
Abstract
Introduzione
La terapia antiretrovirale (ART) ha trasformato l’infezione da HIV da una malattia uniformemente mortale in una condizione cronica che dura tutta la vita, salvando milioni di vite. Tuttavia, l’interruzione dell’ART porta a un rapido rimbalzo virale in poche settimane a causa della persistenza della latenza provirale in rare cellule T CD4+ a riposo a lunga durata e possibilmente in macrofagi tissutali(Honeycutt et al., 2017). La latenza dell’HIV è definita come la presenza di un genoma provirale trascrizionalmente silenzioso, ma replicabile, ma compatibile con la replicazione. La latenza permette alle cellule infette di eludere sia i meccanismi di clearance immunitaria sia l’ART attualmente disponibile, che si basa esclusivamente sull’eliminazione del virus che si replica attivamente.
Un approccio ampiamente studiato per eliminare l’HIV latente è la strategia “shock and kill”, che consiste nel forzare la riattivazione dei provirus latenti (fase di “shock”) con l’uso di agenti invertitori della latenza (LRA), mantenendo l’ART per prevenire le infezioni de novo. Successivamente, la riattivazione dell’espressione dell’HIV esporrebbe tali cellule (cellule scioccate) all’uccisione per effetto citopatico virale e alla clearance immunitaria (fase di “uccisione”). Una varietà di LRA sono state esplorate in vitro ed ex vivo, con solo pochi candidati in fase avanzata di sperimentazione in studi clinici pilota sull’uomo. L’uso di inibitori dell’istone deacetilasi (HDACi: vorinostat, panobinostat, romidepsina e disulfiram) in studi clinici ha dimostrato un aumento della produzione di RNA associato alle cellule e/o della viremia plasmatica dopo somministrazione in vivo(Archin et al., 2012a; Elliott et al., 2015; Elliott et al., 2014; Rasmussen et al., 2014; Søgaard et al., 2015). Tuttavia, nessuno di questi interventi da solo è riuscito a ridurre significativamente le dimensioni del serbatoio latente dell’HIV(Rasmussen e Lewin, 2016).
Diversi ostacoli possono spiegare il fallimento degli LRA, come recensito in(Margolis et al., 2016; Rasmussen et al., 2016). Tuttavia, la sfida più grande fino ad oggi è la nostra incapacità di quantificare con precisione le dimensioni del serbatoio. La quantificazione assoluta (numero di cellule) del serbatoio latente in vivo (ed ex vivo) è stata finora tecnicamente impossibile. I saggi più sensibili, più rapidi e più semplici per misurare la prevalenza di cellule infette da HIV sono quelli basati sulla PCR, che quantificano le trascrizioni totali o integrate del DNA o dell’RNA dell’HIV. Tuttavia, questi test sovrastimano sostanzialmente il numero di cellule latenti infette, a causa della predominanza di genomi di DNA HIV difettosi in vivo(Bruner et al., 2016; Ho et al., 2013). Il miglior test attualmente disponibile per misurare il serbatoio latente è il test di crescita virale relativamente ingombrante (VOA), che si basa sulla quantificazione del numero di cellule T CD4+ a riposo che producono il virus infettivo dopo un singolo ciclo di massima attivazione delle cellule T in vitro. Dopo diverse settimane di coltura, l’escrescenza virale viene valutata con un test ELISA per l’antigene HIV-1 p24 o un test PCR per l’HIV-1 RNA nel supernatante di coltura. È importante notare che il numero di cellule latente infette rilevate nella VOA è 300 volte inferiore al numero di cellule T CD4+ a riposo che ospitano i provirus rilevabili dalla PCR.
Questo affidamento su un singolo ciclo di attivazione delle cellule T probabilmente stima erroneamente il serbatoio virale per due ragioni. In primo luogo, la scoperta di provirus intatti non indotti indica che la dimensione del serbatoio latente può essere molto maggiore di quanto si pensasse in precedenza: gli autori stimano che il numero possa essere almeno 60 volte superiore alle stime basate sulla VOA(Ho et al., 2013; Sanyal et al., 2017). Questo lavoro e quello di altri(Chen et al., 2017) evidenziano la natura eterogenea della latenza dell’HIV e suggeriscono che la riattivazione dell’HIV è un processo stocastico che riattiva solo una piccola frazione di virus latenti in un dato momento(Dar et al., 2012; Ho et al., 2013; Singh et al., 2010; Weinberger et al., 2005). In secondo luogo, la capacità dei provirus difettosi di essere trascritti e tradotti in vivo(Pollack et al., 2017): questo studio dimostra che, sebbene i provirus difettosi non possano produrre particelle infettive, essi esprimono RNA virale e proteine, che possono essere rilevabili da qualsiasi antigene p24 o test PCR utilizzati per la quantificazione della dimensione del serbatoio.
Pertanto, i saggi attuali sottovalutano il numero effettivo di cellule latenti infette, sia in vivo che ex vivo, e la dimensione reale del serbatoio dell’HIV deve ancora essere determinata. Pertanto, è stato difficile giudicare il potenziale degli esperimenti LRA in vitro (modelli primari di latenza), ex-vivo (campioni di pazienti) e in vivo (trial clinico).
La latenza dell’HIV è un processo complesso e multifattoriale (esaminato in[Dahabieh et al., 2015]). La sua costituzione e il suo mantenimento dipendono da: (a) fattori virali, come l’integrase che interagisce in modo specifico con le proteine cellulari, compreso il LEDGF, (b) fattori trans-acting(ad esempio, fattori di trascrizione) e la loro regolazione da parte dello stato di attivazione delle cellule T e gli stimoli ambientali che queste cellule ricevono, e (c) meccanismi cis-acting, come l’ambiente locale cromatina nel sito di integrazione del virus nel genoma. Recenti evidenze hanno anche evidenziato l’associazione di specifici siti di integrazione dell’HIV-1 con l’espansione clonale delle cellule latenti infette (rivista in[Maldarelli, 2016]).
Il ruolo del sito di integrazione dell’HIV nel genoma cellulare nello stabilire e mantenere la latenza dell’HIV è rimasto controverso. Mentre i primi studi hanno scoperto che il sito di integrazione dell’HIV influisce sia sull’ingresso nella latenza(Chen et al., 2017; Jordan et al., 2003; Jordan et al., 2001), sia sulla risposta virale agli LRA(Chen et al., 2017), altri studi non sono riusciti a trovare un ruolo significativo dei siti di integrazione nel regolare il destino dell’infezione da HIV(Dahabieh et al., 2014; Sherrill-Mix et al., 2013).
In questo studio, abbiamo utilizzato un nuovo virus reporter a doppio colore, HIVGKO, per indagare il potenziale di riattivazione di vari LRA nella popolazione latente pura. Abbiamo scoperto che la latenza è eterogenea e che solo una piccola frazione (<5%) delle cellule latenti infette viene riattivata dagli LRA. Dimostriamo anche che sia la localizzazione genomica che il contesto della cromatina del sito di integrazione influenzano il destino dell’infezione da HIV e l’inversione della latenza virale.
Risultati
Un reporter di seconda generazione a doppia fluorescenza HIV-1 (HIVGKO) per studiare la latenza
Il nostro laboratorio ha segnalato lo sviluppo di un virus a doppio marchio (DuoFluoI) in cui eGFP è sotto il controllo del promotore dell’HIV-1 nel 5′ LTR e mCherry è sotto il controllo del fattore di allungamento cellulare un promotore alfa (EF1α) (Calvaneseet al., 2013). Tuttavia, abbiamo notato che il modello è stato limitato da un modesto numero di cellule latentemente infette (<1%) generate indipendentemente dall’input virale (Figura 1-figure supplement 1A-1C), così come un’alta percentuale di cellule produttivamente infette in cui il promotore costitutivo EF1α non era attivo (GFP +, mCherry-).
Per affrontare questi problemi, che sospettavamo fossero dovuti alla ricombinazione tra le regioni 20-30 bp di omologia a N- e C-termini delle proteine fluorescenti adiacenti (eGFP e mCherry) (Salamangoet al., 2013), abbiamo generato una nuova versione del virus a doppia marcatura (HIVGKO), contenente un eGFP a commutazione di codice (csGFP) e una distinta proteina fluorescente non correlata mKO2 sotto il controllo di EF1α (Figura 1A). In primo luogo, la titolazione dell’input di HIVGKO ha rivelato che le cellule infette in modo produttivo e latente sono aumentate proporzionalmente all’aumento del virus di input(Figura 1B e Figura 1-figure supplement 1), a differenza del DuoFluoI originale (Figura 1-figure supplement 1). In secondo luogo, il confronto tra le cellule T CD4+ primarie CD4+ infette da HIVGKO o il DuoFluoI originale ha rivelato un aumento delle cellule infette a doppia positività (csGFP+ mKO2+) nelle cellule infette da HIVGKO(Figura 1C). Una piccola percentuale di cellule csGFP+ mKO2- era ancora visibile nelle cellule infette da HIVGKO. Abbiamo generato un virus HIVGKO privo della regione promotrice U3 del 3′LTR (ΔU3-GKO), con il risultato di un virus integrato privo della regione 5′ HIV U3. Questo è stato associato ad una soppressione della trascrizione dell’HIV e ad un’inversione del rapporto di latenza (rapporti latente/produttivo = 0,34 per HIVGKO-WT-LTR e 8,8 per HIVGKO-ΔU3-3‘LTR – Figura 1D ). Infine, per caratterizzare ulteriormente le popolazioni costituenti le cellule infette, le cellule doppio-negative, le cellule latenti e produttivamente infette sono state ordinate utilizzando FACS e analizzate per l’mRNA virale e il contenuto proteico.(Figure 1E e F, Figura 1 – dati fonte 1). Come previsto, le cellule produttivamente infette (csGFP+) hanno espresso quantità più elevate di mRNA virale e proteine virali, ma le cellule latentemente infette (csGFP- mKO2+) avevano quantità molto piccole di mRNA virale e nessuna proteina virale rilevabile.
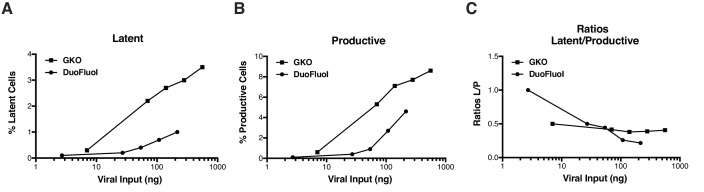
Figura 1-figure supplemento 1.Figura 1. Seconda generazione di doppia fluorescenza HIV-1 reporter, HIVGKO per quantificare la latenza stabile.Taqman RT-qPCR analisi RT-qPCR di unspliced (USA), giuntato singolarmente (SS), e moltiplicare giuntato (MS) HIV-1 mRNA HIV-1 nelle popolazioni non infette, doppio negativo, latente e produttivo.Taqman RT-qPCR analisi Taqman RT-qPCR di mRNA HIV-1 non spliced (US), singolamente giuntati (SS), e moltiplicare giuntati (MS) HIV-1 mRNA nelle popolazioni non infette, doppia negazione, latenti e produttive.(A) Rappresentazione schematica della prima (in alto: HIVDuoFluoI) e della seconda generazione (in basso: HIVGKO) di reporter HIV-1 con doppia marcatura.(B) Esperimento rappresentativo della titolazione del virus HIVGKO in cellule T CD4+ primarie attivate (4 giorni dopo l’infezione). Le cellule T CD4+ primarie CD4+ sono state attivate con perle αCD3/CD28 + 20 U/mL IL-2 per 3 giorni prima dell’infezione con diverse quantità di HIVGKO (input, ng/p24) e analizzate con citometria a flusso 4 giorni dopo l’infezione.(C) Confronto dei profili di infezione da HIVDuoFluoI e HIVGKO mediante citometria a flusso nelle cellule T CD4+ primarie attivate (4 giorni dopo l’infezione). Le cellule sono state trattate come in(B).(D) Confronto di GKO-WT-LTR e GKO-ΔU3 3’LTR profili di infezione da citometria a flusso in cellule trattate come in (B).(E, F) Le cellule primarie CD4 + T sono state trattate come in(B). A 4 giorni dopo l’infezione, sono state ordinate le cellule infette in modo doppio negativo, produttivamente infette e latente, e(E) l’RNA totale isolato da ogni popolazione è stato sottoposto ad analisi Taqman RT-qPCR (Fonte dati – Figura 1). Unspliced (US), singolamente giuntato (SS), e multiplo giuntato (MS) HIV-1 mRNA sono stati quantificati rispetto al GAPDH cellulare.(F) Analisi Western blot di ogni popolazione.10.7554/eLife.34655.004Figure 1-source dati 1.Taqman RT-qPCR analisi di unspliced (US), singolamente giuntato (SS), e moltiplicare giuntato (MS) HIV-1 mRNAs HIV-1 nelle popolazioni non infette, doppio negativo, latente e produttivo.l’esperimento è dettagliata nel testo principale e la figura 1 leggenda.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 1.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 1.La titolazione dell’input di HIVGKO ha rivelato che le cellule infette latenti e produttive sono aumentate proporzionalmente all’aumento dell’input virale(Figura 1-figure supplement 1A e B), come riflesso dai rapporti costanti di latente/produttivo (Figura 1-figure supplement 1C). Al contrario, l’aumento dell’input di HIVDuoFluoI non porta ad un aumento proporzionale delle popolazioni latenti e produttive(Figura 1-figure supplement 1A e B), come riflesso dai rapporti latente/produttivo (Figura 1-figure supplement 1C). In breve, le cellule T CD4+ sono state purificate dal sangue di un donatore rappresentativo sano e attivate per 72 ore con perline αCD3/CD28 e 100 U/ml IL-2 prima dell’infezione con diluizioni seriali di HIVGKOo HIVDuoFluoI. Quattro giorni dopo l’infezione, la percentuale di cellule latenti (csGFP- mKO2+, Figura 1-figure supplement 1A) e cellule produttive (csGFP+, Figura 1-figure supplement 1AB) sono state quantificate dal FACS sia per i virus a doppia fluorescenza, e sono stati calcolati i rapporti di popolazioni latenti rispetto a quelle produttive (Figura 1-figuresupplement 1C).
