
Abstract
Introduzione
Nell’ultimo decennio, i progressi nella trascrittomica ad alto rendimento e nelle analisi dell’occupazione del DNA hanno fornito preziose intuizioni su come i fattori di trascrizione (TF) regolano le decisioni sul destino delle cellule e determinano l’identità delle cellule. È diventato chiaro che durante lo sviluppo embrionale, l’espressione delle principali TF è strettamente regolata, ed è stato dimostrato che l’espressione combinatoria di un numero relativamente limitato di TF è sufficiente per stabilire (e potenzialmente cambiare) l’identità e la differenziazione cellulare attraverso la loro azione sulle reti di regolazione genica sottostanti (GRN) (Iwafuchi-Doi e Zaret, 2016). In questo contesto, sono state utilizzate l’analisi trascrittomica di massa e il sequenziamento di immunoprecipitazione della cromatina (ChIP-seq) per descrivere le interazioni delle TF, fornendo così modelli GRN (Dunn et al., 2014; Mullen et al., 2011). Recentemente, queste tecniche sono state utilizzate con successo per studiare il sistema ematopoietico embrionale in un modello di differenziazione in vitro delle cellule staminali embrionali (ESC) (Goode et al., 2016).
Tuttavia, l’uso di popolazioni di cellule di massa costituisce un’importante limitazione nei nostri sforzi per comprendere appieno i GRN. Anche se la trascrittomica di massa può rivelare correlazioni geniche globali cruciali tra stati cellulari semi-stabili, non può risolvere le interazioni geniche più sottili che si verificano in complessi stati di transizione. Inoltre, l’uso di un approccio di tipo bulk rende difficile dedurre le conseguenze dirette sul paesaggio trascrizionale su cui agiscono queste TF.
Queste limitazioni possono essere superate con l’uso di approcci a cella singola. Negli ultimi cinque anni, sono stati raggiunti enormi progressi tecnici nel campo. L’espressione genica può essere efficacemente valutata a livello monocellulare, rendendo possibile distinguere le sottopopolazioni all’interno di tessuti e colture cellulari (Kolodziejczyk et al., 2015; Scialdone et al., 2016). La trascrittomica monocellulare è stata precedentemente utilizzata per svelare complesse transizioni evolutive come la gastrulazione (Scialdone et al., 2016), dimostrando che è possibile determinare combinazioni di TF che sono espresse a livello monocellulare con il progredire della differenziazione cellulare.
Nel presente lavoro abbiamo applicato approcci di trascrittomica monocellulare all’ontogenesi del sistema sanguigno nell’embrione del topo. I GRN che sono coinvolti nella produzione dello stelo ematopoietico e delle cellule progenitrici (HSPC) all’origine di tutte le cellule del sangue non sono ben compresi. Durante l’embriogenesi del topo, HSPCs sono generati principalmente nel sacco vitellino (YS), da E7-E7.5, e nella regione aorta-gonad-mesonefros (AGM), da E10.5. Essi emergono dalle cellule endoteliali che formano la vascolarizzazione, attraverso un processo chiamato transizione da endoteliale a ematopoietico (EHT) (Boisset et al., 2010; Choi et al., 1998; Eilken et al., 2009; Lancrin et al., 2009; Zovein et al., 2008). È stato proposto che un heptad di sette TF (Gata2, Runx1, Erg, Fli1, Lmo2, Lyl1 e Tal1) noti per essere essenziali per lo sviluppo del sangue formano un complesso trascrizionale potenzialmente coinvolto nella generazione di HSPC. Questa proposta si basava sull’analisi in massa ChIP-seq del legame di queste sette TF agli elementi regolatori di 927 geni della linea cellulare HPC7 (Wilson et al., 2010). Tuttavia, non vi era alcuna prova diretta che tutte e sette le sette TF fossero espresse insieme a livello monocellulare durante l’embriogenesi o che i target heptad giocassero un ruolo cruciale nello sviluppo. Impiegando l’analisi trascrittomica monocellulare e un sistema di differenziazione delle CSE in vitro che dà origine alle cellule del sangue, abbiamo affrontato queste domande e fornito nuovi spunti sulla formazione di HSPC da cellule endoteliali. I nostri dati mostrano che durante l’EHT, due serie di TF hanno effetti opposti che permettono una differenziazione adeguata. Attraverso la transizione, Erg e Fli1 supportano il destino delle cellule endoteliali mentre Runx1 e Gata2 promuovono quello ematopoietico. Inaspettatamente, abbiamo scoperto che il fattore di trascrizione endoteliale Fli1 potrebbe acquisire una funzione ematopoietica quando espresso insieme al regolatore master ematopoietico Runx1. Questo lavoro dimostra che l’analisi GRN basata su dati trascrittomici monocellulari può evidenziare gli aspetti biologici che mancano ai metodi classici di massa, sottolineando la potenza degli approcci monocellulari nella comprensione delle complesse transizioni evolutive.
Risultati
I fattori chiave di trascrizione identificano una popolazione intermedia tra le cellule endoteliali e le cellule del sangue durante l’EHT in vivo e in vitro
Abbiamo eseguito RT-PCR quantitativa monocellulare (sc-q-RT-PCR) su 95 geni associati a cellule ematopoietiche, endoteliali (Endo), e la muscolatura liscia vascolare (VSM) (file supplementare 1). Singole cellule sono state ordinate da YS e AGM sezionate da embrioni di topo a E9, E10.5 e E11. Per arricchire per le cellule sottoposte a EHT, abbiamo selezionato le cellule utilizzando sia il marcatore endoteliale VE-caderina (VE-Cad) e il marcatore ematopoietico CD41 (Figura 1-figure supplementi 1 e 2). Utilizzando clustering gerarchico, queste cellule potrebbero essere separati in tre gruppi principali in entrambi i tessuti: Le cellule endo, Pre-HSPCs, che hanno espresso sia i geni ematopoietici che endoteliali (Taoudi et al., 2008), e HSPCs (Figura 1A e B, Figura 1-figure supplements 1 e 2).
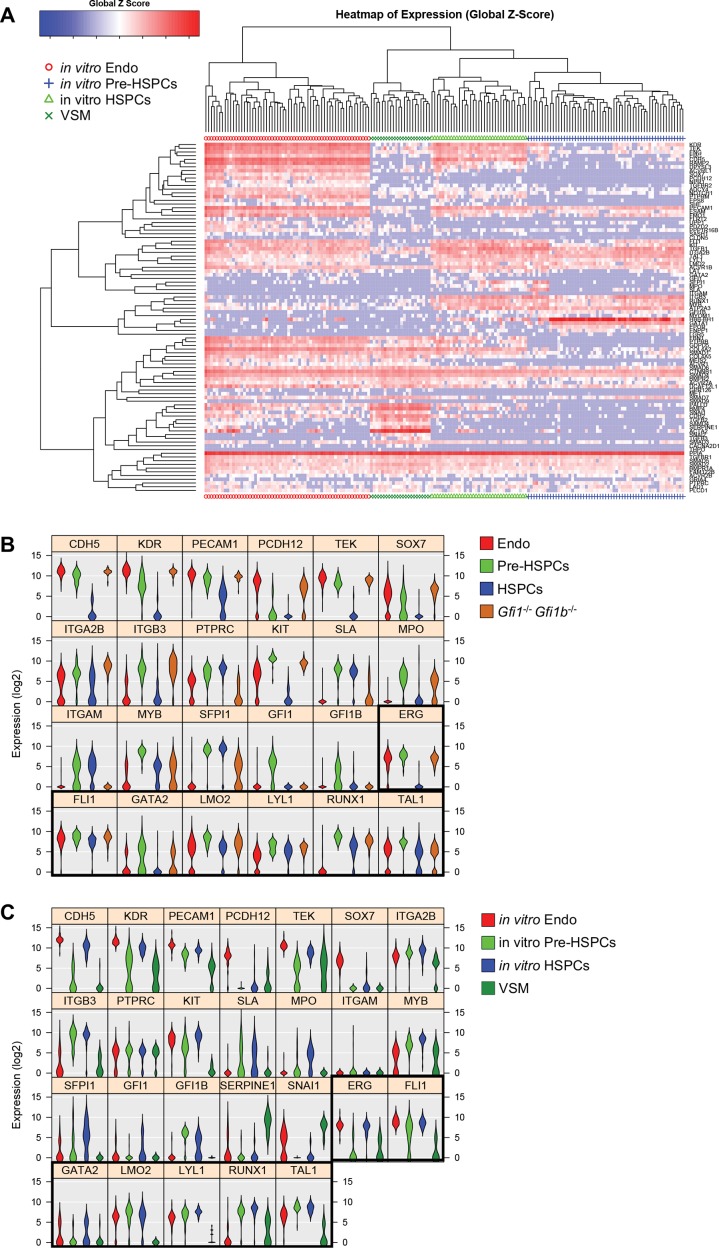
Figura 1-figura supplemento 6.Analisi in vivo e in vitro EHT da single-cell qRT-PCR identificato l’espressione di TF chiave nella fase di Pre-HSPC.analisi di clustering gerarchico di AGM VE-Cad + cellule.analisi di clustering gerarchico di YS VE-Cad + cellule.Analisi del tempo-corso di EHT nel AGM e YS.Comparazione di sottopopolazioni EHT da AGM e YS.Analisi di clustering gerarchico di YS Gfi1-/- Gfi1b-/- cellule.Analisi di clustering gerarchico di CSE differenziati in vitro.(A) Grafico di analisi dei componenti principali (PCA) che mostra le celle isolate a E9, E10,5 e E11 da AGM e YS secondo i cluster di campioni indicati (SC). Per ogni tessuto, ogni punto temporale rappresenta un esperimento. (B) PCA grafico che mostra il Gfi1-/-Gfi1b–/- GFP+ celle YS che si mescolano con il gruppo Pre-HSPCs (ellisse nera). Si noti che l’asse PC2 è stato invertito. (C) Flusso di lavoro sperimentale utilizzato per differenziare i CSE in vitro in cellule del sangue. BL-CFC, cellule che formano una colonia di esplosione; EB, corpo embrionario; FACS, selezione delle cellule attivata a fluorescenza. (D) Trama PCA che mostra i quattro gruppi di cellule provenienti da CSE differenziate in vitro. (E) Heatmap che mostra l’espressione media di endoteliale (blu), geni ematopoietici (rosso) e sette chiave TFs (nero) nei quattro gruppi trovati in vivo. Il rettangolo nero evidenzia l’espressione delle sette TF. (F) Heatmap che mostra l’espressione media dei geni endoteliali (blu), ematopoietici (rosso), vascolari muscolari lisci (viola) e sette chiave TFs (nero) nei quattro gruppi trovati dopo la differenziazione in vitro delle CSE. Il rettangolo nero evidenzia l’espressione delle sette TF. Vedi anche la figura 1-figure supplements 1-6, file supplementare 1, 2, 3 e 11.(A) I grafici FACS dell’espressione VE-Cad e CD41 nella regione AGM a E9, E10.5 ed E11. Diversi embrioni (sette per E9, 13 per E10.5 e 9 per E11) sono stati raggruppati per ogni punto temporale e tre sottopopolazioni sono state ordinate a cella singola: VE-Cad+ CD41- , VE-Cad+ CD41Medium e VE-Cad+ CD41High. (B) Analisi gerarchica del clustering fatta con dati sc-q-RT-PCR da cellule AGM isolate in 1A. Sono stati definiti tre gruppi principali. Vedi anche il file supplementare 11.(A) Trame FACS di espressione VE-Cad e CD41 nella regione YS a E9, E10.5 e E11. Diversi embrioni (sette per E9, 13 per E10.5 e 9 per E11) sono stati messi in comune per ogni punto temporale e tre sottopopolazioni sono state ordinate a cella singola: VE-Cad+ CD41- , VE-Cad+ CD41Medium e VE-Cad+ CD41High. (B) Analisi gerarchica del clustering fatta con dati sc-q-RT-PCR da cellule YS isolate in 2A. Sono stati definiti tre gruppi principali. Vedi anche il file supplementare 11.Trame di dispersione che mostrano l’espressione media dei geni endoteliali rispetto all’espressione media dei geni ematopoietici nei punti di sviluppo indicati.Vedi anche il file supplementare 11.(A) Studio di correlazione eseguito sull’espressione genica media per i gruppi evidenziati in Figura 1A, Figura 1-figure supplements 1B e 2B. (B) Analisi di clustering gerarchico fatto con sc-q-RT-PCR dati di sc-q-RT-PCR da cellule AGM e YS presentati in Figura 1-figure supplements 1B e 2B. Vedi anche il file supplementare 11.(A) FACS trama di espressione delle GFP nella regione del sacco vitellino Gfi1-/- Gfi1b-/- regione del sacco vitellino a E9.5. Singole cellule che esprimono altamente GFP sono state isolate e studiate con sc-q-RT-PCR. (B) Analisi gerarchica di clustering fatto con sc-q-RT-PCR dati da cellule del sacco vitellino isolato in (A). Vedi anche il file supplementare 11.(A) Analisi gerarchica del clustering fatta con dati sc-q-RT-PCR da CSE differenziati in vitro. Sono stati definiti quattro cluster principali. (B) Violino trame che mostrano l’espressione dei 24 geni mostrati in Figura 1E. Sette fattori di trascrizione chiave sono evidenziati con un rettangolo nero. (C) Trame per violino che mostrano l’espressione dei 26 geni mostrati in Figura 1F. Sette fattori di trascrizione chiave sono evidenziati con un rettangolo nero. Vedere anche il file supplementare 11.
Mentre la frequenza di ogni popolazione di cellule differivano tra tessuti e punti temporali (Figura 1-figure supplement 3), i gruppi erano più fortemente correlati (Pearson r > 0,9) alle loro controparti rispetto ad altri gruppi nello stesso tessuto e raggruppati insieme utilizzando l’analisi dei componenti principali (PCA) e clustering gerarchico (Figura 1-figure supplement 4 e Figura 1A).
Inoltre, E9.5 YS Gfi1-/–Gfi1b–/– cellule, che non sono in grado di completare l’EHT a causa della mancanza dei due repressori trascrizionali Gfi1 e Gfi1b (Lancrin et al., 2012), raggruppati insieme ai Pre-HSPC, rafforzando l’idea che questa popolazione è un passo intermedio essenziale tra Endo e HSPC (Figura 1-figure supplement 5 e Figura 1B).
EHT può essere ricapitolato in vitro utilizzando il modello di differenziazione delle CSE (Huber, 2010). Abbiamo eseguito una differenziazione di 3,25 giorni seguita dall’isolamento delle cellule Flk1+, arricchite in cellule che formano una colonia di esplosione (BL-CFC) (Choi et al., 1998), che sono state coltivate per altri 1,5 giorni prima di eseguire sc-q-RT-PCR utilizzando gli stessi 95 geni (Figura 1C). Clustering gerarchico identificato quattro gruppi, corrispondenti a Endo, HSPCs, e Pre-HSPCs trovato nella vascolarizzazione embrionale e un quarto gruppo caratterizzato da un’elevata espressione di Acta2 (actina del muscolo liscio) e Serpine1 e da una bassa espressione dei geni endoteliali ed ematopoietici, che abbiamo identificato come VSM (Figura 1-figure supplemento 6A e Figura 1D).
In entrambi i sistemi in vivo e in vitro, la caratteristica più notevole della popolazione Pre-HSPCs è la co-espressione di sette geni che codificano per i fattori di trascrizione associati all’emopoiesi: Erg, Fli1, Tal1, Lyl1, Lmo2, Runx1 e Gata2 (Figura 1E e F, Figura 1-figure supplement 6B e C), che sono stati proposti per lavorare insieme come un complesso (Wilson et al., 2010). Al contrario, le cellule Endo espresso solo Erg, Fli1, Lmo2 e Tal1 mentre HSPCs espresso Fli1, Lmo2, Lyl1, Runx1 e Tal1. Ciò suggerisce che queste sette TF potrebbero essere importanti per stabilire e/o mantenere l’identità di tipo cellulare della popolazione dei Pre-HSPC.
Figura 1-figura supplemento 6.Figura 1. Analisi in vivo e in vitro EHT da single-cell qRT-PCR identificato l’espressione di TF chiave nella fase di Pre-HSPC.analisi di clustering gerarchico di AGM VE-Cad cellule +.analisi di clustering gerarchico di YS VE-Cad cellule +.Analisi del tempo-corso di EHT nel AGM e YS.Comparazione di sottopopolazioni EHT da AGM e YS.Analisi di clustering gerarchico di YS Gfi1-/- Gfi1b-/- cellule.Analisi di clustering gerarchico di CSE differenziati in vitro.(A) Principal component analysis (PCA) plot che mostra le cellule isolate a E9, E10.5 e E11 da AGM e YS secondo i cluster di campioni indicati (SC). Per ogni tessuto, ogni punto di tempo rappresenta un esperimento. (B) PCA grafico che mostra il Gfi1-/-Gfi1b–/- GFP+ celle YS che si mescolano con il gruppo Pre-HSPCs (ellisse nera). Si noti che l’asse PC2 è stato invertito. (C) Flusso di lavoro sperimentale utilizzato per differenziare i CSE in vitro in cellule del sangue. BL-CFC, cellule che formano una colonia di esplosione; EB, corpo embrionario; FACS, selezione delle cellule attivata a fluorescenza. (D) Trama PCA che mostra i quattro gruppi di cellule provenienti da CSE differenziate in vitro. (E) Heatmap che mostra l’espressione media di endoteliale (blu), geni ematopoietici (rosso) e sette chiave TFs (nero) nei quattro gruppi trovati in vivo. Il rettangolo nero evidenzia l’espressione delle sette TF. (F) Heatmap che mostra l’espressione media dei geni endoteliali (blu), ematopoietici (rosso), vascolari muscolari lisci (viola) e sette chiave TFs (nero) nei quattro gruppi trovati dopo la differenziazione in vitro delle CSE. Il rettangolo nero evidenzia l’espressione delle sette TF. Vedi anche la figura 1-figure supplements 1-6, file supplementare 1, 2, 3 e 11.(A) FACS trame di espressione VE-Cad e CD41 nella regione AGM a E9, E10.5 e E11. Diversi embrioni (sette per E9, 13 per E10.5 e 9 per E11) sono stati messi in comune per ogni punto di tempo e tre sottopopolazioni sono stati ordinati a singola cellula: VE-Cad+ CD41- , VE-Cad+ CD41Medium e VE-Cad+ CD41High. (B) Analisi gerarchica del clustering fatta con dati sc-q-RT-PCR da cellule AGM isolate in 1A. Sono stati definiti tre gruppi principali. Vedi anche il file supplementare 11.(A) Trame FACS di espressione VE-Cad e CD41 nella regione YS a E9, E10.5 e E11. Diversi embrioni (sette per E9, 13 per E10.5 e 9 per E11) sono stati messi in comune per ogni punto temporale e tre sottopopolazioni sono state ordinate a cella singola: VE-Cad+ CD41- , VE-Cad+ CD41Medium e VE-Cad+ CD41High. (B) Analisi gerarchica del clustering fatta con dati sc-q-RT-PCR da cellule YS isolate in 2A. Sono stati definiti tre gruppi principali. Vedi anche il file supplementare 11.Spargere trame che mostrano l’espressione media dei geni endoteliali rispetto all’espressione media dei geni ematopoietici nei punti di sviluppo indicati.Vedi anche il file supplementare 11.(A) Studio di correlazione eseguito sull’espressione genica media per i gruppi evidenziati in Figura 1A, Figura 1-figure supplements 1B e 2B. (B) Analisi di clustering gerarchica fatta con sc-q-RT-PCR dati di sc-q-RT-PCR da cellule AGM e YS presentati in Figura 1-figure supplements 1B e 2B. Vedi anche il file supplementare 11.(A) Trama FACS dell’espressione delle GFP nella regione del sacco vitellino Gfi1-/- Gfi1b-/- a E9.5. Singole cellule che esprimono elevate GFP sono state isolate e studiate con sc-q-RT-PCR. (B) Analisi gerarchica di clustering fatto con sc-q-RT-PCR dati da cellule del sacco vitellino isolato in (A). Vedi anche il file supplementare 11.(A) Analisi gerarchica del clustering fatta con i dati sc-q-RT-PCR da CSE differenziati in vitro. Sono stati definiti quattro cluster principali. (B) Violino trame che mostrano l’espressione dei 24 geni mostrati in Figura 1E. Sette fattori di trascrizione chiave sono evidenziati con un rettangolo nero. (C) Trame per violino che mostrano l’espressione dei 26 geni mostrati in Figura 1F. Sette fattori di trascrizione chiave sono evidenziati con un rettangolo nero. Vedere anche il file supplementare 11.
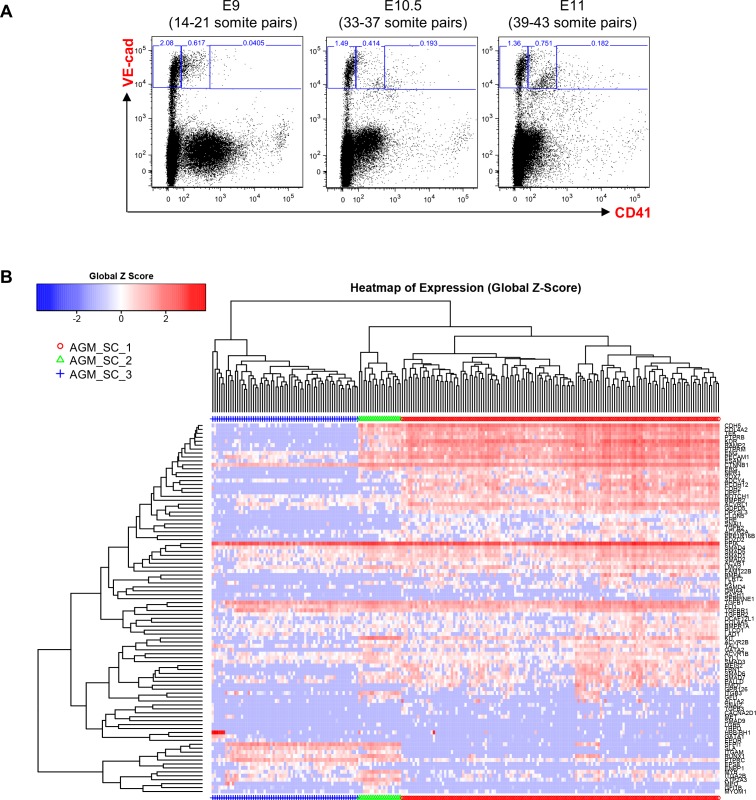
Figura 1-figura supplemento 1.Analisi gerarchica del clustering delle celle AGM VE-Cad+.(A) Trame FACS di espressione VE-Cad e CD41 nella regione AGM a E9, E10.5 e E11. Diversi embrioni (sette per E9, 13 per E10.5 e 9 per E11) sono stati messi in comune per ogni punto temporale e tre sottopopolazioni sono state ordinate a cella singola: VE-Cad+ CD41- , VE-Cad+ CD41Medium e VE-Cad+ CD41High. (B) Analisi gerarchica del clustering fatta con dati sc-q-RT-PCR da cellule AGM isolate in 1A. Sono stati definiti tre gruppi principali. Vedi anche il file supplementare 11.
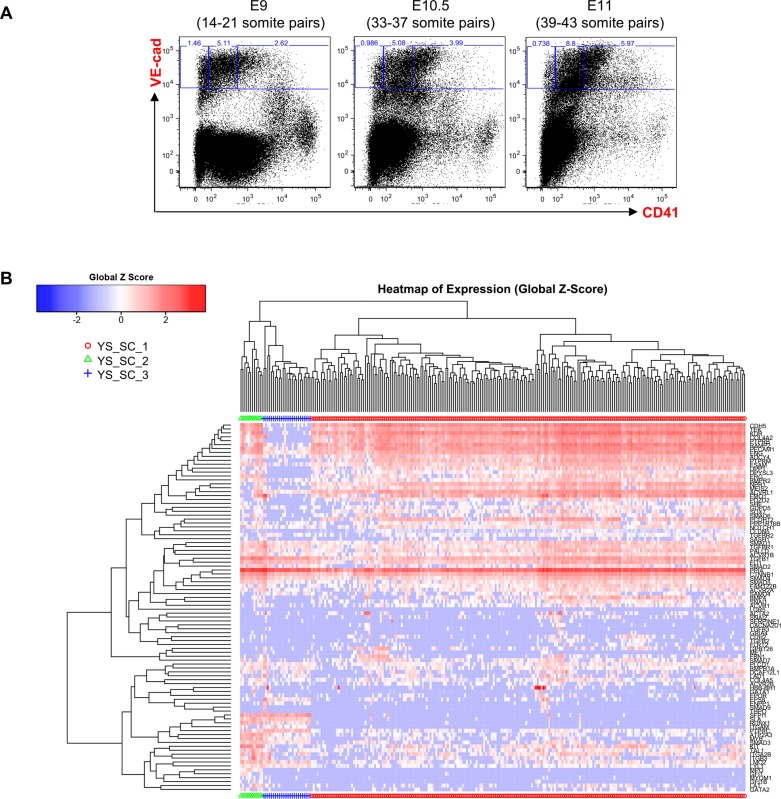
Figura 1-figure supplement 2.Figura 1—supplemento di figura 2. Analisi gerarchica del clustering delle cellule YS VE-Cad+.(A) FACS trame di VE-Cad e CD41 espressione nella regione YS a E9, E10.5 e E11. Diversi embrioni (sette per E9, 13 per E10.5 e 9 per E11) sono stati messi in comune per ogni punto di tempo e tre sottopopolazioni sono stati ordinati a singola cellula: VE-Cad+ CD41- , VE-Cad+ CD41Medium e VE-Cad+ CD41High. (B) Analisi gerarchica del clustering fatta con dati sc-q-RT-PCR da cellule YS isolate in 2A. Sono stati definiti tre gruppi principali. Vedi anche il file supplementare 11.
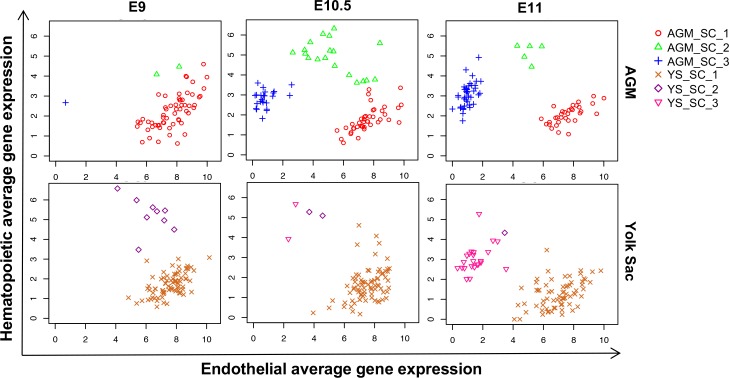
Figura 1-figure supplemento 3.Analisi del corso del tempo di EHT nell’AGM e YS.Trame di dispersione che mostrano l’espressione media dei geni endoteliali rispetto all’espressione media dei geni ematopoietici nei punti di sviluppo indicati.Vedi anche il file supplementare 11.

Figura 1-figure supplemento 4.Figura 1— supplemento di figura 4. Confronto di sottopopolazioni EHT da AGM e YS.(A) Studio di correlazione eseguita sulla espressione genica media per i gruppi evidenziati in Figura 1A, Figura 1-figure integratori 1B e 2B. (B) Analisi di clustering gerarchico fatto con sc-q-RT-PCR dati da cellule AGM e YS presentati in Figura 1-figure integratori 1B e 2B. Vedi anche il file supplementare 11.
