
Introduzione
Il dolore è il sintomo più comune presentato dai pazienti nei reparti di emergenza degli ospedali, in particolare nel contesto dei traumi. Si stima che meno della metà dei pazienti riceva analgesici entro 1 ora dall’arrivo[1- 3]. Nonostante l’ampia gamma di metodi terapeutici disponibili, la loro rapida somministrazione è ostacolata dalla disponibilità delle condizioni materiali necessarie. Le 3 priorità per il trattamento del dolore acuto d’emergenza sono l’attuazione il più presto possibile, l’inizio rapido dell’azione, e abbastanza potente da raggiungere un livello di dolore il più basso possibile. La morfina endovenosa (IVM) è l’analgesico forte più comune, ma è associata ad una chiara limitazione temporale legata alla necessità di ottenere una via d’accesso. Le vie inalatorie, intramuscolari e sublinguali sono state testate per molti anni con molecole gassose o formulazioni adattate[4- 7]. La via intranasale (IN) fornisce una facile somministrazione con un rapido tempo di azione di picco dovuto all’elevata vascolarizzazione e all’assenza del metabolismo di primo passaggio[8- 10]. Molte molecole sono state valutate, ma devono essere ad alte concentrazioni (piccolo volume vaporizzato) e liposolubili per essere efficaci. Nonostante gli studi sul fentanil abbiano mostrato risultati incoraggianti e un’adeguata farmacocinetica IN-to-sistemica[10- 12], il suo uso è ancora scarso nella pratica di emergenza. Il sufentanil ha il vantaggio di essere un oppioide più forte, ben noto ai medici di emergenza e agli intensivisti, e disponibile in alta concentrazione, con un noto tempo di azione rapida di picco (circa 5 minuti) per via endovenosa (IV)[13]. Tuttavia, l’azione analgesica a breve termine del sufentanil somministrato per via nasale in caso di dolore acuto grave è ancora poco valutata[14-17]. Oltre a studi non randomizzati e a marchio aperto[14,15], 2 studi recenti hanno valutato IN sufentanil (INS) in buone condizioni metodologiche. Lemoel et al.[16] hanno confrontato l’INS con l’analgesia venosa, ma senza un comparatore standardizzato: l’analgesia venosa non includeva sistematicamente un oppiaceo. Sin et al.[17] hanno condotto uno studio a bassa potenza (30 pazienti) e non hanno mostrato alcuna differenza nell’analgesia, ma lo studio non è stato progettato per dimostrare la non inferiorità. Il nostro obiettivo era quindi quello di valutare la non inferiorità dell’effetto analgesico e la sicurezza dell’INS titolato rispetto all’IVM titolato somministrato a pazienti con traumi non mortali.
Metodi
Progettazione dello studio e partecipanti
Lo studio ALGOFINE è stato progettato come uno studio multicentrico, in doppio cieco, randomizzato, controllato e non inferiorità. Il protocollo completo per questo trial è disponibile come testo S1. Pazienti adulti (dai 18 ai 75 anni) che presentano un dolore traumatico autovalutato come ≥6/10 su una scala numerica di valutazione del dolore (NRS) sono stati reclutati al triage in 6 reparti di emergenza ospedalieri (Grenoble Alpes University Hospital [Grenoble], Metropole-Savoie Hospital [Chambery], Centre Hospitalier Annecy Genevois [Annecy], Albertville-Ospedale di Moutiers [Albertville], Ospedale di Saint-Jean-de-Maurienne [Saint-Jean-de-Maurienne], e Ospedale Voiron [Voiron]). I pazienti non sono stati iscritti se al triage hanno presentato 1 dei seguenti criteri: dolore di origine non traumatica; insufficienza respiratoria, renale o epatica cronica; qualsiasi storia di tossicodipendenza; patologia del seno medica o chirurgica passata; saturazione di ossigeno < 90%; pressione sanguigna sistolica < 90 mm Hg; lesione cerebrale traumatica con un punteggio della Scala del Coma di Glasgow < 14; allergia agli oppioidi; trauma facciale; paziente incapace di capire o di autovalutare usando una NRS; somministrazione di oppiacei entro 6 ore prima dell’ammissione; o peso > 100 kg. Le informazioni relative alle procedure, a possibili eventi avversi o inefficacia e alla privacy dei dati sono state fornite ai partecipanti prima di ottenere il consenso firmato. In particolare, la partecipazione allo studio è stata proposta durante i primi minuti di triage, data l’urgenza del loro bisogno di sollievo dal dolore. Sono stati informati che, in considerazione del loro livello di dolore, l’IVM era normalmente indicata, e sarebbe stata offerta dal medico responsabile. Una spiegazione riguardo al sufentanil, alla somministrazione di IN e ai principi di randomizzazione e di cecità è stata data mentre l’infermiera responsabile ha ottenuto la via venosa necessaria anche se il paziente si rifiutava di partecipare.
Era previsto il reclutamento di un gruppo di pazienti in un ambiente preospedaliero da parte di un’unità mobile di emergenza. Essi non sono inclusi nell’attuale popolazione dello studio e i risultati di questo studio ausiliario prestabilito non sono qui descritti (su richiesta dei revisori).
Lo studio è stato avviato e condotto secondo i principi della Dichiarazione di Helsinki. Il protocollo (EudraCT 2013-001665-16; ClinicalTrials.gov NCT02095366) è stato approvato dal comitato etico regionale il 15 maggio 2013 (Comitato etico dei centri di ricerca clinica di Rhone-Alpes Auvergne CECIC IRB n. 2013-001665-16), dalla commissione nazionale per le libertà e la protezione dei dati (Commission Nationale de l’Informatique et des Libertés) e dall’agenzia nazionale per la sicurezza dei farmaci (Agence Nationale de Sécurité du Médicament et des Produits de Santé). Tutte le informazioni riguardanti le procedure, i rischi e la privacy dei dati sono state rese note ai partecipanti prima di ottenere il consenso firmato. Questo studio è riportato secondo la linea guida CONSORT (Consolidated Standards of Reporting Trials) (vedi lista di controllo S1 CONSORT ).