Sulla base di tutti questi risultati, la seconda generazione di reporter di doppia fluorescenza, HIVGKO, è in grado di quantificare più accuratamente le infezioni latenti nelle cellule T CD4+ primarie rispetto a HIVDuoFluoI, e quindi permette l’identificazione e la purificazione di un numero maggiore di cellule latenti infette. Utilizzando la citometria a flusso, possiamo determinare l’infezione e la produttività dell’HIV delle singole cellule e contemporaneamente controllare la vitalità cellulare.
Figura 1-figura supplemento 1.Figura 1. Seconda generazione di reporter HIV-1 a doppia fluorescenza, HIVGKO per quantificare la latenza stabile.Taqman RT-qPCR analisi RT-qPCR di mRNA di HIV-1 non infetti, doppi negativi, latenti e produttivi.Taqman RT-qPCR analisi Taqman RT-qPCR di mRNA HIV-1 non spliced (US), singolamente giuntati (SS), e moltiplicare giuntati (MS) HIV-1 mRNA nelle popolazioni non infette, doppia negazione, latenti e produttive.(A) Rappresentazione schematica della prima (in alto: HIVDuoFluoI) e della seconda generazione (in basso: HIVGKO) di reporter HIV-1 con doppia marcatura.(B) Esperimento rappresentativo della titolazione del virus HIVGKO in cellule T CD4+ primarie attivate (4 giorni dopo l’infezione). Le cellule T CD4+ primarie CD4+ sono state attivate con perle αCD3/CD28 + 20 U/mL IL-2 per 3 giorni prima dell’infezione con diverse quantità di HIVGKO (input, ng/p24) e analizzate con citometria a flusso 4 giorni dopo l’infezione.(C) Confronto dei profili di infezione da HIVDuoFluoI e HIVGKO mediante citometria a flusso nelle cellule T CD4+ primarie attivate (4 giorni dopo l’infezione). Le cellule sono state trattate come in(B).(D) Confronto di GKO-WT-LTR e GKO-ΔU3 3’LTR profili di infezione da citometria a flusso in cellule trattate come in (B).(E, F) Le cellule primarie CD4 + T sono state trattate come in(B). A 4 giorni dopo l’infezione, sono state ordinate le cellule infette in modo doppio negativo, produttivamente infette e latente, e(E) l’RNA totale isolato da ogni popolazione è stato sottoposto ad analisi Taqman RT-qPCR (Fonte dati – Figura 1). Unspliced (US), singolamente giuntato (SS), e multiplo giuntato (MS) HIV-1 mRNA sono stati quantificati rispetto al GAPDH cellulare.(F) Analisi Western blot di ogni popolazione.10.7554/eLife.34655.004Figure 1-source dati 1.Taqman RT-qPCR analisi di unspliced (US), singolamente giuntato (SS), e moltiplicare giuntato (MS) HIV-1 mRNAs HIV-1 nelle popolazioni non infette, doppio negativo, latente e produttivo.l’esperimento è dettagliata nel testo principale e la figura 1 leggenda.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 1.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 1.La titolazione dell’input di HIVGKO ha rivelato che le cellule infette latenti e produttive sono aumentate proporzionalmente all’aumento dell’input virale(Figura 1-figure supplement 1A e B), come riflesso dai rapporti costanti di latente/produttivo (Figura 1-figure supplement 1C). Al contrario, l’aumento dell’input di HIVDuoFluoI non porta ad un aumento proporzionale delle popolazioni latenti e produttive(Figura 1-figure supplement 1A e B), come riflesso dai rapporti latente/produttivo (Figura 1-figure supplement 1C). In breve, le cellule T CD4+ sono state purificate dal sangue di un donatore rappresentativo sano e attivate per 72 ore con perline αCD3/CD28 e 100 U/ml IL-2 prima dell’infezione con diluizioni seriali di HIVGKOo HIVDuoFluoI. Quattro giorni dopo l’infezione, la percentuale di cellule latenti (csGFP- mKO2+, Figura 1-figure supplement 1A) e cellule produttive (csGFP+, Figura 1-figure supplement 1AB) sono state quantificate dal FACS sia per i virus a doppia fluorescenza, e sono stati calcolati i rapporti di popolazioni latenti rispetto a quelle produttive (Figura 1-figuresupplement 1C).
Figura 1-figure supplement 1.Confronto tra HIVGKO e HIVDuoFluoI.La titolazione dell’input di HIVGKO ha rivelato che le cellule infette latenti e produttive sono aumentate proporzionalmente all’aumento dell’input virale(Figura 1-figure supplement 1A e B), come riflesso da rapporti stabili di latente/produttivo (Figura 1-figure supplement 1C). Al contrario, l’aumento dell’input di HIVDuoFluoI non porta ad un aumento proporzionale delle popolazioni latenti e produttive(Figura 1-figure supplement 1A e B), come riflesso dai rapporti latente/produttivo (Figura 1-figure supplement 1C). In breve, le cellule T CD4+ sono state purificate dal sangue di un donatore rappresentativo sano e attivate per 72 ore con perline αCD3/CD28 e 100 U/ml IL-2 prima dell’infezione con diluizioni seriali di HIVGKOo HIVDuoFluoI. Quattro giorni dopo l’infezione, la percentuale di cellule latenti (csGFP- mKO2+, Figura 1-figure supplement 1A) e cellule produttive (csGFP+, Figura 1-figure supplement 1AB) sono state quantificate dal FACS sia per i virus a doppia fluorescenza, e sono stati calcolati i rapporti di popolazioni latenti rispetto a quelle produttive (Figura 1-figuresupplement 1C).
Correlazione tra l’efficacia dell’LRA in campioni di pazienti affetti da HIV e l’attività nelle cellule latenti dell’HIVGKO
Successivamente, abbiamo valutato la riattivazione dell’HIVGKO latente nelle cellule T CD4+ primarie da parte degli LRA, e l’abbiamo confrontata con la capacità degli stessi LRA di invertire la latenza nelle cellule T CD4+ isolate da individui affetti da HIV. Abbiamo testato i seguenti LRA: (a) l’inibitore dell’istone deacetilasi (HDACi) panobinostat(Rasmussen et al., 2013), (b) l’inibitore della proteina 4 (BRD4) contenente bromodominio JQ1, che agisce attraverso il fattore di allungamento positivo della trascrizione (P-TEFb)(Banerjee et al., 2012; Boehm et al., 2013; Filippakopoulos et al., 2010; Li et al., 2013; Zhu et al., 2012), e (c) l’attivatore PKC, la briostatina-1(del Real et al., 2004; Mehla et al., 2010). La riattivazione virale mediata da questi LRA è stata confrontata con il trattamento delle cellule T CD4+ con αCD3/CD28 (Spinaet al., 2013). Diversi studi hanno mostrato effetti sinergici quando si combinano diversi LRA(Darcis et al., 2015; Jiang et al., 2015; Laird et al., 2015; Martínez-Bonet et al., 2015), quindi abbiamo anche testato la briostatina-1 in combinazione con panobinostat o JQ1. I farmaci sono stati utilizzati a concentrazioni che in precedenza si sono dimostrate efficaci per invertire la latenza in altri sistemi modello(Archin et al., 2012a; Bullen et al., 2014; Laird et al., 2015; Spina et al., 2013).
Per misurare la riattivazione da parte degli LRA in campioni di pazienti, abbiamo trattato 5 milioni di cellule T CD4+ a riposo purificate da quattro individui infetti da HIV su ART soppressiva (caratteristiche dei partecipanti nella Tabella 1) con singoli LRA, combinazioni di questi, o con il solo veicolo per 24 ore. L’efficacia degli LRA è stata valutata utilizzando un saggio basato sulla PCR, misurando i livelli di HIV-1 RNA intracellulare utilizzando primer e una sonda che rileva la sequenza 3′ comune a tutti gli mRNA HIV-1 correttamente terminati (Bullen etal., 2014). Degli LRA testati singolarmente, nessuno ha mostrato un effetto statisticamente significativo (n=4 – Figura 2A, Figura 2 dati fonte 1). È importante notare che il controllo positivo dell’attivazione delle cellule T, αCD3/CD28 (24,4 volte, Figura 2A), ha mostrato un valore di induzione di piega previsto (da 10 a 100 volte l’aumento dell’RNA dell’HIV nei PBMC [Bullenet al., 2014; Darcis et al., 2015; Laird et al., 2015 ]). Combinazioni di agonista PKC agonista briostatina-1 con JQ1 o con panobinostato (fold-incrementi di 126,2- e 320,8 volte, rispettivamente, Figura 2A), sono stati molto più efficace di briostatina-1, JQ1 o panobinostato solo (fold-incrementi di 6,8, 1,7- e 2,9 volte, rispettivamente, Figura 3A), e anche maggiore di T-cellula attivazione con αCD3/CD28. Questa osservazione è coerente con i rapporti precedenti(Darcis et al., 2015; Jiang et al., 2015; Laird et al., 2015; Martínez-Bonet et al., 2015).
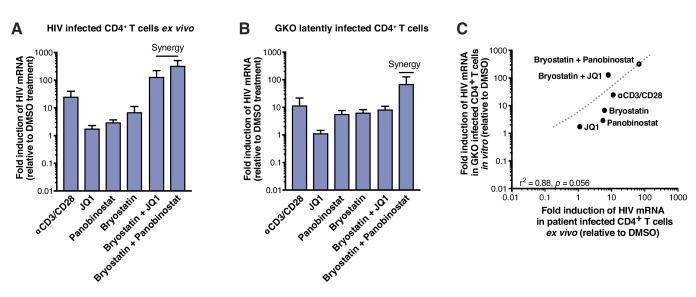
Figura 2 dati fonte 1.Figura 2—dati fonte 1. L’efficacia degli LRA nei campioni dei pazienti è prevista dall’attività nelle cellule latenti infette da HIVGKO.livelli intracellulari di HIV-1 mRNA in rCD4s, ottenuti da individui infetti, o nelle cellule T CD4+ infette da HIVGKO.livelli intracellulari di HIV-1 mRNA in rCD4s, ottenuti da individui infetti, o nelle cellule T CD4+ infette da HIVGKO.(A) Livelli intracellulari di HIV-1 mRNA in rCD4s, ottenuti da individui infetti e trattati ex vivo con un singolo LRA o una combinazione di due LRA per 24 ore in presenza di raltegravir, presentati come induzione di piega rispetto al controllo DMSO. (n = 4, media +SEM) (Figura 2dati fonte1).(B) Livelli intracellulari di HIV-1 mRNA nelle cellule T CD4+ latente infette da HIVGKO e trattate con un singolo LRA o una combinazione di due LRA per 6 ore in presenza di raltegravir, presentati come induzione di piega rispetto al controllo DMSO. (n = 3 (donatori diversi), media +SEM, test t accoppiato) (Figura 2dati fonte1).(C) Correlazione tra i livelli intracellulari di HIV-1 mRNA quantificati in 6 ore stimolate da HIVGKO e le cellule T CD4+ latente infette da HIV da donatori diversi, o 24 ore stimolate da rCD4 da pazienti infetti da HIV, con un singolo LRA o una combinazione di due LRA in presenza di raltegravir.10.7554/eLife.34655.006Cifra dati a 2 fonti 1.Livelli intracellulari di HIV-1 mRNA in rCD4s, ottenuti da individui infetti, o in CD4+ T-cellule T HIVGKOlatente infette da HIVGKO.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 2.L’esperimento è descritto in dettaglio nel testo principale e nella legenda della Figura 2.L’esperimento è descritto in dettaglio nel testo principale e nella legenda della Figura 2.
Figura 3 – Dati sorgente 1.La percentuale di cellule GFP+ viene mostrata dopo la stimolazione delle cellule T CD4+ latenti con LRA e la percentuale di cellule vive per ogni trattamento farmacologico.(A) Schema della procedura sperimentale con le cellule T CD4+ primarie. In breve, le cellule T CD4+ sono state purificate dal sangue di donatori sani e attivate per 72 ore con perline αCD3/CD28 e 100 U/ml IL-2 prima dell’infezione da HIVGKO. Cinque giorni dopo l’infezione, le cellule latentemente infette (csGFP- mKO2+) sono state selezionate, rimesse in coltura durante la notte e stimolate con diversi LRA in presenza di raltegravir per 24 ore prima di eseguire l’analisi FACS.(B) Percentuale di cellule GFP + è mostrato dopo la stimolazione di cellule T CD4 + latente infetti CD4 + con LRA (n = 4 (diversi donatori), media +SEM, accoppiato t-test) (Figura3-dati fonte1).(C) Istogramma grafico della percentuale di cellule vive per ogni trattamento farmacologico (n = 3 (diversi donatori), media + SEM, t-test abbinato) (Figura 3-dati fonte1). p-valore: *p<0.05, **p<0.01 relativo al DMSO.10.7554/eLife.34655.008Figure 3-source data 1.Percentuale di cellule GFP + è mostrato dopo la stimolazione di cellule T CD4 + latente infetti CD4 + con LRA così come le cellule in percentuale di cellule vive per ogni trattamento farmacologico . L’esperimento è dettagliato nel testo principale e nella legenda della Figura 3.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 3.
| ID del campo di applicazione | Età | Sesso | Etnia | Conteggio CD4 | Durata dell’infezione (anni) | Regime ART | Durata di ART (anni) | Peak reporter VL (copie/ml-1) |
|---|---|---|---|---|---|---|---|---|
| 1597 | 56 | M | Misto | 469 | 19 | RPV/TDF/FTC | 5 | 45734 |
| 2147 | 59 | M | Asiatico | 597 | 28 | RPV/TDF/FTC | 23 | 374000 |
| 2461 | 62 | M | Bianco | 664 | 32 | RPV/TCV | 19 | 20000 |
| 3162 | 54 | M | Bianco | 734 | 29 | RTV, DRV, ABC/TCV/3TC | 20 | 171000 |
Gli stessi LRA e le stesse combinazioni sono stati successivamente testati dopo l’infezione delle cellule umane CD4+T in vitro con HIVGKO. Misurazione di HIV-1 mRNA intracellulare in HIVGKO cellule latenti infette da HIVGKO ha mostrato una induzione di latenza prevista piega in risposta a αCD3/CD28 (11,3 volte, Figura 2B, Figura 2 dati fonte 1). In secondo luogo, JQ1, panobinostat, e la sola briostatina-1 ha causato una limitata riattivazione dell’HIV latente (aumento della piega di 1,1-, 5,6- e 6,2 volte, rispettivamente, Figura 2B), come osservato nei campioni dei pazienti. Infine, abbiamo osservato una bassa sinergia nella combinazione di briostatina e JQ1 (aumento di 8 volte), ma un’elevata sinergia tra briostatina e panobinostat (aumento di 67,3 volte). Questi dati insieme dimostrano che HIVGKO imita da vicino in vitro ciò che si osserva nei campioni dei pazienti ex vivo (tasso di correlazione r2 =0,88, p=0,0056 – Figura 2C), e convalidano la robustezza e l’affidabilità del reporter HIV a doppia fioritura come modello per studiare la latenza dell’HIV-1.