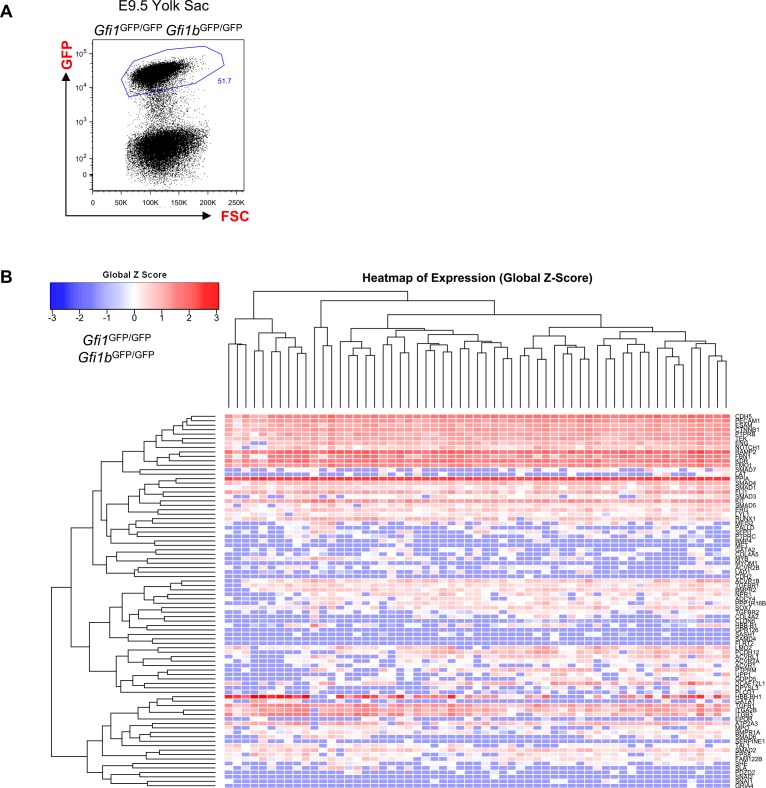
Figura 1-figura supplemento 5.Analisi gerarchica di clustering di YS Gfi1-/- Gfi1b-/- celle.(A) FACS trama di espressione GFP nella Gfi1-/- Gfi1b-/- regione del sacco vitellino a E9,5. Singole cellule che esprimono altamente GFP sono stati isolati e studiati con sc-q-RT-PCR. (B) Analisi gerarchica di clustering fatto con sc-q-RT-PCR dati da cellule del sacco vitellino isolato in (A). Vedi anche il file supplementare 11.
Figura 1-figura supplemento 6.Analisi gerarchica di clustering di CSE differenziati in vitro.(A) Analisi gerarchica di clustering fatto con sc-q-RT-PCR dati provenienti da CSE differenziati in vitro. Sono stati definiti quattro cluster principali. (B) Violino trame che mostrano l’espressione dei 24 geni mostrati in Figura 1E. Sette fattori di trascrizione chiave sono evidenziati con un rettangolo nero. (C) Trame per violino che mostrano l’espressione dei 26 geni mostrati in Figura 1F. Sette fattori di trascrizione chiave sono evidenziati con un rettangolo nero. Vedere anche il file supplementare 11.
Sovraespressione simultanea di Erg, Fli1, Tal1, Lyl1, Lmo2, Runx1, Cbfb e Gata2 durante la differenziazione dell’emangioblasto porta alla formazione di una popolazione simile a quella dei Pre-HSPC
La coespressione delle sette TF nelle popolazioni dei Pre-HSPC in vivo e in vitro ci ha spinto a chiederci se la coespressione di questi geni possa essere collegata all’identità dei Pre-HSPC. Per determinare l’effetto dell’espressione di questi sette fattori a livello monocellulare, abbiamo stabilito una linea cellulare ES inducibile in cui tutti e sette i geni e Cbfb, una proteina essenziale per il legame e la funzione del DNA Runx1 (Wang et al., 1996; Tahirov et al., 2001), potrebbero essere espressi simultaneamente a seguito dell’aggiunta di doxiciciclina (dox). Abbiamo generato una linea cellulare ES inducibile (i8TFs) in cui le otto sequenze di codifica sono state collegate tra loro da sequenze T2A (Figura 2A). Questa strategia sfrutta il meccanismo del “salto ribosomiale” del peptide T2A virale (Donnelly et al., 2001) per consentire la produzione di otto proteine da una singola trascrizione. L’espressione di tutte e otto le proteine è stata convalidata da western blot dopo 24 ore di trattamento dox in coltura ESC (Figura 2B). Come controllo, abbiamo creato una linea ESC, che non contiene alcun nuovo cDNA (Empty), ma è altrimenti identica alla linea i8TFs.
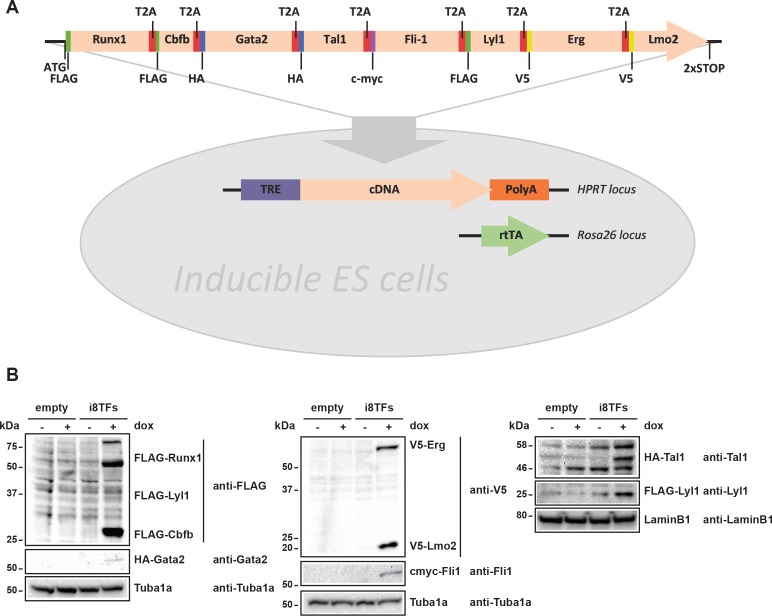
Figura 2.Generazione di una linea di ESC doxiciclina inducibile per l’espressione simultanea degli otto fattori di trascrizione.(A) Schema che mostra la generazione della linea i8TFs ESC. (B) Western blot che mostra l’espressione proteica degli otto TF dopo il trattamento con doxiciclina (dox) nella linea Empty e nella linea i8TFs ESC. Vedi anche il file supplementare 11.
Abbiamo studiato l’effetto della sovraespressione delle otto TF durante la differenziazione dell’emangioblasto. Dopo tre giorni di coltura EB, le cellule Flk1+ sono state differenziate per un giorno in coltura BL-CFC, in quel momento le cellule hanno perso la loro identità mesodermica e hanno prodotto cellule muscolari lisce endoteliali e vascolari. Dox è stato aggiunto a questo punto di tempo, e le cellule sono state coltivate per altri due giorni. Le cellule sono stati ripresi ogni 15 minuti per 48 ore e sono stati raccolti alla fine della cultura per la citometria a flusso. La linea cellulare vuota non ha mostrato alcuna differenza importante, né in termini di marcatori della superficie cellulare né in termini di morfologia cellulare dopo il trattamento con dox. Al contrario, abbiamo notato un cambiamento drammatico in seguito all’attivazione delle otto TF quando quasi tutte le cellule hanno sviluppato lo stesso fenotipo (VE-Cad+ CD41+ e cKit+ CD41+ a seconda della colorazione) (Figura 3A). Questo cambiamento è stato accompagnato da una marcata diminuzione del numero di cellule rotonde (Figura 3B e Figura 3-figure supplement 1). L’espressione simultanea delle otto TF non ha cambiato la frequenza delle cellule viventi (Figura 3C) ma c’è stata una diminuzione del 40% del numero di cellule in fase S, suggerendo che l’espressione delle otto TF ha un impatto negativo sulla proliferazione cellulare (Figura 3D).
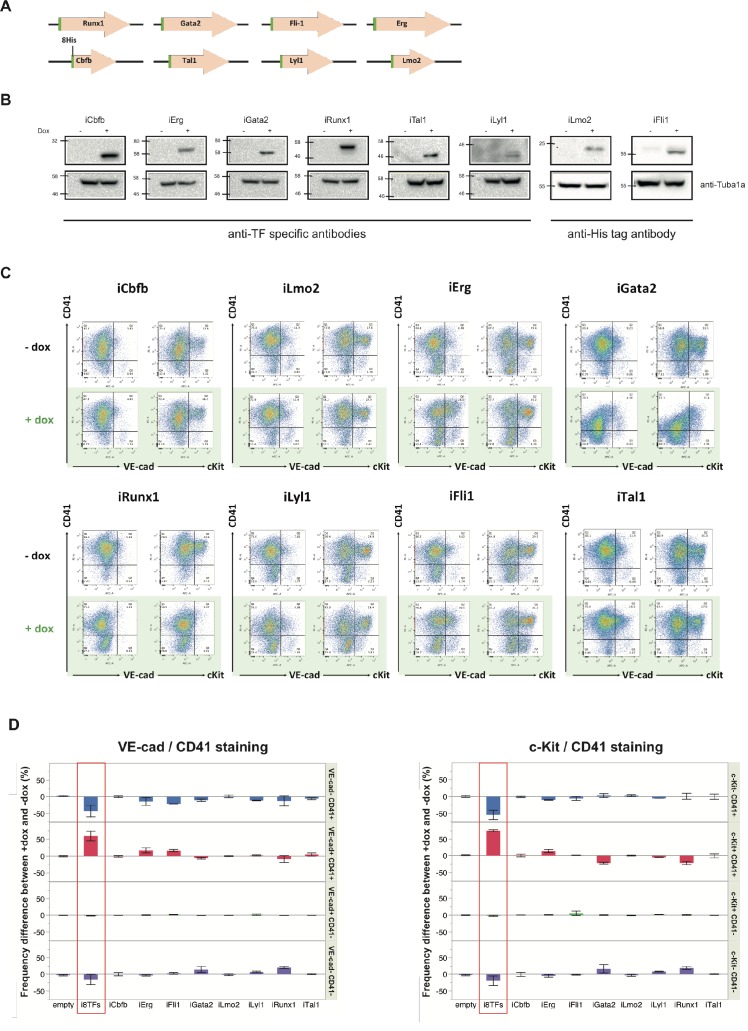
Figura 3-figure supplement 5.La differenziazione ematopoietica della linea di ESC i8TFs evidenzia il ruolo degli otto fattori di trascrizione nella generazione di Pre-HSPCs.immagini di microscopia della cultura BL-CFC i8TFs dopo uno, due e tre giorni di cultura BL-CFC in condizioni -dox e +dox.confronto della risposta delle popolazioni eVSM ed Endo all’induzione dei grafici 8TFs.PCA di dati microarray.analisi dei dati microarray.analisi dei dati microarray.analisi delle linee di ESC i1TF.(A) Rappresentativo FACS trame di espressione VE-Cad, cKit e CD41 dopo 3 giorni di cultura BL-CFC delle linee ESC indicate (n = 3). (B) Grafici che mostrano il numero medio di celle rotonde contate per fotogramma (n = 3) in un corso di tempo di 48 ore per le linee ESC indicate. Le barre di errore rappresentano le deviazioni standard. (C) Barra grafica che indica la frequenza delle cellule viventi non apoptotiche per le linee di ESC indicate. Le barre di errore rappresentano le deviazioni standard (n = 3). (D) Barra grafica che indica la frequenza delle celle cicliche per le linee di ESC indicate. Le barre di errore rappresentano le deviazioni standard (n = 3). (E) Barra grafica che mostra il numero di colonie ematopoietiche dopo aver replicato le cellule in un saggio di unità di colonia-formulazione dalle condizioni indicate. Le barre di errore rappresentano la deviazione standard (n = 4). ns, non significativo. ** p-valore<0,01 (test t accoppiato a due code). * p-valore<0,05 (test t accoppiato a due code). Si veda anche la figura 3-figure supplement 1 a 5, file supplementare 1, 4, 5 e 11.La barra della scala corrisponde a 200 μm. Vedi anche il file supplementare 11.(A) Trama FACS di VE-Cad e CD41 espressione al giorno 1 della cultura BL-CFC. I rettangoli evidenziano la muscolatura liscia vascolare arricchita (eVSM) e le popolazioni endoteliali (Endo). (B) Analisi gerarchica di clustering fatta con sc-q-RT-PCR dati da VE-Cad + CD41- (Endo) e VE-Cad- CD41- (eVSM) popolazioni. I rettangoli rossi indicano le cellule della popolazione eVSM che esprimono i marcatori endoteliali (ad esempio Ramp2, Sox7, Kdr). (C) FACS trame di VE-Cad, cKit e CD41 espressione della muscolatura liscia vascolare arricchita (eVSM) e popolazioni endoteliali (Endo) dalla linea cellulare i8TFs dopo 48 ore in cultura HE. (D) Bar graph che mostra il numero di colonie ematopoietiche dopo aver replicato le cellule in una colonia-formulazione-unità di saggio dalle condizioni indicate. Le barre di errore rappresentano le deviazioni standard (n = 2). Vedere anche il file supplementare 11.(A) Trama PCA che mostra i risultati delle analisi microarray dell’eVSM dopo 24 ore di trattamento dox sulla linea cellulare vuota. Le ellissi indicano i diversi gruppi di campioni. (B) PCA grafico che mostra i risultati delle analisi microarray dalle condizioni indicate dopo 24 ore di trattamento dox sulla linea i8TFs ESC. I numeri indicano il numero di geni espressi in modo significativamente diverso tra le due condizioni (p-valore<0,05). Le ellissi indicano i diversi gruppi di campioni. Vedi anche il file supplementare 11.(A) GO analisi di arricchimento termine dei geni espressi in modo differenziato tra -dox e +dox condizioni per ogni popolazione. (B) diagrammi di Venn che mostrano i geni espressi in modo differenziato per entrambe le popolazioni insieme con i target genici precedentemente identificati da ChIP-seq analisi (Wilson et al., 2010). (C) Elenco dei 26 geni target heptad espressi in modo differenziato all’induzione delle otto TF sia nella popolazione Endo che in quella eVSM. Vedi anche il file supplementare 11.(A) Schema che mostra gli otto costrutti utilizzati per realizzare le linee i1TF ESC. (B) Western blot che mostra l’espressione proteica degli otto fattori di trascrizione dopo il trattamento con doxiciclina per ciascuna delle linee i1TF ESC. (C) rappresentativo FACS trame di VE-Cad, cKit e CD41 espressione dopo tre giorni di cultura BL-CFC delle linee indicate ESC (n = 3). (D) Grafici a barre che indicano la differenza di frequenza tra le condizioni +dox e -dox per le dieci linee cellulari indicate. Le barre di errore rappresentano le deviazioni standard (n = 3). Vedi anche il file supplementare 11.
Per valutare l’impatto della sovraespressione simultanea di questi otto geni sull’attività di differenziazione ematopoietica, abbiamo eseguito un test di unità di formazione delle colonie (CFU). Abbiamo placcato le cellule di entrambe le condizioni i8TFs – dox e i8TFs + dox in assenza di dox in un mezzo che permette la crescita sia delle cellule mieloidi che di quelle eritroidi. Abbiamo osservato un drammatico aumento del numero di unità di formazione di colonia dalle cellule +dox, suggerendo che l’induzione delle otto TFs aumenta il potenziale di differenziazione ematopoietica (Figura 3E).
La sovraespressione delle otto TF ha portato alla produzione di una maggioranza di cellule VE-Cad+ CD41+ (Figura 3A). Tuttavia, all’inizio del trattamento dox, la maggior parte delle cellule erano VE-Cad- CD41- (Figura 3-figure supplement 2A), che sono stati arricchiti in cellule muscolari lisce vascolari (denominate eVSMs) (Figura 3-figure supplement 2B). Per capire l’impatto dell’espressione delle otto TF sulle due principali popolazioni non ematopoietiche – la popolazione endoteliale VE-Cad+ CD41- (chiamata Endo) e l’eVSM – abbiamo isolato le cellule Endo ed eVSM al giorno 1 della coltura BL-CFC (Figura 3-figure supplement 2A) e le abbiamo coltivate in condizioni di coltura emogenica endoteliale (HE) con o senza dox per 48 ore. Utilizzando la citometria a flusso, abbiamo dimostrato che entrambe le popolazioni sono state colpite dal trattamento con dox e sono diventate quasi interamente VE-Cad+ CD41+ (Figura 3-figure supplement 2C). Inoltre, il test CFU ha mostrato una maggiore capacità di differenziazione ematopoietica per l’Endo +dox rispetto alla condizione -dox, confermando la loro conversione di successo (Figura 3-figure supplement 2D). Al contrario, le cellule eVSM non hanno mostrato un effetto comparabile, e insieme ai dati della citometria a flusso, questa evidenza suggerisce che anche se gli eVSM hanno ottenuto il fenotipo Pre-HSPCs, probabilmente sono rimasti più immaturi delle cellule Endo +dox (Figura 3-figure supplement 2D). Per caratterizzare ulteriormente l’effetto delle otto TFs sulle popolazioni eVSM ed Endo, abbiamo effettuato un’analisi microarray sulle condizioni i8TFs eVSM ed Endo prima (t0) e 24 ore dopo la differenziazione con o senza trattamento dox (+dox e -dox) (file supplementare 5). La linea vuota ESC eVSM è stata utilizzata come controllo. Come previsto, il trattamento dox di queste cellule ha avuto poco effetto sul trascrittoma, come mostrato dalla PCA (Figura 3-figure supplement 3A). Al contrario, l’espressione delle otto TF ha cambiato radicalmente le cellule Endo ed eVSM. È interessante notare che il gruppo eVSM +dox si è raggruppato strettamente alle condizioni Endo -dox e Endo +dox, ma lontano dal gruppo eVSM -dox. L’analisi dei geni espressi in modo differenziale (DEG) ha evidenziato i geni responsabili di questi cambiamenti (Figura 3-figure supplement 3B e Supplementary file 5). L’analisi dell’ontologia genica (GO) dei DEG ha mostrato che la sovraespressione delle otto TF ha portato allaridotta espressione dei geni coinvolti nello sviluppo della vascolarizzazione e del cuore, mentre c’è stato un aumento dell’espressione dei geni legati al sistema immunitario e allo sviluppo della vascolarizzazione per l’eVSM (compatibile con la doppia identità endoteliale-ematopoietica dei Pre-HSPC) e la disintossicazione cellulare così come le vescicole per l’endo (Figura 3-figure supplement 4A). Sorprendentemente, confrontando questi DEG con i 927 bersagli dell’eptad identificati da ChIP-seq, la sovrapposizione è stata di soli 26 geni (Figura 3-figure supplement 4B e C). Questo suggerirebbe che i DEG che abbiamo osservato non erano una diretta conseguenza del complesso di otto TF che si legano agli elementi regolatori di questi geni.
Per scoprire se l’effetto sorprendente delle otto TF è realmente dovuto all’espressione di diversi di questi geni e non all’effetto di un particolare fattore di trascrizione, abbiamo generato otto linee ESC inducibili per ciascuno dei singoli fattori di trascrizione (i1TF) (Figura 3-figure supplement 5A). L’espressione inducibile di ogni TF è stata confermata utilizzando western blot (Figura 3-figure supplement 5B). Utilizzando lo stesso schema sperimentale utilizzato in precedenza per i8TF e linee ESC vuote, abbiamo eseguito i saggi BL-CFC e le analisi FACS con queste otto nuove linee ESC e studiato l’effetto del trattamento dox su ciascuna di esse. Abbiamo scoperto che nessuna delle linee i1TF era qualitativamente simile alle i8TF (Figura 3-figure supplement 5C e D).
Per distinguere meglio le popolazioni cellulari differenziate, abbiamo ripetuto la cultura BL-CFC con il trattamento -dox / +dox per le dieci linee cellulari seguite da sc-q-RT-PCR dei 95 geni utilizzati in precedenza (Figura 4 e file supplementare 6). In totale, 854 cellule sono state elaborate utilizzando sc-q-RT-PCR e superato il controllo di qualità. Sei cluster di cellule biologicamente rilevanti possono essere identificati utilizzando l’analisi gerarchica di clustering, e sono stati assegnati i nomi in base all’espressione dei geni marcatori specifici (Figura 4A). I gruppi Endo, VSM e Pre-HSPCs sono stati definiti come prima (Figura 1). D’altra parte, gli HSPC sono stati suddivisi in tre gruppi: Cellule Progenitrici Eritroidi I e II (EryPCs_I e EryPCs_II, rispettivamente) e Cellule Progenitrici Mieloidi (MyePC). EryPCs_I ed EryPCs_II erano caratterizzate dall’espressione di geni eritroidi come Gata1 e Hbb-bh1. EryPCs_I sembrava essere più immaturo di EryPCs_II perché le cellule esprimono ancora un alto livello di Itgb3 e Pecam1. I MyePCs erano caratterizzati dall’espressione di marcatori mieloidi come Sfpi1 (o Pu.1) e Itgam e dalla mancanza di geni eritroidi.
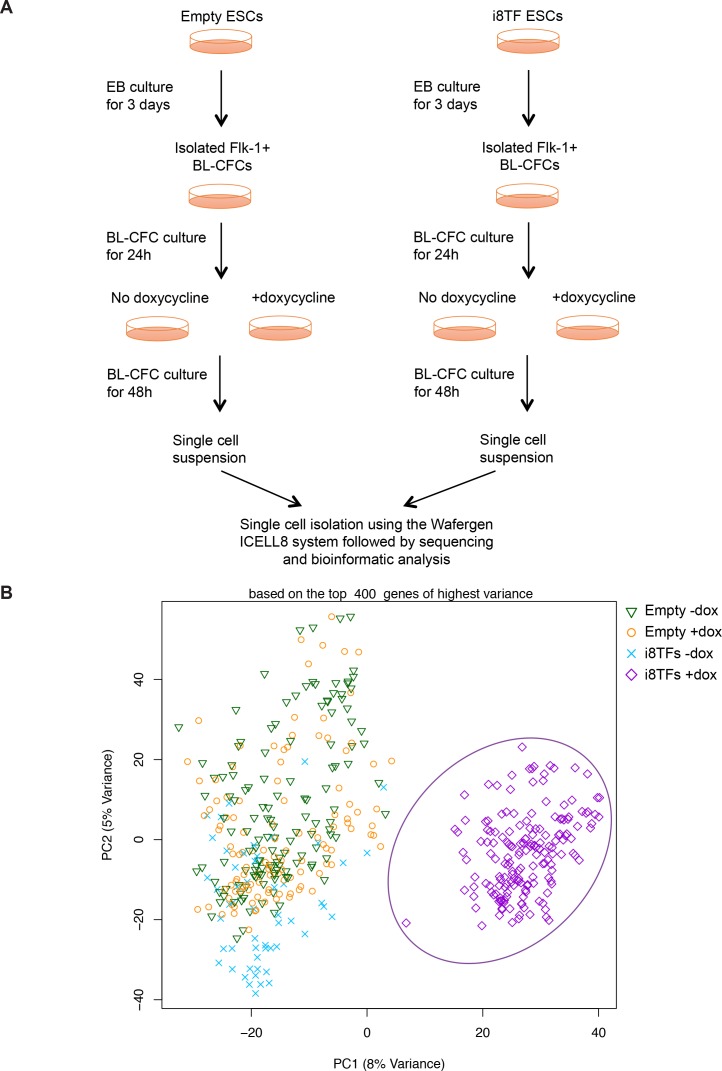
Figura 4-figure supplement 2.Analisi trascrittomica monocellulare delle linee i8TFs e i1TF ESC.Vengono visualizzati sei diagrammi di dispersione corrispondenti alle sei popolazioni definite nella Figura 4.Confronto tra le culture del giorno Empty e i8TFs giorno 3 BL-CFC mediante sequenziamento dell’RNA monocellulare.(A) Clustering gerarchico che mostra i risultati sc-q-RT-PCR per 95 geni sulle dieci linee ESC inducibili dopo tre giorni di coltura BL-CFC. I cluster sono stati definiti secondo l’intersezione della linea rossa punteggiata con il dendrogramma nella parte superiore della mappa termica. (B) Grafici a barre che mostrano il numero di celle in ciascuno dei sei cluster definiti in (A) per le dieci linee di celle. Le stelle indicano differenze significative per le condizioni +dox (vedi file supplementare 6 per i valori p). Vedi anche Figura 4-figure supplements 1 e 2, Supplementary file 1, 6 e 11.Il set di dati è identico a quello della Figura 4. Qui, le frazioni di cellule sono mostrate al posto dei numeri di cellule assoluti. Per la condizione -dox, è stato aggiunto un box plot. Differenze significative tra la condizione -dox e la condizione +dox per ogni linea cellulare e popolazione sono indicate da cerchi rossi come determinato da un modello linearizzato generale (quasi-Poisson). Vedi anche il file supplementare 11.(A) Schema sperimentale seguito per confrontare gli ESC Empty e i8TFs con il sequenziamento dell’RNA monocellulare. (B) Tracciato PCA che mostra l’analisi sc-RNA-seq delle linee cellulari Empty e i8TFs dopo tre giorni di coltura BL-CFC. Ellisse evidenzia i8TFs + dox. Vedere anche il file supplementare 11.
Per ogni cluster, il numero di cellule per condizione è stato calcolato e ogni condizione +dox è stato statisticamente confrontato con tutte le condizioni -dox (Figura 4B e Figura 4-figure supplement 1). Questa analisi ha confermato che dopo l’aggiunta di dox, le cellule i8TFs i8TFs formano una popolazione Pre-HSPCs co-espressione dei geni endoteliali ed ematopoietici, come suggerito dai risultati della citometria a flusso. Al contrario, nessuna delle altre linee cellulari ha presentato un cambiamento così drammatico. L’induzione di Gata2 ha aumentato la frequenza della popolazione EryPCs_II di ~ 4 volte (Figura 4B). D’altra parte, la sovraespressione di Erg e Fli1 ha dato luogo a un po ‘più Pre-HSPCs (Figura 4B). Le altre linee i1TF ESC avevano cambiamenti trascurabili al trattamento dox (Figura 4B e Figura 4-figure supplement 1).
I nostri risultati sc-q-RT-PCR suggeriscono che le cellule erano omogenee dopo l’induzione delle otto TF. Per confermare questa osservazione, abbiamo effettuato il sequenziamento di RNA a cella singola (sc-RNA-seq) su i8TFs -dox, i8TFs +dox, Empty -dox e Empty +dox cellule utilizzando la piattaforma iCELL8 (Figura 4-figure supplement 2A). Anche in questo caso, PCA ha mostrato che i8TFs + cellule dox sono stati chiaramente raggruppati separatamente dalle altre cellule (Figura 4-figure supplemento 2B). Sulla base della varianza PC2, abbiamo notato che c’è meno varianza per le cellule i8TFs +dox rispetto alle altre tre condizioni. Nel complesso, questo suggerirebbe che le cellule i8TFs +dox sono relativamente più omogenee rispetto alle cellule di controllo.
Figura 2.Figura 2. Generazione di una linea di ESC doxiciclina inducibile per l’espressione simultanea degli otto fattori di trascrizione.(A) Schema che mostra la generazione della linea i8TFs ESC. (B) Western blot che mostra l’espressione proteica delle otto TF dopo il trattamento con doxiciclina (dox) nella linea Empty e nella linea i8TFs ESC. Vedi anche il file supplementare 11.