Randomizzazione e mascheramento
Tutto il personale clinico e di ricerca, così come i pazienti, sono stati resi ciechi all’assegnazione del trattamento, ad eccezione dell’infermiere responsabile della preparazione dei trattamenti. I trattamenti sono stati preparati da confezioni randomizzate in una stanza dedicata, e siringhe etichettate solo “IV” e “IN” sono state date allo sperimentatore. Tutti i farmaci dello studio sono stati confezionati in confezioni di prova in cieco da un farmacista della sperimentazione clinica che era cieco agli interventi e agli esiti. La randomizzazione (1:1) in gruppi paralleli è stata eseguita in blocchi di dimensioni casuali e stratificata dal centro utilizzando un software dedicato ospitato nell’ospedale universitario di Grenoble Alpes. La lista di randomizzazione è stata consegnata alla farmacia centrale incaricata di preparare i pacchetti.
Procedure
Al momento dell’inclusione, e a 10 minuti e 20 minuti dopo l’inclusione se la NRS è rimasta >3, ogni paziente ha ricevuto sia una singola dose di agente attivo che una singola dose di placebo attraverso le vie IV e IN. Nel gruppo INS, è stata somministrata una dose iniziale di 0,30 μg/kg di sufentanil (0,15 μg/kg in ogni narice). Ulteriori dosi (0,15 μg/kg) sono state somministrate a 10 e 20 minuti in 1 delle narici se NRS è rimasto >3/10. Una soluzione stock a 50 μg/ml di sufentanil è stata campionata ad ogni somministrazione in base al peso del paziente con una siringa convenzionale da 1 ml e atomizzata con un dispositivo di atomizzazione della mucosa nasale (MAD, Wolfe Tory Medical, Salt Lake City, UT, US). Il MAD è un semplice dispositivo con un puntale Luer lock che è facilmente collegabile alla siringa e fornisce una completa vaporizzazione del contenuto. Il MAD consente procedure in cieco e un dosaggio preciso in base al peso del paziente, poiché viene utilizzata una siringa standard piuttosto che un sistema commerciale pronto all’uso.
Nel gruppo IVM, una dose iniziale di 0,1 mg / kg è stata somministrata, e dosi aggiuntive di 0,05 mg / kg a 10 e 20 minuti se il NRS è rimasto >3/10. In entrambi i gruppi, il placebo era 0,9% di cloruro di sodio, somministrato per via endovenosa o per atomizzazione nasale nello stesso volume che sarebbe stato dato per il trattamento attivo. Durante i primi 30 minuti, il medico responsabile è stato autorizzato ad utilizzare i co-analgesici, ad eccezione dei sedativi e degli oppiacei forti. Dopo una valutazione dell’esito a 30 minuti, il medico responsabile è stato informato della natura e della quantità di farmaco somministrato per aumentare i farmaci, se necessario.
Raccolta dei dati e misure di esito
L’endpoint primario era l’efficacia analgesica definita come una diminuzione del dolore sul NRS tra la prima somministrazione e 30 minuti dopo. Gli endpoint secondari comprendevano l’efficacia dell’analgesia a 10 e 20 minuti definita da una diminuzione dell’NRS e l’incidenza di eventuali eventi avversi gravi e non gravi fino a 4 ore dopo la prima somministrazione. I parametri vitali sono stati raccolti ogni 10 minuti nell’arco di 1 ora, compresi i punteggi della scala di sedazione di Ramsay e della scala di Glasgow Coma. Gli effetti collaterali e i sintomi sono stati registrati durante tutta la procedura, compresi quelli raccolti ponendo domande specifiche al paziente ogni 10 minuti. La soddisfazione del paziente riguardo alla procedura è stata valutata a 30 minuti.
I dati dello studio sono stati raccolti e gestiti utilizzando lo strumento elettronico di acquisizione dati REDCap (Research Electronic Data Capture) ospitato presso l’Ospedale Universitario di Grenoble Alpes[18]. REDCap (Research Electronic Data Capture) è un’applicazione basata sul web per supportare l’acquisizione dei dati, che fornisce l’inserimento di dati convalidati, audit trail per l’elaborazione e l’esportazione dei dati di monitoraggio, procedure di esportazione automatizzate per scaricare i dati su pacchetti statistici di uso comune e procedure per l’importazione dei dati[18].
Analisi statistica
Dimensioni del campione, potenza e metodi statistici
Come studio di non inferiorità, la soglia si basava su una differenza clinicamente significativa di efficacia (NRST30 – NRST0) tra i 2 trattamenti di -1,3 [19,20]. Da studi precedenti è stato stimato che la deviazione standard della riduzione del dolore sarebbe stata di 2,8[21]. Con un campione di 198 pazienti (99 in ogni gruppo), un disegno a 2 gruppi fornirebbe almeno l’80% di potere di rifiutare l’ipotesi nulla (corrispondente ad una perdita di efficacia maggiore o uguale a 1,3[19]), a favore dell’ipotesi alternativa, corrispondente ad un guadagno di efficacia o ad una perdita di efficacia inferiore a 1,3, supponendo che la differenza media attesa fosse 0, la deviazione standard comune 2,8[21], e il livello di significatività 2,5%. La dimensione del campione target è stata quindi fissata a 218 partecipanti (109 in ogni braccio) per tenere conto di potenziali deviazioni del protocollo o interruzioni premature. Il calcolo della dimensione del campione è stato effettuato utilizzando il software NQuery Advisor 7.0 (Statistical Solutions, Boston MA, US).
Analisi dei dati
La non inferiorità è stata determinata sulla base di un test di equivalenza media t su 1 lato (due test su 1 lato t, procedura TOSTT)[22] sulla popolazione per protocollo (endpoint primario: efficacia NRST30 – NRST0) e confermata, per ragioni di sensibilità, sull’intenzione modificata di trattare la popolazione, secondo le raccomandazioni. L’implementazione dell’analisi dell’intenzione modificata di trattare ha richiesto la sostituzione dei dati mancanti; ciò è stato effettuato utilizzando il metodo dell’imputazione multipla. Nel processo di imputazione multipla sono state utilizzate dieci imputazioni dei dati mancanti. Seguendo le raccomandazioni dei revisori, abbiamo eseguito una regressione lineare corretta per il punteggio di base del dolore e la sede.