Figura 2 dati fonte 1.I livelli intracellulari di mRNA HIV-1 negli rCD4, ottenuti da individui infetti, o nelle cellule T CD4+ infette da HIVGKO, o nelle cellule T CD4+ infette da HIVGKO.(A) Livelli intracellulari di HIV-1 mRNA in rCD4s, ottenuti da individui infetti e trattati ex vivo con un singolo LRA o una combinazione di due LRA per 24 ore in presenza di raltegravir, presentati come induzione di piega rispetto al controllo DMSO. (n = 4, media +SEM) (Figura 2dati fonte1).(B) Livelli intracellulari di HIV-1 mRNA nelle cellule T CD4+ latente infette da HIVGKO e trattate con un singolo LRA o una combinazione di due LRA per 6 ore in presenza di raltegravir, presentati come induzione di piega rispetto al controllo DMSO. (n = 3 (donatori diversi), media +SEM, test t accoppiato) (Figura 2dati fonte1).(C) Correlazione tra i livelli intracellulari di HIV-1 mRNA quantificati in 6 ore stimolate da HIVGKO e le cellule T CD4+ latente infette da HIV da donatori diversi, o 24 ore stimolate da rCD4 da pazienti infetti da HIV, con un singolo LRA o una combinazione di due LRA in presenza di raltegravir.10.7554/eLife.34655.006Cifra dati a 2 fonti 1.Livelli intracellulari di HIV-1 mRNA in rCD4s, ottenuti da individui infetti, o in CD4+ T-cellule T HIVGKOlatente infette da HIVGKO.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 2.L’esperimento è descritto in dettaglio nel testo principale e nella legenda della Figura 2.L’esperimento è descritto in dettaglio nel testo principale e nella legenda della Figura 2.
Figura 3 – Dati sorgente 1.La percentuale di cellule GFP+ viene mostrata dopo la stimolazione delle cellule T CD4+ latenti con LRA e la percentuale di cellule vive per ogni trattamento farmacologico.(A) Schema della procedura sperimentale con le cellule T CD4+ primarie. In breve, le cellule T CD4+ sono state purificate dal sangue di donatori sani e attivate per 72 ore con perline αCD3/CD28 e 100 U/ml IL-2 prima dell’infezione da HIVGKO. Cinque giorni dopo l’infezione, le cellule latentemente infette (csGFP- mKO2+) sono state selezionate, rimesse in coltura durante la notte e stimolate con diversi LRA in presenza di raltegravir per 24 ore prima di eseguire l’analisi FACS.(B) Percentuale di cellule GFP + è mostrato dopo la stimolazione di cellule T CD4 + latente infetti CD4 + con LRA (n = 4 (diversi donatori), media +SEM, accoppiato t-test) (Figura3-dati fonte1).(C) Istogramma grafico della percentuale di cellule vive per ogni trattamento farmacologico (n = 3 (diversi donatori), media + SEM, t-test abbinato) (Figura 3-dati fonte1). p-valore: *p<0.05, **p<0.01 relativo al DMSO.10.7554/eLife.34655.008Figure 3-source data 1.Percentuale di cellule GFP + è mostrato dopo la stimolazione di cellule T CD4 + latente infetti CD4 + con LRA così come le cellule in percentuale di cellule vive per ogni trattamento farmacologico . L’esperimento è dettagliato nel testo principale e nella legenda della Figura 3.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 3.
Gli LRA dell’HIV-1 prendono di mira una minoranza di cellule T CD4+ primarie latenti infette da HIV-1
I saggi attuali si sono basati su saggi basati sulla PCR per misurare l’RNA dell’HIV e per valutare l’efficacia di diversi LRA(Figura 2A). L’uso di reporter HIV a doppia fluorescenza, tuttavia, fornisce uno strumento per quantificare direttamente la frazione di cellule che si riattivano.
Per quantificare il numero assoluto di cellule latenti riattivate dopo il trattamento LRA, le cellule T CD4+ primarie CD4+ sono state infettate da HIVGKO e messe in coltura per 5 giorni (in presenza di IL-2) prima di selezionare la popolazione latente pura (GFP-, mKO2+). Le cellule sono state lasciate riposare durante la notte e sono state trattate per 24 ore con i vari LRA (stesse concentrazioni di farmaci come nella Figura 2)(Figura 3A, Figura 3-dati fonte 1). La coltura di cellule T CD4+ primarie CD4+ latenti trattate con DMSO ha prodotto poca riattivazione spontanea (media di quattro esperimenti: 1,4% di cellule GFP+). Inaspettatamente, abbiamo scoperto che nessuno dei singoli LRA o delle loro combinazioni ha riattivato più del 5,5% delle cellule latentemente infette: JQ1 (1,7%) panobinostat (3,7%), briostatina-1 (3%) αCD3/CD28 (4,5%), briostatina-1 e JQ1 (3,3%.). briostatina-1 e panobinostat (5,5%) (Figura 3B).
Piccolo tasso frazionario di riattivazione di latenza non si spiega con la bassa risposta cellulare ai segnali di attivazione
Questi dati evidenziano due fatti importanti: a) la quantificazione dell’HIV RNA associato alle cellule non riflette il numero assoluto di cellule in fase di riattivazione virale, e b) l’HIV RNA associato alle cellule indotto, in risposta a tutti gli agenti invertitori, proviene da una piccola frazione di cellule latenti riattivate. Ciò è stato particolarmente sorprendente con la stimolazione αCD3/CD28, poiché un modello attualmente accettato per la latenza dell’HIV è che lo stato di attivazione delle cellule T determina lo stato trascrizionale del provirus. Il trattamento delle cellule T CD4+ primarie latenti infette con αCD3/CD28 ha stimolato la produzione di HIV in meno del 5% delle cellule, mentre l’altro 95% è rimasto latente, anche se dopo 24 ore di trattamento quasi tutte le cellule avevano upregulated il marcatore precoce di attivazione delle cellule T CD69 (Figura 4-figure supplement 1).
Per escludere ulteriormente la possibilità che le cellule non riattivate latentemente infette (NRLIC) rappresentassero semplicemente una mancanza di risposta efficiente ai segnali di attivazione delle cellule T, abbiamo analizzato i marcatori di attivazione delle cellule T all’interno delle diverse popolazioni (cioè, all’interno delle cellule non infette, non riattivate (NRLIC) e riattivate latentemente infette (RLIC); Figura 4, Figura 4, dati fonte 1). In breve, 72h-stimolato CD4 + cellule T sono state infettate con HIVGKO, e 4 giorni dopo, le cellule GFP- sono stati ordinati, e ha permesso di riposare durante la notte prima della restimolazione con αCD3/CD28. Dopo altre 24 ore, le cellule sono state colorate per i marcatori precoci, intermedi e tardivi di attivazione delle cellule T CD69, CD25 e HLA-DR rispettivamente. Le tre diverse popolazioni, doppio negativo, RLIC e NRLIC, avevano profili simili di cellule T attivate sottoinsiemi di cellule T, come mostrato in Figura 4, ed erano principalmente composti da cellule fortemente attivate (CD69 + / CD25 + / HLA-DR + / -). Abbiamo osservato solo un aumento statisticamente significativo di NRLIC rispetto a RLIC nella popolazione CD69+/CD25-/HLA-DR+, tuttavia questo piccolo aumento in una popolazione relativamente minore è insufficiente a spiegare il basso tasso di riattivazione delle cellule latentemente infette. Nel complesso, il confronto tra popolazioni latenti riattivate e non riattivate ha mostrato poche differenze nel loro stato di attivazione.
Figura 4-figure supplemento 1.Figura 4— figura 1. La riattivazione a basso livello di latenza non è spiegata da risposte cellulari basse ai segnali di attivazione.CD25, CD69 e modelli di marcatori di attivazione HLA-DR tra le cellule latenti infette doppie negative, riattivate (RLIC) e non riattivate (NRLIC).CD25, CD69 e modelli di marcatori di attivazione HLA-DR tra le cellule latenti infette doppie negative, riattivate (RLIC) e non riattivate (NRLIC).24 hr trattamento efficacemente attivare le cellule T CD4+ primarie.I modelli di attivazione delle cellule T tra cellule latenti infette doppie negative, riattivate (RLIC) e non riattivate (NRLIC). In breve, le cellule T CD4+ sono state purificate dal sangue di quattro donatori sani e attivate per 72 ore con perline αCD3/CD28 e 20 U/ml IL-2 prima dell’infezione da HIVGKO. A 4 giorni dopo l’infezione, csGFP- sono stati ordinati, coltivati durante la notte e stimolati con αCD3/CD28 in presenza di raltegravir. A 24 ore dopo il trattamento, le cellule sono state colorate per CD25, CD69, e HLA-DR marcatori di attivazione prima di eseguire l’analisi FACS. (n = 4, media +SEM, accoppiato t-test; *p<0,05; **p<0,01) (Figura 4dati fonte1).10.7554/eLife.34655.012Figure 4 dati sorgente 1.CD25, CD69, e HLA-DR pattern di attivazione marcatori HLA-DR tra doppio negativo, riattivato (RLIC) e non riattivato (NRLIC) cellule latenti infette latente.l’esperimento è dettagliata nel testo principale e la leggenda Figura 4.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 4.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 4.Quantificazione dei marcatori di superficie associati all’attivazione delle cellule T dopo 24 ore di stimolazione. In breve, le cellule T CD4+ sono state purificate dal sangue di due donatori sani e attivate per 72 ore con perline αCD3/CD28 e 100 U/ml IL-2 prima dell’infezione da HIVGKO. Cinque giorni dopo l’infezione, le cellule latenti infette (csGFP- mKO2+) sono state selezionate, coltivate durante la notte e stimolate con αCD3/CD28 o briostatina + panobinostat in presenza di raltegravir. 24 ore dopo il trattamento, le cellule sono state colorate per i marcatori di attivazione CD25 e CD69 prima di eseguire l’analisi FACS.
Figura 4-figure supplemento 1.Figura 4— figura 1. La riattivazione a basso livello di latenza non è spiegata da risposte cellulari basse ai segnali di attivazione.CD25, CD69, e HLA-DR modelli di marcatori di attivazione tra doppio negativo, riattivato (RLIC) e non riattivato (NRLIC) cellule latentemente infette.CD25, CD69, e HLA-DR modelli di marcatori di attivazione tra doppio negativo, riattivato (RLIC) e non riattivato (NRLIC) cellule latentemente infette.24 ore di trattamento in modo efficace attivare cellule T CD4 + primario.I modelli di attivazione delle cellule T tra cellule latenti infette doppie negative, riattivate (RLIC) e non riattivate (NRLIC). In breve, le cellule T CD4+ sono state purificate dal sangue di quattro donatori sani e attivate per 72 ore con perline αCD3/CD28 e 20 U/ml IL-2 prima dell’infezione da HIVGKO. A 4 giorni dopo l’infezione, csGFP- sono stati ordinati, coltivati durante la notte e stimolati con αCD3/CD28 in presenza di raltegravir. A 24 ore dopo il trattamento, le cellule sono state colorate per CD25, CD69, e HLA-DR marcatori di attivazione prima di eseguire l’analisi FACS. (n = 4, media +SEM, accoppiato t-test; *p<0,05; **p<0,01) (Figura 4dati fonte1).10.7554/eLife.34655.012Figure 4 dati sorgente 1.CD25, CD69, e HLA-DR pattern di attivazione marcatori HLA-DR tra doppio negativo, riattivato (RLIC) e non riattivato (NRLIC) cellule latenti infette latente.l’esperimento è dettagliata nel testo principale e la leggenda Figura 4.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 4.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 4.Quantificazione dei marcatori di superficie associati all’attivazione delle cellule T dopo 24 ore di stimolazione. In breve, le cellule T CD4+ sono state purificate dal sangue di due donatori sani e attivate per 72 ore con perline αCD3/CD28 e 100 U/ml IL-2 prima dell’infezione da HIVGKO. Cinque giorni dopo l’infezione, le cellule latenti infette (csGFP- mKO2+) sono state selezionate, coltivate durante la notte e stimolate con αCD3/CD28 o briostatina + panobinostat in presenza di raltegravir. 24 ore dopo il trattamento, le cellule sono state colorate per i marcatori di attivazione CD25 e CD69 prima di eseguire l’analisi FACS.
Figura 4-figure supplemento 1.24 ore di trattamento di 24 ore effettivamente attivare le cellule T CD4 + primario CD4.2. Quantificazione dei marcatori di superficie associati all’attivazione delle cellule T dopo 24 ore di stimolazione. In breve, CD4 + cellule T sono stati purificati dal sangue di due donatori sani e attivati per 72 ore con perline αCD3/CD28 e 100 U / ml IL-2 prima dell’infezione da HIVGKO. Cinque giorni dopo l’infezione, le cellule latenti infette (csGFP- mKO2+) sono state selezionate, coltivate durante la notte e stimolate con αCD3/CD28 o briostatina + panobinostat in presenza di raltegravir. 24 ore dopo il trattamento, le cellule sono state colorate per i marcatori di attivazione CD25 e CD69 prima di eseguire l’analisi FACS.
Siti di integrazione, espressione genica, unità di trascrizione e il destino dell’infezione da HIV
Il ruolo del sito di integrazione dell’HIV nel genoma in latenza rimane un argomento di dibattito(Chen et al., 2017; Dahabieh et al., 2014; Jordan et al., 2003; Jordan et al., 2001; Sherrill-Mix et al., 2013). Per identificare possibili differenze nei siti di integrazione tra genomi HIV riattivati e non riattivati, le cellule T CD4+ primarie CD4+ sono state infettate da HIVGKO. A 5 giorni dall’infezione, le cellule produttivamente infette (GFP+, PIC) sono state selezionate e congelate. La popolazione negativa alle GFP (costituita da una miscela di cellule latenti e non infette) è stata isolata e trattata con αCD3/CD28. 48 ore dopo l’induzione, sono state isolate sia le popolazioni non riattivate (NRLIC) che quelle riattivate (RLIC). Nove biblioteche (tre donatori, tre campioni / donatore: PIC, RLIC, NRLIC) sono stati costruiti da DNA genomico come descritto(Cohn et al., 2015) e analizzati con il sequenziamento ad alto rendimento per individuare i provirus dell’HIV all’interno del genoma umano. Un totale di 1803 siti di integrazione del virus sono stati determinati: 960 integrazioni in PIC, 681 in NRLIC, e 162 in RLIC (Integration Sites Source data).