Figura 3-figure supplemento 5.La differenziazione ematopoietica della linea di ESC i8TFs evidenzia il ruolo degli otto fattori di trascrizione nella generazione di Pre-HSPCs.immagini di microscopia della cultura BL-CFC i8TFs dopo uno, due e tre giorni di cultura BL-CFC in condizioni -dox e +dox.confronto della risposta delle popolazioni eVSM ed Endo all’induzione dei grafici 8TFs.PCA di dati microarray.analisi dei dati microarray.analisi dei dati microarray.analisi delle linee di ESC i1TF.(A) Rappresentativo FACS trame di espressione VE-Cad, cKit e CD41 dopo 3 giorni di cultura BL-CFC delle linee ESC indicate (n = 3). (B) Grafici che mostrano il numero medio di celle rotonde contate per fotogramma (n = 3) in un corso di tempo di 48 ore per le linee ESC indicate. Le barre di errore rappresentano le deviazioni standard. (C) Barra grafica che indica la frequenza delle cellule viventi non apoptotiche per le linee di ESC indicate. Le barre di errore rappresentano le deviazioni standard (n = 3). (D) Barra grafica che indica la frequenza delle celle cicliche per le linee di ESC indicate. Le barre di errore rappresentano le deviazioni standard (n = 3). (E) Barra grafica che mostra il numero di colonie ematopoietiche dopo aver replicato le cellule in un saggio di unità di colonia-formulazione dalle condizioni indicate. Le barre di errore rappresentano la deviazione standard (n = 4). ns, non significativo. ** p-valore<0,01 (test t accoppiato a due code). * p-valore<0,05 (test t accoppiato a due code). Si veda anche la figura 3-figure supplement 1 a 5, file supplementare 1, 4, 5 e 11.La barra della scala corrisponde a 200 μm. Vedi anche il file supplementare 11.(A) Trama FACS dell’espressione VE-Cad e CD41 al primo giorno della cultura BL-CFC. I rettangoli evidenziano le popolazioni di muscolo liscio vascolare arricchito (eVSM) ed endoteliale (Endo). (B) Analisi gerarchica di clustering fatto con sc-q-RT-PCR dati da VE-Cad + CD41- (Endo) e VE-Cad- CD41- (eVSM) popolazioni. I rettangoli rossi indicano le cellule della popolazione eVSM che esprimono i marcatori endoteliali (ad esempio Ramp2, Sox7, Kdr). (C) FACS trame di VE-Cad, cKit e CD41 espressione della muscolatura liscia vascolare arricchita (eVSM) e popolazioni endoteliali (Endo) dalla linea cellulare i8TFs dopo 48 ore in cultura HE. (D) Bar graph che mostra il numero di colonie ematopoietiche dopo aver replicato le cellule in una colonia-formulazione-unità di saggio dalle condizioni indicate. Le barre di errore rappresentano le deviazioni standard (n = 2). Vedere anche il file supplementare 11.(A) Trama PCA che mostra i risultati delle analisi microarray dall’eVSM dopo 24 ore di trattamento dox sulla linea cellulare vuota. Le ellissi indicano i diversi gruppi di campioni. (B) PCA grafico che mostra i risultati delle analisi microarray dalle condizioni indicate dopo 24 ore di trattamento dox sulla linea i8TFs ESC. I numeri indicano il numero di geni espressi in modo significativamente diverso tra le due condizioni (p-valore<0,05). Le ellissi indicano i diversi gruppi di campioni. Vedi anche il file supplementare 11.(A) GO analisi di arricchimento termine dei geni espressi in modo differenziato tra -dox e +dox condizioni per ogni popolazione. (B) diagrammi di Venn che mostrano i geni espressi in modo differenziato per entrambe le popolazioni insieme con i target genici precedentemente identificati da ChIP-seq analisi (Wilson et al., 2010). (C) Elenco dei 26 geni target heptad espressi in modo differenziato all’induzione delle otto TF sia nella popolazione Endo che in quella eVSM. Vedi anche il file supplementare 11.(A) Schema che mostra gli otto costrutti utilizzati per realizzare le linee i1TF ESC. (B) Western blot che mostra l’espressione proteica degli otto fattori di trascrizione dopo il trattamento con doxiciclina per ciascuna delle linee i1TF ESC. (C) rappresentativo FACS trame di VE-Cad, cKit e CD41 espressione dopo tre giorni di cultura BL-CFC delle linee indicate ESC (n = 3). (D) Grafici a barre che indicano la differenza di frequenza tra le condizioni +dox e -dox per le dieci linee cellulari indicate. Le barre di errore rappresentano le deviazioni standard (n = 3). Vedi anche il file supplementare 11.
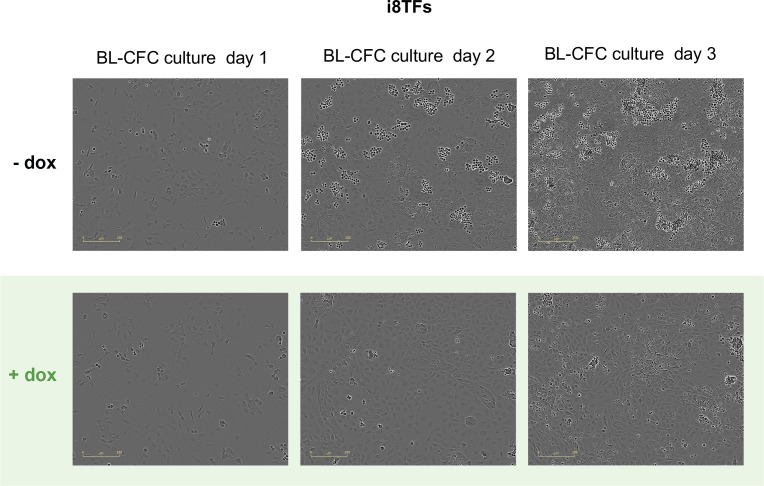
Figura 3-figure supplement 1.Immagini al microscopio della cultura BL-CFC i8TFs dopo uno, due e tre giorni di cultura BL-CFC in condizioni di -dox e +dox.La barra della scala corrisponde a 200 μm. Vedi anche il file supplementare 11.
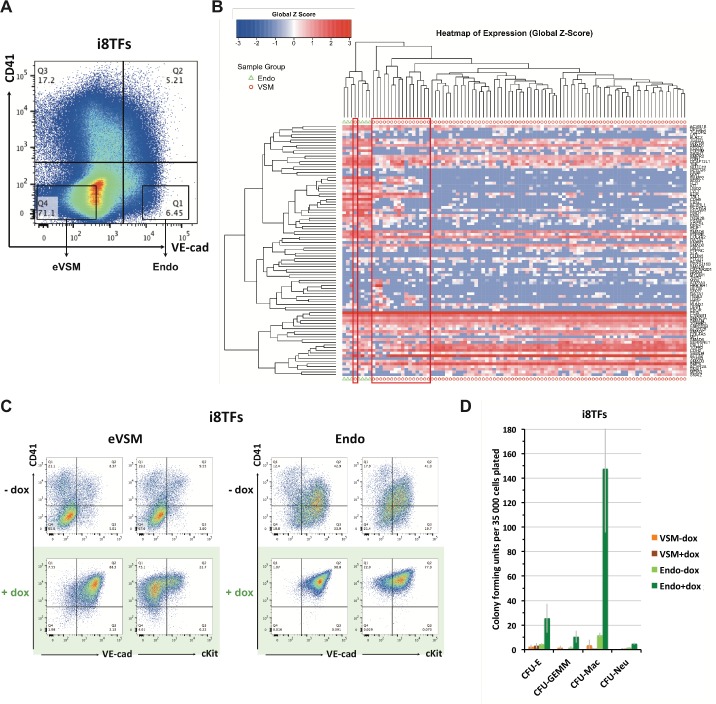
Figura 3-figure supplemento 2.Confronto tra la risposta delle popolazioni eVSM ed Endo all’induzione degli 8TF.(A) Trama FACS di VE-Cad e CD41 espressione al giorno 1 della cultura BL-CFC. I rettangoli evidenziano la muscolatura liscia vascolare arricchita (eVSM) e le popolazioni endoteliali (Endo). (B) Analisi gerarchica di clustering fatto con sc-q-RT-PCR dati da VE-Cad + CD41- (Endo) e VE-Cad- CD41- (eVSM) popolazioni. I rettangoli rossi indicano le cellule della popolazione eVSM che esprimono i marcatori endoteliali (ad esempio Ramp2, Sox7, Kdr). (C) FACS trame di VE-Cad, cKit e CD41 espressione della muscolatura liscia vascolare arricchita (eVSM) e popolazioni endoteliali (Endo) dalla linea cellulare i8TFs dopo 48 ore in cultura HE. (D) Bar graph che mostra il numero di colonie ematopoietiche dopo aver replicato le cellule in una colonia-formulazione-unità di saggio dalle condizioni indicate. Le barre di errore rappresentano le deviazioni standard (n = 2). Vedere anche il file supplementare 11.
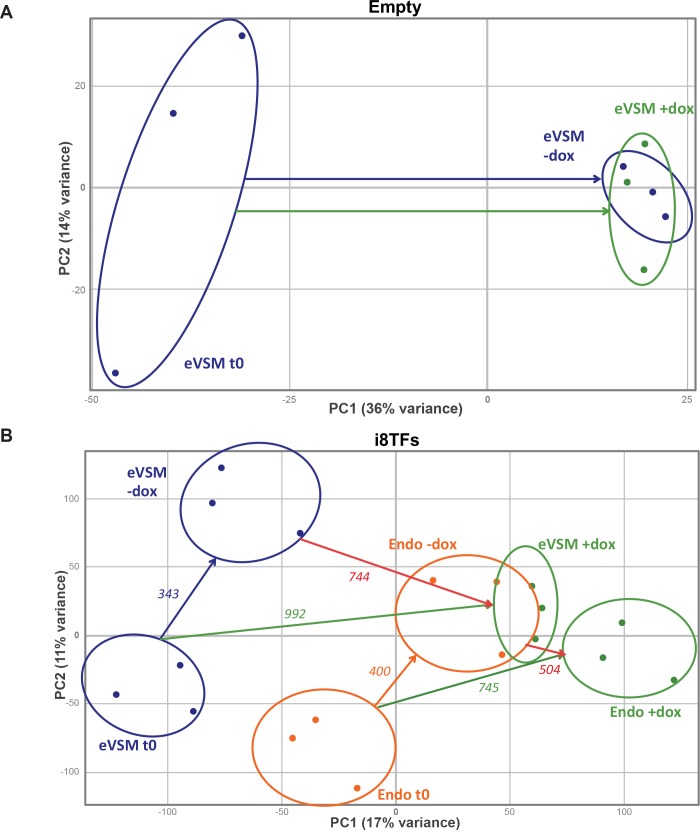
Figura 3-figure supplemento 3.Trame PCA di dati microarray.(A) PCA plot che mostra i risultati delle analisi dei microarray dall’eVSM dopo 24 ore di trattamento dox sulla linea cellulare vuota. Le ellissi indicano i diversi gruppi di campioni. (B) PCA grafico che mostra i risultati delle analisi microarray dalle condizioni indicate dopo 24 ore di trattamento dox sulla linea i8TFs ESC. I numeri indicano il numero di geni espressi in modo significativamente diverso tra le due condizioni (p-valore<0,05). Le ellissi indicano i diversi gruppi di campioni. Vedi anche il file supplementare 11.

Figura 3-figure supplemento 4.Analisi dei dati dei microarray.(A) GO analisi di arricchimento termine dei geni espressi differenzialmente tra le condizioni -dox e +dox per ogni popolazione. (B) diagrammi di Venn che mostrano i geni espressi differenzialmente per entrambe le popolazioni insieme con i target del gene precedentemente identificati da ChIP-seq analisi (Wilson et al., 2010). (C) Elenco dei 26 geni target heptad espressi in modo differenziato all’induzione delle otto TF sia nella popolazione Endo che in quella eVSM. Vedi anche il file supplementare 11.
Figura 3-figure supplement 5.Analisi delle linee i1TF ESC.(A) Schema che mostra gli otto costrutti utilizzati per realizzare le linee i1TF ESC. (B) Western blot che mostra l’espressione proteica degli otto fattori di trascrizione dopo il trattamento con doxiciclina per ciascuna delle linee i1TF ESC. (C) rappresentativo FACS trame di VE-Cad, cKit e CD41 espressione dopo tre giorni di cultura BL-CFC delle linee indicate ESC (n = 3). (D) Grafici a barre che indicano la differenza di frequenza tra le condizioni +dox e -dox per le dieci linee cellulari indicate. Le barre di errore rappresentano le deviazioni standard (n = 3). Vedi anche il file supplementare 11.
Figura 4-figure supplement 2.Figura 4. Figura 4—supplemento alla figura 2. Analisi trascrittomica a cella singola di i8TFs e i1TF linee ESC.Sei trame di dispersione sono visualizzati corrispondenti alle sei popolazioni definite nella Figura 4.Comparison of Empty e i8TFs giorno 3 culture BL-CFC da single-cell RNA sequenziamento.(A) Clustering gerarchico che mostra i risultati sc-q-RT-PCR per 95 geni sulle dieci linee ESC inducibili dopo tre giorni di coltura BL-CFC. I cluster sono stati definiti secondo l’intersezione della linea rossa punteggiata con il dendrogramma nella parte superiore della mappa termica. (B) Grafici a barre che visualizzano il numero di celle in ciascuno dei sei cluster definiti in (A) per le dieci linee di celle. Le stelle indicano differenze significative per le condizioni +dox (vedi file supplementare 6 per i valori p). Vedi anche Figura 4-figure supplements 1 e 2, Supplementary file 1, 6 e 11.Il set di dati è identico a quello della Figura 4. Qui, le frazioni di cellule sono mostrate al posto dei numeri di cellule assoluti. Per la condizione -dox, è stato aggiunto un box plot. Differenze significative tra la condizione -dox e la condizione +dox per ogni linea cellulare e popolazione sono indicate da cerchi rossi come determinato da un modello linearizzato generale (quasi-Poisson). Vedi anche il file supplementare 11.(A) Schema sperimentale seguito per confrontare il Vuoto e i8TFs ESCs con il sequenziamento dell’RNA monocellulare. (B) Tracciato PCA che mostra l’analisi sc-RNA-seq delle linee cellulari Vuoto e i8TFs dopo tre giorni di coltura BL-CFC. Ellisse evidenzia i8TFs + dox. Vedere anche il file supplementare 11.
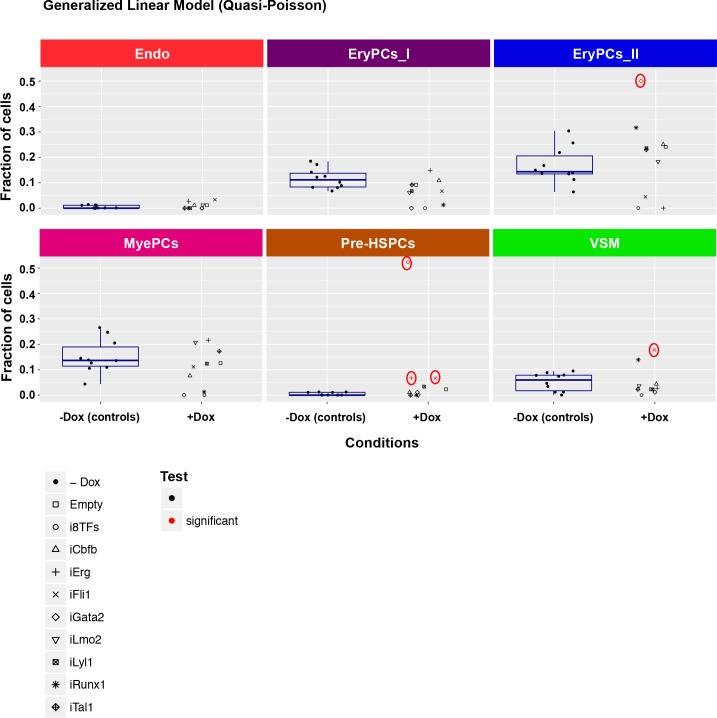
Figura 4-figure supplemento 1.Sono visualizzati sei diagrammi di dispersione corrispondenti alle sei popolazioni definite nella Figura 4.Il set di dati è identico a quello della Figura 4. Qui, le frazioni di cellule sono mostrate al posto dei numeri di cellule assoluti. Per la condizione -dox, è stato aggiunto un box plot. Differenze significative tra la condizione -dox e la condizione +dox per ogni linea cellulare e popolazione sono indicate da cerchi rossi come determinato da un modello linearizzato generale (quasi-Poisson). Vedi anche il file supplementare 11.
Figura 4-figure supplement 2.Figura 4—figura supplemento 2. Confronto tra le colture vuote e i8TFs giorno 3 BL-CFC con sequenziamento RNA monocellulare.(A) Schema sperimentale seguito per confrontare il vuoto e i8TFs ESCs vuoto e i8TFs con il sequenziamento di RNA monocellulare. (B) PCA grafico che mostra sc-RNA-seq analisi delle linee cellulari vuote e i8TFs dopo tre giorni di BL-CFC cultura. Ellisse evidenzia i8TFs + dox. Vedere anche il file supplementare 11.
I Pre-HSPC generati in vitro condividono le somiglianze con i Pre-HSC AGM di tipo I
Per meglio caratterizzare le cellule i8TFs +dox rispetto alle popolazioni embrionali, abbiamo esaminato il modello di espressione delle proteine CD41, CD43 e CD45 insieme al VE-cad. È stato dimostrato che questi marcatori potrebbero definire in modo più preciso diverse fasi intermedie del processo EHT: Pro-HSCs (VE-Cad+ CD41+ CD43+ CD43- CD45- ), Pre-HSCs tipo I (VE-Cad+ CD41+ CD43+ CD45-) e Pre-HSCs tipo II (VE-Cad+ CD41+ CD43+ CD45+) (Rybtsov et al., 2014). I nostri risultati mostrano che circa il 99% delle cellule VE-cad+ nella condizione i8TFs +dox hanno un fenotipo simile ai Pro-HSC (Figura 5A e B). Per verificare questo risultato, abbiamo isolato da E10 AGM le popolazioni Pro-HSCs e Pre-HSCs tipo I ed eseguito sc-q-RT-PCR con il nostro set di 95 geni (Figura 5-figure supplement 1A e B). È interessante notare che la popolazione Pro-HSCs era eterogenea. Anche se tutte le cellule esprimono un alto livello di geni endoteliali, solo la metà esprime geni del sangue come Runx1, Gfi1, Sfpi1 e Myb. Al contrario, i Pre-HSCs di tipo I sono più omogenei. Tutte le cellule esprimono i geni endoteliali, ma ad un livello più basso rispetto ai Pro-HSC. Inoltre, tutte le cellule esprimono i geni ematopoietici (Figura 5-figure supplement 1B). Quando abbiamo analizzato queste cellule insieme con i dati in vivo dalla Figura 1A e le cellule da i8TFs -dox e +dox culture, abbiamo notato che circa la metà dei Pro-HSCs raggruppati con Endo, mentre l’altra parte raggruppati con il Pre-HSPC (Figura 5C). D’altra parte, i Pre-HSCs tipo I raggruppati con i Pre-HSPC. Sorprendentemente, in opposizione al loro fenotipo Pro-HSCs, abbiamo trovato che gli i8TFs +dox erano molto vicini ai Pre-HSCs tipo I (Figura 5C). Ciò è stato confermato quando l’analisi è stata fatta senza le sette TF (Figura 5-figure supplement 1C) e quando i dati qRT-PCR a singola cella delle 1.660 celle delle Figure 1, 4 e 5 sono stati combinati (Figura 5-figure supplement 2).
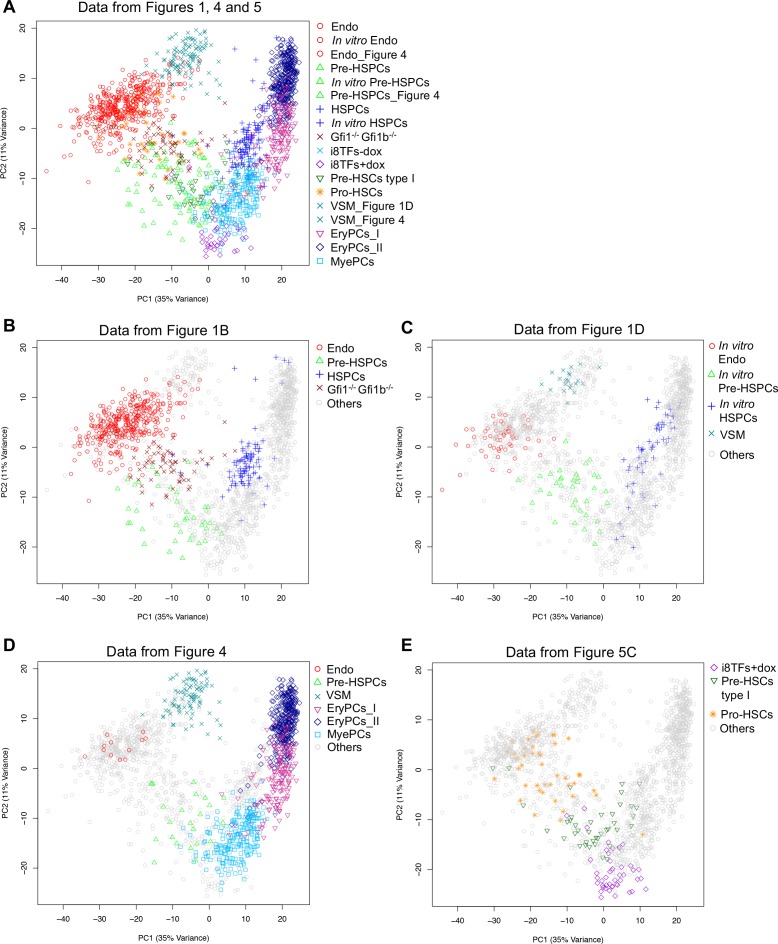
Figura 5-figure supplemento 2.L’induzione di otto TFs dà luogo ad una popolazione di cellule simile a Pre-HSCs tipo I.Hierarchical clustering analysis of Pro-HSCs e Pre-HSCs tipo I.PCA plots confrontando i dati single-cell-q-RT-PCR delle Figure 1, 4 e 5.(A) rappresentativo FACS trame di VE-Cad, CD45, CD43 e CD41 espressione dopo 3 giorni di cultura BL-CFC della linea i8TFs ESC in presenza o assenza di dox (n = 3). (B) Grafici a barre che mostrano la percentuale media delle quattro diverse popolazioni cellulari VE-Cad+ dopo tre giorni di coltura (n = 3). Le barre di errore rappresentano le deviazioni standard. (C) PCA grafico che mostra i risultati sc-q-RT-PCR per 95 geni che combinano le cellule della linea cellulare i8TFs dopo tre giorni di coltura BL-CFC con i risultati di cellule raccolte da wildtype YS e regioni AGM (Figura 1B) e dalla E10 AGM Pro-HSCs e Pre-HSCs tipo I. Si noti che l’asse PC2 è stato invertito. L’ellisse evidenzia i8TFs +dox. Vedere anche la Figura 5-figure supplements 1 e 2, Supplementary file 1, 7, 8 e 11.(A) Trame FACS di espressione VE-Cad, CD45, CD43 e CD41 nella regione AGM a E10 (31-32 coppie di somiti). Sono state isolate singole cellule di popolazioni Pro-HSCs (VE-Cad+ CD41+ CD41+ CD45- CD43-) e Pre-HSCs tipo I (VE-Cad+ CD41+ CD45- CD43+). P9 evidenzia le cellule CD41- CD43- CD43-. (B) Analisi gerarchica del clustering fatta con i dati sc-q-RT-PCR da (A). Endoteliale (Cdh5, Sox7, Kdr e Pcdh12) e geni ematopoietici (Runx1, Gfi1, Sfpi1, Itgam e Sla) sono stati marcati in blu e rosso, rispettivamente. (C) PCA trama PCA che mostra i risultati sc-q-RT-PCR delle stesse cellule come in Figura 5C, ma dopo aver rimosso i geni Runx1, Gata2, Tal1, Fli1, Lyl1, Erg e Lmo2 dai dati di espressione. Si noti che l’asse PC2 è stato invertito. L’ellisse e la freccia rossa evidenziano le cellule i8TFs +dox. Vedere anche il file supplementare 11.(A) Grafico PCA che mostra i risultati qPCR a cella singola di tutte le celle delle Figure 1, 4 e 5 (un totale di 1.660 celle). (B) Stesso grafico PCA della (A) ma con l’evidenziazione delle popolazioni della Figura 1B. (C) Stesso grafico PCA come (A) ma evidenziando le popolazioni dalla Figura 1D. (D) Stesso grafico PCA della (A), ma evidenziando alcune delle popolazioni della Figura 4. (E) Stesso grafico PCA della (A), ma evidenziando alcune delle popolazioni della Figura 5C. Vedere anche il file supplementare 11.
In conclusione, anche se le cellule i8TFs +dox avevano un fenotipo simile a quello delle Pro-HSC, il loro profilo trascrizionale era più simile a quello delle Pre-HSC di tipo I.
Figura 5-figure supplemento 2.L’induzione di otto TFs dà luogo ad una popolazione di cellule simile a Pre-HSCs tipo I.Hierarchical clustering analysis di Pro-HSCs e Pre-HSCs tipo I.PCA plot confrontando i dati single-cell-q-RT-PCR delle Figure 1, 4 e 5.(A) Rappresentativo FACS trame di espressione VE-Cad, CD45, CD43 e CD41 dopo 3 giorni di cultura BL-CFC della linea ESC i8TFs in presenza o meno di dox (n = 3). (B) Grafici a barre che mostrano la percentuale media delle quattro diverse popolazioni cellulari VE-Cad+ dopo tre giorni di coltura (n = 3). Le barre di errore rappresentano le deviazioni standard. (C) PCA grafico che mostra i risultati sc-q-RT-PCR per 95 geni che combinano le cellule della linea cellulare i8TFs dopo tre giorni di coltura BL-CFC con i risultati di cellule raccolte da wildtype YS e regioni AGM (Figura 1B) e dalla E10 AGM Pro-HSCs e Pre-HSCs tipo I. Si noti che l’asse PC2 è stato invertito. L’ellisse evidenzia i8TFs +dox. Vedere anche la Figura 5-figure supplements 1 e 2, Supplementary file 1, 7, 8 e 11.(A) Trame FACS di espressione VE-Cad, CD45, CD43 e CD41 nella regione AGM a E10 (31-32 coppie di somiti). Sono state isolate singole celle di popolazioni Pro-HSC (VE-Cad+ CD41+ CD41+ CD45- CD43-) e Pre-HSC di tipo I (VE-Cad+ CD41+ CD45- CD43+). P9 evidenzia le cellule CD41- CD43- CD43-. (B) Analisi gerarchica del clustering fatta con i dati sc-q-RT-PCR da (A). Endoteliale (Cdh5, Sox7, Kdr e Pcdh12) e geni ematopoietici (Runx1, Gfi1, Sfpi1, Itgam e Sla) sono stati marcati in blu e rosso, rispettivamente. (C) PCA trama PCA che mostra i risultati sc-q-RT-PCR delle stesse cellule come in Figura 5C, ma dopo aver rimosso i geni Runx1, Gata2, Tal1, Fli1, Lyl1, Erg e Lmo2 dai dati di espressione. Si noti che l’asse PC2 è stato invertito. L’ellisse e la freccia rossa evidenziano le cellule i8TFs +dox. Vedere anche il file supplementare 11.(A) Tracciato PCA che mostra i risultati qPCR a cella singola di tutte le celle delle Figure 1, 4 e 5 (un totale di 1.660 celle). (B) Stesso tracciato PCA della (A) ma evidenziando le popolazioni della Figura 1B. (C) Stesso grafico PCA come (A) ma evidenziando le popolazioni dalla Figura 1D. (D) Stesso grafico PCA della (A), ma evidenziando alcune delle popolazioni della Figura 4. (E) Stesso grafico PCA della (A), ma evidenziando alcune delle popolazioni della Figura 5C. Vedere anche il file supplementare 11.