Abbiamo valutato la superiorità del trattamento INS rispetto all’IVM a 30 minuti nell’intenzione modificata di trattare la popolazione utilizzando un test Student t a 2 campioni. L’analisi di superiorità è stata pianificata dopo l’inizio dell’arruolamento del paziente (e prima di qualsiasi deconcentramento), se la non inferiorità è stata soddisfatta. Abbiamo anche confrontato l’efficacia del trattamento INS con il trattamento IVM nei diversi punti temporali (NRST10 – NRST0e NRST20– NRST0) nell‘intenzione di trattare la popolazione utilizzando un test a 2 campioni di Studente t. Abbiamo valutato l’efficacia del trattamento INS e IVM in tempi diversi all’interno di ciascun gruppo (NRST0 contro NRST10 e NRST0 contro NRST20) nella popolazione per protocollo e nell’intenzione modificata di trattare la popolazione utilizzando un test Student t accoppiato (o un test Wilcoxon se necessario). Abbiamo confrontato l’incidenza di eventi avversi gravi e non gravi tra i gruppi utilizzando un test chi-squared, o un test di Fisher esatto se necessario (frequenza prevista inferiore a 5). Abbiamo riassunto la soddisfazione del paziente (scala a 100 punti) come mediana e intervallo interquartile (IQR), confrontandola con un test di Mann-Whitney (il test utilizzato non era previsto prospetticamente nel piano di analisi). Le analisi statistiche sono state effettuate utilizzando il software Stata 13.1 (StataCorp, College Station, TX, US).
Risultati
Caratteristiche del paziente
Tra il 4 novembre 2013 e il 10 aprile 2015 abbiamo valutato un totale di 194 pazienti che presentavano un dolore traumatico acuto per l’idoneità; 157 sono stati inclusi nella coorte del pronto soccorso e randomizzati, e 155 hanno iniziato il trattamento. Diciannove pazienti sono stati esclusi dalle analisi per protocollo, principalmente per sottodosaggio(n = 7) o sovradosaggio(n = 8) (vedi Fig 1 per il diagramma di flusso dettagliato dei pazienti), ottenendo n = 69 per il gruppo IVM e n = 67 per il gruppo INS. Sebbene il reclutamento sia stato più lento del previsto per la durata prevista dello studio, la potenza è rimasta al di sopra dell’80%, con un’efficace deviazione standard nella riduzione del dolore di 2,1 nell’analisi post hoc. I 2 gruppi di trattamento sono stati ben bilanciati rispetto alle caratteristiche di base, con l’eccezione del rapporto maschi/femmine e del numero di pazienti che hanno ricevuto altri analgesici concomitanti (IVM 33% contro INS 22%; Tabella 1). La NRS media al basale (T0) era 7,6 (95% CI 7,3-7,9) nel gruppo IVM e 7,9 (95% CI 7,6-8,2) nel gruppo INS. La dose mediana totale di IVM somministrata era di 12,5 mg (IQR 10,0-15,1), e la dose mediana totale di INS era di 36,0 μg (IQR 30,0-42,7). Dieci minuti dopo la prima somministrazione, 59 (85,5%) pazienti del gruppo IVM hanno ricevuto una seconda dose e 60 (89,6%) pazienti del gruppo INS hanno ricevuto una seconda dose. Venti minuti dopo la prima somministrazione, 42 (60,9%) pazienti del gruppo IVM hanno ricevuto una terza dose e 38 (56,7%) pazienti del gruppo INS hanno ricevuto una terza dose.
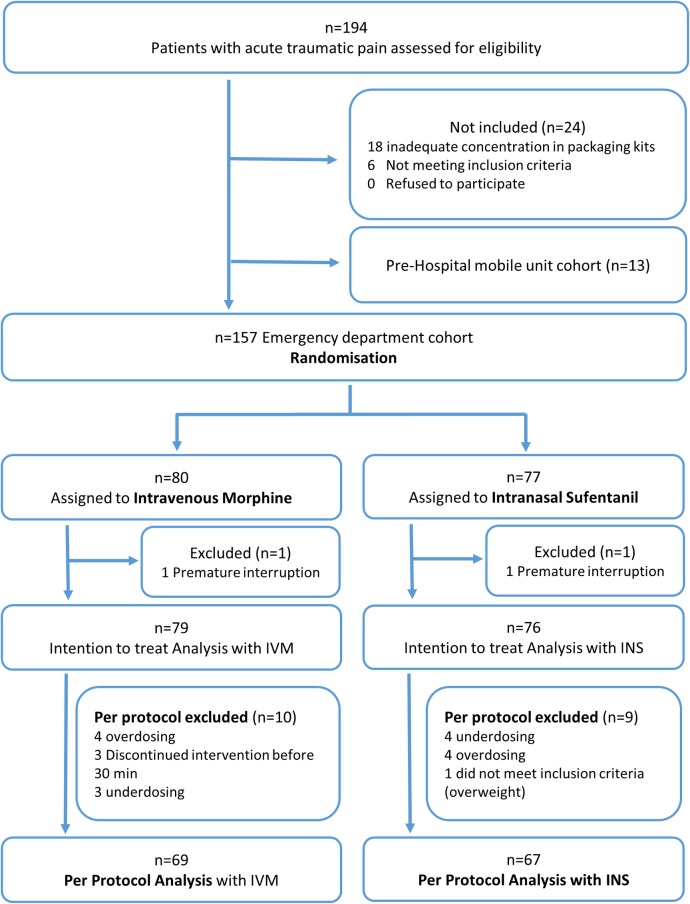
Fig. 1.Iscrizione, randomizzazione e follow-up dei partecipanti allo studio.INS, sufentanil intranasale; IVM, morfina endovenosa.