Per determinare se l’integrazione all’interno dei geni espressi in modo differenziato durante l’attivazione delle cellule T prevedeva il destino di riattivazione dell’infezione, abbiamo confrontato il nostro dataset di integrazione dell’HIV con un dataset pubblicato per l’espressione genica in riposo e attivata (48 ore – αCD3/CD28) CD4+ cellule T da individui sani (Ye etal., 2014). L’analisi ha rivelato che la maggior parte dei provirus latenti indotti da αCD3/CD28 non erano integrati nei geni che rispondono ai segnali di attivazione delle cellule T (Figura 5A e B, Figura 5 – dati fonte 1). È interessante notare che gli eventi di integrazione PIC e RLIC sono stati associati a geni la cui espressione basale era significativamente più alta di quella dei geni mirati in NRLIC, sia nelle cellule T attivate che in quelle a riposo(Figura 5C, Figura 5-dati sorgente 2).
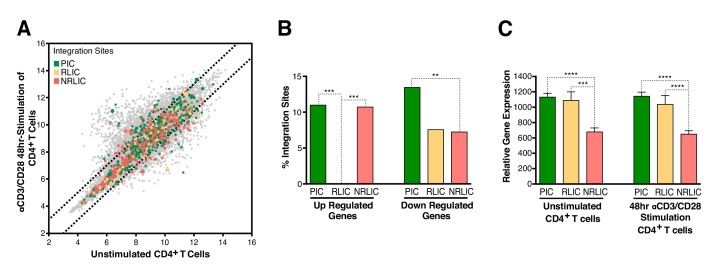
Figura 5-dati fonte 2.Figura 5—dati fonte 2. Espressione relativa dei geni mirati all’integrazione dell’HIV-1 per ogni popolazione, prima o dopo l’attivazione della TCR.Frazione dei siti di integrazione delle diverse popolazioni PIC, RLIC o NRLIC, integrati all’interno di geni la cui espressione è almeno ± due volte espressa in modo differenziale dopo 48 ore di stimolazione αCD3/CD28.Espressione relativa dei geni mirati all’integrazione dell’HIV-1 in PIC, RLIC o NRLIC prima della stimolazione della TCR e dopo 48 ore di stimolazione αCD3/CD28.Frazione di siti di integrazione delle diverse popolazioni PIC, RLIC o NRLIC, integrati all’interno di geni la cui espressione è almeno ± doppia espressione differenziale dopo 48 ore di stimolazione αCD3/CD28.Espressione relativa di geni bersaglio dell’integrazione dell’HIV-1 in PIC, RLIC o NRLIC prima della stimolazione TCR e dopo 48 ore di stimolazione αCD3/CD28.(A) Tabella di dispersione che mostra i cambiamenti primari CD4 + T-cellula T-cellula primaria espressione dei geni dopo 48 ore di stimolazione con perline αCD3/CD28. Siti di integrazione visualizzati al di fuori delle due linee grigie solide sono stati mirati geni la cui espressione è almeno ± due volte differenzialmente espresso dopo 48 ore di stimolazione. La dimensione dei punti di trama può essere diversa, più grande è il punto di trama, più eventi di integrazione sono avvenuti all’interno dello stesso gene.(B) Frazione di siti di integrazione dalle diverse popolazioni PIC, RLIC o NRLIC, integrati all’interno di geni la cui espressione è almeno ± due volte differenzialmente espressa dopo 48 ore di stimolazione αCD3/CD28 (**p<0.01; ***p<0.001; test z a due proporzioni) (Figura 5 – dati fonte 1).(C) Espressione relativa di geni bersaglio dell’integrazione dell’HIV-1 in PIC, RLIC o NRLIC prima della stimolazione TCR e dopo la stimolazione αCD3/CD28 (n = 3, media +SEM, t-test abbinato). ***p<0.001; ****p<0.0001.(Figura 5-dati sorgente 2).10.7554/eLife.34655.014Figure 5-source data 1.Fraction of integration sites from the different populations PIC, RLIC o NRLIC, integrated within genes whose expression is at least ± twofold differenzialmente expressed after 48 hr of αCD3/CD28 stimulation.the experiment is detailed in the main text and Figure 5 legend.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 5.10.7554/eLife.34655.015Figure 5-source data 2.Relative espressione di geni mirati da HIV-1 integrazione in PIC, RLIC o NRLIC prima della stimolazione TCR e dopo 48 ore αCD3/CD28 stimolazione . L’esperimento è dettagliato nel testo principale e nella legenda della Figura 6.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 5.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 6.
Successivamente, abbiamo indagato se diverse regioni genomiche erano associate ad infezioni latenti di HIV-1 produttive, inducibili o non inducibili. In accordo con gli studi precedenti(Cohn et al., 2015; Dahabieh et al., 2014; Maldarelli et al., 2014; Wagner et al., 2014), la maggior parte dei siti di integrazione sono stati trovati all’interno dei geni in ogni popolazione(Figura 6A, Figura 6-dati fonte 1), anche se la proporzione di integrazioni geniche in NRLIC era significativamente inferiore rispetto ai campioni PIC e RLIC. Inoltre, gli eventi di integrazione nelle popolazioni PIC e RLIC erano più frequenti nelle regioni trascritte (64% e 58%, rispettivamente, [somma delle regioni trascritte basse + medie + alte] (Figura 6B),Figura 6-dati fonte 1), mentre queste regioni erano significativamente meno rappresentate nel NRLIC (31%) (Figura 6B). Come previsto, poiché gli introni rappresentano una proporzione molto più grande di geni, gli eventi di integrazione genica sono stati più frequenti negli introni per ogni popolazione (>65%, Figura 6C, Figura 6-dati fonte 1). Infine, l’orientamento virale dei provirus rispetto all’unità trascrizionale non è stato correlato con il destino dell’infezione da HIV (latente vs produttivo) o con la riattivazione o l’assenza di latenza dell’HIV(Figura 6D, Figura 6-dati fonte 1).
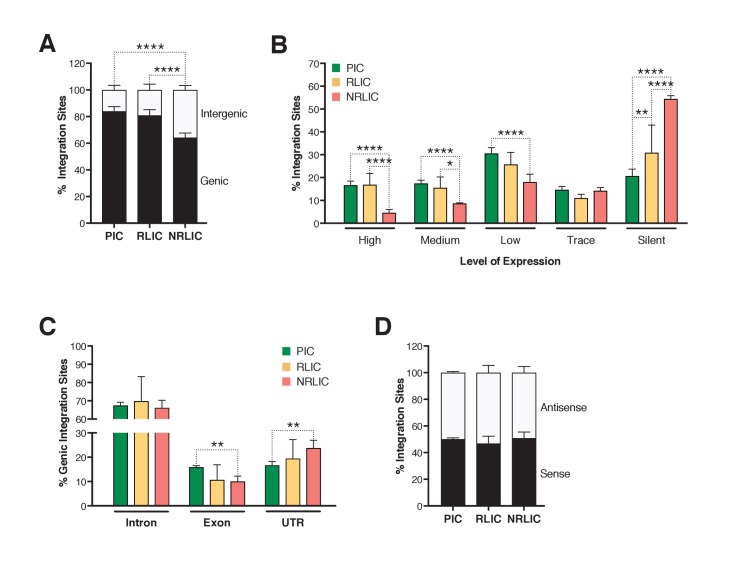
Figura 6 – dati fonte 1.1. Paesaggi di inserimento dell’HIV-1. Percentuale di inserzioni mappate che si trovano in regioni geniche o intergeniche; di siti di integrazione in regioni trascritte con alta, media, bassa espressione, traccia o espressione silenziosa; di siti di integrazione genica unici situati in introni, esoni, UTR o promotori; e orientamento trascrizionale dell’HIV-1 integrato rispetto al gene ospite.Proporzione di inserzioni mappate che si trovano in regioni geniche o intergeniche; di siti di integrazione in regioni trascritte con alta, media, bassa espressione, traccia o silenziosa espressione; di siti di integrazione genica unici situati in introni, esoni, UTR o promotori; e orientamento trascrizionale dell’HIV-1 integrato rispetto al gene ospite.(A) Percentuale di inserzioni mappate che si trovano in regioni geniche o intergeniche.(Figura 6 – dati fonte 1).(B) Percentuale di siti di integrazione in regioni trascritte con alta (top 1/8), media (top 1/4-1/8), bassa espressione (top 1/2-1/4), traccia (bottom 1/2) o silenziosa (0) espressione.(Figura 6 – dati sorgente 1).(C) Percentuale di siti di integrazione genica unici situati in introni, esoni, UTR o promotori.(Figura 6-dati sorgente 1).(D) Orientamento trascrizionale dell’HIV-1 integrato rispetto al gene ospite.(Figura 6-dati fonte 1). p-valore: *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001 utilizzando il test z a due proporzioni.10.7554/eLife.34655.017Figure 6-source data 1.Proporzione di inserzioni mappate che si trovano in regioni geniche o intergeniche; di siti di integrazione in regioni trascritte con alta, media, bassa espressione, traccia o espressione silenziosa; di siti di integrazione genica unici situati in introni, esoni, UTR o promotori; e orientamento trascrizionale dell’HIV-1 integrato rispetto al gene ospite . L’esperimento è dettagliato nel testo principale e nella legenda della Figura 6.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 6.
Figura 5 – Dati di origine 2.Figura 5—dati di fonte 2. Espressione relativa dei geni mirati all’integrazione dell’HIV-1 per ogni popolazione, prima o dopo l’attivazione della TCR.Frazione dei siti di integrazione delle diverse popolazioni PIC, RLIC o NRLIC, integrati all’interno di geni la cui espressione è almeno ± due volte espressa in modo differenziale dopo 48 ore di stimolazione αCD3/CD28.Espressione relativa dei geni mirati all’integrazione dell’HIV-1 in PIC, RLIC o NRLIC prima della stimolazione TCR e dopo 48 ore di stimolazione αCD3/CD28.Frazione di siti di integrazione delle diverse popolazioni PIC, RLIC o NRLIC, integrati all’interno di geni la cui espressione è almeno ± doppia espressione differenziale dopo 48 ore di stimolazione αCD3/CD28.Espressione relativa di geni bersaglio dell’integrazione dell’HIV-1 in PIC, RLIC o NRLIC prima della stimolazione TCR e dopo 48 ore di stimolazione αCD3/CD28.(A) Tabella di dispersione che mostra i cambiamenti primari CD4 + T-cellula T-cellula primaria espressione dei geni dopo 48 ore di stimolazione con perline αCD3/CD28. Siti di integrazione visualizzati al di fuori delle due linee grigie solide sono stati mirati geni la cui espressione è almeno ± due volte differenzialmente espresso dopo 48 ore di stimolazione. La dimensione dei punti di trama può essere diversa, più grande è il punto di trama, più eventi di integrazione sono avvenuti all’interno dello stesso gene.(B) Frazione di siti di integrazione dalle diverse popolazioni PIC, RLIC o NRLIC, integrati all’interno di geni la cui espressione è almeno ± due volte differenzialmente espressa dopo 48 ore di stimolazione αCD3/CD28 (**p<0.01; ***p<0.001; test z a due proporzioni) (Figura 5 – dati fonte 1).(C) Espressione relativa di geni bersaglio dell’integrazione dell’HIV-1 in PIC, RLIC o NRLIC prima della stimolazione TCR e dopo la stimolazione αCD3/CD28 (n = 3, media +SEM, t-test abbinato). ***p<0.001; ****p<0.0001.(Figura 5-dati sorgente 2).10.7554/eLife.34655.014Figure 5-source data 1.Fraction of integration sites from the different populations PIC, RLIC o NRLIC, integrated within genes whose expression is at least ± twofold differenzialmente expressed after 48 hr of αCD3/CD28 stimulation.the experiment is detailed in the main text and Figure 5 legend.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 5.10.7554/eLife.34655.015Figure 5-source data 2.Relative espressione di geni mirati da HIV-1 integrazione in PIC, RLIC o NRLIC prima della stimolazione TCR e dopo 48 ore αCD3/CD28 stimolazione . L’esperimento è dettagliato nel testo principale e nella legenda della Figura 6.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 5.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 6.
Figura 6 – Dati fonte 1.1. Paesaggi di inserimento dell’HIV-1. Percentuale di inserzioni mappate che si trovano in regioni geniche o intergeniche; di siti di integrazione in regioni trascritte con alta, media, bassa espressione, traccia o espressione silenziosa; di siti di integrazione genica unici situati in introni, esoni, UTR o promotori; e orientamento trascrizionale dell’HIV-1 integrato rispetto al gene ospite.Proporzione di inserzioni mappate che si trovano in regioni geniche o intergeniche; di siti di integrazione in regioni trascritte con alta, media, bassa espressione, traccia o silenziosa espressione; di siti di integrazione genica unici situati in introni, esoni, UTR o promotori; e orientamento trascrizionale dell’HIV-1 integrato rispetto al gene ospite.(A) Percentuale di inserzioni mappate che si trovano in regioni geniche o intergeniche.(Figura 6 – dati fonte 1).(B) Percentuale di siti di integrazione in regioni trascritte con alta (top 1/8), media (top 1/4-1/8), bassa espressione (top 1/2-1/4), traccia (bottom 1/2) o silenziosa (0) espressione.(Figura 6 – dati sorgente 1).(C) Percentuale di siti di integrazione genica unici situati in introni, esoni, UTR o promotori.(Figura 6-dati sorgente 1).(D) Orientamento trascrizionale dell’HIV-1 integrato rispetto al gene ospite.(Figura 6-dati fonte 1). p-valore: *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001 utilizzando il test z a due proporzioni.10.7554/eLife.34655.017Figure 6-source data 1.Proporzione di inserzioni mappate che si trovano in regioni geniche o intergeniche; di siti di integrazione in regioni trascritte con alta, media, bassa espressione, traccia o espressione silenziosa; di siti di integrazione genica unici situati in introni, esoni, UTR o promotori; e orientamento trascrizionale dell’HIV-1 integrato rispetto al gene ospite . L’esperimento è dettagliato nel testo principale e nella legenda della Figura 6.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 6.