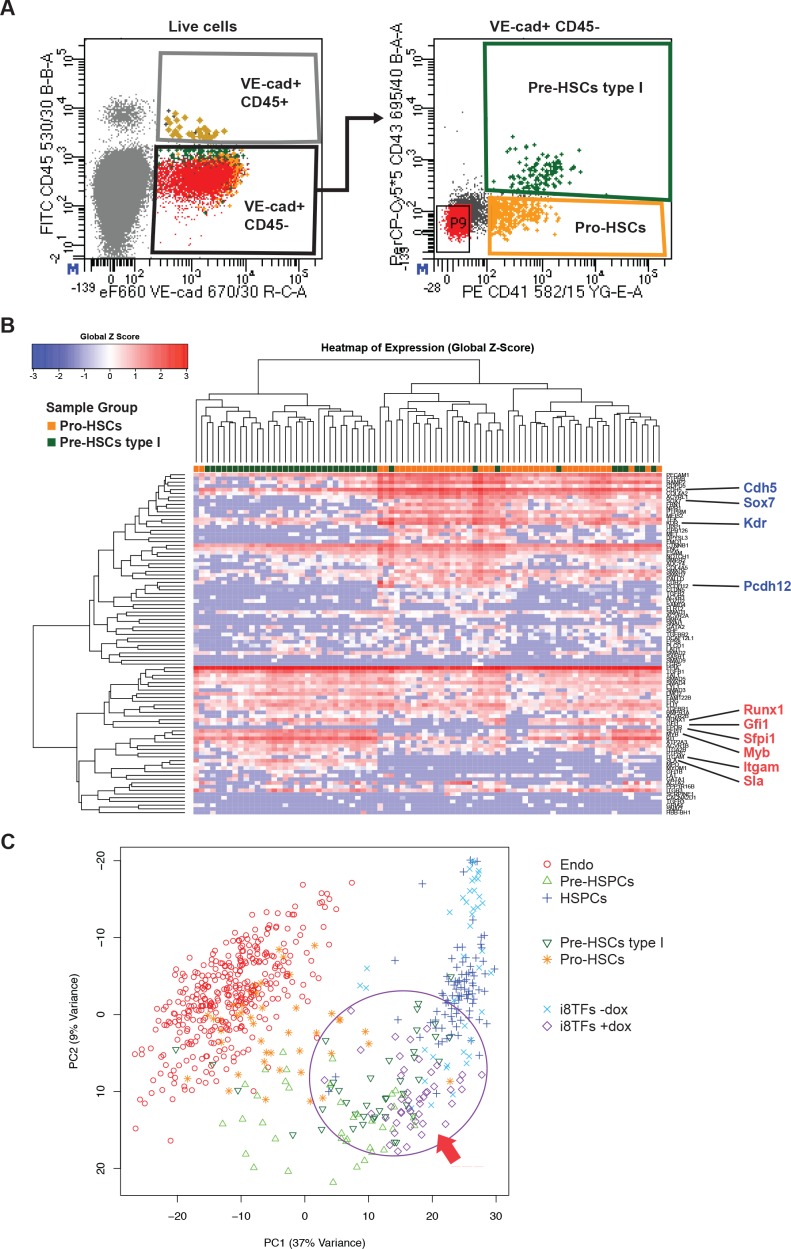
Figura 5-figure supplemento 1.Analisi gerarchica del clustering di Pro-HSC e Pre-HSC di tipo I.(A) FACS trame FACS di VE-Cad, CD45, CD43 e CD41 espressione nella regione AGM a E10 (31-32 coppie di somiti). Singole cellule da Pro-HSCs (VE-Cad + CD41 + CD45 + CD45- CD43-) e Pre-HSCs tipo I (VE-Cad + CD41 + CD45- CD43+) popolazioni sono stati isolati. P9 evidenzia le cellule CD41- CD43- CD43-. (B) Analisi gerarchica del clustering fatta con i dati sc-q-RT-PCR da (A). Endoteliale (Cdh5, Sox7, Kdr e Pcdh12) e geni ematopoietici (Runx1, Gfi1, Sfpi1, Itgam e Sla) sono stati marcati in blu e rosso, rispettivamente. (C) PCA trama PCA che mostra i risultati sc-q-RT-PCR delle stesse cellule come in Figura 5C, ma dopo aver rimosso i geni Runx1, Gata2, Tal1, Fli1, Lyl1, Erg e Lmo2 dai dati di espressione. Si noti che l’asse PC2 è stato invertito. L’ellisse e la freccia rossa evidenziano le cellule i8TFs +dox. Vedere anche il file supplementare 11.
Figura 5-figure supplement 2.Figura 5—supplemento di figura 2. Grafici PCA che confrontano i dati di single-cell-q-RT-PCR delle Figure 1, 4 e 5.(A) Grafico PCA che mostra i risultati qPCR a cella singola di tutte le celle delle Figure 1, 4 e 5 (un totale di 1.660 celle). (B) Stesso grafico PCA come (A) ma evidenziando le popolazioni della Figura 1B. (C) Stesso grafico PCA come (A) ma evidenziando le popolazioni dalla Figura 1D. (D) Stesso grafico PCA della (A), ma evidenziando alcune delle popolazioni della Figura 4. (E) Stesso grafico PCA della (A), ma evidenziando alcune delle popolazioni della Figura 5C. Vedere anche il file supplementare 11.
Identificazione della rete di regolazione genica a partire dai dati di sequenziamento dell’RNA monocellulare
Per determinare come l’induzione delle 8TF da trattamento dox abbia influenzato i GRN con l’utilizzo di monocellule-RNA-seq, abbiamo deciso di concentrarci sulle cellule VSM perché, a differenza della popolazione Endo, non sono naturalmente sottoposte a EHT (Figura 3-figure supplement 2C). Infatti, tra Endo +dox e Endo -dox, c’è stato solo un raddoppio della popolazione VE-Cad+ CD41+. D’altra parte, c’è stato un aumento di dieci volte della popolazione VE-Cad+ CD41+ tra eVSM +dox ed eVSM -dox (Figura 3-figure supplement 2C). Inoltre, questa popolazione mostra il maggior contrasto di espressione genica nei nostri dati microarray al trattamento con dox, facilitando l’identificazione dei GRN controllati dalle otto TF (Figura 3-figure supplement 3B). Come negli esperimenti microarray, le cellule VE-Cad- CD41- sono state ordinate al giorno 1 della coltura BL-CFC e coltivate in assenza o presenza di dox per 24 ore. Successivamente, le cellule delle condizioni -dox e +dox sono state messe in pool con un rapporto 1:1 e messe su un chip Fluidigm C1. Il sistema Fluidigm ha permesso una sensibilità più elevata rispetto al sistema Wafergen, ma non ha permesso l’elaborazione di diverse condizioni e di un gran numero di cellule allo stesso tempo. Il progetto sperimentale in cui le celle venivano mescolate e caricate su un unico chip minimizzava la variabilità tecnica che sarebbe emersa invariabilmente dall’uso di due chip separati (Figura 6-figure supplement 1A). Abbiamo ottenuto due repliche biologiche, e da ogni chip 96 cellule (192 cellule in totale) sono state isolate e sequenziate insieme. Per determinare quali cellule sono state esposte al dox abbiamo usato l’espressione del transgene (Figura 6-figure supplement 1B). Un PCA dei dati di sequenziamento ha mostrato che le cellule che esprimono alti livelli di trascrizioni 8TFs si sono raggruppate separatamente da quelle che esprimono bassi livelli di queste trascrizioni (Figura 6-figure supplement 1C).
Per identificare le relazioni di regolazione per le otto TF, abbiamo eseguito un’analisi di rete utilizzando la misura di correlazione generalizzata della distanza, dcor statistic (Székely et al., 2007), insieme alla sua versione condizionale pdcor (Székely e Rizzo, 2014). Questa misura ha il vantaggio di poter rilevare sia le relazioni lineari che non lineari tra il trascrittoma e i geni del seme. Tuttavia, quando solo le otto TF sono state utilizzate come semi, la rete risultante era molto piccola, con solo cinque delle otto TF rappresentate (non mostrate). Così, abbiamo ampliato l’insieme dei geni del seme per includere Gata1, Gfi1b, Spi1 (o Pu.1), Ldb1 e Cbfa2t3, che sono noti per giocare ruoli importanti nell’emopoiesi. Utilizzando questo insieme esteso di geni, abbiamo ottenuto una rete che illustra come le otto TF interagiscono indirettamente attraverso un percorso di regolazione comune (Ferreira et al., 2005; Goardon et al., 2006; Imperato et al., 2015; Lancrin et al., 2012; Mylona et al., 2013). La direzionalità delle relazioni, correlazione positiva o correlazione negativa, è stata definita utilizzando il segno di un t-test a due code (le coppie di geni con p>0,05 sono state assegnate ad “altro”). Infine, le interazioni tra i geni del seme sono state identificate condizionando i “bersagli” di ogni gene del seme sull’espressione di ogni altro gene del seme. Un’interazione è stata dedotta se le relazioni tra entrambi i geni del seme e un particolare gene ‘target’ aumentavano di forza quando si condizionava l’altro gene del seme.
Quando abbiamo visualizzato la rete risultante, abbiamo tracciato solo i semi che hanno interagito con almeno un altro seme (linee nere) e il gene bersaglio di almeno due interazioni di semi (Figura 6A). Coerentemente con le evidenze della letteratura, il nostro metodo di ricostruzione della rete riporta una stretta interazione tra i fattori del seme, a parte Lbd1, sono tutti inclusi nella rete risultante. Inoltre, osserviamo una rete centrale composta da Erg, Lyl1, Lmo2, Tal1, Gfi1b, e Gata1 con Fli1 e Runx1 alle estremità opposte della periferia. Molti dei geni che sono stati identificati come bersagli dei geni del seme erano anche noti dalla letteratura; per esempio, Gpr56, che è stato upregulated nel nostro esperimento microarray dopo il trattamento dox (Figura 3-figure supplemento 4C), e Nfe2 (Wilson et al., 2010; Woon Kim et al., 2011). Inoltre, molti dei target erano stati precedentemente identificati come espressi in modo differenziato dopo il trattamento dox (Figura 3-figure supplement 4C, file supplementare 5).
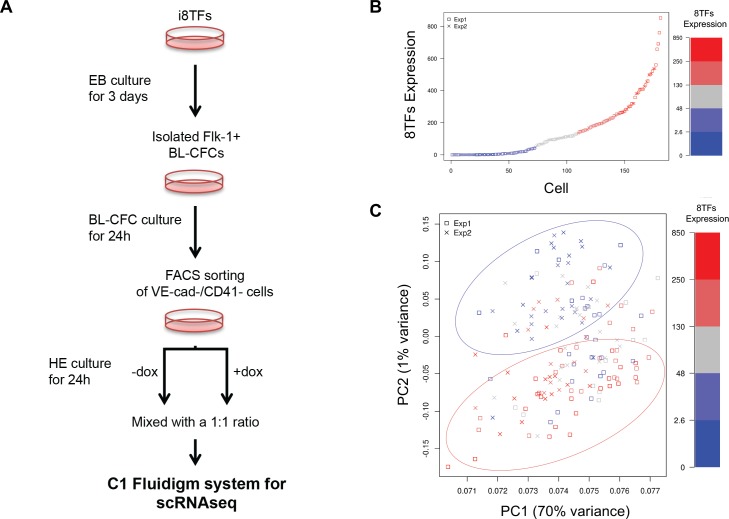
Figura 6-figure supplemento 1.L’analisi trascrittomica a cella singola rivela le reti trascrizionali coinvolte nella generazione di Pre-HSPCs.l’analisi del trascrittoma a cella singola a seguito di sovra-espressione degli 8TFs in celle eVSM.(A) Rete costruita da correlazioni geniche. Gpr56 è evidenziato con un asterisco rosso. (B) Valori di centralità per tutti i geni contenenti un picco eptad entro 1 kb del TSS che sono significativamente al di sopra della mediana. Gpr56 è evidenziato con un asterisco rosso. (C) La parte superiore mostra i dati ChIP-Seq per le sette TF dello studio di Wilson et al. (2010) nel locus Gpr56. L’asse y mostra le letture visualizzate come diagrammi di densità, che sono stati generati da Wilson et al. (2010) e visualizzati utilizzando il software Integrated Genome Browser. I due potenziatori putativi sono evidenziati con rettangoli rossi. La parte inferiore mostra i risultati del test trascrizionale del reporter per due potenziali potenziatori eptadici in locus Gpr56 utilizzando i8TFs ESCs trattati per 24 ore con doxiciciclina. Le barre di errore rappresentano le deviazioni standard (n = 3). Vedere anche la Figura 6-figure supplement 1 e il file supplementare 11.(A) Flusso di lavoro sperimentale utilizzato per differenziare le cellule in vitro eVSM dalla linea i8TFs ESC e per analizzarle da sc-RNA-seq dopo 24 ore di trattamento. (B) Trama di dispersione che mostra l’espressione del costrutto 8TFs tra tutte le cellule dei due replicati. Il rosso indica l’espressione alta, il blu l’espressione bassa e il grigio l’espressione indefinita. L’asse y mostra l’espressione SF-normalizzata colorata da quantile. (C) Tracciato PCA dei risultati sc-RNA-seq. I codici colore sono gli stessi di quelli in (B). Vedere anche il file supplementare 11.
Incoraggiati dai risultati dell’analisi dcor, abbiamo effettuato un’analisi a livello genomico. Poiché Dcor è un metodo di correlazione lenta, era computazionalmente impossibile calcolare la rete globale con esso. Per questo motivo abbiamo utilizzato la correlazione Spearman, un metodo di correlazione molto veloce. Abbiamo combinato i due set di dati e abbiamo mantenuto tutte le correlazioni in cui il valore assoluto della correlazione media era maggiore di 0,25 e in cui il segno era coerente attraverso le normalizzazioni Remove Unwanted Variation (RUV) e Size Factors (SF). Ne è risultata una rete contenente 7.262 geni e 163.474 bordi di cui 123.394 positivi e 40.080 negativi. È importante notare che 11 dei 13 geni dei semi utilizzati nell’analisi precedente sono stati trovati in questa grande rete: Cbfa2t3, Fli1, Gata2, Lmo2, Erg, Tal1, Gfi1b, Gata1, Runx1, Spi1 e Lyl1. Inoltre, tra i 145 geni che contengono picchi di eptad entro 1 kb dal loro TSS, 26 sono stati trovati nella rete, anche se questo non è stato significativamente più di quanto ci si aspettasse per caso (p>0.1, Fisher-exact test).
Per identificare i geni più importanti nella rete, abbiamo calcolato tre diverse misure di centralità (grado, betweenness e eigen) (Figura 6B). Il numero di geni tra gli 11 geni del seme con valori superiori alla mediana era significativamente più alto di quello previsto per tutte e tre le misure (grado p-valore=0,0005, betweenness p-valore=0,006, eigen p-valore=0,03). È interessante notare che, per i 26 obiettivi di eptad, solo il betweenness presentava un numero di obiettivi superiore alla mediana superiore a quello che ci si aspettava per caso (p-valore p=0,04). Questo evidenzia il ruolo centrale atteso dei target di heptad per la loro posizione “nel mezzo” dei principali attori della rete.
Poiché il Grp56 è apparso come uno dei principali candidati nelle nostre due analisi di rete, lo abbiamo scelto per ulteriori esperimenti (Figura 6C). Il Gpr56 ha dimostrato di essere espresso nei cluster ematopoietici dell’aorta (Solaimani Kartalaei et al., 2015) e di essere upregolato lungo l’EHT (Goode et al., 2016). I dati di ChIP-seq (Wilson et al., 2010) mostrano che ci sono due elementi regolatori Gpr56 legati da sette delle otto TF nella linea cellulare HPC7 (Figura 6C). Abbiamo selezionato questi potenziatori putativi e li abbiamo clonati in un plasmide a monte del gene Firefly luciferase reporter. Un test gene reporter nella linea i8TFs ESC ha dimostrato che l’induzione delle otto TF ha portato ad un forte aumento dell’espressione del gene della luciferasi con i due stimolatori Grp56 (Figura 6C). In conclusione, siamo stati in grado di dedurre GRNs sulla base dei dati sc-RNA-seq. Abbiamo identificato gli attori chiave interessati dall’induzione delle otto TF, tra cui il Gpr56 era un importante gene target.
Figura 6-figure supplemento 1.L’analisi trascrittomica a cella singola rivela le reti trascrizionali coinvolte nella generazione di Pre-HSPCs.l’analisi del trascrittoma a cella singola a seguito di sovra-espressione degli 8TFs in celle eVSM.(A) Rete costruita da correlazioni geniche. Gpr56 è evidenziato con un asterisco rosso. (B) Valori di centralità per tutti i geni contenenti un picco eptad entro 1 kb del TSS che sono significativamente al di sopra della mediana. Gpr56 è evidenziato con un asterisco rosso. (C) La parte superiore mostra i dati ChIP-Seq per le sette TF dello studio di Wilson et al. (2010) nel locus Gpr56. L’asse y mostra le letture visualizzate come diagrammi di densità, che sono stati generati da Wilson et al. (2010) e visualizzati utilizzando il software Integrated Genome Browser. I due potenziatori putativi sono evidenziati con rettangoli rossi. La parte inferiore mostra i risultati del test trascrizionale del reporter per due potenziali potenziatori eptadici in locus Gpr56 utilizzando i8TFs ESCs trattati per 24 ore con doxiciciclina. Le barre di errore rappresentano le deviazioni standard (n = 3). Vedere anche la Figura 6-figure supplement 1 e il file supplementare 11.(A) Flusso di lavoro sperimentale utilizzato per differenziare le cellule in vitro eVSM dalla linea i8TFs ESC e per analizzarle da sc-RNA-seq dopo 24 ore di trattamento. (B) Trama di dispersione che mostra l’espressione del costrutto 8TFs tra tutte le cellule dei due replicati. Il rosso indica l’espressione alta, il blu l’espressione bassa e il grigio l’espressione indefinita. L’asse y mostra l’espressione SF-normalizzata colorata da quantile. (C) Tracciato PCA dei risultati sc-RNA-seq. I codici colore sono gli stessi di quelli in (B). Vedere anche il file supplementare 11.
Figura 6-figure supplemento 1.Analisi del trascrittoma a cella singola a seguito di sovra-espressione degli 8TF in celle eVSM.(A) Flusso di lavoro sperimentale utilizzato per differenziare le cellule in vitro eVSM dalla linea i8TFs ESC e per analizzarli da sc-RNA-seq dopo 24 ore di trattamento. (B) Trama di dispersione che mostra l’espressione del costrutto 8TFs tra tutte le cellule dei due replicati. Il rosso indica l’espressione alta, il blu l’espressione bassa e il grigio l’espressione indefinita. L’asse y mostra l’espressione SF-normalizzata colorata da quantile. (C) Tracciato PCA dei risultati sc-RNA-seq. I codici colore sono gli stessi di quelli in (B). Vedere anche il file supplementare 11.
L’analisi della rete di regolazione del gene rivela effetti contrastanti di Runx1 e Fli1
L’espressione simultanea degli otto fattori ha portato alla formazione di una popolazione simile ai Pre-HSPC attraverso la loro azione specifica sui GRN. Non era chiaro, tuttavia, perché le cellule fossero bloccate in questo stadio intermedio normalmente ‘transitorio’ dell’EHT. Per svelare questo, abbiamo applicato la nostra analisi di rete ad un set di dati sc-RNA-seq di cellule endoteliali e HSPC, prima e dopo l’EHT, rispettivamente (Pereira et al., 2016). Abbiamo generato una rete utilizzando gli stessi semi di prima (Figura 6A), e la caratteristica più sorprendente della nuova rete è che Fli1 e Runx1 condividono diversi obiettivi, ma con direzioni opposte di correlazione (Figura 7A). Un’analisi della rete a livello genomico basata sulle correlazioni di Spearman ha mostrato uno schema simile in cui 210 geni sono stati correlati sia a Fli1 che a Runx1 ma in direzioni opposte (Figura 7B e file supplementare 9). Inoltre, l’analisi GO ha rivelato che i geni la cui espressione è positivamente correlata con Fli1 e negativamente correlata con Runx1 (rettangolo giallo) sono arricchiti per i termini sviluppo vascolare, angiogenesi e migrazione cellulare (Figura 7-figure supplemento 1A). D’altra parte, i geni la cui espressione è positivamente correlata con quella di Runx1 e negativamente correlata con quella di Fli1 (rettangolo verde) sono associati al processo del sistema immunitario termine GO (Figura 7-figure supplement 1A). Quindi, Runx1 è più associato con i geni ematopoietici, mentre Fli1 è più associato con i geni vascolari. È interessante notare che i dati di ChIP-seq di Wilson et al. (2010) hanno mostrato che 77 dei 210 sono bersagli diretti sia di Runx1 che di Fli1 (Figura 7-figure supplement 1B e Supplementary file 9). La correlazione osservata opposta potrebbe essere dovuta al fatto che la maggior parte delle cellule endoteliali esprimono solo Fli1 e non Runx1, mentre HSPC co-esprimono Runx1 e Fli1. Vi è infatti un chiaro spostamento nella relativa espressione di Fli1 rispetto a quella di Runx1 tra i due stadi cellulari (Figura 7-figure supplemento 1C). In conclusione, la nostra analisi della rete ha rivelato che Fli1 e Runx1 sono collegati a 210 geni ma con correlazioni opposte, che potrebbero essere legate ai contrastanti schemi di espressione di Fli1 e Runx1 e/o alle opposte proprietà funzionali di queste TF.
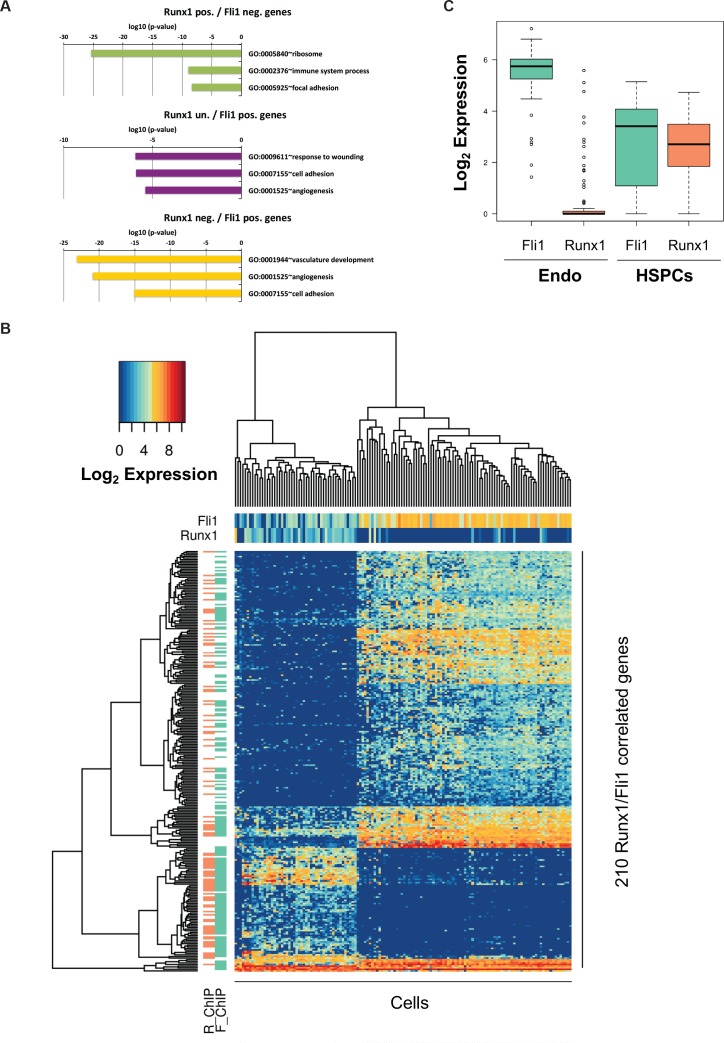
Figura 7-figure supplemento 1.L’analisi del trascrittoma a cella singola suggerisce che Runx1 e Fli1 hanno funzioni opposte durante l’analisi EHT.Bioinformatic del set di dati di Pereira.(A) Rete costruita da correlazioni geniche trovate nel dataset di Peirera (Pereira et al., 2016). (B) Heatmap che mostra le correlazioni Spearman tra i 210 geni trovati per essere correlati a Runx1 e Fli1. L’analisi gerarchica del clustering ci ha dato tre gruppi di geni evidenziati da tre rettangoli di colore diverso. Per ogni gruppo di geni è indicato il termine GO in alto. Vedi anche la figura 7-figure supplement 1 e il file supplementare 9.(A) GO termine arricchimento analisi di arricchimento dei geni correlati con Fli1 e Runx1. Il codice colore corrisponde ai gruppi di geni della Figura 7B. (B) Heatmap che mostra clustering gerarchico dei 210 geni correlati con Fli1 e Runx1. In cima alla heatmap, il livello di espressione di Fli1 e Runx1 è indicato (il gradiente di colore indica il log2 dell’espressione genica). A sinistra della heatmap, è indicata l’occupazione del DNA di Fli1 (F_ChIP) o Runx1 (R_ChIP) sui 210 geni (l’arancione indica i geni target di Runx1 e il verde dei geni target di Fli1). (C) Box plot che mostra l’espressione di Fli1 (verde) e Runx1 (arancione) nelle popolazioni Endo e HSPC isolate da Pereira et al. (2016)). I bordi superiore e inferiore del box corrispondono al primo e al terzo quartile. La linea nera all’interno della scatola rappresenta la mediana. Le linee baffute in alto e in basso segnano rispettivamente i valori massimo e minimo del set di dati.
Figura 7-figure supplement 1.L’analisi del trascrittoma a cella singola suggerisce che Runx1 e Fli1 hanno funzioni opposte durante l’analisi bioinformatica del set di dati di Pereira.(A) Rete costruita da correlazioni geniche trovate nel dataset di Peirera (Pereira et al., 2016). (B) Heatmap che mostra le correlazioni Spearman tra i 210 geni trovati per essere correlati a Runx1 e Fli1. L’analisi gerarchica del clustering ci ha dato tre gruppi di geni evidenziati da tre rettangoli di colore diverso. Per ogni gruppo di geni è indicato il termine GO in alto. Vedi anche la figura 7-figure supplement 1 e il file supplementare 9.(A) GO analisi di arricchimento termine dei geni correlati con Fli1 e Runx1. Il codice colore corrisponde ai cluster di geni della Figura 7B. (B) Heatmap visualizzazione Heatmap clustering gerarchico dei 210 geni correlati con Fli1 e Runx1. In cima alla heatmap, il livello di espressione di Fli1 e Runx1 è indicato (il gradiente di colore indica il log2 dell’espressione genica). A sinistra della heatmap, è indicata l’occupazione del DNA di Fli1 (F_ChIP) o Runx1 (R_ChIP) sui 210 geni (l’arancione indica i geni target di Runx1 e il verde dei geni target di Fli1). (C) Box plot che mostra l’espressione di Fli1 (verde) e Runx1 (arancione) nelle popolazioni Endo e HSPC isolate da Pereira et al. (2016)). I bordi superiore e inferiore del box corrispondono al primo e al terzo quartile. La linea nera all’interno della scatola rappresenta la mediana. Le linee baffute in alto e in basso segnano rispettivamente i valori massimo e minimo del set di dati.