| Caratteristica | IVM(n = 69) | INS(n = 67) |
|---|---|---|
| Caratteristica del paziente | ||
| Età media [IQR] (anni) | 41 [28 a 54] | 38 [30 a 55] |
| Uomini | 40 (58) | 31 (46) |
| Peso medio [IQR] (kg) | 74 [61 a 83] | 70 [60-80] |
| Area traumatologica* | ||
| Responsabile | 2 (2.9) | 1 (1.5) |
| Spalla | 7 (10.1) | 12 (17.9) |
| Braccio/arancia | 12 (17.4) | 9 (13.4) |
| Polso o mano | 17 (24.6) | 8 (11.9) |
| Parete toracica | 3 (4.4) | 5 (7.5) |
| Rachis | 12 (17.4) | 8 (11.9) |
| Bacino/nave | 10 (14.5) | 5 (7.5) |
| Gamba/ginocchio | 8 (11.6) | 13 (19.4) |
| Caviglia o piede | 11 (15.9) | 13 (19.4) |
| Parametri vitali all’inclusione | ||
| Mediana [IQR] HR (al minuto) | 76 [68 a 91] | 74 [da 67 a 84] |
| Mediana [IQR] RR (al minuto) | 16 [15 a 19] | 18 [15-20] |
| Mediana [IQR] SpO2 (%) | 99 [97 a 100] | 99 [97 a 100] |
| Mediana [IQR] MAP (mm Hg) | 98 [91 a 104] | 94 [85 a 103] |
| Mediana [IQR] NRS (/10) | 8 [da 7 a 8] | 8 [da 7 a 9] |
| Co-analgesia* | 23 (33) | 15 (22) |
| Paracetamolo | 22 (32) | 15 (22) |
| Codeine | 2 (3) | 2 (3) |
| Ketoprofene | 4 (6) | 1 (1) |
| Sito di inclusione | ||
| Grenoble (sito nord) | 41 (59.4) | 42 (62.7) |
| Grenoble (sito sud) | 2 (2.9) | 2 (3.0) |
| Saint-Jean-de-Maurienne | 10 (14.5) | 8 (11.9) |
| Annecy | 9 (13.0) | 8 (11.9) |
| Chambery | 4 (5.8) | 3 (4.5) |
| Albertville | 3 (4.3) | 3 (4.5) |
| Voiron | 0 (0) | 1 (1.5) |
Fig. 1.Fig. 1. Iscrizione, randomizzazione e follow-up dei partecipanti allo studio.INS, sufentanil intranasale; IVM, morfina endovenosa.
Endpoint primario (non inferiorità)
La differenza media tra NRS alla prima amministrazione e NRS a 30 minuti è stata di -4,1 (97,5% CI da -4,6 a -3,6) nel gruppo IVM e di -5,2 (97,5% CI da -5,7 a -4,6) nel gruppo INS (per analisi di protocollo). Non inferiorità è stata soddisfatta(p < 0, 001), in quanto l’intervallo di confidenza inferiore del 97,5% di 0,29 è stato maggiore del margine di non inferiorità prespecificato di -1,3 (differenza media nella variazione NRS tra i gruppi 1,11, 97,5% CI 0,29 a 1,93).
Nell’intenzione di trattare l’analisi, la differenza media di NRS tra la prima somministrazione e a 30 minuti era -4,4 (97,5% CI da -4,9 a -3,8) nel gruppo IVM e -5,1 (97,5% CI da -5,7 a -4,5) nel gruppo INS. La non inferiorità è stata soddisfatta(p < 0, 001), in quanto l’intervallo di confidenza inferiore del 97,5% di -0,05 è stato maggiore del margine di non inferiorità predefinito di -1,3 (differenza media della variazione NRS tra i gruppi 0,74, 97,5% CI da -0,05 a 1,54).
Dopo l’aggiustamento per il centro (ospedale) e il livello di base del dolore, la differenza nella riduzione di NRS tra i gruppi è stata di 0,87 (97,5% CI 0,19 a 1,54, p = 0,012).
Punti finali secondari
Superiorità
Nell’analisi di superiorità, l’INS era superiore all’IVM per la riduzione del dolore NRS a 30 minuti nell’intenzione di trattare la popolazione(p = 0,034 per la superiorità), con una differenza clinicamente non significativa nella media NRS tra gruppi di 0,7. Una diminuzione significativa di NRS è stata osservata dopo 10 minuti all’interno di entrambi i gruppi (differenza media -2,7, 95% CI -3,1 a -2,2, p < 0,001, per il gruppo IVM e -2,1, 95% CI -2,5 a -1,7, p < 0.001, per il gruppo INS, intenzione di trattare), così come dopo 20 minuti (differenza media -3,8, 95% IC da -4,2 a -3,3, p < 0,001, per il gruppo IVM e -4,0, 95% CI da -4,5 a -3,5, p < 0,001, per il gruppo INS , intenzione di trattare). In un confronto tra i gruppi, non sono state riscontrate differenze nella riduzione dell’NRS a 10 e 20 minuti. Le variazioni di NRS con il tempo in ogni gruppo sono presentate nella Fig. 2. In un’analisi di sottogruppo, il dolore autovalutato (NRS) era di 3 o inferiore a 30 minuti per 34 (49,3%) pazienti nel gruppo IVM e per 48 (71,6%) nel gruppo INS (differenza assoluta = 22,3%, differenza relativa = 45,2%, p = 0,01; Fig 3). Sedici pazienti in ogni gruppo (23,2% nel gruppo IVM e 23,9% nel gruppo INS) hanno ricevuto i farmaci antidolorifici di salvataggio dal medico responsabile dopo lo sblinamento.