Modifiche della cromatina nel sito di integrazione e latenza dell’HIV
I segni di cromatina, come le modifiche istoniche post-traslazionali (ad esempio, metilazione e acetilazione) e la metilazione del DNA, sono coinvolti nella determinazione e nel mantenimento della latenza dell’HIV-1(De Crignis e Mahmoudi, 2017). Abbiamo esaminato regioni di 500 pb centrate su tutti i siti di integrazione in ogni popolazione per diversi segni di cromatina, confrontando i nostri dati con diverse modificazioni dell’istone e con i set di dati DNaseI ENCODE. Abbiamo prima esaminato le firme di cromatina distinte e predittive, come H3K4me1 (stimolatori attivi), H3K36m3 (regioni trascritte attive), H3K9m3 e H3K27m3 (segni di trascrizione repressivi) (esaminati in[Kumar et al., 2015; Shlyueva et al., 2014]). Tutte e tre le popolazioni presentavano profili distinti, anche se i profili delle infezioni latenti produttive e inducibili apparivano più simili(Figura 7A, Figura 7 – dati fonte 1). L’analisi ha mostrato che gli eventi di integrazione PIC sono stati associati alla cromatina attiva (cioè, geni trascritti – H3K36me3 o stimolatori – H3K4me1), mentre gli eventi di integrazione NRLIC sono apparsi di parte verso l’eterocromatina (H3K27me3 e H3K9me3) e le regioni non accessibili (iposensibilità DNasi).
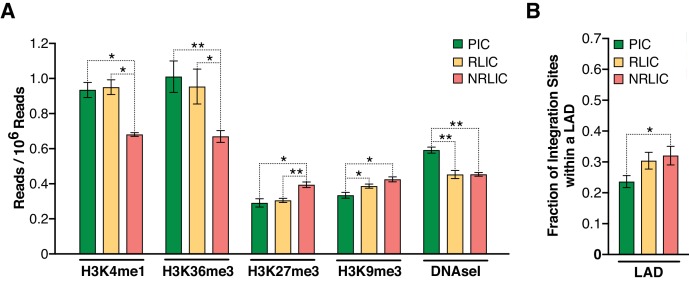
Figura 7 – Dati fonte 1.Figura 7—dati fonte 1. Segni epigenetici e localizzazione nucleare dei siti di integrazione HIV-1.I siti di integrazione HIV-1 per ogni popolazione sono stati analizzati per la presenza di H3K4me1, H3K36m3, H3K9m3, H3K9m3, H3K27m3, accessibilità del DNA, così come la loro localizzazione nucleare.i siti di integrazione HIV-1 per ogni popolazione sono stati analizzati per la presenza di H3K4me1, H3K36m3, H3K9m3, H3K27m3, accessibilità del DNA, così come la loro localizzazione nucleare.(A) 500 bp centrati sui siti di integrazione dell’HIV-1 per ogni popolazione sono stati analizzati per la presenza di H3K4me1 (potenziatori attivi), H3K36m3 (regioni attive trascritte), H3K9m3 e H3K27m3 (segni repressivi di trascrizione), e l’accessibilità del DNA (DNAseI).(Figura 7 – dati fonte 1).(B) Localizzazione nucleare dei siti di integrazione dell’HIV-1. La quantificazione è stata basata all’interno di un LAD (=1) o all’esterno (=0), il che significa che l’asse Y rappresenta la frazione delle integrazioni all’interno di un LAD.(Figura 7 – dati fonte 1).(n = 3-4 donatori ENCODE, media +SEM, t-test abbinato). *p<0,05; **p<0,01; ***p<0,001).10.7554/eLife.34655. 019Figure 7-source data 1.HIV-1 siti di integrazione per ogni popolazione sono stati analizzati per la presenza di H3K4me1, H3K36m3, H3K9m3, H3K27m3, l’accessibilità del DNA, così come la loro localizzazione nucleare.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 7.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 7.
Marini et al. hanno recentemente riportato che l’HIV-1 si integra principalmente alla periferia nucleare(Marini et al., 2015). Abbiamo quindi esaminato la distribuzione topologica dei siti di integrazione di ogni popolazione all’interno del nucleo, confrontando i dati del nostro sito di integrazione con un dataset di domini associati alle lamine (LAD) precedentemente pubblicato(Guelen et al., 2008). I LAD sono costituiti da eterocromatina H3K9me2 e sono presenti alla periferia nucleare. Questa analisi ha mostrato che i siti di integrazione latente sia da RLIC e NRLIC erano in LADs ad un grado significativamente superiore (32% e 30,4%) rispetto alle integrazioni produttive (23,6%) (p<0,05, Figura 7B, Figura 7-source dati 1). Nel complesso, questi dati mostrano caratteristiche simili tra cellule produttivamente infette e cellule latenti inducibili, mentre le cellule latenti non riattivate appaiono distinte dalle altre popolazioni. Questi risultati supportano un ruolo di primo piano per il sito di integrazione e il contesto della cromatina per il destino dell’infezione stessa, così come per l’inversione di latenza.
Figura 7 – Dati fonte 1.I siti di integrazione HIV-1 per ogni popolazione sono stati analizzati per la presenza di H3K4me1, H3K36m3, H3K9m3, H3K9m3, H3K27m3, l’accessibilità del DNA, così come la loro localizzazione nucleare.i siti di integrazione HIV-1 per ogni popolazione sono stati analizzati per la presenza di H3K4me1, H3K36m3, H3K9m3, H3K9m3, H3K27m3, l’accessibilità del DNA, così come la loro localizzazione nucleare.(A) 500 bp centrati sui siti di integrazione dell’HIV-1 per ogni popolazione sono stati analizzati per la presenza di H3K4me1 (potenziatori attivi), H3K36m3 (regioni attive trascritte), H3K9m3 e H3K27m3 (segni repressivi di trascrizione), e l’accessibilità del DNA (DNAseI).(Figura 7 – dati fonte 1).(B) Localizzazione nucleare dei siti di integrazione dell’HIV-1. La quantificazione è stata basata all’interno di un LAD (=1) o all’esterno (=0), il che significa che l’asse Y rappresenta la frazione delle integrazioni all’interno di un LAD.(Figura 7 – dati fonte 1).(n = 3-4 donatori ENCODE, media +SEM, t-test abbinato). *p<0,05; **p<0,01; ***p<0,001).10.7554/eLife.34655. 019Figure 7-source data 1.HIV-1 siti di integrazione per ogni popolazione sono stati analizzati per la presenza di H3K4me1, H3K36m3, H3K9m3, H3K27m3, l’accessibilità del DNA, così come la loro localizzazione nucleare.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 7.L’esperimento è dettagliato nel testo principale e nella legenda della Figura 7.
Discussione
I reporter HIV-1 a due colori sono strumenti unici e potenti(Calvanese et al., 2013; Dahabieh et al., 2013), che permettono l’identificazione e l’isolamento di cellule latenti infette da cellule produttivamente infette e non infette. La latenza si stabilisce molto presto nel corso dell’infezione da HIV-1(Archin et al., 2012b; Chun et al., 1998; Whitney et al., 2014) e, fino all’avvento dei costrutti dual-reporter, nessun modello primario di latenza dell’HIV-1 ha permesso lo studio dell’eterogeneità della latenza in questa fase iniziale. È importante notare che il confronto dei dati ottenuti da diversi modelli di latenza primaria dell’HIV-1 è complicato, poiché alcuni modelli sono più adatti a rilevare l’instaurarsi della latenza (ad esempio, i dual-reporter), mentre altri sono orientati verso il mantenimento della latenza (ad esempio, le cellule T CD4+ con trasduttore Bcl2). L’uso di virus difettosi limita la replicazione dell’HIV a un solo giro e quindi limita la comparsa di virus difettosi(Bruner et al., 2016).
In questo studio, descriviamo e convalidiamo una versione migliorata di HIVDuoFluoI, precedentemente sviluppata nel nostro laboratorio(Calvanese et al., 2013), che consente di ottenere risultati accurati: (a) la quantificazione delle cellule latentemente infette, (b) la purificazione delle cellule latentemente infette e (c) la valutazione della strategia “shock and kill”. I nostri dati evidenziano due fatti importanti: (a) la quantificazione dell’HIV RNA associato alle cellule non riflette il numero di cellule che subiscono una riattivazione virale, e (b) una piccola porzione delle cellule portatrici di provirus latenti (<5%) viene riattivata, anche se gli LRA si rivolgono all’intera popolazione latente. Quindi, anche se le cellule che ospitano il virus riattivato muoiono, questa piccola riduzione rimarrebbe probabilmente non rilevabile quando si quantifica il serbatoio latente in vivo. I nostri dati sono in accordo con i rapporti precedenti, che mostrano che i livelli di produzione cellulare di HIV RNA e virione non sono correlati, e che il numero assoluto di cellule riattivate da αCD3/CD28 è effettivamente limitato a una piccola frazione di cellule latenti infette (Cilloet al., 2014; Sanyal et al. , 2017; Yucha et al., 2017 ). Utilizzando il nostro reporter a doppia fluorescenza, confermiamo questi risultati ed estendiamo queste osservazioni alle combinazioni LRA. Tuttavia, anche se le combinazioni LRA mostrano una sinergia quando si misura l’RNA associato alle cellule dell’HIV, non troviamo tale sinergia a livello delle singole cellule, ma solo un effetto additivo parziale. Il nostro lavoro, così come quello di altri(Cillo et al., 2014; Sanyal et al., 2017; Yucha et al., 2017), dimostra l’importanza dell’analisi di singole cellule quando si tratta di valutare i potenziali LRA. Infatti, è necessario determinare se gli aumenti potenziali di RNA dell’HIV dopo la stimolazione in una popolazione di massa derivano da un piccolo numero di cellule altamente produttive, o da una popolazione più grande ma meno produttiva, in quanto questi due meccanismi hanno probabilmente impatti molto diversi sul serbatoio latente.
I nostri dati evidenziano ulteriormente la natura eterogenea del serbatoio latente(Chen et al., 2017; Ho et al., 2013). Attualmente abbiamo una comprensione limitata del perché alcune cellule latenti infette sono in grado di essere indotte mentre altre non lo sono. È possibile che diversi ambienti della cromatina impongano diversi gradi di repressione trascrizionale sul genoma dell’HIV integrato, con l’HIV latente non riattivabile corrispondente all’ambiente più repressivo.(Chen et al., 2017). Poiché HIVGKO permette di isolare le cellule produttivamente infette e le cellule latenti riattivate da quelle che non si riattivano, offre un’opportunità unica per esplorare l’impatto dell’integrazione dell’HIV sul destino dell’infezione.
Diverse caratteristiche specifiche del sito di integrazione contribuiscono alla latenza, come la struttura della cromatina, compresi i loci adiacenti, ma anche la posizione del provirus nel nucleo(Lusic e Giacca, 2015; Lusic et al., 2013). L’integrazione virale è un processo semi-random(Bushman et al., 2005) in cui l’HIV-1 si integra preferibilmente nei geni attivi(Barr et al., 2006; Bushman et al., 2005; Demeulemeester et al., 2015; Ferris et al., 2010; Han et al., 2004; Lewinski et al., 2006; Mitchell et al., 2004; Schröder et al., 2002; Sowd et al., 2016; Wang et al., 2007). Il LEDGF, uno dei principali fattori di legame della cromatina dell’HIV-1, si lega all’integrasi virale e all’H3K36me3, e in misura minore all’H3K4me1, dirigendo così l’integrazione dell’HIV-1 nelle unità trascrizionali(Daugaard et al., 2012; Eidahl et al., 2013; Pradeepa et al., 2012). Anche il CPSF6, che si lega al capside virale, influenza notevolmente l’integrazione nei geni trascrizionalmente attivi e nelle regioni dell’euchromatina(Sowd et al., 2016), spiegando come l’HIV-1 mantenga la sua integrazione nelle regioni dell’euchromatina del genoma indipendentemente dal LEDGF(Quercioli et al., 2016). Diversi studi hanno caratterizzato i siti di integrazione, tuttavia, queste analisi sono state limitate alle infezioni produttive.
Utilizzando i set di dati di riferimento ENCODE, i nostri dati sono coerenti con i risultati precedenti, dimostrando che l’HIV-1 si rivolge preferibilmente alle regioni trascritte attivamente(Marini et al., 2015; Wang et al., 2007; Chen et al., 2017). Tuttavia, si osserva che i provirus latenti non inducibili sono integrati in misura maggiore nella cromatina silenziata. Inoltre, anche se l’integrazione dell’HIV è normalmente fortemente disfavorita nelle regioni eterocromatiche condensate delle LAD a causa della bassa accessibilità della cromatina, mostriamo che una certa integrazione dell’HIV si verifica nelle LAD quando si utilizza un set di dati pubblicati in precedenza(Guelen et al., 2008; Marini et al., 2015), e che i provirus latenti non prontamente riattivabili sono integrati in misura maggiore nelle LAD.
È importante notare che tra le cellule latenti che possono essere riattivate, identifichiamo una popolazione rara unica tra le cellule latenti che possono essere riattivate. A differenza delle infezioni latenti non inducibili, l’inversione della latenza dei provirus latenti inducibili potrebbe essere spiegata dall’integrazione in un contesto di cromatina aperta, simile ai siti di integrazione per i provirus produttivi, seguita dalla successiva formazione di eterocromatina e dal silenziamento dei provirus. Di conseguenza, i siti di integrazione distinti tra provirus latenti indotti e non indotti evidenziano nuove possibilità di strategie di cura. In effetti, la strategia “shock and kill” mira a riattivare ed eliminare ogni singolo provirus latente replicabile-competente, poiché una singola cellula rimanente che porta un provirus latente inducibile potrebbe, in teoria, riseminare l’infezione. Tuttavia, il nostro studio e altri evidenziano diversi ostacoli significativi alla riuscita dell’implementazione della strategia “shock and kill”. In primo luogo, gli LRA riattivano solo una frazione limitata di provirus latenti. È probabile che alcuni dei provirus non indotti, come quelli integrati negli stimolatori e nelle regioni attive trascrizionali del genoma, si riattivino dopo diversi cicli di attivazione, a causa della natura stocastica dell’attivazione dell’HIV(Dar et al., 2012; Ho et al., 2013; Singh et al., 2010; Weinberger et al., 2005). È anche probabile che le combinazioni di LRA più adatte (due o più LRA) riattivino alcuni dei provirus non indotti integrati nella cromatina silenziata marcata da H3K27me3 e H3K9me3. In effetti, diversi studi hanno dimostrato che l’inibizione farmaceutica dell’H3K27me3 e dell’H3K9me2/3 potrebbe sensibilizzare i provirus latenti agli LRA(Friedman et al., 2011; Nguyen et al., 2017; Tripathy et al., 2015). In secondo luogo, Shan et al. hanno dimostrato che le cellule riattivate latentemente non vengono cancellate a causa degli effetti citopatici o della risposta CTL, il che implica che per ottenere una cura per l’infezione da HIV sono probabilmente necessari approcci immunomodulatori, oltre a LRA più potenti(Shan et al., 2012).