Figura 7-figure supplement 1.Analisi bioinformatica del set di dati di Pereira.(A) GO analisi di arricchimento termine dei geni correlati con Fli1 e Runx1. Il codice colore corrisponde ai gruppi di geni della Figura 7B. (B) Heatmap visualizzazione Heatmap clustering gerarchico dei 210 geni correlati con Fli1 e Runx1. In cima alla heatmap, il livello di espressione di Fli1 e Runx1 è indicato (il gradiente di colore indica il log2 dell’espressione genica). A sinistra della heatmap, è indicata l’occupazione del DNA di Fli1 (F_ChIP) o Runx1 (R_ChIP) sui 210 geni (l’arancione indica i geni target di Runx1 e il verde dei geni target di Fli1). (C) Box plot che mostra l’espressione di Fli1 (verde) e Runx1 (arancione) nelle popolazioni Endo e HSPC isolate da Pereira et al. (2016)). I bordi superiore e inferiore del box corrispondono al primo e al terzo quartile. La linea nera all’interno della scatola rappresenta la mediana. Le linee baffute in alto e in basso segnano rispettivamente i valori massimo e minimo del set di dati.
L’analisi del guadagno di funzione rivela un equilibrio funzionale tra Gata2/Runx1 e Erg/Fli1 TFs durante la transizione da endoteliale a ematopoietico
Per scoprire se Runx1 e Fli1 hanno effetti funzionali opposti sulle decisioni sul destino delle cellule che si verificano durante l’EHT, abbiamo riesaminato la nostra analisi del guadagno di funzione i1TF (Figura 3-figure supplement 5 e Figura 4B). In base a questi risultati, abbiamo identificato due gruppi principali di TF. Il primo include Fli1 ed Erg mentre il secondo include Runx1 e Gata2. Infatti, in condizione +dox, iFli1 e iErg hanno entrambi mostrato un aumento della popolazione Endo così come della popolazione Pre-HSPCs, anche se ad un livello molto più basso che con i8TFs (Figura 4B). Al contrario, le celle iRunx1 e iGata2 hanno mostrato un aumento della frequenza nella popolazione EryPCs_II e una diminuzione della popolazione MyePCs nella condizione +dox (Figura 4B). Questo risultato è coerente con l’ipotesi che alcune delle otto TF abbiano effetti opposti.
Abbiamo poi valutato le conseguenze funzionali della rimozione di Runx1/Gata2 o Erg/Fli1 dal costrutto 8TFs. Abbiamo ipotizzato che la rimozione di Runx1 e Gata2 aumenterebbe il destino delle cellule endoteliali, mentre la rimozione di Erg e Fli1 aumenterebbe il destino delle cellule ematopoietiche. Di conseguenza abbiamo generato due linee di ESC aggiuntive (Figura 8A). La prima, i6TFs, contiene 6TFs che si trovano anche in i8TFs ma è senza Erg e Fli1. La seconda, i5TFs, manca di Runx1, Gata2 così come Cbfb, partner cruciale di Runx1. Abbiamo eseguito la differenziazione ESC di queste linee cellulari nello stesso modo di prima e abbiamo analizzato le conseguenze della sovraespressione di questi due gruppi di TF. È interessante notare che la sovraespressione delle cinque TF ha in parte ricapitolato l’effetto della sovraespressione delle otto TF e ha portato ad un chiaro aumento della frequenza di VE-cad+ CD41+ (Figura 8B). Notevolmente, anche la frequenza della popolazione di VE-Cad+ CD41- è aumentata significativamente, in netto contrasto con quanto osservato con le otto TF (Figura 3A e Figura 8B). Coerentemente con questo risultato, c’è stata una netta diminuzione del numero di cellule rotonde formatesi durante il saggio BL-CFC (Figura 8C). Al contrario, la sovraespressione delle sei TF ha avuto solo un impatto minore sul modello di espressione di VE-cad e CD41 (Figura 8B). Tuttavia, vi è stato un chiaro aumento delle cellule ematopoietiche rotonde come mostrato dall’analisi delle immagini, suggerendo un miglioramento della formazione delle cellule del sangue (Figura 8C). L’impatto osservato dell’induzione sul numero di cellule rotonde è stato confermato in particolare con cellule endoteliali isolate al giorno 1 BL-CFC e coltivate per due giorni in presenza di doxiciciclina in mezzo HE (Figura 8D e Figura 8-figure supplement 1A). Inoltre, sc-q-RT-PCR eseguita sul i5TFs e i6TFs confermato questa dualità (Figura 8E e Figura 8-figure supplement 1B). L’i5TFs ha mostrato una diminuzione dei progenitori ematopoietici EryPCs_I associata ad un aumento della popolazione Endo e Pre-HSPC, mentre l’i6TFs ha mostrato un aumento della popolazione EryPCs_II di progenitori ematopoietici (Figura 8E e Supplemento 10).
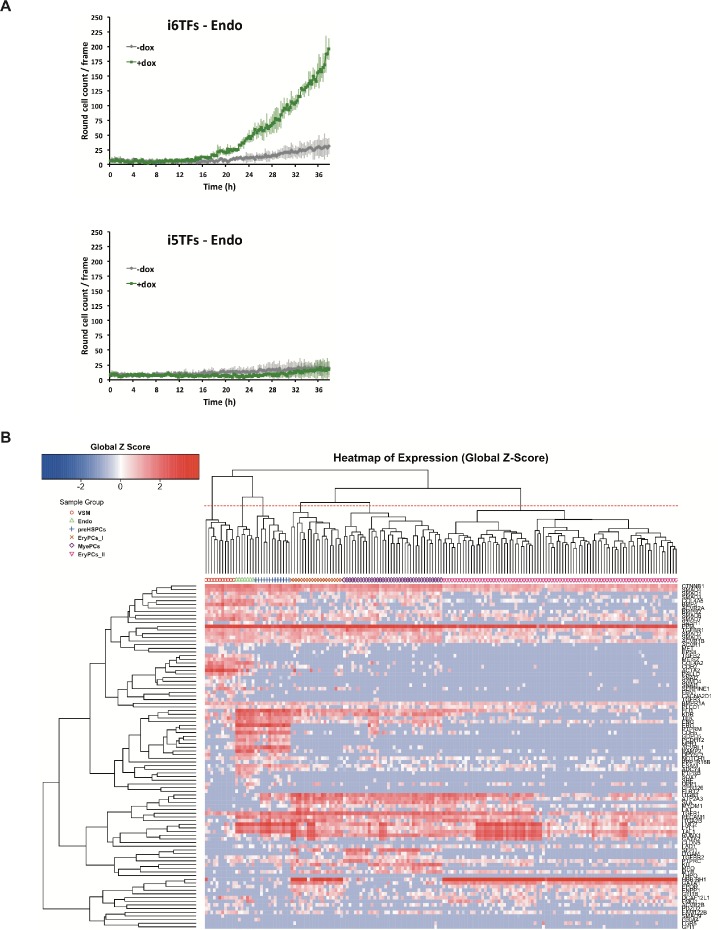
Figura 8-figure supplemento 1.I gruppi di fattori di trascrizione Erg/Fli1 e Runx1/Gata2 hanno funzioni opposte durante l’EHT.Studio del ruolo dei gruppi di fattori di trascrizione Erg/Fli1 e Runx1/Gata2 in EHT.(A) Schema che mostra i costrutti i5TFs e i6TFs usati per generare le due linee di ESC inducibili mancanti Runx1, Gata2 e Cbfb, o Fli1 e Erg (rispettivamente). (B) Trame rappresentative FACS di espressione VE-Cad, cKit e CD41 dopo tre giorni di cultura BL-CFC delle linee ESC indicate (n = 3). (C) Grafici che mostrano il numero medio di celle rotonde contate per fotogramma (n = 3) in un corso di 48 ore per le linee ESC indicate. Le barre di errore rappresentano le deviazioni standard. (D) Immagini rappresentative scattate due giorni dopo la cultura HE di cellule VE-Cad+ CD41- (Endo) ordinate per le linee di cellule indicate. Le cellule rotonde corrispondono alle cellule del sangue. La barra della scala corrisponde a 100 μm. (E) Grafici a barre che visualizzano i risultati sc-q-RT-PCR per le linee di ESC i6TFs e i5TFs. Vedi anche Figura 8-figure supplement 1, Supplementary files 1, 10 e 11.(A) Grafici che mostrano i numeri medi di celle rotonde contate per fotogramma (n = 3) in un corso di tempo di 48 ore di coltura HE per le linee di celle indicate. Le barre di errore rappresentano le deviazioni standard. (B) clustering gerarchico che mostra i risultati sc-q-RT-PCR per 95 geni sulle linee cellulari i5TFs e i6TFs dopo tre giorni di cultura BL-CFC. Sei sottopopolazioni principali sono stati definiti secondo l’intersezione della linea rossa punteggiata con il dendrogramma nella parte superiore della mappa termica. Vedi anche il file supplementare 11.
In conclusione, questi risultati suggeriscono che Fli1 e Erg da un lato e Runx1 e Gata2 dall’altro lato hanno effetti opposti sulle decisioni del destino cellulare nell’emopoiesi che portano alla formazione di Pre-HSPC.
Figura 8-figure supplemento 1.I gruppi Erg/Fli1 e Runx1/Gata2 di fattori di trascrizione hanno funzioni opposte durante EHT.studio del ruolo dei gruppi Erg/Fli1 e Runx1/Gata2 di fattori di trascrizione in EHT.(A) Schema che mostra i costrutti i5TFs e i6TFs usati per generare le due linee di ESC inducibili mancanti Runx1, Gata2 e Cbfb, o Fli1 e Erg (rispettivamente). (B) Trame rappresentative FACS di espressione VE-Cad, cKit e CD41 dopo tre giorni di cultura BL-CFC delle linee ESC indicate (n = 3). (C) Grafici che mostrano il numero medio di celle rotonde contate per fotogramma (n = 3) in un corso di 48 ore per le linee ESC indicate. Le barre di errore rappresentano le deviazioni standard. (D) Immagini rappresentative scattate due giorni dopo la cultura HE di cellule VE-Cad+ CD41- (Endo) ordinate per le linee di cellule indicate. Le cellule rotonde corrispondono alle cellule del sangue. La barra della scala corrisponde a 100 μm. (E) Grafici a barre che visualizzano i risultati sc-q-RT-PCR per le linee di ESC i6TFs e i5TFs. Vedi anche Figura 8-figure supplement 1, Supplementary files 1, 10 e 11.(A) Grafici che mostrano i numeri medi di cellule rotonde contate per fotogramma (n = 3) in un corso di 48 ore di tempo di cultura HE per le linee di cellule indicate. Le barre di errore rappresentano le deviazioni standard. (B) clustering gerarchico che mostra i risultati sc-q-RT-PCR per 95 geni sulle linee cellulari i5TFs e i6TFs dopo tre giorni di cultura BL-CFC. Sei sottopopolazioni principali sono stati definiti secondo l’intersezione della linea rossa punteggiata con il dendrogramma nella parte superiore della mappa termica. Vedi anche il file supplementare 11.
Figura 8-figure supplemento 1.Studio del ruolo dei gruppi Erg/Fli1 e Runx1/Gata2 di fattori di trascrizione in EHT.(A) Grafici che mostrano i numeri medi di cellule rotonde contate per fotogramma (n = 3) in un corso di 48 ore di tempo di cultura HE per le linee di cellule indicate. Le barre di errore rappresentano le deviazioni standard. (B) clustering gerarchico che mostra i risultati sc-q-RT-PCR per 95 geni sulle linee cellulari i5TFs e i6TFs dopo tre giorni di cultura BL-CFC. Sei sottopopolazioni principali sono stati definiti secondo l’intersezione della linea rossa punteggiata con il dendrogramma nella parte superiore della mappa termica. Vedi anche il file supplementare 11.
Discussione
In questo studio, abbiamo utilizzato la trascrittomica monocellulare per definire tre popolazioni principali nella vascolarizzazione embrionale durante la formazione del sangue: cellule endoteliali, cellule staminali pre-ematopoietiche/progenitrici e cellule staminali ematopoietiche/progenitrici. Mentre questi risultati sono stati inizialmente ottenuti con un singolo lotto di embrioni, tutti i modelli chiave sono stati riprodotti in un lotto separato in un ulteriore studio (Morgan Oatley, risultati non pubblicati). Nonostante le lievi differenze temporali, ci sono state forti somiglianze nell’espressione genica tra il sacco vitellino e l’AGM EHT, suggerendo un processo simile in entrambi i siti. Tuttavia, a causa della circolazione sanguigna, non possiamo escludere lo scambio cellulare tra questi due siti.
Le analisi Bulk ChIP-seq hanno suggerito che Erg, Fli1, Tal1, Lyl1, Lmo2, Runx1 e Gata2 lavorano insieme come heptad (Wilson et al., 2010). Qui, mostriamo per la prima volta che i geni heptad sono specificamente coespresi a livello monocellulare all’interno dei Pre-HSPC, e che questa combinazione non è stata trovata in nessun’altra popolazione lungo la transizione. Questa specifica co-espressione può essere conservata nella specie umana, come è stato dimostrato recentemente nel sistema in vitro del modello EHT umano che la popolazione intermedia tra cellule endoteliali e cellule del sangue co-espressiva Runx1, Lyl1, Tal1, Gata2, Erg e Fli1 (Lmo2 non è stato valutato) a livello monocellulare (Guibentif et al., 2017) in modo simile alla popolazione Pre-HSPC che abbiamo descritto. A differenza di Guibentif et al. (2017), abbiamo indagato il significato funzionale della co-espressione di questi fattori di trascrizione. Abbiamo dimostrato che la loro espressione forzata, insieme a quella del Cbfb, durante la differenziazione ematopoietica ha dato origine ai Pre-HSPC in cultura. Questo suggerisce che la coespressione dei geni heptad è necessaria per le cellule per raggiungere la fase transitoria dei Pre-HSPCs prima di perseguire la transizione.
Questi risultati sono coerenti con l’ipotesi dell’esistenza di un complesso proteico heptad. Sorprendentemente, solo il dieci per cento dei geni espressi differenzialmente sono bersagli heptad e solo il dieci per cento dei bersagli heptad sono espressi differenzialmente. Questo suggerisce che i cambiamenti di espressione genica che abbiamo osservato non erano una diretta conseguenza del legame del complesso delle otto TF agli elementi regolatori di questi geni. Ciò implica che la formazione di Pre-HSPCs è probabilmente dovuta ad una combinazione di funzioni individuali che agiscono sui GRNs piuttosto che alla sola azione di un complesso di otto proteine.
Per definire questi GRN, abbiamo sviluppato una pipeline bioinformatica per i dati sc-RNA-seq. Abbiamo caratterizzato le strette relazioni coinvolte nella formazione di Pre-HSPCs, dando così direzionalità alle interazioni proteina-DNA precedentemente identificate da ChIP-seq (Goode et al., 2016; Wilson et al., 2010). Le analisi di rete del dataset di Pereira (Pereira et al., 2016) insieme ai nostri risultati in vitro hanno rivelato una nuova e inaspettata relazione opposta tra le funzioni Runx1 e Fli1. Inoltre, questa rete suggerisce che Gata2 e Lmo2 potrebbero agire come co-regolatori cooperativi con Runx1 e Fli1, rispettivamente (Figura 7A).
Sulla base dei nostri dati e di quanto è noto in letteratura, proponiamo il seguente modello (Figura 9). Inizialmente, il programma di destino delle cellule endoteliali è guidato da Erg e Fli1, entrambi fattori di trascrizione ETS, che è coerente con le nostre analisi funzionali (Figura 3-figure supplement 5D e Figura 4) e gli studi pubblicati prima (Asano et al., 2010; McLaughlin et al., 2001). Inoltre, Wilson et al. (2010) hanno dimostrato che oltre il 90% dei loci target di Fli1 sono anche target di Erg nella linea cellulare HPC7, suggerendo una ridondanza funzionale. Comincia quindi l’espressione di Runx1 e questo – insieme all’espressione di Gata2, che ha funzione pro-ematopoietica – avvia il programma trascrizionale ematopoietico. I dati precedenti supportano questa interazione funzionale come Runx1+/- ::Gata2+/- embrioni eterozigoti composti non sono vitali a causa dei loro gravi difetti ematopoietici, rafforzando la nozione che le due proteine sono coinvolte nello stesso processo biologico (Wilson et al., 2010). In questa fase intermedia di Pre-HSPCs, i geni endoteliali sono progressivamente ridimensionati, come dimostrano i nostri dati. Come Fli1 e l’espressione del gene Erg rimangono stabili tra le fasi Endo e Pre-HSPCs, questo potrebbe essere spiegato da un cambiamento funzionale per questi fattori a favore del destino delle cellule ematopoietiche e una diminuzione della frazione dedicata al mantenimento del programma endoteliale a livello di singola cellula. In particolare, Fli1 può essere coinvolto sia nell’espressione genica endoteliale che ematopoietica a seconda del contesto cellulare. Infatti, in presenza di Runx1, Fli1 potrebbe potenzialmente attivare geni bersaglio comuni come Gfi1 e Gfi1b, che sono coinvolti nella repressione dell’espressione genica endoteliale (Lancrin et al., 2012; Thambyrajah et al., 2016; Wilson et al., 2010). In alternativa (o in concomitanza), Runx1 e Fli1 potrebbero legarsi insieme sui loci del gene endoteliale come mostrato per Sox17 (Lichtinger et al., 2012) e reprimere la loro espressione. D’altra parte, Fli1 potrebbe cambiare la sua funzione su interazione fisica con Runx1, come mostrato durante la differenziazione megacariocitica dove c’è una attivazione sinergica trascrizionale su interazione Fli1-Runx1 (Huang et al., 2009).
Figura 9.Modello per la funzione dinamica dei fattori di trascrizione durante la formazione delle cellule staminali del sangue e delle cellule progenitrici.Modello proposto per la funzione dinamica dei fattori di trascrizione durante la transizione da endoteliale a ematopoietica. La sezione superiore della freccia blu mostra l’equilibrio tra i fattori di trascrizione lungo la transizione. La sezione centrale indica i livelli di espressione genica per i due gruppi di fattori di trascrizione. Nella sezione inferiore della freccia blu è raffigurato un possibile meccanismo per l’interruttore funzionale di Erg/Fli1 in presenza di Runx1/Gata2. Durante la transizione, i loci ematopoietici epigeneticamente chiusi vengono progressivamente aperti e attivati in presenza di Runx1/Gata2 insieme a Fli1/Erg mentre i loci endoteliali vengono progressivamente chiusi attraverso la repressione trascrizionale Gfi1 e Gfi1b. I dati DNA-binding combinati con l’analisi GRN monocellulare suggeriscono una doppia funzione per Fli1 che dipende dal contesto cellulare (vedi testo principale per la descrizione completa).
Alla fine dell’EHT, negli HSPC, il programma endoteliale è completamente soppresso insieme all’espressione di Erg. In questa fase di sviluppo, Fli1 è completamente dedicato al programma del destino delle cellule ematopoietiche e lavora con Runx1 per portare avanti lo sviluppo delle cellule del sangue. Questo è supportato dall’osservazione che quasi tutti i geni target di Runx1 specifici per gli HSPC sono anche legati a Fli1 (file supplementare 9).
In conclusione, il nostro lavoro fornisce un’importante intuizione sul passaggio tra i destini delle cellule endoteliali ed ematopoietiche che è necessario per la formazione delle cellule del sangue. Abbiamo identificato i Pre-HSPC come la fase fondamentale di questo processo, in cui si svolge una competizione tra i programmi di sviluppo a livello di singola cellula. L’espressione dell’eptad delle TF è direttamente collegata a questa fase di transizione. L’equilibrio dell’attività di Erg, Fli1, Gata2 e Runx1 sembra avere il ruolo più importante nell’EHT, mentre l’asse Tal1/Lmo2/Lyl1 potrebbe avere un ruolo essenziale nell’adescamento. Infatti, Tal1 agisce come un repressore di programmi cellulari alternativi come il lignaggio cardiaco ed è attivamente necessario per avviare l’EHT (Lancrin et al., 2009; Org et al., 2015). In questo contesto, il nostro approccio alla trascrittomica monocellulare ci ha permesso di far luce su un cambiamento inaspettato dell’attività di Fli1 come conseguenza dell’espressione Runx1. Questo sembra essere un processo critico per il corretto completamento dell’EHT. Le condizioni che rafforzano l’interazione tra Runx1 e Fli1 probabilmente aiuterebbero a inclinare l’equilibrio verso il destino della cellula ematopoietica. Allo stesso modo, prevenire l’interazione Fli1-Runx1 potrebbe potenzialmente compromettere la formazione di cellule del sangue da endotelio.
Più in generale, una doppia funzione concorrente per un fattore di trascrizione unico potrebbe essere un processo chiave per altre transizioni dello sviluppo come l’epitelio alla transizione mesenchimale. L’approccio bioinformatico che abbiamo utilizzato in questo lavoro potrebbe essere facilmente applicabile allo studio di altri processi di sviluppo e potrebbe aiutare ad affrontare questo tipo di domande. Infine, la discrepanza osservata in questo lavoro tra analisi di massa e analisi monocellulare potrebbe esistere anche in altri studi di complessi TF, che potrebbero indurre i ricercatori in diversi campi a rivedere le ipotesi precedenti utilizzando il nostro approccio.
Figura 9.Modello per la funzione dinamica dei fattori di trascrizione durante la formazione delle cellule staminali del sangue e delle cellule progenitrici.Modello proposto per la funzione dinamica dei fattori di trascrizione durante la transizione da endoteliale a ematopoietica. La sezione superiore della freccia blu mostra l’equilibrio tra i fattori di trascrizione lungo la transizione. La sezione centrale indica i livelli di espressione genica per i due gruppi di fattori di trascrizione. Nella sezione inferiore della freccia blu è raffigurato un possibile meccanismo per l’interruttore funzionale di Erg/Fli1 in presenza di Runx1/Gata2. Durante la transizione, i loci ematopoietici epigeneticamente chiusi vengono progressivamente aperti e attivati in presenza di Runx1/Gata2 insieme a Fli1/Erg mentre i loci endoteliali vengono progressivamente chiusi attraverso la repressione trascrizionale Gfi1 e Gfi1b. I dati DNA-binding combinati con l’analisi GRN monocellulare suggeriscono una doppia funzione per Fli1 che dipende dal contesto cellulare (vedi testo principale per la descrizione completa).
Materiali e metodi
Modello sperimentale e dettagli del soggetto
Animali
C57BL/6 N, Gfi1:GFP (Yücel et al., 2004) e Gfi1b:GFP (Vassen et al., 2007) sono state utilizzate linee di mouse. Gfi1+/GFP e Gfi1b+/GFP sono stati incrociati tra loro per generare animali Gfi1+/GFPGfi1b+/GFP. Questi animali sono stati poi utilizzati per l’accoppiamento a tempo per generare embrioni a doppia uscita Gfi1GFP/GFPGfi1bGFP/GFP/GFP. L’accoppiamento temporizzato è stato impostato e la mattina del rilevamento del tappo vaginale è stato considerato il giorno 0,5. Gli embrioni sono stati messi in scena secondo le proprietà morfologiche. Tutti gli esperimenti sono stati eseguiti in conformità alle linee guida e alle normative definite dalla legislazione europea e italiana (Direttive 2010/63/UE e DLGS 26/2014, rispettivamente). 6. Tutti i topi sono stati allevati e mantenuti presso l’EMBL Rome Mouse Facility in conformità alla legislazione europea e italiana (Direttive UE 634/2010 e DLGS 26/2014, rispettivamente).
Linee cellulari
Generazione di linee di CES inducibili
Tutte le linee ESC inducibili con doxiciclina sono state generate utilizzando il metodo di scambio a cassette inducibili descritto in precedenza (Iacovino et al., 2011; Vargel et al., 2016). Per le linee ESC iCbfb, iRunx1, iLmo2, iGata2 e iFli1, le sequenze di codifica del fattore di trascrizione sono state etichettate in 5′ con un 8His-tag e amplificate mediante PCR da cDNA ESC differenziato. Le sequenze di codifica 8His-tag sono state poi inserite in un pGEM-t-easy prima della clonazione nel plasmide p2lox. Per iTal1, iLyl1 e iErg, le sequenze di codifica 8His-tagged sono state sintetizzate e clonate in un pUC57 Simple dal GenScript Gene Synthesis Service (http://www.genscript.com/ gene_synthesis.html). Le sequenze di codifica 8His-tagged sono state poi clonate dalla pUC57 Simple nel plasmide p2lox. Per ciascuno dei plasmidi p2lox generati, una sequenza Kozak è stata poi aggiunta al codone iniziale mediante clonazione PCR. I plasmidi p2lox successivi sono stati utilizzati per trasfettare la linea ESC A2lox.Cre e per generare la linea ESC a singolo fattore inducibile come descritto in precedenza (Vargel et al., 2016). Per la linea di ESC i8TFs, tre costrutti individuali sono stati sintetizzati e clonati in un plasmide pUC57 dal GenScript Gene Synthesis Service. Il primo costrutto è stato composto dalla sequenza di codifica per V5-Erg-T2A-V5-Lmo2. Construct due era composto dalla sequenza di codifica per HA-Tal1-T2A-cmyc-Fli1-T2A-FLAG-Lyl1-T2A. La costruzione tre era composta dalla sequenza di codifica per FLAG-Runx1-T2A-FLAG-Cbfb-T2A-HA-HA-Gata2-T2A. Tutti e tre i costrutti sono stati affiancati da un codice di avvio e due codici di arresto, insieme ai siti di restrizione adeguati per la clonazione nel plasmide p2lox, al fine di consentirne l’uso individuale. Essi contengono anche siti di restrizione specifici per la loro escissione, l’aggiunta di uno qualsiasi degli altri costrutti, o la rimozione di una qualsiasi delle sequenze di fattori. Ne è stato clonato uno insieme al codice di partenza e due codici di arresto nel plasmide p2lox. Poi, Construct two è stato aggiunto tra il codice di partenza e Construct one per clonazione classica. Infine, Construct three è stato aggiunto tra il codice di partenza e Construct two per ottenere il p2lox-8TFs. Questo plasmide è stato poi trasformato nella linea A2lox.Cre ESC per la generazione degli i8TFs. Per gli i5TFs, il costrutto intermedio p2lox contenente Construct one e Construct two è stato usato per generare la linea ESC. Per gli i6TFs, V5-Erg e cmyc-Fli1 sono stati successivamente eliminati dai p2lox-8TFs mediante clonazione classica per generare i p2lox-6TFs utilizzati per la generazione delle linee di ESC inducibili. La linea di ESC vuota è stata generata utilizzando il plasmide p2lox di base e seguendo la stessa procedura. Per ogni linea di ESC inducibile, è stato selezionato un clone in base all’induzione dell’espressione di trascrizione misurata da qPCR e convalidata dall’analisi western blot.
Identificazione delle linee cellulari
Tutte le linee cellulari utilizzate in questo lavoro erano linee mESC. La linea mESC Runx1+/hCD4 è stata generata nel gruppo di Georges Lacaud dall’autore corrispondente, Christophe Lancrin (Sroczynska et al., 2009a). Tutte le linee mESC di derivazione A2lox.Cre sono state generate dalla linea A2lox.Cre mESC, che è stata regalata da Michael Kyba che l’ha prodotta nel suo laboratorio (Iacovino et al., 2011). Tutte le linee mESC di derivazione A2lox.Cre sono state generate nel nostro laboratorio come descritto nella sezione “Generazione di linee ESC inducibili”. Tutte le linee di CSE avevano una morfologia corretta delle cellule staminali e sono state in grado di dare origine a sangue e cellule endoteliali dopo la differenziazione in vitro (Figura 1D, Figura 3, Figura 3-figure supplementi 1 e 5, Figura 8 e Figura 8-figure supplemento 1A). Tutte le linee mESC inducibili sono state verificate con l’analisi di Western Blot e qPCR (Figura 2, Figura 3-figure supplement 5, Figura 4 e Figura 8-figure supplement 1B).
Tutte le linee cellulari utilizzate in questo manoscritto sono state testate negative per la contaminazione da micoplasma.