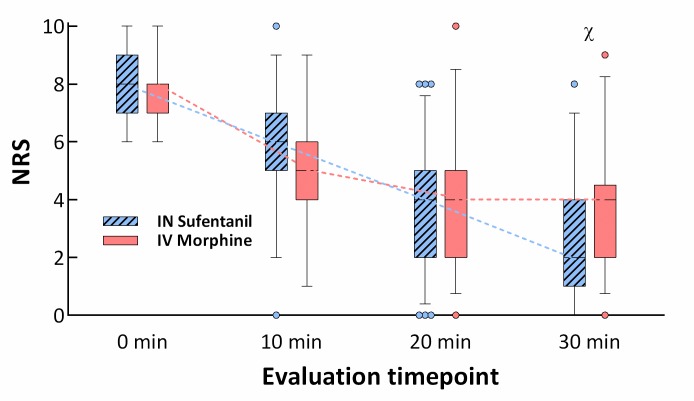
Fig. 2.NRS nei diversi punti temporali per gruppo.La casella si estende dal 25° al 75° percentile, e i baffi vengono tirati giù fino al 5° percentile e fino al 95°. I punti sotto e sopra i baffi sono disegnati come punti individuali. Non sono state osservate differenze significative nei valori NRS tra i gruppi nei punti di 10 e 20 minuti. IN sufentanil non era inferiore e superiore alla morfina per via endovenosa nella riduzione di NRS dalla somministrazione del farmaco a 30 minuti (NRST30 – NRST0). IN, intranasale; IV, endovenosa; NRS, scala numerica di valutazione del dolore.
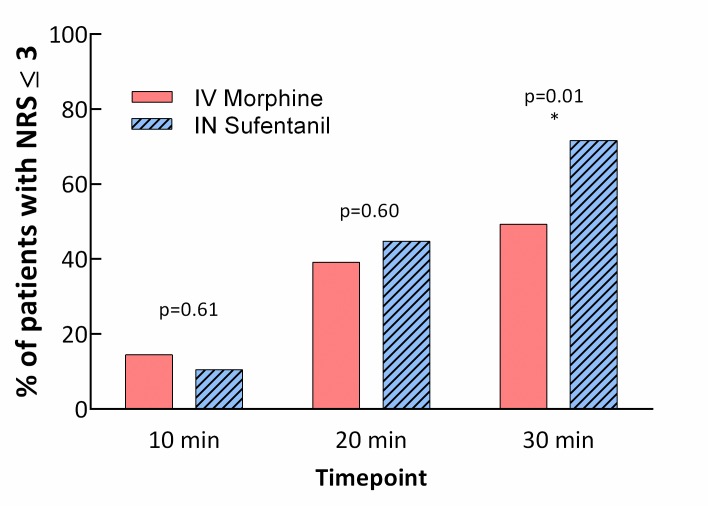
Fig. 3.Pazienti con NRS ≤ 3 nei diversi punti temporali per gruppo.*La NRS dichiarata era 3 o meno a 30 minuti per il 49,3% dei pazienti nel gruppo di morfina IV e 71,6% nel gruppo IN sufentanil(p = 0,01). IN, intranasale; IV, endovenosa; NRS, scala numerica di valutazione del dolore.
Fig. 2.NRS nei diversi punti temporali per gruppo.La casella si estende dal 25° al 75° percentile, e i baffi sono disegnati fino al 5° percentile e fino al 95°. I punti sotto e sopra i baffi sono disegnati come punti individuali. Non sono state osservate differenze significative nei valori NRS tra i gruppi nei punti di 10 e 20 minuti. IN sufentanil non era inferiore e superiore alla morfina per via endovenosa nella riduzione di NRS dalla somministrazione del farmaco a 30 minuti (NRST30 – NRST0). IN, intranasale; IV, endovenosa; NRS, scala numerica di valutazione del dolore.
Fig. 3.Pazienti con NRS ≤ 3 nei diversi punti temporali per gruppo.*La NRS dichiarata era 3 o meno a 30 minuti per il 49,3% dei pazienti nel gruppo di morfina IV e 71,6% nel gruppo IN sufentanil(p = 0,01). IN, intranasale; IV, endovenosa; NRS, scala numerica di valutazione del dolore.
Eventi avversi e soddisfazione del paziente
Non ci sono state differenze statisticamente significative tra i gruppi per quanto riguarda gli eventi avversi lievi o gravi registrati nel protocollo(Tabella 2) o l’intenzione di trattare le popolazioni. Gli eventi avversi più frequenti osservati sono stati vertigini, vampate di calore e nausea o vomito(Tabella 2). L’ipossiemia (SpO2 < 90%) si è verificata in 3 (4,5%) pazienti del gruppo INS e in 1 (1,5%) paziente del gruppo IVM. Bradypnea (frequenza respiratoria < 10/minuto) è stata osservata in 2 (3%) pazienti, entrambi nel gruppo INS. Infine, l’ipotensione è stata riportata in 1 (1,5%) paziente in ogni gruppo. Nessuno dei pazienti ha richiesto naloxone, supporto per la ventilazione o vasopressori. Due pazienti hanno richiesto la somministrazione di ossigeno (3 l/min) per 10 minuti. Il numero di pazienti che hanno avuto bisogno di nuocere era di 17 (95% CI 7-56) per eventi avversi gravi, di cui 33 (95% CI 12-38) per l’ipossiemia e 34 (95% CI 13-53) per la bradipnea.
| Evento o sintomo dichiarato | IVM(n = 69) | INS(n = 67) | p-Valore* |
|---|---|---|---|
| Eventi avversi gravi | 2 (2.9) | 6 (9.0) | 0.16 |
| Ipossiemia (SpO2 < 90%) | 1 (1.5) | 3 (4.5) | 0.36 |
| Ipotensione (SBP < 90 mm Hg) | 1 (1.5) | 1 (1.5) | 1.00 |
| Bradypnea (RR < 10/minuto) | 0 (0) | 2 (3.0) | 0.24 |
| Shock anafilattico | 0 (0) | 0 (0) | — |
| Alterazione della coscienza (Ramsay > 2) | 0 (0) | 0 (0) | — |
| Bradicardia (bpm < 45/minuto) | 0 (0) | 0 (0) | — |
| Uso del naloxone | 0 (0) | 0 (0) | — |
| Eventi avversi di lieve entità | 42 (60.9) | 31 (46.3) | 0.09 |
| Vertigini | 25 (36.2) | 19 (28.4) | 0.33 |
| Vampate di calore | 20 (29.0) | 12 (17.9) | 0.13 |
| Nausea o vomito | 13 (18.8) | 8 (11.9) | 0.27 |
| Cattivo gusto/odore | 3 (4.4) | 2 (3.0) | 1.00 |
| Reazione allergica lieve | 1 (1.5) | 1 (1.5) | 1.00 |
| Epistassi/rinorrea | 1 (1.5) | 0 (0) | 1.00 |
| Allucinazioni | 0 (0) | 0 (0) | — |
La soddisfazione mediana del paziente valutata su una scala di 100 punti alla fine della procedura era di 80 (IQR da 60 a 92,5) nel gruppo IVM e 80 (IQR da 70 a 100) nel gruppo INS (p = 0,34).