In conclusione, l’eterogeneità del serbatoio latente richiede terapie che affrontino i diversi pool di cellule latenti infette. Mentre “shock and kill” potrebbe essere utile per riattivare ed eventualmente eliminare un piccolo sottoinsieme di genomi HIV latenti altamente riattivabili, altri approcci saranno necessari per controllare o eliminare la popolazione meno prontamente riattivabile identificata qui e nei pazienti. Forse, quest’ultima popolazione dovrebbe piuttosto essere “bloccata e bloccata” utilizzando agenti che favoriscono la latenza (LPA), come descritto da diversi gruppi (Besnardet al., 2016; Kessing et al., 2017; Kim et al., 2016; Vranckx et al., 2016 ). Per una cura funzionale, un provirus stabilmente silenziato e non riattivabile è preferibile a una vita di infezione cronica attiva.
Materiali e metodi
Campioni dei pazienti
Sono stati arruolati quattro individui affetti da HIV-1, che hanno soddisfatto i criteri di ART soppressiva, livelli plasmatici di HIV-1 RNA non rilevabili (<50 copie/ml) per un minimo di sei mesi, e con una conta delle cellule T CD4+ di almeno 350 cellule/mm3. I partecipanti sono stati reclutati dalla coorte SCOPE dell’Università della California, San Francisco. Nella Tabella 1 sono riportate in dettaglio le caratteristiche dei partecipanti allo studio.
Costruzione dei plasmidi
Per costruire HIVGKO, la sequenza csGFP è stata progettata e ordinata da Life Technologies. La sequenza è stata ritagliata dal plasmide di Life Technologies con BamHI e XhoI e clonata in DuoFluoI, precedentemente tagliata con gli stessi enzimi (DuoFluoI-csGFP). HIVGKO è stato creato mediante sovrapposizione PCR: csGFP-EF1α (Prodotto 1) è stata amplificata PCR da DuoFluoI utilizzando i primer P1: 5′ per-GATTAGTGAACGGGATCCGGGCACG-3′ e P2: 5′ rev-GGCTTGATCACAGAAACCATGGGTGGGCGACGGTAGGGCGGCGC-3′. mKO2 (Prodotto 2) è stato amplificato con PCR dal plasmide di Brian Webster (gentile dono della Warner Greene) utilizzando i primer P3: 5′ per-GCGGCTACCGGGGTCGCCCCACCATGGGTTTCTGTGTGATCAA
GCC-3′ e P4: 5′ rev-CTCCATGTTTTTTTCCAGGTCCAGGTTCCGGGTCCGGGC CACGGGC-3′. Infine, abbiamo amplificato la sequenza 3’LTR (Prodotto 3) dal plasmide RGH (Dahabiehet al., 2013) utilizzando i primer P5: 5′ per-GCTCGGAGACCTGGAAAAAACATGGAG-3′ e P6: 5′ rev-GTGCCACCTGACGACGTCTAAGAAACC-3′, per aggiungere un frammento contenente il sito di restrizione AatII, al fine di legare la cassetta csGFP-EF1α-mKO2 in pLAI (Peden et al., 1991). Abbiamo poi fatto delle PCR sequenziali: i prodotti 1 e 2 sono stati amplificati usando i primer P1 e P4. Il prodotto PCR (1 + 2) è stato miscelato con il prodotto 3 e la PCR amplificata con P1 e P6 creando così la cassetta completa. La cassetta csGFP-EF1α-mKO2 è stata poi digerita con BamHI e AatII, e clonata in pLAI precedentemente digerita con gli stessi enzimi per creare HIVGKO.
Da notare che il frame di lettura aperto della busta è stato interrotto dall’introduzione di uno spostamento del frame nella posizione 7136 per digestione con KpnI, smussatura e ri-legamento.
Per costruire GKO-∆U3 3’LTR, abbiamo clonato un linker ∆U3 da pTY-EFeGFP (Chang et al., 1999; Cui et al., 1999; Iwakuma et al., 1999; Zolotukhin et al., 1996) nei siti KpnI/SacI del 3′ LTR in HIVGKO.
Produzione di virus
La produzione di HIVGKOe la valutazione degli agenti di inversione della latenza dell’HIV nelle cellule primarie CD4+ T umane sono descritte più dettagliatamente nel Bioprotocollo (Battivellie Verdin, 2018).Le scortevirali di HIVDuoFluoIe HIVGKO pseudotipizzate sono state generate mediante la co-trasformazione (metodo standard di trasfezione del fosfato di calcio) delle cellule HEK293T con un plasmide che codifica l’HIVDuoFluoIo l’HIVGKO,e un plasmide che codifica l’HIV-1 a doppio tropico (pSVIII-92HT593.1). Medio è stato cambiato 6-8 ore post-trasformazione, e supernatanti sono stati raccolti dopo 48 ore, centrifugato (20 min, 2000 rpm, RT), filtrato attraverso una membrana di 0,45 micron per eliminare i detriti cellulari, e poi concentrato da ultracentrifugazione (22.000 g, 2 ore, 4 ° C). I virioni concentrati sono stati risospesi in supporti completi e conservati a -80°C. La concentrazione dei virioni è stata stimata mediante titolazione a p24 utilizzando il saggio FLAQ(Gesner et al., 2014).
Isolamento delle cellule primarie e coltura cellulare
Le cellule T CD4+ sono state estratte da cellule mononucleate di sangue periferico (PBMC) da prodotti di leucoforsi a flusso continuo per centrifugazione con centrifugazione a densità su un gradiente Ficoll-Paque (GE Healthcare Life Sciences, Chicago, IL). I linfociti CD4+ a riposo sono stati arricchiti da deplezione negativa con un kit di isolamento delle cellule T EasySepHuman CD4+ (Stemcell Technologies, Canada). Le cellule sono state coltivate in un mezzo RPMI integrato con il 10% di siero bovino fetale, penicillina/streptomicina e 5 µM di saquinavir.
Le cellule T CD4+ primarie CD4+ sono state purificate da sangue di donatori sani (Blood Centers of the Pacific, San Francisco, CA, e Stanford Blood Center), mediante selezione negativa utilizzando il RosetteSep Human CD4+ T Cell Enrichment Cocktail (StemCell Technologies, Canada). Purificate cellule T CD4 + a riposo CD4 + da HIV-1 o individui sani sono stati coltivati in RPMI 1640 mezzo integrato con 10% FBS, L-glutammina (2 mM), penicillina (50 U / ml), streptomicina (50 mg / ml), e IL-2 (20 a 100 U / ml) (37 ° C, 5% di CO2). Spin-infetto primario CD4 + cellule T sono stati mantenuti nel 50% dei mezzi completi RPMI integrato con IL-2 (20-100 U / ml) e il 50% di supernatante da colture H80 (precedentemente filtrato per rimuovere le cellule) senza perline. Il mezzo è stato reintegrato ogni 2 giorni fino ad un ulteriore esperimento.
HEK293T cellule sono state ottenute da ATCC (senza micoplasma). Alimentatore cellule H80 è stato un regalo gentile da Jonathan Karn. H80 cellule H80 sono state coltivate in RPMI 1640 mezzo integrato con il 10% di siero bovino fetale (FBS), L-glutammina (2 mM), penicillina (50 U / ml), e streptomicina (50 mg / ml) (37 ° C, 5% di CO2). HEK293T cellule sono state coltivate in mezzo DMEM integrato con il 10% FBS, 50 U / ml penicillina, e 50 mg / ml di streptomicina.
Infezione cellulare
Le cellule T CD4+ T purificate isolate da sangue periferico sano sono state stimolate con perle attivanti αCD3/CD28 (Thermofisher, Waltham, MA) ad una concentrazione di 0,5 perle/cellule in presenza di 20-100 U/ml IL-2 (PeproTech, Rocky Hill, NJ) per tre giorni. Tutte le cellule sono state spinoculate con HIVDuoFluoI, HIVGKO o HIV Δ3U-GKO ad una concentrazione di 300 ng di p24 per 1,106 cellule per 2 ore a 2000 rpm a 32°C senza perle di attivazione.
Le cellule infette sono state analizzate mediante citometria a flusso o ordinate 4-5 giorni dopo l’infezione.
Condizioni di trattamento con agente invertitore di latenza
Le cellule T CD4+ sono state stimolate per 24 ore, a meno che non sia stato stabilito diversamente, con agenti invertitori di latenza alle seguenti concentrazioni per tutti i trattamenti singoli e di combinazione: 10 nM briostatina-1, 1 μM JQ1, 30 nM panobinostat, αCD3/CD28 perline attivanti (1 perlina/cellula), o media da soli più 0,1% (v/v) DMSO. Per tutti i trattamenti singoli e combinati, ai media sono stati aggiunti 30 μM Raltregravir (National AIDS Reagent Program). Le concentrazioni sono state scelte sulla base di Laird et al. paper(Laird et al., 2015).
Colorazione, citometria a flusso e selezione cellulare
Le celle della Figura 4 sono state colorate con α-CD69-PE-Cy7 (561928), α-CD25-APC (560987) e α-HLA-DR-PerCP-Cy5.5 (562007) (BD Bioscience, Franklin Lakes, NJ).
Prima di raccogliere i dati utilizzando il FACS LSRII (BD Biosciences, Franklin Lakes, NJ) o il FACS AriaII (BD Biosciences, Franklin Lakes, NJ, Figure 3 e 4), le cellule sono state macchiate con una macchia viola di cellule morte fissabili dal vivo/morte (Thermofisher, Waltham, MA) e fissate con formaldeide al 2%. Le analisi sono state eseguite con il software FlowJo V10.1 (TreeStar).
Ordinamento delle cellule T CD4 + infette CD4 è stata eseguita con un FACS AriaII (BD Biosciences, Franklin Lakes, NJ) sulla base dei loro marcatori di fluorescenza GFP e mKO2 a 4/5 giorni dopo l’infezione, e rimesso in coltura per ulteriori sperimentazioni. Negli esperimenti mostrati nelle figure 2B e 4, abbiamo isolato sia le cellule infette da HIVGKO latente (GFP-, mKO2+, 3%) sia le cellule non infette (csGFP-, mKO2-, 97%) cinque giorni dopo l’infezione, prima di trattare le cellule con LRA.
Nell’esperimento mostrato in figura 3, abbiamo isolato cellule latenti pure (GFP-, mKO2+) cinque giorni dopo l’infezione, prima di trattare questa popolazione pura con LRA.
DNA, RNA ed estrazione di proteine, qPCR e western blot
RNA e proteine(Figura 1B e C) sono stati estratti con il kit PARISTM (Ambion, Thermofisher, Waltham, MA) secondo il protocollo del produttore dagli stessi campioni. RNA è stato retro-trascritto utilizzando primer casuali con la trascrittasi inversa SuperScript II (Thermofisher, Waltham, MA) e qPCR è stata eseguita nel sistema AB7900HT Fast Real-Time PCR System, utilizzando 2X HoTaq Real Time PCR kit (McLab, South San Francisco, CA) e le combinazioni di primer-sonda appropriate descritte in (Calvaneseet al., 2013). La quantificazione per ogni reazione qPCR è stata valutata dall’algoritmo ddCt, relativo al saggio Taq Man GAPDH Hs9999999905_m1. Il contenuto proteico è stato determinato utilizzando il test di Bradford (Bio-Rad, Hercules, CA) e 20 μg sono stati separati per elettroforesi in gel SDS-PAGE al 12%. Le bande sono state rilevate mediante chemiluminescenza (ECL Hyperfilm Amersham, GE Healthcare Life Sciences, Chicago, I) con anticorpi primari anti-Vif, HIV-p24 e α-actina (Sigma, Saint-Louis, MO).
RNA totale(Figura 2A e B) estratto con il kit universale Allprep DNA/RNA/miRNA Universal Kit (Qiagen, Germania) con trattamento della DNAasi a colonna (Qiagen RNase-Free DNase Set, Germania). La sintesi di cDNA è stata eseguita utilizzando SuperScript IV Transcriptasi inversa con una combinazione di esametri casuali e primer oligo-dT (ThermoFisher, Waltham, MA). I livelli relativi di mRNA HIV cellulare sono stati quantificati utilizzando un saggio qPCR TaqMan con primer e sonde descritte in(Bullen et al., 2014) su un sistema PCR in tempo reale QuantStudio 6 Flex Real-Time PCR System (Thermofisher, Waltham, MA). I numeri relativi di copie di mRNA HIV associati alle cellule sono stati determinati in un volume di reazione di 20 μL con 10 μL di 2x TaqMan Universal Master Mix II con UNG (Thermofisher, Waltham, MA), 4 pmol di ogni primer, 4 pmol di sonda, 0,5 μL di trascrittasi inversa e 2,5 μL di cDNA. Le condizioni di ciclo erano di 50°C o 2 min, 95°C per 10 min, poi 60 cicli di 95°C per 15 s e 60°C per 1 min. La PCR in tempo reale è stata eseguita in pozzetti di reazione in triplice copia e il relativo numero di copie di mRNA HIV associato alle cellule è stato normalizzato in equivalenti cellulari utilizzando l’espressione genomica umana GAPDH con qPCR e applicando il metodo comparativo Ct(Livak e Schmittgen, 2001).
