reazione lievi sia per la sintesi organica che per la bonifica ambientale delle materie plastiche eseguita in solventi organici. Il nostro approccio è adatto per una vasta gamma di substrati alcolici. Le condizioni aerobiche hanno permesso l’ossigenazione dei radicali alchilici transitori sulla scissione del legame C-C, che ha portato a nuovi prodotti alcolici che potrebbero subire un’attivazione controllata e a cascata del legame C-C. Abbiamo applicato questa strategia pionieristica a diversi polimeri idrossil-terminati, tra cui il polietilene non biodegradabile e il suo copolimero a blocchi, e abbiamo ottenuto formati e acido formico come prodotti. Sebbene questo studio abbia dimostrato la possibilità di scissione del legame C-C a cascata in substrati di modello a piccole molecole e polimeri di origine commerciale, non si tratta comunque di veri e propri prodotti di consumo in plastica. I nostri attuali sforzi sono diretti ad applicare questo approccio ad una gamma più ampia di polimeri, sia attraverso una prima preelaborazione per renderli solubili in acetonitrile, sia creando un analogo eterogeneo del nostro catalizzatore al vanadio che può operare in altri solventi o in sistemi (foto)-elettrochimici integrati per la produzione di combustibili solari. Così, questo lavoro evidenzia l’utilità dell’attivazione fotocatalitica C-C come strumento sintetico per trasformazioni inusuali di alcoli complessi e per il riutilizzo di inquinanti plastici persistenti in combustibili e materie prime chimiche utilizzando energia rinnovabile. Inoltre, offriamo un progresso concettuale di unificazione della catalisi fotoredox con la valorizzazione dei materiali di scarto nominali e degli inquinanti per creare più valore aggiunto, ma ancora potenzialmente scalabili reazioni alternative per i sistemi AP.
Conflitto di interesse
Una domanda di brevetto che copre parte di questo lavoro sui composti del modello di lignina e sulla lignina da biomassa è stata depositata (11201705500Q PCT-SG) da NTUitive, la società di innovazione e impresa della Nanyang Technological University, Singapore, nominando come inventori S.G., M.Đ., e H.S.S..
Informazioni di supporto
References
- Chem. Soc. Rev.. 2009; 38:25. PubMed
- Annu. Rev. Condens. Matter Phys.. 2011; 2:303.
- Chem. Commun.. 2018; 54:6554.
- Nat. Chem.. 2012; 4:854. PubMed
- Science. 2016; 354:1391. PubMed
- J. Am. Chem. Soc.. 2016; 138:5451. PubMed
- Inorg. Chem.. 2018; 57:2296. PubMed
- Science. 2016; 351:681. PubMed
- Org. Biomol. Chem.. 2014; 12:6059. PubMed
- Science. 2015; 349:1326. PubMed
- Science. 2017; 355:380. PubMed
- Nature. 2016; 536:322. PubMed
- Nature. 2018; 559:83. PubMed
- Nat. Energy. 2017; 2
- Angew. Chem., Int. Ed.. 2010; 49:3791.
- ACS Catal.. 2016; 6:7716.
- ACS Catal.. 2018; 8:2129.
- Science. 2018; 361:171. PubMed
- Nature. 2016; 539:546. PubMed
- J. Am. Chem. Soc.. 2016; 138PubMed
- Angew. Chem., Int. Ed.. 2016; 55
- J. Am. Chem. Soc.. 2013; 135:1823. PubMed
- Angew. Chem., Int. Ed.. 2016; 55
- Angew. Chem., Int. Ed.. 2019; 58:3656.
- J. Am. Chem. Soc.. 2015; 137:3193. PubMed
- J. Am. Chem. Soc.. 2014; 136:9773. PubMed
- J. Am. Chem. Soc.. 2016; 138PubMed
- Angew. Chem., Int. Ed.. 2016; 55:9913.
- J. Am. Chem. Soc.. 2017; 139PubMed
- Acc. Chem. Res.. 2018; 51:1571. PubMed
- J. Am. Chem. Soc.. 2015; 137:8321. PubMed
- J. Am. Chem. Soc.. 2017; 139PubMed
- J. Am. Chem. Soc.. 2017; 139PubMed
- J. Am. Chem. Soc.. 2017; 139PubMed
- Chem. Rev.. 2019; 119:4628. PubMed
- ACS Cent. Sci.. 2017; 3:778. PubMed
- Nat. Catal.. 2018; 1:501.
- Sustainable Energy Fuels. 2018; 2:1905.
- J. Am. Chem. Soc.. 2018; 140PubMed
- J. Am. Chem. Soc.. 2016; 138:9183. PubMed
- Energy Environ. Sci.. 2018; 11:2853.
- Sci. Adv.. 2018; 4PubMed
- Nat. Commun.. 2019; 10:639. PubMed
- Nat. Commun.. 2019; 10:467. PubMed
- Science. 2017; 358:843. PubMed
- Sci. Rep.. 2016; 6PubMed
- Ind. Eng. Chem. Res.. 2012; 51
- Energy Fuels. 2006; 20:754.
- Ind. Eng. Chem. Res.. 2016; 54:9536.
- Science. 2017; 358:872. PubMed
- Science. 2017; 358:868. PubMed
- J. Am. Chem. Soc.. 2019; 141PubMed
- Angew. Chem., Int. Ed.. 2019; 58:3456.
- Appl. Mater. Today. 2019; 15:192.
- Chem. Sci.. 2018; 9:3992. PubMed
- Chem. Sci.. 2015; 6:7130. PubMed
- ACS Catal.. 2017; 7:4682.
- Acc. Chem. Res.. 2017; 50:1702. PubMed
- Organometallics. 2002; 21:1329.
- Organometallics. 2019; 38:1200.
- ACS Catal.. 2017; 7:1340. PubMed
- Nature. 2019; 567:373. PubMed
- Angew. Chem., Int. Ed.. 2016; 55:4361.
- Science. 2015; 347:768. PubMed
- Sci. Adv.. 2017; 3PubMed
- Chem. Eng. News. 2018; 96:31.
- J. Power Sources. 2008; 182:124.
- Catal. Sci. Technol.. 2016; 6:12.
- Energy Environ. Sci.. 2010; 3:1207.
- Appl. Catal.. 1990; 57:1.
Fonte
Gazi S, Đokić M, Chin KF, Ng PR, Soo HS, et al. (2019) Visible Light–Driven Cascade Carbon–Carbon Bond Scission for Organic Transformations and Plastics Recycling. Advanced Science 6(24): 1902020. https://doi.org/10.1002/advs.201902020