Discussione
In questo studio randomizzato, in doppio cieco, che confronta l’analgesia da oppioidi IV e IN nei reparti di emergenza, l’INS era fattibile e non inferiore all’IVM nel ridurre il dolore grave da lesioni traumatiche, a 30 minuti. Inoltre, i risultati suggeriscono che il regime INS (0,30 μg/kg e poi 0,15 μg/kg ogni 10 minuti) è superiore all’IVM (0,1 mg/kg e poi 0,05 mg/kg ogni 10 minuti) a 30 minuti, con una diminuzione mediana di NRS > 5, senza, tuttavia, raggiungere una differenza tra i gruppi nella riduzione di NRS abbastanza grande da essere considerata clinicamente significativa (cioè, una differenza di NRS ≥ 1,3) [19,20]. L’efficacia analgesica dell’INS è stata osservata dopo i primi 10 minuti, con una riduzione media all’interno del gruppo di NRS di 2,1. Gli eventi avversi sono stati frequenti, come previsto per gli oppioidi, ma raramente gravi, senza differenze significative tra i 2 gruppi, nonostante uno squilibrio osservato a scapito degli INS.
Finora, l’uso degli INS è stato studiato principalmente in pazienti palliativi o pediatrici, per lo più in un contesto perioperatorio e in combinazione con altri metodi di sedazione[23- 29]. Solo pochi studi[14-17] hanno testato l’INS per la gestione del dolore acuto nel contesto della pratica di emergenza. Due studi sono stati condotti come prove aperte non randomizzate[14-15], rendendo difficile l’interpretazione dell’efficacia e degli effetti collaterali. In questi 2 studi, 15 e 40 pazienti affetti da lesioni all’estremità distale hanno ricevuto una singola dose di 0,5 μg/kg di INS. Il punteggio medio del dolore a 30 minuti è stato abbassato rispettivamente di 4,3 punti[14] e 4,7[15]. Sono stati riportati alcuni effetti avversi, come disforia (46,6%), nausea (13,3%) e vertigini (7,5%), ma non è stata osservata alcuna apnea. Più recentemente sono stati pubblicati 2 studi controllati monocentrici randomizzati contro placebo[16- 17]. Lemoel et al.[16] hanno somministrato una singola dose di 0,4 μg/kg di INS (o placebo) a 144 pazienti ricoverati al pronto soccorso per una recente (<6 h) lesione isolata di un arto. Ad ogni paziente è stato somministrato il solito trattamento del dolore per via endovenosa, con analgesici multimodali, compresi gli oppiacei per via endovenosa, se necessario. La percentuale di pazienti con un sollievo dal dolore soddisfacente (NRS ≤ 3) è stata migliore nel gruppo del sufentanil (72,2%) rispetto al gruppo di controllo (51,4%). Tuttavia, nel gruppo del sufentanil è stato osservato un tasso più elevato di effetti collaterali (12,5% in più di bradipnea e 24% in più di nausea) rispetto ai controlli. Ciò è probabilmente dovuto alla somministrazione cumulativa di oppioidi nel gruppo attivo, che potrebbe ricevere sia INS che IVM. Sin et al.[17] hanno confrontato la somministrazione di una singola dose di 0,7 μg/kg di INS contro 0,1 mg/kg di IVM in 60 pazienti adulti che si sono presentati al pronto soccorso con dolore acuto. Non hanno trovato alcuna differenza nella NRS tra i 2 gruppi 10 minuti dopo la somministrazione del farmaco. Nonostante l’alta dose di sufentanil che hanno usato, hanno riferito che solo il 7,5% dei casi aveva vertigini, e nessuno aveva disforia o apnea. Tutti questi studi pubblicati hanno preso in considerazione gli effetti di una singola somministrazione di INS. Il nostro protocollo mirava a rispettare il principio della titolazione degli oppioidi, con ripetute somministrazioni di dosi divise. I nostri pazienti hanno ricevuto una dose media totale di 36 μg di sufentanil rispetto alla dose media di 37,7 μg somministrata da Sin et al. [17]. Sorprendentemente, nonostante la nostra strategia di titolazione, abbiamo riportato effetti collaterali più frequenti rispetto a Sin et al. Questo potrebbe essere dovuto al piccolo numero di pazienti che hanno studiato e anche all’attenzione che abbiamo prestato nel raccogliere ogni effetto collaterale nel nostro protocollo di progettazione di non inferiorità.
In effetti, a quanto ci risulta, finora nessuno studio è stato metodologicamente progettato per studiare la non inferiorità dell’INS rispetto all’IVM in termini di efficacia analgesica e di effetti collaterali per i traumi negli adulti, che è la situazione più frequente incontrata dai medici di emergenza. Abbiamo progettato il nostro studio per valutare un oppioide forte e ad azione rapida, sperando di osservare un’azione rapida e chiara che potessimo confrontare con la morfina a partire dai primi 5 minuti. In un protocollo di titolazione paragonabile a quello comunemente usato per la MIV, sia la non inferiorità che la superiorità sono state osservate per l’INS a 30 minuti. I primi 30 minuti sono il tempo normalmente necessario per ottenere una cannula endovenosa; questa durata è più breve del tempo di azione della maggior parte degli altri analgesici, oltre alle terapie inalatorie come il protossido di azoto o il metossiflurano[4,30]. Inoltre, l’effetto dell’INS appare convincente già 5 minuti dopo la somministrazione, con una riduzione del dolore paragonabile a quella ottenuta con la IVM. La durata dell’azione del sufentanil è breve e dipende dalla via di somministrazione utilizzata. Per via endovenosa ha un tempo di dimezzamento di circa 15 minuti; questo tempo di dimezzamento è più lungo per via sublinguale a causa di un ulteriore assorbimento[31,32]. Se somministrato per via intranasale, l’effetto è probabilmente intermedio[10,11], ma rimane breve e si traduce in un’analgesia prevista di circa 45 minuti. Pertanto, l’INS potrebbe essere utile per l’inizio dell’analgesia volta ad alleviare il dolore il più rapidamente possibile. Nel contesto della crescente “epidemia di oppioidi”, questa modalità d’uso potrebbe avere il vantaggio di ridurre la prescrizione di successivi oppioidi orali responsabili della dipendenza [33]. Nel complesso, i nostri risultati sono a favore di un sollievo molto precoce dal dolore durante il triage di emergenza che evita le vie convenzionali. L’uso dell’INS potrebbe diventare un’opzione pragmatica in tutte le situazioni in cui ottenere un accesso venoso è una sfida o ritarda la gestione del dolore. Pertanto, l’INS potrebbe essere un’alternativa all’analgesia per via endovenosa in ambiente preospedaliero, quando l’accesso per via endovenosa non è fattibile o è molto difficile da ottenere.