Manutenzione e differenziazione delle linee di ESC
La crescita e la differenziazione delle CSE sono state effettuate come descritto in precedenza (Sroczynska et al., 2009b). In breve, le CSE sono state mantenute su alimentatori in un mezzo di coltura di CSE in KnockOut DMEM (GIBCO) (integrato con 1% Pen/Strep [GIBCO], 1% L-Glutammina [GIBCO] e 1% NEAA [GIBCO]), 15% FBS (PAA), 0,0024% 1 mg/ml LIF (The Protein Expression and Purification Core Facility, EMBL-Heidelberg) e 0,24% 50 mM 2-mercaptoetanolo (GIBCO). Per la differenziazione, gli ESC sono stati seminati su piastre rivestite di gelatina (0,1% di gelatina [BDH] in PBS per 20 minuti) per un giorno nel normale terreno di coltura degli ESC e poi per un giorno in un terreno simile a quello degli ESC ma contenente IMDM (Lonza) al posto del KnockOut DMEM e senza supplemento NEAA.
Per ottenere corpi embrionali (EB), le cellule sono state successivamente raccolte e coltivate in scatole di Petri ad una densità di 0,3 × 106 cellule per scatola da 10 cm2 con un mezzo EB fatto di IMDM (Lonza) (integrato con 1% Pen/Strep [GIBCO] e 1% L-glutammina [GIBCO]), 0,6% transferrina (Roche, Italia), 0,039% MTG (Sigma) e 50 mg / µl di acido ascorbico (Sigma). Dopo 3-3,25 giorni, gli EB sono stati raccolti e le cellule Flk1+ sono state selezionate utilizzando la tecnologia MACS MicroBead Technology (Miltenyi Biotec). Per la differenziazione degli emangioblasti, le cellule Flk1 + sono state poi coltivate su piastre rivestite di gelatina ad una densità di 0,01 × 106 cellule per cm2 in un mezzo fatto di IMDM (Lonza), 1% Pen/Strep (GIBCO), 1% L-glutammina (GIBCO), 10% FBS (PAA), 0.6% transferrina (Roche), 0,039% MTG (Sigma), 0,5% acido ascorbico (Sigma), 15% D4T supernatante (EMBL-Monterotondo), 0,05% VEGF (10 µg/ml) (R e D) e 0,1% IL-6 (10 µg/ml) (R e D). Durante la differenziazione degli emangioblasti, la popolazione cellulare potrebbe essere ordinata e ulteriormente coltivata in condizioni di coltura emogenica dell’endotelio. A tal fine, le cellule ordinate sono stati seminati su piastre rivestite di gelatina ad una densità di 0,015 × 106 cellule per cm2 in un mezzo specifico fatto di IMDM (Lonza), 1% Pen / Strep (GIBCO), 1% L-glutammina (GIBCO), 10% FBS (PAA), 0.6% transferrina (Roche), 0,039% MTG (Sigma), 0,5% acido ascorbico (Sigma), 0,0024% LIF (EMBL-Heidelberg), 0,25% SCF (20 µg/ml) (R e D), 0,1% oncostatina M (10 µg/ml) (R e D) e 0,01% bFGF/FGF2 (10 µg/ml) (R e D). Per perseguire la differenziazione con un saggio progenitore ematopoietico, le cellule sono state raccolte tre giorni dopo il tipo Flk1 dal tipo di coltura dell’emangioblasto (o l’endotelio emogeno) e placcato ad una densità di 3.3 × 103 cellule per cm2 in un mezzo fatto di IMDM (Lonza), 1% Pen/Strep (GIBCO), 1% L-glutammina (GIBCO), 55% di 0,5 g/ml metilcellulosa (VWR), 15% PDS (Antec), 10% PFMH-II (GIBCO), 0.6% transferrina (Roche), 0,039% MTG (Sigma), 0,5% acido ascorbico (Sigma), 0,5% SCF (20 µg/ml) (R e D), 0,1% IL-3 (25 µg/ml) (R e D), 0,1% GM-CSF (25 µg/ml) (R e D), 0.04% IL-11 (12,5 µg/ml) (R e D), 0,2% EPO (10 µg/ml) (R e D), 0,2% IL-6 (5 µg/ml) (R e D), 0,2% TPO (12,5 µg/ml) (R e D) e 0,05% MCSF (10 µg/ml) (R e D). Le colonie sono state contate per tre repliche dopo 10-12 giorni.
Per il trattamento con doxiciclina della linea inducibile ESC, la doxiciclina (Sigma) è stata aggiunta direttamente al terreno di coltura ad una concentrazione finale di 1 µg/ml per 24 o 48 ore.
Dettagli del metodo
Dissezione embrionale
Le dissezioni del sacco vitellino e dell’AGM sono state eseguite come descritto in precedenza (Bertrand et al., 2005) e Robin e Dzierzak, 2005). In breve, i topi gravidi sono stati uccisi da una dislocazione cervicale tra E9.0 e E11 di gestazione. Sono state raccolte corna uterine; i tessuti materni sono stati rimossi così come la placenta per isolare gli embrioni. Il sacco vitellino è stato strappato delicatamente e separato dall’embrione vero e proprio strappando le arterie ombelicali e vitelline. Poi, le coppie somatiche degli embrioni sono state contate per determinare il loro stadio di sviluppo. Per isolare l’AGM, sono stati rimossi la testa, la coda, le gemme degli arti, gli organi ventrali e i somiti. I campioni di sacco vitellino e di AGM dello stesso stadio di sviluppo embrionale sono stati riuniti nello stesso tubo. Per generare sospensione monocellulare dai tessuti isolati, i campioni di sacca di tuorlo e AGM sono stati sottoposti a trattamento con collagenasi (Sigma) per 30 min. a 37°C. AGM e sospensioni cellulari del sacco vitellino sono stati poi utilizzati per la citometria a flusso.
Analisi Western blot
Le CSE sono state coltivate per due passaggi su gelatina per rimuovere i MEF prima di essere trattate con 1 µg/ml di doxiciclina (Sigma) per 24 ore. Per gli estratti di proteine intere, le cellule sono state raccolte utilizzando TrypLE express (GIBCO), lavate con PBS e lisate in buffer RIPA (Thermo Scientific) per 30 minuti a 4°C. I campioni sono stati poi centrifugati per 5 minuti ad alta velocità e i pellet sono stati scartati. Gli estratti nucleari sono stati preparati utilizzando il kit di estrazione nucleare (ActiveMotif) seguendo le istruzioni del produttore. La concentrazione di proteine è stata misurata utilizzando il kit di analisi delle proteine BCA Pierce (Thermo Scientific) e quantità uguali di campioni di proteine -dox e +dox sono stati caricati su NuPAGE Novex 10% o 12% di gel di proteine Bis-Tris (Life Technologies). La migrazione e il trasferimento a umido su una membrana di nitrocellulosa PROTRAN (PerkinElmer) sono stati eseguiti utilizzando il sistema di elettroforesi a mini celle XCell SureLock di Invitrogen. Le membrane sono state colorate con la soluzione Ponceau S (Sigma) per controllare l’efficienza di trasferimento e le quantità di proteine. Le membrane sono state poi lavate con TBST (TBS [20 mM Tris-Base, 154 mM NaCl, pH 7,5], 0,1% Tween 20) e bloccate con TBST +5% di latte per 45 minuti a RT. Le membrane sono state poi incubate ON a 4°C con anticorpi primari diluiti con TBST +1% di latte come segue: 1:2000 anti-FLAG (Sigma), 1:1000 anti-HA (Sigma), 1:2000 anti-V5 (Life Technologies), 1:5000 anti-Runx1 (Abcam), 1:5000 anti-Cbfb (Abcam), 1:2000 anti-Gata2 (Abcam), 1:2000 anti-Tal1 (Abcam), 1:2500 anti-Fli1 (Abcam), 1:5000 anti-Lyl1 (Abcam), 1:5000 anti-Erg (Abcam), 1:1000 anti-Lmo2 (Abcam), 1:2000 anti-His tag (Abcam), e 1:10000 anti-alfa-tubulina (Sigma) (l’ultimo usato come controllo del carico). Dopo l’incubazione degli anticorpi primari, le membrane sono state lavate tre volte con TBST e incubate per 45 minuti con anticorpi secondari coniugati con HRP diluito in TBST +1% nilk come segue: 1:10000 anti-Mouse HRP (GE Healthcare Life Sciences), 1:10000 anti-Rabbit HRP (Jackson) e 1:10000 anti-Rat HRP (GE Healthcare Life Sciences). Le membrane sono state sviluppate utilizzando l’ECL Prime Western Blotting Reagent di Amersham (GE Healthcare Life Sciences).
Fotografia time-lapse
Le colture di differenziazione cellulare sono state immaginate a partire dal momento dell’aggiunta di doxiciciclina. Le immagini a contrasto di fase sono state scattate con l’IncuCyte HD (Essen Biosciences) all’interno dell’incubatore, ogni 15 minuti, tre aree per pozzetto. I video time-lapse sono stati generati con il software delle Fiji (http://fiji.sc/Fiji) (Schindelin et al., 2012) utilizzando 10 fotogrammi al secondo. Le immagini sono state analizzate dal software CellProfiler (http://www.cellprofiler.org) (Kamentsky et al., 2011) per quantificare il numero di cellule rotonde con una pipeline personalizzata scritta da Christian Tischer dell’EMBL Heidelberg Advanced Light Microscopy Facility. Per ogni punto temporale, la media e la deviazione standard dei conteggi da tutti e tre i punti sono state calcolate per realizzare dei grafici con il software Microsoft Office Excel. Una spiegazione dettagliata su come sono state eseguite la fotografia time-lapse e l’analisi delle immagini è disponibile su Bio-protocollo (Bergiers et al., 2019)
Citometria a flusso e selezione delle cellule
La colorazione è stata effettuata come descritto in precedenza (Sroczynska et al., 2009b) e le analisi sono state eseguite con un FACSCanto (Becton Dickinson) o un FACSAria (Becton Dickinson). I tipi di cellule sono state eseguite con un FACSAria (Becton Dickinson) o utilizzando la selezione magnetica (MACS MicroBead Technology, Miltenyi Biotec) e anti-APC MicroBeads (Miltenyi Biotech). Gli anticorpi monoclonali anti-topo di ratto utilizzati sono stati anti-Topo CD309 (FLK1) APC (Avas12a1, eBioscience), anti-Topo CD41 PE (MWReg30, eBioscience), anti-Topo cKit APC (2B8, eBioscience) e anti-Topo CD144 (VE-Cadherin) eFluor 660 (eBioBV13, eBioscience). I dati FACS sono stati analizzati utilizzando il software FlowJo (Tree Star, Inc.).
Saggio del ciclo cellulare
I test del ciclo cellulare sono stati eseguiti utilizzando il kit Click-iT Plus EdU Alexa Fluor 488 Flow Cytometry Assay di Sonde Molecolari seguendo le istruzioni del produttore. In breve, le cellule dell’emangioblasto, o endotelio emogenico, sono state trattate con 10 µM di EdU per 1 ora. Le cellule sono state poi raccolte e colorate con Ghost Dye Red 780 (Tonbo Biosciences) per la colorazione dal vivo / morto prima della colorazione con anticorpi marcatori di superficie cellulare (anti-Mouse CD41 PE [MWReg30, eBioscience] e anti-Mouse CD144 [VE-Cadherin] eFluor 660 [eBioBV13, eBioscience]). Le cellule macchiate sono state fissate e permeabilizzate e la reazione Click-IT è stata eseguita seguendo le istruzioni del produttore. Infine, le cellule sono state colorate con Hoechst per l’etichettatura del DNA e analizzate con il FACSAria (Becton Dickinson). I dati FACS sono stati analizzati utilizzando il software FlowJo (Tree Star, Inc.).
Saggio di apoptosi
Le cellule provenienti da colture di emangioblasto sono state raccolte e colorate con anti-topo CD41 PE (MWReg30, eBioscience), anti-topo CD144 (VE-Cadherin) eFluor 660 (eBioBV13, eBioscience) e AnnexinV (eBioscience). Infine, le cellule sono state colorate con Sytox Blue per l’etichettatura del DNA e analizzate con il FACSAria (Becton Dickinson). I dati FACS sono stati analizzati con il software FlowJo (Tree Star, Inc.).
Saggio Luciferasi
Le sequenze dei due picchi di eptad trovati nel locus Gpr56 (Wilson et al., 2010) sono state sintetizzate e clonate dal GenScript Gene Synthesis Service in un pGL4.10[luc2] reporter plasmid (Promega), con ulteriori sequenze di 50 bp a monte e a valle per garantire una corretta copertura dei picchi. Il plasmide pGL4.74[hRluc/TK] Renilla reporter plasmid che è stato utilizzato per il controllo dell’efficienza della trasfezione è stato ottenuto da Promega. Gli ESC sono stati placcati su piastre rivestite di gelatina (0,1% di gelatina [BDH] in PBS per 20 minuti) e passati due volte per rimuovere gli alimentatori. Le cellule sono state poi seminate in piastre rivestite di gelatina a 96 pozzetti ad una densità di 5.000 cellule per pozzetto. Il giorno dopo, il mezzo è stato cambiato e le cellule sono state trasfettate con i plasmidi Firefly luciferasi reporter plasmidi insieme con il plasmide Renilla luciferasi reporter utilizzando il kit NanoJuice Transfection (Merck Millipore). Dopo 24 ore, le cellule sono state trattate con doxiciclina (Sigma) per 24 ore e il saggio luciferasi è stato eseguito utilizzando il Dual-Luciferase Reporter Assay System (Promega) e il dispositivo POLARstar Omega (BMG Labtech) seguendo le istruzioni del produttore.
microarray mRNA
Le cellule sono state raccolte e lavate una volta con PBS. L’RNA totale è stato estratto con RNAeasy Plus Micro kit (Qiagen). I campioni sono stati poi testati per la qualità con BioAnalyzer (Agilent) e le loro concentrazioni misurate con Qubit (Life Technologies). Sono stati ibridati su un Affymetrix GeneChip Mouse Gene 2.0 ST Array.
RT-PCR quantitativa monocellulare
Un massimo di 1 giorno prima dell’esperimento, piastre a 96 pozzetti (BioRad) sono state riempite con 5 µl di 2x mix di reazione dal kit CellsDirect One-Step qRT-PCR (Invitrogen). Il giorno dell’esperimento, le cellule sono state raccolte e colorate per l’analisi FACS. Le cellule da Anti-Mouse CD41 PE (MWReg30, eBioscience)/Anti Mouse CD144 (VE-caderina) eFluor 660 (eBioBV13, eBioscience) colorazione sono stati selezionati a cella singola nelle piastre pre-riempite a 96 pozzetti (BioRad) utilizzando il sistema FACSAria (Becton Dickinson). Le piastre sono state congelate a scatto su ghiaccio secco subito dopo la cernita e conservate a -80°C per una settimana al massimo. Per eseguire RT/Specific Target Amplification (RT/STA), 4 µl di una miscela di reazione – composta da 2,8 µl di buffer di risospensione, 0.2 µl di SuperScript III e Platinum Taq polimerasi dal kit CellsDirect One-Step qRT-PCR (Invitrogen) e 1 µl di 500 nM outer primer mix (lista delle sequenze di primer, vedi tabella sotto) – è stato aggiunto ad ogni pozzetto. Le piastre sono stati prima incubati a 50 ° C per 15 min. seguita da incubazione a 95 ° C per 2 min. Poi, sono stati incubati a 95 ° C per 15 s. seguita da incubazione a 60 ° C per 4 min. per 20 cicli. Per preparare le miscele di campioni, 1/5x cDNA diluiti sono stati miscelati con il reagente di carico (Fluidigm) e SsoFastTM EvaGreen Supermix (Bio-Rad). In parallelo, le singole miscele di dosaggio sono state preparate come 5 μM di miscela interna di primer, con tampone di sospensione del DNA e il reagente di carico del dosaggio (Fluidigm). Dopo il priming della matrice dinamica 96,96 IFC (Fluidigm), le miscele del campione e le miscele del saggio sono state messe insieme negli ingressi corrispondenti e caricate nel chip. Infine, il chip è stato eseguito nel sistema Fluidigm Biomark HD utilizzando il software di raccolta dati Biomark e il programma GE96 ×96PCR + Meltv2.pcl.
Per controllare la variazione tecnica delle nostre esecuzioni sc-qRT-PCR, abbiamo utilizzato il reverse cDNA universale trascritto da Random Hexamer: Tessuti normali del mouse (BioChain). Ci ha permesso di rilevare costantemente 93 dei nostri 95 geni di interesse. Un campione senza modello di DNA è stato anche eseguito ogni volta per testare per l’amplificazione PCR non specifica.
La lista dei primer è la seguente:
| Nome del gene | Esterno in avanti | Inversione esterna | Interno in avanti | Inversione interna |
|---|---|---|---|---|
| Acta2 | aggcaccactgaaccctaag | cacagcctgaatagccccacat | ccaaccaccgggagaaaatgac | atggcggggacattgaag |
| Acvr1 | cgagtgctaatgatgatgatgatggctttttttccccc | cgtttcacagtggttttttttttttttttoo | cgattccccggagtgtggaagatga | cgattccccggagtgtggaagatga |
| Acvr1b | atgctggcggccatgaaaacatc | tcgtgatagtcagagacaagccaca | ttggctttttattgctgctgaca | tgggtccccccccccattatc |
| Acvr2a | gctggcaagtctgccaggtgtga | tcagaaatgctttttttgga | acccatggggcaggttttttta | tttatagcaccccccccaactttttoo |
| Acvr2b | gaagggctgctggctagatga | agttgccttttttcgccagcagcagca | tcaattgctactacgacaggaggcagga | aagtacacctggggggggggtttttttttttttc |
| Acvrl1 | cgaattgcccccatcgtgacctcaa | cgtggttttgttggggatatccatccaggta | cgaagtcggcaatgtggctggtcaa | cgaagtcggcaatgtggctggtcaa |
| Adcy4 | cgatggtggcatttttttttttttcccta | cgacattcccaccccacgcctggtggggtc | cgacattcccaccccacgcctggtggggtc | |
| Atp2a3 | cgaccattgtgtggggcagagtagaa | cgtcccagaattgctgtgaggaa | cgagagagggcagggccatctaca | cgagagagggcagggccatctaca |
| Bmp4 | cagccgagccaacactgtga | tgggatgatgctgctgaggttttttga | agtttccatcatcacgaagaacatctgg | gaggaaacgaaaagagcagagagagagc |
| Bmpr1a | acgttcaaagccgaaagccccagccagta | tcgcggatgatgctgccatcaaagaa | acgccggcaggacaatagaatgttgg | acgccggcaggacaatagaatgttgg |
| Bmpr2 | cagacggggggcatggagtat | atgagccagagacggcaagagc | tgcttgtgtgatggatggatttatttatcccaatg | tacccaatcacttttttgtgtggagactca |
| Cacna2d1 | acgccaatggttttagctggtgaca | cgtgattggggggagtgacgttttgaa | acgagcaagttcaatggacaaatgtg | acgagcaagttcaatggacaaatgtg |
| Cdh2 | atcaacaatgagactggacatca | cttccccatgtgtgtggcttttgaa | cactgtgtggcagctggttttttgg | attaacgtatactgttttgcactttttttctctcg |
| Cdh5 | cgacaccatcatcgccaaaagagagagagac | cgtcttagcattattttttttttcggggttcac | cgaggatttggaatggaatcaaatgcacatcacatcg | cgaggatttggaatggaatcaaatgcacatcacatcg |
| Cldn5 | tggccaggtgactgccttttttttt | tgcatgtggcccggggtactg | accacaacatcatcgtgacggcgcccaga | accacggcacgacatccacatccacag |
| Col4a2 | cgaccctggaagccccctggattta | cgtttttttttttttaagtttaagtcccccaaca | cgatggcagggatggatgccttttggt | cgatggcagggatggatgccttttggt |
| Col4a5 | cgaggaggactctctgtgtggattggcta | cgtatgaagggagcggaacgaa | cgattcatgatgatgcatacaagtgcagga | cgattcatgatgatgcatacaagtgcagga |
| Ctnnb1 | aactcctgcacccaccatccatccca | gttccccggcaaaggcatga | tggccttttttgataaaggcaactg | tgctgggggcaaagagggcaag |
| Dcaf12l1 | cgaaggcctccatggggcaactac | cgtggtggggagacccagaaaa | cgagcaggctttttttttaggagcta | cgagcaggctttttttttaggagcta |
| Dpysl3 | cgaccccattgggaaggacaacttccac | cgtcggccccacaaactttttttttca | cgaccatttttgaaggcaccaat | cgaccatttttgaaggcaccaat |
| Eng | gcctgacttttttttggggactccagc | tgctgaccacatgggggctgtcac | gccaggctgaagacacactgacg | tcatgccccgcagctggagtag |
| Enpp1 | cgaccatgaaaggaacggcatca | cgtaattttttttcctggcttcggatgac | cgatcagccggtcccccttttttttt | cgatcagccggtcccccttttttttt |
| Epo | ggttgtgggcagaaggtcccaga | gggacaggttttgccaaact | ctgagtgaaaatattacagtcccaga | tatggggttttttttttccacctcca |
| Epor | tgaagtggacgtgtctcggcag | acagcgaaggtggtagcggcccgt | caaccggggcaggagagggaca | ccgcccccccccggggttttttgcagaa |
| Eps8 | cgaaggctgccatgccatgtttttttcaa | tcgtgtgctgttttttttcctcgccacaaa | acgctcctaatcaatcaccaagtagataggaattatg | acgctcctaatcaatcaccaagtagataggaattatg |
| Erg | tcccgaagctactacggcaaagaa | tttggactgagtgaggtgtgaggtggg | tacaactaactaggccagatttaccttatga | tgtggggggtccccccaggttgat |
| Esam | cgaagttatgtatgtgcaaggggttcaa | cgtcccaacaaaagtgcccacaa | cgacagagtggggggtttgccaagt | cgacagagtggggggtttgccaagt |
| Fam122b | cgaagtttttttttttttttttttoo | cgtaaatgtctcttttggattgagaacaggacac | cgacaaccaccagaggatttttggaaagcaatg | cgacaaccaccagaggatttttggaaagcaatg |
| Fbn1 | acgtggcggggaatgtacaaaca | cgtcagagctgtggtagcagtaacca | cgactgtcagcagcagctacttttttgcaaat | cgactgtcagcagcagctacttttttgcaaat |
| Fli1 | tgctgttttggtcgcaccccccccagttàààà | ttccttgacattcagtcgtgagga | ctcagggaaagttcactgctggccta | tggtctgtatgggggaggttttttgg |
| Flrt2 | cgatgccccctttttttttttttoo | cgtagtgtagagagcagcccctaggaa | cgaccggaccacccttttttttggatt | cgaccggaccacccttttttttggatt |
| Fmo1 | cgaaaccaccacgtgaattacggggta | cgagctccagaagagacaggactca | cgagctccagaagagacaggactca | |
| Gata1 | cctgtgtgcaatgccttttttttttttoo | tgcctgccccctttttttgctgacaa | gtatcacaagatgaatgaatggtcagaacc | cattcgcttttttttttgggggccccatg |
| Gata2 | aagcaagggggggtttttoo | cacaggcattgcacaggtagt | cagaaggaggggagtgtgtc | gcccgtggccatctcgt |
| Gdpd5 | cgaacttttgacaactcccatacc | cgttggggatgatgatgaggaggctgaa | cgatttttttttttttttttttcca | cgatttttttttttttttttttcca |
| Gfi1 | cgagagatgtggcggcaagacc | cgtagcgtgtggatgacctcttttgaa | cgagtgagcctggagagcaacacaa | cgagtgagcctggagagcaacacaa |
| Gfi1b | cgatggacacttaccacttaccactgtgtca | cgaagtgcaacaacaaggtgtgttttttttcc | cgaagtgcaacaacaaggtgtgttttttttcc | |
| Gpr126 | cgactgtgtggcagccacttttcactca | cgtggcagatattccggcacccaata | cgatggagttttgatggatggatttttttcc | cgatggagttttgatggatggatttttttcc |
| Gria4 | cgacgcccccatggggacgaaacta | cgtggccattaccaagacaccatcatcgta | acgatggatccgctggaagaagaaactaga | acgatggatccgctggaagaagaaactaga |
| Hbb-bh1 | gagctgcactgtgacaagcttca | ggggtgtgaattccttttggcaaaa | tggatcctgagaacttcaaggggiiioo ddii tggattttaaggllii | gagtagaaaggacaatcaccaaca |
| Itga2b | ttccaaccaccagccgcttttcacct | tgctcggatccccatccatcaaac | cgacaacagcaaccccagtgttt | gcccacggggctaccgaatatc |
| Itgam | agcaggggtcattcggcctacg | cagctggcttagatgatgcgatgg | gtcggagctctctgcggggact | |
| Itgb3 | tcctccccagctcattgttgatgatgc | aggcaggtggcattgaagga | acgggaaaatccggctctaaa | agtgacttttttttttccccggcggcaggt |
| Kdr | tgtggggcttgatttgatttcacctg | tcgccccacagtcccaggaaag | cactctccaccttttcaaagtttaaagtattttoo | tttcacatccccctttaattttacaattttoo |
| Kit | ctggctctctggacctggatga | cctggctgccaaatctgtgtg | tgctgagcttttttttcctacctaccaggtg | atacaattcttttggaggaggcgaggaa |
| Ragazzo1 | cgacctctttgagaagggagctgtca | cgtcctggggtctcttttggatcca | cgaggccagaaccggcacacagaac | cgaggccagaaccggcacacagaac |
| Lat | cgatgatgctgcctgacagagtagtcc | cgttcactctcaggaacattcacgta | cgactgcccccctttttttttttttoo | cgactgcccccctttttttttttttoo |
| Lgr5 | cgagcaacaacatcatcaggtcaatacc | cgtcaggcaaatgctgaaaagcaaaagca | cgaggagccggggttttcgtagg | cgaggagcgagcggttttcgtagg |
| Lmo2 | tcggccccatcgaaaggaaggaaggggii | gcggtcctattttttatgtttttoo | ctggacccgtgtgaggaacc | gcagccaccacatgtcagca |
| Lyl1 | gctgaagcggcagaccaagccat | gctcacggggttttttttggtgaacact | gtgagctggacttttttttgacg | ccggagccacccctttttttttoo |
| Meis2 | cgacccggtacccttcttcagaagaacagaa | cgttttggtcaatcatcatggggctgcacta | cgagaaacagttagcgcaagagacacac | cgagaaacagttagcgcaagagacacac |
| Incontrato | cgagatcatttgcggggtttttcaa | tcgactcttttgggcatagcatagcgaac | cgagtagttttttttttttatccccggggggggtttttttttttttoo | cgagtagttttttttttttatccccggggggggtttttttttttttoo |
| Mpo | tggcccctagacctgctgaagagag | ttgacacggacaacagattcagattcagc | aagctggccagcccccctgtgtgtgg | gagcaggtgtcaacacatctgtaa |
| Myb | cgagtggcagaaagtgctgaacc | cgttgcttttggggcaataacagaccaac | cgacatcaaaggtccccctggaccaaa | cgacatcaaaggtccccctggaccaaa |
| Myom1 | cgattccccgtgtacgtgtgtgtgtgtgtgtcaa | cgttttccggtcatcatcatccacactcac | acgccccaggcaggcaggttttttggaaag | acgccccaggcaggcaggttttttggaaag |
| Tacca1 | cgaccaaccctgtcaacggcaaa | cgtatttgcctgcgtgtggctcacaa | cgatgcccccctctcggggtaca | cgatgcccccctctcggggtaca |
| Npr1 | cgtcgaaacatccatccagtccagggta | cgagaccaccacccaacatttttttattttoo | cgagaccaccacccaacatttttttattttoo | |
| Palld | cgtttttttttttcctggtggatgatttgaa | cgacttctgaacaatggccaacc | cgacttttttgaacaatggccaacc | |
| Pcdh12 | cgatggggttttttttggcttgcggaac | tcgtgttttttttttggtgggggctggaa | cgaggaacccccggtggagga | cgaggaacccccggtggagga |
| Pdzd2 | cgacaacttttggaaagccccaaac | cgtgtccccatttcggtaccatca | cgaagggggcaacagagtaaaatgaaactcaag | cgaagggggcaacagagtaaaatgaaactcaag |
| Pecam1 | tgcggtggttttttgtcattggag | ctggacatctccacggggttt | gtcatccgccaccccaatttttggcag | tgtttggggtttttttttttttttttttttttttttttttttttc |
| Plcd1 | cgatggcttttttttccagtcctagca | cgtccccacgttatggcggcggacaa | cgatctggcaggcaggcattattatgagatgagatgg | cgatctggcaggcaggcattattatgagatgagatgg |
| Ppia | cgaccgactgtgtggacacagctctaa | cgtagtggagagagagagagattacaggac | cgatttttttttgacttgacttgggcggcatt | cgatttttttttgacttgacttgggcggcatt |
| Ppp1r16b | cgagcgtgtgtggatgtgaaggac | cgtgatgtcctggtggcactgagacta | cgaatggctgggagcctct | cgaatggctgggagcctct |
| Ptprb | acgccaagagcggcaattatatgca | cgttgggcacccccaggacacctttaa | cgaccactccttttcaccgaggaa | cgaccactccttttcaccgaggaa |