Biblioteche di siti di integrazione dell’HIV e analisi computazionale
Le biblioteche dei siti di integrazione dell’HIV e l’analisi computazionale sono state eseguite in collaborazione con Lilian B. Cohn e Israel Tojal Da Silva, come descritto nel loro articolo pubblicato(Cohn et al., 2015), con alcune piccole modifiche aggiunte alla pipeline di analisi computazionale. In primo luogo, abbiamo incluso siti di integrazione con solo una precisa giunzione al genoma ospite. In secondo luogo, per eliminare ogni possibilità di errore di PCR, abbiamo escluso i siti di integrazione identificati entro 100 bp (50 bp a monte e 50 bp a valle) di un motivo di 9 bp identificato nel nostro primer LTR1: TGCCTTGAG. In terzo luogo abbiamo unito i siti di integrazione entro 250 bp e abbiamo contato ogni sito di integrazione come un evento unico. L’elenco dei siti di integrazione per ogni donatore e per ogni popolazione può essere trovato come file di dati sorgente collegato a questo manoscritto (Integration Sites Source data 1).
Set di dati
I dati della cromatina (ChIP-seq) dalle celle CD4+ T sono stati scaricati da ENCODE: H3K4me1 (ENCFF112QDR, ENCFF499NFE, ENCFF989BNS), H3K9me3 (ENCFF044NLN, ENCFF736KRZ, ENCFF844IWD, ENCFF929BPC), H3K27ac (ENCFF618IUD, ENCFF862SKP), H3K27me3 (ENCFF124QDDD, ENCFF298JKA, ENCFF717ODY), H3K36me3 (ENCFF006VTQ, ENCFF169QYM, ENCFF284PKI, ENCFF504OUW), DNAse (GSM665812, GSM665839, GSM701489, GSM701491). I dati sono stati analizzati utilizzando Seqmonk (v0.33, http://www.bioinformatics.bbsrc.ac.uk/projects/ seqmonk/).
Abbiamo calcolato l’espressione (GSM669617) e l’abbondanza del marchio cromatina (i rimanenti set di dati ENCODE) nei siti di integrazione come bins di 500 bp centrati sul sito di integrazione (quantificazione del conteggio di lettura in Seqmonk: tutte le letture non duplicate indipendentemente dal filamento, corrette per milione di letture totali, non trasformate). Le annotazioni genetiche non sono state prese in considerazione. Le soglie per i valori di espressione (1/8 superiore, quarto superiore, metà e superiore a 0) sono state impostate per distinguere cinque diverse categorie, impostate come 1/8 superiore dei valori di espressione (alto), quarto superiore-1/8° (medio), mezzo quarto superiore (basso), metà inferiore ma superiore a 0 (traccia), 0 (silenzioso).
I dati di attivazione delle celle CD4+ T della Figura 5A sono stati scaricati da GEO (GSE60235).
Analisi statistica
La significatività è stata analizzata con il test t-test accoppiato (GraphPad Prism) o con il test delle proporzioni (test standard per la differenza tra le proporzioni), noto anche come test z a due proporzioni(https://www.medcalc.org/calc/comparison_of_proportions.php), e specificato nel manoscritto.
References
- Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, Parker DC, Anderson EM, Kearney MF, Strain MC, Richman DD, Hudgens MG, Bosch RJ, Coffin JM, Eron JJ, Hazuda DJ, Margolis DM. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012a; 487:482-485. DOI | PubMed
- Archin NM, Vaidya NK, Kuruc JD, Liberty AL, Wiegand A, Kearney MF, Cohen MS, Coffin JM, Bosch RJ, Gay CL, Eron JJ, Margolis DM, Perelson AS. Immediate antiviral therapy appears to restrict resting CD4+ cell HIV-1 infection without accelerating the decay of latent infection. PNAS. 2012b; 109:9523-9528. DOI | PubMed
- Banerjee C, Archin N, Michaels D, Belkina AC, Denis GV, Bradner J, Sebastiani P, Margolis DM, Montano M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. Journal of Leukocyte Biology. 2012; 92:1147-1154. DOI | PubMed
- Barr SD, Ciuffi A, Leipzig J, Shinn P, Ecker JR, Bushman FD. HIV integration site selection: targeting in macrophages and the effects of different routes of viral entry. Molecular Therapy. 2006; 14:218-225. DOI | PubMed
- Battivelli E, Verdin E. HIVGKO: a tool to assess HIV-1 latency reversal agents in human primary CD4+ T cells. Bio-Protocol. 2018; 8DOI
- Besnard E, Hakre S, Kampmann M, Lim HW, Hosmane NN, Martin A, Bassik MC, Verschueren E, Battivelli E, Chan J, Svensson JP, Gramatica A, Conrad RJ, Ott M, Greene WC, Krogan NJ, Siliciano RF, Weissman JS, Verdin E. The mTOR complex controls HIV latency. Cell Host & Microbe. 2016; 20:785-797. DOI | PubMed
- Boehm D, Calvanese V, Dar RD, Xing S, Schroeder S, Martins L, Aull K, Li PC, Planelles V, Bradner JE, Zhou MM, Siliciano RF, Weinberger L, Verdin E, Ott M. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle. 2013; 12:452-462. DOI | PubMed
- Bruner KM, Murray AJ, Pollack RA, Soliman MG, Laskey SB, Capoferri AA, Lai J, Strain MC, Lada SM, Hoh R, Ho YC, Richman DD, Deeks SG, Siliciano JD, Siliciano RF. Defective proviruses rapidly accumulate during acute HIV-1 infection. Nature Medicine. 2016; 22:1043-1049. DOI | PubMed
- Bullen CK, Laird GM, Durand CM, Siliciano JD, Siliciano RF. New ex vivo approaches distinguish effective and ineffective single agents for reversing HIV-1 latency in vivo. Nature Medicine. 2014; 20:425-429. DOI | PubMed
- Bushman F, Lewinski M, Ciuffi A, Barr S, Leipzig J, Hannenhalli S, Hoffmann C. Genome-wide analysis of retroviral DNA integration. Nature Reviews Microbiology. 2005; 3:848-858. DOI | PubMed
- Calvanese V, Chavez L, Laurent T, Ding S, Verdin E. Dual-color HIV reporters trace a population of latently infected cells and enable their purification. Virology. 2013; 446:283-292. DOI | PubMed
- Chang LJ, Urlacher V, Iwakuma T, Cui Y, Zucali J. Efficacy and safety analyses of a recombinant human immunodeficiency virus type 1 derived vector system. Gene Therapy. 1999; 6:715-728. DOI | PubMed
- Chen H-C, Martinez JP, Zorita E, Meyerhans A, Filion GJ. Position effects influence HIV latency reversal. Nature Structural & Molecular Biology. 2017; 24:47-54. DOI
- Chun TW, Engel D, Berrey MM, Shea T, Corey L, Fauci AS. Early establishment of a pool of latently infected, resting CD4(+) T cells during primary HIV-1 infection. PNAS. 1998; 95:8869-8873. DOI | PubMed
- Cillo AR, Sobolewski MD, Bosch RJ, Fyne E, Piatak M, Coffin JM, Mellors JW. Quantification of HIV-1 latency reversal in resting CD4+ T cells from patients on suppressive antiretroviral therapy. PNAS. 2014; 111:7078-7083. DOI | PubMed
- Cohn LB, Silva IT, Oliveira TY, Rosales RA, Parrish EH, Learn GH, Hahn BH, Czartoski JL, McElrath MJ, Lehmann C, Klein F, Caskey M, Walker BD, Siliciano JD, Siliciano RF, Jankovic M, Nussenzweig MC. HIV-1 integration landscape during latent and active infection. Cell. 2015; 160:420-432. DOI | PubMed
- Cui Y, Iwakuma T, Chang LJ. Contributions of viral splice sites and cis-regulatory elements to lentivirus vector function. Journal of Virology. 1999; 73:6171-6176. PubMed
- Dahabieh MS, Ooms M, Simon V, Sadowski I. A doubly fluorescent HIV-1 reporter shows that the majority of integrated HIV-1 is latent shortly after infection. Journal of Virology. 2013; 87:4716-4727. DOI | PubMed
- Dahabieh MS, Ooms M, Brumme C, Taylor J, Harrigan PR, Simon V, Sadowski I. Direct non-productive HIV-1 infection in a T-cell line is driven by cellular activation state and NFκB. Retrovirology. 2014; 11DOI | PubMed
- Dahabieh MS, Battivelli E, Verdin E. Understanding HIV latency: the road to an HIV cure. Annual Review of Medicine. 2015; 66:407-421. DOI | PubMed
- Dar RD, Razooky BS, Singh A, Trimeloni TV, McCollum JM, Cox CD, Simpson ML, Weinberger LS. Transcriptional burst frequency and burst size are equally modulated across the human genome. PNAS. 2012; 109:17454-17459. DOI | PubMed
- Darcis G, Kula A, Bouchat S, Fujinaga K, Corazza F, Ait-Ammar A, Delacourt N, Melard A, Kabeya K, Vanhulle C, Van Driessche B, Gatot JS, Cherrier T, Pianowski LF, Gama L, Schwartz C, Vila J, Burny A, Clumeck N, Moutschen M, De Wit S, Peterlin BM, Rouzioux C, Rohr O, Van Lint C. An in-depth comparison of latency-reversing agent combinations in various in vitro and ex vivo hiv-1 latency models identified bryostatin-1+JQ1 and Ingenol-B+JQ1 to potently reactivate viral gene expression. PLoS Pathogens. 2015; 11DOI | PubMed
- Daugaard M, Baude A, Fugger K, Povlsen LK, Beck H, Sørensen CS, Petersen NH, Sorensen PH, Lukas C, Bartek J, Lukas J, Rohde M, Jäättelä M. LEDGF (p75) promotes DNA-end resection and homologous recombination. Nature Structural & Molecular Biology. 2012; 19:803-810. DOI | PubMed
- De Crignis E, Mahmoudi T. The Multifaceted Contributions of Chromatin to HIV-1 Integration, Transcription, and Latency. International Review of Cell and Molecular Biology. 2017; 328:197-252. DOI | PubMed
- del Real G, Jiménez-Baranda S, Mira E, Lacalle RA, Lucas P, Gómez-Moutón C, Alegret M, Peña JM, Rodríguez-Zapata M, Alvarez-Mon M, Martínez-A C, Mañes S. Statins inhibit HIV-1 infection by down-regulating Rho activity. The Journal of Experimental Medicine. 2004; 200:541-547. DOI | PubMed
- Demeulemeester J, De Rijck J, Gijsbers R, Debyser Z. Retroviral integration: Site matters: Mechanisms and consequences of retroviral integration site selection. BioEssays : News and Reviews in Molecular, Cellular and Developmental Biology. 2015; 37:1202-1214. DOI | PubMed
- Eidahl JO, Crowe BL, North JA, McKee CJ, Shkriabai N, Feng L, Plumb M, Graham RL, Gorelick RJ, Hess S, Poirier MG, Foster MP, Kvaratskhelia M. Structural basis for high-affinity binding of LEDGF PWWP to mononucleosomes. Nucleic Acids Research. 2013; 41:3924-3936. DOI | PubMed
- Elliott JH, Wightman F, Solomon A, Ghneim K, Ahlers J, Cameron MJ, Smith MZ, Spelman T, McMahon J, Velayudham P, Brown G, Roney J, Watson J, Prince MH, Hoy JF, Chomont N, Fromentin R, Procopio FA, Zeidan J, Palmer S, Odevall L, Johnstone RW, Martin BP, Sinclair E, Deeks SG, Hazuda DJ, Cameron PU, Sékaly RP, Lewin SR. Activation of HIV transcription with short-course vorinostat in HIV-infected patients on suppressive antiretroviral therapy. PLoS Pathogens. 2014; 10DOI | PubMed
- Elliott JH, McMahon JH, Chang CC, Lee SA, Hartogensis W, Bumpus N, Savic R, Roney J, Hoh R, Solomon A, Piatak M, Gorelick RJ, Lifson J, Bacchetti P, Deeks SG, Lewin SR. Short-term administration of disulfiram for reversal of latent HIV infection: a phase 2 dose-escalation study. The Lancet HIV. 2015; 2:e520-e529. DOI | PubMed
- Ferris AL, Wu X, Hughes CM, Stewart C, Smith SJ, Milne TA, Wang GG, Shun MC, Allis CD, Engelman A, Hughes SH. Lens epithelium-derived growth factor fusion proteins redirect HIV-1 DNA integration. PNAS. 2010; 107:3135-3140. DOI | PubMed
- Filippakopoulos P, Qi J, Picaud S, Shen Y, Smith WB, Fedorov O, Morse EM, Keates T, Hickman TT, Felletar I, Philpott M, Munro S, McKeown MR, Wang Y, Christie AL, West N, Cameron MJ, Schwartz B, Heightman TD, La Thangue N, French CA, Wiest O, Kung AL, Knapp S, Bradner JE. Selective inhibition of BET bromodomains. Nature. 2010; 468:1067-1073. DOI | PubMed
- Friedman J, Cho WK, Chu CK, Keedy KS, Archin NM, Margolis DM, Karn J. Epigenetic silencing of HIV-1 by the histone H3 lysine 27 methyltransferase enhancer of Zeste 2. Journal of Virology. 2011; 85:9078-9089. DOI | PubMed
- Gesner M, Maiti M, Grant R, Cavrois M. Fluorescence-linked antigen quantification (FLAQ) assay for fast quantification of HIV-1 p24Gag. Bio-Protocol. 2014; 4DOI | PubMed
- Guelen L, Pagie L, Brasset E, Meuleman W, Faza MB, Talhout W, Eussen BH, de Klein A, Wessels L, de Laat W, van Steensel B. Domain organization of human chromosomes revealed by mapping of nuclear lamina interactions. Nature. 2008; 453:948-951. DOI | PubMed
- Han Y, Lassen K, Monie D, Sedaghat AR, Shimoji S, Liu X, Pierson TC, Margolick JB, Siliciano RF, Siliciano JD. Resting CD4+ T cells from human immunodeficiency virus type 1 (HIV-1)-infected individuals carry integrated HIV-1 genomes within actively transcribed host genes. Journal of Virology. 2004; 78:6122-6133. DOI | PubMed
- Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DI, Lai J, Blankson JN, Siliciano JD, Siliciano RF. Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell. 2013; 155:540-551. DOI | PubMed