Questo studio randomizzato e in doppio cieco dovrebbe ridurre al minimo la maggior parte dei pregiudizi comuni. Tuttavia, il nostro studio ha una serie di limitazioni. Nonostante l’assegnazione casuale, c’è stato uno squilibrio tra i bracci dello studio nel rapporto maschio/femmina e nei concomitanti analgesici utilizzati. Consentire la co-analgesia solo dopo 30 minuti avrebbe potuto evitare questa distorsione. D’altra parte, i farmaci oppiacei sono generalmente utilizzati come co-analgesici, in combinazione con l’acetaminofene in particolare. Da questo punto di vista, la metodologia scelta è stata più vicina a quanto avviene nella pratica clinica reale. Poiché la co-analgesia è stata utilizzata più spesso nel gruppo IVM, questo squilibrio potrebbe aver ridotto l’efficacia osservata dell’INS rispetto all’IVM, ma non mette in discussione l’interpretazione della non inferiorità. Sebbene il reclutamento sia stato inferiore alle aspettative per la durata prevista dello studio, la potenza è rimasta al di sopra dell’80%, con un’effettiva deviazione standard nella riduzione del dolore inferiore al previsto. Il tasso relativamente basso di inclusione dei partecipanti è dovuto alla difficoltà di implementare uno studio randomizzato durante il triage di pazienti che soffrono di dolore intenso. Infine, lo studio non è stato alimentato per rispondere alla domanda di sicurezza. Sebbene il numero di eventi gravi osservati non fosse statisticamente diverso tra i 2 gruppi di trattamento, abbiamo osservato uno squilibrio tra i gruppi. La conferma della sicurezza richiederebbe uno studio molto più ampio. Gli studi futuri dovranno anche garantire che l’uso dell’INS sia efficace e sicuro in altri contesti di emergenza, come il pre-ospedale o in situazioni difficili, come nel soccorso alpino.
In conclusione, i nostri risultati suggeriscono che la titolazione IN del sufentanil a partire da una dose iniziale di 0,3 μg/kg non è inferiore all’IVM per la riduzione del dolore traumatologico nei primi 30 minuti. Inoltre, i risultati del nostro studio suggeriscono che l’INS può essere superiore all’IVM in questo lasso di tempo, anche se i nostri risultati non hanno mostrato una differenza clinicamente significativa. La via IN, senza la necessità di ottenere una via venosa, può consentire un più rapido inizio di un’efficace analgesia. Sono necessari ulteriori studi più ampi per determinare il profilo di sicurezza dell’INS.
Informazioni di supporto
References
- Sokoloff C, Daoust R, Paquet J, Chauny JM. Is adequate pain relief and time to analgesia associated with emergency department length of stay? A retrospective study. BMJ Open. 2014; 4(3):e004288. DOI | PubMed
- Pines JM, Hollander JE. Emergency department crowding is associated with poor care for patients with severe pain. Ann Emerg Med. 2008; 51(1):1-5. DOI | PubMed
- Hwang U, Richardson L, Livote E, Harris B, Spencer N, Sean Morrison R. Emergency department crowding and decreased quality of pain care. Acad Emerg Med. 2008; 15(12):1248-55. DOI | PubMed
- Ducassé JL, Siksik G, Durand-Béchu M, Couarraze S, Vallé B, Lecoules N. Nitrous oxide for early analgesia in the emergency setting: a randomized, double-blind multicenter prehospital trial. Acad Emerg Med. 2013; 20(2):178-84. DOI | PubMed
- Miner JR, Rafique Z, Minkowitz HS, DiDonato KP, Palmer PP. Sufentanil sublingual tablet 30mcg for moderate-to-severe acute pain in the ED. Am J Emerg Med. 2018; 36(6):954-61. DOI | PubMed
- Jalili M, Fathi M, Moradi-Lakeh M, Zehtabchi S. Sublingual buprenorphine in acute pain management: a double-blind randomized clinical trial. Ann Emerg Med. 2012; 59(4):276-80. DOI | PubMed
- Tveita T, Thoner J, Klepstad P, Dale O, Jystad A, Borchgrevink PC. A controlled comparison between single doses of intravenous and intramuscular morphine with respect to analgesic effects and patient safety. Acta Anaesthesiol Scand. 2008; 52(7):920-5. DOI | PubMed
- Grassin-Delyle S, Buenestado A, Naline E, Faisy C, Blouquit-Laye S, Couderc LJ. Intranasal drug delivery: an efficient and non-invasive route for systemic administration: focus on opioids. Pharmacol Ther. 2012; 134(3):366-79. DOI | PubMed
- Rech MA, Barbas B, Chaney W, Greenhalgh E, Turck C. When to pick the nose: out-of-hospital and emergency department intranasal administration of medications. Ann Emerg Med. 2017; 70(2):203-11. DOI | PubMed
- Helmers JH, Noorduin H, Van Peer A, Van Leeuwen L, Zuurmond WW. Comparison of intravenous and intranasal sufentanil absorption and sedation. Can J Anaesth. 1989; 36(5):494-7. DOI | PubMed
- Haynes G, Brahen NH, Hill HF. Plasma sufentanil concentration after intranasal administration to paediatric outpatients. Can J Anaesth. 1993; 40(3):286. DOI | PubMed
- Borland M, Jacobs I, King B, O’Brien D. A randomized controlled trial comparing intranasal fentanyl to intravenous morphine for managing acute pain in children in the emergency department. Ann Emerg Med. 2007; 49(3):335-40. DOI | PubMed