| Ptprc | ggcttcaaggaacccaggaaata | tgacaataactgtgtggccttttttttttgc | attgctgcacaaggggggggggatg | cagatcatcatcctccagaagtcatcaa |
| Ptprm | cgacagtcctccaacacatca | tcgtgtcctttttttttttagcagtagtcc | cgatcagatgaagtgcgggctgag | cgatcagatgaagtgcgggctgag |
| Rampa 2 | tccccactgaggaggacagtttoo | tccttggacagagtccatgatgcaa | tcaaaagggaagatggaagatggaagactacga | tcttggtacttactcataccagcaaggtaggaca |
| Correrex1 | cgaactactccggcagaactgagaa | cgtactgattgattcagagtgaa | cgaatgctaccctaccggcggggatg | cgaatgctaccctaccggcggggatg |
| Samd4 | cgaacagctccgtccagaagac | cgtactccagcctattgttttgattgatgtca | acgtcgctgccccccgtggcata | acgtcgctgccccccgtggcata |
| Fusciacca1 | cgattgattgatctcactgaggagcccta | tcggccatgttttttggtggcaacatcc | cgactgataagcatggggggggtttttttoo | cgactgataagcatggggggggtttttttoo |
| Serpina1 | aaaaccccggcggcagatcca | cttgttttttccacggccccatga | gatgctatgggattgattcaaagtcaa | ccttttggagagggggcggcggcatga |
| Sfpi1 | gtggggcagcgatggagaaag | tgcagctgtgtgaagtggtttttttttoo | atagcgatcactactattttttotttttttoo | gggaagttttttcaaactcgttggttggttttgg |
| Lei | cgaacatggaaccgtacgatgatgatgca | tcgtctcacagttttttttttttoo | acgtaacagaaatcagagacgccccgtgtggtt | acgtaacagaaatcagagacgccccgtgtggtt |
| Sla | cgacgaatttttttttttcccccccaac | tcgggtggagcacacacacatagcatagac | cgaactggtactactacatctcaccaagg | cgaactggtactactacatctcaccaagg |
| Smad1 | tctcagcccatggacacgaa | caccagtgtttttttttttttttttttttttttoo | atgatggcgcctcctccccactgc | gcaactgcctgaacatctcctc |
| Smad2 | tgctctccaacgttaaccgaaa | tcagcaaacttttttccccaattoo | gccactgtagaaatgacaagaagaca | tgtaatacaagcggcccactccccttttc |
| Smad3 | ccaatgtcaaccggaatgcag | tgaggcactccggcaaagacc | cgtggaacttacaaggcgaca | cccctccgatggtagtagagc |
| Smad4 | ttgcctcaccaccaaaacg | tggaatgcaagctcattgtgtga | ccatttcttcagcaccacccgcccgta | tggccagtaatgtccaggatg |
| Smad5 | aaccatggattcgaggaggctgtg | tgacgtgtcctgtctcggtggtactc | tgagctcaccaagatgtgtacc | gctccccagcccttgacaaa |
| Smad6 | ttctcggttttttttttttttcctccttgac | ttcaccccggagcagtgatga | gtacaagccccactggatctgtccgatt | ggagtttttttggggggggttttt |
| Smad7 | ggaagatcaaccccccggagctg | tgagaaaatccccattgggtatttttatgga | tgtggctgcaacccccccatcac | aaggaggaggagggagagactcta |
| Smad9 | gcacgattcggattcggatgagtttttg | tgccagcggtccatgaagatg | gaagggggggagagcagagt | tctccgatccatccagcagggggtgct |
| Snai1 | cgatctgcacgaccgacctgtgtggaaa | cgtggagcggtcagcaaaagca | cgactctaggccccctggttttgctttt | cgactctaggccccctggttttgctttt |
| Snai2 | cgagcacattcgaacccacaca | cgttgggcagtgagggcaagagaaa | cgattgccttttgtgtgtgtgcaaga | cgattgccttttgtgtgtgtgcaaga |
| Sox7 | agaacccggaccaccacctgcacaac | ccgctctgcctctcatccacat | cggagctcagcaagatgc | ggtctcttttttttgggacagtgactgtcagc |
| Tal1 | accggatgccttttttttccatgatgtt | gcgccggcactactactttttggttttttt | ccaacaacaacaaccgggtgaaga | aggaccaccatcagaaatttccatctccatctca |
| Tek | tccaaaggagaatggcggcagg | tccggattgtttttttttggggttttc | ttccagaacgtgagagaagaacca | |
| Tgfb1 | accccccactgatacggcctga | gcagtgagccggctgaatcgaa | tggctgtctcttttttgacgactcactg | gccctgtgtattccttttttttttttttgg |
| Tgfb2 | ggcatgcccatatctatttatggagttc | cagatccttgggacacacacacagca | gacactcaacacaacacaaagtccaaagtcctca | gggaagcggaagcttttcgggattta |
| Tgfb3 | catgtcactttttcagcccccaat | ctccacggggatggatggattcatct | gagacatactggaaaatgttcatgaggtgtg | cattgtccccactcctttgaatttgaatttga |
| Tgfbr1 | gcagacttggggacttgacttgctgtgtga | catctagaacttcagggggatgt | catgattctgccacagatacaa | ccttttagtgtgcccctacttttttttttttggtgg |
| Tgfbr2 | tggccggctgcatatatcgtcct | gcatttttttttttttttttttccatttcca | tggacggcgccatccgccccagca | atccgacttttgggaacgtg |
| Thpo | ccggacgtcgaccctttttttttttttoo | tgcccctagaatgtcctgtgtgtgc | tgctctgtttttttttttttttttttttttoo | |
| Upp1 | cgagaaggaagagacgtggctctacca | cgtatgaaggtgttttcatccccgggaa | cgattcaaccccccacacacacactagcacacacacac cgattcaacccacacacacacacacacacao | cgattcaaccccccacacacacactagcacacacacac cgattcaacccacacacacacacacacacao |
Sequenziamento RNA a cella singola C1
Dopo aver ottenuto 6.500 cellule in 13 µl di PBS con l’arricchimento FACS, è stato aggiunto Fluidigm Suspension Reagent in rapporto 3:2 e delicatamente miscelato con pipettaggio. Un Fluidigm C1 10-17 uM IFC Chip è stato innescato, 5 µl di miscela di sospensione cellulare è stato aggiunto all’ingresso della cella, e il chip è stato caricato nello strumento C1. L’annotazione della cattura delle cellule è stata effettuata su un microscopio a campo luminoso con ingrandimento 40x e ogni sito catturato è stato notato per avere la cattura di una singola cellula, la cattura di doppiette o detriti. Lisi, trascrizione inversa e reagenti PCR (Clontech Takara) sono stati poi caricati in ingressi appropriati come da layout Fluidigm. I controlli ERCC spike-in (Ambion) sono stati aggiunti ad una diluizione di 1:4000 all’interno della miscela di lisi per ottenere il limite di rilevazione e la normalizzazione nelle analisi a valle. L’IFC è stato caricato nello strumento C1 durante la notte utilizzando l’mRNA-Seq RT e lo script Amp, e ~3 µl di cDNA sono stati raccolti e diluiti in 5 µl di tampone di diluizione del DNA Fluidigm C1. La distribuzione dimensionale e la quantificazione dei singoli campioni di cDNA è stata ottenuta utilizzando un AATI Fragment Analyzer (AATI) e la concentrazione di cDNA è stata diluita a 100 pg/µl in 10 mM Tris HCl pH 8,0 per la preparazione della libreria. La preparazione della biblioteca Illumina è stata effettuata utilizzando il sistema Nextera XT DNA (Illumina), ma i volumi sono stati ridotti secondo il protocollo modificato Fluidigm C1. La tagmentazione è stata eseguita su tutti i 96 campioni utilizzando 1,25 µl di cDNA (125 pg di concentrazione totale) combinato con 2,5 µl di tampone di tagmentazione (TD) e 1,25 µl di Amplicon Tagment Mix (ATM), e incubato per 10 minuti a 55 gradi. Dopo l’incubazione, sono stati aggiunti 1,25 µl di Neutrilize Tagment Mix (NT). Illumina PCR indici di codici a barre sono stati combinati per ottenere 96 combinazioni distinte di codici a barre i7 e i5, e 2,5 µl di indice combinato è stato aggiunto insieme a 3,75 µl di Nextera PCR Mix (NPM). I campioni sono stati poi sottoposti a 12 cicli di PCR secondo il protocollo Illumina. Ogni colonna è stata poi raggruppata in una provetta di PCR a 8 strisce da 0,2 mL e la purificazione di Ampure XP è stata eseguita, dopo di che ogni piscina è stata combinata per ottenere un singolo 96-campione multiplexato pool di 96-campioni di libreria con codice a barre. La libreria di sequenziamento finale è stata quantificata da Qubit (Life Technologies) e la distribuzione delle dimensioni è stata misurata su un BioAnalizzatore Agilent. I campioni sono stati sequenziati con l’Illumina HiSeq 2000.
Wafergen sequenziamento RNA monocellulare
Le cellule sono state colorate con il Cell Viability Imaging kit (Sonde Molecolari), che contiene Hoechst 33342 e ioduro di propidio, e successivamente sono state contate con il Moxi Z Mini Automated Cell Counter (ORFLO). La soluzione cellulare macchiata è stata diluita in una miscela con diluente e inibitore della RNasi (New England Biolabs) a 1 cella/50 nl per l’erogazione sul ICell8-chip (Wafergen) con il MultiSample NanoDispenser (Wafergen). I controlli positivi e negativi sono stati preparati secondo il protocollo ICell8 e dispensati con il MSND nei rispettivi pozzetti del chip. Tutti i pozzetti del chip ICell8 sono stati ripresi con un microscopio a fluorescenza (Olympus). Le immagini sono state analizzate con il software CellSelect (Wafergen). Le singole cellule vive, che sono Hoechst-33342-positive e propidio-iodide negativo, sono state selezionate per la lisi e la trascrizione inversa all’interno del chip ICell8 (200 cellule i8TFs + dox, 150 cellule i8TFs -dox, 150 Empty -dox e 150 Empty +dox). La miscela di reazione RT contenente 5X RT buffer RT, dNTPs, RT e5-oligo (Wafergen), acqua priva di nucleasi, Maxima H Minus RT (Thermo Scientific) e Triton X-100 è stata preparata ed erogata nei nanovalori precedentemente selezionati con singole cellule all’interno. Il chip è stato inserito all’interno di uno SmartChip Cycler (Bio-Rad) modificato per la reazione RT (42°C per 90 min, 85°C per 5 min, 4°C per sempre).
Il cDNA di tutte le singole cellule è stato raccolto insieme e ulteriormente concentrato con il DNA Clean and Concentrator-5 kit (Zymo Research). La reazione dell’esonucleasi I (New England Biolabs) del cDNA (37°C per 30 min, 80°C per 20 min, 4°C per sempre) è stata eseguita all’interno di un termociclatore convenzionale. Successivamente, il cDNA è stato amplificato con l’Advantage 2 PCR Kit (Clontech Takara) contenente tampone, dNTPs, Amp Primer (Wafergen), mix di polimerasi e acqua priva di nucleasi (95°C per 1 min, 18 cicli di 95°C per 15 s, 65°C per 30 s e 68°C per 6 min, seguito da 72°C per 10 min e 4°C per sempre). Il cDNA amplificato è stato purificato con Ampure XP Beads (Beckmann Coulter). La distribuzione della dimensione del cDNA è stata ottenuta con il BioAnalizzatore del DNA ad Alta Sensibilità (Agilent) e la quantificazione è stata eseguita con il Qubit (Life Technologies). La preparazione della libreria Illumina è stata effettuata utilizzando Nextera XT DNA (Illumina). La tagmentazione è stata eseguita in tagment DNA buffer, Amplicon Tagment Mix e 1 ng di cDNA purificato (55°C per 5 min e 10°C per sempre), è stato aggiunto Nextera Tagment Buffer Neutralizza Tagment Buffer (temperatura ambiente per 5 min). Dopo l’incubazione, la miscela di reazione NexteraXT PCR è stata preparata con Nextera PCR Mastermix, i7 Index Primer del Nextera Index Kit (Illumina), Nextera Primer P5 e Tagmented cDNA-NT buffer mix tampone (72°).C per 3 min, 95°C per 30 s, 12 cicli di 95°C per 10 s, 55°C per 30 s e 72°C per 30 s, finale 72°C per 5 min e 10°C per sempre). La purificazione di Ampure XP è stata eseguita con la biblioteca finita. La distribuzione delle dimensioni è stata controllata su un BioAnalizzatore Agilent. I campioni sono stati sequenziati con l’Illumina NextSeq.
Quantificazione e analisi statistica
Analisi dell’arricchimento del termine GO
Tutte le analisi di arricchimento dei termini GO sono state effettuate utilizzando il server web g:Profiler (Reimand et al., 2016).
Analisi citometrica a flusso
Tutti gli esperimenti di citometria a flusso sono stati ripetuti indipendentemente tre volte. Per la Figura 3C e la Figura 3D, il t-test accoppiato è stato eseguito sulle frequenze delle cellule. Per la Figura 3-figure supplement 5D, le frequenze per ogni popolazione sono state tracciate utilizzando il software JMP (SAS). Le barre di errore corrispondono alle deviazioni standard.
Analisi quantitativa dei dati RT-PCR a cella singola
I dati quantitativi monocellulari RT-PCR sono stati prima elaborati con il software Fluidigm Real Time PCR Analysis (soglia di qualità, 0,65; soglia Ct, Auto (Global); e metodo di correzione della linea di base, lineare [derivato]). Il clustering gerarchico e le PCA sono stati eseguiti utilizzando il pacchetto SINGuLAR Analysis toolset (Fluidigm versione 3.5) in R (R Core Team , 2016). Utilizzando questo set di strumenti, i valori Ct sono stati convertiti in log2ex. Il valore Ct è un valore relativo di espressione genica nel dominio log2 (supponendo che l’efficienza della PCR sia uguale a 1). La conversione da Ct a log2ex si basa sulle seguenti formule:
Log2ex = LOD – Ct, se Ct è inferiore al limite di rilevazione (LOD)
Log2ex = 0, se Ct è uguale o superiore a LOD.
Dove il valore predefinito è 24 (il tipico valore Ct per una singola copia di input nel chip Fluidigm). Nel nostro caso, il valore Ct è un’espressione relativa e non ha unità. Quindi, dopo la conversione nel dominio Log2ex, abbiamo etichettato i valori risultanti come ‘Log2 Gene Expression’.
Le analisi gerarchiche di clustering e PCA sono state eseguite con le funzioni HC() e PCA() di questo pacchetto. La funzione HC raggruppa i geni dei cluster con il metodo Pearson Correlation, cioè i geni co-profilati sono raggruppati insieme e i campioni sono raggruppati per distanza euclidea normalizzata (distanza/numero di geni), che rappresenta il cambiamento di piega media. L’analisi HC utilizza il metodo del “collegamento completo” (un metodo dal basso verso l’alto) per trovare cluster simili. L’opzione di visualizzazione ‘global_z_score’ normalizza il valore dell’espressione utilizzando la media globale e la deviazione standard globale.
La media di espressione genica dei geni ematopoietici ed endoteliali è mostrato per ogni singola cellula nella figura 1-figure supplement 3. La media ematopoietica è basata sui geni Epo, Epor, Gata1, Gata2, Gfi1, Gfi1b, Hbb-bh1, Itga2b, Itgam, Itgb3, Kit, Lmo2, Lyl1, Mpo, Myb, Ptprc, Runx1, Sfpi1, Sla, Tal1 e Thpo e la media endoteliale su Cdh5, Cldn5, Eng, Esam, Fbn1, Gpr126, Kdr, Npr1, Pcdh12, Pecam1, Ptprb, Ptprm, Ramp2, Sox7 e Tek.
Un prototipo è stato calcolato per ogni gruppo di cluster di singole celle determinato dal risultato di clustering gerarchico mostrato in Figura 1-figure supplementi 1 e 2. Il prototipo è definito dalla media di tutte le cellule appartenenti al suo cluster. Il coefficiente di correlazione Pearson tra tutti i prototipi a coppie è visualizzato in Figura 1-figure supplemento 4A. Il pacchetto R ComplexHeatmap ComplexHeatmap è stato utilizzato (Gu et al., 2016).
Un modello lineare generalizzato con distribuzione Quasi-Poisson e logaritmo come funzione di collegamento è stato applicato per modellare i dati della tabella di conteggio come mostrato nella Figura 4B. Sono stati definiti sei gruppi di controllo, uno per ogni categoria di cluster di celle come Endo o VSM. Ogni gruppo di controllo comprende 10 campioni e comprende tutte le condizioni -dox. Per ogni categoria di cluster e per ciascuna delle linee cellulari di sovraespressione, il conteggio della condizione +dox è stato confrontato con i conteggi del rispettivo gruppo di controllo. Di questi 60 contrasti statistici (sei categorie di cluster per nove linee di celle di sovraespressione più la linea di cella che porta un vettore vuoto), cinque sono stati identificati per essere significativamente diversi con un valore p corretto<0,05. Il metodo di Benjamini-Hochberg è stato utilizzato per la correzione dei test multipli. Inoltre, il modello lineare generalizzato comprendeva un offset a tassi di modello invece di conteggi. I tassi rappresentano piccole differenze nel numero totale di celle misurate su ciascuna delle piastre del pozzo elaborato. Tutte e cinque le cinque condizioni significativamente diverse sono contrassegnate con una stella nella Figura 4B (vedi file supplementare 6 per i valori p).
analisi dei dati di microarray di mRNA
I dati del microarray mRNA sono stati analizzati utilizzando oligo, filobasi, metodi, biobase, statistiche, ggplot2, gplot, matrixstats, grafici, annotationdbi e pacchetti limma in R (R Core Team , 2016).
C1 analisi dei dati RNAseq monocellulare
Ogni biblioteca è stata messa in sequenza generando 426.065.795 e 440.774.932 paired-end di lettura (di 125 pb). Le letture sono state assegnate al pozzo con il codice a barre corrispondente. All’interno del codice a barre del pozzo sono tollerati fino a due disallineamenti se l’assegnazione è rimasta non ambigua. STAR, versione 2.4.0 (Dobin et al., 2013), ha allineato le letture al genoma del mouse build mm10/GRCm38. Le letture duplicate sono state rimosse usando samtools, versione 0.1.18 (Li et al., 2009). I pozzi con meno di 1 milione di letture sono stati considerati come pozzi vuoti ed esclusi da ulteriori analisi (2 e 1, rispettivamente). Le letture con una qualità di mappatura minima di 30, che mappate su un gene unico, sono state contate utilizzando la versione 1.4.6 di featureCounts (Liao et al., 2014).
I conteggi delle letture sono stati normalizzati utilizzando RUVg, versione 1.0.0 (Risso et al., 2014), utilizzando i controlli ERCC spike-in e i fattori di dimensione (SF) come definiti in DESeq (Anders e Huber, 2010). Sono state escluse le celle con >7% di letture mappate su rRNA (tre in ogni piastra) e che erano outliers sulla base del primo componente principale dopo la normalizzazione del fattore di dimensione. Le restanti cellule contenevano tutte >7.000 geni rilevati e sono state utilizzate in tutte le analisi successive.
Abbiamo effettuato un’analisi di rete utilizzando la misura di correlazione generalizzata della distanza, dcor statistic (Székely et al., 2007), insieme alla sua versione condizionale, pdcor (Székely e Rizzo, 2014). Per ogni chip e per ogni metodo di normalizzazione (quattro configurazioni), tutte le relazioni significative tra il trascrittoma e i geni del seme sono state identificate utilizzando la statistica dcor. I geni del seme utilizzati per questa analisi includevano le 8 TF così come Gata1, Gfi1b, Spi1, Spi1, Ldb1 e Cbfa2t3. I geni dei semi aggiuntivi sono stati selezionati se sono apparsi in reti pubblicate da almeno due diversi autori (Beck et al., 2013; Bonzanni et al., 2013; Moignard et al., 2013; Moignard et al., 2013; Moignard et al., 2015; Tanaka et al., 2012). La direzionalità delle relazioni (correlazione positiva, correlazione negativa o ‘altro’) è stata definita utilizzando un test t a due code, diretto è stato basato sul segno del test e ‘altro’ è stato assegnato a tutti i geni con valore p non corretto>0,05. Per garantire la robustezza, abbiamo incluso solo le relazioni che sono state identificate in almeno due delle quattro configurazioni, e qualsiasi relazione con direzioni contrastanti è stata assegnata ad ‘altro’. Infine, le interazioni tra i geni del seme sono state identificate condizionando i “bersagli” di ogni gene del seme sull’espressione di ogni altro gene del seme. Un’interazione è stata dedotta se le relazioni tra entrambi i geni del seme e un particolare gene ‘target’ aumentavano di forza quando condizionati sull’altro gene del seme.
Per l’analisi della rete Spearman, abbiamo combinato Exp1 ed Exp2 in un’unica matrice di espressione per ogni normalizzazione (SF e RUV). Abbiamo calcolato le correlazioni di Spearman utilizzando la funzione R di base per ogni normalizzazione e abbiamo fatto la media delle correlazioni tra le due normalizzazioni. Abbiamo cambiato tutte le correlazioni con valore assoluto <0.25 (cioè <0.25 e >-0.25) a 0. Abbiamo creato una rete con bordi ponderati tra i geni, correlazioni di 0 = nessun bordo. Abbiamo calcolato la centralità di eigen utilizzando eigen_centrality dal pacchetto igraph R (Bonacich, 1987). Abbiamo preso il valore assoluto di tutti i bordi (trasforma tutte le relazioni in positive) e calcolato la centralità di betweenness (numero di percorsi più brevi che passano attraverso un nodo) usando la betweenness dal pacchetto igraph R (Brandes, 2001). Abbiamo calcolato il grado (numero di bordi attaccati ad un nodo) usando il grado dal pacchetto igraph R. Poi, abbiamo calcolato il valore mediano di ogni misura attraverso tutti i geni della rete. Abbiamo contato il numero di geni nella lista che sono al di sopra della mediana e ne abbiamo testato il significato utilizzando un test binomiale (per definizione il 50% dei geni è al di sopra della mediana).
Analisi dei dati RNAseq monocellulari Wafergen
673 pozzi sul chip Wafergen hanno superato l’ispezione microscopica. Il pooling, la costruzione della biblioteca e il sequenziamento hanno generato 179 milioni di sequenze di paired-end utilizzando il protocollo Illumina Nextera. Si noti che questo sequenziamento più basso riduce notevolmente il tasso di rilevamento delle TF, ma il maggior numero di cellule ha facilitato la valutazione dell’eterogeneità all’interno e tra le popolazioni. La prima lettura della coppia codifica il codice a barre del pozzo e il codice a barre UMI (identificatore molecolare unico), la seconda codifica la fine 3′ della trascrizione. Le letture sono state assegnate al pozzetto con il codice a barre corrispondente. All’interno del codice a barre del pozzo sono tollerati fino a due errori di corrispondenza se l’assegnazione è rimasta non ambigua. Cutadapt ha tagliato le sequenze poly-A, poly-G e adattatore e STAR ha allineato le letture alla costruzione del genoma del mouse mm10 (Dobin et al., 2013; Martin, 2011). FeatureCounts ha determinato tutte le letture in modo specifico in modo che si sovrappongano esattamente con un gene (Liao et al., 2014). I livelli di espressione genica sono calcolati contando il numero di UMI distinte di tutte le letture specifiche del gene (“UMI per gene”), come determinato dai FeatureCounts. Sono state contate solo le letture allineate in modo univoco. Per ridurre ulteriormente l’impatto degli errori di mappatura e di sequenziamento sulla quantificazione, è stata introdotta una fase di correzione e di filtraggio degli errori di codice a barre UMI. Se due o più UMI distinti per gene condividono un codice a barre simile, che differiscono al massimo di uno o due errori di allineamento, allora possono avere origine dallo stesso codice a barre UMI. Il codice a barre UMI viene corretto solo se ha meno del 10% di letture rispetto a quello più abbondante. Questa procedura di correzione inizia con il codice a barre UMI, che è supportato dal maggior numero di letture specifiche del genere, e che determina tutti i codici a barre simili e li corregge, se applicabile. Si riavvia con tutti i codici a barre UMI che non sono stati considerati in una precedente iterazione fino a quando non ne rimane nessuno. In una fase di filtraggio separata, qualsiasi UMI distinto per gene che sia supportato solo da una o due letture allineate viene completamente omesso dal conteggio. Dalla tabella di conteggio risultante, tutti i conteggi provenienti dai 23 pozzetti di controllo sono stati rimossi per la successiva analisi esplorativa. Tutti i pozzetti rimanenti corrispondono a singole cellule. Filtraggio supplementare ha ridotto la sparsità trovato in questo gene per matrice cellulare di UMI conteggi UMI. Solo le cellule con 1000 o più geni con almeno un conteggio UMI e i geni con almeno un conteggio UMI in 10 o più cellule sono stati selezionati, lasciando una matrice di 478 singole cellule e 10.238 geni. Questa matrice è stata ridimensionata e normalizzata con il pacchetto Bioconductor scater (McCarthy et al., 2017). I componenti principali sono stati calcolati per i 400 geni che hanno mostrato la più alta varianza dopo aver sottratto l’andamento della varianza media di tutti i geni. L’analisi esplorativa è stata effettuata utilizzando R (R Core Team , 2016).
Dati e disponibilità del software
Software
Tutti i software sono disponibili gratuitamente o in commercio e sono elencati nella descrizione dei metodi e nella tabella di riferimento del prodotto.
Risorse di dati
Il numero di adesione per tutti i dati sc-RNA-seq e microarray riportati in questo documento è GEO: GSE96986.