- Honeycutt JB, Thayer WO, Baker CE, Ribeiro RM, Lada SM, Cao Y, Cleary RA, Hudgens MG, Richman DD, Garcia JV. HIV persistence in tissue macrophages of humanized myeloid-only mice during antiretroviral therapy. Nature Medicine. 2017; 23:638-643. DOI | PubMed
- Iwakuma T, Cui Y, Chang LJ. Self-inactivating lentiviral vectors with U3 and U5 modifications. Virology. 1999; 261:120-132. DOI | PubMed
- Jiang G, Mendes EA, Kaiser P, Wong DP, Tang Y, Cai I, Fenton A, Melcher GP, Hildreth JE, Thompson GR, Wong JK, Dandekar S. Synergistic reactivation of latent HIV expression by ingenol-3-Angelate, PEP005, Targeted NF-kB signaling in combination with JQ1 Induced p-TEFb Activation. PLoS Pathogens. 2015; 11DOI | PubMed
- Jordan A, Defechereux P, Verdin E. The site of HIV-1 integration in the human genome determines basal transcriptional activity and response to Tat transactivation. The EMBO Journal. 2001; 20:1726-1738. DOI | PubMed
- Jordan A, Bisgrove D, Verdin E. HIV reproducibly establishes a latent infection after acute infection of T cells in vitro. The EMBO Journal. 2003; 22:1868-1877. DOI | PubMed
- Kessing CF, Nixon CC, Li C, Tsai P, Takata H, Mousseau G, Ho PT, Honeycutt JB, Fallahi M, Trautmann L, Garcia JV, Valente ST. In vivo suppression of HIV rebound by didehydro-cortistatin A, a "Block-and-Lock" strategy for HIV-1 treatment. Cell Reports. 2017; 21:600-611. DOI | PubMed
- Kim H, Choi MS, Inn KS, Kim BJ. Inhibition of HIV-1 reactivation by a telomerase-derived peptide in a HSP90-dependent manner. Scientific Reports. 2016; 6DOI | PubMed
- Kumar A, Darcis G, Van Lint C, Herbein G. Epigenetic control of HIV-1 post integration latency: implications for therapy. Clinical Epigenetics. 2015; 7DOI | PubMed
- Laird GM, Bullen CK, Rosenbloom DI, Martin AR, Hill AL, Durand CM, Siliciano JD, Siliciano RF. Ex vivo analysis identifies effective HIV-1 latency-reversing drug combinations. Journal of Clinical Investigation. 2015; 125:1901-1912. DOI | PubMed
- Lewinski MK, Yamashita M, Emerman M, Ciuffi A, Marshall H, Crawford G, Collins F, Shinn P, Leipzig J, Hannenhalli S, Berry CC, Ecker JR, Bushman FD. Retroviral DNA integration: viral and cellular determinants of target-site selection. PLoS Pathogens. 2006; 2DOI | PubMed
- Li Z, Guo J, Wu Y, Zhou Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Research. 2013; 41:277-287. DOI | PubMed
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods. 2001; 25:402-408. DOI | PubMed
- Lusic M, Marini B, Ali H, Lucic B, Luzzati R, Giacca M. Proximity to PML nuclear bodies regulates HIV-1 latency in CD4+ T cells. Cell Host & Microbe. 2013; 13:665-677. DOI | PubMed
- Lusic M, Giacca M. Regulation of HIV-1 latency by chromatin structure and nuclear architecture. Journal of Molecular Biology. 2015; 427:688-694. DOI | PubMed
- Maldarelli F, Wu X, Su L, Simonetti FR, Shao W, Hill S, Spindler J, Ferris AL, Mellors JW, Kearney MF, Coffin JM, Hughes SH. HIV latency. Specific HIV integration sites are linked to clonal expansion and persistence of infected cells. Science. 2014; 345:179-183. DOI | PubMed
- Maldarelli F. The role of HIV integration in viral persistence: no more whistling past the proviral graveyard. Journal of Clinical Investigation. 2016; 126:438-447. DOI | PubMed
- Margolis DM, Garcia JV, Hazuda DJ, Haynes BF. Latency reversal and viral clearance to cure HIV-1. Science. 2016; 353:aaf6517. DOI | PubMed
- Marini B, Kertesz-Farkas A, Ali H, Lucic B, Lisek K, Manganaro L, Pongor S, Luzzati R, Recchia A, Mavilio F, Giacca M, Lusic M. Nuclear architecture dictates HIV-1 integration site selection. Nature. 2015; 521:227-231. DOI | PubMed
- Martínez-Bonet M, Clemente MI, Serramía MJ, Muñoz E, Moreno S, Muñoz-Fernández MÁ. Synergistic activation of latent hiv-1 expression by novel histone deacetylase inhibitors and bryostatin-1. Scientific Reports. 2015; 5DOI | PubMed
- Mehla R, Bivalkar-Mehla S, Zhang R, Handy I, Albrecht H, Giri S, Nagarkatti P, Nagarkatti M, Chauhan A. Bryostatin modulates latent HIV-1 infection via PKC and AMPK signaling but inhibits acute infection in a receptor independent manner. PLoS One. 2010; 5DOI | PubMed
- Mitchell RS, Beitzel BF, Schroder AR, Shinn P, Chen H, Berry CC, Ecker JR, Bushman FD. Retroviral DNA integration: ASLV, HIV, and MLV show distinct target site preferences. PLoS Biology. 2004; 2DOI | PubMed
- Nguyen K, Das B, Dobrowolski C, Karn J. Multiple histone lysine methyltransferases are required for the establishment and maintenance of HIV-1 latency. mBio. 2017; 8DOI | PubMed
- Peden K, Emerman M, Montagnier L. Changes in growth properties on passage in tissue culture of viruses derived from infectious molecular clones of HIV-1LAI, HIV-1MAL, and HIV-1ELI. Virology. 1991; 185:661-672. DOI | PubMed
- Pollack RA, Jones RB, Pertea M, Bruner KM, Martin AR, Thomas AS, Capoferri AA, Beg SA, Huang SH, Karandish S, Hao H, Halper-Stromberg E, Yong PC, Kovacs C, Benko E, Siliciano RF, Ho YC. Defective HIV-1 proviruses are expressed and can be recognized by cytotoxic T lymphocytes, which shape the proviral landscape. Cell Host & Microbe. 2017; 21:494-506. DOI | PubMed
- Pradeepa MM, Sutherland HG, Ule J, Grimes GR, Bickmore WA. Psip1/Ledgf p52 binds methylated histone H3K36 and splicing factors and contributes to the regulation of alternative splicing. PLoS Genetics. 2012; 8DOI | PubMed
- Quercioli V, Di Primio C, Casini A, Mulder LCF, Vranckx LS, Borrenberghs D, Gijsbers R, Debyser Z, Cereseto A. Comparative analysis of HIV-1 and murine leukemia virus three-dimensional nuclear distributions. Journal of Virology. 2016; 90:5205-5209. DOI | PubMed
- Rasmussen TA, Tolstrup M, Winckelmann A, Østergaard L, Søgaard OS. Eliminating the latent HIV reservoir by reactivation strategies: advancing to clinical trials. Human Vaccines & Immunotherapeutics. 2013; 9:790-799. DOI | PubMed
- Rasmussen TA, Tolstrup M, Brinkmann CR, Olesen R, Erikstrup C, Solomon A, Winckelmann A, Palmer S, Dinarello C, Buzon M, Lichterfeld M, Lewin SR, Østergaard L, Søgaard OS. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group, clinical trial. The Lancet HIV. 2014; 1:e13-e21. DOI | PubMed
- Rasmussen TA, Lewin SR. Shocking HIV out of hiding: where are we with clinical trials of latency reversing agents?. Current opinion in HIV and AIDS. 2016; 11:394-401. DOI | PubMed
- Rasmussen TA, Tolstrup M, Søgaard OS. Reversal of latency as part of a cure for HIV-1. Trends in Microbiology. 2016; 24:90-97. DOI | PubMed
- Salamango DJ, Evans DA, Baluyot MF, Furlong JN, Johnson MC. Recombination can lead to spurious results in retroviral transduction with dually fluorescent reporter genes. Journal of Virology. 2013; 87:13900-13903. DOI | PubMed
- Sanyal A, Mailliard RB, Rinaldo CR, Ratner D, Ding M, Chen Y, Zerbato JM, Giacobbi NS, Venkatachari NJ, Patterson BK, Chargin A, Sluis-Cremer N, Gupta P. Novel assay reveals a large, inducible, replication-competent HIV-1 reservoir in resting CD4+ T cells. Nature Medicine. 2017; 23:885-889. DOI | PubMed
- Schröder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002; 110:521-529. DOI | PubMed
- Shan L, Deng K, Shroff NS, Durand CM, Rabi SA, Yang HC, Zhang H, Margolick JB, Blankson JN, Siliciano RF. Stimulation of HIV-1-specific cytolytic T lymphocytes facilitates elimination of latent viral reservoir after virus reactivation. Immunity. 2012; 36:491-501. DOI | PubMed
- Sherrill-Mix S, Lewinski MK, Famiglietti M, Bosque A, Malani N, Ocwieja KE, Berry CC, Looney D, Shan L, Agosto LM, Pace MJ, Siliciano RF, O’Doherty U, Guatelli J, Planelles V, Bushman FD. HIV latency and integration site placement in five cell-based models. Retrovirology. 2013; 10DOI | PubMed
- Shlyueva D, Stampfel G, Stark A. Transcriptional enhancers: from properties to genome-wide predictions. Nature Reviews Genetics. 2014; 15:272-286. DOI | PubMed
- Singh A, Razooky B, Cox CD, Simpson ML, Weinberger LS. Transcriptional bursting from the HIV-1 promoter is a significant source of stochastic noise in HIV-1 gene expression. Biophysical Journal. 2010; 98:L32-L34. DOI | PubMed
- Søgaard OS, Graversen ME, Leth S, Olesen R, Brinkmann CR, Nissen SK, Kjaer AS, Schleimann MH, Denton PW, Hey-Cunningham WJ, Koelsch KK, Pantaleo G, Krogsgaard K, Sommerfelt M, Fromentin R, Chomont N, Rasmussen TA, Østergaard L, Tolstrup M. The depsipeptide romidepsin reverses HIV-1 Latency In Vivo. PLoS Pathogens. 2015; 11DOI | PubMed
- Sowd GA, Serrao E, Wang H, Wang W, Fadel HJ, Poeschla EM, Engelman AN. A critical role for alternative polyadenylation factor CPSF6 in targeting HIV-1 integration to transcriptionally active chromatin. PNAS. 2016; 113:E1054-E1063. DOI | PubMed
- Spina CA, Anderson J, Archin NM, Bosque A, Chan J, Famiglietti M, Greene WC, Kashuba A, Lewin SR, Margolis DM, Mau M, Ruelas D, Saleh S, Shirakawa K, Siliciano RF, Singhania A, Soto PC, Terry VH, Verdin E, Woelk C, Wooden S, Xing S, Planelles V. An in-depth comparison of latent HIV-1 reactivation in multiple cell model systems and resting CD4+ T cells from aviremic patients. PLoS Pathogens. 2013; 9DOI | PubMed
- Tripathy MK, McManamy ME, Burch BD, Archin NM, Margolis DM. H3K27 demethylation at the proviral promoter sensitizes latent HIV to the effects of vorinostat in ex vivo cultures of resting CD4+ T cells. Journal of Virology. 2015; 89:8392-8405. DOI | PubMed
- Vranckx LS, Demeulemeester J, Saleh S, Boll A, Vansant G, Schrijvers R, Weydert C, Battivelli E, Verdin E, Cereseto A, Christ F, Gijsbers R, Debyser Z. LEDGIN-mediated inhibition of integrase-LEDGF/p75 interaction reduces reactivation of residual latent HIV. EBioMedicine. 2016; 8:248-264. DOI | PubMed
- Wagner TA, McLaughlin S, Garg K, Cheung CY, Larsen BB, Styrchak S, Huang HC, Edlefsen PT, Mullins JI, Frenkel LM. HIV latency. Proliferation of cells with HIV integrated into cancer genes contributes to persistent infection. Science. 2014; 345:570-573. DOI | PubMed
- Wang GP, Ciuffi A, Leipzig J, Berry CC, Bushman FD. HIV integration site selection: analysis by massively parallel pyrosequencing reveals association with epigenetic modifications. Genome Research. 2007; 17:1186-1194. DOI | PubMed
- Weinberger LS, Burnett JC, Toettcher JE, Arkin AP, Schaffer DV. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell. 2005; 122:169-182. DOI | PubMed
- Whitney JB, Hill AL, Sanisetty S, Penaloza-MacMaster P, Liu J, Shetty M, Parenteau L, Cabral C, Shields J, Blackmore S, Smith JY, Brinkman AL, Peter LE, Mathew SI, Smith KM, Borducchi EN, Rosenbloom DI, Lewis MG, Hattersley J, Li B, Hesselgesser J, Geleziunas R, Robb ML, Kim JH, Michael NL, Barouch DH. Rapid seeding of the viral reservoir prior to SIV viraemia in rhesus monkeys. Nature. 2014; 512:74-77. DOI | PubMed
- Ye CJ, Feng T, Kwon HK, Raj T, Wilson MT, Asinovski N, McCabe C, Lee MH, Frohlich I, Paik HI, Zaitlen N, Hacohen N, Stranger B, De Jager P, Mathis D, Regev A, Benoist C. Intersection of population variation and autoimmunity genetics in human T cell activation. Science. 2014; 345DOI | PubMed
- Yucha RW, Hobbs KS, Hanhauser E, Hogan LE, Nieves W, Ozen MO, Inci F, York V, Gibson EA, Thanh C, Shafiee H, El Assal R, Kiselinova M, Robles YP, Bae H, Leadabrand KS, Wang S, Deeks SG, Kuritzkes DR, Demirci U, Henrich TJ. High-throughput Characterization of HIV-1 reservoir reactivation using a single-cell-in-droplet PCR assay. EBioMedicine. 2017; 20:217-229. DOI | PubMed
- Zhu J, Gaiha GD, John SP, Pertel T, Chin CR, Gao G, Qu H, Walker BD, Elledge SJ, Brass AL. Reactivation of latent HIV-1 by inhibition of BRD4. Cell Reports. 2012; 2:807-816. DOI | PubMed
- Zolotukhin S, Potter M, Hauswirth WW, Guy J, Muzyczka N. A "humanized" green fluorescent protein cDNA adapted for high-level expression in mammalian cells. Journal of Virology. 1996; 70:4646-4654. PubMed
Fonte
Battivelli E, Dahabieh MS, Abdel-Mohsen M, Svensson JP, Tojal Da Silva I, et al. () Distinct chromatin functional states correlate with HIV latency reactivation in infected primary CD4. eLife 7e34655. https://doi.org/10.7554/eLife.34655