- Scholz J, Steinfath M, Schulz M. Clinical pharmacokinetics of alfentanil, fentanyl and sufentanil. An update. Clin Pharmacokinet. 1996; 31(4):275-92. DOI | PubMed
- Stephen R, Lingenfelter E, Broadwater-Hollifield C, Madsen T. Intranasal sufentanil provides adequate analgesia for emergency department patients with extremity injuries. J Opioid Manag. 2012; 8(4):237-41. DOI | PubMed
- Steenblik J, Goodman M, Davis V, Gee C, Hopkins CL, Stephen R. Intranasal sufentanil for the treatment of acute pain in a winter resort clinic. Am J Emerg Med. 2012; 30(9):1817-21. DOI | PubMed
- Lemoel F, Contenti J, Cibiera C, Rapp J, Occelli C, Levraut J. Intranasal sufentanil given in the emergency department triage zone for severe acute traumatic pain: a randomized double-blind controlled trial. Intern Emerg Med. 2019; 14(4):571-9. DOI | PubMed
- Sin B, Jeffrey I, Halpern Z, Adebayo A, Wing T, Lee AS. Intranasal sufentanil versus intravenous morphine for acute pain in the emergency department: a randomized pilot trial. J Emerg Med. 2019; 56(3):301-7. DOI | PubMed
- Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)—a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009; 42(2):377-81. DOI | PubMed
- Fleischman RJ, Frazer DG, Daya M, Jui J, Newgard CD. Effectiveness and safety of fentanyl compared with morphine for out-of-hospital analgesia. Prehosp Emerg Care. 2010; 14(2):167-75. DOI | PubMed
- Todd KH. Clinical versus statistical significance in the assessment of pain relief. Ann Emerg Med. 1996; 27(4):439-41. PubMed
- Birnbaum A, Esses D, Bijur PE, Holden L, Gallagher EJ. Randomized double-blind placebo-controlled trial of two intravenous morphine dosages (0.10 mg/kg and 0.15 mg/kg) in emergency department patients with moderate to severe acute pain. Ann Emerg Med. 2007; 49(4):445-53. DOI | PubMed
- Schuirmann DJ. A comparison of the two one-sided tests procedure and the power approach for assessing the equivalence of average bioavailability. J Pharmacokinet Biopharm. 1987; 15(6):657-80. PubMed
- Hitt JM, Corcoran T, Michienzi K, Creighton P, Heard C. An evaluation of intranasal sufentanil and dexmedetomidine for pediatric dental sedation. Pharmaceutics. 2014; 6(1):175-84. DOI | PubMed
- Roelofse JA, Shipton EA, de la Harpe CJ, Blignaut RJ. Intranasal sufentanil/midazolam versus ketamine/midazolam for analgesia/sedation in the pediatric population prior to undergoing multiple dental extractions under general anesthesia: a prospective, double-blind, randomized comparison. Anesth Prog. 2004; 51(4):114-21. PubMed
- Bayrak F, Gunday I, Memis D, Turan A. A comparison of oral midazolam, oral tramadol, and intranasal sufentanil premedication in pediatric patients. J Opioid Manag. 2007; 3(2):74-8. PubMed
- Abrams R, Morrison JE, Villasenor A, Hencmann D, Da Fonseca M, Mueller W. Safety and effectiveness of intranasal administration of sedative medications (ketamine, midazolam, or sufentanil) for urgent brief pediatric dental procedures. Anesth Prog. 1993; 40(3):63-6. PubMed
- Zedie N, Amory DW, Wagner BK, O’Hara DA. Comparison of intranasal midazolam and sufentanil premedication in pediatric outpatients. Clin Pharmacol Ther. 1996; 59(3):341-8. DOI | PubMed
- Bates BA, Schutzman SA, Fleisher GR. A comparison of intranasal sufentanil and midazolam to intramuscular meperidine, promethazine, and chlorpromazine for conscious sedation in children. Ann Emerg Med. 1994; 24(4):646-51. PubMed
- Good P, Jackson K, Brumley D, Ashby M. Intranasal sufentanil for cancer-associated breakthrough pain. Palliat Med. 2009; 23(1):54-8. DOI | PubMed
- Coffey F, Dissmann P, Mirza K, Lomax M. Methoxyflurane analgesia in adult patients in the emergency department: a subgroup analysis of a randomized, double-blind, placebo-controlled study (STOP!). Adv Ther. 2016; 33(11):2012-31. DOI | PubMed
- Minkowitz HS, Singla NK, Evashenk MA, Hwang SS, Chiang YK, Hamel LG. Pharmacokinetics of sublingual sufentanil tablets and efficacy and safety in the management of postoperative pain. Reg Anesth Pain Med. 2013; 38(2):131-9. DOI | PubMed
- Willsie SK, Evashenk MA, Hamel LG, Hwang SS, Chiang YK, Palmer PP. Pharmacokinetic properties of single- and repeated-dose sufentanil sublingual tablets in healthy volunteers. Clin Ther. 2015; 37(1):145-55. DOI | PubMed
- Butler MM, Ancona RM, Beauchamp GA, Yamin CK, Winstanley EL, Hart KW. Emergency department prescription opioids as an initial exposure preceding addiction. Ann Emerg Med. 2016; 68(2):202-8. DOI | PubMed
Fonte
Blancher M, Maignan M, Clapé C, Quesada J, Collomb-Muret R, et al. (2019) Intranasal sufentanil versus intravenous morphine for acute severe trauma pain: A double-blind randomized non-inferiority study. PLoS Medicine 16(7): e1002849. https://doi.org/10.1371/journal.pmed.1002849