Tabella di riferimento del prodotto
| Prodotto o risorsa | Fornitore | Numero di riferimento |
|---|---|---|
| Anticorpi | ||
| Anti-6X Il suo anticorpo [HIS.H8]. | Abcam | ab18184 |
| Anti-alfa-Tubulina-Antibody (mouse) | Sigma | T9026 |
| Microsfere anti-APC | Miltenyi Biotec | 130-090-855 |
| Anti-Cbfb Anticorpo | Abcam | ab133600 |
| Anti-Erg Anticorpo | Abcam | ab92513 |
| Anticorpo M2 anti-FLAG | Sigma | F1804 |
| Anticorpo anti-FLI1 [EPR464646]. | Abcam | ab133485 |
| Anticorpo anti-GATA2 [EPR2822(2) | Abcam | ab109241 |
| Anticorpo anti-HA prodotto nel coniglio | Sigma | H6908 |
| Anti-Lmo2 Anticorpo | Abcam | ab91652 |
| Anticorpo anti-Lyl1 [KT43] | Abcam | ab53354 |
| Anti-Mouse CD144 (VE-Cadherin) eFluor 660, eBioBV13 | eBioscienza | 50-1441-80 |
| Anti-Topo CD309 (FLK1) APC, Avas12a1 | eBioscienza | 17-5821-81 |
| Anti-Mouse CD41 PE, MWReg30 | eBioscienza | 12-0411-81 |
| Anti-Topo cKit APC Anticorpo | eBioscienza | 17-1171-82 |
| Anti-Mouse HRP | GE Healthcare Life Sciences | NA931V |
| Anti-Rat HRP | GE Healthcare Life Sciences | NA935V |
| Anti-Runx1 Anticorpo | Abcam | ab92336 |
| Anti-Tal1 Anticorpo | Abcam | ab119754 |
| Anticorpo Anti-V5 | Tecnologie per la vita | 46–0705 |
| Perossidasi AffiniPure Capra Anti-coniglio IgG (H + L) | Jackson | 111-035-144 |
| Prodotti chimici | ||
| 2-mercaptoetanolo | GIBCO | 31350–010 |
| Acido ascorbico | Sigma | A4544 |
| Collagenasi di tipo IA da Clostridium histolyticum liofilizzato in polvere (da soluzione sterile-filtrata), 0,5-5,0 unità FALGPA/mg solido, coltura cellulare testata | Sigma | C9722-50MG |
| D4T surnatante | EMBL-Roma | N/A |
| Doxiciclina | Sigma | D9891 |
| UEB | R e D | 959-ME-010 |
| ERCC RNA Spike-In Mix | Ambion | 4456740 |
| Esonucleasi I | New England Biolabs | M0293L |
| Siero fetale bovino plasma-derivato da plasma povero di piastrine (PDS) | Antech | N/A |
| Siero fetale bovino (FBS) | PAA | A15-102 |
| Fluidigm C1 Tampone di diluizione del DNA | Fluidigm | 100–5317 |
| Fluidigm Reagente di sospensione | Fluidigm | 100–6201 |
| Gelatina | BDH | 440454B |
| Colorante fantasma rosso 780 | Bioscienze Tonbo | 13–0865 |
| GM-CSF | R e D | 425 ML |
| FGF umano di base | R e D | 233-FB-025 |
| IL-11 | R e D | 418 ML |
| IL-3 | R e D | 403 ML |
| IL-6 | R e D | 406 ML |
| IMDM | Lonza | BE12-726F |
| KnockOut DMEM | GIBCO | 10829–018 |
| L-glutammina | GIBCO | 25030–024 |
| LIF | EMBL Heidelberg | N/A |
| Massima H meno trascrittasi inversa | Termo Scientifico | EP0752 |
| MCSF | R e D | 416 ML-010 |
| MEM Soluzione di amminoacidi non essenziali | GIBCO | 11140–035 |
| Metilcellulosa | VWR | 9004-67-5 |
| Monotioglicerolo (MTG) | Sigma | M6145 |
| Oncostatina M | R e D | 495-MO |
| Penicillina-streptomicina | GIBCO | 15140–122 |
| PFMH-II | GIBCO | 12040–077 |
| Buffer RIPA Pierce | Termo Scientifico | 89900 |
| Soluzione Ponceau S | Sigma | P7170 |
| Inibitore RNase | New England Biolabs | M0314S |
| SCF | R e D | 455-MC |
| TPO | R e D | 488-TO-005 |
| Transferrin | Roche (Italia) | 10652202001 |
| TrypLE express | GIBCO | 12605–036 |
| VEGF | R e D | 293-VE |
| Saggi commerciali | ||
| Vantaggio 2 Kit PCR | Clontech Takara | 639207 |
| Amersham ECL Prime Western Blotting Reagent per il rilevamento delle macchie occidentali | GE Healthcare Life Sciences | RPN2232 |
| Allegatoin V-FITC Kit di rilevamento dell’apoptosi | eBioscienza | 88-8005-72 |
| Kit di imaging per la viabilità delle cellule | Sonde molecolari | R37610 |
| CellsDirect One-Step qRT-PCR kit | Invitrogen | 11753100 |
| Kit per il test citometrico a flusso Click-iT Plus EdU Alexa Fluor 488 | Sonde molecolari | C10633 |
| Kit DNA Clean e Concentratore-5 | Ricerca Zymo | D4013 |
| Sistema di analisi del reporter a doppia lievitazione | Promega | E1910 |
| ICell8-chip e kit reagente | Wafergen | 430–000233 |
| Kit di trasfezione NanoJuice | Merck Millipore | 71902 |
| Kit indice Nextera | Illumina | FC-131-1001 |
| Nextera XT DNA Library Prep kit di preparazione della biblioteca del DNA – 24 campioni (esperimento Wafergen) | Illumina | FC-131-1024 |
| Nextera XT DNA Library Prep kit per la preparazione di 96 campioni (esperimento C1) | Illumina | FC-131-1096 |
| Kit di estrazione nucleare | ActiveMotif | 40010 |
| Kit per il dosaggio delle proteine BCA Pierce | Termo Scientifico | 23225 |
| Kit RNAeasy Plus Micro | Qiagen | 74034 |
| Kit SMARTer Ultra Low RNA per il sistema Fluidigm C1, 10 IFC | Clontech Takara | 634833 |
| Dati depositati | ||
| Dati grezzi e analizzati di sequenziamento e microarray | Questo documento | GEO: GSE96986 |
| Dati di sequenziamento grezzi e analizzati | (Goode et al., 2016) | GEO: GSE69101 |
| Dati di sequenziamento grezzi e analizzati | (Pereira et al., 2016) | GEO: GSE54574 |
| Linee cellulari | ||
| Linea A2lox.Cre mESC | (Iacovino et al., 2011) | A2lox.Cre |
| Linea mESC A2lox.empty | Questo documento | Vuoto |
| Linea A2lox.i8TFs mESC | Questo documento | i8TFs |
| Linea A2lox.iCbfb mESC | Questo documento | iCbfb |
| Linea A2lox.iErg mESC | Questo documento | iErg |
| Linea A2lox.iFli1 mESC | Questo documento | iFli1 |
| Linea A2lox.iGata2 mESC | Questo documento | iGata2 |
| Linea A2lox.iLmo2 mESC | Questo documento | iLmo2 |
| Linea A2lox.iLyl1 mESC | Questo documento | iLyl1 |
| Linea A2lox.iRunx1 mESC | Questo documento | iRunx1 |
| Linea A2lox.iTal1 mESC | Questo documento | iTal1 |
| Runx1+/hCD4 | (Sroczynska et al., 2009a) | Runx1+/hCD4 |
| Topi | ||
| C57BL/6N | EMBL Roma | |
| Gfi1b:GFP topi knock-in | (Vassen et al., 2007) | |
| Gfi1:GFP topi knock-in | (Yücel et al., 2004) | |
| DNA ricombinante | ||
| Plasmide: p2lox_5TFs | Questo documento | p2lox_5TFs |
| Plasmide: p2lox_6TFs | Questo documento | p2lox_6TFs |
| Plasmide: p2lox_8TFs | Questo documento | p2lox_8TFs |
| Plasmide: p2lox_vuoto | (Vargel et al., 2016) | p2lox_vuoto |
| Plasmide: p2lox_K_Cbfb | Questo documento | p2lox_K_Cbfb |
| Plasmide: p2lox_K_Erg | Questo documento | p2lox_K_Erg |
| Plasmide: p2lox_K_Fli1 | Questo documento | p2lox_K_Fli1 |
| Plasmide: p2lox_K_Gata2 | Questo documento | p2lox_K_Gata2 |
| Plasmide: p2lox_K_Lmo2 | Questo documento | p2lox_K_Lmo2 |
| Plasmide: p2lox_K_Lyl1 | Questo documento | p2lox_K_Lyl1 |
| Plasmide: p2lox_K_Runx1 | Questo documento | p2lox_K_Runx1 |
| Plasmide: p2lox_K_Tal1 | Questo documento | p2lox_K_Tal1 |
| Plasmide: pGL4.10[luc2] | Promega | E6651 |
| Plasmide: pGL4.74[hRluc/TK] | Promega | E6921 |
| Software e algoritmi | ||
| CellProfiler | (Kamentsky et al., 2011) | http://www.cellprofiler.org |
| Software CellSelect | Wafergen | N/A |
| Figi | (Schindelin et al., 2012) | http://fiji.sc/Fiji |
| FlowJo | Tree Star, Inc. | N/A |
| Software JMP | SAS | N/A |
| Altri | ||
| 96,96 array dinamico IFC | Fluidigm | BMK-M-96,96 |
| Analizzatore di frammenti AATI | AATI | DNF-474 |
| Perline Ampure XP | Beckmann coltro | B23319 |
| BioAnalyzer | Agilent | N/A |
| Sistema Biomark HD | Fluidigm | N/A |
| FACSAria | Becton Dickinson | N/A |
| FACSCanto | Becton Dickinson | N/A |
| Fluidigm C1 10-17 uM Chip IFC | Fluidigm | 100–5760 |
| Microscopio a fluorescenza per l’imaging dei chip Wafergen | Olympus | BX43F |
| Piastre PCR a guscio duro | Bio-Rad | HSP9611 |
| HiSeq 2000 | Illumina | N/A |
| Sistema ICell8 | Wafergen | N/A |
| IncuCyte HD | Essen Bioscienze | N/A |
| Tecnologia MACS MicroBead | Miltenyi Biotec | N/A |
| Moxi Z Mini contacellule automatizzato | ORFLO | MXZ001 |
| NanoDispenser MultiSample | Wafergen | N/A |
| NextSeq | Illumina | N/A |
| NuPAGE 12% Gel Proteici Bis-Tris | Tecnologie per la vita | NP0343BOX |
| Indici con codice a barre PCR | Illumina | FC-131-200 |
| Dispositivo POLARstar Omega | BMG Labtech | N/A |
| Membrana di nitrocellulosa PROTRAN | PerkinElmer | NBA085C |
| Qubit | Tecnologie per la vita | N/A |
| SmartChip Cycler | Bio-Rad | T-100 |
| Universal cDNA Reverse Trascritto da Random Hexamer: Tessuti normali per mouse | BioChain | C4334566-R |
| Sistema di elettroforesi a mini celle XCell SureLock | Invitrogen | N/A |
References
- Anders S, Huber W. Differential expression analysis for sequence count data. Genome Biology. 2010; 11DOI | PubMed
- Asano Y, Stawski L, Hant F, Highland K, Silver R, Szalai G, Watson DK, Trojanowska M. Endothelial Fli1 deficiency impairs vascular homeostasis: a role in scleroderma vasculopathy. The American Journal of Pathology. 2010; 176:1983-1998. DOI | PubMed
- Beck D, Thoms JA, Perera D, Schütte J, Unnikrishnan A, Knezevic K, Kinston SJ, Wilson NK, O’Brien TA, Göttgens B, Wong JW, Pimanda JE. Genome-wide analysis of transcriptional regulators in human HSPCs reveals a densely interconnected network of coding and noncoding genes. Blood. 2013; 122:e12-e22. DOI | PubMed
- Bergiers I, Tischer C, Vargel Bölükbaşı Ö, Lancrin C. Quantification of mouse hematopoietic progenitors’ Formation using Time-lapse microscopy and image analysis. Bio-Protocol. 2019; 9DOI | PubMed
- Bertrand JY, Giroux S, Cumano A, Godin I. Hematopoietic stem cell development during mouse embryogenesis. Methods in Molecular Medicine. 2005; 105:273-288. PubMed
- Boisset JC, van Cappellen W, Andrieu-Soler C, Galjart N, Dzierzak E, Robin C. In vivo imaging of haematopoietic cells emerging from the mouse aortic endothelium. Nature. 2010; 464:116-120. DOI | PubMed
- Bonacich P. Power and centrality: a family of measures. American Journal of Sociology. 1987; 92:1170-1182. DOI
- Bonzanni N, Garg A, Feenstra KA, Schütte J, Kinston S, Miranda-Saavedra D, Heringa J, Xenarios I, Göttgens B. Hard-wired heterogeneity in blood stem cells revealed using a dynamic regulatory network model. Bioinformatics. 2013; 29:i80-i88. DOI | PubMed
- Brandes U. A faster algorithm for betweenness centrality*. The Journal of Mathematical Sociology. 2001; 25:163-177. DOI
- Choi K, Kennedy M, Kazarov A, Papadimitriou JC, Keller G. A common precursor for hematopoietic and endothelial cells. Development. 1998; 125:725-732. PubMed
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013; 29:15-21. DOI | PubMed
- Donnelly ML, Luke G, Mehrotra A, Li X, Hughes LE, Gani D, Ryan MD. Analysis of the aphthovirus 2A/2B polyprotein ‘cleavage’ mechanism indicates not a proteolytic reaction, but a novel translational effect: a putative ribosomal ‘skip’. Journal of General Virology. 2001; 82:1013-1025. DOI | PubMed
- Dunn SJ, Martello G, Yordanov B, Emmott S, Smith AG. Defining an essential transcription factor program for naïve pluripotency. Science. 2014; 344:1156-1160. DOI | PubMed
- Dzierzak E, de Pater E. Regulation of Blood Stem Cell Development. Current Topics in Developmental Biology. 2016; 118:1-20. DOI | PubMed
- Eilken HM, Nishikawa S, Schroeder T. Continuous single-cell imaging of blood generation from haemogenic endothelium. Nature. 2009; 457:896-900. DOI | PubMed
- Ferreira R, Ohneda K, Yamamoto M, Philipsen S. GATA1 function, a paradigm for transcription factors in hematopoiesis. Molecular and Cellular Biology. 2005; 25:1215-1227. DOI | PubMed
- Goardon N, Lambert JA, Rodriguez P, Nissaire P, Herblot S, Thibault P, Dumenil D, Strouboulis J, Romeo PH, Hoang T. ETO2 coordinates cellular proliferation and differentiation during erythropoiesis. The EMBO Journal. 2006; 25:357-366. DOI | PubMed
- Goode DK, Obier N, Vijayabaskar MS, Lie-A-Ling M, Lilly AJ, Hannah R, Lichtinger M, Batta K, Florkowska M, Patel R, Challinor M, Wallace K, Gilmour J, Assi SA, Cauchy P, Hoogenkamp M, Westhead DR, Lacaud G, Kouskoff V, Göttgens B, Bonifer C. Dynamic gene regulatory networks drive hematopoietic specification and differentiation. Developmental Cell. 2016; 36:572-587. DOI | PubMed
- Gu Z, Eils R, Schlesner M. Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics. 2016; 32:2847-2849. DOI | PubMed
- Guibentif C, Rönn RE, Böiers C, Lang S, Saxena S, Soneji S, Enver T, Karlsson G, Woods NB. Single-cell analysis identifies distinct stages of human endothelial-to-hematopoietic transition. Cell Reports. 2017; 19:10-19. DOI | PubMed
- Huang H, Yu M, Akie TE, Moran TB, Woo AJ, Tu N, Waldon Z, Lin YY, Steen H, Cantor AB. Differentiation-dependent interactions between RUNX-1 and FLI-1 during megakaryocyte development. Molecular and Cellular Biology. 2009; 29:4103-4115. DOI | PubMed
- Huber TL. Dissecting hematopoietic differentiation using the embryonic stem cell differentiation model. The International Journal of Developmental Biology. 2010; 54:991-1002. DOI | PubMed
- Iacovino M, Bosnakovski D, Fey H, Rux D, Bajwa G, Mahen E, Mitanoska A, Xu Z, Kyba M. Inducible cassette exchange: a rapid and efficient system enabling conditional gene expression in embryonic stem and primary cells. Stem Cells. 2011; 29:1580-1588. DOI | PubMed
- Imperato MR, Cauchy P, Obier N, Bonifer C. The RUNX1-PU.1 axis in the control of hematopoiesis. International Journal of Hematology. 2015; 101:319-329. DOI | PubMed
- Iwafuchi-Doi M, Zaret KS. Cell fate control by pioneer transcription factors. Development. 2016; 143:1833-1837. DOI | PubMed
- Kamentsky L, Jones TR, Fraser A, Bray MA, Logan DJ, Madden KL, Ljosa V, Rueden C, Eliceiri KW, Carpenter AE. Improved structure, function and compatibility for CellProfiler: modular high-throughput image analysis software. Bioinformatics. 2011; 27:1179-1180. DOI | PubMed
- Kolodziejczyk AA, Kim JK, Tsang JC, Ilicic T, Henriksson J, Natarajan KN, Tuck AC, Gao X, Bühler M, Liu P, Marioni JC, Teichmann SA. Single cell RNA-Sequencing of pluripotent states unlocks modular transcriptional variation. Cell Stem Cell. 2015; 17:471-485. DOI | PubMed
- Lancrin C, Sroczynska P, Stephenson C, Allen T, Kouskoff V, Lacaud G. The haemangioblast generates haematopoietic cells through a haemogenic endothelium stage. Nature. 2009; 457:892-895. DOI | PubMed
- Lancrin C, Mazan M, Stefanska M, Patel R, Lichtinger M, Costa G, Vargel O, Wilson NK, Möröy T, Bonifer C, Göttgens B, Kouskoff V, Lacaud G. GFI1 and GFI1B control the loss of endothelial identity of hemogenic endothelium during hematopoietic commitment. Blood. 2012; 120:314-322. DOI | PubMed
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. The sequence alignment/map format and SAMtools. Bioinformatics. 2009; 25:2078-2079. DOI | PubMed
- Liao Y, Smyth GK, Shi W. featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics. 2014; 30:923-930. DOI | PubMed
- Lichtinger M, Ingram R, Hannah R, Müller D, Clarke D, Assi SA, Lie-A-Ling M, Noailles L, Vijayabaskar MS, Wu M, Tenen DG, Westhead DR, Kouskoff V, Lacaud G, Göttgens B, Bonifer C. RUNX1 reshapes the epigenetic landscape at the onset of haematopoiesis. The EMBO Journal. 2012; 31:4318-4333. DOI | PubMed
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal. 2011; 17:10-12. DOI
- McCarthy DJ, Campbell KR, Lun ATL, Wills QF. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics. 2017; 247DOI
- McLaughlin F, Ludbrook VJ, Cox J, von Carlowitz I, Brown S, Randi AM. Combined genomic and antisense analysis reveals that the transcription factor Erg is implicated in endothelial cell differentiation. Blood. 2001; 98:3332-3339. DOI | PubMed
- Moignard V, Macaulay IC, Swiers G, Buettner F, Schütte J, Calero-Nieto FJ, Kinston S, Joshi A, Hannah R, Theis FJ, Jacobsen SE, de Bruijn MF, Göttgens B. Characterization of transcriptional networks in blood stem and progenitor cells using high-throughput single-cell gene expression analysis. Nature Cell Biology. 2013; 15:363-372. DOI | PubMed
- Moignard V, Woodhouse S, Haghverdi L, Lilly AJ, Tanaka Y, Wilkinson AC, Buettner F, Macaulay IC, Jawaid W, Diamanti E, Nishikawa SI, Piterman N, Kouskoff V, Theis FJ, Fisher J, Göttgens B. Decoding the regulatory network of early blood development from single-cell gene expression measurements. Nature Biotechnology. 2015; 33:269-276. DOI | PubMed
- Mullen AC, Orlando DA, Newman JJ, Lovén J, Kumar RM, Bilodeau S, Reddy J, Guenther MG, DeKoter RP, Young RA. Master transcription factors determine cell-type-specific responses to TGF-β signaling. Cell. 2011; 147:565-576. DOI | PubMed
- Mylona A, Andrieu-Soler C, Thongjuea S, Martella A, Soler E, Jorna R, Hou J, Kockx C, van Ijcken W, Lenhard B, Grosveld F. Genome-wide analysis shows that Ldb1 controls essential hematopoietic genes/pathways in mouse early development and reveals novel players in hematopoiesis. Blood. 2013; 121:2902-2913. DOI | PubMed
- Org T, Duan D, Ferrari R, Montel-Hagen A, Van Handel B, Kerényi MA, Sasidharan R, Rubbi L, Fujiwara Y, Pellegrini M, Orkin SH, Kurdistani SK, Mikkola HK. Scl binds to primed enhancers in mesoderm to regulate hematopoietic and cardiac fate divergence. The EMBO Journal. 2015; 34:759-777. DOI | PubMed
- Padrón-Barthe L, Temiño S, Villa del Campo C, Carramolino L, Isern J, Torres M. Clonal analysis identifies hemogenic endothelium as the source of the blood-endothelial common lineage in the mouse embryo. Blood. 2014; 124:2523-2532. DOI | PubMed
- Pereira CF, Chang B, Gomes A, Bernitz J, Papatsenko D, Niu X, Swiers G, Azzoni E, de Bruijn MF, Schaniel C, Lemischka IR, Moore KA. Hematopoietic reprogramming in vitro informs in vivo identification of hemogenic precursors to definitive hematopoietic stem cells. Developmental Cell. 2016; 36:525-539. DOI | PubMed
- R Core Team . R Foundation for Statistical Computing. 2016.
- Reimand J, Arak T, Adler P, Kolberg L, Reisberg S, Peterson H, Vilo J. g:Profiler-a web server for functional interpretation of gene lists (2016 update). Nucleic Acids Research. 2016; 44:W83-W89. DOI | PubMed
- Risso D, Ngai J, Speed TP, Dudoit S. Normalization of RNA-seq data using factor analysis of control genes or samples. Nature Biotechnology. 2014; 32:896-902. DOI | PubMed
- Robin C, Dzierzak E. Hematopoietic stem cell enrichment from the AGM region of the mouse embryo. Methods in Molecular Medicine. 2005; 105:257-272. PubMed
- Rybtsov S, Batsivari A, Bilotkach K, Paruzina D, Senserrich J, Nerushev O, Medvinsky A. Tracing the origin of the HSC hierarchy reveals an SCF-dependent, IL-3-independent CD43(-) embryonic precursor. Stem Cell Reports. 2014; 3:489-501. DOI | PubMed
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B, Tinevez JY, White DJ, Hartenstein V, Eliceiri K, Tomancak P, Cardona A. Fiji: an open-source platform for biological-image analysis. Nature Methods. 2012; 9:676-682. DOI | PubMed
- Scialdone A, Tanaka Y, Jawaid W, Moignard V, Wilson NK, Macaulay IC, Marioni JC, Göttgens B. Resolving early mesoderm diversification through single-cell expression profiling. Nature. 2016; 535:289-293. DOI | PubMed
- Solaimani Kartalaei P, Yamada-Inagawa T, Vink CS, de Pater E, van der Linden R, Marks-Bluth J, van der Sloot A, van den Hout M, Yokomizo T, van Schaick-Solernó ML, Delwel R, Pimanda JE, van IJcken WF, Dzierzak E. Whole-transcriptome analysis of endothelial to hematopoietic stem cell transition reveals a requirement for Gpr56 in HSC generation. The Journal of Experimental Medicine. 2015; 212:93-106. DOI | PubMed
- Sroczynska P, Lancrin C, Kouskoff V, Lacaud G. The differential activities of Runx1 promoters define milestones during embryonic hematopoiesis. Blood. 2009a; 114:5279-5289. DOI | PubMed
- Sroczynska P, Lancrin C, Pearson S, Kouskoff V, Lacaud G. In vitro differentiation of mouse embryonic stem cells as a model of early hematopoietic development. Methods in Molecular Biology. 2009b; 538:317-334. DOI | PubMed
- Székely GJ, Rizzo ML, Bakirov NK. Measuring and testing dependence by correlation of distances. The Annals of Statistics. 2007; 35:2769-2794. DOI
- Székely GJ, Rizzo ML. Partial distance correlation with methods for dissimilarities. The Annals of Statistics. 2014; 42:2382-2412. DOI
- Tahirov TH, Inoue-Bungo T, Morii H, Fujikawa A, Sasaki M, Kimura K, Shiina M, Sato K, Kumasaka T, Yamamoto M, Ishii S, Ogata K. Structural analyses of DNA recognition by the AML1/Runx-1 Runt domain and its allosteric control by CBFbeta. Cell. 2001; 104:755-767. DOI | PubMed
- Tanaka Y, Joshi A, Wilson NK, Kinston S, Nishikawa S, Göttgens B. The transcriptional programme controlled by Runx1 during early embryonic blood development. Developmental Biology. 2012; 366:404-419. DOI | PubMed
- Taoudi S, Gonneau C, Moore K, Sheridan JM, Blackburn CC, Taylor E, Medvinsky A. Extensive hematopoietic stem cell generation in the AGM region via maturation of VE-cadherin+CD45+ pre-definitive HSCs. Cell Stem Cell. 2008; 3:99-108. DOI | PubMed
- Thambyrajah R, Mazan M, Patel R, Moignard V, Stefanska M, Marinopoulou E, Li Y, Lancrin C, Clapes T, Möröy T, Robin C, Miller C, Cowley S, Göttgens B, Kouskoff V, Lacaud G. GFI1 proteins orchestrate the emergence of haematopoietic stem cells through recruitment of LSD1. Nature Cell Biology. 2016; 18:21-32. DOI | PubMed
- Vargel Ö, Zhang Y, Kosim K, Ganter K, Foehr S, Mardenborough Y, Shvartsman M, Enright AJ, Krijgsveld J, Lancrin C. Activation of the TGFβ pathway impairs endothelial to haematopoietic transition. Scientific Reports. 2016; 6DOI | PubMed
- Vassen L, Okayama T, Möröy T. Gfi1b:green fluorescent protein knock-in mice reveal a dynamic expression pattern of Gfi1b during hematopoiesis that is largely complementary to Gfi1. Blood. 2007; 109:2356-2364. DOI | PubMed
- Wang Q, Stacy T, Miller JD, Lewis AF, Gu TL, Huang X, Bushweller JH, Bories JC, Alt FW, Ryan G, Liu PP, Wynshaw-Boris A, Binder M, Marín-Padilla M, Sharpe AH, Speck NA. The CBFβ subunit is essential for CBFalpha2 (AML1) function in vivo. Cell. 1996; 87:697-708. DOI | PubMed
- Wilson NK, Foster SD, Wang X, Knezevic K, Schütte J, Kaimakis P, Chilarska PM, Kinston S, Ouwehand WH, Dzierzak E, Pimanda JE, de Bruijn MF, Göttgens B. Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell. 2010; 7:532-544. DOI | PubMed
- Woon Kim Y, Kim S, Geun Kim C, Kim A. The distinctive roles of erythroid specific activator GATA-1 and NF-E2 in transcription of the human fetal γ-globin genes. Nucleic Acids Research. 2011; 39:6944-6955. DOI | PubMed
- Yücel R, Kosan C, Heyd F, Möröy T. Gfi1:green fluorescent protein knock-in mutant reveals differential expression and autoregulation of the growth factor independence 1 (Gfi1) gene during lymphocyte development. Journal of Biological Chemistry. 2004; 279:40906-40917. DOI | PubMed
- Zovein AC, Hofmann JJ, Lynch M, French WJ, Turlo KA, Yang Y, Becker MS, Zanetta L, Dejana E, Gasson JC, Tallquist MD, Iruela-Arispe ML. Fate tracing reveals the endothelial origin of hematopoietic stem cells. Cell Stem Cell. 2008; 3:625-636. DOI | PubMed
Fonte
Bergiers I, Andrews T, Vargel Bölükbaşı , Buness A, Janosz E, et al. () Single-cell transcriptomics reveals a new dynamical function of transcription factors during embryonic hematopoiesis. eLife 7e29312. https://doi.org/10.7554/eLife.29312




